Embed Size (px)
Citation preview

1521-0081/68/4/1110–1142$25.00 http://dx.doi.org/10.1124/pr.115.010991PHARMACOLOGICAL REVIEWS Pharmacol Rev 68:1110–1142, October 2016Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
ASSOCIATE EDITOR: PAUL A. INSEL
Proteinases, Their Extracellular Targets, andInflammatory Signaling
Rithwik Ramachandran, Christophe Altier, Katerina Oikonomopoulou, and Morley D. Hollenberg
Inflammation Research Network-Snyder Institute for Chronic Disease, Department of Physiology & Pharmacology (R.R., C.A., M.D.H.) andDepartment of Medicine (M.D.H.),University of Calgary Cumming School of Medicine, Calgary, Alberta, Canada; Department of Pathologyand Laboratory Medicine, Toronto Western Hospital, Toronto, Ontario, Canada (K.O.); and Department of Physiology and Pharmacology,
Western University, London, Ontario, Canada (R.R.)
Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1111I. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1111II. Classification and Overview of Mechanisms Regulating Proteolytic Enzymes . . . . . . . . . . . . . . . . 1112III. Proteinase-Mediated Signaling: Molecular Targets that Trigger the Response . . . . . . . . . . . . . . . 1114
A. Igniting the Inflammatory Response via Proteolytic Signaling. . . . . . . . . . . . . . . . . . . . . . . . . . . 1114B. Molecular Targets. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1114
1. Proteolytic Cascades that Generate Active Inflammatory Polypeptides: TheCoagulation, Complement, Kallikrein-kininogen, and Tissue Kallikrein-kallikrein-Related Peptidase Systems. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1114
2. Complement and Other Interconnected Proteolytic Cascades. . . . . . . . . . . . . . . . . . . . . . . . . 11143. Plasma Kallikrein-kinin and Kininogens: The Kallikrein-kinin System . . . . . . . . . . . . . . . 11164. Kallikrein-Related Peptidases and the Production of Inflammation-Related Peptides . 11165. Generation of Kinins by Kallikrein-Related Peptidase 1. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11176. Interplay between Kallikrein-Related Peptidases, Coagulation Proteinases,
and the Complement Cascade. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1117C. Hormone-like Signaling by Proteinases: Mimicking the Action of Insulin and
Mitogenic Polypeptides. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1117D. Proteinase-Activated G Protein-Coupled Receptors: A New Paradigm for G Protein-
Coupled Receptor Signaling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11181. “Tethered Ligand” Receptor Domains Responsible for Proteinase-Activated
Receptor Activation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11182. Synthetic Receptor-activating Peptides Based on the Tethered Ligands . . . . . . . . . . . . . . 11193. “Noncanonical” Proteinase-Activated Receptor Tethered Ligands Revealed by
Proteinases other than Thrombin and Trypsin. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11194. Regulation of Proteinase-Activated Receptor Signaling: Desensitization,
Internalization, and Intracellular Targeting . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11205. Proteinase-Activated Receptor 1. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11226. Proteinase-Activated Receptor 2. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11237. Proteinase-Activated Receptors 3 and 4 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11238. Effectors that Mediate Proteinase-Activated Receptor Signaling. . . . . . . . . . . . . . . . . . . . . . 11239. Posttranslational Modifications of Proteinase-Activated Receptors and Proteinase-
Activated Receptor Function . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1123E. Endogenous Proteolytic Regulators of Proteinase-Activated Receptor Function and
Proteinase-Activated Receptor Signaling. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11241. Coagulation Cascade Enzymes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1124
Work in the authors’ laboratories that underpins the information in this review is funded by operating grants from the Canadian Institutesfor Health Research (M.D.H., C.A.), Prostate Cancer Canada (M.D.H., C.A., R.R.), the Natural Sciences and Engineering Research Council ofCanada (C.A.), and The Calgary Motorcycle TELUS Ride for Dad and the Prostate Cancer Fight Foundation (M.D.H.).
Address correspondence to: Morley D. Hollenberg, Department of Physiology & Pharmacology, University of Calgary Cumming Schoolof Medicine, 3330 Hospital Drive NW, Calgary AB, Canada T2N 4N1. E-mail: [email protected]
dx.doi.org/10.1124/pr.115.010991.
