Embed Size (px)
Citation preview

PHASE II STUDY OF NILOTINIB EFFICACY IN PIGMENTED VILLO-NODULAR SYNOVITIS/ TENOSYNOVIAL GIANT
CELL TUMOUR (PVNS/TGCT)
CLINICAL STUDY PROTOCOL
Methodology: International, non randomized, open-label, multicentre, phase II study
Sponsor: Centre Léon Bérard (CLB)
Investigator coordinator: Pr Jean-Yves BLAY - CLB
Coordinating centre: Unité de Biostatistique et d’Evaluation des Thérapeutiques (UBET) – CLB
Version: V4
Amendment: Not applicable
Date: 2011/08/25
Sponsor’s trial number: ET2009-095
EudraCT number: 2010-018869-29
N° ID-RCB : A100604-41
AFSSAPS authorization on 29th, July 2010
CPP Sud-Est IV agreement on 8th, June 2010
Confidential
This document contains confidential information which must not be disclosed to anyone other than the recipient investigator(s) and members of the ethics committee. This information should not be used for any purpose other than the evaluation or conduct of the clinical investigation without prior written consent of the sponsor. No part of this document may be reproduced or stored in a retrieval system or transmitted in any other form or by any other means.

PVNS_Protocole_V4_110825 Page 2 of 129
APPROVAL OF THE PROTOCOL
Title PHASE II STUDY OF NILOTINIB IN PIGMENTED VILLO-NODULAR SYNOVITIS/ TENOSYNOVIAL GIANT CELL TUMOUR (PVNS/TGCT)
Version 4
Date 2011/08/25
Investigator coordinator Pr Jean-Yves BLAY - CLB 2011/08/25 Date, Signature
Investigator Date, Signature
Project manager
Dr David PEROL - CLB
2011/08/25 Date, Signature

PVNS_Protocole_V4_110825 Page 3 of 129
STUDY ORGANISATION
CLINICAL AND METHODOLOGICAL REFERENTS
Clinical aspects
Corresponding referent on the clinical aspects of the study is represented by the Coordinating investigator.
� Coordinator investigator
Pr Jean-Yves BLAY – CLB - ℡: + 33 478782757 - �: [email protected]
Methodological aspects
Corresponding referents on methodological aspects are represented by:
� Coordinating centre responsible
David PEROL - UBET, CLB - ℡: + 33 478782752 - �: [email protected]
� Project manager
Séverine GUILLEMAUT – UBET, CLB - ℡: + 33 478782968 - �: [email protected]
� Statistician
Claire CROPET – UBET, CLB - ℡: + 33 478787669 - �: [email protected]
DATA MANAGEMENT
The management and coordination will be performed by:
� Team manager
Nathalie GIRERD - CHAMBAZ – UBET, CLB - ℡: + 33 478785927 - � : [email protected]
� Clinical research technician
Valérie BOURNE-BRANCHU, UBET, CLB - ℡: + 33 478782658 - �: [email protected]
� Clinical research assistant
Anne Claire CADORE, UBET, CLB - ℡: + 33 478782911 - �: [email protected]
ADMINISTRATIVE AND REGULATORY ASPECTS
The administrative and regulatory management will be provided by the Clinical Research Direction:
Zora ABDELBOST - CLB - ℡: + 33 478782775 - �: [email protected]
SAFETY VIGILANCE
Pharmacovigilance data management will be provided by the Lyon Pharmacovigilance Centre, delegated by the sponsor:
Nathalie BERNARD - CRPV - ℡: + 33 472119407 - �: [email protected]
PVNS_Protocole_V4_110825 Page 4 of 129
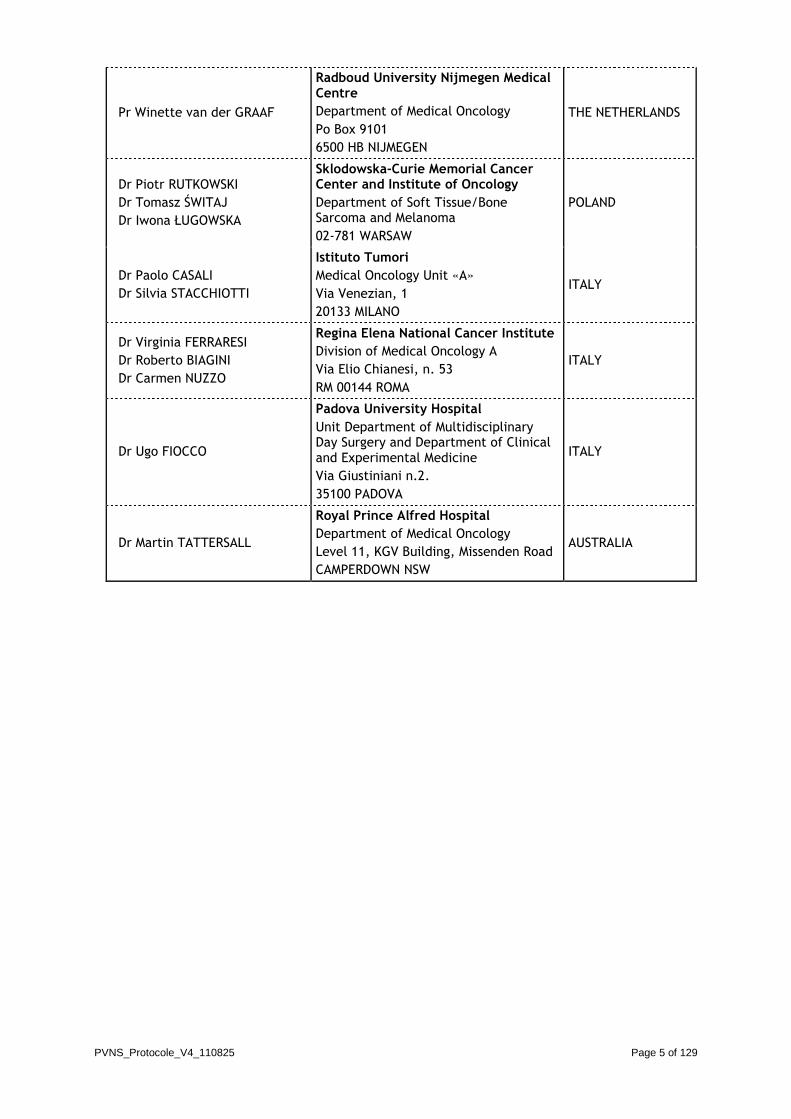
STUDY INVESTIGATORS (potential)
Name Address Country
Pr Jean-Yves BLAY Dr Isabelle RAY-COQUARD Dr Pierre MEEUS Dr Pierre BIRON Dr Philippe CASSIER
Centre Léon Bérard
28 rue Laënnec 69373 LYON Cedex 08
FRANCE
Dr Nicolas PENEL Pr Antoine ADENIS Dr Fabienne WATELLE Dr Marie VANHUYSE Dr Marie-Hélène VIEILLARD
Centre Oscar Lambret Cancérologie Générale 3 rue Frédéric Combemale 59000 LILLE
FRANCE
Dr Antoine ITALIANO Dr Binh BUI NGUYEN
Institut Bergonié
Département d'Oncologie Médicale 229 cours de l'Argonne 33076 BORDEAUX Cedex
FRANCE
Dr François BERTUCCI
Institut Paoli Calmettes Oncologie Médicale 232 bd de Sainte Marguerite 13009 MARSEILLE
FRANCE
Dr Sophie PIPERNO-NEUMANN
Institut Curie Oncologie Médicale 26 rue d'Ulm 75248 PARIS Cedex 05
FRANCE
Dr Christine CHEVREAU Dr Loïc MOUREY Dr Yann BERGE Dr Ewa Anna COTTURA
Institut Claudius Régaud
Oncologie Médicale 20-24 rue du Pont Saint-Pierre 31052 TOULOUSE Cedex
FRANCE
Dr Angela CIOFFI Dr Axel LE CESNE
Institut Gustave Roussy Service d'Oncologie Médicale 39 rue Camille Desmoulins 94805 VILLEJUIF Cedex
FRANCE
Pr Florence DUFFAUD Dr Sébastien SALAS Dr Thanh Khoa HUYNH
Hôpital La Timone Service d'Oncologie Médicale - Pr Favre Bd Jean Moulin 13385 MARSEILLE Cedex 5
FRANCE
Dr Beatrice SEDDON Dr Jeremy WHELAN Dr Sandra J. STRAUSS Dr Palma DILEO
University College Hospital
UCL Hospitals NHS Foundation Trust Department of Oncology 250 Euston Road LONDON NW1 2PG
UNITED KINGDOM
Dr Bass HASSAN
Oxford Cancer Centre
Clinical- Oxford Sarcoma Service University Department of Medical Oncology Churchill Hospital OXFORD OX3 7LJ
UNITED KINGDOM
Dr Hans GELDERBLOM Dr Judith KROEP
Leiden University Medical Center
Department of Clinical Oncology PO Box 9600 - 2300 RC LEIDEN
THE NETHERLANDS
PVNS_Protocole_V4_110825 Page 5 of 129
Pr Winette van der GRAAF
Radboud University Nijmegen Medical Centre
Department of Medical Oncology Po Box 9101 6500 HB NIJMEGEN
THE NETHERLANDS
Dr Piotr RUTKOWSKI Dr Tomasz ŚWITAJ Dr Iwona ŁUGOWSKA
Sklodowska-Curie Memorial Cancer Center and Institute of Oncology
Department of Soft Tissue/Bone Sarcoma and Melanoma 02-781 WARSAW
POLAND
Dr Paolo CASALI Dr Silvia STACCHIOTTI
Istituto Tumori
Medical Oncology Unit «A» Via Venezian, 1 20133 MILANO
ITALY
Dr Virginia FERRARESI Dr Roberto BIAGINI Dr Carmen NUZZO
Regina Elena National Cancer Institute
Division of Medical Oncology A Via Elio Chianesi, n. 53 RM 00144 ROMA
ITALY
Dr Ugo FIOCCO
Padova University Hospital
Unit Department of Multidisciplinary Day Surgery and Department of Clinical and Experimental Medicine Via Giustiniani n.2. 35100 PADOVA
ITALY
Dr Martin TATTERSALL
Royal Prince Alfred Hospital
Department of Medical Oncology Level 11, KGV Building, Missenden Road CAMPERDOWN NSW
AUSTRALIA

PVNS_Protocole_V4_110825 Page 6 of 129
TABLE OF CONTENTS
APPROVAL OF THE PROTOCOL.....................................................................................2
STUDY ORGANISATION ...............................................................................................3
STUDY INVESTIGATORS (POTENTIAL) .............................................................................4
TABLE OF CONTENTS.................................................................................................6
SYNOPSIS................................................................................................................9
LIST OF ABBREVIATIONS........................................................................................... 16
1. BACKGROUND AND STUDY RATIONALE .................................................................. 17
2. STUDY OBJECTIVES .......................................................................................... 19 2.1. PRIMARY OBJECTIVE................................................................................ 19 2.2. SECONDARY OBJECTIVES .......................................................................... 19 2.3. EXPLORATORY OBJECTIVE......................................................................... 19
3. STUDY DESIGN................................................................................................. 20 3.1. EVALUATION CRITERIA ............................................................................. 20 3.2. METHODOLOGY...................................................................................... 20 3.3. BIAS LIMITATION .................................................................................... 21 3.4. ALLOCATION TO TREATMENT GROUPS .......................................................... 21 3.5. STUDY DURATION AND STUDY PERIODS ......................................................... 21 3.6. SOURCE DATA OF THE eCRF ...................................................................... 21
4. STUDY POPULATION ......................................................................................... 22 4.1. INCLUSION CRITERIA ............................................................................... 22 4.2. NON-INCLUSION CRITERIA ......................................................................... 22
5. INVESTIGATIONAL PRODUCTS ............................................................................. 24 5.1. DESCRIPTION, PACKAGING AND LABELLING OF THE TREATMENT UNITS .................. 24
5.1.1. Investigational drug ............................................................... 24 5.1.2. Permitted study drug adjustments ............................................. 24
5.1.2.1. Dosing modifications .............................................. 24 5.1.3. Packaging of the treatment units ............................................... 29
5.1.3.1. Treatment units.................................................... 29 5.1.3.2. Labelling............................................................. 30
5.1.4. Study drug supply and tracking/drug accountability ........................ 30 5.2. TREATMENT COMPLIANCE ......................................................................... 30 5.3. STUDY TREATMENTS STORAGE ................................................................... 30 5.4. SUPPLY TO THE INVESTIGATIONAL CENTRES................................................... 31 5.5. MANAGEMENT IN EACH PARTICIPATING CENTRES ............................................. 31 5.6. PRIOR AND CONCOMITANTS THERAPIES......................................................... 31
5.6.1. Excluded concomitant treatments (Appendix 4) ............................. 31
6. ASSESSMENTS ................................................................................................. 32 6.1. FLOW-CHART OF ASSESSMENTS................................................................... 32 6.2. STUDY PLAN ......................................................................................... 34
6.2.1. Inclusion (visit 1) ................................................................... 34 6.2.1.1. Internet inclusion .................................................. 34
6.2.2. Follow up ............................................................................ 34 6.2.2.1. Biological assessment ............................................. 34 6.2.2.2. Cardiac monitoring ................................................ 34 6.2.2.3. Follow up (visit 2, 3, 4, 5 and 6) ................................ 35 6.2.2.4. End of clinical study (visit 7) .................................... 35
6.3. INVESTIGATIONAL PRODUCTS DELIVERY AND RETURN ....................................... 35

PVNS_Protocole_V4_110825 Page 7 of 129
7. PREMATURE WITHDRAWAL AND EARLY STUDY TERMINATION...................................... 37 7.1. TERMINATION RULES (TEMPORARY OR DEFINITE) ............................................. 37
7.1.1. For patients ......................................................................... 37 7.1.2. Of the study ......................................................................... 37
7.2. STUDY DRUG DISCONTINUATION / PREMATURE WITHDRAWAL AND PATIENT REPLACEMENT....................................................................................... 37 7.2.1. Study drug discontinuation and premature withdrawal .................... 37 7.2.2. Patient replacement............................................................... 38
8. EFFICACY EVALUATION ..................................................................................... 39 8.1. PRIMARY ENDPOINT................................................................................. 39 8.2. SECONDARY ENDPOINTS ........................................................................... 39
9. SAFETY EVALUATION ........................................................................................ 40 9.1. ADVERSE EVENTS (AE).............................................................................. 40
9.1.1. Definitions........................................................................... 40 9.1.2. Adverse events reporting ......................................................... 40 9.1.3. Serious adverse events (SAE) reporting and notification ................... 41
9.2. OTHER SAFETY CRITERIA .......................................................................... 42 9.2.1. Blood analysis ....................................................................... 42 9.2.2. Vital signs............................................................................ 43
10. OTHER EVALUATIONS........................................................................................ 44
11. STATISTICAL METHODS AND SAMPLE SIZE CALCULATION ........................................... 45 11.1. SAMPLE SIZE AND DESIGN.......................................................................... 45 11.2. STATISTICAL METHODS............................................................................. 47
11.2.1. Study populations for analysis ................................................... 47 11.2.2. Efficacy analysis .................................................................... 48
11.2.2.1. Primary endpoint analysis ........................................ 48 11.2.2.2. Secondary endpoints analysis.................................... 48
11.2.3. Safety analysis ...................................................................... 49 11.2.4. Modifications of the statistical analysis planned in the protocol......... 49 11.2.5. Interim analysis..................................................................... 49 11.2.6. Blind code break.................................................................... 49
12. ACCESS TO SOURCE DATA AND DOCUMENTS ........................................................... 50
13. STUDY MANAGEMENT........................................................................................ 51 13.1. COORDINATING CENTRE ........................................................................... 51
14. DATA QUALITY CONTROL................................................................................... 52 14.1. MONITORING......................................................................................... 52 14.2. DATA ENTRY AND DATA MANAGEMENT.......................................................... 53 14.3. CENTRAL REVIEW COMMITTEE .................................................................... 53 14.4. AUDITS AND INSPECTIONS ......................................................................... 54
15. ETHICS .......................................................................................................... 55 15.1. INDEPENDENT ETHICS COMMITTEE (IEC) AND INSTITUTIONAL REVIEW BOARD (IRB) .... 55 15.2. ETHICAL CONDUCT OF THE STUDY – GOOD CLINICAL PRACTICE............................ 55 15.3. PATIENT INFORMATION AND CONSENT .......................................................... 55 15.4. AGENCE FRANCAISE DE SECURITE DES PRODUITS DE SANTE (AFSSAPS).................... 56 15.5. COMITE DE PROTECTION DES PERSONNES ...................................................... 56 15.6. COMITE CONSULTATIF SUR LE TRAITEMENT DE L’INFORMATION EN MATIERE DE
RECHERCHE DANS LE DOMAINE DE LA SANTE (CCTIRS) – COMMISSION NATIONALE DE L’INFORMATIQUE ET DES LIBERTES (CNIL) ..................................................... 56
15.7. SPONSOR RESPONSIBILITY ......................................................................... 56 15.8. INVESTIGATOR REPONSIBILITY.................................................................... 57
16. PROCESSING OF DATA AND STUDY DOCUMENTS ARCHIVING........................................ 58

PVNS_Protocole_V4_110825 Page 8 of 129
17. OTHER CONSIDERATIONS ................................................................................... 59 17.1. CASE REPORT FORM ................................................................................ 59 17.2. AMENDMENTS TO THE PROTOCOL................................................................ 59 17.3. FUNDING AND INSURANCE......................................................................... 59
18. PUBLICATION AND FINAL STUDY REPORT............................................................... 60
19. REFERENCES................................................................................................... 61
20. APPENDICES ................................................................................................... 62

PVNS_Protocole_V4_110825 Page 9 of 129
SYNOPSIS
TITLE Phase II study of nilotinib efficacy in Pigmented Villo-Nodular Synovitis/ Tenosynovial Giant Cell Tumour (PVNS/TGCT)
SHORT TITLE PVNS
THERAPEUTIC INDICATION
Pigmented Villo-Nodular Synovitis / Tenosynovial Giant Cell Tumour (PVNS/TGCT)
SPONSOR Centre Léon Bérard (CLB)
PROTOCOL NUMBER
AFSSAPS Reference: A100604-41 Sponsor n°: ET2009-095 EudraCT N°: 2010-018869-29
VERSION AND DATE
V4 2011/08/25
INVESTIGATOR-COORDINATOR
Pr Jean-Yves BLAY
STUDY DESIGN International, multicentre, non-randomized, open-label phase II clinical trial with a Bayesian design
RATIONALE Pigmented villonodular synovitis (PVNS), also known as tenosynovial giant cell tumour (TGCT), is a rare pathological entity affecting the synovium in young adults. Initially considered as an inflammatory reactive process, recent observations have shown that this disease may actually be a benign neoplastic process with specific genetic alterations. Indeed, a specific translocation, involving the collagen 6A3 gene (on 2q35) and the M-CSF (also known as CSF1) gene (on 1p13), is present in a fraction of tumour cells in PVNS/TGCT. This fusion gene expressed by a fraction of the cells encodes for a fusion protein which attracts non-neoplastic cells expressing M-CSF receptor (macrophages and monocytes), through a paracrine - “landscape”- effect. PVNS/TGCT is usually treated by surgery alone. However, relapses may occur, and re-excision may be needed, sometimes with possible important functional impairment. Nilotinib (Tasigna®) is a phenylaminopyrimidine commercialized by Novartis. Nilotinib has inhibitory properties similar to imatinib on M-CSF receptor pathway. Nilotinib is indicated for the treatment of adults with chronic phase and accelerated phase Philadelphia chromosome positive chronic myelogenous leukaemia (CML) with resistance or intolerance to prior therapy including imatinib. Although a potential contribution of the blockade of other tyrosine kinases by imatinib/nilotinib can not be ruled out, the frequency at which the col6A3/CSF1 fusion gene is observed in PVNS/TGCT as compared to other pathological synovial process strongly suggests that imatinib/nilotinib activity involves M-CSF receptor blockade in this disease, despite recent observation showing limited biological activity of the product of the fusion gene. As a consequence, imatinib and nilotinib are good candidates to induce complete responses in relapsing PVNS/TGCT and may offer an option in patients in whom surgery is not feasible or implies too much risks. The reason for selecting nilotinib as compared to imatinib to treat PVNS in the present study came out from the following considerations: 1) In the limited experience with imatinib in PVNS reported so far the toxicity experienced by some patients was substantial. 2) Nilotinib has a more favourable toxicity profile in particular regarding soft tissue and facial oedema. This may favour a better compliance to the treatment.

