Embed Size (px)
Citation preview

Pentafluorophenyl Copper−Pyridine Complexes: Synthesis,Supramolecular Structures via Cuprophilic and π-StackingInteractions, and Solid-State LuminescenceAmi Doshi,† Anand Sundararaman,† Krishnan Venkatasubbaiah,†,§ Lev N. Zakharov,‡
Arnold L. Rheingold,‡ Mykhaylo Myahkostupov,† Piotr Piotrowiak,† and Frieder Jakle*,†
†Department of Chemistry, Rutgers University-Newark, 73 Warren Street, Newark, New Jersey 07102, United States‡Department of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093, United States
*S Supporting Information
ABSTRACT: The effect of the binding of pyridine ligands to pentafluorophenylcopper, [C6F5Cu]4 (1), on structural features and photophysical properties has beeninvestigated through a combined multinuclear NMR, X-ray crystallography, andphotoluminescence study. Reaction of 1 with 2 equiv of pyridine yields a novelpyridine complex, 3, in which the tetranuclear framework of 1 is retained. Complex 3features a rhombus-shaped tetracopper core with a short diagonal Cu···Cu distance of2.5941(6) Å between the dicoordinate copper centers and a longer distance between thepyridine-coordinated copper centers of 4.178(1) Å. In contrast, treatment of 1 with 4equiv of pyridine results in complete breakdown of the tetranuclear aggregate to give theformally dicoordinate species C6F5Cu(py) (4-H). Reaction of 1 with 2,2′-bipyridineresults in formation of the tricoordinate complex C6F5Cu(2,2′-bipy) (5). Aggregatebreakdown in species 4 and 5 is reflected in a significantly reduced chemical shiftdifference Δδ(19Fmeta/para) and a strong downfield shift of the copper-bound carbonatoms in the 13C NMR spectra in comparison to 3. A dynamic equilibrium is established at ratios of py/C6F5Cu ranging from 0to 2. The solid-state structures of all compounds have been determined by single-crystal X-ray crystallography. Thesupramolecular assembly of complex 3 via arene−arene π-interactions leads to a network structure with solvent-filled channelspropagating through the lattice along the crystallographic c axis. The 2,2′-bipyridine complex 5 also shows π-stacking as thedominant feature in the extended solid-state structure. In contrast, a different mode of supramolecular assembly is found for 4-Hin that cuprophilic interactions lead to assembly into one-dimensional copper chains with equidistant Cu···Cu contacts of2.8924(3) Å. However, the closely related complexes 4-R with methyl or chloro substituents in either the ortho or the paraposition form supramolecular stacks with structural features that, again, are dominated by offset perfluoroarene−areneinteractions with intermolecular plane-to-plane separations of ca. 3.3−3.6 Å. The dicoordinate copper atoms are aligned in one-dimensional chains with alternating short and long Cu(I)···Cu(I) distances of 3.531(1)/3.698(1) Å in 4-pMe, 3.2454(5)/4.2970(5) Å in 4-oMe, 3.521(1)/3.784(1) Å in 4-pCl, and 3.4797(6)/3.8363(6) Å in 4-oCl. Compound 4-H is strongly bluefluorescent at 460 nm in the solid state, but yellow-green fluorescent at 77 K, resulting in an interesting example of luminescencethermochromism. In contrast, the substituted compounds 4-R display strong luminescence only at liquid nitrogen temperature.In all cases, the fluorescence emission band is in the range of ca. 410−425 nm and thus at significantly different energy from thatin 4-H, which strongly suggests that the short Cu···Cu contacts in 4-H give rise to unique luminescence properties.
■ INTRODUCTIONOrganocopper compounds have emerged as an important classof organometallic compounds. They have attracted consid-erable interest from both inorganic and organic chemistsbecause of the rich structural diversity and unusual bondingpatterns, as well as their important roles as intermediates incoupling reactions and as mild and selective reagents andcatalysts in organic synthesis.1−3 An interesting aspect from astructural perspective is that monomeric species RCu remainelusive, whereas well-defined homoleptic organocopper species[RCu]n with varying degrees of aggregation (n = 2−8) havebeen identified in solution and also structurally characterized inthe solid state.2,4,5 Aggregation typically occurs through
bridging of two or more copper centers with an organicmoiety.4,6 The degree of aggregation depends on a number offactors, including steric and electronic effects, and the presenceof external donor ligands.4,7,8 For example, unsubstitutedphenylcopper adopts a polymeric structure and hence isinsoluble in common organic solvents, whereas the presenceof substituents in the ortho position of the aromatic ring leadsto well-defined soluble oligomers as a result of the added steric
Special Issue: Fluorine in Organometallic Chemistry
Received: October 15, 2011Published: December 1, 2011
Article
pubs.acs.org/Organometallics
© 2011 American Chemical Society 1546 dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−1558

bulk.4 Thus, 2,4,6-triisopropylphenylcopper features a tetranu-clear square-planar aggregate due to the presence of stericallydemanding isopropyl groups in the ortho positions of thephenyl ring.9 van Koten and co-workers have demonstratedthat smaller arylcopper aggregates can be stabilized by theintramolecular coordination with chelating amino or alkoxygroups in the ortho position of the aromatic ring.10
It is also well known that treatment of organocopper specieswith strongly coordinating solvents or external ligands can leadto the breakdown of the aggregated structure.11−13 Smallerorganocopper aggregates are obtained, for example, by using N,P, or S compounds as donors. For instance, phenylcopper,which adopts a polymeric structure in the absence of anexternal Lewis base, has been isolated as the dimethylsulfidesolvate [PhCu]4(SMe2)2 with a square-planar tetranuclearcopper(I) core,14 while a monomeric species PhCuL wasobtained in the presence of the sterically demanding tridentateligand l,l,l-tris[(diphenylphosphino)methyl]ethane (triphos).15
However, in general, it is difficult to predict whethermonomeric species or partially aggregated complexes will beformed, and surprisingly, few nonaggregated species RCuLnhave been isolated and structurally characterized. In mostcomplexes, either bulky groups15−18 and/or chelatingligands19−22 enforce the monomeric structure.23 Lewis baseadditives have been related to a beneficial effect of strong donorsolvents, such as DMF, NMP, or HMPA, on the reactivity oforganocopper compounds in organic synthesis, including theiruse in carbocupration, stereoselective conjugate additions, andallylic substitution reactions.2,4,8
We became interested in the coordination behavior andphotophysical properties of pentafluorophenyl copper (1),because we reasoned that the unique combination of stericdemand and the electron-withdrawing effect of the penta-fluorophenyl group should enable us to isolate and structurallycharacterize otherwise inaccessible Lewis base adducts.24
Compound 1 was first introduced by Cairncross and Sheppardin 1968, and they determined the aggregation number inbenzene to be 3.75−3.85 by cryoscopy and to be 3.95 by vaporpressure osmometry, suggesting that a tetranuclear aggregate ispresent in solution.25 Using single-crystal X-ray diffraction, weconfirmed that, in the absence of Lewis bases, 1 adopts atetrameric structure also in the solid state.26 More importantly,we discovered the formation of π-complexes, such as[C6F5Cu]4(toluene)2) (2), in which the tetranuclear aggregatestructure is maintained and investigated the assembly of 1 intooffset supramolecular stacks via multiple Cu···π interactions inthe presence of extended π-systems.26−28 In stark contrast, thepresence of an excess of pyridine results in the breakdown ofthe tetranuclear structure with formation of a strongly blue-luminescent monomeric complex C6F5Cu(py) (py = pyr-idine).29
The aim of the current work was to further investigate thefactors that determine the aggregation behavior of pentafluor-ophenyl copper−pyridine complexes in solution and the solidstate. Special emphasis is given to the role of π-stacking andcuprophilic interactions on the supramolecular assembly andthe photophysical properties.
■ EXPERIMENTAL SECTIONAll reactions and manipulations were carried out under an atmosphereof prepurified nitrogen using either Schlenk techniques or an inert-atmosphere glovebox (Innovative Technologies). Hydrocarbon andchlorinated solvents were purified using a solvent purification system
(Innovative Technologies; alumina/copper columns for hydrocarbonsolvents), and the chlorinated solvents were subsequently distilledfrom CaH2 and degassed via several freeze−pump−thaw cycles.Pentafluorophenyl copper toluene solvate (2) was prepared accordingto a literature procedure.24 Compound 2 was heated to 80 °C undervacuum to yield the solvent-free complex 1 as a colorless whitepowdery solid. The synthesis of C6F5Cu(py) (4-H) has been reportedpreviously.29 2-Picoline, 4-picoline, 2-chloropyridine, 4-chloropyridinehydrochloride, and 2,2′-bipyridine were purchased from ACROS.Pyridine was degassed and distilled from calcium hydride prior to use.2-Picoline, 4-picoline, and 2-chloropyridine were used as received, and4-chloropyridine hydrochloride was treated with 6 M sodiumhydroxide solution and extracted with ether. After removal of etherat 0 °C under vacuum, 4-chloropyridine was dissolved in toluene, andthe solution was degassed via several freeze−pump−thaw cycles priorto use.
The 499.91 MHz 1H, 125.68 MHz 13C, and 470.2 MHz 19F NMRspectra were acquired on a Varian INOVA NMR spectrometerequipped with a 5 mm dual broad-band gradient probe (Nalorac,Varian Inc., Martinez, CA). The 100.5 MHz 13C NMR spectrum wasrecorded on a Varian VXR-S spectrometer. All NMR data wererecorded at ambient temperature. 1H and 13C NMR spectra werereferenced internally to the solvent peaks, and 19F NMR spectra werereferenced externally to α,α′,α″-trifluorotoluene (0.05% in C6D6; δ =−63.73 ppm). The abbreviations Pf and py are used forpentafluorophenyl and pyridyl, respectively.
Variable-temperature solid-state luminescence data were measuredon a Varian Cary Eclipse Fluorescence spectrophotometer equippedwith an Oxford Instruments Cryostat, model Optistat DN. Forphosphorescence measurements, a delay time td of 0.1 ms and a gatetime tg of 1.0 ms were used with excitation and emission slit widths of5/10 nm. For the lifetime measurements, thoroughly degassed sampleswere excited with 5 ns ∼80 mJ pulses at 355 nm (third harmonic of aQ-switched Nd:YAG laser, Quantel, Brilliant). The emission wasdispersed through a monochromator (Oriel M257) and detected witha Hamamatsu R928 photomultiplier. The transients were recordedusing a Tektronix SCD 1000 digital oscilloscope controlled by aLabview subroutine. Fluorescence decays were fitted as singleexponentials using the Igor software package by Wavemetrics, Inc.DFT calculations were performed with the Gaussian 03 program.30
Geometries and electronic properties were calculated by means of theB3LYP hybrid density functional with the basis set of 6-311++G (d,p).The input files and orbital representations were generated withGaussview (scaling radii of 75%). Excitation data were calculated usingTD-DFT (B3LYP).30 Elemental analyses were obtained fromQuantitative Technologies, Inc., Whitehouse, NJ, and Schwarzkopf,Woodside, NY. Melting points and decomposition temperatures weredetermined in sealed capillary tubes and are not corrected.
