Embed Size (px)
Citation preview

TOWARD A DEVELOPMENTAL NEUROBIOLOGYOF AUTISM
Franck Polleux1* and Jean M. Lauder2
1Department of Pharmacology–Neuroscience Center, School of Medicine, University of North Carolina–Chapel Hill,
Chapel Hill, North Carolina2Department of Cell and Developmental Biology, School of Medicine, University of North Carolina–Chapel Hill,
Chapel Hill, North Carolina
Autism is a complex, behaviorally defined, developmental brain dis-order with an estimated prevalence of 1 in 1,000. It is now clear that autismis not a disease, but a syndrome with a strong genetic component. Theetiology of autism is poorly defined both at the cellular and the molecularlevels. Based on the fact that seizure activity is frequently associated withautism and that abnormal evoked potentials have been observed in autisticindividuals in response to tasks that require attention, several investigatorshave recently proposed that autism might be caused by an imbalance be-tween excitation and inhibition in key neural systems including the cortex.Despite considerable ongoing effort toward the identification of chromo-some regions affected in autism and the characterization of many potentialgene candidates, only a few genes have been reproducibly shown to displayspecific mutations that segregate with autism, likely because of the complexpolygenic nature of this syndrome. Among those, several candidate geneshave been shown to control the early patterning and/or the late synapticmaturation of specific neuronal subpopulations controlling the balancebetween excitation and inhibition in the developing cortex and cerebellum.In the present article, we review our current understanding of the develop-mental mechanisms patterning the balance between excitation and inhibi-tion in the context of the neurobiology of autism. © 2004 Wiley-Liss, Inc.MRDD Research Reviews 2004;10:303–317.
Key Words: autism; development; cortex; mouse genetics; serotonin; do-pamine; norepinephrine; glutamate; neurotransmitters; GABA; interneu-rons; patterning; gene expression; neuroligin; neuronal migration; dendrite;synaptogenesis
Autism is a developmental brain disorder characterized bya general inability to form reciprocal social interactions,severe impairment in verbal and nonverbal communica-
tion, and a markedly restricted repertoire of activities and inter-ests. The incidence of autism is currently estimated at 1 in 1,000children [Folstein and Rosen-Sheidley, 2001]. Familial recur-rence of the disorder is 100-fold higher than in the generalpopulation, and the concordance rate among monozygotictwins is estimated at between 70 and 90%, but close to 0% indizygotic twins, thus indicating a strong genetic component tothe disease [reviewed in Veenstra-Vanderweele et al., 2003;Veenstra-VanderWeele and Cook, 2004; see also Wassink et al.,this issue]. Furthermore, the prevalence of autism and Aspergersyndrome are, respectively, 4 and 8 times higher in males thanfemales, strongly suggesting an X-linked genetic component.
The etiology of autism is poorly understood [Piven,1997]. However, the amount and quality of research performedin several fields, including epidemiology, early brain imaging,and genetic identification of candidate chromosomal regions,has led to new hypotheses regarding possible etiologies [Stoks-tad, 2001; Rubenstein and Merzenich, 2003; Belmonte et al.,2004]. Interestingly, upon more detailed clinical examination,approximately 10% of autistic cases reveal association with othergenetic neuropathologies, such as fragile X; tuberous sclerosis,and Rett syndrome. Several other clinical features are frequentlyassociated with autism, such as mental retardation [roughly 70%of cases have an intelligence quotient (IQ) � 70], epilepticseizures (30%), and macrocephaly (larger head circumference)and megencephaly (larger brain volume) [Tuchman, 2003]. Ap-proximately one-third of autistic individuals develop clinicallyapparent seizures and, of those, more than 50% develop “sharp-spike” activity during sleep when recorded by EEG or magne-toencephalography [Lewine et al., 1999; Ballaban-Gil andTuchman, 2000]. Taken together, these observations have re-cently led several investigators to hypothesize that the cortex ofautistic individuals is characterized by an imbalance betweenexcitation and inhibition, leading to hyperexcitability and anunstable activity of cortical networks following normal sensorystimulation [Hussman, 2001; Rubenstein and Merzenich, 2003;Belmonte et al., 2004].
Most interestingly, recent advances in functional imagingin humans, but also in nonhuman primates, have revealed thatrhythmic synchronization of neural discharges in the gammafrequency band (20–60 Hz) may provide the necessary spatialand temporal links that bind together the processing in differentbrain areas to build a coherent percept [Tallon-Baudry and
Grant sponsor: NIMH; Grant number: U54 MH66418 (to JML). Grant sponsor:NIEHS; Grant number: P30ES10126 (to JML). Grant sponsor: NIH/NIDCR; Grantnumber R01 DE13314–05 (to JML). Grant sponsor: NIH-NINDS; Grant number: 1RO1 NS047701–01. (to FP). Grant sponsor: March of Dimes Birth Defects Founda-tion (to FP, JML).*Correspondence to: F. Polleux. E-mail: [email protected] or Jean LauderE-mail: [email protected] 17 November 2004; Accepted 22 November 2004Published online in Wiley InterScience (www.interscience.wiley.com).DOI: 10.1002/mrdd.20044
MENTAL RETARDATION AND DEVELOPMENTAL DISABILITIESRESEARCH REVIEWS 10: 303–317 (2004)
© 2004 Wiley-Liss, Inc.

Bertrand, 1999]. These results suggestthat one particular type of gamma oscil-lation resulting from synchronized neu-ronal activity at the cortical and subcor-tical levels not only plays an essential rolein sensory perception, but also in atten-tion-based cognitive tasks. Importantly,neuronal synchrony in hippocampal,cortical, and thalamic networks has beenshown to be critically dependent on theintegrity of the discharge of interneuronsin relation to the activity of the pyramidalneuron. In the hippocampus, synchro-nous gamma oscillation is considered tooccur through the GABAA receptor–me-diated mutual inhibition among inter-neurons [Buhl et al., 1994; Cobb et al.,1995; Freund and Buzsaki, 1996]. In theneocortex; it is known that a class ofpyramidal neurons termed fast rhythmicbursting cells or “chattering” neuronssynchronize their activities at gamma fre-quencies and are thought to be necessaryfor selective attention or binding process-ing in object recognition [Aoyagi et al.,2003]. Therefore, a slight disruption ofthe balance between excitation and inhi-bition in the cortex could have dramaticconsequences on the function of theneuronal networks underlying percep-tion and attention.
In a very recent study, the brainactivation of a group of high-functioningautistic individuals was measured usingfunctional MRI during sentence com-prehension and the results were com-pared with those of a verbal IQ–matchedcontrol group [Just et al., 2004]. Theirresults show that the functional connec-tivity; i.e., the degree of synchroniza-tion/correlation of brain activation wasconsistently lower for the autistic indi-vidual than for the control participants inthe main cortical areas involved in lan-guage processing [Just et al., 2004]. Thesefindings suggest that the neural basis ofdisordered language in autism entails alower degree of information integrationand synchronization across the large-scalecortical network for language processing[Brock et al., 2002; Just et al., 2004].
Taken together, these results suggestthat the autistic brain might be character-ized by a synchronization deficit during theactivation of cortical networks involved inlanguage processing (and maybe in othersensory modalities or attention) and thatthis synchronization deficit could be theresult of an imbalance between excitationand inhibition [Rubenstein and Mer-zenich, 2003; Belmonte et al., 2004]. Al-though still speculative, this hypothesis isattractive because it is based on functionalstudies. The present review will discusswhy this new hypothesis is especially at-
tractive to describe the pathophysiology ofthe autistic brain in light of recent progressmade in understanding the generation, mi-gration, and differentiation of glutamatergicand GABAergic neurons in the cortex. Wewill also discuss the development of neu-romodulatory systems well known to con-trol the global levels of neuronal excitabilityin the forebrain, including the serotonininputs that have been suspected for a longtime to be altered in the autistic brain.Finally, we review the classes of genes thathave been linked to autism in recent ge-netic studies and discuss several candidategenes in the context of this neurodevelop-mental hypothesis.
NEUROANATOMICALABNORMALITIES IN AUTISM
A few studies using either neuro-anatomical examination of autopsiedbrains of autistic patients or, more re-cently, functional and structural MRIstudies, have revealed three main types ofdefects in (1) the brainstem and cerebel-lum, (2) the limbic system (amygdala andhippocampus), and (3) the cortex[Courchesne, 1997].
Brain Stem and CerebellumAn early anatomical study first re-
vealed that several brainstem nuclei wereeither missing or displayed strong neuro-nal loss, such as the facial motor nuclei orthe superior olive nuclei [Rodier et al.,1996]. A longitudinal study performedon a large set of cases using quantitativestructural imaging also revealed hypopla-sia of brainstem structures [Hashimoto etal., 1995]; however, this set of data prob-ably needs to be replicated to evaluate itsvalidity in light of the progress made indiagnosis of autism.
One of the most reproducible neu-roanatomical findings in autistic brain isthe paucity of Purkinje neurons in thecerebellum [from 35 to 90%; reviewedby Courchesne, 1997]. An early MRIstudy reported a significantly smaller areaof the cerebellar vermis in lobulesVIII–X [Courchesne et al., 1988]. Re-cent MRI studies have also revealed a near40% increase in cerebellar white mattervolume in autistic children compared toage-matched controls [Courchesne et al.,2001]. Subsequent independent reports,however, have failed to replicate the orig-inal observation of hypoplasia of the neo-cerebellar vermis [Piven et al., 1997] andadditional studies of the volume of cerebel-lar cortical volume will be necessary beforefinal conclusions can be drawn about thesize of this structure in autism.
Hippocampus and AmygdalaAn isolated report has revealed a
decreased level of dendritic branching inthe CA1 and CA4 regions of the hip-pocampus of two autistic patients com-pared to two control cases [Raymond etal., 1996]. Other studies have reported anincreased neuronal packing density in thehippocampus, subiculum, mammilarybodies, entorhinal cortex, medial septalnuclei, and several nuclei of the amygdala[reviewed by Courchesne, 1997]; theneuronal networks playing a critical rolein the formation, maintenance, and re-trieval of memory. However, other stud-ies have failed to reproduce the observeddifferences in the packing density ofCA1–CA4 pyramidal neurons [Bailey etal., 1998].
Interestingly, a recent analysis ofbrain morphometric features was per-formed in a large sample of carefully di-agnosed 3- to 4-year-old children withautism spectrum disorder (ASD) com-pared with age-matched control groupsof typically developing (TD) childrenand developmentally delayed (DD) chil-dren [Sparks et al., 2002]. This analysisrevealed increased cortical and cerebellarvolume and also an increased volume ofthe amygdala in autistic children thatcannot be accounted for by a simple in-creased cortical volume. This and an-other recent analysis [Schumann et al.,2004] suggest abnormal brain develop-mental processes affecting the amydgalaearly in the clinical course of autism.
Brain Enlargement in AutismAlthough the defining behavioral
features of autism are present from theearliest ages and change over time withage, perhaps the most compelling biolog-ical argument for autism being a disorderof abnormal brain development comesfrom studies showing increased brainvolume in this disorder. Kanner in hisfirst descriptions of autism [Kanner,1943] noted that 5 of 11 children hedescribed had no obvious dysmorphicfeatures, but did show enlarged head size.Over the years others described this an-ecdotally in studies of dysmorphology[Steg and Rapoport, 1975]. In 1992,Piven et al. reported enlarged midsagittalbrain area [Piven et al., 1992]. A system-atic study of 35 autistic adults and ado-lescents versus 36 controls, to follow upon this initial report, similarly reportedenlarged brain volume and brain tissuevolume [Piven et al., 1995]. More re-cently Courchesne et al. [2001] exam-ined head circumference in a sample of60 boys compared to a nonmentally re-tarded group of controls and reported
304 MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

enlargement of cortical gray and whitematter limited to those in the 2- to4-year-old age group [Courchesne et al.,2001], with an actual decrease in corticalwhite matter in the older 6- to 16-year-old group. Increased white matter in thecerebellum was also noted in thisyounger age group (2–4 years) with de-creased gray matter in the older agegroup. Findings by Sparks et al.[2002]support the finding of increased brainvolume in 45 3- to 4-year olds witheither autism or pervasive developmentaldisorder–not otherwise specified (PDD-NOS) compared to both typically devel-oping and mentally retarded controls,noting increased volume of both the ce-rebral cortex and cerebellum as well asincreased volume of the amygdalae[Sparks et al., 2002]. Finally; increasedbrain volume was found in a large sampleof high-functioning (nonmentally re-tarded) autistic individuals under 12 yearsof age; but not in those over 12 years ofage [Aylward et al., 2002]. Others havereported evidence to suggest enlargementof the caudate nucleus [Sears et al., 1999],amygdalae [Schumann et al., 2004], andhippocampus [Schumann et al., 2004],with a decrease in cross-sectional area ofthe corpus callosum [Piven et al., 1997;Manes et al., 1999; Hardan et al., 2000].The findings from brain MRI studies arealso consistent with systematic large scalesurveys showing that approximately 20%of autistic individuals have macrocephaly(greater than the 98th percentile for headcircumference) that appears not to bepresent at birth [Lainhart et al., 1997;Stevenson, 1997]. More recent prelimi-nary evidence suggests that the increasein head circumference may have its ori-gins in the latter part of the first year ofpostnatal life [Courchesne et al., 2001].
