ReviewObesity and Atherosclerosis: Mechanistic InsightsFina
Lovren, PhD,aHwee Teoh, PhD,a,band Subodh Verma, MD, PhD,
FRCSCaaDivision of Cardiac Surgery, Keenan Research Centre for
Biomedical Science, St Michaels Hospital, University of Toronto,
Toronto, Ontario, CanadabDivision of Endocrinology and Metabolism,
Keenan Research Centre for Biomedical Science, St Michaels
Hospital, University of Toronto, Toronto, Ontario,
CanadaABSTRACTObesityisamultifactorial
chronicdiseasecharacterizedbyanaccu-mulationof visceral
andsubcutaneousfat, whichleadstoapredis-position toward
cardiometabolic diseases. A plethora of mechanisms,including
abnormalities in lipid metabolism, insulin resistance,inammation,
endothelial dysfunction, adipokine imbalance,
andinammasomeactivationhavebeensuggestedtounderlietherela-tionship
between obesity and atherosclerosis. More recent data pointtoward
an emerging role of impaired autophagy and altered
gutmicrobiomehomeostasisaspotentiallycontributingfactors.
Thisre-view provides an overview of this area.R
ESUM
ELob esit eestune maladiechronique multifactoriellecaract eris
eeparune accumulation de graisse visc erale et de graisse
sous-cutan ee, quientraneunepr edispositionaux maladies cardiom
etaboliques. Unepl ethoredem ecanismes, dont lesanomaliesdum
ecanismedesli-pides, linsulinor esistance, linammation,
ladysfonctionendoth elial,led es
equilibredesadipocytokinesetlactivationdelinammasomeontet e sugg er
es pouretayer le lien entre lob esit e et lath eroscl erose.Desdonn
eesplusr ecentessugg eraient commerle emergent lad ecience de
lautophagie et lalt eration de lhom eostasie dumicrobiome
intestinal dtre des facteurs potentiellement contributifs.La pr
esente revue donne un aperu de ce domaine de recherche.Obesityis a
major risk factor for atherosclerotic vasculardis-ease and
cardiometabolic syndrome. Various mechanisms
havebeensuggestedtolinkobesitytoatherosclerosis. Thesearedetailed
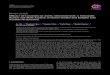
in Table 1 and are reviewed herein.AdipokineImbalanceThe visceral
adipose tissue is a source of numerousadipokines, most of which are
deemed to be proinammatory.Increasing evidence suggests that an
imbalance betweenproinammatory vs anti-inammatory adipokines (such
asadiponectin) might be responsible for the development ofinsulin
resistance and endothelial dysfunction and athero-sclerosis in
patients with obesity (Fig.
1).Adiponectinisthemostabundantanti-inammatoryandvasculoprotective
adipokine secreted by adipose tissues.Numerous studies have to date
demonstrated that plasma
levelsofadiponectinarelowerinpatientswithobesityand/ordia-betes.
Adiponectinimprovesinsulinsensitivitybyincreasingenergy expenditure
and fatty acid oxidation through thephosphorylation of
5-adenosine-monophosphate-activatedprotein kinase, and via an
increase in the expression of perox-isome
proliferator-activatedreceptor-atarget genes suchasCD36,
acyl-coenzyme oxidase, anduncouplingprotein2.2The adiponectin
receptors, AdipoR1 and AdipoR2, areresponsible for mediating the
metabolic actions of adiponectin.Adiponectinexerts its
vasculoprotectiveeffects throughamultitude of pathways. In vitro,
adiponectin induces nitric oxide(NO) productionin human aortic
endothelial cells via activationof the
5-adenosine-monophosphate-activated proteinkinasepathway and
enhancement of endothelial NOsynthase (eNOS)activity.3Adiponectin
additionally suppresses proliferation andsuperoxide generation,
andalsoenhances eNOSactivityinendothelial cells treated with
oxidized low-density lipoprotein.4Adiponectin has also been
demonstrated to attenuate theadhesionof monocytes toendothelial
cells, primarily via amechanism that involves inhibition of tumour
necrosis factor(TNF)-a- and interleukin (IL)-8-induced synthesis of
intercel-lular adhesionmolecule 1(ICAM1), vascular cell
adhesionmolecule 1 (VCAM1), and E-selectin.5Adiponectin
suppressesthe expression of class Amacrophage scavenger receptors
andconsequently reduces foam cell formation and decreases
secre-tion of proinammatory cytokines.6Notably, foam
cellformationis further
reducedbyadiponectin-induceddown-regulationofacyl-coenzymeA:cholesterol
acyltransferase-1inmacrophages, the enzyme that catalyzes the
formation ofcholesteryl esters.7Accordingly, adiponectin limits the
initiationof atherosclerotic plaque formation. Recent data point
towardtheabilityof
adiponectintoprimemonocytedifferentiationtowardtheanti-inammatoryM2macrophagelineage,8andCanadian
Journal of Cardiology 31 (2015) 177e183Received for publication
November 18, 2014. Accepted November 25, 2014.Correspondingauthor:
DrSubodh Verma,Division
ofCardiacSurgery,KeenanResearchCentreforBiomedical Science,
StMichaelsHospital, 30BondSt, Toronto, OntarioM5B1W8, Canada. Tel.:
1-416-864-5997;fax: 1-416-864-5991.E-mail: [email protected] page
181 for disclosure
information.http://dx.doi.org/10.1016/j.cjca.2014.11.0310828-282X/
2015 Canadian Cardiovascular Society. Published by Elsevier Inc.
All rights reserved.that in states of adiponectin deciency (such as
obesity),monocytes might
beprimarilydriventowardtheproathero-sclerotic M1lineage.
