Embed Size (px)
DESCRIPTION
Hasznos segédanyag vegyészeknek.
Citation preview

Created by XMLmind XSL-FO Converter.
ATOMABSZORPCIÓS SPEKTROMETRIA

Created by XMLmind XSL-FO Converter.
ATOMABSZORPCIÓS SPEKTROMETRIA

iii Created by XMLmind XSL-FO Converter.
Tartalom
Előszó ................................................................................................................................................ vi 1. Bevezetés ........................................................................................................................................ 1
1. Köszönetnyilvánítás .............................................................................................................. 1 2. Bevezetés .............................................................................................................................. 1
2. Az atomabszorpciós spektrometria előzményei, kialakulása és fejlődése ...................................... 3 3. Atomspektroszkópia fizikai alapjai ................................................................................................ 5
1. Az atomspektroszkópiai módszerek felosztása és alapelve. .................................................. 5 2. Az atomabszorpciós spektrometria elve ................................................................................ 6 3. Az atom szerkezete és az atomszínképek keletkezése .......................................................... 8 4. A szabad atomok fényelnyelése .......................................................................................... 10 5. Oszcillátor erősség .............................................................................................................. 11 6. Az abszorpciós együttható .................................................................................................. 12 7. A Lambert-Beer törvény .................................................................................................... 14 8. Az alapállapotú és gerjesztett atomok aránya ..................................................................... 16
4. Az atomabszorpciós spektrométerek felépítése, működése .......................................................... 18 1. A spektrométer optikai rendszere ........................................................................................ 18
1.1. Megvilágító fényforrások ....................................................................................... 18 1.2. Az üregkatód lámpa (HCL=hollow cathode lamp,) felépítése és működése ......... 19 1.3. Elektród nélküli kisülési lámpa (electrodeless discharge lamp, EDL) .................... 22 1.4. Egy- és kétfényutas AAS készülékek ..................................................................... 23 1.5. A fényszaggató szerepe .......................................................................................... 25 1.6. Az elemző vonal kiválasztása, mono- és polikromátorok ....................................... 27 1.7. Szimultán készülékek ............................................................................................. 32 1.8. Háttérkorrekciós módszerek ................................................................................... 32
2. A készülékek elektronikus egységei .................................................................................... 38 2.1. Lámpa tápegység .................................................................................................... 38 2.2. Detektorok .............................................................................................................. 38 2.3. Erősítők. A jel/zaj viszony javításának elektronikus módszerei ............................. 42
3. Az adatok kijelzése, gyűjtése, a kijelző módszerek fejlődése ............................................. 45 4. A minta lángatomizációja ................................................................................................... 46
4.1. A láng, mint nagyhőmérsékletű tér jellemzése ....................................................... 46 4.2. Az atomizálásra használt lángok és égők ............................................................... 46 4.3. A lángba juttatott minta termikus és kinetikai folyamatai ...................................... 57 4.4. Elemek atomizációs hatásfoka ................................................................................ 58 4.5. A száraz aeroszol-képződés törvényszerűségei ..................................................... 62
5. Mintabeviteli módszerek a lángba ....................................................................................... 72 5.1. Oldatok mintabevitele ............................................................................................. 72
5.1.1. Pneumatikus porlasztók (PN=pneumatic neblization) ............................... 72 5.1.2. Mintabeviteli sebesség és hatásfok ............................................................ 74 5.1.3. Ultrahangos porlasztás (USN= ultrasonic nebulization) ............................ 75 5.1.4. Hidraulikus nagynyomású porlasztás (HHPN=hydraulic high pressure
nebulization) ........................................................................................................ 76 5.1.5. Elektrotermikus párologtatás (ETV=electrothermal vaporization) ............ 78 5.1.6. Stacionárius és impulzus mintabevitel ....................................................... 78 5.1.7. Flow injection mintabevitel (FI) ................................................................ 82 5.1.8. Folyamatos titráló rendszer ....................................................................... 83
5.2. Szilárd anyagok mintabevitele ................................................................................ 90 5.2.1. Ív- és szikraporlasztás ................................................................................ 90 5.2.2. Lézer abláció .............................................................................................. 96 5.2.3. Elektrotermikus párologtatás ..................................................................... 96
6. Elektrotermikus atomizáció (ETA) ..................................................................................... 99 6.1. Az atomizáló kemencék anyaga ............................................................................. 99 6.2. Az elektrotermikus atomizáló módszer történeti fejlődése ..................................... 99 6.3. A grafitkemencés atomizáló szerkezete, működése .............................................. 103 6.4. A jel/zaj viszony javítása a grafitkemencés módszernél ....................................... 107 6.5. A pirolitikus bevonat és a platform alkalmazása .................................................. 108

ATOMABSZORPCIÓS
SPEKTROMETRIA
iv Created by XMLmind XSL-FO Converter.
6.6. A keresztfűtéses grafitcső ..................................................................................... 109 6.7. Termikus folyamatok és kémiai reakciók a grafitkemencében ............................. 111 6.8. Szilárd minták grafitkemencés elemzése .............................................................. 113
7. A higany hideggőz (CV=cold vapor) technikás atomizációja ........................................... 114 8. Atomizáció hidridképzésen keresztül ................................................................................ 116
5. Zavaró hatások és azok kiküszöbölése ....................................................................................... 119 1. Lángban fellépő zavaró hatások ........................................................................................ 119
1.1. A zavaró hatások különböző szempontú osztályzása ........................................... 119 1.2. Spektrális zavaró hatások ..................................................................................... 119 1.3. Transzport zavarások ............................................................................................ 120 1.4. Oldott anyagok párolgási zavarása ....................................................................... 120 1.5. Gázfázisú kémiai zavaró hatások .......................................................................... 122 1.6. Térbeli eloszlás zavarása ...................................................................................... 125 1.7. Ionos újraeloszlás zavaró hatása ........................................................................... 125 1.8. A zavaró hatások kiküszöbölési módszereit ......................................................... 126 1.9. Grafitkemencében fellépő zavarások kiküszöbölése matrixmódosítókkal ........... 126
6. A kémiai analízis folyamata és teljesítőképessége ..................................................................... 129 1. A kémiai analízis lépései ................................................................................................... 129 2. A módszer teljesítőképessége, érvényesítése (validálása) ................................................. 130
7. Az atomabszorpciós spektrometria alkalmazásai ....................................................................... 132 1. Elemek és minták .............................................................................................................. 132
1.1. A módszerrel vizsgálható elemek és mintatípusok ............................................... 132 1.2. Minta-előkészítési eljárások ................................................................................. 134 1.3. Kiértékelési módszerek ......................................................................................... 137
2. Speciációs analitika ........................................................................................................... 139 2.1. A speciáció fogalma és szükségessége ................................................................. 139 2.2. A speciációs analitikában vizsgált elemek köre .................................................... 140 2.3. Mintatípusok a speciációs analitikában ................................................................ 141 2.4. A speciációs analízis kapcsolt módszerei ............................................................. 141 2.5. Krómspeciációs módszerek .................................................................................. 142
8. Az atomabszorpciós spektrometria jövőbeli fejlődési irányai .................................................... 146 9. Irodalomjegyzék ......................................................................................................................... 150 10. Tárgymutató .............................................................................................................................. 155

v Created by XMLmind XSL-FO Converter.
A táblázatok listája
(1). A gerjesztett és alapállapotú atomok arányának függése a hőmérséklettől [12]. ....................... 17 (2). A lángfotomeriában (FES) és az atomabszorpciós spektrometriában (AAS) gyakrabban használt
lángok paraméterei ........................................................................................................................... 47 (4). A 4. táblázatban foglaltuk össze 28 gyakrabban vizsgált elem kísérletileg meghatározott atomizáció
fokát acetilén – levegő és acetilén – dinitrogén-oxid lángban. ........................................................ 61 . ..................................................................................................................................................... 137 (6). Az eddig leggyakrabban vizsgált elemformák a speciációs analitikában [103] ....................... 141 (7). 7. táblázat. A Cr(VI) különböző on-line elválasztási/dúsítási/atomspektrometriás detektálási
módszerrel kapott kimutatási határai .............................................................................................. 144

vi Created by XMLmind XSL-FO Converter.
Előszó
Az atomabszorpciós spektrometria témakörében az 1960-as évektől napjainkig számos átfogó és jól használható
szakkönyv [1-13] látott napvilágot atomspektroszkópiával foglalkozó kiváló külföldi kutatók tollából. Magyar
nyelven azonban csak egy átfogó munka: John Price atomabszorpciós spektrometria könyvének [6] magyar
fordítása jelent meg a 70-es években [14]. A másik magyar nyelvű kézikönyv Tomcsányi László
Atomabszorpciós praktikuma [15] a 80-as években címéből adódóan egy gyakorlati útmutató ilyen
laboratóriumban dolgozó szakemberek számára. Ezért elmondható, hogy az AAS módszer egészét átfogó könyv
az egyetlen korai fordítást kivéve eddig magyar nyelven nem jelent meg. Egyrészt az előbb említett két könyv a
korlátozott példányszám miatt ma már csak néhány szakkönyvtárban vagy antikváriumban szerezhető be.
Másrészt az AAS módszer az utóbbi 30 évben óriási fejlődésen ment keresztül. Ezért egy új magyar nyelvű
tankönyv megjelenése mindenképpen indokolt és jelentős hiánypótló szerepet tölt be. Az összes magyar
szakközépiskola, főiskola, egyetem kémiával foglalkozó hallgatósága, üzemi, mezőgazdasági,
környezetvédelmi, orvosi, toxikológiai, metallurgiai, geológiai stb. laboratórium kutatói, technikusai,
szakasszisztensei számára fontos egy magyar nyelvű könyv, amely az elméleti alapok mellett a szakterület
legújabb műszaki megoldásait is tartalmazza. És még egy nagyon fontos szempont, hogy a magyar diákok,
hallgatók és szakemberek számára biztosítani kell azt a lehetőséget, hogy megismerjék és alkalmazni tudják a
szakterület magyar nyelvű szavait, szakkifejezéseit. A digitális tankönyv megírásánál ezt is fontos célnak
tekintettem. Örömmel jelenthetem, hogy e tankönyv írása közben került kiadásra Záray Gyula szerkesztésében
„Az elemanalitika korszerű módszerei” című könyv [16], amelyben Lakatos János írt egy fejezetet az
atomabszorpciós spektrometriáról.

1 Created by XMLmind XSL-FO Converter.
1. fejezet - Bevezetés
1. Köszönetnyilvánítás
A digitális tankönyv szerzője megköszöni a Kutatás-fejlesztési Pályázati és Kutatáshasznosítási Irodának, illetve
jogutódjának a Magyar Gazdaságfejlesztési Központnak a könyv megírásához nyújtott anyagi támogatást.
Megköszönöm Karosi Rolandnak az ábrák, animációk, filmek színvonalas elkészítését, a szerkesztési munkákat
pedig Töviskes Ákosnak.
Külön köszönetet mondok Dr. Pap Lajos professzor emeritusznak, a könyv bírálójának, a munka gondos
áttanulmányozásáért és hasznos szakmai és formai tanácsaiért.
A pályázattal kapcsolatos egyéb feladatok elvégzéséért Nagyné Dombi Gizella, Béni Áron, Karosi Roland,
Nagy István és Barcsa Gáborné munkatársaknak mondok köszönetet.
Megköszönöm feleségemnek és családomnak, hogy türelmet tanúsítottak a könyv megírásának ideje alatt.
2. Bevezetés
Az atomabszorpciós spektrometria (AAS) az elemanalitika egyik legelterjedtebb műszeres analitikai módszere.
Magyarországon e műszerek egyetemeken, kutató intézetekben, üzemi, környezetvédelmi, minőségbiztosító és
minőségellenőrző laboratóriumokban mára alapműszernek számítanak. A módszer felfedezésétől és
bevezetésétől, azaz az 1950-es évek közepétől az atomabszorpciós spektrometria igen nagy fejlődésen ment
keresztül. Így mindmáig megtartotta vezető helyét az elemanalízis és újabban a speciációs analitika területén is.
Ennek két jellemző és fontos oka van. Az egyik az, hogy a lángatomabszorpciós spektrometria (FAAS) az egyik
leggyorsabb vizsgáló módszer az összes analitikai eljárás közül. 5-6 másodperc alatt határozható meg egy
oldatban egy elem koncentrációja. A másik igen nagy teljesítményt a grafitkemencés atomabszorpciós
spektrometria (GFAAS) szolgáltatja azzal, hogy az elemanalitikai módszerek közül az egyik legnagyobb
analitikai érzékenységgel rendelkezik. 10─12 – 10─15 gramm, azaz pikogramm – femtogramm tömegű anyag
határozható meg a segítségével. További előnye más hasonló teljesítményű módszerekkel összevetve, hogy
kiemelkedő teljesítőképessége ellenére ára és üzemeltetése viszonylag olcsó.
Az atomabszorpciós spektrométerek száma Magyarországon ezres nagyságrendre tehető. Az egyetemeken,
főiskolákon az analitikai képzés törzsanyagához tartozik. A végzős kémia és vegyész, gyógyszerész,
orvosdiagnosztikai analitikus, környezettudomány szakos hallgatók nagy számban kerülnek atomabszorpciós
laboratóriumokba. Mindezek ellenére eddig magyar nyelvű atomabszorpciós spektrometriáról szóló könyv
csupán az Előszóban említett két mű. Az AAS módszer azóta is igen jelentős fejlődésen ment keresztül. Ezért
fontos hiánypótló anyag az egyetemi, főiskolai hallgatók és a témával mélyebben foglalkozni kívánó
szakemberek számára a korszerű atomabszorpciós spektrometria elveit, a műszerek felépítését, működését és
alkalmazási lehetőségeit ismertető könyv, amely az interneten mindenki számára hozzáférhető.
Miután a műszeres analitika oktatásán és kutatásán belül több mint 30 éve foglalkozom az atomabszorpciós
spektrometria oktatásával, kutatásával és a módszer fejlesztésével, ezeket a tapasztalataimat szívesen osztom
meg jegyzet, könyv formájában a téma iránt érdeklődő szakközvéleménnyel. Felhasználva a jelenleg folyó saját
kutatásainkat és követve a téma legfrissebb nemzetközi irodalmát [12,17], a megírásra kerülő tankönyv a
módszer legkorszerűbb technikai megoldásait is igyekszik bemutatni.
E digitális tankönyv tartalma egyrészt igazodik az eddig elsősorban angol nyelven megjelent szakkönyvek
szerkezetéhez, másrészt megfelel az "Atomabszorpciós spektrometria" speciális előadásom tematikájának,
amelyet közel 30 éve tartok vegyész, kémiatanár, biológus, gyógyszerész, környezettudomány szakos hallgatók
számára.
A tankönyv célja átfogni a módszer teljes vertikumát az elméleti alapoktól a technikai megoldásokon keresztül a
gyakorlati alkalmazásokig. A fizikai alapok, a készülék felépítésének és működésének, az atomizáló rendszerek
tulajdonságainak és a mintabevitel módjának tárgyalása során célom érzékeltetni, hogy milyen lehetőségei,
tartalékai vannak a teljesítőképesség, a jel/zaj viszony javításának. Bemutatom az AAS legkorszerűbb optikai és
elektronikai megoldásait, a mintabevitel és az atomizálás leghatékonyabb és legmegbízhatóbb lehetőségeit.
Mivel tankönyvről van szó, a módszer alkalmazásának elsősorban az elveit mutatom be mind az elemekre, mind

Bevezetés
2 Created by XMLmind XSL-FO Converter.
a mintatípusokra. Az elemek és a mintatípusok egyenkénti tárgyalása aránytalanul megnövelné a könyv
terjedelmét és átvenné a kézikönyvek, recept-gyűjtemények szerepét. Mindenképpen ki kell térni viszont a 21.
század nagy kihívására, a speciációs analitikai alkalmazásra, ahol egy-egy elemnek nemcsak az összes
koncentrációját határozzuk meg, hanem az elem különböző vegyértékű és kötésállapotú formáit külön-külön is.
Ez a kutatási irány még hosszú időre feladatot ad az atomabszorpciós spektrometriának.

3 Created by XMLmind XSL-FO Converter.
2. fejezet - Az atomabszorpciós spektrometria előzményei, kialakulása és fejlődése
A történetírás az optikai spektroszkópia kialakulását általában Isaac Newton (1642–1727) nevével kapcsolja
össze 1672-ben közölt munkája alapján, amelyben azt a vizsgálatát írja le, hogy ha a Nap fényét prizmán
bocsátja át, az különböző színekre bomlik fel. Egy prágai orvos-professzor Joannes Marcus Marci (1595–1667)
azonban már 1648-ban megjelent könyvében máig érvényes magyarázatát adja a szivárvány jelenségének, ami a
Nap fényének a vízcseppeken történő fénytörésén alapszik. Ezért valószínűleg Marcus Marci tekinthető az első
spektroszkópusnak. A spektroszkópia fejlődése szempontjából fontos és tanulságos mozzanat Thomas Melville
közlése, aki sók, tengervíz lángfestését vizsgálva a kibocsátott fényt köralakú nyíláson (aperturán) keresztül
vetítette a prizmára. A prizma túloldalán elhelyezett ernyőn különböző színű korongok jelentek meg. Ahogy
életrajzírói fogalmaznak, ha Melville nem aperturát használt volna, hanem optikai rést és nem hal meg a
következő évben (1753-ban), akkor a spektroszkópia sokkal gyorsabban fejlődhetett volna.
William Wollaston (1766–1828) (a palládium felfedezője) 1802-ben figyelt fel arra, hogy a Nap színképében
fekete vonalak észlelhetők, de ennek nem tulajdonított különösebb jelentőséget. Ezeket a vonalakat Joseph
Fraunhofer (1787–1826) munkássága után Fraunhofer-vonalaknak nevezzük.
1. ábra A Fraunhofer vonalak a Nap színképében
Fraunhofer építette az első mai értelemben vett spektroszkópot és 1815-től részletes vizsgálat alá vette a Nap
színképében e fekete vonalakat. Mintegy 576 vonal hullámhosszát határozta meg és betűjelzésekkel látta el.
Innen maradt fenn a nátrium-D vonal elnevezés is. Fraunhofer kísérletileg arra is rájött, hogy a lángban
juttatott nátrium spektrumában a sárga vonalak ugyanott jelennek meg, ahol a Nap színképében a D fekete
vonalak. David Brewster 1820-ban fejtette ki azt az álláspontját, hogy a Fraunhofer-vonalak a Nap
atmoszférájában végbemenő fényabszorpció következményei. Herschel 1822-ben megállapította, hogy a lángba
helyezett különböző sók spektrumának vizsgálata módot ad arra, hogy az anyagot egyértelműen fel lehessen
ismerni.
Az elméleti áttörést Gustav Robert Kirchhoff (1824–1887) és Robert Wilhelm Bunsen (1811–1899) 1859-ben
megjelent közös cikke jelentette. Ők adták meg a spektrumra vonatkozó ismeretek értelmezését: ha egyes gázok
vagy gőzök atomjaival energiát közlünk, akkor azok a rájuk jellemző vonalas spektrumot bocsátják ki.
Ugyanezek az atomok viszont képesek a kibocsátott fény hullámhosszával egyező hullámhosszú fényt elnyelni,
abszorbeálni. Ezért jelenhetnek meg izzó anyagok (például a Nap) által kibocsátott folytonos színképben fekete
abszorpciós vonalak. Utóbbit Kirchhoff „fordított színképnek” is nevezte.
Ezek a felismerések meggyorsították az új elemek felfedezését színképelemzés alapján. Maga Bunsen és
Kirchhoff 1860-ban két elemet, a rubidiumot és a céziumot, Crookes 1861-ben a talliumot, Reich és Richter
1863-ban az indiumot, L. de Boisbaudran 1875-ben a galliumot, 1870-ben Lockyer a héliumot fedezte fel az
adott elemek színképe alapján. Utóbbival ekkor még a Földön nem is találkoztak, csak a Nap (heliosz)
színképében észlelték, innen kapta a nevét is.
Erre az időszakra egészen az 1950-es évekig az atomemisszió színképelemzés, azaz a lángfotometria illetve az
ugyancsak emissziós módszer, az elektromos ív- és szikra-spektroszkópia fejlődése a jellemző, annak könnyebb

Az atomabszorpciós spektrometria
előzményei, kialakulása és fejlődése
4 Created by XMLmind XSL-FO Converter.
technikai megvalósíthatósága miatt. Ezért, bár Bunsen és Kirchhoff az emissziós módszer mellett az
atomabszorpció elveit is lefektette, az atomabszorpciós spektrometria (AAS) mégis csak közel 100 év után
került bevezetésre. Az AAS módszer bevezetésének fő akadálya az igen kis (0.001 nm) félértékszélességű
vonalakat kibocsátó spektrállámpa, mint megvilágító fényforrás hiánya volt. Ez a hosszabb időszak azonban
fontos előkészülettel telt az AAS módszer gyakorlati kidolgozásához. 1876-ban Gouy kidolgozott egy olyan
koncentrikus pneumatikus porlasztót, amely kis módosításokkal máig is annak biztosítéka, hogy a mintaoldatot
kis cseppméretű aeroszollá alakítva állandó sebességgel, jól reprodukáhatóan juttassuk be a lángba, vagy más
nagyhőmérsékletű térbe (pl. egyenáramú (DCP), mikrohullámú (MIP), induktív csatolású plazmába (ICP)).
Lundegårdh vezette be az előkevert acetilén – levegő lángot és a ködkamrás mintabeviteli módszert, amely
mindmáig a nagy precizitású lángspektrometria alapja. Az atomabszorpciós spektrometria számára
elengedhetetlen vonalas színképet szolgáltató spektrállámpák létrehozásához az alapot az 1800-as évek második
felében a fizikusok által intenzíven tanulmányozott gázkisűlési csövek jelentették. Ilyen gázkisülési cső
katódjának geometriai módosításával Paschen 1916-ban dolgozta ki az első üregkatód lámpát, amelynek a
később tökéletesített, kereskedelmi változata azóta is az AAS elsődleges megvilágító fényforrása.
Bár már 1930-ban (Müller és Pringsheim révén), és később is történtek kísérletek a higany atomabszorpciós
meghatározására, az atomabszorpciós spektrometriás módszer bevezetésének éveként 1955-öt szokták megadni,
amikor Ausztráliában Alan Walsh [18] valamint Hollandiában Alkemade és Milatz [19] egymástól függetlenül
megjelentette az atomabszorpció alapelveit tárgyaló cikkét. Walsh 1952 és 1961 között komoly erőfeszítéseket
tett a módszer elismertetéséért. Munkásságának köszönhetően 1961-ben megépült az első kereskedelmi AAS
készülék, a Perkin Elmer 303 modell. Az első AAS készülékek a lángfotometria addigi tapasztalataira,
vívmányaira építve a minta atomizálására előkevert lángokat használtak, amelybe a mintaoldatokat pneumatikus
porlasztással juttatták be. Boris L’vov azonban már 1959-ben az atomizálás egy új módszerének, a
grafitkemencés atomizálásnak az alapjait vetette meg [20]. Ettől kezdve az AAS módszer két fő irányban
fejlődött tovább. A lángatomabszorpciós spektrometria (FAAS) irányába, amelyik ma az egyik leggyorsabbnak
számító (5–10 minta elemzése percenként) analitikai módszer, valamint a grafitkemencés atomabszorpciós
spektrometria (GFAAS) irányába, amely viszont jelenleg egyike a legnagyobb analitikai érzékenységgel
rendelkező módszereknek; pikogramm-femtogramm tömegű elem határozható meg segítségével. A
lángatomizálás területén a 60-as évek elején nagy gondot jelentett, hogy több fontos elemet (alumínium,
vanádium, szilicium, titán, ritkaföldfémek stb.) acetilén-levegő lángban, annak hőmérséklete és mérsékelt
reduktivitása miatt nem lehet alapállapotú atomokká alakítani. Ezért volt nagyon fontos lépés, hogy 1965-ben
Willis [21] bevezette a dinitrogén-oxid–acetilén lángot, amelynek közel 3000 oC hőmérséklete és igen reduktív
tulajdonsága biztosítja, hogy az említett erős oxidképző elemek is atomizálódjanak.
Az atomabszorpciós analitikában napjainkig tartó fontos fejlesztési lépések közül kiemelhető az úgynevezett
háttérkorrekciós módszerek fejlesztése. Nagy sótartalmú minták atomizációja során a megvilágító fényforrás
fényének gyengülése nemcsak az alapállapotú szabad atomok okozta fényelnyeléstől, hanem a keletkező száraz
aeroszol részecskéken végbemenő fényszóródásból is származik. A háttérkorrekciós módszer célja, hogy ezt a
szórt fényt elválasszuk a tényleges atomos abszorpciótól. A háttérkorrekció legmodernebb és leghatékonyabb
módszerét, nevezetesen a Zeeman-elven alapuló módszert 1969-ben Prugger és Torge [22] szabadalmaztatta.
Ma az újabb kereskedelmi készülékek ezzel a megoldással kerülnek forgalomba.

5 Created by XMLmind XSL-FO Converter.
3. fejezet - Atomspektroszkópia fizikai alapjai
1. Az atomspektroszkópiai módszerek felosztása és alapelve.
Az atomspektroszkópiás módszerek 70-80 elem minőségi és nagy analitikai érzékenységű mennyiségi
meghatározására alkalmas műszeres analitikai eljárások. Közös bennük, hogy a mintában jelenlevő vizsgálni
kívánt elemet szabad atomokká alakítjuk. A szabad atomok létrehozhatók különböző hőmérsékletű lángokkal,
elektromos ívvel, szikrával, egyenáramú, induktív vagy kapacitív csatolású plazmával illetve nagy hőmérsékletű
grafitcsőben. Aszerint, hogy a szabad atomok minőségéről és mennyiségéről hogyan szerzünk adatokat,
beszélünk atomemissziós (AES), atomabszorpciós (AAS) és atomfluoreszcens (AFS) módszerekről. A három
módszer elvi vázlatát a 2. ábrán mutatjuk be.
2.ábra: Az atomemissziós (A), atomabszorpciós (B) és atomfluoreszcens (C) spektrométerek elvi vázlata
Az atomemissziós spektrometriában termikus vagy elektromos energia segítségével a vizsgált elem gerjesztett
atomjait (gerjesztet ionjait) állítjuk elő. E gerjesztett atomok által kisugárzott fény színképéből a jellemző
hullámhosszak alapján állapítjuk meg az adott elem minőségét (minőségi elemzés). Az elem adott
hullámhosszúságú színképvonalának relatív intenzitásából (Irel) pedig annak (c) koncentrációját határozzuk meg
(mennyiségi elemzés). A mért jel és a koncentráció közötti általános összefüggést a Scheibe-Lomakin egyenlet
írja le, ahol K állandó, n pedig anyagszerkezeti tényező, amelynek az értéke 0.5 – 1.5 közé esik. Oldatok
elemzése esetén az n értéke 1 körül van, azaz az elem által adott hullámhosszúságú fény relatív intenzitása
egyenes arányos az elem koncentrációjával.
(1)

Atomspektroszkópia fizikai alapjai
6 Created by XMLmind XSL-FO Converter.
E könyv keretében részletesen tárgyalásra kerülő atomabszorpciós spektrometriában a vizsgálandó elemet
elsősorban termikus energia alkalmazásával alapállapotú szabad atomokká alakítjuk. Az így létrehozott
atomgőzön a vizsgálandó elemre jellemző hosszúságú fénynyalábot bocsátunk keresztül és mérjük a
fényintenzitás csökkenését, amely a későbbiekben részletezett módon, a Lambert-Beer törvény alapján
egyértelmű kapcsolatban áll a fényelnyelést okozó atomok koncentrációjával.
Az atomfluoreszcens spektrometriában ugyancsak alapállapotú szabad atomokat állítunk elő. Ezeket az
atomokat azonban a vizsgált elemre jellemző hullámhosszúságú fénnyel gerjesztjük, majd e fénnyel gerjesztett
atomok által kibocsátott fluoreszcens fény relatív intenzitását (IF) mérjük, amely a (2) szerint arányos az adott
elem c koncentrációjával.
(2)
• Io - a megvilágító fény intenzitása
• ω - a gerjesztő fénynyaláb átmérője
• a - abszorpciós együttható
• c - a vizsgált elem térfogategységben levő alapállapotú atomjainak a száma
• l - az abszorbeálódó fénynyaláb úthossza az atomizáló térben
• φ - fluoreszcens hatásfok
• Ω - az a térszög, amelyen belül a fluoreszcens fény detektálása történik
Az atomspektroszkópiában az elmúlt 20 évben alakult ki az egyik legnagyobb analitikai érzékenységű módszer
az induktív csatolású plazma–tömegspektrometria (ICP-MS), amely esetén az előző módszerektől eltérően a
vizsgált elem ionjait állítjuk elő, amelyeket tömegspektrométerbe juttatva, a mágneses térben az ionok
tömeg/töltés szerint különülnek el egymástól, és az adott ionnyaláb intenzitása arányos az elem
koncentrációjával.
2. Az atomabszorpciós spektrometria elve
Az atomabszorpciós spektrometria elvét a következő lépésekkel lehet jellemezni. A vizsgálandó elemet
alapállapotú szabad atomokká alakítjuk. Az így létrehozott atomgőzön a vizsgálandó elemre jellemző
hosszúságú fénynyalábot bocsátunk keresztül és mérjük a fényintenzitás csökkenését, amely egyértelmű
kapcsolatban áll a fényelnyelést okozó atomok koncentrációjával. A fényintenzitás csökkenésének jellemzésére
szolgáló mérési adatot, amelyet műszeresen észlelünk abszorbanciának (belső transzmissziós sűrűségnek)
nevezzük. Az abszorbancia fogalma egy matematikai művelet eredménye: az adott hullámhosszon mért „Io”
kezdeti fényintenzitás és az elnyelő közegen történő áthaladás utáni „I” csökkent fényintenzitás hányadosának
tízes alapú logaritmusa (3).
(3)
Az abszorbancia és a vizsgált elem koncentrációja közötti összefüggést a Lambert-Beer törvény adja meg (4).
Eszerint
(4)

Atomspektroszkópia fizikai alapjai
7 Created by XMLmind XSL-FO Converter.
ahol az „a” az abszorpciós együttható, a „c” az atomizáló közeg egységnyi térfogatában jelenlevő alapállapotú
atomok száma, az „l” pedig az elnyelő közeg rétegvastagsága, amin a fénynyaláb áthalad. Az abszorpciós
együttható anyagi állandó, amelynek nagysága az adott atom szerkezetétől függ. Ennek nagyságát befolyásolni
nem tudjuk. Az elnyelő közeg rétegvastagságát az atomizáló rendszer geometriai elrendezésével adott határok
(1–10 cm) között változtathatjuk. Az abszorbancia nagyságát legnagyobb mértékben az alapállapotú atomok
koncentrációjának változtatásával befolyásolhatjuk. Az atomabszorpciós módszerek kimutatási képességének
növekedését éppen azzal érjük el, hogy egy adott koncentrációjú minta atomizálása során a fényútban minél
nagyobb atomkoncentrációt hozunk létre.
Az atomabszorpciós spektrometria a vizsgált atomra jellemző hullámhosszúságú elektromágneses sugárzásnak
az alapállapotú atomok által történő elnyelésén alapszik. Azt nevezzük alapállapotú atomnak, amelynek minden
elektronja a kvantummechanika törvényei által megszabott alappályán kering. Ennek megfelelően az
alapállapotú atom nem bocsát ki fényt. Külső fényforrás adott hullámhosszúságú fényét viszont képes elnyelni.
Ezért az atomabszorpciós spektrometriában olyan megvilágító fényforrásra van szükség, amely az adott elemre
jellemző hullámhosszúságú fénysugarat bocsát ki.
A megvilágító fényforrás fényének elnyelése következtében az alapállapotú atom gerjesztett atommá alakul,
azaz legalább egy elektronja egy magasabb energiaszintű pályára ugrik. Az atom ebben a gerjesztett állapotban
igen rövid ideig, 10–9 másodpercig tartózkodik, majd visszaugrik az alappályára. Eközben az atom adott
energiájú foton formájában sugározza ki a két állapot közötti „ΔΕ” energiakülönbséget (1. animáció) (5).
1. animáció
(5)
Az (5) összefüggésben a „h” a Planck-féle hatáskvantum, melynek értéke 6,626·10─34 Js, a „ν” pedig a
kibocsátott fény rezgésszáma. Mértékegysége 1/s. A rezgésszám azoknak a periódusoknak (szinusz
hullámoknak) a száma, amely az adott hullámhosszúságú fény 1 másodperc alatt megtett útjára rámérhető. A
fény 1 másodperc alatt kerekítve 300 000 km utat tesz meg. Ezért a c (300000 km/s) fénysebességből és a
rezgésszámból egyszerűen kiszámolható az emittált fény „λ” hullámhossza (6).
(6)
A fény jellemzésére a rezgésszámhoz hasonló természetű adat a hullámszám (ν*). A hullámszám 1 cm
úthosszra eső hullámok száma. Mértékegysége: 1/cm.
A fényelnyelés jellemzésére az abszorbancián kívül ma már kevéssé használatos mennyiség a T
transzmittancia (átbocsátó képesség) és annak százszorosa, a T% százalékos fényátbocsátó képesség
(transzmisszió %).
(7)
(8)

Atomspektroszkópia fizikai alapjai
8 Created by XMLmind XSL-FO Converter.
Ezek a mennyiségek azt fejezik ki, hogy az abszorbeáló közeg a rajta keresztülhaladó fény intenzitásának
hanyadrészét illetve hány százalékát engedi át. A relatív fényintenzitás csökkenésére ugyancsak használható
mennyiség az α abszorpciófok.
(9)
Az abszorpciófok és az átbocsátóképesség (transzmittancia) összege mindig 1.
(10)
(3.1)
A régi analóg (mutatós) műszereken látszólagos előnyt jelentett a transzmittancia skála alkalmazása, mert ez
lineáris skála volt. De miután a koncentráció nem a transzmittanciával, hanem az abszorbanciával áll lineáris
összefüggésben, a transzmittancia és az abszorpciófok ma már ritkábban használatos.
Megjegyzendő továbbá, hogy a korábbi szakkönyvek az abszorbanciát extinkciónak nevezték és jelölése E volt,
az a abszorpciós együtthatót ε betűvel jelölték és neve extinkciós koefficiens volt. Az IUPAC szerint jelenleg a
vonal vagy sávspecifikus elnyelés elnevezése: abszorbancia, Jele: A.
3. Az atom szerkezete és az atomszínképek keletkezése
Annak idején az elemek emissziós atomszínképe jelentette az alapot az atomok elektronszerkezetének
megfejtéséhez, az elektronhéjak felépítéséhez. A Bohr-féle atommodell szerint az elektronok a nekik megfelelő
alappályákon fény kibocsátás nélkül keringenek. Az ilyen energiaállapotú atomok tekinthetők
alapállapotúaknak. Ha legalább egy elektron magasabb elektronpályára kerül, majd onnan egy alacsonyabbra,
vagy az alappályára visszalép, a pályák közötti energiakülönbségnek megfelelő energiájú (rezgésszámú,
hullámhosszúságú) fotont, fotonokat bocsát ki. Bármelyik pályáról bármelyikre lép vissza az elektron, az mindig
egy adott hullámhosszúságú színképvonal keletkezésével jár.
3. ábra: A hidrogén emissziós színképe látható és ultraibolya tartományban
Legegyszerűbb a hidrogén színképe (3. ábra). A látható tartományban mindössze négy vonala jelenik meg, de
az ultraibolya tartományban a sorozat további, egyre sűrűsödő vonalai figyelhetők meg. 1885-ben Balmer
felfedezte, hogy az alábbi képlettel a vonalak ν* hullámszámai igen pontosan kifejezhetők.
(11)

Atomspektroszkópia fizikai alapjai
9 Created by XMLmind XSL-FO Converter.
ahol R = 109 678 cm─1 és n = 3, 4, 5,…… ∞. Az R állandót (a svéd fizikus neve után) Rydberg-állandónak
nevezzük.
A hidrogén színképének későbbi, részletesebb tanulmányozása a távoli ultraibolya és az infravörös
tartományban további színképszériák felfedezéséhez vezetett. Ezeket a szériákat felfedezőikről Lyman-,
Paschen-, Bracket- és Pfund-szériának nevezték el. Utóbbi sorozatok hullámszámának kiszámításához a (11)
összefüggést annyiban kell változtatni, hogy az első tagban a 22 helyett 12, 32, 42, 52 értékeket kell helyettesíteni.
Így a hidrogén spektrumát kifejező általános képlet
(12)
Az n>m. Az m számok (1–5) azt fejezik ki, hogy az adott szériánál az elektron bármely magasabb pályáról
melyik pályára ugrik vissza. A Balmer-szériánál, például, mindig a 2. pályára. Mint látható, a legegyszerűbb
atom is nagyszámú színképvonallal jellemezhető (4. ábra).
4. ábra: A hidrogén elektronszintjeinek Grotrian diagramja
A hidrogénhez hasonló vonalszériák a többi elem színképében is megfigyelhetők, de a színkép jóval
bonyolultabb, mert az egyes szériák átfedésben vannak egymással. Itt is érvényes, hogy a vonalak hullámszáma
két egész szám függvényének különbségeként fejezhető ki, ahol n>m. E függvények számértékét termeknek
nevezzük.
(13
Atomspektroszkópia fizikai alapjai
10 Created by XMLmind XSL-FO Converter.
A különböző szériák jellegük miatt eltérő neveket kaptak. Az s (sharp), p (principal), d (diffuse), f
(fundamental) elnevezések jól mutatják, hogy őrízték meg az elektronpálya jelölések az eredetileg a színképekre
alkalmazott kifejezéseket.
A elektronpálya átmeneteket a legszemléletesebben az úgynevezett Grotrian diagramokon lehet bemutatni. A
diagram függőleges tengelyén a pályákat jellemző energiákat és az azokhoz tartozó elektronszinteket tüntetjük
fel. Az egyes elektronszintek közti átmeneteket szimbolizáló nyilakra pedig az adott átmentek által megjelenő
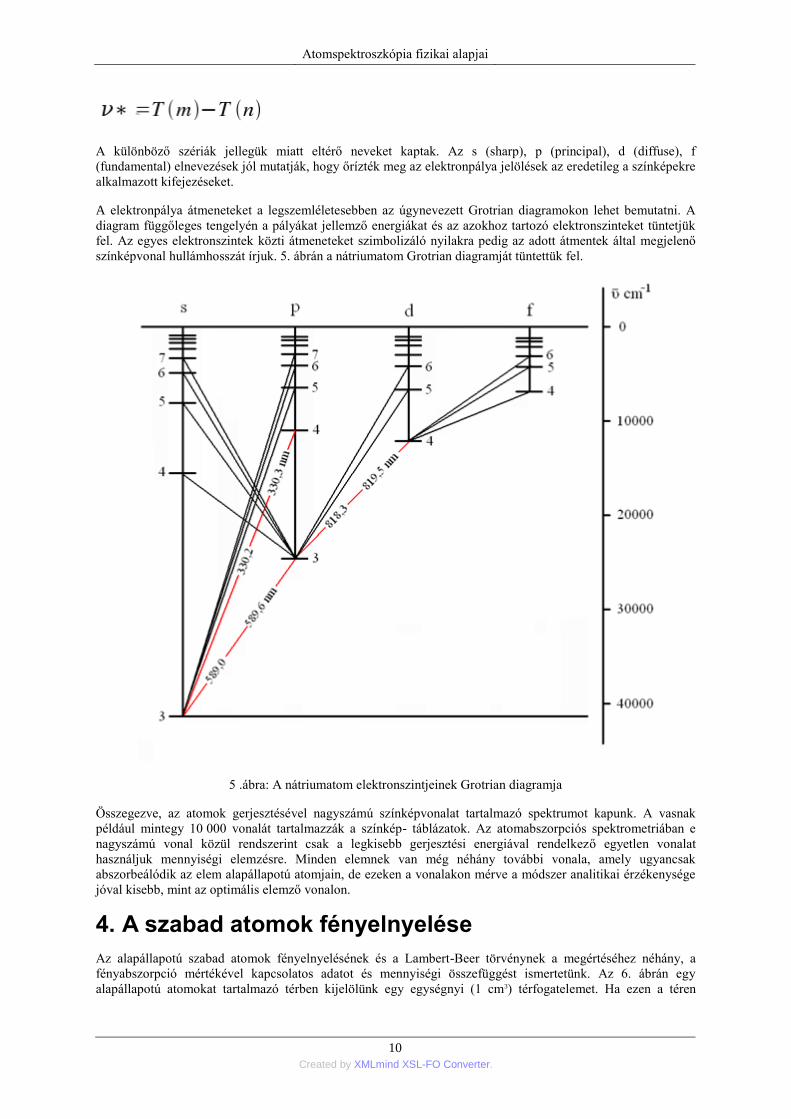
színképvonal hullámhosszát írjuk. 5. ábrán a nátriumatom Grotrian diagramját tüntettük fel.
5 .ábra: A nátriumatom elektronszintjeinek Grotrian diagramja
Összegezve, az atomok gerjesztésével nagyszámú színképvonalat tartalmazó spektrumot kapunk. A vasnak
például mintegy 10 000 vonalát tartalmazzák a színkép- táblázatok. Az atomabszorpciós spektrometriában e
nagyszámú vonal közül rendszerint csak a legkisebb gerjesztési energiával rendelkező egyetlen vonalat
használjuk mennyiségi elemzésre. Minden elemnek van még néhány további vonala, amely ugyancsak
abszorbeálódik az elem alapállapotú atomjain, de ezeken a vonalakon mérve a módszer analitikai érzékenysége
jóval kisebb, mint az optimális elemző vonalon.
4. A szabad atomok fényelnyelése
Az alapállapotú szabad atomok fényelnyelésének és a Lambert-Beer törvénynek a megértéséhez néhány, a
fényabszorpció mértékével kapcsolatos adatot és mennyiségi összefüggést ismertetünk. Az 6. ábrán egy
alapállapotú atomokat tartalmazó térben kijelölünk egy egységnyi (1 cm3) térfogatelemet. Ha ezen a téren

Atomspektroszkópia fizikai alapjai
11 Created by XMLmind XSL-FO Converter.
fénynyalábot bocsátunk keresztül, akkor a kiválasztott egységnyi térelemben a sugárzó energia összegét a
sugárzás sűrűségének nevezzük és ρ-val jelöljük.
6 .ábra: Az atomgőzben kiválasztott térelem (sárga kocka)
Ha ez az összes energia adott rezgésszámú (hullámhosszúságú) sugárzó energiára vonatkozik, akkor sugárzás
spektrális sűrűségéről (ρν) beszélünk. Az 6. ábrán ábrázolt térben az atomok elnyelik ennek a sugárzó
energiának egy részét. Az egységnyi térfogatban másodpercenként elnyelődő teljes energia: Eabs.
(14)
Az (14) összefüggésben szereplő mennyiségek az alábbiak:
• e az elektron töltése
• me az elektron tömege
• ρν a sugárzás spektrális sűrűsége
• f abszorpciós oszcillátor erősség
Ha az egységnyi térelemben minden alapállapotú szabad atom elnyelne egy fotont, akkor az összes elnyelt
energia Eabs = n۰ h۰ν lenne. De miután nem minden atom nyel el fotont, az (14) összefüggésben a Bik۰ ρν
szorzat azoknak az atomoknak a hányadát jelenti az összes atomhoz képest, amelyek egységnyi idő alatt h۰νik
fotont nyelnek el [s-1]. Előbbiekből következik, hogy az adott térelemben az elnyelt energia mértéke egyrészt
egy elemről elemre változó atomi állandótól, az abszorpció (Bik) Einstein-féle valószínűségétől, másrészt a
sugárzás (ρν) spektrális sűrűségétől függ.
5. Oszcillátor erősség
Az atomot tekinthetjük egy kényszerített elektromos dipólus oszcillátornak is. Egyik pólusa a pozitív töltésű
mag a lezárt elektronhéjakkal. A másik az előző pólussal egyenlő nagyságú, de ellentétes, azaz negatív töltésű
külső elektron, vagy elektronok. Ez az elektron, vagy elektronok az atom külső és belső pályái között mozognak
(oszcillálnak). Ezt az oszcillációt kényszeríti ki az elektronok energiájával egyező nagyságú elektromágneses
sugárzás (adott hullámhosszúságú fénynyaláb), amely az adott térelemen áthalad. Ha tehát az atomot klasszikus
harmonikus oszcillátorként kezeljük, akkor az elektrodinamika törvényei szerint megadható, hogy egy
klasszikus harmonikus oszcillátor egységnyi idő alatt mekkora Eabs energiát nyel el:

Atomspektroszkópia fizikai alapjai
12 Created by XMLmind XSL-FO Converter.
(15)
n az alapállapotú szabad atomok száma
Bik az abszorpció Einstein-féle valószínűsége
ρν a sugárzás spektrális sűrűsége
h۰ν a foton energiája
Az f oszcillátor erősség egy dimenzió nélküli szám, amely megadja a klasszikus szabad oszcillátorok effektív
számát, amely adott elektronátmeneteknél kifejezi az atom abszorpcióra való képességét.
Az (14) és (15) összefüggésekből kifejezhetjük az oszcillátor erősségét.
(16)
(17)
A (17) összefüggésből látható, hogy az oszcillátor erősség közvetlen összefüggésben áll az i és k elektronpályák
közötti átmenethez tartozó abszorpció Bik Einstein-féle valószínűségével.
6. Az abszorpciós együttható
Az atomabszorpció mértéke szempontjából igen fontos adat az abszorpciós együttható. Ennek értékét az
alábbiak szerint vezetjük le. ρν sugárzás sűrűségű párhuzamos fénynyalábot vezessünk át egységnyi felületű, dl
rétegvastagságú homogén eloszlású szabad alapállapotú atomgőzön (7. ábra). Az energia, ami ezen az egységnyi
felületen időegység alatt áthalad, azt nevezzük a sugárzás fluxus sűrűségének, amelyet Iν-vel jelölünk. A c a
fénysebesség.
(18)

Atomspektroszkópia fizikai alapjai
13 Created by XMLmind XSL-FO Converter.
7. ábra: Alapállapotú atomokat tartalmazó homogén elnyelő réteg megvilágítása adott hullámhosszúságú
fénynyalábbal
Mivel ebben az esetben nem egységnyi térfogatról van szó, e térelemben az alapállapotú atomok száma sem n,
hanem csak n·dl. Erre az esetre felírva az (14) összefüggést:
(19)
Az (19) összefüggés szerint Eabs az adott térfogatban (7. ábra) egységnyi idő alatt elnyelt energia nagysága.
Ezzel egyidejűleg ezen a rétegen halad át az Iν = ρν · c (18) energia. Utóbbi energia nagyságából kiszámítható az
időegység alatt áthaladt fotonok száma, mivel egy foton energiája h·ν.
(20)
Most bevezetünk egy új fogalmat, az atom effektív keresztmetszetét. Ha ez nagyobb érték, az azt fejezi ki,
hogy az adott elem atomjai a fotonokat nagyobb valószínűséggel képesek abszorbeálni, mint egy másik elem
atomjai. Jelöljük ezt az effektív keresztmetszetet κik-val. A vizsgált rétegben az összes atom effektív
keresztmetszete, azaz a teljes abszorbeáló keresztmetszet:
(21)
A (20) és (21) alapján a rétegben elnyelődő összes fotonok száma
(22)

Atomspektroszkópia fizikai alapjai
14 Created by XMLmind XSL-FO Converter.
A vizsgált rétegben az elnyelődött fotonok energiáját úgy kapjuk meg, hogy az (22) összefüggést megszorozuk
h ۰ ν -vel, egy foton energiájával.
(23)
(19) és (23)-ből következően összefüggést találunk az effektív keresztmetszet és az abszorpció Einstein-féle
valószínűsége (25), valamint az oszcillátor erőssége (26) között.
(24)
(25)
(17) és (25)-ből következik:
Az eddigiekből látható, hogy az abszorpciós együttható, mint elnyelési keresztmetszet, az atomi állandóktól,
elektronszerkezeti adatoktól függ. Az, hogy ténylegesen keresztmetszet jellegű a dimenziója, az a Lambert-Beer
törvényből következik is. Mivel az A abszorbancia dimenzió nélküli érték,
(27)
ha az úthossz [l] = cm , a koncentráció [c] = cm-3, akkor az [a] = κ · n abszorpciós együttható mértékegysége
valóban [a] = cm2.
7. A Lambert-Beer törvény
Az előzőekben a térfogategységben elnyelt fényenergiáról esett szó. A gyakorlatban azonban a sugárzás fluxus
sűrűségét Iν-t szoktuk mérni, azaz azt az energiát, amely a fénynyalábra merőleges egységnyi felületen
időegység alatt áthalad. A sugárzás spektrális sűrűsége és fluxus sűrűsége között az összefüggést, ( Iν = ρν · c)
már korábban bemutattuk (18). Most a korábbi összefüggések felhasználásával levezetjük a Lambert-Beer
törvényt. Legyen dIν egy végtelen kis változás az Iν sugárzás spektrális fluxus sűrűségében. Az (25) összefüggés
és a 6. ábra alapján a térelemben elnyelődött energia egyenlő a megvilágító fényfluxus sűrűségének
csökkenésével (28).
(28)

Atomspektroszkópia fizikai alapjai
15 Created by XMLmind XSL-FO Converter.
(29)
Az alapállapotú szabad atomok homogén eloszlása esetén a κ és az n értéke független az l rétegvastagságtól
illetve úthossztól. Ezért az egyenlet integrálható az l rétegvastagság mentén.
30
(31)
Behelyettesítve a szélső értékeket, mely szerint l=0 értéknél Iν = Iν,0 , a következő logaritmikus összefüggést
kapjuk:
(32)
Ha ezt tízes alapú logaritmusos formára hozzuk, megkapjuk a Lambert-Beer törvényt
(33)
(34)
(35)

Atomspektroszkópia fizikai alapjai
16 Created by XMLmind XSL-FO Converter.
8. Az alapállapotú és gerjesztett atomok aránya
A szabad atomoknak két energiaállapot (i és k szint) közötti megoszlása adott hőmérsékleten termikus
egyensúly alapján valósul meg. Egy kiválasztott hőmérsékleten az i és k energiaszinten előforduló atomok
számát az alábbiak szerint adhatjuk meg:
(36)
(37)
ahol
• ni és nk - az adott energiaszinten levő atomok száma
• Ei és Ek - az adott szintekhez tartozó energia nagysága
• c - Ei és Ek-tól független állandó
• k - Boltzmann állandó
• T - abszolút hőmérséklet
• gi és gk - statisztikus súlyok
A gi és gk statisztikus súlyok is elektronszerkezeti állandók, az adott (i és k szintű) energiaállapot felépítéséhez
szükséges Zeeman és hiperfinom atomi szintek számát adják meg.
A két energiaszinthez tartozó atomszám arányát felírva az alábbi összefüggést kapjuk.
(38)
Ha a (38) összefüggésben az i szintet az alapállapotú atomi szintnek tekintjük, amelynek az energiaértéke Ei = 0,
az összefüggés az alábbiak szerint módosul.
(39)
A 0 indexszel az alapállapothoz, 1 indexszel pedig a gerjesztett szinthez tartozó adatokat jelöltük.
2000-4000 K hőmérsékleten, amely tartományban az atomabszorpciós spektrometriás módszerrel lángban és
grafitkemencében az alapállapotú szabad atomok előállítása történik, a gerjesztett és az alapállapotú atomok
Atomspektroszkópia fizikai alapjai
17 Created by XMLmind XSL-FO Converter.
aránya elemenként változik, de a legkönnyebben gerjeszthető elemnél is alig haladja meg az alapállapotú
atomok 1%-át. Az 1. táblázatban néhány elemnek az n1/n0 értékeit tüntettük fel különböző hőmérsékleten.
(40)
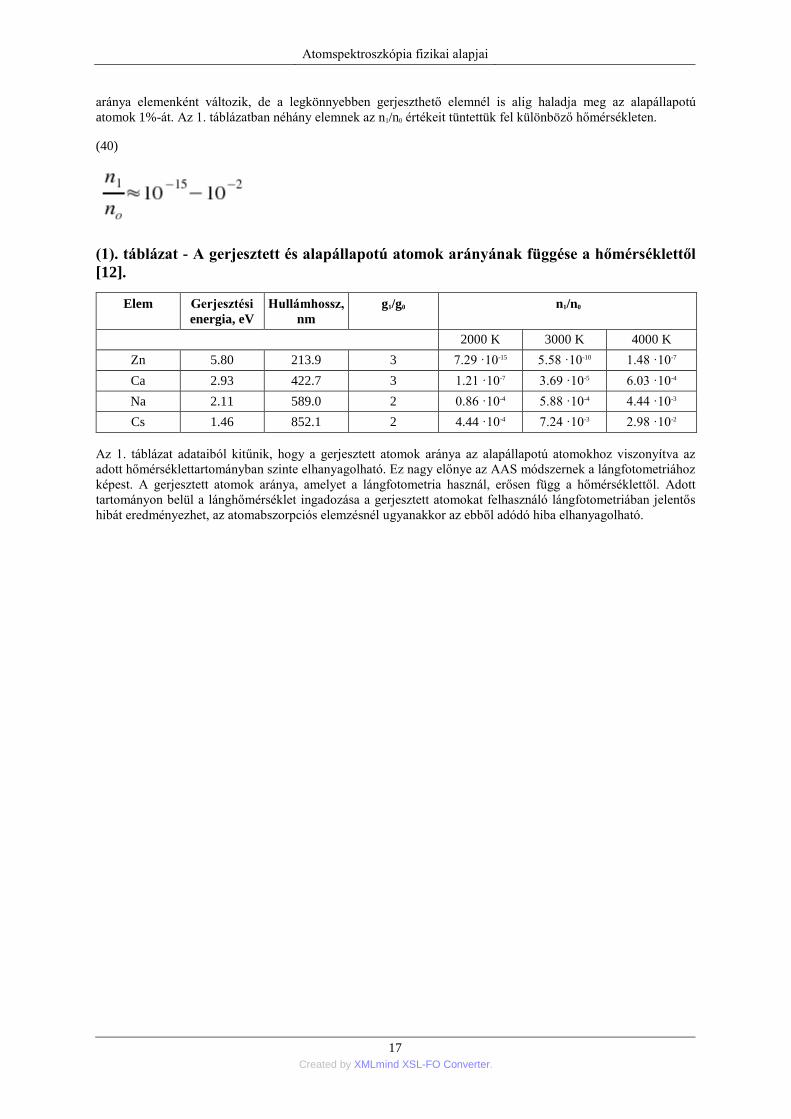
(1). táblázat - A gerjesztett és alapállapotú atomok arányának függése a hőmérséklettől
[12].
Elem Gerjesztési
energia, eV Hullámhossz,
nm g1/g0 n1/n0
2000 K 3000 K 4000 K
Zn 5.80 213.9 3 7.29 ·10-15 5.58 ·10-10 1.48 ·10-7
Ca 2.93 422.7 3 1.21 ·10-7 3.69 ·10-5 6.03 ·10-4
Na 2.11 589.0 2 0.86 ·10-4 5.88 ·10-4 4.44 ·10-3
Cs 1.46 852.1 2 4.44 ·10-4 7.24 ·10-3 2.98 ·10-2
Az 1. táblázat adataiból kitűnik, hogy a gerjesztett atomok aránya az alapállapotú atomokhoz viszonyítva az
adott hőmérséklettartományban szinte elhanyagolható. Ez nagy előnye az AAS módszernek a lángfotometriához
képest. A gerjesztett atomok aránya, amelyet a lángfotometria használ, erősen függ a hőmérséklettől. Adott
tartományon belül a lánghőmérséklet ingadozása a gerjesztett atomokat felhasználó lángfotometriában jelentős
hibát eredményezhet, az atomabszorpciós elemzésnél ugyanakkor az ebből adódó hiba elhanyagolható.

18 Created by XMLmind XSL-FO Converter.
4. fejezet - Az atomabszorpciós spektrométerek felépítése, működése
Az atomabszorpciós spektrométerek felépítését a 8. ábrán látható sematikus ábrán mutatjuk be. A készülékek
optikai rendszere a megvilágító fényforrástól a detektorig tart. Ezt követi a jelfeldolgozó elektronika, majd a
mérési adatokat megjelenítő kijelző rendszer. A megvilágító fényforrás útjában helyezkedik el az alapállapotú
atomgőzöket előállító atomizáló egység, ahova a vizsgálandó mintát a mintabeviteli egység segítségével
juttatjuk be.
8. ábra: Az atomabszorpciós spektrométer felépítésének elvi vázlata
1. A spektrométer optikai rendszere
Az optikai rendszer a megvilágító fényforrással kezdődik. A fényforrás fénye az úgynevezett szaggatón, majd a
vizsgált minta atomgőzein átjutva a fényfelbontó egységbe, a monokromátorba jut, ahol megtörténik az elemző
vonal kiválasztása. A kiválasztott hullámhosszúságú vonal a monokromátor kilépő résén átjutva a detektorra
esik, ahol a fényenergia elektromos energiává alakul. Az AAS készülékek igen fontos optikai egysége a
háttérkorrekciós rendszer. Az alábbiakban ezeket az optikai egységeket mutatjuk be részleteiben.
1.1. Megvilágító fényforrások
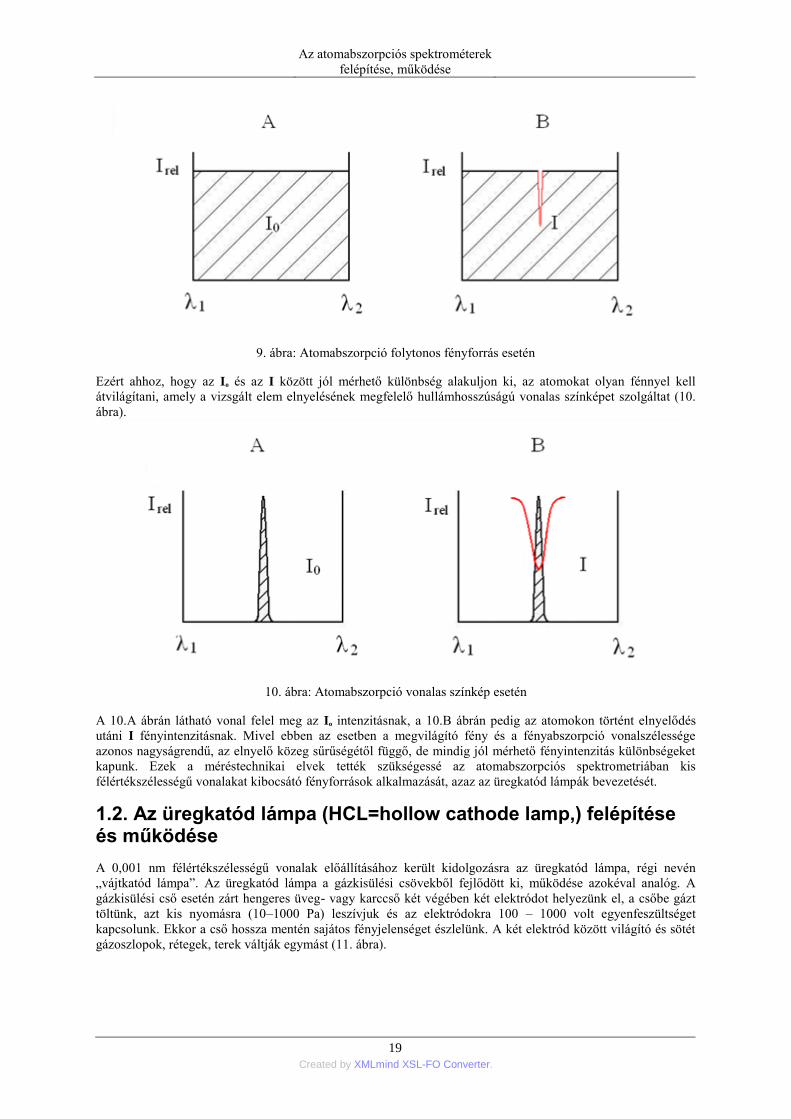
Kérdés, hogy miért nem alkalmas az AAS elemzésekhez folytonos fényforrás, volfram vagy deutérium lámpa,
amelyet a spektrofotometriában használnak a minták átvilágítására. A 9.A ábrán látható, hogy egy hagyományos
spektrális felbontóképességű monokromátor kilépő résének λ2–λ1 sávszélessége 1 nm nagyságrendű. Az adott
intenzitású folytonos fény természetesen egyenletesen kitölti a rés teljes szélességét. Ez a réssel határolt terület
felel meg az Io fényintenzitásnak. Ha ennek a folytonos fénynek az útjába adott elem alapállapotú szabad
atomjait juttatjuk, akkor az elem elnyelésének megfelelő hullámhosszon megtörténik a fényabszorpció. A
fényelnyelés azonban csak az atomabszorpciós vonalprofil (0,01 – 0,001 nm) szélességére korlátozódik (9.B
ábra). A megmaradó terület reprezentálja az elnyelő réteg utáni I fényintenzitást. Az 9.A és 9.B ábrán látható,
ilyen minimális területkülönbség esetén nincs olyan optikai műszer, amelyik különbséget tudna tenni az Io és az
I intenzitás között mivel ez a különbség 0,01 – 0,001 %-nál nem nagyobb.
Az atomabszorpciós spektrométerek
felépítése, működése
19 Created by XMLmind XSL-FO Converter.
9. ábra: Atomabszorpció folytonos fényforrás esetén
Ezért ahhoz, hogy az Io és az I között jól mérhető különbség alakuljon ki, az atomokat olyan fénnyel kell
átvilágítani, amely a vizsgált elem elnyelésének megfelelő hullámhosszúságú vonalas színképet szolgáltat (10.
ábra).
10. ábra: Atomabszorpció vonalas színkép esetén
A 10.A ábrán látható vonal felel meg az Io intenzitásnak, a 10.B ábrán pedig az atomokon történt elnyelődés
utáni I fényintenzitásnak. Mivel ebben az esetben a megvilágító fény és a fényabszorpció vonalszélessége
azonos nagyságrendű, az elnyelő közeg sűrűségétől függő, de mindig jól mérhető fényintenzitás különbségeket
kapunk. Ezek a méréstechnikai elvek tették szükségessé az atomabszorpciós spektrometriában kis
félértékszélességű vonalakat kibocsátó fényforrások alkalmazását, azaz az üregkatód lámpák bevezetését.
1.2. Az üregkatód lámpa (HCL=hollow cathode lamp,) felépítése és működése
A 0,001 nm félértékszélességű vonalak előállításához került kidolgozásra az üregkatód lámpa, régi nevén
„vájtkatód lámpa”. Az üregkatód lámpa a gázkisülési csövekből fejlődött ki, működése azokéval analóg. A
gázkisülési cső esetén zárt hengeres üveg- vagy karccső két végében két elektródot helyezünk el, a csőbe gázt
töltünk, azt kis nyomásra (10–1000 Pa) leszívjuk és az elektródokra 100 – 1000 volt egyenfeszültséget
kapcsolunk. Ekkor a cső hossza mentén sajátos fényjelenséget észlelünk. A két elektród között világító és sötét
gázoszlopok, rétegek, terek váltják egymást (11. ábra).

Az atomabszorpciós spektrométerek
felépítése, működése
20 Created by XMLmind XSL-FO Converter.
11. ábra: Fényjelenség gázkisülési csövekben
E nyomástól és feszültségtől függően váltakozó fényjelenség oka, hogy a katód irányából az anód felé nagy
sebességgel mozgó elektronok a gázrészecskékkel ütközve ionizálják azokat. A nagy méretű pozitív töltésű
gázionok a nagy közepes szabad úthosszuk révén nagy energiával ütköznek viszont a katódba, porlasztva annak
anyagát. Így amíg a többi világító oszlop színképe elsősorban a töltőgázra jellemző, a negatív ködfény, a katód
közelében a katód anyagára jellemző színképet bocsát ki. Ezt figyelembe véve úgy módosították a gázkisülési
csőben a katód geometriáját, hogy a sík felület helyett hengeres üreget képeztek ki, amely üregben a legnagyobb
térfogatot a 12. ábrán látható módon a negatív ködfény tölti ki.
12. ábra: A kisülés szerkezete a katódüregben
A ma forgalomban levő üregkatód lámpák szerkezetét a 13. ábrán mutatjuk be. A lámpa 3-6 cm átmérőjű
hengeres zárt üvegcső. A töltőgáz 100-500 Pa nyomású argon vagy neon. A lámpa ablaka Pyrex üveg, de ha az
elemző vonal hullámhossza az ultraibolya tartományba esik, akkor kvarcablakra van szükség. A lámpa anódja
gyűrű vagy zászló alakú volfram lemez. A hengeres furatos katód, melynek a belső átmérője 2-5 mm, abból az
elemből készül, amilyen elemet vizsgálni akarunk. Ez azt jelenti, hogy minden elem analíziséhez külön lámpára
van szükség. Jól megmunkálható fémek esetén az üreges katód magából a fémből készül. Ha az elem
mechanikailag nem megmunkálható, akkor az elemet vagy vegyületét, például oxidját, valamilyen (pl. kerámia)
hordozóra viszik fel az elektromosan vezető katódüregben. A lámpákat 100–300 volt feszültségen üzemeltetjük.
A fűtőáram nagysága általában 5–20 mA. A lámpa katód- és anód-kivezetése fel nem cserélhető módon
csatlakozik a tápegység aljzatához.

Az atomabszorpciós spektrométerek
felépítése, működése
21 Created by XMLmind XSL-FO Converter.
13. ábra: Az üregkatód lámpa szerkezete
Az üregkatód lámpa működését az 1. animációs ábrán szemléltetjük. Az üregkatód lámpa bekapcsolásakor a
gázkisülési csövek működésének megfelelően a katódból elektronok lépnek ki, amelyek nagy sebességgel az
adód felé tartanak. Az útjukba kerülő nemesgáz atomokat ionizálják. A keletkező pozitív töltésű nemesgázionok
viszont a katód irányába mozogva felgyorsulnak és a katódüregbe csapódva, annak falából a katód anyagának
atomjait teszik szabaddá. A nemesgázionok a továbbiakban már az üregben felhalmozódó alapállapotú
atomokkal ütköznek. Az ionok kinetikus energiájukat az atomoknak átadva gerjesztik azokat. Az üregben
keletkező gerjesztett atomok rekombinációja (azaz elektronjaiknak a magasabb elektronpályákról az alappályára
történő visszalépése) eredményeképpen a katód anyagára jellemző vonalas színképet adó fénynyaláb távozik a
lámpából.
2. animáció Az üregkatód lámpa működése
Az üregkatód lámpa igen kis félértékszélességű vonalas színképet szolgáltat, mert a ritkított gáztérben a vonalak
nyomáskiszélesedése elhanyagolható. Az ionok és atomok intenzív ütközése miatt a színképben nagy gerjesztési
energiájú vonalak is megjelennek anélkül, hogy a lámpa felmelegedne. Ezt a színképet termikus gerjesztéssel
csak több ezer oC hőmérsékleten érhetnénk el. Egy mai kereskedelmi lámpa élettartama, változatlan
fényintenzitást biztosítva 1000-2000 üzemóra.
A lámpagyártó cégek forgalomba hoznak többelemes lámpákat is. Az ilyen lámpa kettőtől akár hat elem
meghatározására is alkalmas lehet E lámpák katódját többféleképpen készítik el. Gyakori megoldás, hogy
fémötvözetet használnak. Ezt akkor érdemes alkalmazni, ha az ötvöző elemek illékonysága közel azonos.
Egyébként az illékonyabb elemre nézve a lámpa élettartama rövidebb lenne. Másik eljárás szerint több gyűrű
alakú fémet préselnek össze üregkatóddá. Ugyancsak állítanak elő többelemes lámpát különböző fémporok
szinterelésével, illetve intermetallikus vegyületek felhasználásával. A többelemes lámpák kevésbé megbízhatók,
mint az egyelemesek. Élettartamuk, stabilitásuk kisebb és esetenként kis felbontású monokromátort alkalmazva
vonalegybeesés, spektrális zavarás is felléphet a többelemes lámpa használata során. Egy fontos érv a
többelemes lámpák mellett az, hogy olcsóbb, mert egy hatelemes lámpa töredékébe kerül hat egyelemes
lámpának.
Már a 60-as évek közepén kidolgozták egy úgynevezett nagyintenzitású lámpa elvét (high-brightness lamp,
boosted lamp). Ilyen lámpák is vannak forgalomban (14. ábra). A nagyintenzitású lámpában az üregkatódon és
az anódon kívül a katód két oldalán két segédelektród helyezkedik el. A segédelektródok között létrejövő
elektron-emisszió a katódüregben katódporlasztással felszabaduló alapállapotú atomok számára egy másodlagos
gerjesztő forrást jelent. Ez a gerjesztési mód nemesgáz ionokkal történő ütközéshez képest kisebb energiájú.
Ezért csak az adott elem elemző vonalának intenzitását növeli, ugyanakkor a töltőgáz vonalainak intenzitását
csökkenti.

Az atomabszorpciós spektrométerek
felépítése, működése
22 Created by XMLmind XSL-FO Converter.
14. ábra: Nagyintenzitású üregkatód lámpa
1.3. Elektród nélküli kisülési lámpa (electrodeless discharge lamp, EDL)
Több elem atomabszorpciós elemző-vonalának hullámhossza az ultraibolya színképtartomány alsó szakaszára
(200 nm közelébe) sőt a vákuum-ultraibolya (200 nm alatti) tartományba esik. Ez azzal jár, hogy az üregkatód
lámpa optimális üzemi paraméterek mellett is kis fényintenzitást biztosít, mert a lámpa és a detektor közötti
körülbelül 1 méteres levegőréteg oxigénje ebben a színkép-tartományban már a fény jelentős részét elnyeli. A
kis fényintenzitás miatt az AAS készülék nagyobb mértékű elektronikus erősítésére van szükség, amely a mért
jel zajszintjét jelentősen megnöveli. Ezzel romlik a jel/zaj viszony, azaz a módszer kimutatási képessége. Ez a
körülmény éppen több olyan környezeti szempontból fontos toxikus nyomelem meghatározását nehezíti meg,
mint arzén, szelén, higany, kadmium, cink stb. E nehézségek kiküszöbölésére fejlesztették ki az úgynevezett
elektród nélküli kisülési lámpákat (EDLs). Az EDL felépítését a 15. ábrán mutatjuk be.
15. ábra: Az elektród nélküli kisülési lámpa (EDL) szerkezete
A lámpa legfontosabb eleme egy zárt kvarcból készült 2-3 cm hosszú 1 cm átmérőjű ampulla, amelybe az elem
illékony vegyületét, leggyakrabban halogenidjét zárják kis nyomású (100 Pa) nemesgáz térbe. A kvarcampulla
köré rádiófrekvenciás tekercset helyeznek, melyre nagyfrekvenciás generátor segítségével 27,12 MHz
frekvenciájú áramot kapcsolnak. Ez a hírközlést nem zavaró ipari frekvencia. A rendszer az induktív csatolású
plazmához (ICP) hasonlóan működik, de az atmoszférikus nyomású ICP-vel szemben a kis nyomású plazma a
zárt ampullában jön létre. Az ampullára kapcsolt energia hatására a benne levő anyag elpárolog, a molekulagőz
atomjaira disszociál, majd az atomok gerjesztődnek. A gerjesztett atomok rekombinációja az elemre jellemző
nagyintenzitású vonalas színkép kibocsátását eredményezi. A kibocsátott fény intenzitása körülbelül 6-10-
szerese a megfelelő üregkatód lámpáénak. Az illékony elemek üregkatód lámpái kevéssé stabilak és az
átlagosnál rövidebb az élettartamuk. Az adott elemek EDL lámpái viszont az intenzív fényt igen egyenletesen
bocsátják ki és igen hosszú a lámpa élettartama. A tárgyalt működési mechanizmus miatt az EDL lámpák üzemi
hőmérséklete körülbelül 80 oC. Ezért alkalmazásukkor gondoskodni kell az érintésvédelmi szabályok
betartásáról.
Az atomabszorpciós spektrométerek
felépítése, működése
23 Created by XMLmind XSL-FO Converter.
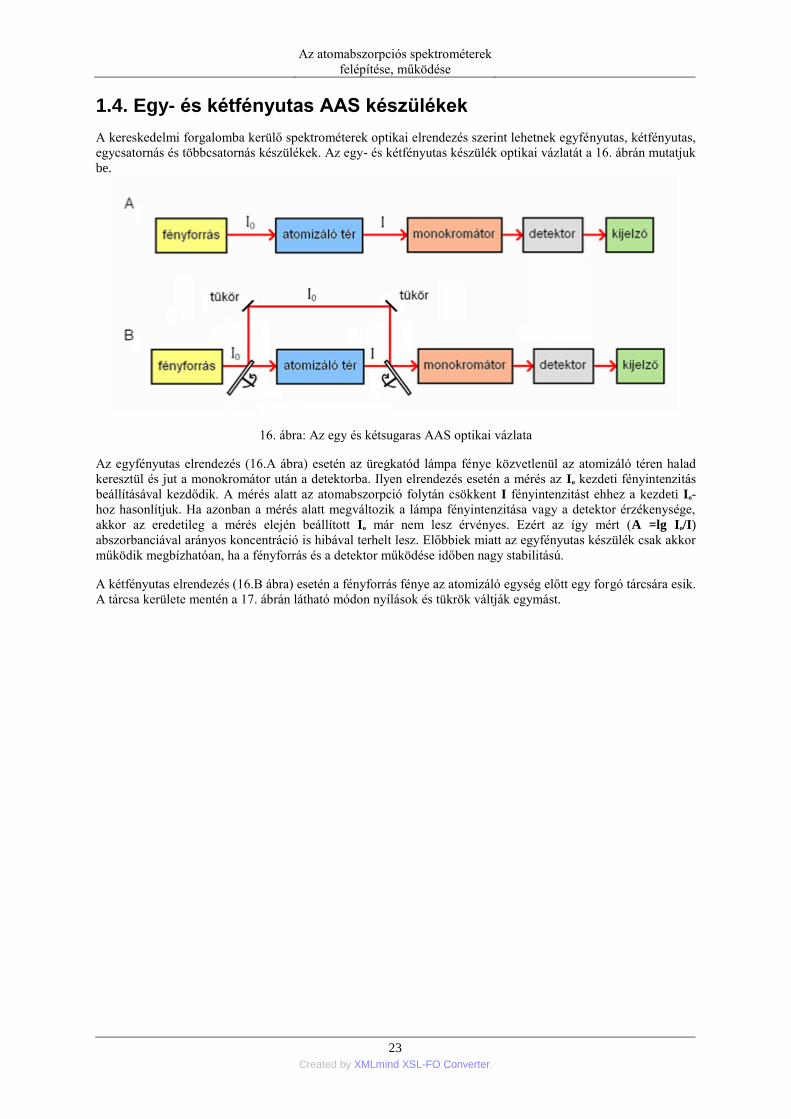
1.4. Egy- és kétfényutas AAS készülékek
A kereskedelmi forgalomba kerülő spektrométerek optikai elrendezés szerint lehetnek egyfényutas, kétfényutas,
egycsatornás és többcsatornás készülékek. Az egy- és kétfényutas készülék optikai vázlatát a 16. ábrán mutatjuk
be.
16. ábra: Az egy és kétsugaras AAS optikai vázlata
Az egyfényutas elrendezés (16.A ábra) esetén az üregkatód lámpa fénye közvetlenül az atomizáló téren halad
keresztül és jut a monokromátor után a detektorba. Ilyen elrendezés esetén a mérés az Io kezdeti fényintenzitás
beállításával kezdődik. A mérés alatt az atomabszorpció folytán csökkent I fényintenzitást ehhez a kezdeti Io-
hoz hasonlítjuk. Ha azonban a mérés alatt megváltozik a lámpa fényintenzitása vagy a detektor érzékenysége,
akkor az eredetileg a mérés elején beállított Io már nem lesz érvényes. Ezért az így mért (A =lg Io/I)
abszorbanciával arányos koncentráció is hibával terhelt lesz. Előbbiek miatt az egyfényutas készülék csak akkor
működik megbízhatóan, ha a fényforrás és a detektor működése időben nagy stabilitású.
A kétfényutas elrendezés (16.B ábra) esetén a fényforrás fénye az atomizáló egység előtt egy forgó tárcsára esik.
A tárcsa kerülete mentén a 17. ábrán látható módon nyílások és tükrök váltják egymást.

