Embed Size (px)
Citation preview

MLAB 1415: HematologyMLAB 1415: HematologyKeri Brophy-MartinezKeri Brophy-Martinez
Thalassemia:Part One
1

Overview Overview Diverse group of congenital disorders which
manifest as anemia of varying degrees. ◦ Can be either homozygous or heterozygous
inheritance
Result of quantitative defective production of one or more globin portion(s) of hemoglobin molecule.
The decreased globin production causes◦ Imbalanced globin chain synthesis◦ Defective hemoglobin production◦ Damage to the RBC
2

Thalassemia DistributionThalassemia Distribution
3

Thalassemia Thalassemia
Results in overall decrease in amount of hemoglobin produced and may induce hemolysis.
Two major types of thalassemia: ◦Alpha (α) - Caused by defect in rate
of synthesis of alpha chains. ◦Beta (β) - Caused by defect in rate of
synthesis in beta chains. May contribute protection against
malaria.4

Review of Hgb StructureReview of Hgb Structure Normal globin genes
◦ Alpha, beta, delta, gamma Form hgb A (97%), hgb A2(2-3%), hgb F (2%)
◦ Epsilon, zeta: in utero◦ Gamma: 3rd trimester until birth◦ Adult hemoglobin composed two alpha and two
beta chains
Thalassemia causes an excess or absence of one of these chains
5

Pathophysiology: Beta Pathophysiology: Beta ThalassemiaThalassemia
α-chain excess unstablePrecipitates within the cell, causes damageMacrophages destroy the damaged RBCs in
the bone marrow, leads to ineffective erythropoiesis
Spleen also removes damaged RBCs, leads to chronic extravascular hemolysis
6

Pathophysiology: Alpha Pathophysiology: Alpha ThalassemiaThalassemia
β-chain excess◦ Unstable◦ Combines to form hgb molecules with 4 β-
chains (Hemoglobin H) Infants: excess gamma chains combine with hgb molecules (Hemoglobin Bart’s)
◦ High oxygen affinity, poor transporter of oxygen
7

Clinical and Laboratory Findings Associated with ThalassemiaClinical and Laboratory Findings Associated with Thalassemia

Clinical FindingsClinical Findings
9

Comparison of Comparison of Hemoglobinopathies and Hemoglobinopathies and ThalassemiasThalassemias
Disease RBC count
Indices RBC Morph Abnormal Hb
Ancestry Retic Count
Hemoglobinopathy
Normocytic
Normochromic
Target cells, sickle cells (HbS),Crystals (HbC)
HbS,HbC, HbE etc
AfricanMediterraneanMiddle EasternAsian
Thalassemia Microcytic
Hypochromic
Target cells, basophilic stippling
HbHHb Bart’s
AfricanMediterraneanAsian
10
Thalassemia: globin chains structurally normalHemoglobinopathies: globin chain is abnormal

Beta Beta ThalassemiaThalassemia
11

Classical Syndromes of Beta Classical Syndromes of Beta ThalassemiaThalassemiaBeta thalassemia minima/ Silent
carrier state – the mildest form of beta thalassemia.
Beta thalassemia minor - heterozygous disorder resulting in mild hypochromic, microcytic hemolytic anemia.
Beta thalassemia intermedia - Severity lies between the minor and major.
Beta thalassemia major - homozygous disorder resulting in severe transfusion-dependent hemolytic anemia.
12

Beta Thalassemia Minor Beta Thalassemia Minor
Caused by heterogenous mutations that affect beta globin synthesis.
Usually presents as mild, asymptomatic hemolytic anemia
Have one normal beta gene and one mutated beta gene.
13
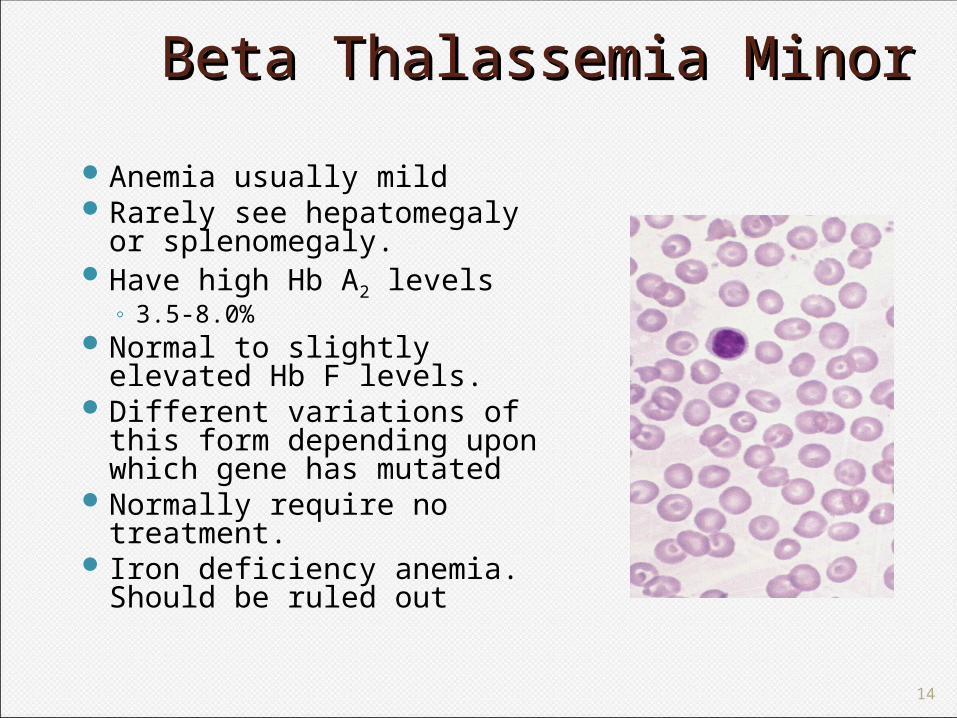
Beta Thalassemia Minor Beta Thalassemia Minor Anemia usually mildRarely see hepatomegaly
or splenomegaly. Have high Hb A2 levels
◦ 3.5-8.0%Normal to slightly
elevated Hb F levels.Different variations of this
form depending upon which gene has mutated
Normally require no treatment.
Iron deficiency anemia. Should be ruled out
14

