Embed Size (px)
Citation preview

Protective Effect of Liver Ischemic Preconditioning on Liver andLung Injury Induced by Hepatic Ischemia-Reperfusion in the Rat
CARMEN PERALTA,1 NEUS PRATS,2 CARME XAUS,1 EMILIO GELPı,1 AND JOAN ROSELLO-CATAFAU1
This study evaluates whether preconditioning couldmodulate the injurious effects of tumor necrosis factor (TNF) onliver and lung following hepatic ischemia-reperfusion (I/R) byinhibiting hepatic postischemic TNF release. The inhibitionof hepatic TNF release from Kupffer cells with gadoliniumchloride (GdCl3) previous to ischemia maintained TNF atcontrol levels, attenuating the increases in transaminases,vascular permeability, and edema associated with hepaticI/R injury. TNF addition reverted this beneficial effect, indicat-ing the implication of the TNF released mainly from Kupffercells in hepatic I/R injury. Preconditioning prevented he-patic TNF increases, thus attenuating the liver injury, whileTNF addition abolished the benefits of preconditioning.Inhibition of nitric oxide (NO) synthesis abolished the effect ofpreconditioning, whereas GdCl3 addition avoided the injuriouseffect of NO inhibition. In addition, NO administration beforeI/R offered similar results to those found in precondition-ing, while TNF addition abolished the benefits of NO. Thus,the effect of preconditioning on TNF release after hepaticI/R is mediated by NO. Inhibition of hepatic TNF releasefrom Kupffer cells with GdCl3 prevented both the increasein plasma TNF and the injurious effect in lung seen afterhepatic I/R, and these effects were reverted with TNFaddition. Preconditioning resulting in reduced hepatic TNFlevels prevented the systemic TNF release, thus reducingthe lung damage following hepatic I/R. However, TNFaddition abolished the protective effect of preconditioningon lung injury. These findings indicate that preconditioningattenuates hepatic postischemic TNF release from Kupffercells, thus probably reducing the liver and lung injuryfollowing hepatic I/R, and that this effect of preconditioningis mediated by NO. (HEPATOLOGY 1999;30:1481-1489.)
Ischemia-reperfusion (I/R) injury is one of the mostimportant causes of graft nonfunction and an importantdeterminant of successful liver transplantation. Primary non-
function of the graft after liver transplantation is alsoassociated with the rapid development of pulmonary insuffi-ciency.1,2 Ischemic preconditioning is an endogenous protec-tive mechanism by which repetitive, short periods of is-chemia, separated by short reperfusions, confer a state ofprotection against subsequent sustained I/R injury.3-5 Thisphenomenon, commonly studied in the heart, has beenrecently described in liver.6,7 However, it is not knownwhether preconditioning is effective only locally (liver) or if itwould also be capable of modulating the injury known toresult from hepatic I/R in a distant organ (lung).
A potential explanation for the association of liver I/Rinjury with pulmonary insufficiency involves liver-derivedcytokine release, following hepatic I/R.8,9 In this regard,recent reports have suggested that tumor necrosis factor a(TNF-a) could be potentially responsible both for the delete-rious effects on the hepatocyte and the damaging effects onpulmonary integrity observed after hepatic I/R.8,9 Recently, astudy of the heart indicated that preconditioning was capableof inhibiting TNF release in myocardium after I/R.10 There-fore, the possibility that hepatic preconditioning could in-hibit hepatic TNF release, reducing liver and lung injury afterhepatic I/R, could also be considered.
Although the mechanisms by which preconditioning couldoffer protection remain unknown, it has been suggested thatorgan protection depends on the release of endothelialsubstances, such as nitric oxide (NO).5,11,12 Recent studieshave demonstrated the implication of NO in liver precondi-tioning and the protective effect of this mediator in front ofthe hepatic injury associated to I/R.6,7,13 Along these lines, ithas been shown that inhibition of NO synthesis with N-nitro-L-arginine methylester (NAME), an L-arginine analogue thatcompetitively blocks nitric oxide synthase, abolished theprotective effect of preconditioning, whereas a NO donor orL-arginine administration to I/R simulated the protectiveeffect of preconditioning on hepatic injury.6,7,13 On the otherhand, the mechanisms by which NO protects from hepaticinjury after I/R have not been established. The existence of arelationship between NO generation and TNF production hasbeen previously demonstrated. In this sense, NO was able toinduce the synthesis of TNF in macrophages, and inhibitionof TNF release by NO has been described in an experimentalmodel of renal ischemia.15,16 To our knowledge, the possibil-ity that NO, implicated in ischemic preconditioning, couldmodulate the injury associated to hepatic I/R through itsaction on TNF release remains to be elucidated.
In the present study, we have designed a series of experi-ments to evaluate whether: 1) preconditioning is capable ofmodulating the lung injury that follows hepatic I/R; 2)preconditioning could modulate the injurious effects of TNF
Abbreviations: I/R, ischemia-reperfusion; TNF; tumor necrosis factor; NAME, N-nitro-L-arginine methylester; GdCl3, gadolinium chloride; PC, preconditioning; AST, aspar-tate transaminase; ALT, alanine transaminase; MPO, myeloperoxidase.
From the 1Department of Medical Bioanalysis, Instituto de Investigaciones Biomedi-cas de Barcelona, Consejo Superior de Investigaciones Cientıficas, Barcelona, Spain; and2Department of Animal Pathology, Veterinary School, Universitat Autonoma de Barce-lona, Bellaterra, Spain.
Received May 26, 1999; accepted September 21, 1999.Supported by the Fondo de Investigaciones de la Seguridad Social (FIS) through the
project 97/2076.Address reprint requests to: Dr. Joan Rosello-Catafau, Department of Medical Bioanalysis,
Instituto de investigaciones Biomedicas de Barcelona, CSIC-IDIBAPS, C/Rosellon 161, 6a y 7a
planta 08036-Barcelona, Spain. E-mail: [email protected]; fax: 34-933638301.Copyright r 1999 by the American Association for the Study of Liver Diseases.0270-9139/99/3006-0020$3.00/0
1481

on liver and lung following hepatic I/R by inhibiting thehepatic postischemic TNF release and; 3) the effect ofpreconditioning on the hepatic postischemic TNF release ismediated by NO.
MATERIALS AND METHODS
Surgical Procedure
The study was performed with male Wistar rats (6 for each group)weighing between 250 and 300 g. All animals (including controls)were anesthetized with urethane (10 mg/kg, intraperitoneally) andplaced in a supine position on a heating pad to maintain bodytemperature between 36°C and 37°C. To induce hepatic ischemia,laparotomy was performed, and the blood supply to the right lobe ofthe liver was interrupted by placement of a bulldog clamp at thelevel of the hepatic artery and portal vein. Reflow was initiated byremoving the clamp.6 The studies were performed in concordancewith the European Union regulations for animal experiments (ECguideline 86/609/CEE). All animals were randomized for groupassignment as follows.
Experimental Design
To study whether ischemic preconditioning could modulate therole of TNF on liver and lung injury after hepatic I/R, the followingexperimental groups were set up:
Group 1. Control: animals subjected to anesthesia and lapa-rotomy.