PVNS_Protocole_V4_110825 Page 10 of 129
PURPOSE The purpose of this study is to explore the efficacy of nilotinib as a treatment of patients with progressive or relapsing pigmented villo-nodular synovitis / tenosynovial giant cell tumour (PVNS/TGCT) who cannot be treated by surgery.
PRIMARY OBJECTIVE
The primary objective of the study will be to determine the efficacy of 12 weeks (3 months) of nilotinib treatment as measured by the non progression rate (Complete response + Partial Response + Stable disease according to Response Evaluation Criteria In Solid Tumours – RECIST version 1.1) in patients with progressive or relapsing PVNS/TGCT who cannot be treated by surgery.
SECONDARY OBJECTIVES
A key secondary objective of the study will be to determine the efficacy of 24 weeks (6 months) of nilotinib treatment as measured by the non progression rate (Complete response + Partial Response + Stable disease according to Response Evaluation Criteria In Solid Tumours – RECIST version 1.1) in patients with progressive or relapsing PVNS/TGCT who cannot be treated by surgery.
This key secondary objective was defined for the purpose of a further analysis (not described in this protocol) which will pool the data of the PVNS study with those of a similar concomitant study conducted in the US and Australia.
The other secondary objectives will be: � To evaluate the efficacy of nilotinib according to: - The objective tumour response rate (Complete response + Partial
Response according to RECIST version 1.1) after 12 weeks of treatment
- The duration of treatment response - The best overall response obtained during the study - The progression-free survival (PFS) - The time to progression (TTP) - The time to treatment failure (TTF) - The proportion of patients with an operable tumour after nilotinib
exposure according to investigator evaluation - The description of concomitant treatments use - The correlation between trough levels of nilotinib and objective
tumour response � To assess the safety of nilotinib for PVNS/TGCT patients
EXPLORATORY OBJECTIVE
An exploratory objective of the study will be to study the relationship between the objective tumour response and the following tumour characteristics (tissues collected in a prior surgery, or by biopsy, upon specific acceptance by the patient; if no tissue is available in the prior surgery, a biopsy will be done at visit 2): � Presence of COL6A3/CSF1 fusion gene � Presence of M-CSF, CSF1R, KIT, PDGFRA and B on immunohistochemistry � Presence of phosphorylated c-fms on tumour samples � Activation of the PI3K/Akt/mTor pathway, presence of activating
mutations of ras, and other potential molecular alterations
ENDPOINTS Primary endpoint
The primary endpoint of the study will be the non progression rate after 12 weeks (3 months) of treatment, based on the response evaluated by CT scan or MRI according to RECIST criteria (RECIST version 1.1) and validated by a central review committee.

PVNS_Protocole_V4_110825 Page 11 of 129
Secondary endpoints
Key secondary efficacy endpoint: � Non progression rate after 24 weeks (6 months) of treatment, based on
the response evaluated by CT scan or MRI according to RECIST criteria (RECIST version 1.1).
Other secondary efficacy endpoints: � Objective tumour response according to RECIST version 1.1 (CR and PR)
after 12 weeks of treatment � Duration of response � Best overall response � Progression-free survival � Time to progression � Time to treatment failure � Non progression rate after 12 weeks of treatment, based on the response
evaluated locally by the investigator in charge using CT scan or MRI and according to RECIST criteria (RECIST version 1.1)
� Proportion of patients with an operable tumour after nilotinib exposure according to investigator evaluation
� Concomitant treatment use during the study � Correlation between trough level of nilotinib at 6 weeks and 12 weeks
and objective tumour response Safety
Safety evaluation will be based on overall safety profile characterized by type, frequency and severity (as graded using NCI-CTCAE V4.0) of adverse events.
SAMPLE SIZE A maximum sample size of 50 patients will be included in the study
STUDY TREATMENT
Nilotinib (Tasigna®)
400 mg twice a day Oral administration Treatment duration: 1 year
POPULATION Inclusion criteria
� Age ≥ 18 years � Histologically confirmed diagnosis of inoperable progressive or relapsing
PVNS/TGCT OR resectable tumour requesting mutilating surgery � Demonstrated progressive disease in the last 12 months � At least one measurable site of disease on MRI/CT scan according to
RECIST criteria (RECIST version 1.1) based on investigator’s assessment � WHO Performance status of 0, 1 or 2 � Adequate organ, electrolyte and marrow function, defined as the
following: serum bilirubin ≤1.5 x ULN, ALT and AST ≤2.5 x ULN, serum creatinine ≤1.5 x ULN or creatinine clearance ≥50 mL/min, absolute neutrophil count (ANC) ≥1.5x109/L, platelets ≥100x109/L, serum lipase ≤1.5 x ULN, magnesium ≥ lower limit of normal (LLN) and potassium ≥ LLN
� Prior adequate physical examination including weight, height, ECOG PS and vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position)
� Signed written informed consent form � Covered by a medical insurance (in countries where applicable)

PVNS_Protocole_V4_110825 Page 12 of 129
Non inclusion criteria
� Pregnant or lactating female or female of child-bearing potential not employing adequate contraception during the study and for up to three months following termination of the study
� Known hypersensitivity to nilotinib or to any of the excipients, galactose intolerance, lactase deficiency or glucose-galactose malabsorbtion prior to enrolment
� Acute or chronic uncontrolled liver disease, or severe renal disease � Impaired cardiac function, including:
- LVEF<50% or below the institutional lower limit of the normal range (whichever is higher) as determined by echocardiogram or MUGA scan
- History or signs of prior myocardial infarction - History of unstable angina - Congenital long QT prolongation - Personal history of unexplained syncope - QTc interval ≥ 450 msec on screening ECG - Other clinically significant heart disease (e.g. bradycardia, congestive
heart failure or uncontrolled hypertension) � Patient with family history of long QT syndrome, of unexplained syncope
or of unexplained sudden death � Patients with severe and/or uncontrolled concurrent medical disease
that in the opinion of the investigator could cause unacceptable safety risks or compromise compliance with the protocol e.g. uncontrolled diabetes, active or uncontrolled infection, history of pancreatitis
� History of non-compliance to medical regimens � Concomitant treatment with medicinal products that induce CYP3A4
(e.g. dexamethasone, phenytoin, carbamazepine, rifampicin, phenobarbital or St. John’s Wort), or that inhibit the CYP3A4 activity (e.g. ketoconazole, itraconazole, voriconazole, erythromycin, clarithromycin, telithromycin)
� Concomitant treatment with warfarin � Concomitant treatment with anti-arrhythmic drug (e. g. amiodarone,
sotalol, disopyramide, quinidine, procainamide) or medication that prolongs the QT interval (e.g. chloroquine, chlorpromazine, domperidone, droperidol, halofantrine, haloperidol, methadone, pentamidine, pimozide, thioridazine)
� Prior treatment with imatinib except if no progression was demonstrated
VISIT SHEDULE AND ASSESSMENT
Inclusion (visit 1)
During the inclusion visit (within 4 weeks prior to inclusion), the investigator will have to: � Inform the patient of the treatment, the objectives and the design of
the study, answer to the patient’s questions and sign with him/her the informed consent form
� Explain to the patient the aim of the biopsy analysis and propose to him/her to perform a biopsy during the second visit of the study. Sign the specific consent form with the patient
� Check inclusion and non-inclusion criteria � Perform a physical examination (including weight, height, ECOG PS and
vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position))
� Indicate the localisation, history and characteristics of tumour � Register accurately family and personal medical history (in particular
cardiac and hepatic medical history, and alcoholism), prior and concomitant treatments

PVNS_Protocole_V4_110825 Page 13 of 129
� Perform for inclusion: - A blood analysis (serum bilirubin, serum lipase, ALT, AST, serum
creatinine or creatinine clearance (using Cockcroft-Gault formula), absolute neutrophil count (ANC), platelets, magnesium and potassium) (within 7 days prior to inclusion)
- A pregnancy test (negative serum pregnancy test) (within 7 days prior to inclusion)
- A cardiac function assessment: • A MUGA scan or echocardiogram to assess the LVEF (within
14 days prior to inclusion) • An electrocardiogram (ECG) - nilotinib may have an
influence on the QT interval (within 14 days prior to inclusion)
� Pass a MRI or CT scan of the tumour (within 4 weeks prior to inclusion) and realize measurement and assessment according to the RECIST criteria (RECIST version 1.1)
The investigator will fill up the upper part of the inclusion form and then proceed to the inclusion via Internet. Treatment
The study drug is nilotinib (Tasigna®). All patients will be administered with nilotinib 400 mg twice a day for one year. The patient will begin the treatment the day of inclusion (or if not possible, at the latest in the next 5 days following the inclusion). The prescribed dose should be swallowed whole with a glass of water. Doses of 400 mg should be administered twice daily approximately 12 hours apart. Patients should not eat within two hours before and one hour after taking nilotinib and need to avoid foods such as grapefruit juice which may inhibit CYP3A4 enzymes. Follow up
Biological assessment Perform blood samples analysis: � A complete blood cells count will be realized every 2 weeks during the
first two months, then once a month or when it is clinically indicated � Ionogram (sodium, potassium, chlorine, calcium), creatinine, urea, AST,
ALT, albumin, bilirubin, and serum lipase) will be realized: - Within 1 or 2 days after beginning of treatment - Within 7 days prior to visit 2, 3, 4, 5 and 6
Cardiac monitoring Perform ECGs for monitoring of cardiac function (in particular QT interval) at: � tmax (3 hours) of 1
st day of treatment � tmax 8 days after beginning of treatment � Each visit of follow up (visit 2, 3, 4, 5 and 6) � End of clinical study (visit 7) Follow up (visit 2, 3, 4, 5 and 6) During the follow-up visits, the investigator will have to: � Perform a physical examination (including weight, ECOG PS and vital
signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position))
� Check for clinical tumour progression

PVNS_Protocole_V4_110825 Page 14 of 129
� Ask for concomitant treatments � Ask for adverse events, check the blood sample analysis results and
adapt the doses of nilotinib if needed � Visit 2 (W6) and 3 (W12): perform nilotinib blood level measurement
before the visit (blood to be sent to central lab, the blood sample be collected at the study centre)
� Pass a MRI or CT scan of the tumour and do the RECIST version 1.1 evaluation
� Perform the biopsy (visit 2 only) if the specific informed consent was signed by the patient and if no tissue is available in the prior surgery
� Schedule the next visit (according to the flow-chart of assessments) End of clinical study (visit 7) During the end-of-study visit, the investigator will have to: � Perform a physical examination (including weight, ECOG PS and vital
signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position))
� Check for clinical tumour progression � Check if the tumour is operable � Check the blood sample analysis results � Ask for concomitant treatments � Ask for adverse events � Pass a MRI or CT scan of the tumour and do the RECIST version 1.1
evaluation � Complete the end of study form
STATISTICS Sample size and design
A maximum sample size of 50 patients will be included in the study. The study will use a Bayesian design with a beta(2,1) as the prior distribution of the non-progression rate at 12 weeks. The distribution will be first updated after the inclusion of 10 patients, and then every 5 patients (i.e. 8 interim analyses at most, given the maximum sample size of 50 patients). A stopping rule for inefficacy will be defined as follows: at each update of the distribution, the trial will stop for inefficacy if there is a high probability (≥0.8) that the estimated non progression rate is lower or equal to p0=30%. In case of no early stopping, once the last group of patients’ outcome is known, we will be able to estimate the final updated probability of success with its 95% credible interval.
Analysis populations
The efficacy analyzable population will include all patients treated with nilotinib during at least 3 weeks and with no major protocol violation. The safety analyzable population will include all patients who received at least one dose of nilotinib and had at least one post-baseline safety assessment. The efficacy analyzable population will be used for all analyses of efficacy and baseline characteristics. The safety analyzable population will be used for safety analyses. Primary endpoint
Central read tumour assessment will be used in the primary efficacy analysis. The 12-week non progression rate will be summarized by a proportion together with its 95% confidence interval.

PVNS_Protocole_V4_110825 Page 15 of 129
Secondary endpoints
The 24-week non progression rate will be analyzed using central read tumour assessment. It will be summarized by a proportion together with its 95% confidence interval. Objective tumour response rate, duration of treatment response, best overall response, PFS, TTP and TTF will be analyzed using central read tumour assessments. Duration of response, PFS, TTP and TTF and will be estimated as a function of time by the Kaplan-Meier method. The relationship between the objective tumour response and tumour characteristics will be tested using univariate analyses. Safety
The assessment of safety will be based mainly on the frequency of adverse events based on the common toxicity criteria (CTC-V3) grade. Descriptive statistics will be provided for characterizing and assessing patient tolerance to treatment
STUDY PERIODS Inclusion: 2 years Whole study duration: 3 years (2 years inclusion + 1 year follow-up) Study duration for 1 patient: 1 year
STUDY SITES 17 centres (FRANCE, UNITED KINGDOM, THE NETHERLAND, ITALY, POLAND, AUSTRALIA)

PVNS_Protocole_V4_110825 Page 16 of 129
LIST OF ABBREVIATIONS
AE Adverse event
AFSSAPS Agence française de sécurité sanitaire des produits de santé
ALT Alanine amino transferase
ANC Absolute neutrophil count
AST Aspartate amino transferase
BID Bis in die (2 times a day)
CML Chronic myeloid leukaemia
COL6A3/CSF1 Collagen type VI alpha-3/ Colony-stimulating factor 1
CLB Centre Léon Bérard
CR Complete response
CRA Clinical research assistant
CSF1R Colony-stimulating factor 1 receptor
CYP Cytochrome P
ECG Electrocardiogram
GCP Good clinical practice
IEC/IRB Independent ethic committee / Institutional review board
LLN Lower limit of normal
LVEF Left ventricular ejection fraction
M-CSF Macrophage colony stimulating factor = CSF1
MUGA MUltipled gated acquisition
MRI Magnetic Resonance Imaging
NCI-CTCAE National Cancer Institute-Common Terminology Criteria for Adverse Events
PDGFR Platelet-derived growth factor receptor
PFS Progression free survival
PR Partial response
PVNS Pigmented villo nodular synovitis
QD Quaque die (once a day)
RECIST Response Evaluation Criteria In Solid Tumours
SAE Serious adverse event
SAP Statistical analysis plan
SD Stable disease
SPC Summary of product characteristics
SSG Scandinavian Sarcoma Group
SUSAR Suspected unexpected serious adverse reaction
TGCT Tenosynovial giant cell tumour
TTF Time to treatment failure
TTP Time to progression
UBET Unité de Biostatistique et d’Evaluation des Thérapeutiques
ULN Upper limit of the norm
V Visit
W Week
WHO World Health Organisation

