Embed Size (px)
Citation preview

Chapter 4
Neurotrophin and Neurotrophin Receptor Involvementin Human Neuroblastoma
Pierdomenico Ruggeri, Antonietta R. Farina,Lucia Cappabianca, Natalia Di Ianni, Marzia Ragone,Stefania Merolle, Alberto Gulino andAndrew R. Mackay
Additional information is available at the end of the chapter
http://dx.doi.org/10.5772/55536
1. Introduction
Neuroblastoma (NB) is an embryonic tumour that originates from cells of the neural crest (NC)arrested in their differentiation at different stages along the sympatho-adrenal lineage and,less frequently, from precursors of sensory neurons [1, 2]. As a consequence, NB can occurthroughout the sympathetic chain from thoracic, abdominal and pelvic sites to the adrenalmedulla, which accounts for the majority of NBs. Consistent with this, NBs exhibit a highdegree of genetic heterogeneity and biological variability, including differences in catechola‐mine expression, according to their differentiation state along the sympathoadrenal lineage,with a small number of primitive midline and spinal NBs that do not secrete catecholaminesconsidered to be of dorsal root sensory origin [1, 2].
Sympathetic nervous system development is orchestrated by neurotrophins (NT) and theirrespective neurotrophin receptors (NTR), which exhibit subtle temporal and spatial changesin expression that are critical for the delamination, migration, proliferation, survival, differ‐entiation and apoptotic programs of NC lineages that form the fully differentiated andfunctional sympathetic nervous system. Not surprisingly NBs, consistent with their origin andparticular differentiation state at the time of transformation, exhibit a variety of differentpatterns of NT and NTR expression. A great deal of research has focussed on characterisingand exploiting these different patterns of expression for potential prognostic and therapeuticbenefit. Recent studies have led to exciting new developments in understanding how block‐ages in sympathetic differentiation promote NB and how NBs utilise different patterns of NT
© 2013 Ruggeri et al.; licensee InTech. This is an open access article distributed under the terms of theCreative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permitsunrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

and NTR expression to select a more malignant, stress-resistant, invasive, genetically unstable,stem cell-like phenotype. Furthermore, they have also identified novel potential therapeutictargets and characterised patterns of NT/NTR expression of value in prognosis and therapeuticchoice. In this chapter therefore, we will review the origins of NB during neural crest migationand sympathetic nervous system development, introduce NTs and NTRs and describe theirroles NC and sympathetic nervous system development, examine patterns of NT/NTRexpression in NB, review their potential roles in regulating spontaneous NB regression andmetastatic NB progression, and discuss potential therapeutic ways to target the NT/NTRsystem in NB.
2. Formation of the neural crest, neural crest cell delamination andmigration
NBs originate from NC cells (NCC) during sympathetic nervous system development. In thissection therefore, we will briefly describe the natural history of neural crest, sensory dorsalroot and sympathetic nervous system development, focussing attention on the sympatho-adrenal neuroblast lineage, which is responsible for generating neuroendocrine chromaffintissues, SIF and ganglion cells, and in particular the adrenal medulla within which the majority(40-50%) of NBs develop [2, 3].
Figure 1. Formation of the Neural Crest and Neural Crest cell migration
During the 3rd week of human embryonic development the intra-embryonic mesodermdifferentiates into paraxial, intermediate and lateral plate portions. The paraxial mesodermorganises into primitive segmented somites and the lateral plate mesoderm splits into somatic(parietal) and splanchnic (visceral) layers. This event occurs in a BMP-induced Notch-dependent “clock” and Wnt-dependent “wave” manner in a rostral to caudal gradient ofFGF [2, 4-6] and results in the simultaneous formation of Somite pairs either side of theforming neural tube, in a head to tail direction along the entire length of the embryo, with
Neuroblastoma48

each new somite forming on the caudal side of an existing somite. Somites further differen‐tiate into dermomyotome and sclerotome structures that will eventually provide the cells forskin, muscle and skeletal formation. Contemporarily, the embryonic neuroectodermundergoes progressive indentation to form the neural groove, neural folds and neural plate.This neurulation process causes the fusion of opposing neural folds at the future uppercervical level, which progresses in both rostral and caudal directions, eventually resulting incontinuity between neural and squamous surface ectoderm. This event separates thepresumptive epidermis from the neural plate, which in turn forms the distinct and sepa‐rate columnar cellular structure of the Neural Tube. Interaction between the neural plate andpresumptive epidermis is regulated by Wnts, BMPs and FGFs and results in mesenchymaltransformation of the epithelial cells that line the margins of the neural fold. These cellsorganise between the epidermis and neural tube to form the transient Neural Crest (NC)embryonic structure [2, 6] (Fig. 1).
NC cells (NCC) delaminate from the NC and migrate initially in a ventrolateral manner andlater in a dorsolateral direction, relative to the somites. Ventrolateral NCC migration occursin chain-like manner [7] between the somites and neural tube and the rostral half of each somite[8]. NCC initially migrate through the inter-somitic boundary before switching to a sclerotomepathway controlled by semaphorin and its receptor neuropilin, with the entire dermomytomerepulsing neuropilin positive trunk NCC [9] (Fig. 2).
Figure 2. Neural Crest cell ventrolateral and dorsolateral migration
Dorsolateral NCC migration occurs between the developing dermis and the dorsal dermo‐myotome boundary [8, 10]. During NCC migration cells receive signals from adjacent struc‐tures that initiate a series of differentiation processes that will eventually lead todifferentiation-commitment and specific cell fates at different locations. This process providesa wide variety of differentiated tissues, including: epidermal pigment cells (melanocytes);dorsal root, sympathetic and parasympathetic ganglia, neurons and plexuses; neuroglial andSchwann cells; endocrine/paracrine cells of the adrenal medulla, carotid body and organ ofZuckerland; cartilage and bones of the facial and ventral skull; corneal endothelium andstroma; tooth papillae; dermis, smooth muscle and adipose tissue of the head and neck;
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
49
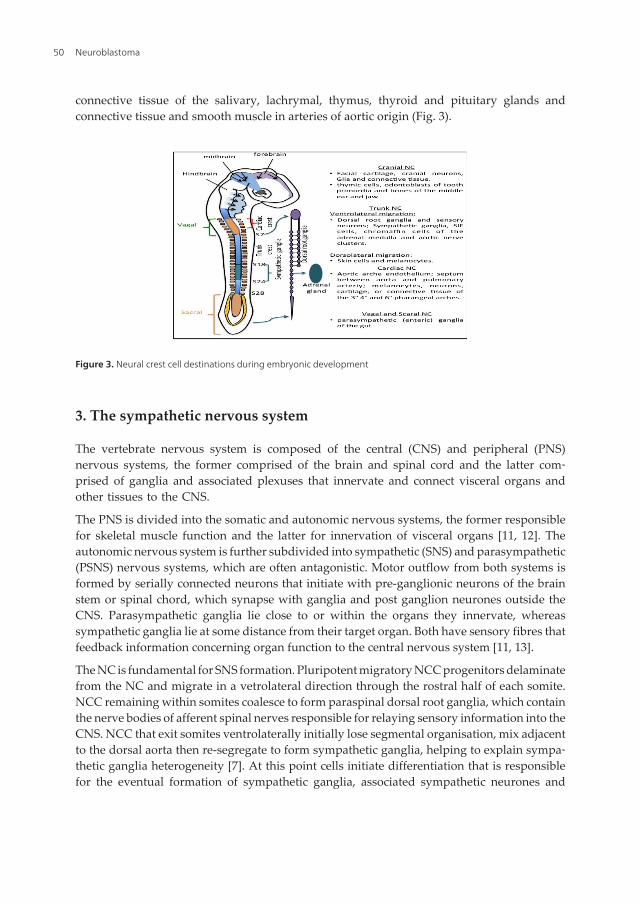
connective tissue of the salivary, lachrymal, thymus, thyroid and pituitary glands andconnective tissue and smooth muscle in arteries of aortic origin (Fig. 3).
Figure 3. Neural crest cell destinations during embryonic development
3. The sympathetic nervous system
The vertebrate nervous system is composed of the central (CNS) and peripheral (PNS)nervous systems, the former comprised of the brain and spinal cord and the latter com‐prised of ganglia and associated plexuses that innervate and connect visceral organs andother tissues to the CNS.
The PNS is divided into the somatic and autonomic nervous systems, the former responsiblefor skeletal muscle function and the latter for innervation of visceral organs [11, 12]. Theautonomic nervous system is further subdivided into sympathetic (SNS) and parasympathetic(PSNS) nervous systems, which are often antagonistic. Motor outflow from both systems isformed by serially connected neurons that initiate with pre-ganglionic neurons of the brainstem or spinal chord, which synapse with ganglia and post ganglion neurones outside theCNS. Parasympathetic ganglia lie close to or within the organs they innervate, whereassympathetic ganglia lie at some distance from their target organ. Both have sensory fibres thatfeedback information concerning organ function to the central nervous system [11, 13].
The NC is fundamental for SNS formation. Pluripotent migratory NCC progenitors delaminatefrom the NC and migrate in a vetrolateral direction through the rostral half of each somite.NCC remaining within somites coalesce to form paraspinal dorsal root ganglia, which containthe nerve bodies of afferent spinal nerves responsible for relaying sensory information into theCNS. NCC that exit somites ventrolaterally initially lose segmental organisation, mix adjacentto the dorsal aorta then re-segregate to form sympathetic ganglia, helping to explain sympa‐thetic ganglia heterogeneity [7]. At this point cells initiate differentiation that is responsiblefor the eventual formation of sympathetic ganglia, associated sympathetic neurones and
Neuroblastoma50
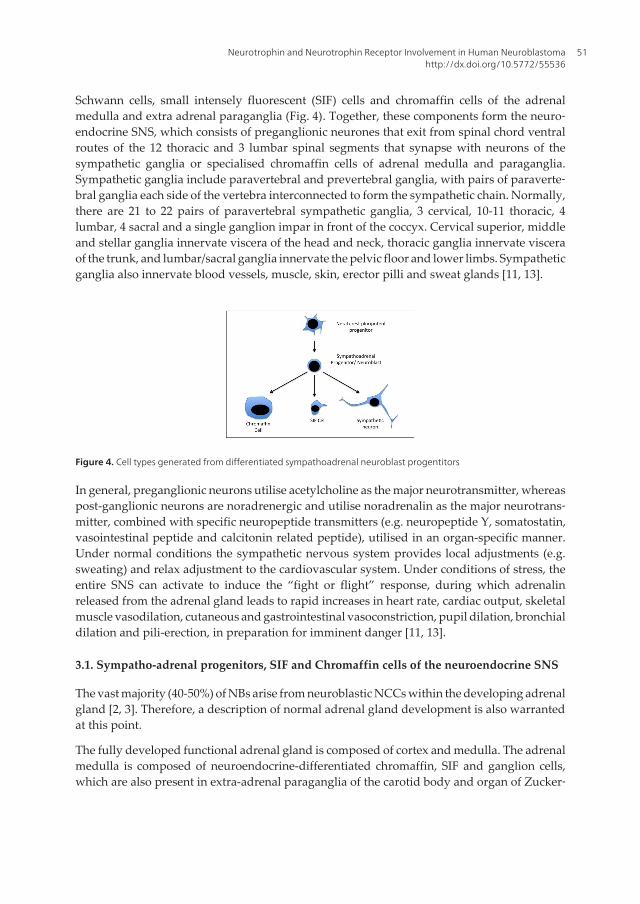
Schwann cells, small intensely fluorescent (SIF) cells and chromaffin cells of the adrenalmedulla and extra adrenal paraganglia (Fig. 4). Together, these components form the neuro‐endocrine SNS, which consists of preganglionic neurones that exit from spinal chord ventralroutes of the 12 thoracic and 3 lumbar spinal segments that synapse with neurons of thesympathetic ganglia or specialised chromaffin cells of adrenal medulla and paraganglia.Sympathetic ganglia include paravertebral and prevertebral ganglia, with pairs of paraverte‐bral ganglia each side of the vertebra interconnected to form the sympathetic chain. Normally,there are 21 to 22 pairs of paravertebral sympathetic ganglia, 3 cervical, 10-11 thoracic, 4lumbar, 4 sacral and a single ganglion impar in front of the coccyx. Cervical superior, middleand stellar ganglia innervate viscera of the head and neck, thoracic ganglia innervate visceraof the trunk, and lumbar/sacral ganglia innervate the pelvic floor and lower limbs. Sympatheticganglia also innervate blood vessels, muscle, skin, erector pilli and sweat glands [11, 13].
Figure 4. Cell types generated from differentiated sympathoadrenal neuroblast progentitors
In general, preganglionic neurons utilise acetylcholine as the major neurotransmitter, whereaspost-ganglionic neurons are noradrenergic and utilise noradrenalin as the major neurotrans‐mitter, combined with specific neuropeptide transmitters (e.g. neuropeptide Y, somatostatin,vasointestinal peptide and calcitonin related peptide), utilised in an organ-specific manner.Under normal conditions the sympathetic nervous system provides local adjustments (e.g.sweating) and relax adjustment to the cardiovascular system. Under conditions of stress, theentire SNS can activate to induce the “fight or flight” response, during which adrenalinreleased from the adrenal gland leads to rapid increases in heart rate, cardiac output, skeletalmuscle vasodilation, cutaneous and gastrointestinal vasoconstriction, pupil dilation, bronchialdilation and pili-erection, in preparation for imminent danger [11, 13].
3.1. Sympatho-adrenal progenitors, SIF and Chromaffin cells of the neuroendocrine SNS
The vast majority (40-50%) of NBs arise from neuroblastic NCCs within the developing adrenalgland [2, 3]. Therefore, a description of normal adrenal gland development is also warrantedat this point.
The fully developed functional adrenal gland is composed of cortex and medulla. The adrenalmedulla is composed of neuroendocrine-differentiated chromaffin, SIF and ganglion cells,which are also present in extra-adrenal paraganglia of the carotid body and organ of Zucker‐
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
51

land [14]. Chromaffin and SIF cells, characterised by their affinity from chromium salts, areclosely related to sympathetic neurons and, like sympathetic neurons, synthesise, store, uptakeand release catecholamines and express enzymes for noradrenalin synthesis including tyrosinehydroxylase (TH) and dopamine-β-hydroxylase (DBH). Unlike sympathetic neurons, chro‐maffin cells also synthesise, store and release adrenalin and retain their capacity proliferatebut do not produce axons or dendrites [15]. Adrenal and extra-adrenal chromaffin tissues, likesympathetic ganglia, are innervated by pre-ganglionic neurones originating from the spinalchord [16]. Chromaffin, SIF and sympathetic neurons exemplify the wide spectrum ofsympathoadrenal cell types that originate from NCC [17].
Chromaffin and SIF cells differentiate from pluripotent NCC progenitors that delaminate fromthe NC at the “adreno-medullary” somite level (somites 18-24 in avian development) [18].These cells migrate ventrolaterally initially between Somite dermomyotome and sclerotomethen through the rostral sclerotome mesenchyme to arrive at para-aortic sites [18-20]. At thesesites, NCC mix, re-segregate and coalesce to form sympathetic ganglia. At the same time, NCCderived from the “adreo-medullar” somite region coalesce adjacent to the adrenal cortexanlage then invade the anlage in considerable numbers, initially in a nerve fibre-independentthen nerve fibre-dependent manner [21], in a Sox transcription factor-dependent manner [22].Once within the adrenal primordium, NCCs form rosettes, nests and nodules along nervefibres, proliferate and initiate pheo-chromoblast differentiation. This process continuesthroughout foetal development and into the neonatal period, providing differentiatedChromaffin and SIF adrenal medulla cell populations. In humans, the gestational periodbetween 17 and 20 weeks is critical for adrenal sympathetic component development, withneuroblastic NCC proliferation peaking during this period in terms of maximal nodule sizeand number, waning thereafter. Neuroblastic nodules tend to disappear during the thirdtrimester and are usually absent at birth. However, nodules that continue to grow and persistinto neonatal life are not infrequent and have been classified as in situ NB. A sizeable numberof these NBs spontaneously regress and are likely, therefore, to represent delayed differentia‐tion in addition to neoplastic transformation [23].
Chromaffin cells, SIF cells and sympathetic neurons develop from catecholaminergic sympa‐thoadrenal (SA) progenitors [18, 24, 25] and their formation involves BMP signalling [18,26-28]. However, the classical concept that a common SA lineage acquires neuronal andcatecholaminergic traits prior to migration to secondary sympathetic ganglia and adrenal sites[24, 25] has now been discounted, as chromaffin cells undergo catecholaminergic differentia‐tion within the adrenal anlage and not within primary sympathetic ganglia, and do not expressneuronal markers at the onset or even following induction of TH expression [29, 30]. Therefore,sympathetic neuronal and chromaffin lineages must separate upstream prior to catecholami‐nergic differentiation, despite evidence of sympathoadrenal marker expression in somemigrating cells [22, 24, 30], and enter the adrenal primordium as undifferentiated Sox10expressing NCC [22, 30]. Indeed, chromaffin and sympathetic neurones originate at the sameaxial level from common NC progenitors but differ in the time of catecholaminergic differen‐tiation [18]. Furthermore, NCC populations migrating to the adrenal anlage and within the
Neuroblastoma52

adrenal medulla exhibit heterogeneity, and consist of SOX10/Phox2B/p75NTR, SOX10/p75NTR and PHox2B/p75NTR sub-populations [22].
3.2. Transcriptional regulation of sympathoadrenal differentiation
Chromaffin and sympathetic neuron differentiation is regulated by BMP-induced transcrip‐tion factors Phox2B, Mash-1, Insm1, Hand2 and Gata 2/3 [31]. Knockout technology hasidentified a fundamental role for Phox2B in chromaffin and sympathetic neuronal differen‐tiation [18], with Phox2B knockout increasing neuron but not chromaffin precursor death.This not only relates to specific cell traits but also differences in environment and migra‐tion [32, 33], and confirms that adrenal anlage are colonised by undifferentiated NCCprogenitors. Knockout technology has also characterised a role for Mash-1 as an accelera‐tor of sympathetic neuronal and chromaffin differentiation [34, 35], a role for Insm-1 as aregulator of catecholamine synthesising enzyme expression and, therefore, endocrinedifferentiation [36, 37], a role for Hand2 in the induction and maintenance of noradrener‐gic differentiation [38, 39] and a role for Gata3 in the differentiation of both sympatheticganglia and chromaffin cells [31, 40].
It has now been confirmed that adrenal cortex glucocorticoids are not responsible for chro‐maffin cell differentiation [41] but they do, however, regulate postnatal chromaffin cellsurvival and phenyl ethanolamine N-methyl transferase expression [30, 42]. The adrenal cortexis also dispensable for chromaffin differentiation, which is also found in extra-adrenalneuroendocrine tissue, but may regulate adrenal chromaffin cell numbers and associatedvascularity [30, 43]. Within the adrenal gland, hypoxia has recently been shown to promotechromaffin/SIF cell differentiation from neuroblasts [44-46].
4. Neurotrophins and neurotrophin receptors in neural crest, sympatheticnervous system and adrenal development
Neurotrophins (NTs) and NT receptors (NTRs) are critical for the development and mainte‐nance of the vertebral CNS and PNS [47-50], NTs and NTRs are also expressed by human NBsand have been implicated in both NB regression and malignant progression. In this section,therefore, we will introduce NTs and NTRs and describe their potential involvement in normalSNS and adrenal development.
4.1. Neurotrophins (NTs)
NTs are a family of growth, differentiation, survival and apoptosis-inducing factors that areinvolved in many aspects of nervous system development, maintenance and function. Theycomprise four structurally related basic 115-130 amino acid containing polypeptides, nervegrowth factor (NGF), brain-derived growth factor (BDNF), and the neurotrophins 3 (NT-3)and 4/5. NGF was first NT to be described and purified from the mouse salivary gland [51].This was followed by the discovery of BDNF, NT-3 and NT-4/5 some 30 years later [52-54].
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
53

NTs exhibit close structural homology, with the exception of NT4/5 that exhibits only 50%homology to the others NTs, and all contain six conserved cysteines that form structurallyimportant disulphide bridges [55, 56]. NTs are expressed by both neuronal and non-neuronalcells as pre-NTs and are converted to pro-NTs upon signal peptide removal. This can occurwithin the endoplasmic reticulum (ER), in which NTs are converted to mature-NTs by furins.Alternatively, NTs are transported to the cell surface and released following signal peptideremoval as pro-NTs. Secreted pro-NTs, which also exhibit biological activity, are converted tomature NTs by enzymes including plasmin and the matrix metalloproteinases MMP-7 andMMP-9 [56-58]. Within the extracellular environment, pro- and mature NTs form homo-dimersand bind specific receptors to induce an array of biological activities, including cell migration,proliferation, survival, differentiation, apoptosis and neuronal synapse/junction plasticity,depending upon the cell population, receptor expression and activation status [57, 60]. Thehuman NGF gene localises to chromosome 1p13.1 [61], the human BDNF gene localises tochromosome 11p13 [62], the human NT-3 gene localises to chromosome 12p13 [62] and thehuman NT4/5 gene localises to chromosome 19q13.3 [63]. Since the discovery of NTs, theirrespective receptors have been identified and many of their roles in nervous system develop‐ment and function have been elucidated.
4.2. NT receptors
4.2.1. Tropomyosin-related kinases TrkA, TrkB and TrkC
The family of NT receptors includes the tropomyosin-related tyrosine kinases TrkA, TrkB andTrkC [64]. TrkA is the preferred receptor of mature NGF but also binds the mature neurotro‐phin NT-3 [64, 65]. Identified following the discovery of the first tumour-associated TrkAoncogene [64, 66, 67], the 25kb human TrkA gene maps to chromosome 1q21-22 and isorganised into 17 exons [68-70]. TrkA proteins are expressed either as the fully splicedgp140kDa TrkAII receptor, alternatively spliced TrkA L0 and L1 variants that exhibit differ‐ential exon 2-4 use [71], the TrkAI variant that exhibits exon 9 skipping [72] or the TrkAIIIvariant, which exhibits in-frame skipping of exons 6 and 7 combined with exon 9 omission [73].TrkA L0 (exons 2, 3 and 4 alternatively spliced) and TrkA L1 (exons 2 and 3 alternativelyspliced) are expressed during rat development [71] as truncated receptors with in-framedeletions of leucine-rich sequences encoded within exons 2-4 [68]. Since, TrkA leucine richsequences may modulate ligand binding [74], these variants may exhibit altered ligand-binding activity similar to analogous alternative TrkB splice variants [75]. TrkAI (exon 9exclusion) and TrkAII (exon 9 inclusion) splice variants [72] are expressed as cell surface trans-membrane receptors and exon 9 omission does not result in ligand-independent receptoractivation. TrkAI and TrkAII variants bind NGF and NT3 [72, 76] but TrkAII exhibits higherlevels of NT-3-mediated activation when co-expressed with the low affinity neurotrophinreceptor CD271/p75NTR [76]. TrkAII is predominantly expressed within the nervous system,whereas TrkAI expression predominates in the thymus [72].
TrkAIII was identified as an unexpected RT-PCR product in primary human NBs [73]. Thisvariant exhibits exon 6 and 7 skipping plus exon 9 omission, resulting in the in-frame deletion
Neuroblastoma54