Physiological concentrations of adipo-nectin signicantly suppress
the proliferation and migration ofhuman aortic smooth muscle cells
induced by platelet-derivedgrowth factor BB in vitro9by directly
binding platelet-derivedgrowth factor-BB and inhibiting growth
factor-stimulatedextracellular signal-regulatedkinase signalling.
These resultssuggest that adiponectinmight also prevent vascular
remodellingin atherosclerosis. In other studies, adiponectin has
been shownto selectively increase the expression of tissue
inhibitor ofmetalloproteinase-1 in human monocyte-derived
macro-phages,10suggestingthat it might favour plaque
stabilization.Via improving vascular function, adiponectin has been
recentlyimplicated in protection against sepsis induced
multiorgandysfunction.11Invivostudies inmice have
conrmedtheantiatherogenic properties of adiponectin.
Adiponectin-decientmice,comparedwithwildtypecontrols,showneo-intimal
thickening and increased proliferation of vascularsmooth muscle
cells after mechanical injury to
arteries.12Adenovirus-mediateddelivery of adiponectininthese
miceconsiderably attenuated the extent of neointimal
proliferation.12Furthermore, treatment of apolipoprotein E-decient
mice(ApoE/)withadiponectin-expressingadenovirusesreducedatherosclerotic
lesion formation compared with control mice.13Takentogether,
thebalanceofpublishedinformationwouldstrongly associate low
adiponectin levels in obesity as a causaland/or permissive basis of
atherosclerotic risk.Resistin is a proinammatory adipokine that is
released inexcess amounts in individuals with visceral adiposity.
Resistinprofoundly upregulates the expression of TNF-a and IL-6
inhuman peripheral blood mononuclear cells.14Human resistinhas been
reported to stimulate the synthesis of proin-ammatory cytokines
such as TNF-a, IL-1, IL-6, andIL-12invariouscell types
throughanuclear
factor-kB-dependentpathway.15Apositivecorrelationbetweenresistinlevelsandvascular
inammation has been demonstrated in obesehumans.16As further
evidence of its proinammatory prole,resistin upregulates the
expression of adhesion moleculesVCAM1andICAM1,
andinduceschemokine(C-Cmotif)ligand 2 and endothelin (ET)-1 release
in human endothelialcells.17Indeed, we demonstratedthat
resistindirectly trig-gered endothelial cell activation by
promoting ET-1 release, inpart by inducing ET-1promoter activity
via the activatorprotein-1site. Furthermore,
resistinupregulatedtheexpres-sion of adhesion molecules and
chemokines and down-regulatedtumor necrosis factor
receptor-associatedfactor-3,aninhibitor of
CD40ligandsignallinginendothelial cells.Therefore,
increasedresistinlevels might be another factorthat contributes to
obesity-induced atherosclerosis.17The adipokine, leptin, has a main
function to control foodintakeandenergyexpenditure.18Levelsof
circulatingleptinuctuate;
theyareincreasedwithoverfeedinganddecreasedwithstarvation.Micewithamutationinthegeneencodingleptin(ob/ob
mice) or the gene encodingthe leptinreceptor(db/db mice) have obese
phenotypes,19and also exhibitincreased atherosclerosis.20Leptin is
considered to be aproinammatory cytokineandis structurallysimilarto
otherproinammatory cytokines such as IL-6, IL-12, and theTable 1.
