Embed Size (px)
Citation preview

Neurobiology of Disease 46 (2012) 234–243
Contents lists available at SciVerse ScienceDirect
Neurobiology of Disease
j ourna l homepage: www.e lsev ie r .com/ locate /ynbd i
Isoflurane anesthesia precipitates tauopathy and upper airways dysfunction inpre-symptomatic Tau.P301L mice: Possible implication forneurodegenerative diseases
Clément Menuet a, Peter Borghgraef b, Nicolas Voituron a, Christian Gestreau a, Lies Gielis b,Herman Devijver b, Mathias Dutschmann c, Fred Van Leuven b, Gérard Hilaire a,⁎a MP3-respiration team, Unité Mixte de Recherche 6231, Centre de Recherche en Neurobiologie et Neurophysiologie de Marseille, Faculté Saint Jérôme, service 362, 13397 Marseille,cedex20, Franceb Experimental Genetics Group, LEGTEGG, Department Human Genetics, KULeuven, Campus Gasthuisberg ON1-06.602, B-3000 Leuven, Belgiumc Institute of Membrane and Systems Biology, Garstang Building, University of Leeds, Leeds LS2 9JT, UK
Abbreviations: AD, Alzheimer's disease; Asp, Airflowtively stained; CSp, Chest Spirogram; KF, Kolliker–FuseNucleus Retroambiguus; PAG, Periaqueductal Gray; RWildtype.⁎ Corresponding author at: MP3-respiration team, Un
Centre de Recherche en Neurobiologie et NeurophysioloJérôme, service 362, 13 Marseille, France. Fax: +33 491
E-mail address: [email protected] (G. HAvailable online on ScienceDirect (www.scienced
0969-9961/$ – see front matter © 2012 Elsevier Inc. Alldoi:10.1016/j.nbd.2012.01.012
a b s t r a c t
a r t i c l e i n f oArticle history:Received 28 October 2011Revised 14 December 2011Accepted 23 January 2012Available online 31 January 2012
Keywords:TauopathyAnesthesiaUpper airwaysMemantineIsofluraneNMDA receptorsMiceBrainstemAlzheimer's disease
The postoperative cognitive decline resulting from volatile anesthesia is gaining acceptance as a major healthproblem. The common anesthetic isoflurane is suspected to precipitate neurodegeneration in Alzheimer'sdisease by unknownmechanisms. We previously validated that 8 month old Tau.P301L mice suffer upper air-ways defects related to tauopathy within the Kolliker–Fuse nucleus that controls upper airways function. Wenow report that isoflurane anesthesia in young, pre-symptomatic Tau.P301L mice triggered precocious upperairways defects and tauopathy in several brainstem nuclei, including the nucleus ambiguus that containsupper airways motor neurons and the Kolliker–Fuse. The prescription drug memantine, identified as anNMDA receptor antagonist, prevented the post-anesthesia upper airways dysfunction and alleviated tauopa-thy in the nucleus ambiguus and Kolliker–Fuse. We further identified protocols of anesthesia in youngTau.P301L mice that mitigated adverse effects of isoflurane anesthesia. Thus, our experimental findings in avalidated mouse model for tauopathy demonstrate the link between isoflurane anesthesia, earlier onset oftauopathy and earlier onset of functional deficits, highlight the implication of NMDA-receptors in the mech-anisms mediating the adverse effects of isoflurane, and potentially identify safer protocols for anesthesia inpatients with tauopathy.
© 2012 Elsevier Inc. All rights reserved.
Introduction
Postoperative cognitive decline is a common phenomenon afteranesthesia with volatile anesthetics such as isoflurane. The problemis gaining additional importance as a secondary health concern intauopathy patients, including Alzheimer's disease (AD) with a possi-ble link between anesthesia and AD onset and progression (Baranovet al., 2009; Bilotta et al., 2010; Pan et al., 2011). AD is characterizedby the formation of extraneuronal plaques of aggregated β-amyloidprotein and intraneuronal tangles of fibrillar aggregates of themicrotubule-associated Tau protein (Brion et al., 1986; Glenner and
Spirogram; AT8+, AT8 posi-; nA, Nucleus Ambiguus; NRA,f, Respiratory frequency; WT,
ité Mixte de Recherche 6231,gie de Marseille, Faculté Saint28 83 97.ilaire).irect.com).
rights reserved.
Wong, 1984; Grundke-Iqbal et al., 1986a, 1986b) that are thought toalter the functioning of neural networks, resulting in cognitive,motor and mental dysfunctions. Tau phosphorylation is reduced bymemantine, a partial NMDA-receptor antagonist, approved for treat-ment of AD (Li et al., 2004; Martinez-Coria et al., 2010; Song et al.,2008).
In transgenic Tau.P301Lmice, amousemodel of tauopathy, we previ-ously reported development of cognitive defects (age 4–6 months),motor and upper airways dysfunction (age 7–9 months), altered ultra-sonic vocalization (8–10 months) and breathing defects leading to pre-mature death (age 9–12 months) (Dutschmann et al., 2010; Menuet etal., 2011a, 2011b; Terwel et al., 2005). From 8months onwards,Tau.P301Lmice develop a paradoxical tendency to upper airways closureduring inspiration that they compensate by producing enlarged respira-tory movements of the chest (Dutschmann et al., 2010). In the sametime-frame, they develop tauopathy within the pontine Kolliker–Fusenucleus (KF) (Dutschmann et al., 2010), a multifunctional network, con-tributing to the off-switch of inspiration (Dutschmann and Herbert,2006; Fung et al., 1994; Ling et al., 1994; Villard et al., 1984), the controlof upper airways (Dutschmann and Herbert, 2006) and the coupling of

235C. Menuet et al. / Neurobiology of Disease 46 (2012) 234–243
respiration with swallowing (Bonis et al., 2011; Gestreau et al., 2005;Kuna and Remmers, 1999; Nunez-Abades et al., 1990), vocalization(Dutschmann and Herbert, 2006), thermoregulation (Bhatnagar andDallman, 1998) and cardiovascular regulations (Dutschmann andHerbert, 1998; Guo et al., 2002; Horiuchi et al., 1999). Thus, the KF tauo-pathy of old Tau.P301L mice correlates well with their upper airwaysdysfunction that may be reminiscent of the well-known oro-pharyngeal problems in tauopathy and Alzheimer's patients (Horner etal., 1994; Humbert et al., 2010; Priefer and Robbins, 1997; Suh et al.,2009; Wada et al., 2001).
Isoflurane anesthesia of Tau.P301L mice, especially coupled withhypothermia, precipitates tau hyper-phosphorylation via poorly un-derstood mechanisms (Planel et al., 2009). Here, we examined the ef-fects of isoflurane anesthesia in young, pre-symptomatic Tau.P301Lmice on upper airways dysfunction and brainstem tauopathy. First,we defined that subjecting young Tau.P301L mice to isoflurane anes-thesia provoked precocious onset of upper airways dysfunction, cor-relating with precocious onset of tauopathy in the KF but also thenucleus ambiguus (nA) that contains upper airways motor neurons.Secondly, we obtained evidence that the adverse effects of isofluraneon upper airways function, nA and KF tauopathy were alleviatedby distinct protocols of anesthesia, including memantine pre-treatment, which highlights implication of NMDA-R in these patho-logical mechanisms. Thus, our results demonstrate the link betweenisoflurane anesthesia, tauopathy and disease onset, at least inTau.P301L mice, and identify possible safer protocols for anesthesiain patients suffering tauopathy.
Material and methods
This work has been carried out in accordance with EU Directive2010/63/EU for animal experiments.
Animals
Experiments were performed in wild type (WT) FVB/N and trans-genic Tau.P301L mice at age 4 and 8 months. Tau.P301L mice wereproduced in the FVB/N genetic background to express the longesthuman tau isoform bearing the P301L mutation (Tau.4R/2N-P301L)under control of the mouse thy1 gene promoter aiming for neuron-specific expression starting in the third postnatal week (Terwel etal., 2005). Tau.P301L mice, genotyped by PCR, were compared withage- and sex-matched control FVB/N mice.
Isoflurane anesthesia protocols
Consistently with previous reports (Planel et al., 2009), youngmice at age 4 months were subjected to 4 hours exposure to isoflur-ane (1-chloro-2,2,2-trifluoroethyl difluoromethyl ether; AErrane;Baxter S.A., Belgium) at 1 MAC (Minimum Alveolar Concentration,1.3% isoflurane) with hyperoxic gas mixture (30% O2 in air insteadof 21% O2). At onset of anesthesia and every following h, we recordedrectal temperature (type T Thermocouple model 5005, Bioblock Sci-entific, France), respiratory frequency (home made constraintgauges), electromyogram (EMG) of chest respiratory muscles andelectrocardiogram (epidermal electrodes inserted within the ribcage). Mice were either kept freely breathing during isoflurane expo-sure (with or without control of body temperature via external heat-er) or were intubated and ventilated. Forced ventilation was adjustedat 100 cycles per min with a volume of 0.4 ml per cycle (SAR-830 ven-tilator, Bioseb, U.S.A.). During forced ventilation, mice received thesame hyperoxic gas mixture as above (30% O2 in air) or a hypoxicgas mixture (10% O2, 90% N2) or a hyperoxic/ hypercapnic gas mix-ture (7% CO2, 30% O2 and 63% N2). A set of WT and Tau.P301L micewas subjected to pre-treatment with the non-competitive NMDA-receptor antagonist memantine (Li et al., 2004; Martinez-Coria et
al., 2010; Song et al., 2008), approved for treatment of AD (Reisberget al., 2003). Memantine hydrochloride (Sigma) was injected intra-peritoneally (20 mg·kg−1) one hour prior to isoflurane anesthesia.
