Embed Size (px)
Citation preview

Modeling of ElectrodeModeling of Electrode Materials for Li-Ion Batteries
M Atanasov J -L Barras LM. Atanasov, J. L. Barras, L. Benco, C. Daul, E. Deiss+
University of Fribourg, Switzerland+Paul Scherrer Institute, Villigen, Switzerland

B tt T h l E l tiBattery Technology Evolution

2500Battery Market
2500
NiCd2000 NiMH
Li-Ion
1500 Total
1000
500
01993 1994 1995 1996 1997 1998 1999 2000
Year

T h l i l A li tiTechnological Applications
• Portable PC ’s• Cellular phonesp• Electrical & hybrid cars• High density power storage
t• etc...

H d it k ?How does it work ?

F t f G d B ttFeatures of a Good Battery
• High voltage• High current densityg y• High cyclability : > 1000 cycles• Cheap
E l i l• Ecological• Safe

E i l d E l i l A t M t i lEconomical and Ecological Aspects : Material
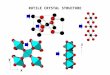
Electrode : - Mn2O4 (spinel) Cheap, non-toxic, high energy density- NiO2 (layered) Expensive, non-toxic, high energy density
+O2 ( aye ed) pe s e, o o c, g e e gy de s y
- CoO2 (layered) Expensive, toxic, high energy density- Rutile (layered) Cheap, non-toxic, low energy density- Anatase (cubic) Cheap non-toxic low energy density- Anatase (cubic) Cheap, non-toxic, low energy density- V2O5 (layered) Toxic
Electrode : Graphite (layered) Cheap non toxic high energy densityElectrode : - Graphite (layered) Cheap, non-toxic, high energy density,safety problems
- Rutile (layered) Cheap, non-toxic, low energy density,f t bl
-
no safety problems- Coke (grains) Cheap, non-toxic, high current density,
safety problems

M d liModeling means :Making predictions and/or descriptions of gphenomena based as much as possible on first principles
This yields :• Basis for targetting new experiments (time and
cost saving)• Optimal design• Optimal design• Analysis of experimental and technical difficulties

M th d lMethodology
• Non empirical calculations of periodic structures
– LAPW, tight-binding• Non empirical calculations of clusters
LCAO MO– LCAO-MO• Semi-empirical calculations of peridodic
structuresstructures– Extended Hückel
• Empirical calculations of extended systems– Molecular mechanics and dynamics
• Engineering modelsFinite elements– Finite elements

LAPW (1) ( Li i d A t d Pl W )LAPW (1) ( Linearized Augmented Plane Waves )
The space is devided in two parts :a) non-overlapping spheresb) the interstitial space
Th LAPW f tiThe LAPW wave functionsin the interstitial space :
ϕ =1
eiknrϕkn=
ωe

LAPW (2)LAPW (2)
The LAPW wave functions inside the spheres :
ϕk = Al ul r, El( )+ Bl Ýul r, El( )[ ]∑ Yl r( )ϕknAlmul r, El( )+ Blm Ý u l r, El( )[ ]
lm∑ Ylm r ( )
Boundary conditions : continuity of the wave function and of its first u ct o a d o ts stderivatives

WIEN97 LAPW C l l tiDensity Functional Theory
WIEN97 LAPW CalculationsDensity Functional Theory
LAPW basis set (WIEN97)
GGA corrections : Perdew Burke Ernzerhof 1
Full periodic Boundary Conditions
GGA corrections : Perdew, Burke, Ernzerhof
Full periodic Boundary Conditions
Brillouin zone average by modified tetrahedron scheme (Blöchl et al.2)3Total energy according Weinert et al.3
References
2 P.E. Blöchl et al., Phys. Rev. B 49, 16223 (1994)3 M Weinert et al Phys Rev B 24 864 (1982)
References1 J.P. Perdew et al., Phys. Rev. Let. 77, 3865 (1996)
3 M. Weinert et al., Phys. Rev. B 24, 864 (1982)

C l l ti f th E D itCalculation of the Energy Density
The discharge is considered as a chemical reaction : LiC6 s( ) + MO2 s( ) → 6C s( ) + LiMO2 s( )6( ) 2 ( ) ( ) 2( )
where M = Metal.
At T = 0, the energy density is given byE G Vd T T, = == −0 0Δ

