Embed Size (px)
Citation preview

Fluid Phase Equilibria, 16 (1984) 361-367 Elsevier Science Publishers B.V.. Amsterdam - Printed in The Netherlands
361
PREDICTION OF THERMODYNAMIC PROPERTIES OF ASSOCIATED SYSTEMS ON THE BASIS OF PROPERTIES OF PURE LIQUIDS. II. EXCESS GIBBS ENERGY AND VAPOUR PRESSURE
A. KSI/liCZAK and H. BUCHOWSKI
Department of Physical Chemistry, Technical University (Politechnika), 00664 Warszawa (Po- land)
(Received July lst, 1983; accepted in revised form November 28th, 1983)
ABSTRACT
Ksiaiczak, A. and Buchowski, H., 1984. Prediction of thermodynamic properties of associated systems on the basis-of properties of pure liquids. II. Excess Gibbs energy and vapour pressure. Fluid Phase Equilibria, 16: 361-367.
Excess Gibbs energies and vapour pressures of 13 mixtures of alcohols or phenols with hydrocarbons have been predicted and compared with experimental values. The agreement is very satisfactory.
INTRODUCTION
In Part I of this series a method for determining thermodynamic associa- tion constants as well as standard enthalpies of association from the vapour pressures of pure alcohols and their homomorphs was presented. The highly satisfactory results obtained without any ajustable parameters suggested adaptation of the concept of homomorphicity for the prediction of excess Gibbs energies and vapour pressures of mixtures of alcohols with hydro- carbons.
EXCESS GIBBS ENERGY
The excess Gibbs energy GE of an alcohol-hydrocarbon mixture is related to the excess chemical potentials pL/E of the alcohol A and of the hydrocarbon B:
GE = x,pLE, +X,/L; (11
where xA and xa are the nominal mole fractions of the two components. We assume that the excess chemical potential of each component can be sep-
0378-3812/84/$03.00 0 1984 Elsevier Science Publishers B.V.

362
arated into two parts: physical (nonspecific)
$ = ,~.=s + $.Ph
the first is due to association, py, and the second to interactions, pTph:
(2)
We adopt the equations given by Kehiaian and Treszczanowicz (1968) for E,ass.
pJ ’
p!?WT= lnh/x,) - +,GG/x~ - (1 + l/K*) ln[(l + K*&)/(l + K*)]
(3)
py/RT = ln( &/xa) - $~n/xn + 1 + rr& - (r/K*) ln(1 + K*+,) (4)
where $A and & = 1 - @A are the nominal volume fractions, with l/& = 1 + LQ/X~; r = V,“/ Vi is the ratio of the molar volumes, and K* = eK” is the volume-fraction association constant determined by the method given in Part I. Equations (3) and (4) are based on the Mecke-Kempter model of association with the Flory-Huggins correction for the entropy due to the size difference between the solute and solvent molecules.
The modified Hildebrand-Scatchard equation (Barton, 1975) is applied for the physical part of the excess chemical potential:
/&E’Ph = J$“(l - $J2DZ J (9
where D = S, - S& 6, being the solubility parameter of the solvent and 6; the partial-solubility parameter of the alcohol related to physical (dispersion) interactions only. Two methods of calculating a’* have been adopted. Either SA is identified with the solubility parameter 6, of the homomorph, or the energy of vaporization of the homomorph is divided by the molar volume of the alcohol: 8’: = ETp/v:. For the computations we adopted the value which gives the larger D. However, the choice of 6; has a very small effect on GE.
