Embed Size (px)
Citation preview

� E – 14-336
Osteoartropatía hipertrofiante
G. Chalès, A. Rouil
La osteoartropatía hipertrofiante (OAH), descrita hace 125 anos por Von Bamberger(1889) y Pierre Marie (1890), es una entidad clínica y radiológica clásicamente secun-daria a enfermedades neoplásicas (síndrome paraneoplásico) o a numerosas afeccionespulmonares, cardiovasculares, digestivas, metabólicas u otras; las principales mani-festaciones afectan al esqueleto (hipocratismo digital, periostosis, artritis con líquidosinovial poco celular de tipo mecánico, acroosteólisis) y la piel (engrosamiento, seborrea,hipersudación). El hipocratismo digital puede medirse objetivamente y los datos de lagammagrafía ósea con tecnecio 99m permiten completar la descripción del síndromeóseo. Generalmente, la OAH se presenta en pacientes con un cáncer de pulmón (de ahíla denominación clásica de OAH néumica). En los pacientes más jóvenes, la OAH puedeser un signo clínico sugestivo de una neumopatía destructiva, de mucoviscidosis o deuna cardiopatía congénita. Sin embargo, el marco etiológico se ha ampliado, en espe-cial en el sector de las causas extratorácicas de OAH. La acumulación de hechos clínicosy la variedad de formas clínicas de la OAH materializan el concepto de disacromeliapropuesto por Bariéty y Coury (1946): existen relaciones de paso entre el hipocratismodigital, la OAH secundaria y la paquidermoperiostosis (PDP), que no es más que unaOAH primitiva. Aunque el conocimiento de la patogenia de la OAH todavía es imperfecto(gran número de observaciones aisladas y anecdóticas, falta de estudios controladosen patología humana y animal, probable existencia de varios mecanismos diferentesresponsables de OAH, denominador común final de los numerosos factores etiológicos),los estudios genéticos recientes de familias afectadas por una OAH primitiva (PDP) handemostrado mutaciones de los genes que codifican el transporte y el catabolismo de lasprostaglandinas (PG), en especial de la E2, lo cual sugiere que la alteración del metabo-lismo de las PGE2 desempena un papel esencial en la patogenia de la OAH. El tratamientode la afección causal es el único tratamiento eficaz de la OAH secundaria. La ausenciade estudios controlados hace difícil la valoración de los tratamientos complementarios(corticoides, mórficos, octreotida, bifosfonatos, bioterapia).© 2014 Elsevier Masson SAS. Todos los derechos reservados.
Palabras clave: Osteoartropatía hipertrofiante; Paquidermoperiostosis; Hipocratismo digital;Periostosis; VEGF; PGE2
Plan
■ Introducción 2■ Síndrome de osteoartropatía hipertrofiante 2
Síndrome acromélico 2Síndrome articular 4Síndrome cutáneo 4Síndrome óseo 4
■ Etiología 5Osteoartropatía hipertrofiante primitivao paquidermoperiostosis 5Osteoartropatía hipertrofiante secundaria 5
■ Osteoartropatía hipertrofiante localizada 6■ Epidemiología 6■ Anatomía patológica 7
Sinovitis 7Periostosis 7Tejidos blandos 7Hipocratismo digital 7
■ Fisiopatología del hipocratismo digital y la periostosis 7Factores neurótropos 7Factores humorales 7Factores plaquetarios 7Factores de crecimiento vasculares 7Prostaglandinas 8
EMC - Aparato locomotor 1Volume 47 > n◦1 > marzo 2014http://dx.doi.org/10.1016/S1286-935X(14)66935-5

E – 14-336 � Osteoartropatía hipertrofiante
■ Datos genéticos en la paquidermoperiostosis 8Mutaciones del gen HPGD 8Mutaciones del gen SLCO2A1 8
■ Diagnóstico 9■ Evolución 10■ Tratamiento 10■ Conclusión 10
� IntroducciónLa osteoartropatía hipertrofiante (OAH) puede definirse
como un conjunto sindrómico, a menudo paraneoplá-sico, que asocia:• un síndrome acromélico que comporta un hipocra-
tismo digital (HD), una hipertrofia de los tejidosblandos y trastornos vasomotores;
• un síndrome articular: artralgias o artritis;• un síndrome cutáneo: hipertrofia de los tejidos blandos;• un síndrome óseo que incluye una periostosis en capas
con hipercaptación en la gammagrafía ósea y unaacroosteólisis.
� Síndromede osteoartropatíahipertrofianteSíndrome acromélicoHipocratismo digital
La primera descripción se atribuye a Hipócrates, de ahísu nombre. Afecta a los dedos de las manos y más rara-mente a los de los pies; asocia:• una deformación de las unas con abombamiento en
«vidrio de reloj» (Fig. 1);• una deformación de la última falange, que se vuelve
bulbosa, ensanchada en «palillo de tambor» (Trom-melschlegel Finger de los autores alemanes) o en«badajo de campana» (clubbing de los autores ingleses)(Fig. 2);
• una hinchazón y una fluctuación de los tejidos blan-dos de la base de la una, que a veces es dolorosa,acompanadas de un eritema periungueal [1].El HD puede medirse objetivamente [2, 3] mediante:
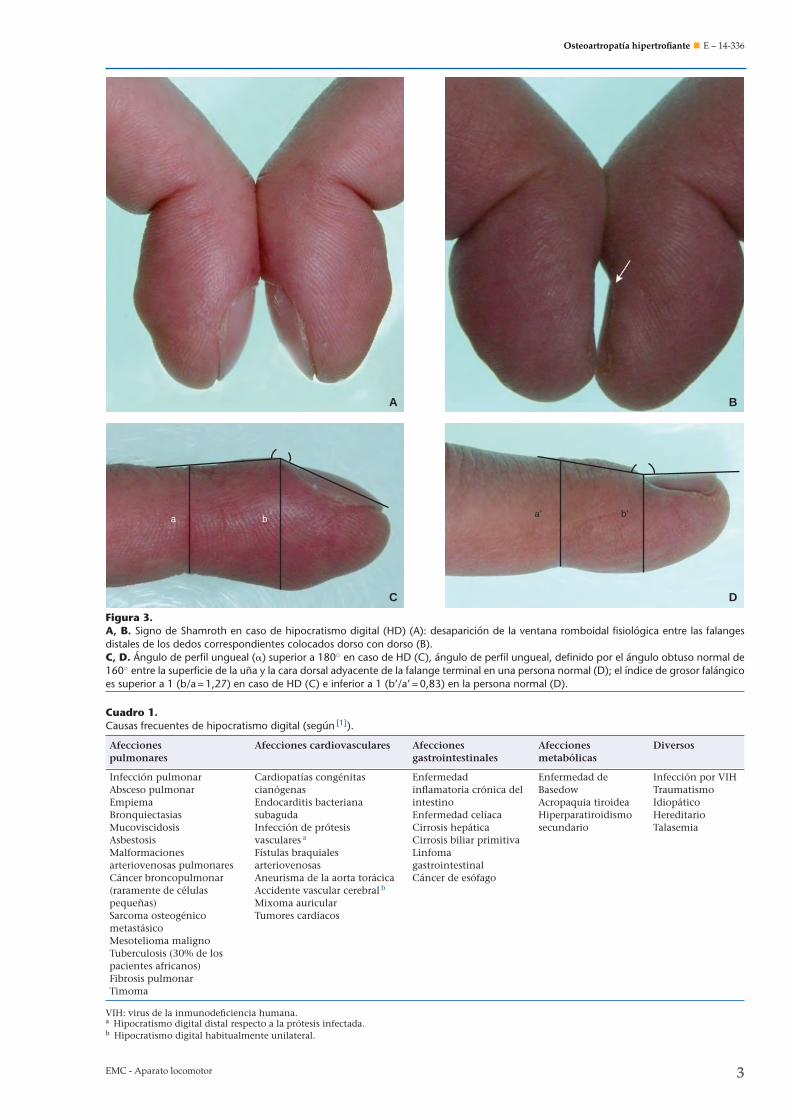
• un aumento del ángulo del perfil ungueal, definido porel ángulo obtuso normal de 160◦ entre la superficie dela una y la cara dorsal adyacente de la falange terminal(Fig. 3); el signo de Shamroth (Fig. 3) permite confirmarque el ángulo del perfil es superior a 180◦; la exac-titud y la reproducibilidad de este signo son satisfac-torios [4];
• un aumento de la relación circunferencia digitalmedida en la cutícula/circunferencia digital de la inter-falángica distal, normalmente inferior a 0,9 (índice degrosor falángico) (Fig. 3).En el HD precoz, el ángulo del perfil ungueal se borra y
se vuelve plano. Esto permite diferenciar el HD del seudo-HD, más a menudo asimétrico [5], de las incurvaciones dela una y de la perionixis, así como del hiperparatiroidismosecundario, relacionado con un hundimiento de los teji-dos blandos debido a la erosión grave de la parte distal dela falange [6].
El HD se observa en el curso de numerosas afeccio-nes pulmonares, cardiovasculares, digestivas, metabólicasu otras [7] (Cuadro 1). Si la causa del HD no es evi-dente, el algoritmo de Spicknall et al puede ser deayuda [6].
El HD puede ser unidigital, en caso de aneurisma de lasubclavia o de la aorta, traumatismo del plexo braquial o
Figura 1. Deformación de las unas con un abombamiento en«vidrio de reloj».
Figura 2. Deformación de la última falange, que se vuelvebulbosa, ensanchada en «palillo de tambor».
del nervio mediano, luxación del hombro, síndrome dePancoast-Tobias, síndrome de Maffucci, gota o sarcoido-sis [8].
Hipertrofia de los tejidos blandosLos dedos de las manos y de los pies son amorcillados;
las manos se ensanchan en «pala»; las munecas formanalmohadillas; los pies y los tobillos se engrosan, en «patade elefante»; la hipertrofia se extiende al tercio inferiorde antebrazos y piernas y da lugar a una infiltraciónedematosa firme con engrosamiento de la piel, a vecesdoloroso [9].
2 EMC - Aparato locomotor
Osteoartropatía hipertrofiante � E – 14-336
A B
C
ba
D
b’a’
Figura 3.A, B. Signo de Shamroth en caso de hipocratismo digital (HD) (A): desaparición de la ventana romboidal fisiológica entre las falangesdistales de los dedos correspondientes colocados dorso con dorso (B).C, D. Ángulo de perfil ungueal (�) superior a 180◦ en caso de HD (C), ángulo de perfil ungueal, definido por el ángulo obtuso normal de160◦ entre la superficie de la una y la cara dorsal adyacente de la falange terminal en una persona normal (D); el índice de grosor falángicoes superior a 1 (b/a = 1,27) en caso de HD (C) e inferior a 1 (b’/a’ = 0,83) en la persona normal (D).
Cuadro 1.Causas frecuentes de hipocratismo digital (según [1]).
