Embed Size (px)
Citation preview

PHYSICAL REVIEW B 98, 235426 (2018)
First-principles calculations of the ultralow thermal conductivityin two-dimensional group-IV selenides
Peng-Fei Liu,1,2,3 Tao Bo,1,2 Juping Xu,1,2 Wen Yin,1,2 Junrong Zhang,1,2 Fangwei Wang,2,4
Olle Eriksson,5 and Bao-Tian Wang1,2,3,*
1Institute of High Energy Physics, Chinese Academy of Sciences (CAS), Beijing 100049, China2Dongguan Neutron Science Center, Dongguan 523803, China
3Collaborative Innovation Center of Extreme Optics, Shanxi University, Taiyuan, Shanxi 030006, China4Beijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100080, China
5Department of Physics and Astronomy, Division of Materials Theory, Uppsala University, Box 516, SE-75120 Uppsala, Sweden
(Received 23 September 2018; revised manuscript received 6 December 2018; published 21 December 2018)
Through first-principles calculations, we report on the phonon-limited transport properties of two-dimensional(2D) hexagonal MSe (M = Ge, Sn, and Pb) compounds, which can be seen as a new family of 2D group-IVselenides established by the isovalent substitutions of germanium and tellurium in layered Ge4Se3Te phase[Angew. Chem. Inter. Edit. 56 10204 (2017)]. We find that 2D PbSe exhibits low values of sound velocity(800–2030 m/s), large Gruneisen parameters (∼1.93), low-lying optical modes (∼20.02 cm−1), and strongoptical-acoustic phonon coupling. These intrinsic properties mainly stem from strong phonon anharmonicity,which greatly suppress the phonon transport and therefore give rise to an ultralow thermal conductivity(∼0.26 W m−1 K−1) for 2D PbSe at room temperature. Our studies may offer perspectives for applications ofthermoelectricity and motivate further experimental efforts to synthesize MSe compounds.
DOI: 10.1103/PhysRevB.98.235426
I. INTRODUCTIONS
Thermoelectricity provides a promising route for convert-ing waste thermal energies into useful electrical energies di-rectly, which is of great significance for energy harvesting. Ingeneral, the conversion efficiency of thermoelectric materialscan be characterized by the well-defined figure of merit [1,2],
ZT = σS2T
κe + κL
, (1)
where S, T, σ , κe, and κL are Seebeck coefficient, temper-ature, electric conductivity, electronic thermal conductivity,and lattice thermal conductivity, respectively. The commonstrategies to increase ZT mainly focus on optimizing electricaltransport property (S, σ ) by band-structure engineering [3]and/or reducing the heat transfer ability (κe + κL) throughalloying and nanostructuring [4,5]. Meanwhile, screening ma-terials with intrinsically low thermal conductivity is anotherimportant trend [6]. This method helps people to simplifycomplex thermoelectric parameters and focus on the optimiza-tion of electrical transport properties alone [7]. Usually, κL ofa crystalline material can be simply evaluated by the Slacktheory [8,9],
κL = AM�3δn1/3
γ 2T, (2)
where A is a physical constant, M is the average mass peratom in the crystal, � is the Debye temperature, δ is the cuberoot of the average volume per atom, n is the number of atoms
*Corresponding author: [email protected]
in the primitive unit cell, and γ is the Gruneisen parameter.Obviously, seeking materials with large average atomic massand strong anharmonicity is crucial for ideal candidates.
Theoretical and experimental studies have revealed thatgroup IV–VI compounds have the strong anharmonicity withlow κL [2,10–15]. For example, PbTe, a representative ofmedium-temperature thermoelectric material, has a very lowthermal conductivity (∼2 W m−1 K−1 at 300 K) [16]. The un-derlying microscopic mechanism mainly stems from its stronganharmonic interaction between the transverse optical andlongitudinal acoustic branches [10]. Ab initio approaches andinelastic neutron-scattering measurements have provided im-portant insights of the softening of the transverse optical modeaffecting the κL [2,13–15]. Similarly in the phosphorene-likeSnSe material, it has attracted intense attention for its highZT (2.6 ± 0.3 at T = 935 K) and the intrinsically ultralowthermal conductivity (0.25 W m−1 K−1) [17]. The intrinsicheat-conducting property in phosphorene-like SnSe has beenrevealed via theoretical calculations and experiment by stronganharmonicity in terms of large Gruneisen parameters. Suchfeatures could be ascribed to the weak Sn-Se atomic bonds[11,12,17]. With the outstanding electrical transport proper-ties [7], the simple material SnSe really surprises the scientificcommunity as a promising thermoelectric material [18–20].