PVNS_Protocole_V4_110825 Page 17 of 129
1. BACKGROUND AND STUDY RATIONALE Pigmented villonodular synovitis (PVNS), also known as tenosynovial giant cell tumour (TGCT), is a rare pathological entity affecting the synovium in young adults. Initially considered as an inflammatory reactive process, recent observations have shown that this disease may actually be a benign neoplastic process with specific genetic alterations. Indeed, a specific t (1;2) translocation, involving the collagen 6A3 gene (on 2q35) and the M-CSF (also known as CSF1) gene (on 1p13), is present in a fraction of tumour cells in PVNS/TGCT. This fusion gene expressed by a fraction of the cells encodes for a fusion protein which attracts non-neoplastic cells expressing M-CSF receptor (macrophages and monocytes), through a paracrine - “landscape” - effect (1-4). Imatinib is a treatment indicated for chronic myeloid leukaemia (CML) and gastrointestinal stromal tumour which block M-CSF receptor activation at therapeutic concentration (5). PVNS/TGCT is usually treated by surgery alone (1). However, relapses may occur, and re-excision may be needed, sometimes with possible important functional impairment. Blay et al. recently reported the case of a patient with recurrent and symptomatic PVNS/TGCT following surgery, in which surgical re-excision would have had important functional consequences (6). In the case report, the patient was treated with imatinib, providing rapid tumour response. A relapse was observed at discontinuation of imatinib, and a secondary response was obtained at imatinib reintroduction. This is the first report of the activity of imatinib in an M-CSF/M-CSF receptor dependent solid tumour. Although a potential contribution of the blockade of other tyrosine kinases by imatinib can not be ruled out, the frequency at which the col6A3/CSF1 fusion gene is observed in PVNS/TGCT (4) as compared to other pathological synovial process strongly suggests that imatinib activity involves M-CSF receptor blockade in this disease, despite recent observation showing limited biological activity of the product of the fusion gene. As a consequence, imatinib is a good candidate to induce complete responses in relapsing PVNS/TGCT and may offer an option in patients in whom surgery is not feasible or implies too much risks. In the first 4 patients treated with imatinib in Lyon, 2 patients interrupted treatment because of poor tolerance (1 interruption at patient request, 1 interruption because of grade 4 liver toxicity). This report has then be confirmed in ASCO 2010 by 2 independent additional publications, including a total of 22 patients, in whom 3 patients experience PR or CR, and only 2 patients experienced PD as best response. The side effects of imatinib, in particular skin toxicity and fatigue was however substantial in some of these patients and 4 additional had to interrupt the treatment and progressed afterwards (7-8). Another phenylaminopyrimidine commercialized by Novartis called nilotinib (Tasigna®) has inhibitory properties similar to imatinib on M-CSF receptor pathway (9). Nilotinib is indicated for the treatment of adults with chronic phase and accelerated phase Philadelphia chromosome positive chronic myelogenous leukaemia (CML) with resistance or intolerance to prior therapy including imatinib. The reason for selecting nilotinib as compared to imatinib came out from different considerations 1) In the limited experience with imatinib in PVNS reported so far the toxicity experienced by some patients was substantial. 2) Nilotinib has a more favourable toxicity profile in particular regarding soft tissue and facial oedema. This may favour a better compliance to the treatment. In this context, it is interesting to set up a clinical study designed to explore the efficacy of nilotinib as a treatment of patients with inoperable PVNS/TGCT. This disease being rare, this clinical trial is a non-randomised open-label and international study. Nilotinib will be administered to patients with progressive or relapsing PVNS/TGCT who cannot be treated by surgery. Patients will receive the medication according to the posology recommended by the summary of product characteristics and used in the treatment of CML (400 mg twice a day). The main benefit anticipated for the patients included in the protocol will be the tumour reduction and the consequent functional improvements. The main risk will be the non-response to the treatment and

PVNS_Protocole_V4_110825 Page 18 of 129
the known adverse effects of nilotinib. In case of secondary effects, doses of nilotinib will be adjusted according to the greatest degree of toxicity. Also, upon specific acceptance of the patients, a biological analysis of the tumour will be conducted by a centralised laboratory to explore the relationship between the tumour response to the treatment and some characteristics of tumours (presence of COL6A3/CSF1 fusion gene, M-CSF and M-CSF receptor and phosphorylated c-fms). Patients will be treated by nilotinib for 1 year. In case of treatment efficacy as assessed by intermediary analyses, maintenance of the treatment upon patients’ acceptance will be considered. This study will be conducted following local legal requirements and according to Good Clinical Practices.

PVNS_Protocole_V4_110825 Page 19 of 129
2. STUDY OBJECTIVES
2.1. PRIMARY OBJECTIVE The primary objective of the study will be to determine the efficacy of 12 weeks (3 months) of nilotinib treatment as measured by the non progression rate (Complete response + Partial Response + Stable disease according to Response Evaluation Criteria In Solid Tumours – RECIST version 1.1) in patients with progressive or relapsing PVNS/TGCT who cannot be treated by surgery.
2.2. SECONDARY OBJECTIVES A key secondary objective will be to determine the efficacy of 24 weeks (6 months) of nilotinib treatment as measured by the non progression rate (Complete response + Partial Response + Stable disease according to Response Evaluation Criteria In Solid Tumours – RECIST version 1.1) in patients with progressive or relapsing PVNS/TGCT who cannot be treated by surgery. This key secondary objective was defined for the purpose of a further analysis (not described in this protocol) which will pool the data of the PVNS study with those of a similar concomitant study conducted in the US and Australia. The other secondary objectives will be:
� To evaluate the efficacy of nilotinib according to:
- The objective tumour response rate (Complete response + Partial Response according to RECIST version 1.1) after 12 weeks of treatment
- The duration of treatment response - The best overall response obtained during the study - The progression-free survival (PFS) - The time to progression (TTP) - The time to treatment failure (TTF) - The proportion of patients with an operable tumour after nilotinib exposure according to
investigator evaluation - The description of concomitant treatments use - The correlation between trough levels of nilotinib and objective tumour response
� To assess the safety of nilotinib for PVNS/TGCT patients
2.3. EXPLORATORY OBJECTIVE An exploratory objective will be to study the relationship between the objective tumour response and the following tumour characteristics (tissues collected in a prior surgery, or by biopsy, upon specific acceptance by the patient):
- Presence of COL6A3/CSF1 fusion gene - Presence of M-CSF, CSF1R, KIT, PDGFRA and B on immunohistochemistry - Presence of phosphorylated c-fms on tumour samples - Activation of the PI3K/Akt/mTor pathway, presence of activating mutations of ras, and other
potential molecular alterations

PVNS_Protocole_V4_110825 Page 20 of 129
3. STUDY DESIGN
3.1. EVALUATION CRITERIA The efficacy of nilotinib will be assessed for most of the criteria using RECIST version 1.1. Tumour will be evaluated at each visit of the study by Magnetic resonance imaging (MRI) or CT scan. All images will be read locally at the site and this interpretation will be used for all clinical decision making. Assessments will be then validated by a central review committee. Centrally reviewed tumour assessments data will be used in all efficacy analysis. Upon patient specific acceptance, tumour specimen obtained from a prior surgery or from a biopsy will be analysed by a centralised laboratory and according to a standardized protocol (Appendix 16).
3.2. METHODOLOGY This is an international, multicentre, non-randomised and open-label phase II clinical study with a Bayesian design (cf. §11.1 for details on the Bayesian design).
A total of 17 study centres (hospitals specialised in the treatment of PVNS/TGCT) located in Europe (France, United Kingdom, The Netherlands, Italy, Poland) and in Australia, will include a maximum of 50 patients.
Patients will be selected among those contacting the study centre for the treatment of PVNS/TGCT according to the inclusion and non-inclusion criteria described above. After being informed of the study and having asked all their questions to the investigator, they will have enough time to decide whether or not they want to be included in the study. It is planned to treat the patients with nilotinib 400 mg twice a day for 1 year. During the study, 6 visits will be performed at 6 weeks, 12 weeks, 18 weeks, 6 months, 9 months and 12 months after day 1 of treatment. A central review committee will review all MRI and CT scans to provide centralized RECIST version 1.1 evaluations for all patients before each interim analysis (cf. §11.1 for details on interim analyses). Interim analyses will be performed after the inclusion of 10 patients, and then every 5 patients (i.e. 8 interim analyses at most, given the maximal sample size of 50 patients).

PVNS_Protocole_V4_110825 Page 21 of 129
Figure 1 Study design
Visit V1 V2 V3 V4 V5 V6 V7
Week 0 6 12 18 24 36 48
Month 6 9 12
Nilotinib (400 mg twice a day)
3.3. BIAS LIMITATION Considering the rarity of PVNS/TGCT it was not possible to randomize the patients vs. an alternative treatment option.
3.4. ALLOCATION TO TREATMENT GROUPS Not applicable.
3.5. STUDY DURATION AND STUDY PERIODS All patients will be administered with nilotinib 400 mg twice a day for one year. Then, the patient’s treatment will be chosen by the investigator. The whole clinical study duration from the first inclusion to the last patient’s end of study visit will be of 3 years (2 years of inclusion + 1 year of study). The end of study is defined as the end of the clinical study of the last patient.
3.6. SOURCE DATA OF THE eCRF Source data will be the patient’s medical files, and results of clinical and paraclinical assessments.
Inclusion: >>>> Informed consent >>>> Physical examination >>>> MRI or CT scan >>>> Blood analysis >>>> Cardiac function assessment >>>> Verification of inclusion and
non-inclusion criteria
V2 to V7: >>>> Physical Examination >>>> MRI or CT scan >>>> Blood analysis >>>> ECG >>>> Adverse events >>>> V2 and V3: nilotinib blood level >>>> V2 only: tumour specimen sampling if
specifically accepted by the patient
Inclusion End of clinical
study

PVNS_Protocole_V4_110825 Page 22 of 129
4. STUDY POPULATION
4.1. INCLUSION CRITERIA Patients will be included in the study if they meet all the following inclusion criteria:
� Age ≥ 18 years
� Histologically confirmed diagnosis of inoperable progressive or relapsing PVNS/TGCT OR resectable tumour requesting mutilating surgery
� Demonstrated progressive disease in the last 12 months
� At least one measurable site of disease on MRI/CT scan according to RECIST criteria (RECIST version 1.1) based on investigator’s assessment
� WHO Performance status of 0, 1 or 2
� Adequate organ, electrolyte and marrow function, defined as the following: serum bilirubin ≤1.5 x ULN, ALT and AST ≤2.5 x ULN, serum creatinine ≤1.5 x ULN or creatinine clearance ≥50 mL/min, absolute neutrophil count (ANC) ≥1.5x109/L, platelets ≥100x109/L, serum lipase ≤1.5 x ULN, magnesium ≥ lower limit of normal (LLN) and potassium ≥ LLN
� Prior adequate physical examination including weight, height, ECOG PS and vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position)
� Signed written informed consent form
� Covered by a medical insurance (in countries where applicable)
4.2. NON-INCLUSION CRITERIA Patients will not be included in the study if they meet any of the following non-inclusion criteria:
� Pregnant or lactating female or female of child-bearing potential not employing adequate contraception during the study and for up to three months following termination of the study
� Known hypersensitivity to nilotinib or to any of the excipients, galactose intolerance, lactase deficiency or glucose-galactose malabsorbtion prior to enrolment
� Acute or chronic uncontrolled liver disease, or severe renal disease
� Impaired cardiac function, including: - LVEF<50% or below the institutional lower limit of the normal range (whichever is higher) as
determined by echocardiogram or MUGA scan - History or signs of prior myocardial infarction - History of unstable angina - Congenital long QT prolongation - Personal history of unexplained syncope - QTc interval ≥ 450 msec on screening ECG - Other clinically significant heart disease (e.g. bradycardia, congestive heart failure or
uncontrolled hypertension)
� Patient with family history of long QT syndrome, of unexplained syncope or of unexplained sudden death
� Patients with severe and/or uncontrolled concurrent medical disease that in the opinion of the investigator could cause unacceptable safety risks or compromise compliance with the protocol e.g. uncontrolled diabetes, active or uncontrolled infection, history of pancreatitis
� History of non-compliance to medical regimens

PVNS_Protocole_V4_110825 Page 23 of 129
� Concomitant treatment with medicinal products that induce CYP3A4 (e.g. dexamethasone, phenytoin, carbamazepine, rifampicin, phenobarbital or St. John’s Wort), or that inhibit the CYP3A4 activity (e.g. ketoconazole, itraconazole, voriconazole, erythromycin, clarithromycin, telithromycin)
� Concomitant treatment with warfarin
� Concomitant treatment with anti-arrhythmic drug (e. g. amiodarone, sotalol, disopyramide, quinidine, procainamide) or medication that prolongs the QT interval (e.g. chloroquine, chlorpromazine, domperidone, droperidol, halofantrine, haloperidol, methadone, pentamidine, pimozide, thioridazine)
� Prior treatment with imatinib except if no progression was demonstrated

PVNS_Protocole_V4_110825 Page 24 of 129
5. INVESTIGATIONAL PRODUCTS
5.1. DESCRIPTION, PACKAGING AND LABELLING OF THE TREATMENT UNITS
5.1.1. Investigational drug The study drug is nilotinib (Tasigna®). The product composition is described hereafter:
Active substance Nilotinib
Excipients
Capsule content: Lactose monohydrate, Crospovidone, Poloxamer 188, Silica colloidal, anhydrous/Colloidal silicon dioxide, Magnesium stearate Capsule shell: Gelatin, Titanium dioxide (E171), Yellow iron oxide (E172) Printing ink: Shellac, Red iron oxide (E172), Soya lecithin (E322)
Dosage 400 mg twice daily
Pharmaceutical form
Hard capsule White to slightly yellowish powder in light yellow opaque gelatin capsules, size 0 with red axial imprint “NVR/TKI”
The patient will begin the treatment the day of inclusion (or if not possible, at the latest in the next 5 days following the inclusion). The prescribed dose should be swallowed whole with a glass of water. Doses of 400 mg should be administered twice daily approximately 12 hours apart. Patients should not eat within two hours before and one hour after taking nilotinib and need to avoid foods such as grapefruit juice which may inhibit CYP3A4 enzymes.
5.1.2. Permitted study drug adjustments For patients who are unable to tolerate the protocol-specified dosing schedule due to drug-related toxicity, certain dose adjustments are permitted in order to keep the patient on study drug. Toxicity will be assessed using the NCI/NIH Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0 grading scale (Appendix 1).
5.1.2.1. Dosing modifications If a treatment interruption is needed and the patient is unable to resume treatment within a period of ≤ 28 days, the patient should be withdrawn from the study. Any changes in dose must be recorded on the eCRF. Dose reduction
� No dose reductions will be performed for haematological toxicities grade 1/2 and non-haematological toxicities grade 1
� For toxicities which are considered by the investigator unlikely to develop into serious or life-threatening events, with the exception of those toxicities outlined in the table hereafter, treatment may be continued at the same dose without reduction or interruption

PVNS_Protocole_V4_110825 Page 25 of 129
Dose reduction steps and criteria for interruption and re-initiation of nilotinib for drug-related toxicities are presented in the table hereafter. Dose re-escalation
Every attempt to re-escalate the dose of study treatment to the initial dose level should be made. This applies to either dose reductions due to haematological or non-haematological toxicities with the exception of dose reductions due to prolongation of QTc interval. The dose should be re-escalated if the following criteria are met at least 28 days after dose reduction:
� All ≥ grade 2 non-haematological toxicities have resolved to ≤ grade 1 or baseline
� All ≥ grade 3 haematological toxicities have resolved to ≤ grade 1 or baseline
� Or alternatively, all ≥ grade 3 haematological and non-haematological toxicities have resolved to ≤ grade 2 and are manageable with supportive therapy
No dose re-escalation is allowed after dose reduction due to QTcF prolongation.
Table 1 Criteria for dose interruption and modification of nilotinib (400 mg bid) treatment for
drug-related toxicity
Haematological toxicity
≥ Grade 3 Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 or baseline, if recovery occurs within 14 days. If toxicity persists for 15-28 days or recurs, hold therapy and resume at next lower dose level after recovery to ≤ grade 1 or baseline: � 400 mg qd If another recurrence is seen at the reduced nilotinib dose of 400 mg qd: � discontinue If recovery to ≤ grade 1 or baseline is greater than 28 days, consult the sponsor
Note: No dose reductions or interruptions will be performed for haematological toxicity of grade 2 or lower. No dose reductions will be performed for grade 3 or 4 anaemia. If the patient develops anaemia, he may be transfused at the discretion of the investigator.
General non-haematological toxicity
Grade 2 (persisting > 7 days with optimal supportive care)
Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 is seen. If toxicity recurs, hold therapy until recovery to ≤ grade 1 is seen and then resume at next lower dose level: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.
≥ Grade 3 Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 is seen. If toxicity recurs, hold therapy until recovery to ≤ grade 1 is seen and then resume nilotinib at next lower dose level: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.

PVNS_Protocole_V4_110825 Page 26 of 129
Serum creatinine
Grade 2 > 1.5 - 3.0 x ULN
Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 is seen. If toxicity recurs, hold therapy until recovery to ≤ grade 1 is seen and then resume at next lower dose level: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.
≥ Grade 3 > 3.0 x ULN
Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 is seen. If toxicity recurs, hold therapy until recovery to ≤ grade 1 is seen and then resume nilotinib at next lower dose level: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.
Hepato-biliary [bilirubin, SGPT(ALT), SGOT (AST)]
Note: If hyperbilirubinemia is due to the indirect component only (i.e. direct bilirubin component ≤1.5 x ULN), and hemolysis as the etiology has been ruled out as per institutional guidelines (e.g. review of peripheral blood smear and haptoglobin determination), and no amylase and/or lipase elevations are seen, nilotinib may be continued at the same dose at the discretion of the investigator.
Grade 2 Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 (or baseline if disease related) is seen. If toxicity recurs, hold therapy until recovery to ≤ grade 1 or baseline is seen and then resume at next lower dose level: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 or baseline is greater than 28 days, the patient must be discontinued from the study.
≥ Grade 3 Hold therapy and resume nilotinib at next lower dose level after recovery to ≤ grade 1 (or baseline if disease related) is seen. � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 or baseline is greater than 28 days, the patient must be discontinued from the study.
Pancreatitis (with abdominal symptoms plus amylase and/or lipase elevation)
Grade 2 Hold therapy and perform abdominal CT with contrast to exclude pancreatic pathology. If CT is positive, continue to hold therapy and repeat CT, at investigator’s discretion. If CT is negative, re-start at the next lower dose level after recovery to ≤ grade 1: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.
Grade 3 Hold therapy and consult the sponsor

PVNS_Protocole_V4_110825 Page 27 of 129
Grade 4 Hold therapy and discontinue patient from the study
Elevated amylase and/or lipase without symptoms
≥ Grade 3 Hold therapy and perform abdominal CT with contrast to exclude pancreatic pathology. If CT is positive, continue to hold therapy and repeat CT, at investigator’s discretion. If CT is negative, re-start nilotinib at the next lower dose level after recovery to ≤ grade 1: � 400 mg qd If toxicity recurs without symptoms: � continue dosing if CT negative, at investigator’s discretion If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.
Diarrhea
Note: Anti-diarrheal medication is recommended at the first sign of loose stools or overt diarrhea. If diarrhea cannot be controlled with optimal anti-diarrheal treatments, take the following actions:
≥ Grade 3 Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 is seen. If toxicity recurs, hold therapy until recovery to ≤ grade 1 is seen and then resume nilotinib at next lower dose level: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.
Vomiting
Note: The use of prophylactic medication for vomiting is not recommended. Antiemetic medication can be used as clinically indicated or in the patient‘s best interest. If nausea and vomiting cannot be controlled with optimal antiemetic treatment take the following actions:
≥ Grade 3 Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 is seen. If toxicity recurs, hold therapy until recovery to ≤ grade 1 is seen and then resume nilotinib at next lower dose level: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.
Skin rash
Note: Institute symptomatic therapy as appropriate. If skin rash does not resolve with optimal treatments, take the following actions:
Grade 2 Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 is seen. If toxicity recurs, hold therapy until recovery to ≤ grade 1 is seen and then resume at next lower dose level: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.