Spectral Data for 1 in CDCl3 and d5-Pyridine. 19F NMR(470.2 MHz, CDCl3, 20 °C): δ = −104.1 (m, 2F, ortho-F), −141.5 (t,J(19F, 19F) = 20 Hz, 1F, para-F), −158.1 (m, 2F, meta-F). 13C NMR(100.5 MHz, CDCl3, 20 °C): δ = 154.7 (dd, J(19F, C) = 244/22 Hz,Pf-C2,6), 145.7 (dm, J(19F, C) = 262 Hz, Pf-C4), 137.2 (dm, J(19F, C)= 260 Hz, Pf-C3,5), 98.7 (t, J(19F, C) = 52 Hz, Pf-C1). 19F NMR(470.2 MHz, C5D5N, 20 °C): δ = −110.6 (m, 2F, ortho-F), −163.4 (t,J(F, F) = 20 Hz, 1F, para-F), −164.1 (m, 2F, meta-F); 13C NMR(125.7 MHz, C5D5N, 25 °C): δ = 150.2 (dd, J(19F, C) = 219 Hz/33Hz, Pf-C2,6), 138.0 (dm, J(19F, C) = 241 Hz, Pf-C4), 136.6 (dm,J(19F, C) = 253 Hz, Pf-C3,5), 130.3 (t, J(19F, C) = 80 Hz, Pf-C1).
Synthesis of [C6F5Cu]4(py)2 (3). Neat pyridine (80 μL, 1.0mmol) was added dropwise to a solution of 1 (0.46 g, 0.50 mmol) inCH2Cl2 (10 mL) at room temperature. Upon addition of pyridine, thesolution turned yellow. The solution was layered with 10 mL ofpentane and stored at −38 °C. Colorless needle-like crystals formedafter 24 h. The crystals contain one molecule of pentane per mainmolecule, but were dried for elemental analysis and for thedetermination of the yield. Yield: 0.44 g (82%). For 3: Tm: 145 °C(dec.). 1H NMR (500 MHz, CDCl3, 20 °C): δ = 8.35 (d, J = 5.0 Hz,4H, Py-H2,6), 7.94 (t, J = 7.5 Hz, 2H, Py-H4), 7.50 (m, 4H, Py-H3,5).19F NMR (470.2 MHz, CDCl3, 20 °C): δ = −108.2 (br m, 8F, ortho-
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581547

F), −151.2 (br, 4F, para-F), −161.2 (br m, 8F, meta-F). 13C NMR(125.7 MHz, CDCl3, 20 °C): δ = 152.1 (d, J(19F, C) = 232 Hz, Pf-C2,6), 148.7 (Py-C2,6), 142.4 (dm, J(19F, C) = 251 Hz, Pf-C4), 139.0(Py-C4), 136.8 (dm, J(19F, C) = 256 Hz, Pf-C3,5), 125.7 (Py-C3,5),106.4 (t, J(19F, C) = 59 Hz, Pf-C1). Elemental analysis forC34H10Cu4F20N2 (1080.61), Calcd: C, 37.79; H, 0.93; N, 2.59%.Found: C, 36.86; H, 0.71; N, 2.68%. The slightly low C value for theelemental analysis is likely due to incomplete combustion as a result ofthe large content of fluorine or the high air sensitivity of the material.Synthesis of C6F5Cu(4-picoline) (4-pMe). Neat 4-picoline
(0.067 g, 0.72 mmol) was added dropwise to a solution of 2 (0.16g, 0.14 mmol) in toluene (5 mL) at room temperature. Upon additionof 4-picoline, an intense yellow color developed. The reaction solutionwas kept at −35 °C for 1 day, and the pure product was isolated fromthe reaction solution as pale yellow crystals. The product obtained waswashed with hexanes and dried under high vacuum. Yield: 0.13 g(72%). For 4-pMe: Tm = 130−134 °C. Tdec = 148−155 °C. 1H NMR(500 MHz, CDCl3, 25 °C): δ = 8.52 (d, J = 6.0 Hz, 2H, Py-H2,6), 7.38(d, J = 6.0 Hz, 2H, Py-H3,5), 2.50 (s, 3H, Me). 19F NMR (470.2 MHz,CDCl3, 25 °C): δ = −112.9 (m, 2F, ortho-F), −160.8 (t, J(19F, 19F) =20 Hz, 1F, para-F), −164.0 (pst, J = 19 Hz, 2F, meta-F). 13C NMR(125.7 MHz, CDCl3, 25 °C): δ = 152.5 (Py-C4), 149.3 (dd, J(19F, C)= 224/32 Hz, Pf-C2,6), 149.4 (Py-C2,6), 138.8 (dm, J(19F, C) = 245Hz, Pf-C4), 136.5 (dm, J(19F, C) = 253 Hz, Pf-C3,5), 126.7 (Py-C3,5),122.2 (t, J(19F, C) = 72 Hz, Pf-C1), 21.8 (Me). Elemental analysis for
C12H7CuF5N (323.73), Calcd: C, 44.52; H, 2.18; N, 4.33%. Found: C,43.21; H, 2.12; N, 3.96%. The slightly low C value for the elementalanalysis is likely due to incomplete combustion as a result of the largecontent of fluorine or the high air sensitivity of the material.
Synthesis of C6F5Cu(2-picoline) (4-oMe). Neat 2-picoline(0.050 g, 0.58 mmol) was added dropwise to a solution of 2 (0.13g, 0.12 mmol) in toluene (5 mL) at room temperature. Upon additionof 2-picoline, an intense yellow color developed. The crude productwas obtained as a colorless microcrystalline solid, which was washedwith hexanes. Yield: 0.13 g (84%). Slow evaporation of toluene at RTgave pale yellow “single crystals” of 4-oMe suitable for X-raydiffraction analysis. For 4-oMe: Tm = 141−144 °C. Tdec = 147−150°C. 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.59 (d, J = 5.0 Hz, 1H,Py-H6), 7.89 (pst, J = 8.0 Hz, 1H, Py-H4), 7.46 (d, J = 8.0 Hz, 1H, Py-H3), 7.38 (pst, J = 7.0 Hz, 1H, Py-H5), 2.93 (s, 3H, Me). 19F NMR(470.2 MHz, CDCl3, 25 °C): δ = −113.1 (m, 2F, ortho-F), −160.9 (t,J(19F, 19F) = 20 Hz, 1F, para-F), −164.0 (m, 2F, meta-F). 13C NMR(125.7 MHz, CDCl3, 25 °C): δ = 159.5 (Py-C2), 149.7 (dd, J(19F, C)= 220 Hz/33 Hz, Pf-C2,6), 149.6 (Py-C6), 139.5 (Py-C4), 138.7 (dm,J(19F, C) = 244 Hz, Pf-C4), 136.4 (dm, J(19F, C) = 253 Hz, Pf-C3,5),125.9 (Py-C3), 122.7 (Py-C5), 122.8 (t, J(19F, C) = 71 Hz, Pf-C1),26.0 (Me). Elemental analysis for C12H7CuF5N (323.73), Calcd: C,44.52; H, 2.18; N, 4.33%. Found: C, 44.38; H, 2.17; N, 4.17%.
Synthesis of C6F5Cu(4-chloropyridine) (4-pCl). A solution of4-chloropyridine (ca. 0.3 mmol) in toluene (5 mL) was added to a
Table 1. Experimental Data for X-ray Diffraction Studies
3 4-pMe 4-oMe 4-pCl 4-oCl 5
empirical formula C39H22Cu4F20N2 C12H7CuF5N C12H7CuF5N C11H4ClCuF5N C11H4ClCuF5N C16H8CuF5N2
formula weight 1152.75 323.73 323.73 344.15 344.15 386.79T, K 218(2) 100(2) 100(2) 100(2) 100(2) 100(2)wavelength, Å 0.71073 0.71073 1.54178 0.71073 1.54178 0.71073cryst syst monoclinic triclinic monoclinic triclinic monoclinic monoclinicspace group C2/c P1 P21/c P1 P21/c P21/ca, Å 20.1931(13) 5.9779(4) 6.10850(10) 5.9903(9) 12.5523(2) 8.7282(9)b, Å 12.4763(9) 7.2228(6) 7.5034(2) 7.2800(10) 7.1538(2) 24.282(3)c, Å 15.8809(11) 13.4698(10) 25.0325(5) 13.0816(19) 24.8910(5) 7.0340(7)α, deg 90 91.634(2) 90 88.434(2) 90 90β, deg 90.6490(10) 90.298(2) 95.5830(10) 86.879(2) 98.5210(10) 112.065(2)γ, deg 90 95.320(2) 90 83.596(2) 90 90V, Å3 4000.7(5) 578.83(8) 1141.91(4) 565.95(14) 2210.46(8) 1381.6(2)Z 4 2 4 2 8 4ρcalcd, g cm−3 1.914 1.857 1.883 2.019 2.068 1.859μ, mm−1 μ(Mo Kα) 2.225 μ(Mo Kα) 1.935 μ(Cu Kα) 3.228 μ(Mo Kα) 2.214 μ(Cu Kα) 5.561 μ(Mo Kα) 1.640F(000) 2264 320 640 336 1344 768cryst size, mm 0.30 × 0.15 × 0.10 0.30 × 0.30 × 0.20 0.25 × 0.21 × 0.17 0.20 × 0.20 × 0.10 0.18 × 0.16 × 0.10 0.25 × 0.19 × 0.10limiting indices −25 ≤ h ≤ 25 −7 ≤ h ≤ 7 −7 ≤ h ≤ 7 −7 ≤ h ≤ 7 −14 ≤ h ≤ 14 −11 ≤ h ≤ 11
−15 ≤ k ≤ 15 −9 ≤ k ≤ 9 −7 ≤ k ≤ 8 −9 ≤ k ≤ 9 −8 ≤ k ≤ 8 −31 ≤ k ≤ 25−20 ≤ l ≤ 19 −17 ≤ l ≤ 17 −29 ≤ l ≤ 29 −16 ≤ l ≤ 16 −29 ≤ l ≤ 28 −9 ≤ l ≤ 8
θ range, deg 1.92−27.00 1.51−27.49 3.55−65.01 2.82−27.53 5.42−64.44 1.68−27.50reflns collected 13688 4730 8950 4782 13171 8470independent reflns 4364 2463 1918 2449 3539 3127absorptioncorrection
semiempirical fromequivalents
semiempirical fromequivalents
semiempirical fromequivalents
semiempirical fromequivalents
semiempirical fromequivalents
semiempirical fromequivalents
refinement method full-matrix least-squares on F2
full-matrix least-squares on F2
full-matrix least-squares on F2
full-matrix least-squares on F2
full-matrix least-squares on F2
full-matrix least-squares on F2
data/restraints/parameters
4364/4/302 2463/0/172 1918/0/174 2449/0/172 3539/0/344 3127/0/249
goodness-of-fit onF2
1.048 1.064 1.215 1.074 1.136 0.967
final R indices [I >2σ(I)]a
R1 = 0.0324 R1 = 0.0422 R1 = 0.0250 R1 = 0.0275 R1 = 0.0338 R1 = 0.0272wR2 = 0.0938 wR2 = 0.1168 wR2 = 0.0686 wR2 = 0.0750 wR2 = 0.0893 wR2 = 0.0628
R indices (alldata)a
R1 = 0.0410 R1 = 0.0464 R1 = 0.0253 wR2 = 0.0296 R1 = 0.0370 R1 = 0.0368wR2 = 0.0991 wR2 = 0.1207 wR2 = 0.0688 wR2 = 0.0767 wR2 = 0.0927 wR2 = 0.0649
peak/hole (e Å−3) 0.612/−0.217 1.379/−0.721 0.322/−0.255 0.769/−0.420 0.575/−0.346 0.384/−0.236aR1 = ∑∥Fo| − |Fc∥/∑|Fo|; wR2 = {∑[w(Fo
2 − Fc2)2]/∑[w(Fo
2)2]}1/2.