Afeccionespulmonares
Afecciones cardiovasculares Afeccionesgastrointestinales
Afeccionesmetabólicas
Diversos
Infección pulmonarAbsceso pulmonarEmpiemaBronquiectasiasMucoviscidosisAsbestosisMalformacionesarteriovenosas pulmonaresCáncer broncopulmonar(raramente de célulaspequenas)Sarcoma osteogénicometastásicoMesotelioma malignoTuberculosis (30% de lospacientes africanos)Fibrosis pulmonarTimoma
Cardiopatías congénitascianógenasEndocarditis bacterianasubagudaInfección de prótesisvasculares a
Fístulas braquialesarteriovenosasAneurisma de la aorta torácicaAccidente vascular cerebral b
Mixoma auricularTumores cardíacos
Enfermedadinflamatoria crónica delintestinoEnfermedad celíacaCirrosis hepáticaCirrosis biliar primitivaLinfomagastrointestinalCáncer de esófago
Enfermedad deBasedowAcropaquia tiroideaHiperparatiroidismosecundario
Infección por VIHTraumatismoIdiopáticoHereditarioTalasemia
VIH: virus de la inmunodeficiencia humana.a Hipocratismo digital distal respecto a la prótesis infectada.b Hipocratismo digital habitualmente unilateral.
EMC - Aparato locomotor 3

E – 14-336 � Osteoartropatía hipertrofiante
El macizo craneofacial y el tronco generalmente estánrespetados. La ginecomastia es rara, tardía, bilateral y másfrecuente en el cáncer broncopulmonar primitivo [10].
Trastornos vasomotoresLas extremidades pueden presentar cianosis, aumento
del calor local con sensación de calor en los extre-mos, dilatación venosa superficia, crisis de hipersudación,disestesias y parestesias [11].
Síndrome articularLas artropatías de la OAH se manifiestan por artral-
gias simples, de ritmo mecánico o inflamatorio, o porverdaderas artritis. Afectan simétricamente a las gran-des articulaciones distales (tobillos, rodillas, munecas ycodos) [12], pero también a las pequenas articulaciones demanos y pies.
El cuadro de poliartritis subaguda o crónica de aspectoreumatoideo es frecuente (más del 50%) y a veces planteaun problema diagnóstico [13] con otros reumatismos [14].
Las articulaciones pueden estar aumentadas de tamanodebido o bien a un edema periarticular o bien a underrame intraarticular de fórmula mecánica que contrastacon el síndrome inflamatorio de laboratorio [15].
Síndrome cutáneoEl síndrome cutáneo no es privilegio de la OAH pri-
mitiva, porque existen paquidermoperiostosis (PDP) sinsignos cutáneos [16] y el síndrome cutáneo puede obser-varse en la OAH secundaria [17], en la que, sin embargo,predomina la afectación ósea sobre la cutánea.
El acné o foliculitis, la hipersudación (manos, pies,grandes pliegues) y la seborrea se deben a una disfunciónde las glándulas cutáneas. La cara presenta una hipertro-fia y un engrosamiento de la piel (paquidermia) que ledan un aspecto arrugado responsable de la ampliación delos rasgos del rostro (pliegues nasogenianos profundos,hipertrofia de los párpados que da lugar a ptosis); en elcuero cabelludo, existen repliegues cutáneos profundos,cerebriformes (cutis verticis gyrata) [18–20].
Síndrome óseoPeriostosis
La periostosis es la «huella» de la OAH [21], pero consti-tuye un elemento que no es necesario ni suficiente parael diagnóstico, sobre todo porque a veces está ausente yno aparece hasta 6 meses después de la evolución de laOAH. La periostosis puede ser asintomática o traducirsepor dolor profundo e incluso sensación de quemadura enlas extremidades.
Se trata de una proliferación perióstica que rodea el ini-cio de las partes proximales y distales de las diáfisis de loshuesos largos, con frecuencia decreciente: tibia-peroné,cúbito-radio, fémur, húmero, metacarpianos, metatarsia-nos y falanges [21]. Se extiende a las zonas de inserción delas membranas interóseas y los tendones (huesos de losantebrazos y las piernas) hasta la metáfisis.
La epífisis se ve afectada [22] en un tercio de las cardio-patías congénitas cianógenas (CCC). Más raramente, seobserva una afectación de la clavícula, la escápula, lascostillas, el cráneo y la columna vertebral.
Existe una relación directa entre la duración de la OAHy el número de huesos afectados [23] y, sobre todo, entre laextensión y la morfología de la periostosis: al inicio, apa-rece un revestimiento opaco, lineal, regular y delgado y,después, poco a poco más grueso y separado de la diáfisispor un fino ribete claro (ribete osteoperióstico) (Fig. 4); laaposición de capas finas sucesivas da lugar a un aspectolaminar, hojaldrado y festoneado con zonas irregulares de
Figura 4. Ribete osteoperióstico paralelo a la cortical de lafalange del pulgar (osteoartropatía hipertrofiante).
Figura 5. Periostosis de contorno ondulado, inflado, que sefusiona con la cortical del extremo inferior del fémur (osteoar-tropatía hipertrofiante).
periostosis de contorno ondulado, abotargado en «cerafundida», que se adhiere y se fusiona con la cortical, pro-gresivamente engrosada a lo largo de la evolución de laOAH (Fig. 5).
AcroosteólisisLas falanges terminales pueden sufrir remodelaciones
óseas, con un primer estadio de hipertrofia y prolifera-ción ósea (espolón, espátula, mata de hierba), seguidasmás tarde por una acroosteólisis más marcada cuando seasocia a una periostosis de los huesos largos [22]. La osteóli-sis de las falanges distales se observa en la OAH secundaria,pero también en la primitiva [24].
Pruebas de imagenLa gammagrafía ósea con tecnecio 99m es un método
sensible que permite una detección más precoz de laOAH que la radiografía [21] y pone de manifiesto locali-zaciones desconocidas (maxilares, escápulas, clavículas,
4 EMC - Aparato locomotor

Osteoartropatía hipertrofiante � E – 14-336
rótulas). La afectación se extiende también a la parte pro-ximal y a la parte distal de los huesos largos, exceptoal húmero. De 1.226 pacientes con cáncer de pulmón,55 (4,5%) presentaban una hipercaptación gammagráficaperióstica, pero sólo 10 (0,8%) tenían un HD y artritis(tríada completa) [25].
La hipercaptación gammagráfica es simétrica y regular,más marcada en los miembros inferiores, o bien difundeen doble línea a lo largo de las corticales de las diáfi-sis, o bien se localiza en la región periarticular; a veceses asimétrica, pero de localización distal periférica, convisualización de una fina reacción perióstica. Las anoma-lías gammagráficas no son específicas de la OAH y debenhacer pensar en todas las causas de reacción perióstica [21].
La gammagrafía ósea es útil en la evaluación de la efica-cia del tratamiento: las anomalías pueden desaparecer enmenos de 2 meses antes que los signos radiográficos [26];su reaparición puede deberse a la recidiva de la afeccióncausal.
La tomografía por emisión de positrones acoplada atomografía computarizada (PET-TC) permite identificarno sólo la OAH, sino también su causa tumoral [27].
La ecografía permite medir el grosor de los tejidos blan-dos bajo la una y otros parámetros [28].
La resonancia magnética (RM) muestra la reacciónperióstica, pero sobre todo permite comprender las modi-ficaciones de los tejidos blandos: edema muscular y septalcon hinchazón de los tejidos blandos que rodean los hue-sos largos, en contacto con la cortical (hipersenal en T2,secuencia de recuperación de la inversión en tiempo corto[STIR]) pero sin afectación del hueso, compatible conun proceso inflamatorio, probablemente muy vasculari-zado [29].
� EtiologíaLa clasificación de la OAH permite distinguir una forma
primitiva (PDP) y formas secundarias (Fig. 6) [30].
Osteoartropatía hipertrofianteprimitiva o paquidermoperiostosis
La PDP u OAH primitiva [20] se caracteriza por un engro-samiento de la piel y una sudación excesiva (PDP) [18, 31],un cierre retardado de las suturas craneanas (osteoartropa-tía craneal) [32, 33] y una persistencia del conducto arteriosopersistente [30]. Se ha confirmado una transmisión auto-sómica dominante en alrededor del 50% de las familias,con una variabilidad clínica marcada inter e intrafamiliar.Se ha sugerido una herencia autosómica recesiva en lasfamilias con consanguinidad.
Los signos dermatológicos de la OAH primitiva tambiénpueden aparecer en la OAH secundaria a enfermedadespulmonares (acantosis palmar, triple palms o paqui-dermatoglifia) [10]. La revisión de las publicaciones de125 pacientes en 1988 [15] muestra un predominio mas-culino (nueve varones por cada mujer), la existencia deantecedentes familiares en el 38% de los casos, de HD enel 89%, de artrosinovitis en el 41% y de periostosis en el97% de los casos. Esta última es más hipertrofiante que encapas a causa de la duración de la evolución, 15 anos demedia.
Osteoartropatía hipertrofiantesecundaria
La OAH secundaria se observa en numerosas afecciones,de origen torácico o extratorácico.
Causas torácicasLa OAH suele ser secundaria a neoplasias pulmonares
(80% de los casos), tanto si se trata de un cáncer primitivode pulmón, sobre todo de células no pequenas [10, 25, 34],como secundario.
Las metástasis pulmonares se asocian con menosfrecuencia a la OAH. En una serie de 41 casos recopi-lados por Yacoub et al [35], las metástasis procedían de
Osteoartropatía hipertrofiante
PrimitivaPaquidermoperiostosis
Secundaria
Generalizada Localizada
Hemiplejia AneurismaInfección
(prótesis vascular)Canalarterial
Pulmonar IntestinalHepáticoCardíaco Mediastínico Otros
De origen
MucoviscidosisFibrosisprimitiva
Infeccionescrónicas
Cáncer (primitivoo metastásico)
FístulaarteriovenosaMesotelioma
Tumorespleuralesbenignos
CCCEndocarditisinfecciosa
CirrosisCáncer
BasedowTalasemia
HemopatíasmalignasSíndromePOEMS
Otros
Cáncer deesófagoAcalasiaTimoma
CrohnCáncerRCH
InfeccionescrónicasLaxantesPoliposis
Figura 6. Clasificación de la osteoar-tropatía hipertrofiante (OAH) (según [30],modificada). CCC: cardiopatías congénitascianógenas; RCH: rectocolitis hemorrágica;POEMS: polineuropatía, organomegalia,endocrinopatía, gammapatía monoclonaly signos cutáneos.
EMC - Aparato locomotor 5

E – 14-336 � Osteoartropatía hipertrofiante
osteosarcoma (30%), de fibrosarcoma (17%), de cáncerde útero (12,7%), de cavum (19%) o de tumores de otrosórganos (21,2%).
Los mesoteliomas pleurales (o tumores fibrosos) sonraros, pero se asocian a la OAH en el 10-20% de loscasos [36], sobre todo en su forma benigna [37].