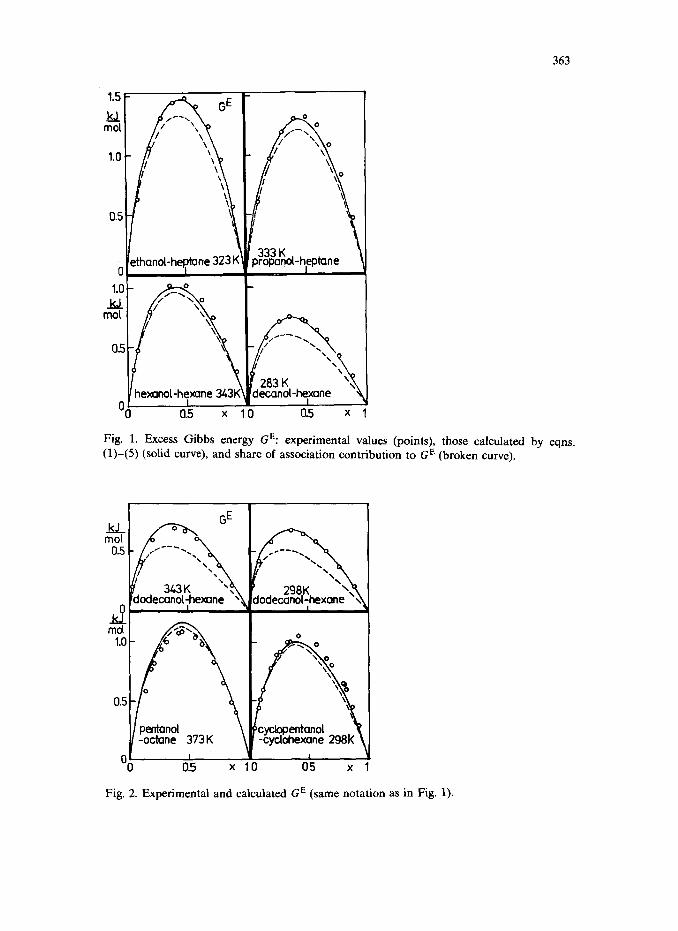
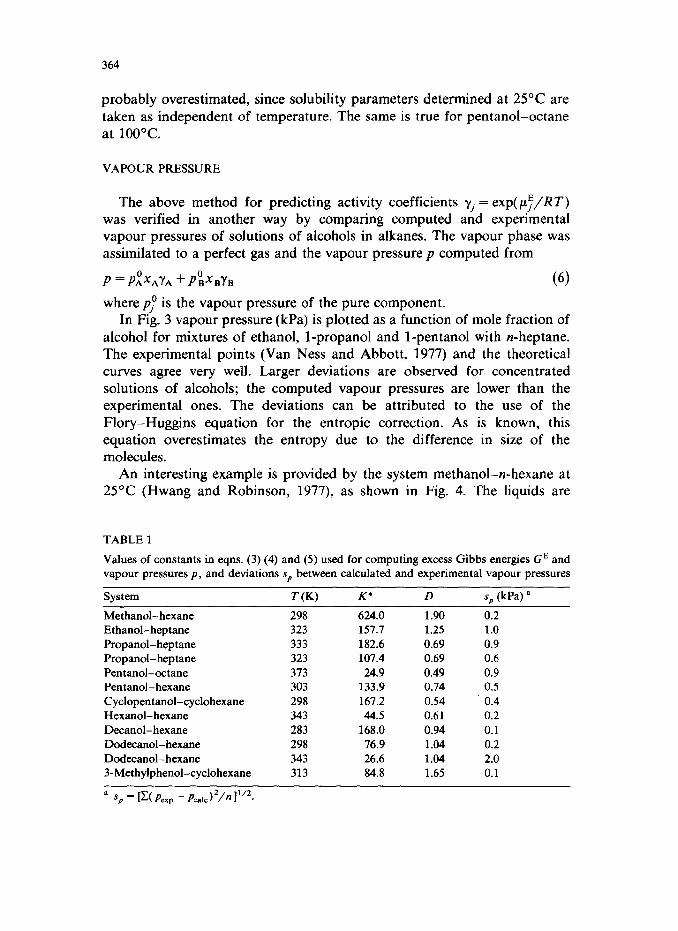
In Figs. 1 and 2 excess Gibbs energies computed by the above method are compared with experimental GE data taken from the literature (Van Ness and Abbott, 1977; Wieczorek and Stecki, 1978; Wieczorek, 1978, 1979; Sipowska and Wieczorek, 1980; Treszczanowicz and Treszczanowicz, 1979; Benson et al., 1974). Points refer to experimental GE values, solid curves to computed ones; the broken lines represent the share of association. The values of K* and D used for computing GE and p are given in Table 1. The curves, although calculated without any adjustable parameters, agree very well with the experimental excess Gibbs energies for all eight systems studied. As expected, the share due to physical interactions increases with increasing length of the hydrocarbon chain of the alcohol. The computed GE value for a mixture of dodecanol with hexane at 70°C (Fig. 2) is a little higher than the experimental one, because the effect of dispersion forces is
363
0 0
c-1
/I ’ \ \ 0
\
/
\ \
/\
/ \O \ \ \
i
I
1 0.5 x
Fig. 1. Excess Gibbs energy GE: experimental values (points), those calculated by eqns. (l)-(5) (solid curve), and share of association contribution to GE (broken curve).