Group 2. I/R: animals subjected to 90 minutes of right-lobehepatic ischemia, followed by 90 minutes of reperfusion.
Group 3. I/R 1 gadolinium chloride (GdCl3): animals subjectedto 90 minutes of ischemia were treated with GdCl3 (10 mg/kg,intravenously) to inactivate Kupffer cells, 48 hours and 24 hoursbefore ischemia.16
Group 4. I/R 1 GdCl3 1 TNF: animals subjected to 90 minutes ofischemia were treated with GdCl3 (10 mg/kg, intravenously) 48hours and 24 hours before ischemia and TNF administration(4 µg/kg, intravenously),17 5 minutes before ischemia.
Group 5. Preconditioning (PC): before the ischemic period (as ingroup 2), animals were subjected to 10 minutes of ischemia and10 minutes of reperfusion.
Group 6. PC 1 TNF: animals subjected to 90 minutes of ischemiawith previous preconditioning (as in group 5) were treated withTNF (4 µg/kg, intravenously) 5 minutes before preconditioning.
To evaluate the effect of NO on the TNF release after hepatic I/R,the following experimental groups were set up:
Group 7. PC 1 NAME: animals subjected to 90 minutes ofischemia with previous preconditioning (as in group 5) were treatedwith L-NAME (10 mg/kg, intravenously)6 5 minutes before precon-ditioning.
Group 8. PC 1 GdCl3 1 NAME): animals subjected to 90minutes of ischemia with previous preconditioning (as in group 5)were treated with GdCl3 (10 mg/kg) 48 hours and 24 hours beforepreconditioning and with L-NAME (10 mg/kg, intravenously) 5minutes before preconditioning.
Group 9. I/R 1 NO donor: animals subjected to 90 minutes ofischemia were treated with the NO-donor spermine NONOate (10mg/kg in PBS [pH 7.4] intravenously)6 10 minutes before ischemia.
Group 10. I/R 1 NO 1 TNF: animals subjected to 90 minutes ofischemia were treated with the NO-donor spermine NONOate (10mg/kg in PBS [pH 7.4] intravenously) and TNF (4 µg/kg, intrave-nously) 10 minutes and 5 minutes, respectively, before ischemia.
Biochemical Determinations
Hepatic Tissue and Plasma TNF Assay. Tissues were homogenized in10 volumes of 50 mmol/L phosphate buffer (pH 6), and aftercentrifugation at 4,000g, the supernatants were frozen at 220°C andsaved for measurement of TNF levels.18 Two milliliters of blood
obtained from the cava vein was centrifuged to obtain plasma, and100-µL aliquots of plasma were saved for measurement of TNFlevels. Hepatic tissue and plasma TNF levels were measured using acommercial kit of Rat Tumor Necrosis Factor-Alpha (ra-TNF-a)Immunoassay (Biosource International, Camarillo, CA).
Hepatic Injury. Evaluation of hepatic injury was performed bydeterminations of aspartate transaminase (AST) and alanine trans-aminase (ALT) in 200-µL samples of plasma (obtained as describedabove) using a commercial kit from Boehringer Mannheim (Munich,Germany).
Liver Permeability Analysis. Vascular permeability was estimatedusing the Evans Blue method.19 Animals received 20 mg/kg of EvansBlue by cava vein injection 15 minutes before killing. The liversamples were added to 10 volumes of deionized formamide andincubated to room temperature for 24 hours. The Evans Blueextracted from tissue was quantitated by spectrophotometric analy-sis and compared the results with standards of known concentra-tions.
Hepatic Edema. After resection, hepatic samples were weighed andthen placed in a drying oven at 55°C until a constant weight wasobtained. In this determination, hepatic edema is represented by anincrease in the wet-to-dry weight ratio.20
Lung Myeloperoxidase Assay. Myeloperoxidase (MPO) has beenused as a marker of pulmonary neutrophil accumulation andactivation.9,21 MPO activity was measured photometrically employ-ing 3,38,5,58-tetramethylbenzidine as a substrate.22 Samples weremacerated with 0.5% hexadecyltrimethylammonium bromide in 50mmol/L phosphate buffer (pH 6). Homogenates were then disruptedfor 30-second sonication and subsequently snap-frozen in dry iceand thawed on three consecutive occasions. Samples were incubatedat 60°C for 2 hours and then spun down at 4,000g for 12 minutes.Supernatants were collected for MPO assay. Enzyme activity wasassessed photometrically. The assay mixture consisted in 20 µLsupernatant, 10 µL tetramethylbenzidine (final concentration, 1.6mmol/L) dissolved in dimethylsulfoxide, and 70 µL H2O2 (finalconcentration, 3.0 mmol/L) diluted in 80 mmol/L phosphate buffer(pH 5.4). An enzyme unit is defined as the amount of enzyme thatproduces an increase of 1 absorbance unit per minute.
Total protein concentration in homogenates was determinedusing a commercial kit (Bio Rad, Munich, Germany).
Histology
Liver and lung samples were taken for histopathology. These werefixed in 10% neutral buffered formalin, embedded in paraplast, and5-µm sections were stained with hematoxylin-eosin according tostandard procedures. Sections were evaluated by light-microscopicexamination.
Statistical Analysis
Data are expressed as means 6 SEM and were statisticallyevaluated by ANOVA, followed by the Student-Newman-Keuls test.An associated probability of P , .05 was considered to be signifi-cant.
