Embed Size (px)
Citation preview

ACTA
UNIVERSITATIS
UPSALIENSIS
UPPSALA
2007
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 297
Pancreatic Islet Transplantation
Modifications of Islet Properties to Improve GraftSurvival
SANJA CABRIC
ISSN 1651-6206ISBN 978-91-554-7039-5urn:nbn:se:uu:diva-8333

���������� �������� �� ������ �������� � �� �������� ������� � ������������������������������ ��� ���������������� ��� ������� ������ �������� !� ���! ���"#$% &� �'� ������ & ���� & ('����'� ) ������ & *������+, -'� �������� .��� ��������� � /.����',
��������
0������ /, ���!, (�������� 1���� -����������, *��&������ & 1���� (�������� � 1�����2��&� /�������, 3��� ����������� ���������, ������� ��� � ���� ����� � � ����������� ������� �� �� ������� � � ����� �"!, 4� ��, ������, 1/56 "!78"$8%%98!�:"8%,
����� �'� ���� ������ ������� ����� ����������� '�� ����� � ������ �������� &� ��������� $ ��������, -'� ������� ������ & ����� ����'�� .��' �'� ��;������� &� ������ &���������� ��� � ��'���� ����� ���������� '�� ������� ������� �'� ��������� & �'��������',-'� ������ ��� �&���� �� �'� ����� ��� �'� ����� ���� �� �� ������ � �'� ���� �'�
���&��� ������ '�� ��� �'. � �� ������� �� �'� ����� ���8�������� �&�������������� )15*1�+� .'��' �� �'��������<�� �� �������� �� �������� �������� �� .����� �������� �&������� �� �'� ������, 1���� �����������<��� �� � �����;��� �������� ���� &��'� ��8���� &���� & �'� ���������� ���&�� .'��' ��� ��������� �� ������� �� �'�15*1�,1 �'�� �'����� .� '��� ������� ��� ���������� &� ����������� �'� �&&���� & �'� 15*1�
�� &���������� ����� �����������<���,/������� �'������ & �'� 15*1� ��� ��������� ��������� .��' � �������� ���� &
�������, =� �'���&�� ��������� ���������� ���������� &� �������� �'� ������ ���� ������������, =� ����������� ���� � �������� ������ �'�� � '��' ����� & ���������� ������� & �'� ���������� '����� ���� �� ������ � '��� ������, 3 ����������������' � ������� �'� 15*1� .�� �������� � .'��' ���������� ������������'����� �������� .��� �������� � �'� ����� ���&���, -'�� ���'�;�� ����� �&&������ �������� �'� 15*1� � ��' � � ���� ��� �� ���� �� � �������� ����� ���� &����� �����������, 3 �������� ��'��� & ���'����� ����� � �'� '�����8����� ��������&��� .�� ����������� �� .�� �'� �������� & �'� '����� ������� � ��� �'� �������&����� >?2 �� 2 @ �'��� ������� '��� ������� ���������� &� �'� �����������<���������,-'� ����� & �'� .�� � �'�� �'���� �������� �'�� ������� & �'� ����� ���&��� �� �
���������� ���������� � �������� �'����� �� � �������� &� �������� �'� 15*1�, *�������'� ���� ���'�;��� �� �� ������� � ����� �����������<��� �� ������ �'�����&���� & �'� ���������� ������, ���������� ������� ����� ��������� �� ����&����.��� ���� ����� ����������� � ��� �&&������ �������� �� ������� �'� ����� & �������.'�� �������� �� �� �����,
� ������ ��������� ����� ������������ ������ & A����'��� �������� ���������15*1�� '������ '������ ��.�' &����� ���������
����� ������ � ���� �� � !����� "������ ��� �������� #����� $%�� ��%����%��� � ������� ���� ����� �&'()*+) ������� �� � �
B /��� 0����� ���!
1//6 $4%$84��41/56 "!78"$8%%98!�:"8%��#�#��#��#����87::: )'���#CC��,��,��C������D��E��#�#��#��#����87:::+

To my parents


List of papers
This thesis is based on the following papers, which will be referred to in the text by their Roman numerals: I Cabric S, Elgue G, Nilsson B, Korsgren O, Schmidt P. Cell Trans-
plantation 2006; 15 (8-9): 759-671. Adenovirus-mediated expres-sion of the anticoagulant hirudin in human islets: A tool to make the islets biocompatible to blood.
II Cabric S, Eich T, Sanchez J, Nilsson B, Korsgren O, Larsson R. Submitted manuscript. A new method for incorporating functional heparin onto the surface of islets of Langerhans.
III Cabric S, Sanchez J, Eich T, Lundgren T, Foss A, Felldin M, Källen
R, Salmela K, Tibell A, Tufveson G, Larsson R, Korsgren O, Nils-son B. Diabetes2 56: 2008-2015, 2007. Islet surface heparinization prevents the instant blood-mediated inflammatory reaction in islet transplantation.
IV Cabric S, Sanchez J, Johansson U, Larsson R, Nilsson B, Korsgren O, Magnusson PU. Manuscript. Anchoring of growth factors to sur-face-immobilized heparin on pancreatic islets; Implications for stimulating islet angiogenesis.
Reprints were made with permission from the publishers
1Copyright © 2006 Cognizant Communication Corporation 2Copyright © 2007 American Diabetes Association


CONTENTS
INTRODUCTION ........................................................................................13 Type 1 diabetes mellitus...........................................................................13
Transplantation as a cure for type 1 diabetes.......................................14 Hemostasis ...............................................................................................15
Primary hemostasis – formation of the primary platelet plug..............16 Secondary hemostasis – formation of fibrin ........................................17 Tertiary hemostasis – formation of plasmin ........................................20 Endogenous inhibitors of hemostasis ..................................................20 Anticoagulant drugs.............................................................................21 Heparin-coated surfaces – immobilized heparin .................................23
The IBMIR ...............................................................................................23 Angiogenesis ............................................................................................24
Endothelial cell growth factors ............................................................25 Revascularization of transplanted islets...............................................26
AIMS OF THE STUDY ...............................................................................27 General aims.............................................................................................27 Specific aims ............................................................................................27
Paper I..................................................................................................27 Paper II ................................................................................................27 Paper III ...............................................................................................27 Paper IV...............................................................................................28
STUDY DESIGN AND METHODS............................................................29 Ethics........................................................................................................29 Preparation and culture of islets and ECs.................................................29
Human islets (Papers I-IV) ..................................................................29 Adult porcine islets (Paper III) ............................................................29 Rodent islets (Paper III).......................................................................30 ECs (Paper IV).....................................................................................30
Modification of islets ...............................................................................30 Adenoviral vectors and transduction procedure (Paper I) ...................30 Heparinization procedure (Paper II-IV)...............................................30 EC coating of islets of Langerhans (Paper IV) ....................................31
Detection/analysis of modification procedures ........................................31 Hirudin detection (Paper I) ..................................................................31

Quartz crystal microbalance with dissipation monitoring (QCM-D) (Paper II, IV)........................................................................................31 Large particle flow cytometry (Paper II) .............................................31 Confocal microscopy (Papers II-IV)....................................................32
In vitro blood experimental models..........................................................32 The Chandler loop model (Paper II) ....................................................32 Tubing loops as a model (Paper III) ....................................................32 Considerations of generated data (Papers II-III)..................................33 Clotting time (ReoRox 4) (Paper I) .....................................................33
Animals and transplantation procedures ..................................................33 Transplantation of human islets to athymic (nu/nu) mice (Paper I) ....33 Syngeneic islet transplantation to alloxan diabetic mice (Paper III) ...34 Intraportal allotransplantation of adult porcine islets (Paper III).........34
Analytical procedures...............................................................................34 Blood and plasma analysis (Paper II-III) .............................................34 Immunohistochemistry (Papers I, III)..................................................34
Statistics ...................................................................................................35
RESULTS AND DISCUSSION ...................................................................36 Strategies to prevent the IBMIR (Papers I-III).........................................36 Strategies to induce revascularization (Paper IV) ....................................36 Paper I: Adenovirus-Mediated Expression of the Anti-coagulant Hirudin in Human Islets: A Tool to Make the Islets Biocompatible to Blood ......37
Expression of secretory hirudin in human islets after adenoviral transduction .........................................................................................37 Effects of islet-derived hirudin on coagulation in human blood..........38 Alternatives to adenoviral transduction ...............................................39
Paper II: A New Method for Incorporating Functional Heparin onto the Surface of Islets of Langerhans ................................................................39
Analysis and visualization of the heparinization procedure ................39 Functional capacity of bound heparin..................................................40
Paper III: Islet Surface Heparinization Prevents the Instant Blood-Mediated Inflammatory Reaction in Islet Transplantation.......................40
Islet functionality after heparinization procedure ................................40 Effect of islet heparinization in the in vitro blood loop model ............41 Intraportal allotransplantation of API ..................................................41 Heparinization of other cells/tissues ....................................................42
Paper IV: Anchoring of Growth Factors to Surface-Immobilized Heparin on Pancreatic Islets; Implications for Stimulating Islet Angiogenesis .....42
Binding of growth factors to immobilized heparin conjugate .............42 ECs on heparinized surfaces................................................................43 Coating of pancreatic islets by ECs .....................................................44 Syngenic islet transplantation into C57BL/6J mice.............................44

CONCLUSIONS ..........................................................................................46 Paper I ......................................................................................................46 Paper II .....................................................................................................46 Paper III....................................................................................................46 Paper IV ...................................................................................................46
ACKNOWLEDGMENTS ............................................................................47
REFERENCES .............................................................................................50


ABBREVIATIONS
APC Activated protein C API Adult porcine islets AT CHS CHS-CT D DTI EC ECM ELISA F FGF FGFR fq HIT HLA IBMIR IEQ iFVIIa IL LMWH MCP-1 PAF PAI PF4 PS PVC QCM SNL T1DM TAFI TAT TF TFP TFPI tPA
Antithrombin Corline heparin surface CHS on cells and tissue Damping Direct thrombin inhibitor Endothelial cell Extracellular matrix Enzyme-linked immunosorbent assay Coagulation factor Fibroblast growth factor FGF receptor Frequency Heparin-induced thrombocytopenia Human leukocyte antigen Instant blood mediated inflammatory reaction Islet equivalent Inactivated recombinant coagulation factor VIIa Interleukin Low molecular weight heparin Monocyte chemotactic protein 1 Platelet activating factor Plasminogen activator inhibitor Platelet factor 4 Phosphatidylserine Polyvinyl chloride Quartz crystal microbalance with dissipation Sulfo-NHS-LC-biotin Type 1 diabetes mellitus Thrombin activatable fibrinolysis inhibitor Thrombin-antithrombin complex Tissue factor Tetrafluorophenyl polyethylene oxide-biotin Tissue factor pathway inhibitor Tissue plasminogen activator

UFH VEGF VEGFR vWf
Unfractionated heparin Vascular endothelial growth factor VEGF receptor Von Willebrand factor

13
INTRODUCTION
The significant advances made in recent years in the field of pancreatic islet transplantation have helped to make the procedure a viable alternative for patients with brittle type 1 diabetes. However, the application of this ap-proach is still hampered by the fact that only a limited number of pancreata are available for transplantation and that a continuing need exists to use pan-creata from multiple donors in order for patients to achieve normoglycemia.
The apparent loss of islets that occurs during the early post-transplantation period has been extensively analyzed in an attempt to iden-tify the mechanism(s) involved and to develop strategies to improve the transplantation procedure. A number of features, including coagulation acti-vation and subsequent insulin dumping, have consistently been observed after clinical islet transplantation in patients receiving islets via infusion into the portal vein.
These undesirable features are characteristic of the instant blood-mediated inflammatory reaction (IBMIR), which is a likely cause of both the loss of transplanted tissue and the intraportal thrombosis that can be associated with clinical islet transplantation. Islet revascularization, which is partially influ-enced by the severity of the IBMIR, is a subsequent step that is critical for the long-term survival of the transplanted graft.
This thesis explores strategies for circumventing the IBMIR and facilitat-ing the revascularization process as a means of improving islet engraftment; the successful development of such strategies should ultimately increase the number of patients who are able to benefit from this therapy.
Type 1 diabetes mellitus Type 1 diabetes mellitus (T1DM) is a chronic disorder that results from autoimmune destruction of the insulin-producing pancreatic ß-cells, leading to uncontrolled high blood glucose levels (1). The prevalence of diabetes varies considerably among various ethnic and regional populations, but it is expected to affect 5 million worldwide by the year 2010 and 30 million by 2030 (2, 3). The differences in incidence can largely be explained by the prevailing susceptibility genes for T1DM in racially distinct populations, but diet and other environmental factors are probably also important (4, 5).

