Embed Size (px)
Citation preview

SHORT AND INTERMEDIATE TERM FOLLOW UP OF ALL
INFANTS DIAGNOSED WITH PULMONARY ATRESIA AND
VENTRICULAR SEPTAL DEFECT - A FIVE YEAR FOLLOW UP
STUDY
PROJECT REPORT
Submitted during the course of DM Cardiology by
DR. PRIYADARSHINI. A
Trainee
DEPARTMENT OF CARDIOLOGY
Jan 2014 – Dec 2016
SREE CHITRA TIRUNAL INSTITUTE FOR MEDICAL
SCIENCES AND TECHNOLOGY, TRIVANDRUM, KERALA,

DECLARATION
I, Dr. Priyadarshini A, hereby declare that the project in this book, titled
“Short and intermediate term follow up of all infants diagnosed with pulmonary
atresia and ventricular septal defect - A five year follow up study” was
undertaken by me under the supervision of the faculty, Department of
Cardiology, Sree Chitra Tirunal Institute for Medical Sciences and Technology.
Trivandrum Dr Priyadarshini A
3/10/16 DM Cardiology trainee
FORWARDED
The candidate, Priyadarshini A, has carried out the minimum required
project.
Thiruvananthapuram Prof. (Dr )Ajit Kumar VK
3/10/16 Head of Department of Cardiology

CERTIFICATE
This is to certify that this thesis titled “Short and intermediate term follow up of
all infants diagnosed with pulmonary atresia and ventricular septal defect - a
five year follow up study” has been prepared by Dr. Priyadarshini. A, DM
Cardiology Resident, Department of Cardiology at Sree Chitra Tirunal Institute
for Medical Sciences & Technology, Thiruvananthapuram. She has shown keen
interest in preparing this project.
GUIDE
Dr. Krishnamoorthy.K.M
Professor, Department of Cardiology
SCTIMST, Thiruvananthapuram
CO GUIDE
Dr. Deepa S. Kumar
Assistant Professor, Department of Cardiology
SCTIMST, Thiruvananthapuram

Dedicated to my
teachers, family and
friends

TABLE OF CONTENTS
Abstract i
Introduction 1
Aims and Objectives 6
Review of literature 8
Materials and methods 17
Statistical analysis 19
Results 20
Discussion 42
Conclusion 48
Limitation 49
References 50
Annexure 54

ABBREVIATIONS
VSD – Ventricular septal defect
PA – Pulmonary atresia
MAPCA – Major Aorto-pulmonary collaterals
PDA - Patent ductus arteriosus
RVOT – Right ventricular outflow tract
CT – Computed tomography
MRI – Magnetic resonance imaging
TOF – Tetralogy of Fallot
DRPA – Diameter of right pulmonary artery at the hilum
DLPA – Diameter of left pulmonary artery at the hilum
DDTAO- Diameter of descending thoracic Aorta at the diaphragm

i
ABSTRACT:
Introduction: Ventricular septal defect- pulmonary atresia (VSD-PA)
encompasses a spectrum of anomalies ranging from short to long segment
pulmonary atresia and duct dependent or MAPCA dependent pulmonary
circulation or a combination of both. From the therapeutic standpoint, surgical
treatment could vary from as simple as VSD closure with RVOT reconstruction
to as complicated as multi-staged unifocalisation. More over children may
require interim palliative procedures in the form of ductal stenting, Aorto-
pulmonary shunt or Cavo-pulmonary anastomosis. There is paucity of data from
the Indian subcontinent regarding the outcomes in this cohort of patients.
Aim: To study variations in pulmonary anatomy, natural history, time to
intervention, complications, outcomes and determinants of mortality in infants
with VSD-PA.
Methodology: All infants (<1 year) registered at our institute between January
2011 and December 2015 with the diagnosis of VSD-PA were enrolled.
Children with complex anatomy like Single ventricle, transposition, AV canal
defects etc were excluded from the study. Deaths were classified according to
the time period in which attrition occurred, T1- before any palliation (Aorto-
pulmonary shunt, PDA stenting, BDG)/ definitive surgery was done, T2- in the

ii
interstage between palliation and definitive repair, T3- postoperative period and
T4- late after definitive repair
Results: A total of 108 infants were included in the study. There was an almost
equal gender distribution- 51% were male and 49% were female. Mean age at
presentation was 1.65 months (range 0 – 11 months) and mean duration of
follow up was 18.9 months. 73% had confluent pulmonary anatomy and long
segment pulmonary atresia was more common. Majority of patients (55.6%)
had a ductal dependence, 25.9% had MAPCA dependence and 18.5% had a
combination of both. Right arch was seen in 25.9 % of the children. Mean Mc
Goon index was 1.26. 7 patients were diagnosed with Di George syndrome. The
average birth weight was 2.64 kg and 12 % were born preterm. There was
history of maternal diabetes in 17%. The mean lowest saturation on follow up
was 73.4 %. 32.4% required prostaglandin support. Imaging in the form of CT/
MR was required in 52% of the population. Imaging was very useful in
MAPCA dependent pulmonary circulation to plan surgery. 6 required ductal
stenting, 37 required Aorto-pulmonary shunt and 4 required BDG. Average age
at initial palliation was 6.24 months. Average time to palliation after diagnosis
was 4.81 months. Out of the 43 patients, who underwent palliative procedures,
13 (29%) proceeded to definitive repair. There were 32 deaths in toto – 19 in
the T1 period. 2 children were lost in the inter-stage period- T2. ICR was done
in 13 patients with the following concomitant procedures- homograft repair was

iii
done with intracardiac repair in 4 patients, 7 children did not require conduit -
only RVOT reconstruction sufficed and primary unifocalisation with intra
cardiac repair was done in 3 children. 10 children died in the post operative
period and one death was late- 6 months after surgery. Branch PA plasty was
concomitantly done in majority (69.2%) of the patients. Pulmonary anatomy-
length of atresia, was statistically significant as a predictor of mortality
(p=0.023). Survival at 1, 2 and 4 years were 70.3%, 68.2% and 65%
respectively.
Conclusion: Despite early diagnosis, mortality associated with VSD-PA
remains high with most of the deaths occurring in the T1 period, before any
palliation or corrective surgery is done. Surveillance and weekly saturation
monitoring may avert the catastrophes associated with worsening hypoxemia.
Keywords: Ventricular septal defect- pulmonary atresia, natural history,
outcomes, predictors of mortality.

1
INTRODUCTION :
Pulmonary atresia with ventricular septal defect is a rare congenital
anomaly, constitutes about 2% of congenital heart diseases , is considered to be the
most severe form of Tetralogy of Fallot and occurs at a frequency of 0.07 per 1,000
live births ( Baltimore Washington study)(1). Anatomically, the right ventricular
outflow tract ends blindly, right ventricular stroke volume is directed towards the
Aorta through a large ventricular septal defect. Pulmonary arterial architecture is
complex, ranging from duct dependent pulmonary circulation to a pulmonary
arterial tree entirely fed by major Aorto-pulmonary collaterals (MAPCAs).
.VSD-PA can be classified into 3 types based on source of pulmonary blood flow.
A classification (2) has been proposed by the Congenital Heart Surgeons Society
(CHSS) based on pulmonary blood flow and is detailed below:
Type A: Native PAs present, pulmonary vascular supply through PDA and no APC
/ MAPCAs
Type B: Native PAs and APC/MAPCAs present
Type C: No native PAs, pulmonary blood supply through APC/MAPCAs only.

2
Figure 1
Types of VSD-PA based on source of pulmonary blood flow
Furthermore, pulmonary atresia can be categorized based on the length of
the atretic segment – short or long. Pulmonary arterial anatomy could also be
classified based on the confluence as confluent or non-confluent pulmonary
anatomy.
In VSD-PA, PDA originates in the majority from the undersurface of the
arch (67%) or from the undersurface of the innominate artery (33%). PDA is
associated with confluent PAs, in as high as 80%. When PDA is present, PAs are
confluent in 80% of cases. Most patients with absent PDA, have non confluent
PAs.