RESULTS
TNF Release in Hepatic Tissue and Plasma After Hepatic I/R. Asignificant increase in hepatic TNF levels was found after 90minutes of reperfusion (Fig. 1). To evaluate whether Kupffercells are the main source of this TNF release, the cells wereinactivated with GdCl3. As shown in Fig. 1, GdCl3 treatment(I/R 1 GdCl3) prevented the significant increase in hepaticTNF levels observed after hepatic I/R. Ischemic precondition-ing, induced by 10 minutes of ischemia, followed by 10minutes of reperfusion, before sustained ischemia leads tohepatic TNF values comparable with those found in thecontrol group. The administration of TNF (4 µg/kg), both tothe I/R group pretreated with GdCl3 (I/R 1 GdCl3 1 TNF)
1482 PERALTA ET AL. HEPATOLOGY December 1999
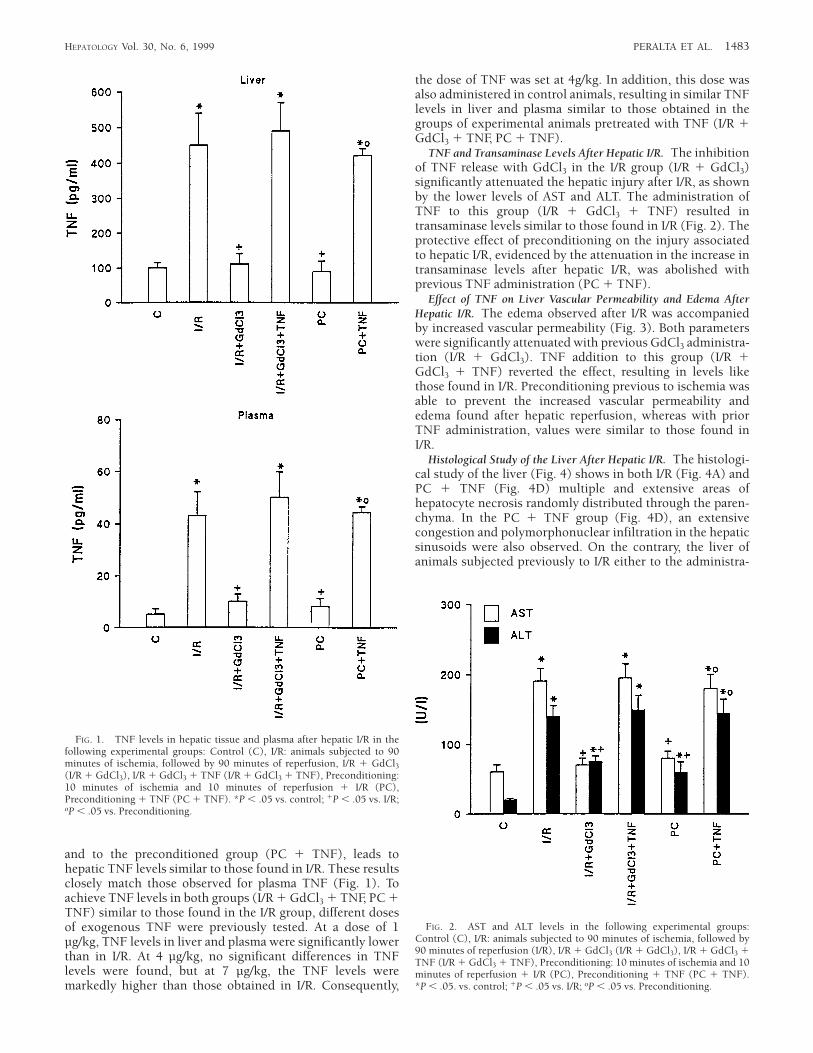
and to the preconditioned group (PC 1 TNF), leads tohepatic TNF levels similar to those found in I/R. These resultsclosely match those observed for plasma TNF (Fig. 1). Toachieve TNF levels in both groups (I/R 1 GdCl3 1 TNF, PC 1TNF) similar to those found in the I/R group, different dosesof exogenous TNF were previously tested. At a dose of 1µg/kg, TNF levels in liver and plasma were significantly lowerthan in I/R. At 4 µg/kg, no significant differences in TNFlevels were found, but at 7 µg/kg, the TNF levels weremarkedly higher than those obtained in I/R. Consequently,
the dose of TNF was set at 4g/kg. In addition, this dose wasalso administered in control animals, resulting in similar TNFlevels in liver and plasma similar to those obtained in thegroups of experimental animals pretreated with TNF (I/R 1GdCl3 1 TNF, PC 1 TNF).
TNF and Transaminase Levels After Hepatic I/R. The inhibitionof TNF release with GdCl3 in the I/R group (I/R 1 GdCl3)significantly attenuated the hepatic injury after I/R, as shownby the lower levels of AST and ALT. The administration ofTNF to this group (I/R 1 GdCl3 1 TNF) resulted intransaminase levels similar to those found in I/R (Fig. 2). Theprotective effect of preconditioning on the injury associatedto hepatic I/R, evidenced by the attenuation in the increase intransaminase levels after hepatic I/R, was abolished withprevious TNF administration (PC 1 TNF).
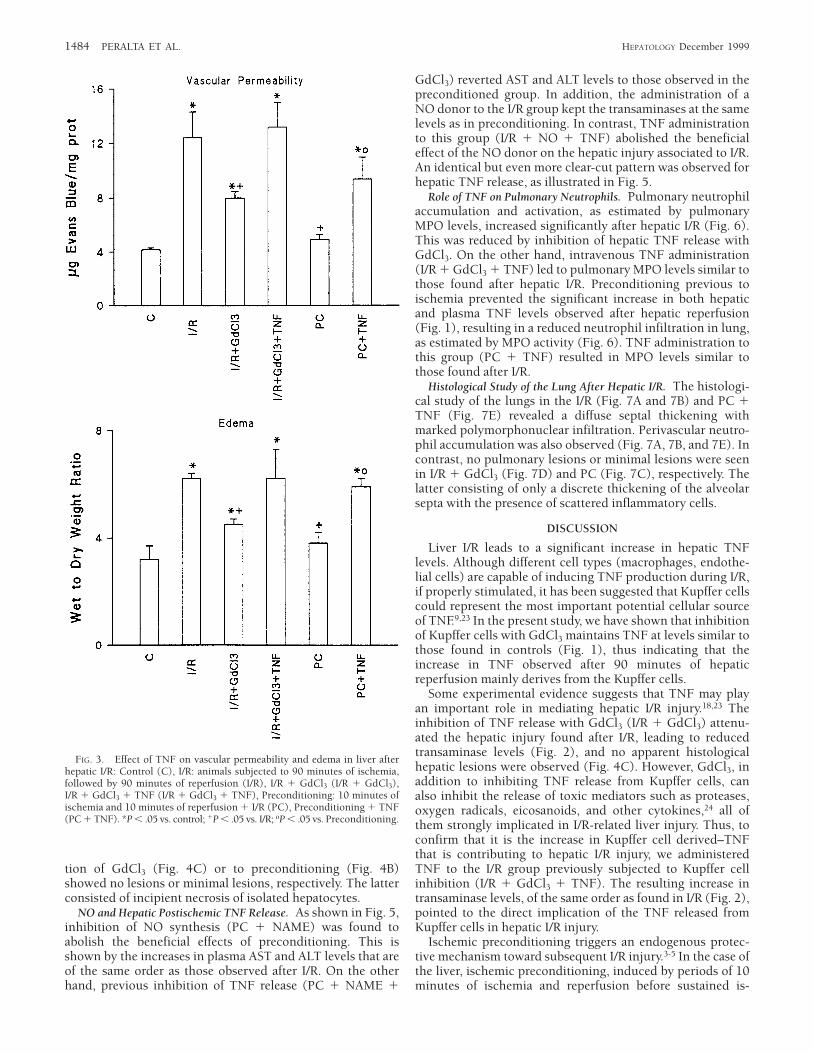
Effect of TNF on Liver Vascular Permeability and Edema AfterHepatic I/R. The edema observed after I/R was accompaniedby increased vascular permeability (Fig. 3). Both parameterswere significantly attenuated with previous GdCl3 administra-tion (I/R 1 GdCl3). TNF addition to this group (I/R 1GdCl3 1 TNF) reverted the effect, resulting in levels likethose found in I/R. Preconditioning previous to ischemia wasable to prevent the increased vascular permeability andedema found after hepatic reperfusion, whereas with priorTNF administration, values were similar to those found inI/R.
