Embed Size (px)
Citation preview

643
Cerebral Resuscitation and Traumatic Brain Injury
CHAPTER 31
CHAPTER OUTLINELearning ObjectivesIntroductionMechanisms of Brain Injury
Primary Brain InjurySecondary Brain InjuryGlobal Cerebral Ischemia and Reperfusion
Neurointensive Care MonitoringNon-invasive MonitoringIntracranial PressureCerebral Perfusion PressureCerebral Blood FlowTranscranial Doppler UltrasonographyCerebral Metabolic MonitoringBrain Tissue OximetryCerebral MicrodialysisEEGComputed TomographyMagnetic Resonance Imaging/Spectroscopy
Clinical Management GuidelinesTraumatic Brain InjuryCardiac Arrest
Clinical OutcomesTraumatic Brain InjuryCardiac Arrest
Final CommentsReview QuestionsAnswersSuggested Readings
LEARNING OBJECTIVES Defi ne the difference between primary and secondary ■
brain injury Describe the cascade of events that occurs with global ■
cerebral ischemia and reperfusion In this context, identify possible strategies for attenuating ■
the poor outcomes associated global cerebral anoxia Describe the modalities currently available for clinical ■
monitoring in brain injured patients and outline indications for the use of:
Intracranial Pressure Monitoring ■
Jugular Bulb Blood Sampling ■
Radiographic studies ■
EEG ■
Review the literature with regard to therapeutic trials in ■
global cerebral ischemia Detail the outcomes associated with global cerebral ■
ischemia Discuss the care of a patient with hypoxic ischemic ■
encephalopathy Describe the initial evaluation of a patient with closed ■
head injury Presumptive neck injury ■
Glasgow Coma Scale Score (and modifi cation for ■
children) Assessment for other injuries ■
Discuss the management and its associated rationale of a ■
pediatric patient with a severe head injury including respiratory, hematologic and nutritional issues Discuss the outcome of moderate and severe head injury ■
in the pediatric patient
ERICKA L. FINK , PATRICK M. KOCHANEK, AND ROBERT S. B. CLARK
INTRODUCTION
The need for improved cerebral resuscitation was born out of progress – advancements in resuscitation, intensive care, and rehabilitation have decreased mortality associated with brain injury in infants and children, but increased the number living with disability. Indeed morbidity, and mortality for that matter, remains unacceptably high. To date, cerebral resus-citation after ischemic and traumatic brain injury (TBI) remains largely supportive, although many promising therapies are being explored at the bench. Now the challenge is to move

644 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
therapies to the bedside, to assist patients in attaining improved outcomes and quality of life. In the interim, optimizing patient care management in the prehospital setting, emergency department, pediatric intensive care unit (PICU), and rehabilitation facilities, is our greatest opportunity for improving outcome in these pediatric patients.
MECHANISMS OF BRAIN INJURY
Acute brain injury, such as that seen after ischemia and trauma, involves both primary and secondary injury (Fig. 31-1 ). After cerebral ischemia, primary injury relates to profound and irreversible ischemia to the point of cellular membrane failure, swelling, and lysis prior to reperfusion. Morphologically, this form of cell death is referred to as necrosis. After TBI, primary injury refers to direct mechanical disruption of brain parenchyma. Since this also includes disruption of the cerebrovasculature, ischemic necrosis can also contribute to pri-mary injury after TBI. In both instances, secondary injury refers to damage produced by a cascade of biochemical, cellular and molecular events, and involves both the endogenous evolution of damage within the brain and the effects of extra-cerebral insults, in particular hypotension and hypoxemia.
Primary Brain Injury Primary injury encompasses what is generally accepted to be damage to the point of where death of the tissue is inevitable. This can be caused by direct mechanical injury from compres-sion or shearing, in the case of trauma. It can also be caused by interruption of cerebral blood
ISCHEMIA TRAUMA
PrimaryInjury
SecondaryInjury
Interruption of blood flowLoss of ionic gradientsMitochondrial failure
Loss of membrane integrityRelease of intracellular contents
ExcitotoxicityCell swelling
Necrosis
Apoptosis
Mechanical somal and axonal damageVascular disruption
Secondary ischemia (hypoperfusion, hypoxemia)Cerebral edema
ExcitotoxicityElevated [Ca2+]I
Free radical damageLoss of trophic factors and survival signals
Mitochondrial failureRelease of death signals from mitochondria
Death receptor-ligand interactionsInflammation
FIGURE 31-1
Cascade of cellular events resulting in primary and second-ary brain injury after ischemia or trauma

645 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
fl ow without reperfusion, or reperfusion beyond the time from which the cell is still salvage-able. The latter results in mitochondrial failure and energy depletion with loss of ionic gradients and membrane pumps. Intracellular contents are increased for calcium and other ions, excit-atory amino acids, proteases, lipases, and nucleases. In suffi cient intracellular concentrations, these substances in and of themselves result in cell death. In suffi cient extracellular concentra-tions, liberation of non-compartmentalized intracellular contents may trigger either primary or secondary injury of adjacent cells, expanding the volume of primary tissue damage.
Because the majority of primary brain injury is necrotic in nature, it has been felt that necrotic cell death is irreversible. However, necrotic cell death may be amenable to pharma-cological treatment. One class of agents, poly(ADP-ribose) polymerase (PARP) inhibitors, have been reported to be effective in preventing both necrotic and apoptotic cell death after brain injury via reducing energy failure. Cyclosporin A may represent another agent that can attenuate necrotic cell death since recent studies suggest that it can reduce contusion volume when administered after experimental TBI. The therapeutic window for treating necrosis, and possibly a portion of primary injury, is likely extremely narrow.
Secondary Brain Injury Secondary brain injury represents the evolution of tissue damage after the primary insult. This is of obvious importance given that it is generally felt that secondary injury represents a target occurring within a clinically relevant therapeutic window, and as such, prevention of second-ary injury represents an area of intense research effort. The duration of this therapeutic win-dow remains unclear, and varies depending upon the type of brain insult. In focal cerebral ischemia, studies have shown that lesions tend to enlarge within the fi rst 12 h after injury. After global ischemia, such as after cardiac arrest, lesions appear later, in selectively vulner-able brain regions including the CA1 hippocampus, basal ganglia, layers 3 and 5 of the cortex, and cerebellum, and include apoptotic cell death peaking 24–72 h after injury. Cell death after TBI has been documented in autopsy specimens as early as 2 h post-injury, demonstrated by eosinophilic neurons, axonal swelling, glial swelling, and PMN infi ltration. Apoptotic cell death after TBI has been shown to evolve over 24–72 h in certain brain regions such as the cerebral cortex and hippocampus. In other brain regions cell death is more protracted, for example neuronal death in the thalamus evolves over a period of weeks after TBI.
While many mechanisms contribute to secondary brain injury and recovery after trauma and ischemia, including free radical damage, disturbances in calcium homeostasis, cell death cascades including apoptosis, mitochondrial failure, axonal injury, autophagy, and infl am-mation, the following discussion focuses on mechanisms related to currently applied ICU, emergency department, and prehospital treatment strategies. Namely, ischemia, excitotoxic-ity, and cerebral edema.
Ischemia
Numerous mechanisms contribute to secondary injury after acute brain injury (Fig. 31-1 ), including delayed ischemia, either direct from hypoperfusion and/or hypoxemia, or relative from hypermetabolism (e.g. during prolonged seizures). Cerebral edema can also result in a vicious cycle of compression ischemia, something that can be seen at the bedside when intracranial pressure (ICP) approaches mean arterial pressure (MAP). Even mild-moderate ICP spikes may cause local ischemia as evidenced by increases in local concentrations of lactate and excitatory amino acids. These increases can be ameliorated by treating ICP spikes with pentobarbital. Cerebral blood fl ow (CBF) values indicative of hypoperfusion, generally defi ned as below an ischemic threshold of 20 mL/100 g brain tissue/min, have been shown to occur in both adults and children after TBI, correlating with poor outcome. Using co-registered maps of CBF, cerebral metabolic rate for oxygen (CMRO 2 ), and oxygen extrac-tion fraction (OEF) suggests that the ischemic threshold after TBI in humans may be 15 mL/100 g brain tissue/min, differing from the threshold of 20 mL/100 g brain tissue/min defi ned in stroke patients. Of compelling interest, many studies have shown that the most obvious causes of secondary ischemia, hypotension and hypoxemia, are strongly associated
Primary brain injury occurs immediately and often results in necrotic cell death, while second-ary injury evolves over hours to days – representing a therapeutic target.
After cerebral ischemia selectively vulnerable brain areas include the CA1 hippocampus, basal ganglia, layers 3 and 5 of the cortex, and cerebellum.

646 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
with unfavorable outcome if occurring early after TBI in both adults and children. These studies strongly support, albeit not causally, correction of hypotension and hypoxemia in patients after acute brain injury. Other evidence implicating undesirable consequences of secondary insult is based on studies measuring the oxygen saturation in jugular venous bulb blood (SjVO 2 ). A single episode of jugular venous desaturation (SjvO2 <50% for more than 10 min) is strongly associated with poor neurological outcome in adult patients after severe TBI. These were intensive care unit (ICU) related incidents, and were often concurrent with systemic hypotension or hypoxemia, or sustained elevation in ICP.
Numerous mechanisms may underlie the early posttraumatic hypoperfusion. These involve not only mechanical disruption of blood vessels and macro and microthrombi, but also an imbalance in the ratio of endogenous vasoconstricters vs. vasodilators. For example, after trauma there are reductions in the vasodilatory response to nitric oxide (NO), cGMP, cAMP, and prostanoids and/or increases in vasoconstrictive substances such as endothelin-1. Furthermore, in areas of hemorrhage rapid metabolism of the potent vasodilators NO to nitrate/nitrite and adenosine by adenosine deaminase released from lysed erythrocytes occurs. Whether or not augmenting post-insult CBF after TBI or ischemia represents a clini-cally relevant therapeutic target remains to be seen; however, pre-clinical studies, for exam-ple using L -arginine the substrate for NO production or endothelin-1 antagonists have shown promise in experimental models of brain injury.
Ischemia may also occur when CBF is not below the ischemic threshold, under conditions of increased metabolic demands – “relative ischemia”. Increases in metabolic demands, related to profound excitation and excess glutamate, as refl ected by increases in brain tissue and CSF lac-tate, early after TBI have been reported. A common example where this may occur is during post-insult seizures, which may have a prevalence of 30% in pediatric patients after TBI and 20–30% after adult cardiac arrest. As such, prophylactic administration of antiepiletics, such as levtiracetaram or fosphenytoin, should be considered. Alternatively, continuous EEG monitoring can be utilized, particularly when muscle relaxants are required as part of the ICU management.
Excitotoxicity
Excitoxicity describes the process by which supraphysiologic amounts of glutamate and other excitatory amino acids produce neuronal damage. Typically, a highly regulated bal-ance between excitatory and inhibitory inputs results in normal neurological function. After brain injury, such as from trauma or ischemia, this balance is altered and results in a pre-dominance of excitatory amino acids, primarily glutamate. Systemic administration of toxic levels of glutamate can produce neuronal death in vivo . Glutamate concentrations above the threshold for producing neuronal damage are well described after ischemia and TBI. Increases in glutamate and other excitatory amino acids have been reported in CSF from infants and children after TBI vs. controls, and there is an independent association between increased CSF glutamate and infl icted injury from child abuse.
Pretreatment with glutamate receptor antagonists such as phencyclidine and MK-801 improve neurological outcome after ischemia and TBI in laboratory animals. Other anti-excitotoxic strategies reported to be effective in pre-clinical studies include magnesium, hypothermia, and pentobarbital. Disappointingly, clinical trials with anti-excitotoxic thera-pies have been unsuccessful. This may be related to study designs where therapies have been applied to all patients with TBI rather than those with excitotoxicity, treatments initiated too late (excitotoxicity may present a narrow therapeutic window), or undesirable specifi c or nonspecifi c effects of the agents tested. Advances in therapeutic drug monitoring for brain injured patients, particularly measurement of CSF or extracellular fl uid (ECF) levels of glu-tamate (and other excitatory amino acids) may help in this regard.
Cerebral Edema
Brain swelling is a hallmark feature after severe brain injury and results in the development of intracranial hypertension, which can have devastating consequences. A contributory pathologic role for brain swelling is much better established after TBI compared with
Cerebral ischemia can be due to hypoperfusion and/or hypermetabolism.
Post-TBI hypoperfusion can be caused by vascular injury, thrombi, vascular dysregulation, and compression ischemia.
Incidence of seizures after TBI or cardiac arrest can approach 30%.
Brain swelling is a hallmark feature of severe TBI, and is refl ected clinically as intracranial hypertension.