3
Majority (65%) of VSD-PA patients present to a cardiac center during
infancy. The remainder presents later because of sufficient pulmonary blood flow
which lead to clinically undetectable cyanosis during the early months of life.
Clinical presentation is in the form of cyanosis (50%), heart failure (25%) or
murmur with mild cyanosis with or without failure to thrive (3). Patients with PA +
VSD, with or without MAPCAs, have been described as having less than a 50%
chance of being alive at 10 years of age. PA + VSD patients with MAPCAs,
instead, have had only a 40% chance being alive at 10 years of age, and a 20%
chance being alive at the age of 30.
Echocardiography is a useful modality for assessment of source of
pulmonary blood flow. It is very useful in delineating central pulmonary anatomy.
Origin of MAPCAs can also be ascertained from echocardiography. But other
modalities of imaging like CT and MRI are useful for making out the exact
anatomy of MAPCAs. Color flow Doppler was accurate in determining the
presence or absence, the side and the origin of the collateral channels in all
patients, with the correct number being determined in 12 (67%) of 18 patients
studied by Acherman et al (4). "Wash-in" to the hilar pulmonary arteries
(retrograde color flow) was seen in 12 (92%) of 13 patients with collateral
channels and confluent pulmonary arteries.

4
Mc Goon ratio is calculated by dividing the sum of the diameters of RPA (at
the level of crossing the lateral margin of vertebral column on angiogram) and
LPA (just proximal to its upper lobe branch), divided by the diameter of aorta at
the level above the diaphragm [DRPA /DDTAO)+( DLPA / DDTAO)]. An
average value of 2.1 was noted in normal subjects. Ratio above 1.2 is associated
with acceptable postoperative RV systolic pressure in Tetralogy of Fallot. Ratio
below 0.8 is deemed inadequate for complete repair of PA – VSD. VSD closure is
deferred in such patients at the time of repair or they underwent Aorto-pulmonary
shunt procedure as first stage (5,6) However, this ratio tends to overestimate the
adequacy of the size of PAs since this is derived using the diameter of descending
thoracic aorta at the level of diaphragm which is frequently smaller in patients with
PA-VSD.
Nakata index was developed to provide prognostication regarding surgical
outcomes in patients with diminutive pulmonary arteries. In tetralogy, all patients
with a PAI (Pulmonary Artery Index) over 100 mm2/BSA could undergo
correction safely (7).Nakata PA index is calculated from the diameter of PAs
measured immediately proximal to the origin of upper lobe branches of the
respective branch PAs. The sum of the cross sectional area (CSA) of right and left
PAs is divided by the body surface area of the patient [Nakata index = CSA of

5
RPA (mm2) + CSA of LPA (mm2)/ BSA (m2)]. A Nakata index of >150 mm2/m2
is acceptable for complete repair without prior palliative shunt (8).
Surgical treatment varies from correction akin to Tetralogy of Fallot- VSD
closure and RVOT reconstruction to complex multi-staged procedures like
unifocalisation. Many children require some intervention in the neonatal period,
especially in the duct dependent group, coinciding in time with closure of ductus.
Palliative procedure for worsening cyanosis could be in the form of ductal stenting
or Aorto-pulmonary shunt. In children older than 3-6 months, a Bidirectional
Glenn could also be performed to improve pulmonary blood flow.
Most of the attrition occurs during the first year of life, with various studies
documenting rates between 25-40%. Non cardiac anomalies could contribute to
death as in the case of Di George syndrome with immunodeficiency. Cyanotic
spells and sudden cardiac death are important causes of mortality apart from the
peri operative deaths.
Overall surgical outcomes are improving over the last few decades (9),with
greater number achieving complete repair. Presence of MAPCA s has been seen in
many studies to be a negative predictive factor for complete repair (10).

6
AIMS AND OBJECTIVES:
VSD - pulmonary atresia commonly referred to as TOF with pulmonary
atresia is a fairly common congenital heart disease with presentation varying from
cyanosis in the neonatal period to later in life. There is paucity of data with regard
to natural history and treatment outcomes of this order from our part of the world,
where percutaneous intervention in the form of ductal stenting is burgeoning.
Initial palliation in the form of ductal stenting or Aorto-pulmonary shunt is usually
required for alleviation of symptoms brought out by ductal closure. But there is a
lot of attrition in this period prior to initial palliation. There are very few centres in
the country which attempt unifocalisation, and the data on outcome of such
procedures is scarce. Compared to the West, where pulmonary rehabilitation is
started at a much earlier age (first 3 months of life), there is a lag in the time to
unifocalisation, whether such delays affect outcomes have not been looked at.
It could be hypothesized that regular saturation monitoring and surveillance
during this period could help improve mortality in this period. MAPCA dependent
pulmonary circulation forms a complicated cohort with respect to surgical
management, where unifocalisation followed by intracardiac repair is warranted
over multiple steps. Some children may require MAPCA coiling when a segment
of lung is supplies by both native pulmonary arteries as well as MAPCAs- dual
blood supply. There is a subgroup of this cohort, where central pulmonary arteries

7
are entirely absent or very atretic, precluding any form of surgical repair and are
left for medical follow up after cardiac imaging.
The real world scenario in our part of the world is enigmatous and a careful
study to observe the natural history and modified history after palliative and
corrective surgery would give us a fair idea as regards to the prognostics and what
to expect regarding outcome in our settings. It would also help us pick up
vulnerable periods in the natural history of this heart disorder so as to strengthen
surveillance to avert mishaps.

8
REVIEW OF LITERATURE:
Pulmonary atresia with ventricular septal defect (PA + VSD) has an
incidence between 4.2 and 10 per 100 000 live births (11,12). Anatomically, the
right ventricular outflow tract ends blindly, right ventricular stroke volume is
directed towards the Aorta through a large ventricular septal defect. Pulmonary
arterial architecture is complex, ranging from duct dependent pulmonary
circulation to a pulmonary arterial tree entirely fed by Aorto-pulmonary collaterals.
Maternal diabetes, phenylketonuria, exposure to drugs like retinoic acid and
trimethadone are associated with increased risk of conotruncal defects. Diabetic
women have a 10 -fold increased risk of giving birth to a child with PA-VSD.
Ryan et al, reported strong association between 22q11 deletion and PA-VSD, their
series showed a 10% prevalence of PA-VSD in patients with this deletion(13).
Aortopulmonary collaterals , right aortic arch and aberrant subclavian artery
occurred more commonly in patients with PA-VSD and 22q11 deletion(14). Brach
pulmonary arteries were smaller and hence clinical outcomes were poorer in this
subgroup of patients (15).
In a postmortem study by Liao et al (16), heart and lung specimens in 31
cases of PA-VSD were studied. Main source of pulmonary blood flow could be the
ductus arteriosus, major collateral arteries or diffuse small pleural arterial plexus
coexisting with either ductus arteriosus or major collateral arteries. The ductus

9
arteriosus and major collateral arteries did not coexist in the same lung in these
cases. Confluent central pulmonary arteries were present in 22 (71%) of the 31
cases, involving 7 (58%) of the 12 cases of ductus arteriosus, 14 (70%) of the 20
cases with major collateral arteries and 1 case with an aorticopulmonary window.
The pulmonary trunk (atretic or patent) was identifiable in 24 (77%) of the 31
cases. A lung or lungs that connected to a ductus (or ligamentum) had a complete
and unifocal intrapulmonary arterial distribution (without arborization
abnormalities). Major collateral blood supply was frequently multifocal and
associated with arborization abnormalities. The size of the central pulmonary
arteries was not related to the type of arterial blood source but seemed to be related
to the amount of blood flow actually reaching the vessels.
A combination of high resolution two dimensional echocardiographic
imaging and color flow Doppler study permits good appreciation of both the
mediastinal pulmonary arteries as well as their blood supply in patients with VSD-
PA. In a study by Acherman et al (17), 42 patients aged a few hours to 19 months
were prospectively assessed by echocardiography. A patent arterial duct was
correctly identified as the sole source of pulmonary blood supply in 23 patients,
whereas Aorto-pulmonary collateral channels were detected in 19, with one of
these having a small patent arterial duct and collateral channels. The patent arterial
duct originated from the undersurface of the aorta in 16 (67%) of 24 patients and