La patología mediastínica está representada por loslinfomas (enfermedad de Hodgkin y linfomas no Hodg-kin) [38], los timomas [39] y los tumores diafragmáticos [21],neurogénicos y cardíacos [11].
La patología broncopulmonar no tumoral, en especialla tuberculosis, y las supuraciones crónicas, bronquiales,pulmonares o pleurales han disminuido su frecuen-cia como causas de OAH desde la aparición de laantibioticoterapia, con algunas excepciones [40]. Las infec-ciones oportunistas (Pneumocystis carinii) en el marcodel síndrome de la inmunodeficiencia humana (SIDA) [41]
pueden ser responsables de una OAH reversible despuésdel tratamiento, al igual que la propia infección por elvirus de la inmunodeficiencia humana (VIH). La muco-viscidosis y otras fibrosis pulmonares pueden complicarsecon un HD y una OAH (cf infra).
La patología cardiovascular incluye la endocarditisinfecciosa subaguda [42] excepcionalmente relacionadacon la OAH, y las CCC.
La patología esofágica raramente se asocia a la OAH,tanto si es benigna (megaesófago, dilatación del cardiascon un tubo de Célestin, leiomioma) como maligna [43].La invasión mediastino-pleuro-pulmonar es rara. En lasformas malignas, la OAH a menudo sigue a la apariciónde signos de estenosis esofágica.
Causas extratorácicasLa patología neoplásica ocupa aquí un lugar reducido,
al contrario que en las afecciones torácicas.El cáncer de cavum se acompana de OAH en el 5% de
los casos, con frecuencia asociado a metástasis pulmona-res [44]. El cáncer de tiroides se relaciona excepcionalmentecon la OAH [45]. Los tumores malignos del tubo digestivotambién se acompanan raramente de OAH: estómago [46],intestino delgado y colon [11], así como el cáncer dehígado [47], de vías biliares y de páncreas [48], incluso enausencia de metástasis pulmonares.
Los demás cánceres asociados a la OAH son el cáncer derinón [49], de mama [50] y el melanoma [51].
Las enteropatías inflamatorias crónicas raramente serelacionan con la OAH, aunque pueden observarse, gene-ralmente en forma de un HD aislado; también se hadescrito una periostosis (o periostitis) en capas en laenfermedad de Crohn, que se manifiesta por dolorperiarticular intenso y permanente [52]. Las demás causasentéricas de OAH incluyen la enfermedad de Whip-ple [53], la colitis seudomembranosa, el abuso de laxantesy la poliposis familiar con HD aislado o asociado a unaperiostosis [54].
Las hepatopatías pueden ser responsables de OAH [55, 56].La asociación HD-hipoxia en un paciente con unahepatopatía crónica debe hacer sospechar un síndromehepatopulmonar, que aparece en el 4-29% de los pacien-tes cirróticos [57]. La OAH puede mejorar con el trasplantehepático [58], asociarse a un rechazo del trasplante hepá-tico o incluso aparecer tras un trasplante, lo cual ofreceun campo de reflexión patogénica.
La OAH del nino se describe en unas 100 observaciones,con la misma distribución de frecuencia que en el adultoentre las causas extratorácicas y torácicas.
A veces, resulta ilusorio separar la OAH secundaria de laprimitiva debido a las numerosas formas intermedias: PDPsin signo cutáneo [16], PDP «secundaria» a una leucemiamielomonoblástica, OAH «aparentemente» primitiva [59].Numerosos autores consideran que el HD, la OAH secun-daria y la PDP son expresiones fenotípicas diferentes deuna disacromelia determinada genéticamente.
� Osteoartropatíahipertrofiante localizada
La OAH localizada en uno o dos miembros es rara y serelaciona con un cortocircuito derecha-izquierda o indicala infección de una prótesis [60]; a menudo se trata de pró-tesis vascular infectada de la bifurcación aortoilíaca, casisiempre fistulizada a las vías digestivas. En este caso, laOAH aparece de 3 meses a 10 anos después de la coloca-ción de la prótesis de dacrón, con artropatía y periostosisdiafisaria constantes del miembro o miembros dolorososy, a veces, hipocratismo de los dedos de los pies.
La OAH precede «providencialmente» al diagnóstico defístula aortodigestiva, que se manifiesta por fiebre y/o unahemorragia digestiva, a menudo recidivante, exteriori-zada o no, con frecuencia mortal en el 50% de los casos. Eltratamiento medicoquirúrgico debe ser precoz, teniendoen cuenta la dificultad de detectar la fistula mediante prue-bas complementarias.
� EpidemiologíaNo existe ningún estudio epidemiológico de la OAH
desde la descripción del HD 400 anos a.C. La asociaciónentre OAH y enfermedades cardiopulmonares se conocedesde 1890. Las únicas referencias son los estudios de gru-pos de pacientes aportados por las publicaciones, en losque se describen 115 observaciones de 1947 a 1990 [61].Estos últimos 20 anos, se han publicado varios centenaresde observaciones.
El HD, entre los 1.511 ingresos durante un ano en unservicio de medicina interna, se observó en el 1% de loscasos, relacionado con una enfermedad concreta en el40% de los casos [62]. La OAH afecta a individuos de todaslas razas, con una relación varón/mujer similar, entre los55-75 anos [63].
La incidencia y la prevalencia de la OAH primitiva (PDP)se desconocen [20, 61], pero la mayoría de pacientes con unaPDP son varones, con una relación varón/mujer de 9/1; laPDP es claramente una enfermedad familiar [20, 31].
En cuanto a la asociación OAH-cáncer broncopulmo-nar, tres estudios aportan precisiones sobre la frecuenciade la OAH secundaria; el primero muestra una frecuen-cia de la OAH en el cáncer broncopulmonar del 3%(9 de 280) [64]; el segundo, retrospectivo, realizado con1.226 pacientes seguidos por un cáncer broncopulmonar,muestra la existencia de una hipercaptación gammagrá-fica cortical sugestiva de OAH en el 4,5%, un HD yartralgias en el 0,8% de los casos [25]; el tercero, referente a2.625 cánceres de pulmón explorados mediante gamma-grafía ósea, muestra una frecuencia de OAH del 0,72% [34].En otras series publicadas, la prevalencia de la OAH es máselevada en el cáncer de pulmón primitivo, entre el 4 y el32% [39].
En las cardiopatías congénitas, la frecuencia de la OAHse considera baja, excepto en un estudio prospectivo deMartínez-Lavín et al [65] en el que estaban afectados el 31%de 32 pacientes mexicanos, de 7-45 anos de edad (mediade 16,3 ± 8,8 anos).
En la mucoviscidosis, la OAH se observa en la adoles-cencia o en la edad adulta en el 4-15% de los casos. La OAHcon periostosis se observa en las formas más evoluciona-das y graves de la enfermedad, con una tasa de mortalidadmás elevada. La rareza de la OAH se opone a la frecuenciadel HD [66].
En la fibrosis intersticial pulmonar, se constata la mismaoposición entre frecuencia del HD (50-60% de los casos),mientras que sólo se han descrito siete observaciones deOAH secundaria a una fibrosis pulmonar [39].
La tuberculosis pulmonar observada en África seacompana de una OAH en un tercio de los casos [67], sobretodo en las formas destructivas graves [68].
6 EMC - Aparato locomotor

Osteoartropatía hipertrofiante � E – 14-336
En las enteropatías inflamatorias, se observa un HD enel 38% de los casos de enfermedad de Crohn y en un 15%de la rectocolitis hemorrágica, y se detecta una correlacióncon la actividad de la enfermedad [52].
En diversas hepatopatías, dos estudios prospectivosponen de manifiesto la frecuencia del HD. El primeroreúne 128 hepatopatías crónicas, entre ellas 74 cirrosisbiliares primitivas (CBP), 24 hepatitis crónicas activasy 30 hepatopatías diversas. La frecuencia de HD y deperiostosis era destacable en estos tres grupos, respecti-vamente del 24 y 35%, 29 y 29%, 23 y 40%. El HDse asociaba intensamente a la periostosis en la CBP,mientras que la periostosis aparecía con frecuencia enausencia de HD en las otras hepatopatías crónicas [55];el segundo confirma la frecuencia de la OAH en la CBP(38%) y su carácter asintomático, pero senala la frecuen-cia de afectación de los metacarpianos y la rareza del HD(23%) [56].
� Anatomía patológicaSinovitis
Al microscopio óptico, a veces se observa una sinovitisproliferativa crónica moderada. Se asocia a un edema, uninfiltrado linfoplasmocítico perivascular moderado y unengrosamiento de la pared de los vasos [61].
Al microscopio electrónico, se sospecha una microan-giopatía sinovial ante la dilatación de los capilares y lasvénulas, que contienen amasijos de plaquetas, restos celu-lares y material lipídico; el revestimiento endotelial estáinterrumpido; se observan depósitos densos (cuerpos deWeibel-Palade) en las paredes de los capilares, que seencuentran engrosadas. La naturaleza de estos depósitoses controvertida: hemoglobina, enzimas, fibrinógeno. Labúsqueda por inmunofluorescencia de los depósitos deinmunoglobulinas (Ig) o de fracciones del complementoes negativa [61, 69].
PeriostosisEl periostio es inicialmente el asiento de un edema
y una hiperplasia vascular (neoangiogénesis), con uninfiltrado linfoplasmocítico. Secundariamente, la prolife-ración osteoblástica da lugar a tejido osteoide en la carainterna del periostio, separado de la cortical por un espa-cio esponjoso; la mineralización del osteoide conduce a lavisibilidad de las capas concéntricas (periostosis en capas),con un sistema haversiano de disposición radiada perpen-dicular a la diáfisis [11, 61].
La hipertrofia ósea predomina en los pacientes concáncer pulmonar, mientras que la acroosteólisis es másfrecuente en los pacientes con cardiopatías congéni-tas cianógenas y de OAH primitiva [61], en relacióncon la capacidad de las prostaglandinas E2 (PGE2)de estimular la actividad de los osteoblastos y lososteoclastos [70].
Tejidos blandosLos tejidos blandos, en especial el pulpejo digital, se ven
invadidos por un tejido conjuntivo joven, infiltrado deedema, con una dilatación capilar visible por capilarosco-pia. El engrosamiento de la piel y la paquidermia se debena una alteración de las células mesenquimatosas [71].
Hipocratismo digitalEl HD no sólo se debe a una hipertrofia de los tejidos
blandos, sino también a un proceso de remodelación óseade la falange subyacente.
� Fisiopatologíadel hipocratismo digitaly la periostosis
La relación entre HD y OAH se ha establecido medianteel estudio de los pacientes con cardiopatías congénitas,todos los cuales padecen un HD y un tercio desarrollauna OAH [72]. Se ha observado un aumento de los mar-cadores de remodelación ósea en algunos pacientes [73] yel aumento de la reabsorción ósea está relacionado conun aumento de la concentración de interleucina 6 (IL-6) y del ligando del receptor activador del factor nuclear(NF)-kappa-B [74].
