Embed Size (px)
Citation preview

Molecular Epidemiology of Outbreak-Associated and Wild-Waterfowl-Derived Newcastle Disease Virus Strains in Finland, Including a NovelClass I Genotype
Erika Lindh,a Christine Ek-Kommonen,b Veli-Matti Väänänen,c Jukka Alasaari,a Antti Vaheri,a,d Olli Vapalahti,a,d,e
and Anita Huovilainenb
Department of Virology, Haartman Institute, Faculty of Medicine, University of Helsinki, Helsinki, Finlanda; Research and Laboratory Department, Veterinary Virology,Finnish Food Safety Authority Evira, Helsinki, Finlandb; Department of Forest Sciences, Faculty of Agriculture and Forestry, University of Helsinki, Helsinki, Finlandc;Department of Virology and Immunology, Helsinki University Central Hospital Laboratory, HUSLAB, Helsinki, Finlandd; and Department of Basic Veterinary Sciences,Faculty of Veterinary Medicine, University of Helsinki, Helsinki, Finlande
Newcastle disease (ND) is a highly contagious, severe disease of poultry caused by pathogenic strains of Newcastle disease virus(NDV; or avian paramyxovirus-1). NDV is endemic in wild birds worldwide and one of the economically most important poultrypathogens. Most of the published strains are outbreak-associated strains, while the apathogenic NDV strains that occur in wildbirds, posing a constant threat to poultry with their capability to convert into more virulent forms, have remained less studied.We screened for NDV RNA in cloacal and oropharyngeal samples from wild waterfowl in Finland during the years 2006 to 2010:39 of 715 birds were positive (prevalence, 5.5%). The partial or full-length F genes of 37 strains were sequenced for phylogeneticpurposes. We also characterized viruses derived from three NDV outbreaks in Finland and discuss the relationships betweenthese outbreak-associated and the wild-bird-associated strains. We found that all waterfowl NDV isolates were lentogenicstrains of class I or class II genotype I. We also isolated a genetically distinct class I strain (teal/Finland/13111/2008) groupingphylogenetically together with only strain HIECK87191, isolated in Northern Ireland in 1987. Together they seem to form anovel class I genotype genetically differing from other known NDVs by at least 12%.
Type 1 avian paramyxovirus (APMV-1), commonly known asNewcastle disease virus (NDV), is a highly significant avian
pathogen, with a strong impact on both commercial and backyardpoultry. It has caused three major panzootics during the past 80years. The genus Avulavirus, a member of the negative-sense RNAvirus family Paramyxoviridae, is composed of the antigenicallydistinct avian paramyxovirus serotypes 1 to 11 (8, 19, 32). Thename NDV is in some contexts reserved exclusively for velogenicpoultry-derived strains (defined by the World Organization forAnimal Health, OIE).
The fusion protein encoded by the F gene mediates the fusionbetween the viral envelope and the host cell membrane. The pre-cursor F0 is cleaved by host cell proteases to F1 and F2 beforegaining its functional activity (34, 39). The amino acid sequence ofthe cleavage site is considered to be a major pathogenicity factor,determining the protease specificity, and thereby the tissue tro-pism, of a given strain. NDVs are categorized as velogenic, meso-genic, and lentogenic depending on the severity of the disease theycause. Lentogens cause a mild or subclinical infection of the respi-ratory tract. Velogens, which are cleaved by proteases of the furinfamily allowing for systemic replication, affect the gastrointestinaltract and/or the nervous system, causing high mortality. Thepathogenicity can be assessed by either an intracerebral pathoge-nicity index (ICPI) with a cutoff value of 0.7 for velogenic strainsor molecularly by the cleavage site sequence of the fusion protein.Multiple basic amino acids at the C terminus of the F2 protein andphenylalanine at residue 117 of the F1 protein are indicative forhigh pathogenicity (OIE).
Within NDV, two clades can be distinguished (9): class I vi-ruses, also designated group H or lineage 6 (3, 6, 14), comprise atleast nine genotypes (23). They are almost exclusively lentogenic
strains that have been found among waterfowl and at live-birdmarkets (35). Class II includes almost all of the velogenic strainsand the majority of all published strains, as they are primarilyresponsible for the infections in poultry and pet birds (9). Class IIviruses are further divided into genotypes I to XI (used in thisstudy) (7, 17, 29–31, 40) or genetic lineages 1 to 6 (3). The geno-types of class II are separated as early (genotypes I to IV and IX)and recent or late (other genotypes, isolated after the 1960s). Thedivision is characterized by different genome sizes, 15,186 nucle-otides (nt) and 15,192 nt, respectively (10). A distinct sublineage(VIb) of class II viruses comprises the pigeon paramyxoviruses(PPMV-1). The group consists of a globally diverse range ofstrains and has further been divided into several sublineages (2).They were first seen in the Middle East in racing pigeons in the1980s and have since then spread to all parts of the world (20).Endemic among feral and domestic pigeons, PPMV-1 is able tospread to and cause severe disease in wild and domestic bird pop-ulations, including commercial poultry (25).
Roughly, the NDV ecology can be divided into two host sys-tems: wild waterfowl harboring the lentogenic strains and poultrywhere velogenic outbreaks are manifested. The ecology of NDVs
Received 31 May 2012 Returned for modification 17 July 2012Accepted 2 September 2012
Published ahead of print 12 September 2012
Address correspondence to Erika Lindh, [email protected], orAnita Huovilainen, [email protected].
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JCM.01427-12
3664 jcm.asm.org Journal of Clinical Microbiology p. 3664–3673 November 2012 Volume 50 Number 11
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from

in wild birds appears to be in many aspects similar to low-patho-genic avian influenza viruses (AIV), which are naturally hosted bywild waterfowl, although it is far less studied (21, 42). The highlypathogenic strains of both viruses seem to arise in a similar wayduring replication in high-density poultry populations, when mu-tations are accumulated at the fusion protein cleavage site (36,37, 41).
The great intra- and intergenotype diversity of NDVs and theircontinuing evolution pose a diagnostic challenge, e.g., as demon-strated by class I viruses which are escaping detection by manyconventional assays (24, 33). Constant monitoring of circulatingviruses is required for evaluation of vaccine compatibility.
Here, we report our results from screening NDV from wildwaterfowl during the years of 2006 to 2010 in Finland. The viruseswere sequenced for phylogeny and to assess their pathogenicity bythe deduced amino acid sequence of the fusion protein cleavagesite. We also report three outbreaks that occurred in pheasants(Phasianus colchicus) in 2003, turkeys (Meleagris) in 2004, andhobby pigeons (Columba sp.) in 2008. Fourteen Finnish strainsisolated before 2006 have been previously described in 2001 byHuovilainen et al. and in 2008 by Lindh et al. (18, 27). Our furtheraim was to elucidate the role of wild waterfowl in the ecology ofNDV and the relationships between the wild-bird-derived andoutbreak-derived NDVs described in this study.