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581548

solution of 2 (0.13 g, 0.12 mmol) in toluene, and the resulting darkyellow mixture was kept at −35 °C for 1 day. Crystallization fromtoluene gave pale yellow crystals of 4-pCl, which were also used for X-ray diffraction analysis. Yield: 0.11 g (68%). For 4-pCl: Tm = 143−145°C. Tdec = 147−150 °C. 1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.59(d, J = 6.5 Hz, 2H, Py-H2,6), 7.60 (d, J = 6.5 Hz, 2H, Py-H3,5). 19FNMR (470.2 MHz, CDCl3, 25 °C): δ = −112.6 (m, 2F, ortho-F),−159.9 (t, J(19F, 19F) = 18 Hz, 1F, para-F), −163.7 (m, 2F, meta-F).13C NMR (125.7 MHz, CDCl3, 25 °C): δ = 150.7 (Py-C2,6), 149.9(dd, J(19F, C) = 222 Hz/27 Hz, Pf-C2,6), 148.5 (Py-C4), 139.0 (dm,J(19F, C) = 245 Hz, Pf-C4), 136.5 (dm, J(19F, C) = 253 Hz, Pf-C3,5),126.6 (Py-C3,5), 121.3 (t, J(19F, C) = 75 Hz, Pf-C1). Elementalanalysis for C11H4ClCuF5N (344.15), Calcd: C, 38.39; H, 1.17; N,4.07%. Found: C, 37.53; H, 1.17; N, 4.01%. The slightly low C valuefor the elemental analysis is likely due to incomplete combustion as aresult of the large content of fluorine or the high air sensitivity of thematerial.Synthesis of C6F5Cu(2-chloropyridine) (4-oCl). Neat 2-chlor-
opyridine (0.056 g, 0.49 mmol) was added dropwise to a solution of 2(0.11 g, 0.099 mmol) in toluene (5 mL) at room temperature. Uponaddition of 2-chloropyridine, an intense yellow color developed. Thesolution was kept at −35 °C for a day, which yielded light yellowfeathery crystals. Yield: 0.12 g (88%). Slow evaporation of toluene atRT gave pale yellow “single crystals” of 4-oCl suitable for X-raydiffraction analysis. For 4-oCl: Tm = 123−127 °C. Tdec = 130−133 °C.1H NMR (500 MHz, CDCl3, 25 °C): δ = 8.5 (d, J = 5.0 Hz, 1H, Py-H6), 7.90 (pst, J = 8.0 Hz, 1H, Py-H4), 7.55 (d, J = 8.5 Hz, 1H, Py-H3), 7.46 (pst, J = 6.0 Hz, 1H, Py-H5). 19F NMR (470.2 MHz,CDCl3, 25 °C): δ = −110.5 (br, 2F, ortho-F), −156.3 (br, 1F, para-F),−162.9 (br, 2F, meta-F). 13C NMR (125.7 MHz, CDCl3, 25 °C): δ =151.7 (Py-C2), 150.9 (d, J(19F, C) = 228 Hz/28 Hz, Pf-C2,6), 150.4(Py-C6), 140.8 (Py-C4), 140.4 (dm, J(19F, C) = 249 Hz, Pf-C4), 136.5(dm, J(19F, C) = 254 Hz, Pf-C3,5), 125.9 (Py-C3), 123.6 (Py-C5),115.0 (t, J(19F, C) = 67 Hz, Pf-C1). Elemental analysis forC11H4ClCuF5N (344.15), Calcd: C, 38.39; H, 1.17; N, 4.07%.Found: C, 38.25; H, 1.07; N, 4.02%.Synthesis of C6F5Cu(2,2′-bipy) (5). A solution of 2,2′-bipyridine
(712 mg, 4.56 mmol) in THF (10 mL) was added dropwise to asolution of 1 (1.00 g, 1.08 mmol) in THF (20 mL) at roomtemperature. Upon addition, the color of the solution became intensereddish brown. This solution was stored at −38 °C for 24 h. Orange-red, needle-like crystals formed and were isolated by decantation of themother liquor and dried under high vacuum for 30 min. Yield: 1.18 g(70%). For 5: Tm: = 90 °C (dec.). 1H NMR (500 MHz, CD2Cl2, 20°C): δ = 8.91 (br, 2H, bipy-H6,6′), 8.16 (d, J = 7.0 Hz, 2H, bipy-H3,3′), 8.04 (pst, 2H, bipy-H4,4′), 7.59 (br, 2H, bipy-H5,5′). 19F NMR(470.2 MHz, CD2Cl2, 20 °C): δ = −111.1 (m, 2F, ortho-F), −164.3 (t,J(19F, 19F) = 20 Hz, 1F, para-F), −165.1 (m, 2F, meta-F). 13C NMR(100.5 MHz, THF-d8, 20 °C): δ = 153.7 (bipy-C2,2′), 150.7 (bipy-C6,6′), 150.2 (dd, J(19F, C) = 226/32 Hz, Pf-C2,6), 139.6 (bipy),138.1 (dm, J(19F, C) = 244 Hz, Pf-C4), 136.7 (dm, J(19F, C) = 255Hz, Pf-C3,5), 128.9 (t, J(19F, C) = 83 Hz, Pf-C1), 127.1 (bipy), 122.4(bipy). Elemental analysis for C16H8CuF5N2 (386.79), Calcd: C,49.68; H, 2.08; N, 7.24%. Found: C, 49.64; H, 1.59; N, 7.21%.Crystal Structure Determinations. X-ray diffraction intensities
were collected at 100 and 218 K (3) on a Bruker Apex (Kα Moradiation, λ = 0.71073 Å) or Apex 2 (Kα Cu radiation, λ = 1.54178 Å)diffractometers. For all structures, SADABS (Sheldrick, G. M. SADABS(2.01): Bruker/Siemens Area Detector Absorption Correction Program;Bruker AXS: Madison, WI, 1998) absorption correction was used.Non-hydrogen atoms were refined with anisotropic displacementcoefficients, and hydrogen atoms were treated as an idealizedcontribution. A solvent pentane molecule in 3 is highly disorderedabout a 2-fold axis. This solvent pentane molecule was refined asdisordered over two positions and with restrictions. The standard C−C bond lengths were used as targets in the refinement forcorresponding C−C distances, and similar carbon atoms were refinedwith the same thermal parameters. H atoms in the disordered pentanemolecule were not taken into consideration. Crystallographic data anddetails of data collections and refinements of the structures are given in
Table 1. The X-ray structures of 3, 4-pMe, 4-oMe, 4-pCl, 4-oCl, and 5have been deposited at the Cambridge Crystallographic Data Center(CCDC) as supplementary publications nos. CCDC-852837 to852842. Copies of the data can be obtained free of charge onapplication to CCDC, 12 Union Road, Cambridge CB2 1EZ, U.K.(Fax: (+44) 1223-336-033. E-mail: [email protected]).
■ RESULTS AND DISCUSSIONSynthesis. [C6F5Cu]4 (1) was prepared according to a
literature procedure by Sheppard and Cairncross25 andrecrystallized from toluene to give the toluene solvate[C6F5Cu]4(toluene)2 (2). Compound 2 was either useddirectly or after drying in high vacuum at 80 °C to removethe toluene solvent molecules, yielding the base-free, purifiedspecies 1. Addition of 2 equiv of pyridine to 1 in CH2Cl2 at RTresulted in a light yellow solution, from which colorless needle-like crystals of 3 were collected in 82% yield after layering withhexanes at −38 °C (Scheme 1). When 2 was treated in toluene
or CH2Cl2 with an excess of different pyridine derivatives, air-sensitive colorless-to-pale yellow needle-like crystals ofcompounds 4-R (4-H, 4-pMe, 4-oMe, 4-pCl, 4-oCl) wereisolated after recrystallization from toluene at −38 °C in highyield. The red-colored bipyridine species 5 was obtained uponcrystallization of an equimolar mixture of 1 and 2,2′-bipyridinein THF solution at −38 °C. All complexes were fullycharacterized by 1H, 13C, and 19F NMR spectroscopies andelemental analysis, and the solid-state structures weredetermined by single-crystal X-ray diffraction. The pyridinecomplexes 3 and 4-R are thermally stable to about 125−150°C, whereas compound 5 decomposes at ca. 90 °C. Acomparison with the reported decomposition temperature forthe dioxane complex31 of 1 (200−220 °C) suggests thatcoordination of pyridine ligands leads to destabilization of thepentafluorophenyl copper complex. This is consistent withearly reports that the isolation and crystallographic character-ization of organocopper pyridine complexes is complicated bytheir relatively low thermal stability.32 We found that thepyridine complexes can be stored at low temperature (−20 °C)under a nitrogen atmosphere for extended periods of time.However, upon exposure to air, they quickly decompose, asevidenced by a rapid color change to greenish-brown.