PVNS_Protocole_V4_110825 Page 28 of 129
≥ Grade 3 Hold therapy and resume nilotinib at 400 mg bid after recovery to ≤ grade 1 is seen. If toxicity recurs, hold therapy until recovery to ≤ grade 1 is seen and then resume nilotinib at next lower dose level: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.
Cardiac QTc prolongation
Note: In case of abnormality of ECG, a cardiologist opinion must be requested.
QTcF > 480 msec Hold dosing when an ECG with a QTcF > 480 msec by automated reading is identified at the site: � Perform an analysis of serum potassium and magnesium, and if below
lower limit of normal, correct with supplements to within normal limits � Concomitant medication usage must be reviewed � Compliance with correct dose and administration of nilotinib must be
checked � Perform a repeat ECG within one hour of the first QTcF of > 480 msec � If QTcF remains > 480 msec, repeat ECG as clinically indicated, but at
least once a day until the QTcF returns to < 480 msec
If QTcF > 480 msec the following actions should be taken: Direct patient to suspend nilotinib dosing and have the following testing performed as soon as possible: � Repeat ECG � Perform an analysis of serum potassium and magnesium, and if below
lower limit of normal, correct with supplements to within normal limits � Review concomitant medication usage � Check compliance with correct dose and administration of nilotinib � If QTcF remains > 480 msec, repeat ECG as clinically indicated, but at
least once a day until the QTcF returns to < 480 msec
Dosing guidelines: A cardiologist opinion must be requested before any re-exposure after an interruption of the treatment due to QTcF > 480 msec. ECGs must be repeated after dose re-start for all patients who had therapy held due to QTcF > 480 msec: � One to be performed at tmax (3 hours) � One to be performed at tmax 8 days Nilotinib may be restarted, at the same dose, if a reason for elevation of QTcF is identified (other than nilotinib) and corrected so that QTcF returns to < 450 msec and to within 20 msec of baseline within 2 weeks. If no reason for elevation of QTcF has been identified and QTcF returns to < 450 msec and to within 20 msec of baseline within 2 weeks, study drug may be restarted at the same dose. If QTcF again exceeds 450 msec upon re-exposure with study drug, guidance shall be sought from the sponsor. As a general principle: if upon re-exposure with study drug, QTcF again exceeds 450 msec, the dose of the study drug has to be changed as follows: � Nilotinib 400 mg bid: dose reduction to 400 mg qd � Nilotinib 400 mg qd: discontinue If upon re-exposure with study drug QTcF again exceeds 480 msec, the study drug should be discontinued. In this case, an ECG must be realized once a day. If the QTcF is repeated and remains for more than 2 weeks > 480 msec nilotinib has to be discontinued. If the QTcF is repeated and if it remains for more than 2 weeks more than 20

PVNS_Protocole_V4_110825 Page 29 of 129
msec greater than baseline or between 450 msec and 480 msec, the dose of the study drug has to be changed as follows: � Nilotinib 400 mg bid: dose reduction to 400 mg qd � Nilotinib 400 mg qd: discontinue
Note: Additional testing ECGs must be repeated 8 days (± 48 hours) after dose re-start for all patients who had therapy held due to QTcF > 480 msec. Repeat QTcF elevations: If QTcF of > 480 msec recurs, the patient is to be discontinued.
QTcF ≥ 500 msec Perform a repeat ECG until QTcF < 500 msec Nilotinib has to be discontinued definitively
LVEF < 45% Investigators should follow the NCI CTCAEv.4 criteria to adjust or discontinue doses of nilotinib therapy with regard to grade 2, 3, or 4 LV function changes which are deemed to be related to study drug. � For grade 2 (asymptomatic, resting LVEF < 50 - 40%), close monitoring
with a follow-up echocardiogram within 4 weeks is recommended � For grade 3 events (symptomatic CHF responsive to intervention; LVEF <
40 - 20%) follow guidance for cardiac “other” described below � For grade 4 (refractory CHF or poorly controlled; LVEF < 20%) discontinue
patient
Cardiac “other” (such as unstable angina)
Grade 2 or Grade 3 Hold therapy and resume nilotinib at next lower dose level after recovery to ≤ grade 1 is seen: � 400 mg qd If another recurrence is seen: � discontinue If recovery to ≤ grade 1 is greater than 28 days, the patient must be discontinued from the study.
Grade 4 Hold therapy and discontinue patient from study.
Serum hypophosphatemia
Grade 2 Continue nilotinib at 400 mg bid and start phosphate supplementation.
≥ Grade 3 Hold therapy, start phosphate supplementation and resume nilotinib at 400 mg bid after recovery to ≤ grade 2 is seen. If grade 3-4 hypophosphatemia recurs despite supplementation, hold therapy again until recovery to ≤ grade 2 is seen and then resume nilotinib at next lower dose: � 400 mg qd If another recurrence of grade 3-4 hypophosphatemia is seen consult sponsor.
5.1.3. Packaging of the treatment units
5.1.3.1. Treatment units The treatment will be packed as boxes of 112 capsules (4x28) of 200 mg in PVC/aluminium blisters that will be provided to the patient at each visit. Packaging for clinical trial will be performed by the sponsor (by service provider) (except for non-European countries that organize themselves packaging for clinical trial according to the legal obligations of their country) according to good practices guidelines.

PVNS_Protocole_V4_110825 Page 30 of 129
5.1.3.2. Labelling
Medication labels will comply with the legal requirements of each country and be printed in the local language. The storage conditions for study drug will be described on the medication label. Since this trial is an open label, non-randomized trial, the study treatment will not have to be specifically produced for the study. The name of the sponsor and the study references of the study will be added on the usual commercialized packaging. Copies of these labels will be kept in the sponsor central file. Labelling for clinical trial will be performed by the sponsor (by service provider) according to good practices guidelines (except for non-European countries that organize themselves labelling for clinical trial according to the legal obligations of their country).
5.1.4. Study drug supply and tracking/drug accountability Study drugs will be provided by Novartis for the 1st year of treatment. Study drugs must be received by a designated person at the study site, handled and stored safely and properly, and kept in a secured location to which only the pharmacist have access. Upon receipt, the study drugs should be stored according to the instructions specified on the drug labels. Clinical supplies are to be dispensed only in accordance with the protocol. Medication labels will be in the local language and comply with the legal requirements of each country. They will include storage conditions for the drug, the drug name and dose but no information about the patient. The investigator must maintain an accurate record of the shipment and dispensing of study drug in a drug accountability ledger. Drug accountability will be noted by the ARC during site visits and at the completion of the trial. Patients will be asked to return all unused study drug and packaging at the end of the study or at the time of study drug discontinuation. At the end of the study, the sponsor will be responsible for ensuring the destruction of non-used clinical supplies. This destruction could be performed by hospital pharmacists in compliance with legal rules in force with the prior agreement of the sponsor. A destruction certificate will be delivered to the sponsor.
5.2. TREATMENT COMPLIANCE No measurement of compliance is planned during the study. Only dates of first and last study treatment intake will be recorded. Nilotinib trough blood levels measurement will be determined after 6 and 12 weeks of treatment. The sample will be performed just before nilotinib intake (residual) and 12 hours (+/- 2h) after the last intake of the drug if BID.
5.3. STUDY TREATMENTS STORAGE The study products will be stored according to SPC requirements below 30°C, in a dry place with restricted access.

PVNS_Protocole_V4_110825 Page 31 of 129
5.4. SUPPLY TO THE INVESTIGATIONAL CENTRES The sponsor (by service provider) will be responsible for transport and supply to each participating institution. A service provider will make the supply of these pharmacies on behalf of laboratory Novartis (Appendix 11) (except for non-European countries that organize themselves transport and supply of treatment in each institution according to the legal obligations of their country).
5.5. MANAGEMENT IN EACH PARTICIPATING CENTRES The pharmacy of each participating institution will be responsible for reception, storage, dispensation, management and destruction of the investigational drug supplied.
5.6. PRIOR AND CONCOMITANTS THERAPIES Concomitant medications corresponding to excluded concomitant treatments as described in section 5.6.1., will have to be recorded in the case report form, with trade name (generic name), route, formulation, dosing scheme, the indication and start and stop dates of administration. Any change during the study should be documented as well.
5.6.1. Excluded concomitant treatments (Appendix 4) Nilotinib is a substrate for CYP3A4/5 and is a competitive inhibitor of CYP3A4/5, CYP2D6 and CYP2C9. Because of the possible risk of either reduced activity or enhanced toxicity of the concomitant medication and/or nilotinib, drugs known to interact with the same CYP450 isoenzymes (CYP2D6 and CYP3A4) as nilotinib should be used with caution. Patients using concomitant medications known to be metabolized by these cytochrome P450 enzymes (e. g. cisapride, pimozide, quinidine, bepridil, ergotamine, dihydroergotamine) will not be excluded from the study. However, the patients must be carefully monitored for potentiation of toxicity due to individual concomitant medication. Consideration should be given to using alternative agents with less potential for interaction with nilotinib. The use of warfarin sodium (e.g. Coumadin®, Marevan®, Warfilone®, Varfine®, Aldocumar®, Tedicumar®, Waran®, Jantoven®, Panwarfin®, Sofarin®) is discouraged during the trial since warfarin is metabolized through the CYP450 system. Patients are not permitted to take warfarin (Coumadin® or Coumadine®) at trial entry. As an alternative, therapeutic anticoagulation may be accomplished using low-molecular weight heparin (e.g. Lovenox®) or heparin. Therefore, if a thrombo-embolic event occurs, patients should be preferably started on anti-coagulation with low-molecular weight heparin. Nilotinib should be used with caution in patients who have or may develop prolongation of QT, including those patients taking anti-arrhythmic medicinal products such as amiodarone, disopyramide, procainamide, quinidine and sotalol or other medicinal products that may lead to QT prolongation such as chloroquine, halofantrine, clarithromycin, haloperidol and methadone. Concomitant therapy with anti-arrhythmic medicinal products (such as amiodarone, disopyramide, procainamide, quinidine and sotalol) or other medicinal products that may lead to QT prolongation (such as chloroquine, chlorpromazine, domperidone, droperidol, halofantrine, haloperidol, methadone, pentamidine, pimozide, thioridazine, clarithromycin) should be discouraged during the trial or introduced with great caution only in the absence of an alternative therapy. The investigator should instruct the patient to notify the study site about any new medications he/she takes after the start of the study drug.

PVNS_Protocole_V4_110825 Page 32 of 129
6. ASSESSMENTS
6.1. FLOW-CHART OF ASSESSMENTS Flow chart of assessments is presented hereafter.

PVNS_Protocole_V4_110825 Page 33 of 129
Clinical study
Visit Number
Week/ Month
Visit 1
Inclusion
W0
Visit 2
W6
Visit 3
W12
Visit 4
W18
Visit 5
M6
Visit 6
M9
Visit 7
M12
Informed consent signed ����
Inclusion / non-inclusion criteria ����
Physical examination (including weight, height, ECOG PS and vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position)) ����
Localisation, history and characteristics of tumour ����
Family and personal medical history (in particular cardiac and hepatic medical history, and alcoholism) ����
Blood analysis (serum bilirubin, serum lipase, ALT, AST, serum creatinine or creatinine clearance, ANC, platelets, magnesium and potassium) ����
Pregnancy test ����
Cardiac function assessment (Echocardiogram or MUGA scan, ECG) ����
Prior and concomitant treatments ���� ���� ���� ���� ���� ���� ����
Vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position) ���� ���� ���� ���� ���� ���� ����
MRI / CT scan ���� ���� ���� ���� ���� ���� ����
Investigator and central RECIST version 1.1 assessment ���� ���� ���� ���� ���� ���� ����
Physical examination (including weight, ECOG PS and vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position)), check for tumour progression, ECG**
���� ���� ���� ���� ���� ����
Blood analysis (complete blood cells count ***, ionogram (sodium, potassium, chlorine, calcium), creatinine, urea, AST, ALT, albumin and bilirubin, serum lipase) ���� ���� ���� ���� ���� ����
Adverse events ���� ���� ���� ���� ���� ����
Use of Nilotinib (Yes/No), dose of nilotinib ���� ���� ���� ���� ���� ����
Nilotinib trough blood level measurement ���� ����
Tumour biopsy ���� *
Patient with operable tumour (Yes/No) ����
Patient wishes to continue nilotinib (Yes/No) ����
* If the specific informed consent was signed by the patient
** One to be performed at tmax (3 hours) of 1st day of treatment, one to be performed at tmax 8 days after beginning of treatment
***Every 2 weeks during the first two months, then once a month or when it is clinically justified

PVNS_Protocole_V4_110825 Page 34 of 129
6.2. STUDY PLAN
6.2.1. Inclusion (visit 1) During the inclusion visit (within 4 weeks prior to inclusion), the investigator will have to:
� Inform the patient of the treatment, the objectives and the design of the study, answer to the patient’s questions and sign with him/her the informed consent form
� Explain to the patient the aim of the biopsy analysis and propose to him/her to perform a biopsy during the second visit of the study. Sign the specific consent form with the patient
� Check inclusion and non-inclusion criteria
� Perform a physical examination (including weight, height, ECOG PS and vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position))
� Indicate the localisation, history and characteristics of tumour
� Register accurately family and personal medical history (in particular cardiac and hepatic medical history, and alcoholism), prior and concomitant treatments
� Perform for inclusion: - A blood analysis (serum bilirubin, serum lipase, ALT, AST, serum creatinine or creatinine
clearance (using Cockcroft-Gault formula), absolute neutrophil count (ANC), platelets, magnesium and potassium) (within 7 days prior to inclusion)
- A pregnancy test (negative serum pregnancy test) (within 7 days prior to inclusion) - A cardiac function assessment:
• A MUGA scan or echocardiogram to assess the LVEF (within 14 days prior to inclusion) • An electrocardiogram (ECG) - nilotinib may have an influence on the QT interval
(within 14 days prior to inclusion)
� Pass a MRI or CT scan of the tumour (within 4 weeks prior to inclusion) and realize measurement and assessment according to the RECIST criteria (RECIST version 1.1)
6.2.1.1. Internet inclusion The investigator will fill up the upper part of the inclusion form (Appendix 9), and then proceed to the inclusion via Internet (Appendix 10).
6.2.2. Follow up
6.2.2.1. Biological assessment
� Perform blood samples analysis: - A complete blood cells count will be realized every 2 weeks during the first two months, then
once a month or when it is clinically indicated - Ionogram (sodium, potassium, chlorine, calcium), creatinine, urea, AST, ALT, albumin, bilirubin,
and serum lipase will be realized: • Within 1 or 2 days after beginning of treatment • Within 7 days prior to visit 2, 3, 4, 5 and 6
6.2.2.2. Cardiac monitoring
� Perform ECGs for monitoring of cardiac function (in particular QT interval) at: - tmax (3 hours) of 1
st day of treatment

PVNS_Protocole_V4_110825 Page 35 of 129
- tmax 8 days after beginning of treatment - Each visit of follow up (visit 2, 3, 4, 5 and 6) - End of clinical study (visit 7)
6.2.2.3. Follow up (visit 2, 3, 4, 5 and 6) During the follow-up visits, the investigator will have to:
� Perform a physical examination (including weight, ECOG PS and vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position))
� Check for clinical tumour progression
� Ask for concomitant treatments
� Ask for adverse events, check the blood sample analysis results and adapt the doses of nilotinib if needed
� Visit 2 (W6) and 3 (W12): perform nilotinib blood level measurement before the visit (blood to be sent to central lab, the blood sample be collected at the study centre)
� Pass a MRI or CT scan of the tumour and do the RECIST version 1.1 evaluation
� Perform the biopsy (visit 2 only) if the specific informed consent was signed by the patient and if no tissue is available in the prior surgery
� Schedule the next visit (according to the flow-chart of assessments) 5 visits will be performed at 6 weeks, 12 weeks, 18 weeks, 6 months, and 9 months after day 1 of treatment. Nilotinib trough blood levels measurement will be determined after 6 and 12 weeks of treatment. The sample will be performed just before nilotinib intake (residual) and 12 hours (+/- 2h) after the last intake of the drug if BID (Appendix 13).
6.2.2.4. End of clinical study (visit 7) During the end-of-study visit, the investigator will have to:
� Perform a physical examination (including weight, ECOG PS and vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position))
� Check for clinical tumour progression
� Check if the tumour is operable
� Check the blood sample analysis results
� Ask for concomitant treatments
� Ask for adverse events
� Pass a MRI or CT scan of the tumour and do the RECIST version 1.1 evaluation
� Complete the end of study form The end-of-study visit will be performed at 12 months after the beginning of treatment.
6.3. INVESTIGATIONAL PRODUCTS DELIVERY AND RETURN The investigator is responsible of the storage and delivery of the investigational products and will respect the local regulation requirements concerning pharmaceutical products detention.

PVNS_Protocole_V4_110825 Page 36 of 129
The investigator is authorized to delegate a person to be responsible for reception, storage, dispensation, management and destruction of the investigational drug supplied such as the centre pharmacist. Upon receipt of the treatment units, this person will have to check the quantity of units as well as their packaging, and send a confirmation page to the sponsor (Appendix 12). During the study, this person will be responsible for counting the delivered, used, unused, and returned units and to fill-in the corresponding form which will contain the following items:
� Patient identification initial (1st letter of surname + “ – “ + 1st letter of first name or 1st letter of surname + two 1st letter of first name) and patient number
� Date of delivery and quantity of treatment
� Date and quantity of treatment returned At the end of the study, the units left as well as the account form will be sent back to the sponsor. A copy of this list will be kept in the investigator file. The study treatment should never be used outwards this protocol.