Suggested mechanisms of obesity-induced atherosclerosisEndothelial
dysfunctionAdipokine imbalanceVascular inammationMacrophage NLRP3
inammasome activationAltered gut microbiomeLoss of
autophagicuxOxidative stressFigure 1. Anti- and proinammatory
adipokines. AT, angiotensin II; CRP, C-reactive protein; EC,
endothelial cell; ET-1, endothelin-1; ICAM, intercellularadhesion
molecule; IL, interleukin; MCP-1, monocyte chemoattractant protein
1; MMP, matrix metalloproteinase; NO, nitric oxide; oxLDL, oxidized
low-density lipoproteins; PAI-1, plasminogen activator inhibitor-1;
SMC, smooth muscle cell; TNF, tumour necrosis factor; VCAM,
vascular cell adhesionmolecule. Reproduced from Lau et al.1with
permission from American Physiological Society. Copyright 2014, The
American Physiological Society.178 Canadian Journal of
CardiologyVolume 31 2015granulocyte colony stimulating
factor.18Leptin modulates theT-cell immune response, stimulates the
proliferationof T-helper cells, and increases production of
proinammatorycytokines byregulatingdifferent immune
cells.21Increasedproductionofleptin,therefore,modulatescaloricintakeandatherosclerotic
susceptibility.ImpairedAutophagyAccumulating data suggest that loss
of autophagy might bean important determinant of atherosclerosis,
particularly withaging.22-25One theory that is rapidly gaining
popularity is thatloss of autophagy might be a central
mechanismthrough whichcardiometabolic diseases arise. An intact
autophagic machineryis essential in limiting lipid uptake by
macrophages. Autophagy-relatedprotein7(ATG7) is a critical
autophagygene andmacrophage specic ATG7-decient mice exhibit
increasedsusceptibility toward atherosclerosis25via an increase
inmacrophage low-density lipoprotein uptake and foam cell
for-mation. Autophagic uxiscompromisedinobesityandthemetabolic
syndrome. Ina recent seminal paper, mice withglobal
haploinsufciencyof ATG7(Atg7/mice) didnotshowmetabolicabnormalities
but developeddiabetes whencrossed with ob/ob mice. Atg7/-ob/ob mice
showaggravatedinsulin resistance with increased lipid content and
inamma-tory changes, suggesting that autophagy
haploinsufciencyimpairstheadaptiveresponsetometabolicstress.26Further-more,
intracellular lipid content and insulin resistance after
lipidloading are increased as a result of autophagy insufciency,
andthere is increased inammasome activation in
Atg7/-ob/obmice.26These results suggest that systemic autophagy
insuf-ciencycouldbeafactor intheprogressionfromobesitytodiabetes,
and associated inammation and atherosclerosis.OxidativeStress Links
Obesity toAtherosclerosisObesity is strongly associated with an
increase in systemicoxidative stress. The extent of fat
accumulation in obesehumans closely correlates with the markers of
oxidativestress.27Similarly, the oxidative stress is augmented in
plasmaand adipose tissue fromobese mice.27Increased
oxidantproduction, through quenching NO, has been linked
toincreased atherosclerosis susceptibility.Oxidative stress results
fromanimbalance
betweentheproductionoffreeradicalsandantioxidantsystems.
Increaseinreactiveoxygenspecies(ROS)productionbytheadiposetissueinobesityisaccompaniedwithanincreaseinnicotin-amide
adenine dinucleotide phosphate (NADPH)
oxidaseexpressionsuggestingthataugmentedNADPHoxidasecon-tributes to
increased ROS production in adipose
tissue.27Impairedexpressionofantioxidativeenzymes,suchas
super-oxidedismutase, glutathioneperoxidase, andcatalaseinadi-pose
tissue lead to increased ROS production.27Furthermore,in cultured
adipocytes, it has been shown that increased levelsof
fattyacidsincreaseoxidativestressandaugmentthepro-ductionof
proinammatorycytokines. Inobesemice, treat-ment with NADPH oxidase
inhibitor reduced ROSproductioninadipose
tissueandstrikinglyaugmentedadi-ponectin production, attenuated
proinammatory cytokinerelease, and improved insulin-sensitivity and
hepatic steatosis.Gut
MicrobiomeasaLinkBetweenObesityandAtherosclerosis?The resident gut
microbiota have crucial roles in hostfunctioning, andsubsequently
inhealthanddisease. Mostbacterial
speciesinthehumanandmousegutbelongtothephyla Bacteroidetes and
Firmicutes and to a lesser extentbacterial phyla, suchas
Actinobacteria, Proteobacteria, andVerrucomicrobia, andmethanogenic
archaea, mainlyMeth-anobrevibacter smithii.28The gut microbiota
possess a varietyof functional properties and affect host
physiology within andoutside the gut.Someoftheessential
functionsofthegutmicrobiotaarenormal development and homeostasis of
the immune systeminthe gut, modulationof epithelial cell
proliferation,
pro-tectionagainstpathogenicbacteria,andmodulationofvillusarchitecture
and angiogenesis within the intestine.29Studies inhumans and mice
have shown that altered gut microbiota areassociatedwithseveral
diseases, includingobesity, diabetes,inammation, and
atherosclerosis.Therst report of gut microbial difference between
obeseand lean phenotypeswas reported in leptin-decient
(ob/ob)mice.30This study showed differences in gut
microbialcomposition with fewer phylumBacteroidetes and
morephylumFirmicutes inobeseob/obmicethanintheir leanlittermates.