of amino acids 192-284 that encode the D4 extracellular immunoglobulin-like domain, severalfunctional N-glycosylation sites and introduces a valine substitution at the novel exon 5/8splice junction [73]. In addition to being expressed by primary human NBs, TrkAIII is alsodevelopmentally regulated and is detected from stages E13-E18 of mouse embryonic devel‐opment and is also expressed by immature thymocytes within the developing thymus [73,77]. Unlike fully spliced TrkA receptors, TrkAIII is not expressed at the cell surface but isretained within intracellular membranes of the endoplasmic reticulum (ER), GN and ER/GNintermediate compartment (ERGIC) [73, 78, 79], within which it exhibits interphase-restrictedspontaneous ligand-independent activation [73, 78, 79].
TrkB is the preferred receptor of BDNF but also binds NT-4/5 [65, 80-82]. The 590kb humanTrkB gene maps to chromosome 9q22 and contains 24 exons [70, 83]. In addition to fully splicedgp145kDa TrkB, eight TrkB variant isoforms have been described, including a gp95kDa C-terminal truncated receptor that lacks the tyrosine kinase and Shc binding domains; a C-terminal truncated receptor that lacks the tyrosine kinase domain but retains the Shc bindingsite; a C-terminal truncated receptor that lacks exons 23 and 24 but retains tyrosine kinaseactivity and four N-terminal truncated receptors that exclude combinations of exons 1-5 andupstream signal sequence [75, 83-85]. The TrkB gene has also been reported to encode up to100 different transcripts ranging from 0.7-9kb, at least 36 of which can be translated intofunctional TrkB proteins [85-87]. Both full length and C-terminal truncated TrkB receptors areexpressed in the brain and share 100% extracellular domain homology, consisting of 5 highlyglycosylated extracellular binding domains (D1-5) [75, 85, 86].
TrkC binds NT-3 and no other NT [88]. The 387kb human TrkC gene maps to chromosome15q25 and is organised into 18 exons [70] and six TrkC isoforms have been described. Inaddition to the fully spliced gp145kDa receptor, these isoforms include C14/K2, C25/K3 andC39 variants which contain 14, 25 and 39 additional amino acid insertions between kinasesubdomains VII and VIII, downstream of the TDYYR motif of the putative Trk receptor familyautophosphorylation site [89] and NC1/T1 and NC-2/T2 non-catalytic variants truncated inthe tyrosine kinase domain by short C-terminal sequences [90-92]. Full-length TrkC receptorsare expressed during development, whereas truncated receptors predominate in later life inpost mitotic cerebellar granule neurons and young stem cell-derived differentiated neuronsbut not in proliferating neural stem cells. TrkC NC1/T1 and NC2/T2 variants do not supportNT-induced neuritogenesis, suggesting that TrkC variants could exert different roles duringnervous system development [90, 93].
4.2.2. CD271/p75 neurotrophin receptor
The p75 neurotrophin receptor (CD271/p75NTR) is a member of the tumour necrosis factor(TNFR)/FAS receptor superfamily and binds all NTs in pro-form with high affinity and matureNGF with low affinity [94, 95]. The 3.4kb CD271/p75NTR gene is organised into 5 exons andmaps to chromosome 17q21-q22 [96]. In addition to the fully spliced 75kDa CD271/p75NTR
receptor, a truncated alternative s-p75NTR splice variant has been described that is devoid ofexon III. S-p75NTR lacks the NT binding domain, does not bind NTs and is expressed by severalneural tissues [97]. The fully spliced CD271/p75NTR extracellular-domain contains four 40-
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
55

amino acid repeats with 6 cysteine residues at conserved positions that are required for NTbinding, a serine/threonine-rich region, a single transmembrane domain and a 155-amino acidcytoplasmic domain, which does not exhibit catalytic activity. CD271/p75NTR acts either as anindependent NT receptor, a NT receptor complex with Sortilin or a co-receptor for TrkA, andis involved in regulating death, differentiation or survival signals [94, 98]. The CD271/P75NTR
receptor is devoid of intrinsic catalytic activity, indicating that signalling from this receptormust depend upon intracellular interactors [99, 100].
4.2.3. Sortilin
Sortilin is a member of the Vps10p domain-containing transmembrane proteins that binds bothmature NGF and the neurotrophins NGF, BDNF and NT-3 in pro-form [98, 101, 102]. The 7kbhuman Sortilin gene localises to chromosome 1p13.3 and is expressed as a gp95-100kdaglycoprotein [103, 104]. Sortilin co-expression with CD271/p75NTR results in the formation of aco-receptor complex that augments affinity for proNGF and acts principally as an inducer ofapoptosis [105].
4.3. NT receptor structure and ligand binding
All three Trk receptors share significant sequence homology and a conserved domain organ‐ization. This organization comprises from N-terminus to C-terminus of five extracellulardomains, a transmembrane region and the intracellular kinase domain.
Figure 5. NT receptor structure and ligand binding domains
The first three extracellular domains consist of a leucine-rich region (D-2) flanked by twocysteine-rich regions (D-1 and D-3), and domains 4 and 5 are immunoglobulin-like domains.Studies on TrkB and TrkC have shown that D-5 is sufficient for the binding of ligands and isresponsible for binding specificity [106-109], although the D-4 domain, leucines and cysteineclusters may regulate ligand binding [55, 73]. Receptor transmembrane and juxta-membraneregions are critical for signal internalisation and transduction. The intracellular tyrosine-richcarboxyl terminal cytoplasmic domain exhibits tyrosine kinase activity upon ligand-mediatedactivation and is responsible for propagating post-receptor signal transduction [74, 107,110-114]. The immunoglobulin-like D4 and D5 domains stabilise receptors in monomeric form
Neuroblastoma56

and prevent spontaneous receptor oligomerisation and activation. Deletions, chimericreceptors and point mutations that disrupt the structure of the first (D4) and second (D5)immunoglobulin-like domains result in ligand-independent spontaneous receptor activationand the acquisition of oncogenic activity [73, 110, 115] (Fig. 5).
CD271/p75NTR receptors modulate the affinity and enhance the specificity of TrkA for NGF,and TrkB for BDNF, with optimal affinity reflecting the ratio of Trk to CD271/p75NTR receptors[116-118]. In contrast, CD271/p75NTR reduces TrkAI activity in response to NT-3 and TrkBactivity in response to NT-3 and NT-4/5 [76, 119, 120]. The CD271/p75NTR receptor analogueneurotrophin-related homolog-2 (NRH2) that is expressed by neural cells, also interacts withTrkA to promote high affinity NGF binding [121].
4.4. NT receptor signalling
In the absence of ligand, Trk receptors are maintained as inactive oligomers [120], concentratedwithin caveolin and cholesterol-containing cell membrane caveolae invaginations, which alsocontain components of the Ras signalling pathway [122]. Receptor oligomers are maintainedin an inactive state by mature extracellular domain N-glycosylation, intact D4 and D5 domainsand by receptor-associated protein tyrosine phosphatases (PTPases) [110, 123-126]. Uponligand binding, oligomeric Trk receptors dimerize, alter their conformation and acquiretyrosine kinase activity, facilitated by temporary inactivation of receptor-associated PTPases,which results in auto- and trans-phosphorylation of receptor tyrosine residues Y490, Y670,Y674/675, Y751 and Y785, in TrkA and their equivalents in TrkB and TrkC. These tyrosines actas phosphorylation-dependent binding sites for a variety of signalling proteins, including theadapters Shc and FRS-2; Grb-2 and SOS; the IP3K subunit p85α and PLCγ. These interactions,which are modulated by CD271/p75NTR, provide avenues for signal transduction through RAS/MAPK, IP3K/Akt/NF-κB and PKC pathways that mediate NT effects upon migration, prolif‐eration, survival, differentiation and apoptosis [73, 111, 127-141]. Cell surface Trk localisationand NT-mediated Trk activation also involves interaction with the heat shock protein chap‐erone Hsp90 [78].
Figure 6. Trk receptor signalling and outcome
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
57

Trk receptors activated by NTs use two main pathways to activate MAPKs. The first pathwayinvolves Shc, Grb-2, SOS, Ras and Raf, and the second pathway involves CrkL, Rap and Raf[142, 143]. Trk activation of MAPK is now considered to depend not only upon the phos‐phorylated Trk Y490 tyrosine residue [144, 145] but also the ankyrin repeat-rich membranespanning protein ARMS, acting through CrkL [146, 147]. MAPKs activate CREB transcriptionfactors to promote differentiation and survival [148-150]. Trk activation of PI3K/Akt signallingoccurs through Shc/Grb-2 and Gab-1 and induces pro-survival signals [73, 151, 152], resultingfrom the phosphorylation of Bad and activates the pro-survival transcription factor NF-κB [73,153, 154]. PLCγ is activated as a consequence of being recruited to the phosphorylated Trktyrosine Y785 [73, 132] and provides additional differentiation and survival signals thatinvolve MAPK [155] (Fig. 6).
The alterative TrkAIII splice variant, in contrast to other Trk receptors (see above), is notexpressed at the cell surface but accumulates within intracellular membranes. IntracellularTrkAIII does not bind extracellular NTs and is prone to spontaneous ligand-independentintracellular activation [73, 78, 79]. In contrast to ligand activated cell surface TrkA signalling,spontaneously active TrkAIII signals through PI3K/Akt/NF-κB but not Ras/MAPK, resultingin increased survival and the induction/maintenance of a stem cell-like undifferentiatedphenotype [73, 78, 79, 156] (Fig. 6).
An additional feature of TrkA receptors is retrograde transport signalling within the cell. Thisdepends upon receptor/ligand interaction, internalisation and retrograde transport of activat‐ed receptors, resulting in signal transduction within the cell body. Sympathetic neurons mostdramatically illustrate this activity, with retrograde transport of NGF-activated TrkA occur‐ring along the axonal length to the neuronal cell body. This phenomenon involves ubiquitinmediated receptor internalisation through interaction with CD271/p75NTR and TRAF6, receptorendocytosis within clatherin-coated vesicles and receptor endocytosis facilitated by theendocytosis inducing protein EHD4/Pincher [157-159]. In addition, immature Trk receptorsalso localise to intracellular membranes of the Golgi Network (GN) and can be trans-activatedby agonists of the G-protein linked A2A adenosine receptors, potentially through the non-receptor tyrosine kinase Src [160, 161], providing evidence for intracellular neurotrophin-independent Trk activation. Post receptor signal transduction from GN-associated TrkAdiffers from cell surface-activated TrkA, by signalling through IP3K/Akt but not RAS/MAPK,which results in NF-κB transcription factor activation, inducing a more stress-resistantphenotype, not dissimilar to that induced by the intracellular alternative TrkAIII splice variant[73, 124, 160]. TrkA localisation to the GN may not only reflect transient passage of de-novosynthesised receptors but also alterations in receptor extracellular domain N-glycosylation andfolding.
CD271/p75NTR receptors regulate cell survival, apoptosis, differentiation and proliferation.CD271/p75NTR is a positive modulator of Trk-mediated survival, and within this context, it islikely that CD271/p75NTR does not directly bind NTs in competition with Trks [162] but acts asa co-receptor, interacting with Trk dimers ligated to active NTs, refining receptor specificity(e.g. increasing specificity for NGF, while restricting NT-3 binding) [163]. This may beresponsible for shifting NT dependence during development coincident with CD271/p75NTR
Neuroblastoma58

expression, which is exemplified by the shift from NT-3/TrkC to NGF/TrkA dependenceobserved during SNS development [164]. CD271/p75NTR may also influence Trk signalling bybinding of the Shc adapter, which also binds to activated Trk, to augment or inhibit Trksignalling [165, 166], and in Trk-complexed form may result in different signalling to that fromTrk dimers alone [147], resulting in differences in capacity to complete differentiation pro‐grams [167] (Fig. 7).
As a pro-apoptotic receptor CD271/p75NTR also exhibits Trk-independent activity. Thecytoplasmic tail of CD271/p75NTR contains death domains and its role in apoptosis has beenclearly demonstrated in CD271/p75NTR exon 3 knockout mice [168]. CD271/p75NTR exon 3knockout mice combined with TrkA knockout mice have highlighted the dual function forCD271/p75NTR in refining innervation and eliminating neuronal excess during early develop‐ment and later in neuronal survival [169, 170]. Apoptosis induced by CD271/p75NTR involvesJNK, phosphorylated c-jun, p53, Bad, Bim and activated caspases [168, 169, 171-174]. Apoptosisinduced by CD271/p75NTR may also involve β-secretase-mediated release of the intra-cyto‐plasmic domain, its subsequent nuclear transport and potential involvement in transcriptionalregulation, together with TRAF6, NRAGE, NADE, NRIF and SC-1. TRAF6 interaction withNRIF has been implicated in the generation of death signals through the activation of JNK[169, 175]. NRAGE interaction with CD271/p75NTR is involved in inducing cell death throughJNK and caspase activation, and is blocked by TrkA [176]. A role for NADE in CD271/p75NTR–mediated apoptosis, involving NGF but not BDNF or NT-3, has been reported [177], whereasCD271/p75NTR interaction with SC-1 has been implicated in cell cycle arrest via transcriptionalrepression of cyclins [178] (Fig. 7). Further advances in the understanding of this effect havecome with the observation that inactive pro-form NT precursors bind CD271/p75NTR receptorswith high affinity and trigger apoptosis at far lower concentrations than active counterparts,which bind with low affinity (Lee et al., 2001). Up to 60% of NTs released by cells are proform[56]. Indeed proNGF induces death in CD271/p75NTR expressing cells, highlighting an oppositeeffect to activated NGF in cells, including sympathetic neurones [56]. The capacity of proNGFto activate CD271/p75NTR but not TrkA is now known to depend upon Sortilin, a 95kDa member
Figure 7. CD271/p75NTR receptor signalling and outcome
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
59

of the Vps10p-domain receptor family [98, 101]. In this interaction, complexes between CD271/p75NTR and Sortilin are augmented by proNGF, which simultaneously binds both receptors toinduce apoptosis. Thus Sortilin acts as an essential co-receptor capable of switching cells thatco-express TrkA and CD271/p75NTR from survival to apoptosis.
CD271/p75NTR receptors, therefore, promote either survival or death in response to NTs,depending upon NT status and the cellular context. Survival through CD271/p75NTR receptorsinvolves NF-κB activated through TRFA6, p62, Interleukin-1 receptor-associated kinase IRAKand receptor interacting protein RIP2 [179]. CD271/p75NTR promotion of axon growth involvesneurotrophin-mediated dissociation of axonal growth inhibitory complexes betweenCD271/p75NTR and the G-protein Rho [180]. Furthermore, the proteolytic shedding of cellsurface CD271/p75NTR releases an intracellular domain that moves to the nucleus and may actas a transcription factor [181].
4.5. Trks A and C are dependence receptors
A classical concept is that NT activation of Trk receptors inhibits default apoptotic programsto promote NT-dependent survival [134]. This concept is considered to involve PI3K/Akt/NF-κB signalling and induction of Bcl-2 inhibitor of mitochondrial apoptosis. In this mechanism,NT depletion results in the turning-off of PI3K/Akt signalling, which reduces Bad phosphor‐ylation and releases it from the chaperone 14-3-3. This results in Bcl-2 and Bcl-XL sequestration[182, 183], reduces FOX03A phosphorylation resulting in nuclear translocation, induces pro-apoptotic FAS, Trail, Puma and BIM transcription [184-188], abrogates CREB and NF-κBsurvival signals [189, 190] and activates pro-apoptotic JNK, inducing BIM expression [188],which together trigger apoptosis (Fig. 8).
Figure 8. TrkA and TrkC receptors and Apoptosis
Recently, however, both TrkA and TrkC have also been characterised as true dependencereceptors. In one study, TrkC but not TrkA or TrkB triggered apoptosis in the absence of NT-3in a variety of cell lines by an activated caspase-dependent cleavage mechanism, releasing apro-apoptotic intracellular TrkC domain capable of inducing caspase-9 dependent death [191].
Neuroblastoma60

In a separate study, TrkA and TrkC but not TrkB induced apoptosis in neurons differentiatedfrom stable transfected embryonic stem cells and promoted loss of all TrkA and TrkC but notTrkB transfected cells with associated loss of nervous system at E13.5 during mouse embryonicdevelopment, through a CD271/p75NTR -mediated mechanism, in which CD271/p75NTR isrecruited as a “hired killer” [192]. Therefore in the absence of ligand, TrkC acts directly as adeath receptor and TrkA death receptor activity appears to depend upon CD271/p75NTR,whereas TrkB does not exhibit death receptor activity (Fig. 8)
5. NTs and NTRs in sympathetic nervous system development
5.1. TrkC and NT–3
TrkC is the only NT receptor expressed during early embryogenesis. During avian develop‐ment TrkC expression coincides with neurulation and is detected in both neural tube andneural plate anlage [193, 194]. TrkC is also expressed in hindbrain rhombomeres 3 and 5. This,however, does not associate with lateral NCC migration, suggesting that either TrkC positiveNCC cells die prior to NCC migration or that they migrate away from these regions. NT-3expression is low at this time and the recent characterisation of TrkC as pro-apoptotic de‐pendency receptor, supports the former hypothesis [191, 192]. Neither TrkC nor NT-3 knock‐out prevent neurulation but do result in neuronal loss from sympathetic ganglia [195-197],indicating that TrkC/NT-3 interactions are not required for neurulation but are required forlater stages of SNS development. Consistent with this, the NT-3 protein is detected at laterdevelopmental stages. There have been no reports concerning the expression of alternativeTrkC isoforms during early development.
During PNS formation, TrkC is expressed by neurogenic pre-migratory and migrating NCCsubsets [194, 198, 199] and TrkC/NT-3 interactions are required prior to NCC arrival atdestination [200]. Indeed, NT-3 acts as a NCC survival factor and promotes NCC proliferationin the presence of somites [201]. Furthermore, somites express NT-3 during this period [198,202], sympathetic neuroblasts and neurons also express TrkC and NT-3, NT-3 is expressed bynon-neuronal sympathetic cells [194, 199, 203], and NT-3 and TrkC expression during this timeis stimulated by neuroregulin, PDGF and CNTF [204]. NT-3 in sympathetic tissues increasesmature neuron numbers by promoting the survival of proliferating neuroblast and theirsubsequent differentiation, without directly effecting proliferation [205]. This temporary effectsubsequently declines with a switch to NGF-dependence [206], associated with reduced TrkCexpression and the induction of TrkA and later CD271/p75NTR expression [204, 207]. NT-3continues to be expressed by both sympathetic neural and non-neural cells [198, 199, 204], byadult non-neural cells [208] and TrkA expression is regulated in part by NT-3 [207]. Therefore,NT-3 acts as both a survival and differentiating factor through TrkA, eventually renderingdifferentiating post-mitotic neurons dependent upon NGF produced by effector tissues. NT-3,at this stage, acts as an autocrine interim and not peripherally derived paracrine factor,corroborated by the lack of target innervation at this time [207]. In support of this, NT-3knockout mice exhibit sympathectomy [106, 197, 209-212] caused principally by neuroblast
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
61

apoptosis, which is partially rescued by exogenous NGF [212]. In the adult, NT-3 continues tobe expressed by a wide range of tissues [202, 203, 213, 214] and, together with NGF, continuesto be important for post-natal sympathetic neuron survival [214, 215]. Consistent with this,exogenous NT-3 promotes target organ sympathetic innervation in NT-3 knockout animals[211, 212], suggesting that the switch from NT-3 to NGF dependence observed in sympatheticneurons in vitro [216] does not actually occur in vivo. This may relate to environmentaldifferences, corroborated by the mitogenic effect of NT-3 on neuroblasts in vitro [201] but notin vivo [217], the susceptibility of TrkC transcription to environmental factors and also by thecapacity of NT-3 to bind and activate TrkA receptors, and in particular TrkAII [76]. This helpsto explain how NT-3 rescues NGF-dependent neurons from NGF depletion and vice versa andis consistent with the characterisation of TrkA as a functional NT-3 receptor in vivo. However,one difference between these two NTs is that exogenous NGF but not NT-3 induces sympa‐thetic ganglia hyperplasia [218].
NT-3 released from effector tissues and acting through TrkA also promotes sympatheticinnervation of target organs [202, 213, 214, 219, 220]. In support of this, effector tissue elimi‐nation induces the death of innervating neurons, which cannot be completely reversed byexogenous NGF alone, and adult sympathetic neurons expressing TrkA are immunoreactivefor both NGF and NT-3 [49, 221, 222]. Therefore, NT-3 plays an important role throughoutsympathetic neuron life-cycle from neuroblast to neuron, acting initially through the TrkCreceptor as an autocrine/paracrine factor stimulator of migration and survival in proliferatingsympathetic neuroblasts and later as a paracrine promoter of sympathetic neuron differentia‐tion, survival and target organ innervation acting through the TrkA receptor. CD271/p75NTR
is also required for optimal neurotrophin sensitivity since CD271/p75NTR deficient dorsal rootand sympathetic neurons exhibit reduced sensitivity to NGF [223].
During sympathoadrenal development, progenitors switch from being dependent upon NT3and TrkC to dependence upon NGF and TrkA, through an intermediate stage of combinedTrkA and TrkC expression. In murine thoracic sympathetic ganglia TrkC expression alone isdetected at E14-15, whereas both TrkA and TrkC expression are detected at E16.5-17 and onlyTrkA at E19.5 [224]. Interestingly, sympathetic chromaffin tissues of the adrenal medulla andparaganglia, which form in parallel to sympathetic ganglia, exhibit differences in NT receptorexpression consistent with upstream progenitor separation. This difference is characterised bythe expression of TrkC but not TrkA by NCCs migrating into the adrenal anlage, at times whenTrkC expression is lost in associated with the induction of TrkA expression by NCCs withinsympathetic ganglia [225].
5.2. TrkA and NGF
Unlike TrkC, TrkA is not expressed during neurulation, NC development or NCC dorsolat‐eral or vetrolateral migration. In rodent development, TrkA is detected at E12.5 within sensorycranial and spinal dorsal root ganglia and subsequently in the paravertebral sympatheticganglia [226]. NGF is expressed during the mid-stage of development initiating within CNSstructures then within PNS structures at later stages of development [227, 228]. Within thedeveloping adrenal gland NGF exhibits a brief period of post-natal expression, whereas NT-3
Neuroblastoma62

is expressed by both the developing and adult adrenal gland [229]. This suggests that TrkA/NGF interactions are of transient importance in adrenal gland development. Consistent withthis, both TrkA and NGF knockout mice exhibit a relatively normal adrenal medullachromaffin cell content, although cholinergic innervation of pre-ganglionic origin is lost inTrkA knockout animals [230], and both chromaffin and SIF cells express TrkA but do notdepend upon NGF for survival. Normal NCC progenitors entering the developing adrenalglands express TrkC and begin to express TrkA upon seeding under the influence of theadrenal environment. This event may depend upon NT-3 and/or NT4/5 expressed by theadrenal cortex anlage, with subsequent chromaffin/SIF differentiation and survival regulat‐ed by these NTs. In contrast, sympathetic neurones in paravertebral sympathetic ganglia,despite their common origin, express TrkA and require NGF for their development,differentiation and survival [229-231]. In support of this, NGF neutralising antibodies do notdelay adrenal development nor induce chromaffin cell degeneration [232]. Differencesbetween human and rodent adrenal development include observations that the adult ratadrenal cortex but not medulla express TrkB or TrkC [229, 230], whereas TrkA immunoreac‐tivity is restricted to the adrenal cortex and TrkC immunoreactivity to the adrenal medullawith no TrkB immunoreactivity detected in the human adult adrenal glands [233]. Interest‐ingly, stress induces a massive release of NGF from salivary glands, which targets adrenalchromaffin cells inducing marked adrenal medullary hyperplasia and catecholaminesynthesis through enhanced TH and BDH expression [234-236]. In chromaffin tissues,sympathoadrenal cells of the carotid body express NGF and TrkA, providing an autocrine/paracrine mechanism [237]. Pre-natal and post-natal differentiating and differentiatedchromaffin cells express TrkA mRNA within the adrenal medulla [238], which increases withdevelopment, at times when NGF expression is all but absent [229]. TrkA knockout elimi‐nates the acetylcholine positive component but does not influence chromaffin content of theadrenal medulla [230], indicating that chromaffin cells, unlike their sympathetic neuronalcousins, do not depend upon NGF/TrkA interactions for survival [232, 239]. Chromaffin cellsdo, however, respond to NGF with acute hyperplasia [235] and eventual neuronal differen‐tiation [240, 241]. In rodents, immature sympathoblasts within sympathetic ganglia cellsexpress TrkA from E14 onwards and express CD271/p75NTR from E16 to birth, in associa‐tion with acquisition of NGF-responsiveness [242]. Differentiated neurons within sympathet‐ic ganglia express TrkA but not NGF [208].
5.3. TrkB, BDNF and NT4/5
TrkB, like TrkA, is also not expressed during neurulation but is expressed by motor progenitorsin hindbrain rhombomere 2 at later stages 9-10 and 12, during avian development, either sideof the floor plate in the caudal midbrain, extending through the hindbrain and into the spinalchord [193]. Alternative TrkB splice variant expression has not been assessed during earlydevelopment.
Following neurulation, TrkB expression is detected within motor neuron progenitors of theventral neural tube and corresponds to BDNF expression by elements within dorsal neuraltube, which coordinate motor neuron development [243]. Consistent with this, both TrkB and
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
63