MATERIALS AND METHODSSample overview. Wild migratory waterfowl were sampled during theyears of 2006 to 2010. The samples were collected by voluntary huntersand staff of the Finnish Game and Fisheries Research Institute, as part ofan active surveillance program of AIV and NDV. Samples were collectedfrom a total of 715 birds, from four main areas in Central and SouthernFinland, where waterfowl nest in high numbers (16, 22). In addition,positive samples derived as part of passive surveillance (dead and clinicallyaffected birds) and from NDV outbreaks were included in our study.
In 2003, NDV with an ICPI of 0.5 was isolated from a flock of pheas-ants that were partly kept outdoors. The pheasants were reared for release.Hatched chicks were kept in the holding for 5 days prior to release. Duringthis time, a slightly higher mortality was seen (�10%), and the flock wasdestroyed when NDV was confirmed in two birds. The affected popula-tion consisted of 750 pheasants, 15 chickens, and 1,600 newly hatchedchicks. This holding had traded birds with another backyard poultryholding.
During routine health controls in June 2004, antibodies to NDV weredetected in western Finland in a flock of 12,000 fattening turkeys, al-though no clinical signs of disease were apparent. Consequently, NDVwas isolated with an ICPI of 1.6. The birds were culled and the outbreakwas controlled according to current regulations. Protection and surveil-lance zones were set around the infected premises, and all poultry withinwere tested serologically but remained seronegative. Wild waterfowl weresuspected to be the source of this outbreak. At the same time, an outbreakby a highly similar virus was reported in Sweden. No connection wasfound, nor were the sources of the outbreaks confirmed at that time.
In 2008, NDV was confirmed in 6 dovecotes within 6 days. NDV wasinitially found in dovecote A, with 54 hobby pigeons, after the death ofseveral birds. When the contacts were traced, NDV was found in dovecoteB, with over 300 birds of several different species. Their contacts wereagain traced, and on days 3 and 4 two small dovecotes, C and D, which hadbeen in contact with dovecote B, were found infected. The next day, coteE, which had been in contact with cote D, was found NDV positive. On thesixth day, dovecote F was found positive for NDV after contact with cotesA and B at a pigeon show. All 482 affected birds from the six affected coteswere culled. The dovecotes had been in contact by trade of birds or relatedproducts and during shows. All their contacts were investigated by mo-
lecular methods but no further NDV-positive cotes were found. All poul-try holdings within close proximity with the infected cotes were also in-vestigated, but no affected birds were found.
Sample handling. Cloacal and oropharyngeal swabs from wild birdswere collected using flocked nylon swabs and UTM (universal transportmedium) (Copan). Samples were kept at room temperature or chilled butwere not frozen until they reached the lab. At the lab, samples were cen-trifuged and supplied with additional antibiotics and stored at �80°C.
Organ suspensions from outbreak-associated birds and birds frompassive surveillance were prepared for PCR studies and positives wereconfirmed by virus isolation in embryonated eggs (12).
RNA extraction and RT-PCR. RNA was extracted from swab samplesor allantoic fluids using a QIAmp viral RNA minikit (Qiagen). For thedetection of NDV RNA, two different PCRs were used: an F gene-target-ing reverse transcription (RT)-PCR for class II viruses, described by Seal etal. (35) and adjusted to comprise class I viruses by Huovilainen et al. (18),was used for studying samples collected in 2006 to 2009. A recently devel-oped L gene-targeting real-time RT-PCR by Fuller et al. (13) was intro-duced in our lab in 2010 and used for studying samples from that year. AllPCR-positive samples were subjected to virus isolation attempts and/orsequencing. Shortly, swab specimens were injected into 8- to 10-day-oldembryonated chicken eggs, and allantoic fluids were harvested from eggswhen embryos appeared dead or dying. The remaining eggs were har-vested 6 days postinoculation and subjected to a second passage. Thecollected allantoic fluids were tested for hemagglutinating activity.
Serotyping and sequencing. Distinction between class I and IIAPMV-1 was done using the previously described F gene RT-PCRs (18,35). Primers for the sequencing of the whole F gene spanning the N- andC-terminal noncoding regions were designed for both classes (Table 1).An �600-nt region including the fusion protein cleavage site was se-quenced for all the strains. The full-length F gene of the class I viruses andthe causative agents of recent outbreaks were also sequenced.
Amplifications of target sequences were done using the OneStep RT-PCR kit (Qiagen). Reaction mixes were prepared with 10 �l 5� PCRbuffer, 2 �l deoxynucleoside triphosphates (dNTPs) (10 mM), 1 �l ofeach primer (50 pmol/�l), 2 �l enzyme mix, 0.4 �l RNase inhibitor (20U/�l), and 5 to 10 �l template RNA and filled up to 50 �l with H2O. Thefollowing thermal amplification cycle conditions were applied: 50°C for30 min, followed by 94°C for 15 min, followed by 45 cycles of 94°C for 1min, 48°C for 1 min, and 72°C for 1 min, followed by 72°C for 4 min.
Samples were prepared for sequencing by gel extraction by theQIAquick gel extraction kit or PCR purification by the MiniElute PCRpurification spin kit (both from Qiagen). The sequencing reactions were
TABLE 1 Primers used for amplification of the full-length NDV F genes
Class Primer Sequence Position
I s1Hf TTG CCA AAT ACA AYC CGT TCA A 5= �188s1 h GTG GTT ACY GAC TCT TGG ATT 3= 322s2Hf ACA ACC CTC CTT GCC CC 5= 264s2 h GGG AGA GTC ACC TGT ATA C 3= 868s3Hf GAC TCG CAA ACT CAG CTT CT 5= 828s3 h AGC TTG CTG TTA CTC TCT TCT A 3= 1462s4Hf ATC ACC TTG CGC CTT AGC 5= 1293s4 h AGT GCA ACT TGG CTG ACC 3= 1896
II s1f CTG TCG CAG TGA CYG CTA 5= �248s1r CCCT GTC TGR GAT GAG GT 3= 176s2f ACA GGG TCA ATC ATA GTC AAG 5= 171s2r CCC AAG AGT TGA GTC TGT G 3= 850s3f TAG CTG GTG GNA ATA TGG ATT A 5= 721s3r ATC RAA TTC CCC ACT GAG C 3= 1322s4f ATA TCG CAA AAY TAT GGA GAA GC 5= 1221s4r GCY TCT CTT TCN TCA TTC TCT A 3= 1917
Molecular Epidemiology of NDV in Northern Europe
November 2012 Volume 50 Number 11 jcm.asm.org 3665
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from

performed by FIMM SeqLab (Institute for Molecular Medicine Finland,Helsinki, Finland).