14
T1DM is an autoimmune disease that typically occurs at an early stage in life and is characterized by a destruction of the insulin-producing �-cells in the pancreatic islets. Several �-cell autoantigens, macrophages, dendritic cells, and B- and T- lymphocytes have been shown to be involved in the pathogenesis of T1DM. The process begins with an infiltration of inflamma-tory cells into the islets, resulting in targeted destruction of the insulin-producing �-cells (1, 6, 7). Subsequently, the infiltrating cells vanish, and what remains are fibrotic islets that lack capacity to produce insulin. T1DM usually progresses for several years before it becomes clinically apparent (8).
The current treatment for the disease is daily injections of insulin, involv-ing intensive monitoring of blood glucose levels. Over- or under-treatment can lead to life-threatening states of either insulin coma or hyperglycemic coma. Despite strict glucose control and considerable improvement in diabe-tes care in recent decades, the disease is progressive and may lead to numer-ous secondary complications, including retinopathy, vasculopathy, neph-ropathy and neuropathy. Thus, insulin treatment should be considered a treat-ment and not a cure for T1DM.
Transplantation as a cure for type 1 diabetes The only current way to consistently achieve physiological control of blood glucose levels is transplantation of whole pancreas or isolated pancreatic islets. Whole organ pancreas transplantation performed in combination with kidney transplantation, leads to sustained normoglyceamia and insulin inde-pendence in the vast majority of recipients, with graft survival as high as 80% at 5 years (9, 10). However, whole pancreas transplantation requires major surgery and is associated with relatively high morbidity and mortality (11). In contrast, islet cell transplantation is a much less invasive procedure, in which the islets are injected percutaneously into the liver via the portal vein, with a much smaller risk and lower levels of morbidity (12). Islet trans-plantation can therefore be applied at an earlier stage of the disease, as a means of avoiding secondary complications, and can also be offered to pa-tients who would otherwise not sustain whole pancreas transplantation.
However, the apparent advantages of restoring physiological blood glu-cose metabolism have to be balanced against life-long treatment with immu-nosuppressive drugs, which increase the risk for infection and certain forms of tumor and are associated with organ toxicity. For this reason, transplant candidates are currently selected from among those patients with unstable T1DM or those who have already received a donated kidney (and are on an immunosuppressive regimen in any case).
Despite the fact that islet transplantation offers several advantages over pancreas transplantation, only approximately 10% of patients transplanted with allogeneic islets obtain prolonged insulin independence (13, 14). The main reason has been the low graft survival rates associated with islet trans-

15
plantation. However, several breakthroughs during the past years have led to a dramatic increase in the number of islet transplantations worldwide. Never-theless, the procedure in most cases requires the use of islets isolated from several donor pancreata to produce insulin independence in the patient (14, 15).
The reason for this obvious difference in successful outcome between whole pancreas and islet transplantation cannot be explained by insufficient islet mass alone. The fate of the islet graft is determined by many factors, some of which are relevant to whole pancreas grafts, (i.e., acute cellular re-jection, recurrence of disease and ischemia), but also by others that appear to be specific to the nonvascularized islet transplants, including low transplant cell mass and reduced viability, as well as nonspecific inflammation at the site of the implant (16). Islet transplantation is performed by injecting the islets into the portal vein, thus embolizing them into the liver. In this respect, islets can be considered to be foreign particles that expose constituents in-compatible with blood to the cascade system, such as collagen and matrix molecules (17, 18). Moreover, whole pancreas transplants have an intact endogenous vascular system in which microvascular blood flow is directly reinstated after the transplantation procedure, thereby achieving an immedi-ate establishment of blood perfusion. The islets, on the other hand, become avascular after the preparatory collagenase digestion prior to transplantation. In this context, an advantage of intraportal transplantation is its presumed association with faster induction of angiogenesis of dispersed islets, when compared to islets transplanted in clusters in a less-vascularized environment (e.g., under the kidney capsule or the skin) (19).
Previous findings have demonstrated that the islets induce an acute in-flammatory reaction when they come into contact with whole blood in vitro. This reaction, the IBMIR, is a critical key factor in the notable failure of islets from a single donor to achieve insulin independence after implantation into the portal vein (20). Recent studies have shown that islets produce and secrete tissue factor (TF), the physiological trigger of the extrinsic pathway of coagulation (21). These findings have provided further insight into the nature of the IBMIR and offer several new strategies for improving the out-come of clinical islet transplantation.
Hemostasis Hemostasis is the spontaneous arrest of bleeding from an injured blood ves-sel. Normal hemostasis is dependent upon a complex interaction of plasma coagulation and fibrinolytic proteins, platelets and the blood vasculature. Vascular endothelial cells are the key mediators of hemostasis because of their role in regulating both blood clotting and bleeding. Platelets and clot-

16
ting factors circulating in the blood interact with damaged endothelial cells (ECs) to ultimately form a fibrin-platelet clot and promote hemostasis (22).
Hemostasis can be divided into three stages: primary hemostasis, which is defined as the formation of the platelet plug; secondary hemostasis, defined as the formation of fibrin through the coagulation cascade; and tertiary he-mostasis, defined as the formation of plasmin for breakdown of the clot. One should bear in mind, however, that in vivo all three processes occur simulta-neously and not sequentially.
Primary hemostasis – formation of the primary platelet plug Primary hemostasis is defined as the formation of the primary platelet plug, which is created when platelets adhere to injured surfaces and aggregate to reduce or abolish blood loss. Platelet response to vascular injury includes adhesion, activation, aggregation and activation of the coagulation cascade (23, 24).
Platelet adhesion Circulating platelets do not adhere to normal endothelium or to each other, since the normal endothelium prevents hemostasis by providing a physical barrier. In addition, the endothelium synthesizes antithrombotic cell surface components such as nitric oxide, prostacyclin, thrombomodulin, heparin-like glycosaminoglycan (i.e., heparan sulfate), tissue plasminogen activator, and adenosine triphosphatase. Also, the electrical charges of endothelial mem-brane proteins repel circulatory coagulation proteins (25). In order for hemo-stasis to occur, platelets must slow down to stop at sites of vascular damage and adhere to exposed collagen and von Willebrand factor (vWf) on the subendothelial matrix (26). The vWf, which binds to the adhesive platelet membrane receptor, glycoprotein Ib-IX (GPIb-IX), mediates the initial adhe-sion of platelets to the vascular subendothelium and is released by both the ECs and the platelets (27).
Platelet activation Platelets adhere to collagen exposed by subendothelium and become acti-vated. Once activated platelets alter their shape and undergo a conforma-tional change in the platelet surface glycoprotein receptor GP IIb/IIIa, bind-ing sites for fibrinogen, fibronectin, vitronection and vWf become exposed. This results in further spreading and allow for platelet aggregation (28). Upon activation, the platelets release factors to promote vasoconstriction (e.g., serotonin, thromboxane A2), which retards extravascular blood loss, slows local blood flow and enhances the adherence of platelets to exposed subendothelial surfaces and the activation of the coagulation process (29, 30). In addition, after platelet activation, ADP, a platelet agonist, is released. ADP is predicted to be the most prominent amplifier of initial platelet activa-

17
tion (31). This activation is enhanced by the generation of thrombin through the coagulation cascade (32). Furthermore, platelets secrete platelet activat-ing factor (PAF), which is a potent platelet-aggregating agonist (33).
Platelet aggregation The process of platelet aggregation is the final phase of primary hemostasis and is mainly mediated by fibrinogen, which binds to GP IIb/IIIa on adjacent platelets (28). This aggregation leads to the formation of the primary platelet plug, which must be stabilized by the formation of fibrin.
In addition, phosphatidylserine (PS), a phospholipid, is translocated to the outer layer of the platelet membrane, providing binding sites for activated coagulation factors (34, 35). Membrane PS is the major determinant of the binding site for FX-activating and prothrombinase complexes at the surface of human platelets (36).
Moreover, glycoproteins such as P-selectin are upregulated, mediating platelet binding to neutrophils and monocytes (37). Leukocytes are able to attach to platelets, which are immobilized on the subendothelium, in a P-selectin-dependent manner (38).
Secondary hemostasis – formation of fibrin Secondary hemostasis is defined as the formation of fibrin through the co-agulation cascade. This process involves circulating coagulation factors, which act as enzymes, cofactors, calcium, and platelets, which provide a source of phospholipids and a binding surface upon which the coagulation cascade can proceed. Activation of one of the clotting factors sequentially activates another in a series of reactions, ultimately yielding cross-linked fibrin. The classical coagulation cascade is traditionally separated into three pathways: TF pathway (extrinsic), contact activation pathway (intrinsic) and common pathway.
Classical cascade model of coagulation
TF pathway The TF pathway, the main pathway for the initiation of coagulation, involves the complex of TF and FVIIa (39), which activates FX in the presence of calcium (40-42). Under normal conditions, TF is not exposed to blood, but it is highly expressed on a variety of extravascular cells and also in richly vas-cularized tissues (43, 44). However, TF expression can be induced in cells that are in contact with blood (e.g., ECs, monocytes) after cytokine- or gram-negative bacterial endotoxin stimulation, in which ECs and monocytes are converted to a procoagulant phenotype in response to the cell-surface ex-pression of TF (41). Circulating TF has been shown to exist in the blood in the form of TF-bearing microparticles, which are either active or can be acti-

18
vated (45). Increased levels of plasma TF have been noted in patients with myocardial infarction, unstable angina or diabetes (46-48).
Contact activation pathway The contact activation pathway involves high molecular weight kininogen, prekallikerin, and FXII, FXI, FIX and FVIII and is initiated by foreign nega-tively charged surfaces such as collagen. For a long time the role of FXII in initiating coagulation in vivo has been questioned, since people and animals with FXII deficiency do not show signs of hemorrhage. Recent studies have shown that FXII is required for the formation and stabilization of platelet-rich thrombi, and this observation suggests that in vivo FXII-mediated acti-vation of coagulation is involved (49). FVIIIa acts as a cofactor, together with calcium and platelet phospholipids, for the FIXa-mediated activation of FX.
Common pathway The extrinsic and intrinsic pathways converge at the activation of FX, which then activates prothrombin, leading to the production of thrombin, which converts fibrinogen to fibrin in the presence of FVa and calcium. Both path-ways follow the same course after the activation of FX and this final path-way has been named common pathway.
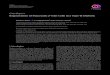
Cell-based model of coagulation The concept of coagulation as a cascade system describes the biochemical interactions of the coagulation factors but does not completely explain the mechanisms leading to hemostasis in vivo. Therefore, a cell-based model was proposed, according to which coagulation takes place on different cell surfaces in three overlapping steps: initiation, amplification and propagation (42) (Fig. 1).
Initiation of coagulation An injury in the vessel wall allows plasma to come in contact with TF-bearing extravascular cells. The initial event is the binding of plasma FVII to TF, forming an FVIIa/TF complex that activates small amounts of FX and FIX. FXa is rapidly inhibited by tissue factor pathway inhibitor (TFPI) or antithrombin (AT) if it leaves the protected environment of the cell surface. However, FXa that remains on the cell surface can combine with plasma FVa to produce small amounts of thrombin (50, 51). In contrast, FIXa can move from the TF-bearing cell on which it was formed to a nearby platelet or to another cell surface, since it is not inhibited by TFPI and it is inhibited much more slowly than FXa by AT (Fig. 1A).
19
Amplification of coagulation Amplification of coagulation occurs as the process moves from the TF-bearing cell to the platelet, which provides an optimal surface for the genera-tion of large amounts of thrombin (51). Small amounts of thrombin gener-ated by the extrinsic pathway (on TF-bearing cells) amplify the signal by increasing platelet adhesion, fully activating the platelets (32) and activating FV, FVIII and FXI (52-54) (Fig. 1B).
VIII
VIIIVIIaTFPI
TF
Tissue factor bearing cell
VIIa
TF
Tissue factor bearing cell
TFVIIa
VIIaIX
IXa
XXaVa
II
IIa
TF
Tissue factor bearing cell
TF
VIIa
XaVa
II
IIa
PlateletV
V
Va
XI
XIa Activatedplatelet
Va
XIa
VIII/vWF VIIIa + free vWF
Activatedplatelet
Va
XIa
IXa
IXaIX
IX
Xa
XII
IIa
XaVIIIa
VIIIa
A
B
C
VIII
VIIIVIIaTFPI
TF
Tissue factor bearing cell
VIIa
TF
Tissue factor bearing cell
TFVIIa
VIIaIX
IXa
XXaVa
II
IIa
TF
Tissue factor bearing cell
TF
VIIa
XaVa
II
IIa
PlateletV
V
Va
XI
XIa Activatedplatelet
Va
XIa
VIII/vWF VIIIa + free vWF
Activatedplatelet
Va
XIa
IXa
IXaIX
IX
Xa
XII
IIa
XaVIIIa
VIIIa
A
B
C
Figure 1. The cell-based model of hemostasis. A. Initiation, B. Amplification, C. Propagation. Figure modified from M. Hoffman, Blood Reviews 2003.
Propagation of coagulation During the propagation phase, the prothrombinase and tenase complexes (see below) are assembled on the platelet surface, where large-scale throm-bin generation can take place (55) (Fig. 1C). Platelets express high-affinity binding sites for FIX, FX and FXI (56-58). Since FIXa can diffuse to the platelet surface from its activation site on the TF-bearing cell without being rapidly inhibited by the AT, it forms an FVIIIa/FIXa tenase complex to-gether with calcium and PS on the platelets (59). In addition, plasma FXI can bind to activated platelets, facilitating its activation by thrombin (53). The FXIa can then provide additional FIXa directly on the platelet surface. The FIXa/FVIIIa complexes activate FX on the platelet surface where the result-ing FXa can move directly into a complex with its cofactor, FVa. The plate-let surface FXa/FVa complexes, which together with calcium and PS form a

20
prothrombinase complex (60), produce a burst of thrombin, which in turn cleaves fibrinogen to fibrin monomers. The monomers are then polymerized to form the fibrin polymers that are necessary to form a fibrin clot. Thrombin also activates FXIII, which together with calcium serves to stabilize fibrin by forming intrachain covalent bonds.
Tertiary hemostasis – formation of plasmin Tertiary hemostasis is defined as the formation of plasmin, which is the main enzyme responsible for the dissolution of the fibrin matrix of thrombi.
At the same time that the coagulation cascade is activated, tissue plasmi-nogen activator (tPA) is released from the endothelial cells. tPA binds to plasminogen within the clot, converting it to plasmin. In the absence of fi-brin, tPA is an inefficient activator of plasminogen, thus limiting the produc-tion of plasmin to the site of thrombus formation; however, once it is bound to fibrin, the activation is greatly enhanced (61). In addition, studies have shown that activated FXII can activate plasminogen into plasmin directly (62), a process that is potentiated by negatively charged surfaces (63).
Endogenous inhibitors of hemostasis Inhibitors are important, as they serve to limit hemostasis to the site of vessel injury and to prevent thrombotic occlusion in adjacent normal areas of the vasculature, a situation that could lead to pathological thrombosis.
In primary hemostasis, the naturally occurring inhibitors of platelet func-tion are prostacyclin and nitric oxide, which are released by ECs (25). In secondary hemostasis, the most important natural anticoagulant is AT. AT inhibits many activated coagulation proteins, including thrombin, FIXa, FXa, FXIa and FXIIa, but these reactions are slow in the absence of heparin (64). With heparin present, the rate of inhibition is accelerated about 1000-fold; this acceleration is produced in vivo by degranulated mast cells (65) or basophils (66) and heparan sulfate (67), a glycosaminglycan that, like hepa-rin, contains a specific AT-binding pentasacharide sequence (68, 69). Most of the heparan sulfate is located on the abluminal surface of the endothelium and is exposed only when the vessel lining is damaged (70). Nevertheless, the small amounts of proteoglycan located on the luminal surface may help to render the intact endothelium nonthrombogenic. Another key inhibitor is TFPI (71), which rapidly inhibits the TF pathway, allowing this pathway to generate only small amounts of thrombin. Thrombin is another anticoagu-lant. Once bound to thrombomodulin on the surface of endothelial cells, it undergoes a conformational change that converts it from a procoagulant enzyme to a potent activator of protein C, a vitamin K-dependent protein. Activated protein C (APC) serves as an anticoagulant by inactivating FVa and FVIIIa, thereby attenuating thrombin generation (72). Protein S, another