Az atomabszorpciós spektrométerek
felépítése, működése
24 Created by XMLmind XSL-FO Converter.
17. ábra: Forgó tárcsa (szaggató)
Amikor a lámpa fénye a forgó tárcsa nyílására esik, a fénynyaláb az atomizáló téren (lángon, grafitkemencén)
halad keresztül, ott létrejön az I csökkent fényintenzitás. Amikor a lámpa fénye a forgó tárcsa tükrére esik,
akkor egy másik tükörre verődve az atomizáló tér mellett halad el fénygyengülés nélkül. Ez felel meg az
aktuális Io-nak. Az atomizáló tér után az időben váltakozva érkező Io és I pályája egy félig áteresztő tükör
segítségével egyesül. A két fénynyaláb felváltva jut be a monokromátorba. Miután a tárcsát szinkronmotor
forgatja és felváltva 3 tükör és 3 nyílás van rajta, másodpercenként (3x50=)150-szer van tükör és nyílás a
fényútban, (1:150=)6,7 milliszekundumonként kapunk egy-egy aktuális Io/I adatpárt. Ha a lámpa intenzitása és a
detektor érzékenysége változik is időben, azzal, hogy minden I adathoz az adott pillanatban érvényes Io-t
hasonlítjuk, a mérési hiba kiküszöbölődik. A kétfényutas elrendezés előbb vázolt előnye mellett viszont
hátránya, hogy több tükröt és féligáteresztő tükröt is igényel, ami viszont fényveszteséggel jár az egyfényutas
készülékhez képest. Emiatt a kétfényutas készüléknek nagyobb elektronikus erősítésre van szüksége, ami
viszont az elektronikus zaj növekedésével járhat.
Az előzőekben részletezett előnyök és hátrányok miatt a kereskedelemben ma is egyidejűleg hoznak forgalomba
egy- és kétfényutas készülékeket még ugyanazon gyártó cég esetén is.
Az atomabszorpciós spektrométerek alapvetően egycsatornás készülékek. Ez azt jelenti, hogy egyszerre egy
lámpával, egy hullámhosszon egy elemet lehet meghatározni. Időről időre felmerül viszont annak az igénye,
hogy ha nem is minden elemet, de adott elemcsoportot egyszerre lehessen meghatározni AAS módszerrel. Ilyen
törekvések folytán kerülnek kereskedelmi forgalomban többcsatornás, szimultán AAS készülékek is. Egy
többcsatornás spektrométer optikai vázlata a 18. ábrán látható.

Az atomabszorpciós spektrométerek
felépítése, működése
25 Created by XMLmind XSL-FO Converter.
18. ábra: Többcsatornás atomabszorpciós spektrométer optikai vázlata
A többcsatornás készülékek azért nem terjedtek el széles körben, mert a több csatorna több monokromátort,
vagy polikromátort és annyi detektort igényel, ahány elemet akarunk egyidejűleg meghatározni. Ezek
megnövelik a készülék előállítási árát. Ugyanakkor nem lehet tetszőlegesen összeválogatni az egyidejűleg
vizsgálható elemeket, mert ha nagyon eltérő az elemek atomizációs sajátsága, nem tudjuk egyidejűleg
mindegyik számára az optimális feltételeket biztosítani.
1.5. A fényszaggató szerepe
Az atomabszorpciós elemzések során az atomizáló térben (lángban, grafitkemencében) a vizsgálni kívánt elem
alapállapotú szabad atomjain kívül egy összetett mintában számos egyéb komponens van jelen. Ezek a kísérő
anyagok ugyanúgy részt vesznek az atomizáció során a hőfolyamatokban, mint maga a vizsgált elem. A kísérő
anyagok molekulái, a teljesen el nem párolgott izzó aeroszol, vagy a grafitcsőről leváló izzó szénrészecskék, de
maguk a lánggázok molekulái, gyökei, vagy az izzó grafitcső fala nagy hőmérsékleten fényt bocsátanak ki. Ha
az így kibocsátott fény a vizsgált elem elemző vonalának hullámhosszán is mérhető, jelentős befolyással lehet a
műszer által mért Io/I viszonyra. Az atomizáló térben keletkező atomok koncentrációjával arányos
fényintenzitás csökkenését kifejező I értékhez ugyanis hozzáadódik a kísérő anyagok minőségétől és
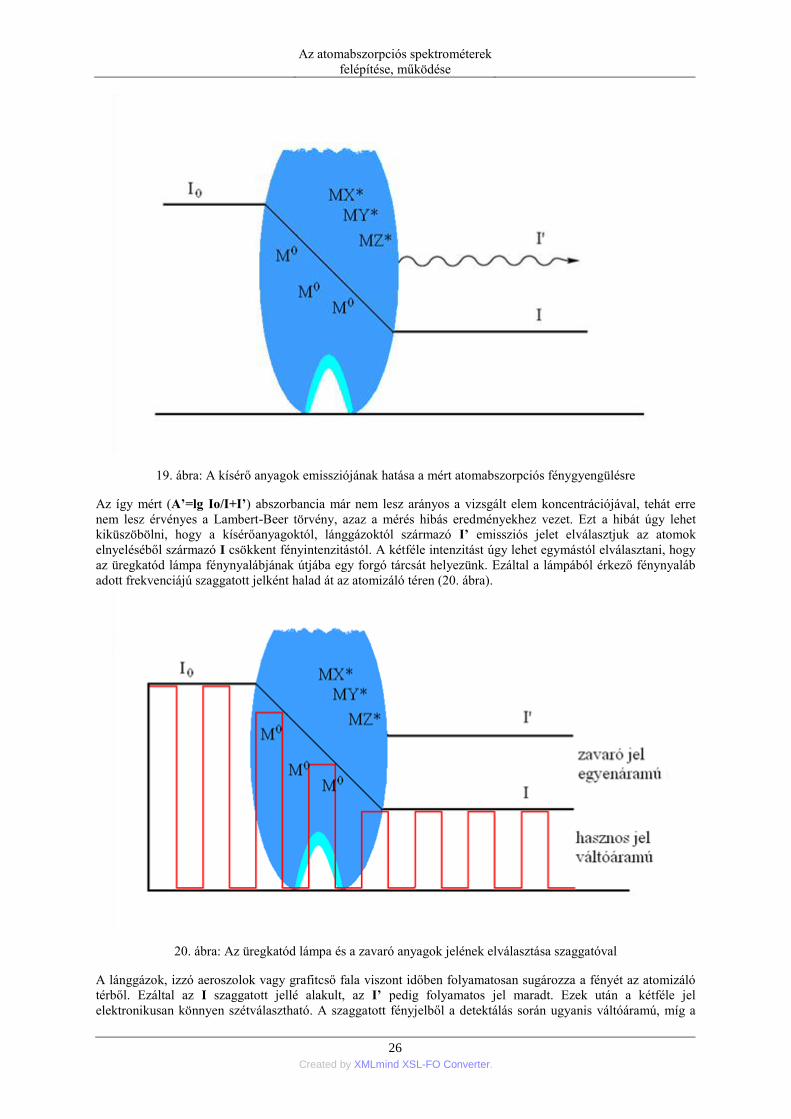
koncentrációjától függő, a vizsgált elem koncentrációjától független I’ emissziós jelintenzitás (19. ábra).
Az atomabszorpciós spektrométerek
felépítése, működése
26 Created by XMLmind XSL-FO Converter.
19. ábra: A kísérő anyagok emissziójának hatása a mért atomabszorpciós fénygyengülésre
Az így mért (A’=lg Io/I+I’) abszorbancia már nem lesz arányos a vizsgált elem koncentrációjával, tehát erre
nem lesz érvényes a Lambert-Beer törvény, azaz a mérés hibás eredményekhez vezet. Ezt a hibát úgy lehet
kiküszöbölni, hogy a kísérőanyagoktól, lánggázoktól származó I’ emissziós jelet elválasztjuk az atomok
elnyeléséből származó I csökkent fényintenzitástól. A kétféle intenzitást úgy lehet egymástól elválasztani, hogy
az üregkatód lámpa fénynyalábjának útjába egy forgó tárcsát helyezünk. Ezáltal a lámpából érkező fénynyaláb
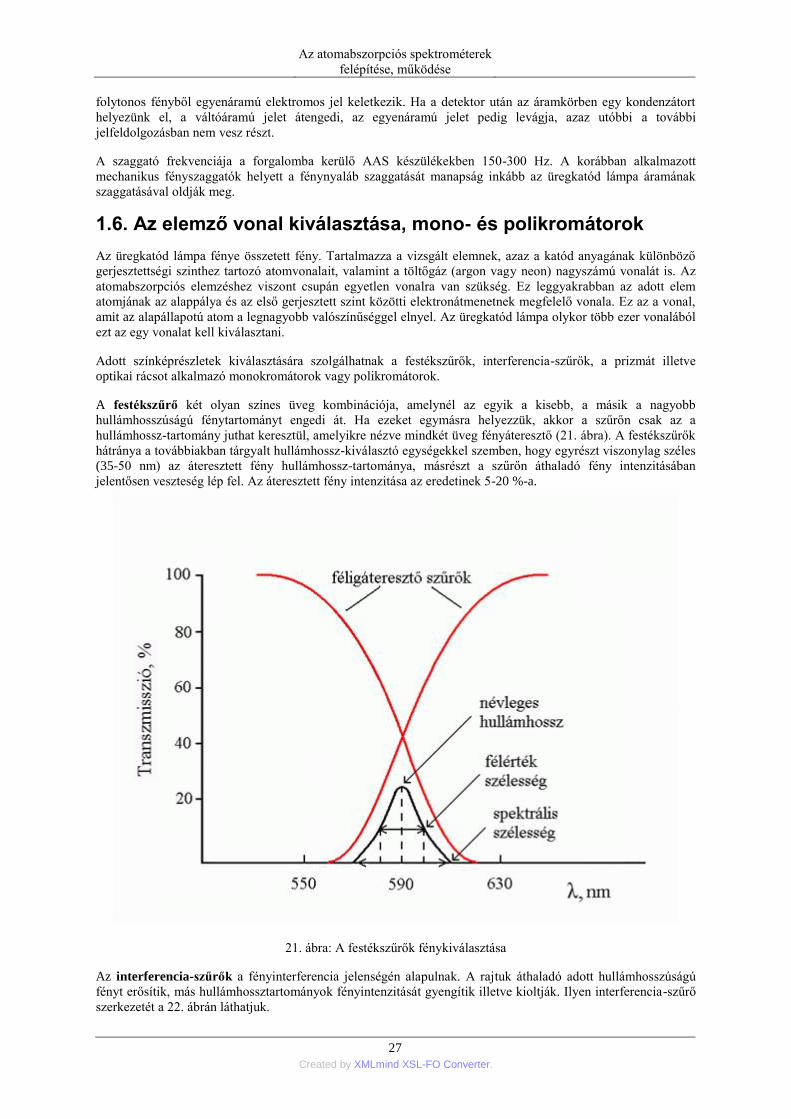
adott frekvenciájú szaggatott jelként halad át az atomizáló téren (20. ábra).
20. ábra: Az üregkatód lámpa és a zavaró anyagok jelének elválasztása szaggatóval
A lánggázok, izzó aeroszolok vagy grafitcső fala viszont időben folyamatosan sugározza a fényét az atomizáló
térből. Ezáltal az I szaggatott jellé alakult, az I’ pedig folyamatos jel maradt. Ezek után a kétféle jel
elektronikusan könnyen szétválasztható. A szaggatott fényjelből a detektálás során ugyanis váltóáramú, míg a
Az atomabszorpciós spektrométerek
felépítése, működése
27 Created by XMLmind XSL-FO Converter.
folytonos fényből egyenáramú elektromos jel keletkezik. Ha a detektor után az áramkörben egy kondenzátort
helyezünk el, a váltóáramú jelet átengedi, az egyenáramú jelet pedig levágja, azaz utóbbi a további
jelfeldolgozásban nem vesz részt.
A szaggató frekvenciája a forgalomba kerülő AAS készülékekben 150-300 Hz. A korábban alkalmazott
mechanikus fényszaggatók helyett a fénynyaláb szaggatását manapság inkább az üregkatód lámpa áramának
szaggatásával oldják meg.
1.6. Az elemző vonal kiválasztása, mono- és polikromátorok
Az üregkatód lámpa fénye összetett fény. Tartalmazza a vizsgált elemnek, azaz a katód anyagának különböző
gerjesztettségi szinthez tartozó atomvonalait, valamint a töltőgáz (argon vagy neon) nagyszámú vonalát is. Az
atomabszorpciós elemzéshez viszont csupán egyetlen vonalra van szükség. Ez leggyakrabban az adott elem
atomjának az alappálya és az első gerjesztett szint közötti elektronátmenetnek megfelelő vonala. Ez az a vonal,
amit az alapállapotú atom a legnagyobb valószínűséggel elnyel. Az üregkatód lámpa olykor több ezer vonalából
ezt az egy vonalat kell kiválasztani.
Adott színképrészletek kiválasztására szolgálhatnak a festékszűrők, interferencia-szűrők, a prizmát illetve
optikai rácsot alkalmazó monokromátorok vagy polikromátorok.
A festékszűrő két olyan színes üveg kombinációja, amelynél az egyik a kisebb, a másik a nagyobb
hullámhosszúságú fénytartományt engedi át. Ha ezeket egymásra helyezzük, akkor a szűrőn csak az a
hullámhossz-tartomány juthat keresztül, amelyikre nézve mindkét üveg fényáteresztő (21. ábra). A festékszűrők
hátránya a továbbiakban tárgyalt hullámhossz-kiválasztó egységekkel szemben, hogy egyrészt viszonylag széles
(35-50 nm) az áteresztett fény hullámhossz-tartománya, másrészt a szűrőn áthaladó fény intenzitásában
jelentősen veszteség lép fel. Az áteresztett fény intenzitása az eredetinek 5-20 %-a.
21. ábra: A festékszűrők fénykiválasztása
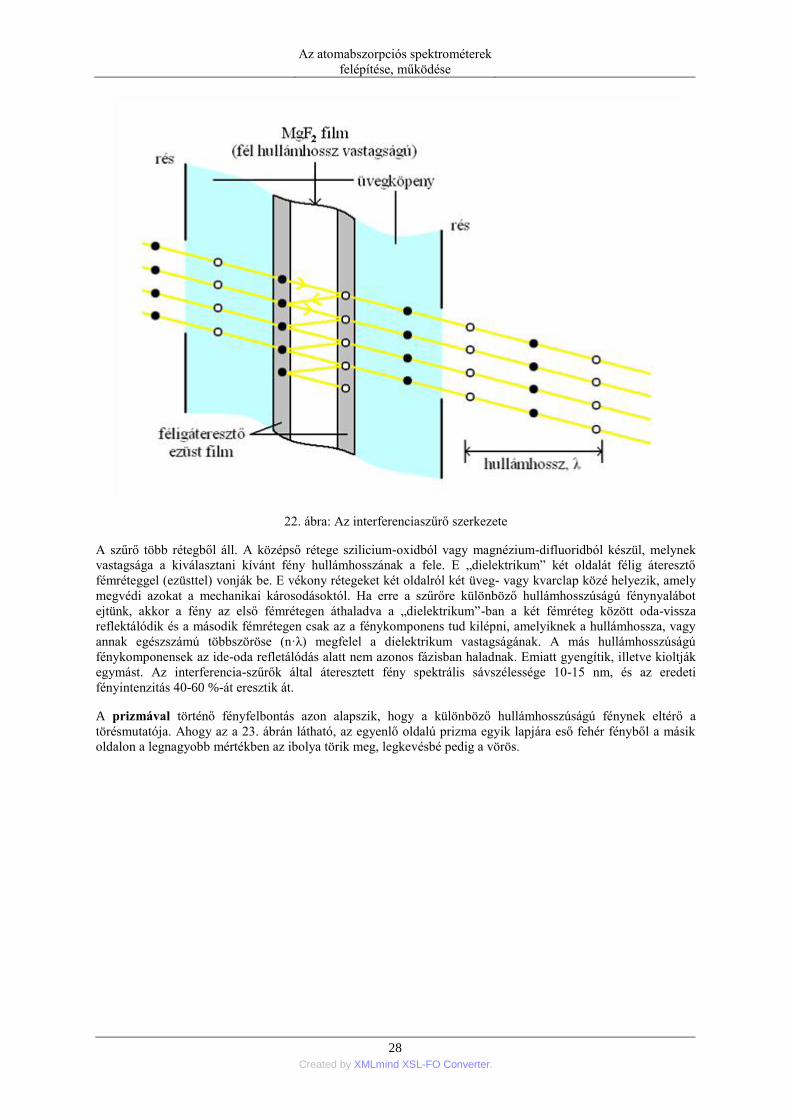
Az interferencia-szűrők a fényinterferencia jelenségén alapulnak. A rajtuk áthaladó adott hullámhosszúságú
fényt erősítik, más hullámhossztartományok fényintenzitását gyengítik illetve kioltják. Ilyen interferencia-szűrő
szerkezetét a 22. ábrán láthatjuk.
Az atomabszorpciós spektrométerek
felépítése, működése
28 Created by XMLmind XSL-FO Converter.
22. ábra: Az interferenciaszűrő szerkezete
A szűrő több rétegből áll. A középső rétege szilicium-oxidból vagy magnézium-difluoridból készül, melynek
vastagsága a kiválasztani kívánt fény hullámhosszának a fele. E „dielektrikum” két oldalát félig áteresztő
fémréteggel (ezüsttel) vonják be. E vékony rétegeket két oldalról két üveg- vagy kvarclap közé helyezik, amely
megvédi azokat a mechanikai károsodásoktól. Ha erre a szűrőre különböző hullámhosszúságú fénynyalábot
ejtünk, akkor a fény az első fémrétegen áthaladva a „dielektrikum”-ban a két fémréteg között oda-vissza
reflektálódik és a második fémrétegen csak az a fénykomponens tud kilépni, amelyiknek a hullámhossza, vagy
annak egészszámú többszöröse (n·λ) megfelel a dielektrikum vastagságának. A más hullámhosszúságú
fénykomponensek az ide-oda refletálódás alatt nem azonos fázisban haladnak. Emiatt gyengítik, illetve kioltják
egymást. Az interferencia-szűrők által áteresztett fény spektrális sávszélessége 10-15 nm, és az eredeti
fényintenzitás 40-60 %-át eresztik át.
A prizmával történő fényfelbontás azon alapszik, hogy a különböző hullámhosszúságú fénynek eltérő a
törésmutatója. Ahogy az a 23. ábrán látható, az egyenlő oldalú prizma egyik lapjára eső fehér fényből a másik
oldalon a legnagyobb mértékben az ibolya törik meg, legkevésbé pedig a vörös.

Az atomabszorpciós spektrométerek
felépítése, működése
29 Created by XMLmind XSL-FO Converter.
23. ábra: A prizmás fényfelbontás
A prizma anyagát a vizsgált színképtartomány szabja meg. Mivel az AAS módszernél az elemző vonalak
hullámhossza 190-860 nm tartományba esik, elsősorban kvarcból készült prizmát alkalmazunk. A prizmák nagy
erénye, hogy a felbontott színképnek nagy a fényereje. Ez azért van, mert az optikai rácsokkal ellentétben a
fényenergia nem oszlik meg a különböző színkép-rendek között. A prizmás fényfelbontás hátránya viszont,
hogy a fényfelbontó képesség, azaz a diszperzió mértéke erősen függ a hullámhossztól. Egy kvarcprizma esetén,
például, két színképvonal 200 nm körül, azaz az ultraibolya alsó tartományaiban különül el a legnagyobb
mértékben egymástól. A hullámhossz növekedésével az azonos Δλ távolságú két vonal a színképben egyre
közelebb kerül egymáshoz.
Az optikai rács a ma legmodernebbnek számító fényfelbontó egység. Eredeti formájában annak idején
üveglapra párhuzamos barázdákat karcoltak. A barázdált lapon áteső fény a fényinterferencia jelensége miatt
színképére bomlott, a különböző hullámhosszúságú fénykomponensek különböző irányban haladtak tovább. A
ma használatos optikai rácsok reflexiós rácsok. A nagy fényfelbontás érdekében nagyszámú (1000-4000
vonal/mm) párhuzamos barázdát alakítanak ki egy nagy reflexiójú tükröző felületen, amit vagy fémből
készítenek, vagy más anyag (üveg, kvarc) felületén fém-rápárlást alkalmaznak.

Az atomabszorpciós spektrométerek
felépítése, működése
30 Created by XMLmind XSL-FO Converter.
24. ábra: Fényfelbontás optikai ráccsal
A 24. ábrán látható módon az erre a felületre eső összetett fény úgy bomlik fel, hogy a visszaverődő fény
különböző komponensei különböző irányban, eltérő szögben verődnek vissza. A fényfelbontás a
fényinterferencia jelenségén alapszik. Az adott hullámhosszúságú fény a barázdák okozta útkülönbség miatt
adott reflexiós szögben erősítik, más szögben kioltják egymást. A 24. ábrán az is látható, hogy a barázdák
nagyszámuk ellenére szabályos profillal rendelkeznek. A barázdák lejtésszöge az úgynevezett csillogási szög,
amely megszabja, hogy az adott rácsnak mely színképtartományban a legnagyobb a fényereje. Ma ezeket a
pontos szögállású rácsokat különleges mikrotechnikával, kristálynövesztéssel alakítják ki.
Az optikai rácsok fényfelbontása, diszperziója a prizmákkal ellentétben a hullámhossz függvényében
egyenletes. A fényinterferencia törvényéből adódóan a rácson adott irányban nemcsak egy hullámhosszúságú
fény, hanem annak n·λ szerint az egészszámú többszörösei is megjelennek. Optikai rács esetén ezek a
többszörösök alkotják a különböző színképrendeket. Az egyre növekvő sorszámú színképrendek egyre kisebb
fényerejűek, de egyre nagyobb fényfelbontást biztosítanak. Ahhoz, hogy az n·λ összetett fényből a megfelelő
hullámhosszúságú színképvonalat kiválaszthassuk, egy nagy áteresztőképességű festékszűrőt alkalmazhatunk
(25. ábra).
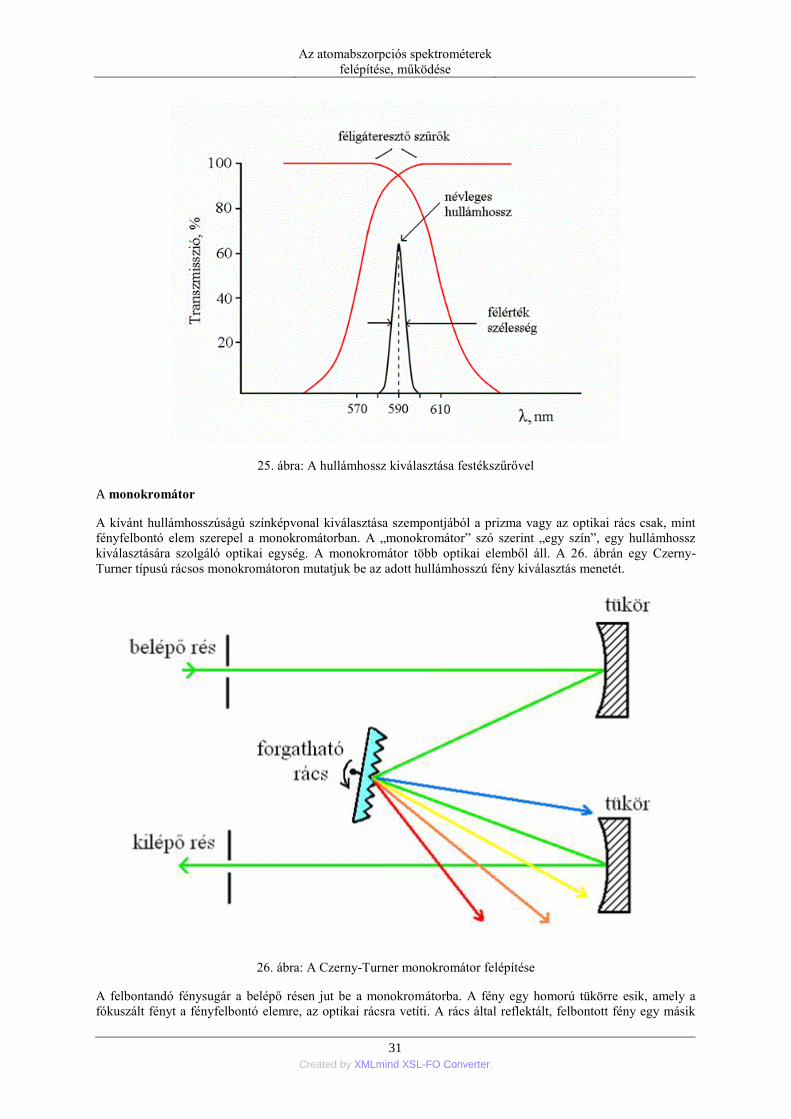
Az atomabszorpciós spektrométerek
felépítése, működése
31 Created by XMLmind XSL-FO Converter.
25. ábra: A hullámhossz kiválasztása festékszűrővel
A monokromátor
A kívánt hullámhosszúságú színképvonal kiválasztása szempontjából a prizma vagy az optikai rács csak, mint
fényfelbontó elem szerepel a monokromátorban. A „monokromátor” szó szerint „egy szín”, egy hullámhossz
kiválasztására szolgáló optikai egység. A monokromátor több optikai elemből áll. A 26. ábrán egy Czerny-
Turner típusú rácsos monokromátoron mutatjuk be az adott hullámhosszú fény kiválasztás menetét.
26. ábra: A Czerny-Turner monokromátor felépítése
A felbontandó fénysugár a belépő résen jut be a monokromátorba. A fény egy homorú tükörre esik, amely a
fókuszált fényt a fényfelbontó elemre, az optikai rácsra vetíti. A rács által reflektált, felbontott fény egy másik

Az atomabszorpciós spektrométerek
felépítése, működése
32 Created by XMLmind XSL-FO Converter.
homorú tükörre jut, amelyikről az ugyancsak fókuszált fény a kilépő résre, onnan pedig a detektorra kerül. Az
optikai rács adott pozíciója esetén a felbontott fénynek csak az a hullámhosszúságú eleme jut a kilépő résre,
illetve a detektorra, amelyik éppen a kilépő réssel szemben álló második tükör középpontjára esik. Ahhoz hogy
bármely elem elemző vonalát ki lehessen választani, az optikai rácsot tengelye körül mozgatjuk. Így a rács a
szögállásától függően más-más hullámhosszú vonalat vetít a tükörre, azon keresztül a kilépő résre.
Az atomabszorpciós készülékekben használt monokromátortól nem kívánunk meg nagy felbontást. Az
üregkatód lámpák fénye ugyanis vonalas szerkezetű. A lámpa által emittált egymástól jól elkülönülő
színképvonalak félértékszélessége 1-3 pm (pikométer), az abszorpciós vonalaké pedig 2-5 pm.
Bár az AAS készülék nem kíván különleges monokromátort, de a készülék teljesítőképességét jelentősen
befolyásolja a monokromátor belépő és kilépő résének a szélessége. A belépő rést (20-50 μm) általában a gyár
előre beállítja. Azt a felhasználó általában nem tudja szabályozni. A kilépő rést viszont a vizsgált elem, illetve
üregkatód lámpa optikai tulajdonságaitól függően a mérést végző analitikus célszerűen változtathatja. Abban az
esetben ugyanis, amikor az elemző vonal közelében más vonalak jelentkeznek az üregkatód lámpa
spektrumában, akkor a kilépő rést olyan szűkre célszerű választani, hogy csak az elemző vonal jusson át a résen
a detektorra. Ha viszont az elemző vonal 1-2 nm környezetében nincs más vonal, akkor célszerű nagyra
választani a kilépő rést, mert így a detektorra jutó fényenergia nagyobb, a készülék fényerősebb, kisebb
elektromos erősítésre van szükség, azaz a jel/zaj viszony kedvezőbb.
1.7. Szimultán készülékek
Az atomabszorpciós spektrometriás módszer jellegzetes vonása, hogy alap-összeállításban egyszerre csak egy
elem koncentrációja határozható meg a készülékkel. A műszer monokromátorával egyszerre ugyanis csak egy
hullámhossz választható ki. Számos analitikai laboratóriumban igény van arra, hogy egyszerre több elemet
tudjon meghatározni ugyanabból a mintából. Ez az igény hívta életre a több elem egyidejű elemzésére alkalmas
készülék-megoldásokat. E készülékek közös elve, hogy több (4-8) lámpa fényét tükrök segítségével közös
nyalábként vezetjük át az atomizáló téren (lángon, grafitkemencén), majd prizmával vagy optikai ráccsal a fényt
felbontjuk. Az így kapott színképből kiválasztjuk az elemek elemző vonalait, amelyek után annyi fényérzékelő
detektort helyezünk el, ahány elemet egyidejűleg meghatározni akarunk. A szimultán működésű készülék
elrendezését a 18. ábrán mutattuk be.
A szimultán AAS készülékek egyidejűleg vizsgált elemcsoportjait az adott laboratórium igényei határozzák meg.
Azzal viszont számolni kell, hogy az atomizálás körülményeit nem lehet egyidejűleg optimálni minden elemre.
Kompromisszumos lángparamétereket illetve a grafitkemencénél kompromisszumos fűtési programot kell
alkalmazni, valamint kerülni kell az igen eltérő termikus és kémiai sajátságú elemek egyidejű mérését.
A szimultán AAS elemzés külön fejezetét jelenti az utóbbi években bevezetett folytonos fényforrással működő
AAS módszer, amelyről részletesebben a 9. fejezetben teszek említést.
1.8. Háttérkorrekciós módszerek
Az atomabszorpciós spektrometriában igen jelentős zavaró hatást okoz az a jelenség, hogy a megvilágító
fényforrás intenzitásának csökkenését nem csak az atomgőzök okozta fényelnyelés, hanem a fényútba kerülő
szilárd részecskéken történő fényszóródás is okozza. A fényszóródás akkor lép fel fokozott mértékben, amikor
nagy sótartalmú, vagy nagy szerves anyag tartalmú mintát lángban, de még inkább ha grafitkemencében
atomizáljuk. Ekkor ugyanis az atomizálás során a nagy koncentrációban jelen levő kísérőanyagnak hosszabb
tartózkodási időre lenne szükség ahhoz, hogy atomos állapotba kerüljön. A fényútban száraz aeroszolként
(füstként) jelenlevő matrix igen jelentős fényintenzitás csökkenést eredményezhet. Ez a fénygyengülés
hozzáadódik az atomok okozta fényelnyeléshez, ami egy teljesen torz abszorbancia-változást eredményez, mert
ekkor a mért abszorbancia nem a vizsgált elem koncentrációjával lesz arányos.
Az előbbi fényszóródás okozta optikai zavaró hatás kiküszöbölését háttérkorrekciós módszernek nevezzük.
A háttérkorrekció céljából eddig négy módszert alkalmaztak:
1. Kétvonalas módszer
2. Deutérium lámpás háttérkorrekció
3. Zeeman háttérkorrekció
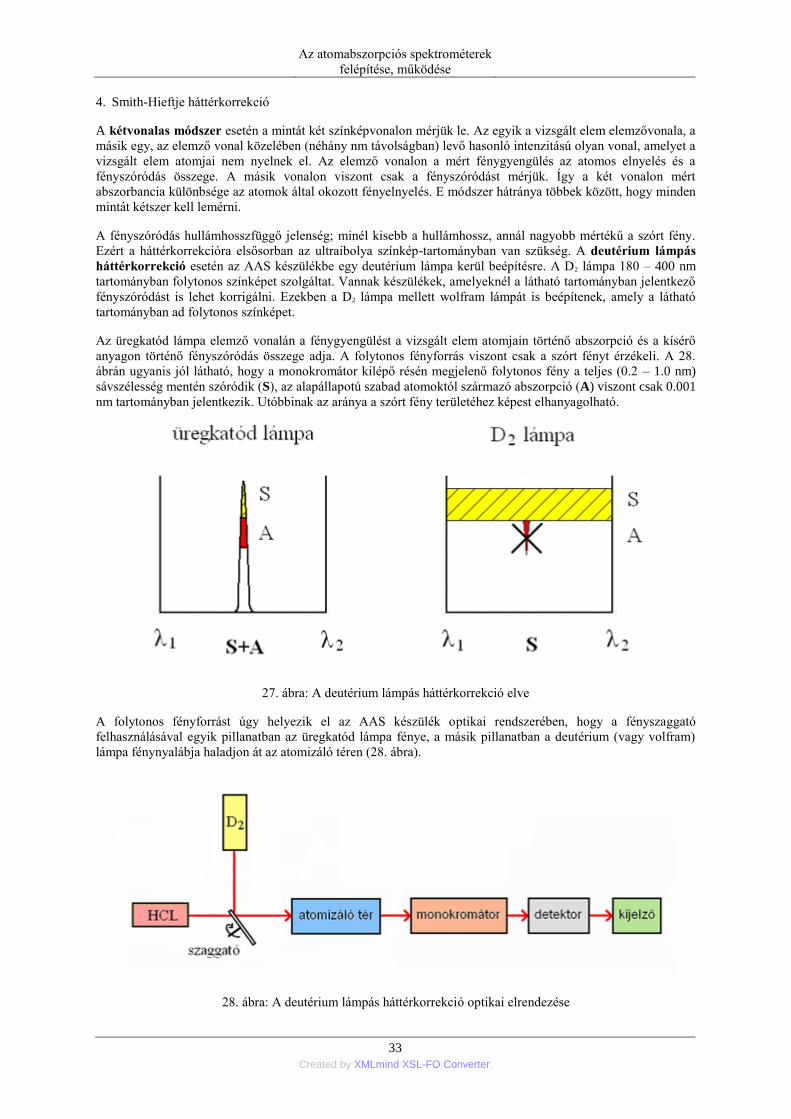
Az atomabszorpciós spektrométerek
felépítése, működése
33 Created by XMLmind XSL-FO Converter.
4. Smith-Hieftje háttérkorrekció
A kétvonalas módszer esetén a mintát két színképvonalon mérjük le. Az egyik a vizsgált elem elemzővonala, a
másik egy, az elemző vonal közelében (néhány nm távolságban) levő hasonló intenzitású olyan vonal, amelyet a
vizsgált elem atomjai nem nyelnek el. Az elemző vonalon a mért fénygyengülés az atomos elnyelés és a
fényszóródás összege. A másik vonalon viszont csak a fényszóródást mérjük. Így a két vonalon mért
abszorbancia különbsége az atomok által okozott fényelnyelés. E módszer hátránya többek között, hogy minden
mintát kétszer kell lemérni.
A fényszóródás hullámhosszfüggő jelenség; minél kisebb a hullámhossz, annál nagyobb mértékű a szórt fény.
Ezért a háttérkorrekcióra elsősorban az ultraibolya színkép-tartományban van szükség. A deutérium lámpás
háttérkorrekció esetén az AAS készülékbe egy deutérium lámpa kerül beépítésre. A D2 lámpa 180 – 400 nm
tartományban folytonos színképet szolgáltat. Vannak készülékek, amelyeknél a látható tartományban jelentkező
fényszóródást is lehet korrigálni. Ezekben a D2 lámpa mellett wolfram lámpát is beépítenek, amely a látható
tartományban ad folytonos színképet.
Az üregkatód lámpa elemző vonalán a fénygyengülést a vizsgált elem atomjain történő abszorpció és a kísérő
anyagon történő fényszóródás összege adja. A folytonos fényforrás viszont csak a szórt fényt érzékeli. A 28.
ábrán ugyanis jól látható, hogy a monokromátor kilépő résén megjelenő folytonos fény a teljes (0.2 – 1.0 nm)
sávszélesség mentén szóródik (S), az alapállapotú szabad atomoktól származó abszorpció (A) viszont csak 0.001
nm tartományban jelentkezik. Utóbbinak az aránya a szórt fény területéhez képest elhanyagolható.
27. ábra: A deutérium lámpás háttérkorrekció elve
A folytonos fényforrást úgy helyezik el az AAS készülék optikai rendszerében, hogy a fényszaggató
felhasználásával egyik pillanatban az üregkatód lámpa fénye, a másik pillanatban a deutérium (vagy volfram)
lámpa fénynyalábja haladjon át az atomizáló téren (28. ábra).
28. ábra: A deutérium lámpás háttérkorrekció optikai elrendezése

Az atomabszorpciós spektrométerek
felépítése, működése
34 Created by XMLmind XSL-FO Converter.
Az elektronika a két egymás után keletkező abszorpciós jelet egymásból kivonja. Így a készülék közvetlenül a
korrigált, a szórt fénytől elválasztott abszorbancia értéket jelzi ki. Ahhoz azonban, hogy a háttérkorrekciónak ez
a módja pontos legyen, két feltételnek kell teljesülnie. A folytonos fényforrást úgy kell beállítani, hogy annak a
fénynyalábja az üregkatód lámpáéval pontosan azonos tengelyben és azonos átmérőjű nyalábként haladjon át az
atomizáló téren. Csak így biztosított, hogy a szórt fényt ugyanazokban a térelemekben korrigáljuk, ahol az
üregkatód lámpa fénye áthaladt. A másik igen fontos feltétel, hogy a folytonos fényforrás intenzitása tökéletesen
egyező legyen az üregkatód lámpa elemző vonalának intenzitásával. Ezt a deutérium lámpa szabályzott
fénygyengítésével és elektronikus szabályzással oldják meg. Többek között ezeket a nehézségeket küszöböli a
ma legmodernebbnek számító Zeeman háttérkorrekciós módszer
A Zeeman háttérkorrekciós módszer a Zeeman effektusnak nevezett jelenségen alapszik, mely szerint mágneses
térben a színképvonalak az eredeti vonal helyéhez képest szimmetrikusan felhasadnak. Ezt felhasználva,
váltóárammal működő elektromágneses térben, amikor a térerő nulla, az elemző vonalon egyidejűleg mérjük az
atomabszorpciót és a fényszóródást, amikor pedig hat a mágneses tér, az elemző vonal mellett csak a szórt fény
kerül mérésre.
A váltóáramú mágnes frekvenciája 120 Hz, a mágneses térerő 10 kG nagyságrendű. A 29. ábra szerint, amikor a
mágneses térerő nem hat, azaz a mágneses kvantumszám nulla (Mj=0), akkor például a Cd 228.80 nm
hullámhosszúságú vonala, amely 5s–5p átmenetnek felel meg, egy színképvonalat ad. A mágneses erőtér
hatására az úgynevezett normális Zeeman effektus szerint ez az 5p szint három energiaszintre: ΔMj = ─1, 0, és
+1 szintekre hasad (29. ábra).
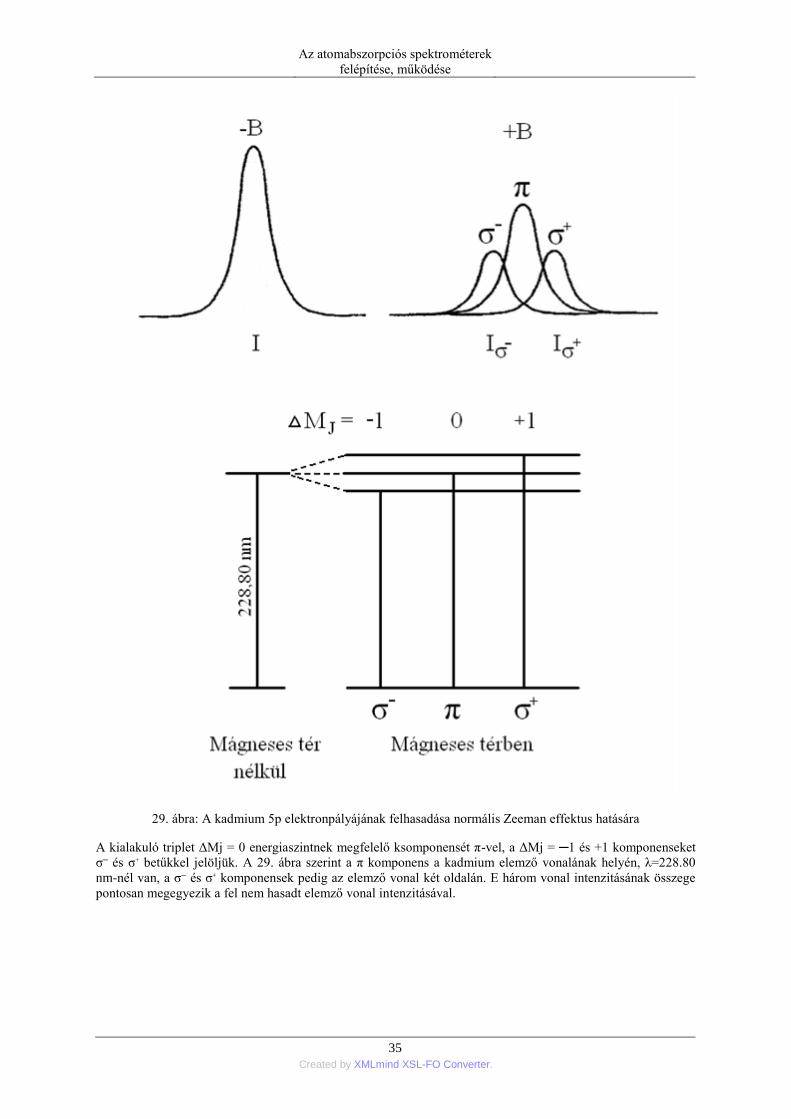
Az atomabszorpciós spektrométerek
felépítése, működése
35 Created by XMLmind XSL-FO Converter.
29. ábra: A kadmium 5p elektronpályájának felhasadása normális Zeeman effektus hatására
A kialakuló triplet ΔMj = 0 energiaszintnek megfelelő ksomponensét π-vel, a ΔMj = ─1 és +1 komponenseket
σ─ és σ+ betűkkel jelöljük. A 29. ábra szerint a π komponens a kadmium elemző vonalának helyén, λ=228.80
nm-nél van, a σ─ és σ+ komponensek pedig az elemző vonal két oldalán. E három vonal intenzitásának összege
pontosan megegyezik a fel nem hasadt elemző vonal intenzitásával.

Az atomabszorpciós spektrométerek
felépítése, működése
36 Created by XMLmind XSL-FO Converter.
30. ábra: A kadmium 228,80 nm-es elemző vonalának felhasadása normális Zeeman effektus hatására A
keresztirányú (transzverzális), B hosszirányú (longitudinális) mágneses térben
A forgó mágneses teret el lehet helyezni a fényforrás, azaz az üregkatód lámpa köré. Ezt a megoldást illusztrálja
a 30. ábra. A gyakorlatban azonban nem a fényforrás, hanem az atomizáló tér, elsősorban a grafitkemence köré
helyezik a mágneses teret. Így nem az emissziós, hanem az abszorpciós vonal hasad fel. Ezt a technikai
megoldást fordított Zemann effektusnak nevezzük. Az atomizáló tér körül a megvilágító fénynyaláb irányához
viszonyítva a mágneses tér iránya lehet merőleges (keresztirányú, transzverzális) vagy azzal párhuzamos
(longitudinális). A longitudinális Zeeman rendszer előnye, hogy ebben az esetben a π-komponens hiányzik a
felhasadt spektrumból (30.B ábra). A transzverzális üzemmód esetén (30.A ábra) úgynevezett polarizátorra van
szükség, hogy a π-komponenst eltávolíthassuk a színképből, ami fénygyengüléssel jár.
A Zeeman háttérkorrekció jelenti a legtökéletesebb módját annak, hogy a ténylegesen atomabszorpció okozta
fényintenzitás-csökkenést elválaszthassuk a fényszóródásból és a molekulák fényelnyeléséből származó
fénygyengüléstől. A tökéletes háttérkorrekció egyik oka, hogy a fénynyaláb a korrekció alatt az atomizáló
egységnek pontosan ugyanazon a térelemein halad át. A korrekció az elemző vonal mindkét oldalán történik.
Ezért ha az elemző vonal két oldalán különböző a háttér, ezzel a módszerrel ezt is korrekcióba lehet venni.
Előbbiek folytán a Zeeman háttérkorrekcióval rendelkező atomabszorpciós készülékek a hasznos jelnél két
nagyságrenddel nagyobb háttérjelet is korrigálni tudnak. Az ilyen háttérkorrekcióval rendelkező készüléknek,
amelyet gyakran ZAAS-nek rövidítenek az ára másfélszerese a hagyományos AAS készülékekének.
A Smith-Hieftje háttérkorrekciós módszer az emissziós színképelemzésben jól ismert önabszorpció, más
néven vonal-visszafordulás jelenségét használja fel. Az önabszorpció jelensége olyankor lép fel, amikor egy
nagyhőmérsékletű tér belső tartományaiban egy elemnek a gerjesztett atomjai, a külső köpenyében pedig
ugyanannak az elemnek az alapállapotú atomjai vannak jelen. Ilyenkor a belső térből kisugárzott fényt részben
vagy teljes egészében elnyelik a külső térben levő alapállapotú atomok. Ez a magyarázata a Nap színképében
látható Fraunhofer-vonalaknak is. Ugyanez a jelenség lép fel az elektromos ívben. A gerjesztett atomok ugyanis
az ív legbelső zónáiban keletkeznek. A külső hidegebb zónákban viszont alapállapotú atomok vannak jelen.
Ezért az elemek kis gerjesztési energiájú vonalait mennyiségi színképelemzésre nem célszerű használni, mert a
koncentráció növekedésével, nagyobb koncentrációknál az elemző vonal intenzitása nemcsak nem növekszik,
hanem még csökken is. Ezért nevezik a jelenséget vonal-visszafordulásnak. A 31. ábrán mutatjuk be, hogy a
koncentráció növekedésével hogyan szélesedik ki egy színképvonal, és hogyan változik a vonal intenzitása az
elemző vonal maximumának megfelelő hullámhosszon.
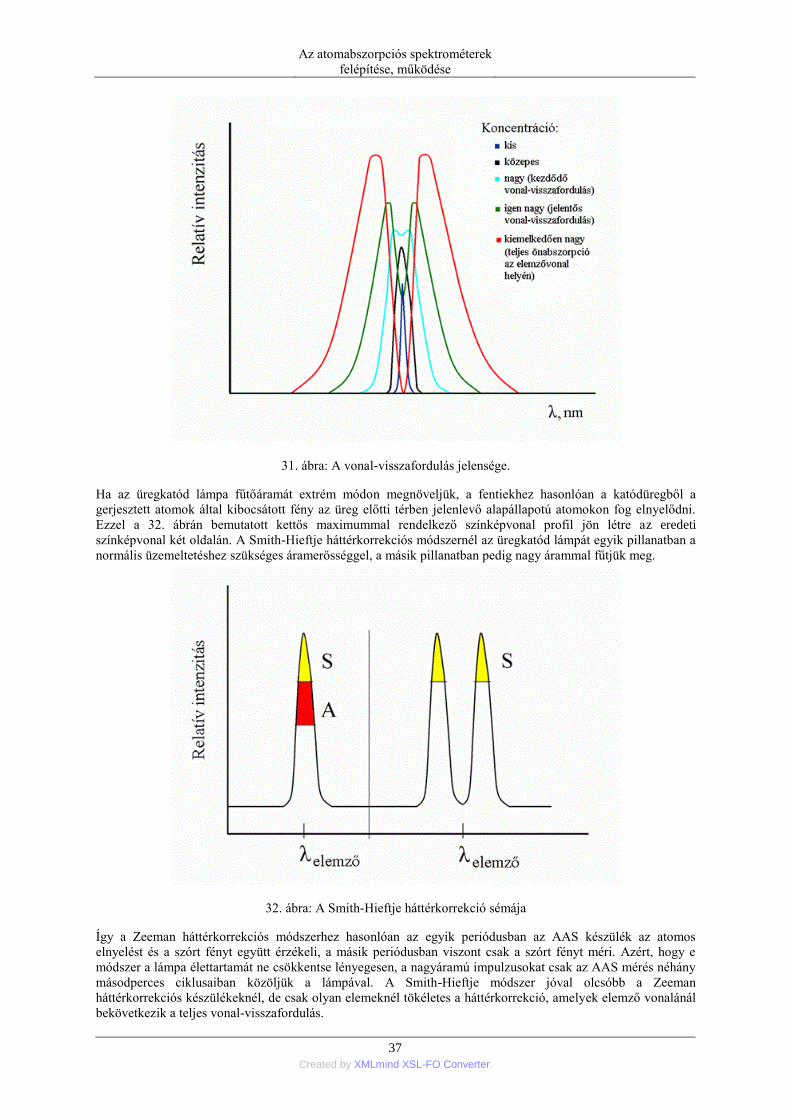
Az atomabszorpciós spektrométerek
felépítése, működése
37 Created by XMLmind XSL-FO Converter.
31. ábra: A vonal-visszafordulás jelensége.
Ha az üregkatód lámpa fűtőáramát extrém módon megnöveljük, a fentiekhez hasonlóan a katódüregből a
gerjesztett atomok által kibocsátott fény az üreg előtti térben jelenlevő alapállapotú atomokon fog elnyelődni.
Ezzel a 32. ábrán bemutatott kettős maximummal rendelkező színképvonal profil jön létre az eredeti
színképvonal két oldalán. A Smith-Hieftje háttérkorrekciós módszernél az üregkatód lámpát egyik pillanatban a
normális üzemeltetéshez szükséges áramerősséggel, a másik pillanatban pedig nagy árammal fűtjük meg.
32. ábra: A Smith-Hieftje háttérkorrekció sémája
Így a Zeeman háttérkorrekciós módszerhez hasonlóan az egyik periódusban az AAS készülék az atomos
elnyelést és a szórt fényt együtt érzékeli, a másik periódusban viszont csak a szórt fényt méri. Azért, hogy e
módszer a lámpa élettartamát ne csökkentse lényegesen, a nagyáramú impulzusokat csak az AAS mérés néhány
másodperces ciklusaiban közöljük a lámpával. A Smith-Hieftje módszer jóval olcsóbb a Zeeman
háttérkorrekciós készülékeknél, de csak olyan elemeknél tökéletes a háttérkorrekció, amelyek elemző vonalánál
bekövetkezik a teljes vonal-visszafordulás.

Az atomabszorpciós spektrométerek
felépítése, működése
38 Created by XMLmind XSL-FO Converter.
2. A készülékek elektronikus egységei
2.1. Lámpa tápegység
Az üregkatód lámpák stabil működéséhez stabilizált egyenáramú tápegységre van szükség. Különösen fontos a
stabil működés az egysugaras AAS készülékeknél, ahol a megbízható elemzés nagyban függ a megvilágító
fénynyaláb hosszúidejű stabilitásától. A tápegység általában 300 volt feszültséget és 1 – 50 mA áramerősséget
biztosít a lámpák működéséhez. Az újabb gyártmányú AAS készülékek a fény szaggatását nem mechanikusan,
forgó szaggatóval (chopperrel) oldják meg, hanem elektronikusan, a lámpaáramnak általában 300 Hz
frekvenciával történő modulálásával. Ezt a feladatot is a lámpa tápegysége oldja meg.
Az elektród nélküli kisülési lámpák működtetéséhez külön nagyfrekvenciás tápegységre van szükség.
Tápegységként ilyen célra az orvosi diatermiás készülékekhez hasonló források bizonyultak alkalmasnak,
amelyek 27 MHz körüli frekvencián 200 W teljesítményt tudnak leadni. Az EDL lámpáknak megfelelő illesztő
rendszerrel kell rendelkezniük, hogy a teljesítmény bármelyik EDL lámpára kicsatolható legyen.
2.2. Detektorok
A műszeres analitika optikai módszereinél a detektor feladata, hogy a fényenergiát elektromos energiává
alakítsa. Mivel az atomabszorpciós spektrometriában az alkalmazott elemző vonal ugyan nagy intenzitású, de a
szélessége 0.001 nm nagyságrendű, fényenergiája viszonylag kicsi. Ezért a molekulaspektroszkópiában,
ultraibolya/látható (UV/VIS) spektrofotometriában alkalmazott fotocellák atomabszorpciós vonalak
detektálására nem alkalmasak. Az AAS módszer kialakulásától kezdve a jellemző detektor a fotoelektron-
sokszorozó (PMT=photo multiplier tube) (33. ábra). A fotoelektron-sokszorozó egy vákuumozott zárt kvarccső,
amelyben egy fotokatód, egy anód és köztük változó számú (7-15) úgynevezett dinód helyezkedik el. Az
egymás utáni dinódokra a katódhoz képest egyre pozitívabb feszültséget kapcsolnak. A legpozitívabb elektród
az anód. A fotoelektron-sokszorozóval szemben a követelmény, hogy széles színkép-tartományban (az arzén
193,7 nm-es vonalától a cézium 852,1 nm-es vonaláig) képes legyen a fényenergiát jól kezelhető elektromos
jellé alakítani. Az elektronsokszorozó spektrális érzékenysége elsősorban a katódbevonat fényérzékeny
anyagától függ. A katódbevonat alkálifémek antimonnal, arzénnel, galliummal és/vagy ezüsttel alkotott változó
arányú ötvözetei, amelyeket ha fény éri, elektronok lépnek ki belőlük.
33. ábra: A fotoelektron sokszorozó vázlata
A fény hatására a katódból kilépő elektronok a katódnál pozitívabb legközelebbi dinódba ütköznek. Ez az
ütközés olyan sebességű, hogy a dinódból 2-5-szörös mennyiségű szekunder elektron lép ki. Ezek az elektronok
a következő még pozitívabb dinód felé száguldanak, amelyből tovább többszörözött elektronkilépés történik. Ez
így folytatódik az anódig. A korszerű elektronsokszorozókkal így általában 108 és 109 nagyságrendű az erősítés,
kivételesen azonban még 1012 nagyságrendű erősítés is elérhető.
Az anódról az elektronokat egy nagy ellenálláson keresztül vezetjük a földbe. Ezáltal a nagy ellenállás két
pontja között jelentős feszültség mérhető. A fotoelektron sokszorozó erősítési tényezőjét a dinódok
feszültségének változtatásával befolyásolhatjuk.

Az atomabszorpciós spektrométerek
felépítése, működése
39 Created by XMLmind XSL-FO Converter.
34. ábra A fény átalakítása (Uki) feszültségjellé fotoelektron sokszorozóval
A legújabb kereskedelmi AAS készülékekben a fotoelektron sokszorozó helyett úgynevezett szilárdtest
detektorokat alkalmaznak a fény elektromos jellé átalakítása céljából. A szilárdtest detektorok kifejlesztését a
sok komponens egyidejű (szimultán) elemzésére alkalmas optikai műszerek gyártásának igénye ösztönözte.
Ilyenek például a gyors kémiai reakciók követésére alkalmas spektrofotométerek (diódasoros
spektrofotométerek), vagy a szimultán elemanalízisre alkalmas induktív csatolású plazma optikai emissziós
spektrométerek (ICP-OES). A szilárdtest detektorok fontos erénye, hogy igen kis, akár 10─4 mm2 (10 μm oldalú
négyzet) méretben is előállíthatók. Ez a méret biztosítja, hogy a fényérzékelő diódákat diódasorokká és
kétdimenziós felületekké alakítsuk, amely lehetővé teszi, hogy a teljes atomszínképet vagy molekulaspektrumot
nagy felbontásban egyidejűleg érzékelhessünk. Utóbbi elrendezések elnevezése a fotodiódasor (photodiode
array – PDA), töltés csatolt detektorok (charge-coupled device – CCD), illetve töltés injektálásos detektorok
(charge-injection device – CID). A kétdimenziós CCD rendszerről a 9. fejezetben a nagyfelbontású folytonos
fényforrással működő AAS (HR-CS-AAS) készüléknél teszünk említést.
A hagyományos egyelemes (szekvens) AAS készülékekben a fotoelektron sokszorozóhoz hasonlóan egy
szilárdtest detektort alkalmaznak. A szilárdtest detektorok fő alkotója a kristályos szilícium félvezető. A
szilícium a IV. csoport eleme, így négy vegyérték-elektronja van. A szilícium kristályban ez a négy elektron
további négy szilícium atom elektronjaival kombinálódva négy kovalens kötést hoz létre. Szobahőmérsékleten
elegendő hőmozgás hatására ebben a szerkezetben időnként felszabaduló elektron a kötött állapotból, szabadon
mozog a kristályban. Az elektron termikus gerjesztése visszahagy egy pozitív töltésű helyet, amit lyuknak
nevezünk, és az elektronhoz hasonlóan mozog. A lyuk mozgásának a mechanizmusa lépésszerű. Egy kötött
elektron a szomszédos szilícium atomtól beugrik az elektronhiányos helybe (a lyukba). Ezáltal létrehoz egy
másik pozitív lyukat az eredeti helyén. Így a vezetés a félvezetőben az elektronok és a lyukak ellentétes irányú
mozgása.
A szilícium vezetőképességét jelentős mértékben növelhetjük, ha adalékoljuk („szennyezzük”), azaz ellenőrzött,
kis mennyiségű (kb. 1 μg/g) V. vagy III. főcsoportbeli elemet oszlatunk el homogénen a kristályban. Ha például
a kristályt V. csoportbeli arzénnel szennyezzük, akkor annak öt elektronjából négy létesít kovalens kötést, és az
ötödik szabadon marad az áramvezetés számára (35A. ábra). Ha a szilíciumot a III. csoportbeli galliummal
adalékoljuk, annak csak 3 vegyérték elektronja van és létesít egy lyukat, ami ugyancsak növeli a
vezetőképességet (35B. ábra). Az a félvezető, amely nem kötött elektronokat tartalmaz, n-típusú félvezető,
amelyik fölöslegben tartalmaz lyukakat (pozitív töltéseket), az a p-típusú félvezető. Az n-típusúban az elektron,
a p-típusúban a lyuk a töltéshordozó.
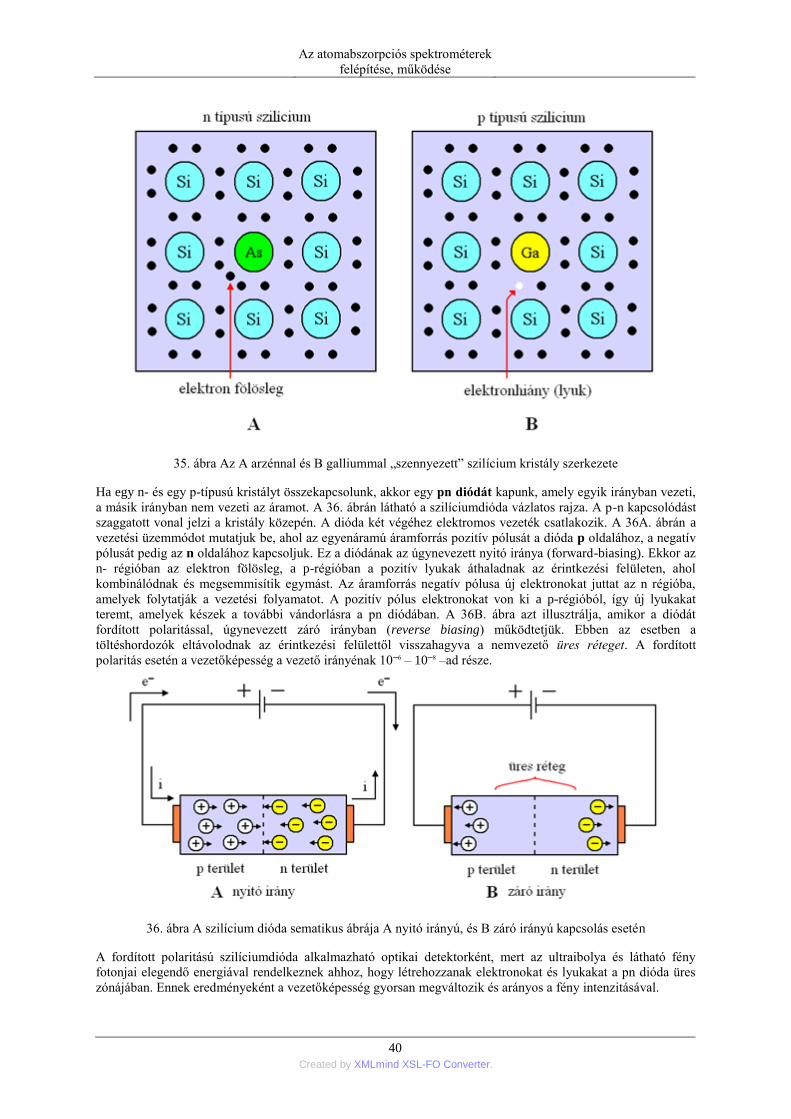
Az atomabszorpciós spektrométerek
felépítése, működése
40 Created by XMLmind XSL-FO Converter.
35. ábra Az A arzénnal és B galliummal „szennyezett” szilícium kristály szerkezete
Ha egy n- és egy p-típusú kristályt összekapcsolunk, akkor egy pn diódát kapunk, amely egyik irányban vezeti,
a másik irányban nem vezeti az áramot. A 36. ábrán látható a szilíciumdióda vázlatos rajza. A p-n kapcsolódást
szaggatott vonal jelzi a kristály közepén. A dióda két végéhez elektromos vezeték csatlakozik. A 36A. ábrán a
vezetési üzemmódot mutatjuk be, ahol az egyenáramú áramforrás pozitív pólusát a dióda p oldalához, a negatív
pólusát pedig az n oldalához kapcsoljuk. Ez a diódának az úgynevezett nyitó iránya (forward-biasing). Ekkor az
n- régióban az elektron fölösleg, a p-régióban a pozitív lyukak áthaladnak az érintkezési felületen, ahol
kombinálódnak és megsemmisítik egymást. Az áramforrás negatív pólusa új elektronokat juttat az n régióba,
amelyek folytatják a vezetési folyamatot. A pozitív pólus elektronokat von ki a p-régióból, így új lyukakat
teremt, amelyek készek a további vándorlásra a pn diódában. A 36B. ábra azt illusztrálja, amikor a diódát
fordított polaritással, úgynevezett záró irányban (reverse biasing) működtetjük. Ebben az esetben a
töltéshordozók eltávolodnak az érintkezési felülettől visszahagyva a nemvezető üres réteget. A fordított
polaritás esetén a vezetőképesség a vezető irányénak 10─6 – 10─8 –ad része.
36. ábra A szilícium dióda sematikus ábrája A nyitó irányú, és B záró irányú kapcsolás esetén
A fordított polaritású szilíciumdióda alkalmazható optikai detektorként, mert az ultraibolya és látható fény
fotonjai elegendő energiával rendelkeznek ahhoz, hogy létrehozzanak elektronokat és lyukakat a pn dióda üres
zónájában. Ennek eredményeként a vezetőképesség gyorsan megváltozik és arányos a fény intenzitásával.
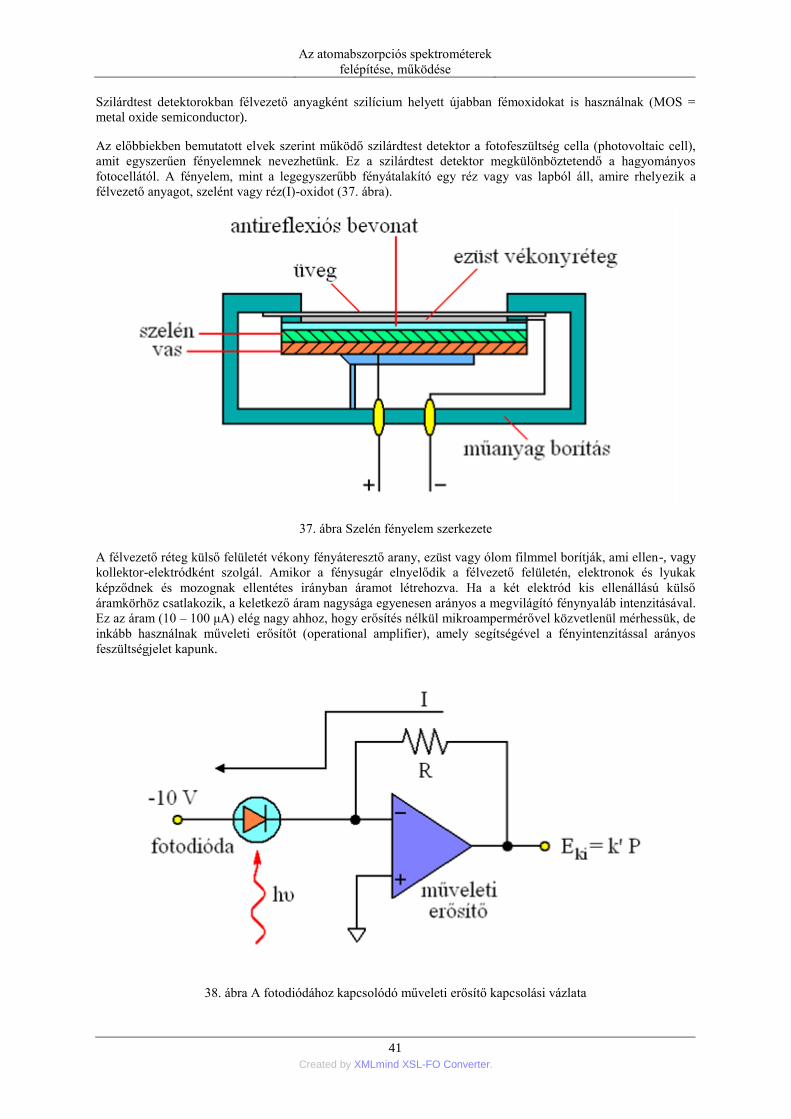
Az atomabszorpciós spektrométerek
felépítése, működése
41 Created by XMLmind XSL-FO Converter.
Szilárdtest detektorokban félvezető anyagként szilícium helyett újabban fémoxidokat is használnak (MOS =
metal oxide semiconductor).
Az előbbiekben bemutatott elvek szerint működő szilárdtest detektor a fotofeszültség cella (photovoltaic cell),
amit egyszerűen fényelemnek nevezhetünk. Ez a szilárdtest detektor megkülönböztetendő a hagyományos
fotocellától. A fényelem, mint a legegyszerűbb fényátalakító egy réz vagy vas lapból áll, amire rhelyezik a
félvezető anyagot, szelént vagy réz(I)-oxidot (37. ábra).
37. ábra Szelén fényelem szerkezete
A félvezető réteg külső felületét vékony fényáteresztő arany, ezüst vagy ólom filmmel borítják, ami ellen-, vagy
kollektor-elektródként szolgál. Amikor a fénysugár elnyelődik a félvezető felületén, elektronok és lyukak
képződnek és mozognak ellentétes irányban áramot létrehozva. Ha a két elektród kis ellenállású külső
áramkörhöz csatlakozik, a keletkező áram nagysága egyenesen arányos a megvilágító fénynyaláb intenzitásával.
Ez az áram (10 – 100 μA) elég nagy ahhoz, hogy erősítés nélkül mikroampermérővel közvetlenül mérhessük, de
inkább használnak műveleti erősítőt (operational amplifier), amely segítségével a fényintenzitással arányos
feszültségjelet kapunk.
38. ábra A fotodiódához kapcsolódó műveleti erősítő kapcsolási vázlata

Az atomabszorpciós spektrométerek
felépítése, működése
42 Created by XMLmind XSL-FO Converter.
A záró irányú (reverse-biased) fotodiódát fénnyel megvilágítva I áramerősség keletkezik. A műveleti erősítőnek
igen nagy a bemeneti ellenállása. Ezért a dióda árama az R ellenálláson tud áthaladni. A kimenő feszültség
könnyen számítható az Ohm törvényből. Eout = – IR. Mivel az áram a diódára eső P fényenergiával arányos, I =
kP, ahol a k állandó, és Eout = – IR = – kPR = Eout = – K’P . A műveleti erősítő kimenetéhez kapcsolt voltmérő
közvetlenül méri a fényenergiával arányos feszültséget.
A tipikus fényelem maximális érzékenysége 550 nm körül van. A válaszjel maximum 10%-kal csökken 350 és
750 nm-nél. A fotocella egy zavartűrő, olcsó detektor a látható fény tartományban, és előnye, hogy nem igényel
külső áramforrást. Érzékenyítő rétegekkel (például foszforral) bevonva az ultraibolya tartományban is
használhatók. Az újabb fejlesztésekre jellemző a növekvő érzékenység és az alacsony zajszint.
2.3. Erősítők. A jel/zaj viszony javításának elektronikus módszerei
Az AAS készülék elektronikája, mielőtt az abszorbancia értéket kijelezné, több fontos feladatot lát el. Erősíti a
jelet, a Lambert-Beer törvénynek megfelelően egy osztókör segítségével elosztja a fényabszorpció előtti
fényintenzitásnak megfelelő feszültségjelet az abszorpció utáni fényintenzitás feszültségjelével, majd e
hányados egy logaritmikus erősítőbe kerül, ahonnan megkapjuk az abszorbancia értékét. A fényforrás és az
optikai elrendezésekhez hasonlóan az elektronikának egyik fontos feladata, hogy a saját eszközeivel minél
kisebbre szorítsa a műszer zajszintjét, azaz minél nagyobb legyen a jel/zaj viszony.
Az elektronikus rendszerben ─ a detektort is beleértve ─ többféle zajtípus lép fel. Ilyen az 50 vagy 60 Hz-es
hálózati frekvencia („hálózati brumm”), a kisfrekvenciás áramingadozások, a termikus elektronok okozta
sötétáram és az elektron ütközések okozta „sörét zaj”, mint nagyobb frekvenciájú zajok. Ezek és még további
zajok csökkentésére, kiszűrésére két igen hatékony elektronikai megoldás született. Az egyik a hangolt erősítő, a
másik a szinkron vagy „lock-in” erősítő.
Hangolt erősítő
Az egyszerű elektronikus erősítő nem tesz különbséget a hasznos jel és az analitikai szempontból káros zaj
között. Ezért ha ilyen erősítőt használnánk, a jel és a zaj viszonya nem változna az erősítés után sem. Ahhoz
hogy a jel/zaj viszonyban javulás következzen be, olyan erősítőre van szükség, amely a hasznos jelet növeli, a
zajokat, vagy azok jelentős részét viszont nem erősíti. Ezt a feladatot végzi el a hangolt erősítő. A hangolt
erősítő két fontos eleme az alul- (39. A ábra) és a felül-áteresztő (39. B ábra) elektronikus szűrő.
39. ábra Alul- (A) és felüláteresztő (B) elektromos szűrő
Az 39.A ábrán bemutatott szűrő azért alul-áteresztő, mert nagy feszültséget kis frekvencia esetén kapunk. A C
kondenzátor jelenlétében a kimenő feszültség ugyanis annál kisebb, minél nagyobb az áthaladó áram
frekvenciája. A 39.B ábrán szereplő áramkör azért felül-áteresztő, mert nagy kilépő feszültséget nagy frekvencia
esetén kapunk. Az R ellenálláson ugyanis azért tud nagy feszültség esni, mert a C kondenzátoron a nagy
frekvenciás áram jut el az R ellenálláshoz. Ha ezt a két szűrőt egymással kombináljuk és egy váltóáramú
erősítőhöz kapcsoljuk a 40. ábrán látható hangolt erősítőhöz jutunk, amelynek az erősítése adott frekvencia-
tartományra korlátozódik. Sem az ennél nagyobb, sem az ennél kisebb frekvenciájú jeleket nem erősíti. Ez a
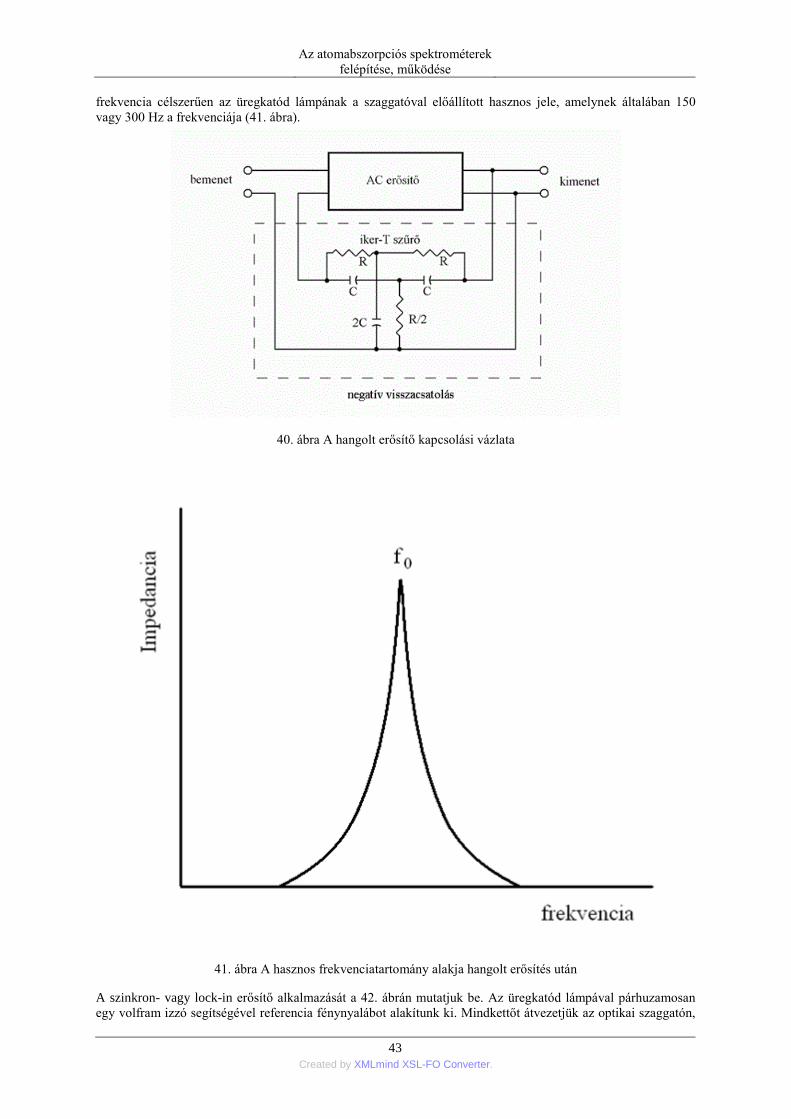
Az atomabszorpciós spektrométerek
felépítése, működése
43 Created by XMLmind XSL-FO Converter.
frekvencia célszerűen az üregkatód lámpának a szaggatóval előállított hasznos jele, amelynek általában 150
vagy 300 Hz a frekvenciája (41. ábra).
40. ábra A hangolt erősítő kapcsolási vázlata
41. ábra A hasznos frekvenciatartomány alakja hangolt erősítés után
A szinkron- vagy lock-in erősítő alkalmazását a 42. ábrán mutatjuk be. Az üregkatód lámpával párhuzamosan
egy volfram izzó segítségével referencia fénynyalábot alakítunk ki. Mindkettőt átvezetjük az optikai szaggatón,
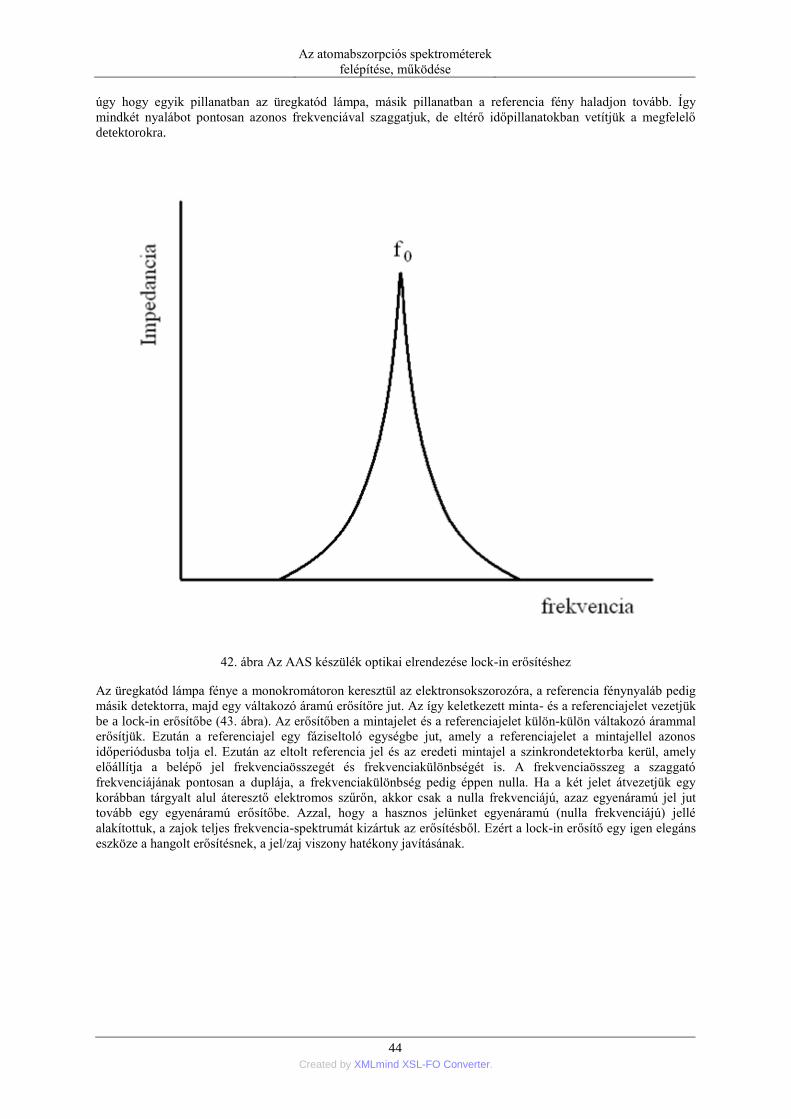
Az atomabszorpciós spektrométerek
felépítése, működése
44 Created by XMLmind XSL-FO Converter.
úgy hogy egyik pillanatban az üregkatód lámpa, másik pillanatban a referencia fény haladjon tovább. Így
mindkét nyalábot pontosan azonos frekvenciával szaggatjuk, de eltérő időpillanatokban vetítjük a megfelelő
detektorokra.
42. ábra Az AAS készülék optikai elrendezése lock-in erősítéshez
Az üregkatód lámpa fénye a monokromátoron keresztül az elektronsokszorozóra, a referencia fénynyaláb pedig
másik detektorra, majd egy váltakozó áramú erősítőre jut. Az így keletkezett minta- és a referenciajelet vezetjük
be a lock-in erősítőbe (43. ábra). Az erősítőben a mintajelet és a referenciajelet külön-külön váltakozó árammal
erősítjük. Ezután a referenciajel egy fáziseltoló egységbe jut, amely a referenciajelet a mintajellel azonos
időperiódusba tolja el. Ezután az eltolt referencia jel és az eredeti mintajel a szinkrondetektorba kerül, amely
előállítja a belépő jel frekvenciaösszegét és frekvenciakülönbségét is. A frekvenciaösszeg a szaggató
frekvenciájának pontosan a duplája, a frekvenciakülönbség pedig éppen nulla. Ha a két jelet átvezetjük egy
korábban tárgyalt alul áteresztő elektromos szűrőn, akkor csak a nulla frekvenciájú, azaz egyenáramú jel jut
tovább egy egyenáramú erősítőbe. Azzal, hogy a hasznos jelünket egyenáramú (nulla frekvenciájú) jellé
alakítottuk, a zajok teljes frekvencia-spektrumát kizártuk az erősítésből. Ezért a lock-in erősítő egy igen elegáns
eszköze a hangolt erősítésnek, a jel/zaj viszony hatékony javításának.