Scheme 1. Synthesis of Pentafluorophenyl Copper−PyridineComplexes
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581549
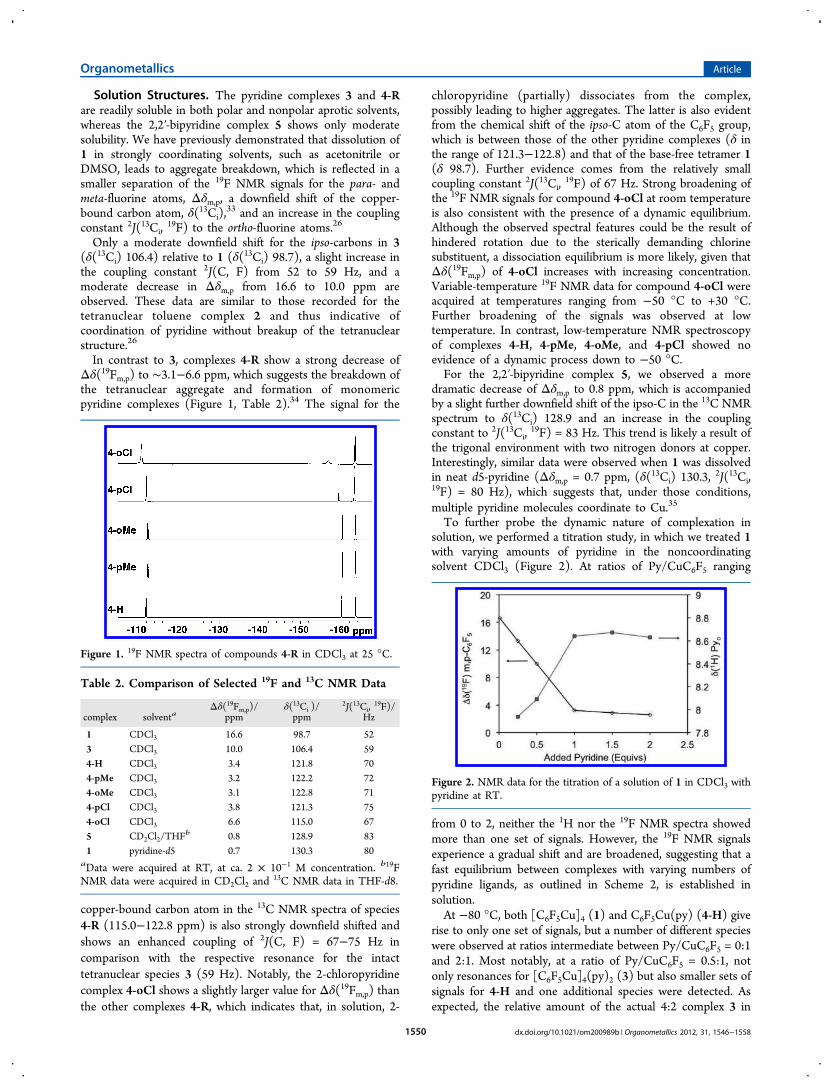
Solution Structures. The pyridine complexes 3 and 4-Rare readily soluble in both polar and nonpolar aprotic solvents,whereas the 2,2′-bipyridine complex 5 shows only moderatesolubility. We have previously demonstrated that dissolution of1 in strongly coordinating solvents, such as acetonitrile orDMSO, leads to aggregate breakdown, which is reflected in asmaller separation of the 19F NMR signals for the para- andmeta-fluorine atoms, Δδm,p, a downfield shift of the copper-bound carbon atom, δ(13Ci),
33 and an increase in the couplingconstant 2J(13Ci,
19F) to the ortho-fluorine atoms.26
Only a moderate downfield shift for the ipso-carbons in 3(δ(13Ci) 106.4) relative to 1 (δ(13Ci) 98.7), a slight increase inthe coupling constant 2J(C, F) from 52 to 59 Hz, and amoderate decrease in Δδm,p from 16.6 to 10.0 ppm areobserved. These data are similar to those recorded for thetetranuclear toluene complex 2 and thus indicative ofcoordination of pyridine without breakup of the tetranuclearstructure.26
In contrast to 3, complexes 4-R show a strong decrease ofΔδ(19Fm,p) to ∼3.1−6.6 ppm, which suggests the breakdown ofthe tetranuclear aggregate and formation of monomericpyridine complexes (Figure 1, Table 2).34 The signal for the
copper-bound carbon atom in the 13C NMR spectra of species4-R (115.0−122.8 ppm) is also strongly downfield shifted andshows an enhanced coupling of 2J(C, F) = 67−75 Hz incomparison with the respective resonance for the intacttetranuclear species 3 (59 Hz). Notably, the 2-chloropyridinecomplex 4-oCl shows a slightly larger value for Δδ(19Fm,p) thanthe other complexes 4-R, which indicates that, in solution, 2-
chloropyridine (partially) dissociates from the complex,possibly leading to higher aggregates. The latter is also evidentfrom the chemical shift of the ipso-C atom of the C6F5 group,which is between those of the other pyridine complexes (δ inthe range of 121.3−122.8) and that of the base-free tetramer 1(δ 98.7). Further evidence comes from the relatively smallcoupling constant 2J(13Ci,
19F) of 67 Hz. Strong broadening ofthe 19F NMR signals for compound 4-oCl at room temperatureis also consistent with the presence of a dynamic equilibrium.Although the observed spectral features could be the result ofhindered rotation due to the sterically demanding chlorinesubstituent, a dissociation equilibrium is more likely, given thatΔδ(19Fm,p) of 4-oCl increases with increasing concentration.Variable-temperature 19F NMR data for compound 4-oCl wereacquired at temperatures ranging from −50 °C to +30 °C.Further broadening of the signals was observed at lowtemperature. In contrast, low-temperature NMR spectroscopyof complexes 4-H, 4-pMe, 4-oMe, and 4-pCl showed noevidence of a dynamic process down to −50 °C.For the 2,2′-bipyridine complex 5, we observed a more
dramatic decrease of Δδm,p to 0.8 ppm, which is accompaniedby a slight further downfield shift of the ipso-C in the 13C NMRspectrum to δ(13Ci) 128.9 and an increase in the couplingconstant to 2J(13Ci,
19F) = 83 Hz. This trend is likely a result ofthe trigonal environment with two nitrogen donors at copper.Interestingly, similar data were observed when 1 was dissolvedin neat d5-pyridine (Δδm,p = 0.7 ppm, (δ(13Ci) 130.3, 2J(13Ci,19F) = 80 Hz), which suggests that, under those conditions,multiple pyridine molecules coordinate to Cu.35
To further probe the dynamic nature of complexation insolution, we performed a titration study, in which we treated 1with varying amounts of pyridine in the noncoordinatingsolvent CDCl3 (Figure 2). At ratios of Py/CuC6F5 ranging
from 0 to 2, neither the 1H nor the 19F NMR spectra showedmore than one set of signals. However, the 19F NMR signalsexperience a gradual shift and are broadened, suggesting that afast equilibrium between complexes with varying numbers ofpyridine ligands, as outlined in Scheme 2, is established insolution.At −80 °C, both [C6F5Cu]4 (1) and C6F5Cu(py) (4-H) give
rise to only one set of signals, but a number of different specieswere observed at ratios intermediate between Py/CuC6F5 = 0:1and 2:1. Most notably, at a ratio of Py/CuC6F5 = 0.5:1, notonly resonances for [C6F5Cu]4(py)2 (3) but also smaller sets ofsignals for 4-H and one additional species were detected. Asexpected, the relative amount of the actual 4:2 complex 3 in
Figure 1. 19F NMR spectra of compounds 4-R in CDCl3 at 25 °C.
Table 2. Comparison of Selected 19F and 13C NMR Data
complex solventaΔδ(19Fm,p)/
ppmδ(13Ci )/ppm
2J(13Ci,19F)/
Hz
1 CDCl3 16.6 98.7 523 CDCl3 10.0 106.4 594-H CDCl3 3.4 121.8 704-pMe CDCl3 3.2 122.2 724-oMe CDCl3 3.1 122.8 714-pCl CDCl3 3.8 121.3 754-oCl CDCl3 6.6 115.0 675 CD2Cl2/THF
b 0.8 128.9 831 pyridine-d5 0.7 130.3 80aData were acquired at RT, at ca. 2 × 10−1 M concentration. b19FNMR data were acquired in CD2Cl2 and
13C NMR data in THF-d8.
Figure 2. NMR data for the titration of a solution of 1 in CDCl3 withpyridine at RT.
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581550

solution diminishes as the ratio of Py/CuC6F5 is eitherincreased or decreased. The amount of the third species,which shows broad signals even at −80 °C (δ(19F) −105.1,−147.2, −158.8; Δδm,p = 11.6 ppm), significantly increases at asmaller ratio of Py/CuC6F5 = 0.25:1. On the basis of theseobservations, we tentatively assign this species to a 4:1 complex[C6F5Cu]4(py).Solid-State Structures. A single-crystal X-ray diffraction
study of 3 confirms that the tetranuclear copper aggregateremained intact, while two pyridine molecules are coordinatedto opposite copper centers (Figure 3). Importantly, the
structural parameters are different from those of 1 and otherhomoleptic tetranuclear organocopper complexes that lackinteractions with donor molecules.26 Indeed, the structure ismore similar to that of the toluene complex 2, except for thatthe four copper atoms in 3 lie in a plane rather than adopting abutterfly arrangement as in the structure of 2 (2; interplanarangle Cu1Cu3Cu2)//Cu1ACu3Cu2 = 37.7°). The pentafluor-ophenyl groups are positioned alternately above and below theCu4 plane, which leads to strong puckering of the eight-membered Cu4C4 ring in 3. The separation between oppositeCu centers is much smaller for Cu2···Cu3 with 2.5940(6) Å
than for Cu1···Cu1A with 4.178(1) Å. This is a feature that ischaracteristic of tetranuclear copper species in which twocopper atoms are coordinated by donor ligands.13,14,26,27,36,37
The Cu−C distances at Cu1 and Cu1A (average = 2.061(3) Å)in 3 are significantly longer than those at Cu2 and Cu3 (average= 1.974(2) Å), indicating a highly unsymmetrical bridgingmode of the aryl groups similar to that in 2. This type of bondalternation is less pronounced14,37 or not observed at all13,36 inorganocopper sulfide complexes. As previously suggested byvan Koten and co-workers for other donor-supportedtetranuclear arylcopper species,4 the long copper−carbonbonds at Cu1 support a description of 3 as a contact ion pairconsisting of two [(C6F5)2Cu]
− cuprate anions bridged by twopyridine-stabilized copper cations via the ipso-carbon atoms ofthe pentafluorophenyl rings.All complexes 4-R were also subjected to X-ray diffraction
analysis. Single crystals of 4-oMe and 4-oCl were grown viaslow solvent evaporation from a mixture of toluene andhexanes, whereas needle-like crystals of 4-H, 4-pMe, and 4-pClwere obtained from the reaction mixture at low temperature.Structure plots are presented in Figure 4, and selectedgeometric parameters are listed in Table 3. The crystalstructure of 4-H has been communicated previously, but thedata are provided in Table 3 for comparison purposes.29 Themolecular structures of complexes 4-R confirm the formation ofmononuclear dicoordinated species C6F5Cu(L) in the solidstate with a linear or nearly linear coordination geometry atcopper (N−Cu−C from 174.66(8) to 178.54(6)°).The Cu−C bonds of all complexes are within a narrow range
from 1.887(2) to 1.901(3) Å, and thus considerably shorterthan those found for the tetranuclear species 3 (1.955(2)−2.076(2) Å), which shows bridging rather than terminal arylgroups. The distances are similar to those of other dicoordinateorganocopper−ligand complexes, such as [(C6H2-2,4,6-t-Bu3)-Cu(Me2S)],
17 with Cu−C = 1.886(3) Å, a related dimericcomplex containing a chelating oxazolinyl group (Cu−C =1.899(5) Å),22 and N-heterocyclic carbene−copper(I) com-plexes.38 The copper−nitrogen distances (1.897(2)−1.917(2)Å) are significantly shorter than the ones in 3 (2.023(2) Å), butcomparable to those in the oxazolinyl complex mentionedabove (Cu−N = 1.902(4) Å)22 and a related dimericpyridylalkyl species [2-(SiMe3)2C(Cu)C5H4N]2
19 (Cu−N =1.910(3) Å). Copper−nitrogen distances in dicoordinatecopper cations [py2Cu]
+ are also found in the same range(1.86−1.96 Å).39,40 Noteworthy is that the Cu−N bond lengthsfor the two independent molecules of 4-oCl (1.909(2),1.917(2) Å) are slightly longer than those in the othercomplexes, suggesting weaker coordination of the pyridineligand to Cu. The latter is consistent with the conclusionsdrawn from the solution NMR studies of 4-oCl.