21
vitamin K-dependent protein synthesized in ECs, megakarocytes and hepa-tocytes, facilitates the action of APC (72). In tertiary hemostasis there are a variety of inhibitors of fibrinolysis, including thrombin-activatable fibrinoly-sis inhibitor (TAFI), which prevents the binding of plasminogen to fibrin, thus inhibiting its conversion to plasmin (73). Additional inhibitors of fibri-nolysis include alpha2-antiplasmin and plasminogen activator inhibitor (PAI). Alpha2-antiplasmin binds to free plasmin and causes its removal by the monocyte-macrophage system, thus preventing widespread fibrinolysis. Inhibitors of tPA (PAI-1 and PAI-2), on the other hand, are released by ECs and limit plasmin generation by binding to tPA (74).
Anticoagulant drugs
Established anticoagulants – warfarin and heparins The currently available anticoagulants, vitamin K antagonists (warfarin) and the heparins, both unfractionated heparin (UFH) and low-molecular weight heparin (LMWH), belong to a group of indirect thrombin inhibitors, which block the generation and action of thrombin either by activating naturally occurring thrombin inhibitors or by inhibiting specific factors in the coagula-tion system that subsequently affect thrombin generation or activity. War-farin attenuates thrombin generation by modulating the vitamin K cycle, thereby inhibiting posttranslational modification of the vitamin K-dependent clotting factors (i.e., prothrombin, FVII, FIX and FX). As a result, vitamin K-dependent clotting factors exhibit reduced capacity to bind calcium and are no longer functional.
Warfarin can be administrated orally, making it the agent of choice for long-term management of thromboembolic conditions, but frequent coagula-tion monitoring is necessary because of its unpredictable anticoagulant ef-fect, as the result of food and drug interactions and its narrow therapeutic window (75).
Heparins act as anticoagulants by activating AT and accelerating the rate at which it inhibits thrombin and activated FX and can only be administrated parenterally (64, 76). UFH is a mixture of sulphated polysaccharides, where the molecular weight varies between 1,800 and 30,000 Da. UFH, like war-farin, is unpredictable because it binds to plasma proteins, the levels of which vary between individuals; hence, coagulation monitoring is also re-quired with UFH, but not with LMWH because of its relatively low level of plasma protein binding (76). Moreover, UFH is neutralized by platelet factor 4 (PF4) released from activated platelets, thereby reducing the UFH activity. Furthermore, antibodies directed against the UFH/PF4 complex can trigger heparin-induced thrombocytopenia (HIT) (77, 78). LMWHs are produced by controlled depolymerization of heparin with an average molecular weight of 5000 Da. They have a more specific inhibition of primarily factor Xa. Al-

22
though LMWH overcomes many of the pharmacodynamic limitations of UFH, neither LMWH nor UFH completely inactivates thrombin once it is bound to fibrin (79).
New anticoagulants The limitations of the currently available anticoagulants, including an ele-vated risk of bleeding and the inconvenience posed by the need for routine coagulation monitoring and/or parenteral administration, have led to the development of new, improved agents designed to target specific coagula-tion enzymes or steps in the coagulation pathway (e.g., FXa inhibitors, FIXa inhibitors, modulators of protein C pathway - inactivating FVa and FVIIIa, inhibitors of the FVIIa/TF pathway, direct thrombin inhibitors) (Fig. 2). Di-rect thrombin inhibitors (DTIs) such as hirudin and melagatran achieve their effect by directly binding to the thrombin molecule, thereby preventing its interaction with different substrates (80, 81). Also, they are capable of inhib-iting both fibrin-bound and fluid-phase thrombin. Furthermore, they exhibit little binding to plasma proteins (81). Hirudin, for instance, forms an irre-versible tight complex with thrombin, resulting in potent and specific inhibi-tion (82), but it is therefore also associated with an increased risk of bleed-ing. One common drawback of these DTIs is that there are no available anti-dotes. Unfortunately, manufacturer of melagatran, AstraZeneca, announced in 2006 that it would not be marketed because of hepatotoxicity that oc-curred during clinical trials (83).
Current anticoagulants are highly effective and relatively safe; however, current strategies for long-term anticoagulant therapy are limited by the re-quirement for parenteral administration (e.g., LMWHs) and frequent coagu-lation monitoring, as well as their variable dose requirements (e.g., UFH) and unpredictable effect on coagulation (e.g., vitamin K antagonists). There-fore, successful future anticoagulant therapies should ideally be oral, be dosed once daily and not require dose adjustment, and exhibit few or no interactions and minimal long-term side effects. However, the development of a single agent with all the desired properties has proved to be a consider-able challenge.

23
Step in coagulation Coagulation pathway Anticoagulant
Initiation TF/VIIa
X IX
IXa VIIIa
Xa Va
Thrombin
Fibrin
Thrombin generation
Thrombin activity
Tissue factor pathwayinhibitors: rTFPI; FVIIa inhibitors; TFmAb
FIXa inhibitors:FIXa inhibitor; FIXamAb
Protein C pathway:APC; thrombomodulin
FXa inhibitors:FXa inhibitor; pentasacharide
Thrombin inhibitors:Hirudin; Melagatran
Step in coagulation Coagulation pathway Anticoagulant
Initiation TF/VIIa
X IX
IXa VIIIa
Xa Va
Thrombin
Fibrin
Thrombin generation
Thrombin activity
Tissue factor pathwayinhibitors: rTFPI; FVIIa inhibitors; TFmAb
FIXa inhibitors:FIXa inhibitor; FIXamAb
Protein C pathway:APC; thrombomodulin
FXa inhibitors:FXa inhibitor; pentasacharide
Thrombin inhibitors:Hirudin; Melagatran
Figure 2. Targeting the coagulation system: new anticoagulants. Figure modified from J. Hirsh, Blood 2005.
Heparin-coated surfaces – immobilized heparin In the past few decades, many different methods of heparin immobilization have been developed for use in coating artificial surfaces with heparin in order to create a substrate that mimics the endothelium with regard to blood compatibility. In the studies described here we used the Corline Heparin Surface (CHS), involving a macromolecular heparin conjugate that is com-posed of a carrier chain, a linear polyamine with a molecular weight of 55,000 Da, to which a large number of heparin molecules (typically 70) have been covalently linked (84). The specificity of the covalent linkages ensures that the AT-binding pentasacharide sequence of heparin is left intact. This surface is stable, with no leakage of heparin into the blood (85). In in vitro experiments, this surface has been shown to be effective in inhibiting both complement and coagulation activation and in reducing platelet activation and adhesion (86-90). In a clinical study on coronary bypass patients, it was demonstrated that activation of coagulation, which normally occurs during bypass despite systemic administration of heparin, could be reduced by ap-plying the CHS to the complete bypass system (85, 91).
The IBMIR Previous studies by Bennet et al. (20, 92) have described a thrombotic reac-tion, the instant blood-mediated inflammatory reaction (IBMIR), that is trig-gered when islets are exposed to human blood. The IBMIR is characterized by an activation of the coagulation and complement systems and a rapid binding of activated platelets to the islet surface. The effects of this reaction

24
terminate rapidly (within 15 minutes) with the islets becoming surrounded by clots and infiltrated with leukocytes (20). This reaction has been observed in both an experimental in vitro model and an allogeneic in vivo pig model (20, 92). The IBMIR has been described as a key factor in both islet loss after islet transplantation and the intraportal thrombosis associated with this procedure (21). In addition to the direct damage caused by the IBMIR, the ongoing inflammation also enhances antigen presentation, evoking a specific immune response involving B and T cells. Studies demonstrating that islets express and synthesize TF, the physiological activator of coagulation (21) have led to a better understanding of how the clotting cascade system is trig-gered in IBMIR and offer new strategies to prevent the IBMIR.
Angiogenesis During development, new blood vessels originate from endothelial precursor cells, angioblasts, by a process called vasculogenesis (93), or from pre-existing blood vessels by angiogenesis (94).
In adult, angiogenesis is a fundamental process in wound healing and re-production. Under these conditions, angiogenesis is highly regulated: It is turned on for a short period and then completely inhibited. Unregulated an-giogenesis may result in a variety of pathological conditions, such as dia-betic retinopathy, rheumatoid arthritis and tumor growth (94).
The most extensively studied mechanism of angiogenesis is angiogenic sprouting (95, 96). In sprouting angiogenesis, ECs that are integrated in the vessel wall digest the surrounding basal lamina and start to migrate toward a gradient of chemotactic substances in response to a stimulus provided by growth factors, among which vascular endothelial growth factor (VEGF) is predominant (97, 98). The initial vasodilation of existing vessels, largely in response to nitric oxide (99), is accompanied by an increase in permeability and the degradation of the surrounding extracellular matrix (ECM), which allows activated and proliferating ECs to migrate and form a lumen. ECM is degraded by various proteases, including matrix metalloproteases, which are released during inflammation and as a result of VEGF receptor (VEGFR) activation (100). Following the proteolytic degradation of the ECM, ECs begin to migrate through the degraded matrix toward the source of the VEGF. This migratory response is followed by the proliferating ECs in re-sponse to stimulation by a variety of growth factors, some of which are re-leased from the degraded ECM (98). The migrating ECs organize into a solid cord which subsequently acquires a lumen (101). Further stabilization of the new capillaries requires the recruitment of pericytes and smooth muscle cells, which is regulated by platelet-derived growth factor produced by the ECs (102). Once new vessels have assembled, the ECs become remarkably

25
resistant to exogenous factors and are quiescent, persisting for years without survival factors.
Although the level of proliferation of ECs in the adult is very low when compared to many other cell types in the body, with a turnover time that can exceed 1000 days, these cells are clearly essential for life and reproduction. Tissue development, wound healing, and ovulation are all accompanied by angiogenesis. Because angiogenesis is crucial for so many important physio-logical functions, it must be carefully controlled if a healthy state is to be maintained.
Endothelial cell growth factors VEGF and fibroblast growth factor (FGF) are angiogenic growth factors that induce the cellular responses necessary for the formation of new capillaries: digestion of the basement membrane and migration, proliferation, and differ-entiation of the ECs (103).
The VEGF family consists of a group of glycoproteins, VEGF-A (or VEGF), -B, -C, -D,-E and placental growth factor. VEGF-A plays an impor-tant role in angiogenesis. Alternative exon splicing of the VEGF-A gene produces a range of VEGF-A isoforms differing in total amino acid number, with the predominant heparin-binding form being the 165-amino acid mole-cule, which is secreted by a broad variety cells (104). The FGF family con-sist today of 23 heparin-binding polypeptides (105), of which FGF-2 is most extensively studied; this growth factor is expressed at low levels in almost all organs and tissues (106).
VEGFs and FGFs mediate their biological effects via specific binding to cell surface-expressed receptors equipped with tyrosine kinase activity, trig-gering an intracellular signal cascade that ultimately results in the prolifera-tion and migration of ECs. The biologic effects of VEGF-A are mediated by two high-affinity tyrosine kinase receptors that are predominantly expressed on ECs: VEGFR-1 (Flt-1) and VEGFR-2 (Flk-1) (107). In contrast, FGF-2 mainly exerts its effects through five tyrosine kinase FGF-receptors (FGF-R) 1-5, exhibiting the highest affinity for FGFR-1 (108).
In order to become stabilized, these receptors and growth factors must in-teract with glycosaminoglycans such as heparan sulfate proteoglycans, which are present on the surface of most cells and in the ECM (109, 110). It has also been suggested that the binding of growth factors to heparan sulfate proteoglycans results in protection of growth factors from inactivation in the extracellular environment (111), and it may also serve as a reservoir of growth factors (112) that can be released from the cell surface in a regulated manner (e.g., by the action of heparanases) (113).

26
Revascularization of transplanted islets Endogenous pancreatic islets have a dense glomerular-like network of capil-laries (114). During the process of islet isolation, this supporting capillary network is disrupted by collagenase digestion. Transplanted islets can par-tially compensate for the resulting reduction in the number of microvessels through the diffusion of oxygen and nutrients from surrounding blood (115); however, in order to reestablish an adequate microcirculation that supplies sufficient nutritional blood supply, transplanted islets must undergo a proc-ess of angiogenesis and revascularization.
Experimental studies have shown that intra-graft blood vessels can be seen as early as 3 days after transplantation and that the vascularization of the islet grafts is complete within 10-14 days, yielding a glomerular network of capillaries similar to that observed in endogenous islets (116, 117). It is likely that islet grafts become revascularized from recipient vessels and ECs (118); however, the exact origin of the newly formed blood vessels within the islet grafts has yet not been completely clarified. Recent studies have suggested that both donor and recipient ECs contribute to the revasculariza-tion process (119, 120). In line with this proposal, soluble factors released from the devascularized and hypoxic islets are likely to be of major impor-tance in this context and several reports have noted that VEGF (121-123) and FGF (121, 124, 125), in particular, contribute significantly to the process of revascularization of transplanted islets. In these studies, a variety of inter-ventions have been investigated, either singly or in combination, in an at-tempt to improve islet revascularization: transfection of islets with VEGF (122), exposure of islets to VEGF (121) or FGF (121, 124), and exposure of islets before transplantation to a brief period of hypoxia in order to induce expression of VEGF (126, 127).
Despite the intense level of research that has been conducted in the islet transplantation field, many questions remain unanswered concerning the process of islet revascularization. It is therefore of great importance to de-velop novel strategies to accelerate the process of islet graft vascularization, with the ultimate goal of increasing the success rate of islet transplantation.

27
AIMS OF THE STUDY
General aims At a time when the demand for islet transplantation is increasing, we are experiencing an overwhelming human organ deficit, and much of the re-search in the field needs to be focused on increasing the number of islets that survive the first crucial days after transplantation. At present, it is not un-common for a patient to require islets from up to four different donors to achieve normoglycemia. To greatly increase the number of diabetic patients who can be offered this cure, clinically applicable strategies for preventing the IBMIR and inducing angiogenesis must be established. As a means of achieving this aim, different ex vivo islet manipulations should be evaluated, since these approaches offer potential advantages over systemic treatment in terms of a lowered risk of severe side effects related to the imbalance in he-mostasis and increased risk of bleeding.
Specific aims
Paper I The aim of this study was to examine the possibility of using adenoviral vectors to induce transgenic expression of the specific thrombin inhibitor hirudin in human islets and to study the functional effects of the resulting product.
Paper II The aim of this study was to evaluate the possibility of modifying pancreatic islets by attaching functional heparin conjugate to their surface in order to render the islet graft blood biocompatible.
Paper III The aim of this study was to examine the effects of this approach on the IBMIR, using heparinized islets in in vitro and in vivo models.