1110
by guest on Decem
ber 16, 2021D
ownloaded from

2. Kallikrein-Related Peptidases and Proteinase-Activated Receptor Signaling . . . . . . . . . . 11253. Neutrophil Proteinases as Regulators of Proteinase-Activated Receptor Function . . . . . 11264. Matrix Metalloproteinases as Proteinase-Activated Receptor Regulators . . . . . . . . . . . . . 11275. Cathepsins, Proteinase-Activated Receptor 2 Activation, and Inflammatory Pain . . . . . 1127
F. Proteinase-Activated Receptor Regulation by Pathogen and Allergen-DerivedProteinases: Enzymes from Cockroaches, Molds, Dust Mites, and Trypanosomes . . . . . . . . 1128
IV. Adhesion G Protein-Coupled Receptors: A Proteinase-Activated Receptor-like TetheredLigand Signaling Mechanism Driven by Receptors with Intrinsic Proteinase Activity . . . . . . . . 1128
V. Proteolytic Regulation of Ion Channels . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1129A. Proteolytic Activation of L-type Calcium Channels . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1129B. Proteolytic Cleavage of the Amiloride-sensitive Epithelial Na+ Channel (ENaC/
Degenerin Family): Epithelial Na+ Channel Activation by Proteolytic Enzymes . . . . . . . . . . 1131C. Proteolytic Cleavage of the Acid-Sensing Ion Channels. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1132D. Proteolytic Regulation of Voltage-Gated Ion Channels by b-Secretase and ɣ-Secretase . . . 1132
VI. Therapeutic Targeting of the Proteolytic Signaling System. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1133VII. Future Perspectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1135
A. Targeting the Receptor. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1135B. Targeting the Proteinase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1135C. Potential Use of Broad-Spectrum Enzyme Inhibitors in a Restricted Setting over a
Limited Time Frame: A Heretical Approach to Consider . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1135Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1136References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1136
Abstract——Given that over 2% of the human genomecodes for proteolytic enzymes and their inhibitors, it isnot surprising that proteinases serve many physiologic-pathophysiological roles. In this context, we providean overview of proteolytic mechanisms regulatinginflammation, with a focus on cell signaling stimulatedby the generation of inflammatory peptides; activationof the proteinase-activated receptor (PAR) family of Gprotein-coupled receptors (GPCR), with a mechanism incommonwithadhesion-triggeredGPCRs (ADGRs); andbyproteolytic ion channel regulation. Thesemechanismsare
considered in the much wider context that proteolyticmechanisms serve, including the processing ofgrowth factors and their receptors, the regulation ofmatrix-integrin signaling, and the generation andrelease of membrane-tethered receptor ligands. Thesesignaling mechanisms are relevant for inflammatory,neurodegenerative, and cardiovascular diseases as wellas forcancer.Wepropose that the inflammation-triggeringproteinases and their proteolytically generated substratesrepresent attractive therapeutic targets and we discussappropriate targeting strategies.
I. Introduction
Proteolytic enzymes or their inhibitors (e.g., the serpins)represent more than 2% of the human genome (Puenteet al., 2005). Given that large genomic investment, it isnot surprising that proteinases (colloquially termed“proteases”) are used physiologically to serve multiplebiologic functions. Thus, added to their long-recognizedroles as digestive enzymes for nutrient assimilation,proteinases can now be seen as “hormone-like” media-tors that regulate target tissues by both nonreceptorand receptor-mediated mechanisms. Thus, in the set-ting of tissue inflammation triggered by injury orcancer, the activation and inactivation of proteinasesin the extracellular microenvironment can regulate cell
function in an autocrine-paracrine hormone-like man-ner as illustrated in Fig. 1. The central hypothesis thatunderlies this review is therefore that signaling byextracellular microenvironment proteinases caused bycleaving a number of distinct targets plays a major rolein a wide spectrum of inflammatory diseases rang-ing from arthritis to colitis and cancer. These targetsinclude the precursors that yield inflammatory pep-tides, cell membrane receptors including those forinsulin and other growth factors, proteinase-activatedG protein-coupled receptors (PARs) that play a cen-tral role in proteinase-stimulated signaling, integrinand adhesion receptors (ADGRs), and also ion channels(Fig. 2).
ABBREVIATIONS: ACE, angiotensin converting enzyme; ADGR, adhesion G protein-coupled receptors (formerly designated aGPCRs);APC, activated protein-C; ASIC, acid-sensitive ion channel; BACE, b-site of amyloid precursor protein; Der p, Dermatophagoidespteronyssinus; ENaC, epithelial sodium channel; EPCR, endothelial cell protein C receptor; GAIN, GPCR autoproteolysis-inducing domain ofadhesion GPCRs; GPS, GPCR proteolysis site within the GAIN domain; KLK, kallikrein-related peptidase family that includes “prostate-specific antigen”/KLK3; MMP, matrix metalloproteinase; PAR, proteinase-activated receptor (PAR1, PAR2, PAR3, PAR4); PSA, prostate-specific antigen; TIMP, tissue inhibitor of metalloproteinase; TF, tissue factor; TL, tethered ligand that activates PARs upon proteolyticunmasking.