Recently, the single crystal structure of Ge4Se3Te has beenexperimentally refined [21], although the hexagonal phasefor GeSe1−xTex with 0.1 � x � 0.5 was first reported inthe 1960s by Muir and Cashman [22]. Different from theconventional GeSe polymorphs [23], the refined Ge4Se3Tephase crystallizes in the hexagonal space group with a stackof two Q-Ge-Ge-Q layers (with Q being Se and Te in a 3:1ratio) in one unit. This reveals a novel layered material with
2469-9950/2018/98(23)/235426(7) 235426-1 ©2018 American Physical Society
LIU, BO, XU, YIN, ZHANG, WANG, ERIKSSON, AND WANG PHYSICAL REVIEW B 98, 235426 (2018)
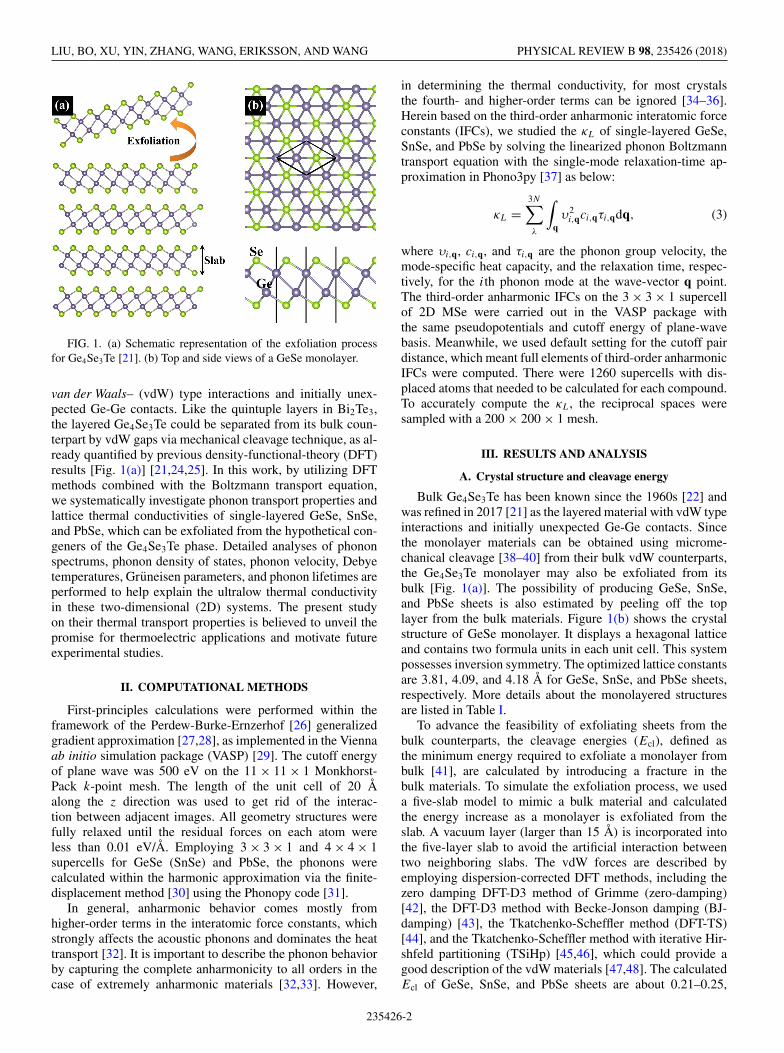
FIG. 1. (a) Schematic representation of the exfoliation processfor Ge4Se3Te [21]. (b) Top and side views of a GeSe monolayer.
van der Waals– (vdW) type interactions and initially unex-pected Ge-Ge contacts. Like the quintuple layers in Bi2Te3,the layered Ge4Se3Te could be separated from its bulk coun-terpart by vdW gaps via mechanical cleavage technique, as al-ready quantified by previous density-functional-theory (DFT)results [Fig. 1(a)] [21,24,25]. In this work, by utilizing DFTmethods combined with the Boltzmann transport equation,we systematically investigate phonon transport properties andlattice thermal conductivities of single-layered GeSe, SnSe,and PbSe, which can be exfoliated from the hypothetical con-geners of the Ge4Se3Te phase. Detailed analyses of phononspectrums, phonon density of states, phonon velocity, Debyetemperatures, Gruneisen parameters, and phonon lifetimes areperformed to help explain the ultralow thermal conductivityin these two-dimensional (2D) systems. The present studyon their thermal transport properties is believed to unveil thepromise for thermoelectric applications and motivate futureexperimental studies.
II. COMPUTATIONAL METHODS
First-principles calculations were performed within theframework of the Perdew-Burke-Ernzerhof [26] generalizedgradient approximation [27,28], as implemented in the Viennaab initio simulation package (VASP) [29]. The cutoff energyof plane wave was 500 eV on the 11 × 11 × 1 Monkhorst-Pack k-point mesh. The length of the unit cell of 20 Åalong the z direction was used to get rid of the interac-tion between adjacent images. All geometry structures werefully relaxed until the residual forces on each atom wereless than 0.01 eV/Å. Employing 3 × 3 × 1 and 4 × 4 × 1supercells for GeSe (SnSe) and PbSe, the phonons werecalculated within the harmonic approximation via the finite-displacement method [30] using the Phonopy code [31].
In general, anharmonic behavior comes mostly fromhigher-order terms in the interatomic force constants, whichstrongly affects the acoustic phonons and dominates the heattransport [32]. It is important to describe the phonon behaviorby capturing the complete anharmonicity to all orders in thecase of extremely anharmonic materials [32,33]. However,
in determining the thermal conductivity, for most crystalsthe fourth- and higher-order terms can be ignored [34–36].Herein based on the third-order anharmonic interatomic forceconstants (IFCs), we studied the κL of single-layered GeSe,SnSe, and PbSe by solving the linearized phonon Boltzmanntransport equation with the single-mode relaxation-time ap-proximation in Phono3py [37] as below:
κL =3N∑λ
∫qυ2
i,qci,qτi,qdq, (3)
where υi,q, ci,q, and τi,q are the phonon group velocity, themode-specific heat capacity, and the relaxation time, respec-tively, for the ith phonon mode at the wave-vector q point.The third-order anharmonic IFCs on the 3 × 3 × 1 supercellof 2D MSe were carried out in the VASP package withthe same pseudopotentials and cutoff energy of plane-wavebasis. Meanwhile, we used default setting for the cutoff pairdistance, which meant full elements of third-order anharmonicIFCs were computed. There were 1260 supercells with dis-placed atoms that needed to be calculated for each compound.To accurately compute the κL, the reciprocal spaces weresampled with a 200 × 200 × 1 mesh.