Fig. 2. Experimental and calculated GE (same notation as in Fig. 1).
364
probably overestimated, since solubility parameters determined at 25°C are taken as independent of temperature. The same is true for pentanol-octane
at 100°C.
VAPOUR PRESSURE
The above method for predicting activity coefficients y, = exp( yT/RT) was verified in another way by comparing computed and experimental vapour pressures of solutions of alcohols in alkanes. The vapour phase was assimilated to a perfect gas and the vapour pressure p computed from
P = PEWA
where p,! is In Fig. 3
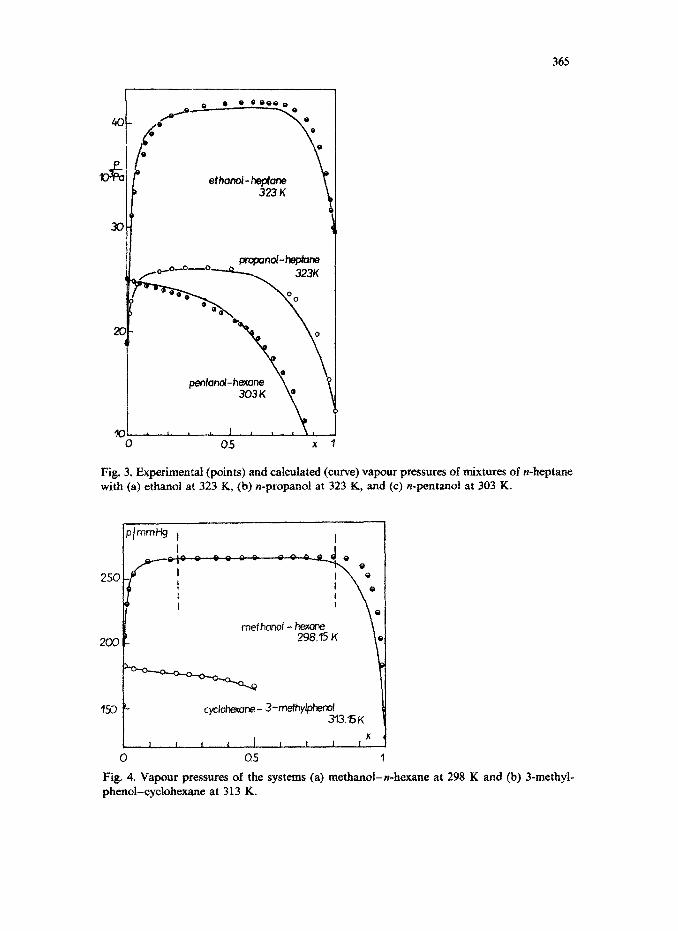
alcohol for
+ Ph3YB (6) the vapour pressure of the pure component. vapour pressure (kPa) is plotted as a function of mole fraction of mixtures of ethanol, 1-propanol and I-pentanol with n-heptane.