Becausebothgroupsof micewerefedthesamediet, theresults
suggestedthat obesitycouldbeduetothedifference in gut microbial
composition. Similarly, a decreaseinBacteroidetes
proportionwithanincrease inFirmicutesproportion was found in obese
human twins.31Furthermore, the transfer of microbiota,
harvestedfromnot only obese mice32but also fromlean or obese
in-dividuals,31into germ-free mice reproduces the
correspondingphenotype demonstrating that the obese phenotype
wastransferred by the microbiota.33It has also been reported
thatobese children already have different gut microbiota
comparedwith lean children.34However, some conictingndings havebeen
observed recently, showing no association35or anopposite
association with an increase in Bacteroidetes in
obeseindividuals.36Obesegutmicrobiotahavebeendemonstratedtoinducechronic
low-grade inammation in the host gut.37,38A
high-fatdietinratsledtochangesinthecompositionofthegutmicrobiotaandactivationoftoll-likereceptor4signallinginthe
gut epithelia and might aggravategastrointestinal inam-mation
associated with the obese phenotype.37Inarecentseminal
reportbyKarlssonet al.,39shotgunsequencing of the gut metagenome
was performed todemonstratethat thegenus Collinsellawas
enrichedinpa-tientswithsymptomaticatherosclerosis,
denedasstenoticatherosclerotic plaques in the carotid artery
leading tocerebrovascularevents,whereasRoseburiaandEubacteriumwere
enriched in healthy control subjects. Furthercharacterization of
the functional capacity of the meta-genomes revealed that patient
gut metagenomes wereenriched in genes encoding peptidoglycan
synthesis anddepleted in phytoene dehydrogenase; patients also
hadreducedserumlevelsof b-carotene. These ndingssuggestthat the gut
metagenome is associated with the inammatorystatus of the host
andpatients withsymptomatic athero-sclerosis harbour characteristic
changes in the gutmetagenome.39Lovren et al. 179Obesity and
Atherosclerosis: Mechanistic InsightsEndothelial DysfunctionAnimal
andhumanstudies have demonstratedimpairedendothelial function in
obesity.40Several mechanisms lead todiminished NO production and
availability in obesity. Theseinclude decreased expression of
eNOS,41,42or increasedinactivationof
eNOS(secondarytoincreasedproductionofROS). Plasma levels of
asymmetric dimethylarginine areincreased in obese patients43and in
obese subjects withmetabolic syndrome. Asymmetric dimethylarginine
serves as astoichiometricinhibitorof
eNOSandmightbeyetanothermechanism through which obesity reduces the
bioavailabilityof NO.In addition to NO, alterations in
endothelium-derivedprostacyclinandthromboxanehavealsobeenimplicatedinobesity.
Obesity increases prostanoid-dependent vasocon-striction and
vascular thromboxane receptor gene expres-sion.44These changes are
likely to promote the developmentof vascular disease, hypertension,
andthrombosis associatedwithobesity.
Animpairedcyclooxygenasepathway, withanincrease inthe release of
the thromboxane A2 metabolitethromboxane B2, has been reported in
microvessels fromobese spontaneous hypertensive rats.45Likewise in
obeseZucker rats, the release of prostacyclinis
attenuated46andactivity of prostacyclin and eNOS in arterial
endothelial cellswas decreasedbecause of oxidation of fatty
acids.47The endotheliumis the source of the potent vasocon-strictor
peptide ET-1. ET-1 augments the vascular actions ofother vasoactive
peptides such as angiotensin-II, norepineph-rine, and serotonin,
participates actively in leukocyte andplateletactivation,
andfacilitatesaprothromboticandproa-therogenic phenotype. Obesityis
associatedwithenhancedET-1-mediated vascular tone. In obesity,
increased ET-1-mediated vasoconstriction contributes to
diminishedendothelium-dependent vasodilation.48Increased
ET-1plasmalevels havebeenfoundinnormotensiveandhyper-tensive obese
subjects.49Additionally, in experimental obesity,there is an
increase in gene and protein expression of ET-1
inthecardiovascularsystem.44ET-1-mediatedvasoconstrictionmight
therefore promote hypertension, atherosclerosis, andthrombosis,
conditions frequently observed in obese
patients.Obesity-inducedvascular dysfunctionis
observedinthemacrovascular andmicrovascular beds. Inhumans,
reducedendothelium-dependent vasodilation to methacholine
wasdemonstrated in the leg microcirculation of obese
patients.50Similarly, a progressive decline inendothelial
functionwasdescribedintheforearmmicrocirculationofoverweightandobesepatients.51Inthesamestudy,intra-arterial
infusionofascorbicacidimprovedacetylcholineresponses,
suggestingarole of oxidative stress in obesity-related endothelial
dysfunc-tion.51Furthermore, small
resistancearteriesfromobesepa-tients are characterized by a
signicantly impairedendothelium-dependent
relaxationcomparedwithleancon-trol
subjects.52,53Thesedatademonstratethatsmall arteriesfrom obese
patients are characterized by a marked endothelialdysfunction as
result of decreased NO availability.Ithasbeen established
thatendothelialcellactivationis acrucial early step in inammation.