III. RESULTS AND ANALYSIS
A. Crystal structure and cleavage energy
Bulk Ge4Se3Te has been known since the 1960s [22] andwas refined in 2017 [21] as the layered material with vdW typeinteractions and initially unexpected Ge-Ge contacts. Sincethe monolayer materials can be obtained using microme-chanical cleavage [38–40] from their bulk vdW counterparts,the Ge4Se3Te monolayer may also be exfoliated from itsbulk [Fig. 1(a)]. The possibility of producing GeSe, SnSe,and PbSe sheets is also estimated by peeling off the toplayer from the bulk materials. Figure 1(b) shows the crystalstructure of GeSe monolayer. It displays a hexagonal latticeand contains two formula units in each unit cell. This systempossesses inversion symmetry. The optimized lattice constantsare 3.81, 4.09, and 4.18 Å for GeSe, SnSe, and PbSe sheets,respectively. More details about the monolayered structuresare listed in Table I.
To advance the feasibility of exfoliating sheets from thebulk counterparts, the cleavage energies (Ecl), defined asthe minimum energy required to exfoliate a monolayer frombulk [41], are calculated by introducing a fracture in thebulk materials. To simulate the exfoliation process, we useda five-slab model to mimic a bulk material and calculatedthe energy increase as a monolayer is exfoliated from theslab. A vacuum layer (larger than 15 Å) is incorporated intothe five-layer slab to avoid the artificial interaction betweentwo neighboring slabs. The vdW forces are described byemploying dispersion-corrected DFT methods, including thezero damping DFT-D3 method of Grimme (zero-damping)[42], the DFT-D3 method with Becke-Jonson damping (BJ-damping) [43], the Tkatchenko-Scheffler method (DFT-TS)[44], and the Tkatchenko-Scheffler method with iterative Hir-shfeld partitioning (TSiHp) [45,46], which could provide agood description of the vdW materials [47,48]. The calculatedEcl of GeSe, SnSe, and PbSe sheets are about 0.21–0.25,
235426-2
FIRST-PRINCIPLES CALCULATIONS OF THE ULTRALOW … PHYSICAL REVIEW B 98, 235426 (2018)
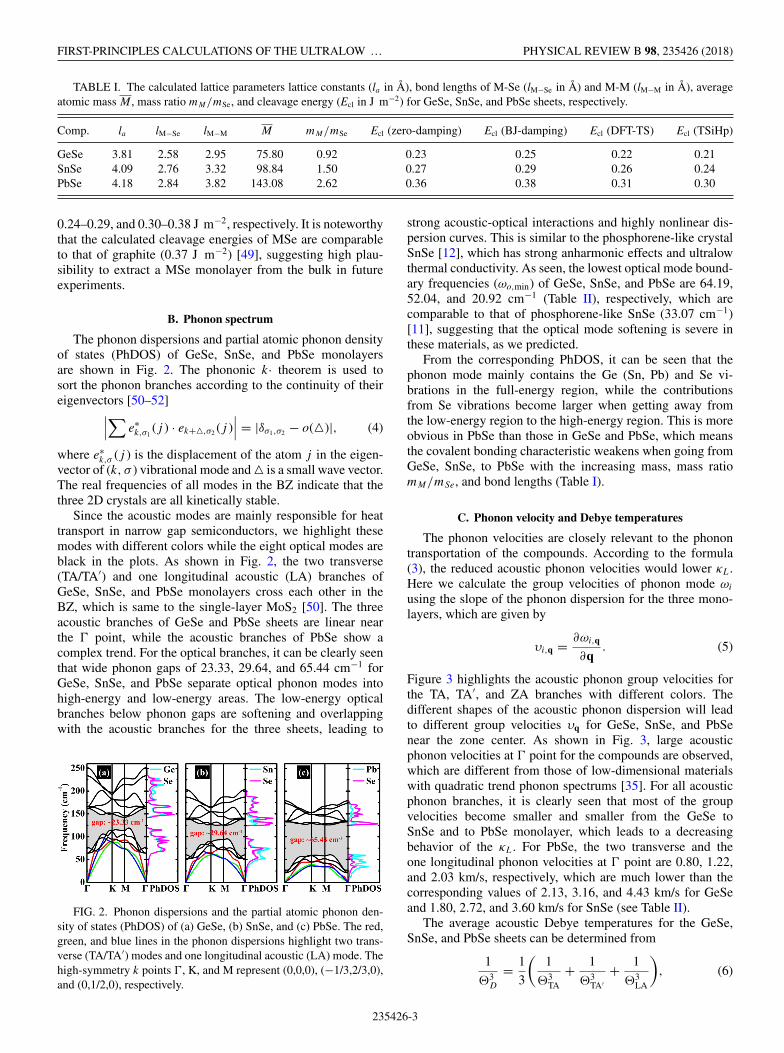
TABLE I. The calculated lattice parameters lattice constants (la in Å), bond lengths of M-Se (lM−Se in Å) and M-M (lM−M in Å), averageatomic mass M , mass ratio mM/mSe, and cleavage energy (Ecl in J m−2) for GeSe, SnSe, and PbSe sheets, respectively.
Comp. la lM−Se lM−M M mM/mSe Ecl (zero-damping) Ecl (BJ-damping) Ecl (DFT-TS) Ecl (TSiHp)
GeSe 3.81 2.58 2.95 75.80 0.92 0.23 0.25 0.22 0.21SnSe 4.09 2.76 3.32 98.84 1.50 0.27 0.29 0.26 0.24PbSe 4.18 2.84 3.82 143.08 2.62 0.36 0.38 0.31 0.30
0.24–0.29, and 0.30–0.38 J m−2, respectively. It is noteworthythat the calculated cleavage energies of MSe are comparableto that of graphite (0.37 J m−2) [49], suggesting high plau-sibility to extract a MSe monolayer from the bulk in futureexperiments.