Az atomabszorpciós spektrométerek
felépítése, működése
45 Created by XMLmind XSL-FO Converter.
43. ábra A lock-in erősítő működési vázlata
3. Az adatok kijelzése, gyűjtése, a kijelző módszerek fejlődése
Az atomabszorpciós spektrométerek fejlődését – más műszeres módszerekhez hasonlóan – leglátványosabban az
adatok kijelzési módjának fejlődésén keresztül követhetjük. A készülékek első generációjára a galvanométeres,
analóg (mutatós) műszerrel történő kijelzés volt a jellemző. A lángatomabszorpciós módszer nagy elemzési
sebessége miatt nagy (1000-3000) mintaszámú sorozatok elemzésekor az adatok leolvasása tükörskálás mutatós
műszerrel igen fárasztó feladat.
Ezért nem sokkal ezután megjelentek először a dekatron csöves, majd a digitális kijelzésű AAS készülékek.
Utóbbi esetben az adatok leolvasása kényelmes, de a nagy szériaelemzés adatainak kiértékelése manuális
grafikus módszerrel továbbra is igen fárasztó feladat maradt volna. A kiértékelés megkönnyítésére több
elektronikus megoldást alkalmaztak a digitális kijelzésű készülékeknél. Ezek közül az egyik legfontosabb a
közvetlen koncentráció kijelzés megoldása. Ennek az a lényege, hogy a műszer úgy végez skálanyújtást,
megváltoztatva az abszorbancia-adatok numerikus értékét, hogy az alapvonalat, azaz az abszorbanciát A=0,000
értéken tartja. Ezáltal tetszés szerint változtatni tudjuk a kalibráló egyenes meredekségét. Ez lehetővé teszi, hogy
egy ismert koncentrációjú kalibráló oldat atomabszorpciós jelének számértékét addig változtatjuk, amíg a
számkijelzőn megjelenő számok éppen megegyeznek a kalibráló oldat koncentrációjával. E beállítást követően
egy ismeretlen koncentrációjú oldatra kapott digit-érték megfelel a koncentráció számértékének. Pontos
koncentrációértéket azonban csak abban a tartományban kapunk, amelyben a kalibráló oldatok abszorbancia
értékei egyenessel közelíthetők. Amennyiben a kalibráló tagok jelei a koncentrációval nem lineárisan változnak,
a közvetlen koncentráció kijelzéséhez előzetesen a kalibráló görbe megfelelő szakaszát elektronikusan
linearizálni kell. A kalibráló görbék tipikus alakja olyan, hogy a nagyobb koncentrációk esetén az abszcissza
felé hajlanak. Ezért az itt alkalmazott elektronikai egység úgy működik, hogy a kis jeleket kevésbé, a
nagyobbakat viszont nagyobb mértékben erősíti. Ennek az úgynevezett görbekorrekciós egység erősítési
tényezőjének az alkalmas beállításával a kalibráló görbe adott szakasza kiegyenesíthető. (A görbülés
mértékének inverz erősítését végzi az elektronika.) Így már az eredetileg görbülő kalibráló esetén is használható
a közvetlen koncentráció kijelzés.
A kijelző módszerek harmadik generációját az AAS készülékbe beépített célszámítógép jelentette. A számítógép
segít a műszerparaméterek beállításában, az adatok gyűjtésében és az eredményeknek a képernyőn történő
megjelenítésében. A számítógépes adatgyűjtés és feldolgozás lehetővé teszi, hogy a kalibráló görbe illesztése
objektív módon, polinomok segítségével történjék. Ezzel növelhető az kiértékelés objektivitása. Ugyancsak
fontos az adatok statisztikai értékelésének lehetősége. A mérésidőt minden adat mérése esetén pontosan be lehet
állítani. Ugyanígy az adott minta mérését tetszés szerint ismételni lehet. Az adat mellett ezért a képernyőn
mindig megjelenik annak szórása, illetve a relatív standard deviációja is. A képernyőn grafikusan is
megjeleníthető a kalibráló görbe és a pontok szórása. Így azonnal észrevehetők az esetleges mérési hibák, amit
újraméréssel gyorsan korrigálni lehet. A képernyőn grafikusan ugyancsak megjeleníthető az egyes mérések
során a jelalakok időbeli alakulása is, amelyek követésével szintén kiszűrhetők a mérési hibák. E harmadik
generációs készülékek könnyen felismerhetők az alapműszer előlapjába épített monitorról. Ez a célszámítógép
azonban csak a műszert szolgálja ki, azon egyéb feladatokat végezni nem lehet.

Az atomabszorpciós spektrométerek
felépítése, működése
46 Created by XMLmind XSL-FO Converter.
A negyedik generációs készülék a személyi számítógép (PC) vezérelt atomabszorpciós spektrométer. Ebben az
esetben a számítógép különálló egység, vezetékkel kapcsolható az alapkészülékhez. Ez a megoldás a korábbi
célszámítógép által elvégzett feladatokat már flexibilisebben, jelenleg a Windows programnyelv alkalmazásával
végzi el és jeleníti meg. A gép az adatgyűjtés közben párhuzamosan más feladatokra is használható.
4. A minta lángatomizációja
Az atomabszorpciós spektrometriás elemzés fő mozzanata a vizsgálandó elem alapállapotú szabad atomokká
alakítása. A rendelkezésre álló gáz, folyékony és szilárd mintákat ezért olyan térbe kell juttatni, ahol a kémiai
kötések felszakadnak, és a vizsgált elem alapállapotú atomgőzei keletkeznek. Ezt az átalakulást általában nagy
hőmérsékleten és megfelelő kémiai környezetben érhetjük el. Atomizáció céljára egyik legrégebben alkalmazott
közeg a láng. A lángot, mint nagyhőmérsékletű gázteret a lángfotometriában már régóta sikerrel alkalmazzák
atomok gerjesztésére. Ezért kézenfekvő volt, hogy az atomabszorpciós spektrometriában is ezt a rendszert
használták először az alapállapotú atomok előállítására. A láng alkalmazása mellett szól, hogy laboratóriumi
körülmények között viszonylag egyszerűen, olcsón nagyhőmérsékletű teret tudunk létrehozni. Ezért a láng
mindmáig az egyik legelterjedtebb atomizáló közeg az atomabszorpciós spektrometriában (lángatomabszorpciós
spektrometria FAAS = flame atomic absorption spectrometry).
Az alapállapotú atomok előállításának másik igen hatékony módszere a fűtött grafitcsőben végzett atomizálás.
Az utóbb elven működő módszer neve grafitkemencés atomabszorpciós spektrometria (GFAAS = graphite
furnace atomic absorption spectrometry). Több elem hatékony atomizálására elterjedt a hidrid-képzésen alapuló
kémiai eljárás. A reakcióelegyből redukcióval felszabaduló hidridet fűtött kvarccsőben hőbontással alakítjuk
alapállapotú atomokká (HG-AAS = hydrid generation atomic absorption spectrometry).
A nagy gőztenziójú higany meghatározásának speciális módja a hideggőz technika (CV-AAS = cold vapor
atomic absorption spectrometry).
4.1. A láng, mint nagyhőmérsékletű tér jellemzése
Adott gázeleggyel szabályzott gázáramok és hőálló, stabil mechanikai felépítésű égőfej esetén jól
reprodukálható hőmérsékletű és kémiai összetételű láng állítható elő. A szilárd és folyékony anyagok égetésével
keletkező lángokat elsősorban fűtési és technológiai célokra használják. Analitikai célra csakis gáz-gáz elegy
égésével előállított lángok jöhetnek szóba. Ahhoz ugyanis, hogy ugyanolyan hőmérsékletű, kémiai összetételű
lángot bármikor elő tudjunk állítani, az éghető és égést tápláló gázok arányát jól szabályozható gáz-
áramlásmérővel lehet biztosítani. Az atomabszorpciós spektrometriának az 1950-es években történt megjelenése
előtti mintegy 100 év során számos különböző összetételű lángot próbáltak ki a lángfotometriában. Az
atomabszorpciós spektrometriánál az általános cél az, hogy olyan közeget alkalmazzunk, amelyben minél több
elemet lehessen atomizálni, és az adott elem alapállapotú atomjai minél nagyobb arányban keletkezzenek.
Ahhoz, hogy megítélhessük, hogy egy-egy láng milyen mértékben felel meg előbbi célnak, a láng több jellemző
adatát kell megvizsgálni. Ilyen paraméternek tekinthetők többek között az égőfej és a láng geometriája, a lángot
létrehozó gázelegy minősége, koncentrációaránya, a láng hőmérséklete, hőmérséklet-eloszlása, a gázelegy
kiáramlási sebessége az égőfejen, a láng égési sebessége, a lángban előforduló gyökök, e gyökök térbeli
eloszlása, a láng kémiai jellegzetessége, redukáló, oxidáló tulajdonsága.
4.2. Az atomizálásra használt lángok és égők
Egy adott geometriájú égőfejen tartósan égő láng két folyamat eredőjeként alakul ki. Az egyik folyamat az égést
tápláló és az éghető gáz kiáramlása az égőfej nyílásán. A másik az ezzel ellentétes folyamat, az égés, amelynek
a frontja a kiáramlással ellentétes irányba halad. Ha a kiáramlási sebesség túl nagy az égési sebességhez képest,
akkor a lángot a kiáramló gázok elfújják. Ha az égési sebesség nagyobb, mint a gázok kiáramlása az égő résén,
akkor úgynevezett visszagyulladás történik. Ekkor az égés az égőfejen keresztül behatol abba a térbe, ahol a
gázokat összekevertük, és a keverék felrobban. A láng folyamatos és biztonságos égésének feltétele, hogy az
égőn a gázok kiáramlási sebessége kissé haladja meg az égés sebességét, hogy kialakuljon a két folyamat
egyensúlya.
A lángspektroszkópiában használatos szénhidrogén-levegő lángok mindegyikét az égést megelőzően össze lehet
keverni egy kamrában, amely közvetlenül kapcsolódik a folyamatos égés feltételeit biztosító geometriájú
égőfejhez. Az ilyen rendszerben működő lángokat előkevert lángoknak nevezzük. Ilyenek például a metán-,
propán-, propán/bután- és az acetilén-levegő lángok.
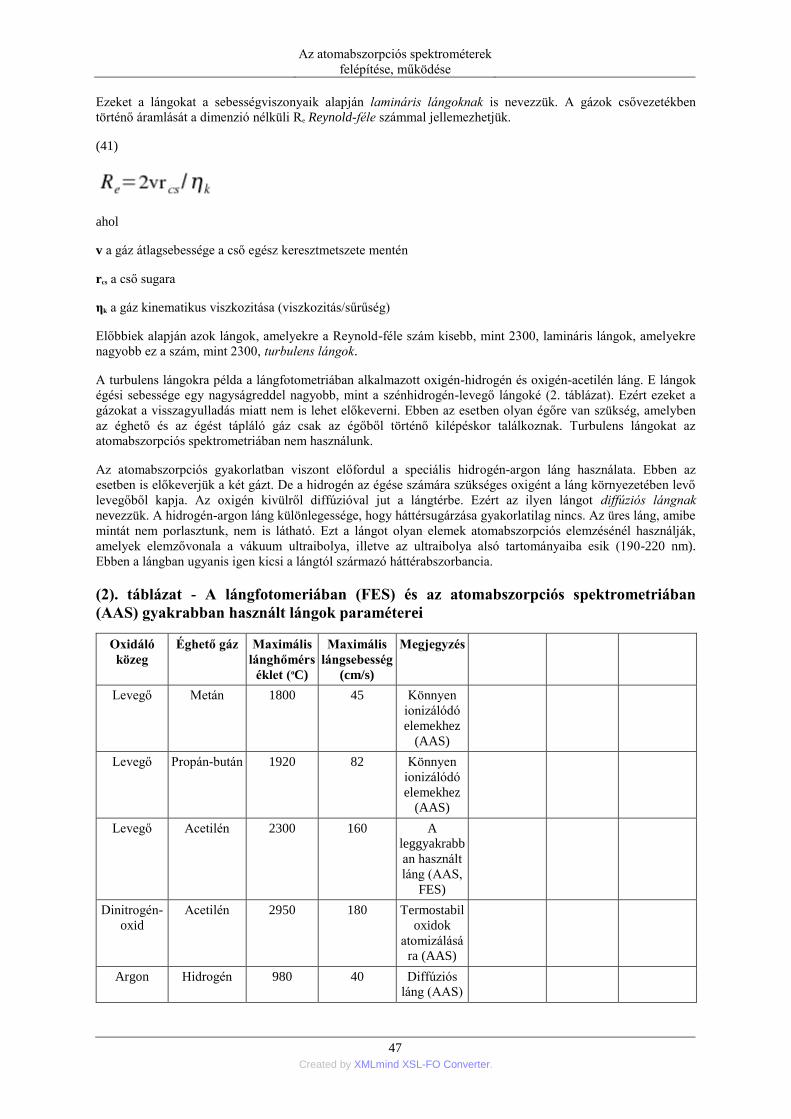
Az atomabszorpciós spektrométerek
felépítése, működése
47 Created by XMLmind XSL-FO Converter.
Ezeket a lángokat a sebességviszonyaik alapján lamináris lángoknak is nevezzük. A gázok csővezetékben
történő áramlását a dimenzió nélküli Re Reynold-féle számmal jellemezhetjük.
(41)
ahol
v a gáz átlagsebessége a cső egész keresztmetszete mentén
rcs a cső sugara
ηk a gáz kinematikus viszkozitása (viszkozitás/sűrűség)
Előbbiek alapján azok lángok, amelyekre a Reynold-féle szám kisebb, mint 2300, lamináris lángok, amelyekre
nagyobb ez a szám, mint 2300, turbulens lángok.
A turbulens lángokra példa a lángfotometriában alkalmazott oxigén-hidrogén és oxigén-acetilén láng. E lángok
égési sebessége egy nagyságreddel nagyobb, mint a szénhidrogén-levegő lángoké (2. táblázat). Ezért ezeket a
gázokat a visszagyulladás miatt nem is lehet előkeverni. Ebben az esetben olyan égőre van szükség, amelyben
az éghető és az égést tápláló gáz csak az égőből történő kilépéskor találkoznak. Turbulens lángokat az
atomabszorpciós spektrometriában nem használunk.
Az atomabszorpciós gyakorlatban viszont előfordul a speciális hidrogén-argon láng használata. Ebben az
esetben is előkeverjük a két gázt. De a hidrogén az égése számára szükséges oxigént a láng környezetében levő
levegőből kapja. Az oxigén kivülről diffúzióval jut a lángtérbe. Ezért az ilyen lángot diffúziós lángnak
nevezzük. A hidrogén-argon láng különlegessége, hogy háttérsugárzása gyakorlatilag nincs. Az üres láng, amibe
mintát nem porlasztunk, nem is látható. Ezt a lángot olyan elemek atomabszorpciós elemzésénél használják,
amelyek elemzővonala a vákuum ultraibolya, illetve az ultraibolya alsó tartományaiba esik (190-220 nm).
Ebben a lángban ugyanis igen kicsi a lángtól származó háttérabszorbancia.
(2). táblázat - A lángfotomeriában (FES) és az atomabszorpciós spektrometriában
(AAS) gyakrabban használt lángok paraméterei
Oxidáló
közeg Éghető gáz Maximális
lánghőmérs
éklet (oC)
Maximális
lángsebesség
(cm/s)
Megjegyzés
Levegő Metán 1800 45 Könnyen
ionizálódó
elemekhez
(AAS)
Levegő Propán-bután 1920 82 Könnyen
ionizálódó
elemekhez
(AAS)
Levegő Acetilén 2300 160 A
leggyakrabb
an használt
láng (AAS,
FES)
Dinitrogén-
oxid Acetilén 2950 180 Termostabil
oxidok
atomizálásá
ra (AAS)
Argon Hidrogén 980 40 Diffúziós
láng (AAS)
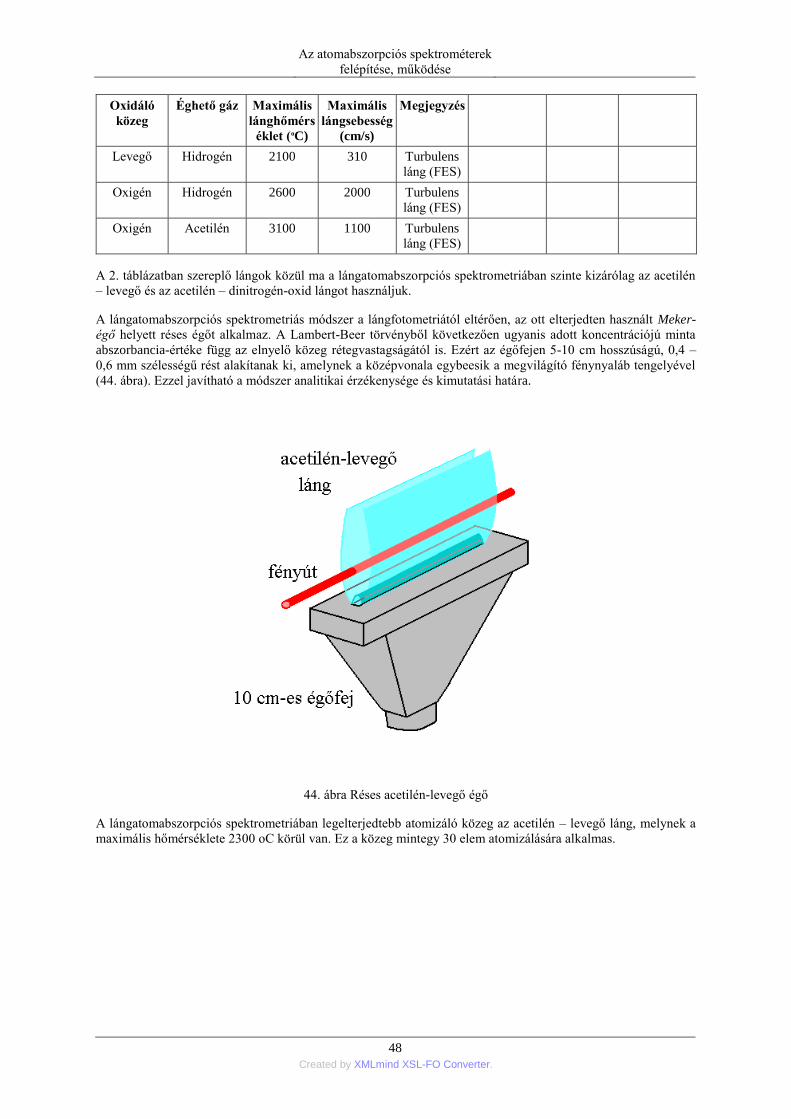
Az atomabszorpciós spektrométerek
felépítése, működése
48 Created by XMLmind XSL-FO Converter.
Oxidáló
közeg Éghető gáz Maximális
lánghőmérs
éklet (oC)
Maximális
lángsebesség
(cm/s)
Megjegyzés
Levegő Hidrogén 2100 310 Turbulens
láng (FES)
Oxigén Hidrogén 2600 2000 Turbulens
láng (FES)
Oxigén Acetilén 3100 1100 Turbulens
láng (FES)
A 2. táblázatban szereplő lángok közül ma a lángatomabszorpciós spektrometriában szinte kizárólag az acetilén
– levegő és az acetilén – dinitrogén-oxid lángot használjuk.
A lángatomabszorpciós spektrometriás módszer a lángfotometriától eltérően, az ott elterjedten használt Meker-
égő helyett réses égőt alkalmaz. A Lambert-Beer törvényből következően ugyanis adott koncentrációjú minta
abszorbancia-értéke függ az elnyelő közeg rétegvastagságától is. Ezért az égőfejen 5-10 cm hosszúságú, 0,4 –
0,6 mm szélességű rést alakítanak ki, amelynek a középvonala egybeesik a megvilágító fénynyaláb tengelyével
(44. ábra). Ezzel javítható a módszer analitikai érzékenysége és kimutatási határa.
44. ábra Réses acetilén-levegő égő
A lángatomabszorpciós spektrometriában legelterjedtebb atomizáló közeg az acetilén – levegő láng, melynek a
maximális hőmérséklete 2300 oC körül van. Ez a közeg mintegy 30 elem atomizálására alkalmas.

Az atomabszorpciós spektrométerek
felépítése, működése
49 Created by XMLmind XSL-FO Converter.
45. ábra Az előkevert acetilén-levegő láng szerkezete
Az acetilén–levegő láng 3 zónára osztható (45. ábra). Az égőfej közvetlen környezetében levő kis kiterjedésű
rész az előzóna. Itt még égési folyamat nem zajlik. Rendeltetése, hogy a lángba jutó nedves aeroszol
bepárlódását elősegítse. A reakciózóna a lángnak az a néhány mm vastagságú rétege, ahol az acetilén
széntartalma szén-dioxiddá, a hidrogén-tartalma pedig vízzé ég el. Mivel az égési folyamat ebben a zónában
megy végbe, e zóna közvetlen környezetében (e zóna fölött) a legnagyobb a láng hőmérséklete. Ebben a
zónában nagy koncentrációban keletkeznek reaktív gyökök, amelyek gerjesztett állapotba kerülnek. Ezért a
reakciózóna minden szénhidrogén lángban – így az acetilén-levegő láng esetén is – intenzív világoskék fénnyel
világít. A reakciózóna további jellegzetessége, hogy ez a térrész a láng úgynevezett nem-egyensúlyi zónája.
Ebben a zónában az égéskor hirtelen képződő gyökök számára a továbbalakuláshoz szükséges energiacserét
akadályozza a hiányzó falhatás. Ezért a reakciózónában az ezen a hőmérsékleten egyensúlyi koncentrációnál
nagyságrendekkel nagyobb gyökkoncentráció halmozódik fel. A gyököknek több milliméter utat kell
megtenniük, mire a termikus egyensúly kialakul a láng utózónájában. A mintának ezen az igen reaktív
reakciózónán kell áthaladnia, ahol a termikus folyamatok mellet a nagy gyökkoncentráció miatt jelentős kémiai
átalakulások is végbemehetnek. A szénhidrogén-levegő lángok reakciózónájában keletkező gyökök a H, O, OH,
CH, CO, C, C2, C3, CN, NO, NH. E gyökök jelentős részét a reakciózóna emissziós színképe alapján is
azonosíthatjuk (46. ábra).
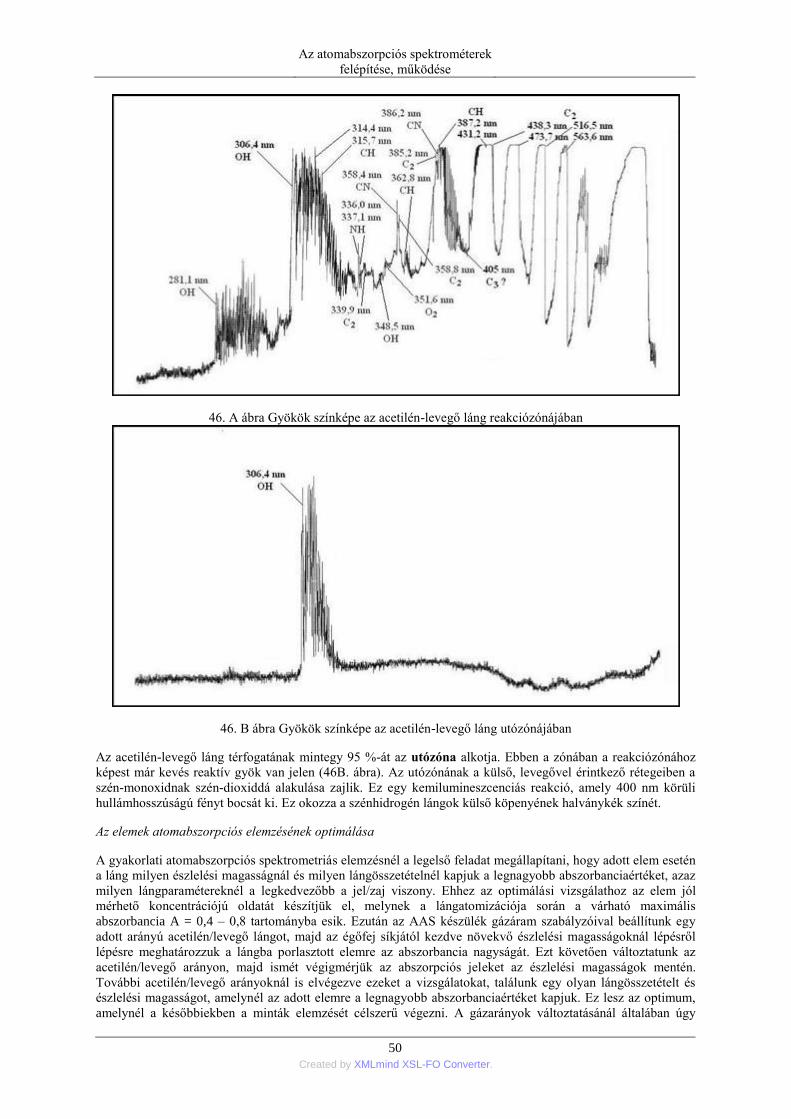
Az atomabszorpciós spektrométerek
felépítése, működése
50 Created by XMLmind XSL-FO Converter.
46. A ábra Gyökök színképe az acetilén-levegő láng reakciózónájában
46. B ábra Gyökök színképe az acetilén-levegő láng utózónájában
Az acetilén-levegő láng térfogatának mintegy 95 %-át az utózóna alkotja. Ebben a zónában a reakciózónához
képest már kevés reaktív gyök van jelen (46B. ábra). Az utózónának a külső, levegővel érintkező rétegeiben a
szén-monoxidnak szén-dioxiddá alakulása zajlik. Ez egy kemilumineszcenciás reakció, amely 400 nm körüli
hullámhosszúságú fényt bocsát ki. Ez okozza a szénhidrogén lángok külső köpenyének halványkék színét.
Az elemek atomabszorpciós elemzésének optimálása
A gyakorlati atomabszorpciós spektrometriás elemzésnél a legelső feladat megállapítani, hogy adott elem esetén
a láng milyen észlelési magasságnál és milyen lángösszetételnél kapjuk a legnagyobb abszorbanciaértéket, azaz
milyen lángparamétereknél a legkedvezőbb a jel/zaj viszony. Ehhez az optimálási vizsgálathoz az elem jól
mérhető koncentrációjú oldatát készítjük el, melynek a lángatomizációja során a várható maximális
abszorbancia A = 0,4 – 0,8 tartományba esik. Ezután az AAS készülék gázáram szabályzóival beállítunk egy
adott arányú acetilén/levegő lángot, majd az égőfej síkjától kezdve növekvő észlelési magasságoknál lépésről
lépésre meghatározzuk a lángba porlasztott elemre az abszorbancia nagyságát. Ezt követően változtatunk az
acetilén/levegő arányon, majd ismét végigmérjük az abszorpciós jeleket az észlelési magasságok mentén.
További acetilén/levegő arányoknál is elvégezve ezeket a vizsgálatokat, találunk egy olyan lángösszetételt és
észlelési magasságot, amelynél az adott elemre a legnagyobb abszorbanciaértéket kapjuk. Ez lesz az optimum,
amelynél a későbbiekben a minták elemzését célszerű végezni. A gázarányok változtatásánál általában úgy
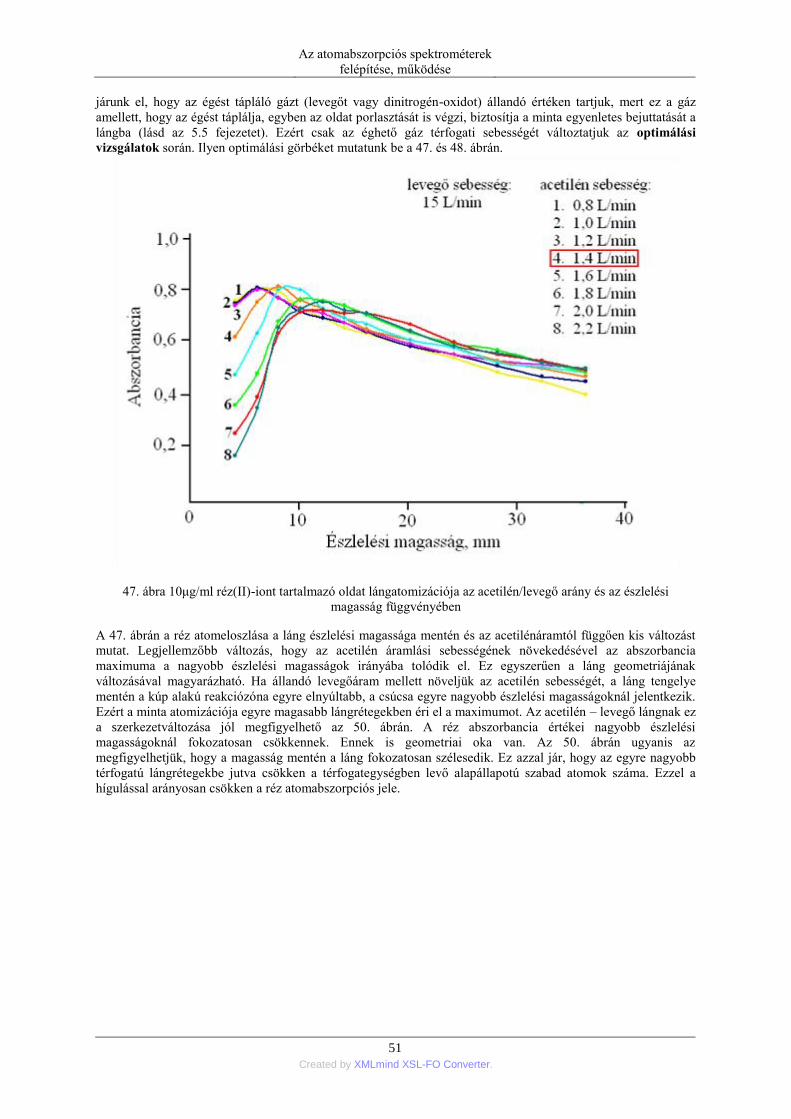
Az atomabszorpciós spektrométerek
felépítése, működése
51 Created by XMLmind XSL-FO Converter.
járunk el, hogy az égést tápláló gázt (levegőt vagy dinitrogén-oxidot) állandó értéken tartjuk, mert ez a gáz
amellett, hogy az égést táplálja, egyben az oldat porlasztását is végzi, biztosítja a minta egyenletes bejuttatását a
lángba (lásd az 5.5 fejezetet). Ezért csak az éghető gáz térfogati sebességét változtatjuk az optimálási
vizsgálatok során. Ilyen optimálási görbéket mutatunk be a 47. és 48. ábrán.
47. ábra 10μg/ml réz(II)-iont tartalmazó oldat lángatomizációja az acetilén/levegő arány és az észlelési
magasság függvényében
A 47. ábrán a réz atomeloszlása a láng észlelési magassága mentén és az acetilénáramtól függően kis változást
mutat. Legjellemzőbb változás, hogy az acetilén áramlási sebességének növekedésével az abszorbancia
maximuma a nagyobb észlelési magasságok irányába tolódik el. Ez egyszerűen a láng geometriájának
változásával magyarázható. Ha állandó levegőáram mellett növeljük az acetilén sebességét, a láng tengelye
mentén a kúp alakú reakciózóna egyre elnyúltabb, a csúcsa egyre nagyobb észlelési magasságoknál jelentkezik.
Ezért a minta atomizációja egyre magasabb lángrétegekben éri el a maximumot. Az acetilén – levegő lángnak ez
a szerkezetváltozása jól megfigyelhető az 50. ábrán. A réz abszorbancia értékei nagyobb észlelési
magasságoknál fokozatosan csökkennek. Ennek is geometriai oka van. Az 50. ábrán ugyanis az
megfigyelhetjük, hogy a magasság mentén a láng fokozatosan szélesedik. Ez azzal jár, hogy az egyre nagyobb
térfogatú lángrétegekbe jutva csökken a térfogategységben levő alapállapotú szabad atomok száma. Ezzel a
hígulással arányosan csökken a réz atomabszorpciós jele.
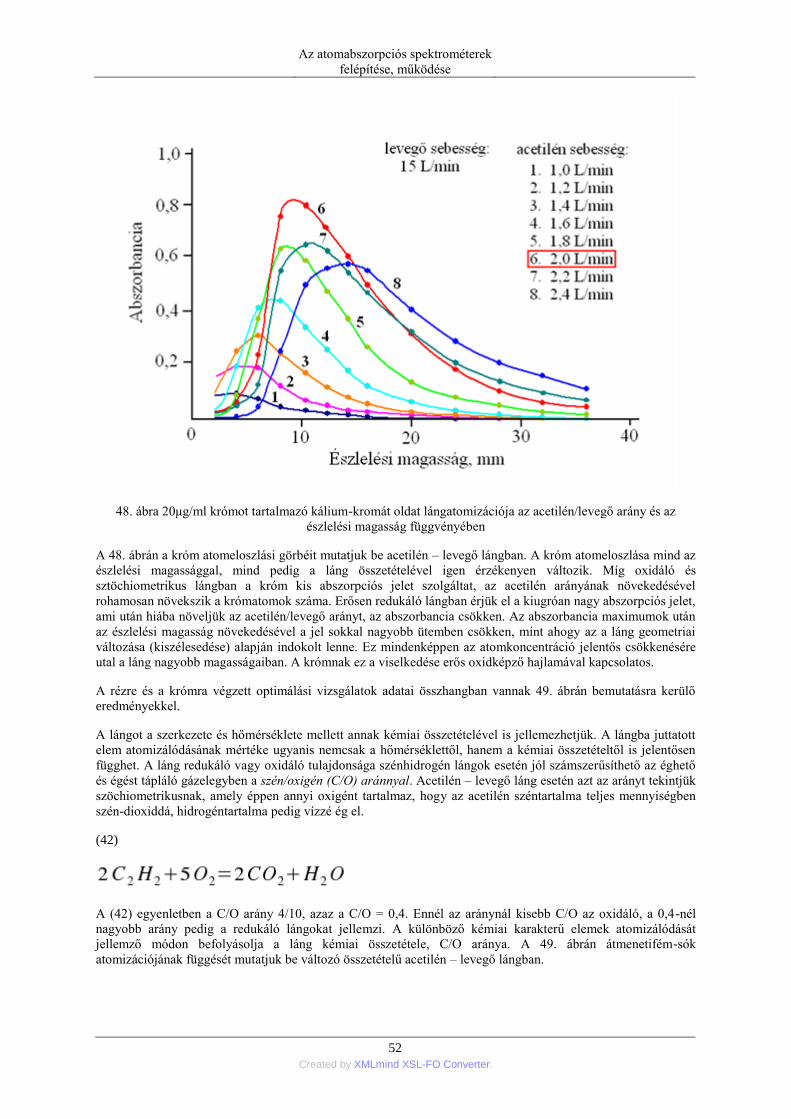
Az atomabszorpciós spektrométerek
felépítése, működése
52 Created by XMLmind XSL-FO Converter.
48. ábra 20μg/ml krómot tartalmazó kálium-kromát oldat lángatomizációja az acetilén/levegő arány és az
észlelési magasság függvényében
A 48. ábrán a króm atomeloszlási görbéit mutatjuk be acetilén – levegő lángban. A króm atomeloszlása mind az
észlelési magassággal, mind pedig a láng összetételével igen érzékenyen változik. Míg oxidáló és
sztöchiometrikus lángban a króm kis abszorpciós jelet szolgáltat, az acetilén arányának növekedésével
rohamosan növekszik a krómatomok száma. Erősen redukáló lángban érjük el a kiugróan nagy abszorpciós jelet,
ami után hiába növeljük az acetilén/levegő arányt, az abszorbancia csökken. Az abszorbancia maximumok után
az észlelési magasság növekedésével a jel sokkal nagyobb ütemben csökken, mint ahogy az a láng geometriai
változása (kiszélesedése) alapján indokolt lenne. Ez mindenképpen az atomkoncentráció jelentős csökkenésére
utal a láng nagyobb magasságaiban. A krómnak ez a viselkedése erős oxidképző hajlamával kapcsolatos.
A rézre és a krómra végzett optimálási vizsgálatok adatai összhangban vannak 49. ábrán bemutatásra kerülő
eredményekkel.
A lángot a szerkezete és hőmérséklete mellett annak kémiai összetételével is jellemezhetjük. A lángba juttatott
elem atomizálódásának mértéke ugyanis nemcsak a hőmérséklettől, hanem a kémiai összetételtől is jelentősen
függhet. A láng redukáló vagy oxidáló tulajdonsága szénhidrogén lángok esetén jól számszerűsíthető az éghető
és égést tápláló gázelegyben a szén/oxigén (C/O) aránnyal. Acetilén – levegő láng esetén azt az arányt tekintjük
szöchiometrikusnak, amely éppen annyi oxigént tartalmaz, hogy az acetilén széntartalma teljes mennyiségben
szén-dioxiddá, hidrogéntartalma pedig vízzé ég el.
(42)
A (42) egyenletben a C/O arány 4/10, azaz a C/O = 0,4. Ennél az aránynál kisebb C/O az oxidáló, a 0,4-nél
nagyobb arány pedig a redukáló lángokat jellemzi. A különböző kémiai karakterű elemek atomizálódását
jellemző módon befolyásolja a láng kémiai összetétele, C/O aránya. A 49. ábrán átmenetifém-sók
atomizációjának függését mutatjuk be változó összetételű acetilén – levegő lángban.
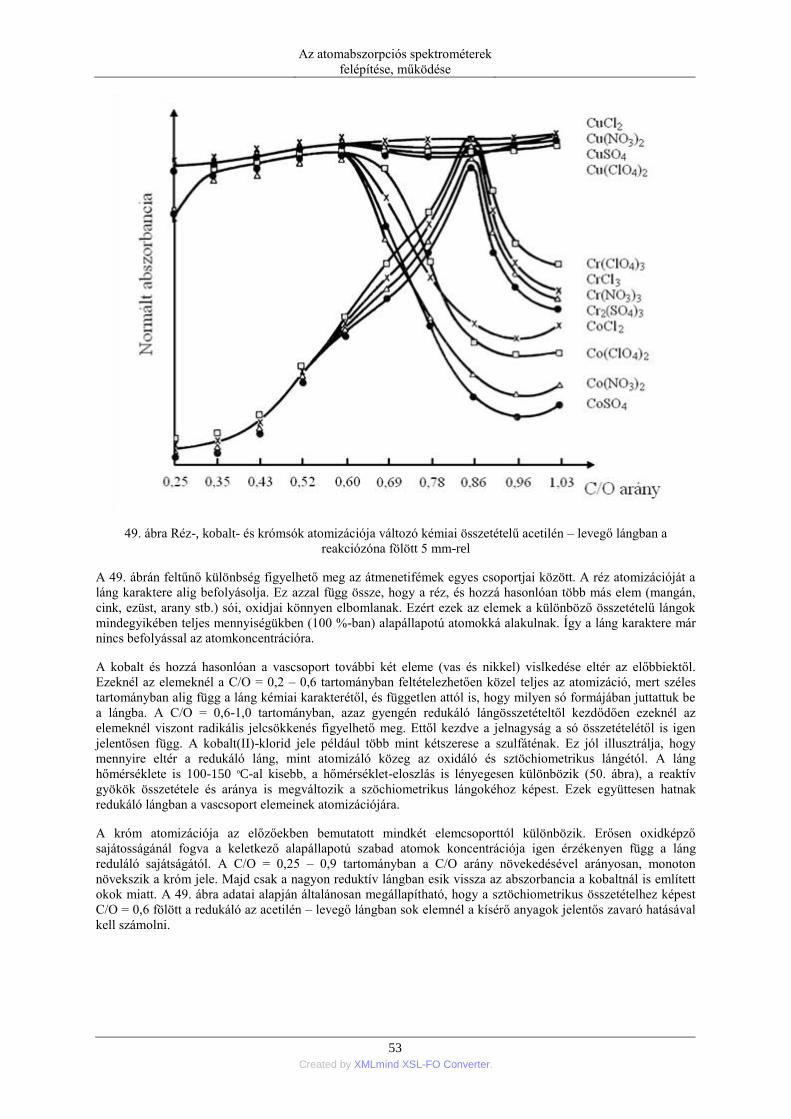
Az atomabszorpciós spektrométerek
felépítése, működése
53 Created by XMLmind XSL-FO Converter.
49. ábra Réz-, kobalt- és krómsók atomizációja változó kémiai összetételű acetilén – levegő lángban a
reakciózóna fölött 5 mm-rel
A 49. ábrán feltűnő különbség figyelhető meg az átmenetifémek egyes csoportjai között. A réz atomizációját a
láng karaktere alig befolyásolja. Ez azzal függ össze, hogy a réz, és hozzá hasonlóan több más elem (mangán,
cink, ezüst, arany stb.) sói, oxidjai könnyen elbomlanak. Ezért ezek az elemek a különböző összetételű lángok
mindegyikében teljes mennyiségükben (100 %-ban) alapállapotú atomokká alakulnak. Így a láng karaktere már
nincs befolyással az atomkoncentrációra.
A kobalt és hozzá hasonlóan a vascsoport további két eleme (vas és nikkel) vislkedése eltér az előbbiektől.
Ezeknél az elemeknél a C/O = 0,2 – 0,6 tartományban feltételezhetően közel teljes az atomizáció, mert széles
tartományban alig függ a láng kémiai karakterétől, és független attól is, hogy milyen só formájában juttattuk be
a lángba. A C/O = 0,6-1,0 tartományban, azaz gyengén redukáló lángösszetételtől kezdődően ezeknél az
elemeknél viszont radikális jelcsökkenés figyelhető meg. Ettől kezdve a jelnagyság a só összetételétől is igen
jelentősen függ. A kobalt(II)-klorid jele például több mint kétszerese a szulfáténak. Ez jól illusztrálja, hogy
mennyire eltér a redukáló láng, mint atomizáló közeg az oxidáló és sztöchiometrikus lángétól. A láng
hőmérséklete is 100-150 oC-al kisebb, a hőmérséklet-eloszlás is lényegesen különbözik (50. ábra), a reaktív
gyökök összetétele és aránya is megváltozik a szöchiometrikus lángokéhoz képest. Ezek együttesen hatnak
redukáló lángban a vascsoport elemeinek atomizációjára.
A króm atomizációja az előzőekben bemutatott mindkét elemcsoporttól különbözik. Erősen oxidképző
sajátosságánál fogva a keletkező alapállapotú szabad atomok koncentrációja igen érzékenyen függ a láng
reduláló sajátságától. A C/O = 0,25 – 0,9 tartományban a C/O arány növekedésével arányosan, monoton
növekszik a króm jele. Majd csak a nagyon reduktív lángban esik vissza az abszorbancia a kobaltnál is említett
okok miatt. A 49. ábra adatai alapján általánosan megállapítható, hogy a sztöchiometrikus összetételhez képest
C/O = 0,6 fölött a redukáló az acetilén – levegő lángban sok elemnél a kísérő anyagok jelentős zavaró hatásával
kell számolni.
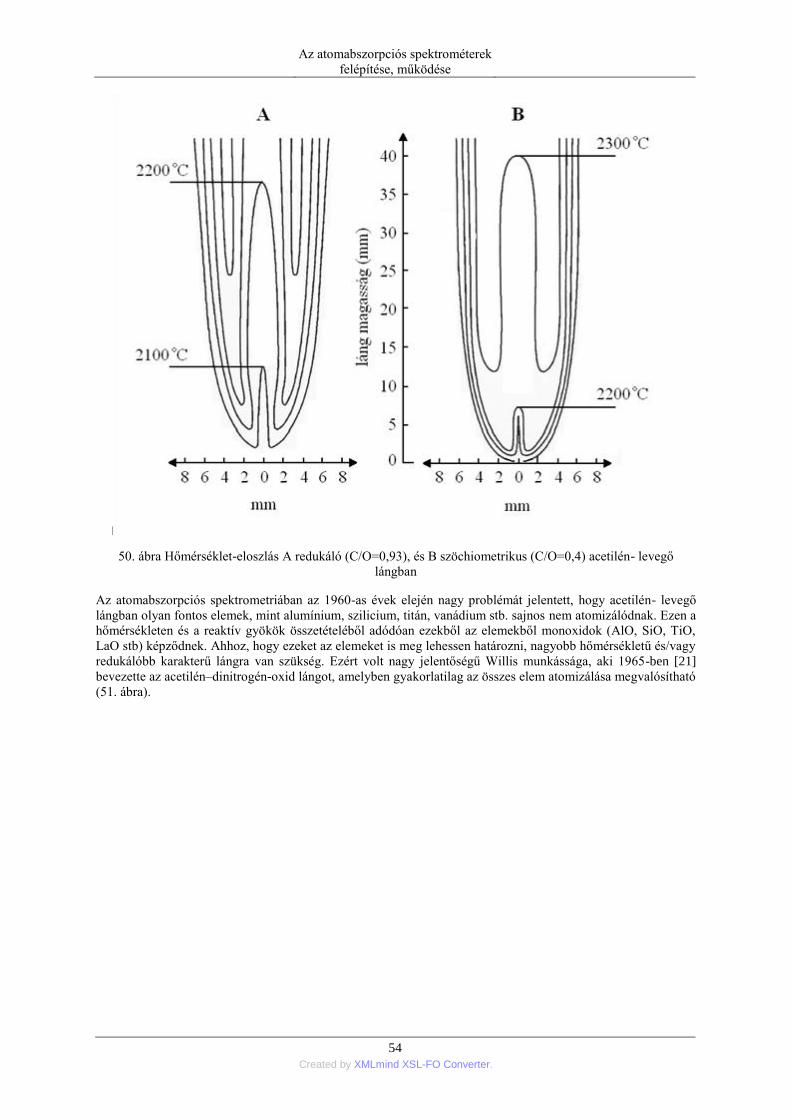
Az atomabszorpciós spektrométerek
felépítése, működése
54 Created by XMLmind XSL-FO Converter.
50. ábra Hőmérséklet-eloszlás A redukáló (C/O=0,93), és B szöchiometrikus (C/O=0,4) acetilén- levegő
lángban
Az atomabszorpciós spektrometriában az 1960-as évek elején nagy problémát jelentett, hogy acetilén- levegő
lángban olyan fontos elemek, mint alumínium, szilicium, titán, vanádium stb. sajnos nem atomizálódnak. Ezen a
hőmérsékleten és a reaktív gyökök összetételéből adódóan ezekből az elemekből monoxidok (AlO, SiO, TiO,
LaO stb) képződnek. Ahhoz, hogy ezeket az elemeket is meg lehessen határozni, nagyobb hőmérsékletű és/vagy
redukálóbb karakterű lángra van szükség. Ezért volt nagy jelentőségű Willis munkássága, aki 1965-ben [21]
bevezette az acetilén–dinitrogén-oxid lángot, amelyben gyakorlatilag az összes elem atomizálása megvalósítható
(51. ábra).

Az atomabszorpciós spektrométerek
felépítése, működése
55 Created by XMLmind XSL-FO Converter.
51. ábra Sztöchiometrikus acetilén – dinitrogén-oxid láng
Az acetilén–levegő lángtól eltérően ennek a lángnak az előzónával együtt négy zónája van. Az alsó, a
reakciózóna a nagyszámú gerjesztett állapotban levő reaktív gyök fényemissziója miatt fehér színű. Fölötte egy
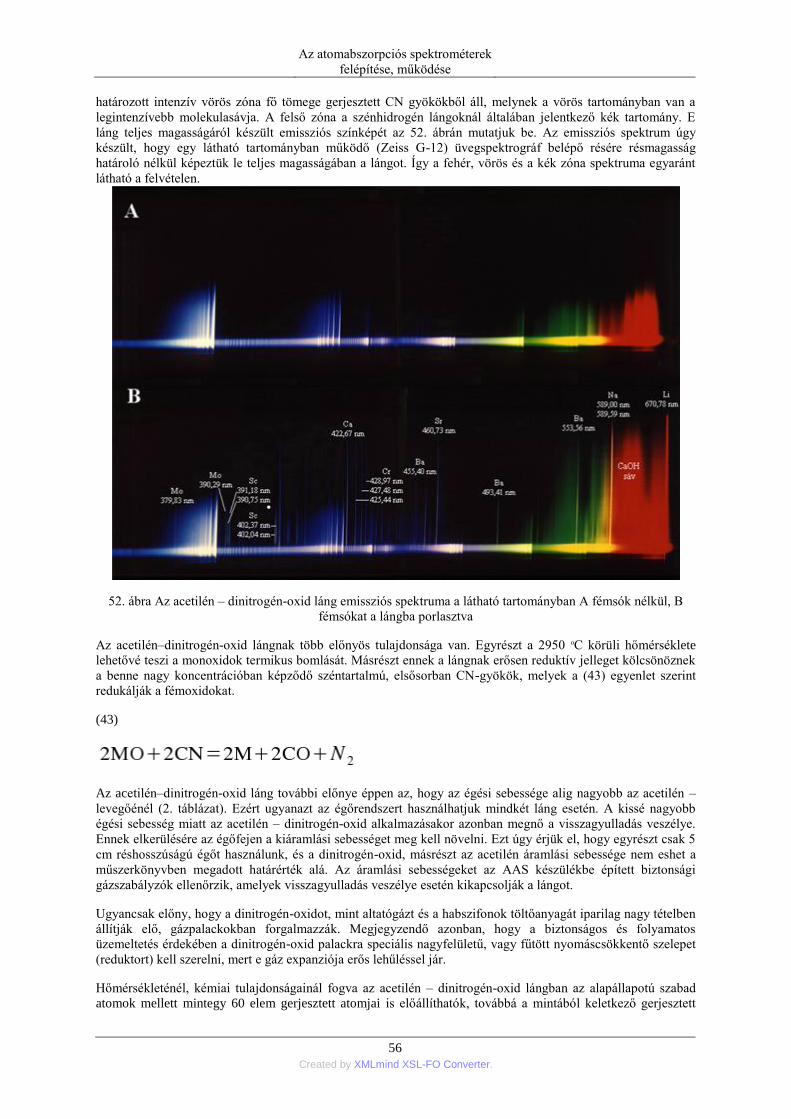
Az atomabszorpciós spektrométerek
felépítése, működése
56 Created by XMLmind XSL-FO Converter.
határozott intenzív vörös zóna fő tömege gerjesztett CN gyökökből áll, melynek a vörös tartományban van a
legintenzívebb molekulasávja. A felső zóna a szénhidrogén lángoknál általában jelentkező kék tartomány. E
láng teljes magasságáról készült emissziós színképét az 52. ábrán mutatjuk be. Az emissziós spektrum úgy
készült, hogy egy látható tartományban működő (Zeiss G-12) üvegspektrográf belépő résére résmagasság
határoló nélkül képeztük le teljes magasságában a lángot. Így a fehér, vörös és a kék zóna spektruma egyaránt
látható a felvételen.
52. ábra Az acetilén – dinitrogén-oxid láng emissziós spektruma a látható tartományban A fémsók nélkül, B
fémsókat a lángba porlasztva
Az acetilén–dinitrogén-oxid lángnak több előnyös tulajdonsága van. Egyrészt a 2950 oC körüli hőmérséklete
lehetővé teszi a monoxidok termikus bomlását. Másrészt ennek a lángnak erősen reduktív jelleget kölcsönöznek
a benne nagy koncentrációban képződő széntartalmú, elsősorban CN-gyökök, melyek a (43) egyenlet szerint
redukálják a fémoxidokat.
(43)
Az acetilén–dinitrogén-oxid láng további előnye éppen az, hogy az égési sebessége alig nagyobb az acetilén –
levegőénél (2. táblázat). Ezért ugyanazt az égőrendszert használhatjuk mindkét láng esetén. A kissé nagyobb
égési sebesség miatt az acetilén – dinitrogén-oxid alkalmazásakor azonban megnő a visszagyulladás veszélye.
Ennek elkerülésére az égőfejen a kiáramlási sebességet meg kell növelni. Ezt úgy érjük el, hogy egyrészt csak 5
cm réshosszúságú égőt használunk, és a dinitrogén-oxid, másrészt az acetilén áramlási sebessége nem eshet a
műszerkönyvben megadott határérték alá. Az áramlási sebességeket az AAS készülékbe épített biztonsági
gázszabályzók ellenőrzik, amelyek visszagyulladás veszélye esetén kikapcsolják a lángot.
Ugyancsak előny, hogy a dinitrogén-oxidot, mint altatógázt és a habszifonok töltőanyagát iparilag nagy tételben
állítják elő, gázpalackokban forgalmazzák. Megjegyzendő azonban, hogy a biztonságos és folyamatos
üzemeltetés érdekében a dinitrogén-oxid palackra speciális nagyfelületű, vagy fűtött nyomáscsökkentő szelepet
(reduktort) kell szerelni, mert e gáz expanziója erős lehűléssel jár.
Hőmérsékleténél, kémiai tulajdonságainál fogva az acetilén – dinitrogén-oxid lángban az alapállapotú szabad
atomok mellett mintegy 60 elem gerjesztett atomjai is előállíthatók, továbbá a mintából keletkező gerjesztett

Az atomabszorpciós spektrométerek
felépítése, működése
57 Created by XMLmind XSL-FO Converter.
molekulák is intenzív fényt bocsátanak ki a láng különböző zónáiban a láng különböző zónáiban a láng
különböző zónáiban. Ezért az acetilén – dinitrogén-oxid lángba juttatott különböző elemek kellően nagy
koncentrációban egymástól eltérő, színpompás lángfestést idéznek elő. Ennek néhány példáját a 3. animációs
ábrán mutatjuk be.
3. animáció Vanádium, mangán, európium, holmium, lutécium és volfram lángfestése acetilén – dinitrogén-oxid
lángban
4.3. A lángba juttatott minta termikus és kinetikai folyamatai
A porlasztókamrából a lángba belépő nedves aeroszol legfontosabb átalakulási folyamatait az 53. ábrán
mutatjuk be.
53. ábra A lángba belépő nedves aeroszol legfontosabb átalakulási folyamatai
Az 5.5. fejezetben ismertetésre kerülő különböző porlasztási módszerekkel a mintaoldatból előállított nedves
aeroszol 5 μm alatti frakciója az égőfejen keresztül lép be a lángba. Az atomabszorpciós módszernél használt
acetilén-levegő láng réses égőjének hőmérséklete üzemi körülmények között 110-120 oC körüli érték. Ezért a
nedves aeroszol bepárlódása már az égő réspofái között elkezdődik, majd a láng előzónájának 4-5 mm
magassága mentén teljessé válik. Az ekkor létrejövő termék az oldószer elpárolgása után visszamaradó
mikrorészecskékből álló szilárd fázis, a száraz aeroszol. A nagyhőmérsékletű lángtérben megkezdődik a szilárd
fázis termikus átalakulása. Ez először kristályvíz vesztését, kis molekulák (HCl, CO2, NOx stb) kilépését jelenti.
A további hőfolyamatok során új szilárd fázis, vagy fázisok is létrejöhetnek. A következő lépés a szilárd fázis
megolvadása, olvadék aeroszol létrejötte. Bizonyos vegyületek esetén (pl. vas(III)-klorid) a szilárd fázis
közvetlenül is szublimálhat molekulagőzökké. Az olvadék aeroszol elpárolgása ugyancsak molekulagőzöket
eredményez. A molekulagőzök egy része gerjesztett állapotba kerül, amelyek termikus egyensúlyban vannak az
alapállapotú molekulákkal. A következő lépés a molekulák homolitikus bomlása alapállapotú szabad atomokká.
Az atomabszorpciós spektrometria szempontjából ez a legfontosabb lépés. Úgy célszerű kiválasztani a
körülményeket, hogy a lángba juttatott elem minél nagyobb arányban alakuljon át alapállapotú atomokká. A
kellően nagy hőmérsékletű lángban az alapállapotú atomok mellett, velük ugyancsak termikus egyensúlyban
gerjesztett atomok, alapállapotú és gerjesztett ionok is keletkeznek. A gerjesztett atomokat és gerjesztett ionokat

Az atomabszorpciós spektrométerek
felépítése, működése
58 Created by XMLmind XSL-FO Converter.
a lángfotometria, azaz a lángemissziós spektrometria (FES=flame emission spectrometry) használja az
elemanalízishez.
Az előzőekben tárgyalt lángfolyamatoknak elsősorban a termikus oldalát hangsúlyoztuk. A lángba juttatott
fémsók azonban nem egy nagyhőmérsékletű inert atmoszférába kerülnek, hanem egy nagyon is aktív, agresszív
kémiai közegbe. A láng ugyanis az égési folyamata során nagyszámú redukáló és oxidáló tulajdonságú
molekulát, gyököt termel. Ezek a gyökök 2000-3000 oC-on igen reaktívak és már a nagy fajlagos felületű száraz
aeroszoltól kezdődően kémiai reakcióba léphetnek a mintakomponensekkel. Ezek a gyökök elősegíthetik az
elem atomizációját, vagy például éppen oxid-, hidroxidképzéssel akadályozhatják az atomok képződését. A
lángalkotók szerepe különösen a láng reakciózónájában nagy jelentőségű. Az égési folyamat, a láng frontja
maga az az 1-2 mm vastagságú reakciózóna, amely szénhidrogén lángok jól látható világoskék zónája. Itt
keletkeznek az 5.4.2. fejezetben már említett gyökök, a H, O, OH, CH, CO, C, C2, C3, CN, NO, NH stb. (46.
ábra). Ezek közül az elemek atomizációja, a fémoxidok redukciója szempontjából elsősorban a H, a C2 és a CN
gyökök a kedvezőek [23]. Az O és OH gyökök viszont a fémoxidok, fémhidroxidok kialakulását segítik. A
kalcium-klorid oldatot acetilén – levegő lángban porlasztva, az ott keletkező alapállapotú és gerjesztett
molekulákat, atomokat és ionokat az 54. ábrán mutatjuk be.
54. ábra A kalcium-klorid oldat termikus és kinetikai átalakulásai acetilén – levegő lángban
4.4. Elemek atomizációs hatásfoka
Az előző fejezet alapján látható, hogy a meghatározni kívánt elem számos termikus és kémiai folyamaton
keresztül jut el a mintaoldattól az alapállatotú szabad atomokig. Figyelembe kell vennünk, hogy alig van olyan
részfolyamat, amelyik során az átalakulás 100 %-os lenne. Ezért egyes részfolyamatokat az átalakulás
hatásfokával (az átalakult hányaddal) jellemezhetjük. Így megítélhetjük, hogy adott koncentrációjú oldatból
végül milyen hányad alakul át mérésre alkalmas alapállapotú atomokká. Ezeket a hatásfok adatokat, mint
számszerűsíthető jelemzőket a 3. táblázat tartalmazza.
3. táblázat A minta lángban lejátszódó folyamatai és tömegárama

Az atomabszorpciós spektrométerek
felépítése, működése
59 Created by XMLmind XSL-FO Converter.
54. kep utani tablazat kepkent megadva
Az 3. táblázatban a betűk jelentése
CA — a vizsgált elem koncentrációja az oldatban (atom/cm3)
F1 — porlasztási sebesség (cm3/s)
εn— porlasztási hatásfok: a vizsgált elem lángba jutó tömege viszonyítva az összes felszívott tömeghez
βs — (a helyileg) bepárolódott hányad: a beszáradt minta aránya az összes lángba jutó mintához
βv — (a helyileg) elpárolgott hányad: a gőzállapotba kerülő minta aránya az összes bepárlódott mintához
βa — (a helyileg) atomizálódott hányad: a szabad atomok aránya az összes gőzállapotba került mintához
εa — atomizálási hatásfok: εa= εn βs βv βa
A „helyileg” kifejezés az észlelési magasság helyén fennálló állapot megjelölésére szolgál.
A 3. táblázatban az anyagátalakulás szempontjából döntő mozzanatnak számít a száraz aeroszol kialakulása. Az
ekkor létrejövő vegyületek kémiai összetétele és termikus sajátossága szabja meg ugyanis a további átalakulások
lépéseit (szilárd- és heterogén fázisú reakciók, olvadás, párolgás, szublimáció, stb.) irányát és sebességét. Az is
tény, hogy épp ezeknek a folyamatoknak, a szilárd fázisban fellépő kölcsönhatásoknak a pontos leírásától állunk
még a legmesszebb.

