Embed Size (px)
Citation preview

T
Carbohydrate Polymers 205 (2019) 27–34
Contents lists available at ScienceDirect
Carbohydrate Polymers
journal homepage: www.elsevier.com/locate/carbpol
PHBV-graft-GMA via reactive extrusion and its use in PHBV/nanocellulose crystal composites
Ting Zhenga,b, Zhan Zhanga, Srishti Shuklaa,c, Shubh Agnihotria,c, Craig M. Clemonsa,d, Srikanth Pillaa,b,e,⁎
a Clemson University, Department of Automotive Engineering, 4 Research Drive, Greenville, SC, 29607, USA b Clemson University, Clemson Composites Center, Greenville, SC, 29607, USA c Department of Mechanical Engineering, Indian Institute of Technology (BHU), Varanasi, Uttar Pradesh, 221005, India d USDA Forest Service, Forest Products Laboratory, Madison, WI, 53726, USA e Clemson University, Department of Material Science and Engineering, Clemson, SC, 29602, USA
A R T I C L E I N F O
Keywords: PHBV Cellulose nanocrystal Extrusion Injection molding Nanocomposite
A B S T R A C T
Reactive extrusion was used for dicumyl peroxide (DCP)-initiated grafting of glycidyl methacrylate (GMA) to poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV). The effects of GMA and DCP content and their weight ratio on the GMA grafting percentage (GP%), the polymer melt viscosity, and the PHBV molecular weight were investigated. FTIR spectroscopy determined that the DCP did indeed initiate GMA grafting. However, the changes in both the zero-shear viscosity (η0) and the molecular weight suggested the existence of crosslinking products in the extruded polymers. A negative correlation between the degree of crystallinity ( χ ) of the PHBV-g-c GMA and the GP% suggested the influence of chain branching on crystallinity. In addition, the GMA content was found as a key factor determining the GP%. The PHBV-g-GMA was used as a matrix polymer in cellulose na-nocomposites to evaluate its effects on CNC dispersion and CNC-matrix adhesion relative to the unmodified PHBV matrix. The SEM images and the change in crystallization temperature suggested enhanced dispersion of CNC in a PHBV-g-GMA matrix. However, little increase in strength properties were found with CNC addition suggesting inadequate stress transfer between the matrix and CNCs.
1. Introduction
PHBV (poly(3-hydroxybutyrate-co-3-hydroxyvalerate)) is a family of bacterially-derived linear polyesters containing randomly arranged 3-hydroxybutyrate (HB) and 3-hydroxyvalerate (HV) segments. Although exhibiting exceptional biocompatibility and biodegradability (Avella, Martuscelli, & Raimo, 2000), this polymer is characterized by brittleness associated with its high glass transition temperature and large spherulites, thus restricting its use as a commodity plastic. This brittleness can be partially mitigated by increasing the content of the HV segment but at the cost of reduced tensile strength and modulus (Avella et al., 2000). When compared with polylactic acid (PLA), a popular benchmark bioplastic, PHBV shows inferior impact strength, tensile strength, stiffness, and a higher price (Koronis, Silva, & Fontul, 2013). Consequently, efforts have been undertaken to improve the characteristics of PHBV, specifically the mechanical properties, while reducing cost. To develop these characteristics, PHBV was reinforced
with renewable and low-cost natural fibers derived from bamboo (Singh, Mohanty, Sugie, Takai, & Hamada, 2008), pineapple (Luo & Netravali, 1999), abaca (Bledzki & Jaszkiewicz, 2010), jute (Bledzki & Jaszkiewicz, 2010), and pine wood (Wei, Stark, & McDonald, 2015) of different dimensions and pre-treatment methods. Improved tensile strength and modulus were observed in some of these fiber-reinforced PHBV composites.
Given that plant-derived fibers are lignocellulosic and usually hy-drophilic in nature, issues of compatibility are of concern when they are compounded with hydrophobic plastics such as PHBV. Incompatibility leads to poor dispersion of the reinforcement and poor stress transfer between the matrix and reinforcing material. To resolve these diffi-culties, strategies have been developed to enhance the polymer matrix-reinforcement adhesion to facilitate stress transfer and thus enhance the mechanical properties of the composites. Typical strategies entail ex-trusion with a free radical initiator (Wei et al., 2015), the application of a fiber surface treatment (Srubar et al., 2012), and the use of coupling
⁎ Corresponding author at: Clemson University, Department of Automotive Engineering, 4 Research Drive, Greenville, SC, 29607, USA. E-mail addresses: [email protected] (T. Zheng), [email protected] (Z. Zhang), [email protected] (S. Shukla),
[email protected] (S. Agnihotri), [email protected] (C.M. Clemons), [email protected] (S. Pilla).
https://doi.org/10.1016/j.carbpol.2018.10.014 Received 1 July 2018; Received in revised form 25 September 2018; Accepted 5 October 2018 Available online 10 October 2018 0144-8617/ © 2018 Published by Elsevier Ltd.

T. Zheng et al. Carbohydrate Polymers 205 (2019) 27–34
agents such as polymer-grafted-maleic anhydride (polymer-g-MA) (Srubar et al., 2012). Of these methods, the polymer-g-MA, which is a common type of coupling agent, is perhaps the most well-known given its facile processing and high stress transfer efficiency. Likewise, the polymer-grafted glycidyl methacrylate (polymer-g-GMA) also exhibits a similar ability to couple reinforcement and matrix (Asgari & Masoomi, 2012). The polymer-g-GMA reacts with polar groups (hydroxyl, car-boxyl, and amine) on the filler surface resulting in a grafting of its polymeric chain to the reinforcement’s polar surface. Examples are abundant in the literature on the use of GMA-grafted polymers of polypropylene (PP) (Jazani et al., 2017), polyethylene (PE) (Santos, Costa, & Pessan, 2018), and polystyrene (PS) (Zhao et al., 2008) as well as commercial products represented by the Joncryl ADR® (BASF) (Al-Itry, Lamnawar, & Maazouz, 2012) and Lotader AX 8840 (Santos et al., 2018). These polymer-g-GMA coupling agents are mainly synthesized by simple reactive extrusion of a polymer, a GMA monomer, and an initiator.