C l l ti f th T t D dCalculation of the Temperature DependanceTotalEWith the temperature dependance, the
Gibbs’ energy is given by :Energy
ΔGT=300 = ΔHT=300 − T ⋅ ΔST=300
But it can be showed that
T ⋅ΔST=300 << ΔH T=300Li positiondistorsion
and so the contribution is given by the ib ti l tvibrational terms
( )ENhvib T
h k T, =
3 υT=0 Equilibrium( )e h k TB −1υ T=0 Equilibrium
Position

Average intercalation voltages forAverage intercalation voltages for M2O4
Average Voltage [V](exp)
CathodeSystem
x parameter for O(exp)
Unit cell a [A](exp)
Unit cell c [A](exp)
Ti2O4
LiTi2O4
Ti :
V2O4V :
Spinels
8.47 Cubic 2.9(3.0)8.45 (8.372) Cubic
0.2664
0.2622 (0.2628)
8 18 Cubic 2 970 2640
Fd3m_
V2O4
LiV2O4
V :
F OF
8.18 Cubic 2.97
8.22 Cubic
0.2640
0.2600
C bi
Mn2O4
LiMn2O4
Mn : 8.134* (8.045) Cubic 3.67(3.9)8.083* (8.247) Cubic
0.2640* (0.2631)
0.2650* (0.2625)
0 2660Fe2O4
LiFe2O4
Fe :
Co2O4
LiCo2O4
Co :
Cubic
... Cubic
7.97 Cubic 4.7
8.04 Cubic
0.2655
0.2640
7.975 0.2660...
...
Ni2O4
LiNi2O4
Ni :
Cu2O4
LiCu2O4
Cu :
8.02 Cubic 3.87
8.12 Cubic
0.2650
0.2645
8.26 Cubic 4.67
8.24 Cubic
0.2655
0.2625
TiO2
LiTiO3
Ti :
Layered
2.99 14.2 1.81
2.93 15.1
0.265
0.255
R3m_
All voltages are calculated (measured) agains metallic lithium anode.*Spin unrestricted calculations

B d St t f M OBand Structure of Mn2O4
fcc Brillouin zone

S i t i t d DOS f M OSpin unrestricted DOS for Mn2O4

S i t i t d DOS f LiM OSpin unrestricted DOS for LiMn2O4

S i t i t d DOS f Li M OSpin unrestricted DOS for Li2Mn2O4(tetragonal distorsion)

El t t ti P t ti l i M OElectrostatic Potential in Mn2O4
In unit cell (008) Hopping Path

Fermi Level Evolution vs. Li IntercalationFermi Level Evolution vs. Li Intercalation

OCV Modeling Using Frozen BandsOCV Modeling Using Frozen Bands
i Li M Ox in LixMn2O4

E i i M d lEngineering Models

S l S t f P ti l Diff ti l E tiSolve Sytem of Partial Differential Equations(Finite Elements)
The following contributions are considered :
• Diffusion of Li in M-oxide grains• Diffusion of Li+ in electrolyte between grains
g
• Diffusion of Li in electrolyte between grains• Electrochemical reaction at grain boundaries• Porosityy• Electrical conduction in electrode• Ion conduction in electrolyte
G f• Grain size distribution of M-oxide

Simulations of Measured PotentialSimulations of Measured Potential Jump Experiments
Adjusted parameters : D = 7.0 10-10 cm2/sk0 =1.3 10-6 cm/s
s/cm
3 ]ns
ity [A
sar
ge d
en
Time [s]
Cha
Time [s]

Illustration of kinetic controlIllustration of kinetic control

Calculated Li+ Concentration in ElectrolyteCalculated Li Concentration in Electrolyte

Hybrid Model of the Li+ Insertion from theHybrid Model of the Li Insertion from the Electrolyte to the Electrode Surface

Li+ Solvent Interaction : In the SolventLi - Solvent Interaction : In the Solvent
E0S l = -qL
2 c (ε -1) (for a spherical cavity)E Solv qLi( )
2R ε(for a spherical cavity)
ε dielectric constant of the solventf
R RSolv(Li+) : radius of the Li+ cavity
E0S l (Li+) = 5 53eV RS l (Li+) = 1 286 ÅE Solv(Li ) 5.53eV RSolv(Li ) 1.286 Å

