Embed Size (px)
Citation preview

Molecular and Cellular Pathobiology
Phosphorylation Regulates c-Myc's Oncogenic Activityin the Mammary Gland
Xiaoyan Wang1, Melissa Cunningham1, Xiaoli Zhang1, Sara Tokarz1, Bryan Laraway1,Megan Troxell2, and Rosalie C. Sears1
AbstractExpression of the c-Myc oncoprotein is affected by conserved threonine 58 (T58) and serine 62 (S62)
phosphorylation sites that help to regulate c-Myc protein stability, and altered ratios of T58 and S62phosphorylation have been observed in human cancer. Here, we report the development of 3 unique c-mycknock-in mice that conditionally express either c-MycWT or the c-MycT58A or c-MycS62A phosphorylation mutantfrom the constitutively active ROSA26 locus in response to Cre recombinase to study the role of thesephosphorylation sites in vivo. Using a mammary-specific Cre model, we found that expression of c-MycWT
resulted in increased mammary gland density, but normal morphology and no tumors at the level expressedfrom the ROSA promoter. In contrast, c-MycT58A expression yielded enhanced mammary gland density,hyperplastic foci, cellular dysplasia, and mammary carcinoma, associated with increased genomic instabilityand suppressed apoptosis relative to c-MycWT. Alternatively, c-MycS62A expression reduced mammary glanddensity relative to control glands, and this was associated with increased genomic instability and normalapoptotic function. Our results indicate that specific activities of c-Myc are differentially affected by T58 and S62phosphorylation. This model provides a robust platform to interrogate the role that these phosphorylation sitesplay in c-Myc function during development and tumorigenesis. Cancer Res; 71(3); 925–36. �2011 AACR.
Introduction
The c-Myc oncoprotein is a pleiotropic transcription factorinvolved in controlling many cellular functions, including cellproliferation, cell growth, and cell differentiation, and path-ways that regulate genome stability and cell death (1). c-Myc isconstitutively highly expressed in most human tumors, andhigh expression of c-Myc in animal models can drive tumor-igenesis (2–4). c-Myc–induced tumorigenesis is tempered byits activation of intrinsic tumor suppressor pathways invol-ving engagement of cell cycle checkpoint and apoptotic celldeath pathways (5). Activation of these intrinsic tumor sup-pressor pathways induces cell death in normal cells (6), but intumor cells these fail-safe mechanisms are bypassed by sec-ondary lesions that likely evolve from genomic instability
associated with high c-Myc expression. Thus, to maintainnormal cell function, c-Myc expression is tightly controlledat the level of transcription, mRNA stability, translation, andprotein stability. In breast cancer, c-MYC gene amplificationhas been reported in approximately 16% of cases andincreased mRNA expression in approximately 22% (7, 8).However, elevated expression of c-Myc protein is reportedin approximately 70% of human breast tumors, arguing for apotentially critical role for posttranslational regulation of c-Myc expression (2, 9, 10).
Phosphorylation of c-Myc at conserved residues serine 62(S62) and threonine 58 (T58) can regulate c-Myc proteinstability in response to mitogen signaling (11). Phosphoryla-tion of S62 by ERK or CDK kinases transiently increases c-Mycstability whereas phosphorylation of T58 by GSK3b triggersdephosphorylation of S62 by protein phosphatase 2A (PP2A),ubiquitination by the SCF-Fbw7 E3 ligase, and proteasomaldegradation (12). The scaffold protein Axin1 coordinatesc-Myc degradation through this pathway (13). Mutations inPP2A subunits, FBW7 and AXIN1 have been reported in manyhuman cancers (14–16), suggesting that this c-Myc degrada-tion pathway can be deregulated in cancer cells leading toaltered levels of T58 and S62 phosphorylation and increased c-Myc stability.
In cell culture experiments, mutation of S62 to alanine(S62A) reduces c-Myc's transforming activity whereas muta-tion of T58 to alanine (T58A) enhances c-Myc's transformingactivity, suggesting that phosphorylation changes at thesesites can affect c-Myc function (17–20). Furthermore,
Authors' Affiliations: 1Molecular and Medical Genetics Department and2Pathology Department, Oregon Health and Sciences University, Portland,Oregon
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Present address for S. Tokarz: Department of Pediatrics, University ofWisconsin at Madison, Madison, WI.
Corresponding Author: Rosalie C. Sears, Molecular and Medical Genet-ics Department, Oregon Health and Sciences University, 3181 S.W. SamJackson Park Rd., Portland, OR. Phone: 503-494-6885; Fax: 503-494-4411. E-mail: [email protected]
doi: 10.1158/0008-5472.CAN-10-1032
�2011 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 925
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

decreased T58 and increased S62 phosphorylation have beenobserved in human cancer cell lines associated with increasedc-Myc protein stability (13, 21). This phosphorylation ratio ispartially mimicked in the c-MycT58A mutant, which has no T58and constitutive high S62 phosphorylation (18, 22, 23).
To carefully address the role of T58 and S62 phosphoryla-tion in c-Myc's activity in vivo, we have generated 3 condi-tional c-myc knock-in mice that express c-MycWT, c-MycT58A,or c-MycS62A (which lacks phosphorylation at both sites dueto their hierarchical nature) from the endogenous ROSA26locus in response to Cre-mediated recombination. In thisstudy, we used Wap-Cre to drive expression in the mammarygland as it provides an elegant system to interrogate multi-ple activities of c-Myc. In normal mammary development, c-Myc expression is important for both pregnancy-associatedproliferation and apoptosis during involution (24, 25). High-level expression of c-Myc can also drive mammary glandtumorigenesis in transgenic mouse models (3, 26, 27). Theextended mammary tumor latencies in these studies suggestthat additional lesions are required for mammary tumor-igenesis. Results from this study indicate that lesions affect-ing phosphorylation at T58 and S62 could contribute totumorigenesis, as the phosphorylation status of these sitessubstantially affected c-Myc function and tumorigenicpotential in the mammary gland.
Methods
Generating RFS-mycWT, RFS-mycT58A, and RFS-mycS62A
miceROSA-Floxed-Stop (RFS)-mycWT, RFS-mycT58A, and RFS-
mycS62A mice were generated using an established geneknock-in strategy (28; see Supplementary Material). Briefly,murine c-mycWT-HA or phosphorylation mutant c-mycT58A-HAor c-mycS62A-HA cDNAs (29) were cloned into targeting vectorsand electroporated into 129 ES cells. Correctly targeted andsequence-confirmed ES clones were injected into C57BL/6blastocysts to obtain chimeric mice. Germline transmissionwas obtained by crossing with C57BL/6 mice to establishhomozygous knock-in RFS-mycWT, RFS-mycT58A, and RFS-mycS62A strains. RFS-mycWT, RFS-mycT58A, and RFS-mycS62A
were crossed with Wap-Cre transgenic mice (C57BL/6),obtained from the National Cancer Institute (NCI) MouseModels of Human Cancer Consortium (MMHCC).