Scheme 2. Proposed Equilibria between Oligonuclear and Mononuclear Arylcopper Complexes
Figure 3. Plot of the molecular structure of 3; a cocrystallizedmolecule of pentane is omitted for clarity. Selected interatomicdistances (Å) and angles (°): Cu(1)−N(1) 2.023(2), Cu(1)−Cu(2)2.4496(4), Cu(1)−Cu(3) 2.4687(4), Cu(2)···Cu(3) 2.5940(6),Cu(1)···Cu(1A) 4.179(1), Cu(1)−C(1) 2.045(3), Cu(1)−C(7)2.077(2), Cu(2)−C(1) 1.991(2), Cu(3)−C(7) 1.956(2), Cu(1)−Cu(2)−Cu(3) 58.528(11), Cu(1)−Cu(3)−Cu(2) 57.812(10),Cu(2)−Cu(1)−Cu(3) 63.660(15), Cu(1)−Cu(2)−Cu(1A)117.06(2), Cu(1)−Cu(3)−Cu(1A) 115.62(2), C(1)−Cu(2)−C(1A)139.40(15), C(7)−Cu(3)−C(7A) 156.64(14).
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581551

The pentafluorophenyl groups and the pyridine rings arecoplanar for 4-H and nearly so for 4-pMe (2.89°) and 4-pCl(2.34°); however, large dihedral angles of 43.12° and 44.21°,respectively, were observed for the two independent moleculesin the complex 4-oCl. The latter is attributed to the high stericdemand of the chloro substituent in the 2-position of thepyridine ring. Interestingly, the methyl group in the 2-positionof 4-oMe leads to only a slight twisting of the two aromaticrings of 5.32°.The crystal structure of 5 also reveals a monomeric species,
but the copper atoms are in an unusual trigonal-planarcoordination geometry3d,18 as a result of chelation of the 2,2′-bipyridine ligand (Figure 5). The Cu−N bond lengths of2.018(2) and 2.082(2) Å are not equivalent, and both distances
are slightly longer than those in the monomeric compounds 4-R, indicating weaker binding in the trigonal geometry incomparison with the typical linear dicoordinate geometry. Theunsymmetric coordination of the bipyridine in 5 furthermanifests itself in an angle C(1)−Cu(1)−N(1) of 148.30(7)°, which is larger than the C(1)−Cu(1)−N(2) angle of131.35(7)°. A more symmetric structure has been reported forthe complex 4-MeOC6F4Cu(phen) (phen = phenanthroline)with Cu−N = 2.0720(18) and 2.0949(19) Å,3d whereas thegold complex ArAu(2,2′-dmphen) (Ar = 2,4,6-(NO2)3C6H2;dmphen = 2,9-dimethyl-1,10-phenanthroline)41 shows muchstronger distortions from trigonal-planar geometry with twovery different Au−N bonds of 2.1363(3) and 2.5753(3) Å. TheCu−C bond of 1.907(2) Å is slightly longer than that in
Figure 4. Plots of the molecular structures of (a) 4-pMe, (b) 4-oMe, (c) 4-pCl, and (d) 4-oCl. Only one of two independent molecules of 4-oCl isshown. Geometric parameters are summarized in Table 3.
Table 3. Comparison of Selected Bond Lengths [Å], Interatomic Distances [Å], and Angles [°] for Complexes 4-R a
4-oCl
4-Hb 4-pMe 4-oMe 4-pCl mol A mol B
Cu1−C1 1.8913(17) 1.891(3) 1.8963(19) 1.8873(19) 1.901(3) 1.898(3)Cu1−N1 1.9022(15) 1.900(2) 1.9072(16) 1.8972(16) 1.917(2) 1.909(2)N1−C7 1.356(2) 1.352(3) 1.353(3) 1.354(2) 1.338(4) 1.340(4)N1−C11 1.346(2) 1.345(3) 1.351(3) 1.345(2) 1.349(4) 1.349(4)C−X (X = Cl, CH3) 1.501(4) 1.498(3) 1.7212(18) 1.727(3) 1.723(3)C1−Cu1−N1 178.54(6) 177.88(8) 174.66(8) 176.34(7) 177.75(11) 176.19(11)Py//Pf 0.0 2.89 5.32 2.34 44.21 43.12Cu1···Cu1A 2.8924(3) 3.531(1) 3.2454(5) 3.521(1) 3.4797(6)Cu1A···Cu1B 2.8924(3) 3.698(1) 4.2970(5) 3.784(1) 3.8363(6)C1−Cu1···Cu1A 89.900(5) 95.89(7) 92.56(7) 98.83(5) 100.14(9)/100.01(9)C1A−Cu1A···Cu1B 89.900(5) 83.27(7) 89.42(7) 77.94(5) 103.85(8)/103.00(9)C1−Cu1···Cu1B 90 96.32(7) 91.44(7) 100.54(5) 90.40(9)/92.45(8)N1−Cu1···Cu1A 90.097(5) 85.24(7) 92.69(5) 82.50(4) 80.31(7)/75.55(7)N1−Cu1···Cu1B 90.097(5) 95.46(7) 85.23(5) 100.21(4) 75.59(7)/80.53(7)N1A−Cu1A···Cu1B 90 84.88(7) 93.89(5) 81.07(7) 93.08(7)/87.04(7)C7−N1−Cu1···Cu1B 90 65.79(20) 60.73(14) 115.12(14) −109.82(20) −109.98(20)C11−N1−Cu1···Cu1B 90 112.49(19) −119.27(15) −62.23(14) 72.85(20) 70.33(02)C2−C1−Cu1···Cu1B 90 −117.29(21) 122.00(18) −119.67(16) −116.40(23) −113.80(22)C6−C1−Cu1···Cu1B 90 65.26(20) −58.19(16) 63.10(14) 63.35(23) 68.76(24)Cu1···Cu1A···Cu1B 179.716(15) 175.16(1) 168.22(1) 170.54(1) 155.80(2)
aSymmetry operations used to generate equivalent Cu atoms for 4-H: −x, −y, z + 1/2; −x, y + 1/2, −z + 1/2; x, −y + 1/2, −z; −x, −y, −z; x, y, −z− 1/2; x, −y − 1/2, z − 1/2; −x, y − 1/2, z. For 4-pMe: (i) −x + 1, −y + 1, −z; (ii) −x + 1, −y + 2, −z; (iii) x, 1 + y, z. For 4-pCl: (i) −x + 2, −y, −z + 1; (ii) −x + 2, −y + 1, −z + 1; (iii) x, 1 + y, z. For 4-oMe: (i) −x + 2, −y + 1, −z + 1; (ii) −x + 2, −y, −z + 1; (iii) x, y − 1, z. For 4-oCl: (i) x −1, y, z; (ii) x − 1, y − 1, z; (iii) 1 + x, 1 + y, z; (iv) 1 + x, y, z; (v) x, y − 1, z; (vi) x, 1 + y, z. bReference 29.
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581552

compounds 4-R, but significantly shorter than that in thetetranuclear species 3.Supramolecular Structures via Cuprophilic and π-
Interactions. All the complexes described in here formextended supramolecular structures in the solid state via eitherπ-stacking interactions or short Cu···Cu contacts as a result ofso-called cuprophilic interactions. Supramolecular assembly ofmolecular materials via π-stacking of arenes and perfluoroar-enes continues to be a topic of much current interest.42,43
Aurophilic interactions, the attractive forces between closed-shell d10 metal ions of gold, have also long been recognized andare now commonly used in supramolecular chemistry.44 Theyare generally considered to be similar in strength to hydrogenbonding interactions and rely, to a large extent, on relativisticeffects. In contrast, attractive interactions between closed-shellCu(I)···Cu(I) pairs have only recently been proposed based onexperimental and theoretical studies, and the relevance andstrength of such interactions remain controversial.40,45,46 Onthe basis of MP2 calculations, Schwerdtfeger et al. concludedthat cuprophilic interactions between neutral pairs [RCuL]2should be attractive by up to −4 kcal mol−1 and that theinteraction potential is very shallow in the range from ca. 2.5 to3.5 Å.47
The complex [C6F5Cu]4(py)2 (3) displays an interestingextended structure, in which intermolecular π-interactions(Py//Py, Pf//Pf, and Py-Pf) lead to channels propagatingalong the crystallographic c axis (Figure 6), which are occupiedby pentane solvent molecules. Whereas short intramolecularCu···Cu contacts (2.5940(6) Å) are observed within the Cu4aggregates, as discussed above, the intermolecular Cu···Cucontacts are very long (>7 Å), precluding the presence of anyCu···Cu interactions.Extended structures that form as a result of π-interactions are
also observed in the crystal structure of the 2,2′-bipyridinecomplex 5 (Figure 7). Both the bipyridyl units and thepentafluorophenyl groups form offset stacks along thecrystallographic c axis. The orientation of the bipyridyl unitsalternates from layer to layer with a 90° offset between pairs ofbipyridyl units. Consequently, only one of the pyridyl rings andthe central five-membered CuN2C2 chelate ring match up withthe respective parts of the adjacent molecule (distance between
centroids = 3.382 Å). The shortest intermolecular C···C andC···N distances fall in the range from 3.3 to 3.6 Å, andinterestingly, they are augmented by a short Cu···C distance of3.323 Å to the meta-pyridyl position.48 The Cu···Cu separationsof 5.631 Å, on the other hand, are very long, which is insofarsurprising as dimers with short Cu···Cu contacts have beenreported for the closely related phenanthroline (phen) complex4-MeOC6F4Cu(phen) (Cu···Cu = 2.5770(6) Å).3d
Inspection of the extended structures of the dicoordinateorganocopper pyridine complexes 4-R reveals, in all cases,chains of Cu atoms that progress throughout the entire crystallattice. However, the stacking direction, and thus theorientation of the chains, as well as the Cu···Cu separationsvary considerably. The parent compound 4-H, which
Figure 5. Plot of the molecular structure of 5. Selected bond lengths(Å) and angles (°): Cu(1)−N(1) 2.017(2), Cu(1)−N(2) 2.082(2),Cu(1)−C(1) 1.907(2), C(11)−C(12) 1.491(3), C(1)−Cu(1)−N(1)148.30(7), C(1)−Cu(1)−N(2) 131.35(7), N(1)−Cu(1)−N(2)79.69(6), C(6)−C(1)−Cu(1) 124.2(1), C(2)−C(1)−Cu(1)123.0(1), (bipy)//(Pf) 46.5, angle sum at Cu 359.34.
Figure 6. View of the extended structure of 3 along thecrystallographic c axis, illustrating the formation of channels that areoccupied by pentane solvent molecules. Only one position of thedisordered pentane molecule is given for clarity.
Figure 7. Two different views of a fragment of the crystal packing of 5showing offset π-stacking along the crystallographic c axis betweenCu(2,2′-bipy) fragments and between the pentafluorophenyl groups.Cu···Cu = 5.631 Å.