Factores neurótroposEn la década de 1970, se incriminó al nervio neumo-
gástrico, dada la desaparición de las artralgias y el edemadespués de vagotomía en las toracotomías [63]; pero estamejoría (aunque sin regresión del HD) también se obser-vaba en las laparoscopias sin disección del nervio.
Factores humoralesLa hipótesis de un factor de crecimiento fibroblástico
normalmente presente en la circulación venosa pero noinactivado por el pulmón podría explicar la aparición deHD/OAH [72]. La concentración plasmática de este factorde crecimiento podría elevarse, bien por un cortocircuitocardíaco derecha-izquierda que evite el pulmón o bienpor una hiperproducción local debida a un tumor u otraenfermedad.
Factores plaquetariosEste factor de crecimiento podría proceder de las pla-
quetas [75], sobre todo teniendo en cuenta que, en lospacientes con un cortocircuito derecha-izquierda, lostrombocitos que escapan a la fragmentación en el pulmónentrarían en la circulación sistémica y llegarían a los teji-dos blandos distales, liberando los factores de crecimientoy provocando la acropaquia [72].
Se ha demostrado un aumento de la concentración delfactor de Von Willebrand en pacientes cianóticos o conuna PDP [76]; esta anomalía refleja la activación local de lasplaquetas y las células endoteliales [20]; estos datos sugie-ren que la activación local de las plaquetas y las célulasendoteliales que induce la liberación de factores de creci-miento tiene un papel clave en el desarrollo de la OAH.
Factores de crecimiento vascularesLa osteogénesis depende en gran medida de la angio-
génesis, por lo que se ha propuesto la intervención defactores angiogénicos.
El factor de crecimiento derivado de las plaquetas(PDGF) podría estar implicado en la patogenia de la OAH;no obstante, los datos son contradictorios, a pesar de queconcentraciones elevadas de PDGF pueden inducir unaproliferación fibroblástica excesiva [50].
El factor de crecimiento endotelial vascular (VEGF), encambio, es un agente angiogénico y osteogénico: en loscultivos celulares, es un potente promotor de la diferen-ciación osteoblástica [77] y podría inducir la proliferaciónperióstica. Es producido por tumores malignos comomecanismo de crecimiento del cáncer. También es una delas citocinas liberadas durante la activación plaquetaria,con efectos importantes sobre el desarrollo de la hiperpla-sia endotelial [78]. Además, el VEGF es un agente inducidopor la hipoxia. Por último, el VEGF parece desempenar unpapel esencial en la patogenia de la enfermedad de Crohn,
EMC - Aparato locomotor 7

E – 14-336 � Osteoartropatía hipertrofiante
la mielofibrosis y el síndrome POEMS (Polineuropatía,Organomegalia, Endocrinopatía, gammapatía Monoclo-nal y Signos cutáneos), lo cual podría explicar el desarrollode estas enfermedades en pacientes con OAH [79].
Así pues, la acción del VEGF en teoría explica,por una parte, cómo diferentes enfermedades neoplá-sicas o hipóxicas pueden inducir una OAH y, porotra parte, la naturaleza de los datos anatomopa-tológicos; la concentración plasmática de VEGF essignificativamente más elevada en la PDP y en la OAHasociada a cáncer de pulmón, comparada con la de loscontroles [80].
ProstaglandinasLas prostaglandinas (PG) podrían estar implicadas en la
patogenia de la OAH primitiva (PDP) [81]. Estudios genéti-cos recientes han demostrado, en familias con probandosafectados por una OAH primitiva (PDP), la existencia demutaciones del gen de la hidroxiprostaglandina deshidro-genasa (HPGD), que codifica la 15-HPGD, que cataboliza laPGE2, sobre todo en el pulmón, lo cual conduce a concen-traciones elevadas de PGE2 supuestamente responsablesdirectas de la PDP [82]. Esta hipótesis se ve confirmada porel hecho de que los pacientes con una hepatopatía tra-tados de manera prolongada con PGE desarrollan signostípicos de OAH y estos síntomas regresan al suspenderel tratamiento [83]. Sin embargo, los antiinflamatorios noesteroideos (AINE), ampliamente utilizados en el trata-miento de la PDP, no han demostrado una acción sobre lareversibilidad de la hipertrofia ósea o de los tejidos blan-dos.
Dado que las PGE2 se metabolizan en el pulmón, lasenfermedades que permiten que la sangre venosa corto-circuite los lechos capilares pulmonares también podríandar lugar a una exposición de las plaquetas con con-centraciones elevadas de este mediador lipídico. Algunosestudios han mostrado que incluso aumentos moderadosde PGE pueden aumentar la activación plaquetaria graciasal receptor EP3 [81].
Las PG podrían actuar como un facilitador de la accióndel VEGF [20]; se ha demostrado que los efectos de las PGsobre el hueso están mediados por el VEGF [84]. Es másdifícil explicar la patogenia de la OAH secundaria por lasPGE2 en las enfermedades que no afectan a los pulmoneso a la circulación pulmonar, como en las enfermedadesinflamatorias crónicas del intestino [81].
No obstante, al enfoque patogénico de la OAH le faltanmodelos experimentales.
� Datos genéticosen la paquidermoperiostosis
Datos recientes [20, 81] sugieren pues que la alteraciónde las concentraciones sistémicas de PGE2 desempenaun papel en la patogenia de la OAH primitiva (PDP).Mutaciones que afectan, por una parte, al gen que codi-fica la 15-HPGD (HPGD), enzima que cataboliza las PGE2
en la célula, y, por otra parte, al gen que codifica eltransportador de la PG (familia de portadores de solu-tos, transportadores de aniones orgánicos, miembro 2A1[SLCO2A1]), que controlan respectivamente la extracciónde PGE2 del medio extracelular y el transporte en la célula,dan lugar a concentraciones elevadas de PGE2 urinarias yse han relacionado con la PDP.
Mutaciones del gen HPGDLa 15-HPGD es la principal enzima de la degrada-
ción de las PG. Por consiguiente, los portadores deuna mutación de la 15-HPGD tienen concentraciones
elevadas crónicas de PGE2 y son incapaces de excretar sumetabolito, el ácido 11-alfa-hidroxi-9,15-dioxo-2,3,4,5-tetranorprostano-1,20-dioico (PGE-M).
En la actualidad, se han identificado 11 mutacionesdiferentes del gen HPGD en pacientes con un HDaislado [85] o una PDP [32, 82, 86–89], con una relaciónvarón/mujer de 1,3/1. Acaba de identificarse una nuevamutación en un turco de 22 anos y su hermana de23 anos, del tipo de una deleción homocigótica 2-bp(c.310 311delCT o p.L104AfsX3) [90].
Todas las mutaciones identificadas, generalmentehomocigóticas (consanguinidad presente en 12 de19 familias), generan una proteína truncada (quecarece del dominio de unión PGE2) o afectan a resi-duos indispensables para la actividad enzimática de la15-HPGD.
Las mutaciones más frecuentes parecen serc.175 176delCT [82, 87, 88] y c.120delA [32, 88], correspon-dientes a deleciones o inserciones no múltiples de tresbases, que dan lugar a una desviación del marco de lecturade las secuencias codificadoras que conduce a la apariciónde un codón de terminación prematuro y a una proteínaincompleta, la mayoría de las veces no funcional [91].Las nuevas mutaciones c.325-1G > C y c.G217 + 1G > Acorresponden a las sustituciones nucleotídicas exónicae intrónica, que dan lugar a una estructura proteicaaberrante.
Todas estas mutaciones pueden considerarse como ale-los nulos que aportan un producto génico no funcional,compatible con los fenotipos clínicos diferentes de todoslos pacientes. La variabilidad referente a la edad de ini-cio y la gravedad de los síntomas quizá es atribuiblea otros factores, en especial al porcentaje de síntesisde las PGE2: los pacientes masculinos con mutacionesHPGD de alelos nulos (homocigóticos y heterocigóticos)tienen concentraciones de PGE2 más elevadas y desarro-llan un HD a una edad más joven que los pacientesfemeninos [82].
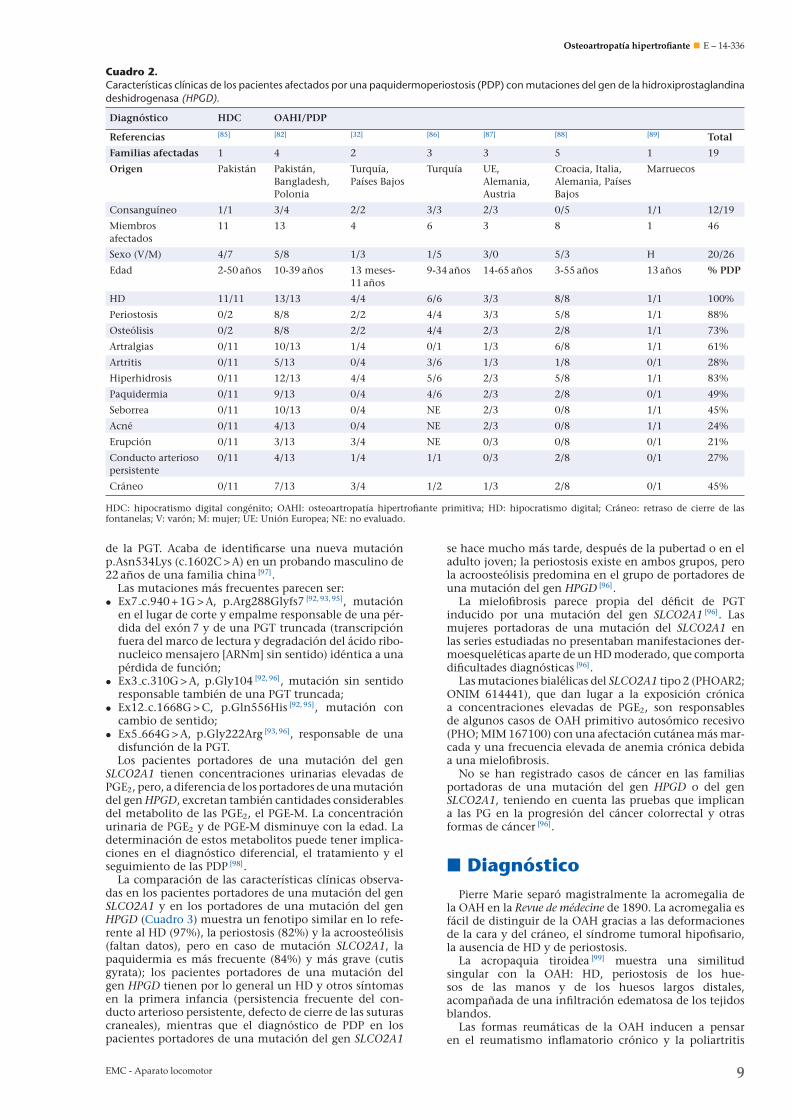
La comparación de las características clínicas observa-das en los pacientes portadores de una mutación del genHPGD (Cuadro 2) muestra un fenotipo que asocia general-mente HD (100%), periostosis (88%), acroosteólisis (78%),artralgias (61%), hiperhidrosis (83%) y afectación cutáneamoderada, puesto que la paquidermia sólo está presenteen el 50% de los casos. El retraso de cierre de las suturascraneanas estaba presente en el 45% y la persistencia delconducto arterioso persistente, en el 27% de los casos. Noexiste correlación evidente entre genotipo y fenotipo.