In summary these reports providefirm support for the idea that the brain inautism is enlarged. There is evidence thatthe enlargement occurs in both gray andwhite matter and occurs in selected re-gions and structures in the brain (with adecrease in the size of some other struc-tures). There is also evidence to suggestthat this enlargement occurs predomi-nantly during early postnatal brain devel-opment. More specific conclusions aboutthe nature of this phenomenon, includ-ing the exact timing or window of brainenlargment, patterns over time in brainstructures, regions, and tissues, and clin-ical correlates, are not yet known.
Cortical Columnar ArchitectureThe finding of brain enlargement
occurring in the early postnatal periodsuggests a range of possibilities about its
pathogenesis. Importantly, this early dif-ference in brain size is not found in adultautistic patients compared to age-matched control brains [Courchesne etal., 2003], suggesting that the initiallyaccelerated rate of cortical growth is fol-lowed by an abnormally slower period ofgrowth [see Piven et al., this issue]. As wewill discuss later, this abnormally rapidgrowth rate detected in cortical grey andwhite matter during the first years of lifein the frontal, temporal, and parietallobes could reflect (1) an increase in den-dritic branching/outgrowth and a corre-spondingly increased rate of synaptogen-esis (grey matter) accompanied by anaccelerated rate of axon myelination(white matter) and/or (2) a reducedelimination of aberrant connections (re-duced dendritic/synaptic pruning). Re-cent work has demonstrated that, duringdevelopment, there is a causal relation-ship between the number of active pre-synaptic afferent inputs and the size andlevel of branching of the correspondingdendritic field harboring postsynapticdensities [reviewed in Cline, 2001; seebelow]. Therefore, one attractive hy-pothesis for the pathogenesis of autism,and potentially other developmental dis-abilities, could be a disregulation of thedevelopmental mechanisms patterningaxonal outgrowth and/or dendritic ar-borizations/synaptic contacts betweenexcitatory and inhibitory neurons in thecortex [Zoghbi, 2003]. The behavioralconsequences of an abnormal balance be-tween excitatory and inhibitory neuro-transmission in the cortex are only start-ing to be explored experimentally[Powell et al., 2003] and are likely to bespecific and complex, especially if a sub-population of cortical interneurons isspecifically reduced or absent as it as beenproposed for schizophrenia [Lewis andLevitt, 2002].
A recent study by Casanova et al.[2002] has provided evidence for abnor-mal patterning of minicolumns in thefrontal and temporal lobes of autistic pa-tients. Interestingly, in humans, corticalminicolumns are the cellular manifesta-tion of the basic columnar organizationof excitatory and inhibitory neurons intocanonical functional units (Fig. 1; [DeFe-lipe et al., 1990; Peters, 1994; Mount-castle, 1997; Jones, 2000]). Functionaland anatomical investigations have en-abled an accurate definition of the cellu-lar composition of these minicolumns.Each column contains an array of pyra-midal neurons in layers 5 and 2/3, char-acterized by bundled apical dendritesreaching layer 1 and pyramidal neuronsin layer 6 with their bundled apical den-
drites reaching layer 4B (Fig. 1A). Thesecond important component of mini-columns is GABAergic interneurons pro-viding powerful and specific inhibitorycontrol in cortical neuronal networks(Fig. 1B). Cortical interneurons can beclassified based on morphological andfunctional criteria into four main classes,each characterized by a precise synaptictargeting of a specific part of the pyrami-dal neuron: (1) large and small basketcells, respectively, making their synapticcontacts onto the cell soma and proximalpart of the apical dendrite of pyramidalneurons within the same layer; (2) doublebouquet cells making their synaptic con-tacts onto more distal parts of the basaldendrites of pyramidal neurons and (3)Chandelier cells making synapses alongthe initial axonal segment of pyramidalneurons; and (4) Cajal-Retzius interneu-rons found in layer I making synapsesonto the distal part of apical dendrites ofcortical pyramidal neurons [reviewed inDeFelipe et al., 1990]. Based on this spe-cialized synaptic targeting, each of theseinterneuronal subpopulations is thoughtto play a specific functional role in thecontrol of synaptic integration, andtherefore, on the propagation of infor-mation through cortical networks[Freund, 2003]. Disorganization of mini-columns could reflect distinct aspects ofan abnormal cortical cytoarchitecture,including: (1) a disrupted laminar distri-bution of a specific subclass of interneu-rons and/or (2) a numerical imbalancebetween pyramidal neurons and inter-neurons and/or (3) a disrupted level ofbranching of the axons and dendrites ofboth pyramidal neurons and interneu-rons. Future investigations are requiredto characterize the number, laminar dis-tribution, and morphology of specificclasses of interneurons in the autistic cor-tex to assess whether this disorder couldbe due to the abnormal patterning of asubpopulation of GABAergic neurons.
Interestingly, an early study byBailey et al. [1998] carefully assessing theclinicopathology of six carefully diag-nosed autistic individuals revealed severalabnormalities that suggest abnormal mi-gration of pyramidal neurons and/or pat-terning of their dendrite outgrowth. Infour of six cases, postmortem examina-tion of the frontal cortex revealed (1) thepresence of ectopic neurons in the whitematter and in layer 1 as well as (2) mi-soriented pyramidal neurons in layer 5with their apical dendrite not orientedtoward layer 1 as in normal cortex and (3)a disorganized cellular organization in su-pragranular layers of areas located in thesuperior temporal gyrus [Bailey et al.,
305MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER
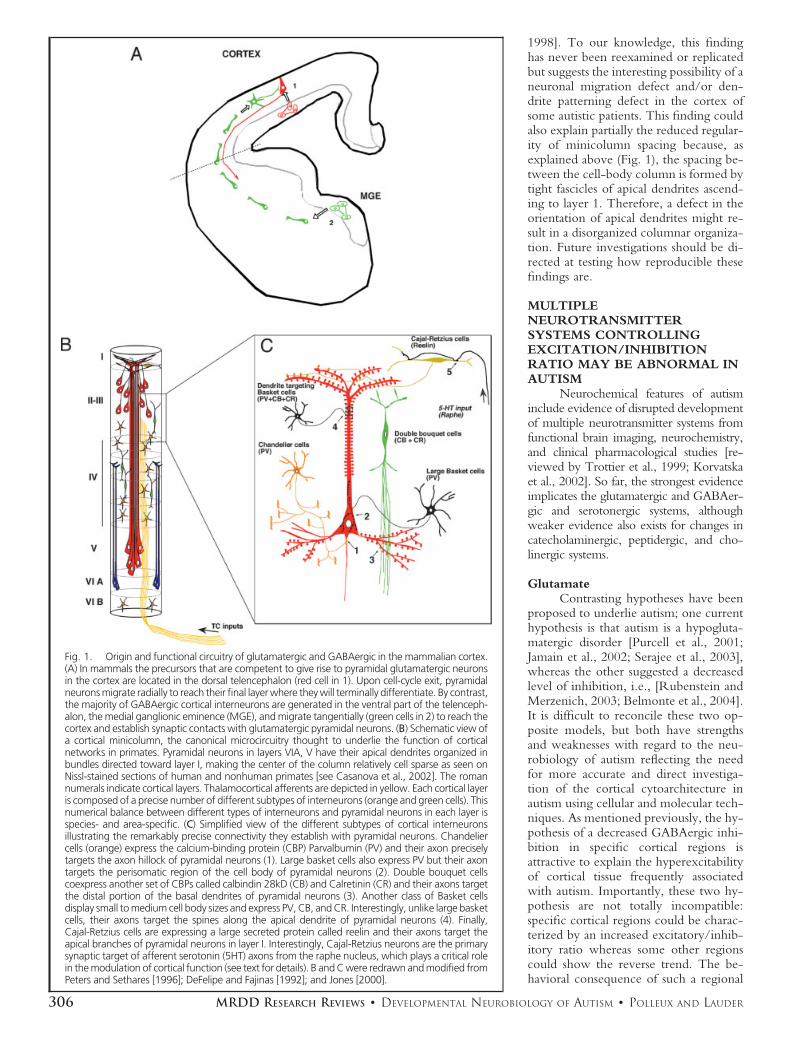
1998]. To our knowledge, this findinghas never been reexamined or replicatedbut suggests the interesting possibility of aneuronal migration defect and/or den-drite patterning defect in the cortex ofsome autistic patients. This finding couldalso explain partially the reduced regular-ity of minicolumn spacing because, asexplained above (Fig. 1), the spacing be-tween the cell-body column is formed bytight fascicles of apical dendrites ascend-ing to layer 1. Therefore, a defect in theorientation of apical dendrites might re-sult in a disorganized columnar organiza-tion. Future investigations should be di-rected at testing how reproducible thesefindings are.
MULTIPLENEUROTRANSMITTERSYSTEMS CONTROLLINGEXCITATION/INHIBITIONRATIO MAY BE ABNORMAL INAUTISM
Neurochemical features of autisminclude evidence of disrupted developmentof multiple neurotransmitter systems fromfunctional brain imaging, neurochemistry,and clinical pharmacological studies [re-viewed by Trottier et al., 1999; Korvatskaet al., 2002]. So far, the strongest evidenceimplicates the glutamatergic and GABAer-gic and serotonergic systems, althoughweaker evidence also exists for changes incatecholaminergic, peptidergic, and cho-linergic systems.
GlutamateContrasting hypotheses have been
proposed to underlie autism; one currenthypothesis is that autism is a hypogluta-matergic disorder [Purcell et al., 2001;Jamain et al., 2002; Serajee et al., 2003],whereas the other suggested a decreasedlevel of inhibition, i.e., [Rubenstein andMerzenich, 2003; Belmonte et al., 2004].It is difficult to reconcile these two op-posite models, but both have strengthsand weaknesses with regard to the neu-robiology of autism reflecting the needfor more accurate and direct investiga-tion of the cortical cytoarchitecture inautism using cellular and molecular tech-niques. As mentioned previously, the hy-pothesis of a decreased GABAergic inhi-bition in specific cortical regions isattractive to explain the hyperexcitabilityof cortical tissue frequently associatedwith autism. Importantly, these two hy-pothesis are not totally incompatible:specific cortical regions could be charac-terized by an increased excitatory/inhib-itory ratio whereas some other regionscould show the reverse trend. The be-havioral consequence of such a regional
Fig. 1. Origin and functional circuitry of glutamatergic and GABAergic in the mammalian cortex.(A) In mammals the precursors that are competent to give rise to pyramidal glutamatergic neuronsin the cortex are located in the dorsal telencephalon (red cell in 1). Upon cell-cycle exit, pyramidalneurons migrate radially to reach their final layer where they will terminally differentiate. By contrast,the majority of GABAergic cortical interneurons are generated in the ventral part of the telenceph-alon, the medial ganglionic eminence (MGE), and migrate tangentially (green cells in 2) to reach thecortex and establish synaptic contacts with glutamatergic pyramidal neurons. (B) Schematic view ofa cortical minicolumn, the canonical microcircuitry thought to underlie the function of corticalnetworks in primates. Pyramidal neurons in layers VIA, V have their apical dendrites organized inbundles directed toward layer I, making the center of the column relatively cell sparse as seen onNissl-stained sections of human and nonhuman primates [see Casanova et al., 2002]. The romannumerals indicate cortical layers. Thalamocortical afferents are depicted in yellow. Each cortical layeris composed of a precise number of different subtypes of interneurons (orange and green cells). Thisnumerical balance between different types of interneurons and pyramidal neurons in each layer isspecies- and area-specific. (C) Simplified view of the different subtypes of cortical interneuronsillustrating the remarkably precise connectivity they establish with pyramidal neurons. Chandeliercells (orange) express the calcium-binding protein (CBP) Parvalbumin (PV) and their axon preciselytargets the axon hillock of pyramidal neurons (1). Large basket cells also express PV but their axontargets the perisomatic region of the cell body of pyramidal neurons (2). Double bouquet cellscoexpress another set of CBPs called calbindin 28kD (CB) and Calretinin (CR) and their axons targetthe distal portion of the basal dendrites of pyramidal neurons (3). Another class of Basket cellsdisplay small to medium cell body sizes and express PV, CB, and CR. Interestingly, unlike large basketcells, their axons target the spines along the apical dendrite of pyramidal neurons (4). Finally,Cajal-Retzius cells are expressing a large secreted protein called reelin and their axons target theapical branches of pyramidal neurons in layer I. Interestingly, Cajal-Retzius neurons are the primarysynaptic target of afferent serotonin (5HT) axons from the raphe nucleus, which plays a critical rolein the modulation of cortical function (see text for details). B and C were redrawn and modified fromPeters and Sethares [1996]; DeFelipe and Fajinas [1992]; and Jones [2000].
306 MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

imbalance will be important to assess us-ing new genetic animal models [Levitt etal., 2004].
The reduction in global levels ofglutamate signaling might occur by over-activation of excitatory receptors on cor-tical GABA interneurons, such as thoseof the 5-HT2A receptor subtype, leadingto pronounced depression of the excita-tory glutamatergic circuitry [Carlsson,1998]. This possibility is supported by therelative efficacy of combined treatmentwith partial glutamate agonists and5-HT2A receptor antagonists (e.g., ris-peridone) in autism as well as in hypo-glutamatergic animal models [Carlsson etal., 1998, 1999; Nilsson et al., 2001].Recent evidence also suggests involve-ment of polymorphisms in genes encod-ing both metabotropic and ionotropicglutamate receptors in autism [Jamain etal., 2002; Serajee et al., 2003]. Recently,a postmortem study has reported elevatedexpression of the glutamate transporter inautistic brain [Purcell et al., 2001].