However, possible crosslinking reactions during grafting of GMA to PHBV renders the process more complex than a simple graft onto vinyl polymers such as PP, PE and PS. During melt processing, the GMA moiety may also react with hydroxyl/carboxyl/amine groups if they appear in the matrix polymer. Polymer containing such groups include polyesters, polycarbonates, polyurethanes, and polyamides, such as polyethylene terephthalate (PET) (Asgari & Masoomi, 2012), PLA (Al-Itry et al., 2012), poly(butylene adipate-co-terephthalate) (PBAT) (Al-Itry et al., 2012), poly(propylene carbonate) (PPC) (Li, Sun, & Zhang, 2012), and PA6 (Polyamide 6) (Villalobos, Awojulu, Greeley, Turco, & Deeter, 2006). Linear PHBV also has GMA-reactive hydroxyl and car-boxyl groups at its chain ends. Additionally, free-radical initiators such as DCP could also induce polymer chain branching through hydrogen abstraction, as shown in Fig. 1. Effects of the initiator type and con-centration, and the reaction time on the DCP-induced crosslinking re-action have been documented (Wei & McDonald, 2015; Yu & Zhu, 1999). Furthermore, DCP-induce β-scission and other side reactions occur simultaneously during melt extrusion (Fig. 1), all of which greatly complicate the PHBV-g-GMA melt-grafting procedure. However, very little research has been undertaken to determine this process. Although the PHBV-g-GMA has been synthesized by a pre-determined protocol, how the processing parameters (e.g., initiator (DCP) content, GMA content, and the GMA/DCP ratio) affect that grafting remain unknown. This lack of information prompted our systematic investigation of the GMA grafting reaction, the results of which are detailed here.
In this study, we also prepare PHBV/cellulose nanocrystal (CNC) composites to investigate the use of the prepared PHBV-g-GMA as a
coupling agent. CNCs are rod-shaped, highly crystalline celluloses produced by removal of the amorphous portions of the native cellulose of plants or other sources by acid hydrolysis (Peng, Ellingham, Sabo, Clemons, & Turng, 2015). The superior stiffness, modest aspect ratio (3–5 nm in wide, 50–500 nm in length (Moon, Martini, Nairn, Simonsen, & Youngblood, 2011)), large surface area, and low density of CNCs could make them useful as a reinforcing material (Rusli & Eichhorn, 2008; Šturcová, Davies, & Eichhorn, 2005). However, the high hydrophilicity of CNC makes uniform dispersion in and good ad-hesion with hydrophobic polymer matrices such as PHBV challenging, thus hindering its application as a reinforcing agent. For example, Du et al. (2017) found that direct compounding of CNCs in PHBV led to reductions in strength properties. While several researchers (Ten, Turtle, Bahr, Jiang, & Wolcott, 2010; Yu, Qin, Yan, & Yao, 2014, 2014; Yu & Yao, 2016) have shown improvements in the strength of PHBV with CNC addition, these improvements were found in solvent-cast films, where the development of a fiber network likely plays a large role in the increases rather than stress transfer between the CNC and matrix. However, solvent casting is generally slower and more expensive than melt processing approaches and often involves toxic solvents, all of which make it less commercially attractive. Jiang, Morelius, Zhang, Wolcott, and Holbery, (2008) directly compared solvent casting and melt processing approaches to PHBV-CNC composites and concluded that melt processing did not produce composites with the same level of dispersion and mechanical properties as solvent–blended composites, even when the CNCs were coated with polyethylene glycol to help prevent particle agglomeration. The authors further concluded that a coupling agent that would graft hydrophobic chains to the CNCs may be an effective alternative. Here we explore PHBV-g-GMA as just such a coupling agent.
2. Experimental
2.1. Materials
PHBV (ENMATTM, Y1000 P) with 3% HV was purchased from Tianan biopolymer (Ningbo, China) with a specific gravity of 1.25. Glycidyl methacrylate (GMA, 779342, Sigma), dicumyl peroxide (DCP, 98%, 81401057, Alfa Aesar), and trichloroacetic acid (TCA, 99% Alfa Aesar) were commercially available. The CNC aqueous suspension (12.2 wt. %) was prepared by the USDA Forest Service, Forest Products Laboratory (Madison, WI).
Fig. 1. Schematic pathways of reactions involved in PHBV GMA reactive extrusion.
28

T. Zheng et al. Carbohydrate Polymers 205 (2019) 27–34
Table 1 Composition of GMA-PHBV reactive extrusion.
GMA/DCP ratio → 2.5:1 5:1 10:1 20:1 30:1 40:1
DCP+GMA =2% P1 P4 P7 P10 P13 P16 DCP+GMA =5% P2 P5 P8 P11 P14 P17 DCP+GMA =10% P3 P6 P9 P12 P15 P18
2.2. Preparation of PHBV-g-GMA
PHBV-g-GMA was prepared by reactive extrusion of GMA and PHBV in the presence of DCP as an initiator using a 32-mm, 36 L/D co-ro-tating, twin-screw extruder (D-TEX extruder; Davis Standard, Pawcatuck, CT) and related gravimetric feed system (Accurate, Whitewater, WI). PHBV was pre-dried at 90 °C for 12 h prior to extru-sion. The DCP was dissolved in the GMA and then pumped via a peri-static pump into the barrel. The barrel temperature profile was set at 135, 141, 146, 154, 157, 157, 160, and 160 from the feed zone to the die. A 50 rpm screw rotation speed used in this study led to a 110 s dwell time, which is approximately 1/4 of the DCP half-life (t1/2, 5–8 min at 160 °C) (Dannenberg, Jordan, & Cole, 1958). This residence time ensured the effective decomposition of DCP while also reducing the degradation of PHBV during extrusion (Dhar, Tarafder, Kumar, & Katiyar, 2016). These processing parameters led to a throughput of approximately 4.5 kg/h. A two-factor (GMA/DCP ratio and PHBV wt. %), multilevel experiment was used. A total of 18 samples (P1 to P18) and a neat PHBV control (P0) were extruded. These sample composi-tions are listed in Table 1.