647 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
cerebral ischemia, however, cerebral edema may certainly also play a detrimental role in subthreshold ischemia. Cerebral swelling and accompanying intracranial hypertension con-tributes to secondary damage in at least two ways. As discussed above, intracranial hyperten-sion can compromise cerebral perfusion through small arteries, arterioles, and capillaries leading to secondary ischemia, essentially producing intracranial “compartment syndrome”. In addition, under conditions of extremely high or rapidly increasing ICP it can produce the devastating consequences of deformation through herniation syndromes compressing major arteries choking off blood supply. Intracranial hypertension results from an increase in intrac-ranial volume from one of many potential sources. After TBI, mechanical injury and hemor-rhage can produce epidural, subdural or parenchymal hematoma formation – under most conditions this is addressed by immediate surgical evacuation. There are also several other important mechanisms related to both trauma and ischemia involved in the development of intracranial hypertension. These mechanisms are particularly relevant to intensivists, as they are key targets of currently available clinical interventions. These are related to brain swell-ing from vasogenic edema, cytotoxic edema, an increase in tissue osmolar load, or vascular dysregulation with swelling secondary to an increase in cerebral blood volume (CBV).
Data suggest that brain swelling after severe TBI results from cytotoxic rather than vasogenic edema. Diffusion-weighted MRI can localize the increase in brain water after dif-fuse TBI in laboratory animals where a decrease in the apparent diffuse coeffi cient after injury suggested predominantly cellular swelling, rather than vasogenic edema, contributing to intracranial hypertension. In human studies, increases in brain water were generally associated with reduced (not increased) CBV. Thus, cytotoxic edema and cellular swelling, rather than increased CBV, appears to be the predominant contributor to cerebral swelling after TBI.
There appear to be at least fi ve mechanisms for edema formation in the injured brain. In addition to increasing CBV, vasogenic edema may result in increased tissue edema in the extracellular space as a result of increased oncotic pressure, particularly in combination with blood-brain barrier (BBB) disruption. Astrocyte swelling occurs during the uptake of sub-stances such excessive glutamate and sodium in response to injury. Glutamate uptake is coupled to glucose utilization via a sodium/potassium ATPase, with sodium and water accu-mulation in astrocytes. Disturbances in aquaporin proteins forming water channels may also result in excessive water accumulation in astrocytes. Swelling of neurons and other cells can also result from ischemia- or trauma-induced ionic pump failure. Finally, osmolar swelling may also contribute to edema formation in the extracellular space, particularly in contusions, secondary to sequestration of osmogenic substances such as ions, proteins, and drugs such as mannitol extravasated at a time when the BBB is disrupted, forming subsequent to rees-tablishment of the BBB. As such, the role of BBB in the development of posttraumatic edema may be dynamic – particularly in the setting of cerebral contusion. One possibility is that as macromolecules from dying or dead cells are degraded within injured brain, the osmolar load in the contused tissue increases. When the BBB reconstitutes a considerable osmolar driving force for the local accumulation of water develops, resulting in the marked swelling so often seen in and around cerebral contusions.
In some cases, particularly in younger children, increases in CBV can be seen after TBI and contribute to intracranial hypertension. When an increase in CBV is seen, it may be in response to local increases in cerebral glucose utilization. In regions with increases in gluta-mate levels, increases in glucose metabolism are observed because astrocyte uptake of gluta-mate is coupled to glycolysis rather than oxidative metabolism. Global oxidative metabolism is generally depressed by ~50% in comatose victims of severe TBI and ischemic injury in the ICU. In contrast, global glucose metabolism is minimally depressed or occasionally increased in subgroups of adult TBI patients who eventually are classifi ed as having a poor outcome.
Aggressive monitoring and early recognition and treatment of physiologic derangements can prevent or lessen the impact of secondary brain injury, these include hypoxemia, hyper/hypocarbia, hyperthermia, seizures, hypotension, hyper/hypoglycemia, and intracranial hypertension. Notably, all of these derangements have direct effects on CBV, edema, and/or the balance between CBF and metabolism that may ultimately result in intracranial hyper-tension, and all of which represent principal targets for titration of supportive and ICP-directed therapies in the ICU. These are discussed in more detail below.
Vasogenic and cytotoxic edema can both contribute to intracranial hypertension after TBI.
Vasogenic edema related to increased CBV may be a more prominent feature in children vs. adults after TBI.
Prevention of secondary injury requires early recognition and treatment of hypotension, hypoxemia, hyper/hypocarbia, hyperthermia, seizures, hyper/hypoglycemia, and intracranial hypertension.

648 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
Global Cerebral Ischemia and Reperfusion Since most of the discussion of secondary insult above relates primarily to TBI or focal ischemia, a brief discussion of events related to global ischemia, e.g. as a consequence of cardiac or cardiorespiratory arrest is warranted. Essentially all of the currently available therapeutic strategies for patients with acute injury target reversibly injured or uninjured brain. For example, an intervention such as osmotherapy does nothing for irreversibly dam-aged brain at the necrotic core of a contusion after TBI or infarct after stroke, but reducing subsequent swelling of damaged tissue may reduce damage to surrounding reversibly injured tissue – often referred to as “penumbra”, or prevent damage to brain distant from the site of initial injury. Obviously after a severe global ischemic insult, the entire brain is at risk for irreversible damage and therefore pathologic processes and treatment strategies differ com-pared with focal insults.
No- or low-fl ow states, seen during asystole or the period leading up to asystole, respec-tively, result in cellular hypoxia and depletion of energy substrates. Consciousness is lost and oxygen stores are exhausted 20 s after normothermic cardiac arrest while glucose and adenosine triphosphate (ATP) stores are lost within 5 min. After cardiac arrest, no fl ow dura-tions of ³ 5 min are associated with cerebral ischemia. Post-ischemia, a transient cerebral hyperemia occurs typically within the fi rst several minutes after restoration of spontaneous circulation, which may be followed by protracted global hypoperfusion lasting several days. This hypoperfusion may represent a therapeutic target, e.g. induced hypertension during the period of reperfusion has been shown to improve outcome in dogs after cardiac arrest, and hypertension is associated with more favorable outcome in humans after cardiac arrest.
Reperfusion injury after return of spontaneous circulation (ROSC) involves a compli-cated cascade of events that begins with membrane depolarization, calcium infl ux, NMDA activation, glutamate release, acidosis, mitochondrial dysfunction, and activation of lipases, proteases, and nucleases. This sets the stage for reoxygenation injury with cascades that involve oxygen and nitrogen radicals, iron, catecholamines and other excitatory amino acids, calcium overload, poly (ADP-ribose) polymerase activation, energy failure, mitochondrial damage, and DNA fragmentation. These processes, regulated by several signaling pathways, can ultimately lead to cell death.
Variable degrees of cerebral swelling occur, and ICP is increased compared with normal, but occurrences above 20 mm Hg are probably less common except after prolonged isch-emia and severe injury. This may be related to several differences between TBI and focal ischemia versus global ischemia. Two possibilities include differences in BBB disruption between TBI and global ischemia. BBB disruption is a predominant feature of TBI, but is generally felt to be minimal or at least very brief after global ischemia. Another factor may be the relative contribution of vasogenic edema after severe TBI versus global ischemia. These differences, ischemic injury to all areas of the cerebrum, cerebellum, and brainstem, and other factors account for the profound differences in outcome seen clinically, with far less favorable outcome seen after prolonged global ischemia compared with TBI, focal intracerebral hemorrhage, or focal ischemia.
NEUROINTENSIVE CARE MONITORING
In the era of modern intensive care, invasive monitoring is generally considered standard of care. This includes continuous monitoring of arterial blood pressure, central venous pres-sure, temperature, carbon dioxide, and oxygenation, and frequent assessment of electro-lytes, serum osmolarity, and arterial blood gases. Although no modality specifi c for “neuro” monitoring is yet to be validated by large, randomized, clinical trials, monitoring of ICP for TBI is considered standard of care. More contemporary neuromonitors are now rou-tinely used in many centers, all with strengths and weaknesses. These monitors if inter-preted correctly provide adjunctive information that may be useful for optimizing care of the brain injured patient, via detection of secondary brain injury or precipitators of secondary brain injury, assisting in applying treatment strategies, and/or evaluating the effectiveness of
Typical early events after cardiac arrest: low fl ow – no fl ow – cellular hypoxia – loss of consciousness – loss of glucose and ATP stores within 5 min – ROSC – transient hyperemia – low fl ow.
Glasgow Coma Scale score Eye opening 4 Spontaneous 3 To speech 2 To pain 1 None Verbal response 5 Oriented 4 Confused 3 Inappropriate words 2 Grunting 1 None Motor response 6 Follows commands 5 Localizes pain 4 Withdraws to pain 3 Abnormal fl exion 2 Abnormal extension 1 Flaccid