Genotyping and detection of recombinationRFS-mycWT, RFS-mycT58A, and RFS-myc S62A knock-in mice
and the presence of Wap-Cre were identified by PCR analysisusing tail DNA and specific primer sets (see SupplementaryMaterial). Cre-mediated recombination in RFS-myc (WT,T58A, S62A)/Wap-Cre mice was detected by PCR analysisand specified primers (see Supplementary Material).
RNA analysisRNA was isolated from mammary glands using TRIzol
reagent (Invitrogen) according to manufacturer's protocol.cDNA was prepared from isolated RNA and analyzed byPCR or quantitative PCR (see Supplementary Material).
Antibodies and Western blottingAntibodies are listed in Supplementary Material. Mouse
mammary gland samples were lysed by homogenizing in EBCbuffer with protease and phosphatase inhibitors, and subjectto Western analysis (see Supplementary Material).
Quantification and statisticsWestern blots were visualized and quantified using LI-COR
Odyssey Infrared software version 1.2, which is linear over 4orders of magnitude. Statistical significance was determinedby Student's t test.
Cycloheximide half-lifePrimary mouse embryo fibroblasts (MEF) or mammary
epithelial cells (MEC; see Supplementary Material; 1–2 pas-sages) were infected with Ad-Cre for 48 hours and then starvedin 0.1% FBS for another 48 hours. Cells were treated withcycloheximide and harvested at each time point (see Supple-mentary Material). HA-tagged WT, T58A, or S62A Myc wasimmunoprecipitated with HA antibody and Western blottedwith Y69 Myc antibody.
Pathologic assessmentMice were anesthetized with avertin, sacrificed, and tissues
were fixed in 10% formalin, embedded in paraffin, and 5-mmthick sections were stained with Mayer's hematoxylin andeosin (H&E). Pathologic findings were classified according tothe MMHCC NIH/NCI by Dr. Troxell (30).
Apoptosis assayApoptosis was detected by terminal deoxynucleotidyltrans-
ferase–mediated dUTP nick end labeling (TUNEL) stainingusing the Apo Tag kit (Chemicon International) and thepercentage of apoptotic cells was determined in 10 randomfields per slide.
Whole mount analysis of mammary glandMammary gland tissues were dissected, spread on glass
slides, and fixed immediately with Carnoy's Fixative. Tissueswere hydrated with serially diluted ethanol and stained with0.6% carmine solution overnight at 4�C. Tissues were clearedof lipid with xylene and mounted with permount.
Chromosome spreads and centrosome stainingPrimary mammary epithelial cells, 80% confluent, were
used for centrosome detection, and chromosome spreadsafter colcemid treatment (see Supplementary Material). Chro-mosome spreads were stained with Giemsa. Centrosomeswere detected by immunofluorescence for pericentrin. G-banding was done by the OHSU Cytogenetics Laboratory.
Results
Generation of conditional RFS-mycWT, RFS-mycT58A, andRFS-mycS62A mice
To study the effects of altering c-Myc phosphorylationat T58 and S62 on c-Myc's activity in vivo, we utilized 2phosphorylation mutants: c-MycT58A, which lacks T58
Wang et al.
Cancer Res; 71(3) February 1, 2011 Cancer Research926
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

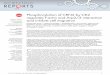
phosphorylation and has constitutively high S62 phosphoryla-tion, as it is resistant to PP2A-mediated dephosphorylation(20); and c-MycS62A, which lacks phosphorylation at both sitesdue to their hierarchical nature (Supplementary Fig. S1A). Weused an established gene knock-in strategy to insert HA-tagged cDNAs, either murine wild-type c-mycWT, or phosphor-ylation site mutants c-mycT58A or c-mycS62A, preceded by atranscription stop sequence flanked by loxP sites, into theconstitutively active ROSA26 gene locus (Fig. 1A; Supplemen-tary Fig. S1B). The ROSA26 locus ubiquitously drives geneexpression in all embryonic and adult tissues; however, due tothe transcription stop sequence, the knock-in c-myc genes willnot be expressed unless Cre recombinase is present. We haveestablished homozygous breeding pairs for each of thesestrains, termed Rosa-Floxed-Stop (RFS)-mycWT, RFS-mycT58A,and RFS-mycS62A (Supplementary Fig. S1C). Primary MEFsfrom each strain were infected with Ad-Cre and nuclearexpression of HA-tagged c-Myc was verified by immunofluor-escence (Supplementary Fig. S2A). The mean half-life of c-MycWT, c-MycT58A, and c-MycS62A in the primary MEFs was19.5, 63.0, and 44.5 minutes, respectively, under serum-starvedconditions (Supplementary Fig. S2B). We did note variabilityin the half-life of the S62A mutant. Because we do not knowwhat E3 ligase targets this mutant, the reason for this varia-bility is currently unknown.
Initial characterization of RFS-mycWT/Wap-Cre,RFS-mycT58A/Wap-Cre, and RFS-mycS62A/Wap-Cre miceTo address the consequences of altering c-Myc T58 and S62
phosphorylation on c-Myc activity in the mammary gland, wecrossed our 3 RFS myc strains with WAP (whey acidic pro-tein)-Cre transgenic mice, which express Cre in the secretoryluminal and ductal epithelial cells of the mammary glandduring late pregnancy and lactation (31), and experimentalmice were subjected to 3 consecutive rounds of pregnancy/lactation. This resulted in approximately 50% Cre-mediatedrecombination in the mammary epithelial cells as detected bybreeding with the R26R reporter mouse (SupplementaryFig. S1D). PCR analysis of genomic DNA from various tissuesshowed that the recombined allele was primarily detected inmammary gland from RFS-mycmice with Wap-Cre, but not incontrol mice without Wap-Cre (Supplementary Fig. S1E). Theadvantage of this mouse model is that unlike other c-myctransgenic mouse models in which c-Myc is expressed at veryhigh levels, the ROSA26 promoter is relatively weak andubiquitously drives expression of only 2 copies of theknocked-in c-myc genes at near physiologic levels (Fig. 1B,Total-myc, Pregnancy; ref.32). We observe partial suppressionof the endogenous c-myc gene with expression of ectopicc-MycWT, c-MycT58A, and c-MycS62A in this system (data notshown), consistent with other reports (33, 34). As expected,expression of ectopic c-mycWT, c-mycT58A, and c-mycS62A
mRNA was equivalent and Cre-dependent (Fig. 1B, Ectopic-myc). Expression of ectopic c-MycT58A protein was on averagehigher than c-MycS62A, which was generally higher than c-MycWT (Fig. 1B, HA IP/Myc WB, and Ectopic Myc graph).However, total c-Myc levels measured in the IP input were notdistinguishable across all of the strains during pregnancy,
when the epithelial cells are stimulated to proliferate andendogenous c-Myc protein expression is high (Fig. 1B, Input-Myc and Total Myc graph). This result highlights the physio-logic expression level of this model. We also measured thehalf-life of the ROSA-driven c-Myc proteins in primary MECsunder serum starvation conditions (Fig. 1C). c-MycT58A hadthe longest half-life, followed by c-MycS62A and c-MycWT,consistent with their expression level (Fig. 1B), and consistentwith the MEF data (Supplementary Fig. S2B), although differ-ences in stability were not as pronounced as in the MEFs.
