Embed Size (px)
Citation preview

MODULE 18(03)
TRANSITIONS BETWEEN MOLECULAR STATES
The total energy of a molecule is made up of several components
Et = Ee+Ev+Er
Where the subscripts refer to total, electronic, vibrational, and rotational, respectively.
Here we are ignoring translational and internal nuclear energies which are irrelevant to electric dipole transitions.
If the above equality refers to the ground state, for an excited state we have
E’t = E’e+E’v+E’r

MODULE 18(03)
Absorption of a photon leads to
where x = e, v, r, or all of them.
Typically Er ~ 10 cm-1, Ev ~ 1000 cm-1, and Ee ~ 30,000 cm-1.
Transitions involving only Er give rise to the rotational absorption spectrum in the far-IR region of the
electromagnetic spectrum.
Ev + Er transitions generate the vibrational-rotational spectrum in the mid-IR.
Ee + Ev transitions (Er negligible in energy) generate the electronic-vibrational absorption spectrum in the uv-
vis region.
'x x xE E E

MODULE 18(03)
When an electronic transition occurs the change
in electron charge distribution subjects the
nuclei to a different Coulomb field.
The nuclei respond by changing their vibrational
state.
Transitions involving changes in both electronic and vibrational states are called vibronic transitions
combination of electronic and vibrational states is a
vibronic state.
4
3
2
1
0
1
0
' '1
2e vE E'eE
' '9
2e vE E
' '3
2e vE E
' '5
2e vE E
' '7
2e vE E
3
2e vE E
1
2e vE E
eE

MODULE 18(03)
Recall that the equilibrium arrangement of molecular
entities among a set of energy states is governed by the Boltzmann expression.
At room temperature most molecules will be in the v =
0 vibrational state of the electronic ground state.
The small proportion of molecules in v = 1 give rise to the weak “hot bands” on the low energy side of the
0,0 transition.
4
3
2
1
0
1
0
' '1
2e vE E'eE
' '9
2e vE E
' '3
2e vE E
' '5
2e vE E
' '7
2e vE E
3
2e vE E
1
2e vE E
eE

MODULE 18(03)
The Figure shows that the Photosciences and Spectrometry are connected by the electric dipole
absorption event.
Photoscience focuses on the reactivity of the new states generated; spectrometry focuses on the structural and
bonding features of the ground state.
The (v’ 0) absorption transitions indicated on the diagram explain the appearance of the absorption spectra
of anthracene.
wavelength/nm
280 300 320 340 360 380 400 420 440
ab
sorb
an
ce
0.0
0.2
0.4
0.6
0.8
1.0
anthracene
The “fingers” correspond to transitions from v = 0 of the ground state to the v’ = 1, 2, 3, etc of the upper state.

MODULE 18(03)
The spectra show that many vibronic transitions
are allowed in these molecules.
The question concerning us now is why some
transitions are more allowed than others, i.e. why are the intensities of the transitions different?
This question leads to the Franck-Condon principle.
4
3
2
1
0
10
' '1
2e vE E'eE
' '9
2e vE E
' '3
2e vE E
' '5
2e vE E
' '7
2e vE E
3
2e vE E
1
2e vE E
eE
hot band
wavelength/nm
280 300 320 340 360 380 400 420 440
ab
sorb
an
ce
0.0
0.2
0.4
0.6
0.8
1.0
anthracene

MODULE 18(03)
The Franck-Condon Principle
Nuclei move much more slowly than the much lighter electrons.
This is a fact that we have employed previously in the context of the Born-Oppenheimer Approximation.
Because of this velocity difference, on a classical picture, during an electric dipole transition the electron changes
its orbital before the nuclei have time to respond.
In other words an electric dipole transition (absorption or emission) occurs within a “stationary” nuclear
framework. This is one statement of the Franck-Condon Principle.

MODULE 18(03)
The classical view:
The nuclei spend most of their time at the
minimum position on the ground state
potential energy curve (the lower one).
Thus an electronic transition will start from there and will terminate
where the vertical arrow meets the upper potential energy curve.
Electronic transitions are vertical.
PM
0 20 40 60 80 100 120 140
ENER
GY
0
2
4
6
8
10
12
14

MODULE 18(03)
As nuclear positions remain constant during the
transition the newly formed vibronic state has a nuclear framework that is not that of
v’ = 0.
This situation occurs because the minima of the two curves
are displaced.
Excitation usually results in increased anti-bonding
character (). PM
0 20 40 60 80 100 120 140
ENER
GY
0
2
4
6
8
10
12
14
This weakens the bond (lower bond order) and the force constant is reduced from the ground state value.
DIATOMIC!