Sequence analysis. Sequence analysis was performed on a 400-nt re-gion of class I viruses (nt 125 to 524) and on a 450-nt region (nt 275 to 724)of class II viruses of the NDV F gene. Sequences were edited using eBioXversion 1.5.1 software (www.ebioinformatics.org) and compared to pub-lished strains by BLAST. Reference strains were picked based on BLASTsearch results as well as from representatives for different lineages. Thealignments and phylogenetic analyses were conducted using MEGA soft-ware version 5.0 (38). The trees were generated by using the neighbor-joining algorithm, and alignments were bootstrapped 1,000 times.
Assessment of pathogenicity. The F gene sequences of our strainswere analyzed for pathogenic determinants in the amino acid sequences ofthe fusion protein cleavage sites (amino acids 112 to 117), shown in Tables2 and 3.
The outbreak-associated strains were sent to The Animal Health andVeterinary Laboratories Agency (AHVLA; formerly VLA, Surrey, UnitedKingdom), where ICPI tests were carried out.
GenBank accession numbers. GenBank accession numbers of theFinnish strains described in this study are designated in Tables 2 and 3.
RESULTSPrevalence and sampled species. In total, 715 birds were sampled(Table 4), most of which were wild waterfowl. The most prevalentspecies sampled for active surveillance were mallard (Anas platy-rhynchos) (53%) and Eurasian teal (Anas crecca) (22%), as they arethe most common bagged duck species and nest in high numbersin our study areas (16, 22). The samples were derived from thewhole country, except from the northernmost part of Finland.
TABLE 2 Finnish NDV strains derived through active surveillance
No.GenBankaccession no. Isolate Yr Class Genotype F cleavage siteb CT valued Host
1 EU493453 12136a 2006 II I GKQGRL Not tested Eurasian teal (Anas crecca)2 EU493451 12104a 2006 I 2 ERQERL Not tested Eurasian teal (Anas crecca)3 EU493454 13193a 2006 I 2 ERQERL Not tested Common pochard (Aythya ferina)4 EU493452 12119a 2006 I 2 ERQERL Not tested Eurasian teal (Anas crecca)5 EU493450 12074a 2006 II I GKQGRL Not tested Eurasian teal (Anas crecca)6 JX844028 13111 2008 I New genotype ERQERL Not tested Eurasian teal (Anas crecca)7 JX844037 8789 2009 II I GKQGRL Not tested Mallard (Anas platyrhynchos)8 JX844038 8791 2009 II I GKQGRL Not tested Mallard (Anas platyrhynchos)9 JX844039 8803 2009 II I GKQGRL Not tested Mallard (Anas platyrhynchos)10 JX844040 9067 2009 II I c Not tested Eurasian wigeon (Anas penelope)
11 JX844041 10666 2009 II I GKQGRL Not tested Eurasian teal (Anas crecca)12 JX844042 10668 2009 II I GKQGRL Not tested Eurasian teal (Anas crecca)13 JX844043 8625 2010 II I GKQGRL 35.7 Eurasian wigeon (Anas penelope)14 JX844044 8633 2010 II I GKQGRL 30.0 Eurasian teal (Anas crecca)15 JX844045 8637 2010 II I GKQGRL 35.9 Eurasian teal (Anas crecca)16 JX844046 8641 2010 II I GKQGRL 39.0 Northern shoveler (Anas clypeata)17 JX844047 8645 2010 II I GKQGRL 34.6 Eurasian wigeon (Anas penelope)18 JX844048 8768 2010 II I EKQGRL 21.8 Mallard (Anas platyrhynchos)19 JX844049 8776 2010 II I EKQGRL 35.4 Eurasian teal (Anas crecca)20 c 8778 2010 II I EKQGRL 32.2 Eurasian teal (Anas crecca)
21 c 9125 2010 II I GKQGRL 38.2 Mallard (Anas platyrhynchos)22 JX844050 9127 2010 II I GKQGRL 37.0 Mallard (Anas platyrhynchos)23 JX844051 9129 2010 II I GKQGRL 25.9 Mallard (Anas platyrhynchos)24 JX844052 9143 2010 II I GKQGRL 26.1 Mallard (Anas platyrhynchos)25 JX844030 9147 2010 I 2 ERQERL 41.2 Mallard (Anas platyrhynchos)26 JX844053 9149 2010 II I GKQGRL 30.3 Mallard (Anas platyrhynchos)27 JX844054 9167 2010 II I GKQGRL 27.4 Mallard (Anas platyrhynchos)28 c 9171 2010 c c c 35.3 Mallard (Anas platyrhynchos)29 c 9185 2010 II I GKQGRL 35.7 Eurasian teal (Anas crecca)30 JX844055 9197 2010 II I GKQGRL 25.5 Eurasian teal (Anas crecca)
31 JX844056 9360 2010 II I GKQGRL 33.4 Mallard (Anas platyrhynchos)32 JX844057 10539 2010 II I EKQGRL 27.0 Eurasian teal (Anas crecca)33 JX844058 10541 2010 II I EKQGRL 32.0 Mallard (Anas platyrhynchos)34 JX844059 12376 2010 II I GKQGRL 30.2 Eurasian teal (Anas crecca)35 JX844060 13349 2010 II I GKQGRL 35.3 Mallard (Anas platyrhynchos)36 JX844029 13355 2010 I 2 ERQERL 38.8 Mallard (Anas platyrhynchos)37 JX844061 13357 2010 II I GKQGRL 32.9 Mallard (Anas platyrhynchos)38 JX844062 15549 2010 II I GKQGRL 32.2 Mallard (Anas platyrhynchos)39 JX844063 15551 2010 II I GKQGRL 32.0 Eurasian teal (Anas crecca)a Published by Lindh et al. (27).b Amino acids 112 to 117.c Not enough sequence obtained.d Analyzed by L gene real-time RT-PCR.
Lindh et al.
3666 jcm.asm.org Journal of Clinical Microbiology
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from

All together, 39 NDV strains were found in the active surveil-lance study. F gene RT-PCR (used for samples collected prior to2010) or real-time L gene RT-PCR (samples from 2010) positivesamples were confirmed to be NDV by either sequencing and/orby isolation in embryonated eggs with hemagglutination activity.All samples which yielded cycle threshold (CT) values of �45 inthe real-time RT-PCR assay were considered potentially positiveand were subject to further verification by the other methodsmentioned above (Table 4). Most (30/32) samples that were con-firmed class II positive in this study had CT values under 37, whichwas used as a threshold by Fuller et al. (13), while our class I virusesyielded CT values over 41 (Table 2). The high CT values of two classII viruses were probably due to poor sample quality, whereas theclass I viruses with high CT values were readily detected by theconventional RT-PCR.