2.3. Preparation of Polymer/CNC composites
A two-step process was used to prepare the composites: premixing by solvent-casting followed by melt-compounding. First, a certain amount of CNC was transferred from its aqueous suspension to the anhydrous acetone by a repeated (4×) washing protocol including centrifuging-decanting-redispersing in acetone to completely remove the water. The CNC precipitate obtained from the last decanting step was then re-suspended in chloroform, mixed with polymer, and then refluxed for 30-min. This mixture was then cast into a silicon mold and the chloroform was evaporated at 23 . A Wiley mill (Arthur H. Thomas Co.) equipped with a 10# mesh screen was used to pulverize the cast CNC/polymer, which was then vacuum-dried at 70 for 4 h. The material was then injection molded using a 15 mL micro-extruder/ injection molding system (Xplore system, DSM, Xplore Instrument). The material was extruded for 5 min at a barrel temperature profile of 160–180–180 (feed zone, middle zone, die) and a screw speed of 50 rpm. The melt was then transferred to the micro-injection molder and standard type-V tensile specimens were inject molded using a barrel temperature of 170 , a mold temperature of 60 °C, injection and holding pressures of 5.5 bar, and a holding time of 15 s. As a reference, composites were also manufactured via a direct compounding route in which the freeze-dried CNC powder was mixed with neat PHBV and then fed into the micro-extruder directly without solvent-blending or adding any coupling agent.
2.4. Grafting percentage (GP%) determination
The grafting percentage determination was based upon Young’s study (Yang, Dominici, Fortunati, Kenny, & Puglia, 2015) with minor modifications. Specifically, 1.5 g of material were first refluxed in 30 mL chloroform for 35 min and subsequently precipitated by 250 mL ethanol. The precipitant was washed several times to remove the re-sidual homopolymerized GMA (polyGMA) and GMA oligomers fol-lowed by Buchner funnel recovery, and then vacuum-drying at 60 °C for 24 h. The GP%, which is defined as the wt.% of GMA in PHBV-g-GMA
was quantified by a standard titration. The purified polymer (0.50 g) was refluxed with 30 mL chloroform with 5.0 mL 0.1 M trichloroacetic acid (TCA)-chloroform solution for 90 min to complete the ring-open reaction between the TCA and the GMA. The resultant solution was titrated immediately with 0.05 M NaOH-methanol solution with four drops of 0.1% phenolphthalein-ethanol solution as the pH indicator. Any color remaining for 30 s indicated the endpoint of the titration. The grafted GMA was calculated from the following equation:
GP%(%) = (142.15×VTCA×c) / (10×m)
Where VTCA represent the consumed volume of TCA (mL); c the con-centration of the TCA solution (M) taken as 0.1; 142.15 is the molecular weight of the GMA; and m the weight of the sample (g).
2.5. Structure and property characterization
FTIR was performed to determine composition using a Thermo-Nicolet Magna 550 FTIR spectrometer in combination with a Thermo-SpectraTech Foundation Series Diamond ATR accessory with a 50° angle of incidence. Dried PHBV and PHBV-g-GMA samples were de-termined in absorbance mode from 4000 to 400 cm−1 at a resolution of 2 cm−1. Scanning electron microscopy (SEM) via a Hitachi-S4800 was used to investigate the microstructure of the fracture cross-section of the composite. Samples were sputter-coated with Pt prior to imaging. Differential Scanning Calorimeter (DSC, Q2000, TA instruments) was used to investigate the thermal behavior of the composites. Under ni-trogen flow, samples (∼5 mg) were subjected to heating/cooling/ heating cycles (−50 to 200 to −50 to 200 ) at the rate of 10 /min. The crystallinity degree ( χ ) was calculated byc
0χ (%) = ΔH /ΔH × 100/a where the ΔHm is the melting enthalpy,c m m ΔHm
0 =146 J/g is the ΔHm of 100% crystalline (Gogolewski, Jovanovic, Perren, Dillon, & Hughes, 1993), and a the weight fraction of the plastic. Both the ΔHm and melting temperatures (Tm) were derived from the second-heating DSC scans. Rheological tests were performed on a closed cavity rheometer (RPA Elite, TA instruments). The linear vis-coelastic region was determined via strain sweep (0.01–5.12 °, at fre-quency of 1 Hz). A 1.4% strain amplitude was selected and used for the subsequent frequency sweep. The frequency sweep was performed at frequencies ranging from 0.022 to 50 Hz (0.138–314 rad/s) at 175 . A complex viscosity curve fitting with the Carreau-Yasuda model was used to determine the zero-shear viscosity (η0) (Ansari, Zisis, Hatzikiriakos, & Mitsoulis, 2012). Gel permeation chromatography (GPC, Waters GPC) was used to determine the number-average mole-cular weight (Mn), weight-average molecular weight (Mw) and poly-dispersity index (PDI) followed by signal-detection via RI in-strumentation. Samples were prepared as 0.5 wt. % in chloroform and dissolved at an elevated temperature (50 ) for 4 days, followed by filtration (0.2 μm PTFE) prior to GPC loading. Mechanical properties (i.e. tensile strength, elongation at break, and Young’s modulus of the extruded composites) were examined per ASTM D638-10. An Instron universal testing machine (Model 1125, Instron) with a load cell of 500 N at a cross-head speed of 1 mm/min with the 25 mm grip se-paration was used for this examination.