However, the role ofoverweight and obesity, and, inparticular, the
associationbetween specic fat depots and endothelial activation,
has
notbeenfullydetermined.Ithasbeenshownthatthereleaseoffreefattyacidbyadiposetissuedirectlyinducedendothelialdysfunction.54Subjects
withupperbodyobesityhavehigherlevels of free fatty acid than
subjects with obesity in the lowerextremities,55thus suggesting
that visceral fat has a moreprominent effect on endothelial
dysfunction than fat stored inother sites of the body. Furthermore,
infusion of free fatty
acidatconcentrationssimilartothoseobservedinobesesubjectsincreased
plasma ICAM1 and VCAM1 levels in healthy sub-jects, thus indicating
a direct effect of free fatty acid on endo-thelial cell
activation.56Similar ndings have already beenreported in animal
models of obesity in which surgical removalof visceral fat resulted
in decreased concentrations of free fattyacid in rat
plasma,57,58suggesting that visceral adiposity playsan important
role in the regulation of free fatty acid levels. Theremoval of
abdominal fat in humans by liposuction leads to adecrease
inconcentrations of free fatty
acid.59Conversely,removalofonlysubcutaneousfatfailsto
showanyimprove-mentininammationordecreaseinfreefattyacidconcen-trations60linking
visceral fat and free fatty acid levels.Children and adolescents
with obesity-related
hypertensionhaveaheightenedsystemicinammatoryresponsewithevi-dence
of endothelial activation relative to normotensive
obesesubjects.61Recently, it has been shown that adiposity
isassociatedwithhigherlevelsofinsulinresistance,
E-selectin,andVCAM1eveninhealthy normal-weight childrenat ayoung
age of 2-3 years, and such associations are evident
eveninchildrenwithrelatively lowlevels of adiposity.62Thesestudies
thereforestronglyarguethat overweight andobesityare associated with
endothelial activation and it is possible thatadiposityin the
visceralregionmightbe a stronger predictorof endothelial activation
than overall adiposity.InammasomeActivationinObesity
andAtherosclerosisInammationmightbeoneofthemostimportantcom-monthreadstolinkobesitywithatherosclerosis.63Accumu-lating
data now suggest that the nucleotide-bindingoligomerization
domain-like receptor family pyrin domaincontaining 3(NLRP3)
inammasome,largely within macro-phages, might be stimulatedby
various cardiovascular riskfactors, includingcrystalline
cholesterol. NLRP3inamma-someactivationisresponsiblefortheeventual
conversionofpro-IL-1b into IL-1b. During obesity, circulating
levels of freefatty acids are increased and lipids such as ceramide
andpalmitate activate the cells of the innate immune system
andtrigger inammasome activation.64Lipopolysaccharide-primed
macrophages stimulated with ceramide displayNLRP3-dependent
caspase-1activationandleadtoproduc-tion of IL-1b at relatively low
levels.65More recently, anotherlipid,
thesaturatedfattyacidpalmitate, has beenshowntoactivate the NLRP3
inammasome.66Palmitate is one of themost abundant free fatty acids
in plasma and is highlyincreasedinobesity. Several studies
demonstratedthat theNLRP3 inammasome in obesity is an important
mechanismthat participates in the development of insulin
resistance.65-67Likewise,theNLRP3inammasomehasbeendemonstratedto be
essential in the development of atherosclerosis,68makingit a very
attractive common mechanism (and potential target)of
cardiometabolic risk.Obesity-induced inammation has been linked to
theactivation ofadipose tissuemacrophages,T cells,andB cells180
Canadian Journal of CardiologyVolume 31 2015within fat
deposits.69The mechanisms that regulate theactivationof these
immune cells inadipose tissue are stilllargely unknown, however, a
recent study found that
obesityitselfinducedtheassemblyoftheNLRP3inammasomeinadipose tissue
macrophages.65Moreover,
inammasome-mediatedcaspase-1activationinadiposetissueandliverhasbeenshowntoimpairinsulinsignallingandglucosehomeo-stasis.67In
free-feeding mice given a normal chowdiet,increased expression of
NLRP3 and IL-1b in visceral
adiposetissuehasbeenfoundtocorrelatedirectlywithbodyweightandadiposity.
DirectinvolvementofNLRP3inobesityhasbeen also conrmed in NLRP3
gene-decient mice fed a highfat diet. Inthese mice,
caspase-1activationandpro-IL-1bexpressioninadipose tissue, andloss
of
serumIL-18pro-ductionwereprofoundlyreducedcomparedwiththeirwildtype
counterparts. Moreover, NLRP3- and
caspase-1-decientmicearemoreprotectedfromhigh-fat
diet-inducedinsulinresistance. Similar observations were reported
in humans.Weight loss inobese diabetic patients is
associatedwithadecrease
inNLRP3andIL-1bexpressioninsubcutaneousadipose
tissue.65,67ConclusionsObesity leads to increased cardiovascular
risk
throughvariousmechanisms.Althoughitisimpossibletodissectouttheuniquemechanisticcontributionofobesity(vstheasso-ciatedfeatures
of the metabolic syndrome) towardathero-thrombotic risk, several
important themes have emerged.Theserelatetoalterationsinadipokines,
inammation, andinammasome activation, gut microbiota, oxidative
stress,
andendothelialdysfunction.Themostcompellingcontemporarymechanismthatmighthelptieall