The NDV-positive active surveillance samples (Table 2) de-rived most often from Eurasian teals (10.3% of sampled teals,statistically significantly more often than from other sampledspecies in this study; P � 0.0092), mallards (4.8% of sam-
pled mallards), Eurasian wigeons (Anas penelope; 5% of sam-pled wigeons), a Northern shoveler (Anas clypeata), and a com-mon pochard (Aythya ferina). The yearly prevalence of NDVamong wild waterfowl varied between 0.5% in 2008 to 11.8% in2010. In passive surveillance, up to 100 samples were collectedper year, and only one NDV-positive sample was detected dur-ing the study period. This passive surveillance sample was froma dead razorbill (Alca torda), which was initially suspected to beinfected with AIV but found positive for NDV. All the birdssampled for active surveillance appeared clinically healthy, al-though five birds (four Eurasian teals and a northern shoveler)were infected by both lentogenic NDV and low-pathogenicavian influenza virus. No velogenic strains were found from theactive surveillance samples.
Characterization of the detected NDV strains. Sequence dataof the F gene cleavage site was gained from all but three of thePCR-positive active surveillance samples, probably because ofpoor sample quality. All the waterfowl-derived strains expressedthe lentogenic fusion protein cleavage site motifs: GKQGRL,EKQGRL, or ERQERL (Table 2). Outbreak-associated isolatesfrom 2004 and 2008 and the feral pigeon strain from 2009 pos-sessed cleavage site motifs typical for velogenic strains: turkey/Finland/5789/2004, RRQRRF; domestic pigeon/Finland/17557/2008, RRQKRF; feral pigeon/Finland/15475/2009, KRQKRF.Pheasant/Finland/8036/2003 had the lentogenic motif GKQGRL(Table 3).
The wild waterfowl-derived viruses were all lentogenic virusesof class I or class II genotype I. The 31 class II wild-waterfowl-derived samples, collected during 2006, 2009, and 2010, were atleast 96.5% identical, but most were over 98% identical at thenucleotide level (comparison of the 450-nt region). On a few oc-casions, fully identical viruses were isolated 4 years apart from thesame location. As many of our class II strains analyzed in this studyare very similar, a few representatives were selected for phyloge-netic purposes in Fig. 1, while the relationships between all Finn-ish strains are depicted in Fig. 2.
Our class I strains (collected in 1997, 2006, 2008, 2009, and
TABLE 3 Finnish NDV strains derived from outbreaks and through passive surveillance
No.GenBankaccession no. Isolate Yr Class Genotype F cleavage siteb ICPI Host
1 AY034794 Fin-69a 1969 II IV RRQRRF NTd Williow grouse (Lagopus lagopus)2 AY034795 Fin-70a 1970 II IV RRQRRF NT Chicken (Gallus gallus)3 AY034796 Fin-92a 1992 II VI RRQKRF 1.4 Feral pigeon (Columba livia)4 AY034797 Fin-96aa 1996 II VI RRKKRF 1.32 Feral pigeon (Columba livia)5 AY034798 Fin-96ba 1996 II VII RRQRRF 1.38 Goosander (Mergus merganser)6 AY034799 Fin-96ca 1996 II VI RRKKRF 1.4 Feral pigeon (Columba livia)7 AY034800 Fin-96da 1996 II VI RRKKRF 1.4 Feral pigeon (Columba livia)8 AY034801 Fin-97a 1997 I 2 ERQERL 0.17 Mallard (Anas platyrhynchos)9 JX844031 7775 2003 II I GKQGRL 0.5 Pheasant (Phasianus colchius)10 JX844032 8036 2003 II I GKQGRL 0.5 Pheasant (Phasianus colchius)11 JX844033 5789 2004 II VII RRQRRF 1.6 Turkey (Melagris)12 JX844034 6985 2008 II VI RRQKRF NT Domestic pigeon (Columba livia domestica)13 JX844035 17557 2008 II VI RRQKRF NT Domestic pigeon (Columba livia domestica)14 JX844036 15475 2009 II VI KRQKRF NT Feral pigeon (Columbia livia)15 c 4913 2010 II Not typed c NT Razorbill (Alca torda)a Published by Huovilainen et al. (18).b Amino acids 112 to 117.c Not enough sequence obtained.d NT, test not done.
TABLE 4 Active surveillance sample size and number of NDV positivesper year
YrNo. ofsamples
No. of PCRpositives
No. ofisolations
Class (no. ofNDV-positivesamples in class)
2006 115 5a 4 I (3)II (2)
2008 182 1a 1 I (1)2009 189 6a 6 II (6)2010 229 27b 5c I (2)
II (24)Untyped (1)
Total 715 39 (5.5%) 16 (�41%) I (6), II (32)a Screened by F gene RT PCR.b Screened by L gene real-time RT-PCR.c Isolation attempted from only a part of the samples.
Molecular Epidemiology of NDV in Northern Europe
November 2012 Volume 50 Number 11 jcm.asm.org 3667
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from
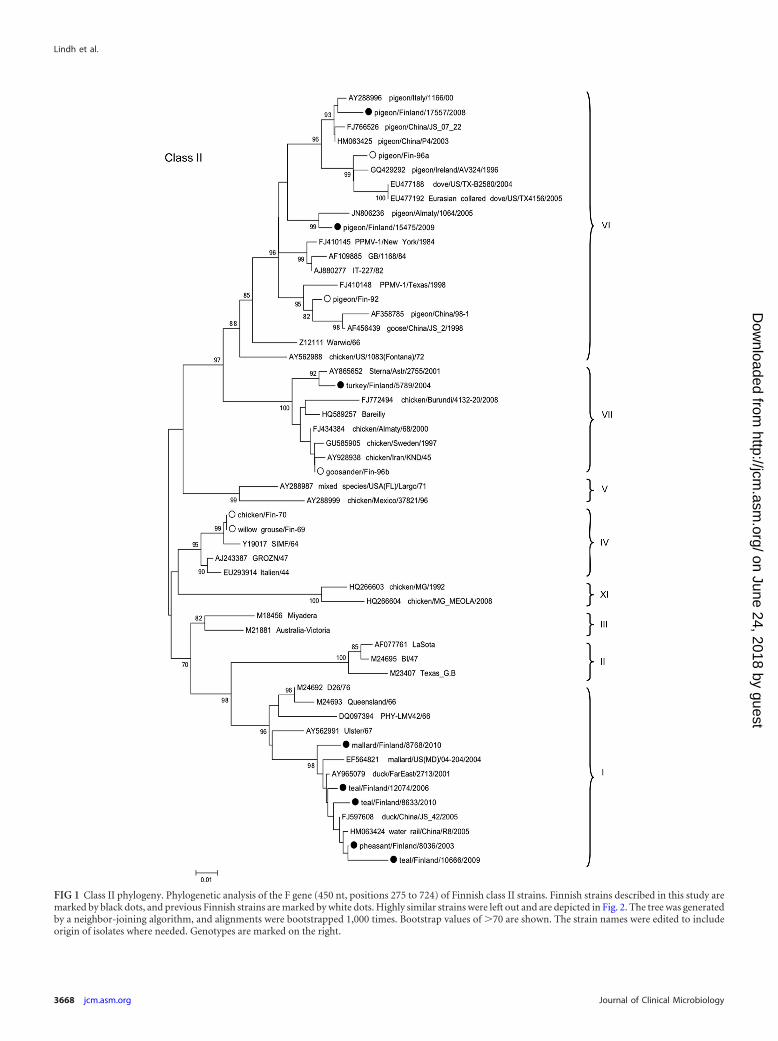
FIG 1 Class II phylogeny. Phylogenetic analysis of the F gene (450 nt, positions 275 to 724) of Finnish class II strains. Finnish strains described in this study aremarked by black dots, and previous Finnish strains are marked by white dots. Highly similar strains were left out and are depicted in Fig. 2. The tree was generatedby a neighbor-joining algorithm, and alignments were bootstrapped 1,000 times. Bootstrap values of �70 are shown. The strain names were edited to includeorigin of isolates where needed. Genotypes are marked on the right.
