Embed Size (px)
Citation preview

Lawrence Hunter, Ph.D.Director, Computational Bioscience ProgramUniversity of Colorado School of Medicine
[email protected]://compbio.uchsc.edu/Hunter
Protein Structure Prediction

Protein structure
• Most proteins will fold spontaneously in water, soamino acid sequence alone should be enough todetermine protein structure
• However, the physics are daunting:– 20,000+ protein atoms, plus equal amounts of water–Many non-local interactions–Can takes seconds (most chemical reactions take place
~1012 --1,000,000,000,000x faster)
• Empirical determinations of protein structure areadvancing rapidly.
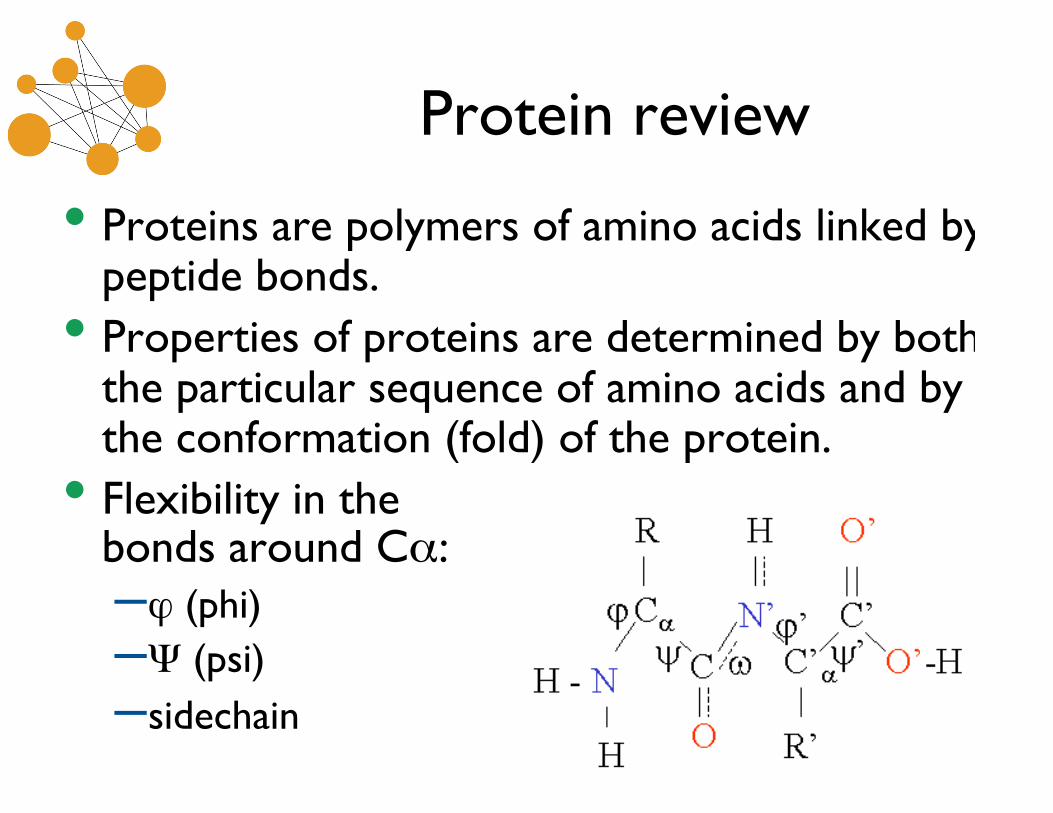
Protein review
• Proteins are polymers of amino acids linked bypeptide bonds.
• Properties of proteins are determined by boththe particular sequence of amino acids and bythe conformation (fold) of the protein.
• Flexibility in thebonds around Cα:–ϕ (phi)–Ψ (psi)–sidechain