10
from the base of the brachiocephalic trunk in 7 (33%) of 24. All patients with a
patent ductus as the sole source of pulmonary blood supply had confluent
pulmonary arteries. Non-confluent pulmonary arteries were present in six patients,
with all but one having Aorto-pulmonary collateral channels as the sole source of
pulmonary flow. Aorto-pulmonary collateral channels were direct in 17 (89%) of
19 patients, whereas in 2 (11%) of 19, both direct and indirect collateral channels
were present. Color flow Doppler study was accurate in determining the presence
or absence, the side and the origin of the collateral channels in all patients, with the
correct number being determined in 12 (67%) of 18.
Imaging in the form of DSCT (dual source computed tomography) has been
evaluated for pick up rate of MAPCAs in comparison to ECHO
(echocardiography) and CA (catheterization) in a study by Yin et al (18) .The
diagnostic rate of the aortopulmonary collateral vessels was 100% (14/14) for
DSCT, 92.9% (13/14) for CA (p = 0.995 in comparison with DSCT), and 35.7%
(5/14) for ECHO (p = 0.001 in comparison with DSCT). Similarly gadolinium
enhanced magnetic resonance angiography (MRA) has been compared with
cathetersation and has been found that it is a simple, fast, and accurate technique
by which to delineate pulmonary arterial anatomy and can be a reliable
noninvasive alternative to cineangiography. MRA can provide required

11
information to plan surgical strategy in infants and children and at times obviate
the need for catheterization (19).
Leonard et al (20), investigated the natural and unnatural history of patients
with pulmonary atresia. 129 cardiac malformations with
congenital pulmonary atresia were identified from 601,635 live births (21.4/100
000): 29 had PA-IVS, 60 had PA-VSD, and 40 had complex pulmonary atresia.
Total mortality was 72/129 (56%), with 15 deaths in the first week and 49 in the
first year. There were 23 surgical deaths, 33 hospital deaths (not related to
surgery), and 16 sudden deaths, 12 of which remained unexplained. The sudden
death rate was 29/1000 patient years of follow up. Of the 57 survivors, 39% have
exercise ability I or II and 61% III or IV. Definitive surgical repair produced better
exercise ability. The authors concluded that early mortality is high in all types
of pulmonary atresia, although survival has improved in recent years. Most
children who have not undergone definitive repair have significant exercise
limitation on follow up.
In a review of 185 patients by Amark et al (21) , 118 patients had simple
PA-VSD and 53 patients had PAVSD with major aortopulmonary collateral
arteries (MAPCAs). Overall survival from initial operation was 71% at 10 years.
Risk factors for death after initial operation included younger age, earlier birth
cohort, fewer bronchopulmonary segments supplied by native pulmonary arteries,

12
and initial placement of a systemic-pulmonary artery (SP) shunt.
Bronchopulmonary arterial supply is an important determinant of mortality,
achievement of definitive repair, and post-repair reoperation.
Treatment strategy in these patients depend on pulmonary blood flow.
Patients with duct dependent circulation usually have well developed confluent
pulmonary arteries and go for biventricular repair akin to patients with Tetralogy
of Fallot. Some centres adopt staged procedures for duct dependent circulation
with systemic pulmonary shunt being a prelude to the intra-cardiac repair. In a
series of 86 patients with duct dependent PA-VSD reported by Alsoufi et al (22),
33% underwent primary repair whereas 67% underwent initial palliation with
systemic pulmonary shunt. Early and late results were compared between the 2
groups. On multivariable analysis, risk factors for mortality were genetic or extra-
cardiac malformations (hazard ratio [HR], 2.8), post-operative extracorporeal
membrane oxygenation (ECMO) (HR, 4.0). Freedom from right ventricular
outflow tract reoperation after achievement of repair was 63.2% at 8 years (52.4%
for BVR versus 70.2% for BTS, p [ 0.170). On multivariable analysis, risk factors
for reoperation were the use of conduit (HR, 8.7) and prematurity (HR, 2.8).
Iyer et al (23) , reported intermediate term outcomes of 58 patients with
VSD-PA, hypoplastic pulmonary arteries with arborization defects, and major
aortopulmonary collaterals between 1979-89. A total of 121 staging procedures

13
were performed with an overall mortality of 10.3%. One hundred thirty-four major
collaterals were either ligated or transplanted. Thirty patients eventually underwent
hemodynamic repair with an early mortality of 3.3% and late mortality of 10.0%.
Twenty-six current survivors of repair remain clinically well after a mean follow-
up of 3.6 years.
Patients with MAPCA dependent pulmonary circulation in lieu of
unfavorable anatomy undergo staged repair more often whereas some centres
advocate single stage unifocalisation. Reddy et al (24), analysed early and
intermediate outcomes after unifocalisation in 85 patients with pulmonary atresia,
VSD, and MAPCAs (median age, 7 months). Complete 1-stage unifocalization
and intracardiac repair were performed through a midline approach in 56 patients,
whereas 23 underwent unifocalization in a single stage with the VSD left open, and
6 underwent staged unifocalization through sequential thoracotomies. There were 9
early deaths. During follow-up (1 to 69 months), there were 7 late deaths. Actuarial
survival was 80% at 3 years. Among early survivors, actuarial survival with
complete repair was 88% at 2 years. Reintervention on the neo–pulmonary arteries
was performed in 24 patients.
Kaskinen et al (25), in a recent review of 109 patients over a period of 37
years, observed that achievement of repair and initial size of true central
pulmonary arteries affect survival of patients with PA + VSD. Presence of

14
MAPCAs did not affect survival but reduced the chance at complete repair.
Palliative surgery may have a role in treatment of PA + VSD because the size of
pulmonary arteries increased after placement of Aorto–pulmonary shunt. In
addition, subtotal repair by a RV–pulmonary artery connection and septal
fenestration improved survival over extracardiac palliation.
Shimazaki et al (26), catheterized 21 patients after surgical repair od VSD-
PA and MAPCA dependent pulmonary circulation. They found that the mean and
systolic pulmonary arterial pressure, as well as the pulmonary vasculature
resistance, were abnormally high in 60% of the patients. 33% of the patients were
found to have pulmonary arterial segments that were not connected to the central
and unbranched hilar portions of the right and left pulmonary arteries. The mean
pulmonary artery pressure and the pulmonary vasculature resistance were
correlated (inversely) with the number of centrally connected pulmonary arterial
segments. The pulmonary vasculature resistance per pulmonary arterial segment
was also inversely correlated with the number of centrally connected pulmonary
arterial segments. This highlights the fact that the more the number of pulmonary
segments connected to the RV after unifocalisation, the better are the right sided
pressures.
Twenty-five patients over a period of 6 years between 2003- 08, were
studied by Liava`a et al (27) and analyzed a protocol of neonatal rehabilitation of

15
hypoplastic PAs in the management of VSD-PA and major aortopulmonary
collateral arteries (MAPCAs). This group did not believe in the school of thought
of MAPCA translocation, rather believed in promotion of native PA growth,
promoted by Aorto-pulmonary shunt. At a 18 months follow up, 12 of 20 patients
had undergone complete repair. There was a significant increase in PA growth
after the initial palliative shunt. The median RV/ LV pressure was 0.66. This group
concluded that rehabilitation of hypoplastic native pulmonary arteries by a
neonatal shunting regimen, without MAPCA translocation, for pulmonary atresia,
VSD, and MAPCAs, provides encouraging results with excellent early survival.
A protocol was proposed by Malhotra et al (28), the group which analyzed
462 patients with VSD-PA and MAPCAs over 15 years. Median age at operation
was 7.7 months, ranging from 10 days to 39 years. Complete single-stage
unifocalization was achieved in 76% of patients. Intracardiac repair was possible at
initial operation in 56%. At 5 years, 90% of patients were completely repaired,
indicating that most patients who did not achieve initial intracardiac repair
eventually did. No factor was predictive of mortality apart from the birth cohort in
this study. The following figure is illustrative of the study flow and their protocol.

16
Fig 1
Study flow
Many studies (29-32) have shown that unfavorable long-term outcomes
from staged approaches can be largely attributed to the significant loss of lung
segments, and development of pulmonary vascular disease, as a result of prolonged
uncontrolled flow through collaterals because of delayed or deferred completion
unifocalization. The most important factor in improving the long-term prognosis of
these patients is not the achievement of intracardiac repair alone, but the
achievement of intracardiac repair with a low RV/LV pressure ratio.