Lindh et al.
3668 jcm.asm.org Journal of Clinical Microbiology
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from
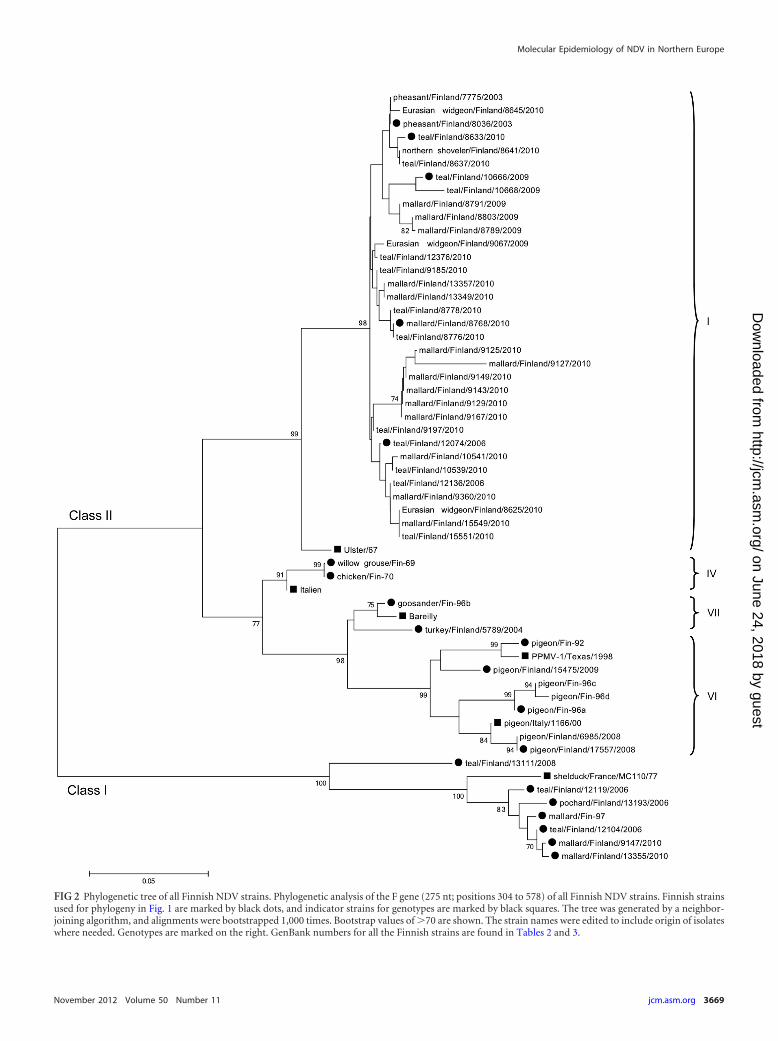
FIG 2 Phylogenetic tree of all Finnish NDV strains. Phylogenetic analysis of the F gene (275 nt; positions 304 to 578) of all Finnish NDV strains. Finnish strainsused for phylogeny in Fig. 1 are marked by black dots, and indicator strains for genotypes are marked by black squares. The tree was generated by a neighbor-joining algorithm, and alignments were bootstrapped 1,000 times. Bootstrap values of �70 are shown. The strain names were edited to include origin of isolateswhere needed. Genotypes are marked on the right. GenBank numbers for all the Finnish strains are found in Tables 2 and 3.
Molecular Epidemiology of NDV in Northern Europe
November 2012 Volume 50 Number 11 jcm.asm.org 3669
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from

2010) were 97.5% to 99.5% identical and clustered phylogeneti-cally with strains of the class I genotype 2 (by analysis of 400 nt ofthe F gene; Fig. 3). An exception was the strain teal/Finland/13111/2008, which differs by �13% of the other Finnish strains. Teal/Finland/13111/2008 is only up to 88% identical at the nucleotidelevel with other class I viruses, with a single exception: strainHIECK87191 (or Espey, AY135759) isolated from a chicken inIreland in 1987 (3). By phylogenetic analysis, these two strainscluster independently from all other published strains and form anew genotype, which based on the grouping made by Kim et al.
(23) would form a 10th genotype. The amino acid sequence ofteal/Finland/13111/2008 and HIECK87191 diverges by 7% ormore from other class I strains. Interestingly, teal/Finland/13111/2008 has a highly variable 20-amino-acid motif, 9YPCILTLTSAV-VTLTLMTG28, in the N-terminal region of the fusion protein,preceding the cleavage site (Fig. 4). A BLAST search of this nucle-otide and amino acid sequence identified a homologous domainsolely in strain HIECK87191. The distinct N-terminal sequencewas left out from phylogenetic analyses and alignments in order tonot confer with the results.
FIG 3 Class I phylogeny. Phylogenetic analysis of the F gene (400 nt; positions 125 to 524) of Finnish class I strains. The Finnish strains described in this studyare marked by black dots and a previous isolate by a white dot. The tree was generated by a neighbor-joining algorithm, and alignments were bootstrapped 1,000times. Bootstrap values of �70 are shown. The strain names were edited to include origin of isolates where needed. Genotypes (according to Kim et al. [24]) aremarked on the right.
Lindh et al.
3670 jcm.asm.org Journal of Clinical Microbiology
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from

Of the outbreak-associated strains, strain pheasant/Finland/8036/03 was found highly identical with some of our wild water-fowl-derived strains, differing by as little as 0.6%. It also clusteredphylogenetically with strains from ducks and wild birds in Chinaand Russia, which shared 99% sequence identity (Fig. 1).
Strain turkey/Finland/5789/04 clustered with the genotypeVIIb viruses and was most closely related (99% identical) to astrain isolated from a shorebird (little tern; Sterna albifrons;GenBank accession no. AY865652) in Russia 3 years earlier. It alsoclustered phylogenetically with an earlier Finnish isolate (Fin-96;97% nucleotide identity) and poultry-derived isolates from Iran(1995), Sweden (1997), and Russia (2000) (96 to 97% nucleotideidentities) (Fig. 1).