Proteinase-Mediated Signaling and Inflammation 1111

II. Classification and Overview of MechanismsRegulating Proteolytic Enzymes
Proteolytic enzymes represent a diverse set of pro-teins, ranging in size from relatively small 20-kDaenzymes that fold to generate the catalytic pocket(e.g., cysteine cathepsins) to large multidomain com-plexes (e.g., the 26S proteasome ;2000 kDa). Based ontheir different catalytic mechanisms, proteinases aredivided into five major classes named for the key amino
acid (or metal ion) involved in peptide bond cleavage(Fig. 2). These classes include the serine, threonine,cysteine, aspartic, and metalloproteinases (see theMEROPs database: http://merops.sanger.ac.uk/). Ofthese, the serine, cysteine, and metalloproteinase fam-ilies are large, with over a 100 members in each class,whereas the threonine and aspartic proteinase familiesare much smaller, with fewer than 30 members each(Lopez-Otin and Bond, 2008). Of relevance to the themeof proteinase-mediated signaling, each of the differentproteinase classes can cleave the same target protein atdistinct sites, and thus each enzyme can have distinctsignaling properties although the target protein mightbe the same one. Given the chaos that unrestrictedproteolysis could cause in biologic systems, proteinasesare under strict regulatory control to ensure that theiraction occurs only in the appropriate context and onlyto a tightly limited extent. This issue is of particu-lar importance in the setting of inflammation, whereextensive proteolysis can occur. Thus endogenous pro-teinase inhibitors are as important for regulating sig-naling as are the proteinases themselves.
A number of mechanisms have evolved to regulateproteinase function. The first step in the regulation ofproteinases occurs at the transcriptional level. Whereasproteasomal and lysosomal enzymes that are involvedin general protein turnover are ubiquitously expressed,other proteinases exhibit significant restriction in theirtissue and cellular distribution and in the timing oftheir expression. The selective expression of the
Fig. 2. Proteolytic enzyme families and their targets that generate cell signaling. The pie chart illustrates the different families of proteinases, sortedaccording to their catalytic mechanisms with the areas reflecting their relative abundance in the genome. The seven different kinds of targets thatgenerate cell signaling as discussed in the text are listed. Those mechanisms highlighted in blue text are more extensively discussed than the otherprocesses.
Fig. 1. Microenvironment proteases, inflammation, and cancer: Invadingtissue inflammatory and tumor cells along with stromal cells secreteproteases into the tissue microenvironment that in turn drive signalingvia the mechanisms outlined in this review to cause inflammatoryresponses, cytokine production, cell division, invasion, and cell migration(large red arrows). Proteinases and proteolytic fragments can be releasedinto plasma and urine to serve as disease biomarkers (large blue arrows).
1112 Ramachandran et al.

neutrophil enzyme cathepsin-G in azurophillic granules(Salvesen et al., 1987; Conus and Simon, 2008) is anexample of such selective transcriptional regulation ofproteinase expression. In the context of inflammation,transcriptional regulation of both the proteinases andtheir inhibitors can also occur. For instance, a number ofmatrix metalloproteinases (MMPs) are reported to beupregulated during inflammation in a nuclear factor-kB-dependent manner (Bond et al., 1998, 2001; Trivediet al., 2006). Furthermore, the inhibitors of the MMPs(tissue inhibitors of matrix metalloproteinases, TIMPs)are also subject to change in inflammatory settings.Another important mechanism by which proteinase
function can be restricted to its site of action is throughthe synthesis of inactive zymogens, which requirelimited local proteolysis to be active on the final tar-get substrate. The activation of zymogens can occurthrough autocatalysis, as is the case for a number ofcysteine proteinases (Turk et al., 2001), or through thesequential “cascade” cleavage of a series of proteinasezymogens to generate active enzymes. This kind ofproteolytic cascade, which results in a considerableamplification of the initial proteolytic signal, is bestexemplified by the coagulation cascade that leadsultimately to the conversion of prothrombin to activethrombin (Davie and Ratnoff, 1964; Macfarlane, 1964).Both the complement and tissue kallikrein enzymecascades generate comparable proteinase-triggeredamplification networks (Yoon et al., 2007; Ricklinet al., 2010).In addition to these regulatory mechanisms for acti-