ofthesefacetstogetherisinammation, and signalling via the NLRP3
inammasome.DisclosuresThe authors have no conicts of interest to
disclose.References1. Lau DC, Dhillon B, Yan H, Szmitko PE, Verma
S. Adipokines:molecular links between obesity and atheroslcerosis.
Am J Physiol HeartCirc Physiol 2005;288:H2031-41.2. Yamauchi T,
Kadowaki T. Adiponectin receptor as a key player in
healthylongevity and obesity-related diseases. Cell Metab
2013;17:185-96.3. Chen H, Montagnani M, Funahashi T, Shimomura I,
Quon MJ.Adiponectin stimulates production of nitric oxide in
vascular endothelialcells. J Biol Chem 2003;278:45021-6.4.
Motoshima H, Wu X, Mahadev K, Goldstein BJ. Adiponectin
suppressesproliferationandsuperoxidegenerationandenhanceseNOSactivityinendothelial
cells treated with oxidized LDL. BiochemBiophys ResComm
2004;315:264-71.5. Ouchi N, Kihara S, Arita Y, et al. Novel
modulator for endothelialadhesion molecules: adipocyte-derived
plasma protein adiponectin.Circulation 1999;100:2473-6.6. Ouchi N,
KiharaS, AritaY, et al. Adipocyte-derivedplasmaprotein,adiponectin,
suppresses lipid accumulation and class A scavenger
receptorexpression in human monocyte-derived macrophages.
Circulation2001;103:1057-63.7. Furukawa K, Hori M, Ouchi N, et al.
Adiponectin down-regulates acyl-coenzymeA:cholesterol
acyltransferase-1inculturedhumanmonocyte-derived macrophages.
Biochem Biophys Res Comm 2004;317:831-6.8. Lovren F, Pan Y, Quan A,
et al. Adiponectin primes human monocytesinto alternative
anti-inammatory M2 macrophages. Am J Physiol HeartCirc Physiol
2010;299:H656-63.9. Arita Y, Kihara S, Ouchi N, et al.
Adipocyte-derivedplasma
proteinadiponectinactsasaplatelet-derivedgrowthfactor-BB-bindingproteinand
regulates growth factor-induced common postreceptor signal
invascular smooth muscle cell. Circulation 2002;105:2893-8.10.
Kumada M, Kihara S, Ouchi N, et al. Adiponectin specically
increasedtissue inhibitor of metalloproteinase-1 through
interleukin-10 expressionin human macrophages. Circulation
2004;109:2046-9.11. TeohH, QuanA, BangKW, et al.
Adiponectindeciencypromotesendothelial activation and profoundly
exacerbates sepsis-related mortality.Am J Physiol Endocrinol Metab
2008;295:E658-64.12. MatsudaM, ShimomuraI, SataM, et al. Roleof
adiponectininpre-venting vascular stenosis. The missing link of
adipo-vascular axis. J BiolChem 2002;277:37487-91.13.
OkamotoY,KiharaS,OuchiN,etal.Adiponectinreducesatheroscle-rosis in
apolipoprotein E-decient mice. Circulation 2002;106:2767-70.14.
Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A. Resistin,
anadipokine with potent proinammatory properties. J
Immunol2005;174:5789-95.15. Silswal N, Singh AK, Aruna B, et al.
Human resistin stimulates the
pro-inammatorycytokinesTNF-alphaandIL-12inmacrophagesbyNF-kappaB-dependent
pathway. BiochemBiophys Res Comm2005;334:1092-101.16. Choi HY, Kim
S, Yang SJ, et al. Association of adiponectin, resistin,
andvascular inammation: analysis with18F-uorodeoxyglucose
positronemission tomography. Arterioscler Thromb Vasc Biol
2011;31:944-9.17. VermaS, Li SH, WangCH, et al. Resistinpromotes
endothelial cellactivation:furtherevidenceofadipokine-endothelial
interaction.Circu-lation 2003;108:736-40.18. LaCavaA, MatareseG.
Theweight of leptininimmunity. Nat RevImmunol 2004;4:371-9.19.
FriedmanJM, HalaasJL. Leptinandtheregulationofbodyweightinmammals.
Nature 1998;395:763-70.20. Wu KK, Huan Y. Diabetic atherosclerosis
mouse models. Atherosclerosis2007;191:241-9.21.
LordGM,MatareseG,HowardJK,etal.LeptinmodulatestheT-cellimmuneresponseandreverses
starvation-inducedimmunosuppression.Nature 1998;394:897-901.22.