B. Phonon spectrum
The phonon dispersions and partial atomic phonon densityof states (PhDOS) of GeSe, SnSe, and PbSe monolayersare shown in Fig. 2. The phononic k· theorem is used tosort the phonon branches according to the continuity of theireigenvectors [50–52]∣∣∣∑ e∗
k,σ1(j ) · ek+�,σ2 (j )
∣∣∣ = |δσ1,σ2 − o(�)|, (4)
where e∗k,σ (j ) is the displacement of the atom j in the eigen-
vector of (k, σ ) vibrational mode and � is a small wave vector.The real frequencies of all modes in the BZ indicate that thethree 2D crystals are all kinetically stable.
Since the acoustic modes are mainly responsible for heattransport in narrow gap semiconductors, we highlight thesemodes with different colors while the eight optical modes areblack in the plots. As shown in Fig. 2, the two transverse(TA/TA′) and one longitudinal acoustic (LA) branches ofGeSe, SnSe, and PbSe monolayers cross each other in theBZ, which is same to the single-layer MoS2 [50]. The threeacoustic branches of GeSe and PbSe sheets are linear nearthe � point, while the acoustic branches of PbSe show acomplex trend. For the optical branches, it can be clearly seenthat wide phonon gaps of 23.33, 29.64, and 65.44 cm−1 forGeSe, SnSe, and PbSe separate optical phonon modes intohigh-energy and low-energy areas. The low-energy opticalbranches below phonon gaps are softening and overlappingwith the acoustic branches for the three sheets, leading to
FIG. 2. Phonon dispersions and the partial atomic phonon den-sity of states (PhDOS) of (a) GeSe, (b) SnSe, and (c) PbSe. The red,green, and blue lines in the phonon dispersions highlight two trans-verse (TA/TA′) modes and one longitudinal acoustic (LA) mode. Thehigh-symmetry k points �, K, and M represent (0,0,0), (−1/3,2/3,0),and (0,1/2,0), respectively.
strong acoustic-optical interactions and highly nonlinear dis-persion curves. This is similar to the phosphorene-like crystalSnSe [12], which has strong anharmonic effects and ultralowthermal conductivity. As seen, the lowest optical mode bound-ary frequencies (ωo,min) of GeSe, SnSe, and PbSe are 64.19,52.04, and 20.92 cm−1 (Table II), respectively, which arecomparable to that of phosphorene-like SnSe (33.07 cm−1)[11], suggesting that the optical mode softening is severe inthese materials, as we predicted.
From the corresponding PhDOS, it can be seen that thephonon mode mainly contains the Ge (Sn, Pb) and Se vi-brations in the full-energy region, while the contributionsfrom Se vibrations become larger when getting away fromthe low-energy region to the high-energy region. This is moreobvious in PbSe than those in GeSe and PbSe, which meansthe covalent bonding characteristic weakens when going fromGeSe, SnSe, to PbSe with the increasing mass, mass ratiomM/mSe, and bond lengths (Table I).
C. Phonon velocity and Debye temperatures
The phonon velocities are closely relevant to the phonontransportation of the compounds. According to the formula(3), the reduced acoustic phonon velocities would lower κL.Here we calculate the group velocities of phonon mode ωi
using the slope of the phonon dispersion for the three mono-layers, which are given by
υi,q = ∂ωi,q
∂q. (5)
Figure 3 highlights the acoustic phonon group velocities forthe TA, TA′, and ZA branches with different colors. Thedifferent shapes of the acoustic phonon dispersion will leadto different group velocities υq for GeSe, SnSe, and PbSenear the zone center. As shown in Fig. 3, large acousticphonon velocities at � point for the compounds are observed,which are different from those of low-dimensional materialswith quadratic trend phonon spectrums [35]. For all acousticphonon branches, it is clearly seen that most of the groupvelocities become smaller and smaller from the GeSe toSnSe and to PbSe monolayer, which leads to a decreasingbehavior of the κL. For PbSe, the two transverse and theone longitudinal phonon velocities at � point are 0.80, 1.22,and 2.03 km/s, respectively, which are much lower than thecorresponding values of 2.13, 3.16, and 4.43 km/s for GeSeand 1.80, 2.72, and 3.60 km/s for SnSe (see Table II).
The average acoustic Debye temperatures for the GeSe,SnSe, and PbSe sheets can be determined from
1
�3D
= 1
3
(1
�3TA
+ 1
�3TA′
+ 1
�3LA
), (6)
235426-3

LIU, BO, XU, YIN, ZHANG, WANG, ERIKSSON, AND WANG PHYSICAL REVIEW B 98, 235426 (2018)
TABLE II. Summary of lowest optical frequency ωo,min in cm−1 at the � point, average transverse (TA/TA′) and longitudinal (LA)Gruneisen parameters (γ TA/TA′/LA), Debye temperatures (�TA/TA′/LA in K), and phonon velocities (υTA/TA′/LA in km s−1) for GeSe, SnSe,and PbSe sheets, respectively.
Comp. ωo,min γ TA γ TA′ γ LA �TA �TA′ �LA υTA υTA′ υLA
GeSe 64.19 0.92 0.68 0.60 126.68 137.34 136.45 2.13 3.16 4.43SnSe 52.04 0.97 1.29 0.68 87.52 96.95 105.60 1.80 2.72 3.60PbSe 20.92 1.79 1.72 2.72 46.43 57.56 70.49 0.80 1.22 2.03
where �i for each acoustic branch i (i = TA, TA′, and LA) isdefined as [53]
�i = ωi,max/kB, (7)
where ωi,max is the largest acoustic frequency of the ithacoustic mode and kB is Boltzmann constant. The Debyetemperature usually reflects the magnitude of phonon velocity[54]: The larger the phonon velocity, the higher the Debyetemperature. The average Debye temperature �D for PbSe iscalculated to be 54.94 K, much lower than those of 133.13 Kfor GeSe and 95.56 K for SnSe. This is consistent with thefact that the acoustic phonon velocities of PbSe are lower thanthose of GeSe and SnSe (Table II). The Debye temperatures ofthese 2D compounds are much lower than those of graphene[55], silicene [35], and phosphorene [56]. The extremely lowDebye temperature and slow speed of phonon may lead to anultralow κL in PbSe sheets according to Slack’s theory [8].