Plethysmographic analysis of breathing parameters
As previously reported (Dutschmann et al., 2010; Menuet et al.,2011a), whole-body plethysmography was used to measure respira-tory frequency (Rf, expressed in cycles per min, c·min−1) in con-scious, unrestrained mice and double-chamber plethysmography torecord the chest respiratory movements in the body chamber(Chest Spirogram, CSp) and the resulting airflow in the head chamber(Airflow Spirogram, ASp) (Fig. 1A). A reduced ASp vs. an increasedCSp resulting in ASp/CSp ratiob1 was indicative of upper airwaysdysfunction impairing the chest respiratory movement to produceadequate airflow. To minimize stress, the mice were habituated tothe plethysmograph chamber before the recording sessions.
Biochemical analysis of blood parameters
Values of pH, pO2 and pCO2 levels (mm of Hg) were measured invenous mixed blood samples of WT and Tau-P301L mice at age4 months as previously reported (Menuet et al., 2011a). After tailblood vessels dilatation with hot water (40–42 °C) and small incisionat the tip of the tail, 100 μL blood samples were collected in plasticcapillaries with electrolyte-balanced heparin (Radiometer, 70 IU hep-arin per mL) and analyzed with ABL 80 Flex analyzer (Radiometer,France). Measurements were performed in different mice eitherprior to (control) or after 4 hours isoflurane anesthesia or at 7 dayspost-isoflurane.
AT8 immunohistochemistry
After mouse anesthesia (Nembutal; 120 mg/kg, i.p.) and transcar-diac perfusion (ice-cold saline, 2 ml/min, 2 min), brainstems were re-moved, fixed (4% paraformaldehyde), and stored (0.1% sodium azidein PBS at 4 °C) until sectioning (40 μm coronal vibratome sections).Briefly, the following procedures were performed (Dutschmann etal., 2010; Menuet et al., 2011a; Terwel et al., 2005, 2008): rinsing(PBS; 15 min with 1.5% H2O2 in 50% methanol/PBS), blockade ofnon-specific binding sites (10% fetal calf serum, 0.1% Triton X-100 inPBS), incubation with primary monoclonal antibodies AT8 (mouseanti-AT8, 1/2500) and AT100 (mouse anti-AT100, 1/1400), 1 hour in-cubation with the secondary goat antimouse IgG antiserum coupledto PAP (1:500 in blocking buffer; Dako), 5 min incubation in 50 mMTris HCl, pH 7.6, enzymatic staining with 3,3′-diaminobenzidine(0.5 mg/ml), 0.3%H2O2 in 50 mM Tris HCl, pH 7.6. Sections werecounterstained with hematoxylin, ethanol dehydrated, delipidatedin xylol and mounted for microscopic analysis of AT8 immunoreactiv-ity. AT8 is a monoclonal antibody specifically directed against phos-phorylated human protein tau at epitopes pS198/pS202pS/pS205(Innogenetics, Gent, Belgium), possibly on the border between phys-iology and pathology (Dutschmann et al., 2010; Menuet et al., 2011a,2011b; Terwel et al., 2005, 2008). AT8 immunoreactivity is the classi-cal method to “stage” tauopathy in human brain (Braak and Braak,1994).
Histological analysis
For a given mouse, about 140–150 consecutive coronal sectionswere cut (from about 0.5 mm caudal to the pyramidal decussation toabout 5 mm more rostral) and one section every three was stainedwith AT8. As previously in terminal Tau.P301L mice (Menuet et al.,2011a), we either counted AT8 positively stained (AT8+) neurons ormeasured the AT8 optical density to quantify AT8 staining in differentstructures defined from neuroanatomical points in mouse atlas
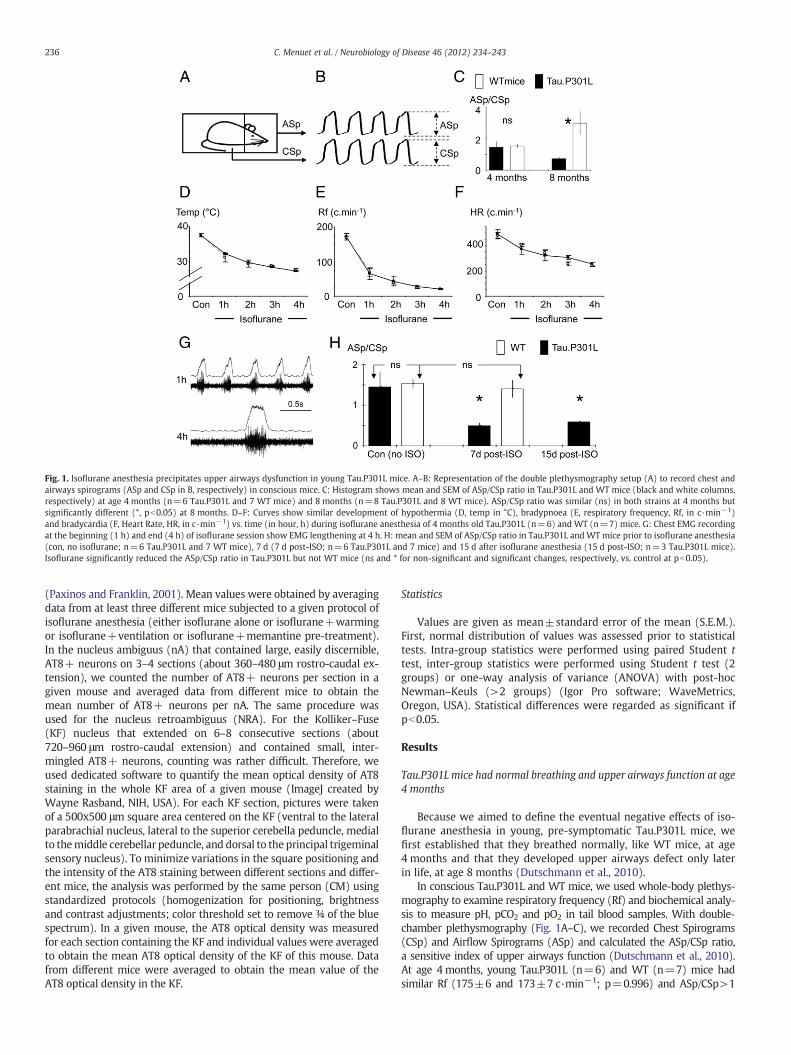
Fig. 1. Isoflurane anesthesia precipitates upper airways dysfunction in young Tau.P301L mice. A–B: Representation of the double plethysmography setup (A) to record chest andairways spirograms (ASp and CSp in B, respectively) in conscious mice. C: Histogram shows mean and SEM of ASp/CSp ratio in Tau.P301L and WT mice (black and white columns,respectively) at age 4 months (n=6 Tau.P301L and 7 WT mice) and 8 months (n=8 Tau.P301L and 8 WT mice). ASp/CSp ratio was similar (ns) in both strains at 4 months butsignificantly different (*, pb0.05) at 8 months. D–F: Curves show similar development of hypothermia (D, temp in °C), bradypnoea (E, respiratory frequency, Rf, in c·min−1)and bradycardia (F, Heart Rate, HR, in c·min−1) vs. time (in hour, h) during isoflurane anesthesia of 4 months old Tau.P301L (n=6) and WT (n=7) mice. G: Chest EMG recordingat the beginning (1 h) and end (4 h) of isoflurane session show EMG lengthening at 4 h. H: mean and SEM of ASp/CSp ratio in Tau.P301L andWTmice prior to isoflurane anesthesia(con, no isoflurane; n=6 Tau.P301L and 7 WT mice), 7 d (7 d post-ISO; n=6 Tau.P301L and 7 mice) and 15 d after isoflurane anesthesia (15 d post-ISO; n=3 Tau.P301L mice).Isoflurane significantly reduced the ASp/CSp ratio in Tau.P301L but not WT mice (ns and * for non-significant and significant changes, respectively, vs. control at pb0.05).