Developmental Origin ofGlutamatergic Cortical Neurons
In mammals, the vast majority ofglutamatergic pyramidal neurons aregenerated in the dorsal telencephalon byprecursors in the ventricular and subven-tricular zones during embryonic devel-opment. In rodents, these neurons aregenerated during the second week of ges-tation (E11 to E17 in mouse or E13 toE19 in rat). In nonhuman primates, cor-tical neurons are generated from E40 andE100 approximately [Rakic, 1972]. Inhumans, the generation of cortical neu-rons is thought to occur between the 6th
and the 13th week of gestation based onhistological appearance of VZ and SVZmitotic figures [reviewed in Sidman andRakic, 1973]. Upon their last mitosis inthe germinal pseudoepithelium lining theventricles, glutamatergic neurons migratealong the radial glial scaffold to accumu-late in the cortical plate in an inside-firstoutside-last manner, so that neurons ininfragranular layers 6 and 5 are generatedfirst, before neurons populating more su-perficial layers 4, 3, and 2 [Rakic, 1972].
Recent progress in the analysis ofthe genetic mechanisms underlying thegeneration of cortical neurons has re-vealed that cortical glutamatergic neu-rons are generated by dorsal precursorsunder the control of bHLH transcriptionfactors, Neurogenin1 and 2 (Ngn1, Ngn2)and as well as Pax6 and Emx2 [reviewedin Schuurmans and Guillemot, 2002]. In-terestingly, Ngn1 and Ngn2 are involvedin the specification of the glutamatergicneurotransmitter phenotype and single
and double knockout mice for Ngn1/2show a striking phenotype, where thevast majority of cortical neurons expressGABA as a neurotransmitter instead ofglutamate [Fode et al., 2000; Schuurmanset al., 2004].
Interestingly, in other parts of thenervous system, for example, the spinalcord, expression of a specific neurotrans-mitter is partially determined by a combi-natorial expression of transcription factors[reviewed in Jessell, 2000]. However, a re-cent report demonstrates that the level ofspontaneous activity and the frequency ofintracellular Ca2� elevations in developingneural circuitry can have dramatic long-term consequences on the number of neu-rons expressing a given type of neurotrans-mitter [Borodinsky et al., 2004]. Thisimportant finding raises the possibility thattranscription factors restrict the type ofneurotransmitter that a neuronal precursorcan express but that the level of spontane-ous activity may play an important role infine tuning the final number of neuronsexpressing a given neurotransmitter pheno-type [Spitzer et al., 2004]. Therefore, thebalance between the number of neuronsexpressing glutamate and GABA is proba-bly the results of complex interactions be-tween intrinsic (transcription factor expres-sion) and extrinsic (spontaneous activityand calcium signaling) activity during de-velopment.
GABAAs discussed earlier, another neu-
rochemical abnormality hypothesized toexplain the pathophisiology of autism issuppressed GABAergic inhibition [Hus-sman, 2001; Rubenstein and Merzenich,2003; Belmonte et al., 2004], which maybe due, at least in part, to reduced ex-pression of GAD65 and GAD67 [Fatemiet al., 2002], the two isoforms of theGABA biosynthetic enzyme glutamatedecarboxylase. Reduced GABAergicneurotransmission may also be caused bydeletional mutations in chromosome15q11-q13, the locus of genes encodingsubunits of the GABAA receptor(GABRB3, GABRA5, and GABRG3).In Angelman’s syndrome (AS), such de-letions lead to reduced expression ofGABAA/BZD receptors [Holopainen etal., 2001a, 2001b; Luddens et al., 1995].AS patients exhibit a behavioral pheno-type consistent with compromisedGABAergic inhibitory neurotransmis-sion, including hyperactivity, hyperkine-sis, seizures, and sleep disturbances as wellas severe mental retardation, lack of mo-tor coordination, craniofacial abnormali-ties, and autism [Holopainen et al.,2001a, 2001b]. As discussed in the pre-
vious section, much work needs to bedone to explore (1) whether specificGABAergic neuron subpopulations aremisplaced or absent in specific corticalregions, (2) whether there is a globalnumerical decrease of the number ofGABAergic interneuron per unit of cor-tical microcolumns, or (3) whether thenumber and localization of cortical inter-neurons is normal in autism but charac-terized by a functional deficit in GABAexpression at the synaptic level as recentlyshown in another neurodevelopmentalpathology, schizophrenia [Lewis andLevitt, 2002].
Developmental Origin ofGABAergic Cortical Interneurons
In rodents, the vast majority ofcortical interneurons are generated in ap-proximately the same time window ascortical pyramidal neurons [E11 to E17in the mouse; E13 to E19 in the rat;Miller, 1985, 1986]. However, corticalinterneurons are generated by a distinctset of precursors located in the ventraltelencephalon, more specifically in themedial and caudal parts of the ganglioniceminence [Fig. 1A; MGE and CGE, re-spectively; Anderson et al., 1997; Tama-maki et al., 1997; Lavdas et al., 1999;Wichterle et al., 2001; Nery et al., 2002;Polleux et al., 2002]. At the genetic level,the generation of GABAergic cortical in-terneurons is under the control of distincttranscription factors: the bHLH tran-scription factor Mash1 and the homeodo-main-containing transcription factorsDlx1 and Dlx2 [Anderson et al., 1997;Casarosa et al., 1999]. Interestingly, in-terneurons generated in the ventral tel-encephalon have to undergo a “longjourney” to reach their dorsal destinationin the cortex through a tangential modeof migration, which, by definition, doesnot involve fasciculation along the radialglial scaffold until they reach their finaldestination [Marin and Rubenstein,2001].
Recently, experiments performedin human telencephalic tissue haveshown that, unlike in rodents, a specificsubset of dorsal progenitors seems to becompetent to produce a subpopulation ofcortical interneurons in a Mash1-depen-dent manner [Letinic et al., 2002]. Thissuggests that, in humans, a subpopulationof cortical dividing precursors are directlyinvolved in the generation of more than60% of cortical GABAergic interneurons.Although some differences might existbetween the mode of generation of cor-tical interneurons in rodents and pri-mates, the important concept emerging isthat (1) specific sets of precursors along
307MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

the ventro-dorsal axis of the telencepha-lon are specialized with regard to theircompetence to generate glutamatergic orGABAergic cortical neurons and (2) theyundergo a drastically different migrationpathway to invade the cortex and to dif-ferentiate in the appropriate layer.
Interestingly, several extracellularcues have been found to control the mo-tility of cortical interneurons migratingfrom the basal forebrain to the cortex.Among them are two types of ligandsthat activate receptor tyrosine kinases(RTK) expressed by migrating interneu-rons: hepatocyte growth factor (HGF),which activates the MET RTK, andNT4, which binds TRK [Powell et al.,2001; Polleux et al., 2002]. Mice carryinga targeted mutation of the gene encodingurokinase plasminogen activator (uPAR;a component of HGF activation) andknockout mice for trkB both show areduced number of cortical interneuronscompared to controls at late embryonic/early postnatal stages [Powell et al., 2001;Polleux et al., 2002]. Interestingly,uPAR knockout mice display a regionaldefect in which approximately 50% ofcalbindin-positive cortical interneuronsare absent from frontal and parietal cor-tical areas [reviewed in Levitt et al.,2004]. The uPAR knockout mice displaybehavioral deficits ranging from scatteredand frequent seizures and, more interest-ingly, increased anxiety-like behaviors[Powell et al., 2003]. The extent towhich this mouse model recapitulatessome behavioral traits characterizing au-tism remains to be explored, but this atleast provides evidence that a mutation ina single gene controlling cortical inter-neuron migration and differentiation canhave complex consequences on mousebehavior [Levitt et al., 2004]. In sum-mary, another important concept regard-ing the link between interneuronal mi-gration and the potential etiology ofautism (and other neurodevelopmentaldisorders potentially) is the fact that thislong-range migration of cortical inter-neurons (several millimeters in humans)might be more vulnerable than radial mi-gration of pyramidal neurons to geneticor environmental alterations during earlydevelopment because of the relativecomplexity of the cell–cell interactionsinvolved in tangential migration [Marinand Rubenstein, 2001; Levitt et al.,2004].
SerotoninOne feature of autism is high levels
of serotonin (5-HT) in blood platelets[hyperserotonemia; Anderson et al.,1990]. To what extent such elevated
plasma levels reflect a change in centralbrain levels remains to be explored. In-terestingly, polymorphisms in the 5-HTtransporter (5-HTT) promoter havebeen proposed as an underlying cause ofautism [Cook et al., 1997; Klauck et al.,1997]. This is presently a controversialtopic, as several studies have recentlyprovided conflicting evidence of a poten-tial link between 5-HTT and autism[Anderson et al., 1990; Maestrini et al.,1999; Persico et al., 2000; Yirmiya et al.,2001; Betancur et al., 2002; Persico et al.,2002; Coutinho et al., 2004]. Interest-ingly, finding evidence for transmissionof polymorphic alleles of the 5-HTTmay depend on the extent of social andcommunication deficits in the autismproband, suggesting that the alleles maymodify severity of autistic traits ratherthan conveying risk for autism per se[Tordjman et al., 2001]. Further supportfor 5-HTT comes from functional MRIstudies showing that individuals with oneor two copies of a short allele of the genehad greater neuronal activity than con-trols in the amygdala, a brain region im-plicated in autism [Hariri et al., 2002;Hariri and Weinberger, 2003]. In addi-tion, multiple single polymorphisms intryptophan 2,3 diooxygenase (TDO2),the rate-limiting enzyme in L-tryptophancatabolism, have been associated with au-tism. Such mutations could increase lev-els of 5-HT throughout the body andbrain [Nabi et al., 2004].
The developing serotonergic sys-tem may be dysregulated in autism. Brain5-HT synthesis is normally high in youngchildren, followed by a gradual decline toadulthood. This dynamic is disrupted inautism, such that 5-HT levels are initiallylower than normal, but gradually increaseto a greater extent than adult levels by 2to 15 years of age [Chugani et al., 1999;Chugani, 2002]. Positron emission to-mography (PET) imaging with radiola-belled L-tryptophan has demonstratedasymmetric 5-HT synthesis in the den-tate-thalamo-cortical pathway of autisticboys [Chugani et al., 1997; Chugani,2002]. Consequences of elevated levels of5-HT in the developing somatosensorysystem have been analyzed in a variety ofanimal models, providing evidence fordisruption in the formation of thalamo-cortical sensory circuits [reviewed by Luoet al., 2003]. This is not all that surpris-ing, since accumulated evidence shows5-HT to be a critical regulator of keyevents in neural and glial development,including cell proliferation, differentia-tion, migration, apoptosis, and synapto-genesis [reviewed by Lauder, 1990, 1993;Whitaker-Azmitia, 2001].
Continued interest in the seroto-nergic system comes from the relativeefficacy of serotonergic drugs in thesymptomatic treatment of some cases ofautism [Volkmar, 2001]. Pharmacologi-cal treatment studies in autism are com-plicated by various factors, including atremendous range of syndrome expres-sion, a lack of robust animal models ofthe disorder, and various methodologicalproblems. Theories have tended to fol-low treatments, and various neurochem-ical systems have been the focus of study.Treatments developed are effective rela-tive to certain disabling symptoms, but“core” problems (e.g., deficits in socialrelatedness and communication) appearless responsive to medications. Amongthese pharmacotherapies, especially inchildren and adolescents, include selec-tive serotonin reuptake inhibitors (SS-RIs), 5-HT2A receptor antagonists, tricy-clic antidepressants, or mixed dopamine/5-HT receptor antagonists [Carlsson,1998; Carlsson et al., 1999; McDougleand Posey, 2002; Veenstra-VanderWeeleet al., 2000]. Although the reasons un-derlying the efficacy of these drugs ispoorly understood, it is possible that suchtreatments may counteract developmen-tal defects in serotonergic pathwaysand/or abnormal dynamics of 5-HT syn-thesis, catabolism, or transport.
Dysregulation of the developingserotonergic system could occur by var-ious mechanisms, including mutations ingenes encoding transcription factors in-volved in specification and patterning of5-HT receptors or neurons (discussedbelow). Another possibility is that alteredexpression of genes regulating cholesterolbiosynthetic or metabolic pathwayscould compromise functioning of keyplayers in the serotonergic system, such asthe 5-HTT [Scanlon et al., 2001],VMAT2, MAO-A, or 5-HT receptors[Hillbrand et al., 2000]. This possibility issupported by hypertrophic developmentof raphe 5-HT neurons in mice withtargeted disruption of Dhcr7, a gene en-coding the last enzyme in cholesterolbiosynthesis, which is also mutated inSmith-Lemli-Opitz Syndrome [Waage-Baudet et al., 2003].
CatecholaminesEvidence for possible involvement
of dopamine and norepinephrine in au-tism comes from evidence of decreasedactivity of dopamine �-hydroxylase(DBH) and increased levels of norepi-nephrine in serum from autistic childrenand their parents [Lake et al., 1977] andchanges in catecholamine metabolites insuch children [Martineau et al., 1994].
308 MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

Recently, the DBH gene on chromo-some 9q34 has been investigated as apossible candidate locus using an affectedsib-pair method. Although no associationwas found for polymorphic alleles in sib-pairs, there was a significantly higher fre-quency in mothers, raising the possibilitythat polymorphisms in DBH could in-crease the risk for having an autistic child[Robinson et al., 2001]. Recent evidencefor maternal modifier effects (where thematernal genotype affects the fetal phe-notype) at DBH and MAO-A loci [Joneset al., 2004] provides support for this ideaand suggests changes in the interuterineenvironment as a possible risk factor.This is consistent with evidence that bothtyrosine hydroxylase (TH) and DBH arerequired for prenatal development ofmouse embryos [Thomas et al., 1995,1998; Roffler-Tarlov and Rios, 2001;Portbury et al., 2003].