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581553

crystallizes in the Pbcm space group, adopts a very unusualstructure in that the copper atoms in 4-H are arranged in one-dimensional chains with Cu···Cu distances of 2.8924(3) Å.29
These Cu···Cu distances are among the very shortest reportedfor unsupported Cu(I)···Cu(I) contacts (Figure 8).49 All atoms,including the hydrogen atoms, reside on a crystallographicmirror plane. Consequently, all copper atoms are equidistantand the direction of the chain is perpendicular to the plane ofthe molecules. Importantly, the ligands in adjacent RCuLfragments adopt a staggered conformation, thereby avoidingany π-stacking and allowing the molecules to approach veryclosely. Hence, aggregation leads to supramolecular structuresthat are unprecedented in organocopper chemistry.50,51
Intrigued by these unusual observations, we set out to furtherstudy the effects of placing electron-donating Me groups andelectron-withdrawing Cl substituents at the ortho and parapositions of the pyridyl ring on the packing of complexesC6F5Cu(py-R) (4-R). Surprisingly, in all cases, a packingpattern that is very different from that of 4-H was observed.Compounds 4-R form offset stacks that progress along thecrystallographic b axis with the RCuL monomers aligned insuch a way that the C6F5 groups undergo π-interactions withthe pyridyl moieties of the adjacent molecules and vice versa(Figure 8). In each stack, the successive molecules adopt aneclipsed, rather than a staggered, arrangement and are offset sothat the resulting stacks are tilted by an angle of α = 23.9° for(4-pMe), 26.8° for (4-oMe), 24.7° for (4-pCl), and 21.2° for(4-oCl). This tilting is the result of lateral slippage of thearomatic groups, a phenomenon that is commonly encountered
for stacks involving perfluoroarene−arene interactions (seeFigure S1, Supporting Information).43,46,52 The tilt angle wasmeasured using the vector perpendicular to the planecontaining the pentafluorophenyl ring. The Cu···Cu distancesbetween adjacent molecules alternate, thereby leading toformation of dimers with relatively short Cu···Cu contacts of3.245−3.531 Å that are, in turn, connected through longerCu···Cu contacts ranging from 3.698 to 4.297 Å to form infiniteCu chains. The alternation is most pronounced for 4-oMe withCu···Cu distances of 3.245 and 4.297 Å, respectively.Noteworthy is also that, for 4-oMe, relatively short contactsof 3.245 Å coincide with the largest deviation from linearcoordination geometry at copper (C1−Cu1−N1 = 174.66(8)°).For 4-oCl, the formation of a zigzag Cu chain is apparent and
reflected in a relatively small angle Cu1···Cu1A···Cu1B of155.79(2)°. The latter is likely related to the presence of thesterically demanding 2-chloro substituent, which also leads totilting of the pyridyl and C6F5 groups with respect to eachother.From the crystallographic studies, we conclude that, unlike
the parent complex 4-H, the substituted pyridine complexes 4-R show multiple face-to-face pentafluoroarene−arene π-interactions. On the basis of the Cu···Cu contacts of2.8924(3) Å in 4-H, which are significantly shorter than thesum of the van der Waals radii of two Cu(I) centers of 3.78 Åand close to the sum of the covalent radii of 2.64 Å,53 thepresence of cuprophilic interactions is likely to play a significantrole. Indeed, recent theoretical studies on 4-H are in agreement
Figure 8. Plots of the extended structures of 4-R (stacking direction for 4-H is the crystallographic c axis; for all substituted derivatives, it is the baxis). (a) View sideways, from left to right: 4-H, 4-pMe, 4-pCl, 4-oMe, 4-oCl. (b) View along the crystallographic a axis: 4-H, 4-pMe, 4-pCl, 4-oMe.View along the crystallographic c axis: 4-oCl. For 4-H, the Cu chain is slightly tilted toward the front to show three-dimensionality. (c) Space fillingillustration of the tilting of the stacks.
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581554
with this assessment.54 The Cu···Cu separations for complexes4-R are comparative much larger (>3.2 Å), and cuprophilicinteractions are expected to be weaker. The supramolecularassembly in those complexes is, to a large extent, governed byperfluoroarene−arene π-interactions, which appears to preventthe Cu centers in neighboring molecules from approachingmore closely.46
Photophysical Properties. Intrigued by the observationof infinite copper chains from the X-ray data, we decided toinvestigate the photoluminescence properties of complexes 4-Rin the solid state. The parent compound 4-H displays strongblue fluorescence (λmax = 460 nm, τ = 30 ns) in the solid stateat RT upon excitation at λ = 330 nm. This band is alsoobserved in phosphorescence mode, possibly as a result of adelayed fluorescence effect. The 460 nm band is accompaniedby a weaker long-lived emission band (τ = 70 μs) that extendsto ca. 700 nm (Figure 9). The emission wavelength was found
to be virtually independent of the excitation wavelength (275−375 nm), indicating that, although the excitation pathway is thesame for the two bands, they decay independently. Theluminescence properties of 4-H proved to be stronglytemperature-dependent, and upon cooling, the emission colorchanges gradually from blue to a bright yellow-green color. Thisso-called luminescence thermochromism55 was traced back toan increase in the intensity of the band at 580 nm relative tothat at 460 nm (Figure 9). The latter all but disappears at 77 K,revealing a weak shoulder band at ca. 500 nm. On the basis ofthe luminescence lifetimes, the emission at 460 nm is assignedto fluorescence from a metal-to-ligand charge transfer (MLCT)state, whereas the band at 580 nm is likely due to emissionfrom a lower-lying triplet excited state.56 Because the twoemissive states are separated by more than 4000 cm−1, that is,20 times the value of kBT at room temperature, it is more likely
that the observed delayed fluorescence is caused by triplet−triplet annihilation (T1 + T1 → S1 + S0) rather than byconventional repopulation of the singlet excited state. Thismechanism, which is common for polymers, solids, andconcentrated solutions, would also account, at least in part,for the increased singlet emission at high temperatures. Tripletmigration or “hopping” across the solid is faster at roomtemperature than at 77 K and facilitates the annihilationprocess.57
In stark contrast to the observed strong blue fluorescence of4-H, among the substituted derivatives, only 4-pMe proved tobe (weakly) emissive in the solid state at room temperature.Upon excitation at 325 nm, 4-pMe displayed a weak yellow-green luminescence with a maximum at 558 nm. At 77 K, allsubstituted compounds exhibited a band in the range of 411−425 nm (Figure 10a). Again, these bands correspond to decay
from singlet excited states as they are absent when theexperiment was performed in phosphorescence mode. How-ever, they all are significantly higher energy than that of 4-H(475 nm at 77 K). No clear correlation with the electroniceffects of the pyridine substituent was observed, suggesting thatthe transition is most likely not simply due to a pyridine-basedMLCT state, but rather involves orbitals centered at Cu and/orthe C6F5 moiety. In the case of 4-pMe, the band at 411 nm isaccompanied by a broad emission at ca. 600 nm, which is redshifted from the room-temperature band at 558 nm.Theoretical calculations by Yang and Su and co-workers
predict an emission at 449 nm for C6F5Cu(py) due to chargetransfer from the pyridine-centered LUMO to the copper-centered HOMO-2.54 Interestingly, they also calculated theemission spectra for a dimeric species and found that theemission maximum shifted from ca. 460 nm at a Cu···Cuseparation of 3.8 Å to 550 nm at a Cu···Cu separation of 2.8 Å.
Figure 9. (a) Variable-temperature solid-state fluorescence and (b)phosphorescence spectra of 4-H (λexc = 330 nm).
Figure 10. (a) Solid-state fluorescence and (b) phosphorescencespectra of compounds 4-H (λexc = 330 nm) and 4-R (λexc = 325 nm) at77 K.
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581555

Taking into consideration the theoretical results by Yang andSu and co-workers,54 our observations might suggest that thelarger Cu···Cu separations in the substituted derivatives 4-R incomparison with those in 4-H result in a higher energyemission due to a smaller contribution of intermolecular orbitalmixing between Cu centers in neighboring molecules.The fluorescence features discussed above are accompanied
by decay from a triplet excited state that results in emissionmaxima in the range from 545 to 565 nm for the substitutedspecies 4-R and, for 4-oMe, a feature at 425 nm that isattributed to delayed fluorescence (Figure 10b). Again, wenotice that the phosphorescence for 4-H is significantly morebathochromic than that for any of the other derivatives,independent of whether electron-withdrawing or electron-donating substituents are attached to the pyridine ligand. Onthe basis of our observations, it is clear that the luminescencefeatures for compound 4-H are distinct from those of all otherderivatives, indicating that the unique solid-state packing withshort Cu···Cu separations affects the photophysical properties,presumably due to mixing of Cu-centered orbitals ofneighboring molecules. There also appears to be a correlationbetween short Cu···Cu contacts and the observation of delayedfluorescence via triplet−triplet annihilation, which is mostprominent for 4-H (Cu polymer with Cu···Cu = 2.892 Å) andfor 4-oMe (Cu dimers with Cu···Cu = 3.245 Å).
■ CONCLUSIONSWe have demonstrated that, depending on the amount ofpyridine as an external ligand added to solutions of [C6F5Cu]4(1), either a tetranuclear species [C6F5Cu]4(py)2 (3) or amononuclear species C6F5Cu(py) (4-H) can be isolated. NMRdata for a complex with 2,2′-bipyridine (5) are strikingly similarto those recorded for solutions of 1 in d5-pyridine, indicatingthat, in neat pyridine, more than one ligand is bound to Cu andmost likely a tricoordinate species of the type C6F5Cu(py)2 ispresent.The solid-state structures of these compounds, determined
by single-crystal X-ray crystallography, reveal different elementsof supramolecular assembly. Arene−arene π-interactions in 3lead to a network structure with channels propagating throughthe lattice along the crystallographic c axis. Similarly, the 2,2′-bipyridine complex 5 shows formation of offset π-stacks as thedominant feature in the extended solid-state structure. Incontrast, the supramolecular assembly of 4-H is based oncuprophilic interactions and leads to one-dimensional copperchains with equidistant Cu···Cu contacts of 2.8924(3) Å. Tofurther investigate this highly unusual structural motif, weprepared a series of complexes, in which the pyridine ring issubstituted in the ortho or para position with an electron-donating methyl group or an electron-withdrawing chlorosubstituent. Again, offset supramolecular stacks are formed withinfinite Cu···Cu chains. However, the Cu···Cu distances aresignificantly longer in all cases. We attribute this difference tothe presence of offset perfluoroarene−arene interactions withintermolecular plane-to-plane separations of ca. 3.3−3.6 Å,which ultimately limit how close the Cu centers can approacheach other (Cu···Cu in the range of 3.2454(5)−4.1970(5) Å).For 4-H, no π-stacking is observed, as neighboring moleculesare oriented at an angle of 90° relative to each other.Compound 4-H is strongly blue fluorescent at 460 nm in thesolid state at room temperature, but shows yellow-greenluminescence at 77 K, giving rise to luminescence thermo-chromism. In contrast, the substituted compounds display
strong luminescence only at liquid nitrogen temperature. In allcases, the fluorescence emission band is in the range of ca.410−425 nm and thus at significantly different energy from thatin 4-H. Triplet−triplet annihilation phenomena are observedfor compounds 4-H and 4-oMe, suggesting that the shorterCu···Cu contacts in these compounds give rise to uniqueluminescence properties.