Domestic pigeon/Finland/17557/2008, isolated during a re-cent outbreak among hobby pigeons, and feral pigeon/Finland/15475/2009 clustered phylogenetically together with genotypeVIb strains. The domestic pigeon isolates were genetically close tostrain P4 and other Chinese PPMV-1 strains (up to 99% nucleo-tide identity), while the feral pigeon isolate was 98% identical witha Kazakhstanian strain but reached only 96% identity with allother strains of clade VIb (Fig. 1).
DISCUSSION
NDV is a common pathogen of birds, and infections have beenreported in at least 236 species (21). Czeglédi and colleagues (10)speculated that class I and class II genotype I are ancestral repre-sentatives of NDV maintained by their natural hosts, the wildwaterfowl. The recent genotypes, including most velogenicstrains, are thought to have arisen in the 20th century through theinvolvement of a secondary host, the chickens. Because of the eco-nomic impact of NDV on the poultry industry, most of the liter-ature is focused on strains derived from commercial flocks. Less isknown about the NDV strains circulating in the nature, their evo-lution, and the role of different host species.
Studies of NDV among wild birds in North America and Chinahave recently been published (9, 23). Similar information on prev-alence and molecular epidemiology of NDV in wild birds in Eu-rope has been scarce, although outbreaks among poultry are fre-quently reported. In our study, the yearly prevalence of NDVranged from 0.5% to 11.8%. The highest prevalence was recordedin 2010 when a new real-time RT-PCR method was introduced. Asimilar, around 10% yearly prevalence of AIV has been recordedin our laboratory (27). Although sampling procedures variedslightly from year to year and relied on voluntary collectors, iso-lation and sequencing success was high for PCR-positive samples.
Most of the strains from waterfowl in Northern Europe foundduring this study were class II genotype I viruses. Our strains ap-pear highly related to Chinese strains that appear endemic in bothlive-bird markets and in wild-waterfowl populations (9, 28).These two areas are connected by the Eurasian migratory flyway.The genotype I viruses isolated between 2006 and 2010 from fourmain sampling sites situated 150 to 500 km apart showed overallvery little variation. The strains were highly identical and showedrather site-specific than species-specific similarities, manifested asspecific nucleotide signatures at given locations. This is not unex-pected, since mallards and Eurasian teals are nesting in the sameareas all over Finland. The observed geographic clustering might,however, be related to sample relatedness in terms of samplingtime and host population.
Class II genotype I viruses are generally lentogenic, but a viru-lent strain that was the causative agent of a disease outbreak inAustralia in 1998 is thought to have arisen from a lentogenic pro-genitor (15). The change from lentogenic NDV to velogenic NDVduring favorable conditions, e.g., replication in dense poultrypopulations, continues to be a major concern. Factors favoringevolution of virulence are (i) the requirement of only a few muta-tions in the F gene and (ii) the circulation of a high load of diverselentogenic NDVs in the wild-bird reservoir (33). Some of the ge-notype I derivatives, e.g., Ulster2C/67 and Queensland/V4, areused as live vaccines in many countries. Billions of doses of theselow-virulence vaccines are used annually, some of which leak intothe environment and might be encountered in wild birds.
Six of 39 wild-bird-derived strains were class I NDV. Notably,teal//Finland/13111/2008 and HIECK87191 strains both differ by�12% from all other published class I NDV strains and harbor adistinct N-terminal motif. Phylogenetic analysis further definesthese two strains as the sole members of a distinct genotype ofNDV, provisionally named here class I genotype 10. Class I viruseshave previously been found in waterfowl and live-bird markets.Although generally regarded as lentogenic strains, a class I strainhas at least on one occasion been associated with a velogenic out-break among poultry (5). Shengqing et al. and Tsunekuni et al.assessed the potential of wild-waterfowl-derived lentogenic class INDV to become velogenic by serial passaging of goose/Alaska415/91. They showed that during 14 passages in chicken, seven aminoacid substitutions arose, leading to a shift to a highly virulentvariant (37, 41).
Class I viruses have been neglected in many screening studies,mainly because of poor detection rates by conventional RT-PCR-based screening assays, designed for class II NDV (23). Kim et al.
FIG 4 Alignment of the fusion protein amino acids 1 to 40. Amino acid sequence of the fusion protein, showing the distinct motif of teal/Finland/13111/2008and HIECK87191 at positions 9 to 28. The region is distinct between class I and class II genotypes. GenBank numbers are found in Fig. 1 and 3.
Molecular Epidemiology of NDV in Northern Europe
November 2012 Volume 50 Number 11 jcm.asm.org 3671
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from

calculated that a commonly used M gene assay (43) probe sitediverged from class I nucleotide sequences by at least 25%, failingto detect them (24). The F gene RT-PCR protocols used in Finlandwere updated to include class I viruses after the first Finnish classI virus isolation in 1997 (18). The use of a rapid real-time multi-plex RT-PCR assay for screening clinical samples is much wel-comed in diagnostic laboratories. However, attention is needed toreassure that also more divergent strains are detected, e.g., by ver-ifying inconclusive results by isolation attempts or by comple-menting PCR assays. It should be kept in mind that a sole CT valuedoes not provide sufficient diagnostic value and that the conven-tional methods are needed to assess other properties of the de-tected strains. The real-time RT-PCR assay used here seems tohave an increased sensitivity compared to our previous methods,judged by the higher number NDV PCR positives in 2010, but asnoted by the developers, this method is less sensitive for the detec-tion of class I viruses, as demonstrated by higher CT values (13).
Finland is located on the Western European flyway, and a bigproportion of hatch-year dabbling ducks wintering in West andCentral Europe have been found to originate in Southern Finland(16). Interestingly, while a clear phylogenetic separation can beseen between North American and Eurasian AIV strains, our wild-waterfowl-derived strains were closely related to strains that wereisolated on the other side of the Atlantic Ocean. This might reflectthe involvement of different host species, with different migratorypatterns, than the common hosts of AIV. Trade of birds might alsoplay a part in the transatlantic exchange of strains. There are noduck species which regularly migrate across the Atlantic sea, al-though many ducks, including American green-winged teal (Anascarolensis), stray to the European side of the Atlantic, where theyshare the same wintering grounds with Eurasian teals. These twospecies are able to reproduce together and could serve as thebridge between North American and European NDV. This wouldfurther indicate that lentogenic NDV has only very mild or noeffect on the fitness of its waterfowl hosts, who are able to migrateacross the Atlantic Ocean while infected. The prevalence of NDVin Eurasian teal was found to be significantly high in this study,which could further indicate that this species has a pronouncedrole in the ecology of NDV in Northern Europe.