Developmental Origin ofMonoamine Neurons
In the vertebrate embryo, midbraindopamine (DA) neurons, hindbrain5-HT neurons of the raphe nuclei, andnoradrenergic (NA) neurons of the locuscoeruleus (LC) are generated adjacent tothe midhindbrain boundary (MHB) oristhmus, an organizing center induced bynetworks of transcription factors and se-creted signals [Ye et al., 2001]. DA neu-rons are generated rostral to the MHB, inresponse to intersecting signals, sonichedgehog (Shh), from the floorplate, andFgf8 from the MHB. 5-HT neuronsform caudal to the MHB in response tothe same two signals, but preceded by anearlier signal, Fgf4, from the primitivestreak [Ye et al., 1998; Hynes andRosenthal, 1999]. NA neurons of the LCare also generated caudal to the MHB, inresponse to signaling by Shh, Fgf8, andBMPs [Crossley et al., 1996; Morin et al.,1997; Guo et al., 1999; Vogel-Hopkerand Rohrer, 2002; Jaszai et al., 2003;Lam et al., 2003; Eddison et al., 2004]. Asdiscussed below, specification of neuro-transmitter phenotypes of MA neuronsrequires a complex signaling throughnetworks of transcription factors, whichis only beginning to be elucidated[Briscoe et al., 1999; Pattyn et al., 1999,2000, 2004]. However, it is already clearthat these transcriptional networks mustinclude En1, En2, Wnt1, and Lmx1b[Smidt et al., 2003].
AcetylcholineSince an early study reported evi-
dence of neuropathology in basal forebrainof autistics, the location of cholinergic neu-rons innervating the cortex, this neuro-
transmitter system has received little atten-tion in the field of autism. Recently,however, the forebrain cholinergic systemhas come under closer scrutiny, with sev-eral studies utilizing neurochemical, immu-nocytochemical, or molecular biologicaltechniques, providing evidence of abnor-mal expression of cholinergic receptors incortex and cerebellum in autism. The firststudy reported lower levels of muscarinicand nicotinic cholinergic receptors in pari-etal and frontal cortices, together with ele-vated levels of the trophic factor BDNF inbasal forebrain [Perry et al., 2001]. A sub-sequent study found evidence for decreasedligand binding to �3/�4/�2 nicotinic re-ceptors in cerebellum [Lee et al., 2002].Recently; reduced expression of �4�2 nic-otinic receptor subunits and receptor bind-ing in parietal cortex of autistic brains, to-gether with increased �7 receptor bindingin the cerebellum, was reported [Martin-Ruiz et al., 2004]. Because the level ofevidence implicating the cholinergic cir-cuitry in autism is still poor at the presenttime, we will not review in detail themechanisms patterning development ofthis neurotransmitter system.
CASE REVIEW OF SPECIFICCANDIDATE GENES FORAUTISMThe concept that emerges from analyzingthe current literature is that autism obvi-ously constitutes a very heterogeneousneuropathology at the genetic level,which probably means that there aremany gene mutations that could inde-pendently lead to an “autistic” brain atthe functional level. In Table 1A–C, wesummarize all of the existing evidencefrom gene linkage and association studiesfor genes associated with autism. Thesetables represent candidate genes identi-fied as confirmed targets in familial formsof autism as well as other genes that aresimply candidates within genetic loci im-plicated in autism through linkage andassociation studies. In the following sec-tions we perform a case review of severalof the most interesting candidates classi-fied into two categories: (1) genes in-volved in the early patterning of specificregions of the CNS or specific neuronalsubpopulations and (2) genes involved inthe synapses assembly of specific neuronalcircuits.
Genes Patterning the CentralNervous System (CNS)
Engrailed and cerebellar neurodevelopmentaldefects
Recently, association studies haveprovided evidence for Engrailed-2 (En2) as
a susceptibility locus for autistic spectrumdisorder [Gharani et al., 2004]. The highlyconserved homeodomain-proteins EN-GRAILED-1 and ENGRAILED-2 arespecifically expressed in the MHB regionduring distinct periods of embryogenesis.This spatio-temporal pattern of engrailedgene expression coincides with the gener-ation of cerebellar precursor cells, whichhas led to the suggestion that they may beimportant for establishing correct cell num-bers in the cerebellum [Kuemerle et al.,1997; Baader et al., 1998]. In support ofthis view; disruption of En-1 in mice resultsin a deletion of the cerebellar anlage andmidbrain [Wurst et al., 1994] and perinataldeath of these mice. En2 knockout miceare viable but show reduced cerebellar sizeand mildly abnormal foliation [Millen et al.,1994]. Given the likely functional redun-dancy of the two engrailed genes, En1 maycompensate for the loss of En2, thus ex-plaining the mild phenotypic changes inEn2 knockout mice [Joyner et al., 1991],especially since En2 is expressed in cerebel-lar Purkinje cells (PCs) throughout embry-onic development but is down-regulated inthese cells after birth.
Engrailed and differentiation of dopaminergicneurons
It has recently been shown thatEn1 and En2 are essential for the main-tenance of DA neurons in the substantianigra (SN) and ventral tegmentum (VT)[Simon et al., 2003]. In the mouse, bothengrailed genes are expressed in mesen-cephalic DA (mDA) neurons from E11to adulthood. In engrailed 1/2 doublemutants, mDA neurons are normally in-duced and express several differentiationmarkers, but then fail to mature and arelost by E14.
The association study reportingEn2 as a susceptibility locus for autismspectrum disorder [Gharani et al., 2004],together with the phenotype observed inEn1/2 knockout mice [Simon et al.,2003], correlates well with the well-doc-umented cerebellar phenotype in autism,including cerebellar hypoplasia and de-creased number of PCs (see Neuroana-tomical Abnormalities in Autism). Fur-thermore, the role of Engrailed in thepatterning of dopaminergic neurons isalso compatible with some findings im-plicating a dopaminergic defect in autism[Ernst et al., 1997].
Wnt is a patterning gene also involved inactivity-dependent dendritic development
Several reports have provided evi-dence for Wnt2 as a susceptibility genefor autism [Wassink et al., 2001; how-ever, see also McCoy et al., 2002]. Sev-
309MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

eral mutations affecting the Wnt2 codingsequence were initially found to segre-gate with autism in families containingmultiple affected individuals [Wassink etal., 2001].
The Wingless-Int (Wnt) genes encodea large family of cysteine-rich secreted gly-coproteins that regulate diverse cell behav-iors during embryonic development [re-viewed in Wodarz and Nusse, 1998].Nineteen Wnt genes have been identifiedin the murine and human genome. Thesecreted Wnt signals positively through theFrizzled family of receptors and can be an-tagonized by secreted Frizzled-related pro-
teins (sFRPs) as one of several unrelatedprotein families that negatively regulate re-ceptor binding by Wnts.
In the developing mammalian brain,Wnts have been implicated in the pattern-ing of the CNS along the antero-posterioraxis, including patterning of the telenceph-alon, diencephalon, and mesencephalon.The signaling mechanisms underlying Wntfunction during CNS patterning includemany cytoplasmic proteins, including Di-shevelled and �-catenin. Specifically, over-expression of �-catenin increases corticalsize by positively regulating the generationof neural precursors [Chenn and Walsh,
2002], while loss-of-function mutations inindividual Wnts cause deletions or malfor-mations of distinct brain regions [Brault etal., 2001; Lyuksyutova et al., 2003; Zhouet al., 2004]. Spatial and temporal expres-sion patterns of WNT genes show strikingsimilarity between human and mouse, sug-gesting that the developmental roles ofthese genes may be highly conserved[Grove et al., 1998; Abu-Khalil et al.,2004].
Recent investigation of the roleof Wnt signaling during neuronal dif-ferentiation has revealed a surprisinglate role in the regulation of activity-
Table 1A. Autism Candidate Genes
Marker Symbol Description
Chromo-somes X–2
ReferencesChromosome
GDB:191610 MAOA monoamine oxidase A Xp11.4-p11.3 Cohen et al., 2003Hs.438877 NLGN3 Neuroligin 3 Xq13.1 Jamain et al., 2003KIAA1260 NLGN4 Neuroligin 4 Xp22.33 Jamain et al., 2003PMC153509P1 AGTR2 angiotensin II receptor, type 2 Xq22-q23 Thomas et al., 1999;
Vervoort et al.,2002
DXS7069 MECP2 methyl CpG binding protein 2 (Rett syndrome) Xq28 Muhle et al., 2004;Longo et al., 2004
DXS1047 MRXS11 mental retardation, X-linked, syndromic 11 Xq26-q27 Liu et al., 2001; Shaoet al., 2003
DXS6789 PTOS2 Ptosis, hereditary congenital 2 Xq24-q27.1 Liu et al., 2001; Shaoet al., 2003
DXS6789 CHDS3 Coronary heart disease, susceptibility to, 3 Xq23-q26 Liu et al., 2001; Shaoet al., 2003
DXS6789 MTBS Mycobacterium tuberculosis, susceptibility to infection by Xq Liu et al., 2001; Shaoet al., 2003
D1S1675 GGAA20F08 NA (moderate effect) 1p12 Risch et al., 1999D1S1656
D1S534SCZD9 schizophrenia disorder 9 1q21-1q22 Buxbaum et al.,
2004; Risch et al.,1999
D1S2141 KCNK2 potassium channel, subfamily K, member 2 1q41 Buxbaum et al., 2004G65049 DISC1 disrupted in schizophrenia 1 1q42.1 Buxbaum et al., 2004D1S3462 SLEB1 systemic lupus erythematosus susceptibility 1 1q41-q42 Buxbaum et al., 2004D1S547 KMO kynurenine 3-monooxygenase (kynurenine 3-hydroxylase) 1q42-q44 Buxbaum et al., 2004WI-20654 KCNK1 potassium channel, subfamily K, member 1 1q42-q43 Buxbaum et al., 2004606058 cAMP-GEFII cAMP-regulated guanine nucleotide exchange factor 1
(also mutations in TBR-1; GAD1; DLX1; DLX2; CHN1;ATF2; HOXD1, NEUROD1)
2q21-q33 Bacchelli et al., 2003
D2S142 PSDA Phrase speech delay, autism-related 2q Buxbaum et al., 2004SHGC-82894 SCN2A2 sodium channel, voltage-gated, type II, alpha 2 2q23-q24 Weiss et al., 2003RH79252 SCN3A sodium channel, voltage-gated, type III, alpha 2q24 Weiss et al., 2003A006O14 SCN1A sodium channel, voltage-gated, type I, alpha 2q24.3 Weiss et al., 2003D2S364 MPRM Myopathy, proximal, with early respiratory muscle involvement
(Edstr2q24-q31 Buxbaum et al., 2004
D2S2188 MMDK Mesomelic dysplasia, Kantaputra type 2q24-q32 IMGSAC, 2001D2S2716 CHRNA1 cholinergic receptor, nicotinic, alpha polypeptide 1 (muscle) 2q24-q32 Agulhon et al., 1999;
Martin-Ruiz, etal., 2004
G54269 AGC1 mitochondrial aspartate-glutamate carrier protein 2q24-q33 Ramoz et al., 2004G16421 INPP1 inositol polyphosphate-1-phosphatase 2q32 Serajee et al., 2003RH68995 ERBB4 receptor for NDF/heregulin 2q33.3-q34 Pescucci et al., 2003RH66377 ADAM23 disintegrin and metalloproteinase domain 23 2q33 Pescucci et al., 2003RH15615 KLF7 Kruppel-like factor 7 2q33.3-q34 Pescucci et al., 2003RH65625 GPR1 G protein-coupled receptor 1 2q33.3 Pescucci et al., 2003PMC16008P2 FZD5 frizzled homolog 5 2q33-q34 Pescucci et al., 2003G62667 MAP2 microtubule-associated protein 2 2q34-q35 Pescucci et al., 2003D2S2128 NRP2 neuropilin 2 2q33.3 Pescucci et al., 2003D2S2634 CREB1 cAMP responsive element binding protein 1 2q34 Pescucci et al., 2003
310 MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

dependent arborization of dendrites inhippocampal pyramidal neurons. Yuand Malenka [2003] have shown thatoverexpression of �-catenin (and othermembers of the cadherin/catenin com-plex) enhances dendritic arborization,whereas sequestering endogenous�-catenin causes a decrease in dendriticbranch tip number and prevents theenhancement of dendritic morphogen-esis caused by neural activity [Yu and
Malenka, 2003]. This study also re-vealed that the release of Wnt, whichoccurs during normal neuronal devel-opment, is enhanced by manipulationsthat mimic increased activity and thatWnt contributes to the effects of neuralactivity on dendritic arborization.These results show that �-catenin is animportant mediator of dendritic mor-phogenesis and that Wnt/-catenin sig-naling is likely to be important for activ-
ity-dependent dendritic differentiation[Yu and Malenka, 2003].