17
MATERIAL AND METHODS:
A total of 108 children were enrolled in the study. Infants registered in our
hospital from January 1 st 2011 to December 31 st 2015 were enrolled in the study.
They were followed up until 30 th June 2015. Data of those, who were lost to
follow up before 30 th June 2015 were analysed till the point, they were followed
up either through a hospital visit or telephonic enquiry. Institute Ethics committee
clearance was obtained prior to commencement of the study. Data was collected
using detailed proforma and data collection by telephone was systematically made
when there was a lack of follow up over a prolonged period. In the event of
mortality outside hospital, details regarding the heralding events were obtained bt
telephonic conversation.
Inclusion criteria:
All children < 1 year with a diagnosis of VSD-PA (Ventricular septal defect-
pulmonary atresia), attending the outpatient department at this institute were
enrolled in the study. Children who had undergone intervention outside , but
subsequently registered in our hospital were also included if their age at first
contact with our institute, was less than 1 year.

18
Exclusion criteria:
Pulmonary atresia associated with complex heart disease were excluded –
eg. Transposition, double outlet right ventricle, AV canal defect etc. Any child
beyond beyond the age of one year at the time of first contact were excluded.
Mortality data:
Mortality data were segregated depending on the time interval at which
death occurred and we introduced the following novel nosology to depict the
timeframe in which it occurred.
“T1” is defined as the period prior to any palliative procedure, in a child who has
undergone direct total correction – it is the time before the first surgical procedure.
“T2”is defined as the period between initial palliative procedure and final
corrective procedure, in children who have undergone an initial palliative
procedure
“T3” is defined as a death in the peri procedural period, regardless of whether the
procedure is palliative or definitive. All deaths till discharge following a procedure
were included in this category.
“T4” is defined as a death following a corrective period at any time beyond
hospital discharge after the procedure.

19
STATISTICAL ANALYSIS:
The data was analyzed by the principal investigator. All data was handled
with care to maintain patient confidentiality. Records were maintained in both
computer and paper formats. Data was tabulated in Microsoft excel sheet.
Descriptive summaries are presented as frequencies and percentages, for
categorical data, and as means and standard deviations for continuous variables.
Continuous variables are compared using Student’s t test or Mann-Whitney U test
as appropriate. Discrete variables are compared using Chi- square test. Kaplan-
Meier survival analyses was performed to evaluate differences in mortality among
various sub-groups. Univariate analysis for predictors of mortality was done by
Chi square test and multivariate analysis was done by binary logistic regression
model. Cox proportional hazard model was done for multivariate analysis of
survival and Log rank test was used for univariate analysis of survival. All
statistical analyses were performed using the SPSS statistical software package
(release 17.0, SPSS Inc.; Chicago,Ill).

20
RESULTS:
Baseline demographic data:
Among the 108 children, who were enrolled, 55 (50.9%) were male and 53
(49.1%) were female, suggesting an equal gender preponderance. The mean age at
diagnosis was 1.65 +/- 2.61 months. Infants presented as early as soon after birth
on the first day of life to as late as 11 months. There was tendency to later
diagnosis in MAPCA dependent pulmonary circulation. The mean duration of
follow up was 18.98 +/- 18.54 months (range 0.7 – 66 months).
There was history of maternal diabetes in 17 infants (15.7%). The average
birthweight of the entire cohort was 2.64 +/- 0.46 kg. 13 infants (12%) were born
preterm. 32.4% of the infants required prostaglandin either orally or parenterally
due to desaturation associated with issues in ductal patency. Lowest saturation
during the follow up period on an average was 73.4 +/- 7.8 %. 48.1% (n=52)
infants required some form of intervention on follow up.

21
Table 1:
Demographic data
N = 108
Age at diagnosis (Mean +/ SD)
1.65 +/- 2.61 months
Follow up (Mean +/ SD) 18.98 +/- 18.54 months
Gender Male – 55 (51%)
Female – 53 (49%)
Birth weight (Mean +/ SD)
2.64 +/- 0.46 kg
Prematurity 13 (12 %)
Maternal risk factors Gestational diabetes - 17
Hypertension - 7
Hypothyroidism - 5
Imaging Total – 56 (52%)
CT – 26 (24%)
MRI – 30 (28%)
Lowest oxygen saturation on follow up (Mean +/ SD)
73.4 +/- 7.8 %
Time to saturation nadir All – 6.85 months
Duct dependent - 4.35 months
MAPCA dependent – 9.83 months
Both – 8.62 months
Prostaglandin 35 (32.4 %)
Associated cardiac Situs ambiguous- 5

22
anomalies PAPVC – 4
LPA stenosis – 3
Corrected transposition – 3
Retro aortic innominate vein – 3
Coronary anomalies - 3
Criss cross heart – 1
Interrupted IVC- 1
Aberrant LSCA- 1
Asymmetric septal hypertrophy – 1
Dextrocardia -1
Bovine arch - 1
Associated extra cardiac anomalies
Di George syndrome – 7
Trisomy 21- 1
Anorectal malformation – 2
Renal anomalies- 2
CTEV-1
Cleft palate – 1
Annular pancreas - 1
Associated cardiac and extra cardiac anomalies:
14 children had bilateral SVC. Other common cardiac anomalies seen in
children with VSD pulmonary atresia were situs ambiguous (n=5), partial
anomalous pulmonary venous connection (n=4), branch PA stenosis (n=3),
corrected transposition (n=3) and retro aortic innominate vein (n=3). Other less

23
common anomalies seen in the cohort were criss cross heart, interrupted inferior
vena cava and aberrant left subclavian artery, Seven children were antenatally
diagnosed to have pulmonary atresia.
Di George syndrome was the commonest syndrome observed in this cohort
(n=7). 22 q11 deletion was proven genetically in only in 4 patients, the others had
facies characteristic of the syndrome with or without hypocalcemia and stigmata of
immunodeficiency. Trisomy 21 was the only chromosomal aneuploidy seen in this
study. Other associated extra cardiac anomalies were renal, anorectal or gut
related. Cleft palate and congenital talipes equinovarus were each seen in 1 patient.
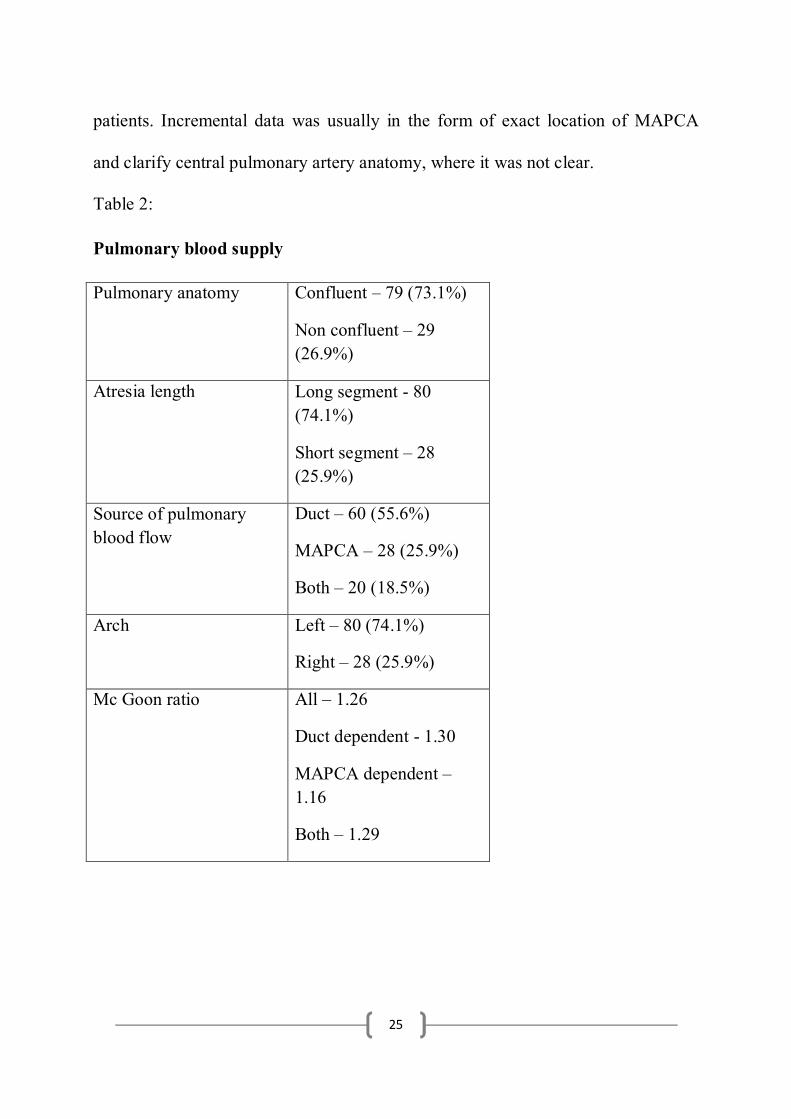
Pulmonary blood flow and anatomy:
73.1% (n=79) of the infants had confluent pulmonary arterial anatomy,
whereas 26.9% had non-confluent anatomy. 80 infants (74.1%) had long segment
pulmonary atresia and 28 of them had short segment pulmonary atresia. 36 of the
53 (66%) female infants had confluent pulmonary arterial anatomy and 41 of the
53 (77.3%) had long segment pulmonary atresia, thereby making confluent long
segment atresia the predominant phenotype in the female cohort. 43 of the 55
(78.2%) male infants had confluent anatomy and 39 of the 55 (70.9%) male infants
had long segment pulmonary atresia.