MODULE 18(03)
The quantum version of previous.
Quantization (boundary conditions) imposes a zero point
energy on the oscillator and leads to wavefunctions as
shown.
Probability distributions (are plotted on the PE curves
Otherwise the electronic state functions are the same as in
above figure.
The v = 0 function has maximum probability at Req as
classical concept.
The Quantum Viewpoint
internuclear distance/pm
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150
pote
ntia
l ene
rgy/
10-1
9 J
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
internuclear distance/pm
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150
pote
ntia
l ene
rgy/
10-1
9 J
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15

MODULE 18(03)
A vertical transition from v = 0 brings the system to an
internuclear distance where there is significant amplitude
in the v’ = 2 density, but much less in the lower two.
internuclear distance/pm
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150
pote
ntia
l ene
rgy/
10-1
9 J
0
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
A vertical shift causes the vibrational wavefunction to undergo the least amount of change.
Thus we might expect the 20 vibronic transition to be more intense than the 00 or the 10.

MODULE 18(03)
We need to calculate the transition dipole moments for
These transitions will each have a dipole moment, expressed as
The first term in the sum is due to the electrons and the second is due to the nuclei of atomic number Z.
Born-Oppenheimer allows the separation of vibronic wavefunctions into electronic and nuclear parts
R is a parameter and each nuclear arrangement (value of R) has its own electronic eigenvalue.
0,0 1',0 ' , 1',1' , 1', 2 '
i S S e Ni S
e r e Z R
, ( ; ) ( )vv r R R

MODULE 18(03)
In the final term of the sum the integral in brackets is zero because the two electronic states are mutually orthogonal (both eigenfunctions of the same hamiltonian operator.
The integral in brackets in the first term in the sum is over the electron coordinates only and it is the transition dipole moment for the electronic transition when the
nuclei have coordinates R.
To a good approx. this integral is constant for small displacements from Req; we replace it by the (constant)
matrix element
* *' '
* *' '
* *' '
ˆ' ' ( ; ) ( )( ) ( ; ) ( )
( ) ( ; ) ( ; ) ( )
( ) ( ; ) ( ; ) ( )
v e N v e N
v e e v N
v N e v N
v v r R R r R R d d
R r R r R d R d
R r R r R d R d
'

MODULE 18(03)
S(v’,v) is the overlap integral between the two vibrational states that are connected by the electronic transition.
The transition dipole moment of the most intense v’ v transition is the one having the largest S value.
This corresponds to the transition in which the upper vibrational state has the largest probability distribution at
the internuclear separation of the ground state.
*' ' 'ˆ' ' ( ) ( ) ( ', )v v Nv v R R d S v v
( ', ) 'S v v v v

MODULE 18(03)
Recalling the anthracene spectrum, we now understand that the different intensities of
the different vibronic bands corresponds to the different
overlap integrals between v = 0 and the v’ states.
Normally several vibrational states have similar S values and transitions to all of them are observed in the spectral
envelope.
wavelength/nm
280 300 320 340 360 380 400 420 440
ab
sorb
an
ce
0.0
0.2
0.4
0.6
0.8
1.0
anthracene
We observe a progression of transitions.
The relative intensities of the lines increase as the square of the transition moment dipoles and thus to the Franck-
Condon factors2( ', )S v v

MODULE 18(03)
A Problem: Two vibronic states have the same force constant but their Req values differ by R. Find a
relationship between the relative intensity of the 0-0 transition and R.
The Solution: The intensity of the band is proportional to
so we need to calculate the R dependence of the overlap integral using two harmonic oscillator wavefunctions, one
centered at x = 0 and one at x = R.
The functions are
2(0,0)S
2 21/ 4 1/ 4
/ 2 ( ) / 20 0'( ) ( )x x Rx e x e
1/ 2( ) /k