236 C. Menuet et al. / Neurobiology of Disease 46 (2012) 234–243
(Paxinos and Franklin, 2001). Mean values were obtained by averagingdata from at least three different mice subjected to a given protocol ofisoflurane anesthesia (either isoflurane alone or isoflurane+warmingor isoflurane+ventilation or isoflurane+memantine pre-treatment).In the nucleus ambiguus (nA) that contained large, easily discernible,AT8+ neurons on 3–4 sections (about 360–480 μm rostro-caudal ex-tension), we counted the number of AT8+ neurons per section in agiven mouse and averaged data from different mice to obtain themean number of AT8+ neurons per nA. The same procedure wasused for the nucleus retroambiguus (NRA). For the Kolliker–Fuse(KF) nucleus that extended on 6–8 consecutive sections (about720–960 μm rostro-caudal extension) and contained small, inter-mingled AT8+ neurons, counting was rather difficult. Therefore, weused dedicated software to quantify the mean optical density of AT8staining in the whole KF area of a given mouse (ImageJ created byWayne Rasband, NIH, USA). For each KF section, pictures were takenof a 500x500 μm square area centered on the KF (ventral to the lateralparabrachial nucleus, lateral to the superior cerebella peduncle, medialto themiddle cerebellar peduncle, and dorsal to the principal trigeminalsensory nucleus). To minimize variations in the square positioning andthe intensity of the AT8 staining between different sections and differ-ent mice, the analysis was performed by the same person (CM) usingstandardized protocols (homogenization for positioning, brightnessand contrast adjustments; color threshold set to remove ¾ of the bluespectrum). In a given mouse, the AT8 optical density was measuredfor each section containing the KF and individual values were averagedto obtain the mean AT8 optical density of the KF of this mouse. Datafrom different mice were averaged to obtain the mean value of theAT8 optical density in the KF.
Statistics
Values are given as mean±standard error of the mean (S.E.M.).First, normal distribution of values was assessed prior to statisticaltests. Intra-group statistics were performed using paired Student ttest, inter-group statistics were performed using Student t test (2groups) or one-way analysis of variance (ANOVA) with post-hocNewman–Keuls (>2 groups) (Igor Pro software; WaveMetrics,Oregon, USA). Statistical differences were regarded as significant ifpb0.05.
Results
Tau.P301L mice had normal breathing and upper airways function at age4 months
Because we aimed to define the eventual negative effects of iso-flurane anesthesia in young, pre-symptomatic Tau.P301L mice, wefirst established that they breathed normally, like WT mice, at age4 months and that they developed upper airways defect only laterin life, at age 8 months (Dutschmann et al., 2010).
In conscious Tau.P301L and WT mice, we used whole-body plethys-mography to examine respiratory frequency (Rf) and biochemical analy-sis to measure pH, pCO2 and pO2 in tail blood samples. With double-chamber plethysmography (Fig. 1A–C), we recorded Chest Spirograms(CSp) and Airflow Spirograms (ASp) and calculated the ASp/CSp ratio,a sensitive index of upper airways function (Dutschmann et al., 2010).At age 4 months, young Tau.P301L (n=6) and WT (n=7) mice hadsimilar Rf (175±6 and 173±7 c·min−1; p=0.996) and ASp/CSp>1

237C. Menuet et al. / Neurobiology of Disease 46 (2012) 234–243
(1.45±0.37 and 1.53±0.11; p=0.425). Analysis of tail blood samplesrevealed similar pH, pCO2 and pO2 values (Table 1). At age 8 months(Fig. 1C), old Tau.P301L (n=8) and WT (n=8) mice still had similarRf (151±5 and 157±4 c·min−1; p=0.212) but ASp/CSp ratio was sig-nificantly reduced in old Tau.P301L compared to old WT mice (0.74±0.09 and 2.88±0.74; p=0.006).
We conclude that the brainstem respiratory network of Tau.P301Lmice produces normal Rf and motor drives to chest and upper air-ways muscles at age 4 months, and normal Rf but altered motordrives at age 8 months, consistent with upper airways defect(Dutschmann et al., 2010).
Isoflurane anesthesia of young Tau.P301L mice precipitated upper air-ways defect
Having demonstrated that young Tau.P301Lmice had normal upperairways function, we went on to analyze the effects of isoflurane anes-thesia at age 4 months. Young Tau.P301L and WT mice were anesthe-tized for 4 h by exposure to isoflurane (1.3% in 30% O2). Isofluraneanesthesia progressively lowered body temperature, heart rate andRf, resulting in similar hypothermia, bradycardia and bradypnoea inTau.P301L and WT mice (Fig. 1D–F). In both genotypes, bradypnoeawas severe at the 4th hour of isoflurane (20±1 and 21±1 c·min−1
for Tau.P301L and WT mice, respectively; p=0.262) and developedwith an abnormal pattern of discharge of chest inspiratory muscles(chest EMG). Initially during anesthesia, chest EMG showed a classicalramp-pattern with a progressive increase in amplitude during inspira-tion until maximum, and a rapid decline at the inspiratory off-switch(Fig. 1G, upper traces). After 4 h of anesthesia, the chest EMG normallyincreased at the beginning of inspiration to reach the maximum, butremained at the plateau level for about 100 ms (Fig. 1G, lower traces),leading to a significant lengthening of the duration of the chest EMG.Consistent with a previous report in WT mice (Saab et al., 2010),blood gas analysis revealed that isoflurane exposure did not induce sig-nificant hypoxia, hypercapnia and acidosis in Tau.P301L and WT mice(Table 1).
All mice recovered well from the 4 h isoflurane session. When ana-lyzed again 7 d later, they showed similar Rf (177±8 and 183±9 c·min−1; p=0.444), pH, pO2 and pCO2 (Table 1). Unexpectedly,double-chamber plethysmography revealed upper airways defect inthe young Tau.P301L mice, which was never observed in WT mice.Seven days post-isoflurane, Tau.P301L mice produced enlarged chestrespiratory movements (large CSp) but weak airflow (weak ASp),which resulted in ASp/CSpb1 (Fig. 1H). The ASp/CSp of isoflurane-exposed Tau.P301L mice was significantly reduced (0.50±0.06;n=6) compared to either isoflurane-exposed WT mice (1.40±0.21;
Table 1Blood parameters of Tau.P301L and WT mice prior to, during and after isoflurane anesthesi(WT) mice prior to, during (4th h) and 7 days (7 d) after isoflurane anesthesia (n, number obetween Tau.P301L vs. WT mice (Tau vs. WT), Tau.P301L or WT mice prior to isoflurane anedays vs. 4 h after isoflurane anesthesia (7 d vs. 4th h), Tau.P301L or WT mice 7 days after v
Conditions Strain pH
Prior to isofluraneanesthesia
WT (n=8) 7.1Tau vs. WT nsTau (n=8) 7.2
4th h of isofluraneanesthesia
WT (n=4) 7.2Tau vs. WT nsTau (n=4) 7.2WT (prior to vs. 4th h) nsTau (prior to vs. 4th h) ns
7 d post isofluraneanesthesia
WT (n=4) 7.2Tau vs. WT nsTau (n=4) 7.2WT (7 d vs. 4th h) nsTau (7 d vs. 4th h) nsWT (7 d vs. prior to) nsTau (7 d vs. prior to) ns
n=7; p=0.011) or age-matched Tau.P301L mice never subjected toisoflurane (1.45±0.37; n=6; p=0.014). The ASp/CSp of isoflurane-exposed Tau.P301L mice at 4 months was similar to that of symptom-atic Tau.P301L mice at 8 months (0.74±0.09; n=8; p=0.042). Thus,subjecting young Tau.P301L mice to isoflurane anesthesia precipitatedthe upper airways defect they normally developed 4 months later.
We went on to define whether the upper airways defect of youngTau.P301L mice post-isoflurane was either permanent or only tran-sient. Tau.P301L mice were subjected to the identical isoflurane ses-sion, but were analyzed at 15 d post-isoflurane. Their ASp/CSpcompared to that of control mice was still significantly reduced(0.59±0.09; n=3; p=0.013) (Fig. 1H).
We concluded that young Tau.P301L mice exhibited a severeupper airways defect after 4 h isoflurane and that this functional def-icit lasted for a prolonged period following termination of anesthesia.
Isoflurane anesthesia protocols and onset of upper airways defect
We therefore went on to examine the eventual contributions ofhypothermia, bradypnoea and bradycardia in the precocious onsetof upper airways defect. We defined first whether counteracting thehypothermia accompanying isoflurane anesthesia counteracted theonset of upper airways defect. We used the same 4 h isoflurane pro-tocol as before, but the body temperature of the young mice was con-trolled by external heating.
After 4 h isoflurane anesthesia with controlled body-temperature,Tau.P301L mice had normal temperature, less severe bradycardia andbradypnoea (Fig. 2A–C, Warm) and normal pattern of chest EMG(Fig. 2D1). All Tau.P301L mice recovered well from the isoflurane ses-sion with controlled body-temperature. At 7 d post-isoflurane(Fig. 2E, ISO+Warm), their ASp/CSp compared to control was notsignificantly reduced (1.11±0.15; n=6; p=0.451).
We conclude that maintaining normothermia during isofluraneanesthesia of Tau.P301L mice prevented the abnormal breathing pat-tern during anesthesia and alleviated the upper airways sequel subse-quent to the isoflurane.