3. Results and discussion
3.1. Free-radical grafting of GMA onto PHBV
PHBV-g-GMA was synthesized via reactive extrusion with two pos-sible crosslinking side reactions. The crosslinking can be induced by either the DCP or the grafted GMA pendant, (pathways I and II in Fig. 1). Pathway I is supported by earlier reported evidence of DCP-induced PHB long chain branching and chain entanglement during re-active extrusion (Wei & McDonald, 2015). Pathway II is characterized by a reaction of the grafted GMA pendant with the end group of
29
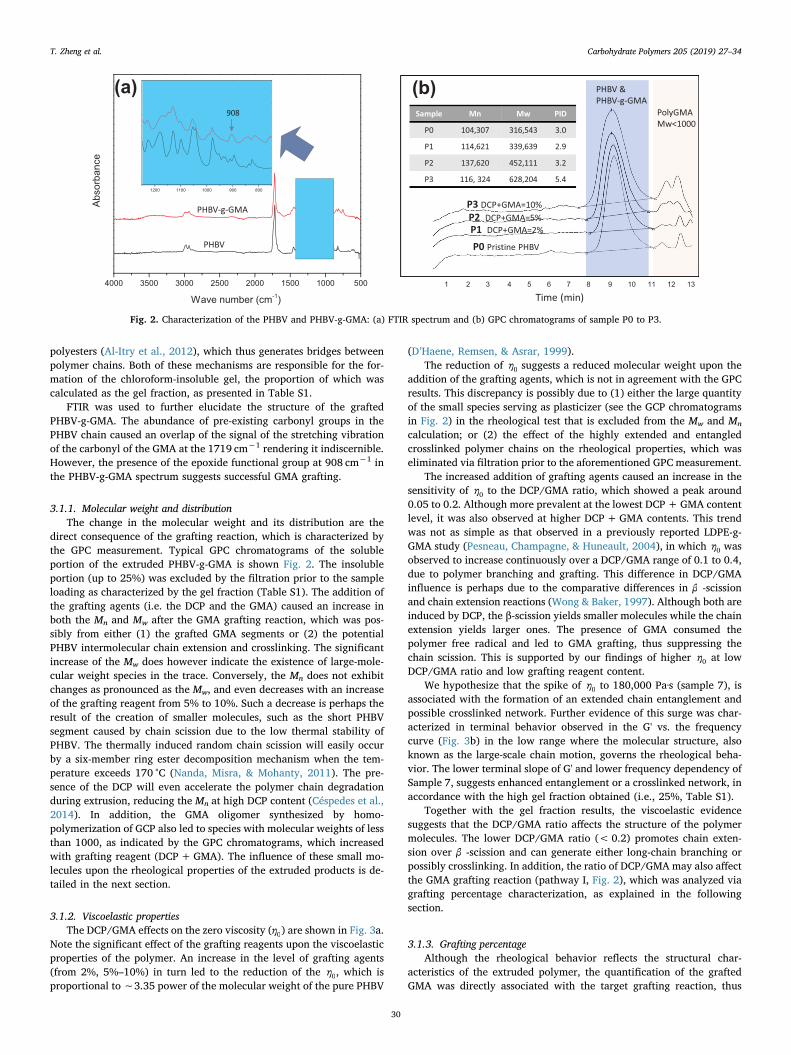
T. Zheng et al. Carbohydrate Polymers 205 (2019) 27–34
Fig. 2. Characterization of the PHBV and PHBV-g-GMA: (a) FTIR spectrum and (b) GPC chromatograms of sample P0 to P3.
polyesters (Al-Itry et al., 2012), which thus generates bridges between polymer chains. Both of these mechanisms are responsible for the for-mation of the chloroform-insoluble gel, the proportion of which was calculated as the gel fraction, as presented in Table S1.
FTIR was used to further elucidate the structure of the grafted PHBV-g-GMA. The abundance of pre-existing carbonyl groups in the PHBV chain caused an overlap of the signal of the stretching vibration of the carbonyl of the GMA at the 1719 cm−1 rendering it indiscernible. However, the presence of the epoxide functional group at 908 cm−1 in the PHBV-g-GMA spectrum suggests successful GMA grafting.
3.1.1. Molecular weight and distribution The change in the molecular weight and its distribution are the
direct consequence of the grafting reaction, which is characterized by the GPC measurement. Typical GPC chromatograms of the soluble portion of the extruded PHBV-g-GMA is shown Fig. 2. The insoluble portion (up to 25%) was excluded by the filtration prior to the sample loading as characterized by the gel fraction (Table S1). The addition of the grafting agents (i.e. the DCP and the GMA) caused an increase in both the Mn and Mw after the GMA grafting reaction, which was pos-sibly from either (1) the grafted GMA segments or (2) the potential PHBV intermolecular chain extension and crosslinking. The significant increase of the Mw does however indicate the existence of large-mole-cular weight species in the trace. Conversely, the Mn does not exhibit changes as pronounced as the Mw, and even decreases with an increase of the grafting reagent from 5% to 10%. Such a decrease is perhaps the result of the creation of smaller molecules, such as the short PHBV segment caused by chain scission due to the low thermal stability of PHBV. The thermally induced random chain scission will easily occur by a six-member ring ester decomposition mechanism when the tem-perature exceeds 170 °C (Nanda, Misra, & Mohanty, 2011). The pre-sence of the DCP will even accelerate the polymer chain degradation during extrusion, reducing the Mn at high DCP content (Céspedes et al., 2014). In addition, the GMA oligomer synthesized by homo-polymerization of GCP also led to species with molecular weights of less than 1000, as indicated by the GPC chromatograms, which increased with grafting reagent (DCP + GMA). The influence of these small mo-lecules upon the rheological properties of the extruded products is de-tailed in the next section.