BDNF knockout mice exhibit the loss of motor and sensory neurons from dorsal root, trige‐minal, nodose/petrosal, vestibular, and geniculate ganglia [244].
During SNS development, TrkB exhibits expression restricted to sub-populations of pre-ganglionic cells [230, 245], and sympathoblasts within coalescing sympathetic ganglia, whichexhibit transient TrkB expression prior to differentiating [225]. Sympathoblasts that expressTrkB within coalescing sympathetic ganglia are non-proliferating but do proliferate inresponse to BDNF in vitro, suggesting that the concentration of BDNF within coalescingsympathetic ganglia is sub-threshold at this time [225]. Pre-ganglionic cells respond to BDNFexpressed and release by effector tissues, resulting in pre-ganglionic innervation [230]. Withinthe adrenal gland, chromaffin cells express NT4/5 but not TrkB, which is weekly expressed bythe adrenal cortex, providing a neurotrophic source for extra-adrenal TrkB expressing pre-ganglionic neurons located in spinal chord segments T7-T10. Thes cells use adrenal medullaryNT-4/5 to project axons into the adrenal medulla in a TrkB-dependent manner [230]. BDNF,on the other hand, is expressed by sympathetic neurones and regulates sympathetic synapticcomplexity [246].
The fact that NT4/5 but not TrkB is expressed within the developing adrenal medulla [230, 238,247] has prompted hypotheses that medullary NT-4/5 may also ligate and activate TrkAreceptors expressed by adrenal medullary neuroblasts and chromaffin cells [63, 80, 229, 230].However, adrenal medullary chromaffin tissues do exhibit rapid stress-induced TrkB expres‐sion, which facilitates the adrenal catecholamine response to stress-induced elevation of bloodbourn BDNF [248].
5.4. CD271/P75NTR
CD271/p75NTR is a neural crest marker that is expressed by NC crest stem cells during earlydevelopment, by NC stem cells in peripheral neural tissues during late development after NCCmigration has ceased, and by nerve associated post natal and adult NC stem cells [249]. CD271/p75NTR expressing adult NC stem cells have been identified as a potential origin for adulttumours of the PNS and NC, including adult NB [249, 250]. Within the human foetal adrenalmedulla, CD271/p75NTR immunoreactivity is detected in nerve fibres and primitive neuroblastclusters, and in the adult adrenal medulla is detected in nerve fibres, ganglion cells andconnective tissue cells of septi but not chromaffin cells [251, 252]. CD271/p75NTR is required fornormal sympathetic neuronal death and the death of damaged neurons [253-255]. CD271/p75NTR knockout alters synapses within sympathetic ganglia and reduces sympathetic targetorgan innervation, consistent with its function in enhancing NT-responsiveness [223, 256].
6. Neurotrophins and neurotrophin receptors in human neuroblastoma
6.1. TrkA and NGF expression in NB
The cloning of the TrkA receptor in 1991 [113] initiated the study of TrkA expression inhuman NBs [257]. This initial report detected an inverse relationship between TrkA mRNA
Neuroblastoma64

levels and N-myc amplification and expression, with low to no TrkA mRNA expressionassociated with poor prognosis. This salient study not only implicated Nmyc in the repres‐sion of TrkA expression but also reported moderate to high TrkA expression in non-Nmycamplified disease. The inverse relationship between Nmyc amplification and expression, lowTrkA expression and advanced stage disease has now been confirmed by many studies, andit is generally accepted that low TrkA expression combined with Nmyc amplification andexpression characterises unfavourable NB and carries poor prognosis [156, 257-270]. Insupport of this, NBs that form in the root ganglia of Nmyc transgenic mice also exhibitreduced TrkA expression [271]. Nmyc amplified NBs, however, also exhibit heterogeneity[272] and a small number of these tumours exhibit high TrkA expression and favourablehistology [270], suggesting that the relationship between NMYC and TrkA in NB is not alwaysstraightforward.
Adding to the observation that moderate to high TrkA levels associate with non-Nmycamplified NB [257, 258], Shimada and colleagues extended the clinical relationship betweenTrkA expression in NB to include outcome, prognostic significance, biological relevance andhistopathological status. They reported that TrkA expression could not distinguish prognosticgroups but could distinguish between Nmyc amplified (low TrkA) and non-Nmyc (high TrkA)amplified NB, between Nmyc amplified NB with favourable (high TrkA) and unfavourable(low TrkA) histology, but could not distinguish between non-Nmyc amplified NB withfavourable histology (moderate to high TrkA) and unfavourable histology (moderate to highTrkA) [270]. This contrasts with some reports [257, 258, 264] but not others [263]. Adult NBsare aggressive non-Nmyc amplified tumours that express high TrkA levels and in such bearsimilarity to non-Nmyc amplified paediatric NBs [156, 268, 273].
Low TrkA expression by Nmyc amplified NBs may relate to an origin along the sympathoa‐drenal lineage within non-TrkA expressing NCC subpopulations that colonize coalescingsympathetic ganglia, paraganglia and adrenal medulla anlage during development [225].Alternatively, reduced TrkA expression in Nmyc amplified NBs may occur post transforma‐tion, since Nmyc represses TrkA transcription by promoting TrkA promoter methylation andTrkA promoter methylation is detected in Nmyc amplified NBs [274, 275].
Moderate to high TrkA levels in non Nmyc-amplified NBs may also relate to cellular originwithin undifferentiated TrkA expressing NCC subpopulations of the sympathetic chain andadrenal primordia [225, 276], or may also occur post-transformation, regulated by NTs, growthfactors and/or cytokines [277-279].
Despite elevated TrkA expression in advanced stage non-Nmyc amplified and in a smallsubgroup of Nmyc amplified NBs with favourable histology [270], full length TrkA exhibits atumour suppressor function in NB models, suggesting that defects in TrkA receptor signallingoccur in NB [280]. Consistent with this, TrkA gene transfection in the absence of CD271/p75NTR restores NGF responsiveness to NB cells, inducing either neuronal differentiation,growth arrest and/or apoptosis in response to NGF [73, 281-284]. Differentiation induced byNGF in TrkA transfected NB cells involves insulin growth factor II [285], RET [286], c-Src [287],protein kinase C-ε [288] and Ras/MAPK/Erk signalling [289, 290], and associates with reducedangiogenic factor expression and angiogenesis resulting in reduced tumorigenic activity [291,
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
65

292]. Furthermore, full length TrkA does not promote genetic instability [73, 293] or invasivebehaviour of NB cells [294]. Apoptosis induced by TrkA in NB cells is p53-dependent [295],involves the cerebral cavernous malformation 2 protein, CCM2 [296], ERK and caspase-7, andcan also be augmented by NGF [297]. As stated above, TrkA may also acts as a true dependencyreceptor, recruiting CD271/p75NTR as a hired killer to promote apoptosis in the absence of NGF[192]. TrkA responsiveness and specificity for NTs is optimised by CD271/p75NTR, which in itsown right acts as a Fas-like apoptosis receptor in response to pro-NTs, supporting thehypothesis that NBs that coexpress TrkA and CD271/p75NTR are favourable tumours that carrygood prognosis [298, 299]. It should be noted, however, that metastatic bone marrow NBinfiltrates induced in SCID mice express TrkA [300] and human NB metastatic bone marrowinfiltrates express CD271/p75NTR [301].
Although, TrkA gene rearrangements have not been described in NB, a c.1810 C>T TrkApolymorphism has been detected in approximately 9% of NB, with potential to predict diseaserelapse in non-Nmyc amplified NB [302].
6.2. The alternative TrkAIII splice variant in NB
Anomalies of TrkA expression that do not support an exclusively tumour suppressing rolefor TrkA in NB, include moderate to high TrkA expression reported in non-Nmyc ampli‐fied advanced stage, metastatic unfavourable NBs. These reports may be explained by TrkAIIIexpression [73], an increase in which was originally reported in advanced stage NB [73], andlater confirmed [156, 303, 304]. Recently, TrkAIII expression in a cohort of 500 NBs was foundto be significantly higher in high TrkA expressing unfavourable NBs compared to high TrkAexpressing favourable NBs (p<0.0001) and to correlate with worse prognosis [156]. Further‐more in the latter study, TrkAIII promoted a cancer stem cell NB phenotype [156], helpingto explain high TrkA levels in unfavourable non-Nmyc amplified NB, adult NB and a subsetof relapsing NBs [73, 156, 270, 303, 304]. In support of this, gene-based outcome predictionstudies focussed on exon-specific expression, have identified a TrkA splicing differencebetween stage I and stage IV non-Myc amplified NBs [305, 306], and an exon gene arrayanalysis using TrkAI/II specific primers, excluding TrkAIII, reported to provide a signifi‐cant prognostic and predictive statistical advantage, associating high TrkAI/II expressionwith better prognosis in NB [307].
TrkAIII represents a developmental and stress-regulated TrkA isoform [73, 77] that exhibitsspontaneous ligand-independent activation and oncogenic activity in NB models [73, 78, 79]and promotes a nestin, CD117, CD133 and Sox2 positive NB stem cell phenotype [156]. Incontrast to full length TrkA, TrkAIII does not restore NGF responsiveness to NB cells norinduce NB cell differentiation or apoptosis [73, 78, 79] but interfers with NGF/TrkA signallingthrough Ras/MAPK, augments genetic instability by promoting centrosome amplification [79]and promotes angiogenesis by altering the equilibrium between MMP-9, VEGF and throm‐bospondin, through IP3K/Atk. Together these phenomena promote NB cell xenograft primary[73] and metastatic tumorigenic activity [308]. Furthermore, TrkAIII increases NB cell resist‐ance to stress, doxorubicin and geladanamycin-induced cytotoxicity [73, 78, 79].
Neuroblastoma66

TrkAIII expression in human NB cells is regulated by hypoxic stress [73] and by agents thatpromote stress within the endoplasmic reticulum (unpublished observations). TrkAIIIsignalling through IP3K/Akt but not Ras/MAPK, combined with interference in NGF/TrkAsignalling, would permit tumours to override NT-dependence, whilst promoting survival andstaminality to provide a selective advantage [73, 78, 79]. Therefore, TrkAIII expression in non-Nmyc amplified NBs may parallel the selective advantage provided by BDNF/TrkB in Nmycamplified NBs [309-313], and NT-3/TrkC in a subgroup of advanced stage NBs [314].
It remains to be determined whether TrkAIII can counteract the pro-apoptotic effects of theSortlin-CD271/p75NTR complex in the presence of pro-NTs or prevent CD271/p75NTR-mediatedapoptosis in the absence of NTs.
6.3. CD271/ p75NTR expression in NB
The CD271/p75NTR low affinity nerve growth factor receptor is a neural crest stem cell marker[249, 250, 315] and is expressed by neural crest-derived melanoma and NB cancer stem cells[250, 316, 317]. In a model of non-Nmyc amplified NB cancer staminality, self replicatingCD133, CD271/p75NTR positive clonogenic stem cells produce both a non-malignant fibromus‐cular lineage and a malignant neuronal (N)-type cell lineage defective in terminal neuronaldifferentiation. Although Trk expression in this NB population remains to be determined,CD271/p75NTR positive self-replicating neural stem cells have been shown to express TrkA,TrkAIII, TrkB and TrkC [73, 318].
Consistent with a restricted pattern of CD271/p75NTR expression in NB, primary human NBshave been reported to not express CD271/p75NTR [252, 259, 319] or to express variable levels ofCD271/p75NTR [251, 319], which either correlate [259] or don’t correlate with TrkA expression[259, 264]. Indeed, differences in CD271/p75NTR co-expression with TrkA have been associatedwith survival, with the co-expression of CD271/p75NTR and TrkA in NB associated with a 100%survival probability, TrkA expression in the absence of CD271/p75NTR with a 62.3% (inter‐mediate) survival probability and no TrkA or p75NTR expression with a 0% probability ofsurvival [259]. Consistent with this, a lack of CD271/p75NTR expression has been reported inNmyc amplified and undifferentiated NB [252, 319, 321] and high CD271/p75NTR expressionreported in more favourable differentiating NBs, ganglioneuromas and ganglioneuroblasto‐mas [251, 252, 299, 320]. However, despite the general concept that high level CD271/p75NTR
expression associates with favourable NB behaviour and outcome [259, 264, 322], CD271/p75NTR expression characterises GD2 positive stage IV metastatic bone marrow NB infiltrates[301] and aggressive adult NBs [268, 323].
Consistent with a general association with favourable NB, CD271/p75NTR exhibits a tumoursuppressor role in NB models, promoting differentiation, apoptosis and reducing tumorigenicactivity [299, 324, 325]. Differentiation promoted by CD271/p75NTR depends upon the molec‐ular context and may involve an IP3K-Akt-mediated BcL-X-dependent survival pathway [326,327] or a TrkA-dependent pathway, in which CD271/p75NTR plays a subtle but critical role inoptimising and prolonging NGF-mediated TrkA activation [328-331]. Indeed, mutation ofCD271/p75NTR within a TrkA context results in proliferation and not differentiation in responseto NGF [332]. Coexpression studies in NB cells have also indicated that, in response to NTs,
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
67

CD271/p75NTR alone induces mild differentiation, TrkA alone causes a more marked differen‐tiation and coexpression an even more marked and rapid differentiation [333].
CD271/p75NTR can acts as either an anti-apoptotic or pro-apoptotic factor, depending uponthe molecular context. At low TrkA to CD271/p75NTR ratios the anti-apoptotic activity of NGFrequires binding to CD271/p75NTR, whilst at higher TrkA to CD271/p75NTR ratios involves amechanism independent of CD271/p75NTR binding [334]. Conversely, NGF also inducesapoptosis in NB cells with a high CD271/p75NTR to TrkA ratios [335]. In the absence of NTsand TrkA, CD271/p75NTR induces apoptosis and inhibits NB tumorigenic activity [299,336-338]. It has been reported that apoptosis, induced by non-ligated CD271/p75NTR isinhibited by non-ligated TrkA but this may reflect spontaneous activation of overex‐pressed TrkA [337]. Furthermore, agents such as prion proteins activate CD271/p75NTR topromote apoptosis in NB cells via NF-κB [339]. In the absence of spontaneous TrkA activationand NT expression, however, the coexpression of CD271/p75NTR and TrkA promotes moremarked apoptosis [333]. In the presence of BDNF CD271/p75NTR interaction with TrkBpromotes NB cell proliferation and survival, through RAS/MAPK and PI3K/AKT/NF-κB [322].These reports suggests that CD271/p75NTR is a pivotal regulator of the disparate behaviourof TrkA and TrkB expressing NBs, exhibiting capacity to enhance differentiation andapoptotic responses in TrkA expressing NBs and enhance proliferation and survival responsesin TrkB expressing NBs, by increasing receptor sensitivity to low NT concentrations andblocking responses to promiscuous NTs.
CD271/p75NTR also interacts with Sortilin and other proteins, complicating potential responsesto both pro- and active NTs. The CD271/p75NTR-Sortilin co-receptor complex augments affinityfor proNGF and induces apoptosis [105, 340]. Furthermore, CD271/p75NTR also interacts withNRIF, TRAF, NRAGE and MAGE proteins to promote apoptosis [340-342].
With respect to the regulation of CD271/p75NTR expression in NB, Nmyc acts as a transcrip‐tional repressor of CD271/p75NTR expression by promoting promoter methylation [274]. Thiseffect can be reversed by HDAC inhibitors, resulting in the resoration of NGF-mediatedapoptosis [274]. This novel pathway, detected in Nmyc amplified NB, may help to explainthe inverse relationship between CD271/p75NTR and Nmyc expression detected in humanNmyc amplified NBs and in root ganglia NBs in Nmyc transgenic mice [271, 343]. Thehistone methyltransferase EZH2A has also been reported to repress CD271/p75NTR provid‐ing an additional Nmyc-independent CD271/p75NTR transcriptional repressing mechanismthat may contribute to the genesis and maintenance of undifferentiated CD271/p75NTR
negative NBs [344].
At the therapeutic level, CD271/p75NTR protects NCC and NB cells from apoptosis induced byantimitotic agents [345], and histone deacetylase inhibitors induce NB cell apoptosis andrestore CD271/p75NTR and TrkA expression [274, 346].
6.4. TrkB and BDNF in NB
Fully spliced TrkB is expressed by a subpopulation of Nmyc amplified NBs [311, 347-349].Despite observations that Nmyc alone is insufficient to induce TrkB expression [348], TrkB
Neuroblastoma68

expression in NB exhibits a positive correlation with Nmyc amplification and expression [309,311, 347, 348, 350]. TrkB expression is stimulated by activated c-erbA in NB cells, unveiling apotential oncogenic receptor tyrosine kinase-mediated mechanism for promoting TrkBexpression [351]. Aggressive unfavourable Nmyc amplified NBs also express BDNF, whichwhen coexpressed with TrkB provides an autocrine/paracrine survival mechanism in tissuesthat do not express NTs [309-313]. Recently, BDNF variants encoding exons 4, 6 and 9 havebeen associated with unfavourable NB outcome [312, 313].
TrkB expression by sympathoblasts subpopulations during SNS development provides apotential origin for TrkB expressing NBs. However, this population proliferates in responseto BDNF in vitro but does express BDNF in vivo [225, 245], suggesting that BDNF expressionmay be acquired at a later stage. TrkB transcription in NB cells is also up regulated by hypoxiainducible factor-1, providing a potential epigenetic mechanism through which tumour-associated hypoxia could augment TrkB expression [352].
In contrast to signals from NGF-activated TrkA, which induces NB cell differentiation andgrowth arrest [73, 324, 353, 354], BDNF activation of TrkB induces partial differentiation in theabsence of growth arrest, through Ret tyrosine kinase [354-357]. BDNF activation of TrkBincreases NB cell survival [358], resistance to chemotherapeutic agents [358-363], augmentsinvasive capacity [294] in cooperation with c-Met [364] and galectin-1 [365], promotes angio‐genesis and angiogenic factor expression [292, 350, 366], augments genetic instability [293](Schulte et al., 2008) and increases metastatic behaviour by inhibiting anoikis [367]. In contrast,NB cells expressing truncated TrkB lacking the tyrosine kinase domain, display a moredifferentiated phenotype [311] and this receptor is more frequently detected in ganglioneur‐oblastomas and ganglioneuromas. Consistent with this, truncated TrkB overexpression in NBcells promotes differentiation suggesting that this receptor variant promotes a more benignphenotype [368]. Oxidative stress up-regulates the expression of full length TrkB relative tothe truncated isoform, providing an additional epigenetic mechanism for regulating TrkBinvolvement in NB [369].
6.5. TrkC and NT3 in NB
TrkC is expressed by migrating NCC progenitors, sympathoblasts and sympathetic neurons[194, 201, 203], providing many potential origins for TrkC expressing NBs. High level TrkCexpression in low stage NBs is associated with favourable outcome (309, 310, 349, 353, 370-372],and is often accompanied by TrkA expression [257, 258, 309]. Recently, however, a subset ofadvanced stage IV NBs has been identified that exhibit high level NT-3 and TrkC co-expression,providing an autocrine/paracrine survival and proliferation mechanism for selecting theseNBs in tissues that do not express NT-3 [314]. This expression pattern bears close similarity tomigrating, proliferating NCC sympathoblasts prior to sympathetic neuronal differentiation,which also coexpress NT-3 and TrkC [194, 201, 203], identifying a potential origin for this NBsubset. TrkC expression in NB, like that of TrkA, inversely correlates with Nmyc amplificationand expression, and Nmyc amplified NBs either do not express TrkC at all, or express truncatedTrkC [371, 372]. With the exception of NBs that coexpress NT3 and TrkC [314], the co-expression of TrkC, TrkA and CD271/p75NTR in NB carries the best prognosis and associates
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
69

more frequently with spontaneous regression, differentiation and chemo-responsiveness [100,258, 259, 333, 370, 371]. The TrkC gene, however, encodes multiple NT-3 receptors with distinctbiological properties and substrate specificities [89, 373] and, although TrkC gene rearrange‐ments in NB have not been reported, the effect of differential TrkC isoform expression in NBremains to be elucidated.
Association between high TrkC expression and favourable NB outcome, in the absence of NT-3[333, 371], is consistent with pro-apoptotic TrkC dependency receptor function, whichpromotes apoptosis in the absence of CD271/p75NTR and NT expression [191, 192]. Further‐more, NT-3 activation of TrkC induces NB cell differentiation [374] and the co-expression ofTrkC with CD271/p75NTR lowers tumorigenic potential and tumour growth [375] but mayprotect NB cells from doxorubicin and cisplatin cytotoxicity [375].
With respect to the transcriptional regulation of TrkC, Nmyc silencing increases TrkC expres‐sion in human NB cells [376], corroborating the inverse relationship reported for TrkCexpression and Nmyc amplification [371,372]. TrkC expression, furthermore, is abrogated bythe activation of c-erbA, providing a potential oncogenic tyrosine kinase-mediated mechanismfor repressing TrkC expression in NB [351]. Retinoic acid induces TrkC expression in humanNB cells, restoring NT-3-dependent differentiation [152]. Retinoids also induce the expressionof microRNAs-9, 125a and 125b that repress truncated kinase domain-deleted TrkC, resultingin altered growth and highlighting a role for the truncated TrkC receptor in the regulation ofNB growth and differentiation [377]. MiR-151-3p represses full length TrkC expression,whereas miRs-128, 485-3p, 765 and 768-5p repress truncated TrkC expression in NB cells [378],indicating that full length and truncated TrkC receptors are regulated by different miRs,linking NT-mediated processes to miR expression in NB.
6.6. General considerations on NT and NTR expression patterns in NB
The concept that different NT and NTR receptor expression profiles characterise NB subsetsand that these differences are involved in divergent NB behaviour and therapeutic suscepti‐bility, continues to evolve with potential to improve prognosis and therapeutic choice, whilstidentifying novel potential therapeutic targets.
The hypothesis that high TrkA, high TrkC and/or high CD271/p75NTR expression alwaysassociate with low disease stage and better prognosis in NB is clearly not the case. Moderateto high levels of TrkA, TrkC and/or CD271/p75NTR can also characterise advanced stage andrelapsing non-Nmyc amplified NBs and a subset of Nmyc amplified NB with favourablehistology (see section 6.1). However, high TrkB expression appears to distinguish advancedstage Nmyc amplified from non-Nmyc amplified NB and carries poor prognosis associatedwith potential therapeutic resistance (see section 6.4). It is also now apparent that NTRs canbe expressed as different isoforms with altered biological activity and can interact with oneother and with a variety of ancillary proteins to modulate function (see section 6.3), compli‐cating prognosis and potential therapeutic outcome, as outlined below (Fig. 9).
Neuroblastoma70
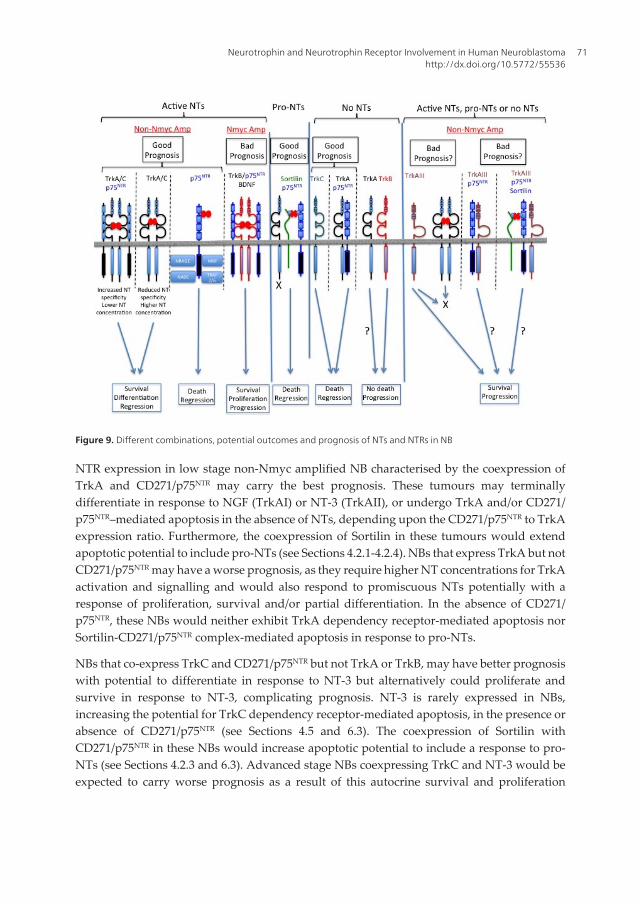
Figure 9. Different combinations, potential outcomes and prognosis of NTs and NTRs in NB
NTR expression in low stage non-Nmyc amplified NB characterised by the coexpression ofTrkA and CD271/p75NTR may carry the best prognosis. These tumours may terminallydifferentiate in response to NGF (TrkAI) or NT-3 (TrkAII), or undergo TrkA and/or CD271/p75NTR–mediated apoptosis in the absence of NTs, depending upon the CD271/p75NTR to TrkAexpression ratio. Furthermore, the coexpression of Sortilin in these tumours would extendapoptotic potential to include pro-NTs (see Sections 4.2.1-4.2.4). NBs that express TrkA but notCD271/p75NTR may have a worse prognosis, as they require higher NT concentrations for TrkAactivation and signalling and would also respond to promiscuous NTs potentially with aresponse of proliferation, survival and/or partial differentiation. In the absence of CD271/p75NTR, these NBs would neither exhibit TrkA dependency receptor-mediated apoptosis norSortilin-CD271/p75NTR complex-mediated apoptosis in response to pro-NTs.
NBs that co-express TrkC and CD271/p75NTR but not TrkA or TrkB, may have better prognosiswith potential to differentiate in response to NT-3 but alternatively could proliferate andsurvive in response to NT-3, complicating prognosis. NT-3 is rarely expressed in NBs,increasing the potential for TrkC dependency receptor-mediated apoptosis, in the presence orabsence of CD271/p75NTR (see Sections 4.5 and 6.3). The coexpression of Sortilin withCD271/p75NTR in these NBs would increase apoptotic potential to include a response to pro-NTs (see Sections 4.2.3 and 6.3). Advanced stage NBs coexpressing TrkC and NT-3 would beexpected to carry worse prognosis as a result of this autocrine survival and proliferation
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
71