Az atomabszorpciós spektrométerek
felépítése, működése
60 Created by XMLmind XSL-FO Converter.
E téma elméletével foglalkozó közlemények [5, 24-30], amelyek nagyobb része a 70-es években jelent meg,
speciális kísérleti technikát alkalmazva és körültekintő matematikai modellezéssel közelítik meg a reális
rendszereket. Alkalmas egyszerűsítésekkel ezekből az eredményekből az alábbi általános következtetések
vonhatók le.
Induljunk ki egy párolgó szilárd aeroszol részecske és környezete szerkezetéből.
55. ábra A párolgó szilárd aeroszol részecske és paraméterei
Az 55. ábrán a betűk jelentése:
λ - a vizsgált elem atomjainak, molekuláinak közepes szabad úthossza, amely megfelel a telített gőznyomású
felületi réteg vastagságának
ps - a minta parciális nyomása a telített rétegben
pg - a minta parciális nyomása a lánggázokban, pg<<ps
Ts - a részecske felületi hőmérséklete
Tg - a lánggázok hőmérséklete
Az 55. ábrán bemutatott gömbszimmetrikus részecske, paraméterei alapján a lángban alapvetően kétféle
mechanizmus szerint párologhat:
a. Ha a részecske gőznyomása kicsi, akkor a részecske hőmérséklete (Ts) gyorsan eléri a Tg lánggázok
hőmérsékletét. Ilyenkor anyagátadás–szabályozott párolgás áll fenn.
b. Ha a részecske gőznyomása nagy, azaz Tg>Ts. Ekkor hőátadás – szabályozott párolgás folyik.
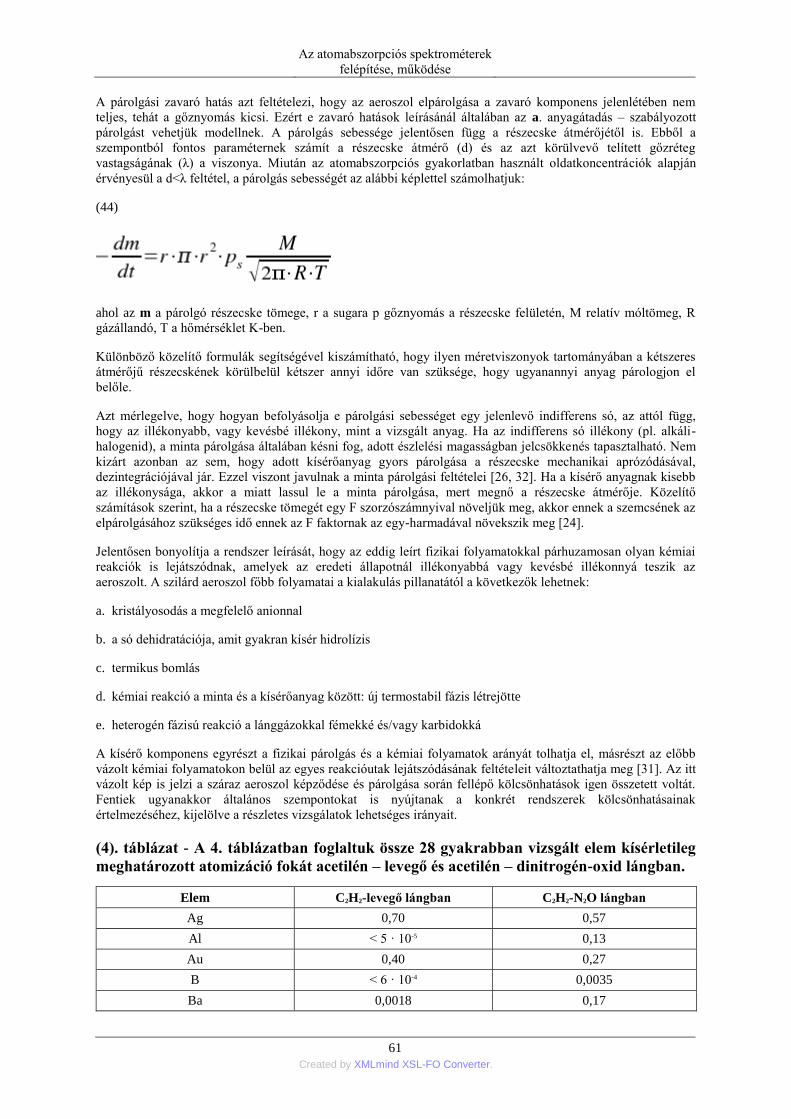
Az atomabszorpciós spektrométerek
felépítése, működése
61 Created by XMLmind XSL-FO Converter.
A párolgási zavaró hatás azt feltételezi, hogy az aeroszol elpárolgása a zavaró komponens jelenlétében nem
teljes, tehát a gőznyomás kicsi. Ezért e zavaró hatások leírásánál általában az a. anyagátadás – szabályozott
párolgást vehetjük modellnek. A párolgás sebessége jelentősen függ a részecske átmérőjétől is. Ebből a
szempontból fontos paraméternek számít a részecske átmérő (d) és az azt körülvevő telített gőzréteg
vastagságának (λ) a viszonya. Miután az atomabszorpciós gyakorlatban használt oldatkoncentrációk alapján
érvényesül a d<λ feltétel, a párolgás sebességét az alábbi képlettel számolhatjuk:
(44)
ahol az m a párolgó részecske tömege, r a sugara p gőznyomás a részecske felületén, M relatív móltömeg, R
gázállandó, T a hőmérséklet K-ben.
Különböző közelítő formulák segítségével kiszámítható, hogy ilyen méretviszonyok tartományában a kétszeres
átmérőjű részecskének körülbelül kétszer annyi időre van szüksége, hogy ugyanannyi anyag párologjon el
belőle.
Azt mérlegelve, hogy hogyan befolyásolja e párolgási sebességet egy jelenlevő indifferens só, az attól függ,
hogy az illékonyabb, vagy kevésbé illékony, mint a vizsgált anyag. Ha az indifferens só illékony (pl. alkáli-
halogenid), a minta párolgása általában késni fog, adott észlelési magasságban jelcsökkenés tapasztalható. Nem
kizárt azonban az sem, hogy adott kísérőanyag gyors párolgása a részecske mechanikai aprózódásával,
dezintegrációjával jár. Ezzel viszont javulnak a minta párolgási feltételei [26, 32]. Ha a kísérő anyagnak kisebb
az illékonysága, akkor a miatt lassul le a minta párolgása, mert megnő a részecske átmérője. Közelítő
számítások szerint, ha a részecske tömegét egy F szorzószámnyival növeljük meg, akkor ennek a szemcsének az
elpárolgásához szükséges idő ennek az F faktornak az egy-harmadával növekszik meg [24].
Jelentősen bonyolítja a rendszer leírását, hogy az eddig leírt fizikai folyamatokkal párhuzamosan olyan kémiai
reakciók is lejátszódnak, amelyek az eredeti állapotnál illékonyabbá vagy kevésbé illékonnyá teszik az
aeroszolt. A szilárd aeroszol főbb folyamatai a kialakulás pillanatától a következők lehetnek:
a. kristályosodás a megfelelő anionnal
b. a só dehidratációja, amit gyakran kísér hidrolízis
c. termikus bomlás
d. kémiai reakció a minta és a kísérőanyag között: új termostabil fázis létrejötte
e. heterogén fázisú reakció a lánggázokkal fémekké és/vagy karbidokká
A kísérő komponens egyrészt a fizikai párolgás és a kémiai folyamatok arányát tolhatja el, másrészt az előbb
vázolt kémiai folyamatokon belül az egyes reakcióutak lejátszódásának feltételeit változtathatja meg [31]. Az itt
vázolt kép is jelzi a száraz aeroszol képződése és párolgása során fellépő kölcsönhatások igen összetett voltát.
Fentiek ugyanakkor általános szempontokat is nyújtanak a konkrét rendszerek kölcsönhatásainak
értelmezéséhez, kijelölve a részletes vizsgálatok lehetséges irányait.
(4). táblázat - A 4. táblázatban foglaltuk össze 28 gyakrabban vizsgált elem kísérletileg
meghatározott atomizáció fokát acetilén – levegő és acetilén – dinitrogén-oxid lángban.
Elem C2H2-levegő lángban C2H2-N2O lángban
Ag 0,70 0,57
Al < 5 · 10-5 0,13
Au 0,40 0,27
B < 6 · 10-4 0,0035
Ba 0,0018 0,17
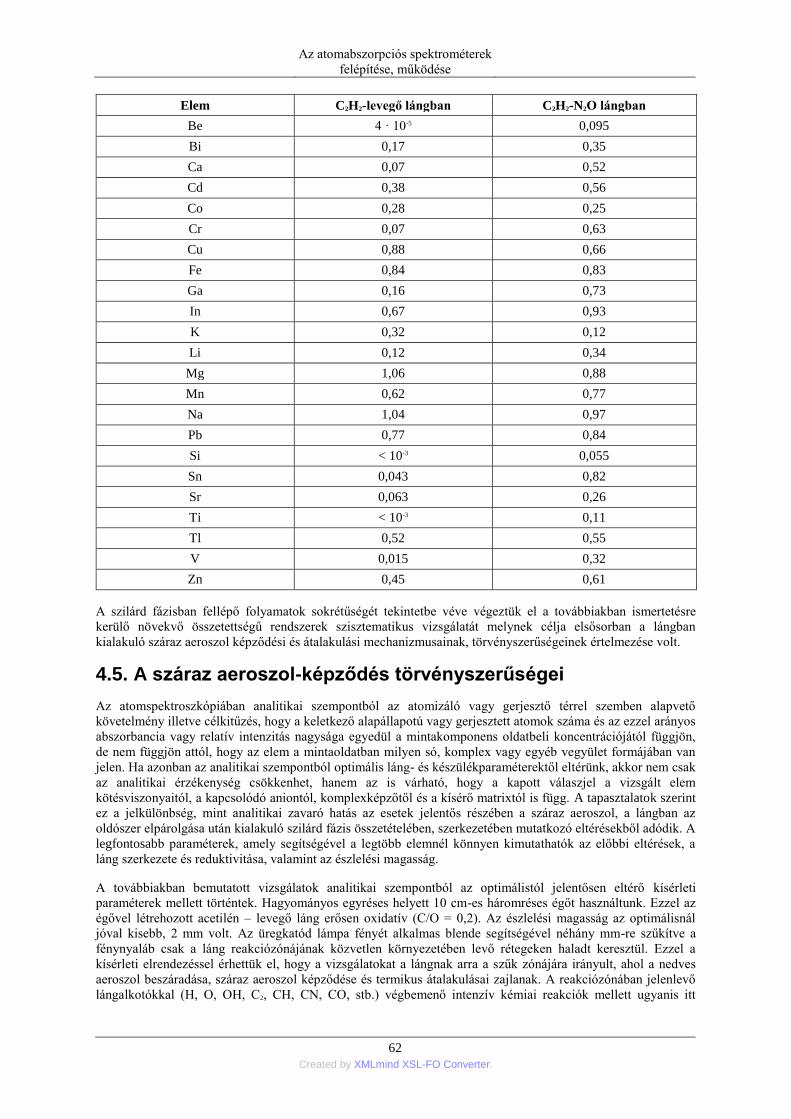
Az atomabszorpciós spektrométerek
felépítése, működése
62 Created by XMLmind XSL-FO Converter.
Elem C2H2-levegő lángban C2H2-N2O lángban
Be 4 · 10-5 0,095
Bi 0,17 0,35
Ca 0,07 0,52
Cd 0,38 0,56
Co 0,28 0,25
Cr 0,07 0,63
Cu 0,88 0,66
Fe 0,84 0,83
Ga 0,16 0,73
In 0,67 0,93
K 0,32 0,12
Li 0,12 0,34
Mg 1,06 0,88
Mn 0,62 0,77
Na 1,04 0,97
Pb 0,77 0,84
Si < 10-3 0,055
Sn 0,043 0,82
Sr 0,063 0,26
Ti < 10-3 0,11
Tl 0,52 0,55
V 0,015 0,32
Zn 0,45 0,61
A szilárd fázisban fellépő folyamatok sokrétűségét tekintetbe véve végeztük el a továbbiakban ismertetésre
kerülő növekvő összetettségű rendszerek szisztematikus vizsgálatát melynek célja elsősorban a lángban
kialakuló száraz aeroszol képződési és átalakulási mechanizmusainak, törvényszerűségeinek értelmezése volt.
4.5. A száraz aeroszol-képződés törvényszerűségei
Az atomspektroszkópiában analitikai szempontból az atomizáló vagy gerjesztő térrel szemben alapvető
követelmény illetve célkitűzés, hogy a keletkező alapállapotú vagy gerjesztett atomok száma és az ezzel arányos
abszorbancia vagy relatív intenzitás nagysága egyedül a mintakomponens oldatbeli koncentrációjától függjön,
de nem függjön attól, hogy az elem a mintaoldatban milyen só, komplex vagy egyéb vegyület formájában van
jelen. Ha azonban az analitikai szempontból optimális láng- és készülékparaméterektől eltérünk, akkor nem csak
az analitikai érzékenység csökkenhet, hanem az is várható, hogy a kapott válaszjel a vizsgált elem
kötésviszonyaitól, a kapcsolódó aniontól, komplexképzőtől és a kísérő matrixtól is függ. A tapasztalatok szerint
ez a jelkülönbség, mint analitikai zavaró hatás az esetek jelentős részében a száraz aeroszol, a lángban az
oldószer elpárolgása után kialakuló szilárd fázis összetételében, szerkezetében mutatkozó eltérésekből adódik. A
legfontosabb paraméterek, amely segítségével a legtöbb elemnél könnyen kimutathatók az előbbi eltérések, a
láng szerkezete és reduktivitása, valamint az észlelési magasság.
A továbbiakban bemutatott vizsgálatok analitikai szempontból az optimálistól jelentősen eltérő kísérleti
paraméterek mellett történtek. Hagyományos egyréses helyett 10 cm-es háromréses égőt használtunk. Ezzel az
égővel létrehozott acetilén – levegő láng erősen oxidatív (C/O = 0,2). Az észlelési magasság az optimálisnál
jóval kisebb, 2 mm volt. Az üregkatód lámpa fényét alkalmas blende segítségével néhány mm-re szűkítve a
fénynyaláb csak a láng reakciózónájának közvetlen környezetében levő rétegeken haladt keresztül. Ezzel a
kísérleti elrendezéssel érhettük el, hogy a vizsgálatokat a lángnak arra a szűk zónájára irányult, ahol a nedves
aeroszol beszáradása, száraz aeroszol képződése és termikus átalakulásai zajlanak. A reakciózónában jelenlevő
lángalkotókkal (H, O, OH, C2, CH, CN, CO, stb.) végbemenő intenzív kémiai reakciók mellett ugyanis itt
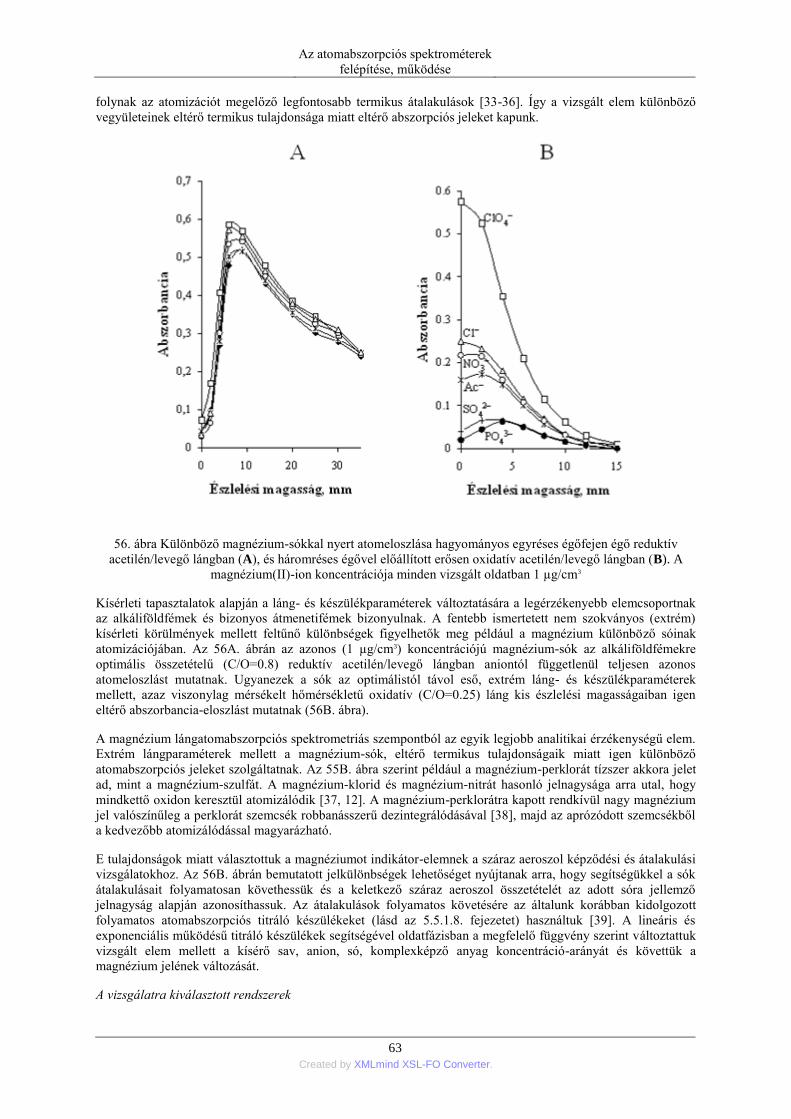
Az atomabszorpciós spektrométerek
felépítése, működése
63 Created by XMLmind XSL-FO Converter.
folynak az atomizációt megelőző legfontosabb termikus átalakulások [33-36]. Így a vizsgált elem különböző
vegyületeinek eltérő termikus tulajdonsága miatt eltérő abszorpciós jeleket kapunk.
56. ábra Különböző magnézium-sókkal nyert atomeloszlása hagyományos egyréses égőfejen égő reduktív
acetilén/levegő lángban (A), és háromréses égővel előállított erősen oxidatív acetilén/levegő lángban (B). A
magnézium(II)-ion koncentrációja minden vizsgált oldatban 1 µg/cm3
Kísérleti tapasztalatok alapján a láng- és készülékparaméterek változtatására a legérzékenyebb elemcsoportnak
az alkáliföldfémek és bizonyos átmenetifémek bizonyulnak. A fentebb ismertetett nem szokványos (extrém)
kísérleti körülmények mellett feltűnő különbségek figyelhetők meg például a magnézium különböző sóinak
atomizációjában. Az 56A. ábrán az azonos (1 µg/cm3) koncentrációjú magnézium-sók az alkáliföldfémekre
optimális összetételű (C/O=0.8) reduktív acetilén/levegő lángban aniontól függetlenül teljesen azonos
atomeloszlást mutatnak. Ugyanezek a sók az optimálistól távol eső, extrém láng- és készülékparaméterek
mellett, azaz viszonylag mérsékelt hőmérsékletű oxidatív (C/O=0.25) láng kis észlelési magasságaiban igen
eltérő abszorbancia-eloszlást mutatnak (56B. ábra).
A magnézium lángatomabszorpciós spektrometriás szempontból az egyik legjobb analitikai érzékenységű elem.
Extrém lángparaméterek mellett a magnézium-sók, eltérő termikus tulajdonságaik miatt igen különböző
atomabszorpciós jeleket szolgáltatnak. Az 55B. ábra szerint például a magnézium-perklorát tízszer akkora jelet
ad, mint a magnézium-szulfát. A magnézium-klorid és magnézium-nitrát hasonló jelnagysága arra utal, hogy
mindkettő oxidon keresztül atomizálódik [37, 12]. A magnézium-perklorátra kapott rendkívül nagy magnézium
jel valószínűleg a perklorát szemcsék robbanásszerű dezintegrálódásával [38], majd az aprózódott szemcsékből
a kedvezőbb atomizálódással magyarázható.
E tulajdonságok miatt választottuk a magnéziumot indikátor-elemnek a száraz aeroszol képződési és átalakulási
vizsgálatokhoz. Az 56B. ábrán bemutatott jelkülönbségek lehetőséget nyújtanak arra, hogy segítségükkel a sók
átalakulásait folyamatosan követhessük és a keletkező száraz aeroszol összetételét az adott sóra jellemző
jelnagyság alapján azonosíthassuk. Az átalakulások folyamatos követésére az általunk korábban kidolgozott
folyamatos atomabszorpciós titráló készülékeket (lásd az 5.5.1.8. fejezetet) használtuk [39]. A lineáris és
exponenciális működésű titráló készülékek segítségével oldatfázisban a megfelelő függvény szerint változtattuk
vizsgált elem mellett a kísérő sav, anion, só, komplexképző anyag koncentráció-arányát és követtük a
magnézium jelének változását.
A vizsgálatra kiválasztott rendszerek
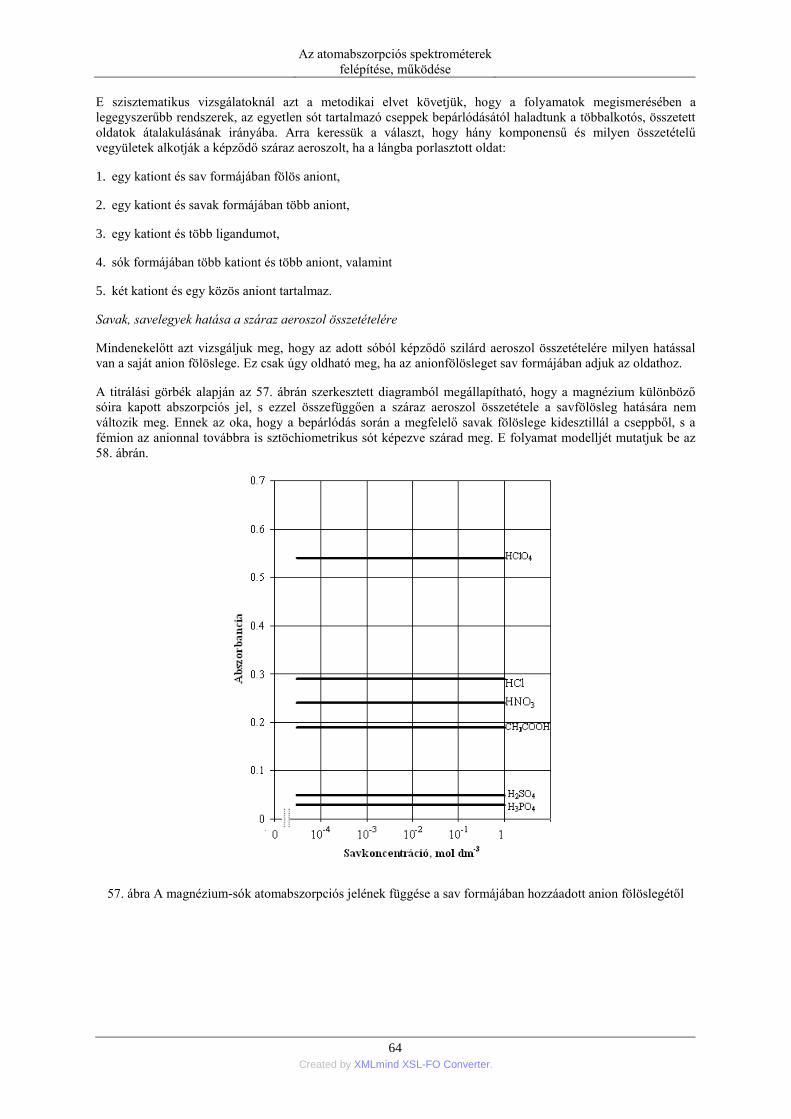
Az atomabszorpciós spektrométerek
felépítése, működése
64 Created by XMLmind XSL-FO Converter.
E szisztematikus vizsgálatoknál azt a metodikai elvet követjük, hogy a folyamatok megismerésében a
legegyszerűbb rendszerek, az egyetlen sót tartalmazó cseppek bepárlódásától haladtunk a többalkotós, összetett
oldatok átalakulásának irányába. Arra keressük a választ, hogy hány komponensű és milyen összetételű
vegyületek alkotják a képződő száraz aeroszolt, ha a lángba porlasztott oldat:
1. egy kationt és sav formájában fölös aniont,
2. egy kationt és savak formájában több aniont,
3. egy kationt és több ligandumot,
4. sók formájában több kationt és több aniont, valamint
5. két kationt és egy közös aniont tartalmaz.
Savak, savelegyek hatása a száraz aeroszol összetételére
Mindenekelőtt azt vizsgáljuk meg, hogy az adott sóból képződő szilárd aeroszol összetételére milyen hatással
van a saját anion fölöslege. Ez csak úgy oldható meg, ha az anionfölösleget sav formájában adjuk az oldathoz.
A titrálási görbék alapján az 57. ábrán szerkesztett diagramból megállapítható, hogy a magnézium különböző
sóira kapott abszorpciós jel, s ezzel összefüggően a száraz aeroszol összetétele a savfölösleg hatására nem
változik meg. Ennek az oka, hogy a bepárlódás során a megfelelő savak fölöslege kidesztillál a cseppből, s a
fémion az anionnal továbbra is sztöchiometrikus sót képezve szárad meg. E folyamat modelljét mutatjuk be az
58. ábrán.
57. ábra A magnézium-sók atomabszorpciós jelének függése a sav formájában hozzáadott anion fölöslegétől
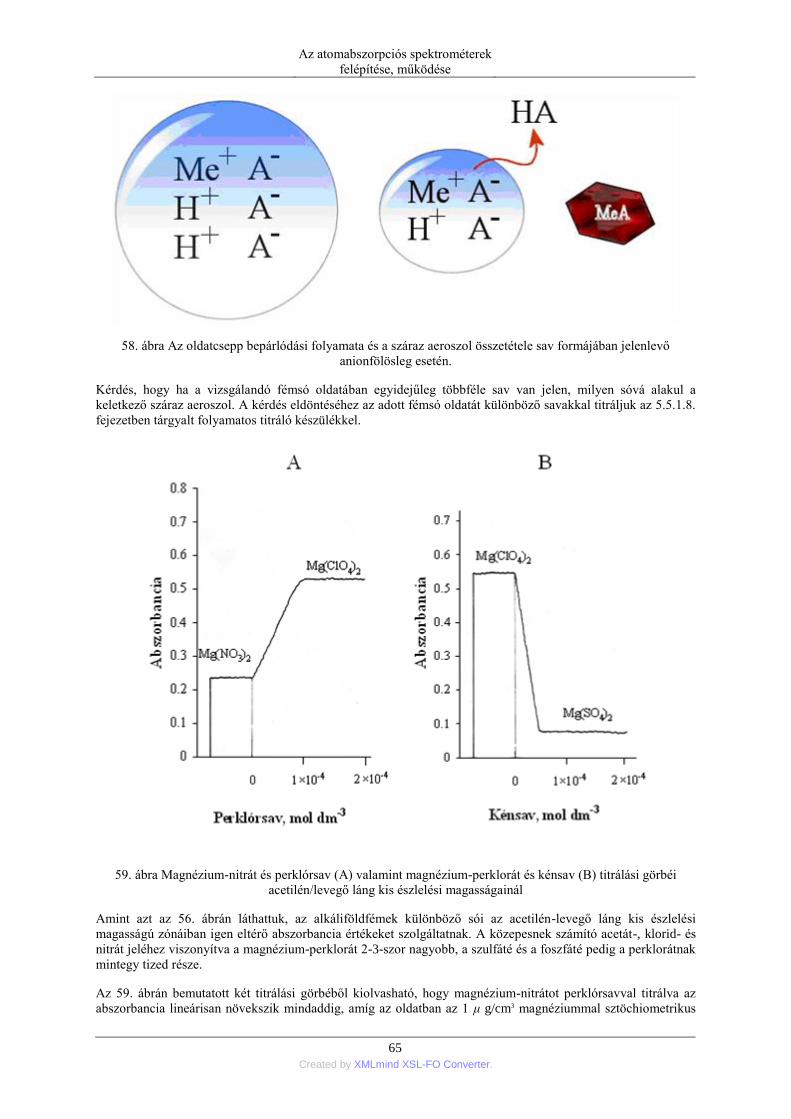
Az atomabszorpciós spektrométerek
felépítése, működése
65 Created by XMLmind XSL-FO Converter.
58. ábra Az oldatcsepp bepárlódási folyamata és a száraz aeroszol összetétele sav formájában jelenlevő
anionfölösleg esetén.
Kérdés, hogy ha a vizsgálandó fémsó oldatában egyidejűleg többféle sav van jelen, milyen sóvá alakul a
keletkező száraz aeroszol. A kérdés eldöntéséhez az adott fémsó oldatát különböző savakkal titráljuk az 5.5.1.8.
fejezetben tárgyalt folyamatos titráló készülékkel.
59. ábra Magnézium-nitrát és perklórsav (A) valamint magnézium-perklorát és kénsav (B) titrálási görbéi
acetilén/levegő láng kis észlelési magasságainál
Amint azt az 56. ábrán láthattuk, az alkáliföldfémek különböző sói az acetilén-levegő láng kis észlelési
magasságú zónáiban igen eltérő abszorbancia értékeket szolgáltatnak. A közepesnek számító acetát-, klorid- és
nitrát jeléhez viszonyítva a magnézium-perklorát 2-3-szor nagyobb, a szulfáté és a foszfáté pedig a perklorátnak
mintegy tized része.
Az 59. ábrán bemutatott két titrálási görbéből kiolvasható, hogy magnézium-nitrátot perklórsavval titrálva az
abszorbancia lineárisan növekszik mindaddig, amíg az oldatban az 1 μ g/cm3 magnéziummal sztöchiometrikus
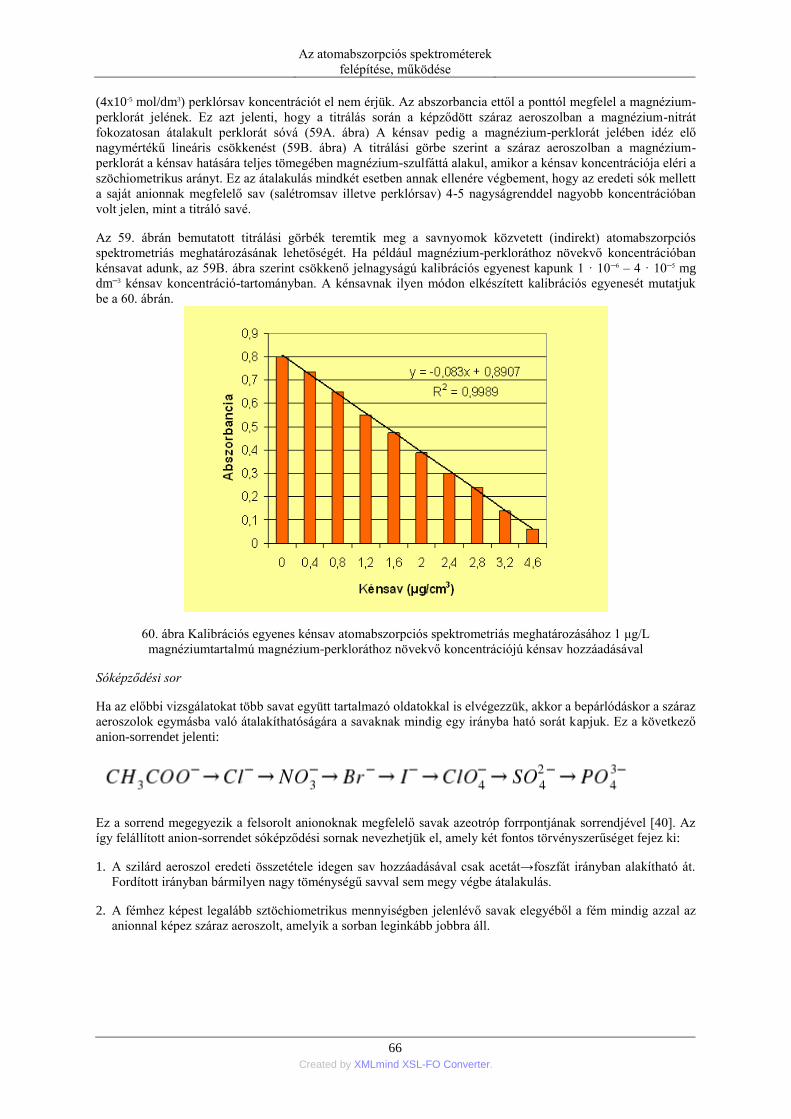
Az atomabszorpciós spektrométerek
felépítése, működése
66 Created by XMLmind XSL-FO Converter.
(4x10-5 mol/dm3) perklórsav koncentrációt el nem érjük. Az abszorbancia ettől a ponttól megfelel a magnézium-
perklorát jelének. Ez azt jelenti, hogy a titrálás során a képződött száraz aeroszolban a magnézium-nitrát
fokozatosan átalakult perklorát sóvá (59A. ábra) A kénsav pedig a magnézium-perklorát jelében idéz elő
nagymértékű lineáris csökkenést (59B. ábra) A titrálási görbe szerint a száraz aeroszolban a magnézium-
perklorát a kénsav hatására teljes tömegében magnézium-szulfáttá alakul, amikor a kénsav koncentrációja eléri a
szöchiometrikus arányt. Ez az átalakulás mindkét esetben annak ellenére végbement, hogy az eredeti sók mellett
a saját anionnak megfelelő sav (salétromsav illetve perklórsav) 4-5 nagyságrenddel nagyobb koncentrációban
volt jelen, mint a titráló savé.
Az 59. ábrán bemutatott titrálási görbék teremtik meg a savnyomok közvetett (indirekt) atomabszorpciós
spektrometriás meghatározásának lehetőségét. Ha például magnézium-perkloráthoz növekvő koncentrációban
kénsavat adunk, az 59B. ábra szerint csökkenő jelnagyságú kalibrációs egyenest kapunk 1 · 10─6 – 4 · 10─5 mg
dm─3 kénsav koncentráció-tartományban. A kénsavnak ilyen módon elkészített kalibrációs egyenesét mutatjuk
be a 60. ábrán.
60. ábra Kalibrációs egyenes kénsav atomabszorpciós spektrometriás meghatározásához 1 μg/L
magnéziumtartalmú magnézium-perkloráthoz növekvő koncentrációjú kénsav hozzáadásával
Sóképződési sor
Ha az előbbi vizsgálatokat több savat együtt tartalmazó oldatokkal is elvégezzük, akkor a bepárlódáskor a száraz
aeroszolok egymásba való átalakíthatóságára a savaknak mindig egy irányba ható sorát kapjuk. Ez a következő
anion-sorrendet jelenti:
Ez a sorrend megegyezik a felsorolt anionoknak megfelelő savak azeotróp forrpontjának sorrendjével [40]. Az
így felállított anion-sorrendet sóképződési sornak nevezhetjük el, amely két fontos törvényszerűséget fejez ki:
1. A szilárd aeroszol eredeti összetétele idegen sav hozzáadásával csak acetát→foszfát irányban alakítható át.
Fordított irányban bármilyen nagy töménységű savval sem megy végbe átalakulás.
2. A fémhez képest legalább sztöchiometrikus mennyiségben jelenlévő savak elegyéből a fém mindig azzal az
anionnal képez száraz aeroszolt, amelyik a sorban leginkább jobbra áll.
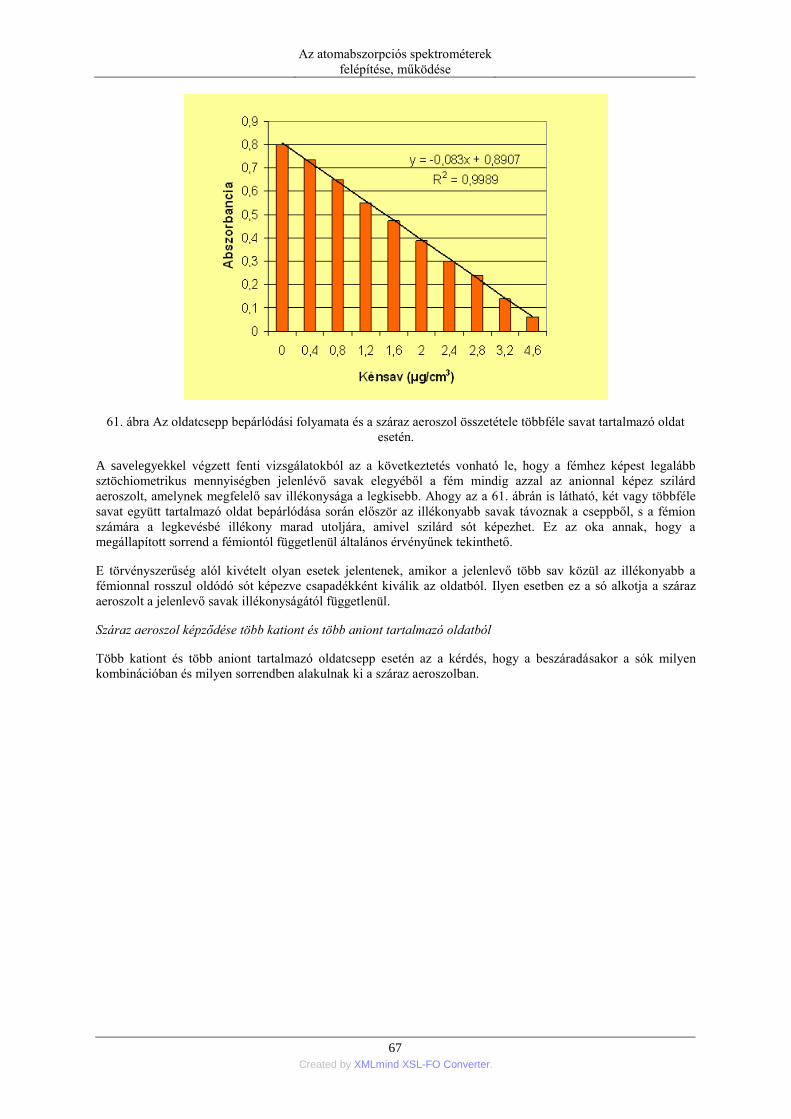
Az atomabszorpciós spektrométerek
felépítése, működése
67 Created by XMLmind XSL-FO Converter.
61. ábra Az oldatcsepp bepárlódási folyamata és a száraz aeroszol összetétele többféle savat tartalmazó oldat
esetén.
A savelegyekkel végzett fenti vizsgálatokból az a következtetés vonható le, hogy a fémhez képest legalább
sztöchiometrikus mennyiségben jelenlévő savak elegyéből a fém mindig azzal az anionnal képez szilárd
aeroszolt, amelynek megfelelő sav illékonysága a legkisebb. Ahogy az a 61. ábrán is látható, két vagy többféle
savat együtt tartalmazó oldat bepárlódása során először az illékonyabb savak távoznak a cseppből, s a fémion
számára a legkevésbé illékony marad utoljára, amivel szilárd sót képezhet. Ez az oka annak, hogy a
megállapított sorrend a fémiontól függetlenül általános érvényűnek tekinthető.
E törvényszerűség alól kivételt olyan esetek jelentenek, amikor a jelenlevő több sav közül az illékonyabb a
fémionnal rosszul oldódó sót képezve csapadékként kiválik az oldatból. Ilyen esetben ez a só alkotja a száraz
aeroszolt a jelenlevő savak illékonyságától függetlenül.
Száraz aeroszol képződése több kationt és több aniont tartalmazó oldatból
Több kationt és több aniont tartalmazó oldatcsepp esetén az a kérdés, hogy a beszáradásakor a sók milyen
kombinációban és milyen sorrendben alakulnak ki a száraz aeroszolban.
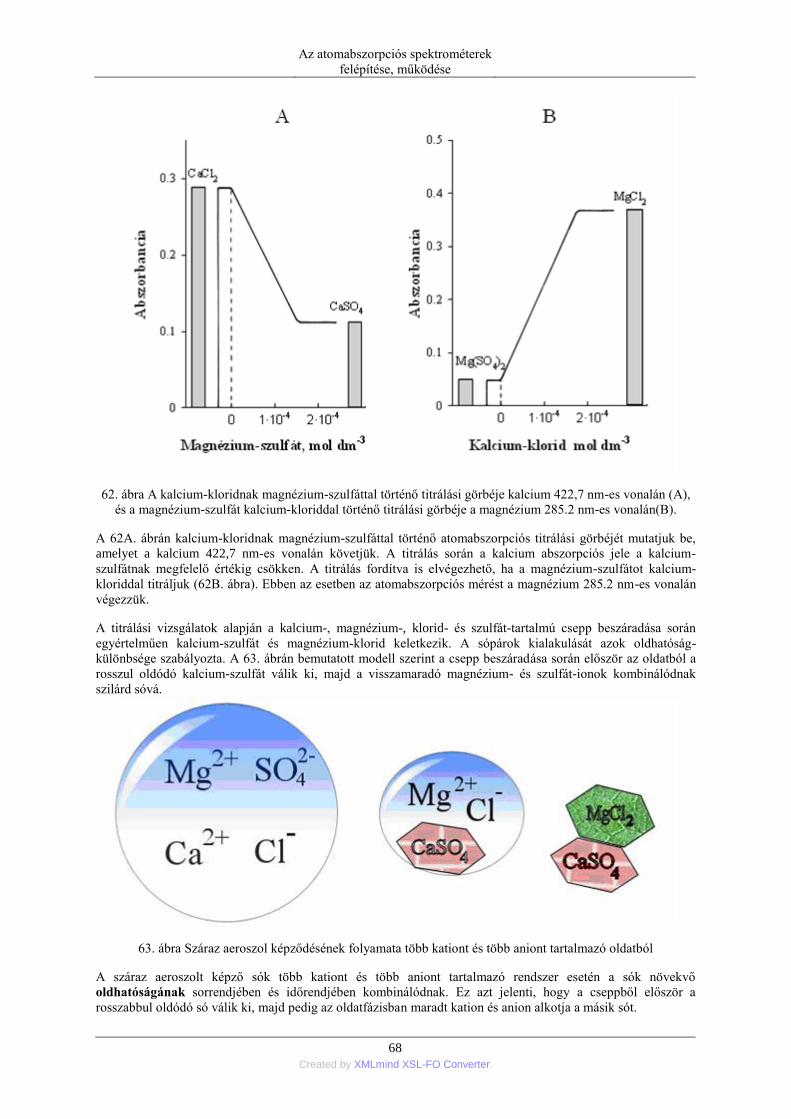
Az atomabszorpciós spektrométerek
felépítése, működése
68 Created by XMLmind XSL-FO Converter.
62. ábra A kalcium-kloridnak magnézium-szulfáttal történő titrálási görbéje kalcium 422,7 nm-es vonalán (A),
és a magnézium-szulfát kalcium-kloriddal történő titrálási görbéje a magnézium 285.2 nm-es vonalán(B).
A 62A. ábrán kalcium-kloridnak magnézium-szulfáttal történő atomabszorpciós titrálási görbéjét mutatjuk be,
amelyet a kalcium 422,7 nm-es vonalán követjük. A titrálás során a kalcium abszorpciós jele a kalcium-
szulfátnak megfelelő értékig csökken. A titrálás fordítva is elvégezhető, ha a magnézium-szulfátot kalcium-
kloriddal titráljuk (62B. ábra). Ebben az esetben az atomabszorpciós mérést a magnézium 285.2 nm-es vonalán
végezzük.
A titrálási vizsgálatok alapján a kalcium-, magnézium-, klorid- és szulfát-tartalmú csepp beszáradása során
egyértelműen kalcium-szulfát és magnézium-klorid keletkezik. A sópárok kialakulását azok oldhatóság-
különbsége szabályozta. A 63. ábrán bemutatott modell szerint a csepp beszáradása során először az oldatból a
rosszul oldódó kalcium-szulfát válik ki, majd a visszamaradó magnézium- és szulfát-ionok kombinálódnak
szilárd sóvá.
63. ábra Száraz aeroszol képződésének folyamata több kationt és több aniont tartalmazó oldatból
A száraz aeroszolt képző sók több kationt és több aniont tartalmazó rendszer esetén a sók növekvő
oldhatóságának sorrendjében és időrendjében kombinálódnak. Ez azt jelenti, hogy a cseppből először a
rosszabbul oldódó só válik ki, majd pedig az oldatfázisban maradt kation és anion alkotja a másik sót.
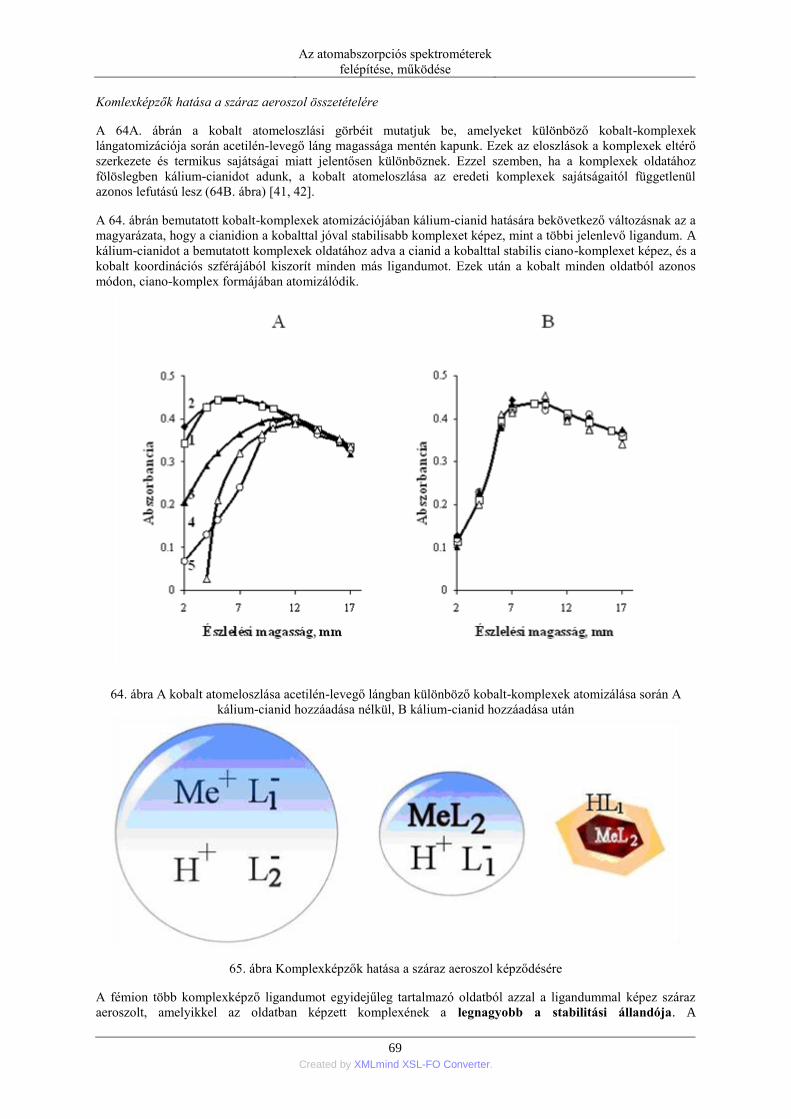
Az atomabszorpciós spektrométerek
felépítése, működése
69 Created by XMLmind XSL-FO Converter.
Komlexképzők hatása a száraz aeroszol összetételére
A 64A. ábrán a kobalt atomeloszlási görbéit mutatjuk be, amelyeket különböző kobalt-komplexek
lángatomizációja során acetilén-levegő láng magassága mentén kapunk. Ezek az eloszlások a komplexek eltérő
szerkezete és termikus sajátságai miatt jelentősen különböznek. Ezzel szemben, ha a komplexek oldatához
fölöslegben kálium-cianidot adunk, a kobalt atomeloszlása az eredeti komplexek sajátságaitól függetlenül
azonos lefutású lesz (64B. ábra) [41, 42].
A 64. ábrán bemutatott kobalt-komplexek atomizációjában kálium-cianid hatására bekövetkező változásnak az a
magyarázata, hogy a cianidion a kobalttal jóval stabilisabb komplexet képez, mint a többi jelenlevő ligandum. A
kálium-cianidot a bemutatott komplexek oldatához adva a cianid a kobalttal stabilis ciano-komplexet képez, és a
kobalt koordinációs szférájából kiszorít minden más ligandumot. Ezek után a kobalt minden oldatból azonos
módon, ciano-komplex formájában atomizálódik.
64. ábra A kobalt atomeloszlása acetilén-levegő lángban különböző kobalt-komplexek atomizálása során A
kálium-cianid hozzáadása nélkül, B kálium-cianid hozzáadása után
65. ábra Komplexképzők hatása a száraz aeroszol képződésére
A fémion több komplexképző ligandumot egyidejűleg tartalmazó oldatból azzal a ligandummal képez száraz
aeroszolt, amelyikkel az oldatban képzett komplexének a legnagyobb a stabilitási állandója. A
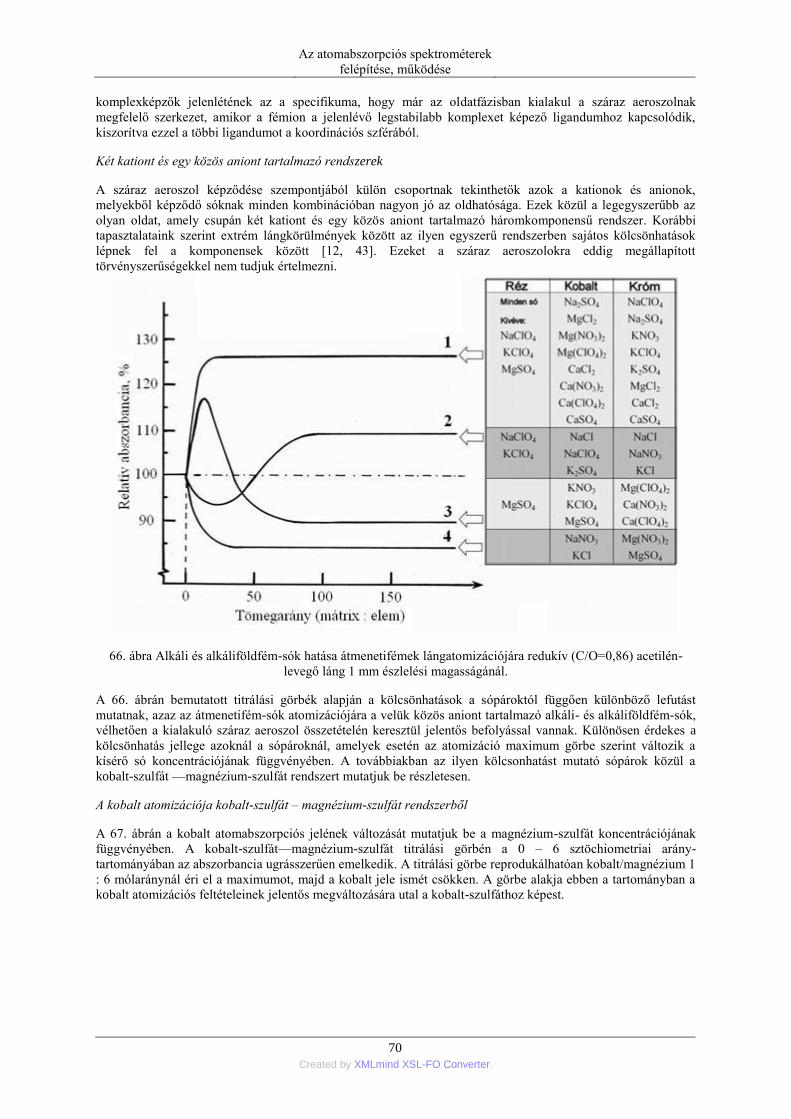
Az atomabszorpciós spektrométerek
felépítése, működése
70 Created by XMLmind XSL-FO Converter.
komplexképzők jelenlétének az a specifikuma, hogy már az oldatfázisban kialakul a száraz aeroszolnak
megfelelő szerkezet, amikor a fémion a jelenlévő legstabilabb komplexet képező ligandumhoz kapcsolódik,
kiszorítva ezzel a többi ligandumot a koordinációs szférából.
Két kationt és egy közös aniont tartalmazó rendszerek
A száraz aeroszol képződése szempontjából külön csoportnak tekinthetők azok a kationok és anionok,
melyekből képződő sóknak minden kombinációban nagyon jó az oldhatósága. Ezek közül a legegyszerűbb az
olyan oldat, amely csupán két kationt és egy közös aniont tartalmazó háromkomponensű rendszer. Korábbi
tapasztalataink szerint extrém lángkörülmények között az ilyen egyszerű rendszerben sajátos kölcsönhatások
lépnek fel a komponensek között [12, 43]. Ezeket a száraz aeroszolokra eddig megállapított
törvényszerűségekkel nem tudjuk értelmezni.
66. ábra Alkáli és alkáliföldfém-sók hatása átmenetifémek lángatomizációjára redukív (C/O=0,86) acetilén-
levegő láng 1 mm észlelési magasságánál.
A 66. ábrán bemutatott titrálási görbék alapján a kölcsönhatások a sópároktól függően különböző lefutást
mutatnak, azaz az átmenetifém-sók atomizációjára a velük közös aniont tartalmazó alkáli- és alkáliföldfém-sók,
vélhetően a kialakuló száraz aeroszol összetételén keresztül jelentős befolyással vannak. Különösen érdekes a
kölcsönhatás jellege azoknál a sópároknál, amelyek esetén az atomizáció maximum görbe szerint változik a
kísérő só koncentrációjának függvényében. A továbbiakban az ilyen kölcsonhatást mutató sópárok közül a
kobalt-szulfát —magnézium-szulfát rendszert mutatjuk be részletesen.
A kobalt atomizációja kobalt-szulfát – magnézium-szulfát rendszerből
A 67. ábrán a kobalt atomabszorpciós jelének változását mutatjuk be a magnézium-szulfát koncentrációjának
függvényében. A kobalt-szulfát—magnézium-szulfát titrálási görbén a 0 – 6 sztöchiometriai arány-
tartományában az abszorbancia ugrásszerűen emelkedik. A titrálási görbe reprodukálhatóan kobalt/magnézium 1
: 6 mólaránynál éri el a maximumot, majd a kobalt jele ismét csökken. A görbe alakja ebben a tartományban a
kobalt atomizációs feltételeinek jelentős megváltozására utal a kobalt-szulfáthoz képest.
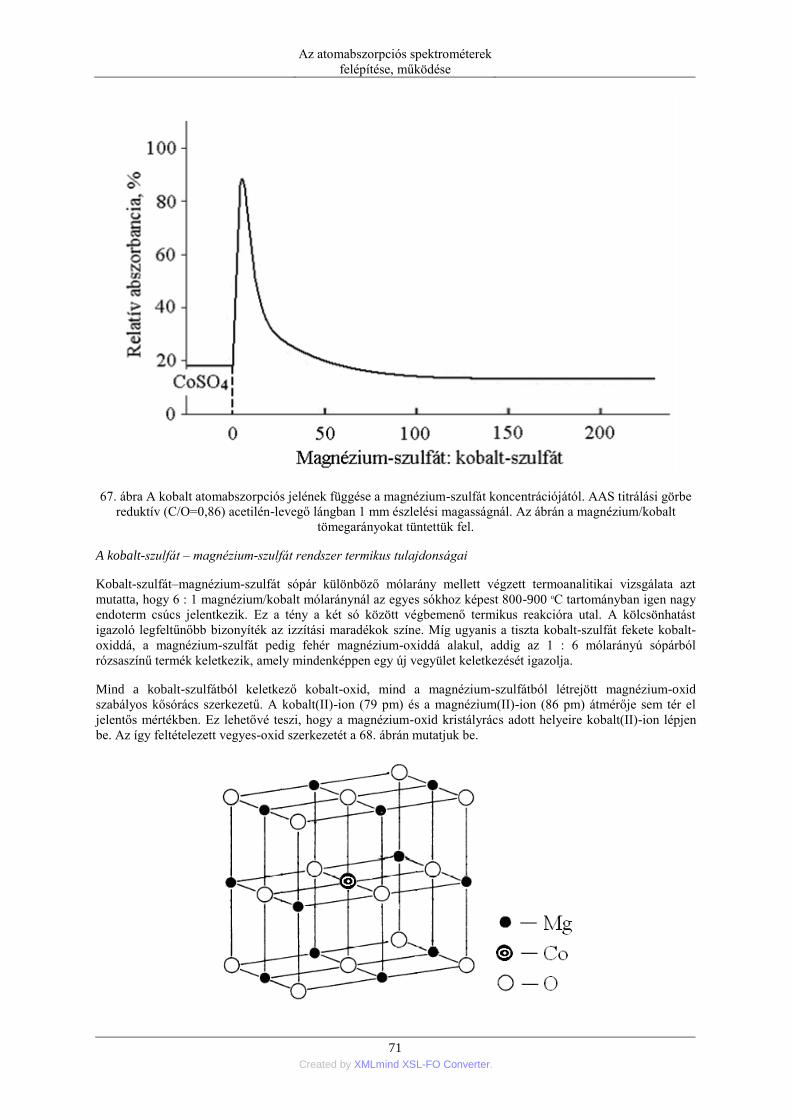
Az atomabszorpciós spektrométerek
felépítése, működése
71 Created by XMLmind XSL-FO Converter.
67. ábra A kobalt atomabszorpciós jelének függése a magnézium-szulfát koncentrációjától. AAS titrálási görbe
reduktív (C/O=0,86) acetilén-levegő lángban 1 mm észlelési magasságnál. Az ábrán a magnézium/kobalt
tömegarányokat tüntettük fel.
A kobalt-szulfát – magnézium-szulfát rendszer termikus tulajdonságai
Kobalt-szulfát–magnézium-szulfát sópár különböző mólarány mellett végzett termoanalitikai vizsgálata azt
mutatta, hogy 6 : 1 magnézium/kobalt mólaránynál az egyes sókhoz képest 800-900 oC tartományban igen nagy
endoterm csúcs jelentkezik. Ez a tény a két só között végbemenő termikus reakcióra utal. A kölcsönhatást
igazoló legfeltűnőbb bizonyíték az izzítási maradékok színe. Míg ugyanis a tiszta kobalt-szulfát fekete kobalt-
oxiddá, a magnézium-szulfát pedig fehér magnézium-oxiddá alakul, addig az 1 : 6 mólarányú sópárból
rózsaszínű termék keletkezik, amely mindenképpen egy új vegyület keletkezését igazolja.
Mind a kobalt-szulfátból keletkező kobalt-oxid, mind a magnézium-szulfátból létrejött magnézium-oxid
szabályos kősórács szerkezetű. A kobalt(II)-ion (79 pm) és a magnézium(II)-ion (86 pm) átmérője sem tér el
jelentős mértékben. Ez lehetővé teszi, hogy a magnézium-oxid kristályrács adott helyeire kobalt(II)-ion lépjen
be. Az így feltételezett vegyes-oxid szerkezetét a 68. ábrán mutatjuk be.

Az atomabszorpciós spektrométerek
felépítése, működése
72 Created by XMLmind XSL-FO Converter.
68. ábra A kobalt-szulfát–magnézium-szulfát sópárból magnézium/kobalt 6:1 mólaránynál nagyhőmérsékleten
(960 oC-on) képződő vegyes-oxid feltételezett szerkezete
Az eddigi vizsgálatok alapján megállapítható, hogy a CoSO4—MgSO4 kölcsönhatás eredménye egy új,
feltételezhetően (Co1/7Mg6/7)O összetételű vegyes-oxid, amelyből a kobalt atomizációja sokkal kedvezőbb, mint
kobalt-oxidból. A kedvezőbb atomizáció magyarázata lehet, hogy a kobalt(II)-ion kisebb ionmérete miatt
könnyebben szabadul ki a vegyes-oxid kötésből, mint a kompaktabb kobalt-oxid rácsból. A 6 : 1-nél nagyobb
koncentráció-arányoknál viszont a magnézium-szulfátból képződő magnézium-oxid mintegy zárványként köti a
vegyes-oxidot, amelyből a kobalt atomizációja ismét fokozatosan egyre kedvezőtlenebb. E hatások eredőjeként
alakulhatott ki a titrálási görbe jellegzetes maximuma (67. ábra).
A fentebb bemutatott esetből is látható, hogy a láng hőmérsékletén sajátos termikus átalakulások mennek végbe,
amelyek adott esetben új fázisok kialakulásával járhatnak.
5. Mintabeviteli módszerek a lángba
A szakirodalomban elterjedt megállapítás, hogy az atomspektrometriás módszerek Achilles-sarka a mintabevitel
[44]. Az atomspektrometriában ugyanis a fő teljesítőképességi mutatók (érzékenység, kimutatási határ,
pontosság, precizitás) döntően függenek attól, hogy milyen a mintabevitel sebessége, hatásfoka, az anyagáram
nagysága, időbeli állandósága. Előbbit kiegészítve, a legfontosabb, hogy az utóbbi jellemzők a kalibráló sorra és
a mintára tökéletesen megegyezzenek. Az atomabszorpciós spektrometriában az idők folyamán számos
mintabeviteli módszer alakult ki, melyeknél a közös törekvés, hogy a vizsgált elem mennyiségi meghatározását
minél jobb jel/zaj viszony mellett valósítsuk meg. A mintabeviteli technikák között a legnagyobb eltéréseket a
minták eltérő halmazállapota eredményezi. Ezért a külön tárgyaljuk a folyadékok, illetve a szilárd minták és
gázok mintabeviteli módszereit. A mintabevitel körülményei természetesen attól is függenek, hogy milyen
atomizáló teret (lángot, grafit vagy kvarckemencét) alkalmazunk.
5.1. Oldatok mintabevitele
Az atomabszorpciós spektrometriában is, mint más analitikai módszereknél a minta egyik leggyakoribb formája
az oldat. Elsősorban a vizes oldat biztosítja a minta könnyű kezelhetőségét és homogenitását. Jelen esetben a fő
kérdés, hogy a minta oldatát milyen módon lehet jól ellenőrizhető módon, egyenletes sebességgel,
reprodukálhatóan bevinni egy olyan 1500-3000 oC hőmérsékletű térbe, mint a láng.
Ez hasonló probléma, mint annak idején a robbanómotor megalkotásánál a benzin egyenletes bejuttatása
dugattyú hengerébe. Ott is és ez esetben is a megoldást az oldatporlasztás jelentette. Ha ugyanis a folyadékokat
igen kis cseppméretű nedves aeroszollá alakítjuk, akkor ezt a ködöt gázárammal egyenletes ütemben vagyunk
képesek szállítani a nagyhőmérsékletű térbe, ahol megtörténnek a minta termikus és kémiai átalakulásai.
A lángatomabszorpciós spektrometriában a mintabevitel legelterjedtebb módja az oldatporlasztás. Az oldatok
porlasztására, azaz a folyadéknak mikroméretű cseppekké alakítására több eltérő eljárás alakult ki. A
legrégebben használt és legelterjedtebb módszer a nagy sebességű gázáram segítségével történő pneumatikus
porlasztás (PN). Ebben az esetben szinte mindig a láng égését tápláló gázt (levegőt, oxigént) használjuk
porlasztógázként. Az utóbbi időben terjedt el a pneumatikusnál nagyobb hatékonyságú ultrahangos porlasztás
(USN), amely szilárd kristály mechanikai rezgését használja fel az oldat diszpergálására. A legújabb és
leghatékonyabb folyadék-porlasztási módszer a hidraulikus nagynyomású porlasztás (HHPN). Az előbbiektől
alapvetően eltérő elvű módszer, amely oldatok mintabevitelére is felhasználható, az elektrotermikus
elpárologtatás (ETV). E mintabeviteli módszerek részleteit a következő alfejezetekben részletezzük.
5.1.1. Pneumatikus porlasztók (PN=pneumatic neblization)
A lángfotometriában majd az atomabszorpciós spektrometriában is a legrégebben használatosak a pneumatikus
porlasztók. Az elmúlt 150 év során a pneumatikus porlasztók igen változatos típusait fejlesztették ki az
atomspektroszkópia számára (szögporlasztók Meinhardt, Babington, V-groove stb.). Ezek legtöbbje ma
elsősorban az induktív csatolású plazma–atomemissziós (ICP-AES) és –tömegspektrometriás (ICP-MS)
módszereknél nyertek alkalmazást. Ma a lángatomabszorpciós spektrométerek szinte mindegyike a gyakorlatban
a koncentrikus pneumatikus porlasztót alkalmazza, melynek szerkezetét a 69. ábrán mutatjuk be.
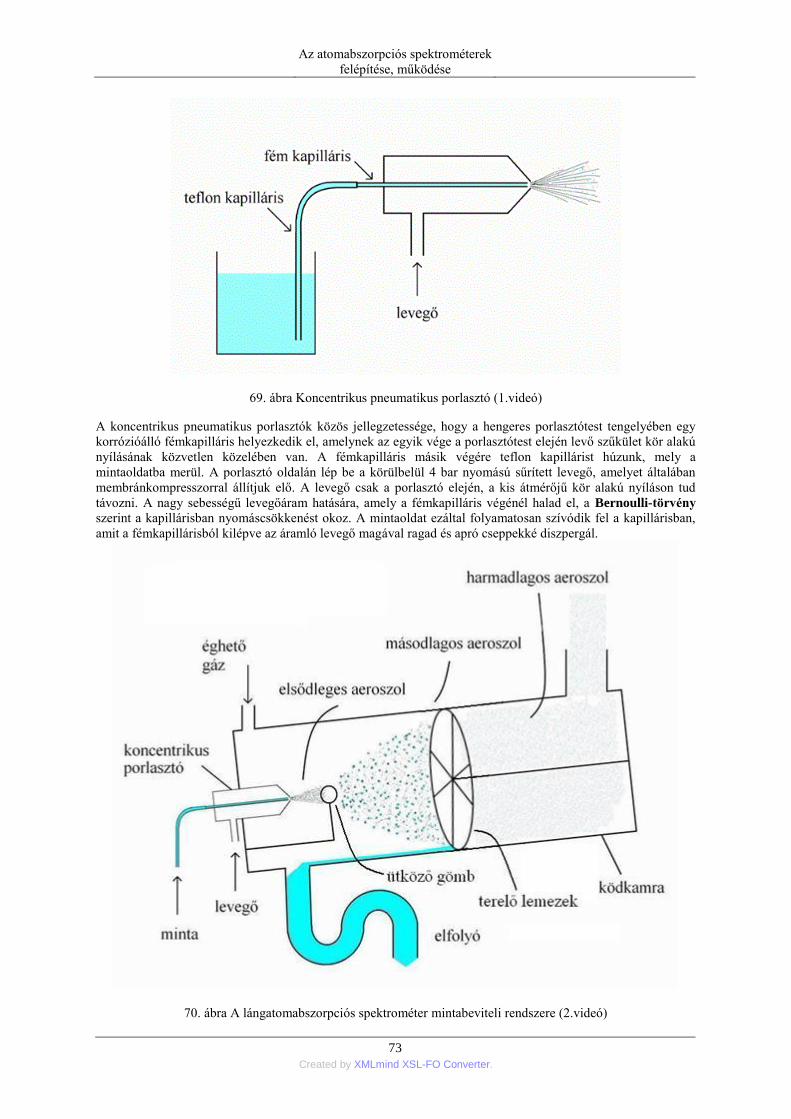
Az atomabszorpciós spektrométerek
felépítése, működése
73 Created by XMLmind XSL-FO Converter.
69. ábra Koncentrikus pneumatikus porlasztó (1.videó)
A koncentrikus pneumatikus porlasztók közös jellegzetessége, hogy a hengeres porlasztótest tengelyében egy
korrózióálló fémkapilláris helyezkedik el, amelynek az egyik vége a porlasztótest elején levő szűkület kör alakú
nyílásának közvetlen közelében van. A fémkapilláris másik végére teflon kapillárist húzunk, mely a
mintaoldatba merül. A porlasztó oldalán lép be a körülbelül 4 bar nyomású sűrített levegő, amelyet általában
membránkompresszorral állítjuk elő. A levegő csak a porlasztó elején, a kis átmérőjű kör alakú nyíláson tud
távozni. A nagy sebességű levegőáram hatására, amely a fémkapilláris végénél halad el, a Bernoulli-törvény
szerint a kapillárisban nyomáscsökkenést okoz. A mintaoldat ezáltal folyamatosan szívódik fel a kapillárisban,
amit a fémkapillárisból kilépve az áramló levegő magával ragad és apró cseppekké diszpergál.
70. ábra A lángatomabszorpciós spektrométer mintabeviteli rendszere (2.videó)

Az atomabszorpciós spektrométerek
felépítése, működése
74 Created by XMLmind XSL-FO Converter.
A mintaoldatból keletkező úgynevezett primer aeroszol igen heterogén cseppméret-eloszlású. Ezért ez az
aeroszol analitikai célra így még alkalmatlan. A porlasztó a porlasztó kamrába (ködkamrába) csatlakozik (70.
ábra). A ködkamrában a porlasztóval szemben rendszerint egy gömb alakú ütközőtestet helyeznek el. Ezen az
ütközőtesten a primer aeroszol nagyobb cseppjei tovább aprózódnak illetve a legnagyobb cseppek leválnak. Az
így kialakuló másodlagos aeroszol cseppeloszlását tovább módosítják a ködkamra közepén elhelyezkedő
terelőlemezek. Ezen a propellerhez hasonló szegmensekből álló lemezrészek között az aeroszolt szállító
porlasztó levegő áramlása megtörik, irányváltásra kényszerül. Ezt az irányváltást csak a kisebb méretű, kisebb
tehetetlenségű cseppek képesek követni. A nagyobb cseppek viszont nekicsapódnak a lemezeknek és
visszafolyva eltávoznak a ködkamra lefolyó nyílásán. A terelőlemezeken átjutó harmadlagos aeroszol átlagos
cseppátmérője 5 μm alá kerül. Ez a cseppfrakció jut be a lángba.
5.1.2. Mintabeviteli sebesség és hatásfok
Analitikai szempontból a folyadékporlasztás két legfontosabb adata a porlasztási (mintabeviteli) sebesség (Fl) és
a porlasztási (mintabeviteli) hatásfok (ε). A porlasztási sebesség a porlasztó által időegység alatt felszívott,
elporlasztott oldat térfogata.
(45)
A folyadékok pneumatikus porlasztásának sebessége elsősorban a folyadék viszkozitásától függ. Vizes oldatok
esetén a mintabeviteli sebesség átlagban 4-6 ml/min. Szerves oldószerek esetén (etanol, izobutil-metil-keton
(IBMK), etil-acetát), amelyeknek a viszkozitása kisebb, mint a vízé, a mintabeviteli sebessége előbbinél jóval
nagyobb is lehet.
A pneumatikus porlasztás sebességének kiszámításánál a Hagen – Poiseuille egyenlet bővített alakját
alkalmazhatjuk [45-48].
(46)
A (46) összefüggésben
• r a kapilláris sugara
• Δp a mintaoldat áramlását biztosító nyomáskülönbség
• η a mintaoldat viszkozitása
• l a kapilláris hossza
• B az oldat csúszási súrlódási együtthatója a kapilláris falán
A mintabeviteli hatásfok (ε) azt fejezi ki, hogy a porlasztó által felszívott oldatnak hány százaléka jut be a
lángba.
(47)
A pneumatikus porlasztás hatásfokát elsősorban a folyadék felületi feszültsége befolyásolja. A porlasztás során
ugyanis a cseppek kialakulása új felületek létrehozása, amihez felületi munkára van szükség. Minél kisebb a
felületi feszültség annál könnyebb új felületet, azaz sok apró cseppet létrehozni. A ködkamra cseppleválasztó
mechanizmusa alapján a porlasztási hatásfok annál nagyobb, minél nagyobb az 5 μm alatti cseppek aránya. A
vizes oldatok pneumatikus porlasztásának a hatásfoka 1 – 10 % közötti érték. Az atomabszorpciós
spektrométerek pneumatikus porlasztóinak hatásfoka általában 5 – 10 %.

Az atomabszorpciós spektrométerek
felépítése, működése
75 Created by XMLmind XSL-FO Converter.
Pneumatikus porlasztókkal előállított aeroszol elsődleges aeroszoljának átlagos cseppméretére Nukiyama és
Tanasawa állapított meg empirikus összefüggést (48), amely jó egyezést mutat a kísérleti adatokkal [49].
(48)
A (48) összefüggésben
• ds a cseppek átlagos úgynevezett Sauter-átmérője (a csepp térfogat/felület aránya)
• vg a porlasztó gáz lineáris áramlási sebessége
• vl a porlasztott folyadék lineáris áramlási sebessége
• γ a mintaoldat felületi feszültsége
• ρ a mintaoldat sűrűsége
• η a mintaoldat viszkozitása
• Fl a mintaoldat térfogati áramlási sebessége
• Fg a porlasztógáz térfogati áramlási sebessége
A porlasztási sebesség (Fl) és a hatásfok szorzata (ε) (49) fejezi ki az anyagáramot, azaz az időegység alatt
lángba jutó oldat térfogatát, ami az elem adott koncentrációjú oldata esetén megfelel a vizsgált elem időegység
alatt lángba jutó tömegének.
(49)
Az atomabszorpciós elemzés pontossága és precizitása szempontjából az a legfontosabb, hogy a mérés alatt a
porlasztási sebesség és a hatásfok szigorúan állandó legyen. Különösen fontos, hogy ezekben az adatokban a
kalibráló oldatok és a minta között ne legyen eltérés. Ha ugyanis a kalibráló oldatok és a minta összetétele
különböző, akkor azok viszkozitásában és felületi feszültségében is jelentős különbségek lehetnek, ami kihat a
porlasztási sebesség és a porlasztási hatásfok, azaz az anyagáram nagyságára. Ezek a követelmények a további
mintabeviteli módszerekre ugyanígy érvényesek.
5.1.3. Ultrahangos porlasztás (USN= ultrasonic nebulization)
Az atomabszorpciós spektrometria bevezetése után, a 60-es években annak érdekében, hogy növelni lehessen a
pneumatikus porlasztás viszonylag szerény mintabeviteli hatásfokát, bevezették az ultrahangos porlasztást.
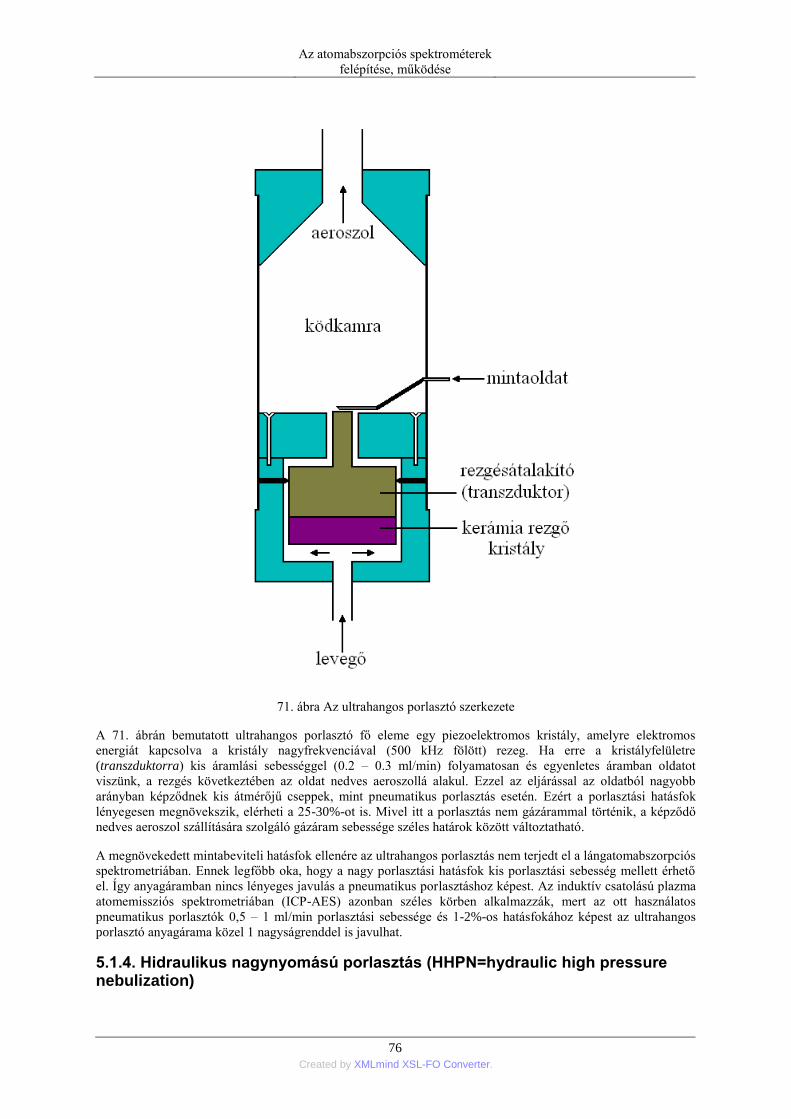
Az atomabszorpciós spektrométerek
felépítése, működése
76 Created by XMLmind XSL-FO Converter.
71. ábra Az ultrahangos porlasztó szerkezete
A 71. ábrán bemutatott ultrahangos porlasztó fő eleme egy piezoelektromos kristály, amelyre elektromos
energiát kapcsolva a kristály nagyfrekvenciával (500 kHz fölött) rezeg. Ha erre a kristályfelületre
(transzduktorra) kis áramlási sebességgel (0.2 – 0.3 ml/min) folyamatosan és egyenletes áramban oldatot
viszünk, a rezgés következtében az oldat nedves aeroszollá alakul. Ezzel az eljárással az oldatból nagyobb
arányban képződnek kis átmérőjű cseppek, mint pneumatikus porlasztás esetén. Ezért a porlasztási hatásfok
lényegesen megnövekszik, elérheti a 25-30%-ot is. Mivel itt a porlasztás nem gázárammal történik, a képződő
nedves aeroszol szállítására szolgáló gázáram sebessége széles határok között változtatható.
A megnövekedett mintabeviteli hatásfok ellenére az ultrahangos porlasztás nem terjedt el a lángatomabszorpciós
spektrometriában. Ennek legfőbb oka, hogy a nagy porlasztási hatásfok kis porlasztási sebesség mellett érhető
el. Így anyagáramban nincs lényeges javulás a pneumatikus porlasztáshoz képest. Az induktív csatolású plazma
atomemissziós spektrometriában (ICP-AES) azonban széles körben alkalmazzák, mert az ott használatos
pneumatikus porlasztók 0,5 – 1 ml/min porlasztási sebessége és 1-2%-os hatásfokához képest az ultrahangos
porlasztó anyagárama közel 1 nagyságrenddel is javulhat.
5.1.4. Hidraulikus nagynyomású porlasztás (HHPN=hydraulic high pressure nebulization)