vating zymogens and terminating proteinase action byautoproteolysis or cleavage by other enzymes, enzymeactivity is regulated further by endogenous enzyme-targeted inhibitors (Rawlings et al., 2004). It is inter-esting to note that relative to the large numbers ofproteinases in the genome, the diversity of endogenousproteinase inhibitors, as outlined in Table 1, is morerestricted (Potempa et al., 1994; Gomez et al., 1997;Gettins, 2002). For example only four endogenousmetalloproteinase inhibitors, TIMP-1, TIMP-2, TIMP-3, and TIMP-4, have been identified for the large familyof MMPs (Gomez et al., 1997; Clark et al., 2008; Khokhaet al., 2013). Similarly, the numbers of cysteine
proteinases exceed the numbers of their endogenousinhibitors, including the cystatins and thyropins (Turket al., 2002; Dubin, 2005; Magister and Kos, 2013).Surprisingly, no endogenous inhibitors are known forthe serine proteinase, tryptase, or the aspartic protein-ases (Turk et al., 2012). The largest number of endog-enous inhibitors is described for those that target serineproteinases, except for tryptase, designated by theacronym, SERPINS (SERine Proteinase INhibitorS)(Potempa et al., 1994; Law et al., 2006). In addition tothese class-specific proteinase inhibitors, a2-macro-globulin is a circulating tetrameric plasma proteinaseinhibitor that forms an enzyme-covalent complex withall four catalytic classes to inhibit a relatively largespectrum of different proteinases (Barrett and Starkey,1973; Barrett, 1981; Rehman et al., 2013). Interestinglyinhibition of an enzyme by a2-macroglobulin may blockproteolysis of large substrates but may still allowcleavage of smaller substrates, such as in the case ofKLK3-alpha-2-macroglobulin or in the case of the alpha-2-macroglobulin-urokinase plasminogen activator com-plex, the latter of which has the ability to retain enzymeactivity to cleave plasminogen, while at the same timebeing resistant to inhibition by the plasminogen activa-tor inhibitor PAI-1 (Christensson et al., 1990; Komissarovet al., 2009). Of note, deficiencies in the actions ofproteinase inhibitors, for example alpha-1-antitrypsin(also designated, alpha1-PI) or the Kazal type relatedinhibitor type 5, lead to a number of diseases likeemphysema and skin inflammation (see Table 1 in Lawet al., 2006; Hovnanian, 2013). Alternatively, the upre-gulation of SerpinA3N and its inhibition of leukocyteelastase in the dorsal root ganglion after nerve injuryappear to be therapeutic in reducing neuropathic pain(Vicuña et al., 2015). That same study showed that theadministration of this serpin can attenuate mechani-cal allodynia in a mouse model of pain. Thus both thedownregulation and upregulation of proteinase inhib-itors can play important physiologic roles in manysettings.
To sum up, the inflammatory milieu can generatemultiple proteinases of five catalytically distinct clas-ses, along with a more restricted number of proteinaseinhibitors (Table 1). These enzymes along with their
TABLE 1Proteinase families and their endogenous inhibitors
Enzyme Family Inhibited Inhibitor Class Comment References
Serine proteinases SERPINs (serineproteinase inhibitors)
Largest number of endogenous inhibitors;deficiencies cause lung and skin disease;upregulation involved in modulatingneuropathic pain
Potempa et al., 1994; Law et al., 2006; Vicuñaet al., 2015
Metalloproteinases(MMPs)
TIMPs 1, 2, 3, and 4 TIMPs elevated in cancer and inflammation;upregulation promotes fibrosis
Clark et al., 2008; Khokha et al., 2013; Arpinoet al., 2015
Cysteine proteinases Cystatins, thyropins Intracellular inhibitors regulate apoptosis Turk et al., 2002; Dubin, 2005; Magister andKos, 2013
General Alpha 2-macroglobulin Alpha 2-M preserves catalytic site function forsmaller substrates
Barrett and Starkey 1973; Barrett, 1981;Rehman et al., 2013
Proteinase-Mediated Signaling and Inflammation 1113

inhibitors will differ from one inflammatory setting toanother and thus their common and/or distinct sub-strate selectivities can influence the inflammatorysignaling process in diverse ways. For that reason,proteinases have long been considered as importanttherapeutic targets for inflammatory disease. However,the complexities of their activation and inhibition, alongwith the common mechanisms of catalysis within afamily have made the design of potent and enzyme-selective inhibitors very challenging. Nonetheless, a keyissue to sort out is which proteinases and whichmolecular targets lead to signaling?