Las mutaciones bialélicas de HPGD tipo 1 (PHOAR1;ONIM 259100), que dan lugar a una exposición crónicaa concentraciones elevadas de PGE2, se asocian a ciertonúmero de casos típicos de OAH primitiva autosómicarecesiva (PHO; MIM 167100) clasificados como PDP uosteoartropatía craneal.
Mutaciones del gen SLCO2A1El gen SLCO2A1 codifica una proteína transportadora
de PG (PGT); la PGT es uno de los miembros del sistemade senalización de los icosanoides, similar al sistema derecaptación de las vesículas sinápticas. En este sistema, laPGT controla la recaptación de las PG en el citoplasma ydespués éstas son metabolizadas por la 15-HPGD. Así pues,la disfunción del gen SLCO2A1 impide la recaptación dela PGE2, lo cual da lugar a una concentración elevada dePGE2 en el suero, la orina y el tejido cutáneo [92].
En la actualidad, se han identificado 28 mutacionesdiferentes del gen SLCO2A1, en 38 pacientes con unaPDP [92–96] procedentes de 28 familias, con una relaciónvarón/mujer de 18,5/1. Todas las mutaciones identifica-das, a menudo homocigóticas (consanguinidad presenteen 13 de 28 familias), generan una proteína truncadao afectan a residuos indispensables para la actividad
8 EMC - Aparato locomotor
Osteoartropatía hipertrofiante � E – 14-336
Cuadro 2.Características clínicas de los pacientes afectados por una paquidermoperiostosis (PDP) con mutaciones del gen de la hidroxiprostaglandinadeshidrogenasa (HPGD).
Diagnóstico HDC OAHI/PDP
Referencias [85] [82] [32] [86] [87] [88] [89] Total
Familias afectadas 1 4 2 3 3 5 1 19
Origen Pakistán Pakistán,Bangladesh,Polonia
Turquía,Países Bajos
Turquía UE,Alemania,Austria
Croacia, Italia,Alemania, PaísesBajos
Marruecos
Consanguíneo 1/1 3/4 2/2 3/3 2/3 0/5 1/1 12/19
Miembrosafectados
11 13 4 6 3 8 1 46
Sexo (V/M) 4/7 5/8 1/3 1/5 3/0 5/3 H 20/26
Edad 2-50 anos 10-39 anos 13 meses-11 anos
9-34 anos 14-65 anos 3-55 anos 13 anos % PDP
HD 11/11 13/13 4/4 6/6 3/3 8/8 1/1 100%
Periostosis 0/2 8/8 2/2 4/4 3/3 5/8 1/1 88%
Osteólisis 0/2 8/8 2/2 4/4 2/3 2/8 1/1 73%
Artralgias 0/11 10/13 1/4 0/1 1/3 6/8 1/1 61%
Artritis 0/11 5/13 0/4 3/6 1/3 1/8 0/1 28%
Hiperhidrosis 0/11 12/13 4/4 5/6 2/3 5/8 1/1 83%
Paquidermia 0/11 9/13 0/4 4/6 2/3 2/8 0/1 49%
Seborrea 0/11 10/13 0/4 NE 2/3 0/8 1/1 45%
Acné 0/11 4/13 0/4 NE 2/3 0/8 1/1 24%
Erupción 0/11 3/13 3/4 NE 0/3 0/8 0/1 21%
Conducto arteriosopersistente
0/11 4/13 1/4 1/1 0/3 2/8 0/1 27%
Cráneo 0/11 7/13 3/4 1/2 1/3 2/8 0/1 45%
HDC: hipocratismo digital congénito; OAHI: osteoartropatía hipertrofiante primitiva; HD: hipocratismo digital; Cráneo: retraso de cierre de lasfontanelas; V: varón; M: mujer; UE: Unión Europea; NE: no evaluado.
de la PGT. Acaba de identificarse una nueva mutaciónp.Asn534Lys (c.1602C > A) en un probando masculino de22 anos de una familia china [97].
Las mutaciones más frecuentes parecen ser:• Ex7 c.940 + 1G > A, p.Arg288Glyfs7 [92, 93, 95], mutación
en el lugar de corte y empalme responsable de una pér-dida del exón 7 y de una PGT truncada (transcripciónfuera del marco de lectura y degradación del ácido ribo-nucleico mensajero [ARNm] sin sentido) idéntica a unapérdida de función;
• Ex3 c.310G > A, p.Gly104 [92, 96], mutación sin sentidoresponsable también de una PGT truncada;
• Ex12 c.1668G > C, p.Gln556His [92, 95], mutación concambio de sentido;
• Ex5 664G > A, p.Gly222Arg [93, 96], responsable de unadisfunción de la PGT.Los pacientes portadores de una mutación del gen
SLCO2A1 tienen concentraciones urinarias elevadas dePGE2, pero, a diferencia de los portadores de una mutacióndel gen HPGD, excretan también cantidades considerablesdel metabolito de las PGE2, el PGE-M. La concentraciónurinaria de PGE2 y de PGE-M disminuye con la edad. Ladeterminación de estos metabolitos puede tener implica-ciones en el diagnóstico diferencial, el tratamiento y elseguimiento de las PDP [98].
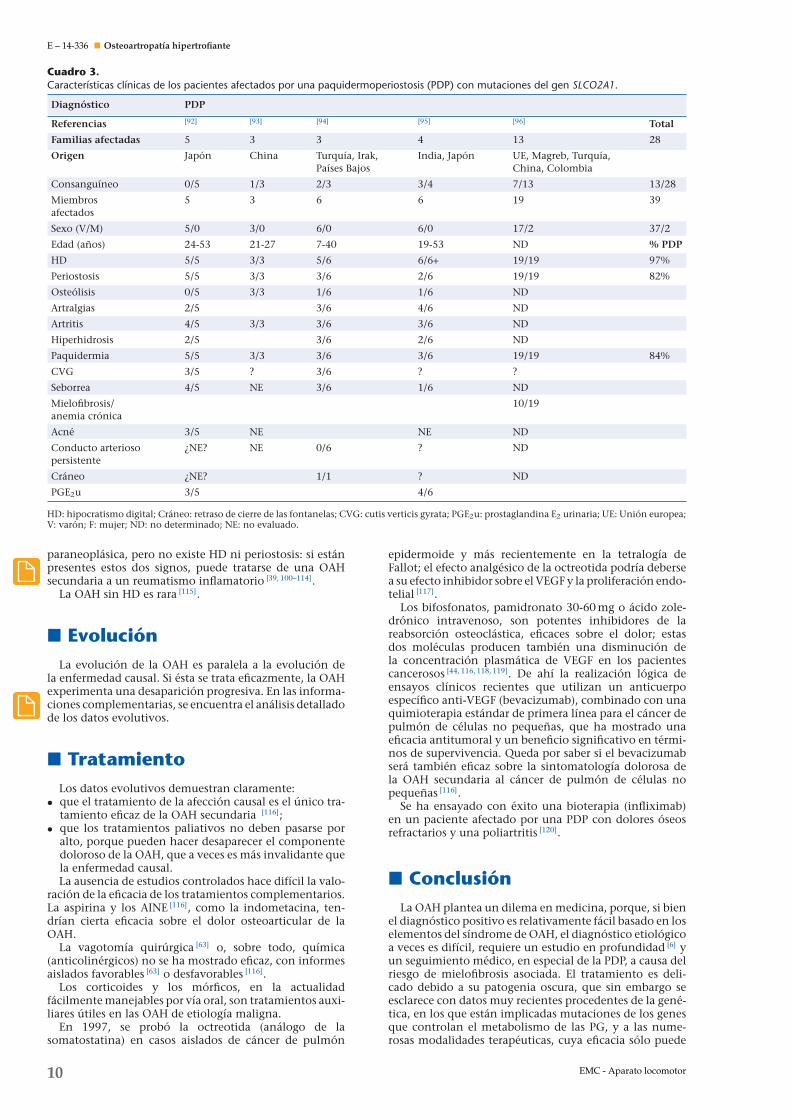
La comparación de las características clínicas observa-das en los pacientes portadores de una mutación del genSLCO2A1 y en los portadores de una mutación del genHPGD (Cuadro 3) muestra un fenotipo similar en lo refe-rente al HD (97%), la periostosis (82%) y la acroosteólisis(faltan datos), pero en caso de mutación SLCO2A1, lapaquidermia es más frecuente (84%) y más grave (cutisgyrata); los pacientes portadores de una mutación delgen HPGD tienen por lo general un HD y otros síntomasen la primera infancia (persistencia frecuente del con-ducto arterioso persistente, defecto de cierre de las suturascraneales), mientras que el diagnóstico de PDP en lospacientes portadores de una mutación del gen SLCO2A1
se hace mucho más tarde, después de la pubertad o en eladulto joven; la periostosis existe en ambos grupos, perola acroosteólisis predomina en el grupo de portadores deuna mutación del gen HPGD [96].
La mielofibrosis parece propia del déficit de PGTinducido por una mutación del gen SLCO2A1 [96]. Lasmujeres portadoras de una mutación del SLCO2A1 enlas series estudiadas no presentaban manifestaciones der-moesqueléticas aparte de un HD moderado, que comportadificultades diagnósticas [96].
Las mutaciones bialélicas del SLCO2A1 tipo 2 (PHOAR2;ONIM 614441), que dan lugar a la exposición crónicaa concentraciones elevadas de PGE2, son responsablesde algunos casos de OAH primitivo autosómico recesivo(PHO; MIM 167100) con una afectación cutánea más mar-cada y una frecuencia elevada de anemia crónica debidaa una mielofibrosis.
No se han registrado casos de cáncer en las familiasportadoras de una mutación del gen HPGD o del genSLCO2A1, teniendo en cuenta las pruebas que implicana las PG en la progresión del cáncer colorrectal y otrasformas de cáncer [96].
� DiagnósticoPierre Marie separó magistralmente la acromegalia de
la OAH en la Revue de médecine de 1890. La acromegalia esfácil de distinguir de la OAH gracias a las deformacionesde la cara y del cráneo, el síndrome tumoral hipofisario,la ausencia de HD y de periostosis.
La acropaquia tiroidea [99] muestra una similitudsingular con la OAH: HD, periostosis de los hue-sos de las manos y de los huesos largos distales,acompanada de una infiltración edematosa de los tejidosblandos.
Las formas reumáticas de la OAH inducen a pensaren el reumatismo inflamatorio crónico y la poliartritis
EMC - Aparato locomotor 9
E – 14-336 � Osteoartropatía hipertrofiante
Cuadro 3.Características clínicas de los pacientes afectados por una paquidermoperiostosis (PDP) con mutaciones del gen SLCO2A1.
