Embed Size (px)
Citation preview

Post-RAMPART Implementation of Midazolam in EMS (PRIME)
A retrospective, observational study of the use of IM midazolam

Study Rationale There is a need to understand the process of implementation of clinical trial
findings in the prehospital setting. In the absence of such understanding the use of effective prehospital treatments may be delayed, increasing morbidity.
There is a need to understand the effectiveness of pre-hospital treatment of status epilepticus and to identify factors that predict treatment failure/success.
Completion of the RAMPART trial provides a unique opportunity and environment to examine prehospital convulsive seizure therapy and changes in treatment.

Study Objectives1. Evaluate prehospital use of midazolam for convulsive seizure before and
after publication of the RAMPART trial results.
2. Explore effectiveness of post-RAMPART EMS drug therapies used in treating convulsive seizure/status epilepticus, specifically examining:
Seizure cessation by emergency department arrival in patients receiving IM midazolam during the post-RAMPART period compared to historical controls from the RAMPART trial.
Seizure cessation by emergency department arrival in post-RAMPART EMS-treated patients by drug and dose.
Frequency and characteristics of pre-hospital treatment-resistant seizures (RAMPART treatment failures).

Hypotheses Objective 1: Publication of the RAMPART trial results did not influence IM
midazolam use between the 12 months preceding publication and the post-publication follow-up period.
Objective 2: IM midazolam administered according to local protocols following RAMPART trial completion was inferior to IM midazolam and IV lorazepam administered during the RAMPART trial in terminating convulsive seizure/status epilepticus prior to emergency department arrival.
Study Design Phase IV, retrospective, observational cohort study with descriptive and
time-series analyses with RAMPART trial enrollees as historical controls as needed.
Subject Population EMS transported patients requiring treatment with benzodiazepines for
convulsive seizure/status epilepticus in the prehospital setting. Adults Children with an estimated body weight of 13 kg or more
Inclusion Criteria EMS delivery of benzodiazepine
Documentation of convulsive seizure activity either preceding or concurrent with benzodiazepine treatment
Subject transported to a hospital where local study personnel have access to subject’s medical record
Exclusion CriteriaMajor trauma as the precipitant of the seizureHypoglycemia (glucose < 60 mg/dl)Known allergy to benzodiazepinesCardiac arrest Bradycardia with a heart rate < 40Treatment as part of another studyKnown pregnancyPrisoner

Primary Endpoint Objective 1: The primary outcome measure is the proportion of EMS
transported patients treated with benzodiazepines for convulsive seizure who received intramuscular midazolam, evaluated monthly
Objective 2: Termination of convulsive seizure activity prior to arrival in the emergency department after an initial prehospital dose of midazolam without the need for a second “rescue” dose of any benzodiazepine by EMS

Secondary Endpoints Treatment concordance with RAMPART clinical trial results. Treatment is considered
concordant if 1) adults and those children with an estimated body weight of more than 40 kg received 10 mg of midazolam intramuscularly or 2) children with an estimated body weight of 13 kg to 40 kg received 5 mg of midazolam intramuscularly.
Frequency of acute recurrence of seizure (either in EMS or ED) Frequency and duration of hospitalization Frequency and duration of ICU admission Frequency of acute endotracheal intubation Clinical, spatial and demographic characteristics of patients without termination of
seizures prior to arrival in the ED and those with recurrent seizures in the prehospital or ED setting (treatment failures).
Treatments This is a retrospective, observational study. No subject treatments are
planned
Data Collection Local study personnel will screen participating EMS systems for
benzodiazepine use using available EMS logs and/or pharmacy data as appropriate for individual sites.
Screen failures will be listed in the appropriate screen log form.
Subjects meeting the inclusion and no exclusion criteria will undergo data collection using the EMS, Emergency Department and Inpatient medical record as required to complete data entry
Copies or access to relevant primary source material from the medical record should be maintained in a secure manner for review on data monitoring visits.
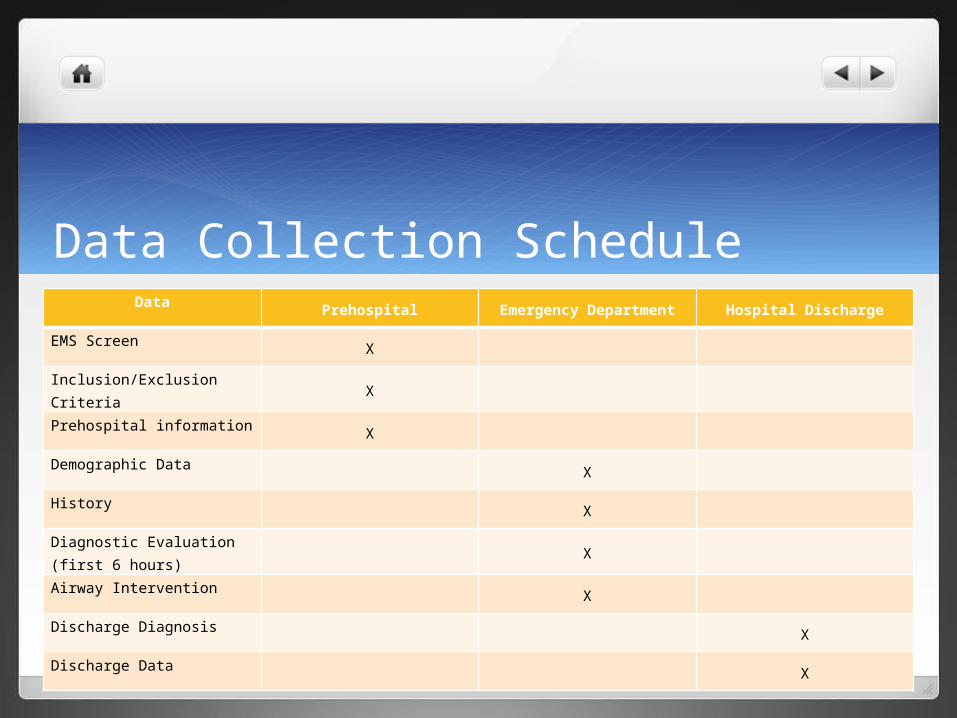
Data Collection ScheduleData
Prehospital Emergency Department Hospital Discharge
EMS ScreenX
Inclusion/Exclusion CriteriaX
Prehospital informationX
Demographic Data X
History X
Diagnostic Evaluation (first 6 hours) X
Airway Intervention X
Discharge Diagnosis X
Discharge Data X
Study Periods The RAMPART trial ended in January 2011. EMS systems reverted to their
standard protocols for the treatment of convulsive seizure following completion of the study.
We estimate the study period will extend 27 months - from February 2011 to May 2013. This will cover three phases;1. Pre-RAMPART result publication phase (Feb 2011 through Jan 2012)
2. Transition phase: the three months surrounding publication of the RAMPART results (Feb 2012 through April 2012)
3. Post-RAMPART result publication phase (May 2012 through April 2013)
Final dates depend on the statistical analysis plan and budget