mechanism that may also extend to NBs expressing TrkC but not NT-3 in tissues that expressNT-3, and could be further optimised by co-expression of CD271/p75NTR (see Section 6.3).
High levels of BDNF and TrkB expression in Nmyc-amplified NBs, in the absence of TrkA,TrkC and CD271/p75NTR, carries the worse prognosis as a result of autocrine/paracrine BDNF-mediated TrkB activation, which would be expected to promote proliferation, survival andmetastatic capacity. Furthermore in the absence of BDNF, TrkB would not be expected topromote apoptosis, as TrkB does not act as a dependence receptor (see Sections 6.4). As withother Trk receptors, TrkB co-expression with CD271/p75NTR would be expected to optimiseNT-specificity and responsiveness, which would be expected to further promote aggressivebahaviour in TrkB expressing NBs.
NBs that express CD271/p75NTR but not Trks may carry better prognosis, as they would beexpected to respond to active NTs with an apoptotic response and if co-expressed with Sortilinin the absence of Trks, would also be expected to exhibit an apoptotic response to pro-NTs,which comprises up to 50% of secreted NTs (see Sections 4.2.2 and 6.3).
Non-Nmyc amplified NBs that express TrkAIII may carry worse prognosis, as spontaneousTrkAIII activation would override NT-dependency, provide a selective growth advantage intissues including those that do not express NTs, promote NB cell stamilality, survival,angiogenesis and genetic instability, resulting in a more tumorigenic, metastatic and stress-resistant phenotype (see Sections 4.2.1 and 6.2). Although it remains to be elucidated whetherTrkAIII may interfere with CD271/p75NTR –mediated apoptosis in the presence or absence ofSortilin, its expression in NB may represent the biological equivalent to BDNF/TrkB expressionin Nmyc amplified NB and TrkC/NT3 expresssion in a subset of advanced stage NBs, as anindicator of poor prognosis.
7. Potential therapeutic approaches
7.1. Trk kinase inhibitors
Trk kinase inhibitors would be more suitable for use in advanced stage Nmyc amplified TrkBexpressing NBs and advanced stage unfavourable non-Nmyc amplified NBs that express theTrkAIII oncogene but may also reduce survival in NBs expressing full length TrkA and TrkCand their corresponding NTs.
Therapeutic Trk kinase inhibitors include the selective Trk kinase inhibitors AZ-23 and AZ623,which inhibit Trk kinase activity at low nanomolar concentrations. AZ-23 has shown efficacyfollowing oral administration in a TrkA-driven mouse allograft NB model [379], whereasAZ623 inhibits BDNF-mediated signalling and NB proliferation, and when combined withtopotecan prolongs the inhibition of tumour regrowth and reduces chemo and radio thera‐peutic resistance [380, 381]. Lestaurtinib (CEP-701) is a small-molecule receptor tyrosine kinaseinhibitor that competitively inhibits ATP binding to the Trk kinase domain at nanomolarconcentrations. This compound not only inhibits the tyrosine kinase activities of full-lengthTrk receptors but also inhibits the kinase activity of the alternative TrkAIII splice variant [73,
Neuroblastoma72

78,79]. Lestaurtinib inhibits NB growth in vitro and in vivo, and substantially enhances theefficacy of conventional chemotherapy, such as 13-cis-retinoic acid, ferenteride and bevacizu‐mab, presumably by inhibiting autocrine TrkB/BDNF [382-384] and/or spontaneous TrkAIIIactivity [73]. Lestaurtinib is an active metabolite of the Trk kinase inhibitor CEP-751, and ismore suitable for clinical trials, as it can be administered orally [384, 385]. These Trk tyrosinekinase inhibitors not only target tumour promoting effects of Trk receptor activation but alsoTrk-mediated chemotherapeutic resistance, which has been attributed not only to TrkB [348,358-363] but also to TrkC [375], fully spliced TrkA [375] and TrkAIII [73, 78, 79]. CEP-701synergises with retinoids in the treatment of NB by inhibiting TrkB activity [386].
Nifutimox, a drug used for years to treat Chagas disease, is also currently in clinical trials forrefractory or relapsed NB, and has been shown to suppress TrkB-mediated Akt activation andinduce caspase-dependent apoptosis of NB cells in vitro and in vivo [387].
7.2. TrkAIII inhibitors
Tyrosine kinase inhibitors K252a and CEP-701 inhibit TrkAIII tyrosine kinase activity. TrkAIIIactivity is also inhibited by the Hsp90 inhibitor geladanamycin and its clinically relevantanalogues 17-AAG and 17-DMAG, and by the ARF inhibitor Brefeldin A (BFA) [78, 79]. CEP701inhibits TrkAIII activity and TrkAIII-induced centrosome amplification at nanomolar concen‐trations, whereas BFA reversibly inhibits spontaneous TrkAIII activation in association withdisruption of the Golgi Network and the endoplasmic reticulum/Golgi Network intermediatecompartment [78, 78]. Geldanamycin and its analogues reversibly inhibit TrkAIII tyrosinekinase activity and reduce proliferation of TrkAIII expressing NB cells in vitro [78]. Inhibitorsof TrkAIII activity, however, do not inhibit TrkAIII expression nor promote TrkAIII elimina‐tion but cause retention within the endoplasmic reticulum, with potential to induce an ERstress response. This may help to explain the high level of resistance to GA-mediated cytotox‐icity exhibited by TrkAIII but not TrkAI transfected NB cells, despite inhibition of TrkAIIIactivity [78, 79]. This suggests that, in addition to other off target effects, reversible TrkAIIItyrosine kinase inhibitors may increase stress-resistance by promoting TrkAIII-ER retentionand inducing an ER stress response. Consistent with this, geldanamycin selects slow growingTrkAIII expressing NB cells from mixed populations, with TrkAIII re-activation post drug-removal, suggesting a mechanism for potential post therapeutic relapse [78]. To counter this,we have also developed a specific peptide nucleic acid (PNA) inhibitor of TrkAIII expressionbased upon the novel exon 5/8 splice junction (TrkAIII PNA conjugate (KKAA)4-GGCCGGGA‐CAC) [78, 79] for use in combination with with TrkAIII tyrosine kinase inhibitors, to maximisetherapeutic efficacy.
7.3. Agents that conserve Trk tyrosine phosphorylation and facilitate signal transduction
TrkA activation and signal transduction is fundamental for NB differentiation and the loss ofTrkA expression or defective activation and/or signalling probably contributes to NB patho‐genesis. Agents that optimise TrkA activation and facilitate subsequent signal transductionmay, therefore, overcome defective TrkA signalling and restore differentiation and/orapoptotic responses to NTs. In this context, a novel cyclophane compound CPPy, with low
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
73

toxicity, has been shown to facilitate NGF-induced TrkA signal transduction through RAS/MAPK and to induce NB differentiation [388]. Since, CD271/p75NTR optimizes TrkA responsesto NTs and augments NT specificity, agents such as CPPy may be particularly useful in NBsthat express TrkA but not CD271/p75NTR.
7.4. DNA methylation and HDACs inhibitors
Recent reports have identified promoter methylation as an important mechanism in thetranscriptional repression of TrkA and CD271/p75NTR in NB [274, 389, 390]. Therapies thatreverse or inhibit DNA methylation may, therefore, be useful in malignant NBs to restore theexpression of favourable NB genes. In support of this, the DNA methylation inhibitor 5-aza-2’-deoxycytidine and histone deacetylase inhibitors 4-phenylbutyrate, trichostatin A andRomidepsin, have been shown to restore TrkA and CD271/p75NTR expression in NB cells,decrease proliferation, reduce tumorigenicity and promote caspase-dependent apoptosis [291,346, 390]. Romidepsin is presently in clinical trials [346].
7.5. Liposome targeting of TrkB expressing cells
Considering the importance of TrkB in advanced stage Nmyc amplified NB, a recent reporthas characterised liposomes that target TrkB expressing cells, providing the opportunity todeliver nanotherapeutic cargos to TrkB expressing cells within NBs [392].
8. Concluding remarks
The complex nature of NT and NTR expression during normal development of the sympatheticnervous system is reflected in the different patterns of NT and NTR expression exhibited byhuman NB, which is consistent with their NCC origin at different stages along the differenti‐ating sympathoadrenal lineage. The different biological potentials of TrkA, TrkB, TrkC,CD271/p75NTR and Sortilin receptors expressed alone or in different combinations, range frompromotion of proliferation and/or differentiation to survival and/or apoptosis and to chemo‐therapeutic resistance. This complexity is increased by the potential of each receptor to beexpressed as a functionally altered alternative splice variant, the recent characterisation ofTrkA and TrkC as true dependency receptors, and the pro-apoptotic behaviour of the CD271/p75NTR -Sortilin complex, providing an exciting array of new potential ways to restore and/ormodulate Trks, CD271/p75NTR and Sortilin behaviour for therapeutic purposes, based uponaccurate characterisation of NT and NTR expression profiles in individual tumours.
Acknowledgements
This work was supported by grants form PRIN, AIRC and the Maugieri Foundation.
Neuroblastoma74

Author details
Pierdomenico Ruggeri1, Antonietta R. Farina1, Lucia Cappabianca1, Natalia Di Ianni1,Marzia Ragone1, Stefania Merolle1, Alberto Gulino2,3 and Andrew R. Mackay1
1 Department of Applied Clinical and Biotechnological Science, University of L’Aquila,Coppito II, L’Aquila, Italy
2 Department of Molecular Medicine, University of Rome “La Sapienza”, Rome, Italy
3 Neuromed Institute, Pozzilli, Italy
References
[1] Voorhess, M. L. & Gardner, L. S. (1961). Urinary excretion of neurepinephrine and 3-methoxy-4-hydroxy mandelic acid by children with neuroblastoma. Journal of Clinicaland Endocrinological Metabolism, 21, 321-355.
[2] Jiang, M.; Stanke, J. & Lahti, J. M. (2011). The connections between neural crest devel‐opment and neuroblastoma. Current Topics in Developmental Biology, 94, 77-127.
[3] Fisher, J. P. H. & Tweddle, D. A. (2012). Neonatal neuroblastoma. Seminars in Fetaland Neonatal Medicine,17,207-215.
[4] Endo, Y.; Osumi, N. & Wakamatsu, Y. (2002). Bimodal functions of Notch-mediatedsignalling are involved in neural crest formation during avian ectoderm develop‐ment. Development, 129, 863-873.
[5] Cornell, R. A. & Eisen, J. S. (2002). Delta/Notch signaling promotes formation of ze‐brafish neural crest by repressing Neurogenin 1 function. Development, 129,2639-2648.
[6] Kalcheim, C. & Burstyn-Cohen, T. (2000). Early stages of neural crest ontogeny: for‐mation and regulation of cell delamination. International Journal of Developmental Biol‐ogy, 49, 105-116.
[7] Kasemeier-Kulesa, J. C.; Kulesa, P. M. & Lefcort, F. (2005). Imaging neural crest celldynamics during formation of dorsal root ganglia and sympathetic ganglia. Develop‐ment, 132, 235-245.
[8] Gammill, L. S. & Roffers-Agarwal, J. (2010). Division of labour during trunk neuralcrest development. Developmental Biology, 344, 555-565.
[9] Schwarz, Q.; Maden, C. H.; Davidson, K. & Ruhrberg, C. (2009). Neuropilin-mediat‐ed neural crest cell guidance is essential to organise sensory neurons into segmenteddorsal root ganglia. Development, 136, 1785-1789.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
75

[10] Anderson, D. J. (2000). Genes, lineages and the neural crest a speculative view. Philo‐sophical Transactions of the Royal Society of Biological Sciences, 355, 953-964.
[11] McCorry, L. K. (2007). Physiology of the autonomic nervous system. American Journalof Pharmaceutical Education, 71, 1-11.
[12] Young, H. M.; Cane, K. N. & Anderson, C. R. (2011). Development of the autonomicnervous system: a comparative view. Autonomic Neuroscience: Basic and Clinical, 165,10-27.
[13] Janig, W. & Habler, H. J. (2000). Specificity in the organisation of the autonomic nerv‐ous system: a basis for precise neural regulation of homeostatic and protective bodyfunctions. Progress in Brain Research, 122, 351-367.
[14] Shepard, D. M. & West, G. B. (1952). Noradrenalin and accessory chromaffin tissue.Nature, 170, 42-43.
[15] Tischler, A. S.; Ruzicka, L. A. & Riseberg, J. C. (1992). Immunocytochemical analysisof chromaffin cell proliferation in vitro. Journal of Histochemistry and Cytochemistry, 40,1043-1045.
[16] Schober, A. & Unsicker, K. (2001). Growth and neurotrophic properties regulatingdevelopment and maintenance of sympathetic preganglionic neurons. InternationalReview of Cytology, 205, 37-76.
[17] Unsicker, K. & Kriegelstein, K. (1996). Growth factors in chromaffin cells. Progress inNeurobiology, 48, 307-324.
[18] Huber, K.; Kalcheim, C. & Unsicker, K. (2009). The development of the chromaffincell lineage from the neural crest. Autonomic Neuroscience: Basic and Clinical, 151,10-16.
[19] Loring, J. F. & Erickson, C. A. (1987). Neural crest cell migratory pathways in thetrunk of the chick embryo. Developmental Biology, 121, 220-236.
[20] Teillet, M. A.; Kalcheim, C, Le Dourain, N. M. (1987). Formation of the dorsal rootganglia in the avian embyo: segmental origin and migratory behaviour of neuralcrest progenitor cells. Developmental Biology, 120, 329-347.
[21] Waring, H (1935). The development of the adrenal gland of the mouse. QuarterlyJournal of Microscopic Science. Vol. LXXVIII.
[22] Reiprich, S.; Stolt, C. C.; Schreiner, S.; Parlato, R. & Wegner, M. (2008). SoxE proteinsare differentially required in mouse adrenal gland development. Molecular and Cellu‐lar Biology, 19, 1575-1586.
[23] Shimada, H. (2005). In situ neuroblastoma: An important concept related to the natu‐ral history of neural crest tumors. Pediatric and Developmental Pathology, 8, 305-306.
Neuroblastoma76

[24] Anderson, D. J. & Axel, R. (1986). A bipotential neuroendocrine precursor whosechoice of cell fate is determined by NGF and glucocorticoids. Cell, 47, 1079-1090.
[25] Anderson, D. J.; Carnahan, J. F.; Michelsohn, A. & Patterson, P. H. (1991). Antibodymarkers identify a common progenitor to sympathetic neurons and chromaffin cellsin vivo and reveal the timing of commitment to neuronal differentiation in the sym‐pathoadrenal lineage. Journal of Neuroscience, 11, 3507-3519.
[26] Reissmann, E.; Ernsberger, U.; Francis-West, P. H.; Rueger, D.; Brickell, P. M. & Rohr‐er, H. (1996). Involvement of bone morphogenetic protein-4 and bone morphogeneticprotein-7 in the differentiation of the adrenergic phenotype in developing sympa‐thetic neurons. Development, 122, 2079-2088.
[27] Shah, N. M.; Groves, A. K. & Anderson, D. J. (1996). Alternative neural crest cell fatesare instructively promoted by TGFbeta superfamily members. Cell, 85, 331-343.
[28] Bilodeau, M. L.; Boulineau, T.; Greulich, J. D.; Hullinger, R. L. & Andrisani, O. M.(2001). Differential expression of sympathoadrenal lineage determining genes andphenotypic markers in cultured primary neural crest cells. In Vitro Cellular and Devel‐opmental Biology: Animal, 37, 185-192.
[29] Ernsberger, U.; Esposito, L.; Partimo, S.; Huber, K.; Franke, A.; Bixby, J. L.; Kalcheim,C. & Unsicker, K. (2005). Expression of neuronal markers suggests heterogeneity ofchick sympathoadrenal cells prior to invasion of the adrenal anlagen. Cell and TissueResearch, 319, 1-13.
[30] Gut, P.; Huber, K.; Lohr, J.; Bruhl, B.; Oberle, S.; Treier, M.; Ernsberger, U.; Kalcheim,C. & Unsicker, K. (2005). Lack of an adrenal cortex in Sf1 mutant mice is compatiblewith the generation of and differentiation of chromaffin cells. Development, 132,4611-4619.
[31] Moriguchi, T.; Lim, K. C. & Engel, J. D. (2007). Transcription factor networks specifysympathetic and adrenal chromaffin cell differentiation. Functional Developmental Em‐bryology, 1, 130-135.
[32] Pattyn, A.; Morin, X.; Cremer, H.; Goridis, C. & Brunet, J. F. (1999). The homeoboxgene Phox2B is essential for the development of autonomic neural crest derivatives.Nature, 399, 366-370.
[33] Brunet, J. F. & Pattyn, A. (2002). Phox2 genes-from patterning to connectivity. CurrentOpinions in Genetic & Development, 12, 435-440.
[34] Guillemont, F.; Lo, L. C.; Johnson, J. E.; Auerbach, A.; Anderson, D. J. & Joyner, A. L.(1993). Mammalian achaete-scute homolog 1 is required for the early development ofolfactory and autonomic neurons. Cell, 75, 463-476.
[35] Hirsch, M. R.; Tiveron, M. C.; Guillemont, E.; Brunet, J. F. & Goridis, C. (1998). Con‐trol of noradrenergic differentiation and Phox2a expression by MASH1 in the centraland peripheral nervous system. Development, 125, 599-608.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
77

[36] Mellitzer, G.; Bonne, S.; Luco, R. F.; Van de Casteele, M.; Lenne-Samuel, N.; Collom‐bat, P.; Mansouri, A.; Lee, J.; Lan, M.; Pipeleers, D.; Nielsen, F. C.; Ferrer, J.; Grad‐wohl, G. & Heimberg, H. (2006). 1A1 in NGN-3-dependent and essential fordifferentiation of the endocrine pancreas. EMBO Journal, 25, 1344-1352.
[37] Gierl, M. S.; Karoulias, N.; Wende, H.; Strehle, M. & Birchmeier, C. (2006). The zincfinger factor Insm1 (IA-1) is essential for the development of pancreatic beta cells andintestinal endocrine cells. Genes and Development, 20, 2465-2478.
[38] Morikawa, Y.; D’Atreaux, F.; Gershon, M. D. & Cserjesi, P. (2007). Hand2 determinesthe noradrenergic phenotype in the mouse sympathetic nervous system. Developmen‐tal Biology, 307, 114-126.
[39] Schmidt, M.; Lin, S.; Pape, M.; Ernsberger, U.; Stanke, M.; Kobayashi, K.; Howard, M.J.; and Rohrer, H. (2009). The bHLH transcription factor Hand2 is essential for themaintenance of noradrenergic properties in differentiated sympathetic neurons. De‐velopmental Biology, 15, 191-200.
[40] Tsarovina, K.; Pattyn, A.; Stubbusch, J.; Muller, F.; van der Wees, J.; Schneider, C.;Brunet, J-F. & Rohrer, H. (2004). Essential role of GATA transcription factors in sym‐pathetic neuron development. Development, 131, 4775-4786.
[41] Finotto,S.; Kriegelstein, K.; Schober, A.; Diemling, F.; Lindner, K.; Bruhl, B.; Beier, K.;Metz, J.; Garcia-Arraras, J. E.; Roig-Lopez, J. L.; Monighan, P.; Schmid, W.; Cole, T. J.;Kellendonk, C.; Tronche, F.; Schutz, G. & Unsicker, K. (1999). Analysis of mice carry‐ing targeted mutations of the glucocorticoid receptor gene argues against an essentialrole for glucocorticoid signalling for generating adrenal chromaffin cells. Develop‐ment,126,2935- 2944.
[42] Parlato, R.; Otto, C.; Tuckermann, J.; Stotz, S.; Kaden, S.; Grone, H. J.; Unsicker, K. &Schutz, G. (2009). Conditional inactivation of glucocorticoid receptor gene in dopa‐mine-beta-hydroxylase cells impairs chromaffin cell survival. Endocrinology, 150,1775-1781.
[43] Lohr, J.; Gut, P.; Karch, N.; Unsicker, K. & Huber, K. (2006). Development of adrenalchromaffin cells in Sf1 heterozygous mice. Cell and Tissue Research, 325, 437-444.
[44] Wassberg, E.; Hedborg, F.; Skoldenberg, E.; Stridsberg, M. & Christofferson, R.(1999). Inhibition of apoptosis induces chromaffin differentiation and apoptosis inneuroblastoma. American Journal of Pathology, 154, 395-403.
[45] Hedborg, F.; Ulleras, E.; Grimelius, L.; Wassberg, E.; Maxwell, P. H.; Hero, B.; Bert‐hold, F.; Schilling, F.; Harms, D.; Sabdstedt, B. & Franklin, G. (2003). Evidence for hy‐poxia-induced neuronal-chromaffin metaplasia in neuroblastoma. FASEB Journal, 17,598-609.
[46] Hedborg, F.; Fischer Colbrie, R.; Ostlin, N.; Sandstedt, B.; Tran, M. G. B. & Maxwell,P. H. (2010). Differentiation in neuroblastoma: diffusion-limited hypoxia induces
Neuroblastoma78

neuro-endocrine secretory protein 55 and other markers of a chromaffin phenotype.PloS ONE 5, e12825.
[47] Bibel, M. & Barde, Y-A. (2000). Neurotrophins: key regulators of cell fate and cellshape in the vertebrate nervous system. Genes and Development, 14, 2919-2937.
[48] Kaplan, D. R. & Miller, F. D. (2000). Neurotrophin signal transduction in the nervoussystem. Current Opinions in Neurobiology, 10, 381-391.
[49] Oppenheim, R. W. (1991). Cell death during development of the nervous system. An‐nual Reviews in Neuroscience, 14, 453-501.
[50] Ernsberger, U. (2009). Role of neurotrophin signalling in the differentiation of neu‐rons from dorsal root ganglia and sympathetic ganglia. Cell and Tissue Research, 336,349-384.
[51] Levi-Montalcini, R. & Brooker, B. (1960). Excessive growth of the sympathetic gan‐glia evoked by a protein isolated from mouse salivary glands. Proceeding of the Na‐tional Acadamy of Science. USA, 46, 373-384.
[52] Leibrock, J.; Lottspeich, F.; Hohn, A.; Hofer, M.; Hengerer, B.; Masiakowski, P.; Thoe‐nen, H. & Barde, Y-A. (1989). Molecular cloning and expression of brain-derived neu‐rotrophic factor. Nature, 341, 149-152.
[53] Ernfors, P. C.; Ibanez, F.; Ebendal, T.; Olson, L. & Persson, H. (1990). Molecular clon‐ing and neurotrophic activities of a protein with similarities to nerve growth factor:developmental and topographical expression in the brain. Proceeding of the NationalAcadamy of Sciences, USA, 87, 5454-5458.
[54] Hallbook, F.; Ibanez, C. F. & Persson, H. (1991). Evolutionary studies of the nervegrowth factor family reveal a novel member abundantly expressed in Xenopus ova‐ry. Neuron, 6, 845-858.
[55] McDonald, N. Q.; Lapatto, R.; Murray-Rust, J.; Gunning, J.; Wlodawaer, A. & Blun‐dell, T. L. (1991). New protein fold revealed by a 2.3-A resolution crystal structure ofnerve growth factor. Nature, 354, 411-414.
[56] Lee, R.; Kermain, P.; Teng, K. K. & Hempstead, B. L. (2001) Regulation of cell surviv‐al by secreted neurotrophins. Science, 294, 1945-1948.
[57] Thoenan, H. (1999). Neurotrophins and neuronal plasticity. Science, 270, 593-598.
[58] Le, A. P. & Friedman, W. J. (2012). Matrix metalloproteinase-7 regulates cleavage ofpro- nerve growth factor and is neuroprotective following Kainic acid-induced seiz‐ures. Journal of Neuroscience, 32, 703-712.
[59] Lu, B. & Figurov, A. (1997). Role of neurotrophins in synapse development and plas‐ticity. Reviews in Neuroscience,8, 1-12
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
79