Diagnóstico PDP
Referencias [92] [93] [94] [95] [96] Total
Familias afectadas 5 3 3 4 13 28
Origen Japón China Turquía, Irak,Países Bajos
India, Japón UE, Magreb, Turquía,China, Colombia
Consanguíneo 0/5 1/3 2/3 3/4 7/13 13/28
Miembrosafectados
5 3 6 6 19 39
Sexo (V/M) 5/0 3/0 6/0 6/0 17/2 37/2
Edad (anos) 24-53 21-27 7-40 19-53 ND % PDP
HD 5/5 3/3 5/6 6/6+ 19/19 97%
Periostosis 5/5 3/3 3/6 2/6 19/19 82%
Osteólisis 0/5 3/3 1/6 1/6 ND
Artralgias 2/5 3/6 4/6 ND
Artritis 4/5 3/3 3/6 3/6 ND
Hiperhidrosis 2/5 3/6 2/6 ND
Paquidermia 5/5 3/3 3/6 3/6 19/19 84%
CVG 3/5 ? 3/6 ? ?
Seborrea 4/5 NE 3/6 1/6 ND
Mielofibrosis/anemia crónica
10/19
Acné 3/5 NE NE ND
Conducto arteriosopersistente
¿NE? NE 0/6 ? ND
Cráneo ¿NE? 1/1 ? ND
PGE2u 3/5 4/6
HD: hipocratismo digital; Cráneo: retraso de cierre de las fontanelas; CVG: cutis verticis gyrata; PGE2u: prostaglandina E2 urinaria; UE: Unión europea;V: varón; F: mujer; ND: no determinado; NE: no evaluado.
paraneoplásica, pero no existe HD ni periostosis: si estánpresentes estos dos signos, puede tratarse de una OAHsecundaria a un reumatismo inflamatorio [39, 100–114].
La OAH sin HD es rara [115].
� EvoluciónLa evolución de la OAH es paralela a la evolución de
la enfermedad causal. Si ésta se trata eficazmente, la OAHexperimenta una desaparición progresiva. En las informa-ciones complementarias, se encuentra el análisis detalladode los datos evolutivos.
� TratamientoLos datos evolutivos demuestran claramente:
• que el tratamiento de la afección causal es el único tra-tamiento eficaz de la OAH secundaria [116];
• que los tratamientos paliativos no deben pasarse poralto, porque pueden hacer desaparecer el componentedoloroso de la OAH, que a veces es más invalidante quela enfermedad causal.La ausencia de estudios controlados hace difícil la valo-
ración de la eficacia de los tratamientos complementarios.La aspirina y los AINE [116], como la indometacina, ten-drían cierta eficacia sobre el dolor osteoarticular de laOAH.
La vagotomía quirúrgica [63] o, sobre todo, química(anticolinérgicos) no se ha mostrado eficaz, con informesaislados favorables [63] o desfavorables [116].
Los corticoides y los mórficos, en la actualidadfácilmente manejables por vía oral, son tratamientos auxi-liares útiles en las OAH de etiología maligna.
En 1997, se probó la octreotida (análogo de lasomatostatina) en casos aislados de cáncer de pulmón
epidermoide y más recientemente en la tetralogía deFallot; el efecto analgésico de la octreotida podría debersea su efecto inhibidor sobre el VEGF y la proliferación endo-telial [117].
Los bifosfonatos, pamidronato 30-60 mg o ácido zole-drónico intravenoso, son potentes inhibidores de lareabsorción osteoclástica, eficaces sobre el dolor; estasdos moléculas producen también una disminución dela concentración plasmática de VEGF en los pacientescancerosos [44, 116, 118, 119]. De ahí la realización lógica deensayos clínicos recientes que utilizan un anticuerpoespecífico anti-VEGF (bevacizumab), combinado con unaquimioterapia estándar de primera línea para el cáncer depulmón de células no pequenas, que ha mostrado unaeficacia antitumoral y un beneficio significativo en térmi-nos de supervivencia. Queda por saber si el bevacizumabserá también eficaz sobre la sintomatología dolorosa dela OAH secundaria al cáncer de pulmón de células nopequenas [116].
Se ha ensayado con éxito una bioterapia (infliximab)en un paciente afectado por una PDP con dolores óseosrefractarios y una poliartritis [120].
� ConclusiónLa OAH plantea un dilema en medicina, porque, si bien
el diagnóstico positivo es relativamente fácil basado en loselementos del síndrome de OAH, el diagnóstico etiológicoa veces es difícil, requiere un estudio en profundidad [6] yun seguimiento médico, en especial de la PDP, a causa delriesgo de mielofibrosis asociada. El tratamiento es deli-cado debido a su patogenia oscura, que sin embargo seesclarece con datos muy recientes procedentes de la gené-tica, en los que están implicadas mutaciones de los genesque controlan el metabolismo de las PG, y a las nume-rosas modalidades terapéuticas, cuya eficacia sólo puede
10 EMC - Aparato locomotor

Osteoartropatía hipertrofiante � E – 14-336
valorarse caso por caso. Es necesario continuar los estudiosde las mutaciones para deducir las correlaciones genotipo-fenotipo que permitirán determinar el valor predictivo delanálisis de las mutaciones de HPGD y SLCO2A1.
� Bibliografía[1] Marrie TJ, Brown N. Clubbing of the digits. Am J Med
2007;120:940–1.[2] Myers KA, Farquhar DR. The rational clinical examina-
tion. Does this patient have clubbing? JAMA 2001;286:341–7.
[3] Capron J, Steichen O. Quantification d’un hippocratisme digi-tal. Presse Med 2008;37:1520–1.
[4] Pallarés-Sanmartín A, Leiro-Fernández V, Cebreiro TL,Botana-Rial M, Fernández-Villar A. Validity and reliabilityof the Schamroth sign for the diagnosis of clubbing. JAMA2010;304:159–61.
[5] Santiago MB, Lima I, Feitosa AC, Braz Ade S, Miranda LG.Pseudoclubbing: is it different from clubbing? Semin ArthritisRheum 2009;38:452–7.
[6] Spicknall KE, Zirwas MJ, English 3rd JC. Clubbing: anupdate on diagnosis, differential diagnosis, pathophysio-logy, and clinical relevance. J Am Acad Dermatol 2005;52:1020–8.
[7] Sarkar M, Mahesh DM, Madabhavi I. Digital clubbing. LungIndia 2012;29:354–62.
[8] Singh A. Unidigital clubbing. Am J Med 2008;121:e15.[9] Jajic Z, Jajic I, Nemcic T. Primary hypertrophic osteoarthro-
pathy: clinical, radiologic, and scintigraphic characteristics.Arch Med Res 2001;32:136–42.
[10] Coury C. L’hippocratisme digital, l’ostéoarthropathie hyper-trophiante et les autres dysacromélies apparentées. Paris:Baillière; 1960.
[11] Altman RD. Hypertrophic osteoarthropathy. En: KoopmanWJ, Moreland LW, editores. Arthritis and allied conditions:a textbook of rheumatology. Philadelphia: LWW; 2005. p.2037–43.
[12] Armstrong DJ, McCausland EM, Wright GD. Hypertrop-hic pulmonary osteoarthropathy (HPOA) (Pierre Marie-Bamberger syndrome): two cases presenting as acuteinflammatory arthritis. Description and review of the litera-ture. Rheumatol Int 2007;27:399–402.
[13] Farhey Y, Luggen M. Seropositive, symmetric polyarthritisin a patient with poorly differentiated lung carcinoma: car-cinomatous polyarthritis, hypertrophic osteoarthropathy, orrheumatoid arthritis? Arthritis Care Res 1998;11:146–9.
[14] Morin F, Marcelli C. Ostéoarthropathie hypertrophiante.EMC (Elsevier Masson SAS, Paris). Appareil locomoteur,14-260-A-10, 2002 : 8 p.
[15] Martinez-Lavin M, Pineda C, Valdez T, Cajigas JC, WeismanM, Gerber N, et al. Primary hypertrophic osteoarthropathy.Semin Arthritis Rheum 1988;17:156–62.
[16] Ben Hamida SK, Kédadi H, Derbel F, Kchir M, BouhaoulaH, Hadj SM, et al. Ostéoarthropathie hypertrophiante primi-tive sans atteinte cutanée (maladie de Currarino). Presse Med2003;32:1455–6.
[17] Cannavò SP, Guarneri C, Borgia F, Vaccaro M. Pierre Marie-Bamberger syndrome (secondary hypertrophic osteoarthro-pathy). Int J Dermatol 2005;44:41–2.
[18] Touraine A, Solente G, Gole L. Un syndrome ostéodermopat-hique : la pachydermie plicaturée avec pachypériostose desextrémités. Presse Med 1935;43:1820–4.
[19] Kabi F, Mkinsi O, Janani S, Raissouni N. Pachydermopérios-tose. À propos d’un cas. Rev Med Interne 2006;27:710–2.
[20] Martinez-Lavin M. Miscellaneous non-inflammatory muscu-loskeletal conditions. Pachydermoperiostosis. Best Pract ResClin Rheumatol 2011;25:727–34.
[21] Resnick D, Enostosis. Hyperostosis, and periostitis. En:Resnick D, editor. Diagnosis of bone and joint disorders.Philadelphia: WB Saunders; 2002. p. 4870–84.
[22] Pineda CJ, Guerra Jr J, Weisman MH, Resnick D, Martinez-Lavin M. The skeletal manifestations of clubbing: astudy in patients with cyanotic congenital heart diseaseand hypertrophic osteoarthropathy. Semin Arthritis Rheum1985;14:263–73.
[23] Pineda CJ, Martinez-Lavin M, Goobar JE, Sartoris DJ,Clopton P, Resnick D. Periostitis in hypertrophic osteoarthro-pathy: relationship to disease duration. AJR Am J Roentgenol1987;148:773–8.
[24] Joseph B, Chacko V. Acro-osteolysis associated withhypertrophic pulmonary osteoarthropathy and pachydermo-periostosis. Radiology 1985;154:343–4.
[25] Izumi M, Takayama K, Yabuuchi H, Abe K, NakanishiY. Incidence of hypertrophic pulmonary osteoarthro-pathy associated with primary lung cancer. Respirology2010;15:809–12.
[26] Albrecht S, Keller A. Postchemotherapeutic reversibility ofhypertrophic osteoarthropathy in a patient with bronchogenicadenocarcinoma. Clin Nucl Med 2003;28:463–6.
[27] Aparici CM, Bains S. Hypertrophic osteoarthropathyseen with NaF18 PET/CT bone imaging. Clin Nucl Med2011;36:928–9.
[28] Roy HS, Wang Z, Ran H, Song W, Zheng Y. Diagnosis ofdigital clubbing by high-frequency ultrasound imaging. Int JDermatol 2013;52:1–5.