24
29 of the 55 male infants (52.8%) had duct dependent pulmonary circulation, 16
(29%) had collateral dependent pulmonary circulation and 10 (18.2%) had
evidence of both. 31 of the 53 female infants (58.4%) had duct dependent
pulmonary circulation, 12 (22.7%) had collateral dependent circulation and 10
(18.9%) had evidence of both. The mean McGoon index in the male cohort was
1.21 and in the female cohort was 1.26. The mean McGoon index in the children
with duct dependent circulation was 1.30, 1.16 in MAPCA dependent group and
was 1.29 in the presence of both.
80 of the 108 infants (74%) had left sided Aortic arch, whereas 28 (26%)
had right arch. Among the patients with collaterals the prevalence of right arch was
41.7%. In the presence of collateral dependent pulmonary circulation (MAPCAs as
the only source of pulmonary blood flow) 71.4% of the infants had right arch.
There were 7 infants diagnosed with DiGeorge syndrome in the study, of which 5
(71.4%) had right arch.
Pulmonary anatomy was assessed by echocardiography and when there was
ambiguity, imaging by CT/ MRI was performed. Most patients with MAPCA
dependent circulation required an additional modality to delineate the origin of
MAPCAs. Imaging in the form of CT was done in 26 children and MRI was
performed in 30 children. Imaging provided incremental data over echo in 33
25
patients. Incremental data was usually in the form of exact location of MAPCA
and clarify central pulmonary artery anatomy, where it was not clear.
Table 2:
Pulmonary blood supply
Pulmonary anatomy Confluent – 79 (73.1%)
Non confluent – 29
(26.9%)
Atresia length Long segment - 80
(74.1%)
Short segment – 28
(25.9%)
Source of pulmonary
blood flow
Duct – 60 (55.6%)
MAPCA – 28 (25.9%)
Both – 20 (18.5%)
Arch Left – 80 (74.1%)
Right – 28 (25.9%)
Mc Goon ratio
All – 1.26
Duct dependent - 1.30
MAPCA dependent –
1.16
Both – 1.29

26
Procedure data:
51 (47.2%) of the 108 patients underwent some procedure – palliative,
corrective or both. Initial palliation was with ductal stenting in 6 patients, with
Aorto-pulmonary shunt in 37 patients and with Cavopulmonary shunt (BDG) in 1
patient. BDG was performed in children after initial palliation in 3 children. Out of
children in the duct dependent pulmonary circulation group, 10 proceeded to
corrective surgery. Whereas in the MAPCA group, 3 children proceeded to
corrective surgery in the form of unifocalisation. Among the 6 children, who
underwent ductal stenting, 2 underwent BDG and 1 underwent total correction.
Among the children, who underwent Aorto-pulmonary shunt, 1 underwent BDG
and 5 underwent total correction. Of the 108 children, who were enrolled in the
study, only 13 underwent total correction, which is in fact the goal of treatment in
every patient. 10 children were kept on medical follow up and any form of surgical
treatment was deferred in them, in view of poor pulmonary anatomy.

27
Fig 2
Study flow
Among the children, who underwent ductal stenting, the commonest stent
size was 3.5 (n=5) and one child received a size 4 mm bare metal stent. None of
them required more than one stent. There were no peri procedural complications in
this subgroup. Left axillary access was the commonest vascular access chosen in
lieu of the ductal anatomy.
Aorto-pulmonary shunt surgery was performed in 37 patients. The commonest
shunt size used was 3.5 mm (n=18), followed by 3 mm (n=12) and 4 mm shunt
was used 7 children only. Blockage of Aorto-pulmonary shunt was seen on follow
VSD-PA
N = 108
INTERVENED
N = 51
PALLIATION
N = 44
ICR
N = 13
PRIMARY ICR
N = 7
ICR AFTER PALLIATION
N= 6
NOT INTERVENED
N = 56
PLANNED FOR
SURGERY
N = 45
SURGERY DEFFERED -DUE TO UNFAVORABLE
ANATOMY
N = 11

28
up in 3 patients, and among them 1 had an acute shunt blockage requiring revision
on the first post0operative day.
Primary ICR was performed in 7 children whereas it was done after an initial
palliation in 6 patients (1 with prior ductal stenting and 5 with prior Aorto-
pulmonary shunt). Unifocalisation was performed in 3 patients, concomitant
branch PA plasty was required in 9, homograft was placed in 4 and VSD patch
fenestration was required in 2 patients.
Surgical site infection (n=3) and ventilator associated pneumonia (n=3) were
the commonest post-operative complications, among all patients who underwent
surgery.
Table 3:
Procedure data
N = 51
Age at procedure (Mean +/ SD) 5.92 +/- 9.84 months
Average duration of “T1” period
(Mean +/ SD)
4.81 +/- 9.06 months
Ductal stenting N = 6
3.5 mm stent – 5
4 mm stent – 1
Left axillary access – 5
Right femoral access - 1

29
BT shunt N = 37
3 mm shunt- 12
3.5 mm shunt – 18
4 mm shunt - 7
Primary BDG 1
Time to ICR (Mean +/ SD) 15.68 +/- 14.98 months
Primary ICR 7
BDG after initial palliation 3
ICR after initial palliation 6
Concomitant procedure Unifocalisation – 3
Homograft – 4
Branch PA plasty – 9
VSD fenestration - 2
Post procedure complications Blocked Aorto-pulmonary shunt – 3
Branch PA stenosis – 3
Surgical site infection – 3
VAP -3
RV hypertension – 2
Severe TR - 1

30
Mortality:
There were a total of 32 deaths in the entire study population, of which 17
were in the duct dependent group, 8 were in the MAPCA dependent group and 7 in
the group with dual pulmonary blood supply. Of the children who died, 16 were
male and 16 were female. Number of deaths before 3 months of age was 10 and 22
died after 3 months of age.
Fig 3
Flow chart showing survival proportions in subgroups based on pulmonary
anatomy
VSD-PA
N= 108
Duct group
N = 60
Survived
N= 43 (71.7%)
Died
N= 17 (28.3%)
MAPCA group
N = 28
Survived
N= 20 (71.5%)
Died
N= 8 (28.5%)
Both group
N = 20
Survived
N= 13 (65%)
Died
N= 7 (35%)

31
Death prior to any procedure referred to in this study as “ T 1” death
numbered 19, 2 in the “ T 2 “ period , defined as period between palliation and
corrective procedures numbered , in the perioperative period ( within 2 weeks of
surgery or till hospital discharge ) referred to as “ T3 “ numbered 10 and late
deaths numbered 1 and was regarded as “T 4” deaths. T1 period had the maximum
number of deaths. Causes of post-operative deaths were as follows- ventricular
dysfunction in 6, RV hypertension in 2 and sepsis in 2.
Table 4:
Mortality data
N = 32
Age at death (Mean +/ SD) 6.25 +/- 7.73 months
Gender Male – 16 (50%)
Female – 16 (50%)
Pulmonary anatomy Duct dependent – 17 (53.1%)
MAPCA dependent – 8 (25%)
Both – 7 (21.9%)
T1 (prior to any palliation/ corrective
surgery)
19 (59.3 %)
T2 (post palliation, prior to corrective
surgery)
2 (6.2 %)

32
T3 (peri operative) 10 (31.2%)
T4 ( late, after corrective surgery) 1(3.1%)
10 children died in the perioperative period, defined as any death prior to
discharge from hospitalization for an interventional procedure. There were no
deaths following ductal stenting. Among the children, who underwent Aorto-
pulmonary shunts, 6 died and the main cause of mortality was ventricular
dysfunction. Most of the children underwent the above procedure as an emergency
following a spell or worsening cyanosis and hence same could be incriminated to
be the cause of the ventricular dysfunction. The cause of deaths post
unifocalisation was suprasystemic right ventricular pressure, reflecting the inability
the ventricle to handle high pulmonary artery pressure, as a consequence of poorly
developed pulmonary arteries, poor arborisation or onset of pulmonary vascular
obstructive disease (PVOD) prior to the procedure in case of hypertensive MAPCA
s. Post complete repair, 1 child died of sepsis and another because of severe RV
dysfunction.