649 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
interventions – i.e. therapeutic drug (or intervention) monitoring. Normal and some critical threshold values for currently used neurointensive care monitors are shown in Table 31-1 .
Non-invasive Monitoring Basic bedside monitoring is also essential for severely brain-injured patients and should include serial Glasgow coma scale (GCS) score and neurological examinations. The neuro-logical examination should also include pupillary and cranial nerve examination. These should be performed by multiple observers including bedside ICU nurses, critical care phy-sicians, neurosurgeons, and/or neurologists. Training of new staff and interval assessment of these simple monitoring measures to reduce inter-observer variability and improve consis-tency and reliability are valuable. Clinical examination is much more imperative when ICP, continuous EEG or other brain monitoring are not utilized.
Intracranial Pressure The sine qua non of neurointensive care monitoring for TBI patients, both adult and pediat-ric, is ICP monitoring. Increased ICP is defi ned as >20 mm Hg (resting normal 5–10 mm Hg). This threshold is the same for both children and adult patients, and is based on a compilation of studies in both identifying 20 mm Hg as a cutoff for identifi cation of poor outcome. Specifi cally, causality was not shown but clear associations were detected between ICP >20 mm Hg and mortality. Importantly, elevated ICP can be inferred, but cannot be defi ni-tively determined clinically, by computerized tomography (CT) scan of the head, or by other currently available non-invasive monitors. Consensus was drawn from adult data regarding which patients are most at risk for raised ICP and should receive ICP monitoring: either GCS score 8 or less, an abnormal CT scan (hematoma, contusion, cerebral edema, compressed basal cisterns), or rapidly declining level of consciousness. Currently available ICP moni-tors, defi ned by where the catheter tip is placed and the mode of pressure measurement, include ventricular, subarachnoid, epidural, subdural, and parenchymal placement and fi beroptic strain gauge and pressure transducer based systems. Advantages of the extraven-tricular drain catheter are low cost, straightforward design (fl uid column and pressure
Glasgow Coma Scale score modifi ed for infants Eye opening 4 Spontaneous 3 To speech 2 To pain 1 None Verbal response 5 Coos, babbles 4 Irritable 3 Cries to pain 2 Moans to pain 1 None Motor response 6 Normal movements 5 Withdraws to touch 4 Withdraws to pain 3 Abnormal fl exion 2 Abnormal extension 1 Flaccid
NORMAL CRITICAL
Cerebral Blood Flow Global 52 mL/100 g/min <18–20 mL/100 g/min Cortical 80 mL/100 g/min
Oxygenation Jugular venous O 2 saturation (SjVO 2 ) 55–71% <50% Arterio-venous O 2 difference (AVDO 2 ) 4.5–8.5 vol% Brain tissue pO 2 (pbtO 2 ) 20–40 mm Hg <8.5–10 mm Hg
Metabolism Cerebral metabolic rate for O 2 (CMRO 2 ) 3.4 mL/100 g/min Cerebral metabolic rate for glucose 0.325 m mol/g/min Cerebral metabolic rate for lactate −0.02 m mol/g/min Extracellular fl uid glucose concentration 1.7 m mol/L Extracellular fl uid lactate concentration 2.9 mmol/L Extracellular fl uid pyruvate concentration 166 mmol/L
Adapted from Robertson and Hlatky ( 2005 ) – see Suggested Readings
TABLE 31-1
VALUES FOR CEREBRAL BLOOD FLOW, OXYGENATION, AND METABOLISM IN ADULTS
650 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
transducer), ease of recalibration, and the capacity to drain CSF as a mode of therapy. Complications regardless of device are rare and include infection (1–10% of cases) and hemorrhage (1–2%). Currently the only major contraindication is severe uncorrected coagu-lopathy. In the era of “goal directed therapy”, it is generally recommended that therapeutic interventions be targeted to achieve an ICP <20 mm Hg.
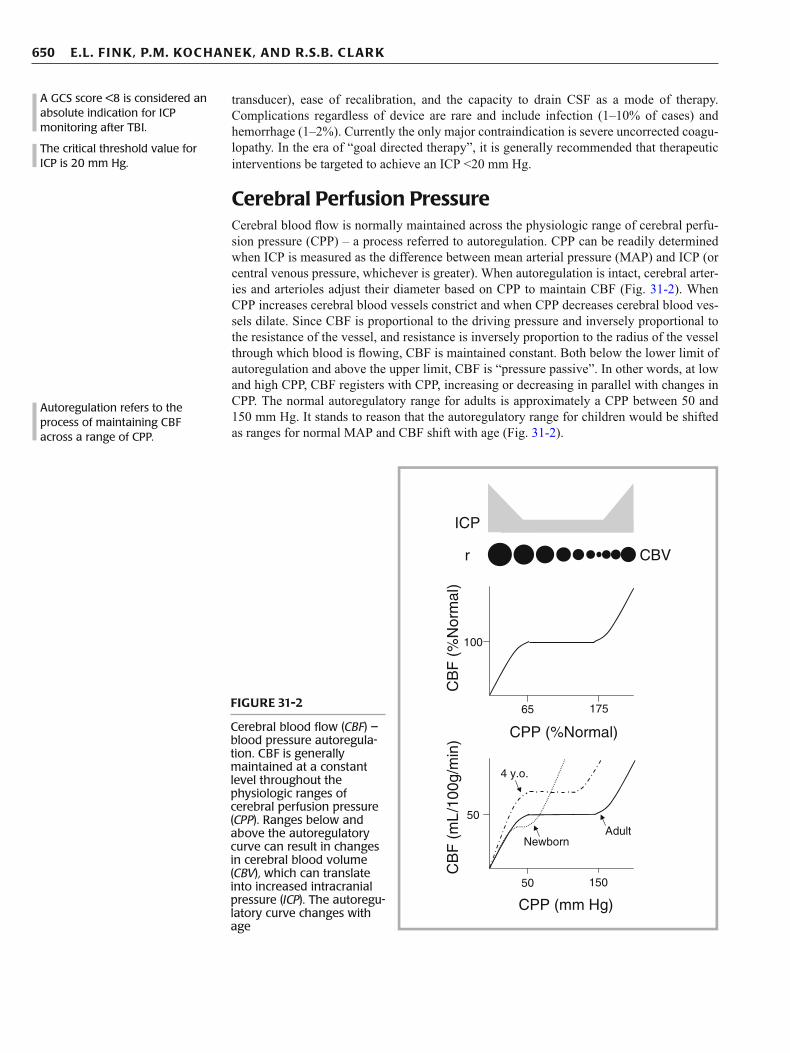
Cerebral Perfusion Pressure Cerebral blood fl ow is normally maintained across the physiologic range of cerebral perfu-sion pressure (CPP) – a process referred to autoregulation. CPP can be readily determined when ICP is measured as the difference between mean arterial pressure (MAP) and ICP (or central venous pressure, whichever is greater). When autoregulation is intact, cerebral arter-ies and arterioles adjust their diameter based on CPP to maintain CBF (Fig. 31-2 ). When CPP increases cerebral blood vessels constrict and when CPP decreases cerebral blood ves-sels dilate. Since CBF is proportional to the driving pressure and inversely proportional to the resistance of the vessel, and resistance is inversely proportion to the radius of the vessel through which blood is fl owing, CBF is maintained constant. Both below the lower limit of autoregulation and above the upper limit, CBF is “pressure passive”. In other words, at low and high CPP, CBF registers with CPP, increasing or decreasing in parallel with changes in CPP. The normal autoregulatory range for adults is approximately a CPP between 50 and 150 mm Hg. It stands to reason that the autoregulatory range for children would be shifted as ranges for normal MAP and CBF shift with age (Fig. 31-2 ).
A GCS score <8 is considered an absolute indication for ICP monitoring after TBI.
The critical threshold value for ICP is 20 mm Hg.
Autoregulation refers to the process of maintaining CBF across a range of CPP.
r
ICP
CBV
CB
F (
%N
orm
al)
100
CPP (%Normal)
CB
F (
mL
/100
g/m
in)
50
CPP (mm Hg)
50 150
Adult
4 y.o.
Newborn
65 175 FIGURE 31-2
Cerebral blood fl ow ( CBF ) – blood pressure autoregula-tion. CBF is generally maintained at a constant level throughout the physiologic ranges of cerebral perfusion pressure ( CPP ). Ranges below and above the autoregulatory curve can result in changes in cerebral blood volume ( CBV ), which can translate into increased intracranial pressure ( ICP ). The autoregu-latory curve changes with age

651 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
The threshold value for critically low CPP after TBI has been estimated to be 60 mm Hg in adult patients. This value is based on accumulated data from multiple studies demonstrat-ing an association between poor outcome and CPP <60 mm Hg. Age-related differences also exist in the specifi city of ICP and CPP in the fi rst 6 h after severe TBI for predicting out-come. Common practice typically assigns CPP thresholds of 60, 50, and 40 mm Hg for adolescents, young children, and infants, respectively; although evaluation of CPP thresh-olds associated with poor outcome support a value of 40 mm Hg for children of all ages. This issue is made complicated by studies showing that hypotension after TBI (to MAP below 50–60 mm Hg even if ICP was normal) is one of the most powerful harbingers of poor out-come, and studies suggesting that CPP thresholds may even be higher in children than in adults, possibly related to higher normative CBF. Low CPP after brain injury can result in regional cerebral ischemia if profound, below the ischemic threshold, and regional ischemia in-and-around injured brain when global CBF is near but above the ischemic threshold. Ischemia as discussed above is an important cause of secondary brain injury. Accordingly, continuous assessment of CPP is a valuable means of detecting risk for secondary brain injury and is useful for its prevention and treatment if it occurs.
Cerebral Blood Flow Cerebral blood fl ow can be measured directly using techniques such as stable Xenon com-puterized tomography (CT), intravenously injected radioactive Xenon with external detec-tors, positron emission tomography (PET), perfusion MRI, or the Kety–Schmidt technique. Up until the late 1990s, the most widely available and user-friendly method for quantifying regional and global CBF values in humans was stable Xenon CT. A mandate by the Food and Drug Administration (FDA) shelved stable Xenon CT until safety could be established, however, resulting in the inability to use Xenon CT studies not directly related to safety evaluation or as part of a research study with informed consent obtained. As such, surrogate measures refl ecting CBF in humans have been more frequently applied.
For adults, global CBF of 50 mL/100 g brain tissue/min is considered normal. Normal values for CBF are age dependent (Fig. 31-3 ) with a nadir in newborn infants of approxi-mately 40 mL/100 g brain tissue/min. CBF peaks around 4 years-of-age to values of approx-imately 80 mL/100 g brain tissue/min, declining to adult values during adolescence. As mentioned above, existing studies support a critical value for low CBF of <20 mL/100 g brain/min for any age patient. Similar to CPP, threshold values may vary depending upon the age of the patient, particularly in the 4–8 year old patient where normal CBF values are more than double the adult norms, and in newborns and very young infants, where CBF values are below adult norms.
Cerebral Perfusion Pressure
CPP MAP ICP= − if CVP > ICP then
CPP MAP CVP= −
Critical CPP after TBI in adults and adolescents, young children, and infants is considered to be 60, 50, and 40 mm Hg, respectively.
Normal CBF is age-dependent, peaking at 4 years-of-age.
Critically low CBF can result in irreversible brain damage is <20 mL/100 g brain/min in adults.
120
Age (y)
2 4 6 8 10 12 14 16 18 20
CB
F (
mL
/100
g/m
in)
0
20
40
60
80
100
?
Ischemic threshold
? FIGURE 31-3
Age dependency of cerebral blood fl ow ( CBF ). It is not known whether the ischemic threshold, defi ned as 20 mL/100 g brain/min in adults, is also age-dependent

652 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
Transcranial Doppler Ultrasonography Cerebral blood fl ow can be indirectly assessed using transcranial Doppler (TCD) ultrasonog-raphy. It is a non-invasive, bedside method that measures middle cerebral arterial blood fl ow velocity as a surrogate for CBF itself. This technique correlates with CBF measured with xenon CT. The non-invasive nature of the technique is its biggest advantage. Disadvantages include the inability to translate CBF velocity to fl ow and possible discrepancies between middle cerebral artery “fl ow” and regional CBF, particularly in heterogeneous insults such as TBI. Changes in CBF velocity can be tracked within individual patients, however, and can be useful particularly when instituting interventions that may affect global CBF, e.g. hyper-ventilation. TCD has been used in assessing stroke risk in patients with sickle cell disease, after TBI to estimate ICP, after birth asphyxia to predict neurological outcome, and it has been demonstrated that abnormal cerebral autoregulation precedes cerebral edema in chil-dren presenting with diabetic ketoacidosis and altered mental status.
Cerebral Metabolic Monitoring Cerebral blood fl ow is normally tightly coupled to metabolism, and can be determined using PET or calculated by the product of CBF and oxygen or substrate (mainly glucose) extraction. For example, cerebral metabolic rate for oxygen can be calculated by the following formula:
2 2CMRO CBF AVDO= × where
2 2 2AVDO CaO SjvO= − For the most part patients in the intensive care unit have arterial oxygen saturations near 100% refl ecting the primary contribution to arterial oxygen content (CaO 2 ). Thus, the arteriovenous oxygen difference (AVDO 2 ) is infl uenced mostly by jugular venous oxygen saturation (SjvO 2 ). Currently, CBF can only be directly measured intermittently, if at all. Accordingly, monitor-ing of SjvO 2 may provide a bedside estimate of the balance of global oxygen delivery and cerebral metabolism. Jugular venous oxygen saturation can be continuously measured with an internal jugular vein fi beroptic catheter placed cephalad into the jugular bulb.
Normal values in adult males range from 55% to 71%, and critical values suggestive of brain tissue ischemia are felt to be <50%. Critically low SjVO 2 can be secondary to increased ICP, systemic hypoxemia, hypotension, anemia, or a combination of these. SjvO 2 <20% can be indicative of irreversible ischemic injury. High SjVO 2 values indicate that cerebral oxygen delivery exceeds consumption, as a result of either increased CBF – referred to as “hypere-mia”, reduced metabolism – an extreme example would be as in the case of brain death, or an uncoupling of CBF and metabolism. Some studies found that SjvO 2 did not substantially infl uence patient management above and beyond systemic ICP monitoring. There are also considerations in terms of laterality, where bilateral placement of jugular venous catheters can often yield signifi cant variance between right and left jugular bulb catheters drawn simul-taneously in the same patient. Major complications in children related to SjvO 2 monitoring are carotid artery puncture, catheter malposition, and bacterial colonization with and without bacteremia. However, SjvO 2 monitoring may be useful in predicting outcome, given that multiple (<2) SjvO 2 desaturations are associated with poor neurological outcome. The occur-rence of high SjvO 2 does not appear to infl uence outcome. Nonetheless, SjvO 2 can provide a useful adjunct to other neurointensive care monitoring devices, particularly in complicated patients where second tier therapies for refractory intracranial hypertension are considered. For example, SjvO 2 monitoring may help direct therapies such as hyperventilation as one can reduce the degree of hyperventilation if SjvO 2 drops below critical thresholds.
Similar to cross-brain utilization of oxygen, cross brain utilization of glucose (and any other metabolites) can also be measured when a jugular venous catheter is in place. Coupled with measurement of CBF, cerebral metabolic rate for glucose (CMRGlu) can be calcu-lated. While the utility of CMRGlu in brain injured patients is far less understood than CMRO 2 , global CMRGlu is generally depressed after TBI and ischemia, similar to CMRO 2 .
TCD non-invasively and indirectly assesses CBF via middle cerebral arterial blood fl ow velocity.
Low SjvO 2 ( <50%) can be secondary to increased ICP, hypoxemia, hypotension, and/or anemia and correlate with poor outcome after TBI.
High SjvO 2 ( > 70%) may represent uncoupling of CBF and metabolism, or “hyperemia”.
2 2CMRO CBF AVDO= ×