D. Gruneisen parameters
The Gruneisen parameter is a quantity describing the an-harmonic interactions of a crystal, which is useful for analyz-ing the physical nature of κL behavior. It can be calculateddirectly from the relationship between phonon frequency andvolume change as below [9]:
γi,q = −d ln ωi,q
d ln V. (8)
Generally, large |γ | implies strong anharmonicity, which ac-cordingly gives rise to low κL. To effectively evaluate the an-harmonicity, we plot the dispersion of the Gruneisen parame-ters of acoustic modes for MSe in Fig. 4. The most noteworthyfeature of these dispersions is the unusually ultrahigh valuesof Gruneisen parameters for PbSe monolayers, particularlyaround the � point, which implies that the acoustic branchesof PbSe monolayer are strongly anharmonic. These large-mode Gruneisen parameters at acoustic frequencies are crucial
FIG. 3. The phonon mode group velocities of (a) GeSe, (b) SnSe,and (c) PbSe. The three acoustic modes are shown as red, green, andblue lines, respectively.
to the phonon transport in materials, because most of the heatis carried by low-frequency acoustic phonons.
The average Gruneisen parameters (γ i) of each acousticdispersion are calculated by [17,53,57]
γ i =∑√
γ 2i,q∑
q. (9)
For GeSe sheets, the average acoustic Gruneisen parametersare γ TA = 0.92, γ TA′ = 0.68, and γ LA = 0.60, which are allless than 1 (Table II). The corresponding values for SnSe are0.97, 1.29, and 0.68, which are larger than those of GeSe butsmaller than the values (1.79, 1.72, and 2.72) of PbSe. Thefurther averages of the acoustic Gruneisens,
γ ′ = 13 (γ TA + γ TA′ + γ LA), (10)
are calculated to be 0.73, 0.98, and 1.93 for GeSe, SnSe, andPbSe, respectively. These values are comparable to the aver-age Gruneisen parameters of low-lattice-thermal-conductivitycompounds, such as layered phosphorene-like SnSe (2.83)[17], cubic PbTe (1.45) [8], Cu3SbSe4 (1.22) [54], andCu3SbSe3 (2.41) [54]. This means MSe sheets also havestrongly anharmonic vibrational properties. In addition, theacoustic γ ′ of PbSe is much larger than those of GeSe andSnSe, indicating that PbSe has even stronger anharmonicityand lower thermal conductivity.
E. Phonon lifetimes
According to the formula (3), the phonon relaxation time isproportional to the κL [37]. It can be solved numerically fromthe phonon linewidth 2�i (ωi ) of the phonon mode:
τi = 1
2�i (ωi ), (11)
where the �i (ω) takes a form analogous to the Fermi goldenrule and fully incorporated scatterings from all of the phonon
FIG. 4. Calculated Gruneisen dispersions for (a) GeSe, (b) SnSe,and (c) PbSe. The three acoustic modes are shown as red, green, andblue lines, respectively.
235426-4
FIRST-PRINCIPLES CALCULATIONS OF THE ULTRALOW … PHYSICAL REVIEW B 98, 235426 (2018)
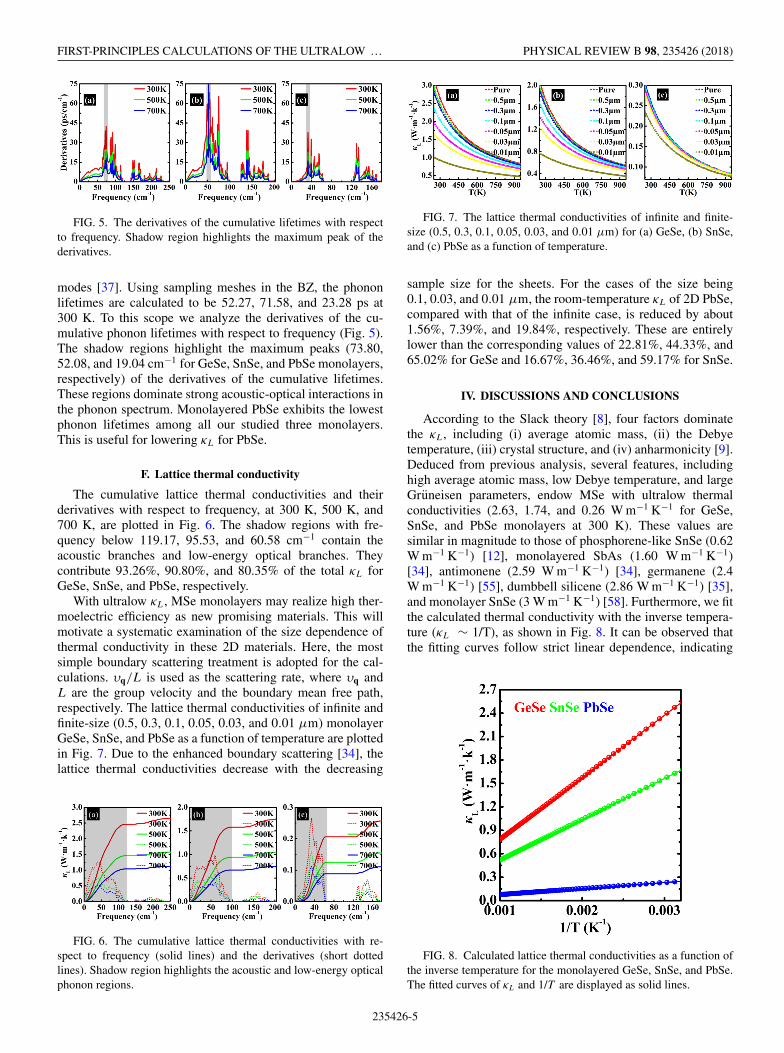
FIG. 5. The derivatives of the cumulative lifetimes with respectto frequency. Shadow region highlights the maximum peak of thederivatives.