[60] Lu, B.; Pang, P. T. & Woo, N. H. (2005). The yin and yang of neurotrophin action.Nature Reviews in Neuroscience, 6, 603-614.
[61] Dracopoli, N. C. & Meisler, M. H. (1990). Mapping the human amylase gene clusteron the proximal short arm of chromosome 1 using a highly informative (CA)n repeat.Genomics, 7, 97-102.
[62] Maisonpierre, P. C.; Le Beau, M. M.; Espinosa, R 3rd.; Ip, N. Y.; Belluscio, L.; de laMonte, S. M.; Squinto, S.; Furth, M. E. & Yancopoulos, G. D. (1991). Human and ratbrain-derived neurotrophic factor and neurotrophin-3 gene structures, distributions,and chromosomal localizations. Genomics, 10, 558-568.
[63] Ip, N. Y.; Ibanez, C. F.; Nye, S. H.; McClain, J.; Jones, P. F.; Gles, R.; Belluscio, L.; LeBeau, M. M.; Espinosa, R 3rd. & Squinto, S. P. (1992). Mammalian neurotrophin-4:structure, chromosomal localization, tissue distribution, and receptor specificity. Pro‐ceeding of the National Academy of Sciences, USA, 89, 3060-3064.
[64] Patapoutian, A. & Reichardt, L. F. (2001). Trk receptors: mediators of neurotrophinaction. Current Opinions in Neurobiology, 11, 272-280.
[65] Klein, R.; Jing, S.; Nanduri, V.; O’Rourke, E. & Barnacid, M. (1991). The trk proto-on‐cogene encodes a receptor for nerve growth factor. Cell, 65, 198-197.
[66] Martin-Zanca, D.; Hughes, S. H. & Barbacid, M. (1986). A human oncogene formedby the fusion of truncated tropomyosin and protein tyrosine kinase sequence. Nature,319, 743-748.
[67] Martin-Zanca, D.; Oskam, R.; Mitra, G.; Copeland, T. & Barbacid, M. (1989). Molecu‐lar and biochemical characterisation of the human Trk oncogene. Molecular and Cellu‐lar Biology, 9, 24-33.
[68] Greco, A.; Villa, R. & Pierotti, M. A. (1996). Genomic organization of the humanNTRK1 gene. Oncogene, 13, 2463-2466.
[69] Weier, H-U. G.; Rhein, A. P.; Shadravan, F.; Collins, C. & Polikoff, D. (1995). Rapidphysical mapping of the human protoncogene (NTRK1) to human chromsome1q21-22 by P1 clone selection, fluorescence in situ hybridisation (FISH), and comput‐er-assisted microscopy. Genomics, 26, 390-393.
[70] Valent, A.; Danglot, G. & Bernheim, A. (1997). Mapping of the tyrosine kinase recep‐tors trkA (NTRK1), trkB (NTRK2) and trkC (NTRK3) to human chromosomes 1q22,9q22 and 15q25 by fluorescence in situ hybridization. Europrean Journal of human ge‐netics, 5, 102-104.
[71] Dubus, P.; Parrens, M.; El-Mokhtari, Y.; Ferrer, J.; Groppi, A. & Merlio, J. P. (2000).Identification of novel TrkA variants with deletions in the leucine-rich motifs of theextracellular domain. Journal of Neuroimmunology, 107, 42-49.
Neuroblastoma80

[72] Barker, P. A.; Lomen-Hoerth, C.; Gensch, E. M.; Meakin, S. O.; Glass, D. J. & Shooter,E. M. (1993). Tissue specific alternative splicing generates two isoforms of the TrkAreceptor. Journal of Biological Chemistry, 268, 15150-15157.
[73] Tacconelli, A., Farina, A. R.; Cappabianca, L.; DeSantis, G.; Tessitore, A.; Vetuschi, A.;Sferra, R.; Rucci, N.; Argenti, B.; Screpanti, I.; Gulino, A. & Mackay, A. R. (2004).TrkA alternative splicing: A regulated tumor-promting switch in human neuroblas‐toma. Cancer Cell, 6, 347-360.
[74] Windisch, J. M.; Marksteiner, R. & Schneider, R. (1995). Nerve growth factor bindingsite on TrkA mapped to a single 24-amino acid rich leucine motif. Journal of BiologicalChemistry, 270, 28133-28136.
[75] Ninkina, N.; Grashchuck, M.; Buchman, V. L. & Davies, A. M. (1997). TrkB variantswith deletions in the leucine-rich motifs of the extracellular domain. Journal of Biologi‐cal Chemistry, 272, 13019-13025.
[76] Clary, D. O.; & Reichardt, L. F. (1994). An alternative splice form of the nerve growthfactor receptor confers an enhanced response to neurotrophin 3. Proceedings of the Na‐tional Academy of Science. USA, 91, 11133-11137.
[77] Tacconelli, A., Farina, A. R.; Cappabianca, L.; Cea, G.; Panella, S.; Chioda, A.; Gallo,R.; Cinque, B.; Sferra, R.; Vetuschi, A.; Campese, A. F.; Screpanti, I.; Gulino, A. &Mackay, A. R. (2007). TrkAIII expression in the thymus. Journal of Neuroimmunology,183, 151-161.
[78] Farina, A. R.; Tacconelli, A.; Cappabianca, L.; Cea, G.; Chioda, A.; Romanelli, A.; Pen‐sato, S.; Pedone, C.; Gulino, A. & Mackay, A. R. (2009). The neuroblastoma tumour-suppressor TrkAI and its oncogenic alternative TrkAIII splice variant exhibitgeldanamycin-sensitive interactions with Hsp90 in human neuroblastoma cells. On‐cogene, 28, 4075-4094.
[79] Farina, A. R.; Tacconelli, A.; Cappabianca, L.; Cea, G.; Pannella, S.; Chioda, A.; Roma‐nelli, A.; Pedone, C.; Gulino, A. & Mackay, A. R. (2009). The TrkAIII splice varianttargets the centrosome and promotes genetic instability. Molecular and Cellular Biolo‐gy, 29, 4812-4830.
[80] Berkemeier, L. R.; Winslow, J. W.; Kaplan, D. R.; Nikolics, K.; Goeddel, D. V. &Rosenthal, A. (1991). Neurotrophin-5: a novel neurotrophic factor that activates Trkand TrkB. Neuron, 7, 857-866.
[81] Glass, D. J.; Nye, S. H.; Hantzopoulos, P.; Macchi, M. J.; Squinto, S. P.; Goldfarb, M. &Yancopoulos, G. D. (1991). TrkB mediates BDNF/NT-3-dependent survival and pro‐liferation in fibroblasts lacking low affinity NGF receptor. Cell, 66, 405-413.
[82] Squinto, S. P.; Stitt, T. N.; Aldrich, T. H.; Davis, S.; Bianco, S. M.; Radziejewski, C.;Glass, D. J.; Masiakowski, P.; Furth, M. E.; Venezuela, D. M.; Distefano, P. S. & Yan‐
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
81

copoulos, G. D. (1991). trkB encodes a functional receptor for brain-derived neurotro‐phic factor and neurotrophin-3 but not nerve growth factor. Cell, 65, 885-893.
[83] Soilov, P.; Castren, E. & Stamm, S. (2002). Analysis of the human TrkB gene genomicorganization reveals novel TrkB isoforms, unusual gene length, and splicing mecha‐nism. Biochemical and Biophysical Research Communications, 290, 1054-1065.
[84] Eide, F. F.; Vining, E. R.; Eide, B. L.; Zang, K.; Wang, X. Y. & Reichardt, L. F. (1996).Naturally occurring truncated TrkB receptors have dominant inhibitory effects onbrain-derived neurotrophic factor signalling. Journal of Neuroscience, 16, 3123-3129.
[85] Luberg, K.; Wong, J.; Weickert, C. S. & Timmusk, T. (2010). Human TrkB gene: novelalternative transcripts, protein isoforms and expression pattern in the prefrontal cere‐bral cortex during postnatal development. Journal of Neurochemistry, 113, 952- 964.
[86] Fenner, B. M. (2012). Truncated TrkB: Beyond a dominant negative receptor. Cytokine& Growth factor Reviews, 23, 15-24.
[87] Middlemas, D. S.; Kihl, B. K.; Zhou, J. F et al., (1999). Brain-derived neurotrophic fac‐tor promotes survival and hemoprotection of human neuroblastoma cells. Journal ofBiological Chemistry, 274, 16451-16460.
[88] Lamballe, F.; Klein, R. & Barbacid, M. (1991). trkC, a new member of the trk family oftyrosine protein kinases, is a receptor for neutotrophin-3. Cell, 66, 967-979.
[89] Lamballe, F.; Tapley, P. & Barbacid, M. (1993). trkC encodes multiple neurotrophin-3receptors with distinct biological properties and biological activities. EMBO Journal,12, 3083-3094.
[90] Tsoulfas, P.; Soppet, D.; Escandon, E.; Tessarollo, L.; Mendoza-Ramirez, J. L.; Rosen‐thal, A.; Nikolics, K. & Parada, L. F. (1993). The rat TrkC locus encodes multiple neu‐rogenic receptors that exhibit differential response to neurotrophin-3 in PC-12 cells.Neuron, 10, 975-990.
[91] Valenzuela, D. M.; Maisonpierre, P. C.; Glass, D. J.; Rojas, E.; Nunez, L.; Kong, Y.;Stitt, Ip, N. Y. & Yancopoulos, G. D. (1993). Alternative forms of rat TrkC with differ‐ent functional capabilities. Neuron, 10, 963-974.
[92] Menn, B.; Timsit, S.; Calothy, G. & Lamballe, F. (1998). Differential expression of trkCcatalytic and noncatalytic isoforms suggests that they act independently or in associ‐ation. Journal of Comparative Neurology, 410, 47-64.
[93] Tsoulfas, P.; Stephens, R. M.; Kaplan, D. R. & Parada, L. F. (1996). trkC isoforms withinsert in the kinase domain show impaired signalling responses. Journal of BiologicalChemistry, 271, 5691-5697.
[94] Liepenish, E.; Llag, L. L.; Otting, G. & Ibanez, C. F. (1997). NMR structure of thedeath domain of the p75 neurotrophin receptor. EMBO Journal, 16, 4999-5005.
Neuroblastoma82

[95] Yano, H. & Chao, M. V. (2000). Neurotrophin receptor structure and function. Phar‐maceutica acta Helvetiae, 74, 253-260.
[96] Wartiovaara, K.; Paavola, P.; Suvanto, P.; Paulin, L.; Saarma, M.; Peltonen, L. & Sario‐la, H. (1997). Exclusion of the p75 neurotrophin receptor gene as a candidate gene forMeckel syndrome. Clinical Dysmorphology, 6, 213-217.
[97] Von Shack, D.; Casademunt, E.; Schweigreiter, R.; Meyer, M.; Bibel, M. & Dechant, G.(2001). Complete ablation of the neurotrophin receptor p75NTR causes defects bothin the nervous and vascular system. Nature Neuroscience, 4, 977-978.
[98] Nykjaer, A.; Lee, R.; Teng, K. K.; Jansen, P.; Madsen, P.; Nielsen, M. S.; Jacobsen, C.;Kliemannel, M.; Schwarz, E.; Willnow, T. E.; Hempstead, B. & Petersen, C. M. (2004).Sortilin is essential for proNGF-induced neuronal cell death. Nature, 427, 843-847.
[99] Zampieri, N. & Chao, M. V. (2004). Structural biology. The p75 NGF receptor ex‐posed. Science, 304, 833-834.
[100] Harel, L.; Costa, B. & Fainzilber, M. (2010). On the death Trk. Developmental Neurobi‐ology, 70, 298-303.
[101] Teng, H. K.; Teng, K. K.; Lee, R.; Wright, S.; Tevar, S.; Almeida, R. D.; Kermani, P.;Torkin, R.; Chen, Z. Y.; Lee, F. S.; Kraemer, R. T.; Nykjaer, A. & Hempstead, B. L.(2005). ProBDNF induces neuronal apoptosis via activation of a receptor complex ofp75NTR and Sortilin. Journal of Neuroscience, 25, 5455-5463.
[102] Yano, H.; Torkin, R.; Martin, L. A.; Chao, M. V. & Teng, K. K. (2009). Proneurotro‐phin-3 is a neuronal apoptotic ligand: evidence for retrograde-directed cell killing.Journal of Neuroscience, 29, 14790-14802.
[103] Petersen, C. M.; Nielsen, M. S.; Nykjaer, A.; Jacobsen, L.; Tommerup, N.; Rasmussen,H. H.; Roigarard, H.; Gliemann, J.; Madsen, P. & Moestrup, S. K. (1997). Molecularidentification of a novel candidate sorting receptor purified from human brain by re‐ceptor-associated protein affinity chromatography. Journal of Biological Chemistry, 272,3599-3605.
[104] Vincent, J-P.; Mazella, J. & Kitabgi, P. (1999). Neurotensin and neurotensin receptors.Trends in Pharmacological Sciences, 20, 302-309.
[105] Schweigreiter, R. (2006). The dual nature of neurotrophins. Bioessays, 28, 583-594.
[106] Schneider, R. & Schweiger, M. (1991). A novel modular mosaic of cell adhesion mor‐ifs in the extracellular domains of the neurogenic trk and trkB tyrosine kinase recep‐tors. Oncogene, 6, 1807-1811.
[107] Holden, P. H.; Asopa, V.; Robertson, A. G.; Clarke, A. R.; Tyler, S.; Bennett, G. S.;Brain, S. D.; Wilcock, G. K.; Allen, S. J.; Smith, S. K. & Dawbarn, D. (1997). Immuno‐globulin-like domains define the nerve growth factor binding site of the trkA recep‐tor. Nature Biotechnology, 15, 668-672.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
83

[108] Perez, P.; Coll, P. M.; Hempstead, B. L.; Martin-Zanca, D. & Chao, M. V. (1995). NGFbinding to the trk tyrosine kinase receptor requires the extracellular immunoglobu‐lin-like domains. Molecular and Cellular Neuroscience, 6, 97-105.
[109] Urfer, R.; Tsoulfas, P.; O’Connell, L. & Presta, L. G. (1997). Specificity determinants inneurotrophin-3 and design of nerve growth factor-based trkC agonists by changingcentral beta strand bundle residues to their neurotrophin-3 analogs. Biochemistry, 36,4775-4781.
[110] Arevalo, J. C.; Conde, B.; Hemstead, B. I.; Chao, M. V.; Martin-Zanca, D. & Perez, P.(2000). TrkA immunoglobulin-like ligand binding domains inhibit spontaneous acti‐vation of the receptor. Molecular and Cellular Biology, 20, 5908-5916.
[111] Peng, X.; Green, L. A.; Kaplan, D. R. & Stephens, R. M. (1995). Deletion of a con‐served juxtamembrane sequence in Trk abolishes NGF-promoted neuritogenesis.Neuron, 15, 395-406.
[112] Monshipouri, M.; Jiang, H. & Lazarovici, P. (2000). NGF stimulation of erk phosphor‐ylation is impaired by a point mutation in the transmembrane domain of the trkA re‐ceptor. Journal of Molecular Neuroscience, 14, 69-76.
[113] Kaplan, D. R.; Martin-Zanca, D. & Prada, L. F. (1991). Tyrosine phosphorylation andtyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature,350, 358-360.
[114] Wiesman, C.; Muller, Y. A. & de Vos, A. M. (2000). Ligand binding sites in Ig-like do‐mains of receptor tyrosine kinases. Journal of Molecular Medicine, 78, 247-260.
[115] Arevalo, J. C.; Conde, B.; Hemstead, B. I.; Chao, M. V.; Martin-Zanca, D. & Perez, P.(2001). A novel mutation within the extracellular domain of TrkA causes constitutivereceptor activation. Oncogene, 20, 1229-1234.
[116] Benedetti, M.; Levi, A. & Chao, M. V. (1993). Differential expression of nerve growthfactors leads to altered binding affinity and neurotrophin responsiveness. Proceedingof the National Academy of Science. USA, 90, 7859-7863.
[117] Mahadeo, D.; Kaplan, L.; Chao, M. V. & Hempstead, B. L. (1994). High affinity nervegrowth factor binding displays a faster rate of association than p140trk binding im‐plications for multi-subunit polypeptide receptors. Journal of Biological Chemistry, 269,6884-6891.
[118] Esposito, D.; Patel, P.; Stephens, R. M.; Perez, P.; Chao, M. V.; Kaplan, D. R. & Hemp‐stead, B. L. (2001). The cytoplasmic and transmembrane domains of the p75 and trkAreceptors regulate high affinity binding to nerve growth factor. Journal of BiologicalChemistry, 276, 32687-32695.
[119] Bibel, H.; Hoppe, E. & Barde, Y-A. (1999). Biochemical and functional interactions be‐tween the neurotrophin receptors trk and p75NTR. EMBO Journal, 18, 616-622.
Neuroblastoma84

[120] Mischel, P. S.; Smith, S. G.; Vining, E. R.; Valletta, J. S.; Mobley, W. C. & Reichardt, L.F. (2001). The extracellular domain of p75NTR is necessary to inhibit neurotrophin-3signaling through TrkA. Journal of Biological Chemistry, 276, 11292-11301.
[121] Murray, S. S.; Perez, P.; Lee, R.; Hempstead, B. L. & Chao, M. V. (2004). A novel p75neurotrophin receptor-related protein, NRH2, regulates nerve growth factor bindingto the TrkA receptor. Journal of Neuroscience, 24, 2742-2749.
[122] Pratcha, G. & Ibanez, C. F. (2002). Lipid rafts and the control of neurotrophic factorsignaling in the nervous system: variations on a theme. Current Opinions in Neurobiol‐ogy, 12, 542-549.
[123] Marsh, H. N.; Dubreuil, C. I.; Quevedo, C.; Lee, A.; Majdan, M.; Walsh, G. S.; Haus‐dorff, S.; Said, F. A.; Zoueva, O.; Kozlowski, M.; Siminovitch, K.; Neel, B. G.; Miller,F. D. & Kaplan, D. R. (2003). SHP-1 negatively regulates neuronal survival by func‐tioning as a TrkA phosphatase. Journal of Cell Biology, 163, 999-1010.
[124] Watson, F. L.; Porcionatto, M. A.; Bhattacharyya, A.; Stiles, C. & Segal, R. C. (1999).TrkA glycosylation regulates localisation and activity. Journal of Neurobiology, 39,323-336.
[125] Ostman, A. & Bohmer, F. D. (2001). Regulation of receptor tyrosine kinase signallingby protein tyrosine phosphatases. Trends in Cell Biology, 11, 258-266.
[126] Sastry, S. K. & Elferink, L. A. (2011). Checks and balances: interplay of RTKs andPTPs in cancer progression. Biochemical Pharmacology http://dx.doi.org/10.1016/j.bcp.2011.06.016.
[127] Kaplan, D. R. & Stephens, R. M. (1994). Neurotrophin signal transduction by the trkreceptor. Journal of Neurobiology, 25, 1404-1417.
[128] Green, L. A. & Kaplan, R. M. (1995). Early events in neurotrophin signaling via Trkand p75 receptors. Current Opinions in Neurobiology, 5, 579-587.
[129] Hallberg, B.; Ashcroft, M.; Loeb, D. M. & Kaplan, R. M. (1998). Nerve factor inducedstimulation of Ras requires Trk interaction with Shc but does not involve phosphoi‐nositol 3-OH kinase. Oncogene, 17, 691-697.
[130] Meakin, S. O.; MacDonald, J. I. S.; Gryz, E. A.; Kubu, C. J. & Verdi, J. M. (1999). Thesignalling adapter FRS-2 competes with Shc for binding to the nerve growth factorreceptor TrkA. A model discriminating between proliferation and differentiation.Journal of Biological Chemistry, 274, 9861-9870.
[131] Cunningham, M. E.; Stephens, R. M.; Kaplan, D. R. & Greene, L. A. (1997). Autophos‐phorylation of activation loop tyrosines regulates signaling by the Trk nerve growthfactor receptor. Journal of Biological Chemistry, 272, 10957-10967.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
85

[132] Obermeier, A.; Haltre, H.; Weismuller, K. H.; Jung, G.; Schlessinger, J. & Ullrich, A.(1993). Tyrosine 785 is a major determinant of Trk-substrate interaction. EMBO Jour‐nal, 12, 933-941.
[133] Obermeier, A.; Bradshaw, R. A.; Seedorf, K.; Choidas, A.; Schlessinger, J. & Ullrich,A. (1994). Neuronal differentiation signals are controlled by nerve growth factor re‐ceptor/Trk binding sites for Shc and PLCγ. EMBO Journal, 13, 1585-1590.
[134] Segal, R. A. & Greenberg, M. E. (1996). Intracellular signalling pathways activated byneurotrophic factors. Annual Reviews in Neuroscience, 19, 463-489.
[135] Yao, R. & Cooper, G. M. (1995). Requirement for phosphoinositol-3 kinase in the pre‐vention of apoptosis by nerve growth factor. Science, 267, 2003-2005.
[136] MacDonald, J. I.; Gryz, E. A.; Kubu, C. J.; Verdi, J. M. & Meakin, S. O. (2000). Directbinding of the signalling adapter protein Grb2 to the activation loop tyrosines on thenerve growth factor receptor tyrosine kinase, TrkA. Journal of Biological Chemistry,275, 18225-18233.
[137] Hagag, N.; Halegoua, S. & Viola, M. (1986). Inhibition of growth factor induced dif‐ferentiation of PC12 cells by microinjection of antibody to ras p21. Nature, 319,680-682.
[138] Hampstead, B. L.; Martin-Zanca, D.; Kaplan, D. R.; Parada, L. F. & Chao, M. V.(1991). High affinity NGF binding requires coexpression of the trk proto-oncogeneand low affinity NGF receptor. Nature, 350, 678-683.
[139] Majdan, M.; Walsh, G. S.; Aloyz, R. & Miller, F. D. (2001). TrkA mediates develop‐mental sympathetic neuron survival by silencing an ongoing p75NTR-mediateddeath signal. Journal of Cell Biology, 155, 1275-1285.
[140] Zaccaro, M. C.; Ivanisevic, L.; Perez, P.; Meakin, S. O. & Saragovi, H. U. (2002). P75coreceptors regulate ligand dependent and ligand independent Trk receptor activa‐tion, in part by altering trk docking subdomains. Journal of Biological Chemistry, 276,31023-31029.
[141] Nykjaer, N.; Willnow, T. E.; & Munk-Peterson, C. (2005). P75NTR-liver or let die.Current Opinions in Neurobiology, 15, 40-57.
[142] York, R. D.; Yao, H.; Dillon, T.; Ellig, C. L.; Eckert, S. P.; McCleskey, E. W. & Stork, P.J. (1998). Rap1 mediates sustained MAP kinase activation induced by nerve growthfactor. Nature, 392, 622-626.
[143] Wu, C.; Lai, C. F. & Mobley, W. C. (2001). Nerve growth factor activates persistentRap1 signaling in endosomes. Journal of Neuroscience, 21, 5406-5416.
[144] Minichiello, L.; Casagrande, F.; Tatche, R. S.; Stucky, C. L.; Postigo, A.; Lewin, G. R.;Davies, A. M. & Klein, R. (1998). Point mutation in trkB causes loss of NT-4-depend‐ent neurons without major effect on diverse BDNF responses. Neuron, 21, 335-345.
Neuroblastoma86