Although differences in expression of the ectopic Mycproteins was not dramatic, histologic examination of mam-mary glands from the RFS-myc/Wap-Cre strains during thethird pregnancy showed significant alterations in the numbersof lobule-alveolar units and mean alveolar area. Specifically,MycS62A-expressing glands showed significantly reduced num-bers of alveoli compared with control, whereas MycT58A hadthe opposite effect, with substantially expanded alveolar areasand the presence of enlarged ducts containing secretion(Fig. 1D and graph). Analysis of proliferation by BrdU incor-poration did not reveal any substantial differences betweenthe strains during pregnancy or early parous stage (data notshown).
Delayed and incomplete mammary gland involutionwith c-MycT58A expression
c-Myc is a potent inducer of apoptosis under growth-limit-ing conditions. Involution of the mammary gland followingweaning represents a period of intense apoptotic activityassociated with tissue remodeling, and c-Myc expressionhas been shown to play an important role in this process(25). We examined the effects of expressing c-MycT58A and c-MycS62A compared with c-MycWT on involution. Histologicanalysis of mammary glands 3 days postweaning showed clearsigns of involution with shrunken alveoli and the reappear-ance of adipose cells in control, MycWT-, and MycS62A-expres-sing mice (Fig. 2A). In contrast, enlarged alveoli withprominent secretions predominated in MycT58A mammaryglands. Analysis of apoptotic cells by TUNEL staining showeda dramatic reduction in apoptosis in MycT58A-expressingmammary glands (Fig. 2B and C). This reduction in apoptosisand impaired involution likely contributed to the expandedlobule-alveolar areas, and the persistence of enlarged secre-tion-filled ducts in MycT58A mammary glands (Fig. 1D). Incontrast, mammary glands expressing MycWT showed anincrease in apoptosis compared with control, consistent withwild-type c-Myc's known apoptotic activity (Fig. 2C). Thus,expression of c-MycT58A, but not c-MycS62A, inhibits the waveof apoptosis in the mammary gland following weaning.
Analysis of mammary glands at 3 days of involution for theexpression of several proteins involved in apoptosis showedthat proapoptotic Bim (both BimEL and BimML) is reduced inMycT58A-expressing mammary glands relative to the otherstrains (Fig. 2D and graphs). No significant change in expres-sion of antiapoptotic Bcl2, or proapoptotic Bax or p53 wasfound across all 4 strains (data not shown). Together, thesedata show that c-MycT58A expression suppresses apoptosis inthe mammary gland at least in part due to downregulation of
c-Myc Phosphorylation Affects Its Activity In Vivo
www.aacrjournals.org Cancer Res; 71(3) February 1, 2011 927
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032
A
B
C
D
Figure 1. Generation and characterization of RFS-mycWT/WAP-Cre, RFS-mycT58A/WAP-Cre, and RFS-mycS62A/WAP-Cre mice. A, knock-in strategy forconditional expression of c-mycWT-HA, c-mycT58A-HA, or c-mycS62A-HA. Arrowheads, loxP sites. tpA is a transcription stop sequence. Inserted c-myc cDNAshave a C-terminal HA tag. Cre recombination activates expression of the inserted c-myc cDNA driven from the ROSA promoter. RFS, ROSA Floxed Stop; seeSupplementary Fig. S1 for more detail. B, expression of ROSA-driven c-Myc. Top, RNA was isolated frommammary glands from the indicated control (ctrl: noWap-Cre) or RFS-myc/WAP-Cre strains (WT, T58A, S62A) at the indicated stages: pregnancy 18 days, lactation 18 days, parous 2months. HA-tagged ectopicand total c-myc mRNA is shown by RT-PCR. Bottom, lysates from mammary glands from 2 mice per indicated strain harvested at pregnancy day 17 wereimmunoprecipitated with anti-HA followed by Western blotting with anti-c-Myc. Input total c-Myc and actin are shown. Data are representative of 4 mice pergenotype. Graphs, quantification of the actin-normalized expression of HA-tagged ectopic and total c-Myc from 3mice per genotype� SD. P values are givenon graphs with significant differences. C, analysis of ectopic c-Myc half-life in primary MECs from the indicated strains. Mean half-life � SD was calculatedfrom 3 independent experiments. CHX, cycloheximide. D, expanded lobule-alveolar areas with c-MycT58A expression. The fourth glands from the indicatedmice on day 17 of the third consecutive pregnancy were analyzed by H&E section staining. Arrowhead, enlarged duct. Scale bars, 50 mm. Data arerepresentative of 4mice per genotype. Mean alveolar area was analyzed by counting 10 areas each section and 3mice per genotype by ImageJ and graphed�SD.
Wang et al.
Cancer Res; 71(3) February 1, 2011 Cancer Research928
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

Bim proteins, which have been shown to play an importantrole in mammary gland apoptosis (35).
Increased chromosomal instability and centrosomeamplification in mammary epithelial cells with alteredc-Myc phosphorylationAberrant c-Myc expression has been associated with
increased genomic instability both in cell culture and invivo, which can contribute to the cancer phenotype (36). Toexamine whether deregulated c-Myc T58 and S62 phosphor-ylation can affect genomic stability, we analyzed and quan-tified chromosome numbers in mitotic spreads from firstpassage primary MECs harvested at 2 months parous. Wefound the highest rates of polyploidy in the MycT58A MECswith a substantial number of cells showing greater thantetraploid karyotypes (Fig. 3A). In addition, G-banding ana-lyses revealed a high degree of aneuploidy in cells expressingboth MycT58A and MycS62A, with 9 of 20 cells analyzed foreach showing abnormalities including nonclonal trisomies,translocations, chromosomal breakages, and various extraand missing chromosomes (Fig. 3B). In contrast, MycWT-expressing cells showed only 4 of 20 cells with abnormalkaryotypes compared with control cells with 2 of 20 abnor-mal cells (data not shown).To examine potential molecular alterations that could
contribute to chromosomal instability, we examined theexpression of several genes important for the spindle assemblycheckpoint (SAC) in 2-month parous mammary glands. Inter-estingly, we found that expression of Bub1b, a key player inthis pathway, was substantially downregulated in MycS62A-expressing mammary glands whereas it was substantiallyupregulated in MycT58A-expressing glands (Fig. 3C). We alsofound that expression of Aurora kinase B, a chromosomalpassenger serine/threonine protein kinase involved in theSAC, was dramatically upregulated with expression of MycT58A
whereas it was substantially downregulated with expression ofMycS62A (Fig. 3C), and Aurora A showed a similar trend, butless dramatic (data not shown). Because precise levels of theseproteins are critical for proper SAC function (37–40), theseresults may partially explain the enhanced genomic instabilitywith MycT58A and MycS62A expression.Abnormalities in the SAC leading to failed cytokinesis can
result in centrosome amplification, as can overexpression ofAurora kinases (38). Immunofluorescent staining for centro-somes revealed clear examples of abnormal numbers ofcentrosomes in MycT58A MECs (Fig. 3D). Quantificationrevealed a high degree of centrosome amplification inMycT58A MECs, compared with MycS62A and MycWT, whichwere still significantly higher than control cells (Fig. 3D,graph). Together, these results reveal a potential for c-Myc–associated genomic instability in our mouse model, andexpression of c-MycT58A and c-MycS62A seem to exacerbatethis in different ways.