modes [37]. Using sampling meshes in the BZ, the phononlifetimes are calculated to be 52.27, 71.58, and 23.28 ps at300 K. To this scope we analyze the derivatives of the cu-mulative phonon lifetimes with respect to frequency (Fig. 5).The shadow regions highlight the maximum peaks (73.80,52.08, and 19.04 cm−1 for GeSe, SnSe, and PbSe monolayers,respectively) of the derivatives of the cumulative lifetimes.These regions dominate strong acoustic-optical interactions inthe phonon spectrum. Monolayered PbSe exhibits the lowestphonon lifetimes among all our studied three monolayers.This is useful for lowering κL for PbSe.
F. Lattice thermal conductivity
The cumulative lattice thermal conductivities and theirderivatives with respect to frequency, at 300 K, 500 K, and700 K, are plotted in Fig. 6. The shadow regions with fre-quency below 119.17, 95.53, and 60.58 cm−1 contain theacoustic branches and low-energy optical branches. Theycontribute 93.26%, 90.80%, and 80.35% of the total κL forGeSe, SnSe, and PbSe, respectively.
With ultralow κL, MSe monolayers may realize high ther-moelectric efficiency as new promising materials. This willmotivate a systematic examination of the size dependence ofthermal conductivity in these 2D materials. Here, the mostsimple boundary scattering treatment is adopted for the cal-culations. υq/L is used as the scattering rate, where υq andL are the group velocity and the boundary mean free path,respectively. The lattice thermal conductivities of infinite andfinite-size (0.5, 0.3, 0.1, 0.05, 0.03, and 0.01 μm) monolayerGeSe, SnSe, and PbSe as a function of temperature are plottedin Fig. 7. Due to the enhanced boundary scattering [34], thelattice thermal conductivities decrease with the decreasing
FIG. 6. The cumulative lattice thermal conductivities with re-spect to frequency (solid lines) and the derivatives (short dottedlines). Shadow region highlights the acoustic and low-energy opticalphonon regions.
FIG. 7. The lattice thermal conductivities of infinite and finite-size (0.5, 0.3, 0.1, 0.05, 0.03, and 0.01 μm) for (a) GeSe, (b) SnSe,and (c) PbSe as a function of temperature.
sample size for the sheets. For the cases of the size being0.1, 0.03, and 0.01 μm, the room-temperature κL of 2D PbSe,compared with that of the infinite case, is reduced by about1.56%, 7.39%, and 19.84%, respectively. These are entirelylower than the corresponding values of 22.81%, 44.33%, and65.02% for GeSe and 16.67%, 36.46%, and 59.17% for SnSe.
IV. DISCUSSIONS AND CONCLUSIONS
According to the Slack theory [8], four factors dominatethe κL, including (i) average atomic mass, (ii) the Debyetemperature, (iii) crystal structure, and (iv) anharmonicity [9].Deduced from previous analysis, several features, includinghigh average atomic mass, low Debye temperature, and largeGruneisen parameters, endow MSe with ultralow thermalconductivities (2.63, 1.74, and 0.26 W m−1 K−1 for GeSe,SnSe, and PbSe monolayers at 300 K). These values aresimilar in magnitude to those of phosphorene-like SnSe (0.62W m−1 K−1) [12], monolayered SbAs (1.60 W m−1 K−1)[34], antimonene (2.59 W m−1 K−1) [34], germanene (2.4W m−1 K−1) [55], dumbbell silicene (2.86 W m−1 K−1) [35],and monolayer SnSe (3 W m−1 K−1) [58]. Furthermore, we fitthe calculated thermal conductivity with the inverse tempera-ture (κL ∼ 1/T), as shown in Fig. 8. It can be observed thatthe fitting curves follow strict linear dependence, indicating
FIG. 8. Calculated lattice thermal conductivities as a function ofthe inverse temperature for the monolayered GeSe, SnSe, and PbSe.The fitted curves of κL and 1/T are displayed as solid lines.
235426-5

LIU, BO, XU, YIN, ZHANG, WANG, ERIKSSON, AND WANG PHYSICAL REVIEW B 98, 235426 (2018)
FIG. 9. The schematic diagram of the degenerate low-lying opti-cal modes at � point for the monolayered MSe. The symbols · and ×denote atoms moving out of and into the plane of the paper.
that the Umklapp scattering is dominant at this temperaturerange, which is also found in other semiconductors such as3D PbSe [59], 3D PbTe [60], and Mg2(Si,Sn) [61].
Previous studies have confirmed that a perfectly flat planestructure enables ultrahigh in-plane thermal conductivity for2D layered materials [36,62]. The buckled structure canreduce the κL by breaking the out-of-plane symmetry, in-creasing anharmonic phonon scattering [55]. In the case ofsandwiched structures of monolayered MoS2, much weakerinteratomic bonds in the nonplanar structures give rise toquite low group velocities and large Gruneisen parameters,which result in remarkably lower thermal conductivities [63].For 2D MSe sheets, the unique structure with the Se-M-M-Se stacking contains buckled and sandwiched configurationssimultaneously. Meanwhile, the absence of of acoustic-opticalgaps greatly enhances the optical-acoustic phonon coupling in2D MSe. DFT-based phonon spectra of the hexagonal phase
at zone center (� point) reveal several soft optical modeswith frequencies induced by the antiparallel movements of thetwo MSe-based planes (highlighted with shadow regions inFig. 9). These features synergistically suppress the κL of 2DMSe sheets.