Az atomabszorpciós spektrométerek
felépítése, működése
77 Created by XMLmind XSL-FO Converter.
Berndt 1988-ban egy teljesen új elven alapuló mintabeviteli eljárást, a hidraulikus nagynyomású porlasztást
vezette be az atomspektrometriában [50]. A HHPN módszer azon alapszik, hogy nagy (100-200 bar) nyomású
folyadékot, oldatot 10-30 μm átmérőjű kör keresztmetszetű furaton átpréselve egy nagy sebességgel áramló 20-
30 cm hosszúságú vékony folyadékszálat kapunk. Ha e folyadékszál útjába egy sima felületű ütközőtestet
helyezünk el, a folyadék nagy része igen kis cseppátmérőjű nedves aeroszollá alakul (72. ábra).
72. ábra A hidraulikus nagynyomású porlasztó fényképe
A hidraulikus nagynyomású porlasztó rendszer összeállítását a 73. ábrán tüntettük fel. A nagynyomású
porlasztófej szerkezete pedig a 74. ábrán látható.
73. ábra A hidraulikus nagynyomású porlasztó rendszer összeállítása
A folyadék számára a nagy nyomást nagynyomású folyadékkromatográfiás (HPLC) pumpával állítjuk elő. A
pumpa folyamatosan egy vivőfolyadékot (vizet vagy más oldószert) továbbít. Ebbe a folyadékáramba
bemérőcsap segítségével injektáljuk a 100-200 μl térfogatú mintaoldatot. Maga a porlasztó egy fémfoglalatban
elhelyezett 2 mm átmérőjű gyémánt lapka, amelyben a porlasztó frontja felé szűkülő kúpos furat található. A
furat szűk részének átmérője, ahol a nagynyomású folyadék kilép a porlasztóból, 10 – 30 μm, optimális esetben
20 μm (74. ábra). Tekintettel a porlasztó kis átmérőjű furatára, a legnagyobb veszélyt a porlasztó eltömődése
jelenti. Ezért a porlasztó előtt a folyadékáram útjába szűrőfoglalatban 3 μm pórusméretű titánhálót kell
elhelyezni. Az ütközőtest fényes felületű 6-8 mm átmérőjű üveggömb, amelynek kb. 15 mm az optimális
távolsága a porlasztó kilépő nyílásától. A fémszennyezések elkerülése érdekében a HPLC csövek, a bemérő
hurok és a bemérőcsap betétje is nyomásálló PEEK (poli-éter-éter-keton) anyagból készül.
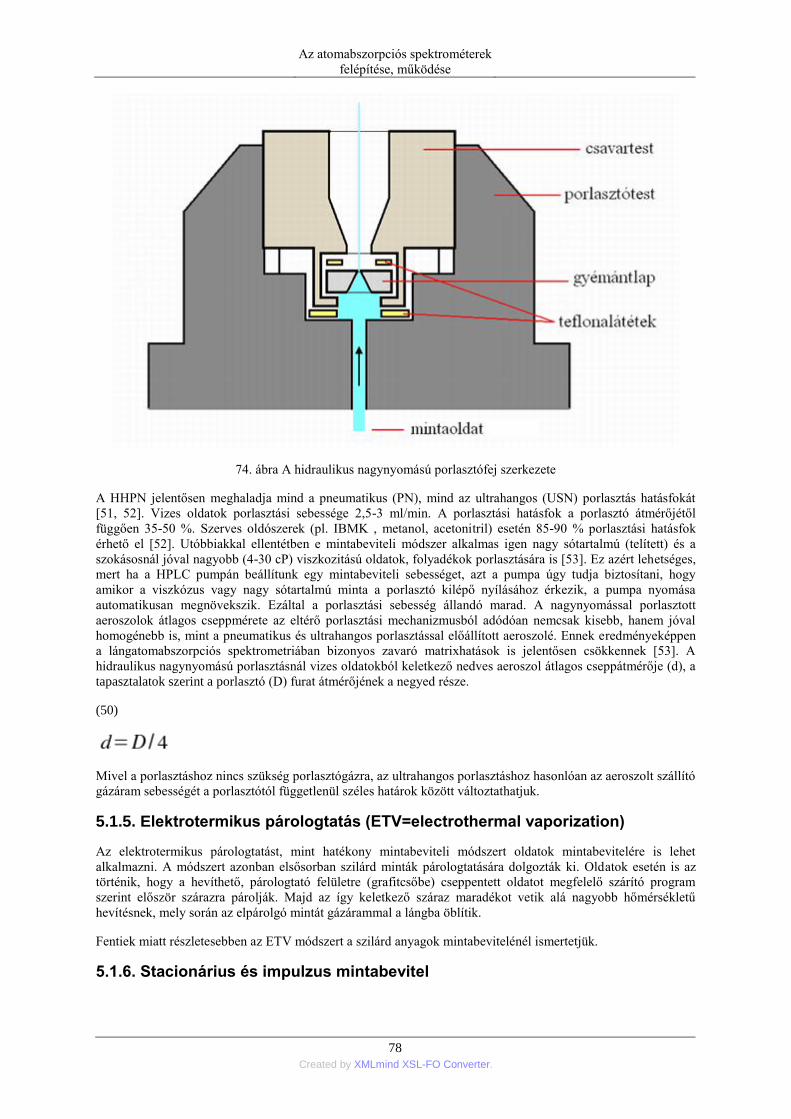
Az atomabszorpciós spektrométerek
felépítése, működése
78 Created by XMLmind XSL-FO Converter.
74. ábra A hidraulikus nagynyomású porlasztófej szerkezete
A HHPN jelentősen meghaladja mind a pneumatikus (PN), mind az ultrahangos (USN) porlasztás hatásfokát
[51, 52]. Vizes oldatok porlasztási sebessége 2,5-3 ml/min. A porlasztási hatásfok a porlasztó átmérőjétől
függően 35-50 %. Szerves oldószerek (pl. IBMK , metanol, acetonitril) esetén 85-90 % porlasztási hatásfok
érhető el [52]. Utóbbiakkal ellentétben e mintabeviteli módszer alkalmas igen nagy sótartalmú (telített) és a
szokásosnál jóval nagyobb (4-30 cP) viszkozitású oldatok, folyadékok porlasztására is [53]. Ez azért lehetséges,
mert ha a HPLC pumpán beállítunk egy mintabeviteli sebességet, azt a pumpa úgy tudja biztosítani, hogy
amikor a viszkózus vagy nagy sótartalmú minta a porlasztó kilépő nyílásához érkezik, a pumpa nyomása
automatikusan megnövekszik. Ezáltal a porlasztási sebesség állandó marad. A nagynyomással porlasztott
aeroszolok átlagos cseppmérete az eltérő porlasztási mechanizmusból adódóan nemcsak kisebb, hanem jóval
homogénebb is, mint a pneumatikus és ultrahangos porlasztással előállított aeroszolé. Ennek eredményeképpen
a lángatomabszorpciós spektrometriában bizonyos zavaró matrixhatások is jelentősen csökkennek [53]. A
hidraulikus nagynyomású porlasztásnál vizes oldatokból keletkező nedves aeroszol átlagos cseppátmérője (d), a
tapasztalatok szerint a porlasztó (D) furat átmérőjének a negyed része.
(50)
Mivel a porlasztáshoz nincs szükség porlasztógázra, az ultrahangos porlasztáshoz hasonlóan az aeroszolt szállító
gázáram sebességét a porlasztótól függetlenül széles határok között változtathatjuk.
5.1.5. Elektrotermikus párologtatás (ETV=electrothermal vaporization)
Az elektrotermikus párologtatást, mint hatékony mintabeviteli módszert oldatok mintabevitelére is lehet
alkalmazni. A módszert azonban elsősorban szilárd minták párologtatására dolgozták ki. Oldatok esetén is az
történik, hogy a hevíthető, párologtató felületre (grafitcsőbe) cseppentett oldatot megfelelő szárító program
szerint először szárazra párolják. Majd az így keletkező száraz maradékot vetik alá nagyobb hőmérsékletű
hevítésnek, mely során az elpárolgó mintát gázárammal a lángba öblítik.
Fentiek miatt részletesebben az ETV módszert a szilárd anyagok mintabevitelénél ismertetjük.
5.1.6. Stacionárius és impulzus mintabevitel
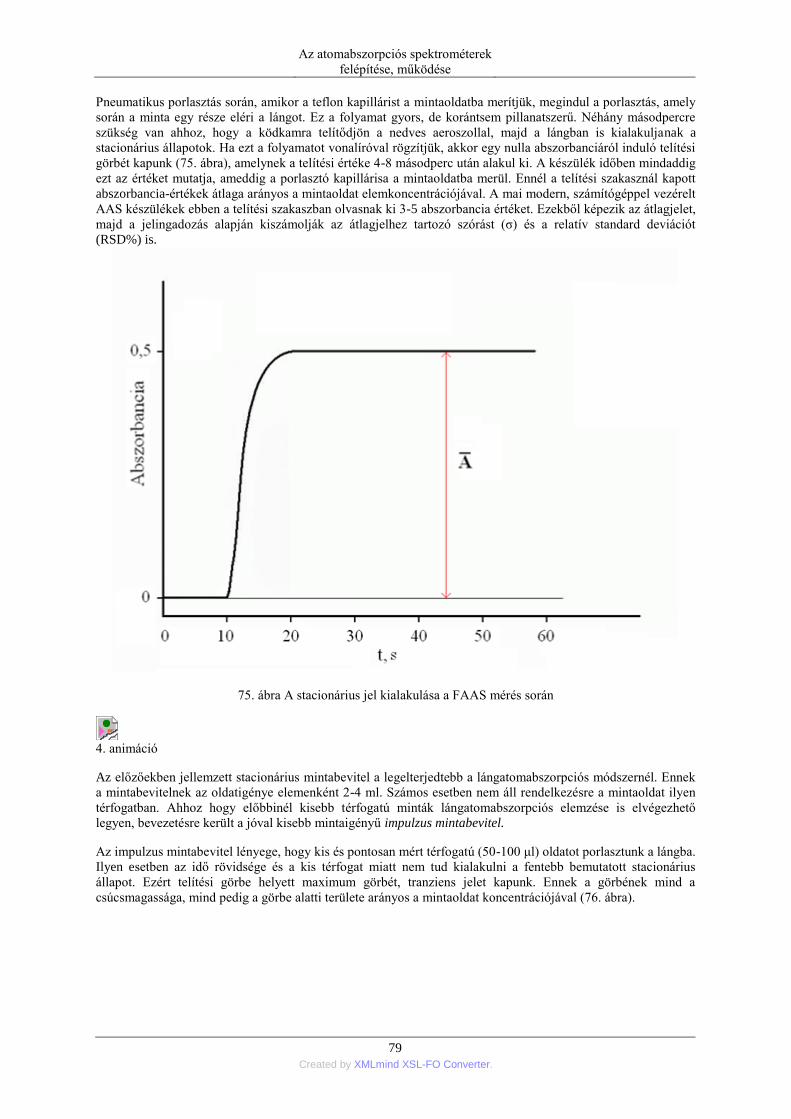
Az atomabszorpciós spektrométerek
felépítése, működése
79 Created by XMLmind XSL-FO Converter.
Pneumatikus porlasztás során, amikor a teflon kapillárist a mintaoldatba merítjük, megindul a porlasztás, amely
során a minta egy része eléri a lángot. Ez a folyamat gyors, de korántsem pillanatszerű. Néhány másodpercre
szükség van ahhoz, hogy a ködkamra telítődjön a nedves aeroszollal, majd a lángban is kialakuljanak a
stacionárius állapotok. Ha ezt a folyamatot vonalíróval rögzítjük, akkor egy nulla abszorbanciáról induló telítési
görbét kapunk (75. ábra), amelynek a telítési értéke 4-8 másodperc után alakul ki. A készülék időben mindaddig
ezt az értéket mutatja, ameddig a porlasztó kapillárisa a mintaoldatba merül. Ennél a telítési szakasznál kapott
abszorbancia-értékek átlaga arányos a mintaoldat elemkoncentrációjával. A mai modern, számítógéppel vezérelt
AAS készülékek ebben a telítési szakaszban olvasnak ki 3-5 abszorbancia értéket. Ezekből képezik az átlagjelet,
majd a jelingadozás alapján kiszámolják az átlagjelhez tartozó szórást (σ) és a relatív standard deviációt
(RSD%) is.
75. ábra A stacionárius jel kialakulása a FAAS mérés során
4. animáció
Az előzőekben jellemzett stacionárius mintabevitel a legelterjedtebb a lángatomabszorpciós módszernél. Ennek
a mintabevitelnek az oldatigénye elemenként 2-4 ml. Számos esetben nem áll rendelkezésre a mintaoldat ilyen
térfogatban. Ahhoz hogy előbbinél kisebb térfogatú minták lángatomabszorpciós elemzése is elvégezhető
legyen, bevezetésre került a jóval kisebb mintaigényű impulzus mintabevitel.
Az impulzus mintabevitel lényege, hogy kis és pontosan mért térfogatú (50-100 μl) oldatot porlasztunk a lángba.
Ilyen esetben az idő rövidsége és a kis térfogat miatt nem tud kialakulni a fentebb bemutatott stacionárius
állapot. Ezért telítési görbe helyett maximum görbét, tranziens jelet kapunk. Ennek a görbének mind a
csúcsmagassága, mind pedig a görbe alatti területe arányos a mintaoldat koncentrációjával (76. ábra).
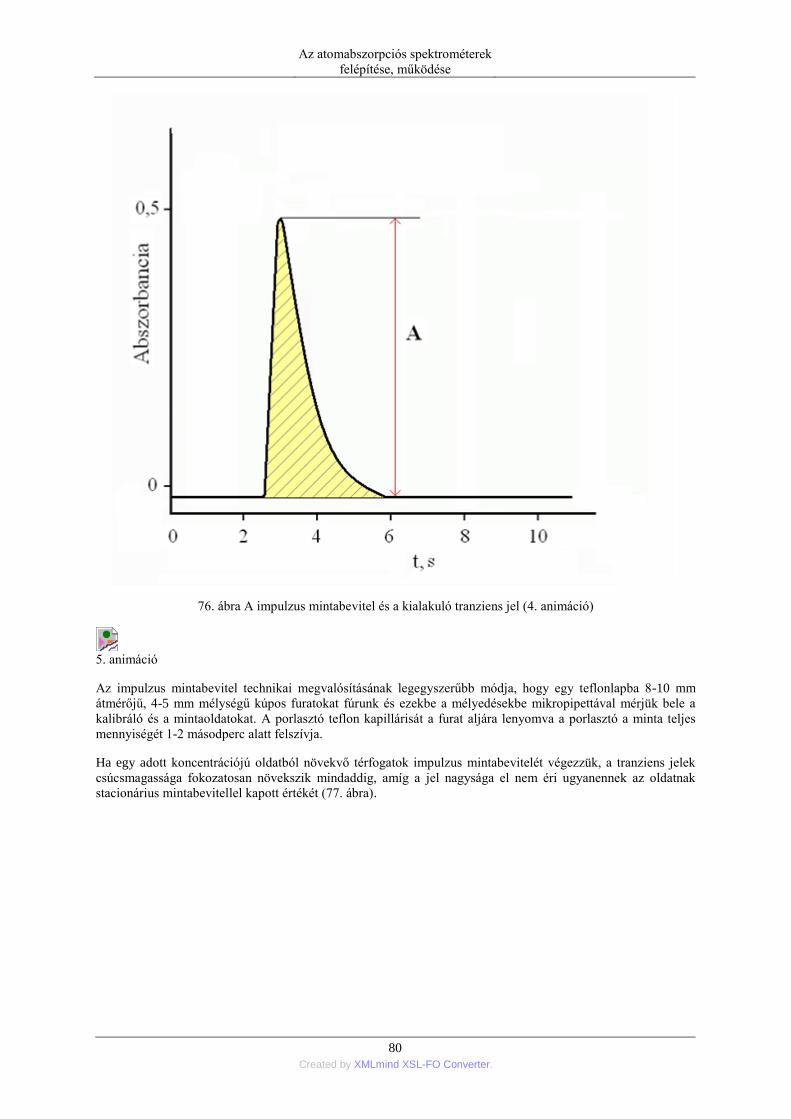
Az atomabszorpciós spektrométerek
felépítése, működése
80 Created by XMLmind XSL-FO Converter.
76. ábra A impulzus mintabevitel és a kialakuló tranziens jel (4. animáció)
5. animáció
Az impulzus mintabevitel technikai megvalósításának legegyszerűbb módja, hogy egy teflonlapba 8-10 mm
átmérőjű, 4-5 mm mélységű kúpos furatokat fúrunk és ezekbe a mélyedésekbe mikropipettával mérjük bele a
kalibráló és a mintaoldatokat. A porlasztó teflon kapillárisát a furat aljára lenyomva a porlasztó a minta teljes
mennyiségét 1-2 másodperc alatt felszívja.
Ha egy adott koncentrációjú oldatból növekvő térfogatok impulzus mintabevitelét végezzük, a tranziens jelek
csúcsmagassága fokozatosan növekszik mindaddig, amíg a jel nagysága el nem éri ugyanennek az oldatnak
stacionárius mintabevitellel kapott értékét (77. ábra).
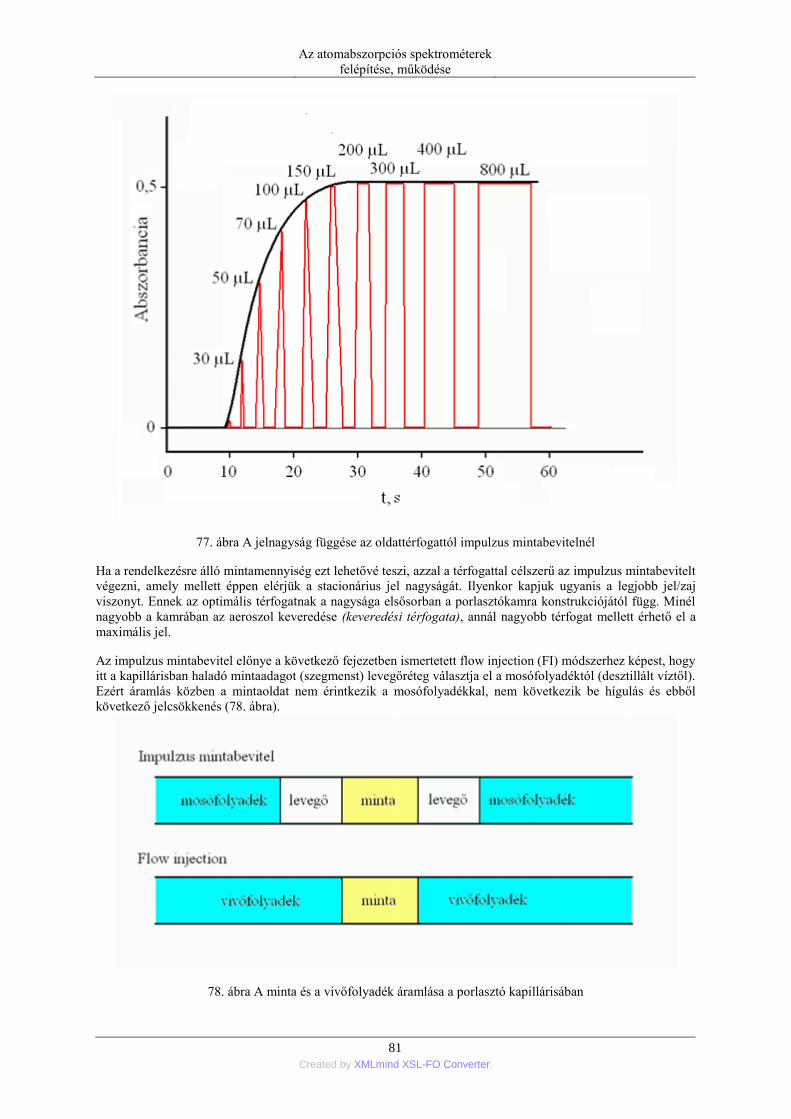
Az atomabszorpciós spektrométerek
felépítése, működése
81 Created by XMLmind XSL-FO Converter.
77. ábra A jelnagyság függése az oldattérfogattól impulzus mintabevitelnél
Ha a rendelkezésre álló mintamennyiség ezt lehetővé teszi, azzal a térfogattal célszerű az impulzus mintabevitelt
végezni, amely mellett éppen elérjük a stacionárius jel nagyságát. Ilyenkor kapjuk ugyanis a legjobb jel/zaj
viszonyt. Ennek az optimális térfogatnak a nagysága elsősorban a porlasztókamra konstrukciójától függ. Minél
nagyobb a kamrában az aeroszol keveredése (keveredési térfogata), annál nagyobb térfogat mellett érhető el a
maximális jel.
Az impulzus mintabevitel előnye a következő fejezetben ismertetett flow injection (FI) módszerhez képest, hogy
itt a kapillárisban haladó mintaadagot (szegmenst) levegőréteg választja el a mosófolyadéktól (desztillált víztől).
Ezért áramlás közben a mintaoldat nem érintkezik a mosófolyadékkal, nem következik be hígulás és ebből
következő jelcsökkenés (78. ábra).
78. ábra A minta és a vivőfolyadék áramlása a porlasztó kapillárisában

Az atomabszorpciós spektrométerek
felépítése, működése
82 Created by XMLmind XSL-FO Converter.
Az impulzus mintabevitel automatizálható. Erre két lehetőség kínálkozik. Vagy a mintatartó edényekbe mért
térfogatú oldatot helyezünk és a mintavevő kapilláris karját úgy állítjuk be, hogy a hajlékony kapilláris éppen
leérjen a mintatartó edények aljára. A másik lehetőség, hogy pontos bemérés nélkül 0,5–1,0 ml térfogatú
mintaoldatokat öntünk a mintatartó edényekbe és a mintavevő automata merülési idejét állítjuk be úgy, hogy
mindig például 100 μL mintát szívjon fel. Mivel ilyen kis oldattérfogat lángba juttatásához csupán 1 – 2
másodpercre van szükség, azaz a mérésidő mintánként néhány másodperc, 1 óra alatt akár 800 minta elemzése
is elvégezhető. Ezért az automatizált impulzus mintabevitellel működő FAAS módszer az egyik leggyorsabb, ha
nem a leggyorsabb technika a műszeres analitikában.
5.1.7. Flow injection mintabevitel (FI)
Ennek az igen széles körben elterjedt mintabeviteli technikának egyelőre nincs pontos magyar elnevezése. A
flow injection lényegét a módszer kidolgozói, Ruzicka és Hansen [54] fogalmazták meg először. A FI az a
technika, amely folyadék mintát injektál mozgó, megfelelő összetételű, nem-szegmentált folyamatosan áramló
vivőfolyadékáramba. Az így bevitt minta a vivőfolyadékban egy zónát képez, amely a detektor felé halad. A
detektor folyamatosan detektálja az abszorbanciát vagy más fizikai paramétert és folyamatosan követi az
átfolyásos cellán a minta változásait. Később, azáltal, hogy a módszert egyre többféle célra alkalmazták Fang
[55] a flow injection más vonatkozásait emelte ki. Szerinte a FI egy nem-kromatográfiás áramlásos technika
mennyiségi analízis céljából, olyan módon, hogy a minta- és reagens-zónákat reprodukálhatóan vezetjük egy
áramló termodinamikailag nem-egyensúlyi rendszerben.
Ahhoz, hogy a mintaoldatot pontosan mért térfogatban juttassunk be egy kapillárisban folyamatosan mozgó
folyadékáramba, bemérőcsapra (valve) és pontos térfogatú mintahurokra (loop) van szükség. A vivő folyadék
egyenletes áramlását a detektor felé perisztaltikus pumpa biztosítja. A mintahurok ismert belső átmérőjű
kapilláris, amelynek a hosszúságával változtatjuk az injektálni kívánt mintatérfogatot. A FI mintabevitelhez
leggyakrabban 50, 100, 200 μL térfogatú mintahurkot használunk, de létezik igen nagynak számító 1, 2 sőt 5
mL- es mintahurok is. A vivőfolyadékot szállító kapilláris és a mintahurok anyaga ─ tekintettel arra, hogy
nyomelem analízisre használjuk ─ fémmentes műanyagból készül. A bemérőcsapnak a mintaoldattal érintkező
bélésanyaga ugyancsak műanyag (teflon, PEEK). A bemérőcsap működését a 79. ábrán mutatjuk be.
79. ábra A bemérőcsap működése a flow injection mintabevitelhez
A FI módszerhez hat csatlakozási ponttal (porttal) rendelkező kétállású bemérőcsapot használunk. A 79. ábrán
látható, hogy a vivőfolyadék az 1. ponton lép be és a 2. ponton távozik. A 3. és 6. pont között kapcsolódik a
csaphoz a mintahurok. Az 5 ponton keresztül tölthető meg a hurok a mintaoldattal. A mintahurokba töltött oldat
fölöslege a 4. ponton távozik a lefolyóba. A bemérőcsap leglényegesebb része két egymáshoz szorosan
illeszkedő, de egymáson elfordulni képes műanyag korong. A két korong egyike a fentebb említett
csatlakozásokat biztosító hat furatot tartalmazza, a hozzá szorosan tapadó másik korong felületén pedig három
vájat található, amelynek mindegyike a két-két szomszédos furatot köti össze. Így az egyik furaton belépő oldat
a szomszédos furaton keresztül akadálytalanul távozhat. A 79A. ábrán a bemérőcsapnak azt az állását mutatjuk
be, amely mellett a huroknak a mintaoldattal történő megtöltése elvégezhető. Látható, hogy ebben az állásban az
egyik vájat az 1. és 2. furatot köti össze. A vivőfolyadék itt halad keresztül a bemérőcsapon. Eközben az 5.
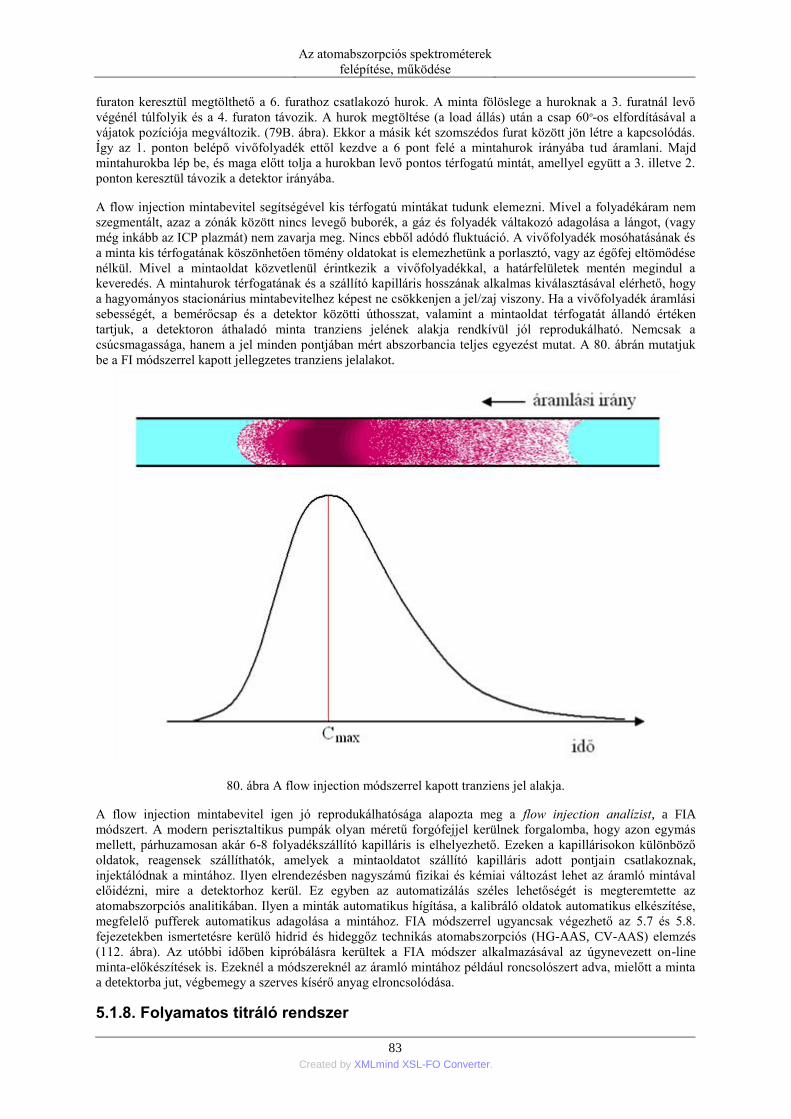
Az atomabszorpciós spektrométerek
felépítése, működése
83 Created by XMLmind XSL-FO Converter.
furaton keresztül megtölthető a 6. furathoz csatlakozó hurok. A minta fölöslege a huroknak a 3. furatnál levő
végénél túlfolyik és a 4. furaton távozik. A hurok megtöltése (a load állás) után a csap 60o-os elfordításával a
vájatok pozíciója megváltozik. (79B. ábra). Ekkor a másik két szomszédos furat között jön létre a kapcsolódás.
Így az 1. ponton belépő vivőfolyadék ettől kezdve a 6 pont felé a mintahurok irányába tud áramlani. Majd
mintahurokba lép be, és maga előtt tolja a hurokban levő pontos térfogatú mintát, amellyel együtt a 3. illetve 2.
ponton keresztül távozik a detektor irányába.
A flow injection mintabevitel segítségével kis térfogatú mintákat tudunk elemezni. Mivel a folyadékáram nem
szegmentált, azaz a zónák között nincs levegő buborék, a gáz és folyadék váltakozó adagolása a lángot, (vagy
még inkább az ICP plazmát) nem zavarja meg. Nincs ebből adódó fluktuáció. A vivőfolyadék mosóhatásának és
a minta kis térfogatának köszönhetően tömény oldatokat is elemezhetünk a porlasztó, vagy az égőfej eltömődése
nélkül. Mivel a mintaoldat közvetlenül érintkezik a vivőfolyadékkal, a határfelületek mentén megindul a
keveredés. A mintahurok térfogatának és a szállító kapilláris hosszának alkalmas kiválasztásával elérhető, hogy
a hagyományos stacionárius mintabevitelhez képest ne csökkenjen a jel/zaj viszony. Ha a vivőfolyadék áramlási
sebességét, a bemérőcsap és a detektor közötti úthosszat, valamint a mintaoldat térfogatát állandó értéken
tartjuk, a detektoron áthaladó minta tranziens jelének alakja rendkívül jól reprodukálható. Nemcsak a
csúcsmagassága, hanem a jel minden pontjában mért abszorbancia teljes egyezést mutat. A 80. ábrán mutatjuk
be a FI módszerrel kapott jellegzetes tranziens jelalakot.
80. ábra A flow injection módszerrel kapott tranziens jel alakja.
A flow injection mintabevitel igen jó reprodukálhatósága alapozta meg a flow injection analízist, a FIA
módszert. A modern perisztaltikus pumpák olyan méretű forgófejjel kerülnek forgalomba, hogy azon egymás
mellett, párhuzamosan akár 6-8 folyadékszállító kapilláris is elhelyezhető. Ezeken a kapillárisokon különböző
oldatok, reagensek szállíthatók, amelyek a mintaoldatot szállító kapilláris adott pontjain csatlakoznak,
injektálódnak a mintához. Ilyen elrendezésben nagyszámú fizikai és kémiai változást lehet az áramló mintával
előidézni, mire a detektorhoz kerül. Ez egyben az automatizálás széles lehetőségét is megteremtette az
atomabszorpciós analitikában. Ilyen a minták automatikus hígítása, a kalibráló oldatok automatikus elkészítése,
megfelelő pufferek automatikus adagolása a mintához. FIA módszerrel ugyancsak végezhető az 5.7 és 5.8.
fejezetekben ismertetésre kerülő hidrid és hideggőz technikás atomabszorpciós (HG-AAS, CV-AAS) elemzés
(112. ábra). Az utóbbi időben kipróbálásra kerültek a FIA módszer alkalmazásával az úgynevezett on-line
minta-előkészítések is. Ezeknél a módszereknél az áramló mintához például roncsolószert adva, mielőtt a minta
a detektorba jut, végbemegy a szerves kísérő anyag elroncsolódása.
5.1.8. Folyamatos titráló rendszer

Az atomabszorpciós spektrométerek
felépítése, működése
84 Created by XMLmind XSL-FO Converter.
A mintabevitel sajátos módszerének tekinthető az az eljárás, amely során változik a lángba jutó komponensek
koncentráció-aránya. Ezzel a módszerrel lehetőség van változó koncentráció-arányok mellett lejátszódó
lángfolyamatok, kölcsönhatások, zavaró hatások folyamatos követésére.
Az atomabszorpciós spektrometriában (AAS) vizsgált elem és a kísérő idegen anyagok (kationok, anionok,
szerves anyagok, sók) lángban fellépő kölcsönhatását hagyományosan úgy tanulmányozzuk, hogy az elem
koncentrációját állandó értéken tartva olyan oldatsorozatot készítünk, amelyben az egyéb komponensek
koncentrációja fokozatosan növekszik. A kölcsönhatásra jellemző görbét úgy nyerjük, hogy a vizsgált elem
abszorbancia-értékét az egyéb alkotók koncentrációjának függvényében ábrázoljuk. A kölcsönhatásokat
lángparaméterektől, illetve a reagáló partnerek minőségétől függően számos esetben szűk koncentráció-
tartományban éles abszorbancia-változások kísérik. E gyors változásokat azonban a hagyományos vizsgálati
módszerrel nem tudjuk pontosan követni. Ezért hiteles kölcsönhatás vagy zavaró hatás görbéket csak úgy
nyerhetünk, ha olyan módszert alkalmazunk, amely lehetővé teszi a vizsgált elemre ható komponens
koncentrációjának folyamatos változását és a mért abszorbancia ezzel egyidejű regisztrálását.
A tapasztalatok szerint a lángban fellépő kölcsönhatások tanulmányozása szempontjából célszerű a reagáló
partnerek koncentrációját lineárisan vagy exponenciálisan változtatni. Ezért olyan készülékeket mutatunk be,
amelyekben a titrálás során e függvénykapcsolatok érvényesülnek. Mindkét bemutatott titráló rendszer közös
vonása, hogy az egyéb tényezőket (térfogatot, koncentrációt) rögzítve a titrálás körülményeit egyetlen
paraméter: az alkalmazott atomabszorpciós spektrométer porlasztási sebessége (Fl) szabja meg.
Lineáris titráló készülék
A 81. ábrán látható a lineáris titráló készülék, amelyben a vizsgált elem állandó koncentrációja mellett a kísérő
komponens (savak, sók, komplexképző ligandumok, összetett matrix) koncentrációja időben közel lineárisan
növekszik. Láthatóan ez két egymáshoz forrasztott üvegedényből áll, melyeket egy 1 mm belső átmérőjű
üvegkapilláris köt össze. A kapillárisban 0,02 – 0,03 cm3 levegőoszlop biztosítja, hogy az oldatok keveredése a
titrálás előtt ne induljon meg.

Az atomabszorpciós spektrométerek
felépítése, működése
85 Created by XMLmind XSL-FO Converter.
81. ábra Lineáris AAS titráló készülék
Mivel a kapilláris hegye 1 mm-re helyezkedik el a titráló oldatot tartalmazó edény aljától, az oldat gyakorlatilag
maradék nélkül átszívható a keverőedényben levő oldatba. A keverőedényből az oldat elszívása a csiszolatos
dugóba gumidugóval rögzített fémkapillárison keresztül történik. A kapilláris belső átmérője 0,3 mm, anyaga
platina–irídium ötvözet, de elkészíthető az injekciós tűk anyagaként használt saválló króm–nikkel acélból is. A
kapillárist az AAS porlasztóval 0,3 mm belső átmérőjű teflon kapilláris köti össze.
A titrálást a következőképpen végezzük: A keverőedénybe a vizsgálandó A komponens oldatából 55 cm mérünk
be. A másik edénybe 20 cm pipettázunk az A + B oldatelegyből, amely az A komponensre nézve pontosan
azonos koncentrációjú, mint a keverőedényben levő oldat. Így az A komponens koncentrációja a titrálás teljes
időtartama alatt nem változik. A mágneses keverő megindítása után a csiszolatos dugót mért időpillanatban úgy
helyezzük bele a keverőedénybe, hogy a csiszolatokon levő két furat egybeessék. Így a keverőedény nyitott
marad, csak az A komponens oldata jut az AAS készülék lángjába. Ekkor a regisztráló az A kölcsönhatásmentes
jelét rögzíti. A porlasztási sebességet pontosan ismerve meghatározott az az időtartam, amely alatt 5 cm3-rel
csökken a keverőedényben az oldat térfogata. Ebben a pillanatban a dugót 90°-kal elfordítva az edény zárttá
válik és megindul a titrálás. A titrálás alatt a B komponens koncentrációja időben az (51) összefüggés szerint
növekszik.
(51)
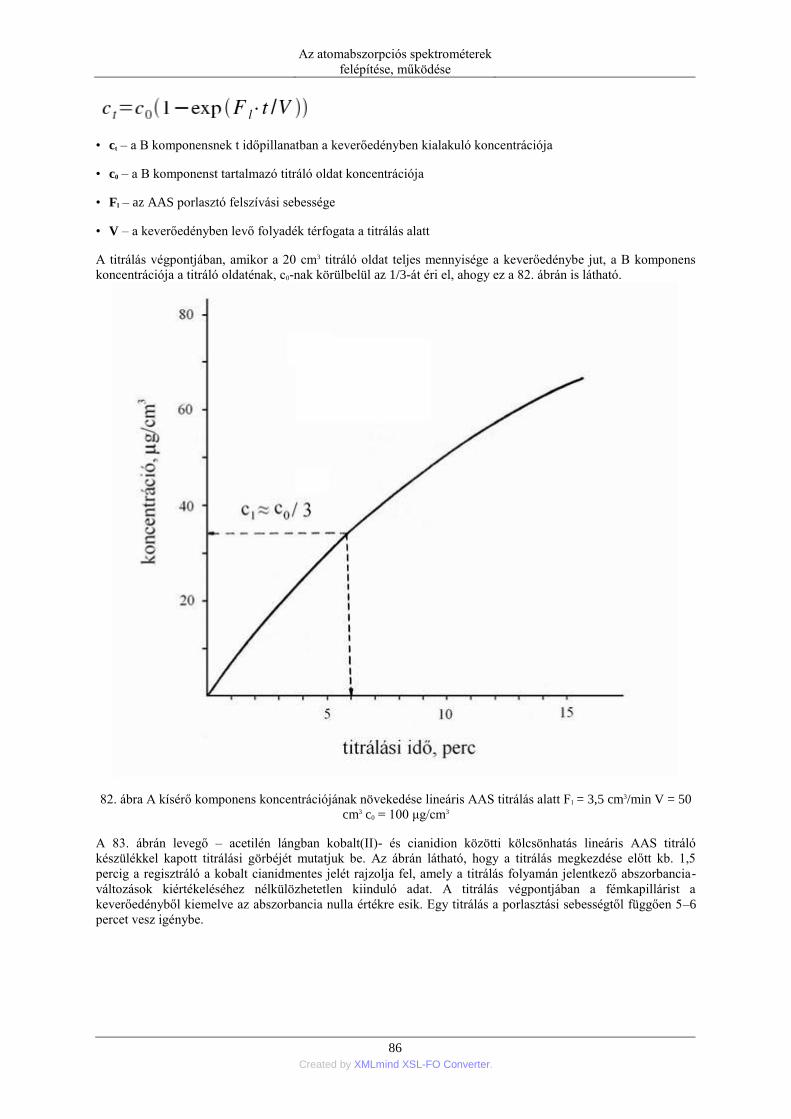
Az atomabszorpciós spektrométerek
felépítése, működése
86 Created by XMLmind XSL-FO Converter.
• ct – a B komponensnek t időpillanatban a keverőedényben kialakuló koncentrációja
• c0 – a B komponenst tartalmazó titráló oldat koncentrációja
• Fl – az AAS porlasztó felszívási sebessége
• V – a keverőedényben levő folyadék térfogata a titrálás alatt
A titrálás végpontjában, amikor a 20 cm3 titráló oldat teljes mennyisége a keverőedénybe jut, a B komponens
koncentrációja a titráló oldaténak, c0-nak körülbelül az 1/3-át éri el, ahogy ez a 82. ábrán is látható.
82. ábra A kísérő komponens koncentrációjának növekedése lineáris AAS titrálás alatt Fl = 3,5 cm3/min V = 50
cm3 c0 = 100 μg/cm3
A 83. ábrán levegő – acetilén lángban kobalt(II)- és cianidion közötti kölcsönhatás lineáris AAS titráló
készülékkel kapott titrálási görbéjét mutatjuk be. Az ábrán látható, hogy a titrálás megkezdése előtt kb. 1,5
percig a regisztráló a kobalt cianidmentes jelét rajzolja fel, amely a titrálás folyamán jelentkező abszorbancia-
változások kiértékeléséhez nélkülözhetetlen kiinduló adat. A titrálás végpontjában a fémkapillárist a
keverőedényből kiemelve az abszorbancia nulla értékre esik. Egy titrálás a porlasztási sebességtől függően 5–6
percet vesz igénybe.
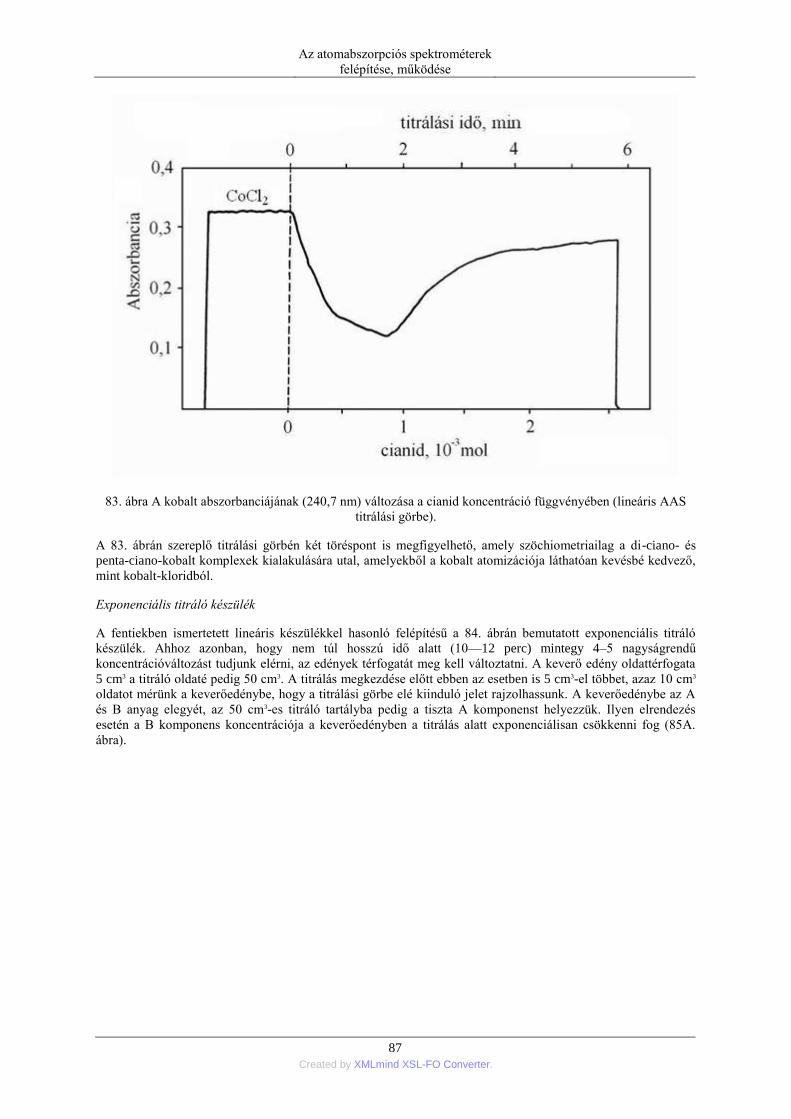
Az atomabszorpciós spektrométerek
felépítése, működése
87 Created by XMLmind XSL-FO Converter.
83. ábra A kobalt abszorbanciájának (240,7 nm) változása a cianid koncentráció függvényében (lineáris AAS
titrálási görbe).
A 83. ábrán szereplő titrálási görbén két töréspont is megfigyelhető, amely szöchiometriailag a di-ciano- és
penta-ciano-kobalt komplexek kialakulására utal, amelyekből a kobalt atomizációja láthatóan kevésbé kedvező,
mint kobalt-kloridból.
Exponenciális titráló készülék
A fentiekben ismertetett lineáris készülékkel hasonló felépítésű a 84. ábrán bemutatott exponenciális titráló
készülék. Ahhoz azonban, hogy nem túl hosszú idő alatt (10—12 perc) mintegy 4–5 nagyságrendű
koncentrációváltozást tudjunk elérni, az edények térfogatát meg kell változtatni. A keverő edény oldattérfogata
5 cm3 a titráló oldaté pedig 50 cm3. A titrálás megkezdése előtt ebben az esetben is 5 cm3-el többet, azaz 10 cm3
oldatot mérünk a keverőedénybe, hogy a titrálási görbe elé kiinduló jelet rajzolhassunk. A keverőedénybe az A
és B anyag elegyét, az 50 cm3-es titráló tartályba pedig a tiszta A komponenst helyezzük. Ilyen elrendezés
esetén a B komponens koncentrációja a keverőedényben a titrálás alatt exponenciálisan csökkenni fog (85A.
ábra).
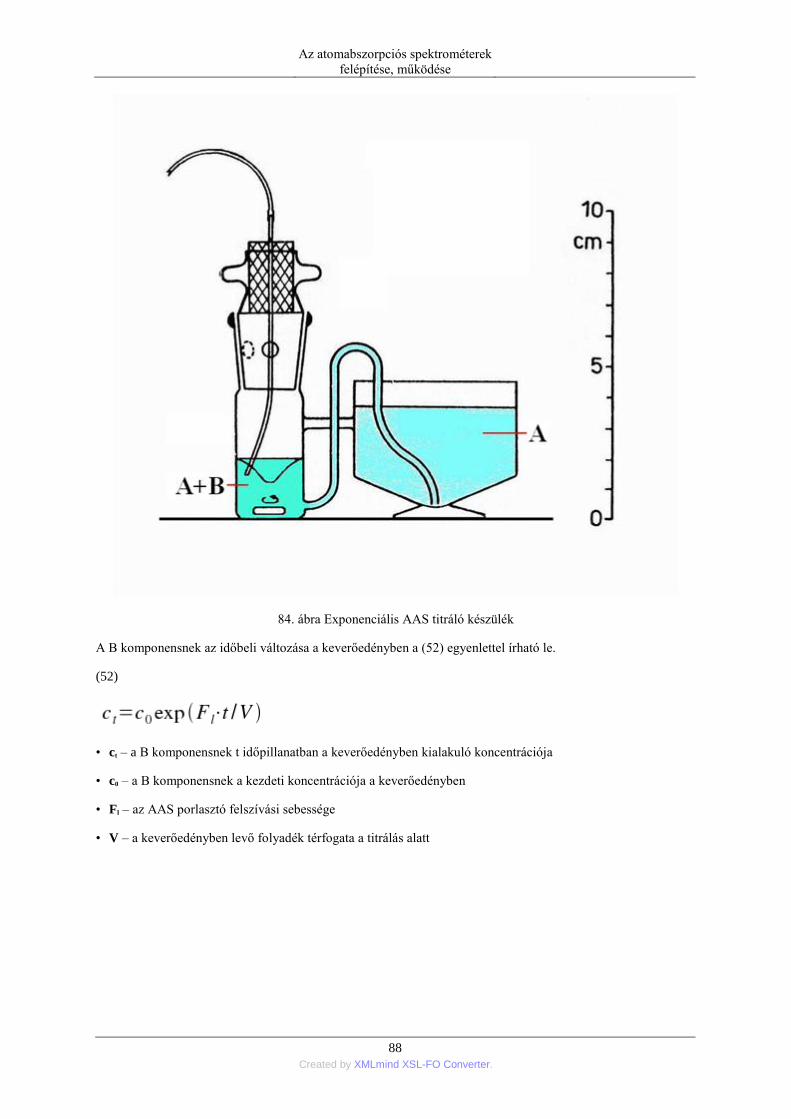
Az atomabszorpciós spektrométerek
felépítése, működése
88 Created by XMLmind XSL-FO Converter.
84. ábra Exponenciális AAS titráló készülék
A B komponensnek az időbeli változása a keverőedényben a (52) egyenlettel írható le.
(52)
• ct – a B komponensnek t időpillanatban a keverőedényben kialakuló koncentrációja
• c0 – a B komponensnek a kezdeti koncentrációja a keverőedényben
• Fl – az AAS porlasztó felszívási sebessége
• V – a keverőedényben levő folyadék térfogata a titrálás alatt
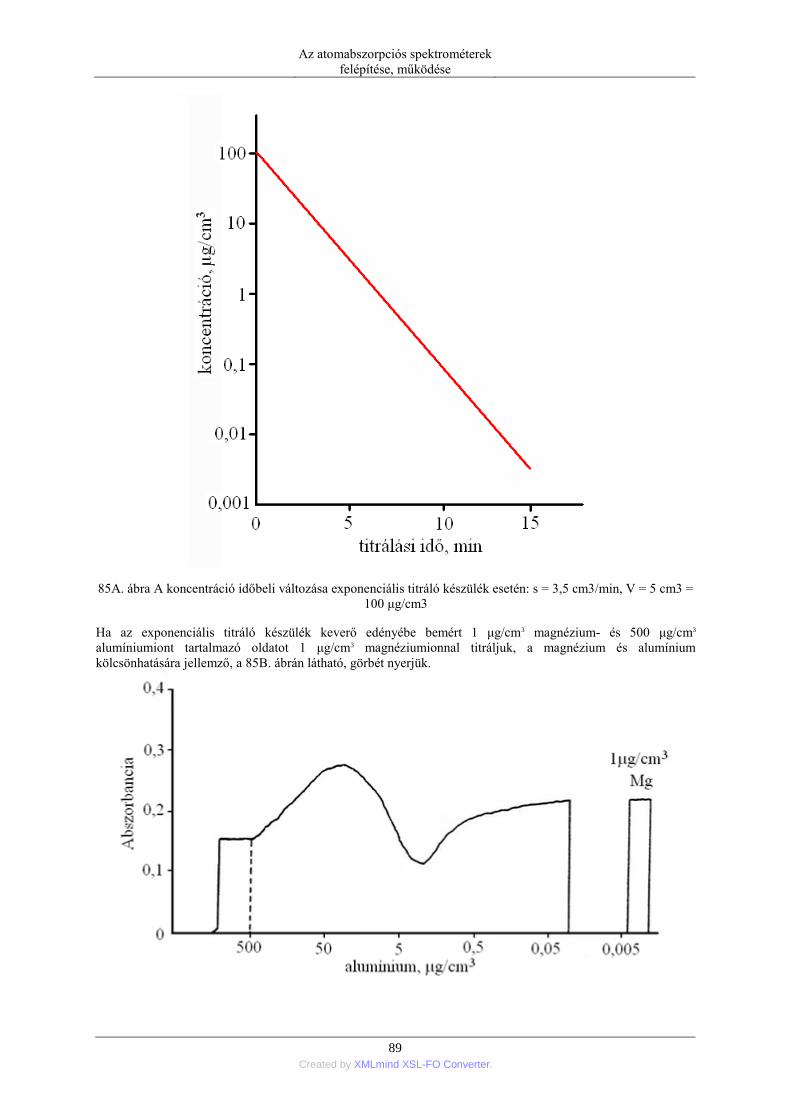
Az atomabszorpciós spektrométerek
felépítése, működése
89 Created by XMLmind XSL-FO Converter.
85A. ábra A koncentráció időbeli változása exponenciális titráló készülék esetén: s = 3,5 cm3/min, V = 5 cm3 =
100 μg/cm3
Ha az exponenciális titráló készülék keverő edényébe bemért 1 μg/cm3 magnézium- és 500 μg/cm3
alumíniumiont tartalmazó oldatot 1 μg/cm3 magnéziumionnal titráljuk, a magnézium és alumínium
kölcsönhatására jellemző, a 85B. ábrán látható, görbét nyerjük.

Az atomabszorpciós spektrométerek
felépítése, működése
90 Created by XMLmind XSL-FO Converter.
85B. ábra A magnézium abszorbanciájának (285,2 nm) változása az alumínium koncentráció függvényében
(exponenciális AAS titrálási görbe)
Mivel utóbbi titrálásnál a kiinduló mérési adat az elegyhez tartozik, a tiszta komponens abszorbancia-értékét a
titrálás után külön vesszük fel. Mint az a 85B. ábrából is kitűnik, az exponenciális titrálás során a regisztrátum
abszcissza tengelyén a koncentrációváltozás közvetlenül logaritmusos léptékben olvasható le. A titrálási görbe
alakjának és a magnézium–alumínium kölcsönhatás mechanizmusának a magyarázatát a 6. fejezetben, a zavaró
hatások keretében tárgyaljuk.
5.2. Szilárd anyagok mintabevitele
Az oldatos mintabevitel mellett az atomabszorpciós spektrometriában is felmerült a szilárd anyagok közvetlen
elemzésének igénye. A szilárd minták elemzése mellett több fontos érv szól. Vannak olyan szilárd minták,
amelyek nagyon nehezen vihetők oldatba. Ezek előkészítése oldatos elemzésre igen idő- és eszközigényes
művelet. A minta az oldatba vitel során az oldáshoz használt vegyszer révén szennyeződhet. Az oldással a
vizsgált elem koncentrációja jelentősen (nagyságrendekkel) csökkenhet, miután a szilárd mintából maximum 1-
2 %-os oldatot lehet készíteni pneumatikus oldatporlasztáshoz. Esetenként a rendelkezésre álló minta
mennyisége igen kicsi, amelynél különösen fontos, hogy oldással ne hígítsuk tovább.
Fenti érvek indokolttá teszik, hogy módszereket dolgozzunk ki szilárd minta atomabszorpciós spektrometriás
elemzésére. E tekintetben különböző technikai megoldást igényel, ha a szilárd mintát lángatomabszorpciós vagy
grafitkemencés atomabszorpciós módszerrel elemezzük. A lángba történő akár folyamatos, akár impulzusszerű
mintabevitel esetén a szilárd mintát olyan alakra kell hozni, hogy az a lángba gázárammal reprodukálhatóan
bevihető legyen. Ennek megfelelően a szilárd minták elemzésére alakultak ki az ív-láng, a lézer ablációs és
elektrotermikus elpárologtatásos módszerek.
5.2.1. Ív- és szikraporlasztás
A szilárd minták esetén is az oldatokhoz hasonló módon meg kell oldani a minta reprodukálható bejuttatását a
lángba. Jones és munkatársai [56, 57] fémötvözetek elemzésére egy olyan aeroszol generátort fejlesztettek ki,
amelyből az egyenáramú ív termékei gázárammal reprodukálhatóan elvezethetők. Az eredetileg spektrográfiás
célra kialakított generátort Winge és munkatársai [58] sikeresen kombinálták atomabszorpciós spektrométerrel,
létrehozva az első ív–láng kombinált AAS eljárást, amely fémötvözetek gyors elemzésére volt alkalmas. Előbbi
kvázi–stacionárius működésű rendszer mellett Kántor és munkatársai [59, 60] nehezen oldatba vihető
nemvezető anyagokra először dolgoztak ki ív-láng kombinált módszert, és elsőként igazolták, hogy nem-
stacionárius párologtatással előállított aeroszolt lángba juttatva, az emissziós vagy abszorpciós jel időintegrálja
arányos az elpárologtatott alkotó tömegével.
A jól szabályzott mintabevitel érdekében a közvetett kombinációjú módszerek terjedtek el. Ezek közös vonása,
hogy az ív- vagy szikraplazma nagy (6000 K) hőmérsékletén a szilárd elektródból kilépő gőzök a hideg (levegő,
nitrogén, argon) vivőgázzal hirtelen érintkezve mikroméretű száraz aeroszollá kondenzálnak. Az így keletkező
száraz aeroszol szemcsemérete olyan, hogy az hosszú úton ülepedés nélkül szállítható. E közvetett
kombinációknál a klasszikusnak számító ív mellett szikrát [61], lézert [62]‚ a fűtött grafit- [63-65] és
kvarckemencét [66, 67] is felhasználják, mint párologtató, aeroszol-előállító egységet. Az utóbbi időben
elterjedő nagyteljesítményű sugárforrások (DCP, ICP, MIP) újabb ösztönzést jelentettek a kombinált eljárások
fejlődésében [68], mivel a lánghoz hasonlóan itt is felvetődik a közönséges oldatoktól eltérő anyagi rendszerek
mintabeviteli problémája.
Példát mutatunk be a nemvezető anyagok egyenáramú ívben furatos grafitelektródból történő elpárologására
valamint fémek és fémötvözetek stacionárius ívporlasztására.
Nemvezető anyagok ívporlasztása
A nemvezető anyagok elemzéséhez a (ásvány, hamvasztási maradék, csont, falevél, szálló és ülepedő por, talaj,
üledék stb.) mintát el kell porítani és 1:1 arányban spektroszkópiai tisztaságú grafitporral keverjük össze. A jól
reprodukálható párolgási feltételek biztosítása érdekében a nemvezető anyag–grafitpor keveréket a Papp és
munkatársai [69, 70] által kidolgozott fenol-formaldehid műgyanta alkoholos oldatával nedvesítjük meg, majd a
keveréket spektroszkópiai tisztaságú grafitelektród furatába tömjük. Az elektródot 1 óra hosszáig 160 oC-on
tartva a gyanta térhálósodik. A hőkezeléssel térhálósított gyanta az íveltetés során megakadályozza a minta
kifúvódását az elektródból, és biztosítja, hogy a minta a töltetből egyenletesen párologjon el. Az így előkészített
elektródokból az egyenáramú íveltetés során az elemek fokozatosan elpárolognak, elfogynak. Ez a folyamat
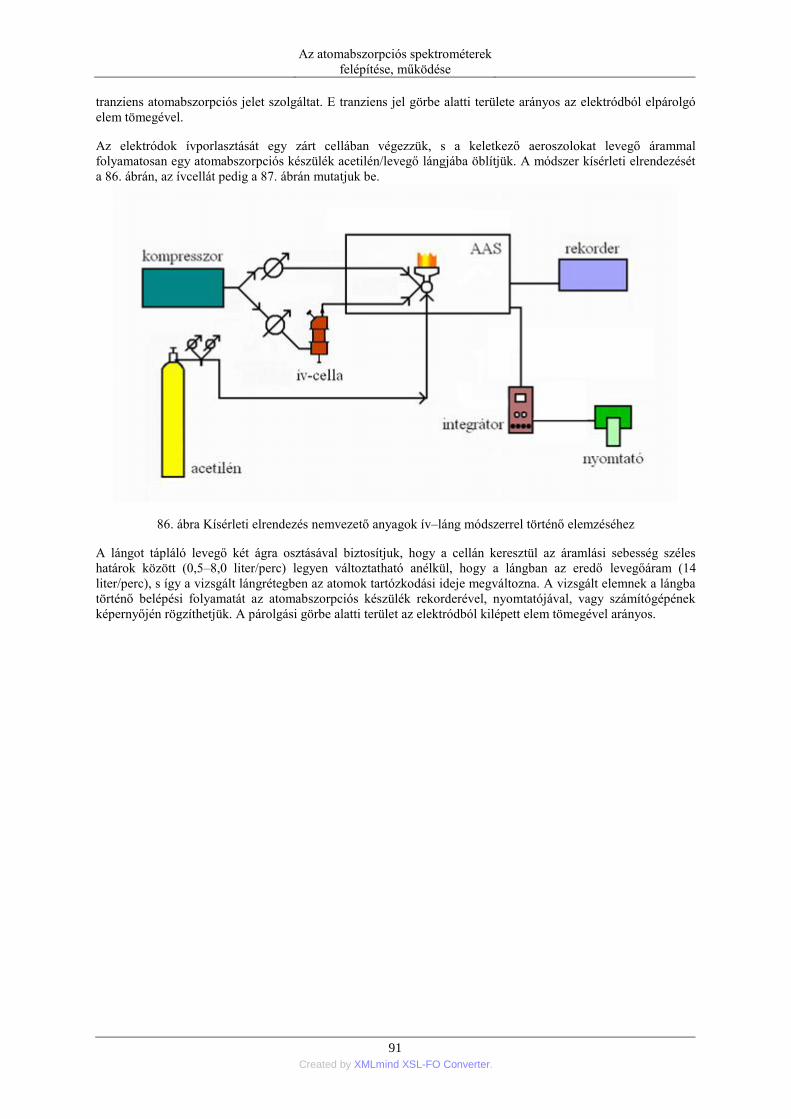
Az atomabszorpciós spektrométerek
felépítése, működése
91 Created by XMLmind XSL-FO Converter.
tranziens atomabszorpciós jelet szolgáltat. E tranziens jel görbe alatti területe arányos az elektródból elpárolgó
elem tömegével.
Az elektródok ívporlasztását egy zárt cellában végezzük, s a keletkező aeroszolokat levegő árammal
folyamatosan egy atomabszorpciós készülék acetilén/levegő lángjába öblítjük. A módszer kísérleti elrendezését
a 86. ábrán, az ívcellát pedig a 87. ábrán mutatjuk be.
86. ábra Kísérleti elrendezés nemvezető anyagok ív–láng módszerrel történő elemzéséhez
A lángot tápláló levegő két ágra osztásával biztosítjuk, hogy a cellán keresztül az áramlási sebesség széles
határok között (0,5–8,0 liter/perc) legyen változtatható anélkül, hogy a lángban az eredő levegőáram (14
liter/perc), s így a vizsgált lángrétegben az atomok tartózkodási ideje megváltozna. A vizsgált elemnek a lángba
történő belépési folyamatát az atomabszorpciós készülék rekorderével, nyomtatójával, vagy számítógépének
képernyőjén rögzíthetjük. A párolgási görbe alatti terület az elektródból kilépett elem tömegével arányos.
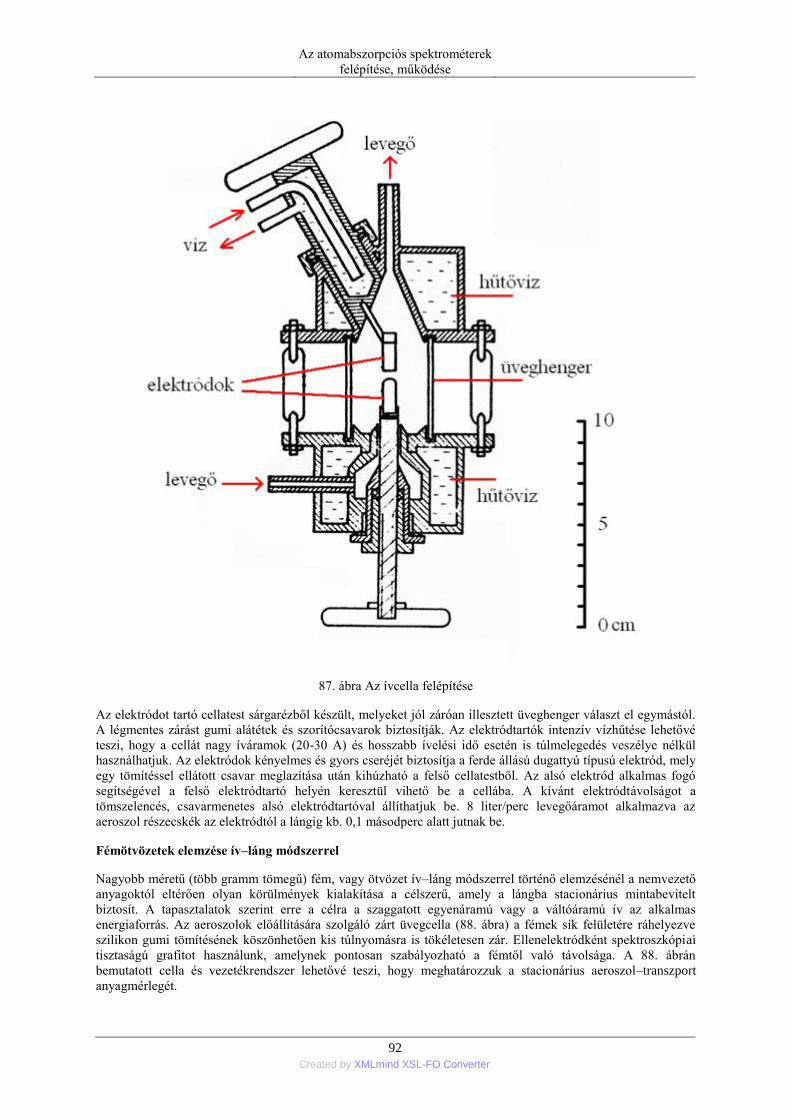
Az atomabszorpciós spektrométerek
felépítése, működése
92 Created by XMLmind XSL-FO Converter.
87. ábra Az ívcella felépítése
Az elektródot tartó cellatest sárgarézből készült, melyeket jól záróan illesztett üveghenger választ el egymástól.
A légmentes zárást gumi alátétek és szorítócsavarok biztosítják. Az elektródtartók intenzív vízhűtése lehetővé
teszi, hogy a cellát nagy íváramok (20-30 A) és hosszabb ívelési idő esetén is túlmelegedés veszélye nélkül
használhatjuk. Az elektródok kényelmes és gyors cseréjét biztosítja a ferde állású dugattyú típusú elektród, mely
egy tömítéssel ellátott csavar meglazítása után kihúzható a felső cellatestből. Az alsó elektród alkalmas fogó
segítségével a felső elektródtartó helyén keresztül vihető be a cellába. A kívánt elektródtávolságot a
tömszelencés, csavarmenetes alsó elektródtartóval állíthatjuk be. 8 liter/perc levegőáramot alkalmazva az
aeroszol részecskék az elektródtól a lángig kb. 0,1 másodperc alatt jutnak be.
Fémötvözetek elemzése ív–láng módszerrel
Nagyobb méretű (több gramm tömegű) fém, vagy ötvözet ív–láng módszerrel történő elemzésénél a nemvezető
anyagoktól eltérően olyan körülmények kialakítása a célszerű, amely a lángba stacionárius mintabevitelt
biztosít. A tapasztalatok szerint erre a célra a szaggatott egyenáramú vagy a váltóáramú ív az alkalmas
energiaforrás. Az aeroszolok előállítására szolgáló zárt üvegcella (88. ábra) a fémek sík felületére ráhelyezve
szilikon gumi tömítésének köszönhetően kis túlnyomásra is tökéletesen zár. Ellenelektródként spektroszkópiai
tisztaságú grafitot használunk, amelynek pontosan szabályozható a fémtől való távolsága. A 88. ábrán
bemutatott cella és vezetékrendszer lehetővé teszi, hogy meghatározzuk a stacionárius aeroszol–transzport
anyagmérlegét.

Az atomabszorpciós spektrométerek
felépítése, működése
93 Created by XMLmind XSL-FO Converter.
88. ábra Üvegcella fémek, fémötvözetek ív–láng módszerrel történő elemzésére és az ívtermékek
transzportfolyamatainak tanulmányozására
A 88. ábrán bemutatott cella és vezetékrendszer lehetővé teszi, hogy meghatározzuk a stacionárius aeroszol–
transzport anyagmérlegét. Az ábrán jelöltek szerint az ívfolyamat során keletkező termékek a rendszerben négy
különböző helyre kerülnek. Az egyébként lángba jutó frakciót 1 μm-es pórusméretű szűrőpapírral kötöttük meg.
Az egyes frakciókat savval leoldva az adott helyekről, meghatározható azok tömege. Fémötvözetek esetén arra
is mód nyílt, hogy az így készített oldatokból megállapítsuk az aeroszol frakciók kémiai összetételét.
Ezekből a vizsgálatokból megállapítható, hogy a néhány igen illékony fémtől és a molibdéntől eltekintve az
ívporlasztás hatásfoka 45—60 % között mozog fémekre és ötvözetekre egyaránt. Az ívporlasztás sebessége
azonban különösen az eltérő termikus sajátságú fémeknél csaknem három nagyságrend különbséget is mutat. A
jelentős, egy nagyságrendet kitevő különbség még az egyébként azonos alapanyagú acélötvözeteknél is fennáll.
Ezek alapján már most megállapíthatjuk, hogy amennyiben fémötvözeteket akarunk ív-láng kombinált AAS
módszerrel elemezni, az optikai emissziós spektroszkópiához hasonlóan gondoskodni kell a módszer pontos
standardizálásáról.
A száraz aeroszolok szemcseméret-eloszlása és morfológiája
A szilárd anyagokat lángba, plazmába leggyakrabban úgy juttatjuk be, hogy elektromomos ívvel, lézerrel,
termikusan stb. a mintát részben vagy egészében elpárologtatjuk. Ezután a gőzöket hideg vivőgázzal
érintkeztetve mikrokristályokká, száraz aeroszollá kondenzáltatjuk. Ezt a száraz aeroszolt a vivőgáz segítségével
a lángba, plazmába öblítjük. A mintabeviteli hatásfok mértéke a keletkezett kondenzációs termékek
szemcseméretétől függ. Ebből a szempontból tanulságos a fémek, ötvözetek ívtermékeinek bemutatni a
szemcseméret-eloszlását és morfológiáját. Az elektronmikroszkópiás vizsgálatok azt mutatják, hogy a polarizált
váltóáramú ívben stacioner feltételek mellett három morfológiailag jelentősen eltérő ívtermék-frakció
keletkezik.
1. típus: l0 – 50 μm átmérőjű, szabálytalan alakú részecskék, amelyek valószínűleg az ív mechanikai, eróziós
folyamata hatására keletkeznek. Ezek az ívcella falán és a vezetékrendszer cellához közel eső részein válnak le
(89. ábra).

Az atomabszorpciós spektrométerek
felépítése, működése
94 Created by XMLmind XSL-FO Converter.
89. ábra Ívcella faláról származó ívtermék mikroszkópikus képe (nikkel 200 x nagyitásban)
2. típus: 1 – 10 μm átmérőjű szabályos gömbök, amelyek az ívből kikerülő fémolvadéknak tekinthetők. Ezeknek
az a része, amelynek átmérője 5μm alatt van, már bejut a lángba (90. ábra).
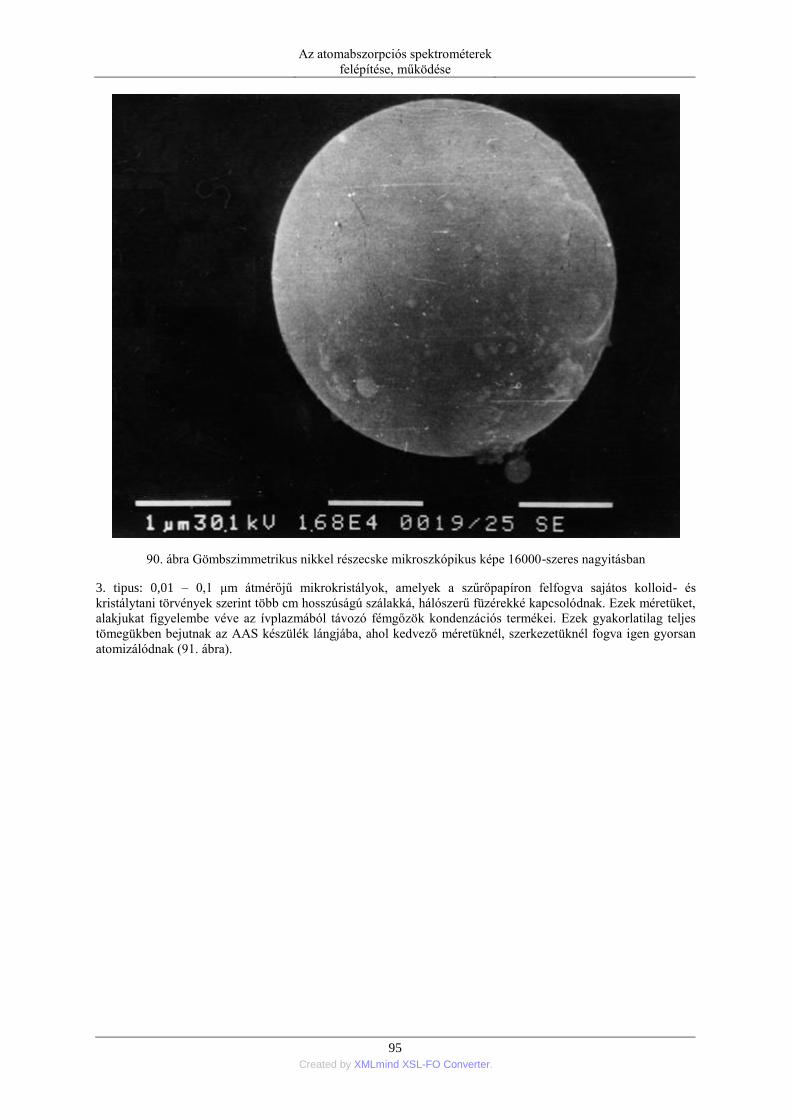
Az atomabszorpciós spektrométerek
felépítése, működése
95 Created by XMLmind XSL-FO Converter.
90. ábra Gömbszimmetrikus nikkel részecske mikroszkópikus képe 16000-szeres nagyitásban
3. tipus: 0,01 – 0,1 μm átmérőjű mikrokristályok, amelyek a szűrőpapíron felfogva sajátos kolloid- és
kristálytani törvények szerint több cm hosszúságú szálakká, hálószerű füzérekké kapcsolódnak. Ezek méretüket,
alakjukat figyelembe véve az ívplazmából távozó fémgőzök kondenzációs termékei. Ezek gyakorlatilag teljes
tömegükben bejutnak az AAS készülék lángjába, ahol kedvező méretüknél, szerkezetüknél fogva igen gyorsan
atomizálódnak (91. ábra).