Histological Study of the Liver After Hepatic I/R. The histologi-cal study of the liver (Fig. 4) shows in both I/R (Fig. 4A) andPC 1 TNF (Fig. 4D) multiple and extensive areas ofhepatocyte necrosis randomly distributed through the paren-chyma. In the PC 1 TNF group (Fig. 4D), an extensivecongestion and polymorphonuclear infiltration in the hepaticsinusoids were also observed. On the contrary, the liver ofanimals subjected previously to I/R either to the administra-
FIG. 1. TNF levels in hepatic tissue and plasma after hepatic I/R in thefollowing experimental groups: Control (C), I/R: animals subjected to 90minutes of ischemia, followed by 90 minutes of reperfusion, I/R 1 GdCl3(I/R 1 GdCl3), I/R 1 GdCl3 1 TNF (I/R 1 GdCl3 1 TNF), Preconditioning:10 minutes of ischemia and 10 minutes of reperfusion 1 I/R (PC),Preconditioning 1 TNF (PC 1 TNF). *P , .05 vs. control; 1P , .05 vs. I/R;oP , .05 vs. Preconditioning.
FIG. 2. AST and ALT levels in the following experimental groups:Control (C), I/R: animals subjected to 90 minutes of ischemia, followed by90 minutes of reperfusion (I/R), I/R 1 GdCl3 (I/R 1 GdCl3), I/R 1 GdCl3 1TNF (I/R 1 GdCl3 1 TNF), Preconditioning: 10 minutes of ischemia and 10minutes of reperfusion 1 I/R (PC), Preconditioning 1 TNF (PC 1 TNF).*P , .05. vs. control; 1P , .05 vs. I/R; oP , .05 vs. Preconditioning.
HEPATOLOGY Vol. 30, No. 6, 1999 PERALTA ET AL. 1483
tion of GdCl3 (Fig. 4C) or to preconditioning (Fig. 4B)showed no lesions or minimal lesions, respectively. The latterconsisted of incipient necrosis of isolated hepatocytes.
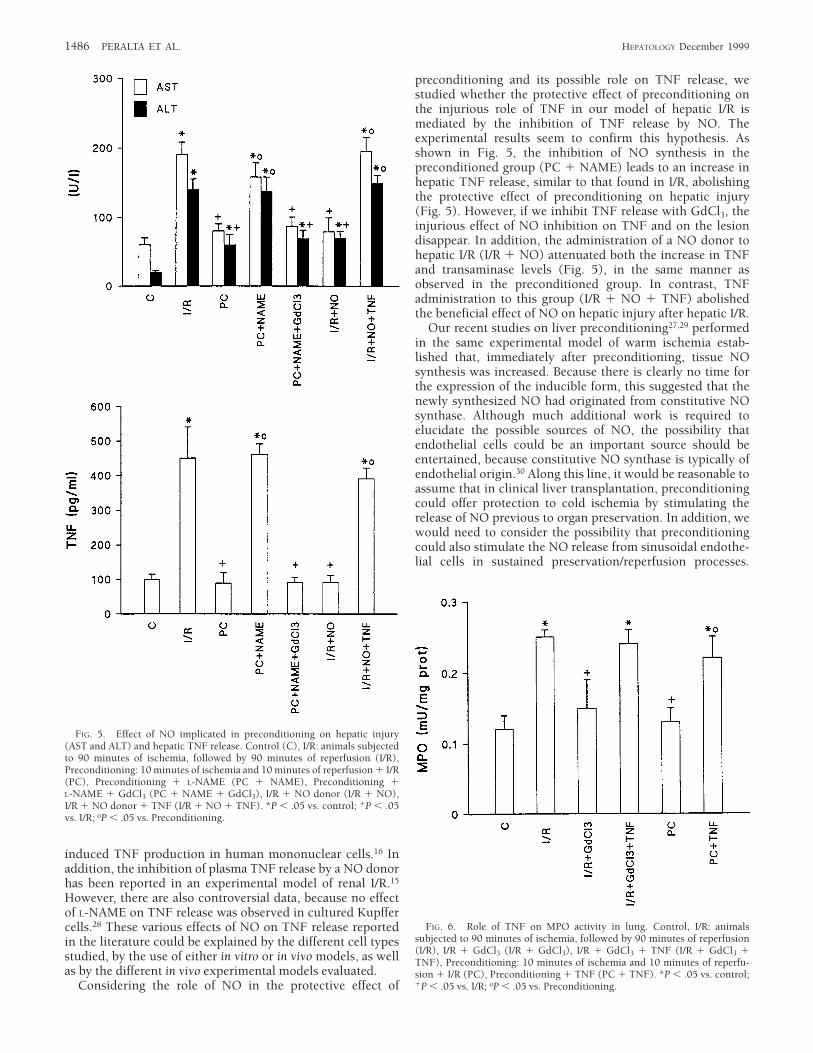
NO and Hepatic Postischemic TNF Release. As shown in Fig. 5,inhibition of NO synthesis (PC 1 NAME) was found toabolish the beneficial effects of preconditioning. This isshown by the increases in plasma AST and ALT levels that areof the same order as those observed after I/R. On the otherhand, previous inhibition of TNF release (PC 1 NAME 1
GdCl3) reverted AST and ALT levels to those observed in thepreconditioned group. In addition, the administration of aNO donor to the I/R group kept the transaminases at the samelevels as in preconditioning. In contrast, TNF administrationto this group (I/R 1 NO 1 TNF) abolished the beneficialeffect of the NO donor on the hepatic injury associated to I/R.An identical but even more clear-cut pattern was observed forhepatic TNF release, as illustrated in Fig. 5.
Role of TNF on Pulmonary Neutrophils. Pulmonary neutrophilaccumulation and activation, as estimated by pulmonaryMPO levels, increased significantly after hepatic I/R (Fig. 6).This was reduced by inhibition of hepatic TNF release withGdCl3. On the other hand, intravenous TNF administration(I/R 1 GdCl3 1 TNF) led to pulmonary MPO levels similar tothose found after hepatic I/R. Preconditioning previous toischemia prevented the significant increase in both hepaticand plasma TNF levels observed after hepatic reperfusion(Fig. 1), resulting in a reduced neutrophil infiltration in lung,as estimated by MPO activity (Fig. 6). TNF administration tothis group (PC 1 TNF) resulted in MPO levels similar tothose found after I/R.