Protein Structure Levels
• Protein structure is described in four levels–Primary structure: amino acid sequence– Secondary structure: local (in sequence) ordering into
• (α)Helices: compressed, corkscrew structures• (β)Strands: extended, nearly straight structures• (β)Sheets: paired strands, reinforced by hydrogen bonds
– parallel (same direction) or antiparallel sheets• Coils, Turns & Loops: changes in direction
–Tertiary structure: global ordering (all angles/atoms)–Quaternary structures: multiple, disconnected amino acid
chains interacting to form a larger structure
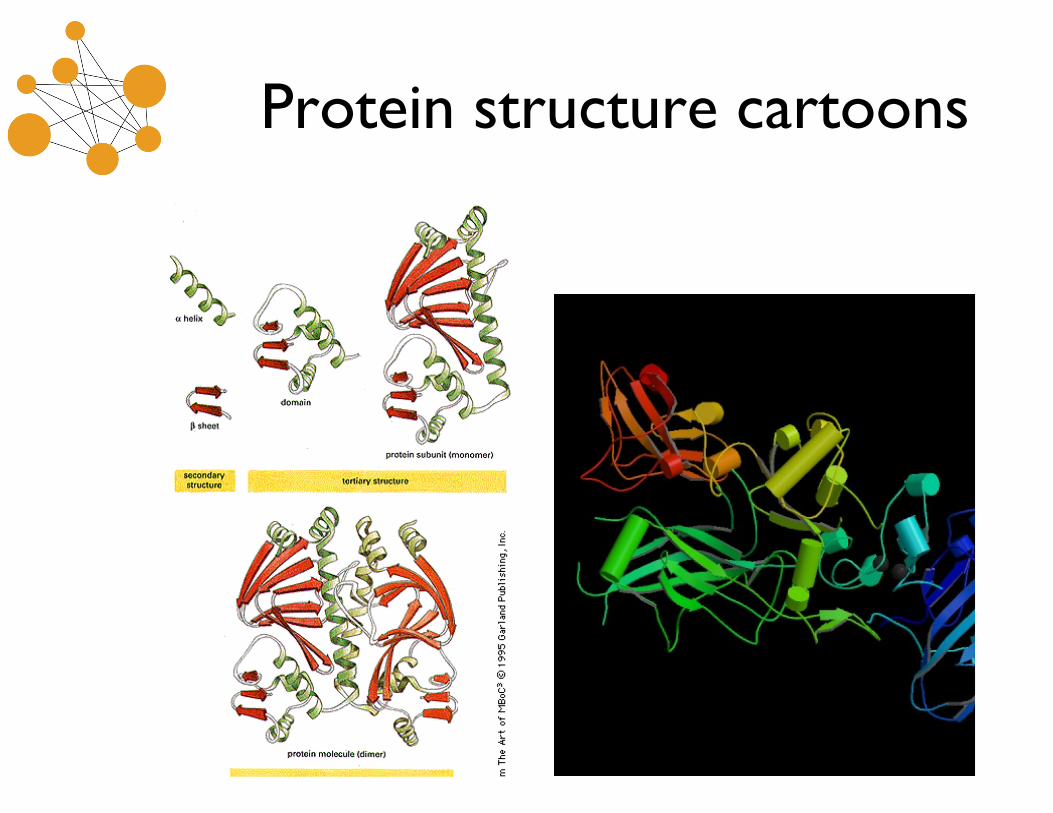
Protein structure cartoons
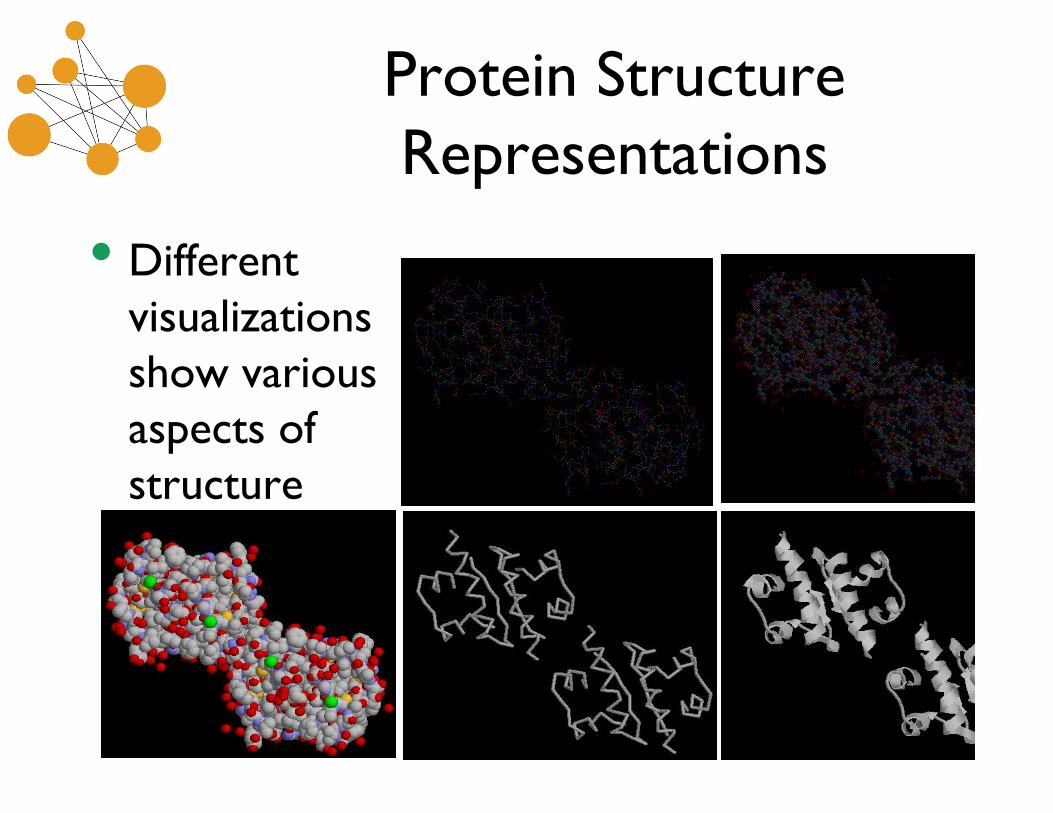
Protein StructureRepresentations
• Differentvisualizationsshow variousaspects ofstructure

Protein Folding
• Proteins are created linearly and then assume theirtertiary structure by “folding.”– Exact mechanism is still unknown–Mechanistic simulations can be illuminating
• Proteins assume the lowest energy structure–Or sometimes an ensemble of low energy structures.
• Hydrophobic collapse drives process• Local (secondary) structure proclivities• Internal stabilizers:
–Hydrogen bonds, disulphide bonds, salt bridges.

Empirical structuredetermination
• Two major experimental methods for determiningprotein structure
• X-ray Crystallography–Requires growing a crystal of the protein
(impossible for some, never easy)
–Diffraction pattern can be inverse-Fourier transformedto characterize electron densities (Phase problem)
• Nuclear Magnetic Resonance (NMR) imaging–Provides distance constraints, but can be hard to find a
corresponding structure–Works only for relatively small proteins (so far)
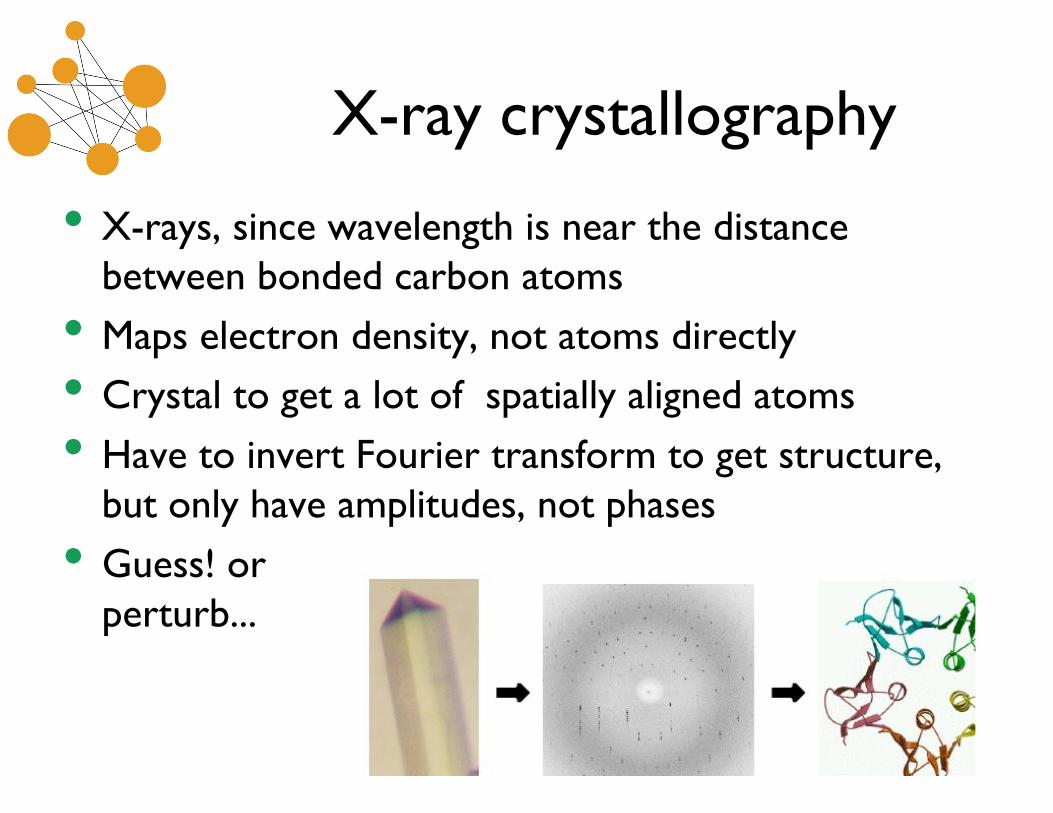
X-ray crystallography
• X-rays, since wavelength is near the distancebetween bonded carbon atoms
• Maps electron density, not atoms directly
• Crystal to get a lot of spatially aligned atoms
• Have to invert Fourier transform to get structure,but only have amplitudes, not phases
• Guess! orperturb...