Az atomabszorpciós spektrométerek
felépítése, működése
96 Created by XMLmind XSL-FO Converter.
91. ábra Szűrőpapíron felfogott ívtermék mikroszkópikus képe (vas 3500-szoros nagyításban)
Az ívtermékekéhez hasonló a morfológiája a lézer ablációs mintabevitelnek is. Az elektrotermikus
elpárologtatás során viszont csak a gőzkondenzációból származó termék alkotja a lángba jutó aeroszolt, mert
utóbbinál eróziós hatások nem lépnek fel.
5.2.2. Lézer abláció
Mossotti és munkatársai [71], majd Vulfson és munkatársai [72] a lézert, mint nagy energiájú nyalábot a szilárd
minták közvetlen atomizálására javasolták, illetve alkalmazták. König és Neumann [73] folytonos ionlézert
alkalmaztak szilárd minták elpárologtatására és AAS meghatározására. Előbbiekhez hasonlóan Ishizuka és
munkatársai [74] acél, bronz és alumínium ötvözetek elpárologtatására használt rubin lézert, és határozott meg
számos ötvözőt a szilárd mintákban.
A lézer ablációt, mint mintabeviteli módszert az ív- és szikraporlasztás modern változatának tekinthetjük. Ebben
az esetben is egy zárt cellában történik a szilárd minta elpárologtatása. Itt is a hideg vivő, szállító gáz
segítségével alakulnak a gőzök mikroméretű száraz aeroszol szemcsékké és szállíthatók az atomizáló térbe. A
szemcsék morfológiájára vonatkozóan is az ívporlasztásnál bemutatott háromféle frakcióval kell számolni.
Különbség ezeknek a frakcióknak az arányában lehet a kísérleti paraméterektől függően.
Ami a lézer abláció külön előnye viszont, hogy a jól fókuszálható lézer fénynyaláb segítségével a szilárd minta
igen kis (30-40 μm) felületi elemét és igen kis (10-30 μm) mélységeit tudjuk elpárologtatni egy-egy vizsgálat
során. Ez lehetővé tesz olyan felületi letapogatásokat (mikro scanning), mikroanalízis elvégzését, amely a
korábbi módszerekkel nem volt megoldható. Ez persze azt is jelenti, hogy az abláció során igen kis (10–100 μg)
anyagmennyiséget párologtatunk el. Ez közel roncsolás-mentes anyagvizsgálatnak tekinthető. De éppen az adott
elemnek a lángba jutó kis tömegárama miatt jelenleg a lézer ablációt a lángatomizációs spektrometria helyett
elsősorban a nagy kimutatási képességű ICP-AES, ICP-MS módszereknél alkalmazzák mintabeviteli célra.
5.2.3. Elektrotermikus párologtatás
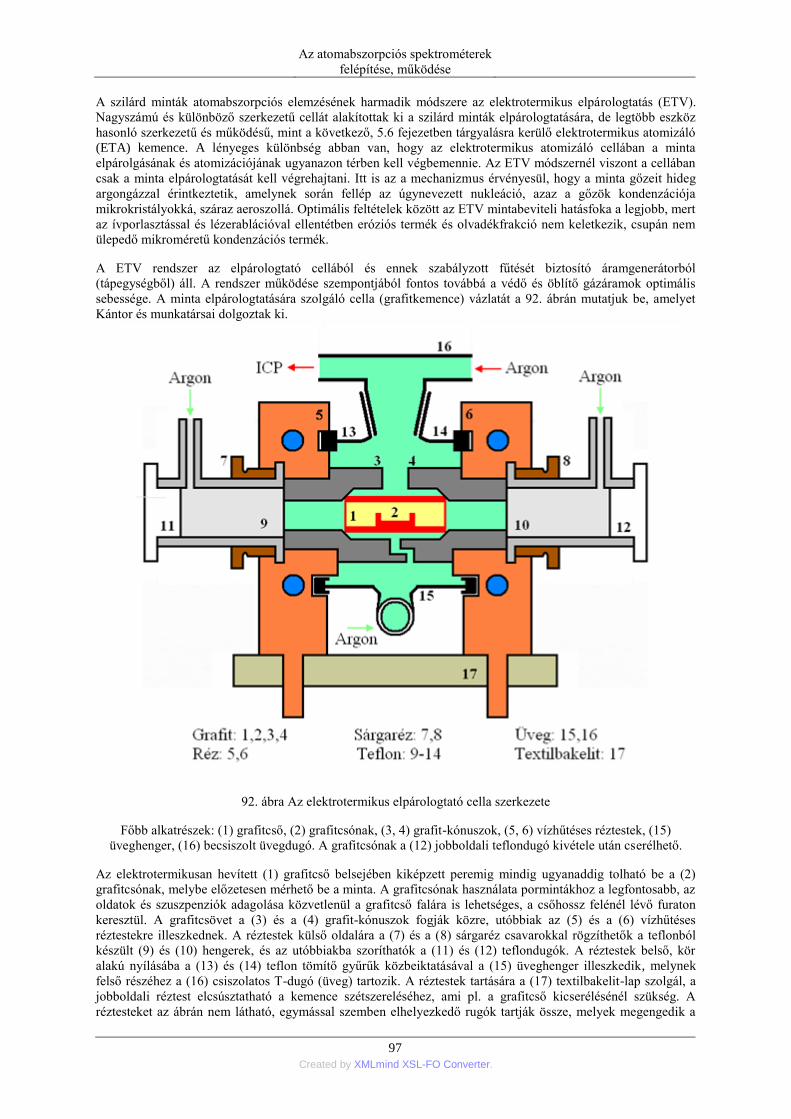
Az atomabszorpciós spektrométerek
felépítése, működése
97 Created by XMLmind XSL-FO Converter.
A szilárd minták atomabszorpciós elemzésének harmadik módszere az elektrotermikus elpárologtatás (ETV).
Nagyszámú és különböző szerkezetű cellát alakítottak ki a szilárd minták elpárologtatására, de legtöbb eszköz
hasonló szerkezetű és működésű, mint a következő, 5.6 fejezetben tárgyalásra kerülő elektrotermikus atomizáló
(ETA) kemence. A lényeges különbség abban van, hogy az elektrotermikus atomizáló cellában a minta
elpárolgásának és atomizációjának ugyanazon térben kell végbemennie. Az ETV módszernél viszont a cellában
csak a minta elpárologtatását kell végrehajtani. Itt is az a mechanizmus érvényesül, hogy a minta gőzeit hideg
argongázzal érintkeztetik, amelynek során fellép az úgynevezett nukleáció, azaz a gőzök kondenzációja
mikrokristályokká, száraz aeroszollá. Optimális feltételek között az ETV mintabeviteli hatásfoka a legjobb, mert
az ívporlasztással és lézerablációval ellentétben eróziós termék és olvadékfrakció nem keletkezik, csupán nem
ülepedő mikroméretű kondenzációs termék.
A ETV rendszer az elpárologtató cellából és ennek szabályzott fűtését biztosító áramgenerátorból
(tápegységből) áll. A rendszer működése szempontjából fontos továbbá a védő és öblítő gázáramok optimális
sebessége. A minta elpárologtatására szolgáló cella (grafitkemence) vázlatát a 92. ábrán mutatjuk be, amelyet
Kántor és munkatársai dolgoztak ki.
92. ábra Az elektrotermikus elpárologtató cella szerkezete
Főbb alkatrészek: (1) grafitcső, (2) grafitcsónak, (3, 4) grafit-kónuszok, (5, 6) vízhűtéses réztestek, (15)
üveghenger, (16) becsiszolt üvegdugó. A grafitcsónak a (12) jobboldali teflondugó kivétele után cserélhető.
Az elektrotermikusan hevített (1) grafitcső belsejében kiképzett peremig mindig ugyanaddig tolható be a (2)
grafitcsónak, melybe előzetesen mérhető be a minta. A grafitcsónak használata pormintákhoz a legfontosabb, az
oldatok és szuszpenziók adagolása közvetlenül a grafitcső falára is lehetséges, a csőhossz felénél lévő furaton
keresztül. A grafitcsövet a (3) és a (4) grafit-kónuszok fogják közre, utóbbiak az (5) és a (6) vízhűtéses
réztestekre illeszkednek. A réztestek külső oldalára a (7) és a (8) sárgaréz csavarokkal rögzíthetők a teflonból
készült (9) és (10) hengerek, és az utóbbiakba szoríthatók a (11) és (12) teflondugók. A réztestek belső, kör
alakú nyílásába a (13) és (14) teflon tömítő gyűrűk közbeiktatásával a (15) üveghenger illeszkedik, melynek
felső részéhez a (16) csiszolatos T-dugó (üveg) tartozik. A réztestek tartására a (17) textilbakelit-lap szolgál, a
jobboldali réztest elcsúsztatható a kemence szétszereléséhez, ami pl. a grafitcső kicserélésénél szükség. A
réztesteket az ábrán nem látható, egymással szemben elhelyezkedő rugók tartják össze, melyek megengedik a

Az atomabszorpciós spektrométerek
felépítése, működése
98 Created by XMLmind XSL-FO Converter.
jobboldali réztest elcsúszását a grafitcső hőtágulásának megfelelően. A grafit-kónuszok olyan kiképzésűek,
hogy nagyrészt leárnyékolják a grafitcső intenzív hősugárzását.
93. ábra Az ETV kemence gázrendszere
Áramforrásként jól alkalmazhatók az eredetileg atomabszorpciós célokra kifejlesztett és forgalmazott
tápforrások. Ezek 10-20 volt tápfeszültség mellett maximálisan 200-250 amper áramerősséggel működnek, és
rendelkeznek a belső és külső kemencegázok áramlási sebességének szabályzásához szükséges eszközökkel.
A grafitkemencét kiszolgáló gázrendszer az 93. ábrán látható.
A grafitkemencéhez négy argongáz bevezetés és az atomabszorpciós spektrométer irányába történő gázkivezetés
tartozik. A grafitcsövön belül, a bal és a jobb oldalról bevezetett argon áramlik (Ar (1) belső kemencegáz), az
üveghengerbe alulról bevezetett argon a grafitcsövön kívül áramlik (Ar (2) külső kemencegáz), utóbbi a levegő
kizárását (a grafitcső oxidációjának kiküszöbölését) szolgálja. A csiszolatos üvegdugó jobboldali csövén
bevezetett hideg argon (Ar (3) segéd vivőgáz) keveredik a felfelé áramló forró mintagőzzel, ami elősegíti a
száraz aeroszol részecskék keletkezését.
Az ETV módszerrel a csónakban elhelyezett szilárd mintákon kívül oldatokat és szuszpenziókat is
elemezhetünk. A fűtési program első lépése ugyanis a szárítás, amely során az oldószer párolog el. Így oldatok
esetében is szilárd fázis marad a csónakban, amelynek a további folyamatai ugyanolyanok, mint a szilárd
mintáké. Az ETV-FAAS módszer kimutatási képessége jelentősen növekszik az oldatos mintabevitelhez képest
a szilárd minták közvetlen elemzésének előnyei miatt és a mintegy 80-90 %-os mintabeviteli hatásfok
következtében. A lézer ablációhoz hasonlóan ma az ETV mintabevitelt is elsősorban az ICP-AES és ICP-MS
módszerrel kombinálják.
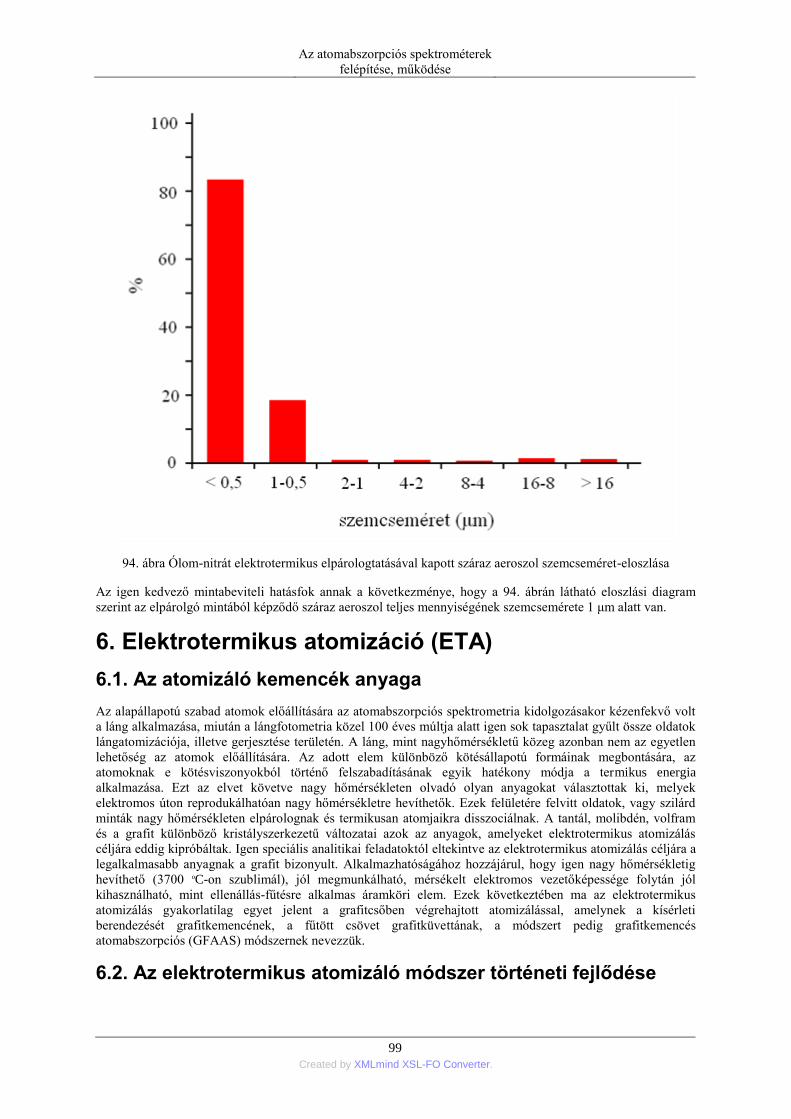
Az atomabszorpciós spektrométerek
felépítése, működése
99 Created by XMLmind XSL-FO Converter.
94. ábra Ólom-nitrát elektrotermikus elpárologtatásával kapott száraz aeroszol szemcseméret-eloszlása
Az igen kedvező mintabeviteli hatásfok annak a következménye, hogy a 94. ábrán látható eloszlási diagram
szerint az elpárolgó mintából képződő száraz aeroszol teljes mennyiségének szemcsemérete 1 μm alatt van.
6. Elektrotermikus atomizáció (ETA)
6.1. Az atomizáló kemencék anyaga
Az alapállapotú szabad atomok előállítására az atomabszorpciós spektrometria kidolgozásakor kézenfekvő volt
a láng alkalmazása, miután a lángfotometria közel 100 éves múltja alatt igen sok tapasztalat gyűlt össze oldatok
lángatomizációja, illetve gerjesztése területén. A láng, mint nagyhőmérsékletű közeg azonban nem az egyetlen
lehetőség az atomok előállítására. Az adott elem különböző kötésállapotú formáinak megbontására, az
atomoknak e kötésviszonyokból történő felszabadításának egyik hatékony módja a termikus energia
alkalmazása. Ezt az elvet követve nagy hőmérsékleten olvadó olyan anyagokat választottak ki, melyek
elektromos úton reprodukálhatóan nagy hőmérsékletre hevíthetők. Ezek felületére felvitt oldatok, vagy szilárd
minták nagy hőmérsékleten elpárolognak és termikusan atomjaikra disszociálnak. A tantál, molibdén, volfram
és a grafit különböző kristályszerkezetű változatai azok az anyagok, amelyeket elektrotermikus atomizálás
céljára eddig kipróbáltak. Igen speciális analitikai feladatoktól eltekintve az elektrotermikus atomizálás céljára a
legalkalmasabb anyagnak a grafit bizonyult. Alkalmazhatóságához hozzájárul, hogy igen nagy hőmérsékletig
hevíthető (3700 oC-on szublimál), jól megmunkálható, mérsékelt elektromos vezetőképessége folytán jól
kihasználható, mint ellenállás-fűtésre alkalmas áramköri elem. Ezek következtében ma az elektrotermikus
atomizálás gyakorlatilag egyet jelent a grafitcsőben végrehajtott atomizálással, amelynek a kísérleti
berendezését grafitkemencének, a fűtött csövet grafitküvettának, a módszert pedig grafitkemencés
atomabszorpciós (GFAAS) módszernek nevezzük.
6.2. Az elektrotermikus atomizáló módszer történeti fejlődése

Az atomabszorpciós spektrométerek
felépítése, működése
100 Created by XMLmind XSL-FO Converter.
Lockyer [75] már 1878-ban kísérleteket végzett fűtött csőben fémgőzök abszorpciós spektrumának
tanulmányozására, King pedig 1908-ban fűtött grafitcsövet használt elemek emisszió spektrumának előállítására
[76]. A grafitkemencés atomabszorpciós spektrometria (GFAAS) kidolgozása és folyamatos fejlesztése azonban
minden kétséget kizáróan Boris L’vov nevéhez fűződik, aki 1959-ben közölte az első cikkét a témában [20].
Ezzel elindította egy új atomabszorpciós spektrometriás módszer fejlesztését, amely jelenleg is az induktív
csatolású plazma tömegspektrometria (ICP-MS) mellett egyik legjobb kimutatási képességű módszernek
tekinthető a nyomelem-analitikában.
Amikor L’vov megépítette az első grafitkemence egységet, már pontosan látta annak szükségességét, hogy csak
akkor tudjuk kihasználni igazán a módszer előnyét, ha a minta olyan rövid idő (τ1) alatt párolog el, hogy a
diffúziós veszteség elhanyagolható. Ennek a csőben való tartózkodási időhöz (τ2) képest a másodperc
törtrészéig kell tartania. Ha ugyanis a minta felhevülése lassú, a keletkező molekulagőzök jelentős része
eltávozik a csőből, mielőtt megtörténne az elem atomizációja.
(53)
Az (53) feltételt L’vov úgy igyekezett biztosítani, hogy a kemencét a mintától függetlenül fűtötte fel. A mintát
egy különálló grafit elektródra helyezte, amely a grafitcső alsó nyílásához illeszkedett. A minta minél gyorsabb
elpárolgása érdekében az elektródot elektromos ívvel, később ohmikusan, Joule-hővel fűtötte fel. A mintát csak
akkor vitte be a grafitcsőbe, amikor az már elérte a kívánt hőmérsékletet. Ezek az izoterm körülmények
biztosítják az optimális atomképződés feltételeit. A L’vov grafitküvetta elvi rajzát a 95. ábrán mutatjuk be.
95. ábra A L’vov-féle grafitküvetta vázlata
A L’vov által kidolgozott elveknek a jelentőségét közel 15 évig nem ismerte fel, és nem vette figyelembe a
szakterület. Ennek többek között az volt az oka, hogy igyekeztek a L’vov-félénél egyszerűbb atomizáló
egységeket kidolgozni.
West [77] például üvegküvettában helyezett el 1-2 mm átmérőjű 20 mm hosszú grafitrúdat két elektród közé
(96. ábra). Mikropipettával 1 μL oldatot cseppentett a rúdra, majd 100 amper áramerősség, 5 volt feszültség
mellett 5-10 s alatt fehér izzásig hevítette a grafitrudat a minta gyors atomizálódása érdekében. Az egyszerű
felépítéssel ellenére azonban hátránya a módszernek a kísérő anyagok zavaró hatása, a nagy hőgradiens a rúd és
a környezet között. A jelnagyság és a zavaró hatás erősen függ attól, hogy az üregkatód lámpa fénynyalábja
milyen távolságra halad az izzó rúd fölött.
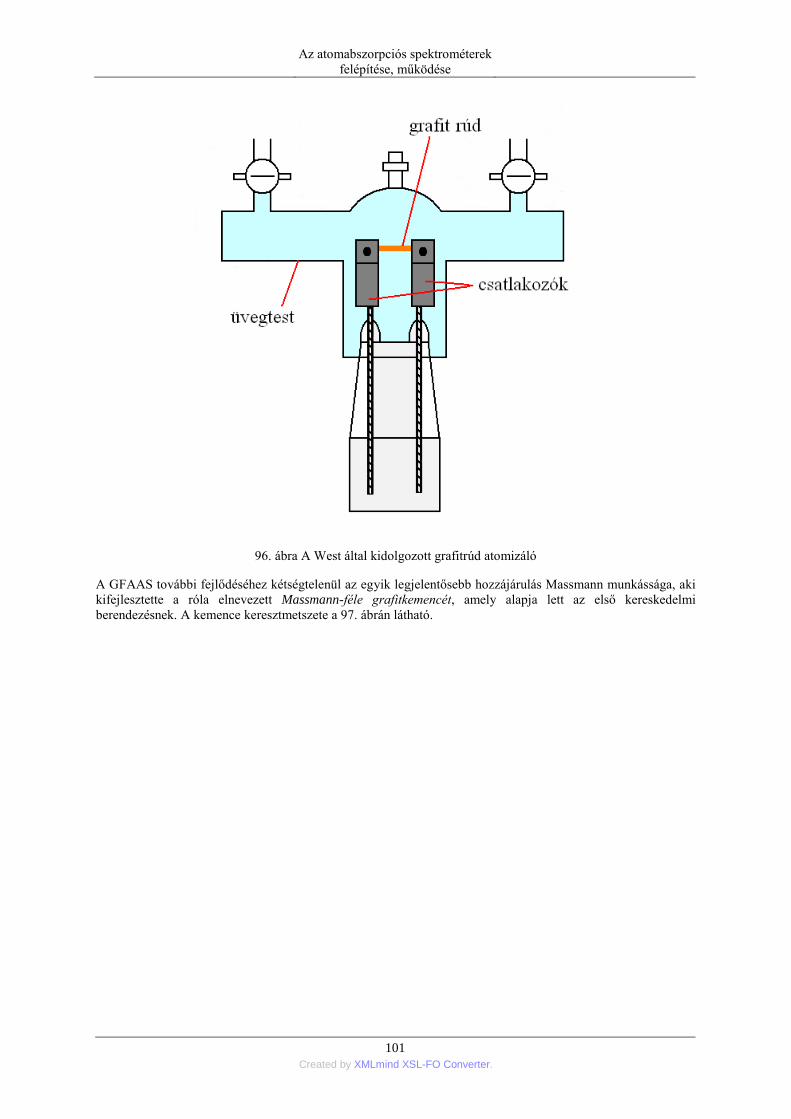
Az atomabszorpciós spektrométerek
felépítése, működése
101 Created by XMLmind XSL-FO Converter.
96. ábra A West által kidolgozott grafitrúd atomizáló
A GFAAS további fejlődéséhez kétségtelenül az egyik legjelentősebb hozzájárulás Massmann munkássága, aki
kifejlesztette a róla elnevezett Massmann-féle grafitkemencét, amely alapja lett az első kereskedelmi
berendezésnek. A kemence keresztmetszete a 97. ábrán látható.
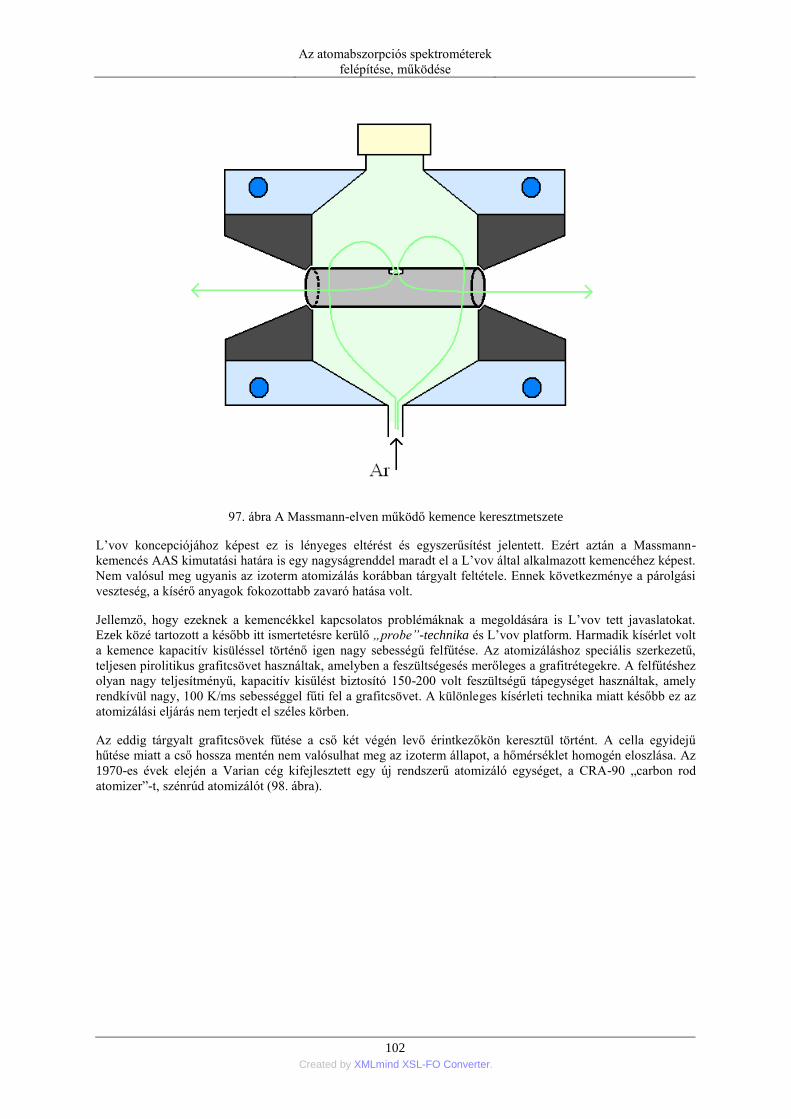
Az atomabszorpciós spektrométerek
felépítése, működése
102 Created by XMLmind XSL-FO Converter.
97. ábra A Massmann-elven működő kemence keresztmetszete
L’vov koncepciójához képest ez is lényeges eltérést és egyszerűsítést jelentett. Ezért aztán a Massmann-
kemencés AAS kimutatási határa is egy nagyságrenddel maradt el a L’vov által alkalmazott kemencéhez képest.
Nem valósul meg ugyanis az izoterm atomizálás korábban tárgyalt feltétele. Ennek következménye a párolgási
veszteség, a kísérő anyagok fokozottabb zavaró hatása volt.
Jellemző, hogy ezeknek a kemencékkel kapcsolatos problémáknak a megoldására is L’vov tett javaslatokat.
Ezek közé tartozott a később itt ismertetésre kerülő „probe”-technika és L’vov platform. Harmadik kísérlet volt
a kemence kapacitív kisüléssel történő igen nagy sebességű felfűtése. Az atomizáláshoz speciális szerkezetű,
teljesen pirolitikus grafitcsövet használtak, amelyben a feszültségesés merőleges a grafitrétegekre. A felfűtéshez
olyan nagy teljesítményű, kapacitív kisűlést biztosító 150-200 volt feszültségű tápegységet használtak, amely
rendkívül nagy, 100 K/ms sebességgel fűti fel a grafitcsövet. A különleges kísérleti technika miatt később ez az
atomizálási eljárás nem terjedt el széles körben.
Az eddig tárgyalt grafitcsövek fűtése a cső két végén levő érintkezőkön keresztül történt. A cella egyidejű
hűtése miatt a cső hossza mentén nem valósulhat meg az izoterm állapot, a hőmérséklet homogén eloszlása. Az
1970-es évek elején a Varian cég kifejlesztett egy új rendszerű atomizáló egységet, a CRA-90 „carbon rod
atomizer”-t, szénrúd atomizálót (98. ábra).

Az atomabszorpciós spektrométerek
felépítése, működése
103 Created by XMLmind XSL-FO Converter.
98. ábra A szénrúd atomizáló küvetta szerkezete
Ahogy ez a 98. ábrán látható, a kis méretű grafitcsövet két oldalról két szénrúd fogja közre. A cső fűtése ezeken
a rudakon keresztül, az eddigiekhez képest keresztirányban történik. Ezzel a megoldással a grafitküvetta
hőmérséklete a cső teljes hossza mentén teljesen azonos lett, azaz megvalósult a térbeli izoterm állapot.
Azonban itt is a grafit belső faláról párolog el a minta, a cső eléggé rövid, a kísérő anyagok zavaró hatása nagy,
és az elemzések nem voltak jó a reprodukálhatók. Mindez azt mutatta, hogy az optimális atomizálási
feltételekhez nem elég a térben izoterm körülmény, időben is biztosítani kell az izoterm körülményeket. A
szénrúd atomizáló korábbi kidolgozása azért tekinthető jelentősnek, mert ez alapozta meg a ma egyik
leghatékonyabbnak számító keresztfűtéses GFAAS kifejlesztését.
6.3. A grafitkemencés atomizáló szerkezete, működése
Egy klasszikus grafitkemence általános felépítését a 97. ábrán mutatjuk be. A grafitkemence központi eleme egy
18-28 mm hosszúságú 5-8 mm belső átmérőjű vízszintesen elhelyezkedő grafitcső. A cső két vége grafitpofákon
keresztül csatlakozik a vízhűtéses zárt cellatesthez, amelynek a két végéhez erősítik a fűtőáram vezetékeit. A
grafitcső falán felül egy 1-1.5 mm átmérőjű furaton lehet a csőbe bejuttatni a mintaoldat 5-40 μL térfogatú
adagjait (3.videó). A cella alsó részén vezetjük be a grafitcső védelmét biztosító argongázt. Az argon körbeveszi
a grafitcsövet, majd a mintaadagoló furaton beáramlik a grafitcső belsejébe és a cső két végén távozik. Ezáltal a
grafitcső külseje, belseje egyaránt védve van az oxigéntől.
A grafitkemence nagy áramerősség továbbítására alkalmas vezetékekkel kapcsolódik egy elektromos
tápegységhez. A ma már számítógéppel vezérelt tápegység a grafitkemence fűtési fokozatainak megfelelően
változó erősségű áramot vezet át a mintát tartalmazó grafitcsövön. A kemence fűtési programjának sémáját az
99. ábrán mutatjuk be.
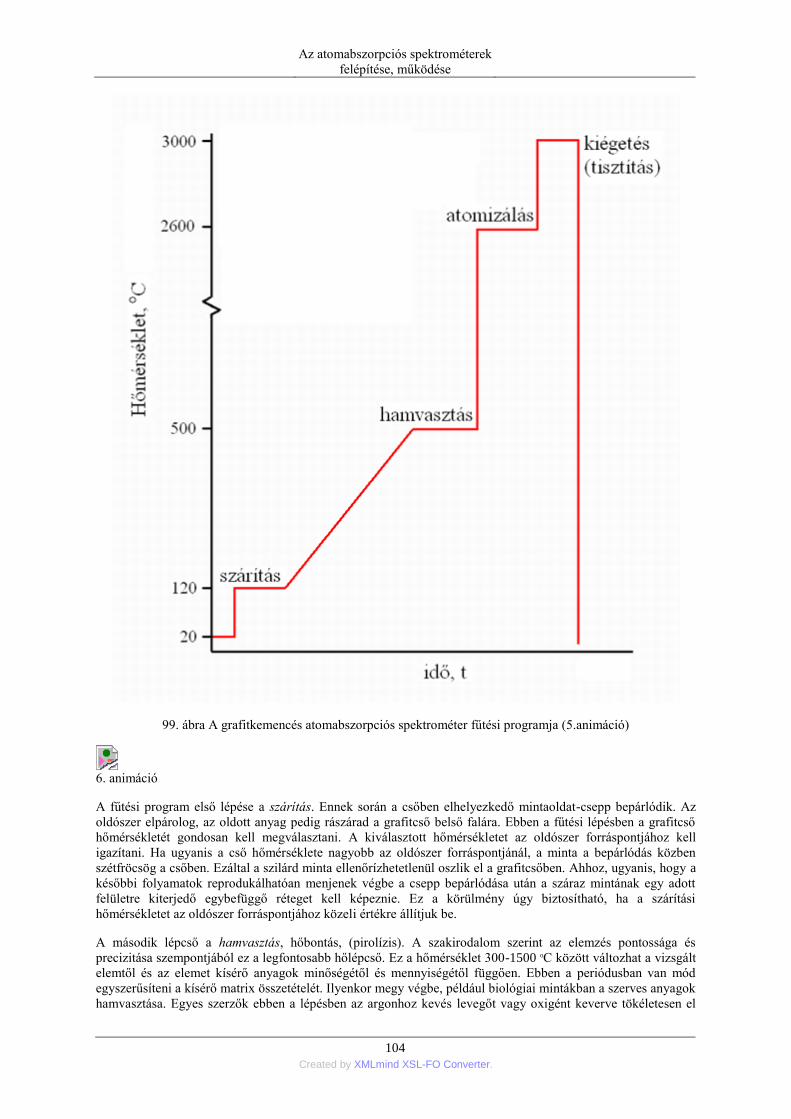
Az atomabszorpciós spektrométerek
felépítése, működése
104 Created by XMLmind XSL-FO Converter.
99. ábra A grafitkemencés atomabszorpciós spektrométer fűtési programja (5.animáció)
6. animáció
A fűtési program első lépése a szárítás. Ennek során a csőben elhelyezkedő mintaoldat-csepp bepárlódik. Az
oldószer elpárolog, az oldott anyag pedig rászárad a grafitcső belső falára. Ebben a fűtési lépésben a grafitcső
hőmérsékletét gondosan kell megválasztani. A kiválasztott hőmérsékletet az oldószer forráspontjához kell
igazítani. Ha ugyanis a cső hőmérséklete nagyobb az oldószer forráspontjánál, a minta a bepárlódás közben
szétfröcsög a csőben. Ezáltal a szilárd minta ellenőrízhetetlenül oszlik el a grafitcsőben. Ahhoz, ugyanis, hogy a
későbbi folyamatok reprodukálhatóan menjenek végbe a csepp bepárlódása után a száraz mintának egy adott
felületre kiterjedő egybefüggő réteget kell képeznie. Ez a körülmény úgy biztosítható, ha a szárítási
hőmérsékletet az oldószer forráspontjához közeli értékre állítjuk be.
A második lépcső a hamvasztás, hőbontás, (pirolízis). A szakirodalom szerint az elemzés pontossága és
precizitása szempontjából ez a legfontosabb hőlépcső. Ez a hőmérséklet 300-1500 oC között változhat a vizsgált
elemtől és az elemet kísérő anyagok minőségétől és mennyiségétől függően. Ebben a periódusban van mód
egyszerűsíteni a kísérő matrix összetételét. Ilyenkor megy végbe, például biológiai mintákban a szerves anyagok
hamvasztása. Egyes szerzők ebben a lépésben az argonhoz kevés levegőt vagy oxigént keverve tökéletesen el

Az atomabszorpciós spektrométerek
felépítése, működése
105 Created by XMLmind XSL-FO Converter.
tudták égetni a széntartalmú anyagokat. Ebben a periódusban lehet kihasználni az úgynevezett matrixmódosítók
szerepét. Ezen anyagok segítségével a kísérő anyagokon például olyan kémiai átalakítást hajthatunk végre,
amely a vizsgált elemhez képest a matrixot jóval illékonyabbá teszi. Ennek segítségével a hamvasztási
hőmérséklet alkalmas megválasztásával a kísérő anyag nagy része vagy egésze ebben a lépésben eltávolítható a
vizsgált elem mellől. A cél az, hogy minél kevesebb kísérő anyag maradjon vissza a csőben azért, hogy a
vizsgálandó elem atomizációját minél kisebb zavaró hatás kísérje. A hamvasztáshoz azt a maximális
hőmérsékletet célszerű kiválasztani, amely mellett a kísérő anyag könnyen el tud távozni, de a vizsgált elem
teljes mennyisége a grafitkemencében marad. Mivel ebben a fűtési lépésben többek között a vizsgált elemnek a
matrixtól történő termikus elválasztása történik, e lépésnél biztosítani kell azt, hogy a fűtés sebessége széles
határok között változtatható, és a minta tulajdonságainak megfelelő értékre legyen beállítható. A kelleténél
gyorsabb fűtés során például a gázfejlődés miatt a minta kifújódhat a csőből, ami ellenőrízhetetlen veszteséghez,
hibás analitikai eredményhez vezet.
A harmadik lépés az atomizáció (4.videó). Ebben a lépésben a két legfontosabb folyamat a hamvasztási
lépésben visszamaradt szilárd fázis elpárologtatása, majd a képződő molekulagőzök termikus disszociációja
alapállapotú atomokká. Ha a grafitcső fűtése nem elég gyors, akkor a molekulagőzök egy része eltávozik a
csőből, mielőtt azok disszociációja végbemenne. Ez jelentős anyagveszteséget, ezzel együtt jelveszteséget
okozna. Ezt figyelembe véve akár 2000 oC/s fűtési sebességet is szokás alkalmazni. Az atomizálási lépésben a
grafitcső hőmérsékletét a vizsgált elem termikus tulajdonságaihoz kell igazítani. Ez a tartomány általában 1200-
2700 oC közötti érték. Ha az atomképződés már alacsonyabb hőmérsékleten végbemegy, azért nem érdemes
nagy atomizálási hőmérsékletet választani, mert az alkalmazott hőmérséklet lényeges befolyással van a cső
élettartamára. Az atomizáció hatékonyságát a későbbiekben tárgyalásra kerülő platform- vagy probe-technikával
tovább lehet javítani.
A grafitkemence fűtési programjának utolsó lépése a kiégetés vagy tisztítás. Ennek során a grafitcsövet rövid
ideig az atomizáció hőmérséketét is meghaladó értékre emeljük azért, hogy ha a mintából az atomizáció után
esetlegesen visszamaradó terméket tökéletesen eltávolíthassuk. Mindezt a memóriaeffektus elkerülése miatt kell
elvégezni; hogy a következő mérésnél a cső ne emlékezzen az előző mintára.
Annak megállapítására, hogy milyen legyen az optimális hamvasztási és atomizálási hőmérséklet, Welz [12]
dolgozott ki egy egyszerű és jól bevált módszert, amely segítségével az optimáláson túl a grafitcsőben
lejátszódó atomizálódás és a zavaró hatások mechanizmusáról is vonhatunk le következtetéseket (100. ábra)
Ezeket az optimálási vizsgálatokat minden elemre és mintatípusra célszerű elvégezni, mielőtt az elemzést
elkezdjük.
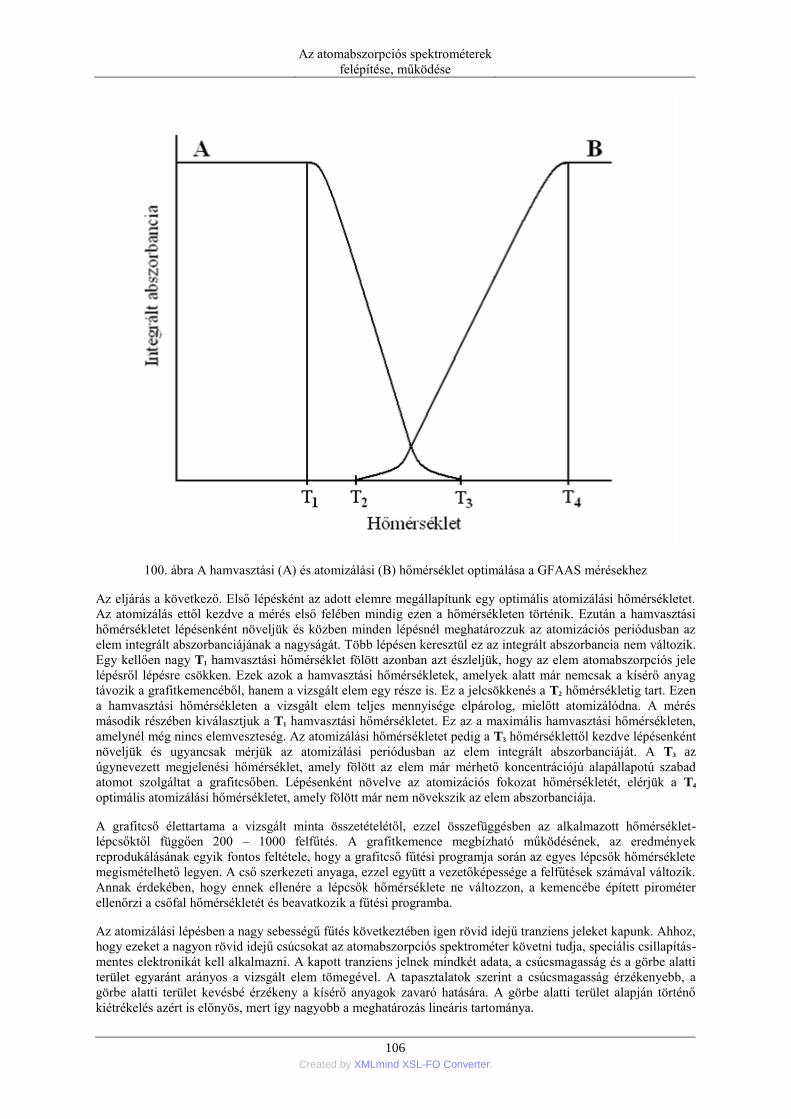
Az atomabszorpciós spektrométerek
felépítése, működése
106 Created by XMLmind XSL-FO Converter.
100. ábra A hamvasztási (A) és atomizálási (B) hőmérséklet optimálása a GFAAS mérésekhez
Az eljárás a következő. Első lépésként az adott elemre megállapítunk egy optimális atomizálási hőmérsékletet.
Az atomizálás ettől kezdve a mérés első felében mindig ezen a hőmérsékleten történik. Ezután a hamvasztási
hőmérsékletet lépésenként növeljük és közben minden lépésnél meghatározzuk az atomizációs periódusban az
elem integrált abszorbanciájának a nagyságát. Több lépésen keresztül ez az integrált abszorbancia nem változik.
Egy kellően nagy T1 hamvasztási hőmérséklet fölött azonban azt észleljük, hogy az elem atomabszorpciós jele
lépésről lépésre csökken. Ezek azok a hamvasztási hőmérsékletek, amelyek alatt már nemcsak a kísérő anyag
távozik a grafitkemencéből, hanem a vizsgált elem egy része is. Ez a jelcsökkenés a T2 hőmérsékletig tart. Ezen
a hamvasztási hőmérsékleten a vizsgált elem teljes mennyisége elpárolog, mielőtt atomizálódna. A mérés
második részében kiválasztjuk a T1 hamvasztási hőmérsékletet. Ez az a maximális hamvasztási hőmérsékleten,
amelynél még nincs elemveszteség. Az atomizálási hőmérsékletet pedig a T3 hőmérséklettől kezdve lépésenként
növeljük és ugyancsak mérjük az atomizálási periódusban az elem integrált abszorbanciáját. A T3 az
úgynevezett megjelenési hőmérséklet, amely fölött az elem már mérhető koncentrációjú alapállapotú szabad
atomot szolgáltat a grafitcsőben. Lépésenként növelve az atomizációs fokozat hőmérsékletét, elérjük a T4
optimális atomizálási hőmérsékletet, amely fölött már nem növekszik az elem abszorbanciája.
A grafitcső élettartama a vizsgált minta összetételétől, ezzel összefüggésben az alkalmazott hőmérséklet-
lépcsőktől függően 200 – 1000 felfűtés. A grafitkemence megbízható működésének, az eredmények
reprodukálásának egyik fontos feltétele, hogy a grafitcső fűtési programja során az egyes lépcsők hőmérséklete
megismételhető legyen. A cső szerkezeti anyaga, ezzel együtt a vezetőképessége a felfűtések számával változik.
Annak érdekében, hogy ennek ellenére a lépcsők hőmérséklete ne változzon, a kemencébe épített pirométer
ellenőrzi a csőfal hőmérsékletét és beavatkozik a fűtési programba.
Az atomizálási lépésben a nagy sebességű fűtés következtében igen rövid idejű tranziens jeleket kapunk. Ahhoz,
hogy ezeket a nagyon rövid idejű csúcsokat az atomabszorpciós spektrométer követni tudja, speciális csillapítás-
mentes elektronikát kell alkalmazni. A kapott tranziens jelnek mindkét adata, a csúcsmagasság és a görbe alatti
terület egyaránt arányos a vizsgált elem tömegével. A tapasztalatok szerint a csúcsmagasság érzékenyebb, a
görbe alatti terület kevésbé érzékeny a kísérő anyagok zavaró hatására. A görbe alatti terület alapján történő
kiétrékelés azért is előnyös, mert így nagyobb a meghatározás lineáris tartománya.

Az atomabszorpciós spektrométerek
felépítése, működése
107 Created by XMLmind XSL-FO Converter.
A láng- és grafitkemencés atomizálás összehasonlítása
Ha összehasonlítjuk a lángatomizációs és a grafitkemencés atomabszorpciós spektrometria atomizációs
körülményeit, azonnal nyilvánvalóvá válik, hogy miért javul a módszer kimutatási határa körülbelül 3
nagyságrendet.
A 101. ábrán a lángot, mint atomizáló közeget ábrázolva jól látható, hogy a fénynyaláb a lángnak csak egy szűk
csatornáját világítja át. Az abszorpcióban csak az adott pillanatban e csatornában tartózkodó atomok vesznek
részt. Ez a zóna a teljes lángtérfogatnak csupán 10 %-a. A többi atom úgy halad át a lángon, hogy nem vesz
részt a jelképzésben. A mintabeviteli hatásfok a lángba ugyancsak 5-10%. Számos elem, a láng oxidáló
tulajdonsága miatt csak részben alakul szabad atomokká, másik része oxid, hidroxid alakban van jelen.
Ugyancsak figyelembe veendő körülmény az atomok tartózkodási ideje a fényútban. Ez a láng esetén
milliszekundum nagyságrendű.
101. ábra A láng és grafitkemence összehasonlítása
Ezek a körülmények a grafitcső esetén jelentősen különböznek. A fénynyaláb a grafitcső teljes térfogatát 100%-
osan kitölti. Ezért a grafitcsőben gyakorlatilag az összes atom részt vesz a fényelnyelésben. A mintának, amit
vizsgálni akarunk, a teljes térfogatát bevisszük a grafitcsőbe. Így a mintabevitel is 100%-os. Az argon
atmoszférában 2500-3000 oC-on izzó grafit igen agresszív közeg a fémoxidok redukciójához. Ezzel javul az
atomizáció hatásfoka is. A minta tartózkodási ideje másodperc, azaz 3 nagyságrenddel nagyobb, mint a lángban.
Ez azt jelenti, hogy az atomok az elemzés ideje alatt többször is részt vesznek a fényelnyelésben.
Fenti alapján könnyen belátható, hogy miért hozott a grafitkemencés atomabszorpciós spektrometria a
lángatomizációhoz képest ilyen nagy mértékű javulást a kimutatási képességben.
6.4. A jel/zaj viszony javítása a grafitkemencés módszernél
Mint minden műszeres analitikai módszernél, így az atomabszorpciós spektrometriában is a nagy analitikai
érzékenység és precíz elemzés feltétele, hogy adott koncentrációjú vizsgált komponensre a zajhoz viszonyítva
minél nagyobb válasz*jelet kapjunk, a zajszint ugyanakkor minél kisebb legyen. A jel/zaj viszony lépésről
lépésre történő javításának technikai körülményeit jól lehet követni a grafitkemencés atomabszorpciós
spektrometriás módszernél is. A grafitkemencés atomabszorpciós spektrometriás módszeren az alapelv
fenntartása mellett végrehajtott kisebb-nagyobb módosítások lényegesen hozzájárultak a teljesítőképesség
javulásához. Az elmúlt 40 év során ezeket a változtatásokat különböző mértékben, különböző módszerekkel és
különböző időben valósították meg az ezzel foglalkozó kutatók és a gyártó cégek.
Az abszorbancia növelésének egyik lehetősége, ha növeljük az alapállapotú atomok tartózkodási idejét a
fényútban. Erre a célra a grafitcső védelmét szolgáló argongáz áramlását az atomizációs lépés néhány
másodperces időtartamára megállítjuk (gázstop). Ugyancsak az atomoknak a grafitcsőből történő gyors
kiürülését akadályozza, azaz a tartózkodási időt növeli, hogy a korábban nyitott csővégek közelébe kvarcablakot
szereltek.

Az atomabszorpciós spektrométerek
felépítése, működése
108 Created by XMLmind XSL-FO Converter.
6.5. A pirolitikus bevonat és a platform alkalmazása
A grafit anyagának, porozitásának fontos szerepe van abban, hogy a grafitcső belső falára cseppentett
mintaoldat milyen arányban és milyen mélységben szívódik be a cső falába. Ez a beszívódott frakció ugyanis
veszteségnek tekinthető, mivel a cső hevítésekor más ütemben, más módon párologhat, mint az a mintarészlet,
amely a felületről távozik. Ez a probléma a GFAAS korai időszakától ismert volt. Az oldatok beszívódását
akadályozó különböző bevonatokkal végzett kísérletek után a végleges megoldást a grafit pirolitikus bevonata
jelentette és jelenti ma is. A ma forgalmazott grafitcsövek mind ezzel a pirolitikus bevonattal rendelkezik. Ez a
bevonat szemmel látható ezüstszürke fémes színt kölcsönöz a grafitnak, amely szerkezetében igen tömör,
kizárja, hogy a mintaoldat bediffundáljon a falba. A pirolitikus bevonat házilag is könnyen készíthető. Ha a
grafitkemencét öblítő védő argongázba metángázt keverünk és azt a felfűtött kemencén átvezetjük, a metán
hőbomlásának eredményeként ez szürke tömör, mikroszkópikus méretű széngömböcskékből álló bevonatot
eredményez a grafit felületén.
A platform jelentősége
A platform jelentősége
Ha pirolitikus grafit csövet használunk, de a mintaoldatot közvetlen a grafitcső belső falára cseppentjük, akkor is
elemveszteséggel kell számolni. A kemence fűtési programjának a szárítást és a hamvasztást követő atomizációs
periódusában következik be a vizsgált elem alapállapotú atomokká alakulása. E viszonylag rövid fűtési lépésben
történik a grafitcső falára szárított és hamvasztott (hőkezelt) mintának az elpárologása, majd a párolgás során
keletkező molekulagőzök atomokra történő disszociációja. Mivel a minta előbb párolog el, mint ahogy
atomizálódna, a gőzök egy része úgy távozik el a kemencéből, hogy nem disszociál atomjaira. A párolgás
kezdetekor ugyanis a kemence hőmérséklete még nem éri el az alapállapotú szabad atomok keletkezéséhez
szükséges hőmérsékletet.
Ezt az el nem hanyagolható párolgási veszteséget úgy lehet csökkenteni, kiküszöbölni, hogy a minta párolgását
késeltetjük addig, míg a grafitcső hőmérséklete el nem éri az elem atomizációs hőmérsékletét. Ennek a
késleltetésnek egyik technikai megoldása az úgynevezett L’vov platform alkalmazása. A L’vov platform egy kis
peremmel ellátott pirolitikus grafitlap, amelyet úgy méreteztek, hogy pontosan beilleszthető a grafitcső alsó
harmadába (102. ábra).
102. ábra A L’vov platform a grafitcsőben
A mintaoldatot erre a grafitlapra (platformra) cseppentjük rá. A kemence fűtőprogramja segítségével ezen a
lapon megy végbe az oldat beszáradása, majd hamvasztása. Amikor az atomizációs lépésben a gyors
felmelegedés elkezdődik, a platform hőmérséklete késni fog a kemence falhőmérsékletéhez képest, mert a
platformnak csak a két éle érintkezik a hevülő grafitcsővel. A rossz hőátadás miatt a platform hőmérséklete csak
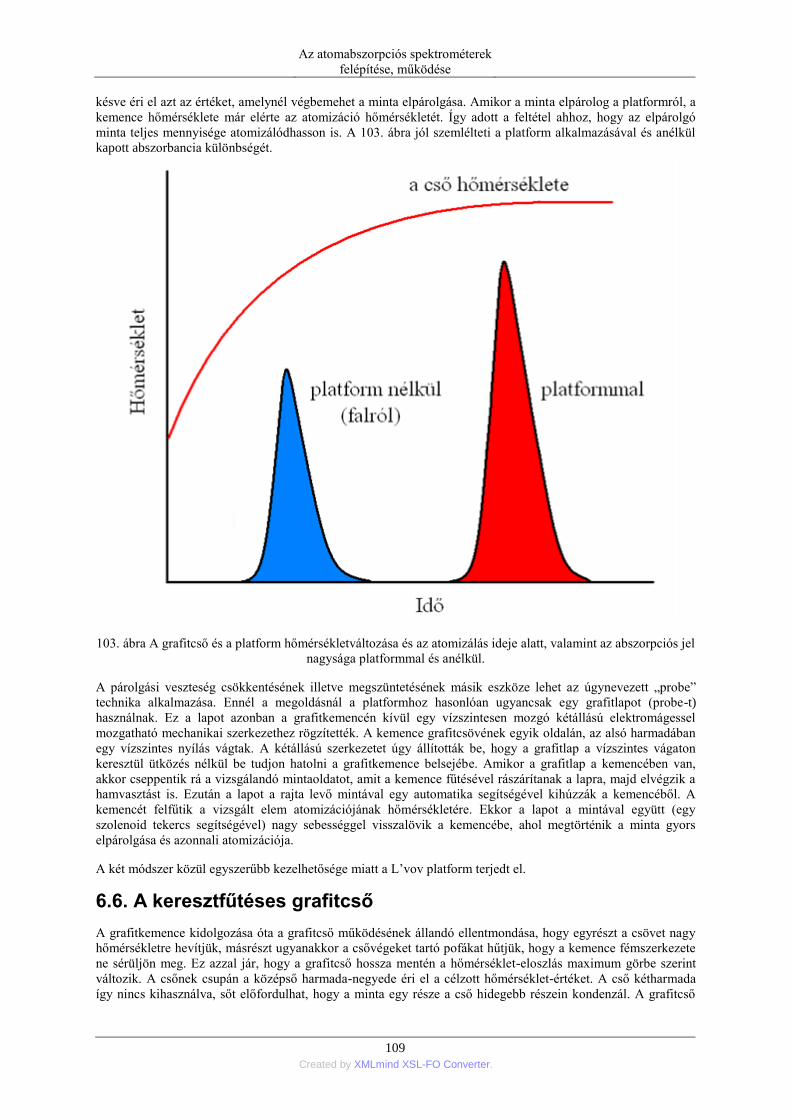
Az atomabszorpciós spektrométerek
felépítése, működése
109 Created by XMLmind XSL-FO Converter.
késve éri el azt az értéket, amelynél végbemehet a minta elpárolgása. Amikor a minta elpárolog a platformról, a
kemence hőmérséklete már elérte az atomizáció hőmérsékletét. Így adott a feltétel ahhoz, hogy az elpárolgó
minta teljes mennyisége atomizálódhasson is. A 103. ábra jól szemlélteti a platform alkalmazásával és anélkül
kapott abszorbancia különbségét.
103. ábra A grafitcső és a platform hőmérsékletváltozása és az atomizálás ideje alatt, valamint az abszorpciós jel
nagysága platformmal és anélkül.
A párolgási veszteség csökkentésének illetve megszüntetésének másik eszköze lehet az úgynevezett „probe”
technika alkalmazása. Ennél a megoldásnál a platformhoz hasonlóan ugyancsak egy grafitlapot (probe-t)
használnak. Ez a lapot azonban a grafitkemencén kívül egy vízszintesen mozgó kétállású elektromágessel
mozgatható mechanikai szerkezethez rögzítették. A kemence grafitcsövének egyik oldalán, az alsó harmadában
egy vízszintes nyílás vágtak. A kétállású szerkezetet úgy állították be, hogy a grafitlap a vízszintes vágaton
keresztül ütközés nélkül be tudjon hatolni a grafitkemence belsejébe. Amikor a grafitlap a kemencében van,
akkor cseppentik rá a vizsgálandó mintaoldatot, amit a kemence fűtésével rászárítanak a lapra, majd elvégzik a
hamvasztást is. Ezután a lapot a rajta levő mintával egy automatika segítségével kihúzzák a kemencéből. A
kemencét felfűtik a vizsgált elem atomizációjának hőmérsékletére. Ekkor a lapot a mintával együtt (egy
szolenoid tekercs segítségével) nagy sebességgel visszalövik a kemencébe, ahol megtörténik a minta gyors
elpárolgása és azonnali atomizációja.
A két módszer közül egyszerűbb kezelhetősége miatt a L’vov platform terjedt el.
6.6. A keresztfűtéses grafitcső
A grafitkemence kidolgozása óta a grafitcső működésének állandó ellentmondása, hogy egyrészt a csövet nagy
hőmérsékletre hevítjük, másrészt ugyanakkor a csővégeket tartó pofákat hűtjük, hogy a kemence fémszerkezete
ne sérüljön meg. Ez azzal jár, hogy a grafitcső hossza mentén a hőmérséklet-eloszlás maximum görbe szerint
változik. A csőnek csupán a középső harmada-negyede éri el a célzott hőmérséklet-értéket. A cső kétharmada
így nincs kihasználva, sőt előfordulhat, hogy a minta egy része a cső hidegebb részein kondenzál. A grafitcső

Az atomabszorpciós spektrométerek
felépítése, működése
110 Created by XMLmind XSL-FO Converter.
hossza mentén a hőmérséklet-eloszlás egyenletesebbé tételét a grafitcső hossza mentén a falvastagság
változtatásával próbálták megoldani (104. ábra).
104. ábra A hőmérséklet-eloszlása hagyományos és keresztfűtéses grafitcső hossza mentén
Az ideális hőmérséklet-eloszlást az jelenti, ha a cső teljes hossza mentén azonos a hőmérséklet. Ezt az eloszlást
is meg lehetett valósítani. Azzal a gyakorlattal kellett szakítani, hogy a fűtőáram és a megvilágító fényforrás
fénynyalábjának iránya megegyezzen. A 105. ábrán látható módon a grafitcső hossztengelyével egyező,
hagyományos fűtésiránytól eltérően az új megoldás szerint a fűtés a cső tengelyére merőlegesen azaz
keresztirányban történik. Ezért ezt a rendszert keresztfűtéses grafitcsőnek nevezik. Ilyen elrendezés esetén a cső
teljes hossza tökéletesen azonos hőmérsékletű.
105. ábra Keresztfűtéses grafitcső
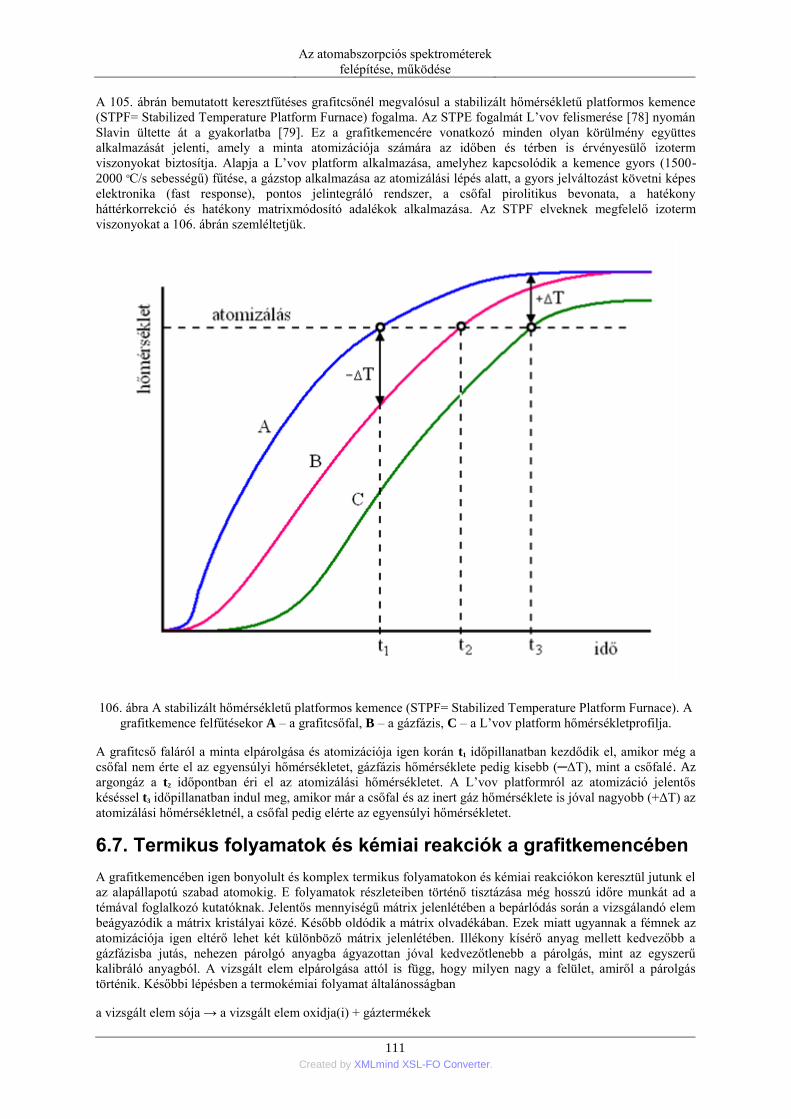
Az atomabszorpciós spektrométerek
felépítése, működése
111 Created by XMLmind XSL-FO Converter.
A 105. ábrán bemutatott keresztfűtéses grafitcsőnél megvalósul a stabilizált hőmérsékletű platformos kemence
(STPF= Stabilized Temperature Platform Furnace) fogalma. Az STPE fogalmát L’vov felismerése [78] nyomán
Slavin ültette át a gyakorlatba [79]. Ez a grafitkemencére vonatkozó minden olyan körülmény együttes
alkalmazását jelenti, amely a minta atomizációja számára az időben és térben is érvényesülő izoterm
viszonyokat biztosítja. Alapja a L’vov platform alkalmazása, amelyhez kapcsolódik a kemence gyors (1500-
2000 oC/s sebességű) fűtése, a gázstop alkalmazása az atomizálási lépés alatt, a gyors jelváltozást követni képes
elektronika (fast response), pontos jelintegráló rendszer, a csőfal pirolitikus bevonata, a hatékony
háttérkorrekció és hatékony matrixmódosító adalékok alkalmazása. Az STPF elveknek megfelelő izoterm
viszonyokat a 106. ábrán szemléltetjük.
106. ábra A stabilizált hőmérsékletű platformos kemence (STPF= Stabilized Temperature Platform Furnace). A
grafitkemence felfűtésekor A – a grafitcsőfal, B – a gázfázis, C – a L’vov platform hőmérsékletprofilja.
A grafitcső faláról a minta elpárolgása és atomizációja igen korán t1 időpillanatban kezdődik el, amikor még a
csőfal nem érte el az egyensúlyi hőmérsékletet, gázfázis hőmérséklete pedig kisebb (─ΔT), mint a csőfalé. Az
argongáz a t2 időpontban éri el az atomizálási hőmérsékletet. A L’vov platformról az atomizáció jelentős
késéssel t3 időpillanatban indul meg, amikor már a csőfal és az inert gáz hőmérséklete is jóval nagyobb (+ΔT) az
atomizálási hőmérsékletnél, a csőfal pedig elérte az egyensúlyi hőmérsékletet.
6.7. Termikus folyamatok és kémiai reakciók a grafitkemencében
A grafitkemencében igen bonyolult és komplex termikus folyamatokon és kémiai reakciókon keresztül jutunk el
az alapállapotú szabad atomokig. E folyamatok részleteiben történő tisztázása még hosszú időre munkát ad a
témával foglalkozó kutatóknak. Jelentős mennyiségű mátrix jelenlétében a bepárlódás során a vizsgálandó elem
beágyazódik a mátrix kristályai közé. Később oldódik a mátrix olvadékában. Ezek miatt ugyannak a fémnek az
atomizációja igen eltérő lehet két különböző mátrix jelenlétében. Illékony kísérő anyag mellett kedvezőbb a
gázfázisba jutás, nehezen párolgó anyagba ágyazottan jóval kedvezőtlenebb a párolgás, mint az egyszerű
kalibráló anyagból. A vizsgált elem elpárolgása attól is függ, hogy milyen nagy a felület, amiről a párolgás
történik. Későbbi lépésben a termokémiai folyamat általánosságban
a vizsgált elem sója → a vizsgált elem oxidja(i) + gáztermékek

Az atomabszorpciós spektrométerek
felépítése, működése
112 Created by XMLmind XSL-FO Converter.
A só anionjától függően nitrogén-oxidok, kén-oxidok, szén-dioxid keletkezhetnek, amelyek jelenléte nem
hanyagolható el a további gázfázisú reakciók szempontjából.
Fontos információval szolgáltak a grafitkemencében lejátszódó folyamatok értelmezéséhez a röngen-diffrakciós,
elektron mikroszkópos, elektron mikropróbás és molekula spektroszkópiás vizsgálatok. Ilyen vizsgálatok
alapján a volt felállítható egy általános séma a 107. ábrán a grafitkemencében lejátszódó főbb folyamatokról
[80].
107.ábra A grafitkemencében lejátszódó atomizációs folyamatok általános vázlata
A konkrét elemek részletes vizsgálata megmutatta, hogy adott esetben a 107. ábrán vázoltak közül mely
folyamatok a meghatározók az atomizáció szempontjából és melyek a kevésbé fontosak. Végig nagy figyelmet
szenteltek a kutatók a szén redukáló szerepének a felderítésére. A pontos redukció mechanizmusának feltárását
nehezíti, hogy legtöbbször csak a végeredményt, a szabad atomok koncentrációjával arányos abszorbanciát
látjuk, a részfolyamatokat nem. Feltételezhetjük, hogy a fémoxidok redukciója a (54) reakció szerint megy
végbe a kemencében.
(54)
A (54) egyenlet szerint felírtakkal ellentétben L’vov és munkatársai [81] alumínium-oxidra és több más
fémoxidra az alábbi kétlépcsős autokatalitikus atomizációs mechanizmust találták érvényesnek (55) (56).
(55)
(56)

Az atomabszorpciós spektrométerek
felépítése, működése
113 Created by XMLmind XSL-FO Converter.
A folyamatok pontos leírását az is nehezíti, hogy az atomizációs folyamatok iránya attól is függ, hogy a
grafitcsőbe cseppentett minta milyen koncentrációjú volt. Ettől függ ugyanis, hogy milyen szerkezetű,
rétegezettségű lesz a szilád fázis az oldószer elpárolgása után a grafitcső falán vagy a platformon.
6.8. Szilárd minták grafitkemencés elemzése
Az oldatok grafitkemencés atomabszorpciós elemzése mellett a lángatomabszorpciós spektrometriához
hasonlóan korán jelentkezett az igény, hogy szilárd mintákat is közvetlenül lehessen elemezni. Ezeknek az
elemzéseknek az előnyei az alábbiak.
1. Az elemzés analitikai érzékenysége nagyobb, mert így a mintát nem kell feloldani. Ezért a vizsgált elem nem
hígul.
2. Jelentősen csökken az elemzési idő, mert jóval rövidebb a minta-előkészítés ideje.
3. Csökken a minta szennyeződésének a veszélye.
4. Csökken az anyagveszteség, ami a minta-előkészítés során lépne fel.
5. Nem kell korrozív és toxikus oldó- és roncsolószerekkel dolgozni.
6. A szilárd mintás elemzés ad lehetőséget a mikro-inhomogenitások vizsgálatára.
Ezekkel az előnyökkel szemben azonban igen sok nehézség lép fel, ha megbízható analitikai eredményeket
akarunk elérni a szilárd minták elemzésével. Néhány ilyen hátrány a következő.
1. Az oldatokhoz képest sokkal nehezebb a szilárd minta pontos bejuttatása a grafitcsőbe.
2. Az oldattal ellentétben minden ismételt mérés új minta bemérését igényli. Így a minta inhomogenitása eleve
növeli az elemzési eredmény szórását.
3. Gyakran igen nehéz az összehasonlító, kalibráló mintasorozat elkészítése, a módszer standardizálása.
4. A kémiai matrixmódosító adalékok kevésbé hatékonyak, mint az oldatminták esetén, mert az adalék nem tud
belsőleg elegyedni a mintával.
5. Nehéz hígítani a szilárd mintát, ha túl nagy az adott elem koncentrációja.
6. A módszer precizitása jellemzően 10 % körül van. A minta tömegének csökkentésével a precizitás tovább
romlik. (A bemérések tömege maximálisan néhány mg lehet.)
7. Jóval nagyobb atomizálási hőmérsékletre van szükség, mint oldatok esetén, ami a grafitcső élettartamát
jelentősen csökkenti
8. Igen nehéz az elemzéshez háttér- (vak) mintát alkalmazni, amelyben minden komponens jelen van, kivéve a
vizsgált elemet.
A szilárd minták bemérésére, a grafitcsőben történő elhelyezésére számos megoldás született [82, 83]. Közös
lényegük, hogy a grafitcsőbe különböző grafit betéteket, csészéket, csónakokat, platformokat stb. helyeznek,
amelyekre/amelyekbe a szilárd mintát mérik, és amelyeket reprodukálható módon a grafitcsőnek mindig azonos
térrészébe (rendszerint a közepébe) helyezik el úgy, hogy a betét és a minta ne akadályozza a fény áthaladását a
csövön. Ezek a betétek a L’vov platformhoz hasonlóan késleltetik a minta felhevülését, elpárolgását a cső
hőmérsékletéhez képest.
A GFAAS elemzésnél a fentebb vázolt nehézségek miatt sokkal inkább elterjedt a szilárd minták szuszpenzió
(slurry) formájában történő analízise. A szuszpenzió elemzésénél kihasználhatók mind a szilárd mintás, mind az
oldatos elemzés előnyei [84]. Ennél a megoldásnál a szilárd mintát gondosan elporítjuk és olyan oldószerben
diszpergáljuk, amelyben a porszemcsék igen lassan ülepednek, stabil szuszpenziót eredményeznek.

Az atomabszorpciós spektrométerek
felépítése, működése
114 Created by XMLmind XSL-FO Converter.
1. A szuszpenzió adagolása a grafitcsőben az oldatokéval azonos módon történik.
2. Az ismételt meghatározáshoz ugyanazt a szuszpenziót használhatjuk.
3. Sok esetben a szuszpenziót vizes kalibráló oldatokkal szemben is kellő pontossággal meg lehet határozni.
4. A matrixmódosítókat a vizes oldatoknál szokásos módon lehet alkalmazni.
5. A szuszpenziót a vizes oldatokhoz hasonlóan lehet hígítani.
6. Szuszpenzió elemzésénél is nehéz háttér- (vak) mintát alkalmazni.
7. Sok esetben igen nehéz homogén és stabil szuszpenziót készíteni.
A szuszpenzió GFAAS elemzésének az egyik legkritikusabb lépése homogén és stabil szuszpenzió elkészítése.
A szuszpenzió ülepedésének megakadályozására kipróbáltak viszkózus folyadékokat (glicerin, nem-ionos
felületaktív, tixotróp anyagokat). A homogenitás biztosításának másik módja a szuszpenzió intenzív keverése. A
különböző keverési módszerek közül a leghatékonyabbnak az ultrahangos keverő bizonyult. Ez a
kereskedelemben is kapható egységnek a keverő karja belenyúlik a GFAAS készülék mintatartó edényébe. Az
ultrahangos keverő automatikusan működésbe lép, mielőtt a GFAAS automata mintaadagoló egység mintavevő
karja a mintavételt végzi. Így a mintavétel ideje alatt biztosítjuk a szuszpenzió homogenitását.
108. ábra Ultrahangos keverő szuszpenziók GFAAS elemzéséhez
7. A higany hideggőz (CV=cold vapor) technikás atomizációja
A higanyt lángatomabszorpciós spektrometriás módszerrel rossz kimutatási határral lehet meghatározni. Mivel a
higany az egyik legtoxikusabb elem, nagyon fontos volt olyan atomizáló módszer kifejlesztése, amelyet
alkalmazva az AAS módszernek jelentősen javítható a kimutatási határa.

Az atomabszorpciós spektrométerek
felépítése, működése
115 Created by XMLmind XSL-FO Converter.
A higany különleges fémes elem, mert viszonylag nagy a gőznyomása, ─ 0,0016 mbar 20 oC-on ─ mely
megfelel 14 mg/m3 atomos higanynak a gőzfázisban. Ebből adódóan a higany közvetlenül elemezhető külön
atomizálás nélkül. Ezért ha a higanyvegyületet elemi higannyá redukáljuk, és azt átvisszük gőzfázisba akkor a
gőzt higany üregkatód lámpával átvilágítva hevítés nélkül, hidegen (20 oC-on) meghatározható a minta
higanytartalma. Ezért nevezik a módszert hideggőz (CV=cold vapor) technikának.
A meghatározás első lépése a mintaoldatban a higanyvegyületek elemi higannyá történő redukciója. A
redukcióhoz a minőségi kémiai analízisből régóta jól ismert Bettendorf-próbát használhatjuk fel. Erősen savas
közegben ón(II)-klorid hatására a higanyvegyületek az alábbi egyenlet szerint elemi higannyá alakulnak.
(57)
Ez a reakció nagyobb (0,1 M) higanykoncentráció mellett fekete csapadékot eredményez, mely a higany
kimutatásának egyik jellemző reakciója. Ha azonban igen kis (10─4 – 10─5 M-os) higanykoncentrációjú oldaton a
reakció közben mérsékelt (1 L/min) sebességgel argon gázt buborékoltatunk keresztül, a higany atomok ─
mielőtt egyesülnének ─, nagy tenziójuk miatt kikerülnek a gázfázisba. Ezt az atomos higanygőzt azután argon
árammal egy 10 cm hosszúságú kvarcablakos küvettába vezetjük, amelyen a higany üregkatód lámpa fényét
átvezetve a higanyatomok számával arányos fényelnyelést mérünk. A higany-meghatározó készülék rajzát a
109. ábrán mutatjuk be.
109. ábra A higany hideggőz technikás meghatározása
A 109. ábrán bemutatott higany meghatározó készülék tartályába 30%-os kénsavban oldott 5%-os ón(II)-
kloridot öntünk. Ebbe a redukáló elegybe adagoljuk szeptumon keresztül a higanytartalmú mintát. A
felszabaduló higanygőzt Mg(ClO4)2-ot tartalmazó szárítócsövön vezetjük át, mielőtt a küvettába kerül. A
kalibráló sorozat felvételéhez elegendő egyetlen higanytartalmú kalibráló oldat, amelyet növekvő térfogatban
adagolunk a redukáló elegyhez.
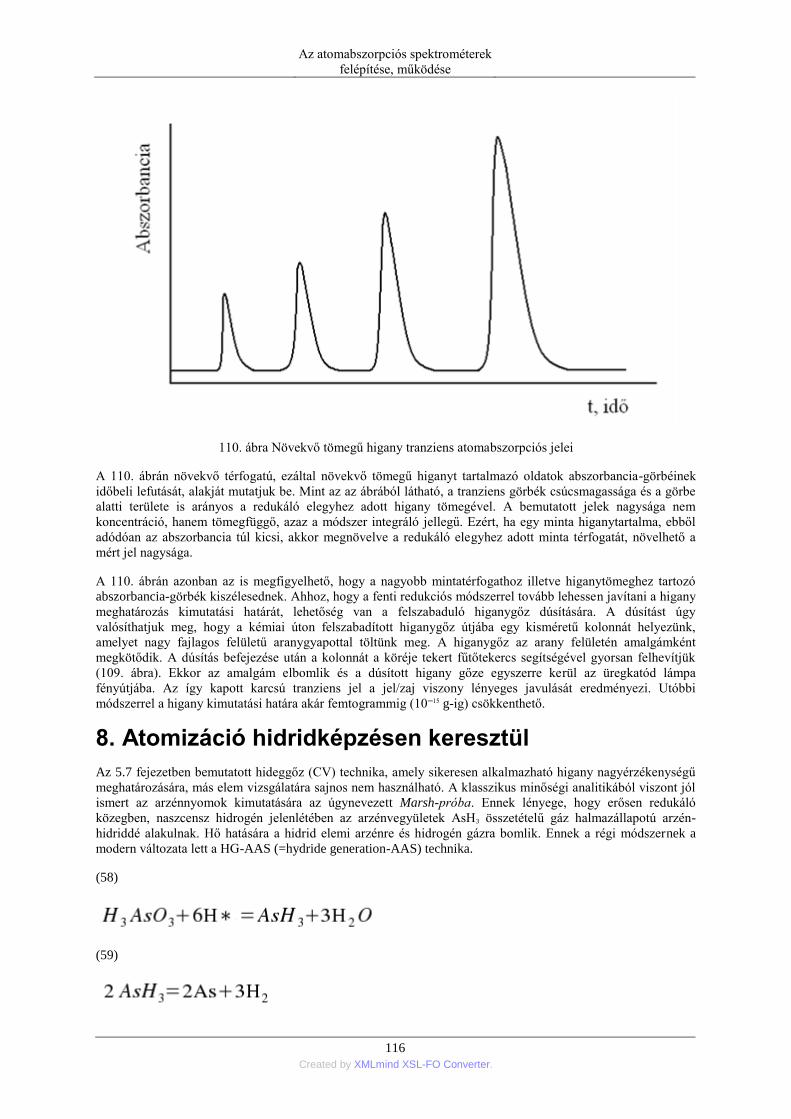
Az atomabszorpciós spektrométerek
felépítése, működése
116 Created by XMLmind XSL-FO Converter.
110. ábra Növekvő tömegű higany tranziens atomabszorpciós jelei
A 110. ábrán növekvő térfogatú, ezáltal növekvő tömegű higanyt tartalmazó oldatok abszorbancia-görbéinek
időbeli lefutását, alakját mutatjuk be. Mint az az ábrából látható, a tranziens görbék csúcsmagassága és a görbe
alatti területe is arányos a redukáló elegyhez adott higany tömegével. A bemutatott jelek nagysága nem
koncentráció, hanem tömegfüggő, azaz a módszer integráló jellegű. Ezért, ha egy minta higanytartalma, ebből
adódóan az abszorbancia túl kicsi, akkor megnövelve a redukáló elegyhez adott minta térfogatát, növelhető a
mért jel nagysága.
A 110. ábrán azonban az is megfigyelhető, hogy a nagyobb mintatérfogathoz illetve higanytömeghez tartozó
abszorbancia-görbék kiszélesednek. Ahhoz, hogy a fenti redukciós módszerrel tovább lehessen javítani a higany
meghatározás kimutatási határát, lehetőség van a felszabaduló higanygőz dúsítására. A dúsítást úgy
valósíthatjuk meg, hogy a kémiai úton felszabadított higanygőz útjába egy kisméretű kolonnát helyezünk,
amelyet nagy fajlagos felületű aranygyapottal töltünk meg. A higanygőz az arany felületén amalgámként
megkötődik. A dúsítás befejezése után a kolonnát a köréje tekert fűtőtekercs segítségével gyorsan felhevítjük
(109. ábra). Ekkor az amalgám elbomlik és a dúsított higany gőze egyszerre kerül az üregkatód lámpa
fényútjába. Az így kapott karcsú tranziens jel a jel/zaj viszony lényeges javulását eredményezi. Utóbbi
módszerrel a higany kimutatási határa akár femtogrammig (10─15 g-ig) csökkenthető.
8. Atomizáció hidridképzésen keresztül
Az 5.7 fejezetben bemutatott hideggőz (CV) technika, amely sikeresen alkalmazható higany nagyérzékenységű
meghatározására, más elem vizsgálatára sajnos nem használható. A klasszikus minőségi analitikából viszont jól
ismert az arzénnyomok kimutatására az úgynevezett Marsh-próba. Ennek lényege, hogy erősen redukáló
közegben, naszcensz hidrogén jelenlétében az arzénvegyületek AsH3 összetételű gáz halmazállapotú arzén-
hidriddé alakulnak. Hő hatására a hidrid elemi arzénre és hidrogén gázra bomlik. Ennek a régi módszernek a
modern változata lett a HG-AAS (=hydride generation-AAS) technika.
(58)
(59)
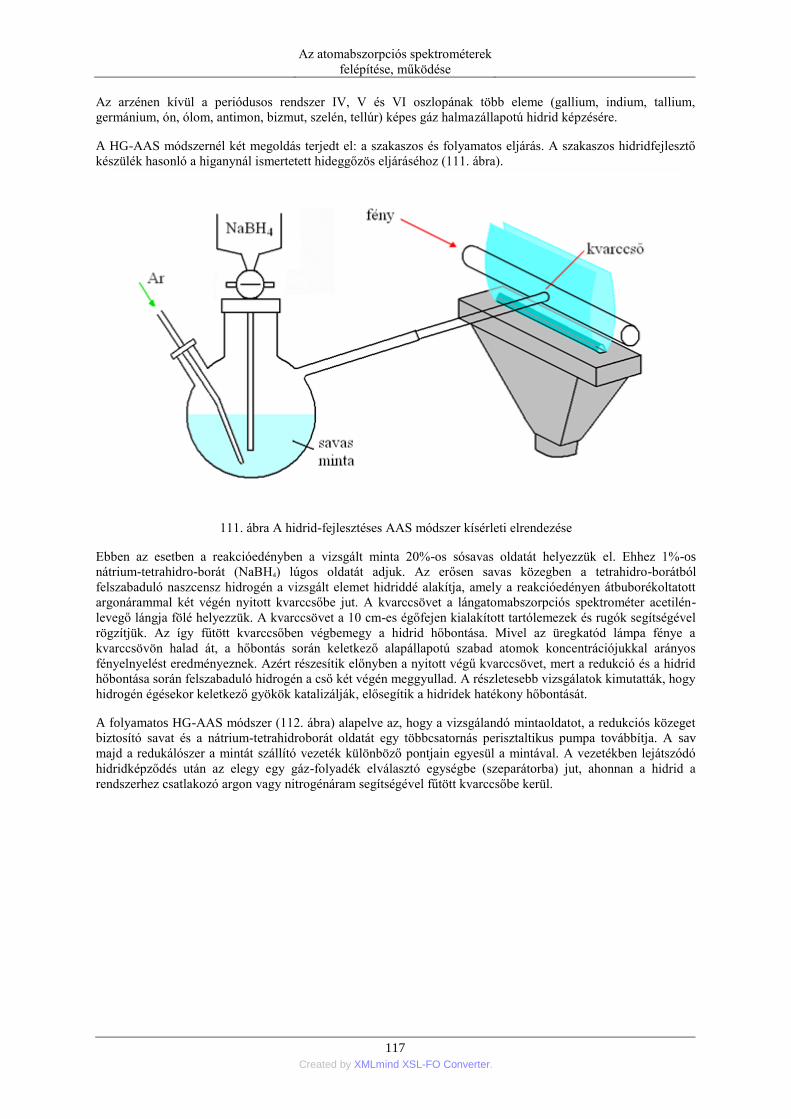
Az atomabszorpciós spektrométerek
felépítése, működése
117 Created by XMLmind XSL-FO Converter.
Az arzénen kívül a periódusos rendszer IV, V és VI oszlopának több eleme (gallium, indium, tallium,
germánium, ón, ólom, antimon, bizmut, szelén, tellúr) képes gáz halmazállapotú hidrid képzésére.
A HG-AAS módszernél két megoldás terjedt el: a szakaszos és folyamatos eljárás. A szakaszos hidridfejlesztő
készülék hasonló a higanynál ismertetett hideggőzös eljáráséhoz (111. ábra).
111. ábra A hidrid-fejlesztéses AAS módszer kísérleti elrendezése
Ebben az esetben a reakcióedényben a vizsgált minta 20%-os sósavas oldatát helyezzük el. Ehhez 1%-os
nátrium-tetrahidro-borát (NaBH4) lúgos oldatát adjuk. Az erősen savas közegben a tetrahidro-borátból
felszabaduló naszcensz hidrogén a vizsgált elemet hidriddé alakítja, amely a reakcióedényen átbuborékoltatott
argonárammal két végén nyitott kvarccsőbe jut. A kvarccsövet a lángatomabszorpciós spektrométer acetilén-
levegő lángja fölé helyezzük. A kvarccsövet a 10 cm-es égőfejen kialakított tartólemezek és rugók segítségével
rögzítjük. Az így fűtött kvarccsőben végbemegy a hidrid hőbontása. Mivel az üregkatód lámpa fénye a
kvarccsövön halad át, a hőbontás során keletkező alapállapotú szabad atomok koncentrációjukkal arányos
fényelnyelést eredményeznek. Azért részesítik előnyben a nyitott végű kvarccsövet, mert a redukció és a hidrid
hőbontása során felszabaduló hidrogén a cső két végén meggyullad. A részletesebb vizsgálatok kimutatták, hogy
hidrogén égésekor keletkező gyökök katalizálják, elősegítik a hidridek hatékony hőbontását.
A folyamatos HG-AAS módszer (112. ábra) alapelve az, hogy a vizsgálandó mintaoldatot, a redukciós közeget
biztosító savat és a nátrium-tetrahidroborát oldatát egy többcsatornás perisztaltikus pumpa továbbítja. A sav
majd a redukálószer a mintát szállító vezeték különböző pontjain egyesül a mintával. A vezetékben lejátszódó
hidridképződés után az elegy egy gáz-folyadék elválasztó egységbe (szeparátorba) jut, ahonnan a hidrid a
rendszerhez csatlakozó argon vagy nitrogénáram segítségével fűtött kvarccsőbe kerül.
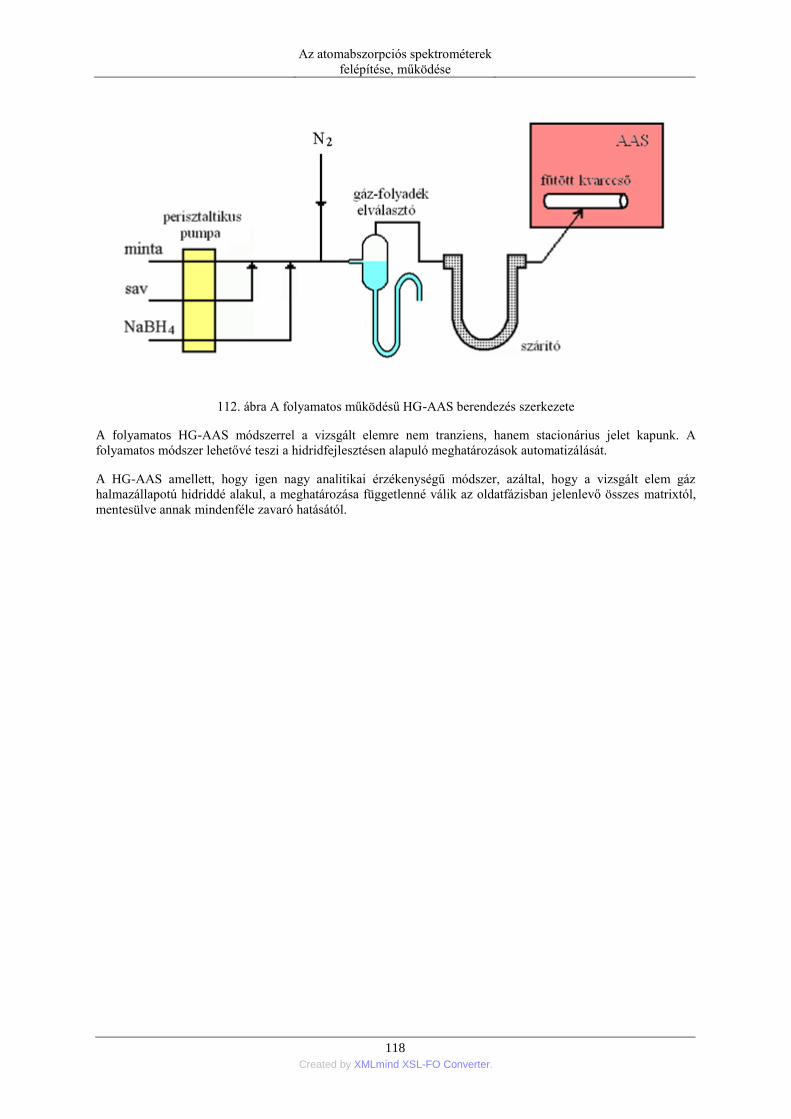
Az atomabszorpciós spektrométerek
felépítése, működése
118 Created by XMLmind XSL-FO Converter.
112. ábra A folyamatos működésű HG-AAS berendezés szerkezete
A folyamatos HG-AAS módszerrel a vizsgált elemre nem tranziens, hanem stacionárius jelet kapunk. A
folyamatos módszer lehetővé teszi a hidridfejlesztésen alapuló meghatározások automatizálását.
A HG-AAS amellett, hogy igen nagy analitikai érzékenységű módszer, azáltal, hogy a vizsgált elem gáz
halmazállapotú hidriddé alakul, a meghatározása függetlenné válik az oldatfázisban jelenlevő összes matrixtól,
mentesülve annak mindenféle zavaró hatásától.