Interestingly, inactivation of Di-shevelled-1 (Dvl1) in the mouse leads toabnormal development of social behav-iors, such as differences in whisker trim-ming, deficits in nest building, reducedhuddling contact during cage sleeping,and subordinate responses in a socialdominance test [Lijam et al., 1997]. No-tably, sensorimotor gating was abnormal
Table 1B. Autism Candidate Genes
Marker Symbol Description
Chromosomes3–13
ReferencesChromosome
D3S3680 KIAA0121 KIAA0121 gene product 3p25.2 IMGSAC 2001D3S3037 AUTS2 Autism, susceptibility to, 2 3q25-27 Auranen et al. 2002D3S3037 AUTS3 Autism, susceptibility to, 3 3q25.1-3q25.2 Auranen et al. 2002D3S2418 DFNA44; FGF12 deafness, autosomal dominant; fibro-
blast growth factor 123q28-29 Risch et al. 1999
D4S2366 HLN2 Huntington-like neurodegenerativedisorder 2
4p15.3 Risch et al. 1999
D4S2366 SLEB3 systemic lupus erythematosus suscepti-bility 3
4p16-15.2 Risch et al. 1999
D4S2366 PPP2R2C protein phosphatase 2 (2A), regulatorysubunit B (PR 52), gamma isoform
4p16.1 Risch et al. 1999
SHGC-50349 GLRB glycine receptor, beta 4q31.3 Ramanthan et al. 2004PMC310777P9 NPY5R neuropeptide Y receptor Y5 4q31-q32 Ramanthan et al. 2004NPY1R_956 NPY1R neuropeptide Y receptor Y1 4q31.3-q32 Ramanthan et al. 2004D4S3293 TDO2 tryptophan 2,3-dioxygenase 4q31-q32 Nabi et al. 2004SHGC-67298 GRIA2 glutamate receptor, ionotropic,
AMPA 24q32-q33 Samanthan et al. 2004
GDB:373727 GRIK2 glutamate receptor, ionotropic, kai-nate 2 (GluR6 kainate receptor)
6q16.3-q21 Jamain et al. 2002
D6S283 IDDM15 insulin-dependent diabetes mellitus 15 6q21 Philippe et al. 1999D7S640 AUTS1 autism susceptibility to, 1 7q Ashley-Koch et al. 1999;
Barrett et al. 1999;IMGSAC 2001
KIAA0442 AUTS2 autism, susceptibility to, 2 7q11.22-q11.23
Sultana et al. 2002
D7S2204 CMT2F Charcot-Marie-Tooth disease, axonal,F
7q11-q21 IMGSAC 1998
D7S3120 RELN reelin 7q22 Hutcheson et al. 2003D7S501-D7S2847 FOXP2, SPCH1; WNT2; RAY1; NRCAM;
SMO; CAV1,2; GPR22; CFTR; MEST/PEG1; MEK2; PAX4
7q22.1-q33 Hutcheson et al. 2003;IMGSAC 2001;Adres 2002
sWSS3754 GMR8 glutamate receptor, metabotropic 8 7q31-q33 Serajee et al. 2003STS-X83368 PIK3CG phosphoinositide-3-kinase, catalytic,
gamma polypeptide7q22.3 Serajee et al. 2003
D7S23 WNT2 wingless-type MMTV integration sitefamily member 2
7q31 Wassinck et al. 2001
D7S684 CHDM Chordoma (malignant tumors derivedfrom notochordal remnants)
7q33 IMGSAC 1998
D7S495 OTSC2 otosclerosis 2 (sclerosis of labyrinthinecapsule; hearing impairment)
7q34-q36 Shao et al. 2003
RH102827 EN2 engrailed homolog 2 7q36 Gharani et al. 2004D9S1826 SPG19 spastic paraplegia 19 (autosomal domi-
nant)9q IMGSAC 2001
GDB:551079 DBH dopamine beta-hydroxylase 9q34 Robinson et al. 2001D9S158 JBTS1 Joubert syndrome 1 9q34.3 IMGSAC 2001D9S1826 DFNB33 deafness, autosomal recessive 33 9q34-qtel IMGSAC 2001D11S2371 SCZD2 schizophrenia disorder 2 11q14-q21 Risch et al. 1999D12S1901 AVPR1A arginine vasopressin receptor 1A 12q14-q15 Wassink et al. 2004608049 AUTS3 Autism, susceptibility to, 3 13q14-q22 Bradford et al. 2001HTR2A-112F HTR2A-2 5-hydroxytryptamine (serotonin) re-
ceptor 2A13q14-q21 Andres et al. 2002;
Veenstra-Vander-Weele et al. 2002
D13S779 SCZD7 schizophrenia disorder 7 13q32 Risch et al. 1999
311MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER
in these mice, as assessed by prepulseinhibition of acoustic and tactile startlereflexes. This interesting behavioral phe-notype has been suggested as a potentialmodel for aspects of several human psy-chiatric disorders, including autism.Given the developmental mechanismsunderlying this interesting behavioralphenotype, it is tempting to speculatethat the Wnt/Dvl1 pathway might con-trol patterning and synaptic assembly anddendritic branching in key cortical andmesencephalic neuronal networks con-trolling complex social behaviors im-paired in autism. Surprisingly, to date,very few studies have explored this pos-sibility, and future investigations shouldtest this hypothesis.
Transcription factors specifying corticalinterneuron phenotype
Aristaless-related proteins (includ-ing Arx) belong to the Pax family ofhomeobox transcription factors, severalmembers of which are known to be in-volved in genetic diseases. A recent studyusing a gene-targeting approach to iden-tify a pivotal role for the Arx transcrip-tion factor during brain development has
demonstrated that this gene controls ap-propriate proliferation of dorsal and ven-tral forebrain neural precursors, but alsothe migration and differentiation ofGABAergic interneurons from the me-dial ganglionic eminence to the cortex[Kitamura et al., 2002]. These findingsprompted these investigators to askwhether any human diseases are causedby ARX inactivation. Their search ledthem to several individuals suffering fromX-linked lissencephaly with abnormalgenitalia (XLAG), who turn out to havemutations in ARX. Interestingly, consis-tent with the findings in mice, pheno-type/genotype studies in humans suggestthat truncating mutations cause X-linkedlissencephaly with abnormal genitalia,and insertion/missense mutations resultin epilepsy and mental retardation with-out cortical dysplasia [Sherr, 2003].Therefore, mutations in the homeoboxgene, ARX, cause a diverse spectrum ofdisorders, including cognitive impair-ment, epilepsy, and, in another group ofpatients, severe cortical malformations.Although the precise prevalence of ARXmutations is unclear, further investiga-tions will undoubtedly test whether dis-
tinct mutations in ARX may cause di-verse forms of mental retardation,epilepsy, or autism in males [Sherr,2003].
Genes Controlling SynapticAssembly and DendriticDevelopment
Neuroligin (NLGN) 3 and 4Recently, the identification of a
frameshift mutation (1186insT) in theNLGN4 gene as well as a cysteine tothreonine substitution in position 451 ofNLGN3 have been reported to segregatewith autism in two independent Swedishfamilies [Jamain et al., 2003]. Subse-quently, another team has confirmed thisfinding and has found a 2-base-pair de-letion in the NLGN4 gene, leading to apremature stop codon, in both autisticand nonautistic mentally retarded males[Laumonnier et al., 2004]. This stronglysuggests that NLGN4 might not only beinvolved in autism, but also in mentalretardation, suggesting that autism andmental retardation might have commongenetic origins.
Table 1C. Autism Candidate Genes
Marker Symbol Description
Chromosomes15–20
ReferencesChromosome
D15S652 OTSC1; LCS1 otosclerosis 1; lymphedema-cholestasis syndrome 1 15q25-q26 Risch et al. 1999D7S530 AUTS1 autism susceptibility 1 15q11-q13 IMGSAC 1998PMC311424P2 HERC2 hect domain and RLD 2 15q13 Smith et al. 2000STS-L08485 GABRA5 gamma-aminobutyric acid (GABA) A receptor, al-
pha 515q11.2-q12 Shao et al. 2003; Longo et
al. 200RH11160 GABRB3 gamma-aminobutyric acid (GABA) A receptor, beta
315q11.2-q12 Shao et al. 2003; Longo et
al. 200PMC24540P1 NDN NECDIN [necdin (mouse) homolog] 15q11-q13 Chibuk et al. 2001D15S10 UBE3A ubiquitin protein ligase E3A (human papilloma vi-
rus E6-associated protein, Angelman syndron15q11-q13 Nurmi et al. 2003
GDB:593257 GABRG3 gamma-aminobutyric acid (GABA) A receptor,gamma 3
15q11-q13 Shao et al. 2003; Long etal. 2004
RH44910 PWCR1 Prader-Willi syndrome chromosome region 1 15q11.2 Veltman et al. 2004RH121309 SNURF SNRPN upstream reading frame 15q12 Mann and Bartolomei 1999GDB:626138 SNRPN small nuclear ribonucleoprotein polypeptide N 15q12 Mann and Bartolomei 1999PMC64493P3 NDNL2/
MAGE-Gnecdin-like 2/Melanoma-associated antigen F1 15q13.1 Chibuk et al. 2001
G10644 PTPN9 protein tyrosine phosphatase, non-receptor type 9 15q23 Smith et al. 2000G06258 SLP-1, hUNC-24 stomatin (EPB72)-like 1 15q24-q25 Smith et al. 2000D16S403 RP22 retinitis pigmentosa 22 (autosomal recessive) 16p12.3-p12.1 Risch et al. 1999GDB:378419 TSC2; PKD1* tuberous sclerosis 2; polycystic kidney disease 1 16p13.3 Serajee et al. 2003D16S407 GRIN2A glutamate receptor, ionotropic, N-methyl D-aspar-
tate 2A16p13.2 IMGSAC 1998
G67509 SSTR5 somatostatin receptor 5 16p13.3 Lauritsen et al. 2003SLC6A4 HTT, HTTINT2 Solute carrier family 6 (neurotransmitter transporter,
serotonin), member 4; brain 5-HT transpo17q11.1-q12 IMGSAC 2001
D17S1298 FIMG1 myasthenia gravis, familial infantile, 1 17p13 Risch et al. 1999D19S587 HSCRS3 Hirschsprung disease, short-segment, 3 19q12 Liu et al. 2001PMC310963P4 APOE apolipoprotein E 19q13.2 Persico et al. 2004D20S482 CHED2 comeal endothelial dystrophy 2 (autosomal reces-
sive)20p13 Risch et al. 1999
312 MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

The Neuroligin gene family wasinitially identified as a component ofpostsynaptic glutamatergic synapses,which have been shown to play a role inthe trans-neuronal signaling that controlssynapse differentiation through bindingof neurexin-� on the presynaptic side[Ichtchenko et al., 1995; Song et al.,1999]. As we were writing this review,Chih and coworkers reported conse-quences of these disease-associated muta-tions on neuroligin function in vitro[Chih et al., 2004]. These authors dem-onstrated that a point mutation in thearginine residue in position 451 and anonsense mutation in aspartate 396 ofNLGN3 and NLGN4, respectively, re-sulted in intracellular retention of themutant proteins. Overexpression ofwildtype NLGN3 and NLGN4 proteinsin hippocampal neurons stimulated theformation of presynaptic terminals,whereas the disease-associated mutationsresulted in loss of this synaptogenic func-tion. These findings suggest that the pre-viously identified mutations in Neuroligingenes are likely to be relevant for theneurodevelopmental defects in autismspectrum disorders and mental retarda-tion, since they impair the function ofsynaptic cell adhesion molecules [Chih etal., 2004].
BDNF and MeCP2Neurotrophins have multiple func-
tions during peripheral and CNS devel-opment, such as controlling neuronalsurvival, target innervation, and synapto-genesis. Neurotrophins are secreted li-gands that exert their biological functionsby binding to a high-affinity receptor,the Trk tyrosine kinase receptor. Aprominent member of this family of neu-rotrophins is brain-derived neurotrophicfactor (BDNF), which plays multipleroles during neuronal differentiation, in-cluding neuronal survival, activity-de-pendent dendritic and axonal out-growth/branching, synapse formation,and neuronal plasticity underlying learn-ing and memory [reviewed in Shieh andGhosh, 1997; Kaplan and Miller, 2000].
In two recent studies monitoringthe plasma levels of different neurotro-phins on large samples of randomlypicked children retrospectively diagnosedwith autism spectrum disorder or mentalretardation without autism, cerebral palsyor age-matched controls revealed thatboth NT4 and BDNF (the two trkBligands) are significantly increased in au-tistic and mentally retarded patients com-pared to controls [Nelson et al., 2001;Miyazaki et al., 2004]. Interestingly,other neurotrophins such as NGF (trkA
ligands) or NT3 (trkC ligands) are un-changed compared to controls. Of courseone should be careful about translatingincreased plasma levels with increasedCNS levels, but these findings suggestthat, during early infancy, trkB ligandsmight specifically be expressed and/orsecreted at higher levels in the CNS ofautistic or mentally retarded childrenfrom a source that remains to be deter-mined (central or peripheral nervous sys-tem?). The effect of BDNF and NT4 onactivity-dependent dendritic outgrowthand branching is well established duringdevelopment [McAllister et al., 1996,1997, 1995] and therefore could be cor-related with the transient, early increaseof brain growth reported in young autis-tic babies [Lainhart et al., 1997;Courchesne et al., 2001, 2003]. Futureinvestigations should test whetherBDNF-mediated signaling is increased inthe early period of postnatal brain devel-opment in autism and whether this cor-relates with a premature increase in den-dritic outgrowth of pyramidal neurons,which could in turn have profound con-sequences on the establishment of corti-cal networks.