We went on to examine whether the abnormal functioning of therespiratory network during isoflurane contributed to the upper air-ways defect. To prevent the abnormal functioning of the respiratorynetwork under isoflurane, we subjected Tau.P301L mice to isofluraneby forced artificial ventilation, with mice breathing the same O2
enriched air mixture as before. The ventilator rhythm was set at theRf of warmed/ISO-exposed mice (100 cycle per min, c·min−1) andthe volume adjusted to 0.4 ml to silent the respiratory network andabolish chest EMG (Fig. 2D2). After 4 h isoflurane, the Tau.P301Lmice were hypothermic and bradycardic (Fig. 2A–C, Vent). All mice
a. Values are mean and SEM of blood parameters of young Tau.P301L (Tau) and FVB/Nf studied mice). Italicized lines show no significant (ns) differences of blood parameterssthesia vs. after 4 h of isoflurane anesthesia (prior to vs. 4th h), Tau.P301L or WT mice 7s. prior to isoflurane anesthesia (7 d vs. prior to).
pCO2 (mm Hg) pO2 (mm Hg)
8±0.02 60.37±4.46 44.88±6.22ns ns
0±0.02 60.63±5.30 51.50±3.548±0.03 44.50±3.48 43.75±4.59
ns ns4±0.02 43.75±4.25 42.50±1.49
ns nsns ns
0±0.04 62.67±2.19 46.33±3.71ns ns
2±0.01 55.00±2.48 58.25±7.06ns nsns nsns nsns ns
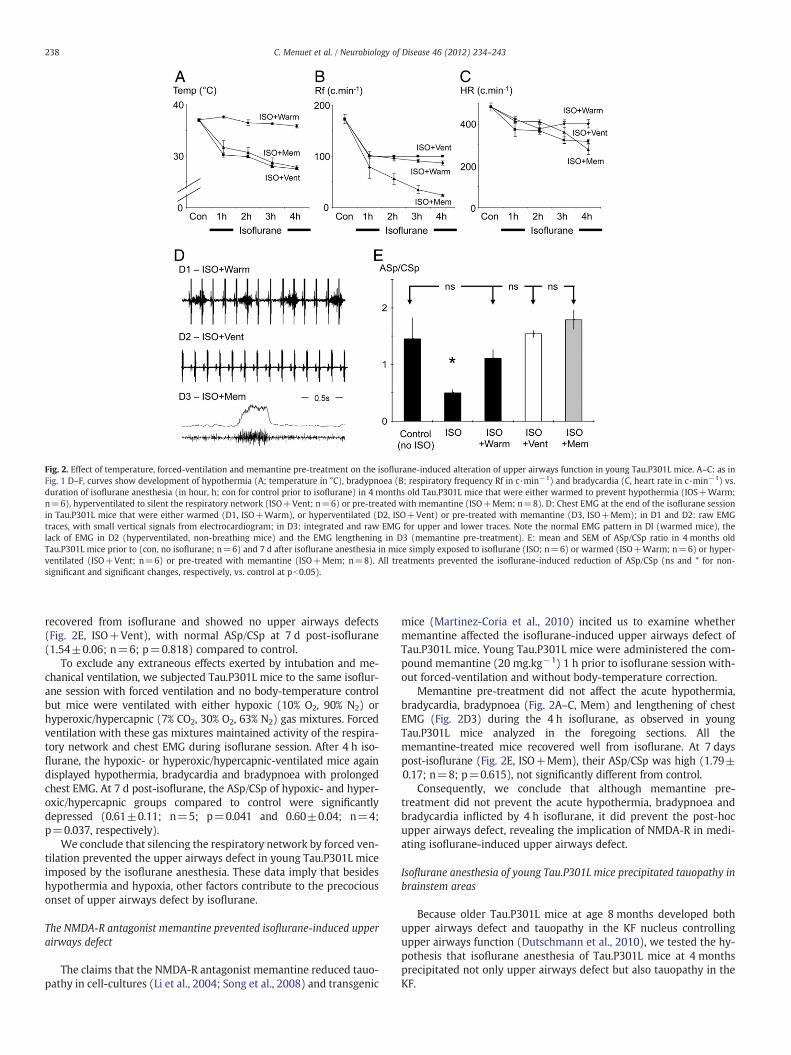
Fig. 2. Effect of temperature, forced-ventilation and memantine pre-treatment on the isoflurane-induced alteration of upper airways function in young Tau.P301L mice. A–C: as inFig. 1 D–F, curves show development of hypothermia (A; temperature in °C), bradypnoea (B; respiratory frequency Rf in c·min−1) and bradycardia (C, heart rate in c·min−1) vs.duration of isoflurane anesthesia (in hour, h; con for control prior to isoflurane) in 4 months old Tau.P301L mice that were either warmed to prevent hypothermia (IOS+Warm;n=6), hyperventilated to silent the respiratory network (ISO+Vent; n=6) or pre-treated with memantine (ISO+Mem; n=8). D: Chest EMG at the end of the isoflurane sessionin Tau.P301L mice that were either warmed (D1, ISO+Warm), or hyperventilated (D2, ISO+Vent) or pre-treated with memantine (D3, ISO+Mem); in D1 and D2: raw EMGtraces, with small vertical signals from electrocardiogram; in D3: integrated and raw EMG for upper and lower traces. Note the normal EMG pattern in Dl (warmed mice), thelack of EMG in D2 (hyperventilated, non-breathing mice) and the EMG lengthening in D3 (memantine pre-treatment). E: mean and SEM of ASp/CSp ratio in 4 months oldTau.P301L mice prior to (con, no isoflurane; n=6) and 7 d after isoflurane anesthesia in mice simply exposed to isoflurane (ISO; n=6) or warmed (ISO+Warm; n=6) or hyper-ventilated (ISO+Vent; n=6) or pre-treated with memantine (ISO+Mem; n=8). All treatments prevented the isoflurane-induced reduction of ASp/CSp (ns and * for non-significant and significant changes, respectively, vs. control at pb0.05).
238 C. Menuet et al. / Neurobiology of Disease 46 (2012) 234–243
recovered from isoflurane and showed no upper airways defects(Fig. 2E, ISO+Vent), with normal ASp/CSp at 7 d post-isoflurane(1.54±0.06; n=6; p=0.818) compared to control.
To exclude any extraneous effects exerted by intubation and me-chanical ventilation, we subjected Tau.P301L mice to the same isoflur-ane session with forced ventilation and no body-temperature controlbut mice were ventilated with either hypoxic (10% O2, 90% N2) orhyperoxic/hypercapnic (7% CO2, 30% O2, 63% N2) gas mixtures. Forcedventilation with these gas mixtures maintained activity of the respira-tory network and chest EMG during isoflurane session. After 4 h iso-flurane, the hypoxic- or hyperoxic/hypercapnic-ventilated mice againdisplayed hypothermia, bradycardia and bradypnoea with prolongedchest EMG. At 7 d post-isoflurane, the ASp/CSp of hypoxic- and hyper-oxic/hypercapnic groups compared to control were significantlydepressed (0.61±0.11; n=5; p=0.041 and 0.60±0.04; n=4;p=0.037, respectively).
We conclude that silencing the respiratory network by forced ven-tilation prevented the upper airways defect in young Tau.P301L miceimposed by the isoflurane anesthesia. These data imply that besideshypothermia and hypoxia, other factors contribute to the precociousonset of upper airways defect by isoflurane.
The NMDA-R antagonist memantine prevented isoflurane-induced upperairways defect
The claims that the NMDA-R antagonist memantine reduced tauo-pathy in cell-cultures (Li et al., 2004; Song et al., 2008) and transgenic
mice (Martinez-Coria et al., 2010) incited us to examine whethermemantine affected the isoflurane-induced upper airways defect ofTau.P301L mice. Young Tau.P301L mice were administered the com-pound memantine (20 mg.kg−1) 1 h prior to isoflurane session with-out forced-ventilation and without body-temperature correction.
Memantine pre-treatment did not affect the acute hypothermia,bradycardia, bradypnoea (Fig. 2A–C, Mem) and lengthening of chestEMG (Fig. 2D3) during the 4 h isoflurane, as observed in youngTau.P301L mice analyzed in the foregoing sections. All thememantine-treated mice recovered well from isoflurane. At 7 dayspost-isoflurane (Fig. 2E, ISO+Mem), their ASp/CSp was high (1.79±0.17; n=8; p=0.615), not significantly different from control.
Consequently, we conclude that although memantine pre-treatment did not prevent the acute hypothermia, bradypnoea andbradycardia inflicted by 4 h isoflurane, it did prevent the post-hocupper airways defect, revealing the implication of NMDA-R in medi-ating isoflurane-induced upper airways defect.
Isoflurane anesthesia of young Tau.P301L mice precipitated tauopathy inbrainstem areas
Because older Tau.P301L mice at age 8 months developed bothupper airways defect and tauopathy in the KF nucleus controllingupper airways function (Dutschmann et al., 2010), we tested the hy-pothesis that isoflurane anesthesia of Tau.P301L mice at 4 monthsprecipitated not only upper airways defect but also tauopathy in theKF.