[145] Postigo, A.; Calella, A. M.; Fritzsch, B.; Knipper, M.; Katz, D.; Eilers, A.; Schimmang,T.; Lewin, G. R.; Klein, K. & Minichiello, L. (2002). Distinct requirements for TrkBand TrkC signalling in target innervation by sensory neurons. Genes and Development,16, 633-645.
[146] Kong, H.; Boulter, J.; Weber, J. L.;lai, C. & Chao, M. V. (2001). An evolutionaily con‐served transmembrane protein that is a novel downstream target of neurotrophinand aphrin receptors. Journal of Neuroscience, 21, 176-185.
[147] Arevalo, J. C.; Yano, H.; Teng, K. K. & Chao, M. V. (2004). A unique pathway for sus‐tained neurotrophin signalling through an akyrin-rich membrane-soanning protein.EMBO Journal, 23, 2358-2368.
[148] Ginty, D. D.; Bonni, A. & Greeberg, M. E. (1994). Nerve growth factor activates a Ras-dependent protein kinase that stimulates c-fos transcription via phosphorylation ofCREB. Cell, 77, 713-725.
[149] Xing, J.; Ginty, D. D. & Greenberg, M. E. (1998). Coupling of the RAS-MAPK path‐way to gene activation by RSK2, a growth factor-regulated CREB kinase. Science, 273,959-963.
[150] Deak, M.; Clifton, A. D.; Lucocq, L. M. & Alessi, D. R. (1998). Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38,and may mediate activation of CREB. EMBO Journal, 17, 4426-4441.
[151] Atwal, J. K.; Massie, B.; Miller, F. D. & Kaplan, D. R. (2000). The TrkB-Shc site signalsneuronal survival and local axon growth via MEK and PI3-kinase. Neuron, 27,265-277.
[152] Ecinas, M.; Iglesias, M.; Llecha, N. & Comella, J. X. (1999). Extracellular-regulatedkinases and phosphoinositol 3-kinase are involved in brain-derived growth factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. Jour‐nal of Neurochemistry, 73, 1409-1421.
[153] Florez, A. I.; Mallon, B. S.; Matsui, T.; Ogawa, W.; Rosenzweig, A.; Okamoto, T. &Macklin, W. B. (2000). Akt-mediated survival of oligodendrocytes induced by neuro‐regulins. Journal of Neuroscience, 20, 7622-7630.
[154] Wooten, M. W.; Vandenplas, M. L.; Seibenhener, M. L.; Geetha, T. & Diaz-Meco, M.T. (2001). Nerve growth factor stimulates tyrosine phosphorylation and activation ofthe atypical protein kinase C’s via a src kinase pathway. Molecular and Cellular Biolo‐gy, 21, 8414-8427.
[155] Corbit, K. C.; Foster, D. A. & Rosner, M. R. (1999). Protein kinase Cdelta mediatesneurogenic but not mitogenic activation of mitogen-activated protein kinase in neu‐ronal cells. Molecular and Cellular Biology, 19, 4209-4218.
[156] Simpson, A. M.; Iyer, R.; Mangino, J. L.; Minturn, J. E.; Zhao, H.; Kolla, V. & Brodeur,G. M. (2012). TrkA3 isoform expression upregulates stem cell markers and correlates
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
87

with worse outcome in neuroblastomas (NBs). Proceedings of the Advances in Neuro‐blastoma Research (2012) (Meeting Abstract POT055), p 164.
[157] Moises, T.; Dreieir, A.; Flohr, S.; Esser, M.; Brauers, E.; Reiss, K.; Merken, D.; Weis, J.& Kruttgen, A. (2007). Tracking TrkA’s trafficking: NGF receptor trafficking controlsNGF receptor signalling. Molecular Neurobiology, 35, 151-159.
[158] Howe, C. L. & Mobley, W. C. (2004). Signaling endosome hypothesis: a cellularmechanism for long distance communication. Journal of Neurobiology, 58, 207-216.2004
[159] Valdez, G.; Akmentin, W.; Philippidou, P.; Kuruvilla, R.; Ginty, D. D. & Halegoua, S.(2005). Pincher-mediated macroendocytosis underlies retrograde signalling by neu‐rotrophin receptors. Journal of Neuroscience, 25, 5236-5247.
[160] Rajagopal, R.; Chen, Z. Y.; Lee, F. S. & Chao, M. V. (2004). Transactivation of Trk neu‐rotrophin receptors by G-protein-coupled receptor ligands occurs on intracellularmembranes. Journal of Neuroscience, 24, 6650-6658.
[161] Shi, G. X.; Jin, L. & Andres, D. A. (2010). Src-dependent TrkA transactivation is re‐quired for pituitary adenylate cyclase- activating polypeptide 38-mediated Rit activa‐tion and neuronal differentiation. Molecular Biology of the Cell, 21, 1597-1608.
[162] He, X. L. & Garcia, K. C. (2004). Structure of nerve growth factor complexed with theshared neurotrophin receptor p75. Science, 304, 870-875.
[163] Huang, E. J. & Reichardt, L. F. (2003). Trk receptors: roles in neuronal signal trans‐duction. Annual Reviews in Biochemistry, 72, 609-642.
[164] Kuruvilla, R.; Zweifel, L. S.; Glebova, N. O.; Lonze, B. E.; Valdez, G.; Ye, H. & Ginty,D. D. (2004). A neurotrophin signaling cascade coordinates sympathetic neuron de‐velopment through differential control of TrkA trafficking and retrograde signaling.Cell 118, 243-255.
[165] Epa, W. R.; Markovska, K. & Barrett, G. L. (2004). The p75 neurotrophin receptor en‐hances TrkA signaling by binding to Shc and augmenting its phosphorylation. Jour‐nal of Neurochemistry, 89, 344-353.
[166] Hannila, S. S.; Lawreance, G. M.; Ross, G. M. & Kawaja, M. D. (2004). TrkA and mito‐gen-activated protein kinase phosphorylation are enhanced in sympathetic neuronslacking functional p75 neurotrophin receptor expression. European Journal of Neuro‐science, 19, 2903-2908.
[167] Lad, S. P.; Peterson, D. A.; Bradshaw, R. A. & Neet, K. E. (2003). Individual and com‐bined effects of TrkA and p75NTR nerve growth factor receptors. A role for high af‐finity receptor sites. Journal of Biological Chemistry, 278, 24808-24817.
Neuroblastoma88

[168] Paul, C. E.; Vereker, E.; Dickson, K. M. & Barker, P. A. (2004). A pro-apoptotic frag‐ment of the p75 neurotrophin receptor is expressed in p75NTR exon IV null mice.Journal of Neuroscience, 24, 1917-1923.
[169] Gentry, J. J.; Barker, P. A. & Carter, B. D. (2004). The p75 neurotrophin receptor: mul‐tiple interactors and numerous functions. Progress in Brain Research, 146, 25-39.
[170] Miller, F. D. & Kaplan, D. R. (2001). Neurotrophin signalling pathways regulatingneuronal apoptosis. Cellular and Molecular Life Sciences, 58, 7879-7887.
[171] Bhakar, A. L.; Howell, J. L.; Paul, C. E.; Salehi, A. H.; Becker, E. B.; Said, F.; Bonni, A.& Barker, P. A. (2003). Apoptosis induced by p75NTR overexpression requires junkinase-dependent phosphorylation of Bad. Journal of Neuroscience, 23, 11373-11381.
[172] Okuno, S.; Saito, A.; Hayashi, T. & Chan, P.H. (2004). The c-Jun N-terminal proteinkinase signaling pathway mediates bax activation and subsequent neuronal apopto‐sis through interaction with bim after transient focal cerebral ischemia. Journal ofNeuroscience, 24, 7879-7887.
[173] Barker, P. A. (2004). P75NTR is positively promiscuous: novel partners and new in‐sights. Neuron, 42, 529-533.
[174] Becker, E. B. E.; Howell, J.; Kodama, Y.; Barker, P. A. & Bonni, A. (2004). Characteri‐sation of the c-Jun N-terminal kinase-BimEL signaling pathway in neuronal apopto‐sis. Journal of Neuroscience, 24, 8762-8770.
[175] Casademunt, E.; Carter, B. D.; Benzel, I.; Frade, J. M.; Dechant, G. & Barde, Y-A.(1999). The zinc finger protein NRIF interacts with the neurotrophin receptorp75(NTR) and participates in programmed cell death. EMBO Journal, 18, 6050-6061.
[176] Salehi, A. H.; Roux, P. P.; Kubu, C. J.; Zeindler, C.; Bhakar, A.; Tannis, L. L.; Verdi, J.M. & Barker, P. A. (2000). RAGE, a novel MAGE protein, interacts with the p75 neu‐rotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron,27. 279-288.
[177] Park, J. A.; Lee, J. Y.; Sato, T. A. & Koh, J. Y. (2000). Co-induction of p75NTR andp75NTR-associated death executor in neurons after zinc exposure in cortical cultureor transient ischemia in the rat. Journal of Neuroscience, 20, 9069-9103.
[178] Chittka, A.; Arevalo, J. C.; Rodriguez-Guzman, M.; Perez, P.; Chao, M. V. & Sendt‐ner, M. (2004). The p75NTR-interacting protein SC1 inhibits cell cycle progression bytranscriptional repression of cyclin E. Journal of Cell Biology, 164, 985-996.
[179] Arevalo, J. C. & Wu, S. H. (2006). Neurotrophin signalling: many exciting surprises.Cell Mol Life Sci 63, 1523-1537.
[180] Yamashito, H.; Avraham, S.; Jiang, S.; Dikic, I. & Avram, H. (1999). The Csk homolo‐gous kinase associates with TrkA receptors and is involved in neurite outgrowth ofPC12 cells. Journal of Biological Chemistry, 274, 15059-15065.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
89

[181] Frade, J. M. (2005). Nuclear translocation of the p75 neurotrophin receptor cytoplas‐mic domain in response to neurotrophin binding. Journal of Neuroscience, 25,1407-1411.
[182] Datta, S. R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y. & Greenberg, M. E.(1997). Akt phosphorylation of BAD couples survival signals to the cell-intrinsicdeath machinary. Cell, 91, 231-241.
[183] Orike, M.; Middleton, G.; Borthwick, E.; Buchman, V. Cowen, T. & Davies, A. M.(2001). Role of PI-3 kinase, Akt and Bcl-2-related proteins in sustaining the survivalof neurotrophic factor-dependent adult sympathetic neurons. Journal of Cell Biology,154, 995-1005.
[184] Brunet, A.; Bonni, A.; Zigmond, A. J.; Lin, M. Z.; Juo, P.; Hu, L. S.; Anderson, M. J.;Arden, K. C.; Blenis, J. & Greenberg, M. E. (1999). Akt promotes cell survaival byphosphorylating and inhibiting a Forkhead transcription factor. Cell, 96, 857-868.
[185] Wyttenbach, A. & Tolkovsky, A. M. (2006). The BH3-only protein Puma is both nec‐essary and sufficient for neuronal apoptosis induced by DNA damage in sympathet‐ic neurons. Journal of Neurochemistry, 96, 1213-11226.
[186] Putcha, G. V.; Moulder, K. L.; Golden, J. P.; Bouillet, P.; Adams, J. A.; Strasser, A. &Johnson, E. M. (2001). Induction of BIM, a proapoptotic BH3-only BCL-2 familymember, is critical for neuronal apoptosis. Neuron, 29, 615-628.
[187] Gilley, J.; Coffer, P. J. & Ham, J. (2003). FOXO transcription factors directly activatebim gene expression and promote apoptosis in sympathetic neurons. Journal of CellBiology, 162, 613-622.
[188] Whitfield, J.; Neame, S. J.; Parquet, L.; Bernard, O. & Ham, J. (2001). Dominant-nega‐tive c-Jun promotes neuronal survival by reducing BIM expression and inhibitingmitochondrial cytochrome c release. Neuron, 29, 629-643.
[189] Du, K. & Mintiminy, M. (1998). CREB is a regulatory target for the protein kinaseAkt/PKB. Journal of Biological Chemistry, 273, 32377-32379.
[190] Riccio, A.; Ahn, S.; Davenport, C. M.; Blendy, J. A. & Ginty, D. D. (1999). Mediationby a CREB family transcription factor of NGF-dependent survival of sympatheticneurons. Science, 286, 2358-2361.
[191] Tauszig-Delamasure, S.; Yu, L-Y.; Cabrera, J. R.; Bouzas-Rodriguez, J.; Mermet-Bouv‐ier, C.; Giux, C.; Bordeux, M-C.; Arumae, U. & Mehlen, P. (2007). The TrkC receptorinduces apoptosis when the dependence receptor meets the neurotrophin paradigm.Proceedings of the National Acadamy of Sciences, USA, 104, 13361-13366.
[192] Nikoletopoulou, V.; Lickert, H.; Frade, J. M.; Rencurel, C.; Giallonardo, P.; Zhang, L.;Bibel, M. & Barde, Y-A. (2010). Neurotrophin receptors TrkA and TrkC cause neuro‐nal death whereas TrkB does not. Nature, 467, 59-64.
Neuroblastoma90

[193] Bernd, P. (2008). The role of neurotrophins during early development. Gene Expres‐sion, 14, 241-250.
[194] Kahane, N. & Kalcheim, C. (1994). Expression of trkC receptor mRNA during devel‐opment of the avian nervous system. Journal of Neurobiology, 25, 571-584.
[195] Chalazonitis, A.; Pham, T. D.; Rothman, T. P.; DiStefano, P. S.; Bothwell, M.; Blair-Flynn, J.; Tassarollo, L. & Gershon, M. D. (2001). Neurotrophin-3 is required for thesurvival-differentiation of subsets of developing enteric neurons. Journal of neuro‐science, 21, 5620-5636.
[196] Ernfors, P.; Kucera, J.; Lee, K. F.; Loring, J. & Jaenisch, R. (1995). Studies on the phys‐iological role of brain-derived neurotrophic factor and neurotrophin-3 in knockoutmice. International Journal of Developmental Biology, 39, 799-807.
[197] Farinas, I.; Jones, K. R.; Backus, C.; Wang, X. Y. & Reichardt, L. F. (1994). Severe sen‐sory and sympathetic deficits in mice lacking neurotrophin-3. Nature, 369, 658-661.
[198] Yao, L.; Zhang, D. & Bernd, P. (1994). The onset of neurotrophin and trk mRNA ex‐pression in early embryonic tissue of the quail. Developmental Biology, 165, 727-730.
[199] Zhang, D.; Yao, L. & Bernd, P. (1994). Expression of trk and neurotrophin mRNA indorsal root and sympathetic ganglia of the quail during development. Journal of Neu‐robiology, 25, 1517-1532.
[200] Henion, P. D.; Garner, A. S.; Large, T. H. & Weston, J. A. (1995). trkC-mediated NT-3signaling is required for the early development of a subpopulation of neurogenicneural crest cells. Developmental Biology,172, 602-613.
[201] Kalcheim, C.; Carmeli, C. & Rosenthal, A. (1992). Neurotrophin-3 is a mitogen forcultured neural crest cells. Proceeding of the National Acadeny of Science. USA, 89,1661-1665.
[202] Maisonpierre, P. C.; Belluscio, L.; Friedman, B.; Alderson, R. F.; Wiegand, S. J.; Furth,M. E.; Lindsay, R. M. & Yancopoulos, G. D. (1990). NT-3, BDNF, and NGF in the de‐veloping rat nervous system: parallel as well as reciprocal petterns of expression.Neuron, 5, 501-509.
[203] Ernfors, P.; Merlio, J. P. & Persson, H. (1992). Cells expressing mRNA for neurotro‐phins and their receptors during embryonic rat development. European Journal ofNeuroscience, 4, 1140-1158.
[204] Verdi, J. M.; Groves, A. K.; Farinas, I.; Jones, K.; Marchionni, M. A.; Reichardt, L. F. &Anderson, D. J. (1996). A reciprocal cell-cell interaction mediated by NT-3 and neure‐gulins controls the early survival and development of sympathetic neuroblasts. Neu‐ron, 16, 515-527.
[205] DiCicco-Bloem, E.; Friedman, W. J. & Black, I. B. (1993). NT-3 stimulates sympatheticneuroblast proliferation by promoting precursor survival. Neuron, 11, 1101-1111.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
91

[206] Levi-Montalcini, R. (1987). The nerve growth factor: thirty-five years later. EMBOJournal, 6, 1145-1154.
[207] Verdi, J. M. & Anderson, D. J. (1994). Neurotrophins regulate sequential changes inneurotrophin receptor expression by sympathetic neuroblasts. Neuron, 13, 1359-1372.
[208] Wetmore, C. & Olson, L. (1995). Neuronal and non neuronal expression of neurotro‐phins and their receptors in sensory and sympathetic ganglia suggest new intercellu‐lar trophic interactions. Journal of Comparative Neurology, 353, 143-159.
[209] Rosenthal, A.; Goeddel, D. V.; Nguyen, T.; Lewis, M.; Shih, A.; Laramee, G. R.; Nikol‐ics, K. & Winslow, J. W. (1990). Primary structure and biological activity of a novelhuman neurotrophic factor. Neuron, 4, 767-773.
[210] Reichardt , L. F. (2006). Neurotrophin-regulated signalling pathways. PhilosophicalTransactions of the Royal Society of London. Series B, Biological Sciences, 361, 1545-1564.
[211] ElShamy, W. M.; Linnarsson, S.; Lee, K. F.; Jaenisch, R. & Enfors, P. (1996a). Prenataland postnatal requirements of NT-3 for sympathetic neuroblast survival and inner‐vation of specific targets. . Development, 122, 491-500.
[212] ElShamy, W. M. & Enfors, P. (1996b). A local action of neurotrophin-3 prevents deathof proliferating sensory neuron precusor cells. Neuron, 16, 963-972.
[213] Schecterson, L. C. & Bothwell, M. (1992). Novel roles for neurotrophins are suggestedby BDNF and NT-3 mRNA expression in developing neurons. Neuron, 9, 449-463.
[214] Zhou, X-F. & Rush, R. A. (1996). Functional roles for neurotrophin-3 in the develop‐ing and mature sympathetic nervous system. Molecular Neurobiology, 13, 185-197.
[215] Francis, N.; Farinas, I.; Brennan, C.; Rivas-Plata, K.; Backus, C.; Reichardt, L. &Landis, S. (1999). NT-3, like NGF, is required for survival of sympathetic neurons,but not their precursors. Developmental Biology, 210, 411-427.
[216] Davies, A. M. (1994). The role of neurotrophins in the developing nervous system.Journal of Neurobiology, 25, 1134-1148.
[217] Ockel, M.; von Schack, D.; Schropel, A.; Dechant, G.; Lewin, G. R. & Barde, Y-A.(1996). Roles of neurotrophin-3 during early development of the peripheral nervoussystem. Philosophical Transactions of the Royal Society: Biological Science, 351, 383-387.
[218] Rush, R. A.; Chie, E.; Liu, D.; Tafreshi, A.; Zettler, C. & Zhou, X. F. (1997). Neurotro‐phic factors are required by mature sympathetic neurons for survival, transmissionand connectivity. Clinical and Experimental Pharmacology and Physiology, 24, 549-555.
[219] Schecterson, L. C. & Bothwell, M. (2009). Neurotrophin receptors: old friends withnew partners. Developmental Neurobiology, 70, 332-338.
Neuroblastoma92

[220] Scarisbrick, I. A.; Jones, E. G. & Isackson, P. J. (1993). Coexpression of mRNAs forNGF, BDNF, and NT-3 in the cardiovascular system of pre- and postnatal rats. Jour‐nal of Neuroscience, 13, 875-893.
[221] Hamberger, V. & Levi-Montalcini, R. (1949). Proliferation, differentiation and degen‐eration in the spinal ganglia of the chick embry under normal and experimental con‐ditions. Journal of Experimental Zoology, 111, 457-501.
[222] Saltis, J. & Rush, R. A. (1995). Effects of nerve growth factor on sympathetic neurondevelopment in normal and limbless chick embryos. International Journal of Develop‐mental Neuroscience, 13, 577-584.
[223] Lee, K. F.; Davies, A. M. & Jaenisch, R. (1994). P75-deficient embryonic dorsal rootsensory and neonatal sympathetic neurons display a decreased sensitivity to NGF.Development, 120, 1027-1033.
[224] Birren, S.; Lo, L. & Anderson, D. J. (1993). Sympathetic neuroblasts undergo a devel‐opmental switch in trophic dependence. Development ,119, 597-610.
[225] Straub, J. A.; Saulnier Sholler, G. L. & Nishi, R. (2007). Embryonic sympathoblaststransiently express TrkB in vivo and proliferate in response to brian-derived neuro‐trophic factor in vitro. BMC Developmental Biology, 7, 1-13.
[226] Elkabes, S.; Dreyfus, C. F.; Schaar, D. G. & Black, I. B. (1994). Embryonic sensory de‐velopment: local expression of neurotrophin-3 and target expression of nerve growthfactor. Journal of Comparative Neurology, 341, 204-213.
[227] Pizzuti, A.; Borsani, G.; Falini, A.; Sidoli, A.; Baralle, F. E.; Scarlato, G. & Silani, V.(1990). Detection of beta-nerve growth factor mRNA in the human fetal brain. BrainResearch, 518, 337-341.
[228] Quartu, M.; Geic, M. & Del Flacco, M. (1997). Neurotrophin-like immunoreactivity inthe human trigeminal ganglion. Neuroreports, 8, 3611-3617.
[229] Schober, A.; Wolf, N.; Huber, K.; Hertel, R.; Krieglstein, K.; Minichiello, L.; Kahane,N.; Widenfalk, J.; Kalcheim, C.; Olsen, L.; Klein, R.; Lewin, G. R. & Unsicker, K.(1998). TrkB and neurotrophin-4 are important for development and maintenance ofsympathetic preganglion neurons innervating the adrenal medulla. Journal of Neuro‐science, 18, 7272-7284.
[230] Schober, A.; Minichiello, L.; Keller, M.; Huber, K.; Layer, P. G.; Roig-Lopez, J. L.; Gar‐cia-Arraras, J. E.; Klein, R. & Unsicker, K. (1997). Reduced acetylcholinesterase(AChE) activity in adrenal medulla and loss of sympathetic preganglionic neurons inTrkA-deficient, but not TrkB-deficient, mice. Journal of Neuroscience, 17, 891-903.
[231] Snider, W. D. (1994). Functions of the neurotrophins during nervous system develop‐ment: what the knockouts are teaching us. Cell, 77, 627-638.
[232] Bode, K.; Hofmann, H. D.; Muller, T. H.; Otten, U.; Schmidt, R. & Unsicker, K. (1986).Effects of pre- and postnatal administration of antibodies to nerve growth factor on
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
93

the morphological and biochemical behaviour of the rat adrenal medulla: a reinvesti‐gation. Brain Research, 392, 139-150.
[233] Shibayama, E. & Koizima, H. (1996). Cellular localisation of the Trk neurotrophin re‐ceptor family in human non-neural tissues. American Journal of Pathology, 148,1807-1818.
[234] Lillien, L. E. & Claude, P. (1985). Nerve growth factor is a mitogen for cultured chro‐maffin cells. Nature, 317, 632-634.
[235] Aloe L.; Alleva, E.; Bohm, A. & Levi-Montalcino, R. (1986). Aggressive behaviour in‐duces release of nerve growth factor from mouse salivary glands. Proceedings of theNational Acadamy of Sciences USA, 83, 6184-6187.
[236] Otten, U.; Schwab, M.; Gangnon, C. & Thoenen, H. (1977). Selective induction of ty‐rosine hydroxylase and beta-hydroxylase by nerve growth factor: comparison be‐tween adrenal medulla and sympathetic ganglia of adult and newborn rats. BrainResearch, 133, 291-303.
[237] Yamamoto, M. & Iseki, S. (2003). Co-expression of NGF and its high-affinity receptortrkA in the rat Carotic body chief cells. Acta Histochem Cytochem 36, 377-383.
[238] Suter-Crazzolara, C.; Lachmund, A.; Arab, S. F. & Unsicker, K. (1996). Expression ofneurotrophins and their receptors in the developing and adult rat adrenal gland. Mo‐lecular Brain Research, 43, 351-355.
[239] Unsicker, K. & Kriegelstein, K. (1996). Growth factors in chromaffin cells. Progress inNeurobiology, 48, 307-324.
[240] Unsicker, K.; Huber, K.; Schutz, G. & Kalcheim, C. (2005). The chromaffin cell and itsdevelopment. Neurochemistry Research, 30, 921-925.
[241] Doupe, A. J.; Landis, S. C. & Patterson, P. H. (1985). Environmental factors in the de‐velopment of neural crest derivatives: glucocorticoids, growth factors, and chromaf‐fin cell plasticity. Journal of Neuroscience, 5, 2119-2142.
[242] Wyatt, S. & Davies, A. M. (1995). Regulation of nerve growth factor receptor gene ex‐pression in sympathetic neurons during development. Journal of Cell Biology, 130,1435-1446.
[243] Jungbluth, S.; Koentges, G. & Lumsden, A. (1997). Coordination of early neural tubedevelopment by BDNF/trkB. Development, 124, 1877-1885.
[244] Pinon, L. G.; Minichiello, L.; Klein, R. & Davies, A. M. (1996). Timing of neuronaldeath in trkA, trkB and trkC mutant embryos reveals developmental changes in sen‐sory neuron dependence on Trk signalling. Development, 122, 3255-3261.
[245] Dixon, J. E. & McKinnon, D. (1994). Expression of trk gene family of neurotrophin re‐ceptors in prevertebral sympathetic ganglia. Brain Research Developmental Brain Re‐search, 77,177-182.
Neuroblastoma94