3.1.2. Viscoelastic properties The DCP/GMA effects on the zero viscosity (η0) are shown in Fig. 3a.
Note the significant effect of the grafting reagents upon the viscoelastic properties of the polymer. An increase in the level of grafting agents (from 2%, 5%–10%) in turn led to the reduction of the η0, which is proportional to ∼3.35 power of the molecular weight of the pure PHBV
(D’Haene, Remsen, & Asrar, 1999). The reduction of η0 suggests a reduced molecular weight upon the
addition of the grafting agents, which is not in agreement with the GPC results. This discrepancy is possibly due to (1) either the large quantity of the small species serving as plasticizer (see the GCP chromatograms in Fig. 2) in the rheological test that is excluded from the Mw and Mn
calculation; or (2) the effect of the highly extended and entangled crosslinked polymer chains on the rheological properties, which was eliminated via filtration prior to the aforementioned GPC measurement.
The increased addition of grafting agents caused an increase in the sensitivity of η0 to the DCP/GMA ratio, which showed a peak around 0.05 to 0.2. Although more prevalent at the lowest DCP + GMA content level, it was also observed at higher DCP + GMA contents. This trend was not as simple as that observed in a previously reported LDPE-g-GMA study (Pesneau, Champagne, & Huneault, 2004), in which η0 was observed to increase continuously over a DCP/GMA range of 0.1 to 0.4, due to polymer branching and grafting. This difference in DCP/GMA influence is perhaps due to the comparative differences in β -scission and chain extension reactions (Wong & Baker, 1997). Although both are induced by DCP, the β-scission yields smaller molecules while the chain extension yields larger ones. The presence of GMA consumed the polymer free radical and led to GMA grafting, thus suppressing the chain scission. This is supported by our findings of higher η0 at low DCP/GMA ratio and low grafting reagent content.
We hypothesize that the spike of η0 to 180,000 Pa·s (sample 7), is associated with the formation of an extended chain entanglement and possible crosslinked network. Further evidence of this surge was char-acterized in terminal behavior observed in the G' vs. the frequency curve (Fig. 3b) in the low range where the molecular structure, also known as the large-scale chain motion, governs the rheological beha-vior. The lower terminal slope of G' and lower frequency dependency of Sample 7, suggests enhanced entanglement or a crosslinked network, in accordance with the high gel fraction obtained (i.e., 25%, Table S1).
Together with the gel fraction results, the viscoelastic evidence suggests that the DCP/GMA ratio affects the structure of the polymer molecules. The lower DCP/GMA ratio (< 0.2) promotes chain exten-sion over β -scission and can generate either long-chain branching or possibly crosslinking. In addition, the ratio of DCP/GMA may also affect the GMA grafting reaction (pathway I, Fig. 2), which was analyzed via grafting percentage characterization, as explained in the following section.
3.1.3. Grafting percentage Although the rheological behavior reflects the structural char-
acteristics of the extruded polymer, the quantification of the grafted GMA was directly associated with the target grafting reaction, thus
30

T. Zheng et al. Carbohydrate Polymers 205 (2019) 27–34
Fig. 3. (a): Zero-shear viscosity as function of DCP/GMA ratio grouped with 2%, 5% and 10% grafting reagent content (neat PHBV has η0 of 364,310 Pa·s). (b) storage modulus vs. frequency.
Fig. 4. Effect of (a) DCP/GMA ratio and (b) GMA content on grafting percentage.
demonstrating the reactivity of the PHBV-g-GMA. The grafting per-centage (GP%) was determined via the acid-based back titration, fol-lowed by an investigation of the effect of the DCP/GMA ratio and the overall amount of grafting reagents on the GP% (See Table S1 and Fig. 4 for details).
The effect of the DCP/GMA ratio on GP% at each grafting (DCP + GMA) reagent level is shown in Fig. 4a. While the differences between each grafting reagent level were large, those within each level were not, nor were their trends consistent. The dependency of the DCP content on GMA content in our experimental design makes it difficult to analyze each one independently. However, perhaps a more useful re-presentation of the data is shown in Fig. 4b, where the data has been grouped into four DCP ranges and plotted versus GMA content. A strong correlation between GP% and GMA content can be readily seen, which is consistent with similar work by others on the synthesis of PLA-g-GMA (Liu, Jiang, & Chen, 2012). Also, at GMA levels above 4 wt.%, we found additional increase in GP% at high DCP levels, although this effect is much smaller than that of GMA at the levels investigated. This marginal impact of the initiator content has also been reported in the preparation of both PP-g-GMA (Li, Ma, & Yang, 2013) and LDPE-g-GMA (Wei, Chionna, Galoppini, & Pracella, 2003).
3.1.4. Grafting effect on crystallinity DSC was used to characterize the thermal behavior of the PHBV-g-
GMA and reveal the effects of the GMA grafting on the crystallization of the polymers. The melting temperature (Tm), the melting enthalpy, and the degree of the crystallinity ( χc ) are summarized in Table S2.
No large differences in the Tm of the pure PHBV and PHBV-g-GMA were observed, regardless of the variability in the GP% (Table S2). However, a continuous decrease in χc with increasing GP% was ob-served regardless of the grafting conditions (Fig. 5). This inverse cor-relation has also been observed in the PCL-g-GMA (Kim, Cho, & Park, 2001) using the BPO as initiators, which can be interpreted as a result of chain branching hindering crystallization. Interestingly, although the inverse correlation between the χ and GP% was clearly observed in the c PHBV-g-GMA, the pristine PHBV did not exhibit the highest χ over its GMA-grafted derivatives, unlike polymers such as PCL (Kim et al., 2001).
3.2. Properties of CNC/PHBV composites
To evaluate the ability of the PHBV-g-GMA to improve CNC dis-persion and CNC-matrix adhesion, composites were prepared from the PHBV-g-GMA of the highest GP% (P9, GP% = 3.14 wt.%) and various CNC contents (1, 3, 5 wt. %). A solvent-casting method was used to blend the composites, followed by injection-molding to prepare tensile specimens. The mechanical properties of the final samples are detailed in Fig. 6 and Table S3.
31
c

T. Zheng et al. Carbohydrate Polymers 205 (2019) 27–34
Fig. 5. Crystallinity as function of grafting percentage.