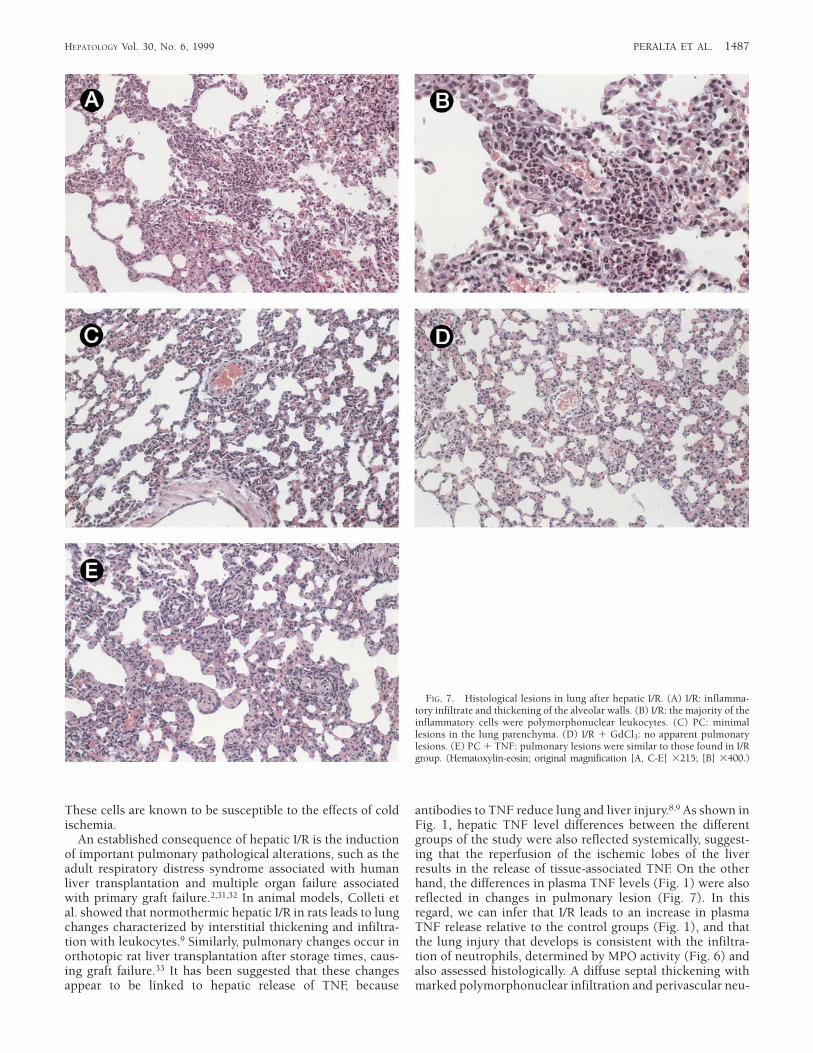
Histological Study of the Lung After Hepatic I/R. The histologi-cal study of the lungs in the I/R (Fig. 7A and 7B) and PC 1TNF (Fig. 7E) revealed a diffuse septal thickening withmarked polymorphonuclear infiltration. Perivascular neutro-phil accumulation was also observed (Fig. 7A, 7B, and 7E). Incontrast, no pulmonary lesions or minimal lesions were seenin I/R 1 GdCl3 (Fig. 7D) and PC (Fig. 7C), respectively. Thelatter consisting of only a discrete thickening of the alveolarsepta with the presence of scattered inflammatory cells.
DISCUSSION
Liver I/R leads to a significant increase in hepatic TNFlevels. Although different cell types (macrophages, endothe-lial cells) are capable of inducing TNF production during I/R,if properly stimulated, it has been suggested that Kupffer cellscould represent the most important potential cellular sourceof TNF.9,23 In the present study, we have shown that inhibitionof Kupffer cells with GdCl3 maintains TNF at levels similar tothose found in controls (Fig. 1), thus indicating that theincrease in TNF observed after 90 minutes of hepaticreperfusion mainly derives from the Kupffer cells.
Some experimental evidence suggests that TNF may playan important role in mediating hepatic I/R injury.18,23 Theinhibition of TNF release with GdCl3 (I/R 1 GdCl3) attenu-ated the hepatic injury found after I/R, leading to reducedtransaminase levels (Fig. 2), and no apparent histologicalhepatic lesions were observed (Fig. 4C). However, GdCl3, inaddition to inhibiting TNF release from Kupffer cells, canalso inhibit the release of toxic mediators such as proteases,oxygen radicals, eicosanoids, and other cytokines,24 all ofthem strongly implicated in I/R-related liver injury. Thus, toconfirm that it is the increase in Kupffer cell derived–TNFthat is contributing to hepatic I/R injury, we administeredTNF to the I/R group previously subjected to Kupffer cellinhibition (I/R 1 GdCl3 1 TNF). The resulting increase intransaminase levels, of the same order as found in I/R (Fig. 2),pointed to the direct implication of the TNF released fromKupffer cells in hepatic I/R injury.
Ischemic preconditioning triggers an endogenous protec-tive mechanism toward subsequent I/R injury.3-5 In the case ofthe liver, ischemic preconditioning, induced by periods of 10minutes of ischemia and reperfusion before sustained is-
FIG. 3. Effect of TNF on vascular permeability and edema in liver afterhepatic I/R: Control (C), I/R: animals subjected to 90 minutes of ischemia,followed by 90 minutes of reperfusion (I/R), I/R 1 GdCl3 (I/R 1 GdCl3),I/R 1 GdCl3 1 TNF (I/R 1 GdCl3 1 TNF), Preconditioning: 10 minutes ofischemia and 10 minutes of reperfusion 1 I/R (PC), Preconditioning 1 TNF(PC 1 TNF). *P , .05 vs. control; 1P , .05 vs. I/R; oP , .05 vs. Preconditioning.