Beta Thalassemia Major/ Beta Thalassemia Major/ Cooley’s anemia Cooley’s anemia
Severe microcytic, hypochromic anemia. ◦ Severe anemia causes marked bone changes
due to expansion of marrow space for increased erythropoiesis.
◦ See characteristic changes in skull, long bones, and hand bones “hair on end”
Detected early in childhood- 6 months- 2 yrs.
Hb A production is reducedHbA2 and Hg F production increased 15

Clinical Findings:Clinical Findings:ββ-Thalassemia Major-Thalassemia Major
Infants◦ Irritability, pallor, failure to
thrive◦ Diarrhea, fever, enlarged
abdomen Severe anemia Cardiac failure Bronze pigmentation of skin Bone changes
◦ Bossing of skull, facial deformities, “hair-on-end” appearance of skull
Hepatosplenomegaly
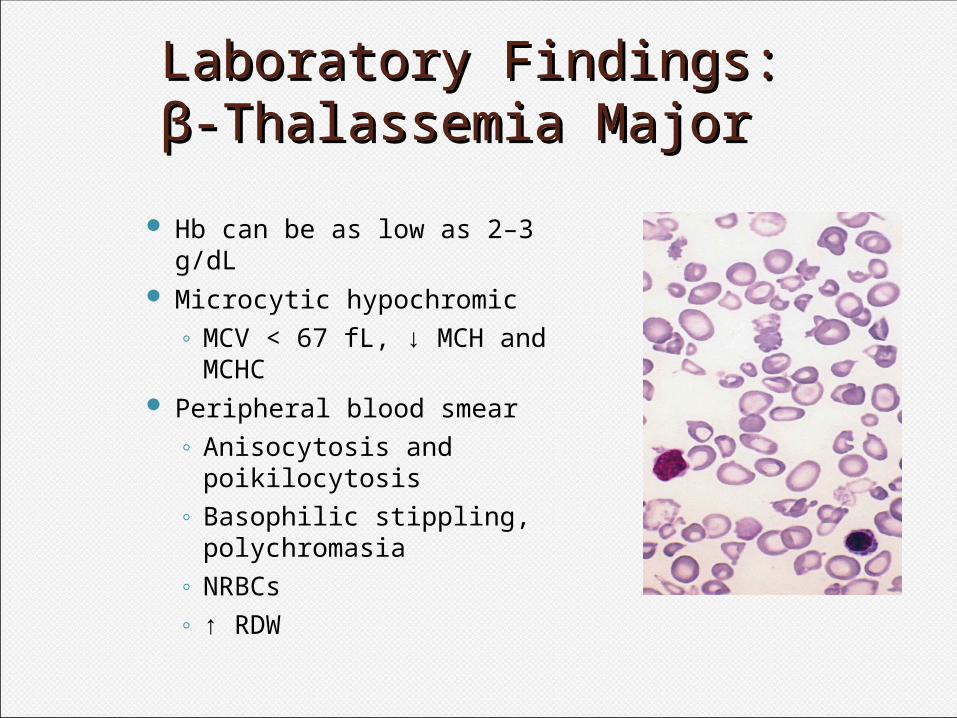
Laboratory Findings:Laboratory Findings:ββ-Thalassemia Major-Thalassemia Major
Hb can be as low as 2–3 g/dL Microcytic hypochromic
◦ MCV < 67 fL, ↓ MCH and MCHC
Peripheral blood smear◦ Anisocytosis and
poikilocytosis◦ Basophilic stippling,
polychromasia◦ NRBCs◦ ↑ RDW

ββ-Thalassemia Major-Thalassemia MajorTreatment
◦Regular transfusions Minimize anemia Suppress ineffective erythropoiesis
◦Iron-chelating agents Reduce excess iron absorption
◦SplenectomyPrognosis
◦Untreated – die during 1st or 2nd decade
◦Hypertransfusion with iron chelation Extend for ≥ 1 decade

Hereditary Persistence of Fetal Hereditary Persistence of Fetal Hemoglobin (HPFH) Hemoglobin (HPFH)
Rare condition characterized by continued synthesis of Hemoglobin F in adult life.
Do not have usual clinical symptoms of thalassemia.
Kleihauer-Betke stain useful tool to identify
19

ReferencesReferences• Harmening, D. M. (2009). Clinical
Hematology and Fundamentals of Hemostasis. Philadelphia: F.A Davis.
• McKenzie, S. B., & Williams, J. L. (2010). Clinical Laboratory Hematology. Upper Saddle River: Pearson Education, Inc.






