119 Created by XMLmind XSL-FO Converter.
5. fejezet - Zavaró hatások és azok kiküszöbölése
Az atomabszorpciós spektrometriát kidolgozásának első éveiben úgy jellemezték, , mint zavaró hatásoktól
mentes módszert. Az 1950-es évekre az atomspektroszkópiában széleskörben elterjedtek az optikai emissziós
spektrometriás módszerek (lángfotométerek, spektroszkópok, spektrográfok, spektrométerek). A
molekulaspektroszkópiában ugyanígy népszerűek voltak a spektrofotométerek. Mindkét módszercsoportnál
azonban sokszor okozott kellemetlenséget, hogy a vizsgált elem vagy molekula színképvonala illetve sávja
egybeesett más kísérő komponens vonalával, sávjával. Ennek az egybeesésnek (koincidenciának), mint
spektrális zavaró hatásnak a kiküszöbölése igen gondos mérlegelést kíván, olykor körülményes korrekciós
feladat elé állította, és állítja ma is az analitikust. Az az üdítő tapasztalat, hogy az atomabszorpciós
spektrometriában gyakorlatilag nincs vonalegybeesés, nincs a korábbi optikai módszerekhez hasonló spektrális
zavarás, eredményezte azt az elhamarkodott kijelentést, hogy az AAS a zavaró hatástól mentes módszer. Később
kiderült, hogy bár a spektrális zavaró hatás valóban minimális, de egyéb zavaró hatások bőségesen jelen vannak
az AAS módszernél is.
1. Lángban fellépő zavaró hatások
1.1. A zavaró hatások különböző szempontú osztályzása
Zavaró hatáson a lángatomabszorpciós spektrometriában az abszorbanciának mindazon szisztematikus
változásait értjük, amelyeket a vizsgálandó elem mellett jelenlévő egyéb (kísérő elemek, matrix, szennyező
komponensek) anyagok okoznak. A zavaró anyag fogalomkörébe azonban nem tartoznak bele a lánggázok, mert
ezek szükségszerűen mindig jelen vannak, mind a minta, mind a kalibráló oldatok esetén. Csak azokat a
szisztematikus változásokat tekinthetjük zavaró hatásnak, amelyek a mérendő oldattal azonos oldószer
összetételű viszonyító (referencia) oldattal történő méréskor lépnek fel [24].
A lángban fellépő zavaró hatások egy régebbi osztályzása inkább formai szempontú volt. Ezt a felosztást főleg a
lángfotometriában alkalmazták. Eszerint a lángban háromféle zavaró hatással kell számolni. Ezek az additív,
arányos és exponenciális zavaró hatás. Az additív a kísérőanyag koncentrációjától függetlenül párhuzamosan
eltolja a kalibráló egyenest. Az arányos zavaró hatás a kísérő anyag koncentrációjával arányos jelváltozást
eredményez, az exponenciális zavarás esetén viszont a kísérő anyag adott koncentráció-tartományában a
jelváltozás hirtelen igen nagy mértékű, valamilyen kémiai kölcsönhatás eredménye.
Később már az atomabszorpciós irodalom [5, 14] a zavaró hatások egyfajta tartalmi szempontú osztályzását
végezte el. Eszerint spektrális, fizikai, gázfázísú és kondenzált fázisú kémiai zavaró hatásokat különböztettek
meg.
A IUPAC(=International Union of Pure and Applied Chemistry) által elfogadott nevezéktan [85] szakított ezzel
az osztályozással, mert a korábbi osztályzás több esetben félreértelmezhetőnek bizonyult. Az új besorolás
szerint a két fő csoport a spektrális és nem-spektrális zavaró hatások.
1.2. Spektrális zavaró hatások
Spektrális zavaró hatás akkor lép fel, ha az analizálandó anyag által elnyelt ill. kibocsátott sugárzást nem tudjuk
teljesen elkülöníteni egyéb sugárzásoktól vagy fénygyengüléstől (vonalátfedés, fényszóródást stb.).
Az atomabszorpciós spektrometriában az olyan spektrális zavaró hatás, amely szerint az egyik elem atomjai egy
másik elem hullámhosszán nyelnének el fényt, igen ritka. Az elemek ajánlott atomabszorpciós elemző vonalai
közül a legközelebbiek is legalább 3-4 nm távolságban vannak egymástól. Az előfordulhat, hogy a másik
elemnek a vizsgált elemhez közel eső más vonala van, de azon a vonalon az elnyelés elhanyagolható mértékű.
Nagyobb az esélye annak, hogy a lángban vagy a grafitcsőben az atomizáció során képződő molekulák sávos
színképének valamely sávalkotó vonala nagyon közel van az atom abszorpciós vonalával vagy egybeesik azzal.
Ennek a spektrális zavarásnak a detektálásában vagy kiküszöbölésében segít a Zeeman vagy a Hieftje
háttérkorrekció (5.1.8. fejezet). A molekulák ilyen típusú zavaró hatásának pontos felmérésére és korrekciójára
az új nagyfelbontású folytonos fényforrással működő készülékek (HR-CS-AAS) (9. fejezet) alkalmasak.

Zavaró hatások és azok
kiküszöbölése
120 Created by XMLmind XSL-FO Converter.
A fényszóródás okozta – szintén spektrális – zavaró hatást ugyancsak az 5.1.8. fejezetben részletesen bemutatott
háttérkorrekciós módszerekkel tudjuk kiküszöbölni.
1.3. Transzport zavarások
A nem-spektrális zavaró hatások közzé azokat a hatásokat soroljuk, amelyek a szabad atomok képződését
befolyásolják. Az osztályozás aszerint történik, hogy a zavaró hatás a szabad atomok előállításának mely
fázisában jelentkezik. Ezek az alábbiak:
A transzport zavarások (transport interferences), amelyek befolyásolják egy adott megfigyelési magasságnál
időegység alatt a vízszintes lángkeresztmetszeten áthaladó minta mennyiségét. Ide tartoznak a porlasztási
sebességet, porlasztási hatásfokot és a bepárlódott (deszolvatált) frakciót befolyásoló tényezők.
Ahogy azt az 5.5. fejezetben részleteztük, a pontos FAAS meghatározás egyik legfontosabb feltétele, hogy a
kalibráló oldat mintabeviteli sebessége, porlasztási (mintabeviteli) hatásfoka pontosan megegyezzen a minta
ugyanezen paramétereivel. Hibát okozhat a kalibráló és a mintaoldat eltérő viszkozitása, ami a mintabeviteli
sebességben okoz különbséget. A szerves oldószerek jelentős hányadának kisebb a viszkozitása, mint a vizes
oldatoké. Ugyancsak eltérhet a kalibráló és mintaoldat felületi feszültsége. Ezáltal különböző lesz a kétféle
oldatból képződő aeroszol cseppméret-eloszlása, amely a mintabeviteli hatásfokot befolyásolja. A hagyományos
híg vizes oldatokéhoz képest csökken a felületi feszültsége a felületaktív anyagokat tartalmazó mintának,
például a szappanoldatnak. Még inkább eltér a vizes oldatokétól számos szerves oldószer felületi feszültsége.
Azonos viszkozitás és felületi feszültség ellenére eltérhet a vizsgált elemnek a lángba jutó hányada, ha az elemet
eltérő illékonyságú oldószerben oldottuk. Az oldatok bepárlódása ugyanis már a ködkamrában megindul. Az
eltérő illékonyságú oldószerekből képződő nedves aeroszol cseppek a porlasztótól a lángig haladva különböző
mértékben párlódnak be. Emiatt eltérő lesz a cseppméret-eloszlás, amely eltérő mintabeviteli hatásfokot
eredményez. Ezért a ködkamrában a bepárlódási folyamat is a transzport zavarások körébe sorolható.
Példaként említhetjük az acetont, melynek a viszkozitása és a felületi feszültsége is lényegesen kisebb a vízénél.
Illékonysága folytán a mintabevitel (a transzportfolyamat) során a cseppek bepárlódásának üteme is eltér a vizes
oldatokétól. Ezek miatt azonos koncentrációjú vizes és acetonos oldatot lángba porlasztva az oldott elem
anyagárama aceton esetén ötszöröse lesz a vizes oldaténak.
1.4. Oldott anyagok párolgási zavarása
A párolgási zavarások (solute volatilization interferences) A vizsgált elem párolgását zavaró hatások a száraz
aeroszol részecskék párolgási sebességének megváltozásából erednek, ha kísérő anyag van jelen. Ez a zavaró
hatás lehet specifikus, ha a vizsgálandó és a zavaró elem egy új fázist (vegyületet) képez, vagy lehet nem-
specifikus, ha a vizsgálandó elem csak szétoszlik, beágyazódik, diszpergálódik a zavaró anyag feleslegében. Ez
a zavarási típus nemcsak jelcsökkenéssel, adott esetben jelnövekedéssel is járhat. Más elnevezés szerint ezek
kondenzált fázisú kölcsönhatások.
Ezek a zavaró hatások a legösszetettebb folyamatok eredménye. Az 5.4.5. fejezetben tárgyalt legegyszerűbbnek
számító két kationt és közös aniont tartalmazó két só esetén is több csoportba sorolhatók a kölcsönhatások (66.
ábra).
Már a lángfotometriából jól ismert a foszfátion zavaró hatása az alkáliföldfémekre. A lángba képződő kalcium-
foszfát a láng hőmérsékletén termikusan igen stabil (Ca2P2O7) kalcium-pirofoszfáttá alakul, amely az acetilén–
levegő lángban csak kis mértékben atomizálódik. Ezért a kalcium-sókhoz fölöslegben adagolt foszfát jelentős
abszorbancia-csökkenéssel jár (113. ábra).
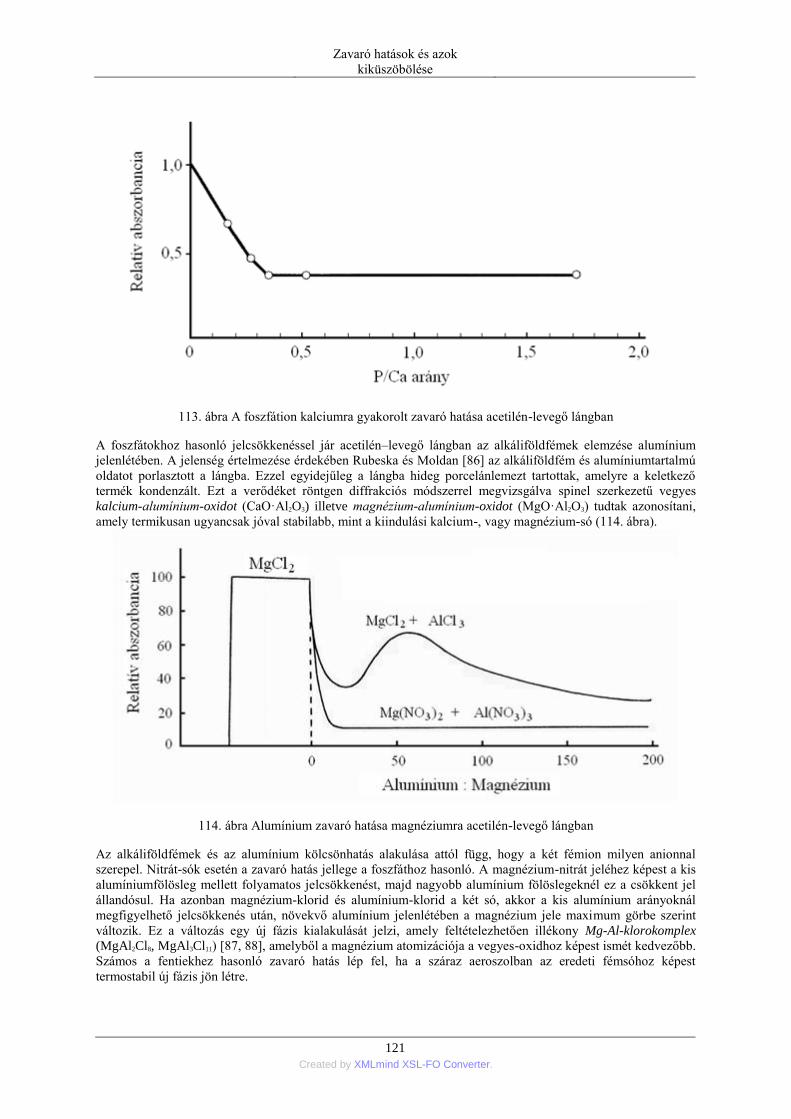
Zavaró hatások és azok
kiküszöbölése
121 Created by XMLmind XSL-FO Converter.
113. ábra A foszfátion kalciumra gyakorolt zavaró hatása acetilén-levegő lángban
A foszfátokhoz hasonló jelcsökkenéssel jár acetilén–levegő lángban az alkáliföldfémek elemzése alumínium
jelenlétében. A jelenség értelmezése érdekében Rubeska és Moldan [86] az alkáliföldfém és alumíniumtartalmú
oldatot porlasztott a lángba. Ezzel egyidejűleg a lángba hideg porcelánlemezt tartottak, amelyre a keletkező
termék kondenzált. Ezt a verődéket röntgen diffrakciós módszerrel megvizsgálva spinel szerkezetű vegyes
kalcium-alumínium-oxidot (CaO·Al2O3) illetve magnézium-alumínium-oxidot (MgO·Al2O3) tudtak azonosítani,
amely termikusan ugyancsak jóval stabilabb, mint a kiindulási kalcium-, vagy magnézium-só (114. ábra).
114. ábra Alumínium zavaró hatása magnéziumra acetilén-levegő lángban
Az alkáliföldfémek és az alumínium kölcsönhatás alakulása attól függ, hogy a két fémion milyen anionnal
szerepel. Nitrát-sók esetén a zavaró hatás jellege a foszfáthoz hasonló. A magnézium-nitrát jeléhez képest a kis
alumíniumfölösleg mellett folyamatos jelcsökkenést, majd nagyobb alumínium fölöslegeknél ez a csökkent jel
állandósul. Ha azonban magnézium-klorid és alumínium-klorid a két só, akkor a kis alumínium arányoknál
megfigyelhető jelcsökkenés után, növekvő alumínium jelenlétében a magnézium jele maximum görbe szerint
változik. Ez a változás egy új fázis kialakulását jelzi, amely feltételezhetően illékony Mg-Al-klorokomplex
(MgAl2Cl8, MgAl3Cl11) [87, 88], amelyből a magnézium atomizációja a vegyes-oxidhoz képest ismét kedvezőbb.
Számos a fentiekhez hasonló zavaró hatás lép fel, ha a száraz aeroszolban az eredeti fémsóhoz képest
termostabil új fázis jön létre.
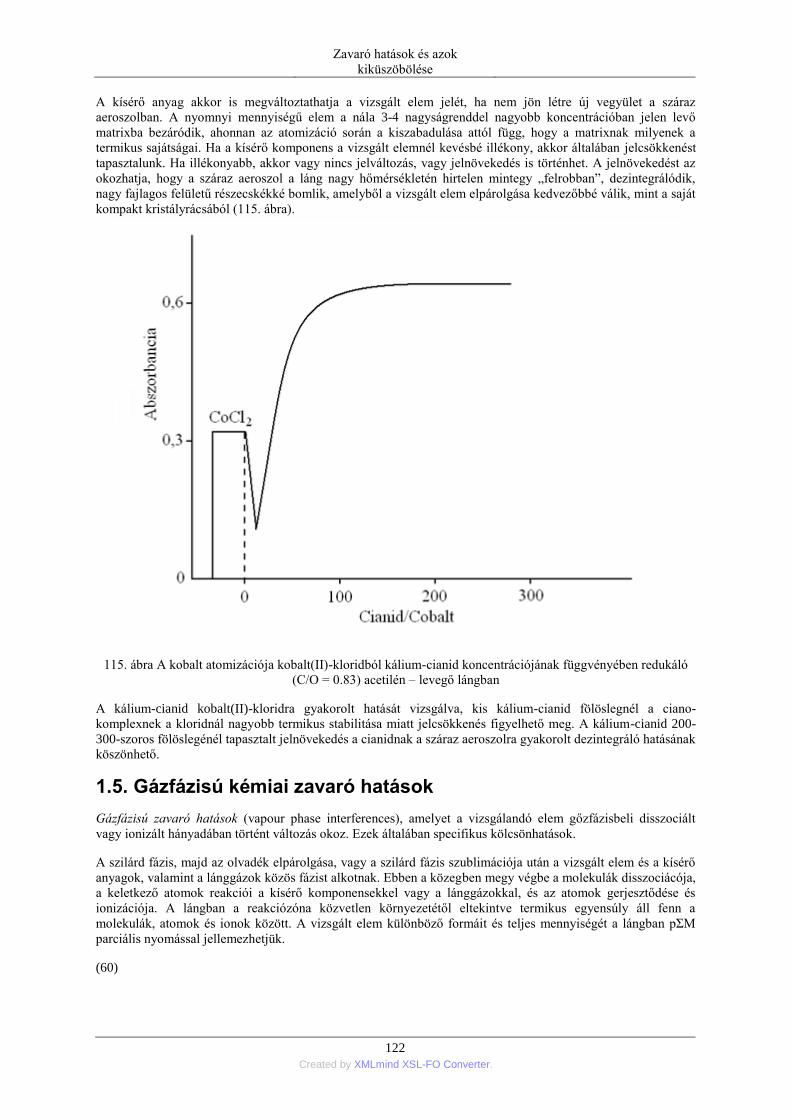
Zavaró hatások és azok
kiküszöbölése
122 Created by XMLmind XSL-FO Converter.
A kísérő anyag akkor is megváltoztathatja a vizsgált elem jelét, ha nem jön létre új vegyület a száraz
aeroszolban. A nyomnyi mennyiségű elem a nála 3-4 nagyságrenddel nagyobb koncentrációban jelen levő
matrixba bezáródik, ahonnan az atomizáció során a kiszabadulása attól függ, hogy a matrixnak milyenek a
termikus sajátságai. Ha a kísérő komponens a vizsgált elemnél kevésbé illékony, akkor általában jelcsökkenést
tapasztalunk. Ha illékonyabb, akkor vagy nincs jelváltozás, vagy jelnövekedés is történhet. A jelnövekedést az
okozhatja, hogy a száraz aeroszol a láng nagy hőmérsékletén hirtelen mintegy „felrobban”, dezintegrálódik,
nagy fajlagos felületű részecskékké bomlik, amelyből a vizsgált elem elpárolgása kedvezőbbé válik, mint a saját
kompakt kristályrácsából (115. ábra).
115. ábra A kobalt atomizációja kobalt(II)-kloridból kálium-cianid koncentrációjának függvényében redukáló
(C/O = 0.83) acetilén – levegő lángban
A kálium-cianid kobalt(II)-kloridra gyakorolt hatását vizsgálva, kis kálium-cianid fölöslegnél a ciano-
komplexnek a kloridnál nagyobb termikus stabilitása miatt jelcsökkenés figyelhető meg. A kálium-cianid 200-
300-szoros fölöslegénél tapasztalt jelnövekedés a cianidnak a száraz aeroszolra gyakorolt dezintegráló hatásának
köszönhető.
1.5. Gázfázisú kémiai zavaró hatások
Gázfázisú zavaró hatások (vapour phase interferences), amelyet a vizsgálandó elem gőzfázisbeli disszociált
vagy ionizált hányadában történt változás okoz. Ezek általában specifikus kölcsönhatások.
A szilárd fázis, majd az olvadék elpárolgása, vagy a szilárd fázis szublimációja után a vizsgált elem és a kísérő
anyagok, valamint a lánggázok közös fázist alkotnak. Ebben a közegben megy végbe a molekulák disszociácója,
a keletkező atomok reakciói a kísérő komponensekkel vagy a lánggázokkal, és az atomok gerjesztődése és
ionizációja. A lángban a reakciózóna közvetlen környezetétől eltekintve termikus egyensúly áll fenn a
molekulák, atomok és ionok között. A vizsgált elem különböző formáit és teljes mennyiségét a lángban pΣM
parciális nyomással jellemezhetjük.
(60)

Zavaró hatások és azok
kiküszöbölése
123 Created by XMLmind XSL-FO Converter.
Az MX a vizsgált elem oldatbeli saját anionnal alkotott molekulája, MY a lángban kialakuló más ellenionnal
képzett molekula, M az atomos, M+ pedig az ionizált állapot. A vizsgált elem pΣM parciális nyomás acetilén-
levegő lángban az oldat c moláris koncentrációjának körülbelül a tízezred része [38].
(61)
A (61) összefüggésből kitűnik, hogy a vizsgált elem parciális nyomása nagyságrendekkel kisebb, mint a
különböző lánggázok parciális nyomása. Ezért az elemnek a lángban fellépő disszociációs egyensúlyát illetve a
lángban fennálló molekuláris állapotát döntően befolyásolják a lánggázok. Az MY forma kialakulásában a
három legfontosabb gázkomponens a H, O és OH gyök. Ezek közül is elsősorban az oxigén a legjelentősebb,
amely a gázfázisú oxidok képződéséért felelős. Ezért is tartjuk a láng egyik legjellemzőbb adatának a szén-
oxigén (C/O) arány. Miután a lánggázok parciális nyomása 3-4 nagyságrenddel nagyobb a vizsgált eleménél, ez
egyben stabilizálja a vizsgált elem molekuláris és atomos formáinak termikus egyensúlyát.
A gázfázisú zavaró hatások tipikus példája az ionizációból következő zavaró hatás. A vizsgált elem a
lángösszetétel és a lánghőmérséklet függvényében részben ionizált állapotba kerül. Az ionizáció mértéke
acetilén-levegő lángban alkálifémeknél a legnagyobb. 10─3 mol/L koncentrációjú oldatok esetén a lítium 10 %-
a, a nátrium 20 %-a, a kálium 60 %-a, a rubidium 75 %-a és a cézium 95 %-a kerül ionizált állapotba. A jóval
nagyobb hőmérsékletű acetilén – dinitrogén-oxid lángban a kalcium 43 %-a, a stroncium 84 %-a, a bárium 88
%-a ionizálódik.
Az ionizáció aránya a lángba porlasztott oldat koncentrációjának is függvénye. Ezért egy kísérőanyag nélküli
alkálifém kalibrációs görbéjének abszorbancia értékei a 116. ábra szerint a növekvő koncentrációval az egyenes
arányosnál nagyobb mértékben növekszenek.
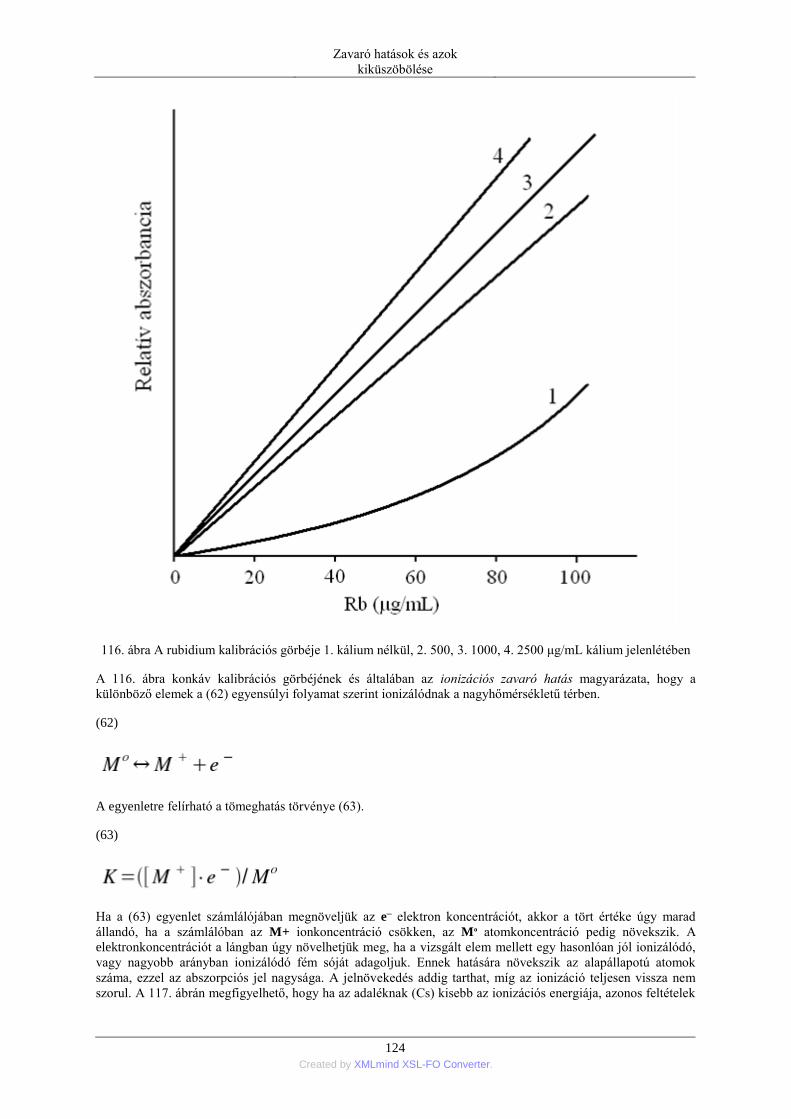
Zavaró hatások és azok
kiküszöbölése
124 Created by XMLmind XSL-FO Converter.
116. ábra A rubidium kalibrációs görbéje 1. kálium nélkül, 2. 500, 3. 1000, 4. 2500 μg/mL kálium jelenlétében
A 116. ábra konkáv kalibrációs görbéjének és általában az ionizációs zavaró hatás magyarázata, hogy a
különböző elemek a (62) egyensúlyi folyamat szerint ionizálódnak a nagyhőmérsékletű térben.
(62)
A egyenletre felírható a tömeghatás törvénye (63).
(63)
Ha a (63) egyenlet számlálójában megnöveljük az e─ elektron koncentrációt, akkor a tört értéke úgy marad
állandó, ha a számlálóban az M+ ionkoncentráció csökken, az Mo atomkoncentráció pedig növekszik. A
elektronkoncentrációt a lángban úgy növelhetjük meg, ha a vizsgált elem mellett egy hasonlóan jól ionizálódó,
vagy nagyobb arányban ionizálódó fém sóját adagoljuk. Ennek hatására növekszik az alapállapotú atomok
száma, ezzel az abszorpciós jel nagysága. A jelnövekedés addig tarthat, míg az ionizáció teljesen vissza nem
szorul. A 117. ábrán megfigyelhető, hogy ha az adaléknak (Cs) kisebb az ionizációs energiája, azonos feltételek
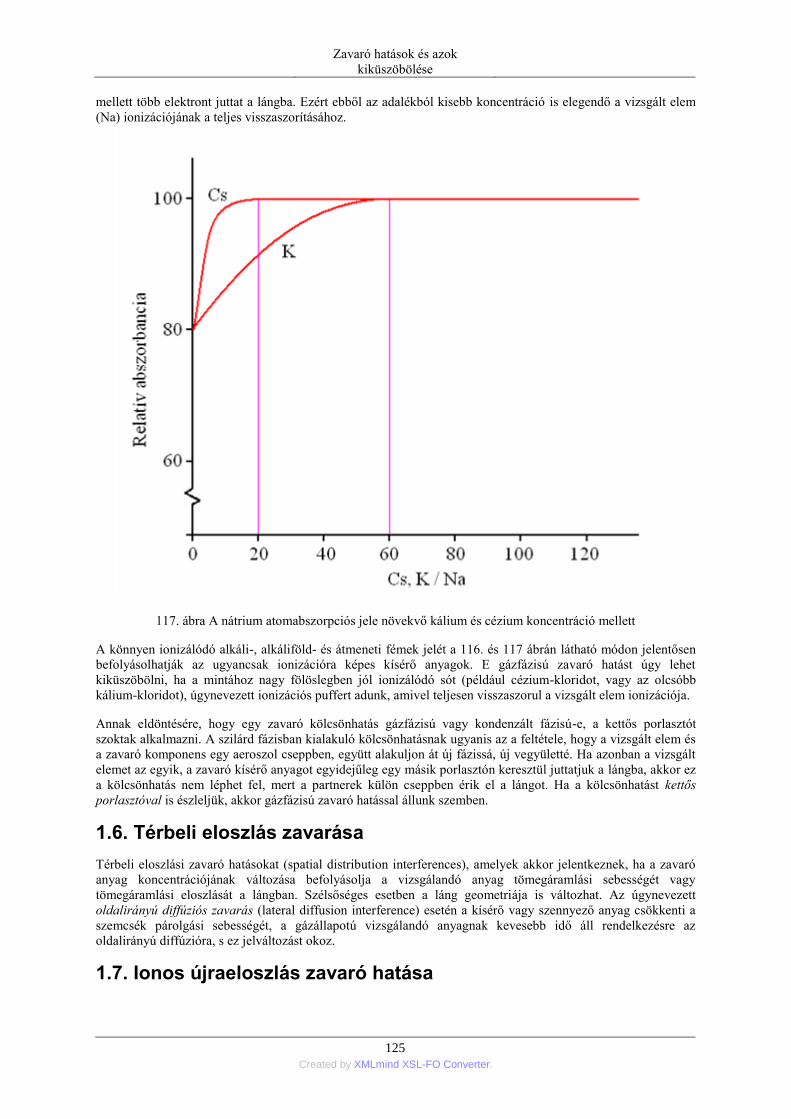
Zavaró hatások és azok
kiküszöbölése
125 Created by XMLmind XSL-FO Converter.
mellett több elektront juttat a lángba. Ezért ebből az adalékból kisebb koncentráció is elegendő a vizsgált elem
(Na) ionizációjának a teljes visszaszorításához.
117. ábra A nátrium atomabszorpciós jele növekvő kálium és cézium koncentráció mellett
A könnyen ionizálódó alkáli-, alkáliföld- és átmeneti fémek jelét a 116. és 117 ábrán látható módon jelentősen
befolyásolhatják az ugyancsak ionizációra képes kísérő anyagok. E gázfázisú zavaró hatást úgy lehet
kiküszöbölni, ha a mintához nagy fölöslegben jól ionizálódó sót (például cézium-kloridot, vagy az olcsóbb
kálium-kloridot), úgynevezett ionizációs puffert adunk, amivel teljesen visszaszorul a vizsgált elem ionizációja.
Annak eldöntésére, hogy egy zavaró kölcsönhatás gázfázisú vagy kondenzált fázisú-e, a kettős porlasztót
szoktak alkalmazni. A szilárd fázisban kialakuló kölcsönhatásnak ugyanis az a feltétele, hogy a vizsgált elem és
a zavaró komponens egy aeroszol cseppben, együtt alakuljon át új fázissá, új vegyületté. Ha azonban a vizsgált
elemet az egyik, a zavaró kísérő anyagot egyidejűleg egy másik porlasztón keresztül juttatjuk a lángba, akkor ez
a kölcsönhatás nem léphet fel, mert a partnerek külön cseppben érik el a lángot. Ha a kölcsönhatást kettős
porlasztóval is észleljük, akkor gázfázisú zavaró hatással állunk szemben.
1.6. Térbeli eloszlás zavarása
Térbeli eloszlási zavaró hatásokat (spatial distribution interferences), amelyek akkor jelentkeznek, ha a zavaró
anyag koncentrációjának változása befolyásolja a vizsgálandó anyag tömegáramlási sebességét vagy
tömegáramlási eloszlását a lángban. Szélsőséges esetben a láng geometriája is változhat. Az úgynevezett
oldalirányú diffúziós zavarás (lateral diffusion interference) esetén a kísérő vagy szennyező anyag csökkenti a
szemcsék párolgási sebességét, a gázállapotú vizsgálandó anyagnak kevesebb idő áll rendelkezésre az
oldalirányú diffúzióra, s ez jelváltozást okoz.
1.7. Ionos újraeloszlás zavaró hatása

Zavaró hatások és azok
kiküszöbölése
126 Created by XMLmind XSL-FO Converter.
Az említett osztályozás óta eltelt időben Browner és munkatársai [89] egy új típusú zavaró hatásról számoltak
be: az úgynevezett. aeroszol ionos újraeloszlás okozta zavaró hatásról. Részletes vizsgálatokból kiderült, hogy a
különböző méretű cseppekben eltér az ionok koncentráció-aránya. Ez a hatás pneumatikus porlasztóknál lép fel,
amely az aeroszol szemcseméretének függvénye. Lényege az, hogy bizonyos ionok az aeroszolban feldúsulnak a
folyadékfázishoz képest. Ez elsősorban az ICP emissziós spektrometriában okoz számottevő zavart. A
lángatomabszorpciós spektrometriában használatos porlasztó rendszerek esetén e hatás kismértékű.
1.8. A zavaró hatások kiküszöbölési módszereit
Fenti zavaró hatások közül a gyakoriságát és nagyságát tekintve legfontosabbak a nem-spektrális zavarások,
azok közül is elsősorban az oldott anyag párolgási és a gőzfázisú zavaró hatások. A nem-spektrális zavaró
hatások által okozott mérési hibák csökkentésére ill. kiküszöbölésére több eljárást alkalmaznak. A
leggyakoribbak a következők:
Standard addíciós e1járás: az elemzendő anyag ismert mennyiségeit adjuk a mintaoldat részleteihez. Az így
készített oldatsorozat abszorbancia értékeit ábrázoljuk a hozzáadott koncentrációk függvényében. A mérési
pontokra egyenest illesztünk. Ennek az egyenesnek és a negatív abszcissza tengely metszéspontja felel meg a
mintaoldat koncentrációjának. Hátránya, hogy csak korlátozott abszorbancia tartományban használható.
Szimulációs technika: a kalibráló oldatokat a mérendő oldathoz hasonló összetételűre készítjük, így a zavaró
hatások mindkét oldatban azonosak lesznek.
Puffer-addíciós technika: valamilyen spektrokémiai puffert adunk mind a minta-, mind a kalibráló oldathoz
azért, hogy a mérés kevésbé legyen érzékeny a zavaró elemek koncentrációjának változására. Ezek az
úgynevezett spektrokémiai pufferek.
— felszabadító adalék (releaser), amely csökkenti a vizsgált elem párolgását zavaró hatásokat azáltal, hogy
vegyületet képez a zavaró ágenssel, s ezzel meggátolja, hogy a mérendő elem termikusan stabil vegyületbe
lépjen. Ennek a felszabadító adaléknak tipikus esete az alkálifém-foszfátok okozta zavaró hatás kiküszöbölése
lantán-klorid adalékkal. A lantán termikusan stabilisabb lantán-foszfátot képez, mint a kalcium. Ezért, ha
kalcium és foszfáttatalmú oldathoz lantán-sót adunk fölöslegben, a lantán leköti a foszfátot és a kalcium zavaró
hatástól mentesen elemezhető [90].
— védő (protektív) adalék, amely kémiailag vegyületet képez az analizálandó anyaggal, így gátolva meg a
zavaró anyag hatását, lehetővé teszi ugyanakkor a vizsgált elem atomizálódását. A különböző komplexképző
ligandumok gyakran zavarják az átmenetifémek atomabszorpciós meghatározását. Ezeket a zavaró hatásokat
úgy lehet kiküszöbölni, ha a mintához fölöslegben kálium-cianidot adunk. Az átmenetifémek ugyanis a
cianiddal adják a legstabilisabb komplexeket. Ezért a mintához adott cianid az egyéb ligandumokat kiszorítja a
fém koordinációs szférájából és mint ciano-komplex atomizálódik, függetlenül attól, hogy a mintában az
átmenetifém eredetileg milyen komplexek formájában volt jelen [91-93].
— ionizációs puffer, amely növeli a szabad elektronkoncentrációt a lánggázokban, így visszaszorul az ionizáció
vagy stabilizálódik az ionizáció foka. Az ionizációs pufferek mechanizmusát részletesebb a 6.1.5. fejezetben
ismertettük.
— párolgást elősegítő adalék, amely növeli a kísérő komponensek illékonyságát, vagy illékonyabb vegyületet
képez a vizsgálandó anyaggal.
— telítő adalék, a mintaoldatokhoz nagy koncentrációban adott zavaró ágens, amellyel a zavaró hatás állandó
szintjét lehet beállítani. Ez a módszer ritkán használható
1.9. Grafitkemencében fellépő zavarások kiküszöbölése matrixmódosítókkal
Amint azt az 5.6.7. fejezetben jeleztük, a grafitkemencében lejátszódó atomizációs folyamatok mértéke, iránya
igen erősen függ a vizgaált elemet kísérő mátrix minőségétől és mennyiségétől. Ezért az így fellépő kondenzált
és gázfázisú zavaró hatások csökkentésére, vagy megszüntetésére olyan adalékanyagokat adunk a mintához,
amely megváltoztatja a kísérő mátrix viselkedését és jelenlétében a vizsgált elem azonos mértékben
atomizálódik a mintából mint a mintával azonos koncetrációjú kalibráló oldatból.

Zavaró hatások és azok
kiküszöbölése
127 Created by XMLmind XSL-FO Converter.
A módosító adalék célja lehet, hogy a kísérő anyagokat eltávolítsuk a vizsgált elem mellől még az atomizáció
előtt. Erre példa a réz GFAAS meghatározása tengervízben. Ediger és munkatársai [94, 95] ammónium-nitrát
adalékot alkalmaztak mátrixmódosítóként. A nem túl illékony nátrium-klorid (olvadáspontja 801 oC,
forráspontja 1413 oC) az ammónium –nitrát hatására nátrium-nitráttá alakul az alábbi egyenlet szerint.
(64)
A keletkező nátrium-nitrát elbomlik 380 oC-on, az ammónium-klorid szublimál 335 oC-on és az ammónium-
nitrát fölöslege pedig 210 oC-on bomlik el. Így minden kísérő anyag eltávolíthatóvá válik kis hőmérsékleten.
Más esetekben az a cél, hogy a vizsgált többnyire illékony elemet kevésbé illékonnyá tegyük, hogy idő előtt ne
távozzon a kemencéből. Nikkel-sóval stabilizálták az arzént, szelént, tellurt, ammónium-hidrogén-foszfáttal a
kadmiumot, ammónium-szulfiddal a higanyt. Erős oxidálószerek, mint perklórsav, vagy salétromsav és
hidrogén-peroxid elegye kedvezően hatnak a germánium és a gallium elemzésére.
A különböző típusú minták elemzésénél újabb és újabb adalékok bizonyultak sikeresnek. Ilyenek a molibdén
vegyületek, kálium-bikromát, magnézium-nitrát. E számos mátrixmódosító adalékról ad részletes áttekintést
Tsalev és munkatársainak összefoglaló munkája [96].
A kezdetektől törekedtek az analitikusok arra, hogy olyan univerzális mátrixmódosítót találjanak, amelyet a
minták és a vízsgált elemek minél szélesebb körére sikerrel lehessen használni. E tekintetben az utóbbi időben
igen hatásosnak tekinthetők a palládium-sók illetve a palládium-só és magnézium-nitrát elegye ( Pd-Mg
módosító adalék) [97].
A palládium mátrixmódosító mechanizmusa a mintákban még a mai napig nincs minden részletében tisztázva.
Feltételezhetően a mátrixmódosító (PdCl2) jelenlétében az előhevítési szakaszban fém állapotba kerülő
palládium interlamelláris formában kötődik meg a grafit felületi rétegeiben, és elegy-vegyületet képez a minta
fémtartalmával. Ezáltal megnöveli annak hőstabilitását, megakadályozza a minta szelén-tartalmának idő előtti
távozását a grafitcellából. Az előbbiek alapján az alábbi mechanizmus valószínűsíthető [98].
1. A szelén-dioxid hidratációjának megakadályozása kb. 400 K-en
(65)
ahol x = 1 vagy 2
2. Elegyvegyület létrejötte >400 K-on
(66)
ahol a (Se−Pd)(ad) asszociátum, mely akkor keletkezik mikor a grafitcső felületén újból adszorbeálódik a
szelén és a palládium.
3. Az elegyvegyült termikus disszociációja 1200 K-on
(67)
4. Szabad szelénatomok keletkezése kb. 1500 K-on

Zavaró hatások és azok
kiküszöbölése
128 Created by XMLmind XSL-FO Converter.
(68)
és 1900 K-on
(69)
A mátrixmódosítók egy további típusa különböző szerves adalékok (aszkorbinsav, citromsav) alkalmazása. E
vegyületek a fémionokkal komplexet képezve álcázzák, megvédik azokat bizonyos kedvezőtlen mátrixhatástól.

129 Created by XMLmind XSL-FO Converter.
6. fejezet - A kémiai analízis folyamata és teljesítőképessége
Az IUPAC az 1990-es évek közepén megfogalmazta, összefoglalta, hogy mivel foglalkozik az analitikai kémia.
Ez a meghatározás eddig így nem jelent meg az analitikai tankönyvekben.
Az analitikai kémia az a tudományág, amely eljárásokat, módszereket, készülékeket és stratégiákat
dolgoz ki annak érdekében, hogy anyagi rendszerek minőségi, mennyiségi összetételéről, szerkezetéről és
energiaállapotáról térbeli és időbeli információkat szerezzen.
1. A kémiai analízis lépései
A kémiai analízis összetett folyamat, amelynek a főbb lépései a 118. ábrán láthatók.
118. ábra A kémiai analízis lépései
A 118. ábrán bemutatott lépésekről általánosan megállapítható, hogy azok szigorúan egymásra épülnek. Ha a
sorban előbb álló lépésben valamilyen hibát követünk el, a további lépések értelmüket vesztik, vagy legalábbis
pontatlan, félrevezető eredményekhez jutunk. Ezért minden lépés egyenrangúan fontos.
• Az 1. Célkitűzés, a probléma megfogalmazása lépésben kell a megoldandó konkrét analitikai feladatot
egyértelműen és pontosan körülhatárolni.
• A 2. Stratégiakészítés lépésben a kitűzött feladathoz igazítva kell megtervezni és kiválasztani az analízis
legcélravezetőbb további lépéseit a mintavételtől a kiértékelésig.
• A 3. Mintavétel az első gyakorlati lépés, amelynél a döntő szempont, hogy egy anyagi rendszerből kivett kis
tömegű, térfogatú minta, amellyel a kémiai analízist végezzük, ugyanolyan összetételű legyen, mint a vizsgált
anyagi rendszer. Az analízis eredményével csak ebben az esetben jellemezhetjük az egész rendszert. A
mintavétel technikai megvalósítása a kitűzött analitikai céltól és a vizsgálni kívánt anyag fizikai és kémiai
tulajdonágaitól egyaránt függ. Ezért mára igen nagy számú mintavételi eljárás található az irodalomban. Az
elkülönített, elemzésre szánt mintának minden esetben az egész rendszer átlagát kell jellemeznie; erre nézve
reprezentatívnak kell lennie.
• A 4. Minta szállítása, tárolása a mintavétel utáni lépés, amely során olyan körülményeket kell biztosítani,
hogy a minta összetétele az elemzés elvégzéséig ne változzon meg. Ennek módját elsősorban a minta típusa
és az elemezni kívánt komponens természete határozza meg.
• Az 5. Mintaelőkészítés a kémiai analízisen belül az a művelet, amely segítségével a mintát olyan alakra
hozzuk, hogy az előzőleg kiválasztott analitikai módszerrel elemezni lehessen. A mintaelőkészítést úgy kell

A kémiai analízis folyamata és
teljesítőképessége
130 Created by XMLmind XSL-FO Converter.
végrehajtani, hogy a vizsgálni kívánt mintakomponensekből ne legyen veszteség, a minta más (idegen)
komponensekkel ne szennyeződjön, azaz a minta minden komponensére megőrízze eredeti összetételét. A
mintaelőkészítés módja egyaránt függ a minta típusától, a vizsgálni kívánt komponensek tulajdonságaitól és a
kiválasztott elemző módszertől.
• A 6. Elemzés lépése az a kémiai, fizikai-kémiai műveletek összessége, amelyek segítségével olyan
érzékszervi észlelésekhez, illetve számszerű vagy számszerűsíthető adatokhoz jutunk, amelyek egyértelmű
összefüggésben vannak a mintában levő komponensek minőségével (mineműségével) és e komponensek
egymáshoz vagy a minta egész tömegéhez, térfogatához viszonyított arányával (koncentrációjával) vagy
abszolút tömegével.
• A 7. Kiértékelés a különböző analitikai módszerekkel kapott érzékszervi (főleg vizuális) észlelések
rendszerezését, illetve számszerű adatok numerikus, grafikus illetve statisztikai vagy egyéb módon történő
feldolgozását jelenti, amelyekből a minta minőségi és mennyiségi összetételét határozzuk meg. Kémiai
analízisnek korábban általában az egész műveletnek ezt a szűkebb értelemben vett két lépését nevezték.
• A 8. Értékelés az előző lépésben kapott analitikai eredmények összevetése a célkitűzésben megfogalmazott
tervekkel, amiért az egész analízis történt. Ekkor válik a kémiai információ felhasználói információvá a
vizsgált anyagról (pl. a gyógyszer megfelelő mennyiségben tartalmazza-e a hatóanyagot, a vizsgált kút vize
alkalmas-e ivásra stb.).
2. A módszer teljesítőképessége, érvényesítése (validálása)
Egy módszer érvényesítése (validálása) az a tevékenység, amely rendszerezett vizsgálatok segítségével
bizonyítja, hogy a módszer teljesítményjellemzői kielégítik az analitikai módszerrel szemben támasztott
követelményeket. Ez azt jelenti, hogy ha egy új módszert dolgozunk ki, akkor úgy lehet bevezetni, ha az alábbi
teljesítményjellemzőket meghatározzuk.
• Szelektivitás és specifitás (Selectivity and specificity)
• Tartomány (Range)
• Linearitás (Linearity)
• Érzékenység (Sensitivity)
• Kimutatási határ (Limit of detection)
• Meghatározási határ (Limit of quantitation)
• Zavartűrés (Ruggedness)
• Pontosság (Accuracy)
• Precizitás (Precision)
Egy módszer szelektivitása arra vonatkozik, hogy a módszer milyen mértékben képes adott alkotók
meghatározására egyéb zavaró komponensek jelenlétében. Azt a módszert, amely a meghatározandó alkotók
egy csoportjára tökéletesen szelektív; specifikusnak nevezzük. A szelektivitás mértékének megállapítása
céljából az elemzéseket különböző mintákkal, a tiszta standard oldatoktól a komplex mátrixokig, el kell végezni.
Minden esetben meg kell határozni a kérdéses alkotó(k) meghatározásának elérhető mértékét és meg kell
állapítani a feltételezett zavaró hatásokat. (A módszer alkalmazhatóságára vonatkozó bármilyen korlátozást a
módszer dokumentációjában rögzíteni kell).
Méréstartomány: A mennyiségi elemzés céljára a módszer méréstartományát az alkotót különböző
koncentrációban tartalmazó minták elemzésével, a válaszjel meghatározásával kell megállapítani, kijelölve azt a
munkatartományt, amelyre az adott feladatnál kielégítő helyesség és precizitás érhető el. Az analitikai
mérőgörbét az alkotót különböző koncentrációban tartalmazó minta elemzési eredményeiből regresszióval
számíthatjuk ki, általában a legkisebb négyzetek módszerének alkalmazásával. A komponens válaszjele és a
koncentráció között nem kell feltétlenül lineáris összefüggésnek lennie, hogy a módszer hatékonyan

A kémiai analízis folyamata és
teljesítőképessége
131 Created by XMLmind XSL-FO Converter.
alkalmazható legyen. A jó linearitású módszerek esetén 5 különböző pont (plusz a vak) általában elégséges az
analitikai mérőgörbe elkészítéséhez. Nem-linearitás esetén több standard alkalmazására van szükség.
Amennyiben a linearitás nem teljesül egy adott eljárás során, a számítás megfelelő algoritmusát meg kell
határozni.
Az analitikai mérőgörbe linearitásán azt értjük, hogy a mérőgörbe adott tartományában, az ún. lineáris
tartományban, adott megbízhatósággal egyenesnek tekinthető. A linearitást a méréstartományt lefedő
koncentrációjú minták elemzésével határozzuk meg. Az eredményekből a legkisebb négyzetek módszerével
számítjuk ki a regressziós egyenest az alkotó koncentrációja függvényében. Előnyös, ha a módszer az
alkalmazni kívánt munkatartományban lineáris, de ez nem feltétlenül követelmény.
Az érzékenység az analitikai mérőgörbe meredeksége, azaz a mért analitikai válaszjelnek a koncentráció vagy
az anyagmennyiség szerinti deriváltja. (Egységnyi koncentrációváltozásra eső válaszjelváltozás.)
Egy alkotó kimutatási határa az a koncentráció, vagy anyagmennyiség, amelyhez tartozó válaszjel értéke
megegyezik a vak minta közepes válaszjelének (σ), és a vak minta válaszjel háromszoros tapasztalati szórásának
összegével (3σ). A minőségi elemzésben elterjedt, hogy több különböző koncentrációjú minta elemzésével
határozzák meg a kimutathatóság pontos határát.
A meghatározási határ az a legkisebb koncentráció, vagy anyagmennyiség, amely még elfogadható
pontossággal és precizitással határozható meg. A meghatározási határ megfelelő standard minta segítségével
állapítható meg. Általában ez az analitikai mérőgörbe legalsó értékelhető pontja. Extrapolációval történő
meghatározás nem fogadható el. A meghatározási határ megadásakor fel kell tüntetni az ehhez elfogadott
helyességi és precizitási követelményt is.
Zavartűrés (Eszköz- és környezetállóság). Ha különböző laboratóriumok ugyanazt a módszert alkalmazzák,
akkor elkerülhetetlenül jelentkeznek olyan apró eltérések, amelyeknek esetleg számottevő hatásuk lehet a
módszer teljesítményére. A módszer zavartűrését úgy vizsgáljuk, hogy szándékosan változtatásokat eszközölünk
a módszer paramétereiben és vizsgáljuk azok következményeit. Számtalan tényezőt kellene vizsgálni, de mivel
többségüknek a hatása elhanyagolhatóan kicsi, több tényező együttes vizsgálatára is lehetőség van. A
zavartűrést általában először a módszerfejlesztő laboratórium vizsgálja meg, mielőtt más laboratóriumok
közreműködésére sor kerülne.
A módszer pontossága a méréstartomány torzítatlanságának (valódiságának) a mértéke. Egy módszer annál
pontosabb, minél kisebb a várható érték és a valódi érték különbsége. Meghatározása megfelelő és megbízható
referenciaanyag elemzésével történhet. Ha megfelelő referenciaanyag nem áll rendelkezésre, a helyesség
becslése úgy is elvégezhető, hogy a mintához kémiai standard anyag ismert mennyiségét adjuk és
meghatározzuk az addíció előtti és utáni analitikai válaszjelértékeket. Az addíció érvénye korlátozott, a
helyesség meghatározására csak a hozzáadást követő koncentrációszakaszra alkalmas. A helyességet egy
(standard) elfogadott módszer vagy több eljárás eredményeivel történő összehasonlítás és/vagy laboratóriumok
közötti körvizsgálatok segítségével is meghatározhatjuk.
A módszer precizitása (szorossága) a kölcsönösen független megismételt vizsgálatok eredményei közötti
egyezés mértéke, rendszerint a tapasztalati szórással kifejezve. Értéke általában függ a komponens
koncentrációjától, ezért ezt a koncentrációfüggést meg kell határozni és dokumentálni kell. A precizitás
különböző módon adható meg, attól függően, hogy meghatározása milyen körülmények között történt. Az
ismételhetőség a precizitás azon fajtája, amely ismételhető körülmények között elvégzett kísérletekre
vonatkozik, vagyis: azonos módszer, azonos anyag, azonos műszer, azonos kezelő, azonos laboratórium. A
reprodukálhatóság a precizitás azon fajtája, amely reprodukálható körülmények között elvégzett kísérletekre
vonatkozik, vagyis: azonos módszer, különböző műszer, különböző kezelő, különböző laboratórium.

132 Created by XMLmind XSL-FO Converter.
7. fejezet - Az atomabszorpciós spektrometria alkalmazásai
1. Elemek és minták
1.1. A módszerrel vizsgálható elemek és mintatípusok
Az atomabszorpciós spektrometriának, mint elemanalitikai módszernek a széleskörű elterjedését egyéb erényei
mellett az is magyarázza, hogy mintegy 70 elem határozható meg segítségével, akár lángatomizációt, akár
grafitkemencés atomizálást alkalmazunk. A tipikusan nem-fémek: nemesgázok, hidrogén, halogének, oxigén,
nitrogén és szén az elemeknek az a köre, amelyeknek nagy a gerjesztési energiájuk. Ezért az elemző vonalaik
hullámhossza a vákuum ultraibolya tartományba esik. A rövid élettartamú transzurán elemeket sem tudjuk
elemezni ezzel a módszerrel. A többi elem atomizációjához elegendő a korábban (5.4.2. fejezetben) bemutatott
két láng, az acetilén – levegő és az acetilén – dinitrogén-oxid. A 119. ábrán tüntettük fel a lángatomabszorpciós
módszerrel meghatározható elemeket és azt, hogy melyik láng alkalmas az atomizációjukra.
119. ábra A lángatomabszorpciós spektrometriás módszerrel meghatározható elemek
Az atomabszorpciós spektrometriás szakkönyvek, kézikönyvek külön fejezetekben tárgyalják azokat a
mintatípusokat, amelyek a mintavétele, a minta-előkészítése illetve elemzése a mintákra jellemző eljárásokat
igényel. Alábbiakban néhány ilyen mintacsoportok listáját közöljük.
1. Vízminták
2. Talajok, üledékek
3. Környezeti minták
4. Élelmiszerek
5. Testfolyadékok és szövetek
6. Biológiai anyagok
7. Kőzetek, ércek, ásványok
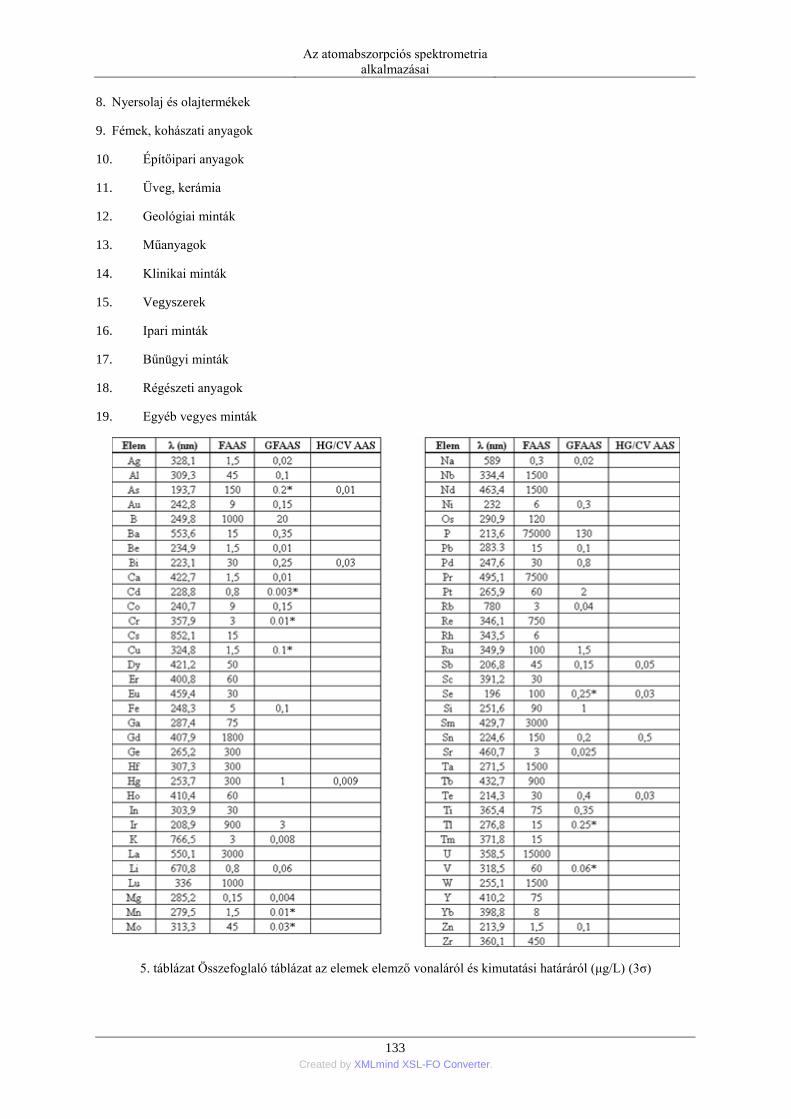
Az atomabszorpciós spektrometria
alkalmazásai
133 Created by XMLmind XSL-FO Converter.
8. Nyersolaj és olajtermékek
9. Fémek, kohászati anyagok
10. Építőipari anyagok
11. Üveg, kerámia
12. Geológiai minták
13. Műanyagok
14. Klinikai minták
15. Vegyszerek
16. Ipari minták
17. Bűnügyi minták
18. Régészeti anyagok
19. Egyéb vegyes minták
5. táblázat Összefoglaló táblázat az elemek elemző vonaláról és kimutatási határáról (μg/L) (3σ)

Az atomabszorpciós spektrometria
alkalmazásai
134 Created by XMLmind XSL-FO Converter.
Megjegyzések: A kimutatási határok mátrixmentes oldatokra vonatkoznak. A GFAAS adatok 50μL
oldattérfogatok, a HG/CV AAS adatok pedig flow injection módszerrel 500 μL oldat elemzése alapján kerültek
kiszámításra. * többelemes AAS készüléken végzett mérés adatai.
1.2. Minta-előkészítési eljárások
A 8.1.1. fejezetben bemutatott számos mintatípus közötti különbség többek között a minta-előkészítés módjában
van. Az AAS analízisben használatos minta előkészítési eljárásokat a biológiai, humánbiológiai minták példáján
mutatjuk be.
Biológiai minták előkészítése nyomelem-analízisre
A biológiai minták közös jellemzője, hogy azok különböző méretű és szerkezetű szervesanyag tartalma mellett
életfontosságú nyomelemeket, bio-katalizátorokat tartalmaznak. Mivel katalizátorokról van szó, ezek
koncentrációja általában nagyon kicsi. Ezek pontos ismerete azért is fontos, mert sok esetben a szükséges
nyomelem-koncentráció és a már mérgezést okozó dózis között nincs nagy különbség. Ugyancsak fontos a
nyomelem-koncentráció meghatározása, ha a szervezetet mérgező hatás éri. Ekkor a toxikus elemek pontos
koncentrációjának meghatározása a feladat. Ezeknek az esszenciális és toxikus elemeknek a biológiai mintákban
történő meghatározása általában nem végezhető el közvetlenül a minták eredeti formájában. A biológiai
mintákban a nyomelemek különböző kémiai kötésben fordulnak elő. A különböző halmazállapotú,
szervesanyag-tartalmú mintákban a szervetlen sókat, nyomelemeket az előkészítési módszerekkel megfelelő
alakra kell hozni ahhoz, hogy AAS módszerrel meghatározhatók legyenek. Ez az esetek túlnyomó többségében
azt jelenti, hogy a szerves anyag teljes mennyiségét eltávolítjuk a mintából, a visszamaradó szervetlen
vegyületeket pedig vízben vagy savban oldható formára hozzuk.
A biológiai minták előkészítésére az alábbi módszereket alkalmazzuk.
I. I.Száraz hamvasztás
II. I.Nedves roncsolás
• Nyílt rendszerben atmoszférikus nyomáson
• Zárt térben acélköpenyes teflon bombában, nagy nyomáson
• Zárt térben, műanyag köpenyes bombában, mikrohullámú energiaközléssel
Száraz hamvasztás
A száraz hamvasztás első lépéseként pontosan lemért tömegű tiszta száraz porcelán izzító tégelyekbe helyezzük
az elemezni kívánt biológiai mintákat. Lemérjük a tömegüket, majd 1 órán át 105 oC-on szárítjuk. Ismét
lemérjük a tömegeket. A tömegcsökkenésből kiszámítjuk a minták nedvességtartalmát. Következő lépésként a
szárított mintákat tartalmazó tégelyeket izzító kemencébe helyezzük, ajtaját gondosan bezárjuk. A 20–1000 oC
között szabályozható hőmérsékletet biztosító kemence például úgynevezett ejtőkengyeles hőfokszabályzóval áll
összeköttetésben. E szabályzó segítségével a kemence hőmérsékletét 20 oC-tól kezdve félóránként 100 fokkal
emeljük 800 oC-ig. Ezzel a kíméletes fűtési sebességgel biztosítjuk, hogy a hőfolyamatok, a kémiai átalakulások
a minta teljes térfogatában lejátszódjanak. A hamvasztási folyamat teljes időtartama alatt a kemence ajtaját
kinyitni nem szabad. Ha ugyanis az ajtót kinyitjuk, akkor nagy mennyiségű oxigén jut a nagy hőmérsékletű
mintához, amitől az lángra lobban. Ezzel azonban a minta felületén a hőmérséklete hirtelen 1500 – 2000 oC-ra
emelkedhet, amivel szabályozatlanná válik a hamvasztás, és komoly mintaveszteséget eredményezhet.
A szabályozott hamvasztás során lejátszódó fő folyamat minden biológiai minta esetén, hogy a szerves
vegyületek széntartalma szén-dioxiddá, hidrogéntartalma pedig vízzé ég el.
(70)
Az izzító kemencében levő mintához a hamvasztás alatt a kemence nyílásain beszivárgó levegővel csak annyi
oxigén jut, hogy az adott hőfokon izzásban legyen és szabályozottan, egyenletesen menjen végbe az oxidáció.