MODULE 18(03)
This is the variation of the FC factor with R, a plot follows
2 2
22
1/ 2/ 2 ( ) / 2
1/ 2( )
2 2
(0,0) 0 0 x x R
R Rx
S e dx
e e dx
21/ 2
axe dxa
2
2(0,0)R
S e
22 ( ) / 2(0,0) RS e
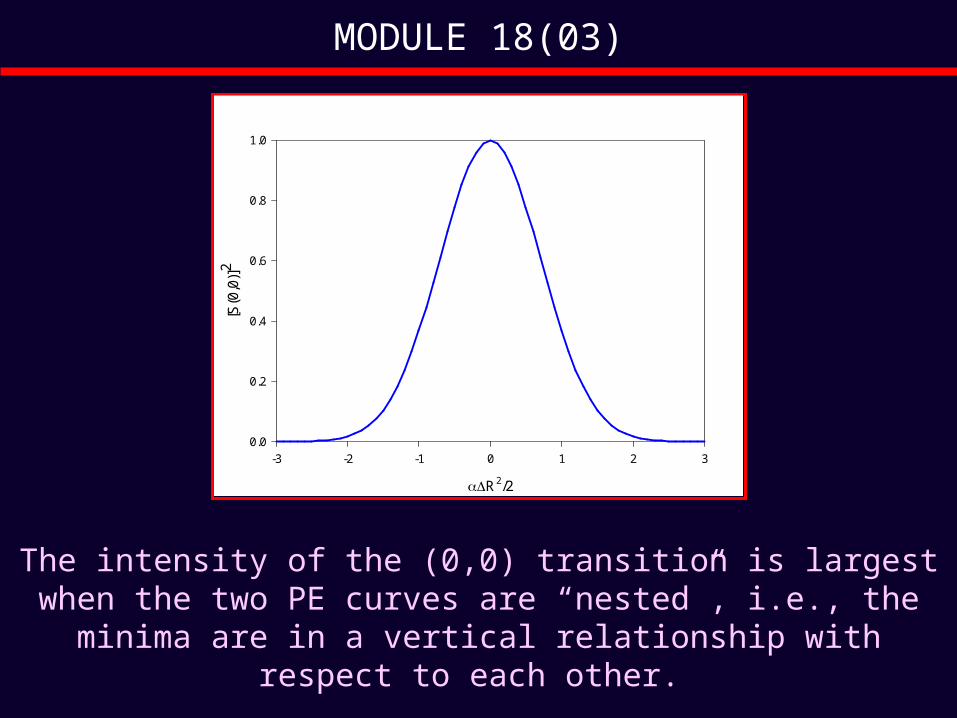
MODULE 18(03)
The intensity of the (0,0) transition is largest when the two PE curves are “nested”, i.e., the minima are in a
vertical relationship with respect to each other.
R2/2
-3 -2 -1 0 1 2 3
[S(0
,0)]
2
0.0
0.2
0.4
0.6
0.8
1.0

MODULE 18(03)
Spectral Bandwidths
Referring to the absorption spectrum, we see that each one of the “fingers” has a significant spectral width.
A rough measurement reveals a full width-half
maximum (FWHM) value of ca 300 cm-1.
wavelength/nm
280 300 320 340 360 380 400 420 440
ab
sorb
an
ce
0.0
0.2
0.4
0.6
0.8
1.0
anthracene
One reason for this large bandwidth is the low resolution of the spectrometer employed in the measurement.
But this is not the sole cause; there are underlying theoretical reasons.

MODULE 18(03)
Our analysis has implied that each of the fingers is
due to a transition between a pair of discreet vibronic
states.
On this basis the observed spectrum should be a “stick”
spectrum composed of a series of narrow lines
separated on the wavelength scale by the
vibrational energy quantum relevant to the upper
electronic state.
Clearly this is not the case.
4
3
2
1
0
10
' '1
2e vE E'eE
' '9
2e vE E
' '3
2e vE E
' '5
2e vE E
' '7
2e vE E
3
2e vE E
1
2e vE E
eE
wavelength/nm
280 300 320 340 360 380 400 420 440
ab
so
rba
nce
0.0
0.2
0.4
0.6
0.8
1.0
anthracene

MODULE 18(03)
One obvious reason for this occurrence of bands rather than lines is the presence of quantized rotational states
which give rise to the P, Q, and R branches in the rotational spectrum.
Every vibronic transition carries with it enough energy to cause molecular rotations to undergo accompanying
changes.
The vapor phase spectrum of benzene at high spectral resolution clearly shows rotational progressions on each
of the vibronic fingers.
Moreover in addition to broadening by low resolution instruments and by rotational transitions, vibronic spectra
are significantly affected by interactions with solvent molecules.

MODULE 18(03)
Solvent-induced effects tend to cause loss of resolution of the sub-structure.
Specific interactions include effects due to solvent polarity, hydrogen bonding, and the presence of aromatic
groups.
These affect the molecular potential energy surface and result in spectral shifts.
The best solvents for minimizing specific solvent interactions are the fluorocarbons, such as C6F14.
Non-specific (universal) interactions occur because a solute molecule occupies a cavity in the solvent and is
surrounded by and buffeted by solvent molecules.
MODULE 18(03)
The solvent is a dielectric medium and it influences solutes via its static and optical (refractive index)
dielectric constants.
A solute molecule has dipoles, either permanent or fluctuating, which set up a reaction field in the
surrounding solvent lowering the energy of the ground state of the solute.
Molecules in fluid solution rotate and translate, so their dipolar interactions fluctuate in time and the reaction field
fluctuates.
A time average of the reaction fields of an ensemble of chromophores in fluid solution thus has an energy
bandwidth and vertical electric dipole transitions will originate from ground states having a (narrow) range of
energies.
Such transitions will therefore carry an intrinsic bandwidth as a result of this homogeneous broadening.





