28
Paper IV The aim of this study was to examine the capacity of an immobilized heparin conjugate to bind the potent angiogenic factors VEGF-A and FGF-2, as a means of attracting ECs in order to induce angiogenesis and revasculariza-tion.

29
STUDY DESIGN AND METHODS
A brief description of the methods used is presented here. More detailed information is given in papers I-IV.
Ethics All human and animal experiments were approved by the Research Ethics Committee of Uppsala University and performed in accordance with local institutional and Swedish national rules and regulations. Human islets were isolated after appropriate consent for multiorgan donation.
Preparation and culture of islets and ECs Human islets (Papers I-IV) Human islets were isolated from human cadaver donors according to modi-fied Ricordi method, followed by purification on a continuous density gradi-ent (128-130). The islet preparations were of good quality but were available for the experimental use because the total islet yield was too low for clinical transplantation. Islets were cultured at 37°C (5% CO2) in CMRL 1066 me-dium supplemented with 10% heat-inactivated human serum. The medium was changed every second day.
Adult porcine islets (Paper III) Adult porcine islets (API) were isolated from the pancreata of adult Land-race pigs according to a modified Ricordi protocol (131). The API were cul-tured at room temperature in CMRL 1066 medium supplemented with 10% heat inactivated pig serum. The medium change was performed every second day.

30
Rodent islets (Paper III) Male inbred C57BL/6J mice served as donors for syngeneic implantation. Pancreatic islets from rodents were prepared by collagenase digestion method. Groups of approximately 150 mouse islets were cultured at 37°C (5% CO2) for 2-3 days.
ECs (Paper IV) Human dermal microvascular ECs obtained from adult foreskin were cul-tured using endothelial cell growth medium MV supplement mix (ECGM MV). The ECs were cultured to 89% confluence in 1% gelatine-coated flasks. The ECs used were from passage 3-12.
Modification of islets Adenoviral vectors and transduction procedure (Paper I) We used a replication-defective E1, E2a and E3 region-deleted adenoviral vector Av3HHV1, coding for secretory hirudin cDNA, or Av3null vector carrying no transgene. The hirudin gene was inserted into the deleted E1 region under the control of the Rous sarcoma virus promoter (RSV) as pre-viously described (132). At the time of transduction, the islets were sedi-mented and then washed in serum-free culture medium. The islets were di-vided into three groups and subsequently transduced with either Av3HHV1 or Av3null or left untreated. Control islets not exposed to adenoviral vector were treated in the same way as the transduced islets.
Heparinization procedure (Paper II-IV) 1. Human, porcine and rodent islets were biotinylated by incubating the is-lets in culture medium containing EZ-LinkTM sulfo-NHS-LC-biotin (biotin SNL) or EZ-LinkTM TFP-PEO-biotin (biotin TFP). The islets were then washed by changing the culture medium three times. In the next step, the islets were incubated in culture medium supplemented with avidin and again washed. Finally, macromolecular conjugates of heparin in culture medium were allowed to bind to the biotin/avidin coating. The islets were finally washed by changing the culture medium three times. 2. The EC surfaces of human pancreatic arteries, obtained from pancreatic grafts to be used for islet isolations, were heparinized using same method. Each artery segment was cut in half; one half was used for heparinization and one as a control. The artery segments were then coated as described above, using the same reagents, washes, and incubation times as described for the islets. The control artery was filled with Ringer’s solution during the

31
heparinization process. Immediately after the immobilization process, both samples were snap-frozen in liquid nitrogen and stored for later analysis.
EC coating of islets of Langerhans (Paper IV) Heparinized or unmodified control human islets were mixed together with 0.25x106 Cell Tracker orange-labeled ECs in 500 μl ECGM MV culture medium and subsequently incubated at 37°C for 2-3 h. After incubation the islets were transferred to Petri dishes and cultured for 24 h.
Detection/analysis of modification procedures Hirudin detection (Paper I) Human islets were transduced with an adenoviral vector encoding secretory hirudin. Islet culture medium was thereafter analyzed for hirudin using an enzyme linked immunosorbent assay (ELISA) and a chromogenic substrate assay based on the thrombin-binding properties of hirudin.
Quartz crystal microbalance with dissipation monitoring (QCM-D) (Paper II, IV) To monitor the various steps in the heparinization procedure and the capacity of AT, VEGF-A or FGF-2 to bind to immobilized heparin a QCM-D tech-nique was used (89). This technique relies on the fact that a mass adsorbed onto the sensor surface of a shear-mode oscillating quartz crystal causes a proportional change in its resonance frequency, fq. Changes in fq reflect the amount of mass deposited onto the surface of the crystal. Sensor crystals (5-MHz), sputtered with stainless steel were used.
Large particle flow cytometry (Paper II) A BioSorter 1000 large particle flow cytometer that can detect and sort large (40-1,500 micron) particles in a continuously flowing stream was used (133). Particles were distinguished by size, optical density and intensity of fluorescence markers. Islets were heparinized using the methods described above. Heparinized islets were detected using AT labeled with the fluoro-chrome Alexa Fluor 488®. In addition, islets from each step of the heparini-zation process were analyzed for background fluorescence. Biotinylated islets were also stained with FITC-labeled streptavidin, and the background fluorescence of the heparinized islets was evaluated using unlabeled AT alone. In addition, heparinized islets marked with fluorochrome labeled AT or avidin, were incubated with coagulation FXa and analyzed.

32
Confocal microscopy (Papers II-IV) The degree of heparinization of the islets and of cryosections of heparinized pancreatic arteries was visualized by confocal microscopy after incubating the tissue with AT labeled with Alexa 488 or with Texas Red conjugated avidin. The binding of VEGF-A to the surface of the heparinized islets was visualized after incubating the islets with VEGF-A labeled using Alexa Fluor 488 Protein Labeling Kit. Propidium iodide was used to stain non-viable cells and DAPI was used to detect cell nuclei. Staining of focal adhe-sions of ECs was performed with mouse anti-paxillin antibody and cytosce-letal actin staining was performed by Texas Red conjugated phalloidin anti-body. Images were acquired with a Zeiss LSM 510 Meta confocal micro-scope equipped with an Axiovert 200 microscope stand.
In vitro blood experimental models
The Chandler loop model (Paper II) For the evaluation of biomaterials in circulating blood, Chandler loop model was used as previously described (87, 89). Pieces of polyvinyl chloride (PVC) tubing furnished with immobilized CHS or CHS-CT (CHS on cells and tissue) or left untreated were used. Each tubing was filled with fresh whole blood and turned into closed circuits using surface heparinized con-nectors (CHS). The tubing loops were rotated vertically in 37°C water bath for 1 h. After incubation, blood samples were collected and analyzed for platelet count and TAT (described below). AT binding capacity was assessed as previously described (89).
Tubing loops as a model (Paper III) Human islets were exposed to fresh human blood using a loop system model designed to resemble a blood vessel (20, 21) which allowed us to study the interaction between blood and islets in a manner similar to that seen in the portal vein during transplantation. In brief, this system consisted of PVC tubing with immobilized heparin on the inner surface. Fresh human blood was added to each loop, and the loops were placed on a rocking device at 37°C. Aliquots of 2μl of either heparinized or nonheparinized islets sus-pended in 100μl culture medium were placed in the loops containing fresh human blood. A control loop was included that had no islets but contained blood supplemented with 100μl culture medium. To monitor the IBMIR, blood samples of 1ml were collected into EDTA-containing tubes before perfusion and at 5, 15, 30, and 60 min after the start of the perfusion for fur-ther analysis (described below).

33
Considerations of generated data (Papers II-III) The values obtained in an in vitro blood loop model are of a higher magni-tude than would be expected in vivo. This difference can be explained by the fact that the same aliquot of blood re-circulates in the loops during the whole experiment, causing an accumulation of coagulation activation products. Furthermore, the heparin-coated inner surface of the tubing does not exhibit the same properties as the vascular endothelium, which possesses other anti-coagulant activities related to expression of thrombomodulin and the activa-tion of fibrinolysis. Also, air enclosed in the tubing further contributes to the activation of the blood. Therefore, the differences between experimental groups are more important than the absolute values when interpreting results obtained from in vitro systems.
Clotting time (ReoRox 4) (Paper I) Clotting time in whole blood after the addition of islet culture supernatants was measured in a four-channel free oscillating rheometer. The rheometer used in this study has four measuring channels embedded in an aluminium block thermostat with temperature control. The sample cup was set into free horizontal oscillation around its vertical axis (11Hz), and the damping (D) and frequency (Fq) of the oscillation were registered. D and Fq are depend-ent on the viscosity and elasticity of the sample. When clotting occurs, the rheological properties of the sample change, the frequency is decreased and damping is increased. Clotting times were identified by the software pro-gram of the instrument as the point of maximal damping, defining a clot.
Animals and transplantation procedures
Transplantation of human islets to athymic (nu/nu) mice (Paper I) Transduced and untreated islets were simultaneously transplanted as separate grafts to C57BL/6J nude (nu/nu) athymic mice under the left kidney capsule as previously described (134). All mice were transplanted with two islet grafts, one composed of transduced islets and one of untreated islets from the same isolation. After 5 weeks, the animals were euthanized and the grafts were harvested with a margin of 5 mm of adjacent kidney tissue and pre-pared for cryosectioning and immunohistological evaluation in light micro-scope.

34
Syngeneic islet transplantation to alloxan diabetic mice (Paper III) Alloxan-diabetic C57BL/6 mice were used as recipients. Diabetes was con-firmed by the presence of hyperglycemia (>25 mmol/L). The number of transplanted islets was chosen on the basis of previous studies in this strain. Cure from diabetes was defined as non-fasting blood glucose concentrations lower than 10 mmol/L. At the end of the experiment, to ensure persistent absence of endogenous pancreatic insulin production, the graft-bearing kid-ney was removed from all animals.
Intraportal allotransplantation of adult porcine islets (Paper III) Five piglets of Swedish mixed country breed, weighing 11- 16 kg, were op-erated on under general anaesthesia. During the experiment, ECG, pulse, blood pressure, central venous pressure and body temperature were continu-ously monitored. A catheter was placed in the superior mesenteric vein, and 7,500 IEQ/kg body weight of adult porcine islets were infused intraportally over 5 min. Blood samples were collected into EDTA tubes before and at 15, 30, and 60 min after islet transplantation. After 60 min, the pigs were eutha-nized, and the livers were excised and the portal system exposed.
Analytical procedures
Blood and plasma analysis (Paper II-III) After incubation in the tubing loop model and intraportal allotransplantation of adult porcine islets, the EDTA blood samples were analyzed for various parameters. Platelet counts were assessed using a cell counter. Plasma con-centrations of thrombin-antithrombin complexes (TAT) and insulin were quantified using a commercially available ELISA kit. The complement acti-vation product C3a was quantified as previously described (135).
Immunohistochemistry (Papers I, III) Transplanted grafts and islets were either embedded in paraffin or snapfro-zen. They were then fixed and subsequently stained for insulin.

35
Statistics All results are presented as mean ± standard error of the mean (SEM). In Paper I, the Student’s paired t-test was used to compare paired data (hirudin expressing islets and controls) in different islet isolations. In Paper III, the in vitro loop studies were evaluated using Friedman’s ANOVA. For evaluation of the in vivo allogeneic porcine islet transplantation experiments, the Mann-Whitney nonparametric test (double tailed) was used.

36
RESULTS AND DISCUSSION
Strategies to prevent the IBMIR (Papers I-III) The fact that the IBMIR also occurs in the clinical setting (21) points to the obvious need to control this reaction. This observation provided the rationale for the work in this thesis, which was focused on assessing potential ap-proaches to counteracting this reaction and thereby overcoming the require-ment for islets from multiple donors to achieve insulin independence. Sev-eral steps in the IBMIR process could serve as targets for pharmacological intervention, as a means of preventing the reaction and thereby promoting successful engraftment of the islets.
Previously, several approaches have already been used in an effort to try to target various aspects of IBMIR, with an in vitro loop model system being used to evaluate the effects of different drugs. These compounds were either added to the blood prior to exposure to the islets, simulating systemic antico-agulant therapies (e.g., melagatran) (136), or used as supplements in the islet culture medium prior to the loop experiments in order to down- regulate TF, as a model for pretreating the islets prior to transplantation (137).
Strategies to modulate the islets prior to the transplantation would offer a valuable complement to systemic anticoagulant treatments, which have ob-vious limitations because of their potentially adverse effects on hemostasis in the recipient. Further, systemic treatment of patients undergoing islet transplantation with anticoagulants is always associated with an increased risk of bleeding since the islets are infused by the transhepatic route. In this thesis we have examined the possibility of inducing the synthesis and secre-tion of biologically active anticoagulant hirudin in human islets using an adenoviral vector (Paper I), as well as using a macromolecular heparin con-jugate to cover the islet surface and thereby reduce the activation of the co-agulation cascade and exposure to various factors that might trigger the in-flammatory response (Papers II-III).
Strategies to induce revascularization (Paper IV) Islet revascularization, which is partially influenced by the severity of the IBMIR, is a subsequent critical step in the long-term survival of the trans-planted graft. If the islets survive the first crucial hours after transplantation,