NMR structure determination
• NMR can detect certain features of hydrogen atoms:–NOESY measures distances between non-bonded H's
within about 5A–COSY and TOCSY described relations through bonds
• Combination of distance and angle constraints, plusknowledge of covalent bonds (amino acid sequence)determines a unique (sometimes) structure.
• Overlapping measurement limits size ~120AA

Why predict protein structure?
• Neither crystallography nor NMR can keeppace with genome sequencing efforts–Only 10566 (3641 with <90% identity) human
proteins in PDB, although growing fast
–Computer scientists love this problem
–Understandable with minimal biology
–Seems like a good discrimination task
• Understand the mechanisms of folding (?)
• First computational Nobel prize?

Kinds of Structure Prediction
• Comparative modelling–Homolog has known structure, which is adjusted for
sequence differences– Energy minimization and molecular dynamics
• Fold recognition–Proteins fall into broad fold classes. Models of folds that
recognize compatible sequences. “Inverse” problem–Predict more than fold class?
• Ab initio or “new fold” prediction–No homologs, not recognized by any fold model

Ab Initio predictions
• Three broad approaches–Molecular dynamics, energy minimization approaches–Empirical “black box” (induce discriminators)–Mechanistic (follow the actual folding path) approaches.
Hybrid between energy and empirical methods.
• Secondary structure predictions–Not tremendously useful nor accurate, but simplest.–Can play a role in tertiary predictors
• Tertiary structure predictions–Best involve a complex mixture of approaches

Energy Minimization
• Many forces act on a protein–Hydrophobic: inside of protein wants to avoid water–Packing: atoms can't be too close, nor too far away–Bond angle/length constraints– Long distance, e.g.
• Electrostatics & Hydrogen bonds• Disulphide bonds• Salt bridges
• Can calculate all of these forces, and minimize• Intractable in general case, but can be useful
Empirical models
• Pose structure prediction as induction task.–What are the inputs and outputs?
–Where do we get enough training data?
–Which induction methods work best?
• Long history in bioinformatics
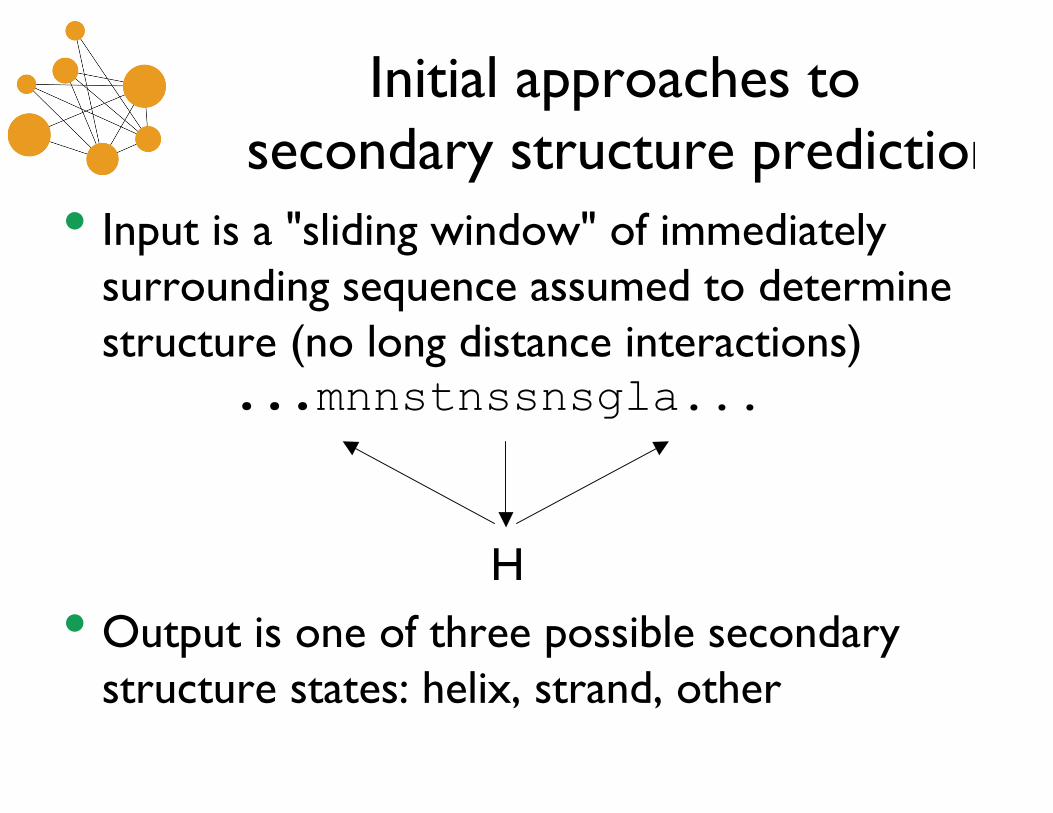
Initial approaches to secondary structure prediction
• Input is a "sliding window" of immediatelysurrounding sequence assumed to determinestructure (no long distance interactions) ...mnnstnssnsgla...
H
• Output is one of three possible secondarystructure states: helix, strand, other