The experimental points (Van Ness and Abbott, 1977) and the theoretical curves agree very well. Larger deviations are observed for concentrated solutions of alcohols; the computed vapour pressures are lower than the experimental ones. The deviations can be attributed to the use of the Flory-Huggins equation for the entropic correction. As is known, this equation overestimates the entropy due to the difference in size of the molecules.
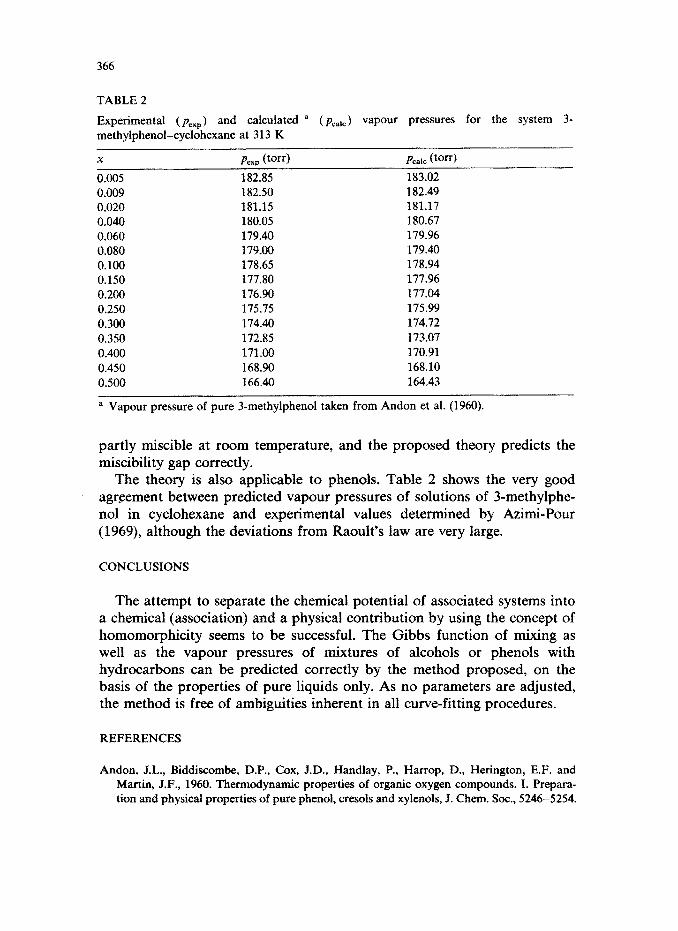
An interesting example is provided by the system methanol-n-hexane at 25°C (Hwang and Robinson, 1977), as shown in Fig. 4. The liquids are
TABLE 1
Values of constants in eqns. (3) (4) and (5) used for computing excess Gibbs energies GE and vapour pressures p, and deviations sp between calculated and experimental vapour pressures
System T(R) K* D s, (kPa) ’
Methanol-hexane 298 624.0 1.90 0.2 Ethanol-heptane 323 157.7 1.25 1.0 Propanol-heptane 333 182.6 0.69 0.9 Propanol-heptane 323 107.4 0.69 0.6 Pentanol-octane 373 24.9 0.49 0.9 Pentanol-hexane 303 133.9 0.74 0.5 Cyclopentanol-cyclohexane 298 167.2 0.54 0.4 Hexanol-hexane 343 44.5 0.61 0.2 Decanol-hexane 283 168.0 0.94 0.1 Dodecanol-hexane 298 76.9 1.04 0.2 Dodecanol-hexane 343 26.6 1.04 2.0 3-Methylphenol-cyclohexane 313 84.8 1.65 0.1
a sp = m Pcxp - P&)‘/~1”‘.
365
Fig. 3. Extents (points) and calculated (curve) vapour pressures of mixtures of n-heptane with (a) ethanol at 323 K, (b) n-propanol at 323 K, and (c) n-pentanoi at 303 K.
250
200
1%
0 05 ?
Fig. 4. Vapour pressures of the systems (a) methanol-n-hexane at 298 K and (b) 3-methyl- phenol-cyclohexane at 313 K.