37
they may still succumb as a result of ischemic damage before they are prop-erly revascularized, a process which takes 7-14 days (116, 138). Soluble factors released from the hypoxic islets are likely to be of major importance in this context.
The heparin coating has the capacity to bind substances with high affinity for heparin, such as the potent angiogenic growth factors VEGF and FGF, which could promote engraftment. Previous experiments using EC coating of the islets (139) as a means of limiting the IBMIR could therefore be taken one step further. Since it is known that heparin has an affinity for many dif-ferent growth factors and is highly negatively charged, one would expect that these properties would lead to improved coverage of the islets with ECs. In this thesis, we have examined the ability of immobilized heparin conju-gate to bind the angiogenic factors VEGF-A and FGF-2 as a means of at-tracting ECs, thereby promoting the angiogenesis of the transplanted islets and ultimately leading to successful revascularization of the graft. Paper I: Adenovirus-Mediated Expression of the Anti-coagulant Hirudin in Human Islets: A Tool to Make the Islets Biocompatible to Blood Expression of secretory hirudin in human islets after adenoviral transduction In Paper I a replication-defective adenoviral vector, Av3HHV1, was used to demonstrate that expression of biologically active hirudin can be induced in intact human islets.
Islet culture medium was analyzed for hirudin using an ELISA and a chromogenic substrate assay based on the thrombin-binding properties of hirudin. Following the transduction, hirudin was first detected after 3 days of culture and the levels then gradually increased and reached a plateau after 6 days. The chromogenic substrate assay reported slightly lower levels of hi-rudin than did the ELISA method. This difference could be explained by the fact that the chromogenic assay measures the level of functional thrombin-binding hirudin present in the sample, while the ELISA measures the total amount of hirudin present. The levels of hirudin/DNA and insulin/DNA were also measured. The level of hirudin content was found to be compara-ble or even higher than the insulin content in terms of both the total amount and on a molar basis. At the same time, the insulin levels in the hirudin-secreting islets were found to be lower than those in untreated control islets. Taken together these findings indicate that additional protein synthesis may have affected the insulin-producing capacity and may also provide an expla-nation for the observed impaired glucose response in hirudin-secreting islets. In this context it should be noted that while only the ß-cells express insulin,

38
hirudin is likely to be expressed in other cell types within the islets and also in contaminating exocrine cells.
Compared to other types of viral vectors, e.g., retroviruses, which induce permanent gene expression, adenoviral transduction will not lead to insertion of DNA into the genome of the infected cell. Hence, infected cells will be expressing the transduced gene for only a limited time (140) and thereby leave the islets unaffected after a few weeks. Moreover, within 1-2 weeks after intraportal transplantation, the islets induce revascularization of recipi-ent origin (19). At this stage, the islets are no longer directly exposed to the blood and therefore protection against coagulation activation is of lesser importance. In terms of safety, another advantage when compared to many other gene therapies is that for the purpose of islet transplantation, only the grafted tissue is exposed to the infective adenovirus in vitro prior to trans-plantation. Consequently, any potential side effects related to the direct pa-tient exposure to the virus are avoided. Also, various strategies to pretreat the islets before transplantation, such as adenoviral gene transfer would offer an advantage over systemic inhibition, since they would have little effect on hemostasis in the recipient.
Effects of islet-derived hirudin on coagulation in human blood Clotting time in whole blood after the addition of islet culture supernatants was measured in a free oscillating rheometer. A delay in the clotting time of approximately 30% was observed in human whole blood after the addition of supernatants taken from Av3HHV1-transduced islets when compared to their corresponding controls.
An increase in clotting time together with hirudin content comparable to the insulin content in transduced islets indicate that systemic effects might be expected in transplanted patients. However, while severe systemic effects could be minimized, the microenvironment around the islets could still cre-ate a substantial concentration gradient that would inhibit coagulation and clot formation in the immediate vicinity of the islets.
Expression of biologically active hirudin from the human islets may con-tribute to a marked protective effect of the graft. Hirudin is a direct thrombin inhibitor that achieves its anticoagulant effect by directly binding to the thrombin molecule, thereby preventing it from interacting with its substrates. Hence, hirudin not only prevents the terminal step of coagulation (fibrin formation) but also inhibits other thrombin-catalyzed reactions, such as the activation of FV, FVIII, FXIII and platelets. The direct inhibitory action of hirudin on thrombin, therefore, indicates that it has valuable therapeutic po-tential as an anticoagulant. Moreover, animal and clinical studies have shown that hirudin has low immunogeneic potential and is well tolerated (141, 142), although repeated treatment with hirudin in rare cases can induce anaphylaxis (143).

39
Alternatives to adenoviral transduction A concern regarding the in vivo use of adenoviral vectors is that infection itself can induce inflammatory and adaptive immune responses (144). In clinical islet transplantation, this effect would counteract the beneficial ef-fects of a transgenic protein expression induced to limit the IBMIR. The use of other types of vectors, e.g., lentiviruses, or recently developed adenoviral vectors with less intrinsic immunogenicity, may circumvent this problem.
Paper II: A New Method for Incorporating Functional Heparin onto the Surface of Islets of Langerhans
Analysis and visualization of the heparinization procedure Since immobilized functional heparin on artificial surfaces, CHS, is associ-ated with high blood compatibility, inhibited coagulation and complement activation and reduced platelet adhesion and activation (86-90), we have now made use of these properties to prevent the IBMIR by applying a coher-ent heparin coating to cell surfaces, CHS-CT. We used a water-soluble mac-romolecular conjugate in which ~70 heparin molecules are covalently at-tached to a carrier backbone (84). The method used in this paper takes ad-vantage of the fact that avidin expresses binding sites for both biotin and heparin (145), allowing the direct binding of heparin to biotin-avidin com-plexes.
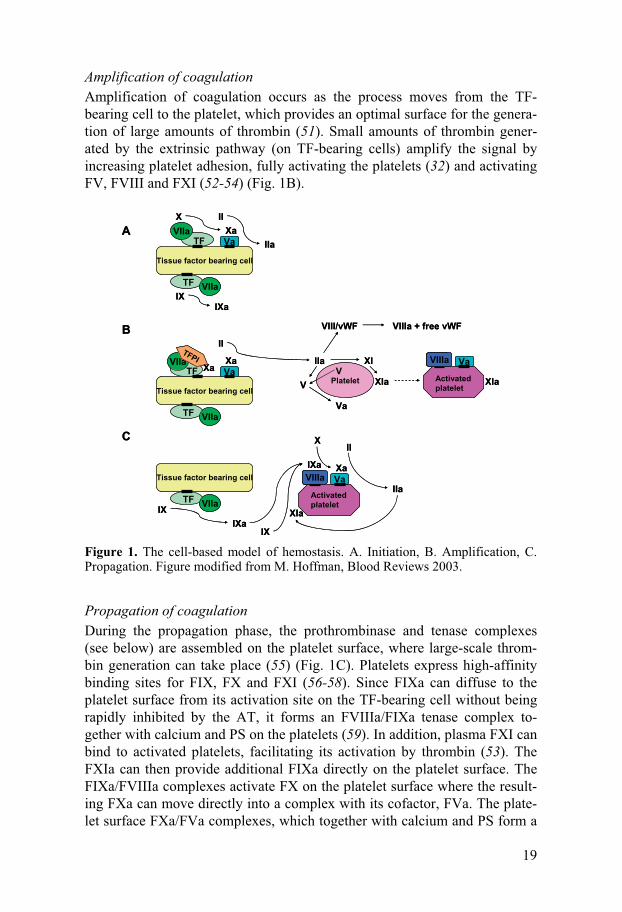
To screen a variety of reagents and strategies for coating surfaces with heparin using biotin and/or avidin, large particle flow cytometry and QCM-D were used. Biotin reagent targeting primary amines (biotin SNL) was tested by flow cytometry and was found to be most efficacious with regard to heparin binding to the islets. The most promising combinations were ana-lyzed in a QCM-D device using human albumin matrix. The results revealed sequential binding of biotin, avidin, heparin complexes, and AT to the sur-face.
Confocal microscopy was used to verify the binding of heparin to human islets and to assess the uniformity of the binding. This analysis of the hepa-rin-coated islets revealed evenly distributed fluorescence, demonstrating that the heparin coat covered the whole islet surface. Also, cross sections of the islets showed that the heparin bound strictly to the surface of the islets, which would be sufficient given that this is where the interaction with the blood occurs during the transplantation. Moreover, since heparin molecules are only conjugated to the islet surface, they should not alter the diffusion properties or impair the dynamics of insulin release. Further, the ligand(s) to which the platelets bind on the islet surface upon activation is (are) still uni-

40
dentified, but collagen, which surrounds human islets (18), is one likely can-didate.
Functional capacity of bound heparin In order to test functional capacity of bound heparin on the islet surface, heparinized islets marked with fluorochrome labeled AT or avidin were in-cubated with FXa and thereafter analyzed by large particle flow cytometry with respect to detection of remaining AT or avidin. Since AT is a natural inhibitor of FXa (64), following incubation with FXa, AT preadsorbed to the heparin coated islets was found to be released from the islets, forming FXa-AT complex. When analyzed, these islets appeared to be identical to heparin coated islets without prior exposure to AT. In contrast, heparinized islets labeled with avidin remained unaffected.
Biocompatibility of CHS-CT and CHS was compared using a modified Chandler loop model. As this assay relies on the use of tubing loops made of PVC, the CHS-CT was prepared onto a layer of adsorbed albumin that served as a biological surface substitute. In the experiments it was shown that CHS-CT was almost equal to using CHS in terms of biocompatibility properties and the effect on coagulation and uptake of AT.
It should be noted that this artificial substitute to the protein composition on the islet surface that the modified Chandler loop model represents is a compromise. Hence, preparation of CHS-CT on the PVC surface may not result in the same coating efficacy as should be the case using islets.
Paper III: Islet Surface Heparinization Prevents the Instant Blood-Mediated Inflammatory Reaction in Islet Transplantation
Islet functionality after heparinization procedure The heparinization approach is distinctly different from coatings such as microencapsulation that not only create a barrier for molecules and cells but also a significant dead space, leading to diffusion barriers for nutrients and secreted products. This supposition was confirmed when human islets were exposed to glucose stimulation in a dynamic perfusion system in which heparinized and non-heparinized islets responded similarly, indicating that insulin secretion was not affected by the heparinization procedure. More-over, long-term graft survival and function, tested by transplantation of islets to diabetic mice revealed successful engraftment and survival of both heparinized and untreated control islets.

41
Effect of islet heparinization in the in vitro blood loop model To assess the ability of heparinized islets to reduce the IBMIR that normally occurs when the untreated islets are exposed to fresh human ABO-compatible blood, we used an in vitro tubing model that mimics the period immediately following islet allotransplantation.
In tubing loops containing non-heparinized islets, macroscopic clotting with cell consumption (platelets) was seen after 60 minutes and was accom-panied by a significant rise in coagulation activation marker (TAT levels). Also, a marked increase in complement activation product C3a was ob-served. In loops containing heparinized islets, there was no macroscopic clotting, and the drop in platelet count and generation of TAT and C3a were attenuated. Since complement activity was decreased in loops containing heparinized islets when compared to non-heparinized islets, we might con-clude that complement activity was dependent on the thrombin generation a trigger pushing the IBMIR reaction forward.
These results, taken together, indicate that heparinization of islets prior to transplantation is a potentially suitable method for the stated purposes and offers appealing approach to preventing the IBMIR that occurs after clinical islet transplantation.
Intraportal allotransplantation of API In order to evaluate the effect of heparin coating on the IBMIR in vivo, we tested the heparinized islets in a porcine allogeneic islet transplantation model. Visible intraluminal macroscopic clotting was found in the portal vein branches of the pigs receiving non-heparinized islets. Moreover, we observed a reduction in the degree of macroscopically visible damage, in-cluding infarcted areas.
During the experiment, blood samples were collected. These retrieved samples were analyzed for coagulation activation (TAT, as a measure of IBMIR), C3a and insulin. In the pigs receiving non-heparinized islets, there was a rise in TAT, a drop in platelet count, release of insulin and an increase in C3a, indicating that the IBMIR had been triggered. In the animals receiv-ing heparinized islets, the increase in TAT, drop in platelet count, release of insulin and increase in C3a were attenuated, indicating that the IBMIR had been reduced.
Immobilization of heparin on the islet surface as described in this thesis potentiates the inhibitory effect of AT and reduces the exposure of the colla-gen and other factors that may be prothrombotic and trigger inflammation. It represents a new approach to reducing the IBMIR without an associated increased risk of bleeding, since the total amount of heparin infused with the islets is calculated to be at most 40 IU. Other side effects are also unlikely,