Why might this work?
• There are local propensities to secondary structuralclasses (largely hydropathy)–Helices: no prolines, sometimes amphipathic (show
alternating hydropathy with period 3.6 residues)– Strands: either alternating hydropathy or ends
hydrophillic and center hydrophobic–Neither: small, polar & flexible residues. Prolines.
• Minimum lengths for secondary structures (heliceslonger than strands)

Early methods
• Chou-Fasman method looked at frequency of eachamino acid in window
• GOR defined an information measureI(S;R) = log[P(S|R)/P(S)]
where S is secondary structure and R is amino acid.Define information gain as:
I(S;R) - I(~S;R)and predict state with highest gain.–How to combine info gain for each element of sliding
window? Independently (just add) or by pairs

How well did they work?
• Not very: Roughly 50-55% accurate on a residue byresidue basis.
• Random prediction that obeyed the observeddistribution of helix/strand/other would be 40%
• Different ways to calculate "correctness"–Needs to be unbiased (especially wrt homology)!–Getting number of helices and strands or order right is
harder than just counting residue by residue (like thedifference between nucleotide and exon level genefinding).

Fancier induction techniques
• Same setup as Chou-Fasman or GOR–Sliding window across amino acid sequence as
input–Three class output (helix/sheet/other)
• Various different induction techniques oversame data, give modest improvements–LDA/QDA–Decision trees–Neural networks
• Best results from neural networks (~ 62%)

Add multiple sequencealignment information
• This is helpful in principle:–insertions/deletions more likely to be coil/turn
–conserved hydropathy more important forprediction than non-conserved.
• GOR method improves 8-9% points (to about64% correct residue by residue).
• Similar improvement for NNs (to ~ 68%)
• SVMs gain a bit more, to about ~70%

But the information isn't there
• Prediction quality has not improved mucheven with huge growth of training data.
• Secondary structure is not completelydetermined by local forces–Long distance interactions do not appear in sliding
window
• Empirical studies show same amino acidsequences can assume multiple secondarystructures.

Mechanistic models
• Move from purely empirical to include someknowledge of folding mechanisms–Compact nature of conformations
• Hydrophobic packing
• Sequences of secondary structures
–Secondary structure predispositions
–Heuristic global energy minimization
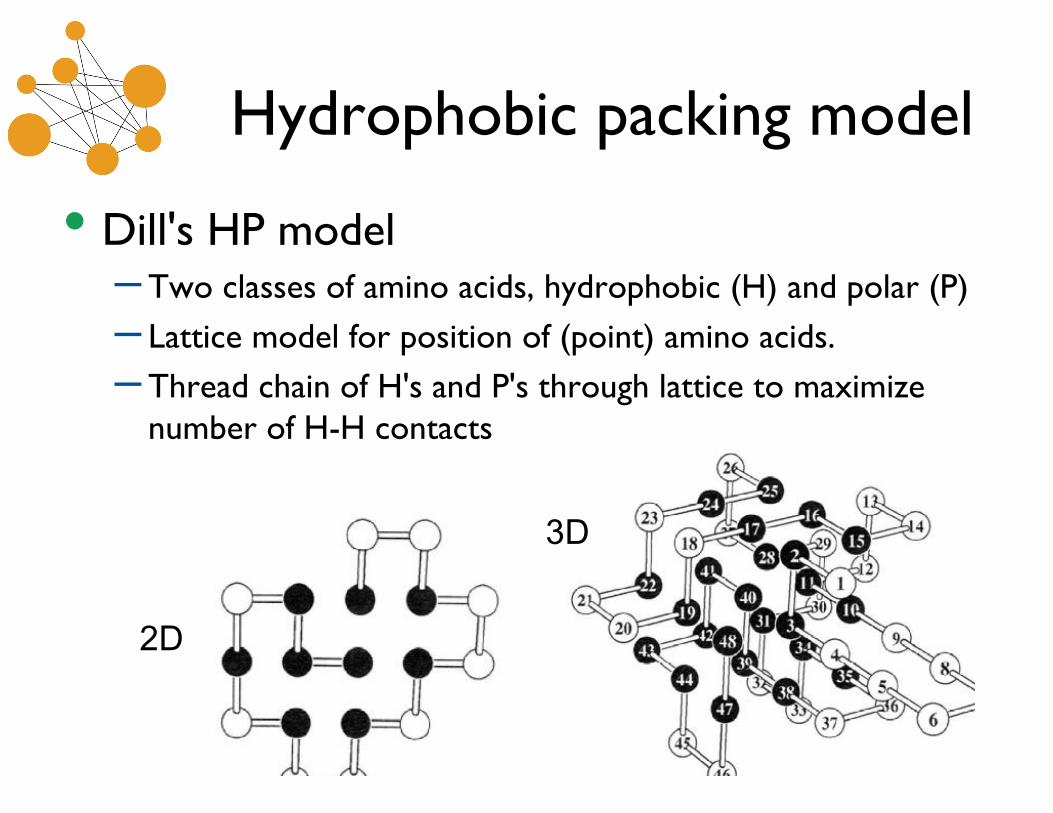
Hydrophobic packing models
• Dill's HP model– Two classes of amino acids, hydrophobic (H) and polar (P)
– Lattice model for position of (point) amino acids.
– Thread chain of H's and P's through lattice to maximizenumber of H-H contacts
2D
3D

But...
• Even the 2D HP packing problem (which iseasier than the 3D one) turns out to be NPcomplete!
• Good approximation results exist.–3/8 of optimal approximation (3D)
–In triangular lattice, algorithm for >60% of optimalpacking
• Other interesting results in the model, e.g.–Which sequences have a single optimal fold?

CASP changed the landscape
• Critical Assessment of Structure Prediction competition.Even numbered years since 1994– Solved, but unpublished structures are posted in May,
predictions due in September, evaluations in December– Various categories
• Relation to existing structures, ab initio, homology, fold, etc.
• Partial vs. Fully automated approaches
– Produces lots of information about what aspects of theproblems are hard, and ends arguments about test sets.
• Results showing steady improvement, and the value ofintegrative approaches.