366
TABLE 2
Experimental (p,..,) and calculated a ( pcalc) vapour pressures for the system 3- methylphenol-cyclohexane at 313 K
X pcxp (torr) Pcdc t -9 0.005 182.85 183.02 0.009 182.50 182.49 0.020 181.15 181.17 0.040 180.05 180.67 0.060 179.40 179.96 0.080 179.00 179.40 0.100 178.65 178.94 0.150 177.80 177.96 0.200 176.90 177.04 0.250 175.75 175.99 0.300 174.40 174.72 0.350 172.85 173.07 0.400 171.00 170.91 0.450 168.90 168.10 0.500 166.40 164.43
a Vapour pressure of pure 3-methylphenol taken from Andon et al. (1960).
partly miscible at room temperature, and the proposed theory predicts the miscibility gap correctly.
The theory is also applicable to phenols. Table 2 shows the very good agreement between predicted vapour pressures of solutions of 3-methylphe- no1 in cyclohexane and experimental values determined by Azimi-Pour (1969), although the deviations from Raoult’s law are very large.
CONCLUSIONS
The attempt to separate the chemical potential of associated systems into a chemical (ass~iation) and a physical cont~bution by using the concept of homomorphicity seems to be successful. The Gibbs function of mixing as well as the vapour pressures of mixtures of alcohols or phenols with hydrocarbons can be predicted correctly by the method proposed, on the basis of the properties of pure liquids only. As no parameters are adjusted, the method is free of ambiguities inherent in all curve-fitting procedures.
REFERENCES
Andon, J.L., Biddiscombe, D.P., Cox, J.D., Handlay, P., Harrop, D., Herington, E.F. and Martin, J.F., 1960. Thermodynamic properties of organic oxygen compounds. I. Prepara- tion and physical properties of pure phenol, cresols and xylenols, J. Chem. Sot., 5246-5254.

361
Azimi-Pour, H., 1969. Etude de solutions du metacresol. Rev. Inst. Fr. Petrol., 25: 17-52. Barton, A.F.M., 1975. Solubility parameters. Chem. Rev., 75: 731-753. Benson, G.C., Anand, SC. and Kiychara, O., 1974. Thermodynamic properties of some
cycloalkane-cycloalkanol systems at 298.15 K. II. J. Chem. Eng. Data, 19: 258-261. Hwang, S. and Robinson, Ir. R.L., 1977. Vapor-liquid equilibria at 25’C for nine
alcohol-hydrocarbon binary systems. J. Chem. Eng. Data, 22: 319-325. Kehiain, H. and Treszczanowicz, A., 1968. Excess free enthalpy of athermal associated
mixtures of the Mecke-Kempter type. Bull. Acad. Polon. Sci., Ser. Sci. Chim., 26: 44-452.
Sipowska, J. and Wieczorek, S., 1980. Vapour pressures and excess Gibbs free energies of (propan-1-ol+ n-heptane) between 278.164 and 304.147 K, J. Chem. Thermodyn., 12: 459-464.
Treszczanowicz, T. and Treszczanowicz, AI., 1979. Vapour-liquid phase equilibria of binary systems formed by pentan-l-01 and alkanes. Bull. Acad. Polon. Sci., Ser. Sci. Chim., 27: 689-695.
Van Ness, H.C. and Abbott, M.N., 1977. Int. Ser. Sel. Data Mixtures, Ser. A, 1: 3. Wieezorek, S., 1978. Vapour pressures and thermodynamic properties of dodecan-1-ol+ n-
hexane between 298.230 and 342.824 K. J. Chem. Thermodyn., 10: 187-194. Wieczorek, S., 1979. Vapour pressures and thermodynamic properties of decan-l-01 + n-hexane
between 283.160 and 333.151 K. J. Chem. Thermodyn., 11: 239-245. Wieczorek, S. and Stecki, J., 1978. Vapour pressures and thermodynamic properties of
hexan-l-01 + n-hexane between 298.230 and 342.824 K. J. Chem. Thermodyn., 10: 177-186.





