1484 PERALTA ET AL. HEPATOLOGY December 1999
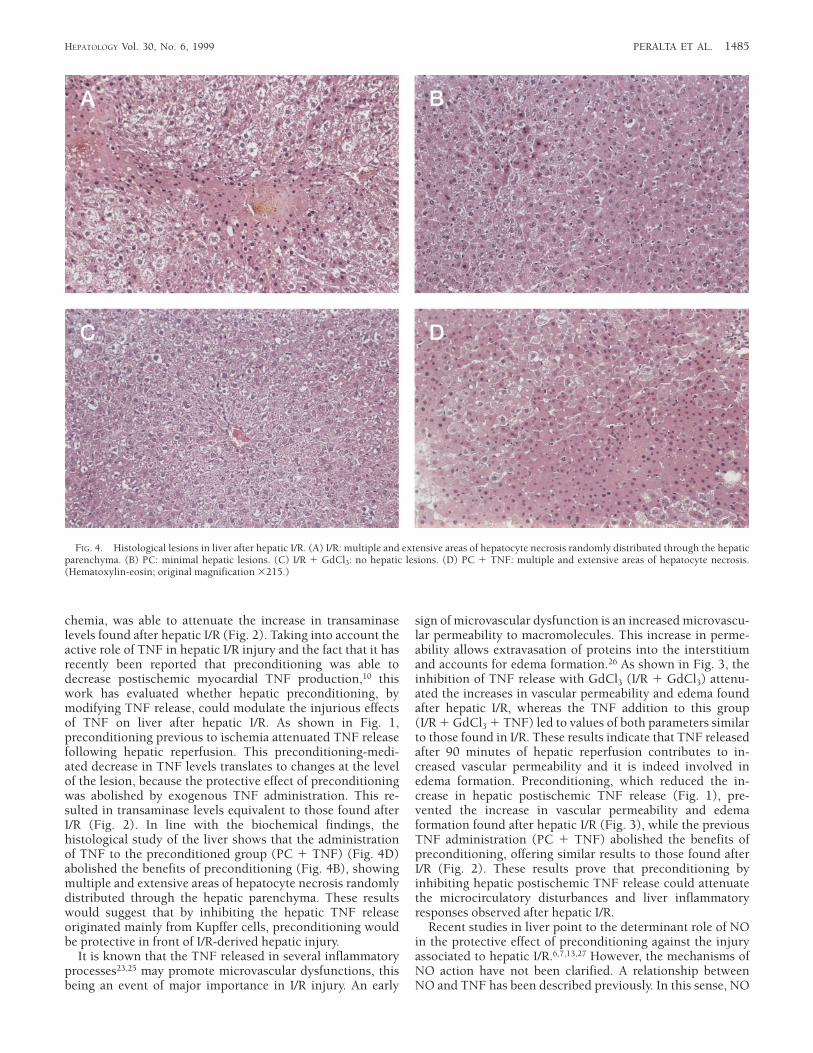
chemia, was able to attenuate the increase in transaminaselevels found after hepatic I/R (Fig. 2). Taking into account theactive role of TNF in hepatic I/R injury and the fact that it hasrecently been reported that preconditioning was able todecrease postischemic myocardial TNF production,10 thiswork has evaluated whether hepatic preconditioning, bymodifying TNF release, could modulate the injurious effectsof TNF on liver after hepatic I/R. As shown in Fig. 1,preconditioning previous to ischemia attenuated TNF releasefollowing hepatic reperfusion. This preconditioning-medi-ated decrease in TNF levels translates to changes at the levelof the lesion, because the protective effect of preconditioningwas abolished by exogenous TNF administration. This re-sulted in transaminase levels equivalent to those found afterI/R (Fig. 2). In line with the biochemical findings, thehistological study of the liver shows that the administrationof TNF to the preconditioned group (PC 1 TNF) (Fig. 4D)abolished the benefits of preconditioning (Fig. 4B), showingmultiple and extensive areas of hepatocyte necrosis randomlydistributed through the hepatic parenchyma. These resultswould suggest that by inhibiting the hepatic TNF releaseoriginated mainly from Kupffer cells, preconditioning wouldbe protective in front of I/R-derived hepatic injury.
It is known that the TNF released in several inflammatoryprocesses23,25 may promote microvascular dysfunctions, thisbeing an event of major importance in I/R injury. An early
sign of microvascular dysfunction is an increased microvascu-lar permeability to macromolecules. This increase in perme-ability allows extravasation of proteins into the interstitiumand accounts for edema formation.26 As shown in Fig. 3, theinhibition of TNF release with GdCl3 (I/R 1 GdCl3) attenu-ated the increases in vascular permeability and edema foundafter hepatic I/R, whereas the TNF addition to this group(I/R 1 GdCl3 1 TNF) led to values of both parameters similarto those found in I/R. These results indicate that TNF releasedafter 90 minutes of hepatic reperfusion contributes to in-creased vascular permeability and it is indeed involved inedema formation. Preconditioning, which reduced the in-crease in hepatic postischemic TNF release (Fig. 1), pre-vented the increase in vascular permeability and edemaformation found after hepatic I/R (Fig. 3), while the previousTNF administration (PC 1 TNF) abolished the benefits ofpreconditioning, offering similar results to those found afterI/R (Fig. 2). These results prove that preconditioning byinhibiting hepatic postischemic TNF release could attenuatethe microcirculatory disturbances and liver inflammatoryresponses observed after hepatic I/R.
Recent studies in liver point to the determinant role of NOin the protective effect of preconditioning against the injuryassociated to hepatic I/R.6,7,13,27 However, the mechanisms ofNO action have not been clarified. A relationship betweenNO and TNF has been described previously. In this sense, NO
FIG. 4. Histological lesions in liver after hepatic I/R. (A) I/R: multiple and extensive areas of hepatocyte necrosis randomly distributed through the hepaticparenchyma. (B) PC: minimal hepatic lesions. (C) I/R 1 GdCl3: no hepatic lesions. (D) PC 1 TNF: multiple and extensive areas of hepatocyte necrosis.(Hematoxylin-eosin; original magnification 3215.)
HEPATOLOGY Vol. 30, No. 6, 1999 PERALTA ET AL. 1485
induced TNF production in human mononuclear cells.16 Inaddition, the inhibition of plasma TNF release by a NO donorhas been reported in an experimental model of renal I/R.15
However, there are also controversial data, because no effectof L-NAME on TNF release was observed in cultured Kupffercells.28 These various effects of NO on TNF release reportedin the literature could be explained by the different cell typesstudied, by the use of either in vitro or in vivo models, as wellas by the different in vivo experimental models evaluated.
Considering the role of NO in the protective effect of
preconditioning and its possible role on TNF release, westudied whether the protective effect of preconditioning onthe injurious role of TNF in our model of hepatic I/R ismediated by the inhibition of TNF release by NO. Theexperimental results seem to confirm this hypothesis. Asshown in Fig. 5, the inhibition of NO synthesis in thepreconditioned group (PC 1 NAME) leads to an increase inhepatic TNF release, similar to that found in I/R, abolishingthe protective effect of preconditioning on hepatic injury(Fig. 5). However, if we inhibit TNF release with GdCl3, theinjurious effect of NO inhibition on TNF and on the lesiondisappear. In addition, the administration of a NO donor tohepatic I/R (I/R 1 NO) attenuated both the increase in TNFand transaminase levels (Fig. 5), in the same manner asobserved in the preconditioned group. In contrast, TNFadministration to this group (I/R 1 NO 1 TNF) abolishedthe beneficial effect of NO on hepatic injury after hepatic I/R.
Our recent studies on liver preconditioning27,29 performedin the same experimental model of warm ischemia estab-lished that, immediately after preconditioning, tissue NOsynthesis was increased. Because there is clearly no time forthe expression of the inducible form, this suggested that thenewly synthesized NO had originated from constitutive NOsynthase. Although much additional work is required toelucidate the possible sources of NO, the possibility thatendothelial cells could be an important source should beentertained, because constitutive NO synthase is typically ofendothelial origin.30 Along this line, it would be reasonable toassume that in clinical liver transplantation, preconditioningcould offer protection to cold ischemia by stimulating therelease of NO previous to organ preservation. In addition, wewould need to consider the possibility that preconditioningcould also stimulate the NO release from sinusoidal endothe-lial cells in sustained preservation/reperfusion processes.
FIG. 5. Effect of NO implicated in preconditioning on hepatic injury(AST and ALT) and hepatic TNF release. Control (C), I/R: animals subjectedto 90 minutes of ischemia, followed by 90 minutes of reperfusion (I/R),Preconditioning: 10 minutes of ischemia and 10 minutes of reperfusion 1 I/R(PC), Preconditioning 1 L-NAME (PC 1 NAME), Preconditioning 1L-NAME 1 GdCl3 (PC 1 NAME 1 GdCl3), I/R 1 NO donor (I/R 1 NO),I/R 1 NO donor 1 TNF (I/R 1 NO 1 TNF). *P , .05 vs. control; 1P , .05vs. I/R; oP , .05 vs. Preconditioning.