Fig. 3. AT8 expression in brainstem of Tau.P301L mice 7 d post-isoflurane. Successive40 μm coronal sections stained with AT8 in a given Tau.P301L mice sacrificed 7 dafter isoflurane anesthesia; right-hand pictures are enlargements of the doted blackbox drawn in the corresponding left-hand pictures; arrows indicate some AT8+ neu-rons. A: caudal medulla; AT8+ neurons were present in the reticular formation (ar-rows), external cuneatus (Cun) and hypoglossal motor nucleus (n12; enlargement inA1) but almost absent in the nucleus tractus solitarius (nTS); A2: n12 at a more caudallevel. B: rostral medulla, note the high density of AT8+ neurons in the nucleus ambi-guus (nA) (enlargement in B1), raphe obscurus (Rob; enlargement in B2), prepositus(pp) and catecholaminergic C2 group (c2). C: pontine level; the KF (enlargement inC1), A6 (A6; enlargement in C2) and trigeminal motor nucleus (Mo5; enlargement inD1) contained high density of AT8+ neurons; note the KF location between the supe-rior cerebellar peduncle (scp) and the principal sensory trigeminal nucleus (Pr5). D:midbrain; the periaqueductal gray (PAG) contained weakly stained AT8+ neurons inthe ventrolateral part (enlargement in D2). Calibration bars: 400 and 200 μm for left-and right-hand pictures, respectively; 100 μm for the KF enlargement in C1.
239C. Menuet et al. / Neurobiology of Disease 46 (2012) 234–243
First in 4 month old Tau.P301L mice that were not subjected toisoflurane (n=3), we performed a detailed immunohistochemicalanalysis of the expression of human mutant protein Tau.P301L phos-phorylated at epitope AT8, the classical method to “stage” tauopathyin human brain (Braak and Braak, 1994). Consistent with our previ-ous report (Dutschmann et al., 2010), we found a diffuse, weak AT8staining without clear tauopathy in the brainstem and midbrain ofyoung Tau.P301L mice.
Conversely, in isoflurane-exposed young Tau.P301L mice (nowarming, no forced ventilation) sacrificed 7 d post-hoc (n=3),marked AT8 signal and tauopathy were evident in several brainstemareas, frequently with a typical pattern of stained somata and pro-cesses similar to the tauopathy previously reported in symptomaticTau.P301L mice at age 8 months (Dutschmann et al., 2010). TheTau.P301L mice post-isoflurane already showed frequent AT8 posi-tively stained (AT8+) neurons at age 4 months. Well stained AT8+neurons were scattered in the reticular formation (arrows inFig. 3A) but their density was high in the KF (Fig. 3C, C1), rapheobscurus (Rob; Fig. 3B, B2), locus coeruleus (A6; Fig. 3C, C2), peria-queductal gray (PAG; Fig. 3D, D2) and nucleus retroambiguus (NRA;Fig. 4C). As previously reported in old and terminal Tau.P301L mice(Dutschmann et al., 2010; Menuet et al., 2011a, 2011b), the nucleustractus solitarius (nTS) and the respiratory-related areas of the ven-tral medulla of post-isoflurane Tau.P301L mice did not show frequentAT8+ neurons, which was consistent with their normal Rf, pH, pCO2and pO2. However, highly frequent, well stained AT8+ neurons werefound in all cranial motor nuclei, including the hypoglossal (n12;Fig. 3A, A1, A2), facial, trigeminal (Mo5; Fig. 3C, D1) and ambiguus(nA; Figs. 3B, B1) nuclei, an AT8 expression profile not observed pre-viously in old Tau.P301L mice (Dutschmann et al., 2010; Menuet etal., 2011a). Because the upper airways muscles are innervated by nAmotor neurons and because nA motor neurons are controlled byNRA, KF and PAG interneurons, the tauopathy in the nA, NRA, KFand PAG network of post-isoflurane Tau.P301L mice correlated wellwith the precocious onset of upper airways defects.
In the three groups of Tau.P301L mice that retained normal upperairways function at 7 d post-isoflurane, AT8 expression was found re-duced in the nA (Fig. 4A) and the KF (Fig. 4B) but not in other brain-stem and midbrain areas. Frequent, well-stained AT8+ neurons wereobserved in the NRA (Fig. 4C), cranial motor nuclei (Fig. 5A–B) andPAG (not shown) of air-ventilated Tau.P301L mice (n=3; Fig. 5A),memantine pre-treated Tau.P301L mice (n=3; Fig. 5B) andtemperature-controlled Tau.P301L mice (n=3; data not shown). Toascertain the reduction of tauopathy in the nA and KF, we countedthe number of AT8+ neurons per section in the nA that containedlarge, tightly packed and easily discernible motor neurons (Fig. 4A)and measured the optical density of the AT8 signal in the KF that con-tained small, intermingled interneurons in a rather extended area(Fig. 4B). In the nA, the number of AT8+ neurons was highly signifi-cantly larger in Tau.P301L mice with upper airway defects post-isoflurane (8.3±2.3 neurons per section) than in air-ventilatedTau.P301L mice during isoflurane anesthesia (0.4±0.4; pb0.001) ormemantine pre-treated Tau.P301L mice (0.4±0.3; pb0.001). In addi-tion, the optical density of the AT8 signal in the KF was significantlylarger in Tau.P301L mice with upper airways defects post-isoflurane(21±1) than in air-ventilated (16±2; p=0.035) and memantine-treated Tau.P301L mice (7±2; pb0.001). No reduction of AT8 immu-noreactivity was observed in other areas with tauopathy, includingthe NRA (Fig. 4C), the hypoglossal motor nucleus (Fig. 5A1, B1), theraphe obscurus (Fig. 5A2, B2), the locus coeruleus (Fig. 5A3, B3), themotor trigeminal nucleus (Fig. 5A4, B4) and the PAG (data notshown). Because the NRA pre-motor neurons project to and controlthe KF interneurons and the nA motor neurons (Subramanian et al.,2008), we further counted AT8+ neurons in the NRA of Tau.P301Lmice with and without upper airways defect post-isoflurane(Fig. 4C). In the NRA, the number of AT8+ neurons was found slightly
but not significantly larger in mice with upper airway defects post-isoflurane (8.1±1.7 neurons per section) than in air-ventilated (5.9±0.9 neurons per section; p=0.08) or memantine pre-treated mice(6.0±1.0 neurons per section; p=0.09).

Fig. 4. Effects of forced-ventilation and memantine pre-treatment on AT8 expression in the brainstem of Tau.P301L mice 7 d post-isoflurane. A: AT8+ neurons in the nA ofTau.P301L mice 7 d post-isoflurane without other treatments (ISO) or with forced-ventilation (ISO+Vent) or with memantine pre-treatment (ISO+Mem). Histogram showsthe mean number (and SEM) of AT8+ neurons in the nA of ISO (n=3), ISO+Vent (n=3) and ISO+Mem (n=3) mice; ventilation or memantine pre-treatment almost zeroedthe number of AT8+ neurons in the nA (*, pb0.05). B: as in A, but for the KF of Tau.P301L mice 7 d post-isoflurane without other treatments (ISO) or with forced-ventilation (ISO+Vent) or with memantine pre-treatment (ISO+Mem). Histogram shows the mean value (and SEM) of AT8+ optical density in the KF of ISO (n=3), ISO+Vent (n=3) and ISO+Mem (n=3) mice; ventilation or memantine pre-treatment significantly reduced (*, pb0.05), but did not abolish the AT8 optical density in the KF. C: as in A, but for the nucleusretroambiguus NRA; ventilation or memantine pre-treatment did not significantly reduce (ns, p>0.05) the number of AT8+ neurons. Calibration bars: 100 μm.
240 C. Menuet et al. / Neurobiology of Disease 46 (2012) 234–243
We conclude first that our combined data from plethysmographyand AT8 immunohistochemistry support a deleterious effect of iso-flurane anesthesia on the onset of the disease in the Tau.P301Lmouse model of tauopathy, second that upper airways defect inTau.P301L mice is closely linked to nA and KF tauopathy, and thirdthat NMDA-R contribute to the deleterious effects of isoflurane.
Discussion
In the Tau.P301L mouse model of tauopathy that develops brain-stem tauopathy and upper airways defect from 8 months onwards(Dutschmann et al., 2010; Menuet et al., 2011a, 2011b), we showthat isoflurane anesthesia at 4 months precociously onsets brainstemtauopathy and upper airways defect. These results demonstrate a linkbetween isoflurane anesthesia, tauopathy and functional defect, iden-tify implication of NMDA-R in the isoflurane adverse effects and sug-gest safer protocols for anesthesia of patients at risks for AD.