42
since conjugation of heparin has been used to produce blood-biocompatible artificial materials.
Heparinization of other cells/tissues The blood-tissue interaction in islet transplantation, described by the IBMIR, shares many features with the potentially harmful interaction observed be-tween recipient blood and the vessel walls of transplanted solid organs. This similarity suggests that the heparin coating technique might prove effective in other types of cells or other tissue or organ transplantation. Therefore, sections of pancreatic artery were heparinized using the same heparinization technique and visualized by confocal microscopy. A distinct fluorescence following the endothelial cell lining could be observed, with sparse distribu-tion into the subendothelium. No fluorescence was detected below the exter-nal elastic membrane or in the muscularis.
Paper IV: Anchoring of Growth Factors to Surface-Immobilized Heparin on Pancreatic Islets; Implications for Stimulating Islet Angiogenesis
Binding of growth factors to immobilized heparin conjugate In order to achieve successful engraftment, the transplanted islets must not only avoid being captured in a clot but also reach the most distal branches of the portal tree and stimulate angiogenesis. In Paper IV, we wanted to look beyond the IBMIR stage to when islets are beginning to engraft after trans-plantation. Since the islets have a delicate vascular tree that is damaged dur-ing the course of the collagenase digestion process and in vitro culture, the revascularization is strongly hampered in the transplant setting. The process of angiogenesis is known to be stimulated by the growth factors VEGF-A and FGF-2 (121-125), which also have a strong affinity for heparin (104, 146).
Using QCM-D and ELISA, we demonstrated an efficient binding of growth factors to immobilized heparin; the binding of VEGF-A was further verified by confocal microscopy. Under our conditions, saturation of the growth factor binding was still not reached, indicating a massive capacity for growth factor storage by the immobilized heparin conjugate. The strong affinity of the VEGF-A for heparin was evidenced by the fact that no VEGF-A release was observed over time in vitro when saline was used as an extrac-tant. In contrast, when immobilized heparin and VEGF-A were incubated with human plasma, a transient release of VEGF-A was detected, most likely
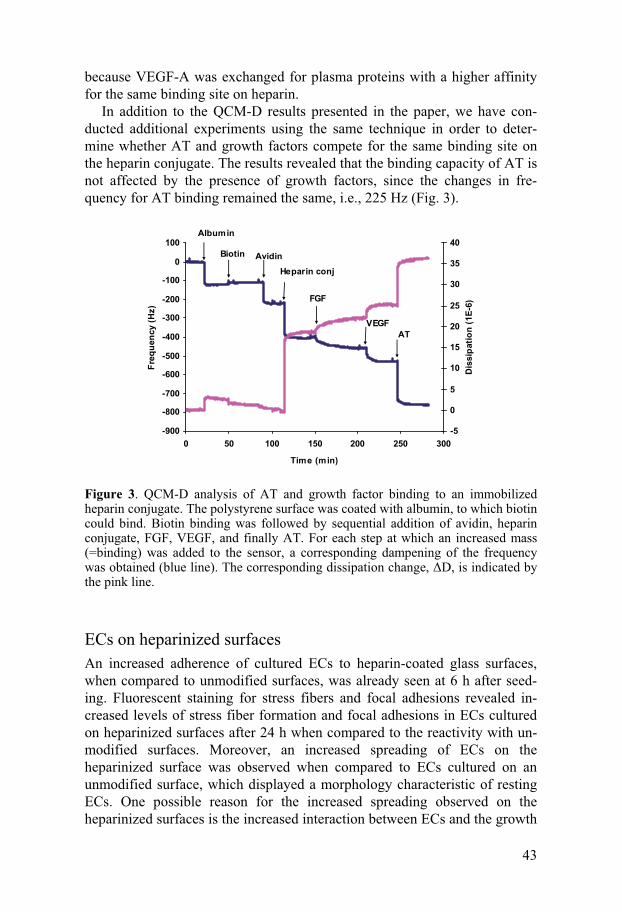
43
because VEGF-A was exchanged for plasma proteins with a higher affinity for the same binding site on heparin.
In addition to the QCM-D results presented in the paper, we have con-ducted additional experiments using the same technique in order to deter-mine whether AT and growth factors compete for the same binding site on the heparin conjugate. The results revealed that the binding capacity of AT is not affected by the presence of growth factors, since the changes in fre-quency for AT binding remained the same, i.e., 225 Hz (Fig. 3).
-900
-800
-700
-600
-500
-400
-300
-200
-100
0
100
0 50 100 150 200 250 300
Time (min)
Freq
uenc
y (H
z)
-5
0
5
10
15
20
25
30
35
40
Dis
sipa
tion
(1E-
6)
Albumin
Biotin AvidinHeparin conj
FGF
VEGFAT
Figure 3. QCM-D analysis of AT and growth factor binding to an immobilized heparin conjugate. The polystyrene surface was coated with albumin, to which biotin could bind. Biotin binding was followed by sequential addition of avidin, heparin conjugate, FGF, VEGF, and finally AT. For each step at which an increased mass (=binding) was added to the sensor, a corresponding dampening of the frequency was obtained (blue line). The corresponding dissipation change, �D, is indicated by the pink line.
ECs on heparinized surfaces An increased adherence of cultured ECs to heparin-coated glass surfaces, when compared to unmodified surfaces, was already seen at 6 h after seed-ing. Fluorescent staining for stress fibers and focal adhesions revealed in-creased levels of stress fiber formation and focal adhesions in ECs cultured on heparinized surfaces after 24 h when compared to the reactivity with un-modified surfaces. Moreover, an increased spreading of ECs on the heparinized surface was observed when compared to ECs cultured on an unmodified surface, which displayed a morphology characteristic of resting ECs. One possible reason for the increased spreading observed on the heparinized surfaces is the increased interaction between ECs and the growth

44
factors present in the medium, which are capable of binding to the immobi-lized heparin on the surface. The increased spreading may ultimately lead to contact inhibition and reduced proliferation. This explanation was verified by a thymidine incorporation assay in which ECs cultured on surfaces with immobilized heparin displayed a lower proliferative capacity before the ad-dition of VEGF-A than did ECs cultured on an untreated surface. This out-come may again reflect the ability of heparin to bind to a variety of matrices and serum proteins, thereby preventing the adsorption of serum components such as fibronectin or cell-synthesized collagen that are needed for normal cell proliferation and spreading. In line with this finding, we observed that ECs cultured on heparin-coated surfaces responded equally as well to exo-genously added VEGF as did ECs on unmodified surfaces. This response may reflect up-regulation of receptors and proteins involved in EC contact regulation, such as VEGFR-2, integrins, and focal adhesion kinase, which is strongly phosphorylated in the presence of VEGF (147-149).
Coating of pancreatic islets by ECs A significantly higher levels of adhesion of ECs to the heparinized islets was observed when compared to non-heparinized islets. This increase was achieved in the absence of additional growth factors. The results could be attributed to the enhanced binding of endogenously secreted growth factors from the islets and growth factors present in the culture medium, leading to the increase in binding of ECs to the islets. In line with this explanation, we reasoned that previous experiments using EC coating of the islets as a means of limiting the IBMIR (139) could be taken one step further by using heparinized islets.
Syngenic islet transplantation into C57BL/6J mice In an in vivo model involving islets transplanted under the kidney capsule of mice, we demonstrated that heparinized islets are more effective than un-modified islets in promoting revascularization in the presence of VEGF-A.
Control islets and heparinized islets with or without VEGF-A were trans-planted as separate grafts under the left kidney capsule of C57BL/6J mice, as previously described. The grafts were retrieved after 1 or 2 weeks, and the degree of islet revascularization was assessed by immunohistochemical staining for the endothelial marker CD31. A difference in the extent of the vessel formation was observed between the heparinized islet grafts (Fig. 4B) and the unmodified control islet grafts (Fig. 4A) as early as 1 week after transplantation, with a higher degree of staining for CD31 being seen in the graft containing heparinized islets. This difference was maintained at 2 weeks, showing a clear difference between control and heparinized islets (Fig. 4D and E respectively). Heparinized grafts treated with VEGF-A also
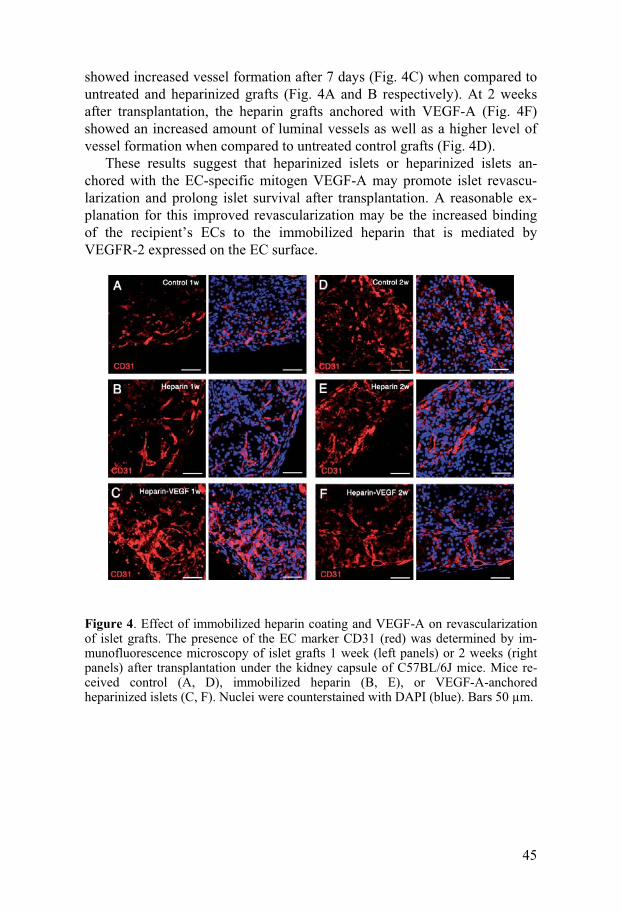
45
showed increased vessel formation after 7 days (Fig. 4C) when compared to untreated and heparinized grafts (Fig. 4A and B respectively). At 2 weeks after transplantation, the heparin grafts anchored with VEGF-A (Fig. 4F) showed an increased amount of luminal vessels as well as a higher level of vessel formation when compared to untreated control grafts (Fig. 4D).
These results suggest that heparinized islets or heparinized islets an-chored with the EC-specific mitogen VEGF-A may promote islet revascu-larization and prolong islet survival after transplantation. A reasonable ex-planation for this improved revascularization may be the increased binding of the recipient’s ECs to the immobilized heparin that is mediated by VEGFR-2 expressed on the EC surface.
Figure 4. Effect of immobilized heparin coating and VEGF-A on revascularization of islet grafts. The presence of the EC marker CD31 (red) was determined by im-munofluorescence microscopy of islet grafts 1 week (left panels) or 2 weeks (right panels) after transplantation under the kidney capsule of C57BL/6J mice. Mice re-ceived control (A, D), immobilized heparin (B, E), or VEGF-A-anchored heparinized islets (C, F). Nuclei were counterstained with DAPI (blue). Bars 50 μm.

46
CONCLUSIONS
Paper I 1. Expression of the specific thrombin inhibitor hirudin can be induced in human islets using adenoviral vectors.
2. A biological effect of the secreted hirudin was demonstrated in terms of a delay in clotting time in human whole blood after the addition of super-natants taken from Av3HHV1-transduced islets.
Paper II Islets of Langerhans can be successfully covered with functional immobi-lized heparin.
Paper III Both in vitro and in vivo models show that pancreatic islets covered with heparin can reduce the IBMIR, with clear implications for clinical islet cell transplantation.
Paper IV Immobilized heparin on the islet surface may be a useful anchor molecule for achieving complete coverage of the islets with growth factors such as VEGF-A and FGF-2, ultimately improving islet revascularization and en-graftment after transplantation.

47
ACKNOWLEDGMENTS
There are many people who have contributed in various ways to the work presented in this thesis and I would like to express my sincere gratitude to all of them. In par-ticular, I would like to thank: Olle Korsgren, my supervisor and the great mind behind the islet research, for giv-ing me the freedom, advice and inspiration. You are a great teacher with a critical eye that has always helped me to improve my work. Your character is truly a testa-ment to the saying “Nothing is impossible!” Bo Nilsson and Peetra Magnusson, my co-supervisors: Bo, I am grateful for all of your help and guidance over the years and for always taking the time despite your busy schedule. Peetra, “Super Duracell Woman”. Thank you, for introducing me to the complicated but yet so amazing world of angiogenesis. I will never forget the day (that went into night) when we had been working until I actually felt sick, while you just kept going. You are a real scientist!
Rolf Larsson: Thank you for sharing your outstanding knowledge about heparin and heparin surfaces and for fast responses to my manuscript writing.
Kristina Nilsson Ekdahl: I am grateful for your door always having been opened to discuss science and you never minding me popping in to ask you about anything.
Javier Sanchez: You have been with me from the beginning of the “heparin era” sharing the ups and downs. It turned out to be quite well at the end!
Graciela “Boluda” Elgue: Mi amor, thank you for your help with everything from never ending ELISA assays to finding errors in my manuscripts.
Corline Systems AB: Jessica Magnusson and Kurt Jansson, thank you for a good cooperation over the years and for giving me an insight into the commercial side of science. My co-authors: Your valuable input has always been appreciated! Blood Donors and the Nordic Network: Without you, the experiments would not have been feasible.

48
Mona Persson, Ann-Sophie Lindberg and Elin Blom Ekberg: I will never ever un-derstand how you can cope with so much paper work! My hat off for you guys! Deborah McClellan: Thank you for your linguistic revision of my manuscripts in-cluding this thesis, for mostly working on short notice and for doing an outstanding job.
Margareta “Bumsan” Engkvist: What would Klinimm be without you? I greatly appreciate the time we shared office, all your help with my experiments and all your support when I was writing on my thesis. I will never be able to thank you enough!
Peter Schmidt: In spite of mutual bullying throughout the years, you are and will remain a good friend – especially in view of your recently developed (or should I say forced�) interest in clothing.
Annika Moëll and Lisa Moberg: Thank you for being such wonderful friends, inside and outside the Klinimm, in good and in less good times. Our conversations over like 3,000 cups of coffee (300,000 for Lisa) and many after-work dinners at Svens-sons made everything so much more fun and easy! Girls - you make me feel �! AnnaKarin Lidehäll: For friendship and mutual pep-talks. I really admire your multi-tasking ability.
Ida Rasmusson: Thank you for introducing me to the world of “färskpressat” and for being so funny (OK, you are a great and enthusiastic scientist too), but I do not know how many times I have been laughing on my way back home from work (peo-ple around must have thought I was crazy) while thinking of your comments to vari-ous things during the day!
Tikomed AB group: Caroline Magnusson (we had lots of fun in Minneapolis – you are the perfect shopping partner!), Jonas Andersson (thank you for always answer-ing my questions on the technical matters), Naomi Forslund (the “immunohisto-chemistry expert”) and Maria Wilén (I will never forget your beautiful pictures from Africa).
The Islet group, including Ulrika Johansson (for sharing your considerable knowl-edge on laboratory techniques and for fun company on our many congress trips), Torsten Eich (for introducing me to the islet heparinization during my UGSBR pro-ject and for responding to my “ask the doctor”-questions), Magnus Ståhle and Nina Mikkola (for covering for my on-call hours during the stressful periods), Andrew Friberg (thanks for taking us to a real American baseball game – I had so much fun even though I will never be able to fully understand the rules though!), Daniel Brandhorst and Heide Brandhorst (for providing me with excellent pig islet prepara-tions), Anna Svensson, Christina Andersson, Viola Selling, José Caballero, Fredrik

49
Carlsson (for making it possible to adjust the strength of the coffee from the ma-chine�), Christian Molnar, …and not to be forgotten; the past members of the Islet group: Henrik Krook (for being a good friend inside and outside the lab), Masafumi Goto (a great scientist who we all miss so much), Megumi Goto (for her smile and humbleness), Anette von Malmborg (for her contagious laughter), Helena Johansson (for good company during many long isolation hours), Hideyuki Yamaya and Linda Holm.
Klinimmare from the C5, where I found “lugnet” (while it lasted�!): Linda Mathsson (for our conversations off-hours at work and for always reminding me of the importance to take time off), Susanne Lindblom (for always, I mean always, helping out – sincerely appreciated!), Jenny Tjernberg (for your quick, funny com-ments on all and everything), Lillemor Funke (for baking the delicious gluten-free cake for me at her place – I appreciate it a lot!), Erik Åhlin, Jennie Bäck, Jonas Lei-jon (for helping me with the QCM), Anna Mattsson, Jaan Hong, Adil Babiker, Osama Hamad, Mohammad Mullazehi and Elisabeth Gustafson (for bringing a fresh perspective on hepatocytes to the islet lab).
Elisabeth Wijkström: I really appreciate all the pep-talks and your concern during nice and less nice times at Klinimm. The present and past members of the GIG-group, greatly supervised by Thomas Tötterman and Magnus Essand, for contributing to the pleasant atmosphere at the lab. Especially thanks to: Helena Dzojic (not only for always smiling, but also for saving me so much time through her invaluable tips on how to prepare the thesis), Ole Forsberg (for wearing such nice clothes�), Björn Carlsson and Wing Cheng (for fun lunch dates, always ending in bizarre – but interesting discussions).
The project workers and the Routine staff at Klinimm, for always being very friendly.
My parents, for always being there for me, for your support, love and guidance. My brother Goran, for being the best brother in the world�. Lars, for all the love and endless support you have given me through the years (de-spite my high maintenance�).