The PHBV-g-GMA without CNCs exhibited a lower tensile strength and modulus and a higher elongation at break compared to the un-modified PHBV. This “brittle to ductile transition” is possibly attribu-table to the reduced crystallinity associated with the polymer chain branching and scission as supported by the GPC and rheological evi-dence. The GMA branches on the PHBV chain prevent the formation of large spherulites, which is responsible for the brittleness of PHBV (Carli, Crespo, & Mauler, 2011; Maiti, Batt, & Giannelis, 2007).
An increased modulus was observed with the incorporation of the CNC (Fig. 6b) in the unmodified PHBV matrix, as is a common ob-servation in CNC nanocomposites (Boujemaoui et al., 2017; Jiang et al., 2008; Kashani Rahimi, Aeinehvand, Kim, & Otaigbe, 2017; Ten et al., 2010). No significant improvements were observed in the tensile strength of either our PHBV or our PHBV-g-GMA with CNC addition (Fig. 6a). The incorporation of CNC, if unmodified, usually reinforces hydrophilic matrices (e.g. PVA (Lee, Clancy, Kontturi, Bismarck, & Shaffer, 2016)), but reduces strength in more hydrophobic matrices
such as PCL (Boujemaoui et al., 2017) and PHBV (Du et al., 2017; Jiang et al., 2008; Ten et al., 2010; Yu, Qin et al., 2014; Yu, Sun et al., 2014; Yu & Yao, 2016) when melt-processed. It was hoped that the PHBV-g-GMA could help improve the situation through covalent bonding be-tween PHBV-g-GMA and CNC. The lack of improvement of the PHBV-g-GMA matrix with CNC is disappointing and suggests poor stress transfer between them. Whether this is due to: 1) still suboptimal (albeit im-proved) CNC dispersion, 2) the limited reinforcing ability the CNCs due to their modest aspect ratio, 3) suboptimal molecular architecture of the PHBV-g-GMA (e.g., too low of a GP% or molecular weight, un-wanted side reactions), or 4) a combination of these factors will require further, focused investigation examining these factors.
The SEM images and DSC thermograms reveal further information regarding the CNC dispersion in the composites. The SEM images of the fracture surfaces of tensile tests are shown in Fig. 6d to i. The CNC composite prepared by direct compounding indicates a rough surface with visible microscale CNC aggregations, as indicated by the circles in Fig. 6g. CNC agglomerates were reduced when the CNC and polymer were premixed by solvent casting prior to extrusion (Fig. 6e and h), and were nearly invisible in the PHBV-g-GMA matrix (Fig. 6f and i). These phenomena support the enhanced CNC dispersion that is possible by (1) additional physical premixing in the solvent (chloroform), and (2) utilizing a PHBV-g-GMA matrix. The premixing may be the causative factor for the similar mechanical behavior exhibited in the PHBV and the PHBV-g-GMA matrices, as shown in Fig. 6a.
The DSC traces of the CNC composites in the matrices of either PHBV or PHBV-g-GMA with varied CNC loadings are illustrated in Fig. 6j, and the extracted parameters are summarized in Table S4. The crystallization temperatures (Tc) of the composites in the PHBV-g-GMA matrix (Fig. 6j bottom) showed a continuous increase with CNC loading. Although an increase in Tc was also observed for composites of the PHBV matrix with initial CNC addition (Fig. 6j upper), further in-creases with additional CNC content was not found. For the composites prepared by direct compounding, no change in Tc over the CNC loading range investigated was observed (Table S4). The CNC can play a dual role in polymer crystallization: acting as a nucleating agent leading to increased Tc, but also reducing the polymer mobility resulting in
Fig. 6. Properties of PHBV/CNC composites: (a) tensile strength, (b) Young’s modulus, and (c)strain at break with varying the CNC content (value and statistical analysis see Table S3); (d) to (h): SEM images of tensile-fractured surfaces (scale bars = 100 μm); (j) DSC thermogram of composites.
32

T. Zheng et al. Carbohydrate Polymers 205 (2019) 27–34
decreased Tc (Kashani Rahimi et al., 2017). The increased Tc in the PHBV-g-GMA composites with CNC addition suggests that the enhanced crystalline kinetics are attributed to heterogeneous nucleation induced by CNC through the creation of additional nucleating sites with CNC loading. On the other hand, the insensitivity of the Tc of the PHBV composites to the CNC concentration suggests the formation of few additional nucleating sites with the addition of CNC, a hypothesis supported by the SEM evidence of CNC aggregation (Fig. 6d, e, g, h).
4. Conclusions
The first objective of this investigation was to establish a reactive extrusion protocol for synthesizing PHBV-g-GMA with a high GMA grafting percentage and few side reactions. Although a crosslinking side-reaction was confirmed by rheological evidence and gel fraction determination, it was suppressed when sufficient GMA was present to consume the polymeric free radical and form GMA grafts. High GMA content and high DCP/GMA ratio facilitated the formation of polymers of less entangled networks. Furthermore, high GMA grafting percentage was directly related to the GMA content but less affected by the initiator (DCP) concentration. GMA grafting led to lower crystallinity, which reduced the brittleness of PHBV. The highest GMA grafting percentage of 3.14% was achieved at 10% GMA + DCP content at the DCP/GMA ratio of 0.1. In the second part of this report, CNCs were used to test the coupling ability of the prepared PHBV-g-GMA by fabricating and evaluating CNC composites made from it or the original PHBV. When the reactive PHBV-g-GMA was used as a matrix, limited reinforcement was observed despite enhanced dispersion relative to the original PHBV matrix due to the CNC surface hydrophobization. To assess whether this lack of reinforcement is due to characteristics of the PHBV-g-GMA (e.g., suboptimal GP%, molecular weight) or the CNC (e.g., modest aspect ratio) or other factors will require further investigation.