Pathological consequences of isoflurane-induced upperairways dysfunction
AD, the most prevalent neurodegenerative disorder in elderly people,is characterized by memory, cognition and behavioral defects. AD is alsoaccompaniedwith breathing, swallowing and language disorders, consis-tently with upper airways dysfunction. At 4 months, young Tau.P301Lmice have normal upper airways function and no brainstem tauopathybut develop upper airways defect after isoflurane anesthesia with tauo-pathy in the KF, NRA and monoaminergic areas, resembling those previ-ously reported in older Tau.P301L mice (Dutschmann et al., 2010;Menuet et al., 2011a). Post-isoflurane, young Tau.P301L mice retain
normal Rf, consistently with the lack of tauopathy in the ventro-lateralmedulla areas that contribute to respiratory rhythm generation, and inthe nTS areas that integrate peripheral regulatory inputs. Conversely toolder Tau.P301L mice, young Tau.P301L mice post-isoflurane show tauo-pathy in all cranial motor nuclei, including the nA. The nAmotor neuronsinnervate upper airways muscles, are driven by inputs from the respira-tory network, the medullary NRA pre-motor neurons, the pontine KF in-terneurons, and further upstream and indirectly, the PAG that controlsthe KF and NRA (Subramanian et al., 2008). As a whole, the nA, NRA,KF and PAG constitute a multifunctional network contributing to respira-tion (Dutschmann and Herbert, 2006), swallowing (Bonis et al., 2011;Gestreau et al., 2005), and vocalization (Subramanian et al., 2008).Thus, the precocious onset of tauopathy within this network correlateswith upper airways defect of Tau.P301L mice and possibly with oro-pharyngeal symptoms of AD. Young Tau.P301L mice post-isofluranemust enhance their chest respiratory movements to compensate airflowreduction caused by paradoxical upper airways narrowing during inspi-ration as demonstrated in old Tau.P301L mice (Dutschmann et al.,2011; Menuet et al., 2011a). This breathing strategy efficiently securestheir pH, pCO2 and pO2 but requires an increased respiratory work-load, which may contribute to their weight loss with aging (Terwel etal., 2005), a predictor of mortality in AD (Hansen et al., 2011). Post-isoflurane, upper airways defect possibly alters swallowing, which mayincrease aspiration risk, a recognized cause of death in AD (Humbert etal., 2010; Suh et al., 2009; Wada et al., 2001). In addition, tauopathy inthe hypoglossal, facial and trigeminal motor neurons highly likely altersmastication, swallowing and food intake (Gestreau et al., 2005). Upperairways defect may facilitate sleep obstructive apneas, which altersbrain oxygenation during sleep, a process possibly contributing to ADevolution (Cooke et al., 2009; Daulatzai, 2010). Finally, upper airways

Fig. 5. Effect of forced-ventilation or memantine pre-treatment on AT8 expression inbrainstem of Tau.P301L mice 7 d post-isoflurane. Coronal brainstem sections (thick-ness 40 μm) stained with AT8 in a given Tau.P301L mice (age 4 months), sacrificed7 d after either isoflurane anesthesia with forced ventilation (A) or isoflurane anes-thesia with memantine pre-treatment (B). Neither forced ventilation nor memantinepre-treatment prevented the isoflurane-induction of frequent AT8+ neurons in thehypoglossal nucleus (A1, A2), raphe obscurus (B1, B2), locus coeruleus (C1, C2) andtrigeminal motor nucleus (D1, D2). Calibration bar: 200 μm.
241C. Menuet et al. / Neurobiology of Disease 46 (2012) 234–243
defect may also affect vocalization in old Tau.P301L mice (Menuet et al.,2011b), which may be reminiscent of language disorders of AD (Rohrerand Schott, 2011). Indeed, isoflurane anesthesia by precipitating tauopa-thy in the network controlling upper airways function may precociouslyinduce distinct deleterious effects in Tau.P301L mice, and possibly inAlzheimer's patients (Baranov et al., 2009).
NMDA mechanisms and isoflurane-induced tauopathy
AD is characterized by extraneuronal plaques and tauopathy. Inyoung Tau.P301L mice, the mechanisms through which isoflurane in-duces tauopathy are poorly understood. First, hypothermia coupledwith isoflurane has been reported to induce tauopathy (Planel et al.,2009). Indeed, we show that preventing hypothermia in anesthetizedTau.P301L mice prevents upper airways defect. However, hyperventi-lation of anesthetized hypothermic Tau.P301L mice also preventsupper airways defect, revealing that hypothermia is not the single,main factor. Hypoxia during anesthesia has been proposed to be cru-cial too (Pan et al., 2011). We show that imposing hypoxia during an-esthesia of forced-ventilated Tau.P301L mice induces upper airways
defect. However, spontaneously breathing Tau.P301L mice were nothypoxic at the end of the isoflurane session, consistent with a previ-ous report in WT mice subjected to isoflurane (Saab et al., 2010). Inaddition, preventing hypoxia in anesthetized Tau.P301L mice byforced-ventilation with hyperoxic/hypercapnic mixture did not pre-vent the onset of upper airways defect. Then, neither hypothermianor hypoxia coupled with isoflurane is the single factor for isofluraneadverse effects. On the other hand, we show that pre-treatment withthe NMDA-R antagonist memantine efficiently alleviates tauopathy inthe nA and KF nuclei and prevents upper airways defect in Tau.P301Lmice, which fully supports implication of NMDA-R.
The nA motor neurons and their KF drivers both express NMDA-R,with an especially high density in the KF where most neurons expressNMDA-R (Guthmann and Herbert, 1999; Yamazaki et al., 2009). TheKF controls the off-switch of inspiration via NMDA-R and their block-ade prolongs inspiration (Fung et al., 1994; Harris and Milsom, 2003;Ling et al., 1994). Pentobarbitone anesthesia depresses or leavesunchanged the activity of most respiratory neurons, but specificallyactivates the KF neurons (Caille et al., 1979). It is worth to note thatnot only isoflurane but also pentobarbitone anesthesia induces ab-normal hyperphosphorylation of tau (Planel et al., 2007; Run et al.,2009). In KF neurons, c-fos expression is induced by anesthesia(Roda et al., 2004), hypothermia (Bhatnagar and Dallman, 1998),cardio-respiratory changes (Dutschmann and Herbert, 1998; Guo etal., 2002; Horiuchi et al., 1999; Teppema et al., 1997), with c-fos andNMDA-R co-expression (Dutschmann and Herbert, 1998;Dutschmann et al., 1998; Guthmann and Herbert, 1999). Thus, duringisoflurane session, anesthesia, hypothermia, bradypnoea and brady-cardia cumulatively activated the KF neurons. On the short term(4 h isoflurane), the massive activation of KF neurons induced exces-sive calcium influx, free radical formation (Chen and Lipton, 2006;Lipton, 2006), excitotoxicity and subsequent KF alteration, asassessed by delayed inspiratory off-switch and prolonged chestEMG in both Tau.P301L and WT mice. On the long term (7 d post-isoflurane), this had no sequels in WT mice but induced KF tauopathyand upper airways defect in Tau.P301L mice, that were both pre-vented by blockade of NMDA-R with memantine pre-treatment.Memantine is a partial NMDA-R antagonist that preferentially blocksexcessive NMDA-R activity (Lipton, 2007). Memantine did not pre-vent the acute hypothermia, bradycardia, bradypnoea induced bythe 4 h isoflurane but possibly reduced the response of KF neuronsto the cumulative actions of anesthesia, hypothermia and cardio-vascular changes, which therefore alleviated KF tauopathy and thesubsequent upper airways defect. Memantine reduces tau hyperpho-sphorylation in cell cultures via inhibition of the inhibitor-2 of proteinphosphatise-2A (Chohan et al., 2006; Li et al., 2004). Indeed, inhibi-tion of protein phosphatise-2A is implicated in tau hyperphosphory-lation after anesthesia in mice (Planel et al., 2007) andexcitotoxicity in neuron cultures (Rahman et al., 2009). Thus, a simi-lar disinhibitory process may also contribute to the memantine bene-ficial effect on the KF of Tau.P301L mice.
That memantine specifically alleviates tauopathy in the nA and KFbut not in other areas is possibly related to different expression pro-files of NMDA-R sub-types, density levels and cellular sites of expres-sion. Suppressing only one of the factors over-activating KF neuronsduring isoflurane (with warming or forced ventilation) significantlyreduces, but does not abolish, the KF tauopathy. Nevertheless, the re-duction of the KF tauopathy is sufficient to prevent upper airways de-fect, which suggests that KF tauopathy must pass beyond a thresholdlevel to induce functional pathology. In the nA, where motor neuronsalso express NMDA-R (Yamazaki et al., 2009), the tauopathy profile isfairly different from that observed in the KF, with a kind of “all ornone” response: tauopathy affects almost all nA motor neurons afterthe simplest isoflurane protocol (no warming, no forced-ventilationand no memantine) but almost none of the nA motor neurons afterisoflurane with forced-ventilation or isoflurane with memantine

242 C. Menuet et al. / Neurobiology of Disease 46 (2012) 234–243
pre-treatment. This may be explained by weak, non-detectable re-duction of isoflurane-induced tauopathy with forced-ventilation ormemantine pre-treatment in the network upstream the nA. Minor re-ductions of tauopathy in the NRA, PAG, raphe, and A6, coupled to themarked reduction of tauopathy in the KF may significantly contributeto protect the nA.
Translation of data obtained in Tau.P301L mice to human anesthesia
In Tau-P301L mice, anesthesia combined with hypothermia andspontaneous ventilation precipitated tauopathy and upper airwaysdefects. These deleterious effects were prevented by normothermiaor by hyperventilation, but not with hypercapnic or hypoxic mix-tures. Although the isoflurane effects may be exacerbated inTau.P301L mice because P301L tau is a more favorable substratethan normal tau for hyperphosphorylation by protein kinases(Alonso et al., 2004), our data obtained in a validated mouse modelfor tauopathy might have some significance for anesthesia in patientssuffering from primary tauopathy but also secondary tauopathies,such as AD.