What could cause this increasedlevel of BDNF expression and/or secre-tion? An interesting possibility lies in theanalysis of the role of MeCP2 in thecontrol of BDNF transcription [Chen etal., 2003]. Mutations in methyl-CpG-binding protein 2 (MeCP2), which en-codes a protein that has been proposed tofunction as a global transcriptional re-pressor, are the cause of Rett syndrome,an X-linked progressive neurological dis-order. Interestingly; Rett syndrome issometime associated with autism. Al-though the selective inactivation ofMeCP2 in neurons is sufficient to confera Rett-like phenotype in mice [Shahba-zian et al., 2002], the specific functions ofMeCP2 in postmitotic neurons are notknown. Chen and coworkers have re-cently shown that MeCP2 binds selec-tively to BDNF promoter III and func-tions to repress expression of the BDNFgene [Shahbazian et al., 2002]. Mem-brane depolarization mimicking sus-tained levels of neuronal activity triggersthe calcium-dependent phosphorylationand release of MeCP2 from BDNF pro-moter III, thereby facilitating transcrip-tion. These studies indicate that MeCP2plays a key role in the control of neuronalactivity–dependent gene regulation andsuggest that the deregulation of this pro-cess may underlie the pathology of Rettsyndrome.
Rett disorder and autism are per-vasive developmental disorders as defined
by the Diagnostic and Statistical Manualof Mental Disorders, Fourth Edition(DSM-IV) and the International Classifi-cation of Diseases, Tenth Revision. Re-cent studies indicate that at least 80% ofRett disorder cases are caused by muta-tions in the MeCP2 gene. Since there issome phenotypic overlap between autis-tic disorder and Rett disorder [Muhle etal., 2004], a recent study aimed at ana-lyzing large groups of females clinicallydiagnosed with autistic disorder for thepresence of mutations in the MeCP2gene. Two females (4%) presenting au-tistic disorder were found to have denovo mutations in MeCP2. These dataprovide additional evidence of variableexpression in the Rett disorder pheno-type and, taken together with the role ofMeCP2 in the control of BDNF tran-scription mentioned above, these findingsuggest a potential mechanism wherebyMeCP2 mutations might represent a riskfactor for the appearance of autismthrough regulating BDNF expressionand potentially dendritic differentiationin the cortex.
CONCLUSIONThe concept that emerges from an-
alyzing the current literature is that au-tism has a strong genetic component, butobviously constitutes a very heteroge-neous neuropathology at the geneticlevel (see Wassink et al., this issue; seealso Table 1A–C). In light of currentevidence, it seems likely that the etiologyof autism involves complex interactionsbetween environmental factors and geneticmutations controlling either (1) the pat-terning of neuronal populations criticalfor the control of inhibition/excitation inthe cortex and/or (2) the synaptic assem-bly of these excitatory and inhibitoryneuronal networks and/or the neuronalnetworks involved in large-scale corticalneuromodulation.
An important concept in develop-mental neurobiology is that genes in-volved in the development of the CNSare extremely pleiotropic: i.e., severalimportant genes involved in the earlypatterning and specification of neuronalsubpopulations also act later in develop-ment to regulate the proper synaptic as-sembly of the same or other neuronalpopulations. Therefore, a mutation inone of the genes reviewed above willundoubtedly have many complex, non-redundant functions during developmentof the CNS that could lead to a divergentand complex neuropathology such as au-tism.
We hope that after reading this re-view the reader will realize that, although
313MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

much work needs to be done in theexploration of the neurodevelopmentalmechanisms that could underlie autism,we now have new directions to explore.A new hypothesis such as the imbalancebetween excitation and inhibition in thecortex is attractive because it is based onfunctional evidence characterizing theautistic brain. The conceptual frameworkand the molecular tools developed in thisemerging field will undoubtedly allowrapid progress in the characterization ofthe neuronal networks underlying thisdevastating developmental neuropathol-ogy. f
REFERENCESAbu-Khalil A, Fu L, Grove EA, et al. 2004. Wnt
genes define distinct boundaries in the devel-oping human brain: Implications for humanforebrain patterning. J Comp Neurol 474:276–288.
Agulhon C, Abitbol M, Bertrand D, Malafosse, A.1999. Localization of mRNA for CHRNA7in human fetal brain. Neuroreport 10:2223–2227
Anderson GM, Horne WC, Chatterjee D, et al.1990. The hyperserotonemia of autism. AnnN Y Acad Sci 600:331–340; discussion 341–332.
Anderson SA, Eisenstat DD, Shi L, et al. 1997.Interneuron migration from basal forebrain toneocortex: Dependence on Dlx genes. Sci-ence 278:474–476.
Andres C. 2002. Molecuar genetics and animalmodels in autistic disorder. Brain Res Bull57:109–119
Aoyagi T, Takekawa T, Fukai T. 2003. Gammarhythmic bursts: Coherence control in net-works of cortical pyramidal neurons. NeuralComput 15:1035–1061.
Ashley-Koch A, Wolpert CM, Menold MM, et al.1999. Genetic studies of autistic disorder andchromosome 7. Genomics 61:227–236.
Auranen M, Vanhala R, Varilo T, et al. 2002. Agenomewide screen for autism-spectrum dis-orders: evidence for a major susceptibility lo-cus on chromosome 3q25-27. Am J HumGenet 71:777–790.
Aylward EH, Minshew NJ, Field K, et al. 2002.Effects of age on brain volume and head cir-cumference in autism. Neurology 59:175–183.
Baader SL, Sanlioglu S, Berrebi AS, et al. 1998.Ectopic overexpression of engrailed-2 in cer-ebellar Purkinje cells causes restricted cell lossand retarded external germinal layer develop-ment at lobule junctions. J Neurosci 18:1763–1773.
Bacchelli E, Blasi F, Biondolillo M, et al. 2003.Screening of nine candidate genes for autismon chromosome 2q reveals rare nonsynony-mous variants in the cAMP-GEFII gene. MolPsychiatry 8:916–924.
Bailey A, Luthert P, Dean A, et al. 1998. A clini-copathological study of autism. Brain 121:889–905.
Ballaban-Gil K, Tuchman R. 2000. Epilepsy andepileptiform EEG: Association with autismand language disorders. Ment Retard DevDisabil Res Rev 6:300–308.
Barrett S, Beck JC, Bernier R, et al. 1999. Anautosomal genomic screen for autism. Collab-orative linkage study of autism. Am J MedGenet 88:609–615.
Belmonte MK, Cook EH, Anderson GM, et al.2004. Autism as a disorder of neural informa-tion processing: Directions for research andtargets for therapy. Mol Psychiatry 9:646–663.
Betancur C, Corbex M, Spielewoy C, et al. 2002.Serotonin transporter gene polymorphismsand hyperserotonemia in autistic disorder.Mol Psychiatry 7:67–71.
Borodinsky LN, Root CM, Cronin JA, et al. 2004.Activity-dependent homeostatic specificationof transmitter expression in embryonic neu-rons. Nature 429:523–530.
Brault V, Moore R, Kutsch S, et al. 2001. Inacti-vation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramaticbrain malformation and failure of craniofacialdevelopment. Development 128:1253–1264.
Briscoe J, Sussel L, Serup P, et al. 1999. Homeoboxgene Nkx2.2 and specification of neuronalidentity by graded Sonic hedgehog signalling.Nature 398:622–627.
Brock J, Brown CC, Boucher J, et al. 2002. Thetemporal binding deficit hypothesis of autism.Dev Psychopathol 14:209–224.
Buhl EH, Halasy K, Somogyi P. 1994. Diversesources of hippocampal unitary inhibitorypostsynaptic potentials and the number ofsynaptic release sites. Nature 368:823–828.
Buxbaum JD, Silverman J, Keddache M, et al.2004. Linkage analysis for autism in subsetfamilies with obsessive-compulsive behaviors:evidence for an autism susceptibility gene onchromosome 1 and further support for sus-ceptibility genes on chromosome 6 and 19.Mol Psychiatry 9:144–150.
Carlsson ML. 1998. Hypothesis: Is infantile autisma hypoglutamatergic disorder? Relevance ofglutamate–serotonin interactions for pharma-cotherapy. J Neural Transm 105:525–535.
Carlsson ML, Martin P, Nilsson M, et al. 1999. The5-HT2A receptor antagonist M100907 ismore effective in counteracting NMDA an-tagonist than dopamine agonist–induced hy-peractivity in mice. J Neural Transm 106:123–129.
Casanova MF, Buxhoeveden DP, Switala AE, et al.2002. Minicolumnar pathology in autism.Neurology 58:428–432.
Casarosa S, Fode C, Guillemot F. 1999. Mash1regulates neurogenesis in the ventral telen-cephalon. Development 126:525–534.
Chen WG, Chang Q, Lin Y, et al. 2003. Derepres-sion of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Sci-ence 302:885–889.
Chenn A, Walsh CA. 2002. Regulation of cerebralcortical size by control of cell cycle exit inneural precursors. Science 297(5580):365–369.
Chibuk TK, Bischof JM, Wevrick R. 2001. Anecdin/MAGE-like gene in the chromosome15 autism susceptibility region: expression,imprinting, and mapping of the human andmouse orthologues. BMC Genet 2:22.
Chih B, Afridi SK, Clark L, et al. 2004. Disorder-associated mutations lead to functional inac-tivation of neuroligins. Hum Mol Genet 13:1471–1477.
Chugani DC. 2002. Role of altered brain serotoninmechanisms in autism. Mol Psychiatry 7Suppl 2:S16–S17.
Chugani DC, Muzik O, Behen M, et al. 1999.Developmental changes in brain serotoninsynthesis capacity in autistic and nonautisticchildren. Ann Neurol 45:287–295.
Chugani DC, Muzik O, Rothermel R et al. 1997.Altered serotonin synthesis in the dentato-
thalamocortical pathway in autistic boys. AnnNeurol 42:666–669.
Cline HT. 2001. Dendritic arbor development andsynaptogenesis. Curr Opin Neurobiol 11:118–126.
Cobb SR, Buhl EH, Halasy K, et al. 1995. Syn-chronization of neuronal activity in hip-pocampus by individual GABAergic inter-neurons. Nature 378:75–78.
Cohen I, Liu X, Schutz C, et al. 2003. Associationof autism severity with a monoamine oxidaseA functional polymorphism. Clin Genet 64:190–197.
Cook EH Jr., Courchesne R, Lord C, et al. 1997.Evidence of linkage between the serotonintransporter and autistic disorder. Mol Psychi-atry 2:247–250.
Courchesne E. 1997. Brainstem, cerebellar and lim-bic neuroanatomical abnormalities in autism.Curr Opin Neurobiol 7:269–278.
Courchesne E, Carper R, Akshoomoff N. 2003.Evidence of brain overgrowth in the first yearof life in autism. JAMA 290:337–344.
Courchesne E, Karns CM, Davis HR, et al. 2001.Unusual brain growth patterns in early life inpatients with autistic disorder: An MRI study.Neurology 57:245–254.
Courchesne E, Yeung-Courchesne R, Press G, etal. 1988. Hypoplasia of cerebellar vermal lob-ules VI and VII in autism. N Engl J Med318:1349–1354.
Coutinho AM, Oliveira G, Morgadinho T, et al.2004. Variants of the serotonin transportergene (SLC6A4) significantly contribute to hy-perserotonemia in autism. Mol Psychiatry9:264–271.
Crossley PH, Martinez S, Martin GR. 1996. Mid-brain development induced by FGF8 in thechick embryo. Nature 380:66–68.
DeFelipe J, Farinas I. 1992. The pyramidal neuronof the cerebral cortex: Morphological andchemical characteristics of the synaptic inputs.Prog Neurobiol 39:563–607.
DeFelipe J, Hendry SH, Hashikawa T, et al. 1990.A microcolumnar structure of monkey cere-bral cortex revealed by immunocytochemicalstudies of double bouquet cell axons. Neuro-science 37:655–673.
Eddison M, Toole L, Bell E, et al. 2004. Segmentalidentity and cerebellar granule cell inductionin rhombomere 1. BMC Biol 2:14.
Ernst M, Zametkin AJ, Matochik JA, et al. 1997.Low medial prefrontal dopaminergic activityin autistic children. Lancet 350:638.
Fatemi SH, Halt AR, Stary JM, et al. 2002. Glu-tamic acid decarboxylase 65 and 67 kDa pro-teins are reduced in autistic parietal and cer-ebellar cortices. Biol Psychiatry 52:805–810.
Fode C, Ma Q, Casarosa S, et al. 2000. A role forneural determination genes in specifying thedorsoventral identity of telencephalic neu-rons. Genes Dev 14:67–80.
Folstein SE, Rosen-Sheidley B. 2001. Genetics ofautism: Complex aetiology for a heteroge-neous disorder. Nat Rev Genet 2:943–955.
Freund TF. 2003. Interneuron diversity series:Rhythm and mood in perisomatic inhibition.Trends Neurosci 26:489–495.
Freund TF, Buzsaki G. 1996. Interneurons of thehippocampus. Hippocampus 6:347–470.
Gharani N, Benayed R, Mancuso V, et al. 2004.Association of the homeobox transcriptionfactor, ENGRAILED 2, 3, with autism spec-trum disorder. Mol Psychiatry 9:474–484.
Grove EA, Tole S, Limon J, et al. 1998. The hemof the embryonic cerebral cortex is defined bythe expression of multiple Wnt genes and iscompromised in Gli3-deficient mice. Devel-opment 125:2315–2325.
314 MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

Guo S, Brush J, Teraoka H, et al. 1999. Develop-ment of noradrenergic neurons in the ze-brafish hindbrain requires BMP, FGF8, andthe homeodomain protein soulless/Phox2a.Neuron 24:555–566.
Hardan AY, Minshew NJ, Keshavan MS. 2000.Corpus callosum size in autism. Neurology55(7):1033–1036.
Hariri AR, Mattay VS, Tessitore A, et al. 2002.Serotonin transporter genetic variation andthe response of the human amygdala. Science297:400–403.
Hariri AR, Weinberger DR. 2003. Functionalneuroimaging of genetic variation in seroto-nergic neurotransmission. Genes Brain Behav2:341–349.