653 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
Since heterogeneous patterns of injury, CBF, and metabolism can occur after TBI, interpret-ing global measurements of CMRGlu, and CMRO 2 for that matter, can be problematic. Regional increases in glucose utilization have been observed in TBI patients using positron emission tomography (PET) scanning, often to values felt to be consistent with “hyperglycolysis”.
In children with severe traumatic brain injury, two or more measurements of SjvO 2 <= 55% were associated with a poor neurologic outcome. Further studies are needed to rec-ommend the use of these variables as a guideline to optimize treatment.
Brain Tissue Oximetry Brain oxygenation can be measured using near infrared spectroscopy (NIRS). The main advantages of NIRS are that the technique is noninvasive and provides a continuous readout. Current disadvantages include relatively limited depth of penetration of the readout (milli-meters), global rather than focal information, and lack of defi nitions in terms of target and critical values. However, measurement of continuous oxygen saturation on the brain surface can provide relative real time alterations in brain oxygenation, which can be useful in terms of titration of therapies. NIRS performed in the fi rst 48 h after neonatal asphyxia measuring cerebral oxygen saturation and fractional tissue oxygen extraction is predictive of outcome at 3 months. NIRS also detected changes in cerebral hemodynamics in pediatric TBI patients, correlating with events such as ICP spikes and seizures.
Measurement of brain tissue oxygen partial pressure (PbtO 2 ) using a separate probe inserted directly into the brain is another invasive way of estimating alterations in CBF and metabolism after brain injury. These fi beroptic catheters measure dissolved oxygen tension, and are also capable of measuring temperature, carbon dioxide and pH. Normal values of PbtO 2 are 20–40 mm Hg, and critical values are 8–10 mm Hg. Studies have reported a cor-relation between critical PbtO 2 values and ischemia detected via PET scan and SjvO 2 moni-toring. Optimal placement of the sensor is somewhat controversial, but valuable information may be gained when the tip of the catheter is placed in viable tissue at risk for irreversible damage, or the penumbra. Alternatively, the sensor could be placed in “normal” brain distant from contusions to provide an estimate of global brain oxygenation. Most experts agree only on avoiding placement of the sensor into the center of a contusion as monitoring PbtO 2 in dead tissue would be misleading. To date, there has been no study examining the utility of PbtO 2 monitoring that focused directly on pediatric patients, although there have been many published reports in primarily adult studies that included children. It does appear based on preliminary work that normobaric-normoxia improves the metabolic milieu after TBI.
Cerebral Microdialysis Cerebral microdialysis uses intermittent or continuous sampling of extracellular fl uid to mea-sure changes in brain chemistry, and is based on the diffusion of water-soluble substances through a semipermeable membrane. It involves insertion of a fi ne catheter in the brain that has a dialysis membrane perfused with physiologic solution at low fl ow provided by a preci-sion pump. Most experience examines molecules which are diffusible below 20,000 Da, how-ever, larger molecules up to 100,000 Da may be measured depending upon the cutoff size of the semipermeable membrane. Assays are employed to analyze dialysate for molecules such as glucose, lactate, neurotransmitters, drugs, and markers of tissue damage and infl ammation. Currently microdialysis in brain is being utilized primarily as a research tool, and its place as a point-of-care monitor in the clinical setting remains to be seen. The potential use of this tool for brain-specifi c therapeutic drug monitoring warrants further investigation.
EEG An electroencephalogram (EEG) is a processed summation of excitatory and inhibitory post-synaptic potentials of cerebral cortex electrical activity, displayed in multiple chan-nels. Information about frequency, amplitude, and location of focal or generalized activity
Global CMRGlu is generally decreased after TBI, although regional increases suggestive of “hyperglycolysis” can be seen.
Brain tissue oxygenation can be measured non-invasively using NIRS.
Brain tissue oxygenation can be measured invasively using a PbtO 2 monitor. Normal values = 20–40 mm Hg in adults.
Brain glucose, lactate, drug levels, and markers of tissue injury can be measured using a microdialysis catheter.

654 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
can be attained using continuous EEG monitoring. This is useful in brain injured patients in terms of detecting non-clinical seizures or seizures in patients receiving muscle relaxants as part of a clinical protocol. Continuous EEG monitoring also has an important role when using high dose barbiturates either for status epilepticus or refractory intracranial hyperten-sion to achieve burst suppression, since administering barbiturates beyond isoelectricity will result in undesirable cardiovascular side effects without further suppression of brain metabolism.
EEG has been used to predict outcome after TBI and cardiac arrest in pediatric patients. In children who survived for 24 h or longer after cardiac arrest a discontinuous EEG, pres-ence of epileptiform spikes, or discharges were associated with poor outcome. Somatosensory evoked potentials (SSEPs) also have been demonstrated to possess adequate sensitivity for predicting unfavorable outcome. Additional assessment of continuous EEG is warranted in the pediatric ICU.
Computed Tomography Head computed tomography (CT) is often the fi rst choice of imaging modality after TBI or cardiac arrest since it is a readily available and rapid test (Fig. 31-4 ). Head CT after TBI is indicated whenever there is a concern for moderate to severe TBI, and has value in detecting skull fractures, extra-axial hematomas, and parenchymal brain injury. Head CT is often in the decision tree in terms of whether or not ICP monitoring may be warranted, and thus greatly assists in patient management. After mild TBI, the prevalence of intracranial CT scan abnormalities is 5–30% in patients presenting with a GCS score ³ 13. About 1% of all patients diagnosed initially with mild TBI require neurosurgical intervention. The reliability of skull fracture detected using plain fi lms to detect intracranial lesions is poor. Relationships between CT scan characteristics and outcome after TBI have been reported, although this can prove diffi cult considering the heterogeneity of intracranial and extracranial injuries, as well as differences in mechanism of injury and development when considering children. There appears to be good correlation between “reversal sign” (diffusely decreased density of cere-bral cortical gray and white matter with a decreased or lost gray/white matter interface in contrast to the relatively greater density of the cerebellum and basal ganglia) and poor out-come in pediatric patients with TBI and hypoxic-ischemic encephalopathy. The utility of a repeat head CT scan within 24–36 h after admission in pediatric patients with moderate to severe TBI is unlikely to yield any change in therapy unless clinically indicated. Indications include signifi cant clinical change and the need to rule new hemorrhage or herniation, and verifi cation of placement of invasive monitoring devices.
After hypoxic-ischemic injury, a normal initial CT scan is common in the majority of comatose patients, and does not reliably predict neurological outcome. On the other hand, an abnormal initial or follow-up CT can indicate higher probability of an unfavorable neuro-logical outcome. Therefore, the role of head CT after cardiac arrest, drowning, or other injuries associated with cerebral ischemia is limited to ruling out trauma or intracerebral hemorrhage as a cause of the cardiac arrest. Perhaps magnetic resonance imaging (MRI) and/or magnetic resonance spectroscopy (MRS) will yield more accurate prognostic infor-mation in patients with hypoxic-ischemic encephalopathy.
Magnetic Resonance Imaging/Spectroscopy Compared with head CT, brain MRI is more sensitive for all brain lesions except skull frac-ture and subarachnoid hemorrhage, but has a signifi cantly longer scanning time, often requires sedation and/or muscle relaxants, and other neuromonitoring devices need to be MRI compatible. MRI after brain injury not only provides structural assessment, but can also be used for the measurement of CBF and cerebral blood volume, and can be used to detect edema and potentially “penumbral” tissue or tissue at risk (Fig. 31-4h ). In hypoxic-ischemic injury, a scoring system has been developed for infants that correlates neurological examina-tion with MRI to predict outcome.
EEG is an essential neuromonitor when using barbiturate-induced coma.
Head CT scan can be “normal” immediately following cardiac arrest.
Brain MRI is more sensitive in detecting ischemic injury.

655 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
The addition of MRS allows for the semiquantifi cation of metabolites such as lactate, pyru-vate, glutamate, phosphocreatine, and N-acetylaspartate. Infants with infl icted TBI have elevated lactate by MRS suggesting hypoxic-ischemic in addition to TBI and these patients had worse neurological outcome. MRS has been used as a predictor of neurological outcome in children after brain injury. MRI in combination with MRS after severe perinatal asphyxia is felt to be powerfully predictive of neurological outcome. Lactate levels can remain ele-vated up to 1 week after asphyxial insult in children.
FIGURE 31-4
Head computerized tomography (CT) and brain magnetic resonance imaging (MRI) studies. ( a ) Diffuse axonal injury in a 14 year old after a motor vehicle accident. CT scan on arrival was negative for intracranial pathology. The patient’s GCS score declined from 10 to 8 warranting this repeat CT scan. There are bilateral multifocal patchy hyper- densities at the gray-white junction and in white matter and right parietal soft tissue swelling. ( b ) A large right thalamic and intraventricular hemorrhage surrounding a mass lesion in a 9 year old who presented with headache and seizures. ( c ) Acute left convexity subdural hematoma with mass effect extending into the interhemispheric fi ssure in an 8 month old. ( d ) Severe, diffuse cerebral edema in a 6 week old. with infl icted TBI. ( e ) Penetrating TBI with foci of intraparenchymal hemorrhage and signifi cant surrounding edema bilaterally. In addition, there is subdural hemorrhage, scattered subarachnoid hemorrhage, and bilateral parietal bone fractures. A right-frontal EVD is in place. ( f ) A 12 year old patient after motorcycle accident who required bilateral frontal craniectomies for refractory intracranial hypertension. There is diffuse edema with ischemic changes and decreased gray-white differentiation. Note the EVD on the right and a PbtO 2 monitor probe on the left. ( g ) Chronic changes after severe hypoxic-ischemic encephalopathy showing signifi cant atrophy and ventricular dilatation. ( h ) Early hypoxic-ischemic enceph-alopathy. Brain MRI 1 week after a 25 min out-of-hospital cardiac arrest in a 17 year old patient showing hyperintense T2 signal within the bilateral occipital lobes, basal ganglia, and perirolandic regions as well as cytotoxic edema in bilateral occipital lobes and putamin consistent with anoxic injury