Chen WQ, Zhong L, Zhang L, et al. Oral rapamycin
attenuatesinammation and enhances stability of atherosclerotic
plaques in rabbitsindependent of serum lipid levels. Br J Pharmacol
2009;156:941-51.23. Hill BG, Haberzettl P, Ahmed Y, Srivastava S,
Bhatnagar A. Unsaturatedlipid peroxidation-derived aldehydes
activate autophagy in vascularsmooth-muscle cells. Biochem J
2008;410:525-34.24. Mueller MA, Beutner F, Teupser D, Ceglarek U,
Thiery J. Prevention ofatherosclerosis by the mTORinhibitor
everolimus inLDLR-/- micedespite severe hypercholesterolemia.
Atherosclerosis 2008;198:39-48.25. OuimetM, FranklinV, MakE, etal.
Autophagyregulatescholesterolefux from macrophage foam cells via
lysosomal acid lipase. Cell Metab2011;13:655-67.Lovren et al.
181Obesity and Atherosclerosis: Mechanistic Insights26. LimYM,
LimH, Hur KY, et al. Systemic autophagy insufciencycompromises
adaptationtometabolicstress andfacilitates progressionfrom obesity
to diabetes. Nat Commun 2014;5:4934.27. Furukawa S, Fujita T,
Shimabukuro M, et al. Increased oxidative stress inobesity and its
impact on metabolic syndrome. J Clin Invest 2004;114:1752-61.28.
Eckburg PB,Bik EM,Bernstein CN,et al.Diversity
ofthehumanin-testinal microbial ora. Science 2005;308:1635-8.29.
Sommer F, Backhed F. The gut microbiotaemasters of host
developmentand physiology. Nat Rev Microbiol 2013;11:227-38.30.
LeyRE, BackhedF, TurnbaughP, et al. Obesityalters gut
microbialecology. Proc Natl Acad Sci U S A 2005;102:11070-5.31.
Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut
microbiomein obese and lean twins. Nature 2009;457:480-4.32.
Vijay-Kumar M, Aitken JD, Carvalho FA, et al. Metabolic syndrome
andaltered gut microbiota in mice lacking Toll-like receptor 5.
Science2010;328:228-31.33. Sokol H, Pigneur B, Watterlot L, et al.
Faecalibacterium prausnitzii is ananti-inammatory commensal
bacteriumidentiedby gut microbiotaanalysis of Crohn disease
patients. Proc Natl Acad Sci U S A 2008;105:16731-6.34. Kalliomaki
M, Collado MC, Salminen S, Isolauri E. Early differences infecal
microbiota composition in children may predict overweight. Am JClin
Nutr 2008;87:534-8.35. Fukuda S, Toh H, Hase K, et al. Bidobacteria
can protect fromenteropathogenic infection through production of
acetate. Nature2011;469:543-7.36. Macfarlane S, Macfarlane GT,
Cummings JH. Review article: prebioticsin the gastrointestinal
tract. Aliment Pharmacol Ther 2006;24:701-14.37. de La Serre CB,
Ellis CL, Lee J, et al. Propensity to high-fat
diet-inducedobesityin ratsisassociatedwith changesin
thegutmicrobiota andgutinammation. AmJ Physiol Gastrointest Liver
Physiol 2010;299:G440-8.38. Cani PD, AmarJ, Iglesias MA, et al.
Metabolicendotoxemiainitiatesobesity and insulin resistance.
Diabetes 2007;56:1761-72.39. Karlsson FH, Tremaroli V, NookaewI, et
al. Gut metagenome inEuropeanwomenwithnormal,
impairedanddiabeticglucosecontrol.Nature 2013;498:99-103.40. Virdis
A, Neves MF, Duranti E, Bernini G, Taddei S.
Microvascularendothelial dysfunctioninobesityandhypertension. Curr
PharmDes2013;19:2382-9.41. Kobayasi R, Akamine EH, Davel AP, et al.
Oxidative stress andin-ammatorymediatorscontributetoendothelial
dysfunctioninhigh-fatdiet-induced obesity in mice. J Hypertens
2010;28:2111-9.42. Sansbury BE, Cummins TD, Tang Y, et al.
Overexpression of
endothelialnitricoxidesynthasepreventsdiet-inducedobesityandregulatesadipo-cyte
phenotype. Circulation Res 2012;111:1176-89.43. Krzyzanowska K,
Mittermayer F, Kopp HP, Wolzt M, Schernthaner G.Weight
lossreducescirculatingasymmetrical
dimethylarginineconcen-trationsinmorbidlyobesewomen.
JClinEndocrinol Metab2004;89:6277-81.44. TraupeT, LangM, GoettschW,
et al. Obesityincreases prostanoid-mediated vasoconstriction and
vascular thromboxane receptor geneexpression. J Hypertens
2002;20:2239-45.45. Mendizabal Y, Llorens S, Nava E. Reactivity of
the aorta and mesentericresistance arteries from the obese
spontaneously hypertensive rat: effectsof glitazones. Am J Physiol
Heart Circ Physiol 2011;301:H1319-30.46. Hodnett BL, DearmanJA,
Carter CB, Hester RL. AttenuatedPGI2synthesis in obese Zucker rats.