■ ASSOCIATED CONTENT*S Supporting InformationIllustration of the offset (slippage) of the π-systems in thestacks of 4-R and results from DFT and TD-DFT calculationson complexes 4-R. This material is available free of charge viathe Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] Address§School of Chemical Sciences, National Institute of ScienceEducation and Research, Institute of Physics Campus,Bhubaneswar-751005, Orissa, India.
■ ACKNOWLEDGMENTSAcknowledgment is made to the National Science Foundationand the Alfred P. Sloan Foundation for support of this research.
■ REFERENCES(1) Krause, N. Modern Organocopper Chemistry; Wiley VCH GmbH:Weinheim, Germany, 2002.(2) Nakamura, E.; Mori, S. Angew. Chem., Int. Ed. 2000, 39, 3750−3771.(3) For recent examples, see: (a) Schaper, F.; Foley, S. R.; Jordan, R.F. J. Am. Chem. Soc. 2004, 126, 2114−2124. (b) Norinder, J.;Baeckvall, J.-E.; Yoshikai, N.; Nakamura, E. Organometallics 2006, 25,2129−2132. (c) Henze, W.; Gartner, T.; Gschwind, R. M. J. Am.Chem. Soc. 2008, 130, 13718−13726. (d) Do, H.-Q.; Kashif Khan, R.M.; Daugulis, O. J. Am. Chem. Soc. 2008, 130, 15185−15192.(e) Herron, J. R.; Ball, Z. T. J. Am. Chem. Soc. 2008, 130, 16486−16487.(4) Jastrzebski, J. T. B. H.; van Koten, G. In Modern OrganocopperChemistry; Krause, N., Ed.; Wiley VCH GmbH: Weinheim, Germany,2002; pp 1−44.(5) (a) van Koten, G.; James, S. L.; Jastrzebski, J. T. B. H. InComprehensive Organometallic Chemistry; Abel, E. W., Stone, F. G. A.,Wilkinson, G., Eds.; Pergamon Press: Oxford, U.K., 1995; Vol. 3,Chapter 2. (b) Hakansson, M.; Eriksson, H.; Jagner, S. Inorg. Chim.Acta 2006, 359, 2519−2524, and references cited therein.(6) Recent examples of heteroaggregates: (a) Venkatasubbaiah, K.;DiPasquale, A. G.; Bolte, M.; Rheingold, A. L.; Jakle, F. Angew. Chem.,Int. Ed. 2006, 45, 6838−6841. (b) Davies, R. P.; Hornauer, S.;Hitchcock, P. B. Angew. Chem., Int. Ed. 2007, 46, 5191−5194.(c) Bomparola, R.; Davies, R. P.; Gray, T.; White, A. J. P.Organometallics 2009, 28, 4632−4635. (d) Venkatasubbaiah, K.;Bolte, M.; Jakle, F. J. Fluorine Chem. 2010, 131, 1247−1251.(e) Stollenz, M.; Gehring, H.; Konstanzer, V.; Fischer, S.; Dechert,S.; Grosse, C.; Meyer, F. Organometallics 2011, 30, 3708−3725.(7) Xie, X.; Auel, C.; Henze, W.; Gschwind, R. M. J. Am. Chem. Soc.2003, 125, 1595−1601.(8) Henze, W.; Vyater, A.; Krause, N.; Gschwind, R. M. J. Am. Chem.Soc. 2005, 127, 17335−17342.(9) Nobel, D.; van Koten, G.; Spek, A. L. Angew. Chem., Int. Ed. Engl.1989, 28, 208−210.(10) For examples of tetranuclear aggregates that display intra-molecular coordination of donor substituents attached to the aromaticmoiety, see: (a) Guss, J. M.; Mason, R.; Sotofte, I.; van Koten, G.;
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581556

Noltes, J. G. J. Chem. Soc., Chem. Commun. 1972, 446−447. (b) vanKoten, G.; Noltes, J. G. J. Organomet. Chem. 1975, 84, 129−138.(c) Wehman, E.; van Koten, G.; Knotter, M.; Spelten, H.; Heijdenrijk,D.; Mak, A. N. S.; Stam, C. H. J. Organomet. Chem. 1987, 325, 293−309.(11) (a) Costa, G.; Camus, A.; Marsich, N.; Gatti, L. J. Organomet.Chem. 1967, 8, 339−346. Ikariya, T.; Yamamoto, A. J. Organomet.Chem. 1974, 72, 145−151. (b) Miyashita, A.; Yamamoto, A. Bull.Chem. Soc. Jpn. 1977, 50, 1102−1108. (c) van Koten, G.; Noltes, J. G.;Spek, A. L. J. Organomet. Chem. 1978, 159, 441−463.(12) Camus, A.; Marsich, N. J. Organomet. Chem. 1970, 21, 249−258.(13) Meyer, E. M.; Gambarotta, S.; Floriani, C.; Chiesi-Villa, A.;Guastini, C. Organometallics 1989, 8, 1067−1079.(14) Olmstead, M. M.; Power, P. P. J. Am. Chem. Soc. 1990, 112,8008−8014.(15) Gambarotta, S.; Strologo, S.; Floriani, C.; Chiesi-Villa, A.;Guastini, C. Organometallics 1984, 3, 1444−1445.(16) (a) Coan, P. S.; Folting, K.; Huffman, J. C.; Caulton, K. G.Organometallics 1989, 8, 2724−2728. (b) Schiemenz, B.; Power, P. P.Organometallics 1996, 15, 958−964. (c) Stein, T.; Lang, H. J.Organomet. Chem. 2002, 664, 142−149.(17) He, X.; Olmstead, M. M.; Power, P. P. J. Am. Chem. Soc. 1992,114, 9668−9670.(18) Janssen, M. D.; Kohler, K.; Herres, M.; Dedieu, A.; Smeets, W. J.J.; Spek, A. L.; Grove, D. M.; Lang, H.; van Koten, G. J. Am. Chem. Soc.1996, 118, 4817−4829.(19) Papasergio, R. I.; Raston, C. L.; White, A. H. J. Chem. Soc., Chem.Commun. 1983, 1419−1420.(20) Papasergio, R. I.; Raston, C. L.; White, A. H. J. Chem. Soc.,Dalton Trans. 1987, 3085−3091.(21) (a) Hitchcock, P. B.; Lappert, M. F.; Layh, M.; Klein, A. J. Chem.Soc., Dalton Trans. 1999, 1455−1459. (b) Wingerter, S.; Gornitzka, H.;Bertrand, G.; Stalke, D. Eur. J. Inorg. Chem. 1999, 173−178. (c) Reiß,P.; Fenske, D. Z. Anorg. Allg. Chem. 2000, 626, 1317−1331. (d) vanden Ancker, T. R.; Bhargava, S. K.; Mohr, F.; Papadopoulos, S.;Raston, C. L.; Skelton, B. W.; White, A. H. J. Chem. Soc., Dalton Trans.2001, 3069−3072. (e) Eaborn, C.; Hill, M. S.; Hitchcock, P. B.; Smith,J. D. J. Chem. Soc., Dalton Trans. 2002, 2467−2472.(22) Wehman, E.; van Koten, G.; Jastrzebski, J. T. B. H.; Rotteveel,M. A.; Stam, C. H. Organometallics 1988, 7, 1477−1485.(23) Nonaggregated dicoordinate copper centers are also encoun-tered in organocuprates; see, for example: (a) Nardin, G.; Randaccio,L.; Zangrando, E. J. Organomet. Chem. 1974, 74, C23−C25.(b) Eaborn, C.; Hitchcock, P. B.; Smith, J. D.; Sullivan, A. C. J.Organomet. Chem. 1984, 263, C23−C25. (c) Hope, H.; Olmstead, M.M.; Power, P. P.; Sandell, J.; Xu, X. J. Am. Chem. Soc. 1985, 107,4337−4338. (d) Kronenburg, C. M. P.; Jastrzebski, J. T. B. H.; vanKoten, G. Polyhedron 2000, 19, 553−555. (e) Boche, G.; Bosold, F.;Marsch, M.; Harms, K. Angew. Chem., Int. Ed. 1998, 37, 1684−1686.(f) John, M.; Auel, C.; Behrens, C.; Marsch, M.; Harms, K.; Bosold, F.;Gschwind, R. M.; Rajamfhanan, M. J.; Boche, G. Chem.Eur. J. 2000,6, 3060−3068.(24) Jakle, F. Dalton Trans. 2007, 2851−2858.(25) (a) Cairncross, A.; Sheppard, W. A. J. Am. Chem. Soc. 1968, 90,2186−2187. (b) Cairncross, A.; Omura, H.; Sheppard, W. A. J. Am.Chem. Soc. 1971, 93, 248−249.(26) Sundararaman, A.; Lalancette, R. A.; Zakharov, L. N.; Rheingold,A. L.; Jakle, F. Organometallics 2003, 22, 3526−3532.(27) Doshi, A.; Venkatasubbaiah, K.; Rheingold, A. L.; Jakle, F. Chem.Commun. 2008, 4264−4266.(28) The structure of monomeric pentafluorophenyl coppersupported by a dialkyne ligand has been reported: Lang, H.;Kohler, K.; Rheinwald, G.; Zsolnai, L.; Buchner, M.; Driess, A.;Huttner, G.; Strahle, J. Organometallics 1999, 18, 598.(29) Sundararaman, A.; Zakharov, L. N.; Rheingold, A. L.; Jakle, F.Chem. Commun. 2005, 1708−1710.(30) Frisch, M. J.; et al. Gaussian 03, revision C.02; Gaussian, Inc.:Wallingford, CT, 2004.