Statistical Analysis: Objective 1 Use of midazolam will be plotted graphically over time by one-month blocks from
February 2011 through April 2013.
Data from the three month transition period will be excluded from the statistical models but included in the visual presentation.
We will assess change in the primary outcome before and after publication of the trial results.
We will fit a time series linear regression to estimate monthly use of intravenous midazolam for convulsive seizure after publication of the RAMPART trial results.
Details of the model and final analysis plan will be provided in the Statistical Analysis Plan (pending)

Ethics IRB review at each participating institution is required to demonstrate
compliance with the conditions set out for the protection of human subjects as compiled in the “Common Rule”, 45 CFR 46 subparts A-D.
Given the retrospective, observational nature of the study we plan to conduct it under a waiver of informed consent.

Waiver of informed consent The research involves no more than minimal risk to the subjects (i.e., the probability
and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests);
The waiver or alteration will not adversely affect the rights and welfare of the subjects;
The research could not practicably be carried out without the waiver or alteration; and
Whenever appropriate, the subjects (including their physicians, as applicable) are provided with additional pertinent information after participation.

Confidentiality and Privacy An important objective of the PRIME study is to characterize treatment
resistant seizures.
It is therefore necessary to evaluate specific elements of protected health information. These include: subject age, treatment date and location, and hospital length of stay information, in order to identify characteristics associated with treatment failures.

HIPAA Waiver of Authorization HIPAA requires that the IRB or a Privacy Board find and document the following
when a waiver of authorization will result in use or disclosure of protected health information (“PHI”) in connection with a research project: No more than a minimal risk to the privacy of individuals; Plan to protect the identifiers; Plan to destroy the identifiers; and Written assurances that the PHI will not be reused or disclosed to any other person or
entity, except as required; The waiver will not adversely affect the privacy rights and the welfare of the individuals; The research could not practicably be conducted without the waiver; and The research could not practicably be conducted without use of the PHI

Quality Assurance The study will be conducted in accordance with the ICH Guidelines for Good Clinical Practice and
all relevant local, national and international regulations.
Hub investigators will provide quality assurance within their Hub spoke complex in a process called Verification. This is independent of Monitoring, which is used to mean only independent external monitoring by the NETT Project Monitor of the Clinical Coordinating Center.
Data quality monitoring is performed continuously. Out of range and logical errors are identified at the time of data entry.
Site visits may be conducted periodically by the Project Monitor(s).
At site visits, the records of a sample of subjects will be reviewed against source documents. The proportion of records to be reviewed will be determined by the Statistical and Data Management Center.
Data Entry Form (draft) file://localhost/Users/phlsctt/Desktop/RAMPART Implementation Study/Rev
iewed Documents/PRIME Data Entry Form 1C DRAFT ps 062212.docx
Payment $1,500 start up payment for participating HUB/Spoke Complexes
$160 / completed CRF, paid quarterly In RAMPART, each Hub/Spoke complex averaged 96 PRIME subjects per year Worth additional $15,360/year, on average, for each Hub/Spoke complex
Large variation in screening between complexes, therefore reimbursement will have to be reviewed quarterly to ensure balance in enrollment and ability to complete needed duration of study for time series analysis
Questions?





