33
Fig 4
Distribution of mortality in the various time intervals
Table 5
Post procedure mortality data:
N=10
Post ductal stenting 0
Post Aorto-pulmonary shunt 6
Ventricular dysfunction – 5
Sepsis- 1
Post BDG 0
Post Unifocalisation + ICR
N = 2
2 ( 66.67%)
Suprasytemic RV pressure - 2
Post ICR 2 (15.38%)
60% 6%
31%
3%
Mortality data of the entire cohort T1
T2
T3
T4

34
N = 13 RV dysfunction – 1
Sepsis, MODS- 1
Among the 44 children, who underwent intervention in the duct dependent
group, 79.5% survived, whereas in the “no intervention” arm only 50% survived.
In the MAPCA dependent group, only one child underwent intervention, in the
form of unifocalisation and that child died, translating into a mortality of 100%.
Despite not undergoing intervention 74.1% of the children in MAPCA dependent
group survived. In the group with dual pulmonary blood supply, 30 % underwent
intervention, of which 50% survived. Among the children with no intervention in
the same group, 71.4% survived.
Figure 5
Flow chart showing survival of various subgroups based on intervention
Total no. of children
n = 108
Duct dependent
n = 60
Intervention
n = 44 (73.3%)
Alive
n = 35 (79.5%)
Died
n = 9(20.5%)
No intervention
n = 16 (26.7%)
Alive
n = 8(50%)
Died
n = 8(50%)
MAPCA dependent
n = 28
intervention
n = 1 (3.5%)
Alive
n = 0
Died
n = 1(100%)
No intervention
n = 27(96.5%)
Alive
n = 20(74.1%)
Died
n = 7(25.9%)
Both
n = 20
Intervention
n =6 ( 30%)
Alive
n = 3 (50%)
Died
n = 3(50%)
No intervention
n = 14 (70%)
Alive
n = 10(71.4%)
Died
n = 4(28.6%)

35
Predictors of mortality:
Analysis was done to assess predictors of mortality. Only length of
pulmonary atresia (long segment) and prematurity were predictive of survival
among the various parameters analysed, with a “p” value of .011 and 0.041
respectively. The other variable nearing significance statistically was prostaglandin
use (p=0.075). Other variables like age at diagnosis, source of pulmonary blood
flow, Mc Goon index, intervention and association with a syndrome were not
predictive of mortality.
Table 6:
Predictors of mortality– univariate analysis
Death
Total χ2 df p Parameter
No Yes
N % N % N %
Gender Male 39 51.3 16 50 55 50.9 0.016 1 0.901 Female 37 48.7 16 50 53 49.1 PA anatomy
Confluent
57
75
22
68.8
79
73.1
0.448
1
0.503
Non confluent 19 25 10 31.3 29 26.9 Long 51 67.1 29 90.6 80 74.1 6.486 1 0.011 Short 25 32.9 3 9.4 28 25.9 Source of PBF
PDA
43
56.6
17
53.1
60
55.6
0.34
2
0.844
MAPCA 20 26.3 8 25 28 25.9 Both 13 17.1 7 21.9 20 18.5 Arch
Right
21
27.6
7
21.9
28
25.9
0.389
1
0.533
Left 55 72.4 25 78.1 80 74.1

36
Prematurity
6
7.9
7
21.9
13
12
4.157 1
0.041
Prostaglandin usage
20 26.3 14 43.8 34 31.5 3.173 1 0.075
Any procedure
38 50 13 40.6 51 47.2 0.794 1 0.373
Palliative procedure
BTS 27 81.8 10 100 37 86 2.113 1 0.146
PDA 6 18.2 0 0 6 14 Intervention vs No intervention
Complete repair 9 11.8 4 12.5 13 12 1.027 2 0.598
Palliation 29 38.2 9 28.1 38 35.2 No intervention 38 50 19 59.4 57 52.8 Mc Goon ratio
<1.2 19 25 9 28.1 28 25.9 0.115 1 0.735
=>1.2 57 75 23 71.9 80 74.1 Age at surgery
<=10 32 84.2 11 84.6 43 84.3 0.001 1 0.972
>10 6 15.8 2 15.4 8 15.7 Associated Syndrome
4 5.3 5 15.6 9 8.3 3.165 1 0.075
In multi variate analysis only long segment pulmonary atresia remained a
significant predictor of death (p = 0.023).

37
Table 7:
Binary logistic regression model for outcome of death as dependent variable
B S.E. Wald df p
Long / short segment
atresia -1.515 .665 5.191 1 .023
Prematurity 1.102 .649 2.888 1 .089
Prostaglandin usage .689 .482 2.044 1 .153
Associated syndrome 1.127 .793 2.018 1 .155
Survival analysis:
Kaplan Meier curves were plotted for the entire cohort and also for various
subgroups like and are shown below. One year survival for the entire cohort was
70.3%, at 2 years it was 68.2% and was 65% at 4 years.

38
Fig 6:
Kaplan Meier curve for survival for the entire population
One year survival for children with duct dependent pulmonary circulation
was 70.9%, in MAPCA dependent circulation was 68.1% and was 67.8% in the
group having both duct and MAPCAs.

39
Fig 7:
Kaplan Meier curve for survival based on source of pulmonary blood flow
Figure showing years of follow up in the x axis and and cumulative survival in the y axis
One year survival in children who underwent Aorto-pulmonary shunt as the
initial palliation was 72.5%, 75.9% in those undergoing any form of palliation

40
(surgical or interventional),76.9 % in those undergoing ICR and 64.6 % in those
not undergoing any form of intervention.
Fig 8:
Survival curves depending on intervention performed

41
0 10 20 30 40 50 60 70 80
Entire cohort
Non MAPCA group
MAPCA group
Complete repair
Palliation group
No intervention group
70
71
68
79
77
65
Key findings - 1 Year survival

42
DISCUSSION:
Both old ((33) (34)) and contemporary (21) series show at MAPCA
dependent circulation occurs in about 25 – 31%. This is in line with our series
where the MAPCA dependent group constituted 25.9 % (n=28)
Data from our study suggests that about three fourths (3/4) of the duct
dependent population required some form of intervention and once intervened,
survival was good, around 80%. On the contrary, in the MAPCA dependent group,
majority of them survived without any intervention. In the mixed group, survival
with intervention was around 50% and without any intervention was 71%. Duct
dependent cohort is the subgroup which needed intervention in infancy to alleviate
worsening cyanosis and the other two subgroups were less vulnerable, though
MAPCA group was better in terms of survival without any intervention, re-
iterating the fact that MAPCA s were a more stable source of pulmonary blood
flow in an infant. The fact that many in the MAPCA group (n= 9) were planned for
an unifocalisation and still had not undergone the same during the period in which,
they were followed up in this study should be borne in mind. In our study,
mortality rate after ICR is 15.38% whereas 2 of the 3 children who underwent
unifocalisation with ICR is 66.67%.