In summary, we report on the phonon thermal properties of2D MSe sheets by using first-principles calculations and solv-ing linearized the phonon Boltzmann transport equation. Cal-culated results show that 2D MSe sheets exhibit low κL, es-pecially for monolayered PbSe. Its value of 0.26 W m−1 K−1
at room temperature is even lower than that of phosphorene-like SnSe (0.62 W m−1 K−1) [12]. Compared to other 2Dmaterials, 2D PbSe shows some intrinsic factors, such as lowvalues of sound velocity, large Gruneisen parameters, softoptical modes, and strong optical-acoustic phonon coupling.All these features can be ascribed to the strong anharmonicityof the unexpected metal contacts between layers. These resultswill provide a new playground for exploring materials forthermal related applications.
ACKNOWLEDGMENTS
We gratefully acknowledged the financial support fromNational Natural Science Foundation of China (Grants No.11675195, No. 11675255, and No. 11634008), NationalKey Basic Research Program (Grant No. 2017YFA0403700),Guangdong Provincial Department of Science and Technol-ogy (Grant No. 2018A0303100013), and China PostdoctoralScience Foundation (Grant No. 2018M641477). The calcu-lations were performed at Supercomputer Centre in ChinaSpallation Neutron Source (CSNS).
[1] A. Majumdar, Science 303, 777 (2004).[2] Y. Lu, T. Sun, and D.-B. Zhang, Phys. Rev. B 97, 174304
(2018).[3] Y. Pei, H. Wang, and G. J. Snyder, Adv. Mater. 24, 6125 (2012).[4] Z. Chen, X. Zhang, and Y. Pei, Adv. Mater. 30, 1705617 (2018).[5] W. Kim, J. Mater. Chem. C 3, 10336 (2015).[6] C. Chang and L.-D. Zhao, Mater. Today Phys. 4, 50 (2018).[7] L.-D. Zhao, C. Chang, G. Tan, and M. G. Kanatzidis, Energy
Environ. Sci. 9, 3044 (2016).[8] G. A. Slack, Solid State Phys. 34, 1 (1979).[9] L. Hou, W.-D. Li, F. Wang, O. Eriksson, and B.-T. Wang, Phys.
Rev. B 96, 235137 (2017).[10] O. Delaire, J. Ma, K. Marty, A. F. May, M. A. McGuire, M.-H.
Du, D. J. Singh, A. Podlesnyak, G. Ehlers, M. D. Lumsden, andB. C. Sales, Nat. Mater. 10, 614 (2011).
[11] C. W. Li, J. Hong, A. F. May, D. Bansal, S. Chi, T. Hong, G.Ehlers, and O. Delaire, Nat. Phys. 11, 1063 (2015).
[12] Y. Xiao, C. Chang, Y. Pei, D. Wu, K. Peng, X. Zhou, S. Gong,J. He, Y. Zhang, Z. Zeng et al., Phys. Rev. B 94, 125203 (2016).
[13] C. Li, O. Hellman, J. Ma, A. F. May, H. B. Cao, X. Chen, A. D.Christianson, G. Ehlers, D. J. Singh, B. C. Sales, and O. Delaire,Phys. Rev. Lett. 112, 175501 (2014).
[14] Y. Chen, X. Ai, and C. A. Marianetti, Phys. Rev. Lett. 113,105501 (2014).
[15] J. W. Doak and C. Wolverton, Phys. Rev. B 86, 144202 (2012).[16] G. A. Akhmedova and D. S. Abdinov, Inorg. Mater. 45, 854
(2009).[17] L. D. Zhao, S. H. Lo, Y. Zhang, H. Sun, G. Tan, C. Uher, C.
Wolverton, V. P. Dravid, and M. G. Kanatzidis, Nature 508, 373(2014).
[18] G. J. Snyder and E. S. Toberer, Nat. Mater. 7, 105 (2008).[19] C. Chang, M. Wu, D. He, Y. Pei, C.-F. Wu, X. Wu, H. Yu, F.
Zhu, K. Wang, Y. Chen, L. Huang, J.-F. Li, J. He, and L.-D.Zhao, Science 360, 778 (2018).
[20] H. Yu, W. Lao, L. Wang, K. Li, and Y. Chen, Phys. Rev. Lett.118, 137002 (2017).
[21] M. Küpers, P. M. Konze, S. Maintz, S. Steinberg, A. M. Mio, O.Cojocaru-Mirédin, M. Zhu, M. Müller, M. Luysberg, J. Mayer,M. Wuttig, and R. Dronskowski, Angew. Chem. Int. Ed. 5610204 (2017).
[22] J. Muir and R. Cashman, J. Phys. Chem. Solids 28, 1009 (1967).[23] A. Kuhn, A. Chevy, and R. Chevalier, Phys. Status Solidi A 31,
469 (1975).[24] B. Sa, Z. Sun, and B. Wu, Nanoscale 8, 1169 (2016).[25] Y. Ma, L. Kou, Y. Dai, and T. Heine, Phys. Rev. B 94, 201104
(2016).[26] G. Kresse and D. Joubert, Phys. Rev. B 59, 1758 (1999).[27] P. E. Blöchl, Phys. Rev. B 50, 17953 (1994).
235426-6

FIRST-PRINCIPLES CALCULATIONS OF THE ULTRALOW … PHYSICAL REVIEW B 98, 235426 (2018)
[28] P. E. Blöchl, O. Jepsen, and O. K. Andersen, Phys. Rev. B 49,16223 (1994).