Pheasants are susceptible to all strains of NDV, usually intro-duced by strains prevalent in poultry and wild birds (1). Isolatesfrom the outbreak among farmed pheasants in 2003 are highlysimilar (over 99% nucleotide identity) to genotype I viruses thathave repeatedly been detected among waterfowl populations inFinland since 2006 and can be considered endemic. The pheasantswere most likely infected through contact with wild birds whilesharing common grounds during part of their rearing or throughtrade with another backyard-poultry holding where the rearedbirds were exposed to wild birds.
During the outbreak in a turkey farm in 2004, similar outbreakscoincided in other Nordic countries. While velogenic strains gener-ally cause acute fatal infections of chickens of all age groups, thisturkey farm showed no symptoms of disease. The only publishedstrain with high similarity (99%) to our outbreak-derived virus is avelogenic isolate from a little stern in Russia. A previous, closely re-lated wild-bird-derived strain, goosander/Fin-96b, was isolated con-currently with outbreaks among poultry in Sweden and Norway andoutbreaks in the British Isles in 1996 to 1997. Genetic analyses wereconsistent with the theory of viral spread by migratory birds, al-though a role of trade of birds or related products could not be ruled
out (4, 26). Apparently, endemic genotype VIIb viruses are reg-ularly introduced to domestic birds where they cause out-breaks. Our isolate (turkey/Finland/5789/04) clustered togetherwith mainly poultry-derived strains, but this is probably a biascaused by the trend to sample and publish sequences of poultryorigin.
Epidemiological investigations did not reveal the source of theoutbreak among hobby pigeons in 2008. Although import wassuspected to be the primary source, genetic analyses of the virusesisolated during the outbreak did not give any further evidence ofthis. Whether the introduction happened through trade of birdsor through unidentified contacts with wild birds remains un-solved, as the closest matches were only up to 98% identical. Al-though the focus is often on protecting poultry populations,transmission of pathogens can also occur in the opposite direc-tion, from domestic birds to wild birds. This should be noted inorder to reduce the spread of PPMV-1 strains.
The Finnish Food Safety Authority has evaluated the effects ofvaccinations for the Finnish poultry industry in 2005 (11). Theconclusion was that vaccination of poultry should remain bannedand that NDV should continuously be prevented in Finland byefficient use of biosecurity measures and effective laboratorydiagnostics. An exception to our nonvaccinating policy is racingpigeons participating in shows and races, where vaccinations witha killed virus are required.
In conclusion, our study shows that lentogenic NDVs of class Iand class II genotype I are endemic among clinically healthy wildwaterfowl in Finland. Because Central and Northwestern Europeare connected by migratory routes of the species analyzed here,our data should apply to Europe in general. Efforts are needed torestrict contacts between wild birds and poultry, as these two hostsystems appear to have a continuous exchange of NDVs. Import,trade, and bird shows are other events that easily allow the spreadof new virulent strains to susceptible populations, unless strictcontrol measures are applied. Identification of new strains escap-ing conventional diagnostic assays and vaccines underlines theneed to continuously update our surveillance systems in terms ofdiagnostic assays, biosecurity, and research, in order to protectboth poultry and wild birds.
ACKNOWLEDGMENTS
We gratefully acknowledge all the volunteer hunters for providing sam-ples for this study and the Finnish Game and Fisheries Research Institutefor their support. Hannu Pöysä, Jorma Autioniemi, Jorma Korhonen,Petri Timonen, and Einari Väyrynen are especially acknowledged for theircontribution to the sample collection. We also thank technical staff mem-bers, especially Merja Hautala and Mia Biström (BVDc), for their contri-bution in the laboratory work. Tarja Sironen is thanked for the expertiseshe provided in phylogenic analyses.
The Animal Health and Veterinary Laboratories Agency (AHVLA;formerly VLA) is acknowledged for assessing the intracerebral pathoge-nicity indexes (ICPI) for strains described in our study.
The research was funded by the Finnish Ministry of Agricultureand Forestry, Helsinki Biomedical Graduate School (HBGS), andVictoriastiftelsen.
REFERENCES1. Aldous EW, Alexander DJ. 2008. Newcastle disease in pheasants
(Phasianus colchicus): a review. Vet. J. 175:181–185.2. Aldous EW, Fuller CM, Mynn JK, Alexander DJ. 2004. A molecular
epidemiological investigation of isolates of the variant avian paramyxovi-
Lindh et al.
3672 jcm.asm.org Journal of Clinical Microbiology
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from

rus type 1 virus (PPMV-1) responsible for the 1978 to present panzootic inpigeons. Avian Pathol. 33:258 –269.
3. Aldous EW, Mynn JK, Banks J, Alexander DJ. 2003. A molecularepidemiological study of avian paramyxovirus type 1 (Newcastle diseasevirus) isolates by phylogenetic analysis of a partial nucleotide sequence ofthe fusion protein gene. Avian Pathol. 32:239 –256.
4. Alexander DJ, et al. 1999. Antigenic and genetic characterisation of New-castle disease viruses isolated from outbreaks in domestic fowl and turkeysin Great Britain during 1997. Vet. Rec. 145:417– 421.
5. Alexander DJ, et al. 1992. Characterisation of an antigenically unusualvirus responsible for two outbreaks of Newcastle disease in the Republic ofIreland in 1990. Vet. Rec. 130:65– 68.
6. Alexander DJ, et al. 1997. Antigenic diversity and similarities detected inavian paramyxovirus type 1 (Newcastle disease virus) isolates usingmonoclonal antibodies. Avian Pathol. 26:399 – 418.
7. Ballagi-Pordany A, Wehmann E, Herczeg J, Belak S, Lomniczi B. 1996.Identification and grouping of Newcastle disease virus strains by restric-tion site analysis of a region from the F gene. Arch. Virol. 141:243–261.
8. Briand FX, Henry A, Massin P, Jestin V. 2012. Complete genomesequence of a novel avian paramyxovirus. J. Virol. 86:7710.
9. Cai S, et al. 2011. Genetic characterization and evolutionary analysis of 4Newcastle disease virus isolate full genomes from waterbirds in SouthChina during 2003–2007. Vet. Microbiol. 152:46 –54.
10. Czegledi A, et al. 2006. Third genome size category of avian paramyxo-virus serotype 1 (Newcastle disease virus) and evolutionary implications.Virus Res. 120:36 – 48.
11. Ek-Kommonen C, Jakava-Viljanen M, Perko-Mäkelä P, Rosengren H,Rossow L. 2005. Impact of vaccination for Newcastle disease in Fin-land—a report. Nat. Vet. Food Res. Inst. Pub. ISSN 1458 – 6878.
12. EU. 1992. Community measures for the control of Newcastle disease.European Union law. Council directive 92/66/EEC. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri�CELEX:31992L0066:EN:NOT.