CASP 6 Categories• Human intervention versus fully automated predictions• Comparative modeling
– A structure exists for a good homolog– Looking for mutations, bond rotations, etc.
• Fold recognition (Homologs)– Distant homolog recognition and adaptation– Looking at loop placement, domain boundaries
• Fold recognition (Analogous)– No homolog, but similar structures in DB– Finding the right model structure
• Ab Initio– No similar structures in DB. Most fundamental problem.
• Other issues– Domain boundaries, disordered regions, residue-residue contacts

CASP Results
• Fully automated methods now nearly as goodas ones with human intervention
• Consensus methods (looking for agreementamong servers) do best overall, but not bymuch and not all the time.
• Consistent best approach is Rosetta fromDavid Baker’s lab
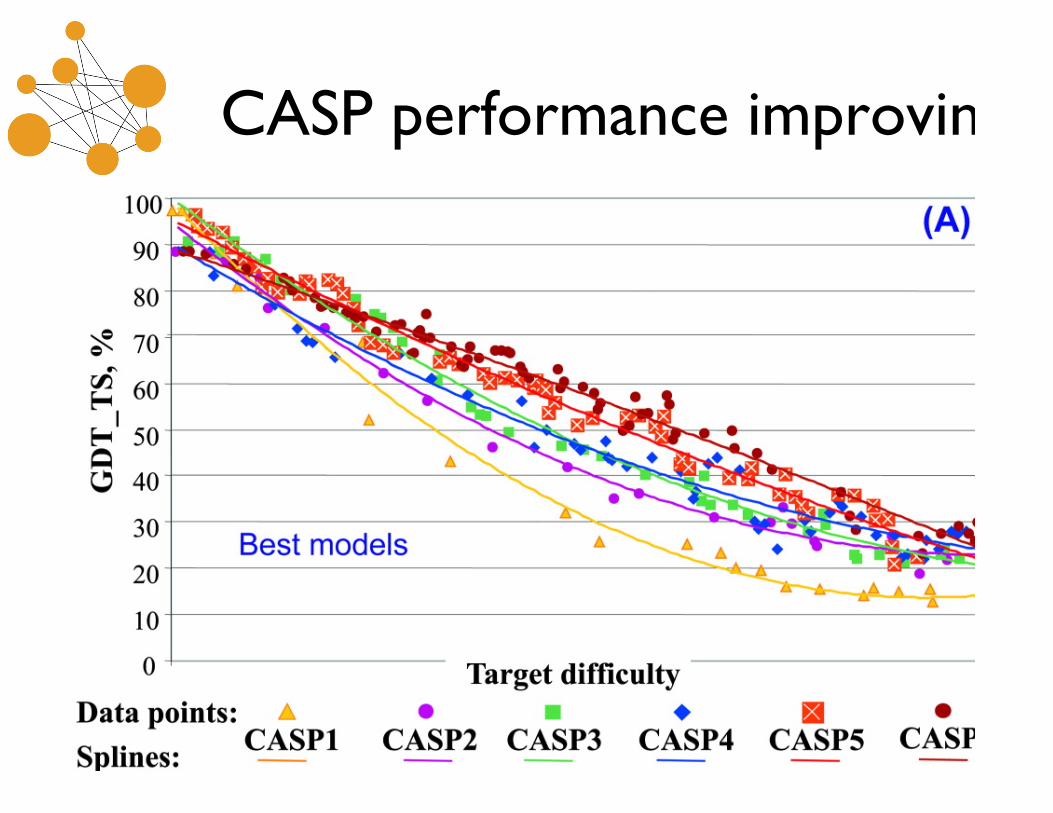
CASP performance improving

Baker: best strategy so far
• Two step process:–Generate a good sized collection of plausible structures
and near-miss bad structures• Requires a good energy function, good optimization approach• Quality of “decoy” (incorrect, but plausible folds) is important
–Build discriminators to separate correct from decoystructures.
• Rosetta (Baker lab) and fully automated Robetta.–Ran away with CASP4, still the best at CASP5 & 6–Robetta almost as good as Rosetta–Outstanding at ab initio, competitive at the rest.

Rosetta approach
• Integrated method– I-Sites: much finer grained substructures than secondary
structures. A library of all consistent structures of shortpolypeptides is defined (taken from PDB)
–Build initial models by assigning I-sites to new amino acidsequence (many possibilities)
–Monte Carlo search through assignments of I-Sites tominimize energy function.
–Use of sophisticated global energy function
• Take good scoring structures, and test them on a“decoy detector”, which looks for high scoring butnon-native structure patterns.

I-Sites
• I-sites are a set of sequence patterns thatstrongly correlate with protein structure atthe local level.
• Ungapped amino acid sequence motifs–Length 3-9 (now longer)–Originally 82 classes (now more)–Defined by amino acid log odds matrix and phi/psi
angles
• Far more detailed than the 3 statehelix/sheet/other local structure models
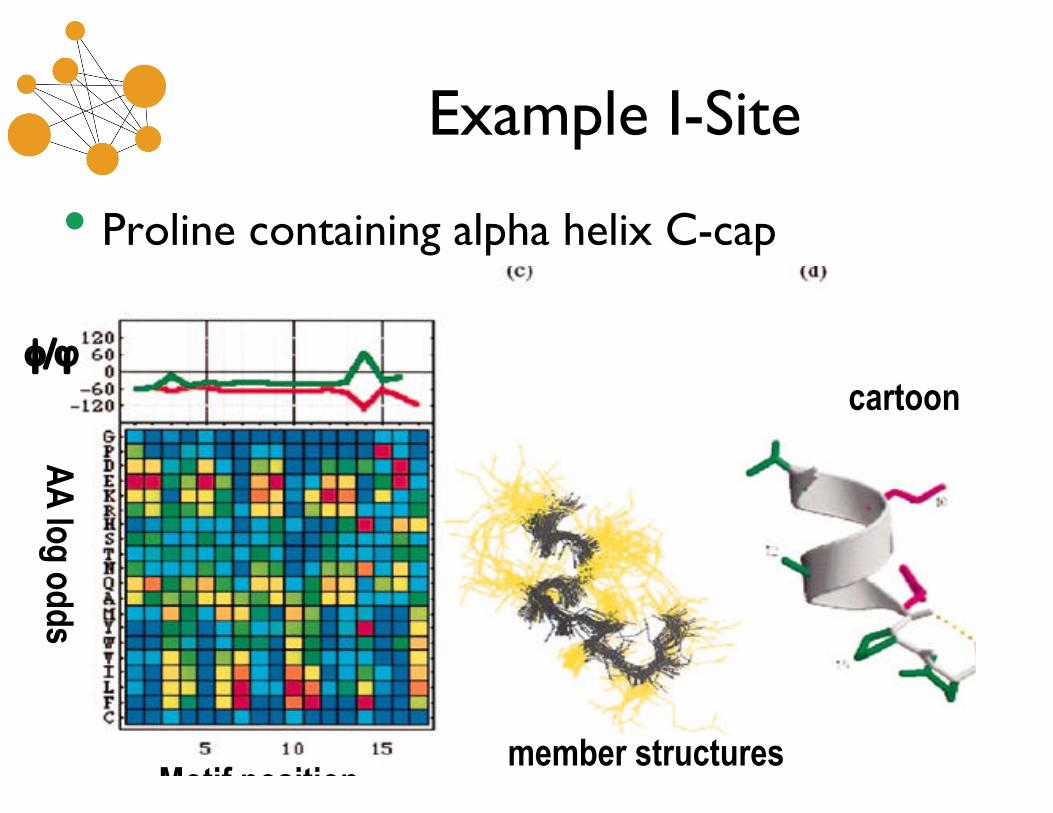
Example I-Site
• Proline containing alpha helix C-cap
φ/ϕ
AA
log odds
Motif position member structures
cartoon