656 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
CLINICAL MANAGEMENT GUIDELINES
Traumatic Brain Injury Acute Management
Management of the patient with TBI begins with the “ABCs” (Airway-Breathing-Circulation) of resuscitation in order to minimize and/or prevent secondary brain injury (Fig. 31-5 ). It is important to fi rst confi rm or obtain an open airway. Cervical spine precau-tions should always be observed, given that approximately 8% of patients with TBI may also have cervical spine injuries. Indications for endotracheal intubation include a GCS score of <8, deteriorating level of consciousness, lack of airway protective refl exes, respi-ratory insuffi ciency or failure, cardiovascular instability, or to facilitate obtaining CT scans. If intubation is warranted, careful choice of medications prior to intubation and the use of cricothyroid pressure (Sellick maneuver) to prevent over-distension of the stomach and aspiration are imperative. Oral (vs. nasal) endotracheal intubation and the use of sedation and neuromuscular blockade are generally recommended for pediatric patients in order to shorten procedure time and increase intubation success rate. Neuromuscular blocking agents and sedatives are indicated for all TBI patients with the exception of those where airway diffi culties are anticipated (such as patients with facial or laryngotracheal trauma) or those presenting in cardiac arrest. The choice of neuromuscular blocking agents and sedatives are typically institution dependent.
Control of oxygenation and ventilation are the next goals in the acute management of TBI patients. The target for PaCO 2 is 35 mm Hg. Typically, CBF is tightly regulated by PaCO 2 – the loss of CO 2 reactivity is seen in only the most severely injured patients. Generally CBF changes by 3% for every mm Hg change in PaCO 2 , increasing as PaCO 2 increases and vice versa (Fig. 31-6 ). Accordingly, changes in CBF can result in increased cerebral blood vol-ume (due to cerebrovasodilation) and increased ICP. If intracranial compliance is low, as may be seen particularly early after TBI, moderate increases in PaCO 2 could result in hernia-tion. In contrast, hyperventilation leading to hypocarbia could result in reduced CBF at a time when CBF is generally low after TBI, with the potential for exacerbating ischemia. Epidemiologic data support maintenance of PaCO 2 above 32 mm Hg, since adult patients after TBI maintained at a PaCO 2 of 32 mm Hg had accelerated neurological recovery
Indications for intubation after TBI include GCS score <8, deteriorating level of conscious-ness, shock, lack of cough or gag, respiratory insuffi ciency, or to facilitate CT scan.
CBF changes by ~3% for every mm Hg change in PaCO 2 .
ABCs
Mini-neuro exam
Emergency Txfor herniationCT scan
PaCO2 32-38, sedation, paralysis if needed
ICP > 20, inadequate CPP
CSF drainage
ICP monitor
Reassess neuro status,cardiorespiratory status
Refractory intracranial hypertension
Barbiturates
Hypothermia
Surgical decompression
Consider
Second Tier Therapies
Osmotherapy(mannitol, hypertonic saline)
Hyperventilation
Induced hypertension
Hyper-osmotherapy
Refractory intracranial hypertensionCombination second tier therapies
FIGURE 31-5
Algorithm for the treatment of severe traumatic brain injury based on the practice and experience at the Children’s Hospital of Pittsburgh
657 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
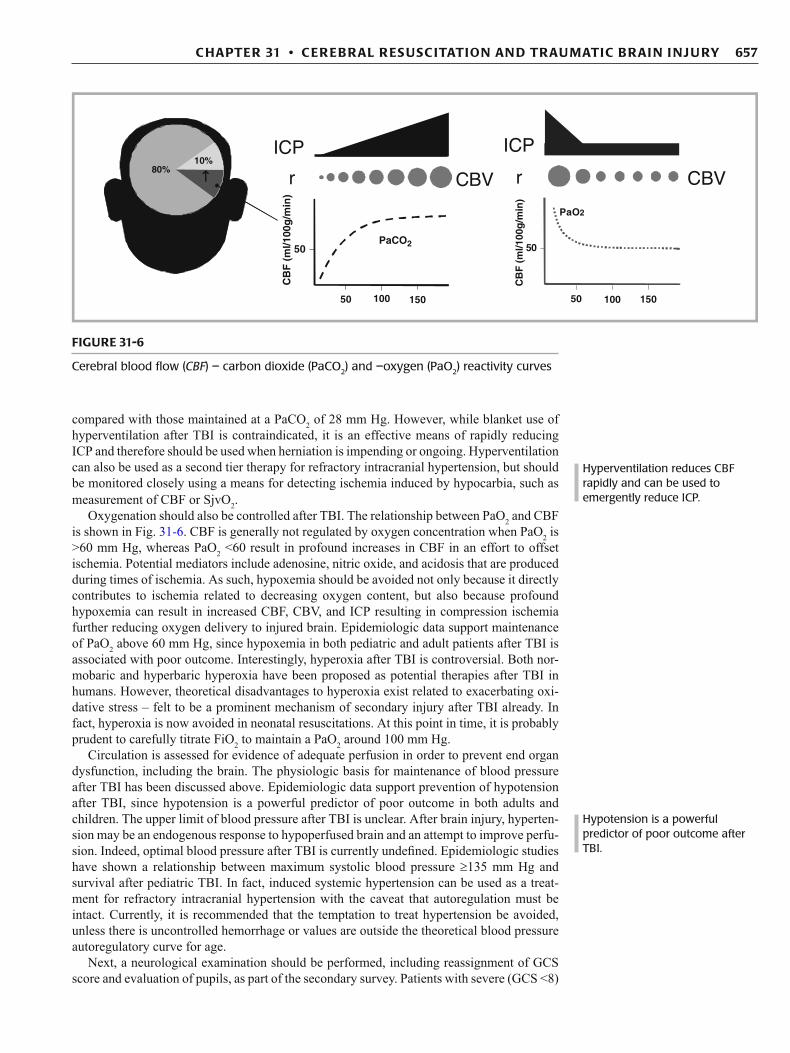
compared with those maintained at a PaCO 2 of 28 mm Hg. However, while blanket use of hyperventilation after TBI is contraindicated, it is an effective means of rapidly reducing ICP and therefore should be used when herniation is impending or ongoing. Hyperventilation can also be used as a second tier therapy for refractory intracranial hypertension, but should be monitored closely using a means for detecting ischemia induced by hypocarbia, such as measurement of CBF or SjvO 2 .
Oxygenation should also be controlled after TBI. The relationship between PaO 2 and CBF is shown in Fig. 31-6 . CBF is generally not regulated by oxygen concentration when PaO 2 is >60 mm Hg, whereas PaO 2 <60 result in profound increases in CBF in an effort to offset ischemia. Potential mediators include adenosine, nitric oxide, and acidosis that are produced during times of ischemia. As such, hypoxemia should be avoided not only because it directly contributes to ischemia related to decreasing oxygen content, but also because profound hypoxemia can result in increased CBF, CBV, and ICP resulting in compression ischemia further reducing oxygen delivery to injured brain. Epidemiologic data support maintenance of PaO 2 above 60 mm Hg, since hypoxemia in both pediatric and adult patients after TBI is associated with poor outcome. Interestingly, hyperoxia after TBI is controversial. Both nor-mobaric and hyperbaric hyperoxia have been proposed as potential therapies after TBI in humans. However, theoretical disadvantages to hyperoxia exist related to exacerbating oxi-dative stress – felt to be a prominent mechanism of secondary injury after TBI already. In fact, hyperoxia is now avoided in neonatal resuscitations. At this point in time, it is probably prudent to carefully titrate FiO 2 to maintain a PaO 2 around 100 mm Hg.
Circulation is assessed for evidence of adequate perfusion in order to prevent end organ dysfunction, including the brain. The physiologic basis for maintenance of blood pressure after TBI has been discussed above. Epidemiologic data support prevention of hypotension after TBI, since hypotension is a powerful predictor of poor outcome in both adults and children. The upper limit of blood pressure after TBI is unclear. After brain injury, hyperten-sion may be an endogenous response to hypoperfused brain and an attempt to improve perfu-sion. Indeed, optimal blood pressure after TBI is currently undefi ned. Epidemiologic studies have shown a relationship between maximum systolic blood pressure ³ 135 mm Hg and survival after pediatric TBI. In fact, induced systemic hypertension can be used as a treat-ment for refractory intracranial hypertension with the caveat that autoregulation must be intact. Currently, it is recommended that the temptation to treat hypertension be avoided, unless there is uncontrolled hemorrhage or values are outside the theoretical blood pressure autoregulatory curve for age.
Next, a neurological examination should be performed, including reassignment of GCS score and evaluation of pupils, as part of the secondary survey. Patients with severe (GCS <8)
Hyperventilation reduces CBF rapidly and can be used to emergently reduce ICP.
Hypotension is a powerful predictor of poor outcome after TBI.
150150 100100 5050
50
CB
F (
ml/1
00g
/min
)
PaO2
r
ICP
CBV80% ↑↑↑↑10%
80%↑↑↑↑
10%
r
ICP
50
CB
F (
ml/1
00g
/min
)
PaCO2
CBV
FIGURE 31-6
Cerebral blood fl ow ( CBF ) – carbon dioxide (PaCO 2 ) and –oxygen (PaO 2 ) reactivity curves

658 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
or moderate (GCS 8–12) TBI should have an immediate neurosurgical consult (ideally neu-rosurgery is part of the trauma response team) followed by a head CT scan without contrast to quickly assess the presence or absence of obvious intracranial trauma. The GCS score can be diffi cult to interpret in the intubated patient, secondary to prevention of speech and requirement for sedative medications. The GCS measures a patient’s ability to understand and follow commands, so it is especially challenging in infants and toddlers and as such has been modifi ed for these age groups. The CHOP infant coma scale (“Infant Face Scale”) attempts to provide a useful neurological test for children under 2 years of age. Serial exami-nations should be undertaken frequently, with interventions aimed at reducing ICP instituted if there are clinical or radiological signs of herniation syndrome, such as decerebrate or decorticate posturing, anisocoria, or Cushing’s triad (bradycardia, hypertension, and Cheyne–Stokes respirations). Patients with moderate TBI should be admitted to an ICU for serial and frequent neurological evaluations, those with severe TBI should be admitted for neurointensive care monitoring and therapeutic interventions if necessary.
Intensive Care Unit Management
Linchpins of neurointensive care management include maintenance of cardiopulmonary parameters, prevention of secondary injury, and optimizing the milieu for potential neuro-logical recovery. Maintenance of adequate oxygenation, targeted ventilation, and blood pressure are paramount. This is facilitated most accurately in severe TBI patients using inva-sive arterial catheters to measure PaO 2 , PaCO 2 , and continuous blood pressure, and central venous catheters to optimize fl uid balance and administer vasoactive medications when nec-essary. PaCO 2 should be maintained between 32 and 40 mm Hg in patients unless hyperven-tilation is being used for refractory intracranial hypertension. PaO 2 should be maintained between 100 and 200 mm Hg in patients unless hyperoxic therapy is being titrated using CMRO 2 , SjvO 2 , NIRS or PbtO 2 monitoring. Fluid balance should be carefully recorded and euvolemia should be the therapeutic goal. Maintenance fl uid composition should consist of an isotonic, or perhaps hypertonic solution, with hypotonic fl uids avoided (of note, lactated Ringer’s solution is mildly hypotonic). This is based on the fact that within hours after TBI the BBB is generally reestablished and that the intact BBB is impermeable to sodium and other ions but permits free fl ow of water across the BBB. Osmotic gradients are established as discussed above, therefore, isotonic or hypertonic fl uids would reduce the osmotic pres-sure for water to transit into the brain (Fig. 31-7 ). A comparison of hypertonic saline as maintenance fl uid for pediatric patients after TBI demonstrated reduced fl uid requirement and need for ICP-directed interventions versus lactated Ringer’s solution without obvious
Cushing’s triad = bradycardia, hypertension, and Cheyne–Stokes respiration.
FIGURE 31-7
Cerebral edema produced by tissue osmolar load after reestab-lishment of the blood-brain barrier