PVNS_Protocole_V4_110825 Page 37 of 129
7. PREMATURE WITHDRAWAL AND EARLY STUDY TERMINATION
7.1. TERMINATION RULES (TEMPORARY OR DEFINITE)
7.1.1. For patients Patients may voluntarily withdraw from the study or be dropped from it at the discretion of the investigator at any time (for example: non-compliance, unacceptable toxicity or disease progression). In case of definite withdrawal, the treatment for these patients will be chosen by the investigator.
7.1.2. Of the study The sponsor could stop the study at any time. In this case, the sponsor will promptly inform all study investigators, the ethics review committee as well as competent authorities of the study stop and of the reasons. Some special circumstances should imply study stop, including ethics or safety reasons such as:
� An unexpected frequency or intensity of adverse events
� A bad patient recruitment, specially regarding to the quality or quantity of collected data In case an investigator wishes to stop the study, he will have to inform the sponsor as soon as possible of his decision, and to explain his reasons. He will also have to collect and validate the data of patients already included in the study.
7.2. STUDY DRUG DISCONTINUATION / PREMATURE WITHDRAWAL AND PATIENT REPLACEMENT
7.2.1. Study drug discontinuation and premature withdrawal The following criteria would be reasons for study drug discontinuation:
� Disease progression
� Adverse event
� Toxicity
� Abnormal laboratory value(s)
� Abnormal test procedure result(s) The following criteria would be reasons for premature withdrawal:
� Consent withdrawal
� Toxicity
� Patient lost to follow up
� Death
� Investigator’s decision In case of study drug discontinuation/early withdrawal, the reason for discontinuation/withdrawal will have to be documented in the eCRF as completely as possible. Patients lost to follow up should be recorded as such on the eCRF.

PVNS_Protocole_V4_110825 Page 38 of 129
In case of study withdrawal after the first intake of the study treatment, a visit will have to be scheduled as soon as possible, at which time all of the assessments listed for the final visit should be performed, especially evaluations related to the study treatment safety. Reasons and circumstances of discontinuation/premature withdrawal will be fully documented both in the eCRF and in the investigator’s files. In case of study drug discontinuation, the patient will still be followed and evaluated in the same way as other patients. In case of study drug discontinuation related to an adverse event, adverse event description and evolution will be documented. The patient will have to be followed by the investigator until recovery or until the event becomes clinically not significant, or until the sponsor decides that the follow-up is not anymore required.
7.2.2. Patient replacement Replacement is planned for patients meeting at least one of the following criteria:
� Patients with at least one major protocol violation
� Patient treated with nilotinib during less than 3 weeks
� Patients prematurely withdrawn from the study before the 12-weeks evaluation of response, with premature withdrawal not related to a toxicity due to nilotinib, and without prior documented progression

PVNS_Protocole_V4_110825 Page 39 of 129
8. EFFICACY EVALUATION
8.1. PRIMARY ENDPOINT The primary endpoint of the study will be the non progression rate after 12 weeks (3 months) of treatment, based on the response evaluated by CT scan or MRI according to RECIST criteria (RECIST version 1.1) and validated by a central review committee.
8.2. SECONDARY ENDPOINTS Key secondary efficacy endpoint:
� Non progression rate after 24 weeks (6 months) of treatment, based on the response evaluated by CT scan or MRI according to RECIST criteria (RECIST version 1.1).
Other secondary efficacy endpoints:
� Objective tumour response according to RECIST version 1.1 (CR and PR) after 12 weeks of treatment
� Duration of response
� Best overall response
� Progression-free survival
� Time to progression
� Time to treatment failure
� Non progression rate after 12 weeks of treatment, based on the response evaluated locally by the investigator in charge using CT scan or MRI and according to RECIST criteria (RECIST version 1.1)
� Proportion of patients with an operable tumour after nilotinib exposure according to investigator evaluation
� Concomitant treatment use during the study
� Correlation between trough level of nilotinib at 6 weeks and 12 weeks and objective tumour response

PVNS_Protocole_V4_110825 Page 40 of 129
9. SAFETY EVALUATION
9.1. ADVERSE EVENTS (AE)
9.1.1. Definitions Adverse event (AE): Any untoward medical occurrence in a patient or clinical trial subject administered a medicinal product and which does not necessarily have a causal relationship with this treatment*. Adverse reaction: All untoward and unintended responses to an investigational medicinal product related to any dose administered*. Serious adverse event (SAE) or Serious adverse reaction: Any untoward medicinal occurrence or effect that at any dose results in death, is life-threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, or is a congenital anomaly or birth defect*. Prolongation of hospitalisation is defined as prolongation of at least 1 day. Unexpected Adverse Reaction: An adverse reaction, the nature or severity of which is not consistent with the applicable product information (e.g. investigator’s brochure for an unauthorised investigational product or summary of product characteristics for an authorised product)*.
9.1.2. Adverse events reporting The investigator should ask the patient at each visit whether or not he/she experienced any unusual medical problem or deterioration of medical condition since the last visit / last documentation by asking a non-specific question, such as “How are you feeling today?” or “How have you been feeling since the last visit?” Adverse events that were ongoing at the conclusion of the previous visit should be queried for resolution or change in severity or seriousness. Any adverse event spontaneously reported by the patient or observed by the investigator will be reported in the patient’s file. Adverse events graded 3-4 (according to CTCAE), or related the study treatment, or serious adverse events will have to be reported in the appropriate section of the eCRF with the following specifications:
� The CTCAE grade
� Its relationship to each study drug (suspected/not suspected)
� Its duration (start and end dates or if continuing at final exam)
� Action taken (no action taken; study drug dosage adjusted/temporarily interrupted; study drug permanently discontinued due to this adverse event; concomitant medication taken; non-drug therapy given; hospitalization/prolonged hospitalization)
� Whether it is serious
* Source: Directive 2001/20/Ec of the European Parliament. * Source: Directive 2001/20/Ec of the European Parliament.

PVNS_Protocole_V4_110825 Page 41 of 129
As far as possible, the adverse event should be described using medical terms (such as hypertension). Any signs or symptoms related to a previously reported diagnosis do not have to be reported unless they represent atypical or extreme manifestations of the disease: in such a case, they have to be reported as separated AE. In case no clear diagnosis was established, every sign or symptom will be recorded on its own. In case of ongoing adverse events, the investigator must follow the patient to resolution of the event or until it is deemed clinically insignificant by the investigator. It is important to distinguish if an AE is severe or serious. The severity is related to the intensity whereas a serious AE is defined by the criteria described in section 9.1.1. A severe AE should not be always considered as serious. For instance, persistent nausea could be considered as severe nausea but not as a SAE. On the contrary, a cerebrovascular accident resulting in a limited invalidity could be considered as an event of mild intensity but would be reported as SAE. AE following misuse or overdose of the study treatment will have to be considered as SAE, even if they don't meet the seriousness criteria. A value outside the normal or reference range in a routine safety assessment, such as clinical laboratory, vital signs or ECG, may signify an adverse event. If the investigator considers the abnormality as corresponding to the definition of an AE to be reported according to the protocol, he should record it on the Adverse Events section of the eCRF. If the findings contribute to a clinical diagnosis (such as hepatitis in case of increased liver enzymes), then this diagnosis should be recorded as an adverse event.
9.1.3. Serious adverse events (SAE) reporting and notification The investigator must report to the sponsor:
� Serious adverse events (SAE), adverse events following misuse or overdose and pregnancies
� Occurring or observed during the course of the study and within 28 days after the last administration of the investigational drug. This have to be done within 24 hours after the investigator becomes aware of the event
� Whatever their relationship to the study treatment SAE occurring more than 28 days after study treatment completion need only be reported if a relationship to the study drug (e. g. secondary malignancy) is suspected by the investigator. In case of SAE, the investigator will have to:
� Fill and sign an SAE report form in English (Appendix 6)
� Collect any additional document necessary for SAE assessment (such as hospitalisation report, biological results)
� Assess the seriousness and causality of the event with the study drug using the causality criteria: unrelated, unlikely, possible, probable, certain (Appendix 5)
� Send the SAE within 24 hours and additional documents by fax to:
The Lyon Pharmacovigilance Centre- ���� + 33 (0)4 72 11 69 85
NB: The Lyon Pharmacovigilance Centre will send the SAE from Italy to the Novartis Italian safety department (fax number: +39 2 96703051). The French Sponsor will receive Safety Information Notice by Novartis France. The sponsor will have the responsibility to send them to the Italian participating site. A complete description and medical diagnosis shall be provided. In case of incomplete information, the investigator will have to provide follow-up information as soon as possible, again using the SAE report form (within 3 days) to the Lyon Pharmacovigilance Center.

PVNS_Protocole_V4_110825 Page 42 of 129
Data concerning SAE follow-up will have to be reported by the investigator within the same period, until complete outcome of the event. The investigator shall supply the sponsor with any additional requested information. All the SAE will be recorded in the eCRF. The following events are not to be considered as SAE:
� Any event requiring short consultation in an hospital
� An external treatment in a hospital emergency service, however, the reason of the treatment initiation has to be reported as SAE
� Hospitalisation (1 night or more) or hospitalisation prolongation for one of the following reasons:
- Planned hospitalisation for routine intervention or treatment as a part of a prior therapy
- Hospitalisation or intervention requested by the protocol
- Hospitalisation for explorations not related to a modification of the patient health
- Hospitalisation for comfort or for social reasons (for example: hospitalisation of an elderly person due to dependence on his partner who was hospitalised)
- Hospitalisation not related to a patient health worsening and not related to the study objectives (for example: plastic surgery)
The sponsor will have to report any suspected unexpected SAR (SUSAR) potentially related to the study treatment to the EMEA (Eudravigilance database), the Competent Authorities and the Ethics Committees within the requested period. The sponsor will also have to inform the Ethics review committee of any SAE with the potential to modify the project (to be adapted for each country). The evaluation of expectedness is based on knowledge of the adverse reaction on the reference document. The reference document will be the Summary of Product Characteristics of Tasigna®. The sponsor will be responsible for reporting of New Safety Issues and Annual safety Reports to Competent Authorities and to Ethics Committees.
9.2. OTHER SAFETY CRITERIA
9.2.1. Blood analysis Blood analysis will be performed during the follow up and will concern the following parameters:
� Complete blood cells count
� Ionogram (sodium, potassium, chlorine, calcium)
� Creatinine
� Urea
� AST
� ALT
� Albumin
� Bilirubin
� Serum lipase

PVNS_Protocole_V4_110825 Page 43 of 129
9.2.2. Vital signs Vital signs (systolic and diastolic blood pressure, heart rate after at least 5 minutes in supine position) and weight should be assessed at each visit of the study.

PVNS_Protocole_V4_110825 Page 44 of 129
10. OTHER EVALUATIONS
An exploratory analysis will be performed to explore the relationship between the objective tumour response and the following tumour characteristics (Appendix 16):
� Presence of COL6A3/CSF1 fusion gene
� Expression of M-CSF, CSF1R, KIT, PDGFRA AND B on immunohistochemistry
� Presence of phosphorylated c-fms on tumour samples
� Activation of the PI3K/Akt/mTor pathway, presence of activating mutations of ras, and other potential molecular alterations
Tissues will be collected in a prior surgery or by biopsy, upon specific acceptance by the patient. If no tissue is available in the prior surgery, a biopsy will be done at visit 2.

PVNS_Protocole_V4_110825 Page 45 of 129
11. STATISTICAL METHODS AND SAMPLE SIZE CALCULATION
A detailed statistical analysis plan (SAP) will be written as a separate document and will be finalised before the database lock.
11.1. SAMPLE SIZE AND DESIGN The primary objective of the study is to evaluate the non progression rate of nilotinib in patients with inoperable PVNS/TGCT, after 12 weeks of treatment. The main goal of this phase II study is not to provide definite evidence of nilotinib efficacy, but rather to be able to reject the drug for further testing in a phase III trial if it has not sufficient promising activity to require further experimentation. Traditional clinical trial design—called frequentist design (e.g. the Fleming design) is based on statistical inferences and has been the bedrock of science within the realm of oncology and in medicine more generally for decades. A great virtue of frequentist design is its rigor and narrow focus on the specific experiment. On the other hand, such designs have some limitations, the major one being their inflexibility. First, the design (in particular the number of interim analyses, which is furthermore limited to restricted revisions) has to be initially defined. Such designs also require the specification of various parameters, including the fixed target response (or non-progression) rate for the new treatment as well as limits on the type I and type II errors. Both design and hypotheses can not be adapted in the course of the study, event if they reveal to be inadequate, and yet the conclusions resulting from such approaches are extremely dependant on them. Thus, when using the frequentist approach, it is necessary to be able to define sound hypotheses (in particular regarding the fixed target response (or non-progression) rate), based on accurate information (from previous studies for example). Only one observation of a response in a single patient is available in the literature about the potential efficacy of nilotinib as a treatment of PVNS/TGCT. Thus, the frequentist approach does not seem to be the most appropriate for the study. We will use instead a Bayesian design which has several advantages in drug development. In particular, allowing for smaller more informative trials, and specifically tied to decision making, the Bayesian design provides a good alternative to frequentist approach as part of the study. Unlike the frequentist approach, in the Bayesian approach all uncertainty is measured by probability. In a Bayesian framework, the response (or non-progression) rate is not treated as a fixed value, but is regarded as a variable with a probability to be determined. This then allows the calculation of the probability that the response (or non-progression) rate lies in a given interval, which enables simpler and more direct interpretation of the results. The first step of the Bayesian approach is to define a prior distribution for the response (or non-progression) rate, which summarises the information on the response (or non-progression) rate prior to observing data. It can be a non informative prior (when no or little information is available), or a more informative one, the incorporation of relevant prior information leading to an increased power. Then, the results of experiments are used to update and refine the distribution. Accumulating results can be assessed at any time, including continually, and predictive probabilities are then updated to become more accurate as the trial progresses. The trial continues until the treatment is shown with high posterior probability to be either promising or not promising (pre-defined stopping rules are applied at each interim analysis), or until a predetermined maximum sample size is reached. In the PVNS study, the Bayesian approach will be used with a maximum sample size set to 50 patients. It was determined by calculating the sample size we would have needed in a Fleming design, considering that the minimum clinical benefit non progression rate considered of interest is p1=50%, with an uninteresting rate of p0=30% and assuming a type I error alpha of 0.05 and 90% power. As part of the study, the probability of success can be modelled based on a beta distribution. Thus, the prior distribution will be a Beta(a,b), where a and b are to be defined (cf. the next paragraph for the specification of a and b). The advantage of this distribution is that it simplifies mathematical
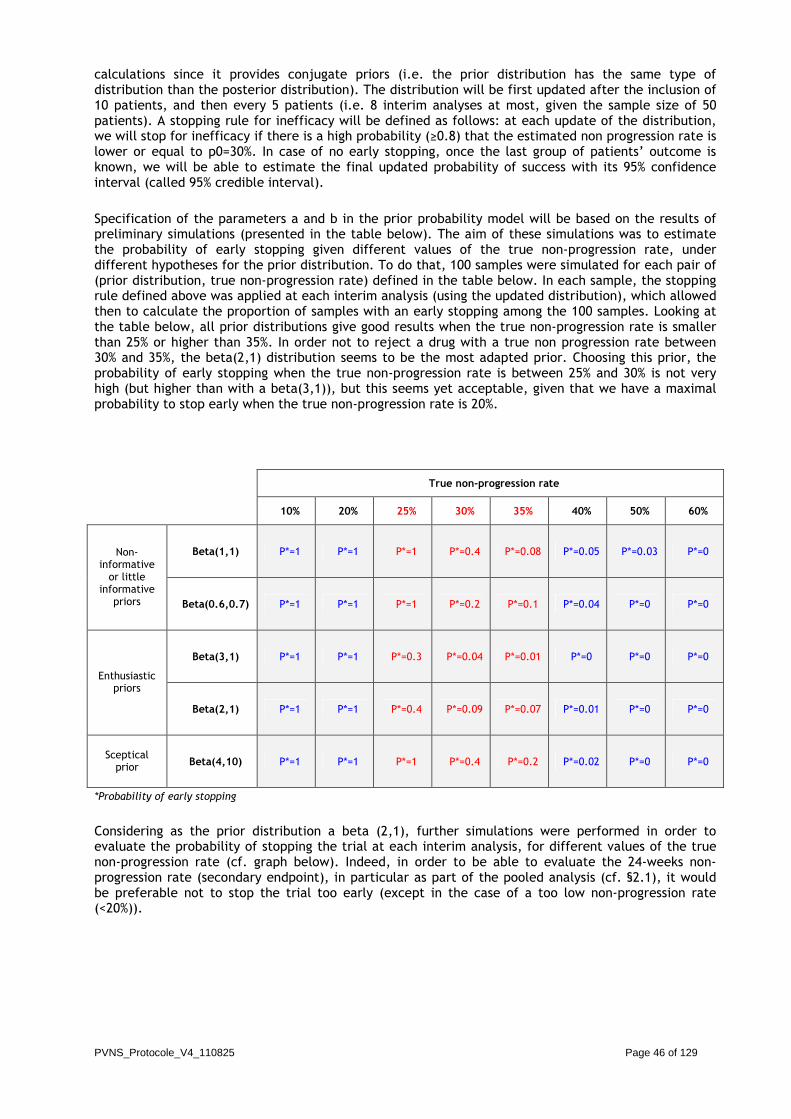
PVNS_Protocole_V4_110825 Page 46 of 129
calculations since it provides conjugate priors (i.e. the prior distribution has the same type of distribution than the posterior distribution). The distribution will be first updated after the inclusion of 10 patients, and then every 5 patients (i.e. 8 interim analyses at most, given the sample size of 50 patients). A stopping rule for inefficacy will be defined as follows: at each update of the distribution, we will stop for inefficacy if there is a high probability (≥0.8) that the estimated non progression rate is lower or equal to p0=30%. In case of no early stopping, once the last group of patients’ outcome is known, we will be able to estimate the final updated probability of success with its 95% confidence interval (called 95% credible interval). Specification of the parameters a and b in the prior probability model will be based on the results of preliminary simulations (presented in the table below). The aim of these simulations was to estimate the probability of early stopping given different values of the true non-progression rate, under different hypotheses for the prior distribution. To do that, 100 samples were simulated for each pair of (prior distribution, true non-progression rate) defined in the table below. In each sample, the stopping rule defined above was applied at each interim analysis (using the updated distribution), which allowed then to calculate the proportion of samples with an early stopping among the 100 samples. Looking at the table below, all prior distributions give good results when the true non-progression rate is smaller than 25% or higher than 35%. In order not to reject a drug with a true non progression rate between 30% and 35%, the beta(2,1) distribution seems to be the most adapted prior. Choosing this prior, the probability of early stopping when the true non-progression rate is between 25% and 30% is not very high (but higher than with a beta(3,1)), but this seems yet acceptable, given that we have a maximal probability to stop early when the true non-progression rate is 20%.
True non-progression rate
10% 20% 25% 30% 35% 40% 50% 60%
Beta(1,1) P*=1 P*=1 P*=1 P*=0.4 P*=0.08 P*=0.05 P*=0.03 P*=0 Non-informative or little
informative priors Beta(0.6,0.7) P*=1 P*=1 P*=1 P*=0.2 P*=0.1 P*=0.04 P*=0 P*=0
Beta(3,1) P*=1 P*=1 P*=0.3 P*=0.04 P*=0.01 P*=0 P*=0 P*=0
Enthusiastic priors
Beta(2,1) P*=1 P*=1 P*=0.4 P*=0.09 P*=0.07 P*=0.01 P*=0 P*=0
Sceptical prior Beta(4,10) P*=1 P*=1 P*=1 P*=0.4 P*=0.2 P*=0.02 P*=0 P*=0
*Probability of early stopping
Considering as the prior distribution a beta (2,1), further simulations were performed in order to evaluate the probability of stopping the trial at each interim analysis, for different values of the true non-progression rate (cf. graph below). Indeed, in order to be able to evaluate the 24-weeks non-progression rate (secondary endpoint), in particular as part of the pooled analysis (cf. §2.1), it would be preferable not to stop the trial too early (except in the case of a too low non-progression rate (<20%)).
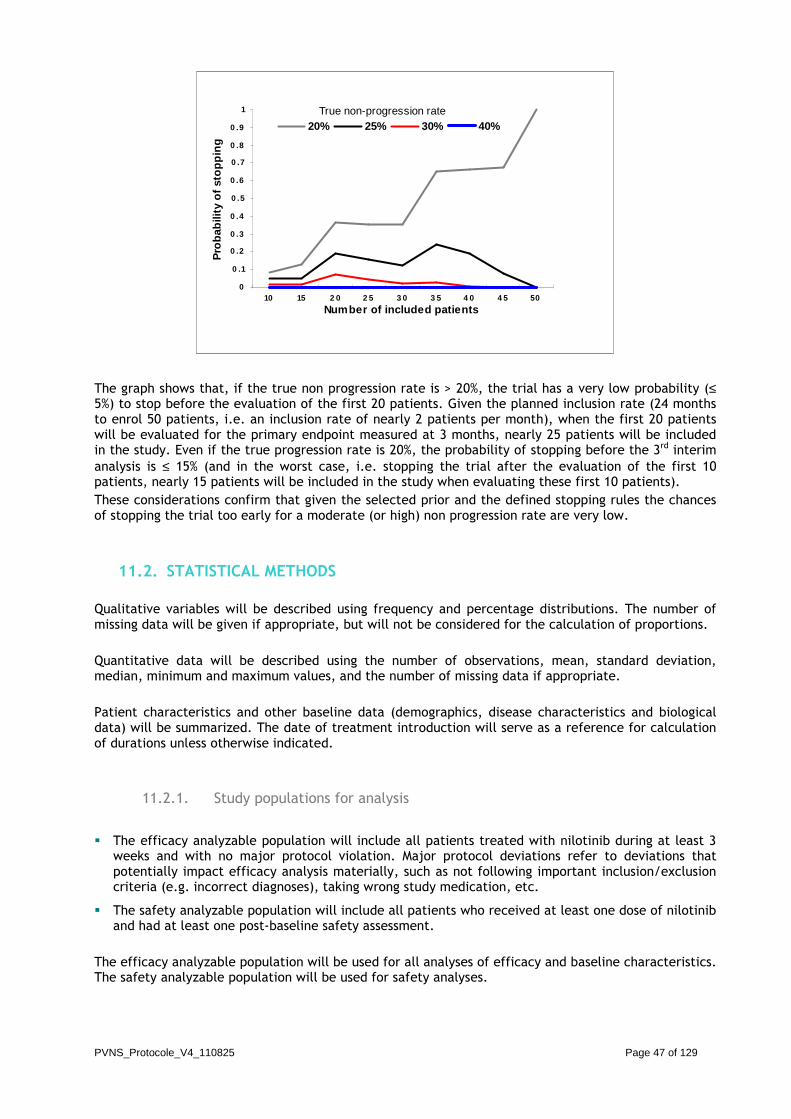
PVNS_Protocole_V4_110825 Page 47 of 129
0
0 .1
0 .2
0 .3
0 .4
0 .5
0 .6
0 .7
0 .8
0 .9
1
10 15 2 0 2 5 3 0 3 5 4 0 4 5 50
Number of included patients
Pro
bab
ility
of
sto
pp
ing
20% 25% 30% 40%True non-progression rate
The graph shows that, if the true non progression rate is > 20%, the trial has a very low probability (≤ 5%) to stop before the evaluation of the first 20 patients. Given the planned inclusion rate (24 months to enrol 50 patients, i.e. an inclusion rate of nearly 2 patients per month), when the first 20 patients will be evaluated for the primary endpoint measured at 3 months, nearly 25 patients will be included in the study. Even if the true progression rate is 20%, the probability of stopping before the 3rd interim analysis is ≤ 15% (and in the worst case, i.e. stopping the trial after the evaluation of the first 10 patients, nearly 15 patients will be included in the study when evaluating these first 10 patients). These considerations confirm that given the selected prior and the defined stopping rules the chances of stopping the trial too early for a moderate (or high) non progression rate are very low.
11.2. STATISTICAL METHODS Qualitative variables will be described using frequency and percentage distributions. The number of missing data will be given if appropriate, but will not be considered for the calculation of proportions. Quantitative data will be described using the number of observations, mean, standard deviation, median, minimum and maximum values, and the number of missing data if appropriate. Patient characteristics and other baseline data (demographics, disease characteristics and biological data) will be summarized. The date of treatment introduction will serve as a reference for calculation of durations unless otherwise indicated.
11.2.1. Study populations for analysis
� The efficacy analyzable population will include all patients treated with nilotinib during at least 3 weeks and with no major protocol violation. Major protocol deviations refer to deviations that potentially impact efficacy analysis materially, such as not following important inclusion/exclusion criteria (e.g. incorrect diagnoses), taking wrong study medication, etc.
� The safety analyzable population will include all patients who received at least one dose of nilotinib and had at least one post-baseline safety assessment.
The efficacy analyzable population will be used for all analyses of efficacy and baseline characteristics. The safety analyzable population will be used for safety analyses.