How I-sites are defined
• Starting from all sequences in PDB at the time– Remove sequences with 25% or greater sequence identity to
compensate for oversampling of certain families
• Cluster all possible subsequences of these structures oflength 3-15.
• For each cluster, define “paradigm” structure.– Remove members that are too far away structurally
– Add new members that are structurally similar
– If can't distinguish well (bimodal scores) between members andnon-members, drop the cluster

I-sites are not unique
• One amino acid subsequence may be compatiblewith several I-sites– I-sites are not defined to be mutually exclusive over
sequence.– Slightly different starting positions or lengths may yield
quite different (even incompatible) I-sites for the samesequence region.
• This has biological relevance–Local predispositions are not determinative or unique–Multiple predispositions are more informative than none.

I-sites pro and con
• Lots of ad hoc fiddling to get I-site library–Distance measure on sequence has two free parameters–Many different structure distance measures tried–K-means clustering (K is free parameter)–Test for bimodal scoring (two more parameters)–Occasional subdivision of an I-Site that seemed to have
two good structures associated with it
• Corresponds reasonably well to existingcrystallographic concepts (e.g. Type II β turns)
• They are more predictable than H/S/C
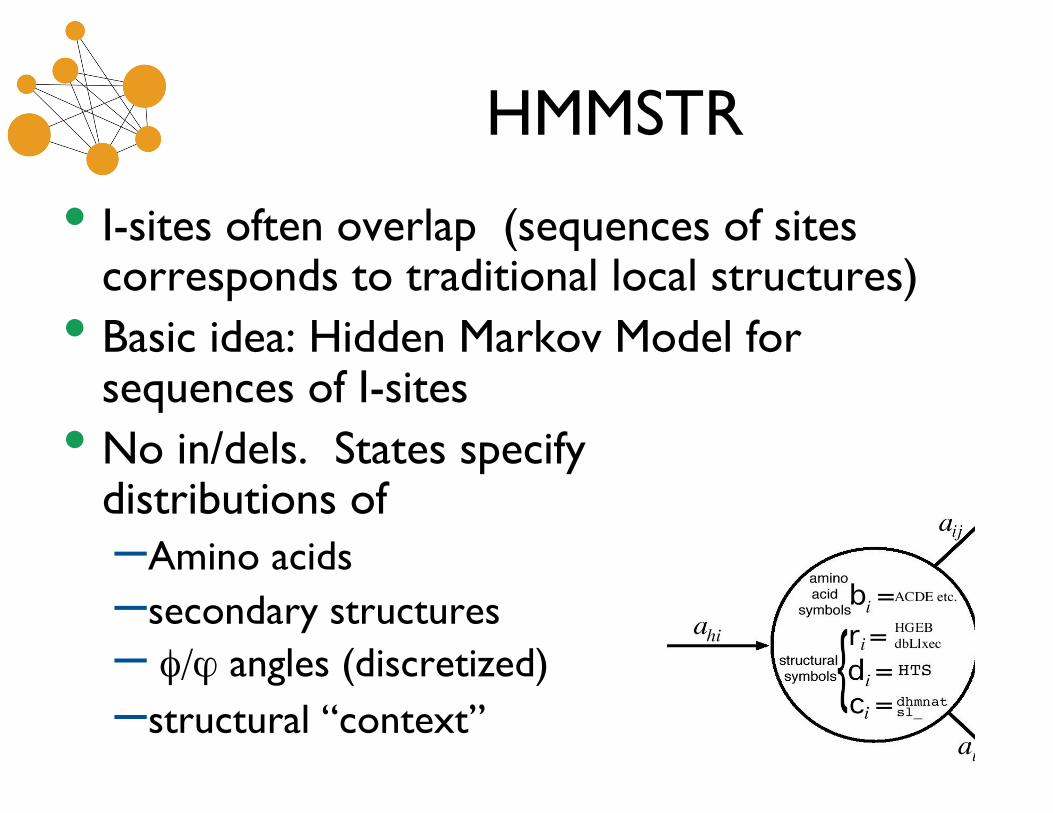
HMMSTR
• I-sites often overlap (sequences of sitescorresponds to traditional local structures)
• Basic idea: Hidden Markov Model forsequences of I-sites
• No in/dels. States specifydistributions of–Amino acids–secondary structures– φ/ϕ angles (discretized)–structural “context”
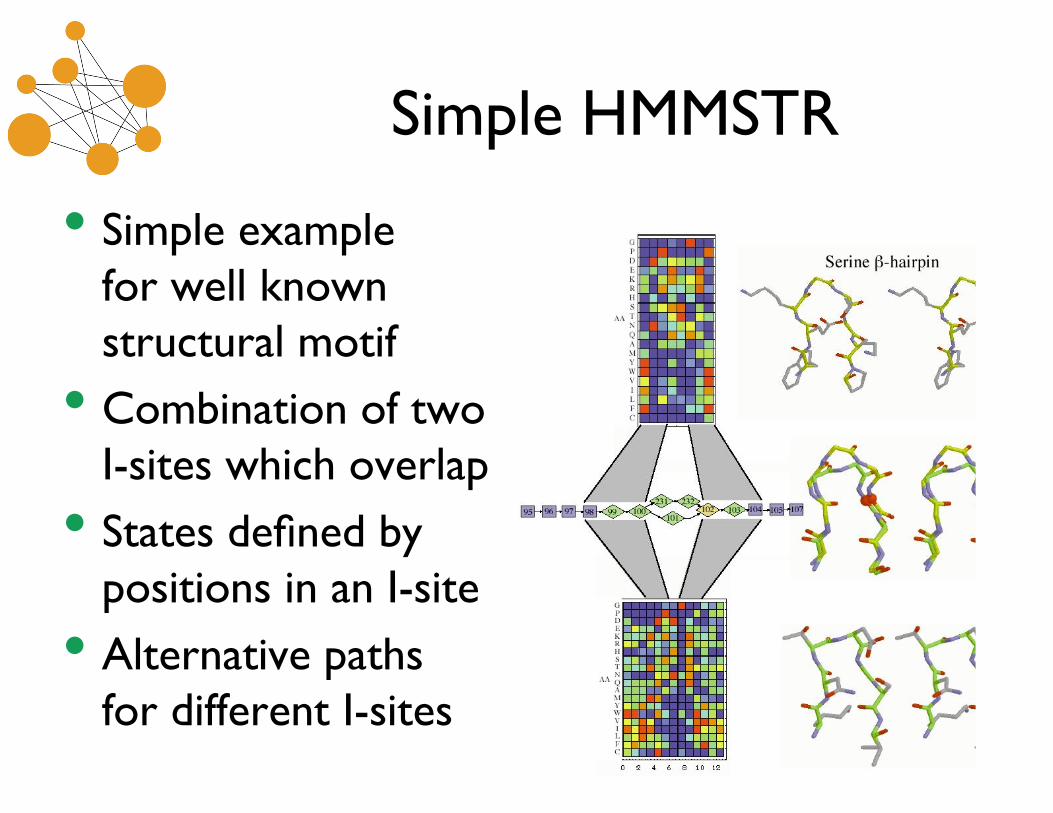
Simple HMMSTR
• Simple examplefor well knownstructural motif
• Combination of twoI-sites which overlap
• States defined bypositions in an I-site
• Alternative pathsfor different I-sites
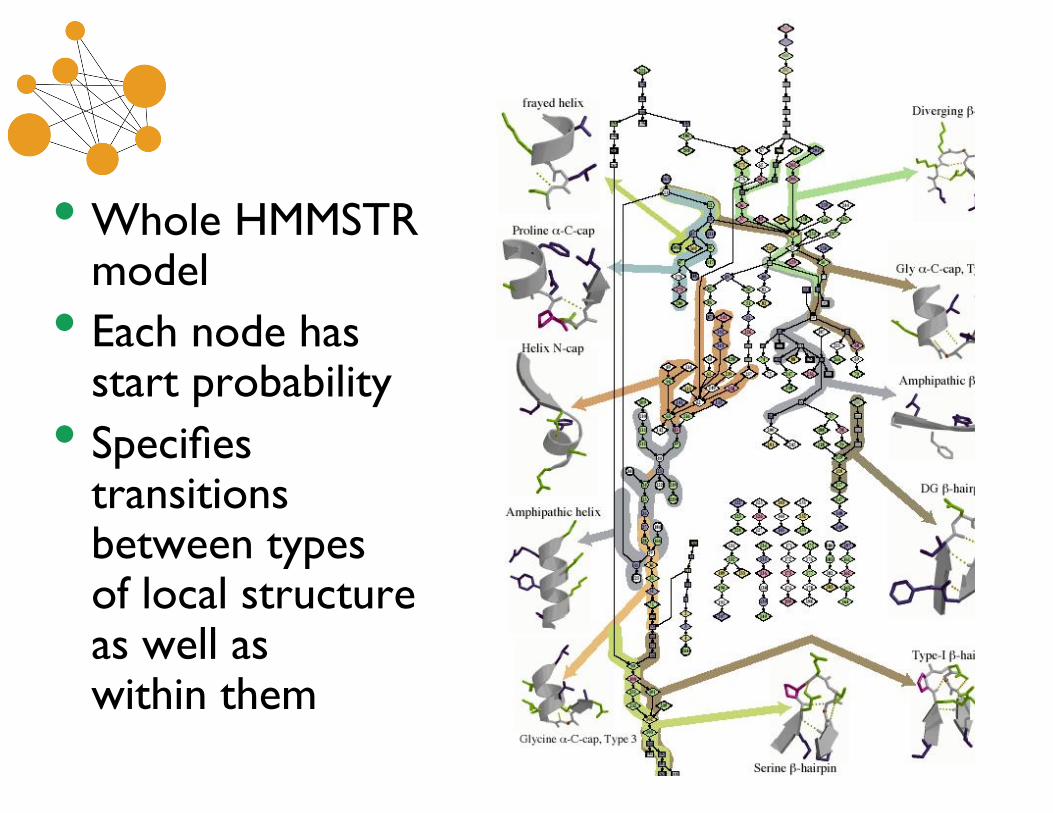
• Whole HMMSTRmodel
• Each node hasstart probability
• Specifiestransitionsbetween typesof local structureas well aswithin them

Training of HMMSTR
• Many ad hoc approaches based on biologicalintuitions–When to merge overlapping states?–Dynamic programming to find likely transitions
between I-sites–Null transition state to connect otherwise
disconnected subtrees.–Model “surgery” adding, splitting and deleting
states.–Structure predictions by “voting” rather than
most probable parse.