659 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
side effects. Regardless, serum electrolyte levels and osmolality should be checked fre-quently, and the patient should be monitored for development of SIADH, cerebral salt wast-ing, and diabetes insipidus. In general, hematocrit should be maintained >30% in severe TBI patients. Transfusion thresholds could potentially be guided by NIRS, SjvO 2 , and/or PbtO 2 . Important nursing issues include elevating the head of the bed, pulmonary toilet for the pre-vention of nosocomial pneumonia, and deep venous thrombosis/pressure sore prevention. Related to head of the bed elevation, ICP is minimized when the head is raised, although CBF and CPP were shown to be similar with or without head elevation. It has been sug-gested that the head of the bed be maintained at 30° in patients at risk for intracranial hyper-tension. Hyperthermia should be treated immediately, as even small (1–2°) increases in brain temperature exacerbate damage after TBI in experimental models.
Administration of dextrose in the absence of hypoglycemia is extremely controversial in the fi rst 24–48 h after TBI. Hyperglycemia is associated with worse outcome after TBI in pediatric patients in a “dose-dependent” fashion. Although cause and effect has not been clearly established, all patients in a clinical study presenting with a serum glucose over 300 mg/dL died. Experimental studies show that hyperglycemia at the time of brain insult can exacerbate brain damage, felt to be related to increased lactic acidosis-induced generation of iron catalyzed free radicals. As such, dextrose is typically avoided in patients with brain injury for approximately 24–48 h unless hypoglycemia and/or ketoacidosis occurs. However, infants, who have immature glycogen stores, may require addition of dextrose to intravenous fl uids sooner than older children. An important caveat is that hypoglycemia in brain injured patients, whether traumatic, ischemic, or excitotoxic in nature, should be avoided at all costs, since hypoglycemia alone can result in catastrophic neurological damage. There is evidence that protocolized glucose control reduces mortality in certain patient populations. In adults with isolated TBI, tight glucose control can reduce mean and maximal ICP, without an effect on CPP, and reduce seizure frequency. In contrast, intensive insulin therapy has the potential to increase lactate/pyruvate ratio, glutamate, and CMRO 2 relative to “normal” glucose man-agement, implying detrimental effects of tight glucose control. It is generally recommended that nutrition begin by 72 h after injury. Enteral feeding is preferred; however, parenteral nutrition can be given with careful attention paid to sodium concentration.
ICP-Directed Therapies
Important for TBI patients is the use of ICP monitoring to enable goal-directed therapy, targeting ICP <20 mm Hg as the “goal”. The fi rst question to ask is, “ why monitor ICP? ” While there is no randomized clinical trial (“Class I evidence”) comparing outcomes in pediatric patients with or without ICP monitoring – and there likely will never be one – as noted above there is a powerful, albeit not causative, relationship between ICP >20 mm Hg and poor outcome after TBI. There also is a profound relationship between evidence of sec-ondary ischemia during incidents of intracranial hypertension. Clinically elevated ICP is also associated with depressed level of consciousness. In the present day, ICP remains the primary target for goal-directed therapy in patients with TBI.
Primary or fi rst-tier therapies for severe TBI patients include sedation and paralysis, whose most important function may be facilitation of patient comfort and control of mechan-ical ventilation. Although there are some theoretical benefi ts to using sedatives related to reducing cerebral metabolism, there are those who feel that the reduction in blood pressure caused by the sedatives may actually negate any potential benefi cial effects and perhaps may be undesirable. While the use of muscle relaxants without sedation, even in patients in coma, may seem inhumane, certain centers apply this in adult TBI patients and report outcomes that are comparable or better than many trauma centers. Current guidelines support use of sedatives and neuromuscular blockade in children after TBI, however, if for nothing else to minimize wide variations in physiologic variables such as heart rate, blood pressure, PaCO 2 , PaO 2 , glucose, and ICP. Intermittent dosing of osmolar agents such as mannitol or hyper-tonic saline are also considered fi rst tier therapies for intracranial hypertension. However, requirement for multiple doses would suggest refractory intracranial hypertension, warrant-ing use of second tier therapies.
Hyperglycemia and hypoglycemia after TBI are associated with worse outcome.
Dextrose is typically avoided until 48 h after TBI unless there is hypoglycemia or ketoacidosis,but data are lacking to support this practice.
660 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
Treatment of refractory intracranial hypertension, defi ned as an ICP >20 mm Hg sus-tained for at least several minutes, is often institution dependent and should be tailored to individual patients. Certainly early, non-selective application of ICP-directed therapies to patients with severe TBI with of any of these second tier therapies (Fig. 31-5 ) in clinical tri-als has resulted either in futility, undesirable effects, or even worse neurological outcome, examples including hypothermia, barbiturates, and hyperventilation. Pros and cons for each of these interventions exist, and like anything else in medicine, should be vigilantly applied, monitored, and titrated for each patient. Interventions deserving additional comment include CSF drainage, hyperosmolar therapy, hypothermia, surgical decompression, and barbiturates.
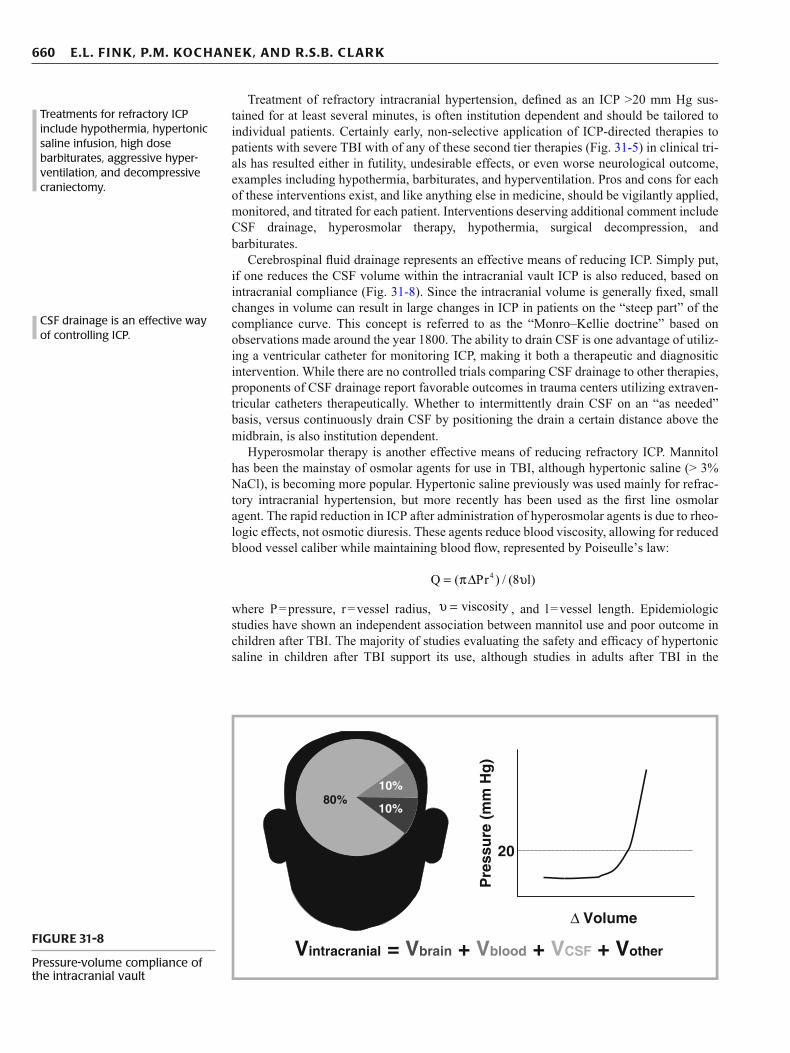
Cerebrospinal fl uid drainage represents an effective means of reducing ICP. Simply put, if one reduces the CSF volume within the intracranial vault ICP is also reduced, based on intracranial compliance (Fig. 31-8 ). Since the intracranial volume is generally fi xed, small changes in volume can result in large changes in ICP in patients on the “steep part” of the compliance curve. This concept is referred to as the “Monro–Kellie doctrine” based on observations made around the year 1800. The ability to drain CSF is one advantage of utiliz-ing a ventricular catheter for monitoring ICP, making it both a therapeutic and diagnositic intervention. While there are no controlled trials comparing CSF drainage to other therapies, proponents of CSF drainage report favorable outcomes in trauma centers utilizing extraven-tricular catheters therapeutically. Whether to intermittently drain CSF on an “as needed” basis, versus continuously drain CSF by positioning the drain a certain distance above the midbrain, is also institution dependent.
Hyperosmolar therapy is another effective means of reducing refractory ICP. Mannitol has been the mainstay of osmolar agents for use in TBI, although hypertonic saline (> 3% NaCl), is becoming more popular. Hypertonic saline previously was used mainly for refrac-tory intracranial hypertension, but more recently has been used as the fi rst line osmolar agent. The rapid reduction in ICP after administration of hyperosmolar agents is due to rheo-logic effects, not osmotic diuresis. These agents reduce blood viscosity, allowing for reduced blood vessel caliber while maintaining blood fl ow, represented by Poiseulle’s law:
4Q ( Pr ) / (8 l)= πΔ υ
where P = pressure, r = vessel radius, viscosityυ = , and l = vessel length. Epidemiologic studies have shown an independent association between mannitol use and poor outcome in children after TBI. The majority of studies evaluating the safety and effi cacy of hypertonic saline in children after TBI support its use, although studies in adults after TBI in the
Treatments for refractory ICP include hypothermia, hypertonic saline infusion, high dose barbiturates, aggressive hyper-ventilation, and decompressive craniectomy.
CSF drainage is an effective way of controlling ICP.
Pre
ssu
re (
mm
Hg
)
20
Δ Volume
80%10%
10%
Vintracranial = Vbrain + Vblood + VCSF + Vother FIGURE 31-8
Pressure-volume compliance of the intracranial vault

661 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
prehospital setting are less convincing. A head to head study examining equimolar doses of mannitol (20%) versus NaCl/dextran (7.5%/6%) suggests that hypertonic saline is more effective in reducing ICP. These factors may result in the replacement of mannitol with hypertonic saline in the future. Most experience after TBI is with the use of 3% NaCl, though higher concentrations, up to 23.4%, are being tested clinically.
Hypothermia is another plausible second tier therapy. Within institutions, hypothermia has been shown to effectively improve neurological outcome after TBI, although multi-center trials did not show effi cacy and even a tendency towards harm. The potential benefi cial effects of hypothermia after TBI include balancing cerebral metabolism and blood fl ow, prevention and/or reduction in cerebral edema, and inhibition of infl ammation and free radical produc-tion. To date, universal application of hypothermia for all patients after TBI cannot be recom-mended. However, hypothermia may be used for treatment of intracranial hypertension as a second tier therapy. Complications of hypothermic therapy such as electrolyte abnormalities, dysrhythmias, and coagulation disturbances should be monitored for and corrected. Recent multi-center studies do not support early application of hypothermia in children after TBI.
Surgical decompression is another effective means of reducing and/or preventing intrac-ranial hypertension. In essence, decompression via unilateral or bilateral craniectomies changes the playing fi eld by increasing intracranial volume and allowing for expansion, throwing the Monroe–Kelly doctrine out the window. Both unilateral and bilateral craniec-tomies have been shown to be effective in reducing ICP in children after TBI. Similar to hypothermia, surgical decompression may be more effective when applied early after TBI as prevention, rather than for treatment during refractory intracranial hypertension.
Barbiturates have been used for decades to treat refractory intracranial hypertension based on the concept that reducing metabolic demands – putting the brain to sleep – can reduce cell death in regions with compromised substrate and oxygen delivery. While barbi-turates certainly reduce cerebral metabolism, this has not yielded improved outcome in patients after TBI. This may be related to undesirable cardiovascular effects of barbiturates, particularly at doses required to achieve burst suppression or “barbiturate coma”. If using high dose barbiturates, monitoring for cardiovascular side effects is essential with immediate correction if/when they occur.
A common theme emerging in terms of choosing second tier therapies for treatment of TBI is that these therapies should be tailored to individual patients, based on physiologic, radiographic, and perhaps demographic data. When using these therapies patients should be rigorously monitored not only for benefi cial effects (ICP), but also for undesirable effects as well. Side effects that may counteract or potentially harm patients, such as hypotension, cerebral ischemia, infection, etc., should be immediately corrected and prevented if possible. These therapies should be tailored to individual patients, based on physiologic, radiographic, and perhaps demographic data.
Cardiac Arrest Acute Management
Similar to treatment of TBI, treatment of the cardiac arrest patient begins with the ABCs of resuscitation. For patients with cardiac arrest, rapid recognition of pulselessness and initiation of cardiopulmonary resuscitation (CPR) improves the likelihood of ROSC, survival, and good neurological outcome. The duration of pulselessness and time required to achieve ROSC are strong predictors of outcome in these patients. In other words, seconds matter and may mean the difference between good and poor outcome. To maximize outcome after out-of-hospital arrests, this mandates a community educated in CPR and rapid response of health care provid-ers. For in-hospital arrests, an organized and effi cient response team is necessary.
Important for pediatric cardiac arrest is establishment of an open airway and ventilation and oxygenation. This has diverged from adult CPR guidelines, where restarting the heart and chest compressions are now the fi rst intervention (“CABs”). We feel that it important to main-tain the ABCs in pediatric cardiac arrests, however, related to differences in etiologies between infants and children versus adults. Infants and children are much more likely to have asphyxia
Hypothermia reduces ICP but has not been shown to improve outcome after TBI in large multi-center trials.
Decompressive craniectomy allows for expansion of edema-tous brain is an effective way of reducing ICP.
Barbiturates decrease brain metabolic demands but have signifi cant undesirable hemodynamic effects.
Second tier therapies for refrac-tory ICP should be tailored to individual patients.
Outcome after cardiac arrest depends upon time to return of spontaneous circulation … seconds matter!