50
REFERENCES
1. Atkinson MA, Maclaren NK. The pathogenesis of insulin-dependent diabetes mellitus. N Engl J Med 1994; 331 (21): 1428.
2. Amos AF, McCarty DJ, Zimmet P. The rising global burden of dia-betes and its complications: estimates and projections to the year 2010. Diabet Med 1997; 14 Suppl 5: S1.
3. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Dia-betes Care 2004; 27 (5): 1047.
4. Knip M. Environmental triggers and determinants of beta-cell auto-immunity and type 1 diabetes. Rev Endocr Metab Disord 2003; 4 (3): 213.
5. Adeghate E, Schattner P, Dunn E. An update on the etiology and epidemiology of diabetes mellitus. Ann N Y Acad Sci 2006; 1084: 1.
6. Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 1965; 14 (10): 619.
7. Foulis AK. C. L. Oakley lecture (1987). The pathogenesis of beta cell destruction in type I (insulin-dependent) diabetes mellitus. J Pathol 1987; 152 (3): 141.
8. Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 1986; 314 (21): 1360.
9. Sollinger HW, Odorico JS, Knechtle SJ, D'Alessandro AM, Kalayoglu M, Pirsch JD. Experience with 500 simultaneous pan-creas-kidney transplants. Ann Surg 1998; 228 (3): 284.
10. Ming CS, Chen ZH. Progress in pancreas transplantation and com-bined pancreas-kidney transplantation. Hepatobiliary Pancreat Dis Int 2007; 6 (1): 17.
11. Sutherland DE, Gruessner AC, Gruessner RW. Pancreas transplanta-tion: a review. Transplant Proc 1998; 30 (5): 1940.
12. Shapiro AM, Ryan EA, Lakey JR. Diabetes. Islet cell transplanta-tion. Lancet 2001; 358 Suppl: S21.
13. Bretzel RG, Hopt UT, Schatz H. Transplantations in patients with diabetes mellitus. Exp Clin Endocrinol Diabetes 2000; 108 (4): 241.
14. Ryan EA, Paty BW, Senior PA, et al. Five-year follow-up after clinical islet transplantation. Diabetes 2005; 54 (7): 2060.

51
15. Shapiro AM, Lakey JR, Ryan EA, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 2000; 343 (4): 230.
16. Berney T, Buhler L, Caulfield A, et al. Transplantation of islets of Langerhans: new developments. Swiss Med Wkly 2001; 131 (47-48): 671.
17. Van Deijnen JH, Van Suylichem PT, Wolters GH, Van Schilfgaarde R. Distribution of collagens type I, type III and type V in the pan-creas of rat, dog, pig and man. Cell Tissue Res 1994; 277 (1): 115.
18. van Suylichem PT, van Deijnen JE, Wolters GH, van Schilfgaarde R. Amount and distribution of collagen in pancreatic tissue of differ-ent species in the perspective of islet isolation procedures. Cell Transplant 1995; 4 (6): 609.
19. Menger MD, Yamauchi J, Vollmar B. Revascularization and micro-circulation of freely grafted islets of Langerhans. World J Surg 2001; 25 (4): 509.
20. Bennet W, Sundberg B, Groth CG, et al. Incompatibility between human blood and isolated islets of Langerhans: a finding with impli-cations for clinical intraportal islet transplantation? Diabetes 1999; 48 (10): 1907.
21. Moberg L, Johansson H, Lukinius A, et al. Production of tissue fac-tor by pancreatic islet cells as a trigger of detrimental thrombotic re-actions in clinical islet transplantation. Lancet 2002; 360 (9350): 2039.
22. Guyton AC HJ. Hemostasis and blood coagulation. Textbook of Medical Physiology. Tenth edition ed. Philadelphia: W.B. Saunders Co., 2000.
23. George JN. Platelets. Lancet 2000; 355 (9214): 1531. 24. de Gaetano G. Historical overview of the role of platelets in hemo-
stasis and thrombosis. Haematologica 2001; 86 (4): 349. 25. Sagripanti A, Carpi A. Antithrombotic and prothrombotic activities
of the vascular endothelium. Biomed Pharmacother 2000; 54 (2): 107.
26. Baumgartner HR. Platelet interaction with collagen fibrils in flowing blood. I. Reaction of human platelets with alpha chymotrypsin-digested subendothelium. Thromb Haemost 1977; 37 (1): 1.
27. Andrews RK, Shen Y, Gardiner EE, Berndt MC. Platelet adhesion receptors and (patho)physiological thrombus formation. Histol Histopathol 2001; 16 (3): 969.
28. Phillips DR, Law D, Scarborough RM. Glycoprotein IIb-IIIa in platelet aggregation: an emerging target for the prevention of acute coronary thrombotic occlusions. Arch Pathol Lab Med 1998; 122 (9): 811.
29. Rao AK, Gabbeta J. Congenital disorders of platelet signal transduc-tion. Arterioscler Thromb Vasc Biol 2000; 20 (2): 285.

52
30. Dale GL, Friese P, Batar P, et al. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell sur-face. Nature 2002; 415 (6868): 175.
31. Gachet C. ADP receptors of platelets and their inhibition. Thromb Haemost 2001; 86 (1): 222.
32. Brass LF. Thrombin and platelet activation. Chest 2003; 124 (3 Suppl): 18S.
33. Hanahan DJ, Weintraub ST. Platelet-activating factor isolation, identification, and assay. Methods Biochem Anal 1985; 31: 195.
34. Comfurius P, Williamson P, Smeets EF, Schlegel RA, Bevers EM, Zwaal RF. Reconstitution of phospholipid scramblase activity from human blood platelets. Biochemistry 1996; 35 (24): 7631.
35. Solum NO. Procoagulant expression in platelets and defects leading to clinical disorders. Arterioscler Thromb Vasc Biol 1999; 19 (12): 2841.
36. Bevers EM, Rosing J, Zwaal RF. Membrane phospholipids are the major determinant of the binding site for factor X activating--and prothrombinase complexes at the surface of human platelets. Agents Actions Suppl 1986; 20: 69.
37. Singbartl K, Forlow SB, Ley K. Platelet, but not endothelial, P-selectin is critical for neutrophil-mediated acute postischemic renal failure. Faseb J 2001; 15 (13): 2337.
38. Furie B, Furie BC, Flaumenhaft R. A journey with platelet P-selectin: the molecular basis of granule secretion, signalling and cell adhesion. Thromb Haemost 2001; 86 (1): 214.
39. Nemerson Y, Repke D. Tissue factor accelerates the activation of coagulation factor VII: the role of a bifunctional coagulation cofac-tor. Thromb Res 1985; 40 (3): 351.
40. Martin DM, Boys CW, Ruf W. Tissue factor: molecular recognition and cofactor function. Faseb J 1995; 9 (10): 852.
41. Zioncheck TF, Roy S, Vehar GA. The cytoplasmic domain of tissue factor is phosphorylated by a protein kinase C-dependent mecha-nism. J Biol Chem 1992; 267 (6): 3561.
42. Hoffman M, Monroe DM, 3rd. A cell-based model of hemostasis. Thromb Haemost 2001; 85 (6): 958.
43. Drake TA, Morrissey JH, Edgington TS. Selective cellular expres-sion of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol 1989; 134 (5): 1087.
44. Fleck RA, Rao LV, Rapaport SI, Varki N. Localization of human tissue factor antigen by immunostaining with monospecific, poly-clonal anti-human tissue factor antibody. Thromb Res 1990; 59 (2): 421.
45. Giesen PL, Rauch U, Bohrmann B, et al. Blood-borne tissue factor: another view of thrombosis. Proc Natl Acad Sci U S A 1999; 96 (5): 2311.

53
46. Suefuji H, Ogawa H, Yasue H, et al. Increased plasma tissue factor levels in acute myocardial infarction. Am Heart J 1997; 134 (2 Pt 1): 253.
47. Zumbach M, Hofmann M, Borcea V, et al. Tissue factor antigen is elevated in patients with microvascular complications of diabetes mellitus. Exp Clin Endocrinol Diabetes 1997; 105 (4): 206.
48. Misumi K, Ogawa H, Yasue H, et al. Comparison of plasma tissue factor levels in unstable and stable angina pectoris. Am J Cardiol 1998; 81 (1): 22.
49. Renne T, Nieswandt B, Gailani D. The intrinsic pathway of coagula-tion is essential for thrombus stability in mice. Blood Cells Mol Dis 2006.
50. Monkovic DD, Tracy PB. Activation of human factor V by factor Xa and thrombin. Biochemistry 1990; 29 (5): 1118.
51. Monroe DM, Hoffman M, Roberts HR. Transmission of a procoagu-lant signal from tissue factor-bearing cell to platelets. Blood Coagul Fibrinolysis 1996; 7 (4): 459.
52. Pieters J, Lindhout T, Hemker HC. In situ-generated thrombin is the only enzyme that effectively activates factor VIII and factor V in thromboplastin-activated plasma. Blood 1989; 74 (3): 1021.
53. Oliver JA, Monroe DM, Roberts HR, Hoffman M. Thrombin acti-vates factor XI on activated platelets in the absence of factor XII. Arterioscler Thromb Vasc Biol 1999; 19 (1): 170.
54. Alberio L, Safa O, Clemetson KJ, Esmon CT, Dale GL. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, colla-gen, and convulxin. Blood 2000; 95 (5): 1694.
55. Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin gen-eration. Arterioscler Thromb Vasc Biol 2002; 22 (9): 1381.
56. Greengard JS, Heeb MJ, Ersdal E, Walsh PN, Griffin JH. Binding of coagulation factor XI to washed human platelets. Biochemistry 1986; 25 (13): 3884.
57. Ahmad SS, Rawala-Sheikh R, Monroe DM, Roberts HR, Walsh PN. Comparative platelet binding and kinetic studies with normal and variant factor IXa molecules. J Biol Chem 1990; 265 (34): 20907.
58. Cirino G, Cicala C, Bucci M, et al. Factor Xa as an interface be-tween coagulation and inflammation. Molecular mimicry of factor Xa association with effector cell protease receptor-1 induces acute inflammation in vivo. J Clin Invest 1997; 99 (10): 2446.
59. Mertens K, Bertina RM. The contribution of Ca2+ and phospholip-ids to the activation of human blood-coagulation Factor X by acti-vated Factor IX. Biochem J 1984; 223 (3): 607.
60. Krishnaswamy S, Jones KC, Mann KG. Prothrombinase complex assembly. Kinetic mechanism of enzyme assembly on phospholipid vesicles. J Biol Chem 1988; 263 (8): 3823.

54
61. Camiolo SM, Thorsen S, Astrup T. Fibrinogenolysis and fibrinolysis with tissue plasminogen activator, urokinase, streptokinase-activated human globulin, and plasmin. Proc Soc Exp Biol Med 1971; 138 (1): 277.
62. Braat EA, Dooijewaard G, Rijken DC. Fibrinolytic properties of activated FXII. Eur J Biochem 1999; 263 (3): 904.
63. Schousboe I. Factor XIIa activation of plasminogen is enhanced by contact activating surfaces and Zn2+. Blood Coagul Fibrinolysis 1997; 8 (2): 97.
64. Rosenberg RD, Bauer KA, Marcum JA. Protease inhibitors of hu-man plasma. Antithrombin-III. "The heparin-antithrombin system". J Med 1985; 16 (1-3): 351.
65. Marcum JA, McKenney JB, Galli SJ, Jackman RW, Rosenberg RD. Anticoagulantly active heparin-like molecules from mast cell-deficient mice. Am J Physiol 1986; 250 (5 Pt 2): H879.
66. Braunsteiner H, Thumb N. [Mast cells, basophils and heparin libera-tion.]. Bibl Haematol 1963; 15: 9.
67. Kojima T, Leone CW, Marchildon GA, Marcum JA, Rosenberg RD. Isolation and characterization of heparan sulfate proteoglycans pro-duced by cloned rat microvascular endothelial cells. J Biol Chem 1992; 267 (7): 4859.
68. Lindahl U, Backstrom G, Thunberg L, Leder IG. Evidence for a 3-O-sulfated D-glucosamine residue in the antithrombin-binding se-quence of heparin. Proc Natl Acad Sci U S A 1980; 77 (11): 6551.
69. Lindahl U, Thunberg L, Backstrom G, Riesenfeld J, Nordling K, Bjork I. Extension and structural variability of the antithrombin-binding sequence in heparin. J Biol Chem 1984; 259 (20): 12368.
70. de Agostini AI, Watkins SC, Slayter HS, Youssoufian H, Rosenberg RD. Localization of anticoagulantly active heparan sulfate pro-teoglycans in vascular endothelium: antithrombin binding on cul-tured endothelial cells and perfused rat aorta. J Cell Biol 1990; 111 (3): 1293.
71. Broze GJ, Jr. Tissue factor pathway inhibitor. Thromb Haemost 1995; 74 (1): 90.
72. Esmon CT. The protein C pathway. Chest 2003; 124 (3 Suppl): 26S. 73. Bajzar L, Manuel R, Nesheim ME. Purification and characterization
of TAFI, a thrombin-activable fibrinolysis inhibitor. J Biol Chem 1995; 270 (24): 14477.
74. Collen D. The plasminogen (fibrinolytic) system. Thromb Haemost 1999; 82 (2): 259.
75. Hirsh J, Dalen J, Anderson DR, et al. Oral anticoagulants: mecha-nism of action, clinical effectiveness, and optimal therapeutic range. Chest 2001; 119 (1 Suppl): 8S.
76. Hirsh J, Warkentin TE, Shaughnessy SG, et al. Heparin and low-molecular-weight heparin: mechanisms of action, pharmacokinetics,