Increasing evidence indicates that surgery with total anesthesiacan cause post-operative cognitive decline (Vanderweyde et al.,2010), while increasing CSF levels of tau biomarkers (Tang et al.,2011), and is suspected to precipitate AD onset and progression(Baranov et al., 2009). With respect to patient ventilation and tem-perature, peri-operative procedures vary. Spontaneous ventilation iscommon for minor surgery, with or without laryngeal airway mask(Froessler et al., 2010), but spontaneous ventilation is uncommonfor major surgery. Spontaneous ventilation is often associated withsome degree of hypercapnia, helping to maintain the respiratory net-work active and to shorten emergence time from anesthesia (Sakataet al., 2007). At variance with clinical practice until the '80s(Benumof, 1986), forced hyperventilation and hypocapnia are no lon-ger an accepted practice, except for limited periods of time to elimi-nate volatile anesthetics. Hypothermia is common during surgery,despite active re-warming of the patient by the anesthesiologist,while mild hypothermia is recommended following cardiac arrest,cardiopulmonary resuscitation and traumatic head injury (Arrich etal., 2010; Dietrich and Bramlett, 2010; Sinclair and Andrews, 2010;Tokutomi et al., 2009). Consequently, we believe that our data indi-cate that anesthesia combined with hypothermia, spontaneous venti-lation and mild hypercapnia may hasten tauopathy in patients thatare either at risk for AD, or present with probable AD.
Conclusions
Using young Tau.P301L mice as experimental model for examin-ing the onset and progression of network dysfunction during neuro-degenerative disease, we show normal upper airways function priorto, but not after isoflurane anesthesia. Indeed, anesthesia of youngTau.P301L mice precociously induced, via NMDA-R, tauopathy in thenetwork controlling upper airways and subsequent functional defect.The efficiency of memantine pre-treatment and distinct protocols ofanesthesia to alleviate KF tauopathy and prevent upper airways dys-function in Tau.P301L mice may offer new, promising issues forsafer anesthesia of Alzheimer's patients.
Acknowledgments
This work was supported by the Unité Mixte de Recherche 6231,the Fonds Wetenschappelijk Onderzoek-Vlaanderen (FWO-Vlaande-ren), Instituut voor Wetenschap en Techniek (IWT), KULeuven-Research Fund (BOF), KULeuven-Research&Development and EECFramework programs FP6 & FP7 and NHLBI. The authors thank LucQuintin, MD, PhD, for his invaluable comments and advices about
the translation of our data obtained in Tau.P301L mice to humananesthesia.
References
Alonso, Adel C., Mederlyova, A., Novak, M., Grundke-Iqbal, I., Iqbal, K., 2004. Promotionof hyperphosphorylation by frontotemporal dementia tau mutations. J. Biol. Chem.279, 34873–34881.
Arrich, J., Holzer, M., Herkner, H., Müllner, M., 2010. Cochrane corner: hypothermia forneuroprotection in adults after cardiopulmonary resuscitation. Anesth. Analg. 110,1239.
Baranov, D., Bickler, P.E., Crosby, G.J., Culley, D.J., Eckenhoff, M.F., Eckenhoff, R.G., et al.,2009. Consensus statement: First International Workshop on Anesthetics andAlzheimer's Disease. Anesth. Analg. 108, 1627–1630.
Benumof, J.L., 1986. Respiratory physiology and respiratory function during anes-thesia, In: Miller, R.D. (Ed.), 2nd ed. Anesthesia, vol. 2. Churchill, Livingstone,pp. 1115–1164.
Bhatnagar, S., Dallman, M., 1998. Neuroanatomical basis for facilitation of hypothalam-ic–pituitary–adrenal responses to a novel stressor after chronic stress. Neurosci-ence 84, 1025–1039.
Bilotta, F., Doronzio, A., Stazi, E., Titi, L., Fodale, V., Di Nino, G., et al., 2010. Postoperativecognitive dysfunction: toward the Alzheimer's Disease pathomechanism hypothesis.J. Alzheimers Dis. 22, 81–89.
Bonis, J.M., Neumueller, S.E., Marshall, B.D., Krause, K.L., Qian, B., Pan, L.G., et al., 2011.The effects of lesions in the dorsolateral pons on the coordination of swallowingand breathing in awake goats. Respir. Physiol. Neurobiol. 175, 272–282.
Braak, H., Braak, E., 1994. Morphological criteria for the recognition of Alzheimer'sDisease and the distribution pattern of cortical changes related to this disorder.Neurobiol. Aging 15, 355–356.
Brion, J.P., Flament-Durand, J., Dustin, P., 1986. Alzheimer's disease and tau proteins.Lancet 2, 1098.
Caille, D., Vibert, J.F., Bertrand, F., Gromysz, H., Hugelin, A., 1979. Pentobarbitone effectson respiration related units; selective depression of bulbopontine reticularneurones. Respir. Physiol. 36, 201–216.
Chen, H., Lipton, S.A., 2006. The chemical biology of clinically tolerated NMDA receptorantagonists. J. Neurochem. 97, 1611–1626.
Chohan, M.O., Khatoon, S., Iqbal, I.G., Iqbal, K., 2006. Involvement of I2PP2A in theabnormal hyperphosphorylation of tau and its reversal by memantine. FEBS Lett.580, 3973–3979.
Cooke, J.R., Ayalon, L., Palmer, B.W., Loredo, J.S., Corey-Bloom, J., Natarajan, L., et al.,2009. Sustained use of CPAP slows deterioration of cognition, sleep, and mood inpatients with Alzheimer's Disease and obstructive sleep apnea: a preliminarystudy. J. Clin. Sleep Med. 5, 305–309.
Daulatzai, M.A., 2010. Early stages of pathogenesis in memory impairment duringnormal senescence and Alzheimer's Disease. J. Alzheimers Dis. 20, 355–367.
Dietrich, W.D., Bramlett, H.M., 2010. The evidence for hypothermia as a neuroprotectantin traumatic brain injury. Neurotherapeutics 7, 43–50.
Dutschmann, M., Herbert, H., 1998. NMDA and GABAA receptors in the rat Kolliker–Fuse area control cardiorespiratory responses evoked by trigeminal ethmoidalnerve stimulation. J. Physiol. (Lond.) 510, 793–804.
Dutschmann, M., Herbert, H., 2006. The Kölliker–Fuse nucleus gates the postinspiratoryphase of the respiratory cycle to control inspiratory off-switch and upper airwayresistance in rat. Eur. J. Neurosci. 24, 1071–1084.
Dutschmann, M., Guthmann, A., Herbert, H., 1998. NMDA receptor subunit NR1-immunoreactivity in the rat pons and brainstem and colocalization with Fosinduced by nasal stimulation. Brain Res. 809, 221–230.
Dutschmann, M., Menuet, C., Stettner, G.M., Gestreau, C., Borghgraef, P., Devijver, H.,et al., 2010. Upper airway dysfunction of Tau-P301L mice correlates with tauopa-thy in midbrain and ponto-medullary brainstem nuclei. J. Neurosci. 30,1810–1821.
Froessler, B., Brommundt, J., Anton, J., Khanduja, R., Kuhlen, R., Rossaint, R., et al., 2010.Spontaneously breathing anesthetized patients with a laryngeal mask airway:positive end-expiratory pressure does not improve oxygen saturation. Anaesthesist59, 1003–1007.
Fung, M.L., Wang, W., St John, W.M., 1994. Involvement of pontile NMDA receptors ininspiratory termination in rat. Respir. Physiol. 96, 177–188.
Gestreau, C., Dutschmann, M., Obled, S., Bianchi, A.L., 2005. Activation of XII motoneuronsand premotor neurons during various oropharyngeal behaviors. Respir. Physiol.Neurobiol. 147, 159–176.
Glenner, G.G., Wong, C.W., 1984. Alzheimer's disease and Down's syndrome: sharing ofa unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun.122, 1131–1135.
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y.C., Zaidi, M.S., Wisniewski, H.M., 1986a.Microtubule-associated protein tau. A component of Alzheimer paired helicalfilaments. J. Biol. Chem. 261, 6084–6089.
Grundke-Iqbal, I., Iqbal, K., Tung, Y.C., Quinlan, M., Wisniewski, H.M., Binder, L.I.,1986b. Abnormal phosphorylation of the microtubule-associated protein tau(tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U. S. A. 83,4913–4917.
Guo, Z., Li, P., Longhurst, J.C., 2002. Central pathways in the pons andmidbrain involvedin cardiac sympathoexcitatory reflexes in cats. Neuroscience 113, 435–447.
Guthmann, A., Herbert, H., 1999. Expression of N-methyl-D-aspartate receptor subunitsin the rat parabrachial and Kölliker–Fuse nuclei and in selected pontomedullarybrainstem nuclei. J. Comp. Neurol. 415, 501–517.

243C. Menuet et al. / Neurobiology of Disease 46 (2012) 234–243
Hansen, M., Waldorff, F.B., Waldemar, G., 2011. Prognostic factors for weight loss over1-year period in patients recently diagnosed with mild Alzheimer disease. AlzheimerDis. Assoc. Disord. 25, 269–275 Available.
Harris, M.B., Milsom, W.K., 2003. Apneusis follows disruption of NMDA-type glutamatereceptors in vagotomized ground squirrels. Respir. Physiol. Neurobiol. 134, 191–207.