Hashimoto T, Tayama M, Murakawa K, et al.1995. Development of the brainstem and cer-ebellum in autistic patients. J Autism DevDisord 25:1–18.
Hillbrand M, Waite BM, Miller DS, et al. 2000.Serum cholesterol concentrations and moodstates in violent psychiatric patients: An expe-rience sampling study. J Behav Med 23:519–529.
Holopainen IE, Kokkonen H, Korpi ER. 2001a.Angelman syndrome: From the phenotype ofa developmental disorder to the genes. Duo-decim 117:383–387.
Holopainen IE, Metsahonkala EL, Kokkonen H, etal. 2001b. Decreased binding of [11C]fluma-zenil in Angelman syndrome patients withGABA(A) receptor beta3 subunit deletions.Ann Neurol 49:110–113.
Hussman JP. 2001. Suppressed GABAergic inhibi-tion as a common factor in suspected etiolo-gies of autism. J Autism Dev Disord 31:247–248.
Hynes M, Rosenthal A. 1999. Specification of do-paminergic and serotonergic neurons in thevertebrate CNS. Curr Opin Neurobiol 9:26–36.
IMGSAC. 1998. A full genome screen for autismwith evidence for linkage to a region onchromsome 7q. International Molecular Ge-netic Study of Autism Consortium. Hum MolGenet 7:571–578.
IMGSAC. 2001. A genomewide screen for autism:strong evidence for linkage to chromosomes2q, 7q, and 16p. Am J Hum Genet 69:570–581.
Ichtchenko K, Hata Y, Nguyen T, et al. 1995.Neuroligin 1: A splice site-specific ligand forbeta-neurexins. Cell 81:435–443.
Jamain S, Betancur C, Quach H, et al. 2002. Link-age and association of the glutamate receptor6 gene with autism. Mol Psychiatry 7:302–310.
Jamain S, Quach H, Betancur C, et al. 2003. Mu-tations of the X-linked genes encoding neu-roligins NLGN3 and NLGN4 are associatedwith autism. Nat Genet 34:27–29.
Jaszai J, Reifers F, Picker A, et al. 2003. Isthmus-to-midbrain transformation in the absence ofmidbrain-hindbrain organizer activity. Devel-opment 130:6611–6623.
Jessell TM. 2000. Neuronal specification in thespinal cord: Inductive signals and transcrip-tional codes. Nat Rev Genet 1:20–29.
Jones EG. 2000. Microcolumns in the cerebral cor-tex. Proc Natl Acad Sci U S A 97:5019–5021.
Jones MB, Palmour RM, Zwaigenbaum L, et al.2004. Modifier effects in autism at theMAO-A and DBH loci. Am J Med Genet126B:58–65.
Joyner AL, Herrup K, Auerbach BA, et al. 1991.Subtle cerebellar phenotype in mice homozy-gous for a targeted deletion of the En-2 ho-meobox. Science 251:1239–1243.
Just MA, Cherkassky VL, Keller TA, et al. 2004.Cortical activation and synchronization dur-ing sentence comprehension in high-func-tioning autism: Evidence of underconnectiv-ity. Brain 127(Pt 8):1811–1821.
Kanner, L. 1943. Autistic disturbances of affectivecontact. Nervous Child. 2:217–250.
Kaplan DR, Miller FD. 2000. Neurotrophin signaltransduction in the nervous system. CurrOpin Neurobiol 10:381–391.
Kitamura K, Yanazawa M, Sugiyama N, et al. 2002.Mutation of ARX causes abnormal develop-ment of forebrain and testes in mice and X-linked lissencephaly with abnormal genitaliain humans. Nat Genet 32:359–369.
Klauck SM, Poustka F, Benner A, et al. 1997.Serotonin transporter (5-HTT) gene variantsassociated with autism?. Hum Mol Genet6:2233–2238.
Korvatska E, Van de Water J, Anders TF, et al.2002. Genetic and immunologic consider-ations in autism. Neurobiol Dis 9:107–125.
Kuemerle B, Zanjani H, Joyner A, et al. 1997.Pattern deformities and cell loss in En-grailed-2 mutant mice suggest two separatepatterning events during cerebellar develop-ment. J Neurosci 17:7881–7889.
Lainhart JE, Piven J, Wzorek M, et al. 1997. Mac-rocephaly in children and adults with autism.J Am Acad Child Adolesc Psychiatry 36:282–290.
Lake CR, Ziegler MG, Murphy DL. 1977. In-creased norepinephrine levels and decreaseddopamine-beta-hydroxylase activity in pri-mary autism. Arch Gen Psychiatry 34:553–556.
Lam CS, Sleptsova-Friedrich I, Munro AD, et al.2003. SHH and FGF8 play distinct roles dur-ing development of noradrenergic neurons inthe locus coeruleus of the zebrafish. Mol CellNeurosci 22:501–515.
Lauder JM. 1993. Neurotransmitters as growth reg-ulatory signals: Role of receptors and secondmessengers. Trends Neurosci 16:233–240.
Lauder JM. 1990. Ontogeny of the serotonergicsystem in the rat: Serotonin as a developmen-tal signal. Ann N Y Acad Sci 600:297–314.
Lauritsen MB, Nyegaard M, Betancur C, et al.2003. Analysis of transmission of novel poly-morphisms in the somatostatin receptor 5(SSTR5) gene in patients with autism. Am JMed Genet 121B:100–104.
Laumonnier F, Bonnet-Brilhault F, Gomot M, etal. 2004. X-linked mental retardation and au-tism are associated with a mutation in theNLGN4 gene, a member of the neuroliginfamily. Am J Hum Genet 74:552–557.
Lavdas AA, Grigoriou M, Pachnis V, et al. 1999. Themedial ganglionic eminence gives rise to a pop-ulation of early neurons in the developing ce-rebral cortex. J Neurosci 19:7881–7888.
Lee M, Martin-Ruiz C, Graham A, et al. 2002.Nicotinic receptor abnormalities in the cere-bellar cortex in autism. Brain 125(Pt 7):1483–1495.
Letinic K, Zoncu R, Rakic P. 2002. Origin ofGABAergic neurons in the human neocortex.Nature 417:645–649.
Levitt P, Eagleson KL, Powell EM. 2004. Regula-tion of neocortical interneuron developmentand the implications for neurodevelopmentaldisorders. Trends Neurosci 27:400–406.
Lewine JD, Andrews R, Chez M, et al. 1999.Magnetoencephalographic patterns of epilep-tiform activity in children with regressive au-tism spectrum disorders. Pediatrics 104(3 Pt1):405–418.
Lewis DA, Levitt P. 2002. Schizophrenia as a dis-order of neurodevelopment. Annu Rev Neu-rosci 25:409–432.
Lijam N, Paylor R, McDonald MP, et al. 1997.Social interaction and sensorimotor gating ab-normalities in mice lacking Dvl1. Cell 90:895–905.
Liu J, Nyholt DR, Magnussen P, et al. 2001. Agenomewide screen for autism susceptibilityloci. Am J Hum Genet 69:327–340.
Longo I, Russo L, Meloni I, et al. 2004. Three Rettpatients with both MECP2 mutation and15q11-13 rearrangements. Eur J Hum Genet12:682–685.
Luddens H, Korpi ER, Seeburg PH. 1995.GABAA/benzodiazepine receptor heteroge-neity: Neurophysiological implications. [Re-view]. Neuropharmacology 34:245–254.
Luo X, Persico A, Lauder J. 2003. Serotonergicregulation of somatosensory cortical develop-ment: Lessons from genetic mouse models.Dev Neurosci 25:173–183.
Lyuksyutova AI, Lu CC, Milanesio N, et al. 2003.Anterior-posterior guidance of commissuralaxons by Wnt-frizzled signaling. Science 302:1984–1988.
Maestrini E, Lai C, Marlow A, et al. 1999. Sero-tonin transporter (5-HTT) and gamma-ami-nobutyric acid receptor subunit beta3(GABRB3) gene polymorphisms are not as-sociated with autism in the IMGSA families.The International Molecular Genetic Study ofAutism Consortium. Am J Med Genet 88:492–496.
Manes F, Piven J, Vrancic D, et al. 1999. An MRIstudy of the corpus callosum and cerebellumin mentally retarded autistic individuals. JNeuropsychiatry Clin Neurosci 11(4):470–474.
Mann MR, Bartolomei MS. 1999. Towards a mo-lecular understanding of Prader-Willi andAngelman syndromes. Hum Mol Genet8:1867–1873.
Marin O, Rubenstein JL. 2001. A long, remarkablejourney: Tangential migration in the telen-cephalon. Nat Rev Neurosci 2:780–790.
Martineau J, Herault J, Petit E, et al. 1994. Cat-echolaminergic metabolism and autism. DevMed Child Neurol 36:688–697.
Martin-Ruiz CM, Lee M, Perry RH, et al. 2004.Molecular analysis of nicotinic receptor ex-pression in autism. Brain Res Mol Brain Res123:81–90.
McAllister AK, Katz LC, Lo DC. 1996. Neurotro-phin regulation of cortical dendritic growthrequires activity. Neuron 17:1057–1064.
McAllister AK, Katz LC, Lo DC. 1997. Opposingroles for endogenous BDNF and NT-3 inregulating cortical dendritic growth. Neuron18:767–778.
McAllister AK, Lo DC, Katz LC. 1995. Neurotro-phins regulate dendritic growth in developingvisual cortex. Neuron 15:791–803.
McCoy PA, Shao Y, Wolpert CM, et al. 2002. Noassociation between the WNT2 gene and au-tistic disorder. Am J Med Genet 114:106–109.
McDougle CJ, Posey D. 2002. Genetics of child-hood disorders: XLIV. Autism, part 3: Psy-chopharmacology of autism. J Am AcadChild Adolesc Psychiatry 41:1380–1383.
Millen KJ, Wurst W, Herrup K, et al. 1994. Ab-normal embryonic cerebellar developmentand patterning of postnatal foliation in twomouse Engrailed-2 mutants. Development120:695–706.
Miller MW. 1985. Cogeneration of retrogradelylabeled corticocortical projection and GABA-
315MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

immunoreactive local circuit neurons in ce-rebral cortex. Brain Res 355:187–192.
Miller MW. 1986. The migration and neurochem-ical differentiation of gamma-aminobutyricacid (GABA)-immunoreactive neurons in ratvisual cortex as demonstrated by a combinedimmunocytochemical-autoradiographic tech-nique. Brain Res 393:41–46.
Miyazaki K, Narita N, Sakuta R, et al. 2004. Serumneurotrophin concentrations in autism andmental retardation: A pilot study. Brain Dev26:292–295.
Morin X, Cremer H, Hirsch MR, et al. 1997.Defects in sensory and autonomic ganglia andabsence of locus coeruleus in mice deficientfor the homeobox gene Phox2a. Neuron 18:411–423.
Mountcastle VB. 1997. The columnar organizationof the neocortex. Brain 120(Pt 4):701–722.
Muhle R, Trentacoste SV, Rapin I. 2004. Thegenetics of autism. Pediatrics 113:e472–e486.
Nabi R, Serajee FJ, Chugani DC, et al. 2004.Association of tryptophan 2,3 dioxygenasegene polymorphism with autism. Am J MedGenet 125B:63–68.
Nelson KB, Grether JK, Croen LA, et al. 2001.Neuropeptides and neurotrophins in neonatalblood of children with autism or mental re-tardation. Ann Neurol 49:597–606.
Nery S, Fishell G, Corbin JG. 2002. The caudalganglionic eminence is a source of distinctcortical and subcortical cell populations. NatNeurosci 5:1279–1287.
Nilsson M, Waters S, Waters N, et al. 2001. Abehavioural pattern analysis of hypoglutama-tergic mice: Effects of four different antipsy-chotic agents. J Neural Transm 108:1181–1196.
Nurmi EL, Amin T, Olson LM, et al. 2003. Denselinkage disequilibrium mapping in the 15q11-q13 maternal expression domain yields evi-dence for association in autism. Mol Psychi-atry 8:570.
Pattyn A, Goridis C, Brunet JF. 2000. Specificationof the central noradrenergic phenotype by thehomeobox gene Phox2b. Mol Cell Neurosci15:235–243.
Pattyn A, Morin X, Cremer H, et al. 1999. Thehomeobox gene Phox2b is essential for thedevelopment of autonomic neural crest deriv-atives. Nature 399:366–370.
Pattyn A, Simplicio N, Van Doorninck JH, et al.2004. Ascl1/Mash1 is required for the devel-opment of central serotonergic neurons. NatNeurosci 7:589–595.
Perry EK, Lee ML, Martin-Ruiz CM, et al. 2001.Cholinergic activity in autism: Abnormalitiesin the cerebral cortex and basal forebrain.Am J Psychiatry 158:1058–1066.
Persico AM, D’Agruma L, Zelante L, et al. 2004.Enhanced APOE2 transmission rates in fam-ilies with autistic probands. Psychiatr Genet14:73–82.
Persico AM, Militerni R, Bravaccio C, et al. 2000.Lack of association between serotonin trans-porter gene promoter variants and autistic dis-order in two ethnically distinct samples. Am JMed Genet 96:123–127.
Persico AM, Pascucci T, Puglisi-Allegra S, et al.2002. Serotonin transporter gene promotervariants do not explain the hyperserotonine-mia in autistic children. Mol Psychiatry7:795–800.
Pescucci C, Meloni I, Bruttini M, et al. 2003.Chromosome 2 deletion encompassing theMAP2 gene in a patient with autism andRett-like features. Clin Genet 64:497–501.