[29] Capelastegui A, Astigarraga E, García-Iturraspe C. MRfindings in pulmonary hypertrophic osteoarthropathy. ClinRadiol 2000;55:72–5.
[30] Martínez-Lavín M. Hypertrophic osteoarthropathy. CurrOpin Rheumatol 1997;9:83–6.
[31] Castori M, Sinibaldi L, Mingarelli R, Lachman RS, RimoinDL, Dallapiccola B. Pachydermoperiostosis: an update. ClinGenet 2005;68:477–86.
[32] Seifert W, Beninde J, Hoffmann K, Lindner TH, Bassir C,Aksu F, et al. HPGD mutations cause cranioosteoarthropathybut not autosomal dominant digital clubbing. Eur J HumGenet 2009;17:1570–6.
[33] Chen X, Zou CC, Dong GP, Liang L, Zhao ZY.Cranioosteoarthropathy: a rare variant of hypertrophicosteoarthropathy. Ir J Med Sci 2012;181:257–61.
[34] Ito T, Goto K, Yoh K, Niho S, Ohmatsu H, Kubota K,et al. Hypertrophic pulmonary osteoarthropathy as a para-neoplastic manifestation of lung cancer. J Thorac Oncol2010;5:976–80.
[35] Yacoub MH, Simon G, Ohnsorge J. Hypertrophic pulmonaryosteoarthropathy in association with pulmonary metastasesfrom extrathoracic tumours. Thorax 1967;22:226–31.
[36] Toelen C, Deleersnijder R, Thomas B. Malignant fibroustumor of the pleura: case report and literature review. ActaChir Belg 2012;112:314–6.
[37] Rakovich G, Laflamme M, Ouellette D, Beauchamp G. Soli-tary fibrous tumour of the pleura: a case report. Can Respir J2010;17:113–4.
[38] Goodyer MJ, Cronin MC, Ketsitlile DG, O’Reilly SP, Moy-lan EJ, Maher MM, et al. Hodgkin’s lymphoma with digitalclubbing. J Clin Oncol 2009;27:e95–6.
[39] Yao Q, Altman RD, Brahn E. Periostitis and hypertrophicpulmonary osteoarthropathy: report of 2 cases and review ofthe literature. Semin Arthritis Rheum 2009;38:458–66.
[40] McNaughton DA, Nguyen BD. AJR teaching file: cavitatedmass with hypertrophic osteoarthropathy. AJR Am J Roent-genol 2007;188(Suppl. 3):S7–9.
[41] Booth TC, Chhaya NC, Bell JR, Holloway BJ. Update onimaging of non-infectious musculoskeletal complications ofHIV infection. Skeletal Radiol 2012;41:1349–63.
[42] Ozdemir B, Sentürk T, Kaderli AA, Kecebas M, Güllülü S,et al. Postoperative regression of clubbing at an unexpectedrate in a patient with aortic and mitral valve replace-ment due to infective endocarditis. Ir J Med Sci 2009;178:351–3.
[43] Wechalekar MD, Kennedy NA, Ahern M, Slavotinek J,Smith MD. Esophageal adenocarcinoma and hypertrop-hic osteoarthropathy with improvement following resec-tion of esophageal cancer. J Clin Rheumatol 2011;17:323–4.
[44] Sonthalia N, Mukherjee K, Saha A, Talukdar A. Treat-ment of hypertrophic osteoarthropathy in the case ofpulmonary metastasis secondary-to-nasopharyngeal carci-noma with zoledronic acid: an enlightening experience. BMJCase Rep 2012;2012.pii:bcr2012006759.
[45] Vico P, Delcorde A, Rahier I, Treille de Grandsaigne S,Famaey JP, Body JJ. Hypertrophic osteoarthropathy and thy-roid cancer. J Rheumatol 1992;19:1153–6.
EMC - Aparato locomotor 11

E – 14-336 � Osteoartropatía hipertrofiante
[46] Ikeda F, Okada H, Mizuno M, Kawamoto H, Okano N, Oka-zaki H, et al. Pachydermoperiostosis associated with juvenilepolyps of the stomach and gastric adenocarcinoma. J Gastro-enterol 2004;39:370–4.
[47] Dettmer M, Itin P, Miny P, Gandhi M, Cathomas G,Willi N. Giant ectopic liver, hepatocellular carcinomaand pachydermia-a rare genetic syndrome? Diagn Pathol2011;6:75.
[48] Elmahou S, Porteau-Cassard L, Zabraniecki L, Peron JM,Fournie B. Ostéoarthropathie hypertrophiante secondaire àune tumeur du pancréas. Presse Med 2001;30:901.
[49] Golimbu C, Marchetta P, Firooznia H, Rafii M. Hypertrophicosteoarthropathy in metastatic renal cell carcinoma. Urology1983;22:669–72.
[50] Landrum ML, Ornstein DL. Hypertrophic osteoarthropathyassociated with metastatic phyllodes tumor. Am J Clin Oncol2003;26:146–50.
[51] Thompson MA, Warner NB, Hwu WJ. Hypertrophicosteoarthropathy associated with metastatic melanoma.Melanoma Res 2005;15:559–61.
[52] Lemann M, Bonnet J, Allez M, Gornet JM, Mariette X.Lésions ostéo-articulaires au cours des maladies inflam-matoires chroniques de l’intestin. Gastroenterol Clin Biol2004;28:D75–82.
[53] Marie I, Levesque H, Levade MH, Cailleux N, LecomteF, Francois A, et al. Hypertrophic osteoarthropathy canindicate recurrence of Whipple’s disease. Arthritis Rheum1999;42:2002–6.
[54] Lamireau T, Olschwang S, Rooryck C, Le Bail B, ChateilJF, Lacombe D. SMAD4 germinal mosaicism in a familywith juvenile polyposis and hypertrophic osteoarthropathy.J Pediatr Gastroenterol Nutr 2005;41:117–20.
[55] Epstein O, Dick R, Sherlock S. Prospective study of periosti-tis and finger clubbing in primary biliary cirrhosis and otherforms of chronic liver disease. Gut 1981;22:203–6.
[56] Mills PR, Vallance R, Birnie G, Quigley EM, Main AN,Morgan RJ, et al. A prospective survey of radiological boneand joint changes in primary biliary cirrhosis. Clin Radiol1981;32:297–302.
[57] Goossens N, Joshi D, O’Grady J. Image of the month. Digi-tal clubbing in association with hepatopulmonary syndrome.Hepatology 2011;53:365–6.
[58] Taillandier J, Alemanni M, Samuel D, Bismuth H. Hepatichypertrophic osteoarthropathy: the value of liver transplanta-tion. Clin Exp Rheumatol 1998;16:80–1.
[59] Jajic Z, Grazio S, Nemcic T, Jajic I. Reactivation of primaryhypertrophic osteoarthropathy by bronchogenic carcinoma.Clin Exp Rheumatol 2001;19:95–7.
[60] Lacoin Q, Gaultier JB, Charmion S, Guichard I, Cathé-bras P. Ostéoarthropathie hypertrophiante et infection deprothèse aortique : deux observations. Rev Med Interne2011;32:432–5.
[61] First International workshop on hypertrophic osteoarthro-pathy. Clin Exp Rheumatol 1992;10(Suppl. 7):1–70.
[62] Vandemergel X, Renneboog B. Prevalence, aetiologies andsignificance of clubbing in a department of general internalmedicine. Eur J Intern Med 2008;19:325–9.
[63] Ooi A, Saad RA, Moorjani N, Amer KM. Effective sympto-matic relief of hypertrophic pulmonary osteoarthropathy byvideo-assisted thoracic surgery truncal vagotomy. Ann Tho-rac Surg 2007;83:684–5.
[64] Rassam JW, Anderson G. Incidence of paramalignantdisorders in bronchogenic carcinoma. Thorax 1975;30:86–90.
[65] Martínez-Lavín M, Bobadilla M, Casanova J, Attié F, Martí-nez M. Hypertrophic osteoarthropathy in cyanotic congenitalheart disease: its prevalence and relationship to bypass of thelung. Arthritis Rheum 1982;25:1186–93.
[66] Botton E, Saraux A, Laselve H, Jousse S, Le Goff P. Muscu-loskeletal manifestations in cystic fibrosis. Joint Bone Spine2003;70:327–35.
[67] Aissa I, Messadi A, Boudaya S, El Mezni F, Kilani T, GhédiraH. Ostéoarthropathie hypertrophique pneumique associéeà la tuberculose pulmonaire. Rev Pneumol Clin 2011;67:101–4.
[68] Reeve PA, Harries AD, Nkhoma WA, Nyangulu DS, WirimaJJ. Clubbing in African patients with pulmonary tuberculosis.Thorax 1987;42:986–7.
[69] Martinez-Lavin M, Pineda C. Digital clubbing and hyper-trophic osteoarthropathy. En: Hochberg M, Silman AJ, SolenJS, Weinblatt ME, Weisman MH, editores. Rheumatology.Edinburgh: Mosby-Elsevier; 2011. p. 1701–5.
[70] Blackwell KA, Raisz LG, Pilbeam CC. Prostaglandinsin bone: bad cop, good cop? Trends Endocrinol Metab2010;21:294–301.
[71] Kabashima K, Sakabe J, Yoshiki R, Tabata Y, Kohno K,Tokura Y. Involvement of Wnt signaling in dermal fibroblasts.Am J Pathol 2010;176:721–32.
[72] Martinez-Lavin M. Exploring the cause of the most ancientclinical sign of medicine: finger clubbing. Semin ArthritisRheum 2007;36:380–5.
[73] Martínez-Ferrer A, Peris P, Alós L, Morales-Ruiz M,Guanabens N. Prostaglandin E2 and bone turnover markersin the evaluation of primary hypertrophic osteoarthropathy(pachydermoperiostosis): a case report. Clin Rheumatol2009;28:1229–33.
[74] Rendina D, De Filippo G, Viceconti R, Soscia E, Sirignano C,Salvatore M, et al. Interleukin (IL)-6 and receptor activator ofnuclear factor (NF)-kappaB ligand (RANKL) are increased inthe serum of a patient with primary pachydermoperiostosis.Scand J Rheumatol 2008;37:225–9.
[75] Dickinson CJ, Martin JF. Megacaryocytes and platelet clumpsas the cause of finger clubbing. Lancet 1987;2:1434–5.
[76] Matucci-Cerinic M, Martinez-Lavin M, Rojo F, Fonseca C,Kahaleh BM. Von Willebrand factor antigen in hypertrophicosteoarthropathy. J Rheumatol 1992;19:765–7.
[77] Midy V, Plouët J. Vasculotropin/vascular endothelial growthfactor induces differentiation in cultured osteoblasts. BiochemBiophys Res Commun 1994;199:380–6.
[78] Atkinson S, Fox SB. Vascular endothelial growth factor(VEGF)-A and platelet-derived growth factor (PDGF) playa central role in the pathogenesis of digital clubbing. J Pathol2004;203:721–8.