Acknowledgments
The authors are grateful to Kimberly Ivey for her assistance in conducting FTIR and GPC tests. The authors would also like to ac-knowledge the financial support by the USDA National Institute of Food and Agriculture, AFRI project [contract#2016-67021-25016], Robert Patrick Jenkins Professorship, and Dean’s Faculty Fellow Professorship. Ting Zheng would like to acknowledge the financial support provided by Cooper-Standard Postdoctoral Fellowship.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.carbpol.2018.10.014.
References
Al-Itry, R., Lamnawar, K., & Maazouz, A. (2012). Improvement of thermal stability, rheological and mechanical properties of PLA, PBAT and their blends by reactive extrusion with functionalized epoxy. Polymer Degradation and Stability, 97, 1898–1914.
Ansari, M., Zisis, T., Hatzikiriakos, S. G., & Mitsoulis, E. (2012). Capillary flow of low-density polyethylene. Polymer Engineering & Science, 52(3), 649–662.
Asgari, M., & Masoomi, M. (2012). Thermal and impact study of PP / PET fibre compo-sites compatibilized with glycidyl methacrylate and maleic anhydride. Composites Part B Engineering, 43(3), 1164–1170.
Avella, M., Martuscelli, E., & Raimo, M. (2000). Properties of blends and composites based on poly(3-hydroxy)butyrate (PHB) and poly(3-hydroxybutyrate-hydro-xyvalerate) (PHBV) copolymers. Journal of Materials Science, 35(3), 523–545.
Bledzki, A. K., & Jaszkiewicz, A. (2010). Mechanical performance of biocomposites based on PLA and PHBV reinforced with natural fibres - A comparative study to PP. Composites Science and Technology, 70(12), 1687–1696.
Boujemaoui, A., Cobo Sanchez, C., Engström, J., Bruce, C., Fogelström, L., Carlmark, A., et al. (2017). Polycaprolactone nanocomposites reinforced with cellulose nanocrys-tals surface-modified via covalent grafting or physisorption: A comparative study. ACS Applied Materials and Interfaces, 9(40), 35305–35318.
Carli, L. N., Crespo, J. S., & Mauler, R. S. (2011). PHBV nanocomposites based on
organomodified montmorillonite and halloysite: The effect of clay type on the mor-phology and thermal and mechanical properties. Composites Part A: Applied Science and Manufacturing, 42(11), 1601–1608.
Céspedes, R. I. N., Gámez, J. F. H., Velázquez, M. G. N., Belmontes, F.Á., De Leõn, R. E. D., Fernández, O. S. R., et al. (2014). Thermoplastic elastomers based on high-density polyethylene, ethylene-propylene-diene terpolymer, and ground tire rubber dyna-mically vulcanized with dicumyl peroxide. Journal of Applied Polymer Science, 131(4), 39901–39909.
D’Haene, P., Remsen, E. E., & Asrar, J. (1999). Preparation and characterization of a branched bacterial polyester. Macromolecules, 32(16), 5229–5235.
Dannenberg, E. M., Jordan, M. E., & Cole, H. M. (1958). Peroxide crosslinked carbon black polyethylene compositions. Journal of Polymer Science, 31(122), 127–153.
Dhar, P., Tarafder, D., Kumar, A., & Katiyar, V. (2016). Thermally recyclable polylactic acid/cellulose nanocrystal films through reactive extrusion process. Polymer, 87, 268–282.
Du, J., Zhao, G., Pan, M., Zhuang, L., Li, D., & Zhang, R. (2017). Crystallization and mechanical properties of reinforced PHBV composites using melt compounding: Effect of CNCs and CNFs. Carbohydrate Polymers, 168, 255–262.
Gogolewski, S., Jovanovic, M., Perren, S. M., Dillon, J. G., & Hughes, M. K. (1993). The effect of melt-processing on the degradation of selected polyhydroxyacids: polylac-tides, polyhydroxybutyrate, and polyhydroxybutyrate-co-valerates. Polymer Degradation and Stability, 40(3), 313–322.
Jazani, O. M., Rastin, H., Formela, K., Hejna, A., Shahbazi, M., Farkiani, B., et al. (2017). An investigation on the role of GMA grafting degree on the efficiency of PET/PP-g-GMA reactive blending: Morphology and mechanical properties. Polymer Bulletin, 74(11), 4483–4497.
Jiang, L., Morelius, E., Zhang, J., Wolcott, M., & Holbery, J. (2008). Study of the poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhisker composites prepared by solution casting and melt processing. Journal of Composite Materials, 42(24), 2629–2645.
Kashani Rahimi, S., Aeinehvand, R., Kim, K., & Otaigbe, J. U. (2017). Structure and biocompatibility of bioabsorbable nanocomposites of aliphatic-aromatic copolyester and cellulose nanocrystals. Biomacromolecules, 18(7), 2179–2194.
Kim, C. H., Cho, K. Y., & Park, J. K. (2001). Grafting of glycidyl methacrylate onto polycaprolactone: Preparation and characterization. Polymer, 42(12), 5135–5142.
Koronis, G., Silva, A., & Fontul, M. (2013). Green composites: A review of adequate materials for automotive applications. Composites Part B: Engineering, 44, 120–127.
Lee, W. J., Clancy, A. J., Kontturi, E., Bismarck, A., & Shaffer, M. S. P. (2016). Strong and stiff: High-performance cellulose Nanocrystal/Poly(vinyl alcohol) composite fibers. ACS Applied Materials & Interfaces, 8(46), 31500–31504.
Li, J., Sun, C. R., & Zhang, X. Q. (2012). Preparation, thermal properties, and morphology of graft copolymers in reactive blends of PHBV and PPC. Polymer Composites, 33(10), 1737–1749.
Li, Z., Ma, Y., & Yang, W. (2013). A facile, green, versatile protocol to prepare poly-propylene-g-poly(methyl methacrylate) copolymer by water-solid phase suspension grafting polymerization using the surface of reactor granule technology poly-propylene granules as reaction loci. Journal of Applied Polymer Science, 129(6), 3170–3177.