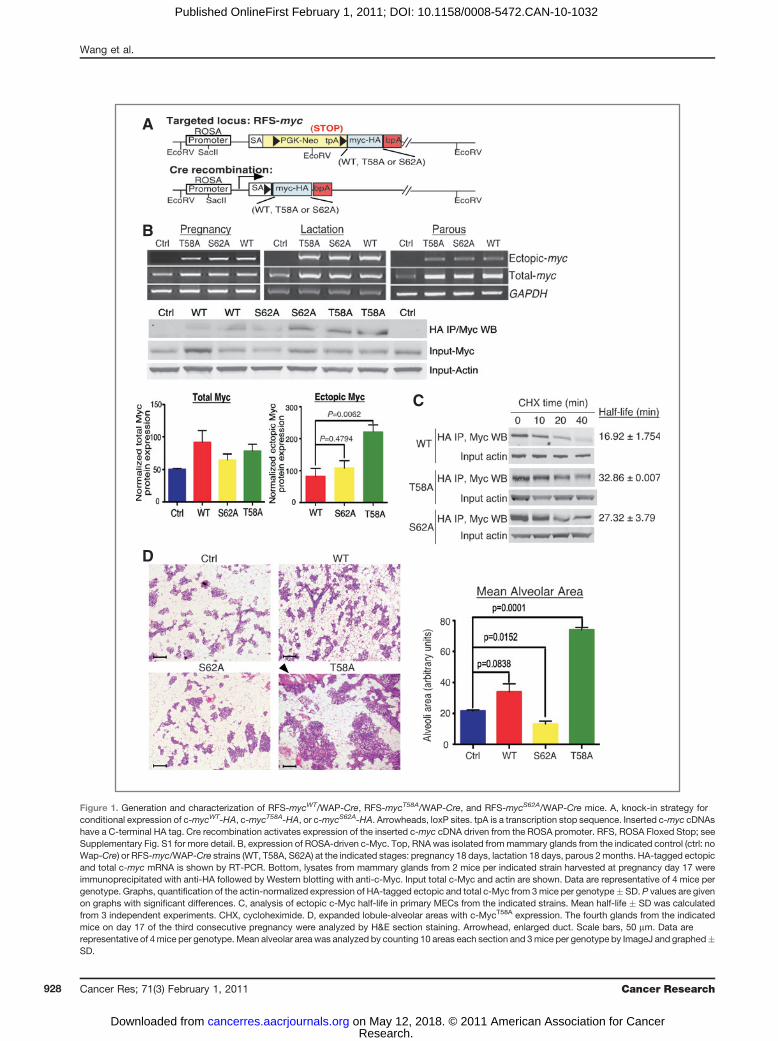
Altered mammary gland morphology with expression ofthe Myc phosphorylation mutantsWe assessed the phenotypic effects of expressing ROSA-
driven c-MycWT, c-MycT58A, and c-MycS62A in the mammary
gland 2 and 5 months parous after the third pregnancy andfound substantial differences in morphology at both timepoints (Fig. 4A; Supplementary Fig. S3). Expression of MycWT
showed a consistent increase in mammary ductal branchingrelative to control mammary glands, whereas MycS62A
showed a reduction, particularly when compared withMycWT. In contrast, expression of MycT58A resulted in dra-matically increased mammary ductal branching and alveolarbudding (Fig. 4A) and the appearance of hyperplastic fociresembling hyperplastic alveolar nodules by 5 months (Sup-plementary Fig. S3A; Fig. 5A). H&E sections revealed normalarchitecture in all strains except MycT58A, in which expandedalveolar regions with increased stromal and epithelial cellswere detected (Fig. 4B; Supplementary Fig. S3B). Meanalveolar area reflected the observed changes in density withexpression of the 3 c-Myc isoforms (Fig. 4C). Further ana-lyses of mammary gland sections from the MycT58A mice at 5to 8 months parous revealed hyperplastic foci (Fig. 5A,middle) with high proliferation (Fig. 5A, right), areas ofdysplasia including atypical nuclei with multiple nucleoli(Fig. 5B, left), disorganized alveolar structures with hyper-chromatic crowded nuclei, loss of polarity, and mitoticfigures (Fig. 5B, middle), and abnormal foci with increasedstromal cells, atypical nuclei, and immune cell infiltrationincluding mast cells (Fig. 5B, right). Loss of polarity andalveolar disorganization was also apparent in costaining forthe luminal epithelial marker Keratin8/18 (green) and myoe-pithelial marker Keratin14 (red), which normally form asingle layer of supporting cells around the polarized luminalepithelial cells (Fig. 5C, left, arrow). Moreover, alveoli inMycT58A glands showed downregulation of E-cadherinexpression (red) compared with alveoli in MycWT glands(Fig. 5C, middle versus right), suggesting loosened adhesionof intercellular junctions in the luminal cells. H&E sectionsand K8/18, K14 staining also revealed ductal hyperplasia(Fig. 5D).