FIG. 6. Role of TNF on MPO activity in lung. Control, I/R: animalssubjected to 90 minutes of ischemia, followed by 90 minutes of reperfusion(I/R), I/R 1 GdCl3 (I/R 1 GdCl3), I/R 1 GdCl3 1 TNF (I/R 1 GdCl3 1TNF), Preconditioning: 10 minutes of ischemia and 10 minutes of reperfu-sion 1 I/R (PC), Preconditioning 1 TNF (PC 1 TNF). *P , .05 vs. control;1P , .05 vs. I/R; oP , .05 vs. Preconditioning.
1486 PERALTA ET AL. HEPATOLOGY December 1999
These cells are known to be susceptible to the effects of coldischemia.
An established consequence of hepatic I/R is the inductionof important pulmonary pathological alterations, such as theadult respiratory distress syndrome associated with humanliver transplantation and multiple organ failure associatedwith primary graft failure.2,31,32 In animal models, Colleti etal. showed that normothermic hepatic I/R in rats leads to lungchanges characterized by interstitial thickening and infiltra-tion with leukocytes.9 Similarly, pulmonary changes occur inorthotopic rat liver transplantation after storage times, caus-ing graft failure.33 It has been suggested that these changesappear to be linked to hepatic release of TNF, because
antibodies to TNF reduce lung and liver injury.8,9 As shown inFig. 1, hepatic TNF level differences between the differentgroups of the study were also reflected systemically, suggest-ing that the reperfusion of the ischemic lobes of the liverresults in the release of tissue-associated TNF. On the otherhand, the differences in plasma TNF levels (Fig. 1) were alsoreflected in changes in pulmonary lesion (Fig. 7). In thisregard, we can infer that I/R leads to an increase in plasmaTNF release relative to the control groups (Fig. 1), and thatthe lung injury that develops is consistent with the infiltra-tion of neutrophils, determined by MPO activity (Fig. 6) andalso assessed histologically. A diffuse septal thickening withmarked polymorphonuclear infiltration and perivascular neu-
FIG. 7. Histological lesions in lung after hepatic I/R. (A) I/R: inflamma-tory infiltrate and thickening of the alveolar walls. (B) I/R: the majority of theinflammatory cells were polymorphonuclear leukocytes. (C) PC: minimallesions in the lung parenchyma. (D) I/R 1 GdCl3: no apparent pulmonarylesions. (E) PC 1 TNF: pulmonary lesions were similar to those found in I/Rgroup. (Hematoxylin-eosin; original magnification [A, C-E] 3215; [B] 3400.)
HEPATOLOGY Vol. 30, No. 6, 1999 PERALTA ET AL. 1487

trophil accumulation was observed in the lung after hepaticI/R (Fig. 7A and 7B).
Our results further suggest that the increase in plasma TNFconsequent to hepatic I/R appears to be mainly the result ofthe liver-derived TNF resulting from Kupffer cell activation.It is known that the Kupffer cell population within the liver isthe largest fixed macrophage population in the body and theliver; therefore, they represent the most important potentialcellular source for TNF.9,23 Along these lines, it is important toconsider that the inactivation of Kupffer cells with GdCl3
(I/R 1 GdCl3) prevented both the increase in plasma TNF(Fig. 1) and the injurious effects in the lung seen after hepaticI/R (Fig. 7A and 7B). Reduced MPO levels (Fig. 6) and noapparent pulmonary lesions were observed (Fig. 7D). Thebeneficial effect of TNF inhibition was clearly revertedafter intravenous TNF administration to this group (I/R 1GdCl3 1 TNF), showing MPO levels similar to those foundafter I/R (Fig. 6).
Thus, new strategies involving alteration or modificationin TNF release may be important for the attenuation of boththe local and the distant injury associated with hepatic I/R. Asshown in Fig. 1, preconditioning prevented the increase inhepatic TNF, thus preventing the release of TNF from theliver into the systemic circulation. The reduction of plasmaTNF levels by preconditioning could represent the distaleffector mechanism of preconditioning. In this regard, he-patic preconditioning attenuated lung damage resulting fromhepatic I/R, as shown by a reduced neutrophil infiltration,measured by MPO (Fig. 6) and the minimal lesions seen inthe lung parenchyma (Fig. 7C). However, the intravenousTNF administration to the preconditioned group (PC 1TNF) abolished the protective effect of hepatic precondition-ing on pulmonary damage, as shown by the increase in MPOlevels (Fig. 6) and pulmonary lesions (Fig. 7E), which werecomparable with those found after hepatic I/R (Fig. 7A and 7B).
In liver transplantation, the preservation-reperfusion inju-ries are still one of the most serious insults that affectpostoperative graft viability and ultimate operative results.1
The present experimental study demonstrates that the ben-efits of organ preconditioning could be of clinical interest.Thus, the stimulation of endogenous NO or the administra-tion of NO donors could protect liver grafts from subsequentI/R injuries. This could afford the possibility of improving theinitial conditions of the organs available for transplantation,as well as to being able to use donor organs, presentlydiscarded on account of their general or local conditions,such as older age, a high grade of fatty replacement, orabnormal liver function tests.34
In conclusion, the findings in our current study indicatethat preconditioning reduced both liver and lung damageassociated with hepatic I/R. Preconditioning, through NO,prevented the increase in hepatic TNF mainly released fromKupffer cells, thus attenuating the liver injury followinghepatic I/R. Also, preconditioning, by reducing hepatic TNFlevels after I/R, could probably prevent the systemic release ofliver-associated TNF, thus attenuating the lung injury conse-quent to hepatic I/R.
REFERENCES
1. Clavien PA, Harvey PRC, Strasberg SM. Preservation and reperfusioninjuries in liver allografts. An overview and synthesis of current studies.Transplantation 1992;53:957-978.
2. Matuschak GM, Rinaldo JE, Pinski MR, Gavaler JS, Van Thiel DH. Effectof end stage liver failure on the incidence and resolution of the adultrespiratory distress syndrome. J Crit Care 1987;2:162-173.
3. Meldrum D, Mitchell MB, Banerjee A, Harken AH. Cardiac precondition-ing. Induction of endogenous tolerance to ischemia-reperfusion injury.Arch Surg 1993;128:1208-1211.
4. Liang-Xiong FU, Kirkeboen KA, Liang QM, Sjorgren KG, Hjalmarson A,Hebekk A. Free radical scavenging enzymes and G protein mediatedreceptor signalling systems in ischaemically preconditioned porcinemyocardium. Cardiovasc Res 1993;27:612-616.
5. Parrat JR. Protection of the heart by ischaemic preconditioning: mecha-nisms and possibilities for pharmacological exploitation. Tips Reviews1994;15:19-25.
6. Peralta C, Hotter G, Closa D, Gelpı E, Bulbena O, Rosello-Catafau J.Protective effect of preconditioning on the injury associated to hepaticischemia-reperfusion: role of nitric oxide and adenosine. HEPATOLOGY
1997;25:934-937.7. Yin DP, Sankary HN, Chong AS, Ma LL, Shen J, Foster P, Williams JW.