PVNS_Protocole_V4_110825 Page 48 of 129
11.2.2. Efficacy analysis
11.2.2.1. Primary endpoint analysis Centrally reviewed tumour assessment will be used in the primary efficacy analysis. Tumour response (non progression) rate will be summarized by a proportion together with its 95% confidence interval. The following rules will be applied for the primary endpoint analysis:
� Patient still treated with nilotinib at the time of the evaluation of the primary endpoint (12 weeks after start date of treatment) and for whom no progression is documented at the 12-week tumour assessment will be considered as “success” (non progressive)
� Patient having stopped nilotinib before the 12-week visit without having started another 2nd line treatment after the stop, and for whom no progression is documented within the 12 weeks after the start of nilotinib will be considered as “success” (non progressive)
� Patient still treated with nilotinib at the time of the evaluation of the primary endpoint (12 weeks after start date of treatment) and for whom a progression is documented at the 12-week tumour assessment will be considered as “failure” (progressive)
� Patient having stopped nilotinib before the 12-week visit and : having started another 2nd line treatment after the stop or for whom a progression is documented within the 12 weeks after the start of treatment will be considered as “failure” (progressive)
� When the investigator’s assessment of the response after 12 weeks of treatment will be available but not validated by the central review committee, the evaluation made by the investigator will be taken into account
11.2.2.2. Secondary endpoints analysis The analysis of the non progression rate after 24 weeks of treatment (key secondary endpoint) will be based on the central read tumour assessment. The 24-week non progression rate will be summarized by a proportion together with its 95% confidence interval. The rules defined for the primary endpoint analysis (cf. above), once adapted with a 24 weeks time-point, will be applied for the analysis of this key secondary endpoint. Objective tumour response rate after 12 weeks of treatment will be determined based on the central read tumour assessment. It will be summarized by a proportion together with its 95% confidence interval. The best overall response obtained during the study will be determined based on the central read tumour assessment and will be described by frequencies and percentages. Progression-free survival (PFS), time to progression (TTP), time to treatment failure (TTF) and duration of response will be analyzed based on the central read tumour assessment, and will be estimated as a function of time by the Kaplan-Meier method. PFS will be measured from the date of treatment onset to the date of event defined as the first documented progression or death due to any cause. Patients with no event at the time of the analysis will be censored at the date of the last adequate tumour assessment. TTP will be measured from the date of treatment onset to the date of event defined as the first documented progression or death due to underlying disease. Patients with no event at the time of the analysis will be censored at the date of the last adequate tumour assessment. TTF will be measured from the date of treatment onset to the earliest date of progression, date of death due to any cause, or date of discontinuation due to reasons other than ‘protocol deviation’ or

PVNS_Protocole_V4_110825 Page 49 of 129
‘administrative problems’. Patients without treatment failure at the time of the analysis will be censored at the date of the last adequate tumour assessment. Duration of response will apply only to patients with an objective tumour response after 12 weeks of treatment. It will be measured from the date of documented objective response (CR or PR) to the date of event defined as the first documented progression or death due to underlying disease. Patients with no event at the time of the analysis will be censored at the date of last adequate tumour assessment. The relationship between the objective tumour response and tumour characteristics will be tested using univariate analyses.
11.2.3. Safety analysis The assessment of safety will be based mainly on the frequency of adverse events based on the common toxicity criteria (CTC-V3) grade. Descriptive statistics will be provided for characterizing and assessing patient tolerance to treatment. Adverse events will be coded according to the MedDRA®.
11.2.4. Modifications of the statistical analysis planned in the protocol If applicable, substantial modifications of the statistical methods described herein will be described in the appropriate part of the final study report (section 9.8 Changes in the conduct of the study or planned analyses).
11.2.5. Interim analysis See section §11.1.
11.2.6. Blind code break Not applicable.

PVNS_Protocole_V4_110825 Page 50 of 129
12. ACCESS TO SOURCE DATA AND DOCUMENTS
To check completeness and reliability of the data compared to patients files, sponsor authorised persons as well as authorities representatives will have access to source documents, including patient’s medical history. This access will be clearly stated in the informed consent form and will be allowed by the patient’s signature. The investigators commit themselves to let the sponsor and the health authorities’ representative have a direct access to source data and source documents. The investigator will grant the CRA access to the subjects' personal medical records for the verification of the proper documentation of study data. French regulations on data protection will be fully observed (the CRA is bound by medical secrecy when comparing the CRFs with the source documents). The investigator will grant the CRA access to the source documents for the fulfilment of his/her duty.

PVNS_Protocole_V4_110825 Page 51 of 129
13. STUDY MANAGEMENT
13.1. COORDINATING CENTRE Project management will be performed by the UBET, CLB.

PVNS_Protocole_V4_110825 Page 52 of 129
14. DATA QUALITY CONTROL
14.1. MONITORING The sponsor will perform the study monitoring and will help the investigator to conduct the study in compliance with the protocol, Good Clinical Practices and local law requirements. The investigator will be available for contacts and visits of the sponsor or its representative regularly at appropriate intervals. During these visits, eCRF and source documents will be checked for completeness and accuracy. No document allowing the identification of the patient will be transmitted outside the study centre. Monitoring visits will also allow analyzing the study progress, to check the existence of signed informed consent, the compliance of patients with inclusion and non-inclusion criteria and serious adverse events source documents. The CRA will have to check the compliance of eCRF data with source documents, as specified in the GCP guidelines. He will ask the investigator to modify any wrong, forgotten or illegible data. All the modifications will be explained (if necessary), dated and signed. If the data are modified by another person than the investigator, the authorisation of this person will be documented. The study will be monitored to verify, in particular:
� The rights and well-being of the trial subjects are protected
� The reported trial data are accurate, complete and verifiable from source documents
� The conduct of the clinical trial is in compliance with the currently approved protocol / the currently approved protocol amendment(s), with ICH-GCP and with applicable regulatory requirements
All investigators agree that the CRA will visit the centre before, during and after completion of the study. The investigator must allow sufficient time for these visits, alternatively the CRA may be provided with other trained staff for assistance during the visits. The investigator will grant the CRA access to the source documents for the fulfilment of his/her duty. The aim and purpose of these visits is, in particular:
� Evaluation of the progress of the study
� Check for compliance with the trial protocol
� Discussion of problems including AEs
� eCRF review for accuracy and completeness
� eCRF validation against source data
� Verification of proper handling and dispensing of the investigational medicinal product A monitoring report will be written on each visit. This will document the progress of the clinical trial and give an account of all problems that occurred (e.g. refusal of inspection). Patient’s medical files should contain at least the following data:
� Inclusion of the patient in the study, including study reference, patient identification number and initials, the treatment number (batch number) and informed consent signature modalities (draft number, date)
� Details about PVNS/TGCT diagnosis and history, medical history and concomitant diseases
� All treatments stopped because of the inclusion in the present study, ongoing or initiated during the study
� Dates of the visits of the study
� All adverse events that occurred during the study

PVNS_Protocole_V4_110825 Page 53 of 129
Source data as defined by ICH-GCP include data such as hospital records, clinical and office charts, laboratory notes, memoranda, subjects' diaries or evaluation checklists, pharmacy dispensing records, recorded data from automated instruments, copies or transcriptions certified after verification as being accurate copies, microfiches, photographic negatives, microfilm or magnetic media, x-rays, and records kept at the pharmacy, at the laboratories and at medico-technical departments involved in the clinical trial.
14.2. DATA ENTRY AND DATA MANAGEMENT Data entry will be performed by investigators on the eCRF. A print out of the electronic sheet must be available at any stage of the study. A printed final copy of the eCRF of each patient will be signed by the Investigator and filed at the Centre. All the data will be computerised to detect missing, out of range and inconsistent data. Self evident corrections will be performed by the data manager and documented according to a validated data management plan. Queries will be made for wrong and missing data. Data clarification forms will be sent to the investigators and will be completed in compliance with source data, initialled, dated and sent back to the UBET in charge of the data management for data entry. A patient file will be validated after no more inconsistency is detected by the program. All data base modifications will be recorded in the audit trail. The database will be submitted to a quality control. Concomitant treatments codification will be performed according to WHO Drug. Adverse events codification will be performed according to MedDRA®. The database will be locked after all queries are solved, and after data review and final validation. Computerized management of collected data will be performed by the clinical research technician (UBET – CLB), using specific tools developed by the CLININFO SA society. The sponsor is the owner of all collected data and reports.
14.3. CENTRAL REVIEW COMMITTEE The central review committee will be composed of independent radiologists and/or oncologists. The presence of the study investigators is optional. This committee will meet to review all radiological assessments in order to evaluate the response according to the RECIST criteria (RECIST version 1.1), and to validate or not the assessment previously made by the investigator. The necessary examinations for this committee are: � CT scan and physical examination at inclusion � CT scan and physical examination at 12 weeks � CT scan and physical examination once the completion of follow up (including the 24 weeks
examination) � CT scan and physical examination between inclusion and W12 are optional These examinations will be sent as soon as possible to the coordinating centre.

PVNS_Protocole_V4_110825 Page 54 of 129
14.4. AUDITS AND INSPECTIONS Audits and inspections may be carried out by the sponsor's quality assurance department or its designee, local authorities, or authorities to whom information on this trial has been submitted. All documents pertinent to the trial must be made available for such inspection after an adequate announcement. Informed consent of patients participating in this study has to include the consent in this access to source documents.

PVNS_Protocole_V4_110825 Page 55 of 129
15. ETHICS
15.1. INDEPENDENT ETHICS COMMITTEE (IEC) AND INSTITUTIONAL REVIEW BOARD (IRB)
The final version of the study protocol, including the final version of the informed consent form, have to received the written approval of the IEC/IRB and competent authority (AFSSAPS in France) before any patient's inclusion in the study. The IEC/IRB and competent authority will be informed of any protocol amendment significantly modifying the study, especially regarding security and study objectives, in accordance with local legislation. A copy of the written approvals will be attached to the protocol.
15.2. ETHICAL CONDUCT OF THE STUDY – GOOD CLINICAL PRACTICE This study will be performed in compliance with the ethical principles of the World Medical Association Declaration of Helsinki (version 2004), as well as the GCP, the local laws and the bioethics policy of the sponsor. The study will be conducted in agreement with:
� The ethical principles of the Declaration of Helsinki
� Good Clinical Practice guidelines issued by the International Conference on harmonisation (ICH-E6, 17/07/96)
� European Directive (2001/20/EC) of April 4th 2001 on clinical trial conduct
� French Public Health Law (n°2004-806) of August 9th 2004
� French law "Loi Informatique et Libertés n°78-17" of January 6th 1978 modified by the law n°2004-801 of August 6th 2004 related to the protection of individuals with regard to the processing of personal data
� French bioethics Law n°2004-800 of August 6th 2004 All the documentation concerning the study has to be sent to the investigator before the study begins for him to confirm his consent to participate in the study. All the study documents will be strictly confidential. The investigator and his staff will be committed to use the study documents exclusively in the scope of the study protocol. They will be bound to confidentiality until the data from the study are published by the sponsor.
15.3. PATIENT INFORMATION AND CONSENT The patient’s informed consent form will be made of 2 original copies. The signed forms will be collected by the investigator before the inclusion of the patient in the study. One copy will be for the patient and one copy for the investigator. The investigator will have to explain and discuss with the patient the study objectives, the potential benefits and risks of the treatment and data confidentiality concerns. Patient will have time to ask all the questions they need to and to consider all the consequences before signing the informed consent.