43
As early as 1978, survival data were published for VSD-PA, with necropsy
data and it was published that survival without surgery was 50% at 1 year and 8%
at 10 years ((35)).
One year survival in our series is 70.3 %. This conforms with other studies,
which also show a similar high rate of attrition in the first year of life. 47.2%
children underwent some form of intervention, whereas 39.8% underwent initial
palliation with ductal stenting or Aorto-pulmonary shunt. Primary ICR rate was 6.4
% (7 among 108 patients). Only 13.6 % of patients, who underwent initial
palliation underwent corrective surgery on follow up of 18 months. Children who
underwent initial palliation (ductal stenting/ Aorto-pulmonary shunt), on an
average underwent corrective surgery in mean period of 15.29 months. On the
other hand, children without any palliation underwent corrective surgery in 16.02
months after their initial diagnosis. The average time to initial palliation was 2.71
months.
In a landmark paper by Amark et al, evaluating the factors associated with
mortality and predicting complete repair, 220 children with ventricular septal
defect and pulmonary atresia, diagnosed over a period of 30 years were followed
up. In their series, definitive repair was attained in 75%. Systemic pulmonary
artery shunt was done in 57%, complete primary repair was done in 31% and RV
ventricular outflow reconstruction in 12 %. After initial palliative shunt, 68.5% of

44
patients underwent complete repair. Risk factors for death after initial operation
included younger age at repair, earlier birth cohort, fewer broncho-pulmonary
segments supplied by native pulmonary arteries, and placement of an Aorto-
pulmonary shunt. 10 - year survival rate in this series after initial surgery was 71%.
Only 15.9% of children in their series did not undergo any form of intervention.
Nearly half of them (n=105) underwent an initial Aorto-pulmonary shunt. They
also found that higher rates of initial complete repair occurred in later birth cohorts
as opposed to earlier ones. 38 children underwent 47 re-operations and 56 patients
underwent 104 catheter based interventions. Catheter based interventions were
more common in the MAPCA group and were done sooner after complete repair at
a median gap of 1.1 years after surgery.
Mortality in the MAPCA group in this series was 39.6%, 55.56 % without
intervention, 33.33 % after unifocalisation and 30.43% in the initially palliated
group.
The above data suggests that though rates of initial palliation with Aorto-
pulmonary shunts in our series is similar to the western data, rates of primary
repair and corrective surgery after initial palliation were much lesser in our cohort,
though our follow up was relatively short term and many children were awaiting
surgical procedures. In our study, we could not find strong predictors of mortality
other than length of pulmonary atresia and prematurity. Since our complete repair

45
rates were low, no meaningful survival analysis of this subgroup was done. No
child in our study, underwent a repeat surgical procedure or catheter based
intervention primarily because of low ICR rates and secondarily due to shorter
follow up compared to historical cohorts. Whereas the population in the series by
Amark et al, represents the modified natural history of VSD-PA; Our study was a
combination of both unmodified and modified natural history in view of lower
rates of complete repair.
In a French study (36), 52 patients with VSD-PA were followed up over a
period of 8.6 years, 26 (50%) were duct dependent and the rest were MAPCA
dependent. Corrective surgery was performed in 14 (53.8%) in the former group
and 9 (34.6%) in the latter group. After a mean follow up of 8.6 years, 71% were
alive, 39 % were corrected and 11.5% were deemed inoperable.
When MAPCA dependent and MAPCA + PDA group are pooled together in
our study, they constitute about 44.5% of the study population, which is similar to
the above population. 18.7% of children the mixed group as described above were
deemed unsuitable for any form of repair in view of poor pulmonary anatomy.
Rates of cardiac catheterization are very low in our study – only 2 patients had
undergone cardiac catheterisation for delineation of MAPCAs. This may be due to
improvement in echo techniques and deployment of additional modalities of
imaging, when clarity was not achieved with echocardiography alone.

46
Though there is debate regarding the approach to managing children with
VSD-PA and MAPCAs, with some groups advocating single stage repair with
unifocalisation and some advocating staged repair with initial Aorto-pulmonary or
RV-PA conduit to promote the growth of PAs, we have only attempted 3 cases of
unifocalisation with intracardiac repair in a single stage procedure. Two of the 3
children aged 7 and 37 months, where unifocalisation with ICR was done died due
to suprasystemic RV pressures despite fenestration.
Among the patients, who died in the T1 period, 10 had sudden death. In 4
children, there is history cyanosis/ spell. Two died of lower respiratory tract
infections and 2 died due to issues related to prematurity. In the T2 period, one
died of lower respiratory tract infection and the other death was sudden.
In another surgical series from India, Murthy et al ((37)) have reported
outcomes of single stage unifocalisation in 124 patients over 13 years. Of these
patients during the initial procedure, 60% underwent complete repair, 21%
underwent only RV-PA conduit and the VSD was left behind whereas in the
remaining 19% only Aorto-pulmonary shunt was accomplished. There were 16
early deaths and 3 late deaths in this series amounting to a total mortality of 15.3%.
The authors concluded that single-stage unifocalization performed at an early age
achieves normalization of physiology with correction of cyanosis or pulmonary
hypertension and attendant complications. Early single-stage unifocalization

47
decreases the number of subsequent surgeries and hospitalizations, and thereby, is
cost effective also compared to a multi staged procedure.
The deaths post single staged unifocalisation with intra cardiac repair in our
series was due to RV hypertension and mandated fenestration of the VSD patch. It
is difficult to conclude at this point in time if a staged unifocalisation would have
been a better option versus an earlier rehabilitation of pulmonary arteries.

48
CONCLUSION:
Survival in a child with VSD-pulmonary atresia in our setting can be
estimated to be around 70% at 12 months
Duct dependent infants are more vulnerable to worsening hypoxemia and
warrant palliative intervention in early infancy
Death most commonly occurs in the ‘T1’ period, prior to any palliation
Only 12% of patients in our study proceeded to complete repair
Only length of pulmonary atresia was a predictor of mortality
Meticulous monitoring of saturation at nearby points of health care may lead
to prompt referral and improve mortality data as majority of deaths happen
before any palliation

49
LIMITATIONS:
The current study is a retrospective analysis of data, some patients were lost
to follow up and hence follow up data are limited to the last hospital visit or
telephonic enquiry.
The average duration of follow up was 18.9 months, which only indicates
the intermediate term follow up of these patients. Long term data remain elusive.
Since it is a medium term follow up, true data of intervention on branch pulmonary
arteries is not represented. Many patients were awaiting unifocalisation or ICR at
the end of study, hence percentage of children undergoing procedure may be an
under-estimate. Longer follow up would give us the true numerator of percentage
of children proceeding to complete repair.

50
REFERENCE:
1. Ferencz C, Rubin JD, McCarter RJ, Brenner JI, Neill CA, Perry LW, et al. Congenital heart disease: prevalence at livebirth. The Baltimore-Washington Infant Study. Am J Epidemiol. 1985 Jan;121(1):31–6.
2. Tchervenkov CI, Roy N. Congenital Heart Surgery Nomenclature and Database Project: pulmonary atresia--ventricular septal defect. Ann Thorac Surg. 2000 Apr;69(4 Suppl):S97-105.
3. Bull K, Somerville J, Ty E, Spiegelhalter D. Presentation and attrition in complex pulmonary atresia. J Am Coll Cardiol. 1995 Feb;25(2):491–9.
4. Acherman RJ, Smallhorn JF, Freedom RM. Echocardiographic assessment of pulmonary blood supply in patients with pulmonary atresia and ventricular septal defect. J Am Coll Cardiol. 1996 Nov 1;28(5):1308–13.
5. Piehler JM, Danielson GK, McGoon DC, Wallace RB, Fulton RE, Mair DD. Management of pulmonary atresia with ventricular septal defect and hypoplastic pulmonary arteries by right ventricular outflow construction. J Thorac Cardiovasc Surg. 1980 Oct;80(4):552–67.
6. Blackstone EH, Kirklin JW, Bertranou EG, Labrosse CJ, Soto B, Bargeron LM. Preoperative prediction from cineangiograms of postrepair right ventricular pressure in tetralogy of Fallot. J Thorac Cardiovasc Surg. 1979 Oct;78(4):542–52.
7. Nakata S, Imai Y, Takanashi Y, Kurosawa H, Tezuka K, Nakazawa M, et al. A new method for the quantitative standardization of cross-sectional areas of the pulmonary arteries in congenital heart diseases with decreased pulmonary blood flow. J Thorac Cardiovasc Surg. 1984 Oct;88(4):610–9.
8. Anonymou Castaneda AR, Jonas RA, Mayer JE, Hanley FL. Cardiac surgery of the neonate and infant. Philadelphia: W.B.Saunders Company; 1994. pp 215-234.
9. Hofbeck M, Sunnegårdh JT, Burrows PE, Moes CA, Lightfoot N, Williams WG, et al. Analysis of survival in patients with pulmonic valve atresia and ventricular septal defect. Am J Cardiol. 1991 Apr 1;67(8):737–43.
10. Warnes CA. Tetralogy of Fallot and pulmonary atresia/ventricular septal defect. Cardiol Clin. 1993 Nov;11(4):643–50.