662 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
as the cause of cardiac arrest, from respiratory failure or shock, as opposed to arrhythmia, which is more prevalent in adults. The initial rhythm in children is more likely to be asystole or bradycardia, with ventricular arrhythmias occurring in perhaps up to 10% of cases of out-of-hospital arrests. The number of ventricular arrhythmia-induced cardiac arrests in children is somewhat higher in in-hospital arrests. That said, reestablishment of circulation should begin immediately after the airway, with effective chest compressions and rapid obtainment of vascular access. Current research efforts include (1) improving the quality of CPR – more effective chest compressions by human or device, (2) defi ning the most effective vasopressor and dose, and (3) developing a implementing healthcare simulation to improve CPR skills.
Intensive Care Unit
Currently, ICU care remains entirely supportive for victims of cardiac arrest (Fig. 31-9 ). While it remains imperative to prevent possible secondary injury from hypotension, hypoxia, seizure, hyperthermia, and hypo/hyperglycemia, there are no unique goal-directed strategies for these patients. ICP directed strategies were applied a few decades ago, but were aban-doned when increased survival without improved neurological outcome was achieved. Recent advances in critical care in general and contemporary neurointensive care monitoring, cou-pled with recent clinical studies in adults and neonates, rekindle hope that strategies may be developed that may someday improve outcome in infants and children after cardiac arrest.
The most promising strategy to date is the use of moderate, induced hypothermia. Improved survival and neurological outcome has been shown in adult patients with ventricular dysrrhyth-mia induced cardiac arrest. Accordingly, hypothermia has been recommended by the American Heart Association as a therapy to decrease the incidence of neurological morbidity in comatose adults after cardiac arrest. Studies have also shown a benefi cial effect of both whole body and selective head (and mild whole body) hypothermia in neonates at risk for hypoxic-ischemic encephalopathy, with an apparent advantage to whole body versus selective head cooling.
Pediatric cardiac arrest is most often caused by asphyxia vs. adult cardiac arrest which is most often caused by cardiac arrhythmias.
Algorithm for Treament of Hypoxic-Ischemic Encephalopathy
CPR
ROSC
ICU
Initial 24−72 hrs
Supportive care• PaCO2 35−40 mm Hg• PaO2 100−150 mm Hg
• Euvolemia
• Avoid hyperthermia• Avoid hypotension
• Treat seizures• Nutrition• Family Support
• Early physical and occupational therapy
• Neuropsychological testing• Advanced neuroimaging (MRI)
Rehabilitation
• Serial neurological examinations• Brain CT• EKG• Toxicology screen• Other laboratory tests• Continuous EEG
Investigation
>72 hours
In-hospital? ConsidereCPR
FIGURE 31-9
Algorithm for the treatment of cerebral hypoxic-ischemic injury based on the practice and experience at the Children’s Hospital of Pittsburgh

663 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
Selective head cooling is also diffi cult to achieve outside of the neonatal period. Extrapolating from these and a plethora of experimental studies showing the effi cacy of induced hypothermia and a good safety profi le, the AHA recommends considering induced hypothermia (32–34°C for 12–24 h) for comatose pediatric patients after cardiac arrest. During hypothermia, attention must be paid to possible adverse events such as electrolyte abnormalities, shivering – which would require administration of muscle relaxants, coagulopathy, or dysrrythmias. Previous concerns about hypothermia reducing the capacity for defi brillation/cardioversion appear unfounded, as a recent experimental study has showed that inducing hypothermia during resus-citation actually increases the likelihood successful cardioversion from ventricular fi brillation. Based on experimental studies, mild hypothermia should be induced as rapidly as possible, ideally in the pre-hospital setting, although all of the multi-center clinical studies cited above show that hypothermia can still be effective when applied up to 6 hours after the arrest. Pilot studies using iced saline to more rapidly achieve hypothermia in adults are promising and do not reveal any signifi cant adverse effects. More research is needed to address the timing, dura-tion, and degree of cooling, optimal re-warming strategies, and which patients stand to benefi t from hypothermia, including infants and children after asphyxial cardiac arrest. Therapeutic Hypothermia after Pediatric Cardiac Arrest (THAPCA) is a multicenter, randomized, con-trolled trial of hypothermia (33 deg C) for 48 hours versus controlled normothermia for neuro-protection after pediatric cardiac arrest and is currently enrolling study subjects.
Some centers are using extracorporeal membrane oxygenation as a rescue therapy during in-hospital pediatric cardiac arrest (ECPR), especially in patients with cardiac etiologies. ECPR can be implemented during active CPR, although it requires signifi cant resources and highly skilled personnel. ECPR is an effective means of reestablishing cardiac output in patients after cardiac arrest, and can be used in combination with hypothermia since bypass enables rapid cooling and maintenance of temperature. ECPR has been shown to have utility for in-hospital CA and implementation can take place during CPR. Although patients may have had long resuscitation times, good neurological outcome is still possible, with survival rate higher in patients with isolated cardiac disease. While the jury remains out, it appears that ECPR can occasionally be effective in select patients refractory to conventional CPR.
Other potential strategies include high-volume continuous veno-venous hemofi ltration, which has been shown to improve neurological outcome in a small non-randomized trial in adults after cardiac arrest. Thrombolytics, one of the only proven treatments for stroke, may represent a logical therapy in some cases, though even in stroke patients they have a narrow therapeutic time window. There is some evidence that thrombolytics may be useful during CPR in patients with cardiac arrest from myocardial infarction or pulmonary embolus. Pilot data suggests showed that a combination of coenzyme Q and mild, induced hypothermia reduces mortality versus hypothermia alone after cardiac arrest. Many other pharmacological strategies have been tried, including magnesium with and without diazepam, calcium channel blockers, barbiturates, and steroids – all without success.
CLINICAL OUTCOMES
Traumatic Brain Injury Trauma is an important cause of death in children throughout the world, and the number one killer of children in the United States. Mortality for severe TBI in children is approximately 24%, with studies fi nding varying effects of age or gender on survival. Outcomes after mild, moderate, and severe TBI in children reveal a strong association between injury severity and outcomes across rehabilitation domains. Males have been found to have worse memory and processing speed when assessed months after injury. As noted above, hyperthermia, hypoten-sion, hyperglycemia, and/or hypoxemia after TBI are associated with poor outcome after TBI.
The GCS has been used to predict outcome in pediatric TBI patients, and although clearly associated with outcome, it is not a sensitive instrument. Modifi cations of the GCS for chil-dren have also been used, including the grimace component of a modifi ed pediatric GCS, found to be more reliable than the verbal component in predicting outcome. Others have
Hypothermia improves neurologi-cal outcome in comatose adults who survive initial cardiac arrest from ventricular arrhythmia and neonates after birth asphyxia.
The American Heart Association lists hypothermia as a class IIb post-resuscitation treatment recommendation after pediatric cardiac arrest.
ECMO may be an effective rescue therapy for select patients.
Trauma is the most common cause of death of children worldwide, and the #1 killer in the United States.

664 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
used the motor component of the GCS, which may be the most reliable and repeatable com-ponent in infants and children after TBI, to demonstrate a linear relationship with survival.
In the pediatric population, TBI is predominately due to blunt trauma and is referred to as closed head injury, but penetrating brain injury also occurs. Outcome after penetrating brain injury is generally poorer versus closed head injury, with a higher mortality; however, good outcomes in survivors are sometimes observed. Children with infl icted TBI from child abuse represent a population unique to pediatric TBI, and have a very poor prognosis overall. This may be due to several factors including age, a contribution of hypoxic-ischemic injury, repeated insults, and/or delay in seeking medical attention. Mortality rates range from 13% to 36% with high morbidity seen in survivors, with approximately half of them classifi ed as having severe neurological damage.
Cardiac Arrest Outcome for infants and children after cardiac arrest remains dismal. The outpatient survival rate for children after cardiac arrest is approximately 8.6%, with over half of the survivors having neurological impairment. Most patients are <1 year of age, with half of these being newly born and having the best survival rate at 36%. Just over half of pediatric cardiac arrest patients are male with equal survival rates between genders. The most prevalent etiologies are sudden infant death syndrome and trauma, which have worse outcomes than those with respiratory and submersion etiologies. Patients suffering witnessed cardiac arrest have better outcomes compared with unwitnessed, but pre-hospital CPR does not necessarily make a difference in survival rate. Out of hospital cardiac arrest patients had better survival if the fi rst assessed rhythm was pulseless electrical activity (24%) or ventricular fi brillation (9%), but most had asystole (67%). Three or more doses of epinephrine or resuscitation >30 min were associated with unfavorable neurological outcome in survivors. Survival rate to hospi-tal discharge is higher for inpatient pediatric cardiac arrest patients versus outpatient cardiac arrest patients (~27%), with approximately 65% of patients in the National Registry of Cardiopulmonary Resuscitation recovering to good neurological outcome. It should be noted that the time to fi rst CPR was <1 min on average with these inpatient arrests.
Patients who have a cardiac arrest in a pediatric ICU have a hospital discharge rate of 13.7%. For CPR durations of <15 min, 15–30 min, and >30 min, the survival rates were 18.6%, 12.2%, and 5.6%, respectively. Only two (5.7%) of 35 patients who had multiple events of cardiac arrest in the ICU survived to discharge. Severity of illness, as measured by the Pediatric Risk of Mortality III score, was found to be a signifi cant predictor of survival.
FINAL COMMENTS
The goal as intensivists is to improve survival and neurological outcome in infants and chil-dren after moderate to severe traumatic or ischemic brain injury. Hopefully, the goal of transferring these patients to a rehabilitation center with the optimal chance for meaningful recovery is achievable. Inpatient rehabilitation for pediatric patients with moderate-severe TBI can result in signifi cant improvements in neurological function. While there is limited information on the effectiveness of rehabilitation in pediatric patients recovering from car-diac arrest, in adults, signifi cant cognitive improvement and decreased dependency in activi-ties of daily living can be achieved. Although health-related quality of life improved over the year, many were unable to return to work. Accordingly, much effort is still required not only to improve the quality and consistency of current management, but also to make progress in terms of innovative breakthrough treatments that make a real difference in improved func-tional outcome in this challenging group of pediatric patients.
Infl icted TBI due to child abuse is associated with extremely poor outcome.
Survival rates after cardiac arrest in children: 12% overall, 24% in-hospital, and 8% out-of-hospital.
Inpatient rehabilitation can improve neurological function after moderate-severe TBI.