PVNS_Protocole_V4_110825 Page 56 of 129
He should be clearly informed that he will be able to withdraw the study at any time without any consequence for the management of his disease. The information notice is presented in appendix 7. An informed consent form is presented in appendix 8. Informed consent form should follow local regulations.
15.4. AGENCE FRANCAISE DE SECURITE DES PRODUITS DE SANTE (AFSSAPS) This study received an authorization by AFSSAPS on the 2010/07/29 (Appendix 21).
15.5. COMITE DE PROTECTION DES PERSONNES This study received a favourable approval by the CPP Sud-Est IV on the 2010/06/08 (Appendix 20).
15.6. COMITE CONSULTATIF SUR LE TRAITEMENT DE L’INFORMATION EN MATIERE DE RECHERCHE DANS LE DOMAINE DE LA SANTE (CCTIRS) – COMMISSION NATIONALE DE L’INFORMATIQUE ET DES LIBERTES (CNIL)
This study already has the authorization of the Comité Consultatif sur le Traitement de l’Information en matière de Recherche dans le domaine de la Santé (CCTIRS) – Commission Nationale de l’Informatique et des Libertés (CNIL): Record Number: 00.S.018 for CCTIRS, and 00-1142 for CNIL.
15.7. SPONSOR RESPONSIBILITY The sponsor is the natural person or legal entity, who initiates the biomedical research on human being, provides the management and check for financing. Sponsor responsibility is:
� To subscribe an insurance (Appendix 19) (except for non-European countries)
� To register the trial and get a EudraCT number
� To submit the protocol to the CPP approval (Appendix 20)
� To submit the protocol to the competent authority AFSSAPS approval (Appendix 21)
� To declare the medical and computerized data collection to the CCTIRS
� To inform directors, investigators and pharmacists in involved centres of the research
� To declare to the competent authority the start and the end of the study
� To send to investigators all information necessary to the research
� To declare to EMEA (Eudravigilance), competent authority (AFSSAPS), CPP and principal investigators each unexpected serious adverse event suspicion in link with the experimental drug
� To communicate the annual security report to the competent authority (AFSSAPS), CPP and principal investigators
� To elaborate the final report of the study
� To inform the competent authority (AFSSAPS), CPP and participants of the results
� To store study main documents during the minimal length as defined by the Good Clinical Practices, i.e. 15 years after the end of the research

PVNS_Protocole_V4_110825 Page 57 of 129
15.8. INVESTIGATOR REPONSIBILITY The investigator is responsible for the organization and quality of the study, notably regarding protocol compliance, data collection and, more generally, Good Clinical Practices. Sponsor, CPP and competent authority approval are required for any modification. Under his responsibility, clinical research technicians provide administrative and logistic support to the study; assist in the auditing of source records and the documentation of CRFs. Principal investigator responsibilities are:
� To send his and others investigators’ signed curriculum vitae with inscription number to the Conseil National de l’Ordre des Médecins (CNOM) to the sponsor
� To identify members of his staff involved in the research and to define their responsibilities
� To start recruitment after sponsor authorization,
� To try to enrol the required number of patient during the defined period of recruitment
� Investigators responsibilities are
- To collect written informed consent form before enrolment of patients
- To make sure that organization and quality of the study are good, notably regarding protocol compliance, data collection and, more generally, Good Clinical Practices, he can be helped by the TRC who perform controls on CRFs
- To complete CRF for each enrolled patient
- To validate collected data in the CRFs
- To date, correct and sign corrections in the CRFs
- To transmit the CRFs to the coordinating centre
- To notify to the sponsor immediately each serious adverse event, excepted those mentioned on the protocol as not requiring immediate notification
- To accept potential visits of control by inspectors mandated by the sponsor or the competent authority
- To make sure that patients anonymity and confidentiality is preserved
- To store data and identification of enrolled patients in accordance with current legislation, for a minimal length of 15 years after the end of the study

PVNS_Protocole_V4_110825 Page 58 of 129
16. PROCESSING OF DATA AND STUDY DOCUMENTS ARCHIVING
Personal patient data will be kept confidential: eCRF or other documents submitted to the sponsor will identify a patient by initials (1 initial for the first name and 1 initial for the last name) and number only. Each investigator will keep in his file a screening Log and a patient identification list (including complete name and date of birth of each patient). To allow compliance with GCP principles, each patient will be asked for consent regarding the access to source documents for monitoring, audits, and inspections. The information notice and the informed consent form have to explain that the study data will be kept in a electronic form, confidentially, in accordance with the French law N°78-17. All the computerised data will be identified by "centre n°/ patient n°/Study code/patient initials". The information notice and the informed consent form will specify that the sponsor representative and the competent authorities will have direct access upon request to the source documents related to the study, including medical antecedents, to check the accuracy of the data. Study documents and records will be kept for at least 15 years after the end of the study by the investigator: this includes codes allowing the identification of patients, medical files and all the source data, the eCRF print out, the queries, the patient's informed consent, the copies of IEC/IRB and competent authorities approval and any other document pertinent to the study. All other documentation relative to the trial (protocol, investigators’ folders, etc…) as well as all other original documents (laboratory results, radiographic pictures, reports of physician-patient consultations and physical examinations, etc…) are confidential material and must be kept in a secured location.

PVNS_Protocole_V4_110825 Page 59 of 129
17. OTHER CONSIDERATIONS
17.1. CASE REPORT FORM All the data concerning the patients will be recorded in the eCRF all along the study. Only the investigator and authorised staff could complete the eCRF which will be filled up online.
17.2. AMENDMENTS TO THE PROTOCOL Any modification of the protocol has to be agreed by the coordinating investigator and the sponsor in the form of a written amendment. Any amendment which modifies significantly the patient's security, the objectives of the study or its scientific quality have to be approved before they could be used.
17.3. FUNDING AND INSURANCE The study will be partially funded by Novartis. Nilotinib will be provided by Novartis for a dose of 400 mg twice a day for 1 year. The sponsor has subscribed an insurance policy in compliance with local laws covering its responsibility for all the participants for any injury that might be caused by the trial (Appendix 19) (except for non-European countries).

PVNS_Protocole_V4_110825 Page 60 of 129
18. PUBLICATION AND FINAL STUDY REPORT
A report will be prepared under the responsibility and according to the standards of the sponsor, provided that all eCRFs have been completed. It will include the study objectives, the methodology, statistical analysis and raw data listings, and the conclusions of the study. It will also include all the AE that occurred during the study and data concerning all the patients included in the study. It will be submitted to the coordinating investigator for acknowledgement and signature. The study report will be prepared and will be available for health authorities within one year after the end of the study. Also, a summary of the study will be sent to IEC/IRB and health authorities. The manuscript of the publication will be prepared within the 6 months following the publication of the final study report by the principal investigator, or upon agreement, by an investigator (selected by ranks of accrual). Investigators are informed that the sponsor reserves all rights to data generated from this study. Written approval from the sponsor must be obtained prior to any publication or presentation of data from this study. The sponsor is not allowed to use investigator's name in any publication without a prior written consent. The investigator is not allowed to use sponsor's name in any publication without a prior written consent. The principal investigator agrees to publish the results. It is mandatory that no publication can be done without the principal investigator and the UBET approval; the sponsor and Novartis support will be mentioned. However, the final decision for the publication of the study will be taken by the principal investigators, statisticians and the sponsor. Any publication or communication (oral or written) will be defined by mutual agreement between the investigators according to international guidelines (http://www.icmje.org/). All the authors who participated actively to the conception of the study, its development and writing of results will be cited, i.e.:
� The principal investigator, the co-investigator, and all investigators who have included and followed patients. The order of citation will be established according to the number of inclusions in the study.
� The contributors of the coordinating centre team (UBET) who participated in the drafting of the protocol.
� The Centre Léon Bérard will be cited as Sponsor of the study.
� Novartis and all the funders will be included in the list of acknowledgments.

PVNS_Protocole_V4_110825 Page 61 of 129
19. REFERENCES
1. Mendenhall WM, Mendenhall CM, Reith JD, Scarborough MT, Gibbs CP, Mendenhall NP. Pigmented
villonodular synovitis. Am J Clin Oncol. 2006; 29: 548-50. 2. Martin RC 2nd, Osborne DL, Edwards MJ, Wrightson W, McMasters KM. Giant cell tumour of tendon
sheath, tenosynovial giant cell tumour, and pigmented villonodular synovitis: defining the presentation, surgical therapy and recurrence. Oncol Rep. 2000 Mar-Apr; 7(2):413-9.
3. West RB, Rubin BP, Miller MA, Subramanian S, Kaygusuz G, Montgomery K, Zhu S, Marinelli RJ, De
Luca A, Downs-Kelly E, Goldblum JR, Corless CL, Brown PO, Gilks CB, Nielsen TO, Huntsman D, van de Rijn M. A landscape effect in tenosynovial giant-cell tumour from activation of CSF1 expression by a translocation in a minority of tumour cells. Proc Natl Acad Sci U S A. 2006; 103: 690-5.
4. Cupp JS, Miller MA, Montgomery KD, Nielsen TO, O'Connell JX, Huntsman D, van de Rijn M, Gilks
CB, West RB. Translocation and expression of CSF1 in pigmented villonodular synovitis, tenosynovial giant cell tumour, rheumatoid arthritis and other reactive synovitides. Am J Surg Pathol. 2007; 31:970-6.
5. Dewar AL, Cambareri AC, Zannettino AC, Miller BL, Doherty KV, Hughes TP, Lyons AB. Macrophage
colony-stimulating factor receptor c-fms is a novel target of imatinib. Blood. 2005; 105:3127-32. 6. Blay JY, El Sayadi H, Thiesse P, Garret J, Ray-Coquard I. Complete response to imatinib in relapsing
pigmented villonodular synovitis/ tenosynovial giant cell tumour (PVNS/TGCT). Ann Oncol. 2008 Apr; 19 (4):821-2.
7. P. A. Cassier, S. Stacchiotti, H. Gelderblom, D. M. Thomas, W. Van Der Graaf, B. M. Seddon, D.
Julien, A. J. Wagner, J. Blay. Imatinib mesylate for the treatment of locally advanced and/or metastatic pigmented villonodular synovitis/tenosynovial giant cell tumor (PVNS/TGCT). J Clin Oncol. 2010 (suppl; abstr 10012); 28:15s. Meeting: 2010 ASCO Annual Meeting Abstract No: 10012
8. V. Ravi, W. Wang, D. M. Araujo, J. A. Ludwig, R. J. Luke, V. O. Lewis, J. C. Trent, R. S. Benjamin,
S. Patel. Imatinib in the treatment of tenosynovial giant-cell tumor and pigmented villonodular synovitis. J Clin Oncol. 2010 (suppl; abstr 10011); 28:15s. Meeting: 2010 ASCO Annual Meeting Abstract No: 10011
9. Brownlow N, Russell AE, Saravanapavan H, Wiesmann M, Murray JM, Manley PW, Dibb NJ.
Comparison of nilotinib and imatinib inhibition of FMS receptor signalling, macrophage production and osteoclastogenesis. Leukaemia. 2008 Mar; 22 (3):649-52.

PVNS_Protocole_V4_110825 Page 62 of 129
20. APPENDICES
Appendix 1 NCI-CTCAE Classification V.04 p 63
Appendix 2 RECIST criteria (RECIST version 1.1) p 64
Appendix 3 Cockcroft and Gault formula p 65
Appendix 4 Contraindicated medications p 66
Appendix 5 Adverse event – Causality assessment p 72
Appendix 6 Adverse event – Serious Adverse Event report form p 73
Appendix 7 Information notice (French and English version) p 74
Appendix 8
Informed consent form (French and English version) Addendum to patient information sheet – consent form (French and English version)
p 88
Appendix 9 Inclusion form p 96
Appendix 10 Inclusion procedure p 98
Appendix 11 Treatment supply to the investigational centre - procedure p 99
Appendix 12 Treatment supply form (French and English version) p 100
Appendix 13 Procedure – Bioassay of nilotinib p 102
Appendix 14 Procedure – Bioassay of nilotinib Sample shipment form
p 103
Appendix 15
Procedure – Bioassay of nilotinib Sample shipment procedure (France, Italy, The Netherlands, Poland, United Kingdom)
p 104
Appendix 16 Procedure – Exploratory analysis p 109
Appendix 17 Procedure – Exploratory analysis Tumor sample shipment form
p 110
Appendix 18 Procedure – Exploratory analysis Tumor sample shipment procedure
p 111
Appendix 19 Insurance attestation (France) p 112
Appendix 20 CPP approval p 113
Appendix 21 AFSSAPS authorization p 118
Appendix 22 Group specific appendix for each country: additional information about trial conduct in foreign country (Competent authority Authorization, Ethic committee approval, insurance attestation…)
p 120

PVNS_Protocole_V4_110825 Page 63 of 129
APPENDIX 1 NCI-CTCAE Classification V4.0 The CTCAE toxicity scale can be viewed electronically or downloaded and printed by visiting the following website:
http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0 Published: May 28, 2009 (v4.03: June 14, 2010)

PVNS_Protocole_V4_110825 Page 64 of 129
APPENDIX 2 RECIST Criteria (RECIST version 1.1) E. Eisenhauer, P. Therasse, J. Bogaerts, L. Schwartz, D. Sargent, R. Ford, J. Dancey, S. Arbuck, S. Gwyther, M. Mooney. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). European Journal of Cancer, 2009 Jan, Volume 45, Issue 2, Pages 228-247

PVNS_Protocole_V4_110825 Page 65 of 129
APPENDIX 3 Cockcroft and Gault formula Cockcroft and Gault formula:
CrCl (ml/min) = [(140 – age (year)) x weight (kg)] / (serum creatinine (mg/ml) x 72) (x 0.85 for females) CrCl: creatinine clearance

PVNS_Protocole_V4_110825 Page 66 of 129
APPENDIX 4 Contraindicated medications Interaction with other medicinal products and other forms of interaction Nilotinib is mainly metabolised in the liver and is also a substrate for the multi-drug efflux pump, P-glycoprotein (P-gp). Therefore, absorption and subsequent elimination of systemically absorbed nilotinib may be influenced by substances that affect CYP3A4 and/or P-gp. Nilotinib is a substrate for CYP3A4 and is a competitive inhibitor of CYP3A4. Nilotinib should be used with caution in patients who have or may develop prolongation of QT. Indicative list of contraindicated medications:

PVNS_Protocole_V4_110825 Page 67 of 129
List of inducers, inhibitors and substrates of P-glycoprotein ((not exhaustive list) Please refer to the following website: http://www.genemedrx.com/PGPtable.php
SUBSTRATES INHIBITORS INDUCERS
actinomycin D alfentanil amitriptyline
aldosterone amiloride amprenavir
alpha-methyldigoxin amiodarone ASA
amiloride amitripyline bromocriptine
amitriptyline astemizole chlorambucil
amoxicillin atovaquone (modest) cisplatin
amprenavir atorvastatin clotrimazole
atorvastatin azelastine colchicine
beta-acetyldigoxin azidopine cyclosporine
bisantrene azithromycin delavirdine
bunitrolol bepidil daunorubicin
carbamazepine biricodar dexamethasone
celiprolol bromocriptine doxorubicin
cetirizine carbamazepine efavirenz
chloroquine Watkins carvedilol erythromycin
chlorpromazine chloroquine etoposide
cimetidine chlorpromazine flurouracil
citalopram clarithromycin hydroxyurea
ciprofloxacin cyclosporin insulin
colchicine cyproheptadine lopinavir-chronic
corticosteroids darunavir methotrexate
cortisol desethylamiodarone mitoxantrone
cyclosporine desipramine morphine
dexamethasone dexniguldipine nefazodone
daunorubicin dexrazoxane nelfinavir
debrisoquine diltiazem nevirapine
dexamethasone dipyridamole nicardipine
digoxin disulfiram nifedipine
digitoxin doxazosin paclitaxel
diltiazem elicridqr phenobarbital
dipyridamole emetine phenothiazines
docetaxel erythromycin phenytoin
domperidone felodipine prazosin
doxepine fenofibrate probenecid
doxorubicin fentanyl reserpine
doxycycline flavonoids retinoid acid
emetine fluoxetine ritonavir (chronic)
enoxacin fluphenazine rifampin
epirubicin fluvoxamine St. John's Wort
erythromycin fucidin tacrolimus
estradiol gallpamil tamoxifen
etoposide glyburide trazodone
fexofenadine gramicidin D verapamil
glyburide grapefruit juice vinblastine
gramicidin D garlic vincristine
grepafloxacin green tea yohimbine

PVNS_Protocole_V4_110825 Page 68 of 129
hydrocortisone haloperidol
imatinib hydrocortisone
indinavir hyroxyzine
irinotecan josamycin
itraconazole ketoconazole
ivermectin imipramine
ketoconazole itraconazole
LAAM ivermectin
L-dopa ketoconazole
lansoprazole laniquidar
levofloxacin lansoprazole
loperamide levothyroxin
lopinavir lidocaine
losartan loperamide
lovastatin lopinavir-acute
maraviroc loratadine
methylprednisolone lovastatin
mibefradil maprotiline
mithramycin mefloquine
mitomycin C methadone
mitoxantrone mibefradil
morphine midazolam
nelfinavir mitomycin C
nicardipine nefazodone
nifedipine nelfinavir
nizatidine nicardipine
nortriptyline nitrendipine
olanazpine nobilitin
ondansetron norverapamil
paclitaxel omeprazole
paroxetine orange juice-Seville
pentazocaine ofloxacin
phenobarbital paroxetine
phenytoin pantoprazole
prednisolone phenothiazines
progesterone piperine
puromycin Watkins pimozide
quetiapine probenecid
quinidine progesterone
ranitidine promethazine
rapamycin propafenone
rifampin propranolol
risperidone quercetin
ritonavir quinacrine
saquinavir quinidine
sertraline quinine
sirolimus reserpine
sparfloxacin ritonavir (initial)
tacrolimus saquinavir
talinolol sertraline
tamoxifen simvastatin