RFS-mycT58A/Wap-Cre mice develop mammary glandtumors
To study potential affects of altering c-Myc phosphoryla-tion on tumorigenesis, we followed cohorts of RFS-mycWT, T58A, and S62A/Wap-Cre and control RFS-myc (noCre) female mice that had undergone 3 cycles of pregnancy(25–28 mice per genotype). Neither the control nor RFS-mycWT/Wap-Cre mice developed mammary gland tumorsout to 2 years, although the latter expressed deregulatedc-Myc. In contrast, RFS-mycT58A/Wap-Cre mice developedmammary adenocarcinomas between 7 and 13 months ofage. These were poorly differentiated carcinomas of solid,cribriform, papillary, and/or adenosquamous architecture,many with necrosis (Fig. 6A and data not shown), and allwith a high proliferation index (Fig. 6B and data not shown).Invasion was indicated by absence or disruption of myope-pithelial smooth muscle actin (SMA), and K14 staining inmost of the tumors (Fig. 6C and data not shown; ref. 41).Several of these were accompanied by in situ carcinoma atthe periphery (data not shown). Moreover, one of thesetumors metastasized to lung, liver, and spleen, and the
c-Myc Phosphorylation Affects Its Activity In Vivo
www.aacrjournals.org Cancer Res; 71(3) February 1, 2011 929
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

A
BC
D
Figure 2. Inhibition of mammary gland involution and apoptosis with c-MycT58A expression. A, the fourth gland from the indicated mice was harvested after12 days of lactation, 3 days postweaning after the third pregnancy, and analyzed by H&E section staining. Scale bars, 50 mm. Data are representativeof 4 to 6 mice per genotype. B, c-MycT58A expression inhibits apoptosis during involution. Mammary gland sections as in A were analyzed by TUNEL assay.Scale bars, 50 mm. C, TUNEL assays as in B were quantified to determine the percent apoptotic epithelial cells. Graph, 4 mice per strain � SD, D, reducedexpression of proapoptotic Bim in mammary glands expressing c-MycT58A. Protein lysates from the indicated mice (2 per genotype) 3 days postweaning wereWestern blotted with the indicated antibodies and quantified. Data are representative of 4 mice per genotype. Average actin-normalized BimEL and BimMLexpression relative to control is graphed � SD.
Wang et al.
Cancer Res; 71(3) February 1, 2011 Cancer Research930
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

metastatic lesions also showed high rates of proliferation(Fig. 6D and data not shown). Of the 28 RFS-mycT58A/Wap-Cre mice, 5 mammary gland tumors developed in 4mice (Table 1). Unfortunately, but of interest, the remainderof the RFS-mycT58A/Wap-Cre cohort succumbed to braintumors with a median survival of approximately 10.5 months(46 weeks; (Table 1). This corresponded with spuriousexpression of Wap-Cre in the newborn mouse brain (Sup-plementary Fig. S4) as previously reported (31). Choroidplexus and pituitary tumors were observed. Interestingly,RFS-mycS62A/Wap-Cre mice also developed the same spec-trum of brain tumors with a median survival of 12.5 months(54 weeks; Table 1). Of the RFS-mycT58A/Wap-Cre and RFS-mycS62A/Wap-Cre mice with pituitary tumors, one each also
developed a mammary tumor. Owing to potential confound-ing effects from prolactin in mice with pituitary hyperplasiaor tumors, any mice exhibiting these features were excludedfrom data presented in this paper. Analysis of the 7 MycT58A
mice that succumbed to choroid plexus tumors withoutevidence of abnormal pituitary, showed that 80% had aty-pical mammary gland changes similar to those shown inFigure 5, suggesting that given more time, these mice mayalso have developed mammary gland tumors (data notshown). In contrast, MycS62A mammary glands from micesuccumbing to choroid plexus tumors only, displayedreduced density similar to that shown in Figure 4 andSupplementary Figure S3 (data not shown). Together, theseresults indicate that phosphorylation of c-Myc at T58 and
Figure 3. Increased genomicinstability with expression of c-Myc phosphorylation mutants. A,mitotic spreads from primaryMECs from the indicated strainswere analyzed for chromosomenumbers. Average percent mitoticcells with diploid (40chromosomes) and hyperdiploidkaryotypes from 3 mice per strain� SD is graphed. B, expression ofc-MycT58A and c-MycS62A inducesaneuploidy. Representative G-banding of mitotic cells fromcontrol mice or mice expressing c-MycT58A or c-MycS62A are shown.Cells were prepared as in A.Twenty mitotic cells wereanalyzed per strain. C, substantialand opposing changes inexpression of spindle checkpointgenes with c-MycT58A and c-MycS62A expression. RNA wasisolated from 2-month parousmammary glands from theindicated strains. Quantitative RT-PCR analysis for the expression ofBub1b and Aurora-B genesrelative to control mice aregraphed � SD. Data represent 4to 5 mice per strain. D, substantialcentrosome amplification withexpression of c-MycT58A.Centrosomes were visualizedby antipericentrinimmunofluorescence staining(green) and DNA by 40,6 diamidino2 phenylindole (DAPI; blue). Cellswere prepared as in A.Representative MycT58A cells areshown. Centrosome amplification(more than 2 per mitotic cell) wasquantified from 3 mice perindicated strains and graphed �SD.
A
C
D
B
Normal centrosomes Abnormal centrosomes
c-Myc Phosphorylation Affects Its Activity In Vivo
www.aacrjournals.org Cancer Res; 71(3) February 1, 2011 931
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032
S62 has profound and differential affects on c-Myc's onco-genic potential in the mammary gland.
Discussion
Although c-MYC is a well-known oncogene, many questionsremain about how deregulation of c-Myc leads to tumorigen-esis. c-Myc is constitutively overexpressed in the majority ofhuman tumors, and it is clear that the expression level of c-Myc helps to determine its oncogenic activity (32, 34). Spe-cifically, deregulated low levels of c-Myc increase cell prolif-eration without engaging tumor suppressor pathways toinduce apoptosis (32). Higher levels of c-Myc activate bothproliferation and apoptosis (3, 32), and high levels of c-Myc areassociated with genomic instability, which can contribute tocancer development (36). c-Myc expression level is in partcontrolled by phosphorylation at T58 and S62 that affect c-Myc stability. In this study, we investigated the consequencesof altering T58 and S62 phosphorylation in vivo for mammarygland development and tumorigenesis. We found that the levelof T58 and S62 phosphorylation can have dramatic effects onspecific activities of c-Myc in the mammary gland. Theseresults have important implications for the oncogenic func-tion of c-Myc with altered phosphorylation that we findexpressed in human breast cancer (Ref. 13; Sears lab, unpub-lished data.