13. Fuller CM, Brodd L, Irvine RM, Alexander DJ, Aldous EW. 2010.Development of an L gene real-time reverse-transcription PCR assay forthe detection of avian paramyxovirus type 1 RNA in clinical samples.Arch. Virol. 155:817– 823.
14. Gould AR, et al. 2003. Newcastle disease virus fusion and haemaggluti-nin-neuraminidase gene motifs as markers for viral lineage. Avian Pathol.32:361–373.
15. Gould AR, et al. 2001. Virulent Newcastle disease in Australia: molecularepidemiological analysis of viruses isolated prior to and during the out-breaks of 1998-2000. Virus Res. 77:51– 60.
16. Gunnarsson G, et al. 2012. Disease dynamics and bird migration—linking mallards Anas platyrhynchos and subtype diversity of the influ-enza A virus in time and space. PLoS One. 7:e35679. doi:10.1371/journal.pone.0035679.
17. Herczeg J, et al. 1999. Two novel genetic groups (VIIb and VIII) respon-sible for recent Newcastle disease outbreaks in Southern Africa, one (VIIb)of which reached Southern Europe. Arch. Virol. 144:2087–2099.
18. Huovilainen A, Ek-Kommone C, Manvell R, Kinnunen L. 2001. Phylo-genetic analysis of avian paramyxovirus 1 strains isolated in Finland. Arch.Virol. 146:1775–1785.
19. ICTV, King A, Lefkowitz E, Adams MI, Carstens EB. 2012. Virustaxonomy: ninth report of the International Committee on Taxonomy ofViruses. Academic Press, London, United Kingdom.
20. Kaleta EF, Alexander DJ, Russell PH. 1985. The first isolation of theavian PMV-1 virus responsible for the current panzootic in pigeons?Avian Pathol. 14:553–557.
21. Kaleta EF, Baldauf C. 1988. Newcastle disease in free-living and pet birds,p 197–246. In Alexander DJ (ed), Newcastle disease. Kluwer AcademicPublishers, Boston, MA.
22. Kauppinen J, Väänänen VM. 1999. Factors affecting changes in water-fowl populations in eutrophic wetlands in the Finnish lake district. Wildl.Biol. 7:73– 81.
23. Kim LM, et al. 2007. Phylogenetic diversity among low-virulence New-castle disease viruses from waterfowl and shorebirds and comparison of
genotype distributions to those of poultry-origin isolates. J. Virol. 81:12641–12653.
24. Kim LM, King DJ, Suarez DL, Wong CW, Afonso CL. 2007. Charac-terization of class I Newcastle disease virus isolates from Hong Kong livebird markets and detection using real-time reverse transcription-PCR. J.Clin. Microbiol. 45:1310 –1314.
25. Kommers GD, King DJ, Seal BS, Brown CC. 2003. Virulence of sixheterogeneous-origin Newcastle disease virus isolates before and after se-quential passages in domestic chickens. Avian Pathol. 32:81–93.
26. Linde AM, et al. 2010. Complete genome characterisation of a Newcastledisease virus isolated during an outbreak in Sweden in 1997. Virus Genes41:165–173.
27. Lindh E, et al. 2008. Orthomyxo-, paramyxo- and flavivirus infections inwild waterfowl in Finland Virol. J. 5:35.
28. Liu X, et al. 2009. Surveillance for avirulent Newcastle disease viruses indomestic ducks (Anas platyrhynchos and Cairina moschata) at live birdmarkets in Eastern China and characterization of the viruses isolated.Avian Pathol. 38:377–391.
29. Liu XF, Wan HQ, Ni XX, Wu YT, Liu WB. 2003. Pathotypical andgenotypical characterization of strains of Newcastle disease virus isolatedfrom outbreaks in chicken and goose flocks in some regions of Chinaduring 1985–2001. Arch. Virol. 148:1387–1403.
30. Lomniczi B, et al. 1998. Newcastle disease outbreaks in recent years inWestern Europe were caused by an old (VI) and a novel genotype (VII).Arch. Virol. 143:49 – 64.
31. Maminiaina OF, et al. 2010. Newcastle disease virus in Madagascar: identi-fication of an original genotype possibly deriving from a died out ancestor ofgenotype IV. PLoS One 5:e13987. doi:10.1371/journal.pone.0013987.
32. Miller PJ, et al. 2010. Evidence for a new avian paramyxovirus serotype 10detected in rockhopper penguins from the Falkland Islands. J. Virol. 84:11496 –11504.
33. Miller PJ, Decanini EL, Afonso CL. 2010. Newcastle disease: evolution ofgenotypes and the related diagnostic challenges. Infect. Genet. Evol. 10:26 –35.
34. Nagai Y, Klenk HD, Rott R. 1976. Proteolytic cleavage of the viralglycoproteins and its significance for the virulence of Newcastle diseasevirus. Virology 72:494 –508.
35. Seal BS, King DJ, Bennett JD. 1995. Characterization of Newcastle dis-ease virus isolates by reverse transcription PCR coupled to direct nucleo-tide sequencing and development of sequence database for pathotype pre-diction and molecular epidemiological analysis. J. Clin. Microbiol. 33:2624 –2630.
36. Senne DA, et al. 1996. Survey of the hemagglutinin (HA) cleavage sitesequence of H5 and H7 avian influenza viruses: amino acid sequence at theHA cleavage site as a marker of pathogenicity potential. Avian Dis. 40:425–437.
37. Shengqing Y, et al. 2002. Generation of velogenic Newcastle diseaseviruses from a nonpathogenic waterfowl isolate by passaging in chickens.Virology 301:206 –211.
38. Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysisusing maximum likelihood, evolutionary distance, and maximum parsi-mony methods. Mol. Biol. Evol. 28:2731–2739.
39. Toyoda T, et al. 1987. Structural comparison of the cleavage-activationsite of the fusion glycoprotein between virulent and avirulent strains ofNewcastle disease virus. Virology 158:242–247.
40. Tsai HJ, et al. 2004. Antigenic and genotypical characterization of New-castle disease viruses isolated in Taiwan between 1969 and 1996. Vet. Mi-crobiol. 104:19 –30.
41. Tsunekuni R, Ito H, Otsuki K, Kida H, Ito T. 2010. Genetic comparisonsbetween lentogenic Newcastle disease virus isolated from waterfowl andvelogenic variants. Virus Genes 40:252–255.
42. Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. 1992.Evolution and ecology of influenza A viruses. Microbiol. Rev. 56:152–179.
43. Wise MG, et al. 2004. Development of a real-time reverse-transcriptionPCR for detection of Newcastle disease virus RNA in clinical samples. J.Clin. Microbiol. 42:329 –338.
Molecular Epidemiology of NDV in Northern Europe
November 2012 Volume 50 Number 11 jcm.asm.org 3673
on June 24, 2018 by guesthttp://jcm
.asm.org/
Dow
nloaded from





