Li+ Solvent Interaction : At the SurfaceLi - Solvent Interaction : At the Surface
ε 12ESolv(r ) = E0Solv (ε-1)
εqLi
2c1 r
2
Rsolv(Li+)
0 R(1 )α
αr
r : 0 R(1-cos ) = rmax2α
ESSolv(Li+) = E0
Solv - Esolv(rmax) = 21 E0
S l (1+cos )2αE Solv(Li ) E Solv Esolv(rmax) 2 E Solv(1 cos )2
Results
Mn2O4 : ESSolv = 0.50 E0
Solv (16c insertion site) α = 180°
ESSolv = 0.79 E0
Solv (8a insertion site) α = 109.47°

Li+ Crystal Interaction
With the host crystal
Li - Crystal Interaction
Li+ - Crystal Electrostatic Closed shellI t ti E Att ti R l i= +
With the host crystal
Interaction Energy Attraction Repulsion+
Fitti f th ff ti h di t b d t t l l tiFitting of the effective charges according to band structure calculations(vide supra)
With the incoming electron
e- σ - antibonding eg of Mn4+ (Li+ - e-) - coupling

ResultsResults
Conditions of the model :Conditions of the model :Mn2O4 : Surface 010Solvent :Water
8a 16c Energy Barrier (red. charges) 0.16 eV 0.95 eVEnergy Barrier (red. charges) 0.16 eV 0.95 eVLi+ solvation energy -3.27 eV -1.46 eVLi+ - e- interaction -4.32 eV -5.06 eV
1)Li+ - e- coupling : 1) reduction of the bulk energy barrier2) increase of surface energy barrier2) increase of surface energy barrier

ResultsResults
Infinite lattice summation
S i fi i l i iS i i fi it l tti tiSemi-nfinite lattice summationSemi-infinite lattice summation
Infinite lattice summation
Octahedral 16c
Semi-infinite lattice summation
Tetraheral 8a

DFT: Heuristic approach
ρ r( )
pp
X-ray diffractionX-ray diffractionρ( )nuclear positions
ρ r( )∫ dr # electrons# electrons
( )⎛ ⎞
ρ r ( )∫ dr # electrons# electrons
∂ρ Rk( )∂Rk
⎛
⎝ ⎜
⎞
⎠ ⎟
Rk =R0
= −2Zkρ R0( )CuspCusp
HΨ = EΨ H Ψ = EΨ

General TheoryExact energy expression
1 Eel = - 12 ∑
i
∫
φi( r
→1 )∇2φi ( r
→1 )d r
→1
+ ∑A
∫
ZA
|R→
A- r→
1| ρ( r
→1 ) d r
→1
12
⎮⎮⌠
ρ( r→
1)ρ( r→
2) d r
→1 d r
→2 2
⌡⎮
| r→
1 - r→
2 |1 2
E + Exc
Parr R G ;Yang W : Density Functional Theory of Atoms AParr,R.G.;Yang,W : Density Functional Theory of Atoms A
Molecules, Oxford University Press, New York 1989

The Kohn-Sham Equationq
φ φhksφi = εiφi
hKS = - 12∇2 + ∑
ZA → →
KS 2 A |R
→A- r
→1|
⎮⌠ ρ( r
→2)
d→
+
⌡⎮⎮ 2
| r→
1 - r→
2 | d r 2 + VXC
⌡
| r 1 r 2 |

Approximate density functional theories for exchange andApproximate density functional theories for exchange and
X
theories for exchange and correlationtheories for exchange and correlation
XαLocal exchange
Xα : Local exchange functional of the homogeneous electron gas
LDALocal exchange +local correlation
LDA: Local exchange functional + local correlationfunctional of the homogeneous electron gas
local correlation
GGA GGA: Same as LDA + “non-local” gradient correctionsLocal exchange +local correlation +gradient corrections
GGA: Same as LDA non local gradient correctionsto exchange and correlation
3rd Generation
3rd Generation of functionals: Same as GGA + instilation of “exact-exchange” and + 2nd derivatives
of functionals of the density corrections