T58 and S62 phosphorylation affects c-Myc activity inthe mammary gland
Previous reports have indicated that mutation of T58 orS62 to alanine affects c-Myc–mediated cell transformation intissue culture in which T58A Myc is more transforming andS62A less transforming compared with c-MycWT (18–20). Inaddition, a study using retrovirally transduced hematopoie-tic stem cells (HSCs) expressing c-MycWT or c-MycT58A
showed more robust lymphomagenesis with c-MycT58A
expression (42). Although these studies all point to a rolefor these phosphorylation sites in regulating c-Myc function,we have found that high expression of c-Myc, whether wild-type or phosphorylation mutant, can swamp out or dilutedifferences in their activity, making interpretation difficult(data not shown). In this regard, expressing c-MycWT, c-MycT58A, or c-MycS62A from the constitutive, but weak, ROSApromoter is an ideal system to study how these phosphor-ylation sites affect specific activities of c-Myc in vivo atphysiologic levels both in development and for tumorigen-esis. We focused on the mammary gland as it is a highlydynamic organ, which undergoes repeated cycles of prolif-eration and apoptosis, and c-Myc is involved in both of theseprocesses (24, 25). We found that expression of c-MycT58A
inhibited the apoptotic program of involution associatedwith reduced proapoptotic Bim expression, similar toMycT58A-expressing HSCs (42), whereas c-MycWT increased
A
BC
Figure 4. Characterization ofmammary glands expressing c-MycWT, c-MycT58A, and c-MycS62A
at 2 months parous. A, the fourthglands from the indicated strainsof mice 2 months parous after thethird pregnancy were analyzed bywhole mount. Solid, darklystaining areas are lymph nodes.Scale bars, 5 mm (top row) and200 mm (bottom row). Imagesshown are representative of 3 to 4mice per genotype. B, H&Esections from mice as in A. Scalebars, 50 mm. Images arerepresentative of 4 mice pergenotype. C, expanded lobule-alveolar areas with c-MycT58A
expression. Mean alveolar areafrom mice as in A was analyzed bycounting 10 areas each H&Esection. Graph, three mice pergenotype � SD.
Wang et al.
Cancer Res; 71(3) February 1, 2011 Cancer Research932
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

apoptosis and did not impair involution even thoughc-MycWT is expressed at lower protein levels in our systemand thus should be less likely to induce apoptosis (32).Moreover, c-MycS62A, which is expressed at intermediatelevels between c-MycWT and c-MycT58A was also competentfor apoptosis and involution. These results point to differ-ential specific activities of c-Myc with altered phosphoryla-tion. However, since expression of the different c-Mycproteins in this system is not equivalent due to differencesin their stability (see Fig. 1), we cannot definitively distin-guish quantitative versus qualitative effects.We also found that changes in the phosphorylation of T58
and S62 affected genomic stability in primary MECs isolatedfrom the mice. Although expression of c-MycWT showed some
increase in genomic instability over control, expression of c-MycT58A substantially increased chromosomal instability asso-ciated with dramatically increased expression of SAC proteins,Bub1b and Aurora B, which are overexpressed in many humancancers associated with chromosomal instability (37, 38).Interestingly, the expression of c-MycS62A was also associatedwith significant chromosomal instability, particularly evidentin the G-banding studies, but it had the opposite effect on theexpression of Bub1b and Aurora B, which showed substan-tially reduced expression. Decreased expression of these pro-teins is also associated with aneuploidy, but generally resultsin an aging phenotype, growth arrest, and apoptosis (39, 40),which could contribute to the reduced mammary glanddensity observed in the MycS62A mice.
Figure 5. Preneoplastic changeswith expression of c-MycT58A at 5to 8 months parous. A,hyperplastic foci in RFS-mycT58A/WAP-Cre mammary glands 5 to8 months parous after the thirdpregnancy. Whole mount, H&Esection, andimmunohistochemistry for BrdUlabeling are shown. Arrow in wholemount, hyperplastic foci. In H&E,dark blue/purple representscalcifications in acinar lumens. B,areas of cellular dysplasia inMycT58A mammary glands. H&Esections from MycT58A mice as inA. Arrows (from left to right),atypical nuclei with multiplenucleoli, mitotic figure, and mastcell. C, alveolar disorganizationand reduced adhesion in MycT58A
mammary glands.Immunofluorescence costainingfor luminal markerK8/18 (green) and myoepithelialmarker K14 (red) in mice as in A.Arrow, disorganized alveolarregion. E-cadherin staining (red),DAPI stained nuclei (blue) inMycT58A (middle) and MycWT
(right) mammary glands. D, ductalhyperplasia with c-MycT58A
expression. H&E staining andimmunofluorescence K8/18(green)/K14 (red) in mice as in A.Dilated ducts with proliferativeepithelium (tufting) are shown. Allwhole mount and H&E images arefrom different MycT58A mice andare representative of 8 RFS-mycT58A/WAP-Cre mice 5 to8 months parous.Immunofluorescence images arerepresentative of 3 mice pergenotype. All scale bars, 50 mm.
A
B
C
D
c-Myc Phosphorylation Affects Its Activity In Vivo
www.aacrjournals.org Cancer Res; 71(3) February 1, 2011 933
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

Analysis of mammary glands over time revealed thatMycWT-expressing mammary glands had increased ductalbranching, but maintained phenotypically normal alveoli,whereas MycT58A-expressing glands had increased ductalbranching, and developed expanded and disorganized alveoli.We speculate that increased genomic instability in the face ofreduced apoptotic potential in the MycT58A mammary glandscontributes to their dysplastic phenotype. In contrast, MycS62A
mammary glands generally showed reduced ductal branchingwith fewer alveoli. In this case, genomic instability in the faceof competent activation of apoptotic programs could lead toelimination of aneuploid cells, potentially explaining the
reduction in mammary gland density in the MycS62A glands.In addition, recent publications report that phosphorylationat S62 is important to inhibit Ras-induced cellular senescenceand that reducing pS62 through elimination of Cdk2 increasesc-Myc–driven senescence (43, 44). Increased cellular senes-cence in the MycS62A-expressing glands could also contri-bute to their reduced density, and future experiments willaddress this. In addition, future ChIP-Seq experiments toidentify global differences in DNA binding among the 3isoforms of c-Myc in MECs from our model may provideinsight into how the T58 and S62 phosphorylation sitesaffect c-Myc function.
A
D
B
C
Figure 6. Expression of c-MycT58A
induces mammaryadenocarcinoma with an averagelatency of 9 months. A, examplesof mammary tumors in MycT58A-expressing glands. Grossphotograph of fifth gland tumor in10-month-old RFS-mycT58A/Wap-Cre mouse (top left), H&E sectionshowing the cribriformarchitecture of same (top right),adenosquamous tumor from 7-month RFS-mycT58A/Wap-Cremouse (bottom left),adenocarcinoma with solidarchitecture in different 10-monthRFS-mycT58A/Wap-Cre mouse(bottom right). B, high proliferationin MycT58A-driven tumor. Ki-67staining of tumor shown in A,bottom right. C, invasive featuresof tumor from MycT58A gland.Immunohistochemistry for SMA innormal duct (top) close to theperiphery of tumor shown in A, topright, and loss of SMA expressionin the tumor (bottom). D,metastasis to liver and lung ofMycT58A-inducedmammary tumorshown in A, bottom right. All scalebars, 50 mm.