Liu, J., Jiang, H., & Chen, L. (2012). Grafting of glycidyl methacrylate onto poly(lactide) and properties of PLA/starch blends compatibilized by the grafted copolymer. Journal of Polymers and the Environment, 20(3), 810–816.
Luo, S., & Netravali, A. N. (1999). Interfacial and mechanical properties of environment-friendly “green” composites made from pineapple fibers and poly (hydroxybutyrate-co-valerate) resin. Journal of Materials Science, 34(15), 3709–3719.
Maiti, P., Batt, C. A., & Giannelis, E. P. (2007). New biodegradable polyhydroxybutyrate/ layered silicate nanocomposites. Biomacromolecules, 8(11), 3393–3400.
Moon, R. J., Martini, A., Nairn, J., Simonsen, J., & Youngblood, J. (2011). Cellulose nanomaterials review: Structure, properties and nanocomposites. Chemical Society Reviews, 40(7), 3941–3994.
Nanda, M. R., Misra, M., & Mohanty, A. K. (2011). The effects of process engineering on the performance of PLA and PHBV blends. Macromolecular Materials and Engineering, 296(8), 719–728.
Peng, J., Ellingham, T., Sabo, R., Clemons, C. M., & Turng, L. S. (2015). Oriented poly-vinyl alcohol films using short cellulose nanofibrils as a reinforcement. Journal of Applied Polymer Science, 132(48) n/a.
Pesneau, I., Champagne, M. F., & Huneault, M. A. (2004). Glycidyl methacrylate-grafted linear low-density polyethylene fabrication and application for polyester/poly-ethylene bonding. Journal of Applied Polymer Science, 91(5), 3180–3191.
Rusli, R., & Eichhorn, S. J. (2008). Determination of the stiffness of cellulose nano-whiskers and the fiber-matrix interface in a nanocomposite using Raman spectro-scopy. Applied Physics Letters, 93, 033111.
Santos, L. G., Costa, L. C., & Pessan, L. A. (2018). Development of biodegradable PLA/PBT nanoblends. Journal of Applied Polymer Science, 135(12), 45951.
Singh, S., Mohanty, A. K., Sugie, T., Takai, Y., & Hamada, H. (2008). Renewable resource based biocomposites from natural fiber and polyhydroxybutyrate-co-valerate (PHBV) bioplastic. Composites Part A: Applied Science and Manufacturing, 39(5), 875–886.
Srubar, W. V., Pilla, S., Wright, Z. C., Ryan, C. A., Greene, J. P., Frank, C. W., et al. (2012). Mechanisms and impact of fiber – matrix compatibilization techniques on the ma-terial characterization of PHBV / oak wood flour engineered biobased composites. Composites Science and Technology, 72(6), 708–715.
Šturcová, A., Davies, G. R., & Eichhorn, S. J. (2005). Elastic modulus and stress-transfer properties of tunicate cellulose whiskers. Biomacromolecules, 6(2), 1055–1061.
Ten, E., Turtle, J., Bahr, D., Jiang, L., & Wolcott, M. (2010). Thermal and mechanical properties of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhiskers composites. Polymer, 51(12), 2652–2660.
Villalobos, M., Awojulu, A., Greeley, T., Turco, G., & Deeter, G. (2006). Oligomeric chain
33

T. Zheng et al. Carbohydrate Polymers 205 (2019) 27–34
extenders for economic reprocessing and recycling of condensation plastics. Energy, 31(15), 3227–3234.
Wei, L., & McDonald, A. G. (2015). Peroxide induced cross-linking by reactive melt processing of two biopolyesters: Poly(3-hydroxybutyrate) and poly(l -lactic acid) to improve their melting processability. Journal of Applied Polymer Science, 132(13).
Wei, L., Stark, N. M., & McDonald, A. G. (2015). Interfacial improvements in bio-composites based on poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hy-droxyvalerate) bioplastics reinforced and grafted with α-cellulose fibers. Greem Chemistry, 17(10), 4800–4814.
Wei, Q., Chionna, D., Galoppini, E., & Pracella, M. (2003). Functionalization of LDPE by melt grafting with glycidyl methacrylate and reactive blending with polyamide-6. Macromolecular Chemistry and Physics, 204(8), 1123–1133.
Wong, B., & Baker, W. E. (1997). Melt rheology of graft modified polypropylene. Polymer, 38(11), 2781–2789.
Yang, W., Dominici, F., Fortunati, E., Kenny, J. M., & Puglia, D. (2015). Melt free radical grafting of glycidyl methacrylate (GMA) onto fully biodegradable poly(lactic) acid films: Effect of cellulose nanocrystals and a masterbatch process. RSC Advances,
5(41), 32350–32357. Yu, H., & Yao, J. (2016). Reinforcing properties of bacterial polyester with different
cellulose nanocrystals via modulating hydrogen bonds. Composites Science and Technology, 136, 53–60.
Yu, Q., & Zhu, S. (1999). Peroxide crosslinking of isotactic and syndiotactic poly-propylene. Polymer, 40(11), 2961–2968.
Yu, H., Qin, Z., Yan, C., & Yao, J. (2014). Green nanocomposites based on functionalized cellulose nanocrystals: A study on the relationship between interfacial interaction and property enhancement. ACS Sustainable Chemistry and Engineering, 2(4), 875–886.
Yu, H., Sun, B., Zhang, D., Chen, G., Yang, X., & Yao, J. (2014). Reinforcement of bio-degradable poly(3-hydroxybutyrate-co-3-hydroxyvalerate) with cellulose nano-crystal/silver nanohybrids as bifunctional nanofillers. Journal of Materials Chemistry B, 2(48), 8479–8489.
Zhao, W., Liu, Y., Feng, Q., Xie, X., Wang, X., & Ye, X. (2008). Dispersion and noncovalent modification of multiwalled carbon nanotubes by various polystyrene‐based poly-mers. Journal of Applied Polymer Science, 109(6), 3525–3532.
34





