III. Proteinase-Mediated Signaling: MolecularTargets that Trigger the Response
A. Igniting the Inflammatory Response viaProteolytic Signaling
Amplified proteolytic signaling in a localized setting isof key importance in triggering an inflammatory responseresulting from tissue injury or from invading infectiousagents. Injury to a finger provides all of us with a familiarexperience of this “hormonal role” of proteinases. Thus,shortly after the immediate “pain response” to the injurydue to the stimulation of sensory nerves, the extrinsic andintrinsic coagulation pathways are activated, resulting inan amplified proteolytic signaling pathway that gener-ates thrombin and other members of the coagulationcascade, including the counterregulatory proteinase,activated protein-C (APC). Shortly thereafter, 1) theblood clots, due to the proteolytic conversion of fibrinogento fibrin and 2) all of the hallmark signs of inflammationare generated:more pain (dolor) and increased blood flow(rubor), resulting in increased warmth at the tissuesurface (calor), increased fluid leakage with swelling(tumor), and subsequently, the scarring that can lead toreduced function (functio laesa).This activation of the coagulation cascade is tightly
linked to triggering of the complement and plasmakallikrein-kininogen systems, both of which representenzymatic cascades that generate inflammatory pep-tides. After these responses generated by enzymes ofthe coagulation, kininogen, and complement cascades,the cellular “shock-troops” of inflammation (neutro-phils) migrate into the site of injury to initiate a slightlydelayed component of the acute inflammatory response.The influx of neutrophils, followed by other leukocytes,leads to proteinase secretion that stimulates furthertissue responses along with the generation of cytokinesin the microenvironment. All of these processes repre-sent the “innate inflammatory response” aimed atcombating the injurious stimulus and initiating theprocess of healing. All of these inflammatory responsesare regulated by proteinase-mediated signaling, involv-ing enzymes of the coagulation, kininogen, and comple-ment cascades and proteinases secreted by the invadingneutrophils and other cells at the site of injury.
B. Molecular Targets
The generic targets for proteinase-mediated signaling(Fig. 2) vary considerably in their protein sequences andanatomic-tissue specific locations. These substratesrange from extracellular matrix components to veryspecific cell surface receptors like the one for insulin.Although the entire repertoire of proteinase-cleavedsubstrates is too large to cover in this review, we wish tohighlight here the ability of proteinases to signal bygenerating extracellular bioactive peptides like thekinins and complement pathway-produced peptides,by activating cell surface receptors, including the in-sulin receptor and the PAR family of G protein-coupledreceptors and by regulating ion channels. Together,these proteolytically triggered mechanisms control awide variety of complex cell behaviors involved in theinflammatory response.
1. Proteolytic Cascades that Generate Active Inflam-matory Polypeptides: The Coagulation, Complement,Kallikrein-kininogen, and Tissue Kallikrein-kallikrein-Related Peptidase Systems. In addition to the well-recognized proteolytic generation of active peptidehormones from precursor polypeptides as alluded toabove, e.g., via the action of prohormone convertases(Steiner et al., 1967; Steiner, 2011; Chretien, 2012), anumber of proteolytic cascades are now known to yieldinflammatory peptides by processing extracellularrather than intracellular substrates, via mechanismsoutlined elsewhere in some detail (Turk et al., 2012).The coagulation cascade (Fig. 3), which triggers cellsignaling via the G protein-coupled proteolytically acti-vated receptor family (PARs), is also linked to two otherplasma-localized proteolytic cascades that generate in-flammatory polypeptide signals: the complement sys-tem (Fig. 4) and the plasma kallikrein-kininogen-kininsystem (Fig. 5). Another more recently recognized cas-cade, that of the tissue kallikrein family, now termedkallikrein-related peptidases (KLKs), is also cominginto sharper focus as a multiproteinase inflammation-related cascade network that can be activated in set-tings of inflammatory disease (Yoon et al., 2007;Sotiropoulou and Pampalakis, 2010; Hovnanian, 2013;de Veer et al., 2014). These interconnected proteolytic“signal amplification” cascades represent key constitu-ents of the innate immune inflammatory responses toinjury, and their signaling is coordinated with that ofthe coagulation cascade (Joseph and Kaplan, 2005;Kaplan and Ghebrehiwet, 2010).
2. Complement and Other Interconnected ProteolyticCascades. The complement proteolytic cascade is well-recognized as a source of inflammatory peptides (Fig. 4).These peptides signal to cells via their peptide-selectiveG protein-coupled receptors (Marceau and Regoli, 2004;Leeb-Lundberg et al., 2005; Alexander et al., 2013).What is now evident is that not only is the complementcascade linked to the coagulation cascade (Fig. 3, top
1114 Ramachandran et al.
left) (Kaplan and Ghebrehiwet, 2010; Oikonomopoulouet al., 2012), but the tissue kallikrein-kallikrein-relatedpeptidase family can also generate active complement-derived peptides in addition to signaling via the PARs(Oikonomopoulou et al., 2006). The KLK-generatedcomplement-derived peptides trigger inflammatory sig-nals via the C3a receptor (Oikonomopoulou et al., 2013).Both the KLKs and the coagulation enzymes can causethe generation of the complement-derived molecules,C3a, C3b, C5a, or C5b, which among other actions canalso, like thrombin, regulate platelet function [aggrega-tion; ADP release (Polley and Nachman, 1983)] andtrigger tissue inflammation via their G protein-coupledreceptor targets, C3AR1 and C5AR1 (Alexander et al.,2013; Klos et al., 2013). This generation of complement-derived inflammatory peptides can thus occur in stepwith the ability of the coagulation cascade enzymes, theplasma kallikrein, and the KLKs to liberate kinins andto activate PARs. It is thus possible to link theactivation of target cells by enzymes of the coagulationpathway with those triggered by the complementsystem along with the kininogen and KLK cascadesystems. A common theme for these proteolytic systems
is that they represent an amplification mechanism forsensing injury because of 1) the large signal magnifica-tion process inherent in a catalytic proteolytic cascadeand 2) the subsequent activation of “hormone-like”receptor-mediated signaling pathways that in turngreatly amplify the initial proteinase-triggered signal.