Beating HMMSTR
• OK, but not great results in predictiveaccuracy.
• Too many alternative paths through themodel, and difficulty choosing between themon the basis of sequence alone.
• Only local information; no global measuresused.
• Rosetta: add global information to I-siteassignments and get a big improvement

Rosetta prediction method
• Define global scoring function that estimatesprobability of a structure given a sequence
• Generate version of I-sites with fixed lengthsubsequences (9 amino acids)–Calculate P(I-Site|sequence) for all sequences and I-sites
• Generate structures by Monte Carlo sampling ofassignments of fixed size I-sites to subsequences
• End up with ensemble of plausible structures

Rosetta Scoring Function
• Global scoring function issues:–Distinguish native-like structures from not. Generation
methods unlikely to produce exact native structure.–“Decoy” testing. Create many structures that are
plausible and not too far from native fold, and try todistinguish these
• Bayesian approach:
• Sequence dependent and sequence independentevaluation of predicted structure.
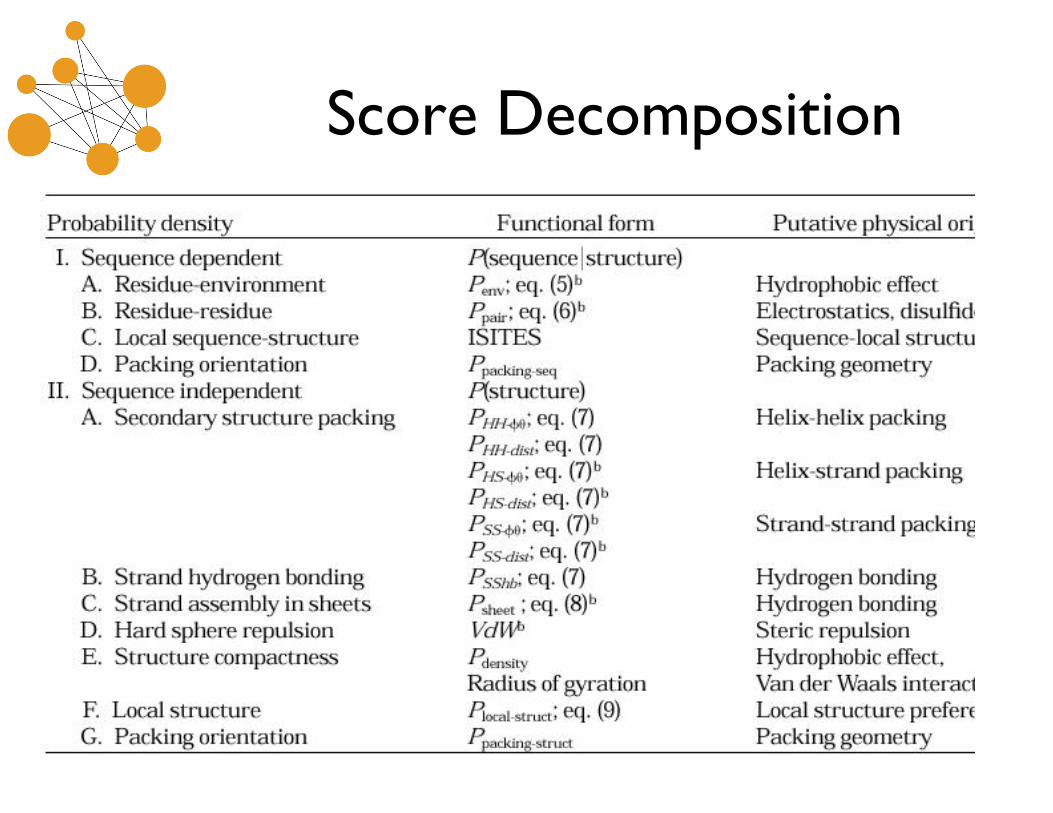
Score Decomposition

Good Performance
• An ab initio target
• Red = correct, Grey = incorrect
• Missed a sheet
• Good overalltopology
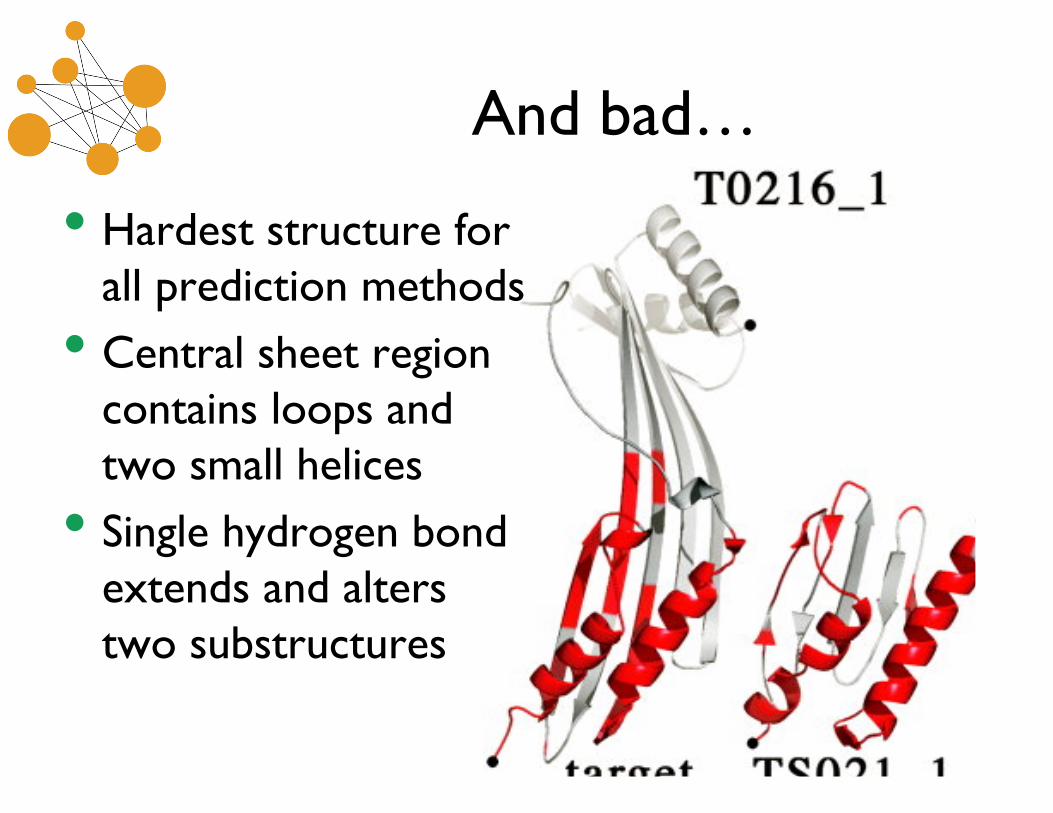
And bad…
• Hardest structure forall prediction methods
• Central sheet regioncontains loops andtwo small helices
• Single hydrogen bondextends and alterstwo substructures






