PVNS_Protocole_V4_110825 Page 69 of 129
taxol spironolactone
teniposide sufentanil
terfenadine tacrolimus
tetracycline tamoxifen
topiramate tariquidar
topotecan telithromycin
triamcinolone terfenadine
trimipramine testosterone
valinomycin tetrabenzine
valspodar thioridazine
vecuronium trifluoperazine
venlafaxine trifluopromazine
verapamil trimipramine
vinblastine valinomycin
vincristine vanadate
vindesine (venlafaxine)
vinorelbine verapamil
FK 506 vinblastine
FK 506
RU(mifepristone)
Valspodar PSC 833
zosuquidar
n-propylquinoline

PVNS_Protocole_V4_110825 Page 70 of 129
List of substrates, inhibitors and inducers of CYP3A4 ((not exhaustive list) Please refer to the following website: http://medicine.iupui.edu/flockhart/table.htm
SUBSTRATES INHIBITORS INDUCERS
Macrolide antibiotics:
Clarithromycin erythromycin NOT azithromycin Telithromycin
Anti-arrhythmics:
quinidine→3-OH
Benzodiazepines:
Alprazolam diazepam→3OH midazolam triazolam
Immune Modulators:
Cyclosporine tacrolimus (FK506)
HIV Antivirals:
Indinavir Ritonavir Saquinavir
Prokinetics:
Cisapride
Antihistamines:
Astemizole Chlorpheniramine
Calcium Channel Blockers:
Amlodipine Diltiazem Felodipine nifedipine nisoldipine nitrendipine verapamil
HMG CoA Reductase Inhibitors:
Atorvastatin Lovastatin NOT pravastatin NOT rosuvastatin Simvastatin
Others:
Aripiprazole Buspirone Gleevec Haloperidol Methadone Pimozide Quinine Sildenafil Tamoxifen Trazodone vincristine
HIV Antivirals: indinavir nelfinavir ritonavir clarithromycin itraconazole ketoconazole nefazodone erythromycin grapefruit juice verapamil diltiazem cimetidine amiodarone NOT azithromycin Fluvoxamine Mibefradil troleandomycin
Carbamazepine phenobarbital phenytoin pioglitazone rifabutin rifampin St. John's wort troglitazone

PVNS_Protocole_V4_110825 Page 71 of 129
List of drugs that prolong the QT Interval (not exhaustive list)
Please refer to the following website: http://www.hrspatients.org/patients/heart_disorders/long_qt_syndrome.asp
Drug and Brand Names Drug Class (Clinical Use)
Arsenic trioxide (Trisenox®) Anti-cancer/Leukemia
Bepridil (Vascor®) Anti-anginal/heart pain
Chlorpromazine (Thorazine®) Anti-psychotic/Anti-emetic/schizophrenia/nausea
Cisapride (Propulsid®) GI stimulant/heartburn
Clarithromycin (Biaxin®) Antibiotic/bacterial infection
Disopyramide (Norpace®) Anti-arrhythmic/abnormal heart rhythm
Dofetilide (Tikosyn®) Anti-arrhythmic/abnormal heart rhythm
Dolasetron (Anzemet®) Anti-nausea/nausea, vomiting
Droperidol (Inapsine®) Sedative; Anti-nausea/anesthesia adjunct, nausea
Erythromycin (E.E.S.® ,Erythrocin®) Antibiotic;GI stimulant/bacterial infection; increase GI motility
Felbamate (Felbatrol®) Anti-convulsant/seizure
Fluoxetine (Prozac®,Sarafem®) Anti-depressant/depression
Foscarnet (Foscavir®) Anti-viral/HIV infection
Fosphenytoin (Cerebyx®) Anti-convulsant/seizure
Gatifloxacin (Tequin®) Antibiotic/bacterial infection
Halofantrine (Halfan®) Anti-malarial/malaria infection
Haloperidol (Haldol®) Anti-psychotic/schizophrenia, agitation
Ibutilide (Corvert®) Anti-arrhythmic/abnormal heart rhythm
Indapamide (Lozol®) Diuretic/stimulate urine & salt loss
Isradipine (Dynacirc®) Anti-hypertensive/high blood pressure
Levofloxacin (Levaquin®) Antibiotic/bacterial infection
Levomethadyl (Orlaam®) Opiate agonist/pain control, narcotic dependence
Mesoridazine (Serentil®) Anti-psychotic/schizophrenia
Moexipril/HCTZ (Uniretic®) Anti-hypertensive/high blood pressure
Moxifloxacin (Avelox®) Antibiotic/bacterial infection
Naratriptan (Amerge®) Serotonin receptor agonist/Migraine treatment
Nicardipine (Cardene®) Anti-hypertensive/high blood pressure
Octreotide (Sandostatin®) Endocrine/acromegaly, carcinoid diarrhea
Paroxetine (Paxil®) Anti-depressant/depression
Pentamidine (NebuPent®,Pentam®) Anti-infective/pneumocystis pneumonia
Pimozide (Orap®) Anti-psychotic/Tourette's tics
Procainamide (Procan® ,Pronestyl®) Anti-arrhythmic/abnormal heart rhythm
Quetiapine (Seroquel®) Anti-psychotic/schizophrenia
Quinidine (Cardioquin®, Quiniglute®) Anti-arrhythmic/abnormal heart rhythm
Risperidone (Risperdal®) Anti-psychotic/schizophrenia
Salmeterol (Serevent®) Sympathomimetic/asthma, COPD
Sertraline (Zoloft®) Anti-depressant/depression
Sotalol (Betapace®) Anti-arrhythmic/abnormal heart rhythm
Sparfloxacin (Zagam®) Antibiotic/bacterial infection
Sumatriptan (Imitrex®) Serotonin receptor agonist/Migraine treatment
Tacrolimus (Prograf®) Immunosuppressant/Immune suppression
Tamoxifen (Nolvadex®) Anti-cancer/breast cancer
Thioridazine(Mellaril®) Anti-psychotic/schizophrenia
Tizanidine (Zanaflex®) Muscle relaxant
Venlafaxine (Effexor®) Anti-depressant/depression
Ziprasidone(Geodon®) Anti-psychotic/schizophrenia
Zolmitriptan (Zomig®) Migraine treatment
Adapted with permission from the Arizona Center for Education and Research on Therapeutics (www.ArizonaCERT.org)

PVNS_Protocole_V4_110825 Page 72 of 129
APPENDIX 5 Adverse event – Causality assessment CERTAIN
A clinical event occurring in a plausible time relationship to drug administration, and which cannot be explained by concurrent disease or other drugs or chemicals. The response to withdrawal of the drug (dechallenge) should be clinically plausible. The event must be definitive pharmacologically or using a satisfactory rechallenge procedure if necessary PROBABLE
A clinical event with a reasonable time sequence to administration of the drug, unlikely to be attributed to concurrent disease or other drugs or chemicals, and which follows a clinically reasonable response on withdrawal (dechallenge). Rechallenge information is not required to fulfil this definition POSSIBLE
A clinical event with a reasonable time sequence to administration of the drug, but which could also be explained by concurrent disease or other drugs or chemicals. Information on drug withdrawal may be lacking or unclear UNLIKELY
A clinical event with a temporal relationship to drug administration which makes a causal relationship improbable, and in which other drugs, chemicals or underlying disease provide plausible explanations UNRELATED
There is no evidence of any causal relationship

PVNS_Protocole_V4_110825 Page 73 of 129
APPENDIX 6 Adverse event – Serious Adverse Event report form

PVNS_Protocole_V4_110825 Page 74 of 129
APPENDIX 7 Information notice
French version

PVNS_Protocole_V4_110825 Page 75 of 129

PVNS_Protocole_V4_110825 Page 76 of 129

PVNS_Protocole_V4_110825 Page 77 of 129

PVNS_Protocole_V4_110825 Page 78 of 129

PVNS_Protocole_V4_110825 Page 79 of 129

PVNS_Protocole_V4_110825 Page 80 of 129

PVNS_Protocole_V4_110825 Page 81 of 129
Information notice
English version

PVNS_Protocole_V4_110825 Page 82 of 129

PVNS_Protocole_V4_110825 Page 83 of 129

PVNS_Protocole_V4_110825 Page 84 of 129

PVNS_Protocole_V4_110825 Page 85 of 129

PVNS_Protocole_V4_110825 Page 86 of 129

PVNS_Protocole_V4_110825 Page 87 of 129

PVNS_Protocole_V4_110825 Page 88 of 129
APPENDIX 8 Informed consent form
French version

PVNS_Protocole_V4_110825 Page 89 of 129

PVNS_Protocole_V4_110825 Page 90 of 129
Informed consent form
English version

PVNS_Protocole_V4_110825 Page 91 of 129

PVNS_Protocole_V4_110825 Page 92 of 129
Addendum to the Information notice – Consent form
French version

PVNS_Protocole_V4_110825 Page 93 of 129

PVNS_Protocole_V4_110825 Page 94 of 129
Addendum to the Information notice – Consent form
English version

PVNS_Protocole_V4_110825 Page 95 of 129

PVNS_Protocole_V4_110825 Page 96 of 129
APPENDIX 9 Inclusion form

PVNS_Protocole_V4_110825 Page 97 of 129

PVNS_Protocole_V4_110825 Page 98 of 129
APPENDIX 10 Inclusion procedure If eligible patient:
� Fill in the inclusion form (Appendix 9)
� Proceed to the inclusion via internet
http://www.pvns-study.net/
� Indicate your username and password communicated by coordinating centre
� Click on “OK”
� Click on “Enter a new patient”
� Enter patient initials (1st letter of last name + - + 1st letter of surname / 1st letter of each last name in case of compound name + 1st letter of surname)
� Click on “SEND”
� Fill in the following information (cf. information collected on the inclusion form):
- Date of birth
- Date of signed informed consent
- Date of treatment start (day 1)
- Patient’s participation in the additional biological study (biopsy) (yes / no)
- Are the inclusion criteria all respected? (yes / no)
- Are the non-inclusion criteria all respected? (yes / no)
- Name of the Investigator who included the patient
� Click on “SAVE & VALIDATE”
� Click on “Yes” to answer to the following question “No more errors are detectable on-line. Do you want to freeze and sign this form?”
The patient is enrolled in PVNS study.
A confirmation of inclusion will be sent to the Investigator (by post mail).

PVNS_Protocole_V4_110825 Page 99 of 129
APPENDIX 11 Treatment supply to the investigational centre – procedure (except for non-European countries)
Once a treatment supply form, “CLINICAL SUPPLIES SHIPMENT REQUEST“ (Appendix 12), has been completed, this form needs to be faxed to the coordinating centre to:
Valérie BOURNE-BRANCHU Fax: +33 (0)4 78 78 27 15
The form must be faxed only from Monday till Wednesday. Plan 5 days between the order and the delivery of the treatment in the centre.

PVNS_Protocole_V4_110825 Page 100 of 129
APPENDIX 12 Treatment supply form
French version

PVNS_Protocole_V4_110825 Page 101 of 129
Treatment supply form
English version

PVNS_Protocole_V4_110825 Page 102 of 129
APPENDIX 13 Procedure – Bioassay of nilotinib
Nilotinib trough blood levels will be determined after 6 and 12 weeks of treatment.
For the determination of nilotinib concentration in serum:
� 5 mL of whole blood will be drawn prior to the patient taking the morning dose of study drug, using an heparinised tube (LiH)
� The tube will be gently shaken by reversals and placed vertically for about 30 minutes at room temperature
� It will then be centrifuged for 10 minutes at approximately 1200 g (3000 rpm for a lot of centrifuger)
� Immediately after centrifugation, the upper serum sample (about 1.5 mL) will be transferred
� Identify each sample with the study name, centre number, patient number and patient initial (e.g. label)
� The sample should be quickly sent at ambient temperature (below 30°C) For the determination of nilotinib concentration in plasma:
� 5 mL of whole blood will be drawn using an heparinised vial just before nilotinib intake (residual) 10h to 14h after last intake if BID
� Transfer plasma in a vial after centrifugation (at least 5 min at 3500 rpm)
� Identify each sample with the study name, centre number, patient number and patient initial (e.g. label)
� The plasma should be quickly sent at ambient temperature (below 30°C)
For the shipment (except for non-European countries):
� Place the samples and the filled out “Nilotinib Monitory Request“ form in a shipping container for diagnostic specimens (UN 3373)
� Fix on the outside of the packaging the specific shipping form “Nilotinib Monitory Request“
� Ship samples via shipment society to:
LABORATOIRE DE PHARMACOLOGIE CLINIQUE ET TOXICOLOGIE CHU Bordeaux - Centre Hospitalier Pellegrin – Tripode
Place Amélie Raba Léon Pr M. MOLIMARD
Plateau Technique 2ème étage - Accueil de Biologie Centralisé 33076 BORDEAUX CEDEX - FRANCE
Tel : +33 (0)5 56 79 59 91 - Fax : +33 (0)5 56 79 47 95 Once a sample shipment form, “Nilotinib Monitory Request“, (Appendix 14) has been completed, this form needs to be faxed to the coordinating centre to:
Valérie BOURNE-BRANCHU Fax: +33 (0)4 78 78 27 15
Any sampling problems (i.e., sample obtained at wrong time, missing sample, sample not obtained) must be noted as a comment to the sample shipment form.

PVNS_Protocole_V4_110825 Page 103 of 129
APPENDIX 14 Procedure – Bioassay of nilotinib
Sample shipment form (except for non-European countries)

PVNS_Protocole_V4_110825 Page 104 of 129
APPENDIX 15 Procedure – Bioassay of nilotinib
Sample shipment procedure - FRANCE

PVNS_Protocole_V4_110825 Page 105 of 129
Procedure – Bioassay of nilotinib
Sample shipment procedure - ITALY

PVNS_Protocole_V4_110825 Page 106 of 129
Procedure – Bioassay of nilotinib
Sample shipment procedure – THE NETHERLAND

PVNS_Protocole_V4_110825 Page 107 of 129
Procedure – Bioassay of nilotinib
Sample shipment procedure – POLAND

PVNS_Protocole_V4_110825 Page 108 of 129
Procedure – Bioassay of nilotinib
Sample shipment procedure – UNITED KINGDOM

PVNS_Protocole_V4_110825 Page 109 of 129
APPENDIX 16 Procedure – Exploratory analysis Tumor biopsy schedule
Tissues will be collected in a prior surgery, or by biopsy, upon specific acceptance by the patient. If no tissue is available in the prior surgery, a biopsy will be done at visit 2. Optional biopsy should be performed at visit 2 only (6 week) if the specific informed consent was signed by the patient. The tumor sample has to be lengthier to perform tissue analysis. Materials and supplies
For collection of this sample use the following materials:
� 16 or 18 gauge biopsy needle
� 2 mL polypropylene screw-cap cryogenic tubes
� 60 mL container
� Tumor sample shipment form (Appendix 17) Core biopsy performing
� The tumor collection processing laboratory has to be notified.
� Core biopsy is performed with the use of local anaesthetic to numb the area where the needle is inserted. A small incision is made in the skin over the lump, and a needle is inserted through the incision
� If possible, please obtain an intact piece of tumor approximately 7-8 cm in length and 1 mm² in diameter (16 gauges)
� Wrap the specimens in gauze that has been lightly dampened with saline. Never immerse a specimen in saline or fixative of any kind before submitting to the pathologist
Tumor biopsy handling
� The sample will be snapping frozen in 3 cryogenic tubes by submerging it into liquid nitrogen
� The specimens have to be labelled with the centre number, patient number, patient identification initial (1st letter of surname + “ – “ + 1st letter of first name or 1st letter of surname + two 1st letter of first name), collection date, and the study number and name
� Complete the tumor sample shipment form (Appendix 17) Shipping
The tumor samples should be shipped with the tumor sample shipment form at study completion. Once a tumor sample shipment form (Appendix 17) has been completed, this form needs to be faxed to the coordinating centre to:
Valérie BOURNE-BRANCHU Fax: +33 (0)4 78 78 27 15
This process needs to be repeated for each tumor sample collection.

PVNS_Protocole_V4_110825 Page 110 of 129
APPENDIX 17 Procedure – Exploratory analysis
Tumor sample shipment form

PVNS_Protocole_V4_110825 Page 111 of 129
APPENDIX 18 Procedure – Exploratory analysis
Tumor sample shipment procedure In progress

PVNS_Protocole_V4_110825 Page 112 of 129
APPENDIX 19 Insurance attestation
FRANCE

PVNS_Protocole_V4_110825 Page 113 of 129
APPENDIX 20 CPP approval (initial + amendment)
(French ethics committee)

PVNS_Protocole_V4_110825 Page 114 of 129

PVNS_Protocole_V4_110825 Page 115 of 129

PVNS_Protocole_V4_110825 Page 116 of 129

PVNS_Protocole_V4_110825 Page 117 of 129

PVNS_Protocole_V4_110825 Page 118 of 129
APPENDIX 21 AFSSAPS authorization (initial + amendment)
(French competent authority)

PVNS_Protocole_V4_110825 Page 119 of 129

PVNS_Protocole_V4_110825 Page 120 of 129
APPENDIX 22 Group specific appendix for any institution: additional information about trial conduct in foreign country
Competent authority authorization, ethic committee approval, insurance attestation…

PVNS_Protocole_V4_110825 Page 121 of 129
APPENDIX 22-a University College Hospital
UCL Hospitals NHS Foundation Trust
LONDON

PVNS_Protocole_V4_110825 Page 122 of 129
APPENDIX 22-b Oxford Cancer Centre
OXFORD

PVNS_Protocole_V4_110825 Page 123 of 129
APPENDIX 22-c Leiden University Medical Center
LEIDEN

PVNS_Protocole_V4_110825 Page 124 of 129
APPENDIX 22-d Radboud University Nijmegen Medical Centre
NIJMEGEN

PVNS_Protocole_V4_110825 Page 125 of 129
APPENDIX 22-e Istituto Nazionale Tumori
MILANO

PVNS_Protocole_V4_110825 Page 126 of 129
APPENDIX 22-f Regina Elena National Cancer Institute
ROMA

PVNS_Protocole_V4_110825 Page 127 of 129
APPENDIX 22-g Sklodowska-Curie Memorial Cancer Center and Institute of Oncology
WARSAW

PVNS_Protocole_V4_110825 Page 128 of 129
APPENDIX 22-h Padova University Hospital
PADOVA

PVNS_Protocole_V4_110825 Page 129 of 129
APPENDIX 22-i Royal Prince Alfred Hospital
CAMPERDOWN





