[246] Causing, C. G.; Gloster, A.; Aloyz, R.; Bamj, S. X.; Chang, E.; Fawcett, J.; Kuchel, G. &Miller, F. D. (1997). Synaptic innervation density is regulated by neuron-derivedBDNF. Neuron, 257-267.
[247] Timmusk, T.; Belluardo, N.; Metsis, M. & Persson, H. (1993). Widespread and devel‐opmentally regulated expression of neurotrophin-4 mRNA in rat brain and peripher‐al tissues. European Journal of Neuroscience, 5, 605-613.
[248] Kondo, Y.; Saruta, J.; To, M.; Shiiki, N.; Sato, C. & Tsukinoki, K. (2010). Expressionand role of the BDNF receptor-TrkB in rat adrenal gland under acute immobilisationstress. Acta Histochemica Cytochemica, 43, 139-147.
[249] Morrison, S. J.; White, P. M.; Zock, C.; & Anderson, D. J. (1999). Prospective identifi‐cation, isolation by flow cytometry, and in vivo self-renewal of multipotent mamma‐lian neural crest stem cells. Cell, 96, 737-749.
[250] Boiko, A, D.; Razorenova, O. V.; van de Rijn, M.; Swetter, S. M.; Johnson, D. L.; Ly, D.P.; Butler, P. D.; Yang, G. P.; Joshua, B.; Kaplan, M. J.; Longaker, M. T. & Weissman, I.L. (2010). Human melanoma initiating cells express neural crest nerve growth factorreceptor CD-271. Nature, 466, 133-137.
[251] Baker, D. L.; Molenaar, W. M.; Trojanowski, J. Q.; Evans, A. E.; Ross, A. H.; Rorke, L.B.; Packer, R. J.; Lee, V. M-Y. & Pleasure, D. (1991). Nerve growth factor receptor ex‐pression in peripheral and central neuroectodermal tumors, other pediatric brain tu‐mors and during development of the adrenal gland. American Journal of Patholology,139, 115-122.
[252] Garin-Chesa, P.; Rettig, W.; Thomson, T. M.; Old, L. J. & Melamed, M. R. (1988). Im‐munohistochemical analysis of nerve growth factor receptor expression in normaland malignant human tissues. Journal of Histochemistry and Cytochemistry, 36, 383-389.
[253] Barrett, G. L.; Georgiou, A.; Ried, K.; Bartlett, P. F. & Leung, D. (1998). Rescue of dor‐sal root sensory neurons by nerve growth factor and neurotrophin-3, but not brain-derived neurotrophic factor or neurotrophin-4, is dependent on the level of the p75neurotrophin receptor. Neuroscience, 85, 1321-1328.
[254] Bamji, S. X.; Majdan, M.; Pozniak, C. D.; Belliveau, D. J.; Aloyz, R.; Kohn, J.; Causing,C. G. & Miller, F. D. (1998). The p75 neurotrophin receptor mediates neuronal apop‐tosis and is essential for naturally occurring sympathetic neuron death. Journal of CellBiology, 140, 911-923.
[255] Ibanez, C. F. & Simi, A. (2012). P75 neurotrophin receptor signalling in nervous sys‐tem injury and degeneration: paradox and opportunity. Trends in Neuroscience, 35,431-440
[256] Lorentz, C. U.; Woodward, W. R.; Tharp, K. & Habecker, B. A. (2011). Altered norepi‐nephrine content and ventricular function in p75NTR-/- mice after myocardial infarc‐tion. Autonomic Neuroscience, 164, 13-19.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
95

[257] Nakagawara, A.; Arima, M.; Azar, C. G.; Scavards, N. J. & Brodeur, G. M. (1992). In‐verse relationship between trk expression and N-myc amplification in human neuro‐blastomas. Cancer Research, 52, 1364-1368.
[258] Nakagawara, A.; Arima-Nakagawara, M.; Scavarda, N. J.; Azar, C. G.; Cantor, A. B.& Brodeur, G. M. (1993). Association between high levels of expression of the trkgene and favourable outcome in human neuroblastoma. New England Journal of Medi‐cine, 328, 847-854.
[259] Kogner, P.; Barbany, G.; Dominici, C.; Castello, C.; Raschella, G. & Persson, H. (1993).Coexpression of messenger RNA for TRK protooncogene and low affinity nervegrowth factor receptor in neuroblastoma with favourable prognosis. Cancer Research,53, 2044-2050.
[260] Cheung, N. –K. V.; Kushner, B. H.; LaQuaglia, M. P.; Kramer, K.; Ambros, P.; Am‐bros, I.; Ladanyi, M.; Eddy, J.; Bonilla, M.-A. & Gerald, W. (1997). Survival from non-stage 4 neuroblastoma without cytotoxic therapy: an analysis of clinical andbiological markers. European Journal of Cancer, 33, 2117-2120.
[261] Combaret, V.; Gross, N.; Lasset, C.; Balmas, K.; Bouvier, R.; Frappaz, D.; Beretta-Brognara, C.; Philip, T.; Favrot, M. C. & Coll, J-L. (1997). Clinical relevance of trkAexpression on neuroblastoma: camparison with Nmyc amplification and CD44 ex‐pression. British Journal of Cancer , 75, 1151-1155.
[262] Matsunaga, T.; Shirasawa, H.; Enomoto, H.; Yoshida, H.; Iwai, J.; Tanabe, M.; Kawa‐mura, K.; Etoh, T. & Ohnuma, N. (1998). Neuronal Src and Trk A protoncogene ex‐pression in neuroblastomas and patient prognosis. International Journal of Cancer(Predictive Oncology), 79, 226-231.
[263] Matsunaga, T.; Shirasawa, H.; Hishiki, T.; Yoshida, H.; Kouchi, K.; Ohtsuka, Y.; Ka‐wamura, K.; Etoh, T. & Ohnuma, N. (2000). Enhanced expression of N-myc messen‐ger RNA in neuroblastoma found by mass screening. Clinical Cancer Research, 6,3199-3204.
[264] Suzuki, T.; Bogenmann, E.; Shimada, H.; Stram, D. & Seeger, R. C. (1993b) Lack ofhigh-affinity nerve growth factor receptors in aggressive neuroblastomas. Journal ofthe National Cancer Institute, 85, 377-384.
[265] Terui, E.; Matsunaga, T. ; Yoshida, , H.; Kouchi, K.; Kuroda, H.; Hishiki, T.; Saito, T.;Yamada, S-I.; Shirasawa, H. & Ohnuma, N. (2005). Shc family expression in neuro‐blastoma: high expression of Shc C is associated with a poor prognosis in advancedneuroblastoma. Clinical Cancer Research, 11, 3280-3287.
[266] Warnat, P.; Oberthuer, A.; Fischer, M.; Westermann, F.; Eils, R. & Brors, B. (2007).Cross-study analysis of gene expression data for intermediate neuroblastoma identi‐fies two biological subtypes. BMC Cancer, 7, 89-100.
Neuroblastoma96

[267] Tajiri, T.; Higashi, M.; Souzaki, R.; Tatsuta, K.; Kinoshita, Y. & Taguchi, T. (2007).Classification of neuroblastomas based on an analysis of the expression of genes re‐lated to prognosis. Journal of Pediatric Surgery, 42, 2046-2049.
[268] Weinreb, I.; Goldstein, D.; Irish, J. & Perez-Ordonez, B. (2009). Expression patterns ofTrk-A, Trk-B, GRP78, and p75NRT in olfactory neuroblastoma. Hum Pathol 40,1330-1335.
[269] Light, J. E.; Koyama, H.; Minturn, J. E.; Ho, R.; Simpson, A. M.; Iyer, R.; Mangino, J.L.; Kolla, V.; London, W. B. & Brodeur, G. M. (2012). Clinical significance of NTRKfamily gene expression in neuroblastomas. Pediatric Blood Cancer, 59, 226-232.
[270] Shimada, H.; Nakagawara, A.; Peters, J.; Wang, H.; Wakamatsu, P- K.; Lukens, J. N.;Matthay, K. K.; Siegel, S. E. & Seeger, R. C. (2004). TrkA expression in peripheral neu‐roblastic tumours. Cancer, 101, 1873-1881.
[271] Cheng, A. J.; Cheng, N. C.; Ford, J.; Smith, J.; Murray, J. E.; Flemming, C.; Lastowska,M.; Jackson, M. S.; Hackett, C. S.; Weiss, W. A.; Marshall, G. M.; Kees, U. R.; Murray,D. N. & Haber, M. (2007). Cell lines from MycN transgenic murine tumours reflectmolecular and biological characteristics of human neuroblastoma. European Journal ofCancer, 43, 1467-1475.
[272] Cohn, S. L.; Look, T. A.; Joshi, V. V.; Holbrook, T.; Salwern, H.; Chagnovich, D.;Chesler, L.; Rowe, S. T.; Valentine, M. B.; Komuro, H.; Castleberry, R. P.; Bpwman, L.C.; Rao, P. V.; Seeger, R. C. & Brodeur, G. M. (1995). Lack of correlation of N-mycgene amplification with prognosis in localised neuroblastoma: a pediatric oncologygroup study. Cancer Research, 55, 721-726.
[273] Comstock, J. M.; Willmore-Payne, C.; Holden, J. A. & Coffin, C. M. (2009). Compositepheochromocytoma: a clinicopathologic and molecular comparison with ordinarypheochromocytoma and neuroblastoma. American Journal of Clinical Phology,132,69-73.
[274] Iraci, N.; Diolati, D.; Papa, A. et al., (2011). A SP1/MIZ1/MYCN repression complexrecruits HDAC1 at the TrkA and p75NTR promoters and effects neuroblastoma ma‐lignancy by inhibiting the cell response to NGF. Cancer Research, 71, 404-412.
[275] Lau, D. T.; Hesson, L. B.; Norris, M. D.; Marshall, G. M.; Haber, M. & Ashton, L. J.(2012). Prognostic significance of promoter DNA methylation in patients with child‐hood neuroblastoma. Clinical Cancer Research, 18, 5690-5700.
[276] De Preter, K.; Vandesompele, J.; Heimann, P.; Yigit, N.; Beckman, S.; Schramm, A.;Eggert, A.; Stallings, R. L.; Benoit, Y.; Renard, M.; De Paepe, A.; Laureys, G.; Pahl‐man, S. & Speleman, F. (2006). Human fetal neuroblasts and neuroblastoma tran‐scriptome analysis confirms neuroblast origin and highlights neuroblastomacandidate genes. Genome Biology, 7 (R84) 3-17.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
97

[277] Lucarelli, E.; Kaplan, D. & Thiele, C. J. (1995). Selective regulation of TrkA and TrkBreceptors by retinoic acid and interferon-γ in human neuroblastoma cells. Journal ofBiological Chemistry, 270, 24725-24731.
[278] Chang, B. B.; Persengiev, S. P.; de Diego, J. G.; Sacristan, M. P.; Martin-Zanca, D. &Kilpatrick, D. L. (1998). Proximal promoter sequences mediate cell specific and ele‐vated expression of favourable prognosis marker TrkA in human neuroblastomacells. Journal of Biological Chemistry, 273, 39-44.
[279] Condello, S.; Caccamo, D.; Curro, M.; Ferlazzo, N.; Parisi, G. & Ientile R. (2008).Transglutaminase and NF-kB interplay during NGF-induced differentiation in neu‐roblastoma cells. Brain Research, 1207, 1-8.
[280] Azar, C. G.; Scavarda, N. J.; Nakagawara, A. & Brodeur, G. M. (1994). Expression andfunction of the nerve growth gactor receptor (TRK-A) in human neuroblastoma celllines. Progress in Clinical and Biological Research, 385, 169-175.
[281] Matsushima H & Bogenmann, E. (1993). Expression of TrkA cDNA in neuroblasto‐mas mediates differentiation in vitro and in vivo. Molecular and Cellular Biology, 13,7447-7456.
[282] Hartman, D. S. & Hertel, C. (1994). Nerve growth factor-induced differentiation inneuroblastoma cells expressing trkA but lacking p75NTR. Journal of Neurochemistry,63, 1261-1270.
[283] Poluha, W.; Poluha, D. K. & Ross, A. H. (1995). TrkA neurogenic receptor regulatesdifferentiation of neuroblastoma cells. Oncogene, 10, 185-189.
[284] Gryz, A. A. & Meakin, S. O. (2003). Acidic substitution of the activation loop tyro‐sines in TrkA supports nerve growth factor-dependent, but not nerve growth factor-independent differentiation and cell cycle arrest in the human neuroblastoma cellline, SY5Y. Oncogene, 22, 8774-8785.
[285] Kim, C. J.; Matsuo, T.; Lee, K-H. & Thiele, C. J. (1999). Up-regulation of insulin-likegrowth factor-II expression is a feature of TrkA but not TrkB activation in SH-SY5Yneuroblastoma cells. American Journal of Pathology, 155, 1661-1670.
[286] Peterson, S. & Bogemann, E. (2004). The RET and TrkA pathways collaborate to regu‐late neuroblastoma differentiation. Oncogene, 23, 213-215.
[287] Tsuruda, A.; Suzuki, S.; Maekawa, T. & Oka, S. (2004). Constitutively active Src facili‐tates NGF-induced phosphorylation of TrkA and causes enhancement of MAPK sig‐nalling in SK-N-MC cells. FEBS Letters, 560, 215-220.
[288] Fagerstrom, S.; Pahlman, S. Gestblom, C. & Nanberg, E. (1996). Protein kinase C-ε isimplicated in neurite outgrowth in differentiating neuroblastoma cells. Cell Growth &Differentiation, 7, 775-785.
Neuroblastoma98

[289] Eggert, A.; Ikegaki, N.; Liu, X-G.; Chou, T. T.; Lee, M. V. & Brodeur, G. M. (2000).Molecular dissection of TrkA signal transduction pathways leading to differentiationin human neuroblastoma cells. Oncogene, 19, 2043-2051.
[290] Olsson, A-K. & Nanberg, E. (2001). A functional role for ERK in gene induction, butnot in neurite outgrowth in differentiating neuroblastoma cells. Experimental Cell Re‐search, 265, 21-30.
[291] Eggert, A.; Grotzer, M. A.; Ikegaki, N.; Liu, X-G.; Evans, A. E. & Brodeur, G. M.(2000). Expression of neurotrophin receptor TrkA inhibits angiogenesis in neuroblas‐toma. Medical and Pediatric Oncology, 35, 569-572.
[292] Eggert, A.; Grotzer, M. A.; Ikegaki, N.; Liu, X. G.; Evans, A. E. & Brodeur, G. M.(2002). Expression of the neurotrophin receptor TrkA down-regulates expression andfunction of angiogenic stimulators in SH-SY5Y neuroblastoma cells. Cancer Research,62, 1802-1808.
[293] Schulte, J. H.; Kuhfittig-Kulle, S.; Klein-Hitpass, L.; Schramm, A.; Baird, D. S. F.;Pfeiffer, P. & Eggert, A. (2008). Expression of the TrkA and TrkB receptor tyrosinekinase alters the double-strand break (DSB) repair capacity of SY5Y neuroblastomacells. DNA Repair, 7, 1757-1764.
[294] Matsumoto, K.; Wada, R. K. & Yamashiro, J. M. (1995). Expression of brain derivedneurotrophic factor and p145TrkB affects survival, differentiation, and invasivenessof human neuroblastoma cells. Cancer Research, 55, 1798-1806.
[295] Lavoie, J-F.; LeSauteur, L.; Kohn, J.; Wong, J.; Furtoss, O.; Thiele, C. J.; Miller, F. D. &Kaplan, D. R. (2005). TrkA induces apoptosis of neuroblastoma cells and does so viaa p53-dependent mechanism. Journal of Biological Chemistry, 280, 29199-29207.
[296] Harel, L.; Costa, B.; Tcherpakov, M.; Zapatka, M.; Oberthuer, A.; Hansford, L. M.;Vojvodic, M.; Levy, Z.; Chen, Z-Y.; Lee, F. S.; Avigad, S.; Yaniv, I.; Shi, L.; Eils, R.;Fischer, M.; Brors, B.; Kaplan, D. R. & Fainzilber, M. (2009). CCM2 mediates deathsignaling by the TrkA receptor tyrosine kinase. Neuron, 63, 585-591.
[297] Jung, E. J. & Kim, D. R. (2008). Apoptotic death in TrkA-overexpressing cells: kineticregulation of ERK phosphorylation and caspase-7 activation. Molecular Cells, 26,12-17.
[298] Kogner, P.; Barbany, G.; Bjork, O.; castello, C.; Donfrancesco, A.; Falkmer, U. G.;Hedborg, F.; Kouvidou, H.; Persson, H.; Raschella et al., (1994). Trk mRNA and lowaffinity nerve growth factor receptor mRNA expression and triploid DNA content infavourable neuroblastoma tumors. Progress in Clinical and Biological Research, 385,137-145.
[299] Schulte, J. H.; Pentek, F.; Hartmann, W.; Schramm, A.; Friedrichs, N.; Ora, I.; Koster,J.; Versteeg, R.; Kirfel, J.; Buettner, R. & Eggert, A. (2009). The low affinity neurotro‐phin receptor, p75, is upregulated in ganglioneuroblastoma/ ganglioneuroma and re‐
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
99

duces tumorigenicity of neuroblastoma in vivo. International Journal of Cancer, 124,2488-2494.
[300] Bogemann, E. (1996). A metastatic neuroblastoma model in SCID mice. InternationalJournal of Cancer, 67, 379-385.
[301] Morandi, F.; Scaruffi, P.; Gallo, F.; Stigliani, S.; Moretti, S.; Bonassi, S.; Gambini, C.;Mazzocco, K.; Fardin, P.; Haupt, R.; Arcamone, G.; Pistoia, V.; Tonini, G. P. & Cor‐rias, M. V. (2012). Bone marrow infiltrating human neuroblastoma cells express highlevels of calprotectin and HLA-G- proteins. PloS ONE, 7, e2pp22
[302] Lipska, B. S.; Drozynska, E.; Scaruffi, P.; Tonini, G. P.; Izycka-Swieszewska, E.; Ziet‐kiewicz, S.; Balcerska, A.; Perek, D.; Chybicka, A.; Biernat, W. & Limon, J. (2009). C.1810C>T polymorphism of NTRK1 gene is associated with reduced survival in neu‐roblastoma patients. BMC Cancer, 9, 436-444.
[303] Cao, F.; Liu, X.; Zhang, L.; Wang, Y. & Zhang, N. (2010). Expression of TrkA spliceisoforms in neuroblastoma and its clinical significance. Chinese Journal of Clinical On‐cology, 37, 1282-1285.
[304] Minturn, J. E.; Evans, A. E.; Villablanca, J. G.; Yanik, G. A.; Park, J. R.; Shusterman, S.;Groshen, S.; Hellriegel, E. T.; Bensen-Kennedy, D.; Matthay, K. K.; Brodeur, G. M. &Maris, J. M. (2011). Phase I trial of laustaurtinib for children with refractory neuro‐blastoma: a new approaches to neuroblastoma therapy consortium study. Cancer Che‐motherapy and Pharmacology, 68, 1057-1065.
[305] Schramm, A.; Vandesompele, J.; Schulte, J. H.; Dreesman, S.; Kaderali, L.; Brors, B.;Eils, R.; Speleman, F. & Eggert, A. (2007). Translating expression profiling into a clini‐cally feasible test to predict neuroblastoma outcome. Clinical Cancer Research, 13,1459-1465.
[306] Guo, X.; Chen, Q-R.; Song, Y. K.; Wei, J. S. & Khan, J. (2011). Exon array analysis re‐veals neuroblastoma tumors have distinct alternative splicing patterns according tostage and MYCN amplification status. BMC Medical Genomics, 4, 35-50.
[307] Hishiki, T.; Saito, T.; Terui, K.; Sato, Y.; Takenouchi, A.; Yahata, E.; Ono, S.; Nakaga‐wara, A.; Kamijo, T.; Nakamura, Y.; Matsunga, T. & Yoshida, H. (2010). Reevaluationof trkA expression as a biological marker of neuroblastoma by high-sensitivity ex‐pression analysis – a study of 106 primary neuroblastomas treated in a single study.Journal of Pediatric Surgery, 45, 2293-2298.
[308] Farina, A. R.; Cappabianca, L.; Ruggeri, P.; Di Ianni, N.; Ragone, M.; Merolla, S.; Gu‐lino, A. & Mackay, A. R. (2012). Alternative TrkA Splicing and neuroblastoma. In:Neuroblastoma-Present and Future (Ed. Hiroyuki Shimada) Intech, Rijeka Croatia,pp111-136.
Neuroblastoma100

[309] Brodeur, G. M.; Nakagawara, A.; Yamashiro, D. J.; Ikegaki, N.; Liu, X. G.; Azar, C. G.;Lee, C. P. & Evans, A. E. (1997). Expression of TrkA, TrkB and TrkC in human neuro‐blastomas. Journal of Neurooncology, 31, 49-55.
[310] Brodeur, G. M; Minturn, J. E.; Ho, R.; Simpson, A. M.; Iyer, R.; Varela, C. R.; Light, J.E.; Kolla, V. & Evans A. E. (2009). Trk expression and inhibition in neuroblastomas.Clinical Cancer Research, 15, 3244-3256.
[311] Nakagawara, A.; Azar, C. G.; Scavarda, N. J. & Brodeur, G. M. (1994). Expression andfunction of Trk-B and BDNF in human neuroblastomas. Molecular and Cellular Biolo‐gy, 14, 759-767.
[312] Aoyama, M.; Sai, K.; Shishikura, T.; Kawamoto, T.; Miyachi, T.; Yokoi, T.; Togari, H.;Wada, Y.; Kato, T. & Nakagawara, A. (2001). Human neuroblastomas with unfavour‐able biologies express high levels of brain-derived neurotrophic factor mRNA and avariety of its variants. Cancer Letters, 164, 51-60.
[313] Baj, G. & Tongiorgi, E. (2008). BDNF splice variants from the second promoter clustersupport cell survival of differentiated neuroblastoma upon cytotoxic stress. Journal ofCell Science, 122, 36-43.
[314] Bouzas-Rodrigues, J.; Cabrera, J. R.; Delloye-Bourgeois, C.; Ichim, G.; Delcros, J-G.;Raquin, M-A.; Rousseau, R.; Combaret, V.; Bénard, J.; Tauszig-Delamasure, S. &Mehlen, P. Neurotrophin-3 production promotes human neuroblastoma cell survivalby inhibiting TrkC-induced apoptosis. Journal of Clinical Investigation, 120, 850-858.
[315] Stemple, D. L. & Anderson, D. J. (1992). Isolation of a stem cell for neurons and gliafrom the mammalian neural crest. Cell, 71, 973-985.
[316] Biagiotti, T.; D’Amico, M.; Marzi, I.; Di Gennaro, P.; Arcangeli, A.; Wanke, E. & Oli‐votto, M. (2006). Cell renewing in neuroblastoma: electrophysiological and immuno‐cytochemical characterization of stem cells and derivatives. Stem Cells, 24, 443-453.
[317] Marzi, I.; D’Amico, M. ; Biagiotti, T.; Giunti, S.; Carbone, M. V.; Fredducci, D.;Wanke, E. & Olivotto, M. (2007). Purging of the neuroblastoma stem cell componentand tumor regression on exposure to hypoxia or cytotoxic treatment. Cancer Research,67, 2402-2407.
[318] Islam, O.; Loo, T. X. & Heese, K. (2009). Brain-derived neurotrophic factor (BDNF)has proliferative effects on neural stem cells through the truncated Trk-B receptor,MAP kinase, Akt, and STAT-3 signaling pathways. Current Neurovascular Reseach, 6,42-53.
[319] Fanburg-Smith, J. C. & Miettinen, M. (2001). Low-affinity nerve growth factor recep‐tor (p75) in Dermatofibrosarcoma Protuberans and other neural tumors: A study of1,150 tumors and fetal and adult normal tissues. Human Pathology, 32, 976-983.
[320] Perosio, P. M. & Brooks, J. J. (1988). Expression of nerve growth factor receptor inparaffin-embedded soft tissue tumors. American Journal of Pathology, 132, 152-160.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
101