Table 1. Tumorigenesis table of RFS-MycWT/Wap-Cre, RFS-MycT58A/Wap-Cre, and RFS-MycS62A/Wap-Cre mice
WT/Cre(n ¼ 27)
T58A/Cre(n ¼ 28)
S62A/Cre(n ¼ 28)
Control(n ¼ 25)
Mice with mammary tumor 0 4 0 0Mean mammary tumor latency 38 weeksMice with brain tumor 0 24 28 0Mean brain tumor latency 46 weeks 54 weeks
NOTE: Cohorts of the indicated numbers of RFS-MycWT/Wap-Cre, RFS-MycT58A/Wap-Cre, and RFS-MycS62A/Wap-Cre or control(RFS-MycWT, T58A, or S62A no Cre) were maintained for 24 months or until moribund with tumor. Numbers of mice with the indicatedtumor types along with mean latency are indicated.
Wang et al.
Cancer Res; 71(3) February 1, 2011 Cancer Research934
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

Phosphorylation at T58 and S62 impacts c-Myctumorigenic potentialMice with mammary gland expression of c-MycT58A, but not
c-MycWT or c-MycS62A, developed mammary carcinoma. Inaddition to increased genomic instability and suppressedapoptosis contributing to this, MycT58A glands showed disor-ganization of alveolar glandular architecture. Studies haveshown that expression of c-Myc does not induce proliferationin polarized epithelial cells in three-dimensional culture, butdisruption of polarity allows c-Myc driven proliferation (45);consistent with our observation of mitotic figures in dysplasticfoci in MycT58A glands. We also observed that c-MycT58A–expressing mammary glands had increased STAT3 activation(data not shown), which is common in human breast cancerand contributes to the disruption of epithelial adhesion andpolarity (46). Thus, disruption of alveolar architecture couldallow for inappropriate proliferation with accompanying geno-mic instability, promoting tumorigenesis in MycT58A glands.In contrast, although c-MycWT–expressing mammary
glands showed increased density, they seemed morphologi-cally normal, suggesting that the deregulated, but nearphysiologic expression of c-MycWT from the ROSA locuswas able to achieve some sort of homeostatic, nontumori-genic balance. This provides a potentially important newmodel for examining collaborative interactions with c-Mycin vivo, which will be important to exploit in future studies.Interestingly, c-MycS62A–expressing mammary glands, whichgenerally showed higher c-Myc protein expression comparedwith c-MycWT glands, had reduced density; nevertheless,expression of c-MycS62A was tumorigenic in the choroidplexus and pituitary. This finding is difficult to reconcile
with both our data showing decreased mammary glanddensity with c-MycS62A expression and recent reports sug-gesting that decreased S62 phosphorylation may preferen-tially drive senescence (43). One possibility is that neuralepithelial tissues are much less sensitive to drivers of apop-tosis and senescence, and in this setting, the increasedgenomic instability with c-MycS62A is protumorigenic. Wealso cannot rule out the possibility that c-MycS62A expressionin the mammary gland could eventually be tumorigenic,similar to the Bub1b�/� mice, which show increased geno-mic instability and aging phenotypes, with cancer predis-position (39).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank our lab colleagues for comments and feedback.
Grant Support
This study was supported by the Department of Defense Breast CancerResearch Program award BC061306 to R.C. Sears, the Susan G. Komen BreastCancer Foundation awards BCTR0201697 and BCTR0706821 to R.C. Sears, andthe NIH award 1 R01 CA129040-01 to R.C. Sears.
The costs of publication of this article were defrayed in part by the paymentof page charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received March 25, 2010; revised November 1, 2010; accepted November 23,2010; published OnlineFirst January 25, 2011.
References1. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer
2008;8:976–90.2. Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human
neoplastic disease. Oncogene 1999;18:3004–16.3. D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody
SE, et al. c-MYC induces mammary tumorigenesis by means of apreferred pathway involving spontaneous Kras2 mutations. Nat Med2001;7:235–9.
4. Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of lifeand death. Nat Rev Cancer 2002;2:764–76.
5. Khan M, Evan G, Pelengaris S. Suppression of Myc-induced apop-tosis in beta cells exposes multiple oncogenic properties of Myc andtriggers carcinogenic progression. Nat Rev Cancer 2002;2:764–76.
6. Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, BrooksM, et al.Induction of apoptosis in fibroblasts by c-myc protein. Cell1992;69:119–28.
7. Bieche I, Laurendeau I, Tozlu S, Olivi M, Vidaud D, Lidereau R, VidaudM. Quantitation of MYC gene expression in sporadic breast tumorswith a real-time reverse transcription-PCR assay. Cancer Res1999;59:2759–65.
8. Deming SL, Nass SJ, Dickson RB, Trock BJ. C-myc amplification inbreast cancer: a meta-analysis of its occurrence and prognosticrelevance. Br J Cancer 2000;83:1688–95.
9. Agnantis NJ, Mahera H, Maounis N, Spandidos DA. Immunohisto-chemical study of ras andmyc oncoproteins in apocrine breast lesionswith and without papillomatosis. Eur J Gynaecol Oncol 1992;13:309–15.
10. Chrzan P, Skokowski J, Karmolinski A, Pawelczyk T. Amplificationof c-myc gene and overexpression of c-Myc protein in breastcancer and adjacent non-neoplastic tissue. Clin Biochem 2001;34:557–62.
11. Sears RC. The life cycle of C-myc: from synthesis to degradation. CellCycle 2004;3:1133–7.
12. Welcker M, Orian A, Jin J, Grim JA, Harper JW, Eisenman RN, ClurmanBE. The Fbw7 tumor suppressor regulates glycogen synthase kinase3 phosphorylation-dependent c-Myc protein degradation. Proc NatlAcad Sci U S A 2004;101:9085–90.
13. Arnold HK, Zhang X, Daniel CJ, Tibbitts D, Escamilla-Powers J, FarrellA, et al. The Axin1 scaffold protein promotes formation of a degrada-tion complex for c-Myc. EMBO J 2009;28:500–12.
14. O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, et al. FBW7mutations in leukemic cells mediate NOTCH pathway activation andresistance to gamma-secretase inhibitors. J Exp Med 2007;204:1813–24.
15. Janssens V, Goris J, Van Hoof C. PP2A: the expected tumor sup-pressor. Curr Opin Genet Dev 2005;15:34–41.
16. Salahshor S, Woodgett JR. The links between axin and carcinogen-esis. J Clin Pathol 2005;58:225–36.
17. Chang DW, Claassen GF, Hann SR, Cole MD. The c-Myc transactiva-tion domain is a direct modulator of apoptotic versus proliferativesignals. Mol Cell Biol 2000;20:4309–19.
18. Pulverer BJ, Fisher C, Vousden K, Littlewood T, Evan G, Woodgett JR.Site-specific modulation of c-Myc cotransformation by residuesphosphorylated in vivo. Oncogene 1994;9:59–70.
c-Myc Phosphorylation Affects Its Activity In Vivo
www.aacrjournals.org Cancer Res; 71(3) February 1, 2011 935
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

19. Thibodeaux CA, Liu X, Disbrow GL, Zhang Y, Rone JD, Haddad BR,Schlegel R. Immortalization and transformation of human mammaryepithelial cells by a tumor-derived Myc mutant. Breast Cancer ResTreat 2009;116:281–94.