Am J Physiol Regul Integr Comp Physiol2009;296:R715-21.47. DuX,
EdelsteinD, Obici S, et al. Insulinresistance reduces
arterialprostacyclin synthase and eNOS activities by increasing
endothelial fattyacid oxidation. J Clin Invest 2006;116:1071-80.48.
WeilBR,WestbyCM,VanGuilderGP,etal.Enhancedendothelin-1systemactivitywithoverweight
andobesity. AmJPhysiol Heart CircPhysiol 2011;301:H689-95.49.
Parrinello G, Scaglione R, Pinto A, et al. Central obesity and
hyperten-sion: the role of plasma endothelin. Am J Hypertens
1996;9:1186-91.50. Steinberg HO, Chaker H, Leaming R, et al.
Obesity/insulin resistance isassociated with endothelial
dysfunction. Implications for the syndrome ofinsulin resistance. J
Clin Invest 1996;97:2601-10.51. PerticoneF, CeravoloR,
CandigliotaM, et al. Obesityandbodyfatdistribution induce
endothelial dysfunction by oxidative stress: protectiveeffect of
vitamin C. Diabetes 2001;50:159-65.52. Grassi G, Seravalle G,
Scopelliti F, et al. Structural and functionalalterations of
subcutaneous small resistance arteries insevere humanobesity.
Obesity 2010;18:92-8.53. Virdis A, Santini F, Colucci R, et al.
Vascular generationof tumornecrosis factor-alpha reduces nitric
oxide availability in small arteries fromvisceral fat of obese
patients. J Am Coll Cardiol 2011;58:238-47.54. Nielsen S, Guo Z,
Johnson CM, Hensrud DD, Jensen MD. Splanchniclipolysis in human
obesity. J Clin Invest 2004;113:1582-8.55. MartinML, JensenMD.
Effects of bodyfat distributiononregionallipolysis in obesity. J
Clin Invest 1991;88:609-13.56. MathewM, TayE, Cusi K.
Elevatedplasmafreefattyacids increasecardiovascular
riskbyinducingplasmabiomarkers of endothelial acti-vation,
myeloperoxidase and PAI-1 in healthy subjects. Cardiovasc Dia-betol
2010;9:9.57. KimYW, KimJY, Lee SK. Surgical removal of visceral fat
decreasesplasma free fatty acid and increases insulin sensitivity
on liver andperipheral tissue in monosodium glutamate (MSG)-obese
rats. J KoreanMed Sci 1999;14:539-45.58. GabrielyI, MaXH, YangXM,
et al. Removal of visceral fat preventsinsulin resistance and
glucose intolerance of aging: an adipokine-mediated process?
Diabetes 2002;51:2951-8.59. YbarraJ, Blanco-VacaF, FernandezS,
etal. Theeffectsofliposuctionremoval of subcutaneous abdominal fat
onlipidmetabolismareinde-pendent of insulin sensitivity in
normal-overweight individuals. ObesitySurg 2008;18:408-14.60. Klein
S, Fontana L, Young VL, et al. Absence of an effect of
liposuctiononinsulinactionandriskfactorsforcoronaryheartdisease.
NEngl JMed 2004;350:2549-57.61. Garanty-BogackaB, SyreniczM,
SyreniczA, et al. Serummarkers ofinammation and endothelial
activation in children with obesity-relatedhypertension. Neuro
Endocrinol Lett 2005;26:242-6.62. CastroC, TracyRP, DeckelbaumRJ,
BaschCE, SheaS. Adiposityisassociatedwithendothelial
activationinhealthy2-3year-oldchildren.J Pediatr Endocrinol Metab
2009;22:905-14.63. Verma S, Gupta M, Ridker PM. Therapeutic
targeting of inammationin atherosclerosis: we are getting closer.
Can J Cardiol 2012;28:619-22.182 Canadian Journal of
CardiologyVolume 31 201564. DeNardoD, LatzE. NLRP3inammasomes
linkinammationandmetabolic disease. Trends Immunol
2011;32:373-9.65. Vandanmagsar B, YoumYH, RavussinA, et al.
TheNLRP3inam-masomeinstigatesobesity-inducedinammationandinsulinresistance.Nat
Med 2011;17:179-88.66. WenH, GrisD, Lei Y, etal.
Fattyacid-inducedNLRP3-ASCinam-masome activation interferes with
insulin signaling. Nat Immunol2011;12:408-15.67. Stienstra
R,Joosten LA, KoenenT, etal.
Theinammasome-mediatedcaspase-1activationcontrolsadipocytedifferentiation
andinsulinsensi-tivity. Cell Metab 2010;12:593-605.68. Duewell P,
Kono H, Rayner KJ, et al. NLRP3 inammasomes
arerequiredforatherogenesisandactivatedbycholesterol crystals.
Nature2010;464:1357-61.69. Odegaard JI, Chawla A. Mechanisms of
macrophage activation inobesity-inducedinsulinresistance. Nat
ClinPract Endocrinol Metab2008;4:619-26.Lovren et al. 183Obesity
and Atherosclerosis: Mechanistic Insights