Az atomabszorpciós spektrometria
alkalmazásai
135 Created by XMLmind XSL-FO Converter.
Az elhamvasztott mintát lehűlés után kivesszük a kemencéből. A visszamaradt fehér vagy enyhén sárga por az
eredeti biológiai mintának csak a szervetlen fő- és nyomkomponenseit tartalmazza elsősorban oxidok
formájában. E maradék tömegét lemérjük, ami az adott mintára ugyancsak jellemző adat. A hamvasztási
maradékot ezután salétromsavban feloldjuk és mérőlombikban adott térfogatra hígítjuk. Az így elkészített
oldatokat használjuk fel a műszeres analízishez.
A száraz hamvasztás módszer előnyös tulajdonságai:
1. A hőfok beállításán túl minta előkészítése nem kíván állandó felügyeletet
2. A minta tömegétől, a porcelán tégelyek méretétől függően nagyszámú (20-25 db) minta előkészítése is
elvégezhető egyidejűleg.
3. A szerves anyag teljes mennyisége eltávozik a mintából
A száraz hamvasztás hátránya:
Az illékony elemek (higany, kadmium, ólom, vas kloridja stb.) jelentős része, vagy teljes mennyisége eltávozik
a mintából. Így a száraz hamvasztással történő előkészítéssel ezekre az elemekre a minta elemzése jelentős
negatív hibával jár. Az előkészítéshez a hőmérsékletet úgy kell megválasztani, hogy a minta meghatározandó
komponenseinek teljes tömege megmaradjon a hamvasztás alatt.
Nedves roncsolás atmoszférikus nyomáson
A pontosan lemért tömegű (0.5 – 1 gramm) biológiai mintát Erlenmeyer-lombikba helyezzük. A mintához 10 ml
koncentrált salétromsavat adunk. A lombikot szabályozható hőmérsékletű homokfürdőre helyezzük, és óvatosan
melegítjük. Az erős nitrózusgőz fejlődés jelzi, hogy a szerves anyag oxidációja folyik. A nedves roncsolás során
a száraz hamvasztáshoz hasonlóan a szerves anyagok az erős oxidáló savak hatására elvileg széndioxiddá és
vízzé alakulnak át. Ha a nitrogén-oxidok fejlődése megszűnik, akkor további 10 ml tömény salétromsavat adunk
a mintához és melegítjük. A mintát lehűtve óvatosan 2-3 ml 30%-os hidrogén-peroxid hozzáadásával segítjük
elő a teljes oxidációt. A lombikban levő folyadékelegyet ezután óvatosan szárazra pároljuk és megfigyeljük,
hogy a lombik aljára száradó maradék nem barnul-e. Ha igen, az azt jelzi, hogy a minta karamellizálódik, azaz
még van szerves anyag tartalma. Ekkor további salétromsav és hidrogén-peroxid adagolással folytatjuk a
roncsolást. Amikor a roncsolás teljessé vált, az óvatosan szárazra párolt maradék az eredeti minta szervetlen
vegyületeit, elsősorban nitrátjait tartalmazza. Ezt a maradékot 0.1 mol/L salétromsavval oldjuk fel és
mérőlombikban adott térfogatra töltjük. Ez az oldat használható fel minőségi és mennyiségi elemzésekre.
Az atmoszférikus nedves roncsolás előnyei:
Minden fő- és nyomelem veszteség nélkül a mintában marad. Nedves roncsolás nem eredményez negatív hibát.
Az atmoszférikus nedves roncsolás hátrányai:
1. A mintaelőkészítés állandó, folyamatos figyelmet igényel.
2. A maró hatású savgőzök miatt a munka kényelmetlen, számos munkavédelmi előírás folyamatos betartását
igényli (jól húzó vegyi fülke, gumikesztyű, védő álarc).
3. A roncsoláshoz használt vegyszerek maguk is szennyezettek bizonyos nyomelemekkel. Ezeket nagyobb
mennyiségben a mintához adagolva jelentősen elszennyezhetjük a mintát. Az eredetinél akár nagyságrenddel
nagyobb koncentrációban kerülhetnek bizonyos elemek a mintához. Emiatt a nedves roncsolással pozitív
hibát követünk el.
Nedves roncsolás zárt térben, teflon bombában
Az atmoszférikus nedves roncsolás hátrányainak (fokozott figyelem, egészségkárosító gázok fejlődése, a
roncsoló szerek szennyező hatása) kiküszöbölésére került kialakításra a teflon bombás roncsolás.
A teflon bomba belső része egy teflon edény teflon kupakkal, amelyet egy nyomásálló acélköpeny vesz körül,
amire menetesen acél kupak csavarható. A kupakban egy acél lemezrugó azt biztosítja, hogy ha a bombában
túlnyomás alakulna ki, a rugó megemelkedik, a bomba kinyílik és megszűnik a nyomás. A savgőzök ilyenkor a

Az atomabszorpciós spektrometria
alkalmazásai
136 Created by XMLmind XSL-FO Converter.
kupakon levő kis furaton távoznak. Ezzel persze éppen a bombában levő minta tönkre megy, de így nem robban
szét a bomba.
A roncsolás során a 2–3 ml folyadékot (például vért, vérszérumot) vagy 0.2–0.5 gramm szilárd mintát a
hengeres teflonedénybe mérjük be. A bemért mintához 3–4 ml koncentrált salétromsavat, 1–2 ml 30 %-os
hidrogén-peroxidot adunk. Ezután a bomba acél kupakját nyomaték-kulccsal rácsavarjuk a testre és a bombát
behelyezzük egy fűtőblokkba, vagy egyszerűen egy szárítószekrénybe. A szárítószekrény hőmérsékletét 150 oC-
ra állítjuk be. A bomba körülbelül 1 óra alatt veszi fel szekrény hőmérsékletét. További 1 óra hosszáig ezen a
hőmérsékleten tartjuk a bombát. Eközben a zárt bombában kb. 100 bar nyomás alakul ki. Ilyen nagy nyomáson
és hőmérsékleten egyrészt a kémiai reakciók sebessége többszörösére nő. Másrészt a mintához adagolt néhány
ml roncsoló anyag is elegendő ahhoz, a szerves anyagok teljes mennyisége elvileg széndioxiddá és vízzé
alakuljon. Kikapcsoljuk a szárító szekrényt és megvárjuk, hogy a bomba teljesen lehűljön. (Ez további 1 órát
vesz igénybe) Ezután (az esetleges kis túlnyomás miatt) óvatosan kinyitjuk a bombát és a tiszta folyadékot
átöntjük mérőlombikba és a bombát utána mosva ioncserélt vízzel jelre töltjük. Ezt az oldatot lehet elemzésre
felhasználni.
A teflon bombás mintaelőkészítés előnyei:
1. Kis térfogatú roncsolószer szükséges, ezért kis mértékű a minta szennyeződése.
2. Kis odafigyelést igényel a művelet.
3. A zárt rendszer folytán mérgező gázok, gőzök távozásával csak esetlegesen ( a szelep „lefújásakor”) kell
számolni.
A teflon bombás mintaelőkészítés hátrányai:
1. A minta előkészítése viszonylag hosszú időt vesz igénybe (3 óra)
2. A bomba jóval drágább, mint egy atmoszférikus roncsoló lombik
Mikrohullámmal elősegített nedves roncsolás
Az acélköpenyes teflon bombában végzett előkészítés hátránya a viszonylag hosszú műveleti idő. A nedves
roncsolás időtartamának jelentős csökkentése érhető el a mikrohullámmal elősegített nedves roncsolás
kidolgozásával. A hosszú előkészítési időt többek között az okozza, hogy a bombában a reakcióelegyet külső
hőenergia közléssel melegítjük a kívánt hőmérsékletre. Mikrohullámú energiaközléssel a minta hatékonyabb és
gyorsabb felmelegítését érhetjük el. A mikrohullámmal elősegített roncsolásnál megtartjuk a zárttérben nyomás
alatt az acélköpenyes teflon bombában végzett eljárás minden előnyét. A változás annyi, hogy az acél köpeny
helyett az acél mechanikai sajátságához, nevezetesen hő- és nyomásállóságához hasonló műanyag köpenybe
helyezzük a teflon edényt. Ez a 100 bar nyomásnak ellenálló műanyag a poli-éter-éter-keton, rövidítése: PEEK.
A minta és roncsoló anyagok bemérése után a PEEK köpenyes bombát lezárjuk. Ezután a lezárt bombát az erre
a célra kifejlesztett mikrohullámú szekrénybe helyezzük. Ez a szekrény annyiban tér el a hagyományos
háztartási mikrohullámú sütőktől, hogy a szekrény közepén egy nagy szilárdságú műanyagból készült forgó test
(rotor) helyezkedik el, amelynek a vájataiba 6 vagy 8 bomba rakható be. A mikrohullámú energia közlése pedig
számítógép vezérléssel történik. A legújabb ilyen bombákba hőmérséklet és nyomásérzékelő szondákat is
elhelyeznek, amelyek segítségével a roncsolás teljes ideje alatt regisztrálható a hőmérséklet és a nyomás
alakulása. A mikrohullámú energiaközlés hatékonysága annak köszönhető, hogy mikrohullámú térben a dipólus
molekulák (jelen esetben a vízmolekulák) nagy sebességű rezgő mozgást végeznek, dörzsölik egymást és ezért a
reakcióelegy gyorsan felmelegszik anélkül, hogy maga az edényzet melegedne. E ma legmodernebbnek számító
módszerrel a minta előkészítés ideje mintegy 20–25 percre csökken.
E módszer egyesíti a minta-előkészítés minden előnyét. Csupán azt lehet hátrányaként felhozni, hogy jóval
drágább minden korábban említett eljárásnál.
Ha a roncsoláshoz alkalmazott nagytisztaságú savak nyomelem-tartalmát tovább kívánjuk csökkenteni, akkor
úgynevezett forrpont alatti (subboiling) desztillálással állíthatunk elő a még nagyobb tisztaságú roncsolószert.
A nyomelem-analitikában használatos koncentrációegységek

Az atomabszorpciós spektrometria
alkalmazásai
137 Created by XMLmind XSL-FO Converter.
A nyomelem-analitikában a kis koncentrációk jellemzésére a mol/dm3 helyett a korábban a fizikában szilárd
mintákra alkalmazott koncentrációegységeket vettük át. Ezeket az egységeket használják a kimutatási határ és a
mérési tartomány meghatározásánál is. Ezek az egységek az alábbiak.
A ppm (parts per million) az a koncentráció, melynél a mintában a vizsgált részecske 1 tömegegysége mellett a
kísérő anyag 1 millió tömegegysége van jelen. Ebből adódóan a ppm = 10-6 g/g. Miután 10-6 g = 1 µg, valamint
híg vizes oldatok esetén 1 g ≈ 1 cm3, így 1 ppm ≈1 µg/cm3. Megjegyzendő, hogy az SI rendszerben továbbra is a
µg/cm3 és annak a kisebb egységei az elfogadott koncentrációegységek.
. táblázat -
1 ppm (parts per million) = 1 µg/g ≈ 1 µg/cm3 1 µg = 10-6 g (mikrogram)
1 ppb (parts per billion) = 1 ng/g ≈ 1 ng/cm3 1 ng = 10-9 g (nanogram)
1 ppt (parts per trillion) = 1 pg/g ≈ 1 pg/cm3 1 pg = 10-12 g (pikogram)
1 ppq (parts per quadrillon) = 1 fg/g ≈ 1 fg/cm3 1 fg = 10-15 g (femtogram)
1.3. Kiértékelési módszerek
Az AAS módszer a legtöbb más műszeres módszerhez hasonlóan relatív analitikai módszer. Ezért a műszert
ismert koncentrációjú anyagokkal kalibrálnunk kell. Ezekre a kalibráló oldatokra, anyagokra kapott
abszorbancia értékeket ábrázoljuk a koncentráció függvényében. Az ismeretlen minták koncentrációját az így
nyert kalibráló vagy mérőgörbe segítségével grafikus úton, illetve a grafikonok számítógépes kiértékelésével
határozzuk meg. Az atomabszorpciós analitikában a két leggyakrabban alkalmazott kiértékelő eljárás az
összehasonlító és a standard addíciós módszer.
Összehasonlító módszer
Az ismeretlen koncentrációjú oldatok mérését megelőzően ismert koncentrációjú törzsoldatból hígítással
oldatsorozatot készítünk az adott elem AAS meghatározásának optimális méréstartományában (az abszorbancia
A = 0,1 – 0,8 közötti érték legyen). Ezeket az oldatokat lemérjük és a mérési adatok alapján szerkesztjük meg az
analitikai mérőgörbét (kalibráló egyenest vagy görbét). A derékszögű koordináta rendszer függőleges tengelyén
(ordinátán) az AAS készülék által mért jelet, jelen esetben az abszorbancia-értékeket, a vízszintes tengelyen
(abszcisszán) pedig a kalibráló oldatok koncentrációját ábrázoljuk. Az ismeretlen koncentrációjú mintára a
műszer válaszjelét, jelen esetben az abszorbancia-értékét az ordináta tengelyről a vízszintesen a mérőgörbére,
onnan pedig merőlegesen az abszcissza tengelyre vetítjük. A vetítő vonal és az abszcissza tengely
metszéspontjában olvasható le a minta koncentrációja. Az összehasonlító módszerrel történő kiértékelést a 120.
ábrán mutatjuk be.
Az összehasonlító módszer pontossága egyszerű, egy komponenst tartalmazó mintánál az elkészített kalibráló
oldatok (kalibráló anyagok) koncentrációjának a pontosságától függ. Ha összetett, többféle kísérő anyagot
tartalmazó mintát vizsgálunk, akkor a meghatározás pontosságának az is feltétele, hogy a kalibráló anyag
tartalmazza, és azonos koncentrációban tartalmazza mindazokat a kísérőanyagokat, amelyek a mintában
előfordulnak. Ezt a feltételt teljesíteni nehéz lenne, de az elérhető, hogy a fontosnak ítélt kísérő komponenseket
átlagkoncentrációban hozzáadjuk a kalibráló oldathoz (anyaghoz) is. Az összehasonlító módszert nagyszámú
minta sorozatelemzése esetén alkalmazzuk.
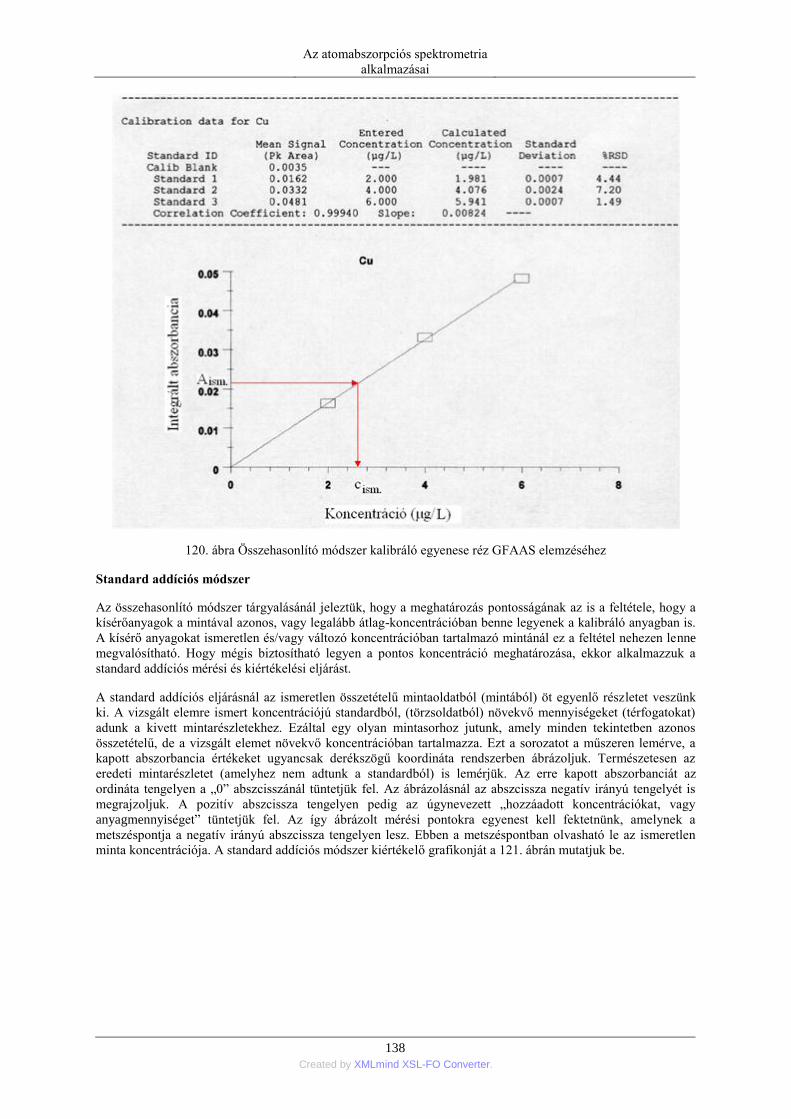
Az atomabszorpciós spektrometria
alkalmazásai
138 Created by XMLmind XSL-FO Converter.
120. ábra Összehasonlító módszer kalibráló egyenese réz GFAAS elemzéséhez
Standard addíciós módszer
Az összehasonlító módszer tárgyalásánál jeleztük, hogy a meghatározás pontosságának az is a feltétele, hogy a
kísérőanyagok a mintával azonos, vagy legalább átlag-koncentrációban benne legyenek a kalibráló anyagban is.
A kísérő anyagokat ismeretlen és/vagy változó koncentrációban tartalmazó mintánál ez a feltétel nehezen lenne
megvalósítható. Hogy mégis biztosítható legyen a pontos koncentráció meghatározása, ekkor alkalmazzuk a
standard addíciós mérési és kiértékelési eljárást.
A standard addíciós eljárásnál az ismeretlen összetételű mintaoldatból (mintából) öt egyenlő részletet veszünk
ki. A vizsgált elemre ismert koncentrációjú standardból, (törzsoldatból) növekvő mennyiségeket (térfogatokat)
adunk a kivett mintarészletekhez. Ezáltal egy olyan mintasorhoz jutunk, amely minden tekintetben azonos
összetételű, de a vizsgált elemet növekvő koncentrációban tartalmazza. Ezt a sorozatot a műszeren lemérve, a
kapott abszorbancia értékeket ugyancsak derékszögű koordináta rendszerben ábrázoljuk. Természetesen az
eredeti mintarészletet (amelyhez nem adtunk a standardból) is lemérjük. Az erre kapott abszorbanciát az
ordináta tengelyen a „0” abszcisszánál tüntetjük fel. Az ábrázolásnál az abszcissza negatív irányú tengelyét is
megrajzoljuk. A pozitív abszcissza tengelyen pedig az úgynevezett „hozzáadott koncentrációkat, vagy
anyagmennyiséget” tüntetjük fel. Az így ábrázolt mérési pontokra egyenest kell fektetnünk, amelynek a
metszéspontja a negatív irányú abszcissza tengelyen lesz. Ebben a metszéspontban olvasható le az ismeretlen
minta koncentrációja. A standard addíciós módszer kiértékelő grafikonját a 121. ábrán mutatjuk be.
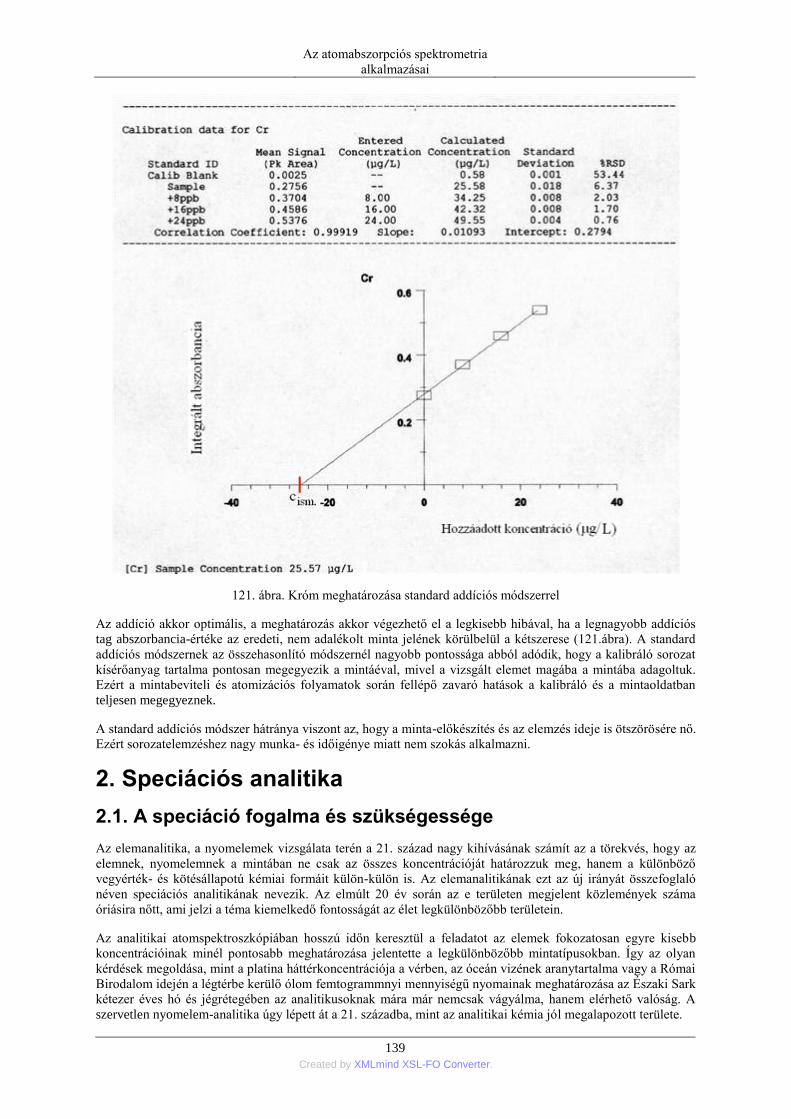
Az atomabszorpciós spektrometria
alkalmazásai
139 Created by XMLmind XSL-FO Converter.
121. ábra. Króm meghatározása standard addíciós módszerrel
Az addíció akkor optimális, a meghatározás akkor végezhető el a legkisebb hibával, ha a legnagyobb addíciós
tag abszorbancia-értéke az eredeti, nem adalékolt minta jelének körülbelül a kétszerese (121.ábra). A standard
addíciós módszernek az összehasonlító módszernél nagyobb pontossága abból adódik, hogy a kalibráló sorozat
kísérőanyag tartalma pontosan megegyezik a mintáéval, mivel a vizsgált elemet magába a mintába adagoltuk.
Ezért a mintabeviteli és atomizációs folyamatok során fellépő zavaró hatások a kalibráló és a mintaoldatban
teljesen megegyeznek.
A standard addíciós módszer hátránya viszont az, hogy a minta-előkészítés és az elemzés ideje is ötszörösére nő.
Ezért sorozatelemzéshez nagy munka- és időigénye miatt nem szokás alkalmazni.
2. Speciációs analitika
2.1. A speciáció fogalma és szükségessége
Az elemanalitika, a nyomelemek vizsgálata terén a 21. század nagy kihívásának számít az a törekvés, hogy az
elemnek, nyomelemnek a mintában ne csak az összes koncentrációját határozzuk meg, hanem a különböző
vegyérték- és kötésállapotú kémiai formáit külön-külön is. Az elemanalitikának ezt az új irányát összefoglaló
néven speciációs analitikának nevezik. Az elmúlt 20 év során az e területen megjelent közlemények száma
óriásira nőtt, ami jelzi a téma kiemelkedő fontosságát az élet legkülönbözőbb területein.
Az analitikai atomspektroszkópiában hosszú időn keresztül a feladatot az elemek fokozatosan egyre kisebb
koncentrációinak minél pontosabb meghatározása jelentette a legkülönbözőbb mintatípusokban. Így az olyan
kérdések megoldása, mint a platina háttérkoncentrációja a vérben, az óceán vizének aranytartalma vagy a Római
Birodalom idején a légtérbe kerülő ólom femtogrammnyi mennyiségű nyomainak meghatározása az Északi Sark
kétezer éves hó és jégrétegében az analitikusoknak mára már nemcsak vágyálma, hanem elérhető valóság. A
szervetlen nyomelem-analitika úgy lépett át a 21. századba, mint az analitikai kémia jól megalapozott területe.

Az atomabszorpciós spektrometria
alkalmazásai
140 Created by XMLmind XSL-FO Converter.
A toxikus vagy esszenciális hatást a biológiában, a humánbiológiában és az orvoslásban gyakorlatilag sohasem
magához az elemhez köthetjük, hanem annak egyszerű akva- komplexeitől bonyolultabb komplexeken keresztül
a változó összetételű elemorganikus vegyületeiig, amelyeknek egymástól jól elkülönülő fizikai, kémiai és
biológiai sajátságai vannak. Egyre több adat igazolja, hogy a toxikus nyomelemeknek kémiai formájuktól
függően igen eltérő élettani hatásuk lehet [99-102]. Esetenként az azonos elem két formája között a toxicitásbeli
különbség nagyságrendekkel eltérő is lehet.
A természetes mintákban előforduló arzénformák közül a szervetlen As(III) és As(V) tekinthető a
legtoxikusabbnak. Az alkilezett formák: monometil-arzonsav (MMA), dimetil-arzinsav (DMA), arzenobetain
(AsBet), arzenocholin (AsCo) az élő szervezetbe jutó szervetlen arzénformák detoxifikálódási termékei,
amelyek ennek megfelelően kevésbé toxikusak. Az arzenocholin és arzenobetain arányának meghatározásával
például becsülni lehet, hogy a biológiai rendszert mikor érte az arzénszennyeződés.
Előbbiekhez hasonlóan igen eltérő a szervetlen higanysók és az alkilezett higanyszármazékok toxicitása.
Utóbbiaké mintegy 100-szorosa a szervetlen higany-vegyületekének. Ez azt jelenti, hogy ha az alkilezett
származék a szervetlen higanynak csupán 1-2%-a, már akkor is többszörösére nő a minta toxicitása. Az
alkilezett higanyszármazékok azért is jelentenek veszélyt az emberre, mert zsíroldhatók lévén át tudnak jutni az
vér-agy-gáton és az agyban komoly idegrendszeri károsodások léphetnek fel. Ma már az is ismert, hogy például
az ónnak mintegy 15-20 különböző toxicitású kémiai formája létezik a természetben és arra is fény derült, hogy
ezek nemcsak az ember által okozott környezetszennyezés termékei, hanem egy részük a környezetben
természetes úton is keletkezhet.
Előbbiekből következően, ha a mintában egy fémnek csak a teljes koncentrációját adjuk meg, ahogy azt eddig a
nyomelem-analitikában tettük, az nemcsak hogy hiányos, de gyakran még félrevezető információval is
szolgálhat a minta tulajdonságairól. Hogy miként jutunk hozzá a fentebb említett molekuláris információkhoz,
jelenleg az egyik legnagyobb kihívásnak számít az analitikai kémiában.
Míg a nyomelemek gyakran közvetlenül meghatározhatók az ismert műszeres optikai, elektroanalitikai,
radioanalitikai módszerekkel, addig a speciációs analízisnél a detektálást mindig meg kell előznie egy
elválasztási lépésnek. Adott esetekben a klasszikus nyomelem-analitika is alkalmaz elválasztást, például
nyomelem dúsítás vagy a zavaró matrixanyag eltávolítása végett. Korábban azonban minden esetben a
nyomelem összes formája együtt és egyidejűleg kerül detektálásra. A speciációs analízis során azonban az
elemezni kívánt nyomelem minden eltérő vegyérték- és kötésformáját egymástól is elválasztjuk, és külön-külön
detektáljuk. Utóbbi feladat sikeres megoldása különböző háttérrel rendelkező kutatókat követel meg: kémikust,
biológust, talaj és üledékkutatót, fizikust és az élelmiszerek, gyógyszerek különböző szempontjait vizsgáló
specialistát, akik tapasztalatának együttes, komplex alkalmazásával lehet eredményt elérni.
2.2. A speciációs analitikában vizsgált elemek köre
A 122. ábrán jelzett elemek az átmeneti, másodfajú és a félfémek csoportjába tartoznak. A speciációs analitika
érdeklődése azért fordult elsősorban ezen elemek felé, mert fontos élettani hatással rendelkeznek, és toxicitásuk
nagyban függ attól, hogy milyen vegyértékűek és milyen kötésállapotban fordulnak elő a természetben és a
biológiai, humánbiológiai mintákban.
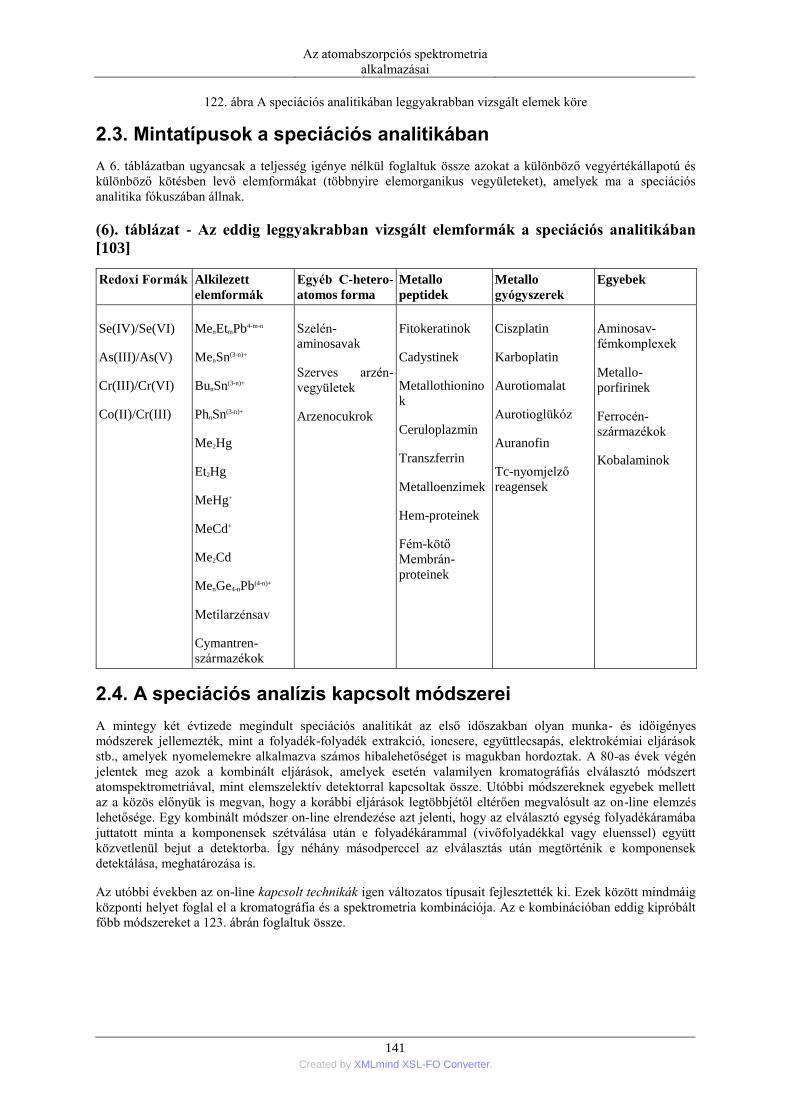
Az atomabszorpciós spektrometria
alkalmazásai
141 Created by XMLmind XSL-FO Converter.
122. ábra A speciációs analitikában leggyakrabban vizsgált elemek köre
2.3. Mintatípusok a speciációs analitikában
A 6. táblázatban ugyancsak a teljesség igénye nélkül foglaltuk össze azokat a különböző vegyértékállapotú és
különböző kötésben levő elemformákat (többnyire elemorganikus vegyületeket), amelyek ma a speciációs
analitika fókuszában állnak.
(6). táblázat - Az eddig leggyakrabban vizsgált elemformák a speciációs analitikában
[103]
Redoxi Formák Alkilezett
elemformák Egyéb C-hetero-
atomos forma Metallo
peptidek Metallo
gyógyszerek Egyebek
Se(IV)/Se(VI)
As(III)/As(V)
Cr(III)/Cr(VI)
Co(II)/Cr(III)
MenEtmPb4-m-n
MenSn(3-n)+
BunSn(3-n)+
PhnSn(3-n)+
Me2Hg
Et2Hg
MeHg+
MeCd+
Me2Cd
MenGe4-nPb(4-n)+
Metilarzénsav
Cymantren-
származékok
Szelén-
aminosavak
Szerves arzén-
vegyületek
Arzenocukrok
Fitokeratinok
Cadystinek
Metallothionino
k
Ceruloplazmin
Transzferrin
Metalloenzimek
Hem-proteinek
Fém-kötő
Membrán-
proteinek
Ciszplatin
Karboplatin
Aurotiomalat
Aurotioglükóz
Auranofin
Tc-nyomjelző
reagensek
Aminosav-
fémkomplexek
Metallo-
porfirinek
Ferrocén-
származékok
Kobalaminok
2.4. A speciációs analízis kapcsolt módszerei
A mintegy két évtizede megindult speciációs analitikát az első időszakban olyan munka- és időigényes
módszerek jellemezték, mint a folyadék-folyadék extrakció, ioncsere, együttlecsapás, elektrokémiai eljárások
stb., amelyek nyomelemekre alkalmazva számos hibalehetőséget is magukban hordoztak. A 80-as évek végén
jelentek meg azok a kombinált eljárások, amelyek esetén valamilyen kromatográfiás elválasztó módszert
atomspektrometriával, mint elemszelektív detektorral kapcsoltak össze. Utóbbi módszereknek egyebek mellett
az a közös előnyük is megvan, hogy a korábbi eljárások legtöbbjétől eltérően megvalósult az on-line elemzés
lehetősége. Egy kombinált módszer on-line elrendezése azt jelenti, hogy az elválasztó egység folyadékáramába
juttatott minta a komponensek szétválása után e folyadékárammal (vivőfolyadékkal vagy eluenssel) együtt
közvetlenül bejut a detektorba. Így néhány másodperccel az elválasztás után megtörténik e komponensek
detektálása, meghatározása is.
Az utóbbi években az on-line kapcsolt technikák igen változatos típusait fejlesztették ki. Ezek között mindmáig
központi helyet foglal el a kromatográfia és a spektrometria kombinációja. Az e kombinációban eddig kipróbált
főbb módszereket a 123. ábrán foglaltuk össze.

Az atomabszorpciós spektrometria
alkalmazásai
142 Created by XMLmind XSL-FO Converter.
123. ábra A speciációs analitikában használt főbb kapcsolt technikák
Egy folyadékkromatográf hagymányosan többek között UV detektorral működik. Az elválasztó oszlopot
követően a spektrofotométer fényútjában elhaladó mintafrakciók (főleg szerves molekulák) fényelnyelése jól
érzékelhető, de az ilyenkor regisztrált kromatogramból nem kaphatunk választ arra, hogy az adott szerves
molekula milyen toxikus vagy esszenciális elemet tart éppen kötésben. Ez az, amiért a kromatográfiás technikák
összekapcsolása az atomspektrometriával (FES, AAS, ICP-AES, ICP-MS módszerekkel) az analitikai
lehetőségek új dimenzióját nyitotta meg.
2.5. Krómspeciációs módszerek
A toxicitásban mutatkozó nagyfokú eltérésekre tipikus példa a króm. A króm a természetben két viszonylag
stabil vegyértékállapotú formában, króm(III) és króm(VI) alakban található. E két forma környezetre, biológiai
rendszerekre gyakorolt élettani hatása teljesen ellentétes. Míg az egyéb, tipikusan toxikus elemnek számító
arzén, higany, kadmium, ólom esetén a különböző vegyérték- és kötésállapotú formák toxicitásában inkább csak
fokozati különbségek jelentkeznek, addig a króm esetén a két forma közül a króm(III) az élő szervezet számára
létfontosságú, a króm(VI) viszont kifejezetten toxikus, rákkeltő hatású. A króm(III) fontos szerepet játszik az
élő szervezet anyagcsere-folyamataiban, fokozza számos enzim aktivitását, és stimuláló hatással van a
koleszterin- és zsírsavszintézisre [104, 105]. Ebből adódóan a króm(III) különböző komplex vegyületek
formájában gyógyszerek, gyógyhatású készítmények alkotójaként kereskedelmi forgalomban is kapható (pl.
króm-pikolinát tabletta, Centrum multivitamin és ásványi anyag tabletta, Béres-csepp, stb.)
A króm(III) és króm(VI) ellentétes élettani hatása miatt különösen fontossá vált olyan analitikai módszerek
kidolgozása, amelyek segítségével a természetes mintákban (pl. ivóvíz, felszíni víz, tengervíz, vérszérum,
vizelet, élelmiszer, gyógyhatású készítmények, stb.) e két vegyértékformát külön-külön meg tudjuk határozni.
A króm(III) és króm(VI) elválasztására és meghatározására a 70-es évektől kezdve számos módszer látott
napvilágot, ezek azonban eléggé munka- és időigényesek, valamint számos hibával terheltek voltak [106-112].
A 80-as évek végére már olyan közlemények is megjelentek, amelyek valamilyen kromatográfiás elválasztó

Az atomabszorpciós spektrometria
alkalmazásai
143 Created by XMLmind XSL-FO Converter.
eljárás és elemszelektív detektor (atomspektrometriás módszer) kombinációját mutatják be [113-117]. E
módszereknek egyebek mellett közös előnyük a korábbi eljárásokkal szemben, hogy megvalósult az on-line
elemzés lehetősége. Ez azt jelenti, hogy az elválasztó egység folyadékáramába juttatott minta a komponensek
elválása után e folyadékárammal együtt közvetlenül bejut a detektorba, így néhány másodperccel az elválasztás
után megtörténhet az elválasztott komponensek atomspektrometriás meghatározása is.
Cr(III)/Cr(VI) elválasztása és dúsítása [118, 119] Az elválasztás azon alapszik, hogy a nagynyomású
folyadékkromatográfiás (HPLC) pumpával továbbított metanoltartalmú eluenshez és a mintahurokba töltött
mintához adott tetrabutil-ammóniumsó (TBA-só) a Cr(VI)-tal ionpár-komplexet képez. A rendszerbe iktatott
C18-as kolonnán a minta Cr(III)-tartalma (az egyéb szervetlen kationokkal, anionokkal együtt) akadálytalanul
áthalad. A Cr(VI)-ionpár-komplex a C18-al való kölcsönhatás miatt viszont határozott késéssel jut keresztül az
oszlopon. Ezért a Cr(III) és Cr(VI) egymástól eltérő időszakaszban jut be a detektorba. Következésképpen a két
vegyértékformára időben jól elkülönülő két csúcsot kapunk, melynek görbe alatti területe illetve
csúcsmagassága arányos a Cr(III) és a Cr(VI) koncentrációjával. Az előbbi elven működő automatizált on-line
krómspeciációs rendszer vázlatát az 124. ábrán, a kapott kromatogramot pedig a 125. ábrán mutatjuk be.
124. ábra Cr(III)/Cr(VI) on-line elválasztására kidolgozott automatikus elemző rendszer
125. ábra 2-2 µg/ml Cr(III)/Cr(VI) FAAS detektálással kapott kromatogramja
Cr(VI) on-line kromatográfiás dúsítását az on-line elválasztó rendszer módosításával alakítottuk ki. A
dúsításnál, a szeparációhoz hasonlóan azt használtuk ki, hogy a Cr(VI) tetrabutil-ammónium (TBA)-sókkal
olyan ionpár-komplexet képez, amely C18 oszlopon megköthető. Eszerint a HPLC pumpa után elhelyezett
nagyobb (2-5 ml-es) mintahurokból a Cr(VI)-TBA komplex az oszlopra jutva ott teljes mennyiségében
megkötődik, ha a vivőfolyadék ecetsavval pH=3-ra beállított desztillált víz. A megkötődés után egy másik
bemérőcsapon keresztül az oszlopról a Cr(VI)-ot 1 ml 60%-os metanollal eluálva a spektrométer
atomizáló/gerjesztő terébe jut. A módszer kísérleti elrendezését az 126. ábrán mutatjuk be.

Az atomabszorpciós spektrometria
alkalmazásai
144 Created by XMLmind XSL-FO Converter.
126. ábra Cr(VI) on-line dúsító és atomspektrometriás meghatározó módszer kísérleti elrendezése
Cr(III) dúsítása KH-ftaláttal [120, 121] A Cr(III) dúsítási módszerhez ugyancsak C18-as fordított fázisú kolonna
használható. A vizsgálatok szerint, ha C18 oszlopra 0.075 mol/L KH-ftalát tartalmú minta (pH=3.8) vizes
oldatát visszük, az oszlopon kialakuló ftalátréteg sajátos szorpciós kölcsönhatás folytán kvantitatívan képes
megkötni az oszlopon áthaladó Cr(III) ionokat. Ezt a kromatográfiás szempontból is újszerűnek számító hatás
használható ki a dúsítási művelethez. 2-5 ml mintaoldatból megkötött Cr(III)-at 1 ml 80 %-os metanol-víz
eleggyel eluálva jut a detektorba. E műveletekhez is az 123. ábrán látható kísérleti elrendezést használtuk.
Cr(VI) szorpciós dúsítása [122, 123] Ez a dúsítási módszer azon az elven alapszik, hogy oldatfázisban előállított
bizonyos fémkomplexek adott pH értéken (például a Cr-PDC, pH=1.9-nél) molekuláris szorpcióval
kvantitatívan megkötődhetnek hidrofób sajátságú felületeken, ilyen anyagból (teflon, polietilén, PEEK) készült
csövek belső falán (szorpciós hurokban). A Cr(VI)-ra szorpciós dúsítással elérhető kimutatási határ javítása
érdekében a dúsításhoz használt szorpciós hurkot bemérőcsap segítségével nagynyomású porlasztó
folyadékáramába iktattuk. Így a szorpciós dúsítást követő elúciós lépés során a detektáláshoz a pneumatikusnál
nagyobb hatékonyságú, nagynyomású (HHPN) mintabevitelt alkalmaztunk. A módszer kísérleti elrendezését a
127. ábrán mutatjuk be.
127. ábra Cr(VI) szorpciós dúsításának és nagynyomású mintabevitellel történő lángatomabszorpciós
spektrometriás detektálásának kísérleti elrendezése
(7). táblázat - 7. táblázat. A Cr(VI) különböző on-line
elválasztási/dúsítási/atomspektrometriás detektálási módszerrel kapott kimutatási
határai
Szeparáló/dúsító eljárás Mintabevitel Detektálási módszer Kimutatási határ (3σ)
ng/ml
Cr(III)/Cr(VI) PN FAAS 80.0
szeparálás HHPN FAAS 20.0
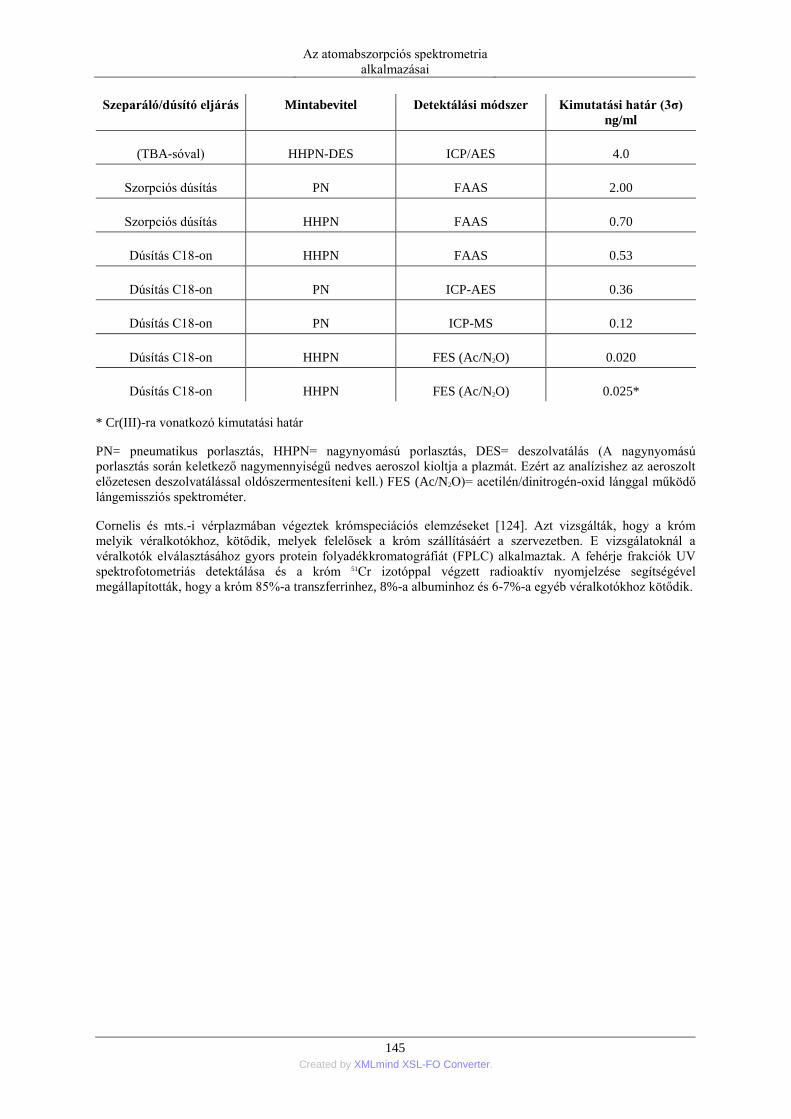
Az atomabszorpciós spektrometria
alkalmazásai
145 Created by XMLmind XSL-FO Converter.
Szeparáló/dúsító eljárás Mintabevitel Detektálási módszer Kimutatási határ (3σ)
ng/ml
(TBA-sóval) HHPN-DES ICP/AES 4.0
Szorpciós dúsítás PN FAAS 2.00
Szorpciós dúsítás HHPN FAAS 0.70
Dúsítás C18-on HHPN FAAS 0.53
Dúsítás C18-on PN ICP-AES 0.36
Dúsítás C18-on PN ICP-MS 0.12
Dúsítás C18-on HHPN FES (Ac/N2O) 0.020
Dúsítás C18-on HHPN FES (Ac/N2O) 0.025*
* Cr(III)-ra vonatkozó kimutatási határ
PN= pneumatikus porlasztás, HHPN= nagynyomású porlasztás, DES= deszolvatálás (A nagynyomású
porlasztás során keletkező nagymennyiségű nedves aeroszol kioltja a plazmát. Ezért az analízishez az aeroszolt
előzetesen deszolvatálással oldószermentesíteni kell.) FES (Ac/N2O)= acetilén/dinitrogén-oxid lánggal működő
lángemissziós spektrométer.
Cornelis és mts.-i vérplazmában végeztek krómspeciációs elemzéseket [124]. Azt vizsgálták, hogy a króm
melyik véralkotókhoz, kötődik, melyek felelősek a króm szállításáért a szervezetben. E vizsgálatoknál a
véralkotók elválasztásához gyors protein folyadékkromatográfiát (FPLC) alkalmaztak. A fehérje frakciók UV
spektrofotometriás detektálása és a króm 51Cr izotóppal végzett radioaktív nyomjelzése segítségével
megállapították, hogy a króm 85%-a transzferrinhez, 8%-a albuminhoz és 6-7%-a egyéb véralkotókhoz kötődik.

146 Created by XMLmind XSL-FO Converter.
8. fejezet - Az atomabszorpciós spektrometria jövőbeli fejlődési irányai
Az elmúlt évtized atomspektroszkópiai irodalmát áttekintve megállapítható, hogy az atomabszorpciós
spektrometria az egyik legelterjedtebb atomspektroszkópiai módszer a rutin analitika területén. A
lángatomabszorpciós (FAAS) módszerről már alig jelennek meg cikkek, mert annyira mindennapos rutin
módszerré vált, hogy teljesítőképességét korlátaival együtt senki nem vitatja. Nem ennyire problémamentes még
mindmáig a grafitkemencés (GFAAS) módszer. A 2000 – 3000 oC-on izzó grafit felületen, inert atmoszférában,
összetett szilárd fázis folyamatai még alapkutatás szintjén is számos kérdést vetnek fel. E folyamatok tisztázása
teszi lehetővé zavaró hatások további csökkentését, az analitikai érzékenység növelését. Az alapkutatási
tapasztalatok teszik lehetővé az abszolút atomabszorpciós spektrometria bevezetését, amely már évtizedek óta
foglalkoztatja a szakembereket. Ugyancsak régóta foglalkoznak az e területen dolgozó szakemberek a szimultán
multielemes AAS megvalósításával, amely törekvést a legutóbbi időben siker koronázta.
Nagy felbontású folytonos fényforrással működő AAS (HR-CS AAS = high resolution continuous source
AAS)
A szimultán AAS elemzés külön fejezetét jelenti az utóbbi években bevezetett folytonos fényforrással működő
AAS módszer, amelyről az első összefoglaló munka 2005-ben jelent meg [17]. Ahogy azt a 5. 1. 1. fejezetben
tárgyaltuk, az atomabszorpciós spektrometria bevezetésekor kiderült, hogy hagyományos felbontású
monokromátort alkalmazva csak akkor tudjuk a módszert analitikai célra használni, ha kis félértékszélességű
vonalakat kibocsátó spektrállámpákat (üregkatód lámpákat) használunk. Ez természetesen azzal járt, hogy
minden elem meghatározásához külön lámpára van szükség. Az AAS készülékeket tervező és gyártó cégek az
elmúlt 50 évben azonban továbbra sem mondtak le arról a lehetőségről, hogy folytonos fényforrással működő
készüléket tervezzenek, amely többek között magában hordozza a szimultán atomabszorpciós elemzés
lehetőségét is, és ezzel kiváltható legyen a mintegy 70, eléggé költséges üregkatód lámpa egyetlen folytonos
fényforrással.
Az új elven működő készülék hatásos működését többek között az is gátolta, hogy a konstruktőrök a
rendelkezésre álló kereskedelmi alkatrészekből, illetve azok módosításaival próbálták összeállítani az új
rendszert. Berlinben viszont az ISAS (Institute for Analytical Science) kutatócsoportja az új elven működő
készülék követelményeiből indult ki, és ehhez dolgozott ki megfelelő egységeket. Ez a készülék három
egységét: (i) a fényforrást, (ii) a fényfelbontó, hullámhossz kiválasztó rendszerét és (iii) detektorát jelenti.
Fényforrás
Olyan fényforrásra van szükség, amely folyamatos és közel egyenletes fényintenzitást biztosít 190 nm-től
kezdődően a teljes UV és látható színképtartományban. A CS-AAS korábbi bevezetésénél az egyik fontos
akadálya épp az volt, hogy a nagy fényerejűnek tartott kereskedelmi nagynyomású xenon lámpa fényintenzitása
több mint egy nagyságrenddel marad el az üregkatód lámpák intenzitásától. Különösen szerény volt a fényereje
a vákuum UV tartományban, ahol például az arzén és szelén meghatározása történik.
Ezen a téren a nagy áttörést az jelentette, hogy a berlini kutatócsoport a hagyományos xenon lámpa szerkezetét
és működési paramétereit a jelen célnak megfelelően megváltoztatta. A hagyományos xenon lámpa diffúz ívét
módosítva, az eddiginél is nagyobb nyomású mikro-ív lámpát fejlesztett ki. A ív kiterjedése kisebb lett mint 1
mm, átmérője 0,2 mm („hot-spot” lámpa). A xenon nyomása hidegen 17 bar, amely üzem közben a
négyszeresére növekszik. A plazma hőmérséklete 10 000 K. Az új lámpa teljesítménye 300 W (20 V, 15 A).
Ezzel a hagyományos xenon lámpához képest a fényintenzitás 100-szorosra nőtt az UV alsó tartományaiban is.
Hullámhossz kiválasztása
A hagyományosan üregkatód lámpával működő AAS készülék rácsos monokromátorának kilépő résén
megjelenő színkép sávszélessége általában 0,2 – 1 nm között változtatható. Ahhoz, hogy a készülék folytonos
fényforrással működtethető legyen, legalább 3 nagyságrenddel kisebb, azaz pikométer nagyságrendű
sávszélességre van szükség.
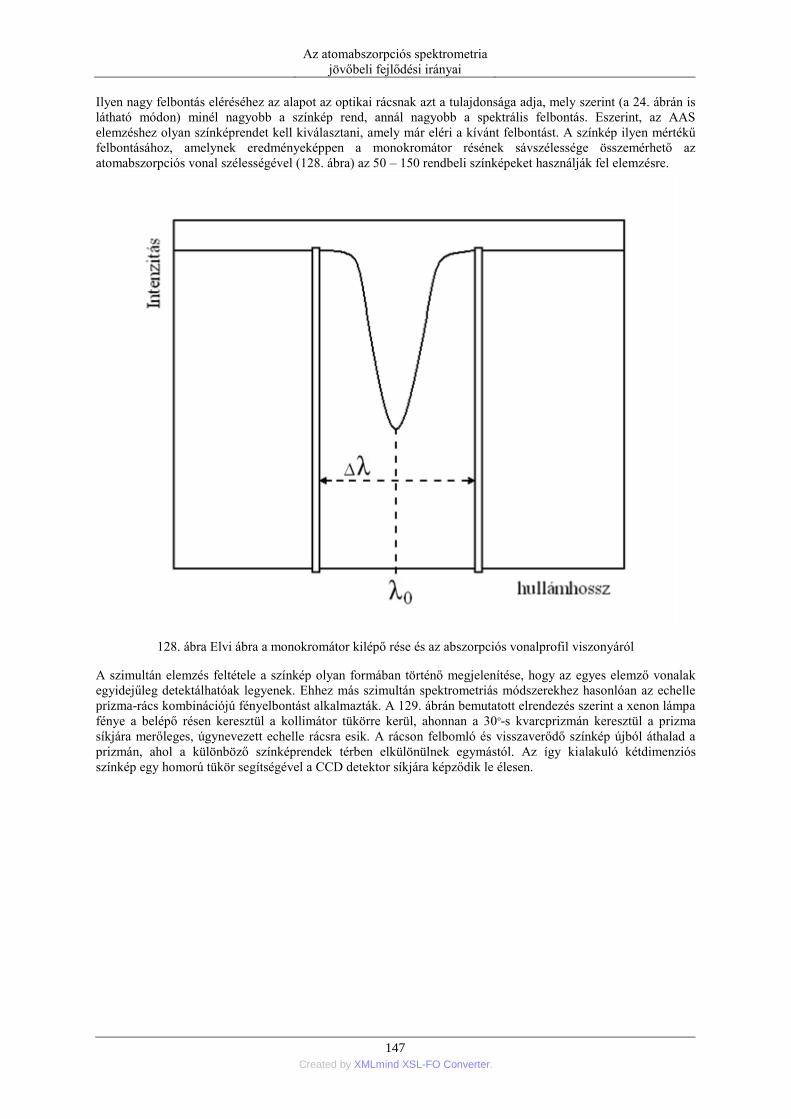
Az atomabszorpciós spektrometria
jövőbeli fejlődési irányai
147 Created by XMLmind XSL-FO Converter.
Ilyen nagy felbontás eléréséhez az alapot az optikai rácsnak azt a tulajdonsága adja, mely szerint (a 24. ábrán is
látható módon) minél nagyobb a színkép rend, annál nagyobb a spektrális felbontás. Eszerint, az AAS
elemzéshez olyan színképrendet kell kiválasztani, amely már eléri a kívánt felbontást. A színkép ilyen mértékű
felbontásához, amelynek eredményeképpen a monokromátor résének sávszélessége összemérhető az
atomabszorpciós vonal szélességével (128. ábra) az 50 – 150 rendbeli színképeket használják fel elemzésre.
128. ábra Elvi ábra a monokromátor kilépő rése és az abszorpciós vonalprofil viszonyáról
A szimultán elemzés feltétele a színkép olyan formában történő megjelenítése, hogy az egyes elemző vonalak
egyidejűleg detektálhatóak legyenek. Ehhez más szimultán spektrometriás módszerekhez hasonlóan az echelle
prizma-rács kombinációjú fényelbontást alkalmazták. A 129. ábrán bemutatott elrendezés szerint a xenon lámpa
fénye a belépő résen keresztül a kollimátor tükörre kerül, ahonnan a 30o-s kvarcprizmán keresztül a prizma
síkjára merőleges, úgynevezett echelle rácsra esik. A rácson felbomló és visszaverődő színkép újból áthalad a
prizmán, ahol a különböző színképrendek térben elkülönülnek egymástól. Az így kialakuló kétdimenziós
színkép egy homorú tükör segítségével a CCD detektor síkjára képződik le élesen.
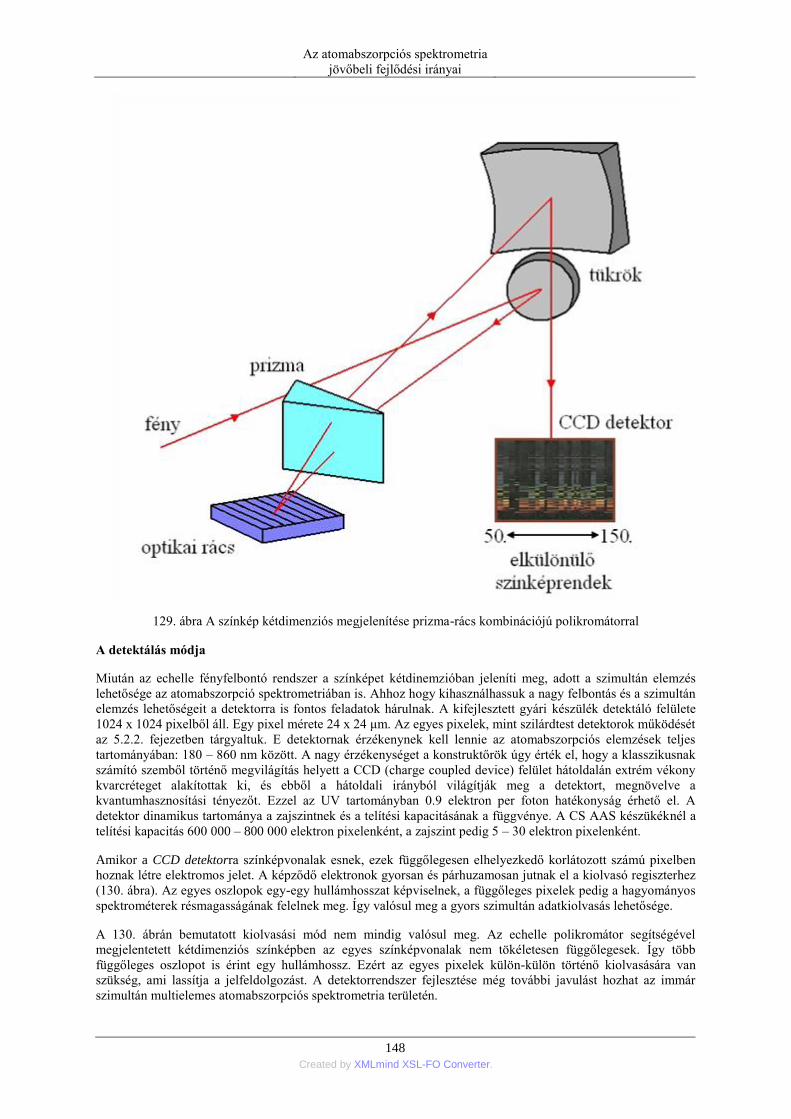
Az atomabszorpciós spektrometria
jövőbeli fejlődési irányai
148 Created by XMLmind XSL-FO Converter.
129. ábra A színkép kétdimenziós megjelenítése prizma-rács kombinációjú polikromátorral
A detektálás módja
Miután az echelle fényfelbontó rendszer a színképet kétdinemzióban jeleníti meg, adott a szimultán elemzés
lehetősége az atomabszorpció spektrometriában is. Ahhoz hogy kihasználhassuk a nagy felbontás és a szimultán
elemzés lehetőségeit a detektorra is fontos feladatok hárulnak. A kifejlesztett gyári készülék detektáló felülete
1024 x 1024 pixelből áll. Egy pixel mérete 24 x 24 μm. Az egyes pixelek, mint szilárdtest detektorok működését
az 5.2.2. fejezetben tárgyaltuk. E detektornak érzékenynek kell lennie az atomabszorpciós elemzések teljes
tartományában: 180 – 860 nm között. A nagy érzékenységet a konstruktőrök úgy érték el, hogy a klasszikusnak
számító szemből történő megvilágítás helyett a CCD (charge coupled device) felület hátoldalán extrém vékony
kvarcréteget alakítottak ki, és ebből a hátoldali irányból világítják meg a detektort, megnövelve a
kvantumhasznosítási tényezőt. Ezzel az UV tartományban 0.9 elektron per foton hatékonyság érhető el. A
detektor dinamikus tartománya a zajszintnek és a telítési kapacitásának a függvénye. A CS AAS készükéknél a
telítési kapacitás 600 000 – 800 000 elektron pixelenként, a zajszint pedig 5 – 30 elektron pixelenként.
Amikor a CCD detektorra színképvonalak esnek, ezek függőlegesen elhelyezkedő korlátozott számú pixelben
hoznak létre elektromos jelet. A képződő elektronok gyorsan és párhuzamosan jutnak el a kiolvasó regiszterhez
(130. ábra). Az egyes oszlopok egy-egy hullámhosszat képviselnek, a függőleges pixelek pedig a hagyományos
spektrométerek résmagasságának felelnek meg. Így valósul meg a gyors szimultán adatkiolvasás lehetősége.
A 130. ábrán bemutatott kiolvasási mód nem mindig valósul meg. Az echelle polikromátor segítségével
megjelentetett kétdimenziós színképben az egyes színképvonalak nem tökéletesen függőlegesek. Így több
függőleges oszlopot is érint egy hullámhossz. Ezért az egyes pixelek külön-külön történő kiolvasására van
szükség, ami lassítja a jelfeldolgozást. A detektorrendszer fejlesztése még további javulást hozhat az immár
szimultán multielemes atomabszorpciós spektrometria területén.
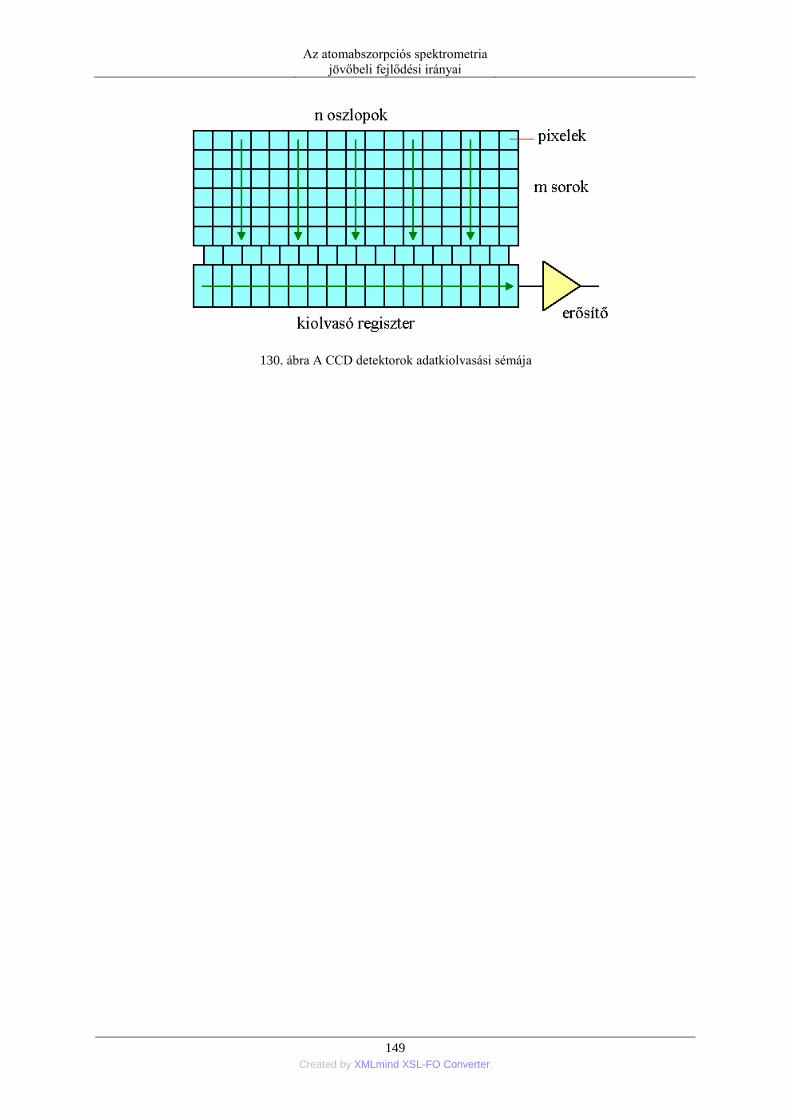
Az atomabszorpciós spektrometria
jövőbeli fejlődési irányai
149 Created by XMLmind XSL-FO Converter.
130. ábra A CCD detektorok adatkiolvasási sémája

150 Created by XMLmind XSL-FO Converter.
9. fejezet - Irodalomjegyzék
1. Slavin, W.: Atomic Absorption Spectroscopy, Interscience, New York, 1968.
2. Ramirez-Muňoz, J.: Atomic-Absorption Spectroscopy, Elsevier, London, 1968.
3. Rubeška, I., Moldan, B.: Atomic Absorption Spectrophotometry, Iliffe Books, London, 1968.
4. L’vov, B.: Atomic Absorption Spectrochemical Analysis, Adam Hilger, London, 1970.
5. Dean, J.A., Rains, T.C. (Eds): Flame Emission and Atomic Absorption Spectrometry I-III, Marcel Dekker,
1969., 1971., 1975.
6. Price, W.J.:Spectral Analysis by Atomic Absorption, Heyden & Sons, London, 1972
7. Alkemade, C.Th.J., Herrmann, R.: Fundamentals of Analytical Flame Spectroscopy, Adam Hilger, Bristol,
1979.
8. Alkemade, C.Th.J., Hollander, T., Snelleman, W., Zeegers, P.J.Th.: Metal Vapours in Flames, Pergamon
Press, Oxford, 1982.
9. Cantle, J.E. (Ed): Atomic Absorption Spectrometry, Elsevier, Amsterdam, 1982.
10. Haswell, S.J. (Ed): Atomic Absorption Spectrometry,. Elsever, Amsterdam, 1991.
11. Ebdon, L., Evans, E.H., Fisher, A., Hill, S.J.: An Introduction to Atomic Absorption Spectroscopy,
Wiley-VCH, New York, 1998.
12. Welz, B., Sperling, M.: Atomic Absorption Spectrometry, Wiley-VCH, Weinheim, 1999.
13. Jackson, K.W.(Ed): Electrothermal Atomization for Analytical Atomic Spectrometry, Wiley & Sons,
Chichester, 1999.
14. Price, W.J.: Atomabszorpciós spektrometria, Műszaki Könyvkiadó, Budapest, 1977.
15. Tomcsányi L.: Atomabszorpciós praktikum, Műszaki Könyvkiadó, Budapest, 1986.
16. Lakatos J.: Atomabszorpciós spektrometria, Fejezet Záray Gyula (szerk.): Az elemanalitika korszerű
módszerei, Akadémiai Kiadó, Budapest, 2006.
17. Welz, B., Becker-Ross, H., Florek, S., Heitmann, U.: High-Resolution Continuum Source AAS, Wiley-
VCH, Weinheim, 2005.
18. Walsh, A., Spectrochim. Acta, 7, 108-117 (1955).
19. Alkemade, C.Th.J., Milatz, J.M.W., Appl. Sci. Res., Sect. B 4, 289-299 (1955).
20. L’vov, B. V., Inzhener.-Fiz. Zhur., Akad. Nauk Belorus. SSR (J. Eng. Phys.[USSR]) 2/2, 44-52 (1959).
21. Willis, J.B., Nature (London) 207, 715-716 (1965).
22. Prugger, H., Torge, R., Patent, Ger. Offen. DE 1,964,469, 1969.
23. Halls, D. J., Spectrochim. Acta, 32B, 221 (1977).
24. Rubeška, I., Musil, J., Prog. Analyt. Atom. Spectrosc., 2, 309 (1979).
25. Holcombe, J. H., Eklung, R. H., Grice, K. E., Anal. Chem., 50, 2097 (1978).
26. Clampitt, N.C., Hieftje, G. M., Anal. Chem., 46, 382 (1974).
27. Bastiaan, G. J., Hieftje, G. M., Anal. Chem., 46, 901 (1974).

Irodalomjegyzék
151 Created by XMLmind XSL-FO Converter.
28. Hieftje, G. M.,Malmstadt, H. V., Anal. Chem., 40, 1860 (1968).
29. Clampitt, N.C., Hieftje, G. M., Anal. Chem., 44, 1211 (1974).
30. Johnston, P. D., Combust. Flame, 18, 373 (1972).
31. Rubeška, I., Methods Phys. Anal., No 3., 61 (1971).
32. Alkemade, C. Th.J., Anal. Chem., 38, 1252 (1966).
33. Galan, L. de, Samaey, G.F., Spectrochim. Acta 25B, 245 (1970).
34. Halls, D. J., Anal. Chim. Acta 88, 69 (1977).
35. Rubeška, I., Pelikánová, M., Spectrochim. Acta 33B, 301 (1978).
36. Chakrabarti, C.L., Singhal, S.P., Spectrochim. Acta, 24B, 663 (1969).
37. Greenwood, N. N., Earnshaw, A.: Chemistry of the Elements 2ed., Butterworth-Heinemann, London,
1997.
38. Alkemade, C. Th. J., Fundamental aspects of decomposition, atomization, and excitation of the sample
in the flame, In: Dean, J. A., T. C. Rains, T. C (Eds), Flame Emission and Atomic Absorption Spectrometry,
I. Theory, Marcel Dekker New York, 1969, pp109-110.
39. Posta J., Lakatos J., Magy. Kém. Folyóirat, 86, 284 (1980).
40. Handbook of Chemistry and Physics, 87th ed., David R. Lide (Ed.), CRC Press, Cleveland, Ohio, 2006.
41. El-Defrawy, M. M. M., Posta, J., Beck, M. T., Anal. Chim. Acta, 102, 185 (1978).
42. El-Defrawy, M. M. M., Posta, J., Beck, M. T., Anal. Chim. Acta, 115, 155 (1980).
43. Posta J., Szűcs L., Acta Chim. Acad. Sci. Hung., 126, 325 (1989).
44. Browner, R. F., Boorn, A. W., Anal. Chem., 56/7, 786A (1984).
45. Winefordner, J. D., Latz, H. W., Anal Chem., 33, 1727 (1961).
46. Dahlquist, R. L., Knoll, J. W., Appl. Spectrosc., 32, 1 (1978).
47. O’Grady, C. E., Marr, I.L., Cresser, M. S., Analyst (London) 109/9, 1183 (1984).
48. Cresser, M.S., Prog. Anal. Spectrosc., 5, 35 (1982).
49. Nukiyama, S., Tanasawa, Y., Trans. Soc. Mech. Eng. (Japan), Report 4, 5 & 6, 1938-1940. Translated
by E. Hope), Defense Research Board, 1950.
50. Berndt, H., Fresenius Z. Anal. Chem., 331, 321 (1988).
51. Posta, J., Berndt, H., Derecskei, B., Anal. Chim. Acta, 262, 261 (1992).
52. Posta J., Gáspár A., Magy. Kém. Folyóirat, 106. 183 (2000).
53. Posta, J., Berndt, H., Spectrochim. Acta, 47, 993 (1992).
54. Ruzicka, J., Hansen, E. H., Flow Injection Analysis, Wiley & Sons, New York, 1981.
55. Fang, Z. –l., Microchem. J. 45, 137 (1992).
56. Jones, J. L., Dalquist, R.L., Hoyt, R.E., Appl. Spectrosc., 25, 628 (1971).
57. Dalquist, R. L., Jones, J. L., Paschen, K. W., US Patent, 3.602.595 (1971).

Irodalomjegyzék
152 Created by XMLmind XSL-FO Converter.
58. Winge, R. K., Fassel, V. S, Kniseley, R.N., Appl. Spectrosc., 25, 636 (1971).
59. Kántor, T., Pungor, E., Proceedings of the 17th Colloquium Spectroscopicum Internationale, Florence,
Italy, 1973, Vol. I, pp 83.
60. Kántor, T., Fodor, P., Pungor, E., Anal. Chim. Acta, 102, 15 (1978).
61. Human, H. G. C., Scott, R. H., Oakes, A. R., West, C. D., Analyst, 101, 265 (1976).
62. Kántor, T., Pólos, l., Fodor, P., Pungor, E., Talanta, 23, 586 (1976).
63. Chapman, J. F., Dale, L. S., Anal. Chim. Acta, 89, 363 (1977).
64. Thomassen J. I. Y., Butler, L. R. P., Radziuk, B., Van Loon, J. C., Anal. Chim. Acta, 110, 1 (1979).
65. Kántor, T., Bezur, L., Pungor, E., Winefordner, J. D., Spectrochim. Acta, 38B, 581 (1983).
66. Skogerboe, R. K., Dick, D. L., Pavlica, D. A., Lichte, F. E., Anal. Chem.,47, 568 (1975).
67. Kántor, T., Bezur, L., Sztatisz, J., Pungor, E., J. Thermal. Anal., 22, 179 (1981).
68. Kántor, T., Spectrochim. Acta, 56B, 1523 (2001).
69. Papp, L., Vilimi, J., Acta Univ. Debrecina, Ser. Phys. et Chim., XVII, 205 (1971).
70. Papp, L., Spectrochim. Acta, 38B, 1203 (1983).
71. Mossotti, V. G., Laqua, K, Hagenah, W. D., Spectrochim. Acta, 23B, 197 (1967).
72. Vulfson, E. K., Karyakin, A. V., Shidlovsky, A. I., Zh. Anal. Khim., 28, 1253 (1973).
73. König, K. –H., Neumann, P., Fresenius Z. Anal. Chem., 279, 337 (1976).
74. Ishizuka, T, Uvamino, Y., Sunahara, H., Anal. Chem., 49, 1340 (1977).
75. Lockyer, J. N.: Studies in Spectrum Analysis, Appleton, London, 1878.
76. King, A.S., Astrophys. J., 28, 300 (1908).
77. West, T. S., Williams, X. K., Anal. Chim. Acta 45, 27 (1969).
78. L’vov, B. V., Spectrochim. Acta, 33B, 153 (1978).
79. Slavin, W., Manning, D. C., Carnrick, G. R., At. Spectrosc., 2/5 137 (1981)
80. Wendl, W.,Fresenius’ Z. Anal. Chem. 323, 726 (1986).
81. L’vov, B. V., Polzik, L. K., Romanova, N. P., Yuzefovskii, A. I., J. Anal. At. Spectrom., 5/3, 163
(1990).
82. Bendicho, C., Loos-Vollebregt, de M. T. C., J. Anal. At. Spectrom., 6/5, 353 (1991).
83. Langmyhr, F. J., Wobetoe, G., Prog. Anal. Spectrosc., 8/3-4, 193 (1985).
84. Stephen, S. C., Littlejohn, D., Ottaway, J.M., Analyst (London), 110/8, 1147 (1985).
85. Pure and Appl. Chem., 45, 105 (1976).
86. Rubeška, I., Moldan, B., Anal. Chim. Acta 37, 421 (1967).
87. Dewing, E. H., Nature, 214, 483 (1967).
88. Dewing, E. H., Metallurgical Transactions, 1, 2169 (1970).

Irodalomjegyzék
153 Created by XMLmind XSL-FO Converter.
89. Borowiec, J.A., Dillard, J. H., Cresser, M. S., Browner, R. F. Anal. Chem., 52, 1054 (1980).
90. Yofe, J., Finkelstein, R., Anal. Chim. Acta, 19, 166 (1958).
91. El-Defrawy, M. M., Posta, J., Beck, M. T., Magy. Kém. Folyóirat, 86, 59 (1980).
92. El-Defrawy, M. M., Posta, J., Beck, M. T., Anal. Chim. Acta, 102, 185 (1978).
93. El-Defrawy, M. M., Posta, J., Beck, M. T., Anal. Chim. Acta, 115, 155 (1980).
94. Ediger, R. D., Peterson, G. E., Kerber, J. D., At. Abszorpt. Newsletter, 13/3, 61 (1974).
95. Ediger, R. D., At. Abszorpt. Newsletter, 14/5, 127 (1975).
96. Tsalev, D. L., Slaveykova, V. I., Mandjukov, P. B., Spectrochimica Acta Rev.,13/3, 225 (1990).
97. Welz, B., Schlemmer, G., Mudakavi, J. R., J. Anal. At. Spectrom. 7/8, 1257 (1992).
98. Styris, D. L., Modifiers in electrotermal atomic absorption spectrometry, In Jackson, K.W.(Ed):
Electrothermal Atomization for Analytical Atomic Spectrometry, Wiley & Sons, Chichester, 1999.
99. Caroli S., (Ed): Element Speciation in Bioinorganic Chemistry. J. Wiley, New York 1996.
100. Ure, A. M., Davidson, C. M., (Eds): Chemical Speciation in the Environment. Chapman and Hall,
London 1995.
101. Berman, E.: Toxic Metals and Their Analysis. Heyden, London 1980.
102. Donard, O. F. X., Ritsema, R.: hyphenated techniques applied to the speciation of organometallic
compounds in the environment. In: Barcelo, D. (Ed): Environmental Analysis. Elsevier, Amsterdam 1993.
103. Lobinski, R., Appl. Spectrosc., 51/7, 259A (1997).
104. Nriagu, J. O., Nieboer, E. (Eds): Chromium in the Natural and Human Environment. J. Wiley, New
York 1988.
105. Katz, S.A., Salem, H.: The Biological and Environmental Chemistry of Chromium. VCH, New York
1994.
106. Pavel, J., Kliment, J., Stoerk, S., Suter, O., Fresenius’ J.Anal. Chem. 321, 587, (1985).
107. Wai, C. M. – Tsay, L. M. – Yu, J. C.: Microchim. Acta Part II. 73, (1987).
108. Miyazaki, A., Barnes, R.M., Anal. Chem. 53, 364 (1981).
109. Isshiki, K., Sohrin, Y., Karatani, H., Nakayama, E., Anal. Chim. Acta 224, 55 (1989).
110. Obiols, J., Devesa, R., Garcia-Berro, J., Serra, J., Int. J. Environ. Anal. Chem. 30, 197 (1987).
111. Batley, G. E., Matousek, J. P., Anal. Chem. 52, 1570 (1980).
112. Issa, R.M., Abdel-Nabey, B.A., Dadek, H., Electrochim. Acta 13, 1827 (1968).
113. Krull, I. S., Panaro, K. W., Gershmann, L. L., J. Chromatogr. Sci. 21, 460 (1983).
114. Syty, A., Christensen, R.G., Rains, T.C., J. Anal. At. Spectrom. 3, 193 (1988).
115. Cox, A. G., Cook, I. G., McLeod, C. W., Analyst 110, 331 (1985).
116. Urasa, I. T., Nam, S.H., J. Chromatogr. Sci. 27, 30 (1989).
117. Sperling, M., Xu, S., Welz, B., Anal. Chem. 64, 3101 (1992).
118. Posta, J., Alimonti, A., Petrucci, F., Caroli, S., Anal. Chim. Acta 325, 185 (1996).

Irodalomjegyzék
154 Created by XMLmind XSL-FO Converter.
119. Posta, J., Berndt, H., Luo, S. K., Schaldach, G., Anal. Chem. 65, 2590 (1993).
120. Gáspár, A., Posta, J., Tóth, R., J. Anal. At. Spectrom. 11, 1067 (1996).
121. Gáspár, A., Posta J., Tóth R., Magy. Kém. Folyóirat 103, 330 (1997).
122. Gáspár, A., Posta, J., Magy. Kém. Folyóirat 102, 503 (1996).
123. Gáspár, A., Posta, J., Sógor, Cs., Magy. Kém. Folyóirat 104, 153 (1998).
124. Cornelis, R, Borguet, F., Dyg, S., Griepink, B., Microchim. Acta 109, 145 (1992).

155 Created by XMLmind XSL-FO Converter.
10. fejezet - Tárgymutató
abszorpciós együttható
abszorpció Einstein-féle valószínűsége
abszorpciófok
abszorpciós együttható
acetilén – dinitrogén-oxid láng
acetilén-levegő láng
Alkemade
alkilezett elemformák
aluláteresztő elektronikus szűrő
analitikai mérőgörbe
anyagáram [75]
anyagátadás–szabályozott párolgás
atom effektív keresztmetszetét
atomemissziós spektrometria
atomfluoreszcens spektrometria
atomizálási hatásfok
atomizáció
atomszínképek
Balmer-széria
bemérőcsap
Berndt
Bernoulli-törvény
Bettendorf-próba
biológiai minták
Boisbaudran
Bunsen
CCD detektor
célkitűzés
célszámítógép
C-hetero-atomos forma
Cr(III)/Cr(VI) elválasztása és dúsítása

Tárgymutató
156 Created by XMLmind XSL-FO Converter.
Crookes
Cr(VI) szorpciós dúsítása
csillogási szög
Czerny-Turner monokromátor
Brewster D.
deutérium lámpás háttérkorrekció
diffúziós láng
digitális kijelzés
égési sebesség
egy- és kétfényutas AAS készülékek
Elektrotermikus atomizáció (ETA)
elektród nélküli kisülési lámpa
elemek atomizációs hatásfoka
elemzés
elektrotermikus párologtatás
előkevert lángok
előzóna
elektrotermikus párologtatás
értékelés
érzékenység (Sensitivity)
exponenciális titráló készülék
felszabadító adalék
felüláteresztő elektronikus szűrő
fényszaggató
festékszűrő
flow injection analízis (FIA)
flow injection
Fordított színkép
fordított Zemann effektus
foszfátion zavaró hatása
folyamatos titráló rendszer
forrpont alatti (subboiling) desztillálás

Tárgymutató
157 Created by XMLmind XSL-FO Converter.
fotoelektron sokszorozó
Fraunhofer J.
gázfázisú zavaró hatások
gázkisülési csövek
Gouy
grafitkemencés atomizáló
grafitrúd atomizáló
Grotrian diagram
Hagen – Poiseuille egyenlet
hamvasztás
hangolt erősítő [42]
harmadlagos aeroszol
háttérkorrekciós módszerek
Herschel
hideggőz (CV=cold vapor) technikás atomizáció
hidraulikus nagynyomású porlasztás
hidrogén színképe [8]
hidrid-fejlesztéses AAS
hőátadás – szabályozott párolgás
hullámhossz
hullámszám
impulzus mintabevitel
interferencia-szűrők [27]
ionizációs puffer
ionos újraeloszlás
ionizációs zavaró hatás
ívcella
ív- és szikraporlasztás
kalcium-alumínium-oxidot
kapcsolt technikák
kettős porlasztó
keresztfűtéses grafitcső

Tárgymutató
158 Created by XMLmind XSL-FO Converter.
keveredési térfogat
kiégetés
kiértékelés
kiértékelési módszerek
kimutatási határ (Limit of detection)
King
ködkamra
közvetlen koncentráció kijelzés
krómspeciációs módszerek
Lambert-Beer törvény
Lambert-Beer törvény
lamináris lángok
lángatomabszorpciós spektrometria FAAS
lézer abláció
linearitás (Linearity)
lineáris titráló készülék
Lockyer
longitudinális Zeeman rendszer
L’vov platform
L’vov
L’vov-féle grafitküvetta
magnézium-alumínium-oxidot
Marsh-próba
Marci M.
másodlagos aeroszol
Massmann-féle grafitkemence
Melville Th.
metallogyógyszerek
meghatározási határ (Limit of quantitation)
megvilágító fényforrások
Meker-égő
metallopeptidek

Tárgymutató
159 Created by XMLmind XSL-FO Converter.
Mg-Al-klorokomplex
mikrohullámmal elősegített nedves roncsolás
Milatz
mintabeviteli hatásfok
minta szállítása, tárolása
minta-előkészítési eljárások
mintaelőkészítés
mintahurok
mintabeviteli módszerek
mintatípusok
mintavétel
minta tartózkodási ideje
módszer teljesítőképessége
monokromátor
Müller
mutatós műszer
műveleti erősítő
nagy felbontású folytonos fényforrással működő AAS
nagyintenzitású üregkatód lámpa
nátrium-D vonal
nemvezető anyagok ívporlasztása
nedves roncsolás atmoszférikus nyomáson
nedves roncsolás zárt térben
Newton I.
normális Zeeman effektus
n-típusú félvezető
oldalirányú diffúzió
optikai rács
optimálási vizsgálatok
Oszcillátor erősség
összehasonlító módszer
Paschen

Tárgymutató
160 Created by XMLmind XSL-FO Converter.
párolgást elősegítő adalék
párolgási zavarások
PC vezérelt atomabszorpciós spektrométer
pirolitikus bevonat
Planck-féle hatáskvantum
pneumatikus porlasztók
pontosság (Accuracy)
porlasztási hatásfok
porlasztási sebesség
porlasztás sebesség
precizitás (Precision)
primer aeroszol
Pringsheim
prizma-rács kombinációjú polikromátor
prizmás fényfelbontás
„probe”-technika
Prugger
p-típusú félvezető
puffer-addíciós technika
reakciózóna
redoxi formák
Reich
réses acetilén-levegő égő
Reynold-féle szám
rezgésszám
Richter
Rydberg-állandó
Sauter-átmérő
Scheibe-Lomakin egyenlet
Smith-Hieftje háttérkorrekciós módszer
sóképződési sor
speciációs analitika

Tárgymutató
161 Created by XMLmind XSL-FO Converter.
spektrális zavaró hatások
standard addíció
standard addíciós módszer
stacionárius mintabevitel
statisztikus súly
stratégiakészítés
sugárzás spektrális sűrűsége
sugárzás sűrűsége
száraz hamvasztás
szárítás
szénrúd atomizáló
szelektivitás és specifitás (Selectivity and specificity)
Szelén fényelem
szén/oxigén (C/O) arány
szilárdtest detektorok
szilícium dióda
szilícium félvezető
szimultán AAS készülékek
színképrendek
színképszériák
szimulációs technika
tartomány (Range)
teflon bomba
telítő adalék
térbeli eloszlás zavarása
terelőlemezek
tetrabutil-ammóniumsó
tisztítás
többcsatornás atomabszorpciós spektrométer
Torge
transzduktor
transzmittancia

Tárgymutató
162 Created by XMLmind XSL-FO Converter.
transzport zavarások
tranziens jel
turbulens lángok
Ultrahangos porlasztás
ultrahangos keverő
univerzális mátrixmódosító
utózóna
üregkatód lámpa
védő (protektív) adalék
vizsgálható elemek
vonal-visszafordulás
Walsh
Wollaston W.
Willis
zavaró hatások
zavartűrés (Ruggedness)
Zeeman háttérkorrekciós módszer