[79] Martinez-Lavin M, Vargas A, Rivera-Vinas M. Hypertrophicosteoarthropathy: a palindrome with a pathogenic connota-tion. Curr Opin Rheumatol 2008;20:88–91.
[80] Silveira LH, Martínez-Lavín M, Pineda C, Fonseca MC,Navarro C, Nava A. Vascular endothelial growth factorand hypertrophic osteoarthropathy. Clin Exp Rheumatol2000;18:57–62.
[81] Coggins KG, Coffman TM, Koller BH. The Hippocratic fin-ger points the blame at PGE2. Nat Genet 2008;40:691–2.
[82] Uppal S, Diggle CP, Carr IM, Fishwick CW, Ahmed M,Ibrahim GH, et al. Mutations in 15-hydroxyprostaglandindehydrogenase cause primary hypertrophic osteoarthropathy.Nat Genet 2008;40:789–93.
[83] Cattral MS, Altraif I, Greig PD, Blendis L, Levy GA. Toxiceffects of intravenous and oral prostaglandin E therapy inpatients with liver disease. Am J Med 1994;97:369–73.
[84] Harada S, Nagy JA, Sullivan KA, Thomas KA, Endo N,Rodan GA, et al. Induction of vascular endothelial growthfactor expression by prostaglandin E2 and E1 in osteoblasts.J Clin Invest 1994;93:2490–6.
[85] Tariq M, Azeem Z, Ali G, Chishti MS, Ahmad W.Mutation in the HPGD gene encoding NAD+ dependent15-hydroxyprostaglandin dehydrogenase underlies isola-ted congenital nail clubbing (ICNC). J Med Genet2009;46:14–20.
[86] Yüksel-Konuk B, Sırmacı A, Ayten GE, Özdemir M, AslanI, Yılmaz-Turay Ü, et al. Homozygous mutations in the15-hydroxyprostaglandin dehydrogenase gene in patientswith primary hypertrophic osteoarthropathy. Rheumatol Int2009;30:39–43.
[87] Bergmann C, Wobser M, Morbach H, Falkenbach A,Wittenhagen D, Lassay L, et al. Primary hypertrophicosteoarthropathy with digital clubbing and palmoplantarhyperhidrosis caused by 15-PGHD/HPGD loss-of-functionmutations. Exp Dermatol 2011;20:531–3.
[88] Diggle CP, Carr IM, Zitt E, Wusik K, Hopkin RJ, Prada CE,et al. Common and recurrent HPGD mutations in Cauca-sian individuals with primary hypertrophic osteoarthropathy.Rheumatology 2010;49:1056–62.
[89] Sinibaldi L, Harifi G, Bottillo I, Iannicelli M, El HassaniS, Brancati F, et al. A novel homozygous splice site muta-tion in the HPGD gene causes mild primary hypertrophicosteoarthropathy. Clin Exp Rheumatol 2010;28:153–7.
12 EMC - Aparato locomotor

Osteoartropatía hipertrofiante � E – 14-336
[90] Erken E, Köroglu C, Yıldız F, Ozer HT, Gülek B, Tolun A.A novel recessive 15-hydroxyprostaglandin dehydrogenasemutation in a family with primary hypertrophic osteoarthro-pathy. Mod Rheumatol 2013. Apr 25. [Epub ahead of print].
[91] Hanna N, Parfait B, Vidaud D, Vidaud M. Mécanismes etconséquences des mutations. Med Sci 2005;21:969–80.
[92] Sasaki T, Niizeki H, Shimizu A, Shiohama A, Hiraki-yama A, Okuyama T, et al. Identification of mutationsin the prostaglandin transporter gene SLCO2A1 and itsphenotype-genotype correlation in Japanese patients withpachydermoperiostosis. J Dermatol Sci 2012;68:36–44.
[93] Zhang Z, Xia W, He J, Zhang Z, Ke Y, Yue H, et al. Exomesequencing identifies SLCO2A1 mutations as a cause ofprimary hypertrophic osteoarthropathy. Am J Hum Genet2012;90:125–32.
[94] Seifert W, Kühnisch J, Tüysüz B, Specker C, Brouwers A,Horn D. Mutations in the prostaglandin transporter encodinggene SLCO2A1 cause primary hypertrophic osteoarthropathyand isolated digital clubbing. Hum Mutat 2012;33:660–4.
[95] Busch J, Frank V, Bachmann N, Otsuka A, Oji V, Metze D,et al. Mutations in the prostaglandin transporter SLCO2A1cause primary hypertrophic osteoarthropathy with digitalclubbing. J Invest Dermatol 2012;132:2473–6.
[96] Diggle CP, Parry DA, Logan CV, Laissue P, Rivera C,Restrepo CM, et al. Prostaglandin transporter mutationscause pachydermoperiostosis with myelofibrosis. Hum Mutat2012;33:1175–81.
[97] Zhang Z, He JW, Fu WZ, Zhang CQ, Zhang ZL. A novel muta-tion in the SLCO2A1 gene in a Chinese family with primaryhypertrophic osteoarthropathy. Gene 2013;521:191–4.
[98] Zhang Z, He JW, Fu WZ, Zhang CQ, Zhang ZL. Mutationsin the SLCO2A1 gene and primary hypertrophic osteoarth-ropathy: a clinical and biochemical characterization. J ClinEndocrinol Metab 2013;98:E923–33.
[99] Fatourechi V. Thyroid dermopathy and acropachy. Best PractRes Clin Endocrinol Metab 2012;26:553–65.
[100] Diamond S, Momeni M. Primary hypertrophic osteoarthro-pathy in a patient with rheumatoid arthritis. J Clin Rheumatol2007;13:242–3.
[101] Shinjo SK, Borba EF, Goncalves CR, Levy-Neto M. Anky-losing spondylitis in a patient with primary hypertrophicosteoarthropathy. J Clin Rheumatol 2007;13:175.
[102] Périchon S, Pagnoux C, Seror R, Dassonville L, MangoukaL, Cohen P, et al. Périostéite au cours des vasculari-tes systémiques nécrosantes : à propos de 4 observationsd’une atteinte méconnue et pourtant évocatrice. Presse Med2010;39:e165–73.
[103] Burson JS, Grana J, Varela J, Atanes A, Galdo F. Lami-nar periostitis and multiple osteonecrosis in systemic lupuserythematosus. Clin Rheumatol 1990;9:535–8.
[104] Kim JE, Kolh EM, Kim DK. Takayasu’s arteritis presen-ting with focal periostitis affecting two limbs. Int J Cardiol1998;67:267–70.
[105] de Lastours V, Lidove O, Lieberherr D, Laissy JP, LebtahiR, Cerceau J, et al. Lower limb hypertrophic osteoarthro-pathy can reveal aortic graft infection in Behcet syndrome.Rheumatology 2006;45:117–8.
[106] Hashmi S, Kaplan D. Asymmetric clubbing as a manifestationof sarcoid bone disease. Am J Med 1992;93:471.
[107] Garcia-Gonzalez A, Weisman MH. The arthritis offamilial Mediterranean fever. Semin Arthritis Rheum1992;22:139–50.
[108] Harris AW, Harding TA, Gaitonde MD, Maxwell JD. Is club-bing a feature of the anti-phospholipid antibody syndrome?Postgrad Med J 1993;69:748–50.
[109] Magazine R, Shetty R, Goneppanavar U, Mohapatra AK.Idiopathic clubbing confined to lower limb digits and idio-pathic pulmonary fibrosis: an unusual association. Case RepPulmonol 2012;2012:684285.
[110] Bourke SJ. Clubbing in hypersensitivity pneumonitis. ArchIntern Med 1991;151:2105–9.
[111] Smahi M, Lakranbi M, Choumi I, Elbiaz M, Amara B, Ben-jelloun MC. Association hippocratisme digital et hydatidosepulmonaire. Rev Mal Respir 2010;27:99–101.
[112] Wald R, Crean A. Differential clubbing and cyanosis in apatient with pulmonary hypertension. CMAJ 2010;182:E380.
[113] Chen L, Mulligan ME. Medication-induced periostitis in lungtransplant patients: periostitis deformans revisited. SkeletalRadiol 2011;40:143–8.
[114] Pracht M, Le Roux C, Kerjouan M, Boucher E, AudrainO, Raoul JL. Clubbing and hypertrophic osteoarthropathyin two patients taking long-term bevacizumab for metastaticcolorectal cancer. J Gastrointest Cancer 2011;42:176–8.
[115] Clarke S, Barnsley L, Peters M, Morgan L, van der Wall H.Hypertrophic pulmonary osteoarthropathy without clubbingof the digits. Skeletal Radiol 2001;30:652–5.
[116] Nguyen S, Hojjati M. Review of current therapies forsecondary hypertrophic pulmonary osteoarthropathy. ClinRheumatol 2011;30:7–13.
[117] Angel-Moreno Maroto A, Martínez-Quintana E, Suárez-Castellano L, Pérez-Arellano JL. Painful hypertrophicosteoarthropathy successfully treated with octreotide. Thepathogenetic role of vascular endothelial growth factor(VEGF). Rheumatology 2005;44:1326–7.
[118] Jayakar BA, Abelson AG, Yao Q. Treatment of hypertrophicosteoarthropathy with zoledronic acid: case report and reviewof the literature. Semin Arthritis Rheum 2011;41:291–6.
[119] Bernardo SG, Emer JJ, Burnett ME, Gordon M. Hypertrophicosteoarthropathy presenting as unilateral cellulitis with suc-cessful treatment using pamidronate disodium. J Clin AesthetDermatol 2012;5:37–46.
[120] da Costa FV, de Magalhães Souza Fialho SC, ZimmermannAF, Neves FS, Werner de Castro GR, Pereira IA. Infli-ximab treatment in pachydermoperiostosis: a rare diseasewithout an effective therapeutic option. J Clin Rheumatol2010;16:183–4.
G. Chalès, Professeur des Universités, praticien hospitalier ([email protected]).Service de rhumatologie, Hôpital sud, Centre hospitalier universitaire de Rennes, 16, boulevard de Bulgarie, BP 90347, 35203 Rennescedex 02, France.Université de Rennes 1, Faculté de médecine, 2, avenue du Professeur-Léon-Bernard, CS 34317, 35043 Rennes cedex, France.
A. Rouil, Interne des Hôpitaux.Service de rhumatologie, Hôpital sud, Centre hospitalier universitaire de Rennes, 16, boulevard de Bulgarie, BP 90347, 35203 Rennescedex 02, France.
Cualquier referencia a este artículo debe incluir la mención del artículo: Chalès G, Rouil A. Osteoartropatía hipertrofiante. EMC - Aparatolocomotor 2014;47(1):1-13 [Artículo E – 14-336].
Disponibles en www.em-consulte.com/es
Algoritmos Ilustracionescomplementarias
Videos/Animaciones
Aspectoslegales
Informaciónal paciente
Informacionescomplementarias
Auto-evaluación
Casoclinico
EMC - Aparato locomotor 13




