(31) Cairncross, A.; Sheppard, W. A.; Wonchoba, E.; Guildford, W.J.; House, C. B.; Coates, R. M. Org. Synth. 1980, 59, 122−131.(32) A similar effect has been reported for bipyridine complexes ofphenylcopper, which are unstable at ambient temperature; see ref 12.(33) See also: Bertz, S. H.; Dabbagh, G.; He, X.; Power, P. P. J. Am.Chem. Soc. 1993, 115, 11640−11641.(34) Values for Δδm,p similar to those for 4-R have been reported forthe related mononuclear gold complexes C6F5Au(3-Pic) (3-Pic = 3-methylpyridine) with Δδm,p = 3.6 and C6F5Au(3-FcPy) (3-FcPy = 3-ferrocenylpyridine) with Δδm,p = 3.5. See: (a) Jones, P. G.; Ahrens, B.Z. Naturforsch., B 1998, 53, 653−662. (b) Barranco, E. M.; Crespo, O.;Gimeno, M. C.; Jones, P. G.; Laguna, A.; Villacampa, M. D. J.Organomet. Chem. 1999, 592, 258−264.(35) The observed spectral trends could also be due to ligandredistribution in solution, a phenomenon that has been observed, forexample, for mesityl copper in the presence of 1,2-bis(diphenylphos-phino)ethane (dppe) and for methyl copper in neat trimethylphos-phine. In those cases, the cuprate salts [CuMes2]
−[Cu(dppe)2]+ (dppe
= 1,2-bis(diphenylphosphino)ethane; Mes = 2,4,6-trimethylphenyl)and [CuMe2]
−[Cu(PMe3)4]+ were structurally characterized:
(a) Leoni, P.; Pasquali, M.; Ghilardi, C. A. J. Chem. Soc., Chem.Commun. 1983, 240−241. (b) Dempsey, D. F.; Girolami, G. S.Organometallics 1988, 7, 1208−1213.(36) Gambarotta, S.; Floriani, C.; Chiesi-Villa, A.; Guastini, C. J.Chem. Soc., Chem. Commun. 1983, 1156−1158.(37) (a) Knotter, D. M.; Smeets, W. J. J.; Spek, A. L.; van Koten, G. J.Am. Chem. Soc. 1990, 112, 5895−5896. (b) Lenders, B.; Grove, D. M.;Smeets, W. J. J.; van der Sluis, P.; Spek, A. L.; van Koten, G.Organometallics 1991, 10, 786−791.(38) Goj, L. A.; Blue, E. D.; Delp, S. A.; Gunnoe, T. B.; Cundari, T.R.; Petersen, J. L. Organometallics 2006, 25, 4097−4104.(39) (a) Engelhardt, L. M.; Pakawatchai, C.; White, A. H.; Healy, P.C. J. Chem. Soc., Dalton Trans. 1985, 117−123. (b) Munakata, M.;Kitagawa, S.; Shimono, H.; Masuda, H. Inorg. Chim. Acta 1989, 158,217−220. (c) Habiyakare, A.; Lucken, E. A. C.; Bernardinelli, G. J.Chem. Soc., Dalton Trans. 1991, 2269−2273. (d) Jin, K.; Huang, X.;Pang, L.; Li, J.; Appel, A.; Wherland, S. Chem. Commun. 2002, 2872−2873.(40) Siemeling, U.; Vorfeld, U.; Neumann, B.; Stammler, H.-G.Chem. Commun. 1997, 1723−1724.(41) Vicente, J.; Arcas, A.; Jones, P. G.; Lautner, J. J. Chem. Soc.,Dalton Trans. 1990, 451−456.(42) (a) Patrick, C. R.; Prosser, G. S. Nature 1960, 187, 1021.(b) Williams, J. H. Acc. Chem. Res. 1993, 26, 593−598. (c) Janiak, C. J.Chem. Soc., Dalton Trans. 2000, 3885−3896. (d) Hunter, C. A.;Lawson, K. R.; Perkins, J.; Urch, C. J. J. Chem. Soc., Perkin Trans. 2001,2, 651−669. (e) Russell, V.; Scudder, M.; Dance, I. J. Chem. Soc.,Dalton Trans. 2001, 789−799. For recent examples, see: (f) Collings,J. C.; Smith, P. S.; Yufit, D. S.; Batsanov, A. S.; Howard, J. A. K.;Marder, T. B. CrystEngComm 2004, 6, 25−28. (g) Watt, S. W.; Dai, C.;Scott, A. J.; Burke, J. M.; Thomas, R. L.; Collings, J. C.; Viney, C.;Clegg, W.; Marder, T. B. Angew. Chem., Int. Ed. 2004, 43, 3061−3063.(h) Collings, J. C.; Roscoe, K. P.; Robins, E. G.; Batsanov, A. S.;Stimson, L. M.; Howard, J. A. K.; Clark, S. J.; Marder, T. B. New J.Chem. 2002, 26, 1740−1746. (i) Martin, E.; Spendley, C.; Mountford,A. J.; Coles, S. J.; Horton, P. N.; Hughes, D. L.; Hursthouse, M. B.;Lancaster, S. J. Organometallics 2008, 27, 1436−1446. (j) Mountford,A. J.; Lancaster, S. J.; Coles, S. J.; Horton, P. N.; Hughes, D. L.;Hursthouse, M. B.; Light, M. E. Organometallics 2006, 25, 3837−3847.(k) Martin, E.; Clegg, W.; Harrington, R. W.; Hughes, D. L.;Hursthouse, M. B.; Male, L.; Lancaster, S. J. Polyhedron 2010, 29,405−413.(43) Smith, C. E.; Smith, P. S.; Thomas, R. L.; Robins, E. G.;Collings, J. C.; Dai, C.; Scott, A. J.; Borwick, S.; Batsanov, A. S.; Watt,S. W.; Clark, S. J.; Viney, C.; Howard, J. A. K.; Clegg, W.; Marder, T.B. J. Mater. Chem. 2004, 14, 413−420.(44) (a) Pyykko, P. Angew. Chem., Int. Ed. 2002, 41, 3573−3578.(b) Codina, A.; Fernandez, E. J.; Jones, P. G.; Laguna, A.; Lopez-de-Luzuriaga, J. M.; Monge, M.; Olmos, M. E.; Perez, J.; Rodriguez, M. A.
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581557

J. Am. Chem. Soc. 2002, 124, 6781−6786. (c) Yang, G.; Raptis, R. G.Inorg. Chem. 2003, 42, 261−263. (d) Gussenhoven, E. M.; Fettinger, J.C.; Pham, D. M.; Malwitz, M. M.; Balch, A. L. J. Am. Chem. Soc. 2005,127, 10838−10839. (e) Omary, M. A.; Elbjeirami, O. J. Am. Chem. Soc.2007, 129, 11384−11393. (f) Schmidbaur, H.; Schier, A. Chem. Soc.Rev. 2008, 37, 1931−1951. (g) Pyykko, P.; Muniz, J.; Wang, C.Chem.Eur. J. 2011, 17, 368−377.(45) (a) Benard, M.; Poblet, J. M. Chem. Commun. 1998, 1179−1180. (b) Che, C.-M.; Mao, Z.; Miskowski, V. M.; Tse, M.-C.; Chan,C.-K.; Cheung, K.-K.; Phillips, D. L.; Leung, K.-H. Angew. Chem., Int.Ed. 2000, 39, 4084−4088. (c) Zheng, S.-L.; Messerschmidt, M.;Coppens, P. Angew. Chem., Int. Ed. 2005, 44, 4614−4617.(d) Petrukhina, M. A.; Hietsoi, O.; Filatov, A. S.; Dubceac, C.Dalton Trans. 2011, 40, 8598−8603. (e) Petrukhina, M. A.; Hietsoi,O.; Dubceac, C.; Filatov, A. S. Chem. Commun. 2011, 47, 6939−6941.(46) Related discussions: Zhang, J.-P.; Wang, Y.-B.; Huang, X.-C.;Lin, Y.-Y.; Chen, X.-M. Chem.Eur. J. 2005, 11, 552−561.(47) Hermann, H. L.; Boche, G.; Schwerdtfeger, P. Chem.Eur. J.2001, 7, 5333−5342.(48) Shorter Cu···π distances have been reported for offset stacks of1 with arenes. See ref 27 and: Lopez, S.; Keller, S. W. Inorg. Chem.1999, 38, 1883−1888.(49) Carvajal, M. A.; Alvarez, S.; Novoa, J. J. Chem.Eur. J. 2004, 10,2117−2132.(50) Linear polymeric copper chains for {[Cu(NH3)2]
+}n(X−)n and
{[Cu2terpy2]2+}n(X
−)2n (terpy = terpyridine) have been reported. Incontrast to these ionic Cu(I) complexes, in which the counterions X−
likely play a significant role in the bonding, the individual units in 4-Hare neutral RCuL fragments. See: (a) Margraf, G.; Bats, J. W.; Bolte,M.; Lerner, H.-W.; Wagner, M. Chem. Commun. 2003, 956−957.(b) Al-Anber, M.; Walfort, B.; Vatsadze, S.; Lang, H. Inorg. Chem.Commun. 2004, 7, 799−802.(51) For a “nonlinear” chain combining intra- and intermolecularcontacts in an organocopper trimer, see: Garcia, F.; Hopkins, A. D.;Kowenicki, R. A.; McPartlin, M.; Rogers, M. C.; Wright, D. S.Organometallics 2004, 23, 3884−3890.(52) (a) Favorable π-interactions are generally to be expected whenelectron-deficient π-systems interact as in our compounds, consistingof C6F5 and substituted pyridyl groups. See refs 42c and 42d. (b) Thesubstituents on the pyridine ring and the CuL fragment attached to thefluorinated benzene ring affect the packing mode, favoring theobserved slipped face-to-face interaction. We expect this to be both asteric and an electronic effect, because the substituents will influencealso the electronic structure, including the quadrupole exerted by eachof the π-systems. This means that, unlike in the C6F6/C6H6 system,offset π-stacks are expected and experimentally observed. (c) Collings,J. C.; Batsanov, A. S.; Howard, J. A. K.; Dickie, D. A.; Clyburne, J. A.C.; Jenkins, H. A.; Marder, T. B. J. Fluorine Chem. 2005, 126, 515−519.(d) Bacchi, S.; Benaglia, M.; Cozzi, F.; Demartin, F.; Filippini, G.;Gavezzotti, A. Chem.Eur. J. 2006, 12, 3538−3546. (e) Hori, A.;Shinohe, A.; Yamasaki, M.; Nishibori, E.; Aoyagi, S.; Sakata, M. Angew.Chem., Int. Ed. 2007, 46, 7617−7620.(53) (a) Batsanov, S. S. J. Mol. Struct. 2011, 990, 63−66.(b) Cordero, B.; Gomez, V.; Platero-Prats, A. E.; Reves, M.;Echeverrıa, J.; Cremades, E.; Barragan, F.; Alvarez, S. Dalton Trans.2008, 2832−2838.(54) Yu, F.; Wu, S.-X.; Geng, Y.; Yang, G.-C.; Su, Z.-M. Theor. Chem.Acc. 2010, 127, 735−742.(55) Dias, H. V. R.; Diyabalanage, H. V. K.; Rawashdeh-Omary, M.A.; Franzman, M. A.; Omary, M. A. J. Am. Chem. Soc. 2003, 125,12072−12073.(56) (a) Yam, V. W.-W.; Lee, W.-K.; Cheung, K. K.; Lee, H.-K.;Leung, W.-P. J. Chem. Soc., Dalton Trans. 1996, 2889−2891. (b) Ford,P. C.; Cariati, E.; Bourassa, J. Chem. Rev. 1999, 99, 3625−3647.(c) Dias, H. V. R.; Diyabalanage, H. V. K.; Eldabaja, M. G.; Elbjeirami,O.; Rawashdeh-Omary, M. A.; Omary, M. A. J. Am. Chem. Soc. 2005,127, 7489−7501. (d) Grimes, T.; Omary, M. A.; Dias, H. V. R.;Cundari, T. R. J. Phys. Chem. A 2006, 110, 5823−5830. (e) Barbieri,A.; Accorsi, G.; Armaroli, N. Chem. Commun. 2008, 2185−2193.
(57) Sternlicht, H.; Nieman, G. C.; Robinson, G. W. J. Chem. Phys.1963, 38, 1326−1335.
Organometallics Article
dx.doi.org/10.1021/om200989b | Organometallics 2012, 31, 1546−15581558






![Charge Transfer Fluorescence in Imine Borane Adducts ... · 1.2.1 Synthesis of the borane reagents. Tris(pentafluorophenyl) borane. Tris(pentafluorophenyl)borane [1]was synthesised](https://img.pdfslide.net/doc/110x75/5ec38c345e3bae053a7989e0/charge-transfer-fluorescence-in-imine-borane-adducts-121-synthesis-of-the.jpg)