The complement network that generates the inflam-matory peptides consists of more than 50 plasma-borneand membrane-bound proteins that form an essentialcomponent of the innate defense system. This networkforms a first line of defense against microbial invadersand exerts important functions in immune surveillanceand homeostasis (Ricklin et al., 2010). Activation of thecomplement system is primarily initiated by pathogen-and damage-associated molecular surface patterns (of-ten abbreviated by the acronyms PAMPs or DAMPs)acting via cell surface receptors. The complement-generated process is commonly categorized under threemajor pathways (classic, lectin, and alternative) ofstepwise proteolytic activation (Fig. 4). However, thereis increasing evidence for direct activation of individualcomplement components by proteinases extrinsic to thecomplement cascade, as part of an intricate crosstalkbetween physiologic effector systems (Le Friec andKemper, 2009; Ricklin et al., 2010). Under this extrinsicinfluence, active complement factors C3a and C5a (alsoknown as anaphylatoxins) and inactive derivatives areknown to be released by the proteolytic action of trypsin(Wetsel and Kolb, 1982; Eggertsen et al., 1985), plasma
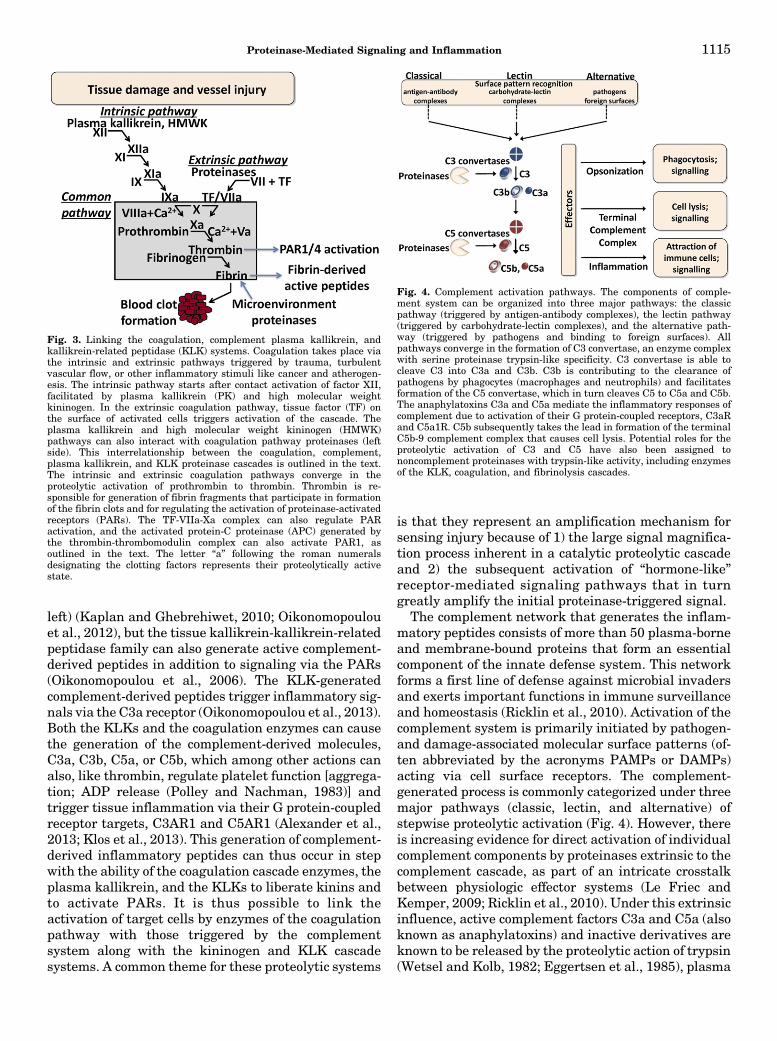
Fig. 3. Linking the coagulation, complement plasma kallikrein, andkallikrein-related peptidase (KLK) systems. Coagulation takes place viathe intrinsic and extrinsic pathways triggered by trauma, turbulentvascular flow, or other inflammatory stimuli like cancer and atherogen-esis. The intrinsic pathway starts after contact activation of factor XII,facilitated by plasma kallikrein (PK) and high molecular weightkininogen. In the extrinsic coagulation pathway, tissue factor (TF) onthe surface of activated cells triggers activation of the cascade. Theplasma kallikrein and high molecular weight kininogen (HMWK)pathways can also interact with coagulation pathway proteinases (leftside). This interrelationship between the coagulation, complement,plasma kallikrein, and KLK proteinase cascades is outlined in the text.The intrinsic and extrinsic coagulation pathways converge in theproteolytic activation of prothrombin to thrombin. Thrombin is re-sponsible for generation of fibrin fragments that participate in formationof the fibrin clots and for regulating the activation of proteinase-activatedreceptors (PARs). The TF-VIIa-Xa complex can also regulate PARactivation, and the activated protein-C proteinase (APC) generated bythe thrombin-thrombomodulin complex can also activate PAR1, asoutlined in the text. The letter “a” following the roman numeralsdesignating the clotting factors represents their proteolytically activestate.