[321] Kimura, N.; Nakamura, M.; Kimura, I. & Nagura, H. (1996). Tissue localisation ofnerve growth factor receptors: TrkA and low-affinity nerve growth factor receptor inneuroblastoma, pheochromocytoma, and retinoblastoma. Endocrine Pathology, 7,281-289.
[322] Ho, R.; Minturn, J. E.; Simpson, A. M.; Iyer, R.; Light, J. E.; Evans, A. E. & Brodeur, G.M. (2011). The effect of P75 on Trk receptors in neuroblastoma. Cancer Letters, 305,76-85.
[323] Zhao, S. P. (2003). Co-expression of TrkA and p75 neurotrophin receptor in extracra‐nial olfactory neuroblastoma cells. Humnan Yi Ke Da Xue Xue Bao, 28, 50-52.
[324] Matsushima, H. & Bogenmann, E. (1994). NGF induces terminal differentiation intrkA expressing neuroblastoma cells in vitro and in vivo. Progress in Clinical and Bio‐logical Research, 385, 177-183.
[325] Ehrehard, P.B.; Ganter, U.; Schmutz, B.; Bauer, J. & Otten, U. (1993). Expression oflow affinity nerve growth factor receptor and TrkB messenger RNA in human SH-SY5Y neuroblastoma cells. FEBS letters, 330, 287-292.
[326] Roux, P. P.; Bhakar, A. L.; Kennedy, T. E. & Barker, P. A. (2001). The p75 neurotro‐phin receptor activates Akt (protein kinase B) through a phosphoinositol 3-kinase-dependent pathway. Journal of Biologcal Chemistry, 276, 23097-23104.
[327] Levrerrier, Y.; Thomas, J.; Mathieu, A. L.; Low, W.; Blanquier, B. & Marvel, J. (1999).Role of PI-3kinase in Bcl-X induction and apoptosis inhibition mediated by IL-3 orIGF in Baf-3 cells. Cell Death & Differentiation, 6, 290-296.
[328] Gargano, N.; Levi, A. & Alema, S. (1997). Modulation of nerve growth factor internal‐isation by direct interaction between p75 and trkA receptors. Journal of NeuroscienceResearch, 50, 1-12.
[329] Wehrman, T.; He, X.; Raab, B.; Dukipatti, A.; Blau, H. & Garcia, K. C. (2007). Structur‐al and mechanistic insights into nerve growth factor interactions with the TrkA andp75 receptors. Neuron, 53, 25-38.
[330] Makkerth, J. P.; Ceni, C.; Auld, D. S.; Vaillancourt, F.; Dorval, G. & Barker, P. A.(2005). P75 neurotrophin receptor reduces ligand-induced Trk receptor ubiquinationand delays Trk receptor internalisation and degradation. EMBO Reports, 6, 936-941.
[331] Zhang, C.; Helmsing, S.; Zagrebelsky, M.; Schirrmann, T.; Marschall, A. L. J.; Schun‐gel, M.; Korte, M.; Hust, M. & Dubel, S. (2012). Suppression of p75 neurotrophin re‐ceptor surface expression with itraantibodies influences Bcl-X mRNA expression andneurite outgrowth in PC12 cells. PloS ONE, 7, e30684.
[332] Ito, H.; Nomoto, H. & Furukawa, S. (2003). Growth arrest of PC12 cells by nervegrowth factor is dependent on the phosphoinositol 3-kinase pathway via p75 neuro‐trophin receptor. Journal of Neuroscience, 72, 211-217.
Neuroblastoma102

[333] Chen, J. & Zhe, X. (2003). Cotransfection of TrkA and p75(NTR) in neuroblastomacell line (IMR32) promotes differentiation and apoptosis of tumor cells. Chinese Medi‐cal Journal, 116, 906-912.
[334] Yan, C.; Liang, Y.; Nylander, K. D.; Wong, J.; Rudavsky, R. M.; Saragovi, H. U. &Schor, N. F. (2002). P75-Nerve growth factor as an antiapoptotic complex: Independ‐ence versus cooperativity in protection from enediyne chemotherapeutic agents. Mo‐lecular Pharmacology, 61, 710-719.
[335] Holub, J. L.; Qui, Y. Y.; Chu, F. & Madonna, M. B. (2011). The role of nerve growthfactor in caspase-dependent apoptosis in human BE(2)C neuroblastoma. Journal of Pe‐diatric Surgery, 46, 1191-1196.
[336] Bunone, G.; Mariotti, A.; Compagni, A.; Morandi, E. & Della Valle, G. (1997). Induc‐tion of apoptosis by p75 neurotrophin receptor in human neuroblastoma cells. Onco‐gene, 14,1463-1470.
[337] Eggert, A.; Sieverts, H.; Ikegaki, X-G. & Brodeur, G. M. (2000). P75 mediated apopto‐sis in neuroblastoma cells is inhibited by expression of TrkA. Medical and PediatricOncology, 35, 573-576.
[338] Giraud, S.; Lautrette, B.; Bessette, B.; Decourt, C.; Mathonnet, M. & Jauberteau, M. O.(2005). Modulation of Fas-induced apoptosis by p75 neurotrophin receptor in a hu‐man neuroblastoma cell line. Apoptosis, 10, 1271-1283.
[339] Bai, Y.; Qiang, Li.; Yang, J.; Zhou.; X.; Yin, X. & Zhao, D. (2008). P75NTR activation ofNF-kB is involved in prP106- 126-induced apoptosis in mouse neuroblastoma cells.Neuroscience Research, 62, 9-14.
[340] Rogers, M-L.; Beare, A.; Zola, H. & Rush, R. A. (2008). CD-271 (P75 neurotrophin re‐ceptor). Journal of Biological Regulation & Homeostatic Agents, 22, 1-6.
[341] Kuwako, K-I.; Taniura, H. & Yoshikawa, K. (2004). Necdin-related MAGE proteinsdifferentially interact with the E2F1 transcription factor and the p75 neurotrophin re‐ceptor. Journal of Biological Chemistry, 279, 1703-1712.
[342] Kenchappa, R. S.; Zampieri, N.; Chao, M. V.; Barker, P. A.; Teng, H. K.; Hempstead,B. L. & Carter, B. D. (2006). A ligand-dependent cleavage of P75 neurotrophin recep‐tor is necessary for NRIF nuclear translocation and apoptosis in sympathetic neu‐rons. Neuron, 50, 219-232.
[343] Christiansen, H.; Christiansen, N. M.; Wagner F et al., (1990). Neuroblastoma-inverserelationship between expression of N-Myc and NGF-R. Oncogene, 5, 437-440.
[344] Wang, C.; Liu, Z.; Woo, C-W.; Li, Z.; Wang, L.; Wei, J. S.; Marquez, V. E.; Bates, S. E.;Jin, Q.; Khan, J.; Ge, K. and Thiele, C. J. (2011). EZH2 mediates epigenetic silencing ofneuroblastoma suppressor genes CASZ1, CLU, RUNX3, and NGFR. Cancer Research,72, 315-324.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
103

[345] Cortazzo, M. H.; Kassis, E. S.; Sproul, K. A. & Schor, N. F. (1996). Nerve growth fac‐tor (NGF)-mediated protection of neural crest cells from antimitotic agent-inducedapoptosis: the role of low-affinity NGF receptor. Journal of Neuroscience, 16, 3895-3899.
[346] Panicker, J.; Li, Z.; McMahon, C.; Sizer, C.; Steadman, K.; Piekarz, R.; Bates, S. E. &Thiele, C. J. (2010). Romidepsin (FK228/depsipeptide) controls growth and inducesapoptosis in neuroblastoma tumor cells. Cell Cycle, 9, 1830-1838.
[347] Borrello, M. G.; Bongarzone, I.; Pierotti, M. A.; Luksch, R.; Gasparini, M.; Collini, P.;Pilotti, S.; Rizzetti, M. G.; Mondellini, P.; De Bernardi, B.; Di Martino, D.; Garaventa,A.; Brisigotti, M. & Tonini, G. P. (1993). Trk and ret proto-oncogene expression in hu‐man neuroblastoma specimens: high frequency of trk expression in non-advancedstages. International Journal of Cancer, 54, 540-545.
[348] Edsjo, A.; Lavinius, E.; Nilsson, H.; Hoehner, J. C.; Simonsson, P.; Culp, L. A.; Mar‐tinsson, T.; Larsson, C.; Pahlman, S. (2003). Expression of trkB in human neuroblasto‐ma in relation to MycN expression and retinoic acid treatment. LaboratoryInvestigation, 83, 813-823.
[349] Fung, W.; Hasan, M. Y.; Loh, A. H.; Chua, J. H.; Yong, M. H.; Knight, L.; Hwang, W.S.; Chan, M. Y.; Seow, W. T.; Jacobsen, A. S. & Chui, C. H. (2011). Gene expression ofTrk neurotrophin receptors in advanced stage neuroblastomas in Singapore - a pilotstudy. Pediatric Hematology and Oncology, 28, 571-578.
[350] Zhang, J.; Zhend, Y.; Wang, Y. & Tong, H. (2010). The studies on the correlation forgene expression of tyrosine-kinase receptors and vascular endothelial growth factorin human neuroblastomas. Journal of Pediatric Hematology and Oncology, 32, 180-184.
[351] Pastor, R.; Bernal, J. & Rodriguez-Pena, A. (1994). Unliganded c-erbA/thyroid hor‐mone receptor induces trkB expression in neuroblastoma cells. Oncogene, 1081-1089.
[352] Martens, L. K.; Kirschner, K. M.; Warnecke, C. & Scholz, H. (2007). Hypoxia-induci‐ble factor-1 (HIF-1) is a transcriptional activator of the TrkB neurotrophin receptorgene. Journal of Biological Chemistry, 282, 14379-14388.
[353] Hoehner, J. C.; Olsen, L.; Sandstedt, B.; Kaplan, D. R. & Pahlman, S. (1995). Associa‐tion of neurotrophin receptor expression and differentiation in human neuroblasto‐ma. American Journal of Pathology, 147, 102-113.
[354] Lucarelli, E.; Kaplan, D. & Thiele, C. J. (1997). Activation of trk-A but not trk-B signaltransduction pathway inhibits growth of neuroblastoma cells. European Journal ofCancer, 33, 2068-2070.
[355] Kaplan, D. R.; Matsumoto, K.; Lucarelli, E. & Thiele, C. J. (1993). Induction of TrkB byretinoic acid mediates biologic responsiveness to BDNF and differentiation of humanneuroblastoma cells. Neuron, 11, 321-331.
Neuroblastoma104

[356] Esposito, C. L.; D’Alessio, A.; de Franciscis, V. & Cerchia, L. (2008). A cross-talk be‐tween TrkB and Ret tyrosine kinases receptors mediates neuroblastoma cells differ‐entiation. PloS ONE,3(2):e1643.doi:10.1371/journal.pone. 001643
[357] Shirohira, H.; Kitaoka, A.; Enjoji, M.; Uno, T. & Nakashima, M. (2012). AM80 inducesneuronal differentiation via increased tropomyosin-related kinase B expression inhuman neuroblastoma SH-SY5Y cell line. Biomedical Research, 33, 291-297.
[358] Middlemas, D. S.; Kihl, B. K.; Zhou, J. & Zhu, X. (1999). Brain-derived neurotrophicfactor promotes survival and chemoprotection of human neuroblastoma cells. Journalof Biological Chemistry, 274, 16451-16460.
[359] Scala, S.; Wosikowski, K.; Giannakakou, P.; Valle, P.; Biedler, J.; Spengler, B. A.; Lu‐carelli, E.; Bates, S. E. & Thiele, C. J. (1996). Brain-derived neurotrophic factor pro‐tects neuroblastoma cells from vinblastine toxicity. Cancer Research, 56, 3737-3742.
[360] Ho, R.; Eggert, A.; Hishiki, T.; Minturn, J. E.; Ikegaki, N.; Foster, P.; Camoratto, A. M.;Evans, A. E. & Brodeur, G. M. (2002). Resistance to chemotherapy mediated by TrkBin neuroblastoma. Cancer Research, 62, 6462-6466.
[361] Jaboin, J.; Kim, C. J.; Kaplan, D. R. & Thiele, C. J. (2002). Brain-derived neurotrophicfactor activation of TrkB protects neuroblastoma cells from chemotherapy-inducedapoptosis via phosphatidylinositol 3’-kinase pathway. Cancer Research, 62, 6756-6763.
[362] Jaboin, J.; Hong, A.; Kim, C. J. & Thiele, C. J. (2003). Cisplatin-induced cytotoxicity isblocked by brain-derived neurotrophic factor activation of TrkB signal transductionpath in neuroblastoma. Cancer Letters, 193, 109-114.
[363] Li, Z. & Thiele, C. J. (2007). Targeting Akt to increase the sensitivity of neuroblastomato chemotherapy: lessons learned from the brain derived neurotrophic factor/TrkBsignal transduction pathway. Expert Opinions in Therapeutic Targets, 11, 1611-1621.
[364] Hecht, M.; Schulte, J. H.; Eggert, A.; Wilting, J. & Schweigerer, L. (2005). The neuro‐trophin receptor TrkB cooperates with c-Met in enhancing neuroblastoma invasive‐ness. Carcinogenesis, 26, 2105-2115.
[365] Cimmino, F.; Schulte, J. H.; Zollo, M.; Koster, J.; Versteeg, R.; Iolascon, A.; Eggert, A.& Schramm, A. (2009). Galectin-1 is a major effector of Trk-B-mediated neuroblasto‐ma aggressiveness. Oncogene, 28, 2015-2023.
[366] Nakamura, K.; Martin, K. C.; Jackson, J. K.; Beppu, K.; Woo, C-W. & Thiele, C. J.(2006). Brain-derived neurotrophic factor activation of TrkB induces vascular endo‐thelial growth factor expression via hypoxia-inducible factor-1a in neuroblastomacells. Cancer Research, 66, 4249-4255.
[367] Geiger, T. R. & Peeper, D. S. (2005). The neurotrophic receptor TrkB in anoikis andmetastasis: a perspective. Cancer Research, 65, 7033-7036.
[368] Haapsalo, A.; Saarelainen, T.; Moshynakov, M.; Arumae, U.; Kiema, T. R.; Saarma,M.; Wong, G. & Castrén, E. (1999). Expression of the naturally occurring truncated
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
105

trkB neurotrophin receptor induces outgrowth of filopodia and processes in neuro‐blastoma cells. Oncogene, 18, 1285-1296.
[369] Olivieri, G.; Otten, U.; Meier, F.; Baysang, G.; Dimitriades-Schmutz, B.; Muller-Spahn, F. & Savaskan, E. (2003). β-Amyloid modulates tyrosine kinase B receptor ex‐pression in SHSY5Y neuroblastoma cells: influence of the antoxidant melatonin.Neuroscience, 120, 659-665.
[370] Ryden, M.; Sehgal, R.; Dominici, C.; Schilling, F. H.; Ibanez, C. F. & Kogner, P. (1996).Expression of mRNA for the neurotrophin receptor TrkC in neuroblastomas with fa‐vourable tumour stage and good prognosis. British Journal of Cancer ,74, 773-779.
[371] Yamashiro, D. J.; Liu, X-G.; Lee, C. P.; Nakagawara, A.; Ikegaki, N.; McGregor, L. M.;Baylin, S. B. & Brodeur, G. M. (1997). Expression and function of Trk-C in favourablehuman neuroblastomas. European Journal of Cancer 33, 2054-2057.
[372] Svensson, T.; Ryden, M.; Schilling, F. H.; Dominici, C.; Sehgal, R.; Ibanez, C. F. &Kogner, P. (1997). Coexpression of mRNA for the full-length neurotrophin receptorTrk-C and trk-A in favourable neuroblastoma. European Journal of Cancer, 33,2058-2063.
[373] Menn, B.; Timsit, S.; Represa, A.; Mateos, S.; Calothy, G. & Lamballe, F. (2000). Spa‐tiotemporal expression of noncatalytic TrkC NC2 isoform during early and late CNSneurogenesis: a comparative study with TrkC catalytic and p75NTR receptors. Euro‐pean Journal of Neuroscience, 12, 3211-3223.
[374] Edsjo, A.; Hallberg, B.; Fagerstrom, S.; Larsson, C.; Axelson, H. & Pahlman, S. (2001).Differences in the early and late responses between neurotrophin-stimulated TrkAand TrkC transfected SH-SY5Y neuroblastoma cells. Cell Growth & Differentiation, 12,39-50.
[375] Bassili, M.; Birman, E.; Schor, N. F. & Saragovi, U. H. (2010). Differential roles of Trkand p75 neurotrophin receptors in tumorigenesis and chemoresistance ex vivo and invivo. Cancer Chemother Pharmacol 65, 1047-1056.
[376] Nara, K.; Kasafuka, T.; Yoneda, A.; Oue, T.; Sangkhathat, S. & Fukuzawa, M. (2007).Silencing MYCN by RNA interference induces growth inhibition, apoptotic activityand cell differentiation in a neuroblastoma cell line with MYCN amplification. Inter‐national Journal of Oncology, 30, 1189-1196.
[377] Laneve, P.; Di Marcotullio, L.; Gioia, U.; Fiori, M. E.; Ferretti, E.; Gulino, A.; Bozzoni,I. & Caffarelli, E. (2007). The interplay between microRNAs and the neurotrophin re‐ceptor tropomyosin-related kinase C controls proliferation of human neuroblastomacells. Proceedings of the National Academy of Science. USA. 104, 7957-7962.
[378] Guidi, M.; Muinos-Gimeno, M.; Kagerbauer, B.; Martl, E.; Estivilli, X. & Espinosa-Parrilla, Y. (2010). Overexpression of miR-128 specifically inhibits the truncated iso‐
Neuroblastoma106

form of NTRK3 and upregulates BCL2 in SH-SY5Y neuroblastoma cells. BMCMolecular Biology, 11, 95.
[379] Thress, K.; Macintyre, T.; Wang, H.; Whitston, D.; Liu, Z. Y.; Hoffmann, E.; Wang, T.;Brown, J. L.; Webster, K.; Omer, C.; Zage, P. E.; Zeng, L. & Zweidler-McKay, P. A.(2009). Identification and preclinical characterisation of AZ-23, a novel, selective, andorally bioavailable inhibitor of the Trk kinase pathway. Molecular Cancer Therapeutics,8, 1818-1827.
[380] Zage, P. E.; Graham, T. C.; Zeng, L.; Fang, W.; Pien, C.; Thress, K.; Omer, C.; Brown,J. L. & Zweidler-McKay, P. A. (2011). The selective Trk inhibitor AZ623 inhibitsbrain-derived neurotrophic factor-mediated neuroblastoma cell proliferation and sig‐nalling and is synergistic with topotecan. Cancer, 117, 1321-1329.
[381] Iyer, R.; Varela, C. R.; Minturn, J. E.; Ho, R.; Simpson, A. M.; Light, J. E.; Evans, A. E.;Zhao, H.; Thress, K.; Brown, J. L. & Brodeur, G. M. (2012). AZ64 inhibits TrkB andenhances the efficacy of chemotherapy and local radiation in neuroblastoma xeno‐grafts. Cancer Chemotherapy and Pharmacology, 10.1007/s00280-012-1879-x
[382] Evans, A. E.; Kisselbach, K. D.; Yamashiro, D. J.; Ikegaki, N.; Camoratto, A. M.; Dio‐nne, C. A. & Brodeur, G. M. (1999). Antitumor activity of CEP-751 (KT-6587) on hu‐man neuroblastoma and medulloblastoma xenografts. Clinical Cancer Research, 5,3594-3602.
[383] Evans, A. E.; Kisselbach, K. D.; Liu, X.; Eggert, A.; Ikegaki, N.; Camoratto, A. M.; Dio‐nne, C. A. & Brodeur, G. M. (2001). Effect of CEP-751 (KT-6587) on neuroblastomaxenografts expressing trkB. Medical Pediatric Oncology, 36, 181-184.
[384] Iyer, R.; Evans, A. E.; Qi, X.; Ho, R.; Minturn, J. E.; Zhao, H.; Balamuth, N.; Maris, J.M. & Brodeur, G. M. (2010). Lestaurtinib enhances the antitumour efficacy of chemo‐therapy in murine xenograft models of neuroblastoma. Clinical Cancer Research, 16,1478-1485.
[385] Minturn, J. E.; Evans, A. E.; Villablanca, J. G.; Yanik, G. A.; Park, J. R.; Shusterman, S.;Groshen, S.; Hellriegel, E. T.; Bensen-Kennedy, D.; Matthay, K. K.; Brodeur, G. M. &Maris, J. M. (2011). Phase I trial of laustaurtinib for children with refractory neuro‐blastoma: a new approaches to neuroblastoma therapy consortium study. Cancer Che‐motherapy and Pharmacology, 68, 1057-1065.
[386] Norris, R. E.; Minturn, J. E.; Brodeur, G. M.; Maris, J. M. & Adamson, P. C. (2011).Preclinical evaluation of lesaurtinib (CEP-701) in combination with retinoids fromneuroblastoma. Cancer Chemotherapy and Pharmacology, 68, 1469-1475.
[387] Saulnier Sholler, G. L.; Brard, L.; Straub, J. A.; Dorf, L.; Illeyne, S.; Koto, K.; Kalkunte,S.; Bosenberg, M.; Ashikaga, T. & Nishi, R. (2009). Nifurtimox induces apoptosis ofneuroblastoma cells in vitro and in vivo. Journal of Pediatric Hematology and Oncology,31, 187-193.
Neurotrophin and Neurotrophin Receptor Involvement in Human Neuroblastomahttp://dx.doi.org/10.5772/55536
107

[388] Yamaguchi, Y.; Tabata, K.; Asami, S.; Miyake, M. & Suzuki, T. (2007). A novel cyclo‐phane compound, CPPy, facilitates NGF-induced TrkA signal transduction and in‐duces cell differentiation in neuroblastoma. Biological and Pharmacological Bullitin, 30,638-643.
[389] Ikegaki, N.; Gotoh, T.; Kung, B.; Riceberg, J. S.; Kim, D. Y.; Zhao, H.; Rappaport, E. F.;Hicks, S. L.; Seeger, R. C. & Tang, X. X. (2007). De novo identification of MIZ-1(ZBTB1) encoding MYC-interacting zinc finger protein as a new favourable neuro‐blastoma gene. Clinical Cancer Research, 13, 6001-6009.
[390] Wang, C.; Liu, Z.; Woo, C. W.; Li, Z.; Wang, L.; Wei, J. S.; Marquez, V. E.; Bates, S. E.;Jin, Q.; Khan, J.; Ge, K. & Thiele, C. J. (2012). EZH2 mediates epigenetic silencing ofneuroblastoma suppressor genes CASZ1, CLU, RUNX3, and NGFR. Cancer Research,72, 315-324.
[391] Tang, X. X.; Robinson, M. E.; Riceberg, J. S.; Kim, D. Y.; Kung, B.; Titus, T. B.; Haya‐shi, S.; Flake, A. W.; Carpentieri, D. & Ikegaki, N. (2005). Favorable neuroblastomagenes and molecular therapeutics of neuroblastoma. Clinical Cancer Research, 10,5837-5844.
Neuroblastoma108





