55
dosing, monitoring, efficacy, and safety. Chest 2001; 119 (1 Suppl): 64S.
77. Greinacher A, Potzsch B, Amiral J, Dummel V, Eichner A, Mueller-Eckhardt C. Heparin-associated thrombocytopenia: isolation of the antibody and characterization of a multimolecular PF4-heparin com-plex as the major antigen. Thromb Haemost 1994; 71 (2): 247.
78. Visentin GP, Ford SE, Scott JP, Aster RH. Antibodies from patients with heparin-induced thrombocytopenia/thrombosis are specific for platelet factor 4 complexed with heparin or bound to endothelial cells. J Clin Invest 1994; 93 (1): 81.
79. Weitz JI, Hudoba M, Massel D, Maraganore J, Hirsh J. Clot-bound thrombin is protected from inhibition by heparin-antithrombin III but is susceptible to inactivation by antithrombin III-independent in-hibitors. J Clin Invest 1990; 86 (2): 385.
80. Weitz JI. A novel approach to thrombin inhibition. Thromb Res 2003; 109 Suppl 1: S17.
81. Di Nisio M, Middeldorp S, Buller HR. Direct thrombin inhibitors. N Engl J Med 2005; 353 (10): 1028.
82. Sohn JH, Kang HA, Rao KJ, et al. Current status of the anticoagu-lant hirudin: its biotechnological production and clinical practice. Appl Microbiol Biotechnol 2001; 57 (5-6): 606.
83. Testa L, Andreotti F, Biondi Zoccai GG, Burzotta F, Bellocci F, Crea F. Ximelagatran/melagatran against conventional anticoagula-tion: a meta-analysis based on 22,639 patients. Int J Cardiol 2007; 122 (2): 117.
84. Kristensen EM, Rensmo H, Larsson R, Siegbahn H. Characteriza-tion of heparin surfaces using photoelectron spectroscopy and quartz crystal microbalance. Biomaterials 2003; 24 (23): 4153.
85. Johnell M, Elgue G, Larsson R, Larsson A, Thelin S, Siegbahn A. Coagulation, fibrinolysis, and cell activation in patients and shed mediastinal blood during coronary artery bypass grafting with a new heparin-coated surface. J Thorac Cardiovasc Surg 2002; 124 (2): 321.
86. Pekna M, Larsson R, Formgren B, Nilsson UR, Nilsson B. Comple-ment activation by polymethyl methacrylate minimized by end-point heparin attachment. Biomaterials 1993; 14 (3): 189.
87. Gong J, Larsson R, Ekdahl KN, Mollnes TE, Nilsson U, Nilsson B. Tubing loops as a model for cardiopulmonary bypass circuits: both the biomaterial and the blood-gas phase interfaces induce comple-ment activation in an in vitro model. J Clin Immunol 1996; 16 (4): 222.
88. Sanchez J, Elgue G, Riesenfeld J, Olsson P. Inhibition of the plasma contact activation system of immobilized heparin: relation to surface density of functional antithrombin binding sites. J Biomed Mater Res 1997; 37 (1): 37.

56
89. Andersson J, Sanchez J, Ekdahl KN, Elgue G, Nilsson B, Larsson R. Optimal heparin surface concentration and antithrombin binding ca-pacity as evaluated with human non-anticoagulated blood in vitro. J Biomed Mater Res A 2003; 67 (2): 458.
90. Christensen K, Larsson R, Emanuelsson H, Elgue G, Larsson A. Improved blood compatibility of a stent graft by combining heparin coating and abciximab. Thromb Res 2005; 115 (3): 245.
91. Johnell M, Elgue G, Thelin S, Larsson R, Siegbahn A. Cell adhesion and tissue factor upregulation in oxygenators used during coronary artery bypass grafting are modified by the Corline Heparin Surface. Scand Cardiovasc J 2002; 36 (6): 351.
92. Bennet W, Sundberg B, Lundgren T, et al. Damage to porcine islets of Langerhans after exposure to human blood in vitro, or after in-traportal transplantation to cynomologus monkeys: protective effects of sCR1 and heparin. Transplantation 2000; 69 (5): 711.
93. Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol 1995; 11: 73.
94. Carmeliet P. Angiogenesis in health and disease. Nat Med 2003; 9 (6): 653.
95. Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol 2002; 248 (2): 307.
96. Uemura A, Kusuhara S, Katsuta H, Nishikawa S. Angiogenesis in the mouse retina: a model system for experimental manipulation. Exp Cell Res 2006; 312 (5): 676.
97. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med 2000; 6 (4): 389.
98. Conway EM, Collen D, Carmeliet P. Molecular mechanisms of blood vessel growth. Cardiovasc Res 2001; 49 (3): 507.
99. Kimura H, Weisz A, Kurashima Y, et al. Hypoxia response element of the human vascular endothelial growth factor gene mediates tran-scriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood 2000; 95 (1): 189.
100. Mignatti P, Rifkin DB. Plasminogen activators and matrix metallo-proteinases in angiogenesis. Enzyme Protein 1996; 49 (1-3): 117.
101. Kamei M, Saunders WB, Bayless KJ, Dye L, Davis GE, Weinstein BM. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature 2006; 442 (7101): 453.
102. Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999; 126 (14): 3047.
103. Dunn IF, Heese O, Black PM. Growth factors in glioma angiogene-sis: FGFs, PDGF, EGF, and TGFs. J Neurooncol 2000; 50 (1-2): 121.

57
104. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med 2003; 9 (6): 669.
105. Yamashita T, Yoshioka M, Itoh N. Identification of a novel fibro-blast growth factor, FGF-23, preferentially expressed in the ventro-lateral thalamic nucleus of the brain. Biochem Biophys Res Com-mun 2000; 277 (2): 494.
106. Folkman J, Klagsbrun M, Sasse J, Wadzinski M, Ingber D, Vlo-davsky I. A heparin-binding angiogenic protein--basic fibroblast growth factor--is stored within basement membrane. Am J Pathol 1988; 130 (2): 393.
107. Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol 2006; 39 (5): 469.
108. Johnson DE, Williams LT. Structural and functional diversity in the FGF receptor multigene family. Adv Cancer Res 1993; 60: 1.
109. Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol 2006; 174 (3): 323.
110. Salmivirta M, Lidholt K, Lindahl U. Heparan sulfate: a piece of information. Faseb J 1996; 10 (11): 1270.
111. Rosengart TK, Johnson WV, Friesel R, Clark R, Maciag T. Heparin protects heparin-binding growth factor-I from proteolytic inactiva-tion in vitro. Biochem Biophys Res Commun 1988; 152 (1): 432.
112. Flaumenhaft R, Moscatelli D, Saksela O, Rifkin DB. Role of ex-tracellular matrix in the action of basic fibroblast growth factor: ma-trix as a source of growth factor for long-term stimulation of plasmi-nogen activator production and DNA synthesis. J Cell Physiol 1989; 140 (1): 75.
113. Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest 2001; 108 (3): 341.
114. Brunicardi FC, Stagner J, Bonner-Weir S, et al. Microcirculation of the islets of Langerhans. Long Beach Veterans Administration Re-gional Medical Education Center Symposium. Diabetes 1996; 45 (4): 385.
115. van Schilfgaarde R, de Vos P. Factors influencing the properties and performance of microcapsules for immunoprotection of pancreatic islets. J Mol Med 1999; 77 (1): 199.
116. Menger MD, Jaeger S, Walter P, Feifel G, Hammersen F, Messmer K. Angiogenesis and hemodynamics of microvasculature of trans-planted islets of Langerhans. Diabetes 1989; 38 Suppl 1: 199.
117. Menger MD, Jager S, Walter P, Hammersen F, Messmer K. A novel technique for studies on the microvasculature of transplanted islets of Langerhans in vivo. Int J Microcirc Clin Exp 1990; 9 (1): 103.

58
118. Vajkoczy P, Olofsson AM, Lehr HA, et al. Histogenesis and ultra-structure of pancreatic islet graft microvasculature. Evidence for graft revascularization by endothelial cells of host origin. Am J Pathol 1995; 146 (6): 1397.
119. Brissova M, Fowler M, Wiebe P, et al. Intraislet endothelial cells contribute to revascularization of transplanted pancreatic islets. Dia-betes 2004; 53 (5): 1318.
120. Nyqvist D, Kohler M, Wahlstedt H, Berggren PO. Donor islet endo-thelial cells participate in formation of functional vessels within pancreatic islet grafts. Diabetes 2005; 54 (8): 2287.
121. Stagner JI, Samols E. Induction of angiogenesis by growth factors: relevance to pancreatic islet transplantation. Exs 1992; 61: 381.
122. Zhang N, Richter A, Suriawinata J, et al. Elevated vascular endothe-lial growth factor production in islets improves islet graft vasculari-zation. Diabetes 2004; 53 (4): 963.
123. Lai Y, Schneider D, Kidszun A, et al. Vascular endothelial growth factor increases functional beta-cell mass by improvement of angio-genesis of isolated human and murine pancreatic islets. Transplanta-tion 2005; 79 (11): 1530.
124. Hayek A, Lopez AD, Beattie GM. Angiogenic peptides in pancreatic islet transplantation to diabetic rats. Transplantation 1990; 50 (6): 931.
125. Hayek A, Beattie GM, Lopez AD, Chen P. The use of digital image processing to quantitate angiogenesis induced by basic fibroblast growth factor and transplanted pancreatic islets. Microvasc Res 1991; 41 (2): 203.
126. Vasir B, Aiello LP, Yoon KH, Quickel RR, Bonner-Weir S, Weir GC. Hypoxia induces vascular endothelial growth factor gene and protein expression in cultured rat islet cells. Diabetes 1998; 47 (12): 1894.
127. Vasir B, Jonas JC, Steil GM, et al. Gene expression of VEGF and its receptors Flk-1/KDR and Flt-1 in cultured and transplanted rat islets. Transplantation 2001; 71 (7): 924.
128. Ricordi C, Lacy PE, Scharp DW. Automated islet isolation from human pancreas. Diabetes 1989; 38 Suppl 1: 140.
129. Brandhorst H, Brandhorst D, Brendel MD, Hering BJ, Bretzel RG. Assessment of intracellular insulin content during all steps of human islet isolation procedure. Cell Transplant 1998; 7 (5): 489.
130. Lakey JR, Warnock GL, Shapiro AM, et al. Intraductal collagenase delivery into the human pancreas using syringe loading or controlled perfusion. Cell Transplant 1999; 8 (3): 285.
131. Ricordi C, Finke EH, Lacy PE. A method for the mass isolation of islets from the adult pig pancreas. Diabetes 1986; 35 (6): 649.

59
132. Gorziglia MI, Kadan MJ, Yei S, et al. Elimination of both E1 and E2 from adenovirus vectors further improves prospects for in vivo hu-man gene therapy. J Virol 1996; 70 (6): 4173.
133. Fernandez LA, Hatch EW, Armann B, et al. Validation of large par-ticle flow cytometry for the analysis and sorting of intact pancreatic islets. Transplantation 2005; 80 (6): 729.
134. Schmidt P, Krook H, Maeda A, Korsgren O, Benda B. A new mur-ine model of islet xenograft rejection: graft destruction is dependent on a major histocompatibility-specific interaction between T-cells and macrophages. Diabetes 2003; 52 (5): 1111.
135. Nilsson Ekdahl K, Nilsson B, Pekna M, Nilsson UR. Generation of iC3 at the interface between blood and gas. Scand J Immunol 1992; 35 (1): 85.
136. Ozmen L, Ekdahl KN, Elgue G, Larsson R, Korsgren O, Nilsson B. Inhibition of thrombin abrogates the instant blood-mediated inflam-matory reaction triggered by isolated human islets: possible applica-tion of the thrombin inhibitor melagatran in clinical islet transplanta-tion. Diabetes 2002; 51 (6): 1779.
137. Moberg L, Olsson A, Berne C, et al. Nicotinamide inhibits tissue factor expression in isolated human pancreatic islets: implications for clinical islet transplantation. Transplantation 2003; 76 (9): 1285.
138. Jansson L, Carlsson PO. Graft vascular function after transplantation of pancreatic islets. Diabetologia 2002; 45 (6): 749.
139. Johansson U, Elgue G, Nilsson B, Korsgren O. Composite islet-endothelial cell grafts: a novel approach to counteract innate immu-nity in islet transplantation. Am J Transplant 2005; 5 (11): 2632.
140. Chengalvala MV, Lubeck MD, Selling BJ, et al. Adenovirus vectors for gene expression. Curr Opin Biotechnol 1991; 2 (5): 718.
141. Bichler J, Gemmerli R, Fritz H. Studies for revealing a possible sen-sitization to hirudin after repeated intravenous injections in baboons. Thromb Res 1991; 61 (1): 39.
142. Close P, Bichler J, Kerry R, et al. Weak allergenicity of recombinant hirudin CGP 39393 (REVASC) in immunocompetent volunteers. The European Hirudin in Thrombosis Group (HIT Group). Coron Artery Dis 1994; 5 (11): 943.
143. Greinacher A, Lubenow N, Eichler P. Anaphylactic and anaphylac-toid reactions associated with lepirudin in patients with heparin-induced thrombocytopenia. Circulation 2003; 108 (17): 2062.
144. McCarter SD, Scott JR, Lee PJ, et al. Cotransfection of heme oxy-genase-1 prevents the acute inflammation elicited by a second ade-novirus. Gene Ther 2003; 10 (19): 1629.
145. Kett WC, Osmond RI, Moe L, Skett SE, Kinnear BF, Coombe DR. Avidin is a heparin-binding protein. Affinity, specificity and struc-tural analysis. Biochim Biophys Acta 2003; 1620 (1-3): 225.

60
146. Schlessinger J, Plotnikov AN, Ibrahimi OA, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell 2000; 6 (3): 743.
147. Gitay-Goren H, Soker S, Vlodavsky I, Neufeld G. The binding of vascular endothelial growth factor to its receptors is dependent on cell surface-associated heparin-like molecules. J Biol Chem 1992; 267 (9): 6093.
148. Wu MH, Guo M, Yuan SY, Granger HJ. Focal adhesion kinase me-diates porcine venular hyperpermeability elicited by vascular endo-thelial growth factor. J Physiol 2003; 552 (Pt 3): 691.
149. Ashikari-Hada S, Habuchi H, Kariya Y, Kimata K. Heparin regu-lates vascular endothelial growth factor165-dependent mitogenic ac-tivity, tube formation, and its receptor phosphorylation of human endothelial cells. Comparison of the effects of heparin and modified heparins. J Biol Chem 2005; 280 (36): 31508.


Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 297
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofMedicine. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Medicine”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-8333
ACTA
UNIVERSITATIS
UPSALIENSIS
UPPSALA
2007