51
11. Report of the New England Regional Infant Cardiac Program. Pediatrics. 1980 Feb;65(2 Pt 2):375–461.
12. Samánek M, Vorísková M. Congenital heart disease among 815,569 children born between 1980 and 1990 and their 15-year survival: a prospective Bohemia survival study. Pediatr Cardiol. 1999 Dec;20(6):411–7.
13. Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997 Oct;34(10):798–804.
14. Momma K, Kondo C, Matsuoka R, Takao A. Cardiac anomalies associated with a chromosome 22q11 deletion in patients with conotruncal anomaly face syndrome. Am J Cardiol. 1996 Sep 1;78(5):591–4.
15. Mahle WT, Crisalli J, Coleman K, Campbell RM, Tam VKH, Vincent RN, et al. Deletion of chromosome 22q11.2 and outcome in patients with pulmonary atresia and ventricular septal defect. Ann Thorac Surg. 2003 Aug;76(2):567–71.
16. Liao PK, Edwards WD, Julsrud PR, Puga FJ, Danielson GK, Feldt RH. Pulmonary blood supply in patients with pulmonary atresia and ventricular septal defect. J Am Coll Cardiol. 1985 Dec;6(6):1343–50.
17. Acherman RJ, Smallhorn JF, Freedom RM. Echocardiographic assessment of pulmonary blood supply in patients with pulmonary atresia and ventricular septal defect. J Am Coll Cardiol. 1996 Nov 1;28(5):1308–13.
18. Yin L, Lu B, Han L, Wu R-Z, Johnson L, Xu Z-Y, et al. Quantitative analysis of pulmonary artery and pulmonary collaterals in preoperative patients with pulmonary artery atresia using dual-source computed tomography. Eur J Radiol. 2011 Sep;79(3):480–5.
19. Srinivas B, Patnaik AN, Rao DS. Gadolinium-enhanced three-dimensional magnetic resonance angiographic assessment of the pulmonary artery anatomy in cyanotic congenital heart disease with pulmonary stenosis or atresia: comparison with cineangiography. Pediatr Cardiol. 2011 Aug;32(6):737–42.
20. Leonard H, Derrick G, O’Sullivan J, Wren C. Natural and unnatural history of pulmonary atresia. Heart Br Card Soc. 2000 Nov;84(5):499–503.
21. Amark KM, Karamlou T, O’Carroll A, MacDonald C, Freedom RM, Yoo S-J, et al. Independent factors associated with mortality, reintervention, and

52
achievement of complete repair in children with pulmonary atresia with ventricular septal defect. J Am Coll Cardiol. 2006 Apr 4;47(7):1448–56.
22. Alsoufi B, Mori M, McCracken C, Williams E, Samai C, Kogon B, et al. Results of Primary Repair Versus Shunt Palliation in Ductal Dependent Infants With Pulmonary Atresia and Ventricular Septal Defect. Ann Thorac Surg. 2015 Aug;100(2):639–46.
23. Iyer KS, Mee RB. Staged repair of pulmonary atresia with ventricular septal defect and major systemic to pulmonary artery collaterals. Ann Thorac Surg. 1991 Jan;51(1):65–72.
24. Reddy VM, McElhinney DB, Amin Z, Moore P, Parry AJ, Teitel DF, et al. Early and intermediate outcomes after repair of pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries: experience with 85 patients. Circulation. 2000 Apr 18;101(15):1826–32.
25. Kaskinen AK, Happonen J-M, Mattila IP, Pitkänen OM. Long-term outcome after treatment of pulmonary atresia with ventricular septal defect: nationwide study of 109 patients born in 1970-2007. Eur J Cardio-Thorac Surg Off J Eur Assoc Cardio-Thorac Surg. 2016 May;49(5):1411–8.
26. Shimazaki Y, Tokuan Y, Lio M, Nakano S, Matsuda H, Blackstone EH, et al. Pulmonary artery pressure and resistance late after repair of tetralogy of Fallot with pulmonary atresia. J Thorac Cardiovasc Surg. 1990 Sep;100(3):425–40.
27. Liava’a M, Brizard CP, Konstantinov IE, Robertson T, Cheung MM, Weintraub R, et al. Pulmonary atresia, ventricular septal defect, and major aortopulmonary collaterals: neonatal pulmonary artery rehabilitation without unifocalization. Ann Thorac Surg. 2012 Jan;93(1):185–91.
28. Malhotra SP, Hanley FL. Surgical management of pulmonary atresia with ventricular septal defect and major aortopulmonary collaterals: a protocol-based approach. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2009;145–51.
29. Puga FJ, Leoni FE, Julsrud PR, Mair DD. Complete repair of pulmonary atresia, ventricular septal defect, and severe peripheral arborization abnormalities of the central pulmonary arteries. Experience with preliminary unifocalization procedures in 38 patients. J Thorac Cardiovasc Surg. 1989 Dec;98(6):1018-1028-1029.

53
30. Metras D, Chetaille P, Kreitmann B, Fraisse A, Ghez O, Riberi A. Pulmonary atresia with ventricular septal defect, extremely hypoplastic pulmonary arteries, major aorto-pulmonary collaterals. Eur J Cardio-Thorac Surg Off J Eur Assoc Cardio-Thorac Surg. 2001 Sep;20(3):590-596-597.
31. Sullivan ID, Wren C, Stark J, de Leval MR, Macartney FJ, Deanfield JE. Surgical unifocalization in pulmonary atresia and ventricular septal defect. A realistic goal? Circulation. 1988 Nov;78(5 Pt 2):III5-13.
32. Sawatari K, Imai Y, Kurosawa H, Isomatsu Y, Momma K. Staged operation for pulmonary atresia and ventricular septal defect with major aortopulmonary collateral arteries. New technique for complete unifocalization. J Thorac Cardiovasc Surg. 1989 Nov;98(5 Pt 1):738–50.
33. Jefferson K, Rees S, Somerville J. Systemic arterial supply to the lungs in pulmonary atresia and its relation to pulmonary artery development. Br Heart J. 1972 Apr;34(4):418–27.
34. Rabinovitch M, Herrera-deLeon V, Castaneda AR, Reid L. Growth and development of the pulmonary vascular bed in patients with tetralogy of Fallot with or without pulmonary atresia. Circulation. 1981 Dec;64(6):1234–49.
35. Bertranou EG, Blackstone EH, Hazelrig JB, Turner ME, Kirklin JW. Life expectancy without surgery in tetralogy of Fallot. Am J Cardiol. 1978 Sep;42(3):458–66.
36. Hadjo A, Jimenez M, Baudet E, Roques X, Laborde N, Srour S, et al. Review of the long-term course of 52 patients with pulmonary atresia and ventricular septal defect. Anatomical and surgical considerations. Eur Heart J. 1995 Nov;16(11):1668–74.
37. Murthy K, Reddy KP, Nagarajan R, Goutami V, Cherian K. Management of ventricular septal defect with pulmonary atresia and major aorto pulmonary collateral arteries: Challenges and controversies. Ann Pediatr Cardiol. 2010;3(2):127–35.

54

55

56

57
Data collection sheet

58

59
Master chart

60
Pliagarism check

61

















![Evaluation of the Adherence to the Discharge Management ... › Cardiology › cardiology-5-1097.pdfattack [TIA], DM, asthma, chronic obstructive pulmonary disease [COPD], peptic ulcer](https://img.pdfslide.net/doc/110x75/60c561f2af4256786a3cb8d8/evaluation-of-the-adherence-to-the-discharge-management-a-cardiology-a-cardiology-5-1097pdf.jpg)