20. Yeh E, CunninghamM, Arnold H, Chasse D, Monteith T, Ivaldi G, et al.A signalling pathway controlling c-Myc degradation that impactsoncogenic transformation of human cells. Nat Cell Biol 2004;6:308–18.
21. Malempati S, Tibbitts D, Cunningham M, Akkari Y, Olson S, Fan G,Sears RC. Aberrant stabilization of c-Myc protein in some lympho-blastic leukemias. Leukemia 2006;20:1572–81.
22. Lutterbach B, Hann SR. Hierarchical phosphorylation at N-terminaltransformation-sensitive sites in c-Myc protein is regulated by mito-gens and in mitosis. Mol Cell Biol 1994;14:5510–22.
23. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. MultipleRas-dependent phosphorylation pathways regulate Myc protein sta-bility. Genes Dev 2000;14:2501–14.
24. Sodir NM, Evan GI. Nursing some sense out of Myc. J Biol2009;8:77.
25. Sutherland KD, Lindeman GJ, Visvader JE. The molecular culpritsunderlying precocious mammary gland involution. J Mammary GlandBiol Neoplasia 2007;12:15–23.
26. Schoenenberger CA, Andres AC, Groner B, Van Der Valk M, LeMeurM, Gerlinger P. Targeted c-myc gene expression in mammary glandsof transgenic mice induces mammary tumours with constitutive milkprotein gene transcription. EMBO J 1988;7:169–75.
27. Stewart TA, Pattengale PK, Leder P. Spontaneous mammary adeno-carcinomas in transgenicmice that carry and expressMTV/myc fusiongenes. Cell 1984;38:627–37.
28. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporterstrain. Nat Genet 1999;21:70–1.
29. Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunitB56alpha associates with c-myc and negatively regulates c-mycaccumulation. Mol Cell Biol 2006;26:2832–44.
30. Cardiff RD, Anver MR, Gusterson BA, Hennighausen L, Jensen RA,Merino MJ, et al. The mammary pathology of genetically engineeredmice: the consensus report and recommendations from the Anna-polis meeting. Oncogene 2000;19:968–88.
31. Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, GarrettL, et al. Cre-mediated gene deletion in the mammary gland. NucleicAcids Res 1997;25:4323–30.
32. Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, BuiDA, Brown-Swigart L, Johnson L, Evan GI. Distinct thresholdsgovern Myc's biological output in vivo. Cancer Cell 2008;14:447–57.
33. Blakely CM, Sintasath L, D’Cruz CM, Hahn KT, Dugan KD, Belka GK,Chodosh LA. Developmental stage determines the effects of MYC inthe mammary epithelium. Development 2005;132:1147–60.
34. Smith DP, Bath ML, Metcalf D, Harris AW, Cory S. MYC levels governhematopoietic tumor type and latency in transgenic mice. Blood2006;108:653–61.
35. Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A,Brugge JS. BIM regulates apoptosis during mammary ductal mor-phogenesis, and its absence reveals alternative cell death mechan-isms. Dev Cell 2007;12:221–34.
36. Felsher DW, Bishop JM. Transient excess of MYC activity can elicitgenomic instability and tumorigenesis. Proc Natl Acad Sci U S A1999;96:3940–4.
37. Scintu M, Vitale R, Prencipe M, Gallo AP, Bonghi L, Valori VM, et al.Genomic instability and increased expression of BUB1B andMAD2L1genes in ductal breast carcinoma. Cancer Lett 2007;254:298–307.
38. Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumouramplified kinase STK15/BTAK induces centrosome amplification,aneuploidy and transformation. Nat Genet 1998;20:189–93.
39. Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S,Kopecka A, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat Genet 2004;36:744–9.
40. HardwickeMA, Oleykowski CA, Plant R,Wang J, Liao Q,Moss K, et al.GSK1070916, a potent Aurora B/C kinase inhibitor with broad anti-tumor activity in tissue culture cells and human tumor xenograftmodels. Mol Cancer Ther 2009;8:1808–17.
41. Sternlicht MD, Barsky SH. The myoepithelial defense: a host defenseagainst cancer. Med Hypotheses 1997;48:37–46.
42. Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA,Cordon-Cardo C, et al. Evasion of the p53 tumour surveillance net-work by tumour-derived MYC mutants. Nature 2005;436:807–11.
43. Campaner S, Doni M, Hydbring P, Verrecchia A, Bianchi L, Sardella D,et al. Cdk2 suppresses cellular senescence induced by the c-myconcogene. Nat Cell Biol 2010;12:54–9;sup 1–14.
44. Hydbring P, Bahram F, Su Y, Tronnersjo S, Hogstrand K, von der LehrN, et al. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc Natl Acad Sci U S A2010;107:58–63.
45. Partanen JI, Nieminen AI, Makela TP, Klefstrom J. Suppression ofoncogenic properties of c-Myc by LKB1-controlled epithelial organi-zation. Proc Natl Acad Sci U S A 2007;104:14694–9.
46. Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G,Giancotti FG. Beta 4 integrin amplifies ErbB2 signaling to promotemammary tumorigenesis. Cell 2006;126:489–502.
Wang et al.
Cancer Res; 71(3) February 1, 2011 Cancer Research936
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032

2011;71:925-936. Published OnlineFirst February 1, 2011.Cancer Res Xiaoyan Wang, Melissa Cunningham, Xiaoli Zhang, et al. Mammary GlandPhosphorylation Regulates c-Myc's Oncogenic Activity in the
Updated version
10.1158/0008-5472.CAN-10-1032doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2011/01/25/0008-5472.CAN-10-1032.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/71/3/925.full#ref-list-1
This article cites 46 articles, 14 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/71/3/925.full#related-urls
This article has been cited by 24 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
SubscriptionsReprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. (CCC)Click on "Request Permissions" which will take you to the Copyright Clearance Center's
.http://cancerres.aacrjournals.org/content/71/3/925To request permission to re-use all or part of this article, use this link
Research. on May 12, 2018. © 2011 American Association for Cancercancerres.aacrjournals.org Downloaded from
Published OnlineFirst February 1, 2011; DOI: 10.1158/0008-5472.CAN-10-1032
![OPEN ACCESS viruses - Semantic Scholar...III alpha (PIKIII α) binds to and regulates th e phosphorylation status of NS 5A [14]. The phosphorylation state likely plays an important](https://img.pdfslide.net/doc/110x75/60f6ad79a1e0ef6b36059fb3/open-access-viruses-semantic-scholar-iii-alpha-pikiii-binds-to-and-regulates.jpg)