Horiuchi, J., Potts, P.D., Polson, J.W., Dampney, R.A., 1999. Distribution of neuronsprojecting to the rostral ventrolateral medullary pressor region that are activatedby sustained hypotension. Neuroscience 89, 1319–1329.
Horner, J., Alberts, M.J., Dawson, D.V., Cook, G.M., 1994. Swallowing in Alzheimerdisease. Alzheimer Dis. Assoc. Disord. 8, 177–189.
Humbert, I.A., McLaren, D.G., Kosmatka, K., Fitzgerald, M., Johnson, S., Porcaro, E., et al.,2010. Early deficits in cortical control of swallowing in Alzheimer disease. J. AlzheimersDis. 19, 1185–1197.
Kuna, S.T., Remmers, J.E., 1999. Premotor input to hypoglossal motoneurons fromKölliker–Fuse neurons in decerebrate cats. Respir. Physiol. 117, 85–95.
Li, L., Sengupta, A., Haque, N., Grundke-Iqbal, I., Iqbal, K., 2004. Memantine inhibits andreverses the Alzheimer type abnormal hyperphosphorylation of tau and associatedneurodegeneration. FEBS Lett. 566, 261–269.
Ling, L., Karius, D.R., Speck, D.F., 1994. Role of N-methyl-D-aspartate receptors in thepontine pneumotaxic mechanism in the cat. J. Appl. Physiol. 76, 1138–1143.
Lipton, S.A., 2006. Paradigm shift in neuroprotection by NMDA receptor blockade:memantine and beyond. Nat. Rev. Drug Discov. 5, 160–170.
Lipton, S.A., 2007. Pathologically-activated therapeutics for neuroprotection: mechanism ofNMDA receptor block bymemantine and S-nitrosylation. Curr. Drug Targets 8, 621–632.
Martinez-Coria, H., Green, K.N., Billings, L.M., Kitazawa, M., Albrecht, M., Rammes, G., et al.,2010. Memantine improves cognition and reduces Alzheimer's-like neuropathology intransgenic mice. Am. J. Pathol. 176, 870–880.
Menuet, C., Borghgraef, P., Matarazzo, V., Gielis, L., Lajard, A.-M., Voituron, N., et al.,2011a. Raphé tauopathy alters serotonin metabolism and breathing activity interminal Tau.P301L mice: possible implications for tauopathies and Alzheimer'sDisease. Respir. Physiol. Neurobiol. 178, 290–303.
Menuet, C., Cazals, Y., Gestreau, C., Borghgraef, P., Gielis, L., Dutschmann, M., et al.,2011b. Age-related impairment of ultrasonic vocalization in Tau.P301L mice:possible implication for progressive language disorders. PLoS One 6 (10), e25770.
Nunez-Abades, P.A., Portillo, F., Pasaro, R., 1990. Characterisation of afferent projections tothe nucleus ambiguus of the rat by means of fluorescent double labelling. J. Anat. 172,1–15.
Pan, C., Xu, Z., Dong, Y., Zhang, Y., Zhang, J., McAuliffe, S., et al., 2011. The potential dualeffects of anesthetic isoflurane on hypoxia-induced caspase-3 activation andincreases in {beta}-site amyloid precursor protein-cleaving enzyme levels. Anesth.Analg. 113, 145–152.
Paxinos, G., Franklin, K.B., 2001. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.Academic Press, Sydney.
Planel, E., Richter, K.E., Nolan, C.E., Finley, J.E., Liu, L., Wen, Y., et al., 2007. Anesthesialeads to tau hyperphosphorylation through inhibition of phosphatase activity byhypothermia. J. Neurosci. 27, 3090–3097.
Planel, E., Bretteville, A., Liu, L., Virag, L., Du, A.L., Yu, W.H., et al., 2009. Acceleration andpersistence of neurofibrillary pathology in a mouse model of tauopathy followinganesthesia. FASEB J. 23, 2595–2604.
Priefer, B.A., Robbins, J., 1997. Eating changes in mild-stage Alzheimer's Disease: a pilotstudy. Dysphagia 12, 212–221.
Rahman, A., Ting, K., Cullen, K.M., Braidy, N., Brew, B.J., Guillemin, G.J., 2009. Theexcitotoxin quinolinic acid induces tau hyperphosphorylation in human neurons.PLoS One 4 (7), e6344.
Reisberg, B., Doody, R., Stöffler, A., Schmitt, F., Ferris, S., Möbius, H.J., 2003. Meman-tine in moderate-to-severe Alzheimer's Disease. N. Engl. J. Med. 348,1333–1341.
Roda, F., Pio, J., Bianchi, A.-L., Gestreau, C., 2004. Effects of anesthetics on hypoglossalnerve discharge and c-Fos expression in brainstem hypoglossal premotor neurons.J. Comp. Neurol. 468, 571–586.
Rohrer, J.D., Schott, J.M., 2011. Primary progressive aphasia — defining genetic andpathological subtypes. Curr. Alzheimer Res. 8, 266–272.
Run, X., Liang, Z., Zhang, L., Iqbal, K., Grundke-Iqbal, I., Gong, C.X., 2009. Anesthesiainduces phosphorylation of tau. J. Alzheimers Dis. 16, 619–626.
Saab, B.J., Maclean, A.J., Kanisek, M., Zurek, A.A., Martin, L.J., Roder, J.C., et al., 2010.Short-term memory impairment after isoflurane in mice is prevented by the α5γ-aminobutyric acid type A receptor inverse agonist L-655,708. Anesthesiology113, 1061–1071.
Sakata, D.J., Gopalakrishnan, N.A., Orr, J.A., White, J.L., Westenskow, D.R., 2007. Hypercap-nic hyperventilation shortens emergence time from isoflurane anesthesia. Anesth.Analg. 104, 587–591.
Sinclair, H.L., Andrews, P.J., 2010. Bench-to-bedside review: hypothermia in traumaticbrain injury. Crit. Care 14 (1), 204.
Song, M.S., Rauw, G., Baker, G.B., Kar, S., 2008. Memantine protects rat cortical culturedneurons against beta-amyloid-induced toxicity by attenuating tau phosphoryla-tion. Eur. J. Neurosci. 28, 1989–2002.
Subramanian, H.H., Balnave, R.J., Holstege, G., 2008. The midbrain periaqueductal graycontrol of respiration. J. Neurosci. 28, 12274–12283.
Suh, M.K., Kim, H., Na, D.L., 2009. Dysphagia in patients with dementia: Alzheimerversus vascular. Alzheimer Dis. Assoc. Disord. 23, 178–184.
Tang, J.X., Baranov, D., Hammond, M., Shaw, L.M., Eckenhoff, M.F., Eckenhoff, R.G., 2011.Human Alzheimer and inflammation biomarkers after anesthesia and surgery.Anesthesiology 115, 727–732.
Teppema, L.J., Veening, J.G., Kranenburg, A., Dahan, A., Berkenbosch, A., Olievier, C.,1997. Expression of c-fos in the rat brainstem after exposure to hypoxia and tonormoxic and hyperoxic hypercapnia. J. Comp. Neurol. 388, 169–190.
Terwel, D., Lasrado, R., Snauwaert, J., Vandeweert, E., Van Haesendonck, C., Borghgraef,P., et al., 2005. Changed conformation of mutant Tau-P301L underlies themoribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2Ntransgenic mice. J. Biol. Chem. 280, 3963–3973.
Terwel, D., Muyllaert, D., Dewachter, I., Borghgraef, P., Croes, S., Devijver, H., et al., 2008.Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am.J. Pathol. 172, 786–798.
Tokutomi, T., Miyagi, T., Takeuchi, Y., Karukaya, T., Katsuki, H., Shigemori, M., 2009.Effect of 35 degrees C hypothermia on intracranial pressure and clinical outcomein patients with severe traumatic brain injury. J. Trauma 66, 166–173.
Vanderweyde, T., Bednar, M.M., Forman, S.A., Wolozin, B., 2010. Iatrogenic risk fac-tors for Alzheimer's disease: surgery and anesthesia. J. Alzheimers Dis. 22,91–104.
Villard, M.F., Caille, D., Hugelin, A., 1984. Dissociation between respiratory phaseswitching and phasic phrenic response on low-intensity stimulation ofpneumotaxic complex and nearby structures. J. Physiol. (Paris) 79, 11–16.
Wada, H., Nakajoh, K., Satoh-Nakagawa, T., Suzuki, T., Ohrui, T., Arai, H., et al., 2001.Risk factors of aspiration pneumonia in Alzheimer's Disease patients. Gerontology47, 271–276.
Yamazaki, H., Ohi, Y., Haji, A., 2009. Mu-opioid and N-methyl-D-aspartate receptorsare localized at laryngeal motoneurons of guinea pigs. Biol. Pharm. Bull. 32,293–296.



















![Volumetric Changes in the Upper Airways after Rapid and Slow … · 2020. 11. 27. · Almuzian et al. [33,34], whose protocol has been followed in the present study. Patients’ airways](https://img.pdfslide.net/doc/110x75/610d75e33c210e65876b4526/volumetric-changes-in-the-upper-airways-after-rapid-and-slow-2020-11-27-almuzian.jpg)