Peters AR. 1994. Primary visual cortex of primates.In: Peters AR, Rockland K. S., editor. Cere-bral Cortex. New York: Plenum. p 1–36.
Peters A, Sethares C. 1996. Myelinated axons andthe pyramidal cell modules in monkey pri-mary visual cortex. J Comp Neurol 365:232–255.
Philippe A, Martinez M, Guilloud-Bataille M, et al.1999. Genome-wide scan for autism suscep-tibility genes. Human Molecular Genetics8:805–812.
Piven J. 1997. The biological basis of autism. CurrOpin Neurobiol 7:708–712.
Piven J. 2004. Preface. Ment Retard Dev DisabilRes Rev 10:219–220
Piven J, Arndt S, Bailey J, et al. 1995. An MRIstudy of brain size in autism. Am J Psychiatry152:1145–1149.
Piven J, Nehme E, Simon J, et al. 1992. Magneticresonance imaging in autism: Measurement ofthe cerebellum, pons, and fourth ventricle.Biol Psychiatry 31:491–504.
Piven J, Saliba K, Bailey J, et al. 1997. An MRIstudy of autism: The cerebellum revisited.Neurology 49:546–551.
Polleux F, Whitford KL, Dijkhuizen PA, et al.2002. Control of cortical interneuron migra-tion by neurotrophins and PI3-kinase signal-ing. Development 129:3147–3160.
Portbury AL, Chandra R, Groelle M, et al. 2003.Catecholamines act via a beta-adrenergic re-ceptor to maintain fetal heart rate and sur-vival. Am J Physiol Heart Circ Physiol 284:H2069–H2077.
Powell EM, Campbell DB, Stanwood GD, et al.2003. Genetic disruption of cortical interneu-ron development causes region- and GABAcell type-specific deficits, epilepsy, and be-havioral dysfunction. J Neurosci 23:622–631.
Powell EM, Mars WM, Levitt P. 2001. Hepatocytegrowth factor/scatter factor is a motogen forinterneurons migrating from the ventral todorsal telencephalon. Neuron 30:79–89.
Purcell AE, Jeon OH, Zimmerman AW, et al.2001. Postmortem brain abnormalities of theglutamate neurotransmitter system in autism.Neurology 57:1618–1628.
Rakic P. 1972. Mode of cell migration to thesuperficial layers of fetal monkey neocortex.J Comp Neurol 145:61–83.
Ramanathan S, Woodroffe A, Flodman PL, et al.2004. A case of autism with an interstitialdeletion on 4q leading to hemizygosity forgenes encoding for glutamine and glycineneurotransmitter receptor sub-units(AMPA2, GLRA3, GLRB) and neuropeptidereceptors NPY1R, NPY5R. BMC MedGenet 5:10.
Ramoz N, Reichert JG, Smith CJ, et al. 2004.Linkage and association of the mitochondrialaspartate/glutamate carrier SLC25A12 genewith autism. Am J Psychiatry 161:662–669.
Raymond GV, Bauman ML, Kemper TL. 1996.Hippocampus in autism: A Golgi analysis.Acta Neuropathol (Berl) 91:117–119. RoscjM. Spiker D, Lotspeich L, et al. 1999. Agenomic screen of autism: evidence for a mul-tilocus etiiology. Am J Hum Genet 65:493–507.
Risch N, Spiker D, Lotspeich L, et al. 1999. Agenomic screen of autism: evidence for a mul-tilocus etiology. Am J Hum Genet 65:493–507.
Robinson PD, Schutz CK, Macciardi F, et al. 2001.Genetically determined low maternal serumdopamine beta-hydroxylase levels and the eti-ology of autism spectrum disorders. Am JMed Genet 100:30–36.
Rodier PM, Ingram JL, Tisdale B, et al. 1996.Embryological origin for autism: Develop-mental anomalies of the cranial nerve motornuclei. J Comp Neurol 370:247–261.
Roffler-Tarlov S, Rios M. 2001. Catecholaminesbefore nervous sctivity. In: Kalverboer AF,Gramsbergen A, editors. Handbook of Brainand Human Development. Dordrecht/Bos-ton/London: Kluwer Academic Publishers. p165–184.
Rubenstein JL, Merzenich MM. 2003. Model ofautism: Increased ratio of excitation/inhibi-tion in key neural systems. Genes Brain Behav2:255–267.
Scanlon SM, Williams DC, Schloss P. 2001. Mem-brane cholesterol modulates serotonin trans-porter activity. Biochemistry 40:10507–10513.
Schumann CM, Hamstra J, Goodlin-Jones BL, etal. 2004. The amygdala is enlarged in childrenbut not adolescents with autism; the hip-pocampus is enlarged at all ages. J Neurosci24:6392–6401.
Schuurmans C, Armant O, Nieto M, et al. 2004.Sequential phases of cortical specification in-volve Neurogenin-dependent and -indepen-dent pathways. Embo J 23:2892–2902.
Schuurmans C, Guillemot F. 2002. Molecularmechanisms underlying cell fate specificationin the developing telencephalon. Curr OpinNeurobiol 12:26–34.
Sears LL, Vest C, Mohamed S, et al. 1999. An MRIstudy of the basal ganglia in autism. ProgNeuropsychopharmacol Biol Psychiatry 23:613–624.
Serajee FJ, Zhong H, Nabi R, et al. 2003. Themetabotropic glutamate receptor 8 gene at7q31: Partial duplication and possible associ-ation with autism. J Med Genet 40:e42.
Shahbazian M, Young J, Yuva-Paylor L, et al.2002. Mice with truncated MeCP2 recapitu-late many Rett syndrome features and displayhyperacetylation of histone H3. Neuron 35:243–254.
Shao Y, Cuccaro ML, Hauser ER, et al. 2003. Finemapping of autistic disorder to chromosome15q11-q13 by use of phenotypic subtypes.Am J Hum Genet 72:539–548.
Sherr EH. 2003. The ARX story (epilepsy, mentalretardation, autism, and cerebral malforma-tions): One gene leads to many phenotypes.Curr Opin Pediatr 15:567–571.
Shieh PB, Ghosh A. 1997. Neurotrophins: Newroles for a seasoned cast. Curr Biol 7:R627–R630.
Sidman RL, Rakic P. 1973. Neuronal migration,with special reference to developing humanbrain: A review. Brain Res 62:1–35.
Simon HH, Bhatt L, Gherbassi D, et al. 2003.Midbrain dopaminergic neurons: Determina-tion of their developmental fate by transcrip-tion factors. Ann N Y Acad Sci 991:36–47.
Smidt MP, Smits SM, Burbach JP. 2003. Molecularmechanisms underlying midbrain dopamineneuron development and function. EurJ Pharmacol 480:75–88.
Smith M, Filipek PA, Wu C, et al. 2000. Analysisof a 1-megabase deletion in 15q22-q23 in anautistic patient: identification of candidategenes for autism and of homologous DNAsegments in 15q22-q23 and 15q11-q13. Am JMed Genet 96:765–770.
Song JY, Ichtchenko K, Sudhof TC, et al. 1999.Neuroligin 1 is a postsynaptic cell-adhesionmolecule of excitatory synapses. Proc NatlAcad Sci U S A 96:1100–1105.
Sparks BF, Friedman SD, Shaw DW, et al. 2002.Brain structural abnormalities in young chil-dren with autism spectrum disorder. Neurol-ogy 59:184–192.
316 MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER

Spitzer NC, Root CM, Borodinsky LN. 2004.Orchestrating neuronal differentiation: Pat-terns of Ca(2�) spikes specify transmitterchoice. Trends Neurosci 27:415–421.
Steg JP, Rapoport JL. 1975. Minor physical anom-alies in normal, neurotic, learning disabled,and severely disturbed children. J AutismChild Schizophr 5:299–307.
Stevenson RE, Schroer RJ, Skinner C, et al. 1997.Autism and macrocephaly. Lancet 349(9067):1744–1745.
Stokstad E. 2001. Development: New hints intothe biological basis of autism. Science 294:34–37.
Sultana R, Yu CE, Yu J, et al. 2002. Identificationof a novel gene on chromosome 7q11.2 in-terrupted by a translocation breakpoint in apair of autistic twins, Genomics 80:129–134.
Tallon-Baudry C, Bertrand O. 1999. Oscillatorygamma activity in humans and its role in objectrepresentation. Trends Cogn Sci 3:151–162.
Tamamaki N, Fujimori KE, Takauji R. 1997. Or-igin and route of tangentially migrating neu-rons in the developing neocortical intermedi-ate zone. J Neurosci 17:8313–8323.
Thomas SA, Marck BT, Palmiter RD, et al. 1998.Restoration of norepinephrine and reversal ofphenotypes in mice lacking dopamine beta-hydroxylase. J Neurochem 70:2468–2476.
Thomas SA, Matsumoto AM, Palmiter RD. 1995.Noradrenaline is essential for mouse fetal de-velopment. Nature 374:643–646.
Thomas NS, Sharp AJ, Browne CE, et al. 1999. Xpdeletions associated with autism in three fe-males. Hum Genet 104:43–48.
Tordjman S, Gutknecht L, Carlier M, et al. 2001.Role of the serotonin transporter gene in thebehavioral expression of autism. Mol Psychi-atry 6:434–439.
Trottier G, Srivastava L, Walker CD. 1999. Etiol-ogy of infantile autism: A review of recentadvances in genetic and neurobiological re-search. J Psychiatry Neurosci 24:103–115.
Tuchman R. 2003. Autism. Neurol Clin 21:915–932, viii.
Veenstra-VanderWeele J, Anderson GM, CookEH Jr. 2000. Pharmacogenetics and the sero-tonin system: Initial studies and future direc-tions. Eur J Pharmacol 410:165–181.
Veenstra-Vanderweele J, Cook E Jr., Lombroso PJ.2003. Genetics of childhood disorders: XLVI.Autism, part 5: Genetics of autism. J Am AcadChild Adolesc Psychiatry 42:116–118.
Veenstra-VanderWeele J, Cook EH. 2004. Molec-ular genetics of autism spectrum disorder.Mol Psychiatry 9:819–832.
Veenstra-VanderWeele J, Kim SJ, Lord C, et al.2002. Transmission disequilibrium studies ofthe serotonin 5-HT2A receptor gene(HTR2A) in autism. Am J Med Genet 114:277–283.
Veltman MW, Thompson RJ, Roberts SE, et al.2004. Prader-Willi Syndrome—a study com-paring deletion and uniparental disomy caseswith reference to autism spectrum disorders.Eur Child Adolesc Psychiatry 13:42–50.
Vervoort VS, Beachem MA, Edwards PS, et al.2002. AGTR2 mutations in X-linked mentalretardation. Science 296:2401–2403.
Vogel-Hopker A, Rohrer H. 2002. The specifica-tion of noradrenergic locus coeruleus (LC)neurones depends on bone morphogeneticproteins (BMPs). Development 129:983–991.
Volkmar FR. 2001. Pharmacological interventionsin autism: Theoretical and practical issues.J Clin Child Psychol 30:80–87.
Waage-Baudet H, Lauder JM, Dehart DB, et al.2003. Abnormal serotonergic development ina mouse model for the Smith-Lemli-Opitzsyndrome: Implications for autism. Int J DevNeurosci 21:451–459.
Wassink TH, Brzustowicz LM, Bartlett CW, Szat-mari P. 2004. The search for autism diseasegenes. Ment Retard Dev Disabil Res Rev10:272–283.
Wassink TH, Piven J, Vieland VJ, et al. 2001.Evidence supporting WNT2 as an autism sus-ceptibility gene. Am J Med Genet 105:406–413.
Whitaker-Azmitia PM. 2001. Serotonin and braindevelopment: Role in human developmentaldiseases. Brain Res Bull 56:479–485.
Wichterle H, Turnbull DH, Nery S, et al. 2001. Inutero fate mapping reveals distinct migratorypathways and fates of neurons born in themammalian basal forebrain. Development128:3759–3771.
Wodarz A, Nusse R. 1998. Mechanisms of Wntsignaling in development. Annu Rev CellDev Biol 14:59–88.
Wurst W, Auerbach AB, Joyner AL. 1994. Multi-ple developmental defects in Engrailed-1 mu-tant mice: An early mid-hindbrain deletionand patterning defects in forelimbs and ster-num. Development 120:2065–2075.
Ye W, Bouchard M, Stone D, et al. 2001. Distinctregulators control the expression of the mid-hindbrain organizer signal FGF8. Nat Neuro-sci 4:1175–1181.
Ye W, Shimamura K, Rubenstein JL, et al. 1998.FGF and Shh signals control dopaminergicand serotonergic cell fate in the anterior neu-ral plate. Cell 93:755–766.
Yirmiya N, Pilowsky T, Nemanov L, et al. 2001.Evidence for an association with the serotonintransporter promoter region polymorphismand autism. Am J Med Genet 105:381–386.
Yu X, Malenka RC. 2003. Beta-catenin is criticalfor dendritic morphogenesis. Nat Neurosci6:1169–1177.
Zhou CJ, Zhao C, Pleasure SJ. 2004. Wnt signalingmutants have decreased dentate granule cellproduction and radial glial scaffolding abnor-malities. J Neurosci 24:121–126.
Zoghbi HY. 2003. Postnatal neurodevelopmentaldisorders: Meeting at the synapse? Science302:826–830.
317MRDD RESEARCH REVIEWS ● DEVELOPMENTAL NEUROBIOLOGY OF AUTISM ● POLLEUX AND LAUDER





