665 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
1. A twelve year old male is involved in a motor vehicle accident. A computerized tomogram of the brain reveals a small subdural hematoma with multiple punctuate hemorrhages scattered throughout the frontal and parietal cortex. On clinical exam, he is intubated and hemodynamically stable. He opens his eyes minimally, but only to a fi rm sternal rub. He does not focus. Prior to the intubation, he moaned but made no discernible words. His pupils are equal and reactive. Which of the follow-ing is the best motor response that should still prompt consid-eration of placement of an intracranial pressure monitor? A. Extension of his lower extremities in response to noxious
stimuli B. Flexion of his upper extremities in response to noxious stimuli C. Moving all extremities spontaneously D. Reaching across midline to resist a noxious stimuli E. Withdrawing from a noxious stimuli
2. A fi ve year old girl was involved in a motor vehicle collision where she sustained injury to her brain with multiple punc-tuate hemorrhages visualized on computer axial tomogram. She also incurred blunt trauma to her abdomen and has a markedly distended abdomen. She has developed marked acute respiratory distress syndrome requiring mechanical ventilation. After placement of a central venous catheter (tip in the superior vena cava), right radial arterial catheter, foley catheter, and intraventricular intracranial pressure monitor, the following data are obtained: Arterial blood pressure: 115 / 67 mm Hg (Mean 83 mm Hg) Central venous pressure: 12 mm Hg Intra-abdominal Pressure: 18 mm Hg Mean Airway Pressure: 18 cm H 2 O Intracranial Pressure: 22 mm Hg The correct value of her cerebral perfusion pressure is which of the following? A. 43 mm Hg B. 61 mm Hg C. 65 mm Hg D. 71 mm Hg E. 93 mm Hg
3. Normal cerebral blood fl ow values (mL/100 g brain/min) are highest at which age of life? A. Newborns B. Two years of age C. Four years of age D. Adolescence E. Adulthood
4. The primary value of head computer axial tomography (CAT) immediately after an event associated with signifi cant cerebral ischemia such as cardiac arrest or near drowning is which of the following? A. A normal initial CAT scan of the brain portends a favorable
prognosis. B. Although likely to be normal, it establishes a baseline for
comparison with future CAT scans. C. An abnormal initial CAT scan indicates a higher probability
of an unfavorable neurological outcome
D. It may be used to assess the possibility of trauma or intracranial hemorrhage as the cause of the cardiac arrest or near-drowning.
E. There is no value to a CAT scan of the brain in this setting.
5. The following graph depicts which of the following relation-ships?
A. The relationship between cerebral blood fl ow and mean arte-rial blood pressure.
B. The relationship between cerebral blood fl ow and the initial Glasgow Coma Scale score
C. The relationship between cerebral blood fl ow and the partial pressure of arterial carbon dioxide.
D. The relationship between cerebral blood fl ow and the partial pressure of arterial oxygen.
E. The relationship between cerebral blood volume and intrac-ranial pressure.
6. The following graph depicts which of the following relation-ships?
A. The relationship between cerebral blood fl ow and mean arte-rial blood pressure.
B. The relationship between cerebral blood fl ow and the initial Glasgow Coma Scale score.
C. The relationship between cerebral blood fl ow and the partial pressure of arterial carbon dioxide.
D. The relationship between cerebral blood fl ow and the partial pressure of arterial oxygen.
E. The relationship between cerebral blood volume and intrac-ranial pressure.
REVIEW QUESTIONS

666 E. L. FI N K , P. M. KOC HAN EK, AN D R. S. B. C LAR K
7. The following graph depicts which of the following relation-ships?
A. The relationship between cerebral blood fl ow and mean arte-rial blood pressure.
B. The relationship between cerebral blood fl ow and the initial Glasgow Coma Scale score.
C. The relationship between cerebral blood fl ow and the partial pressure of arterial carbon dioxide.
D. The relationship between cerebral blood fl ow and the partial pressure of arterial oxygen.
E. The relationship between cerebral blood volume and intrac-ranial pressure.
8. The following graph depicts which of the following relation-ships?
A. The relationship between cerebral blood fl ow and mean arte-rial blood pressure.
B. The relationship between cerebral blood fl ow and the initial Glasgow Coma Scale score.
C. The relationship between cerebral blood fl ow and the partial pressure of arterial carbon dioxide.
D. The relationship between cerebral blood fl ow and the partial pressure of arterial oxygen.
E. The relationship between intracranial pressure and intracra-nial volume
9. In the setting of traumatic brain injury, epidemiologic data support maintenance of the PaO2 above which of the follow-ing values? A. 45 mm Hg B. 60 mm Hg C. 75 mm Hg D. 90 mm Hg E. It varies based on the age of the patient.
10. Which of the following statements regarding the maintenance of body temperature following traumatic brain injury is cur-rently recommended? A. The induction of extreme hyperthermia should be attempted to
optimize neurologic recovery (40–42°C) as fi rst tier therapy. B. The induction of extreme hypothermia should be attempted to
optimize neurologic recovery (28–31°C) as fi rst tier therapy. C. The induction of moderate hyperthermia should be attempted
to optimize neurologic recovery (38.5–39.5°C) as fi rst tier therapy.
D. The induction of moderate hypothermia should be attempted to optimize neurologic recovery (32–34°C) as fi rst tier therapy.
E. The maintenance of normothermia with particular attention to avoid any elevation of temperature should be attempted to optimize neurologic recovery.
1. E 2. B 3. C 4. D 5. C
6. D 7. A 8. E 9. B 10. E
ANSWERS

667 C HAPTER 31 • C ER EB RAL R ES USC ITATION AN D TRAU MATIC B RAI N I NJ U RY
Adelson PD, Clyde B, Kochanek PM, Wisniewski SR, Marion DW, Yonas H. Cerebrovascular response in infants and young children following severe traumatic brain injury: a preliminary report. Pediatr Neurosurg. 1997;26(4):200–7.
Adelson PD, Bratton SL, Carney NA, Chesnut RM, du Coudray HE, Goldstein B, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adoles-cents. Chapter 19 . The role of anti-seizure prophylaxis following severe pediatric traumatic brain injury. Pediatr Crit Care Med. 2003a;4(3 Suppl):S72–5.
Adelson PD, Bratton SL, Carney NA, Chesnut RM, du Coudray HE, Goldstein B, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adoles-cents. Chapter 7. Intracranial pressure monitoring technology. Pediatr Crit Care Med. 2003b;4(3 Suppl):S28–30.
Adelson PD, Bratton SL, Carney NA, Chesnut RM, du Coudray HE, Goldstein B, et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adoles-cents. Chapter 4. Resuscitation of blood pressure and oxygenation and prehospital brain-specifi c therapies for the severe pediatric traumatic brain injury patient. Pediatr Crit Care Med. 2003c;4(3 Suppl):S12–8.
Anderson VA, Catroppa C, Dudgeon P, Morse SA, Haritou F, Rosenfeld JV. Understanding predictors of functional recovery and outcome 30 months following early childhood head injury. Neuropsychology. 2006;20(1):42–57.
Chambers IR, Stobbart L, Jones PA, Kirkham FJ, Marsh M, Mendelow AD, et al. Age-related differences in intracranial pressure and cerebral perfusion pressure in the fi rst 6 hours of monitoring after children’s head injury: association with outcome. Childs Nerv Syst. 2005;21(3):195–9.
Cochran A, Scaife ER, Hansen KW, Downey EC. Hyperglycemia and outcomes from pediatric traumatic brain injury. J Trauma. 2003;55(6):1035–8.
Cooper DJ, Myles PS, McDermott FT, Murray LJ, Laidlaw J, Cooper G, et al. Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury: a randomized controlled trial. JAMA. 2004;291(11):1350–7.
Cunningham AS, Salvador R, Coles JP, Chatfi eld DA, Bradley PG, Johnston AJ, et al. Physiological thresholds for irreversible tissue damage in contusional regions following traumatic brain injury. Brain. 2005;128(Pt 8):1931–42.
Gupta AK. Monitoring the injured brain in the intensive care unit. J Postgrad Med. 2002;48(3):218–25.
Hutchison JS, Ward RE, Lacroix J, Hébert PC, Barnes MA, Bohn DJ, et al. Hypothermia therapy after traumatic brain injury in chil-dren. N Engl J Med. 2008;358(23):2447–56.
Liou AK, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69(2):103–42.
Maas AI, Hukkelhoven CW, Marshall LF, Steyerberg EW. Prediction of outcome in traumatic brain injury with computed tomographic characteristics: a comparison between the computed tomographic classifi cation and combinations of computed tomographic predictors. Neurosurgery. 2005;57(6):1173–82; discussion 1173–82.
Mandel R, Martinot A, Delepoulle F, Lamblin MD, Laureau E, Vallee L, et al. Prediction of outcome after hypoxic-ischemic encephal-opathy: a prospective clinical and electrophysiologic study. J Pediatr. 2002;141(1):45–50.
Morrison WE, Arbelaez JJ, Fackler JC, De Maio A, Paidas CN. Gender and age effects on outcome after pediatric traumatic brain injury. Pediatr Crit Care Med. 2004;5(2):145–51.
Manole MD, Foley LM, Hitchens TK, et al. Magnetic resonance imag-ing assessment of regional cerebral blood fl ow after asphyxial cardiac arrest in immature rats. J Cereb Blood Flow Metab. 2009;29:197–205.
Nadkarni VM, Larkin GL, Peberdy MA, Carey SM, Kaye W, Mancini ME, et al. First documented rhythm and clinical outcome from in-hospital cardiac arrest among children and adults. JAMA. 2006;295(1):50–7.
Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, et al. Clinical trials in head injury. J Neurotrauma. 2002;19(5):503–57.
Perez A, Minces PG, Schnitzler EJ, Agosta GE, Medina SA, Ciraolo CA. Jugular venous oxygen saturation or arteriovenous differ-ence of lactate content and outcome in children with severe trau-matic brain injury. Pediatr Crit Care Med. 2003;4(1):33–8.
Peterson B, Khanna S, Fisher B, Marshall L. Prolonged hypernatremia controls elevated intracranial pressure in head-injured pediatric patients. Crit Care Med. 2000;28(4):1136–43.
Roberts JS, Vavilala MS, Schenkman KA, Shaw D, Martin LD, Lam AM. Cerebral hyperemia and impaired cerebral autoregulation associated with diabetic ketoacidosis in critically ill children. Crit Care Med. 2006;34(8):2217–23.
Robertson CL, Hlatky R. Advanced bedside neuromonitoring. In: Fink MP, Abraham E, Vincent JL, Kochanek PM, editors. Textbook of critical care. 5th ed. Philadelphia: Elsevier Saunders; 2005. p. 287–94.
Ruppel RA, Kochanek PM, Adelson PD, Rose ME, Wisniewski SR, Bell MJ, et al. Excitatory amino acid concentrations in ventricular cere-brospinal fl uid after severe traumatic brain injury in infants and children: the role of child abuse. J Pediatr. 2001;138(1): 18–25.
Ruppel RA, Clark RS, Bayir H, Satchell MA, Kochanek PM. Critical mechanisms of secondary damage after infl icted head injury in infants and children. Neurosurg Clin N Am. 2002;13(2):169–82.
Safar P, Behringer W, Bottiger BW, Sterz F. Cerebral resuscitation poten-tials for cardiac arrest. Crit Care Med. 2002;30(4 Suppl):S140–4.
Siesjo BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1(2):155–85.
Skippen P, Seear M, Poskitt K, Kestle J, Cochrane D, Annich G, et al. Effect of hyperventilation on regional cerebral blood fl ow in head-injured children. Crit Care Med. 1997;25:1402–9.
Soustiel JF, Glenn TC, Shik V, Boscardin J, Mahamid E, Zaaroor M. Monitoring of cerebral blood fl ow and metabolism in traumatic brain injury. J Neurotrauma. 2005;22(9):955–65.
Stiefel MF, Spiotta A, Gracias VH, Garuffe AM, Guillamondegui O, Maloney-Wilensky E, et al. Reduced mortality rate in patients with severe traumatic brain injury treated with brain tissue oxy-gen monitoring. J Neurosurg. 2005;103(5):805–11.
The International Liaison Committee on Resuscitation (ILCOR). The International Liaison Committee on Resuscitation (ILCOR) con-sensus on science with treatment recommendations for pediatric and neonatal patients: pediatric basic and advanced life support. Pediatrics. 2006;117(5):e955–77.
Toet MC, Lemmers PM, van Schelven LJ, van Bel F. Cerebral oxygen-ation and electrical activity after birth asphyxia: their relation to outcome. Pediatrics. 2006;117(2):333–9.
White JR, Farukhi Z, Bull C, Christensen J, Gordon T, Paidas C, et al. Predictors of outcome in severely head-injured children. Crit Care Med. 2001;29(3):534–40.
Young KD, Seidel JS. Pediatric cardiopulmonary resuscitation: a col-lective review. Ann Emerg Med. 1999;33(2):195–205.
SUGGESTED READINGS





