Practical ImplementationPractical Implementation
1∇ + ( )+
ρ r'( )∫ d ' +V ( )( )
⎡ ⎢
⎤ ⎥ Ψ Ψ ( )SolveSolve −
2∇ + v r( )+
ρ( )r − r'∫ dr' +VXC ρ r( )( )
⎣ ⎢
⎦ ⎥ Ψi = εiΨi r( )Solve
Kohn-Sham eqs
Solve Kohn-Sham eqsSham eqs.Sham eqs.Features:Features:LCAO expansionLCAO expansion: STO, GTO, numerical, plane wavesLCAO expansionLCAO expansion: STO, GTO, numerical, plane waves
Coulomb potentialCoulomb potential: solve Poisson’s eq. or fit ρ(r) to a set of one-center auxilliaryCoulomb potentialCoulomb potential: solve Poisson’s eq. or fit ρ(r) to a set of one-center auxilliarya set of one-center auxilliary functionsa set of one-center auxilliary functions
MatrixelementsMatrixelements: accurate numerical integration in the irreducible wedge of the MatrixelementsMatrixelements: accurate numerical integration in the irreducible wedge of the

Methodology based on Approximate DFT
α1966
αMS-X
MS-X : Make use of partial-waves as basis (37). Relatively fast. Good for ionization potentials and excitation energies (10). Total energies unreliable ( 39). No geometry optimization. Full use of symmetry. H l ti i ti t i (53f) M k f ffi tiApproximate DFT
αDV-X : Make use of numerical atomic orbitals or STO's. Avoids Muffin-tin approximation by fit of density (45a).
1966 Has relativistic extension ( 53f). Make use of muffin-tin approximation (38). Developed by K.H. Johnson (37) .
Avoids Muffin tin approximation by fit of density (45a). Accurate total energies (76d). Relativistic extension (53e). Numerical integration of matrix elements by Diophantine integration (40). Developed by Ellis and Painter (40). Extensive improvements by Delley (D-MOL-program) including new integration scheme (46c) and geometry optimization.
1970
DV-X α
HFS-LCAO : Make use of STO's . Accurate potentials (41). Full use of symmetry. Relativistic extensions (53a,b). Highly vectorized (47). Accurate total energies (49). Geometry optimization (54c). Accurate numerical integration (46b). Many auxiliary property programs Pseudo potentials (52a d)
ADF
FRIMOL 1994-
development in progress
1973 -Many auxiliary property programs . Pseudo potentials (52a,d). Embedding procedures (76h). Energy decomposition scheme (72). Developed by Baerends,Snijders,Ravenek,Vernooijs and te Velde (41,53,47,46d)
development in progressDeMon
1976 -
LCGTO-LSD : Make use of GTO's. Fit of exchange-correlation and Coulomb potential (43). Analytical calculation of matrix elements (48b). Accurate energies. Geometry optimization (54b,h). Strongly vectorized (48b). First developed by Dunlap (43) as well as Sambe and Felton (42). Extensive improvements by Salahub and Andzelm (48b) 1976 - (D-GAUSS-program) as well as Rösch (74a). Also work by Pederson (45e) and Painter (45d)
NUMOL NUMOL : Unique basis-set free program (50a,e). Accurate
1982
NUMOL : Unique basis set free program (50a,e). Accurate numerical integration (46a). Efficient generation of Coulomb potential (50c). Geometry optimization. Developed by Becke (50 ).

Modeling the Intercalation Dynamics ofModeling the Intercalation Dynamics of Li+ :Cl t St d U i M l l DFTCluster Study Using Molecular DFT

Vibrational Modes Involved in VibronicVibrational Modes Involved in Vibronic Coupling of the eg(σ*) Electron

Vibronic Coupling Model of the PolaronVibronic Coupling Model of the Polaron

Energy Contour Diagram of the PolaronicThe image cannot be displayed. Your computer may not have enough memory to open the image, or the image may have been corrupted. Restart your computer, and then open the file again. If the red x still appears, you may have to delete the image and then insert it again.
Energy Contour Diagram of the Polaronic Model

Calculated ( ) Electric ConductivityCalculated (-----) Electric Conductivity Using the Polaron Model

Energy Profiles of Li+ Diffusion in BulkEnergy Profiles of Li Diffusion in Bulk Mn2O4

Acknowledgements
Financial Support : Swiss Federal Office for EnergyFinancial Support : Swiss Federal Office for Energy
The Li-Ion Battery Modeling Group :
Erich Deiss,PSI
Claude Daul,Jean-Luc Barras,U i it
Michael Atanasov,Uni ersit
Lubomir Benco,U i i PSIUniversity of
FribourgUniversityof Fribourg
Universityof Fribourg
Universityof Fribourg
Cluster and Intercalation
Band Structure Calculations
Band Structure Calculations
Engineering Models
Project LeaderIntercalation Dynamics
Calculations Calculations Models