Protective effect of ischemic preconditioning on liver preservation-reperfusion injury in rats. Transplantation 1998;66:152-157.
8. Colletti LM, Burtch GD, Remick DG, Kunkel SL, Strieter RM, Guice KS,Oldham KT, et al. Production of tumor necrosis factor alpha and thedevelopment of a pulmonary capillary injury following hepatic ischemia/reperfusion. Transplantation 1989;49:268-273.
9. Colletti LM, Remick DG, Burtch GD, Kunkel SL, Strieter RM, CampbellDA. Role of tumor necrosis factor-a in the pathophysiologic alterationsfollowing hepatic ischemia/reperfusion in the rat. J Clin Invest 1990;85:1936-1943.
10. Meldrum DR, Dinarello CA, Shames BD, Cleveland JC, Cain BS, BanerjeeA, Meng X, et al. Ischemic preconditioning decreases postischemicmyocardial tumor necrosis factor-alpha production. Potential ultimateeffector mechanism of preconditioning. Circulation 1998;98:214-219.
11. Hotter G, Closa D, Prados M, Fernandez-Cruz L, Prats N, Gelpı E,Rosello-Catafau J. Intestinal preconditioning is mediated by a transientincrease in nitric oxide. Biochem Biophys Res Commun 1996;222:27-32.
12. Bilinska M, Maczewski M, Beresewicz A. Donors of nitric oxide mimiceffects of ischaemic preconditioning on reperfusion induced arrhythmiasin isolated rat heart. Mol Cell Biochem 1996;161:265-271.
13. Peralta C, Closa D, Xaus C, Gelpı E, Rosello-Catafau J, Hotter G. Hepaticpreconditioning in rats is defined by a balance of adenosine andxanthine. HEPATOLOGY 1998;28:768-773.
14. Eigler A, Sinha B, Endres S. Nitric oxide-releasing agents enhancecytokine-induced tumor necrosis factor synthesis in human mono-nuclear cell. Biochem Biophys Res Commun 1993;196:494-501.
15. Garcia Criado FJ, Eleno N, Santos Benito F, Valdunciel JJ, Reverte M,Lozano Sanchez FS, Ludena MD, et al. Protective effect of exogenousnitric oxide on the renal function and inflammatory response in a modelof ischemia-reperfusion. Transplantation 1998;66:982-990.
16. Eigler A, Sinha B, Endres S. Nitric oxide–releasing agents enhancecytokine-induced tumor necrosis factor synthesis in human mono-nuclear. Biochem Biophys Res Commun 1993;196:494-501.
17. Klemm P, Warner TD, Hohlfeld T, Corder R, Vane JR. Endothelin 1mediates ex vivo coronary vasoconstriction caused by exogenous andendogenous cytokines. Proc Natl Acad Sci U S A 1995;92:2691-2695.
18. Scales WE, Campbell DA, Green ME, Remick DG. Hepatic ischemia/reperfusion injury: importance of oxidant/tumor necrosis factor interac-tions. Am J Physiol 1994;267:G1122-G1127.
19. Colletti LM, Cortis A, Lukacs N, Kunkel SL, Green M, Strieter RM.Tumor necrosis factor up-regulates intercellular adhesion molecule 1,which is important in the neutrophil-dependent lung and liver injuryassociated with hepatic ischemia and reperfusion in the rat. Shock1998;10:182-191.
20. Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ.Chemokine involvement in hepatic ischemia/reperfusion injury in mice:Roles for macrophage inflammatory protein-2 and KC. HEPATOLOGY
1998;27:1172-1177.21. Schmekel B, Karlsson SE, Linden M, Sundstrom C, Tegner H, Venge P.
Myeloperoxidase in human lung lavage. I. A marker of local neutrophilactivity. Inflammation 1990;14:447-454.
22. Trush MA, Egner PA, Kensler TW. Myeloperoxidase as a biomarker ofskin irritation and inflammation. Fed Chem Toxic 1994;32:143-147.
23. Strieter RM, Kunkel SL, Bone RC. Role of tumor necrosis factor-a indisease states and inflammation. Crit Care Med 1993;21:S447-S463.
24. Wake K, Decker K, Kirn A, Knook DL, McCuskey RS, Bouwens L, Wisse
1488 PERALTA ET AL. HEPATOLOGY December 1999

E. Cell biology and the kinetics of Kupffer cells in the liver. Int Rev Cytol1989;118:173-229.
25. Guice KS, Oldham KT, Remick DG, Kunkel SL, Ward PA. Anti-tumornecrosis factor antibody augments edema formation in caerulein-induced acute pancreatitis, J Surg Res 1991;51:495-499.
26. Noel AA, Hobson RW, Duran WN. Platelet-activating factor and nitricoxide mediate microvascular permeability in ischemia-reperfusion in-jury. Microvasc Res 1996;52:210-220.
27. Peralta C, Hotter G, Closa D, Prats N, Xaus C, Gelpı E, Rosello-Catafau J.The protective role of adenosine in inducing nitric oxide synthesis in ratliver ischemia preconditioning is mediated by activation of adenosine A2
receptors. HEPATOLOGY 1999;29:126-132.28. Stadler J, Harbrecht BG, Di Silvio M, Curran RD, Jordan ML, Simmons
RL, Billiar TR. Endogenous nitric oxide inhibits the synthesis ofcyclooxygenase products and interleukin-6 by rat Kupffer cells. J LeukocBiol 1993;53:165-172.
29. Peralta C, Closa D, Hotter G, Gelpı E, Prats N, Rosello-Catafau J. Liver
ischemic preconditioning is mediated by the inhibitory action of nitricoxide on endothelin. Biochem Biophys Res Commun 1996;229:264-270.
30. Palmer RMJ, Ashton DS, Moncada S. Vascular endothelial cells synthe-size nitric oxide from L-arginine. Nature 1988;33:664-666.
31. Grieg PD, Woolf GM, Sinclair SB, Abecassis M, Strasberg SM, Taylor B,Blendis LM, et al. Treatment of primary liver graft non function withprostaglandin E1. Transplantation 1989;48:447-453.
32. Ringe B, Pinchlmayr R, Lubbe N, Bornscheuer A, Kuse E. Totalhepatectomy as a temporary approach to acute hepatic or primary graftfailure. Transplant Proc 1988;20:552-557.
33. Takei Y, Marzi I, Kauffman FC, Currin RT, Lemasters JJ, Thurman RG.Increase in survival time of liver transplants by protease inhibitors and acalcium channel blocker, nisoldipine. Transplantation 1990;50:14-20.
34. Mor E, Klintmalm GB, Gonwa TA, Solomon H, Holman MJ, Gibbs JF,Watemberg I, et al. The use of marginal donors for liver transplantation.A retrospective study of 365 liver donors. Transplantation 1992;53:383-386.
HEPATOLOGY Vol. 30, No. 6, 1999 PERALTA ET AL. 1489





