Fig. 4. Complement activation pathways. The components of comple-ment system can be organized into three major pathways: the classicpathway (triggered by antigen-antibody complexes), the lectin pathway(triggered by carbohydrate-lectin complexes), and the alternative path-way (triggered by pathogens and binding to foreign surfaces). Allpathways converge in the formation of C3 convertase, an enzyme complexwith serine proteinase trypsin-like specificity. C3 convertase is able tocleave C3 into C3a and C3b. C3b is contributing to the clearance ofpathogens by phagocytes (macrophages and neutrophils) and facilitatesformation of the C5 convertase, which in turn cleaves C5 to C5a and C5b.The anaphylatoxins C3a and C5a mediate the inflammatory responses ofcomplement due to activation of their G protein-coupled receptors, C3aRand C5a1R. C5b subsequently takes the lead in formation of the terminalC5b-9 complement complex that causes cell lysis. Potential roles for theproteolytic activation of C3 and C5 have also been assigned tononcomplement proteinases with trypsin-like activity, including enzymesof the KLK, coagulation, and fibrinolysis cascades.
Proteinase-Mediated Signaling and Inflammation 1115

kallikrein (Wiggins et al., 1981; DiScipio, 1982), compo-nents of the coagulation cascade such as factor Xa andthrombin (Amara et al., 2010), the neutrophil-releasedproteinases elastase and cathepsin-G (Orr et al., 1979),and mast cell tryptase (Fukuoka et al., 2008). Theanaphylatoxins C3a and C5a can signal to cells andtissues mainly via two members of the G protein-coupled receptor family, the C3AR1 and C5AR1 recep-tors (Klos et al., 2009, 2013; Alexander et al., 2013). Notsurprisingly, the downstream signaling initiated by thecomplement system-generated peptides via their GPCRtargets hasmuch in commonwith inflammation-relatedsignaling by the PARs (see below) and kinin-stimulatedreceptors. The recent availability of C3AR1-targetedagonist ligands will enable a more extensive interroga-tion of this area of interest (Reid et al., 2014).3. Plasma Kallikrein-kinin and Kininogens: The
Kallikrein-kinin System. Plasmakallikrein, or “Fletcherfactor,” is a complex serine proteinase, genomically dis-tinct and not to be confused with the other family of“kallikrein” enzymes known as “tissue kallikreins” or,more correctly, kallikrein-related peptidases (KLKs)(Chung et al., 1986; Bhoola et al., 1992). The plasmakallikrein-kinin system is an endogenous proteolyticcascade that catalyzes the release of vasoactive kinins(bradykinin-related peptides) from plasma-located pre-cursors (high and low molecular weight kininogens). Theactive kinins that trigger their G protein-coupled recep-tor targets are implicated in many physiologic and path-ologic processes (Fig. 5).Unlike theKLK family, the plasma kallikrein enzyme
that generates the kinins has a number of functionalprotein domains in addition to its catalytic domainresponsible for substrate cleavage (Chung et al., 1986;Bhoola et al., 1992). The plasma mechanism by whichhigh molecular weight kininogen is processed to brady-kinin and other inflammatory peptides is linked tightly
to the proteinase cascades for coagulation and fibrino-lysis (Joseph and Kaplan, 2005). Thus coagulationFactor XIIa is involved not only in the downstreamconversion of prothrombin to thrombin, but also in theconversion of prekallikrein to plasma kallikrein. As amajor function, plasma kallikrein, a key player in theinnate inflammatory response, catalyses the release ofthe inflammatory peptide bradykinin from a highmolecular weight precursor molecule (high molecularweight kininogen) produced by the liver. In turn,bradykinin triggers signals via the bradykinin Gprotein-coupled receptors (mainly B2, also possiblyB1). There is a longstanding extensive literature towhich the reader is referred (Bhoola et al., 1992;Marceau and Regoli, 2004; Leeb-Lundberg et al., 2005;Moreau et al., 2005; Whalley et al., 2012) dealing bothwith the biochemistry of the kinin-forming proteolyticcascade and with agonists and antagonists for thetarget bradykinin B1 and B2 receptors that convey theinflammatory signals.