[29] G. Kresse and J. Furthmüller, Phys. Rev. B 54, 11169 (1996).[30] S. Baroni, S. De Gironcoli, A. Dal Corso, and P. Giannozzi,
Rev. Mod. Phys. 73, 515 (2001).[31] A. Togo, F. Oba, and I. Tanaka, Phys. Rev. B 78, 134106 (2008).[32] A. H. Romero, E. K. U. Gross, M. J. Verstraete, and O. Hellman,
Phys. Rev. B 91, 214310 (2015).[33] S.-Y. Yue, X. Zhang, G. Qin, S. R. Phillpot, and M. Hu, Phys.
Rev. B 95, 195203 (2017).[34] S.-D. Guo and J.-T. Liu, Phys. Chem. Chem. Phys. 19, 31982
(2017).[35] B. Peng, H. Zhang, H. Shao, Y. Xu, R. Zhang, H. Lu,
D. W. Zhang, and H. Zhu, ACS Appl. Mater. Inter. 8, 20977(2016).
[36] Y. Wang, N. Xu, D. Li, and J. Zhu, Adv. Funct. Mater. 27,1604134 (2016).
[37] A. Togo, L. Chaput, and I. Tanaka, Phys. Rev. B 91, 094306(2015).
[38] Y.-H. Lee, X.-Q. Zhang, W. Zhang, M.-T. Chang, C.-T. Lin,K.-D. Chang, Y.-C. Yu, J. T.-W. Wang, C.-S. Chang, L.-J. Liet al., Adv. Mater. 24, 2320 (2012).
[39] C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone, and S. Ryu,ACS Nano 4, 2695 (2010).
[40] V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano, andJ. N. Coleman, Science 340, 1226419 (2013).
[41] S. Zhao, Z. Li, and J. Yang, J. Am. Chem. Soc. 136, 13313(2014).
[42] S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, J. Chem. Phys.132, 154104 (2010).
[43] S. Grimme, S. Ehrlich, and L. Goerigk, J. Comput. Chem. 32,1456 (2011).
[44] A. Tkatchenko and M. Scheffler, Phys. Rev. Lett. 102 073005(2009).
[45] T. Bucko, S. Lebègue, J. Hafner, and J. G. Ángyán, J. Chem.Theory Comput. 9, 4293 (2013).
[46] T. Bucko, S. Lebègue, J. G. Ángyán, and J. Hafner, J. Chem.Phys. 141, 034114 (2014).
[47] J. M. Skelton, D. Tiana, S. C. Parker, A. Togo, I. Tanaka, andA. Walsh, J. Chem. Phys. 143, 064710 (2015).
[48] J. M. Skelton, L. A. Burton, F. Oba, and A. Walsh, J. Phys.Chem. C 121, 6446 (2017).
[49] R. Zacharia, H. Ulbricht, and T. Hertel, Phys. Rev. B 69, 155406(2004).
[50] L. F. Huang, P. L. Gong, and Z. Zeng, Phys. Rev. B 90, 045409(2014).
[51] L. F. Huang and Z. Zeng, J. Appl. Phys. 113, 083524 (2013).[52] P.-F. Liu, T. Bo, Z. Liu, O. Eriksson, F. Wang, J. Zhao, and B.-T.
Wang, J. Mater. Chem. C 6, 12689 (2018).[53] Y. Zhang, J. Materiomics 2, 237 (2016).[54] H. Shao, X. Tan, T. Hu, G. Q. Liu, J. Jiang, and H. Jiang,
Europhys. Lett. 109, 47004 (2015).[55] B. Peng, D. Zhang, H. Zhang, H. Shao, G. Ni, Y. Zhu, and H.
Zhu, Nanoscale 9, 7397 (2017).[56] B. Peng, H. Zhang, H. Shao, K. Xu, G. Ni, J. Li, H. Zhu, and
C. M. Soukoulis, J. Mater. Chem. A 6, 2018 (2018).[57] B.-T. Wang, J.-J. Zheng, X. Qu, W.-D. Li, and P. Zhang, J.
Alloy. Compd. 628, 267 (2015).[58] L. C. Zhang, G. Qin, W. Z. Fang, H. J. Cui, Q. R. Zheng, Q. B.
Yan, and G. Su, Sci. Rep. 6, 19830 (2016).[59] D. Parker and D. J. Singh, Phys. Rev. B 82, 035204 (2010).[60] L. D. Zhao, H. J. Wu, S. Q. Hao, C. I. Wu, X. Y. Zhou, K.
Biswas, J. Q. He, T. P. Hogan, C. Uher, C. Wolverton, V. P.Dravid, and M. G. Kanatzidis, Energy Environ. Sci. 6, 3346(2013).
[61] J. J. Pulikkotil, D. J. Singh, S. Auluck, M. Saravanan, D. K.Misra, A. Dhar, and R. C. Budhani, Phys. Rev. B 86, 155204(2012).
[62] X. Xu, J. Chen, and B. Li, J. Phys.: Condens. Matter. 28, 483001(2016).
[63] Y. Cai, J. Lan, G. Zhang, and Y.-W. Zhang, Phys. Rev. B 89,035438 (2014).
235426-7





![PHYSICAL REVIEW B98, 155402 (2018) · M. ZUBAIR, M. TAHIR, AND P. VASILOPOULOS PHYSICAL REVIEW B 98, 155402 (2018) Hamiltonian of bilayer WSe 2near the K and K valleys reads as [24,25,27,39]](https://img.pdfslide.net/doc/110x75/6029d619187a7e10d4259542/physical-review-b98-155402-2018-m-zubair-m-tahir-and-p-vasilopoulos-physical.jpg)




![PHYSICAL REVIEW B98, 245413 (2018) - Theoretical Quantum Physics …€¦ · cept of higher-order topological insulators (TIs) [30–41]was put forward, where the usual form of the](https://img.pdfslide.net/doc/110x75/5f33d897ca3c855f137afce7/physical-review-b98-245413-2018-theoretical-quantum-physics-cept-of-higher-order.jpg)


