Given the involvement of this proteolytic system indisorders of pain and inflammation, there are a numberof therapeutic settings in which plasma kallikrein, thekinins, and their receptors are of importance (Marceauand Regoli, 2004; Joseph and Kaplan, 2005; Moreauet al., 2005). Surprisingly, only one of the KLK familymembers, namelyKLK1 (termed tissue kallikrein in theolder literature) is also able to generate active kininsfrom a kininogen precursor.
4. Kallikrein-Related Peptidases and the Productionof Inflammation-Related Peptides. The KLKs, belongto an extensive family of tissue serine proteinases withtrypsin or chymotrypsin-like activity. Biochemically theKLKs are distinct from plasma kallikrein both in struc-ture and substrate specificity (Borgono and Diamandis,2004; Sotiropoulou and Pampalakis, 2010). The bestknown member of this family, now termed KLK3, waspreviously known as an androgen-regulated prostate-specific antigen (PSA). PSA was first discovered in1979 as a unique antigen in human prostatic tissueextracts (Wang et al., 1979, 1981). Although thisantigenic property of PSA was rapidly recognized, theproteolytic activity of human PSA, like that of thecomparable KLK serine proteinase in canine seminalplasma, was not appreciated for another 5 years (Banet al., 1984; Isaacs and Coffey, 1984). PSA/KLK3 is well-recognized as a biomarker for prostate cancer monitor-ing (Stephan et al., 2007; Bryant and Lilja, 2014). It isnow known that this enzyme belongs to a large familyof serine proteinases, which are encoded on chromo-some 19 of the human genome, consisting of 15 distinctenzymes [KLK1 to KLK15 (Borgono and Diamandis,2004; Sotiropoulou and Pampalakis, 2010)]. KLK3/PSA,along with KLKs 7 and 9 are “chymotryptic” in termsof their substrate specificity, whereas the other 12members of the human KLK family have a trypsin-like selectivity (Clements et al., 2001; Yousef and
Fig. 5. Role of kallikreins in kininogen fragmentation. In response totrauma, immune, and proteolytic triggers, plasma and tissue kallikreinsare both potent generators of kinins. Plasma kallikrein is able to releasebradykinin from high molecular weight kininogen (HMWK), whereastissue kallikrein (also known as KLK1) fragments lower molecular weightkininogen (LMWK) to lysyl-bradykinin. The two interconnected kininforms act as agonists of G protein-coupled bradykinin B1 and B2receptors to cause pain, vasodilation, vascular permeability, andchemotaxis.
1116 Ramachandran et al.

Diamandis, 2001). These enzymes, well-recognized asfruitful cancer biomarkers, are now known to be widelyexpressed in many organs and to have a broad impacton cell function (Kuriyama et al., 1980; Yousef andDiamandis, 2001, 2009; Sotiropoulou et al., 2009). Inrodents and other species, gene duplication has resultedin the generation of more than 25 KLK genes, some ofwhich are pseudogenes and are not expressed asfunctional proteinases (Clements, 2008; Lundwall andBrattsand, 2008; Lundwall, 2013). The KLKs areupregulated hormonally (e.g., androgens, estrogens,progestins, vitamin D) in many tissues, mainly ofepithelial origin (Lawrence et al., 2010), and can befound in tumor-derived biologic fluids, such as ascitesfluid and in sera from cancer patients. A wide array ofpotential substrates has been identified whereby KLKsmay affect cell function (Sotiropoulou et al., 2009;Lawrence et al., 2010). In this context, studies donein vitro have singled out proteins of the extracellularmatrix, pro-urokinase-plasminogen activator (pro-uPA),kininogens, growth factor precursors (and bindingproteins), cytokines, insulin-A and -B chains, and otherKLKs, all as potential targets of kallikrein proteolysis,particularly in the setting of cancer progression(Frenette et al., 1997; Takayama et al., 2001; Borgonoand Diamandis, 2004; Lawrence et al., 2010). A recent“degradomic” approach has identified other potentialKLK substrates cleaved in cultured ovarian cancercells by the combined actions of KLKs 4, 5, 6, and7 (Shahinian et al., 2014). Such substrates (e.g., extra-cellular matrix; pro-urokinase-plasminogen activator,growth factor precursors, kininogens, TGFb-1-precur-sor) may well explain some but by no means all of thephysiologic signaling actions of the KLKs, particularlyin the setting of inflammatory disease and cancer. Theprecise mechanisms whereby the KLKs can play anactive role in pathophysiology are not fully understood.Some concrete examples of KLK-dependent signalingare discussed in the following sections.5. Generation of Kinins by Kallikrein-Related Pepti-
dase 1. A concrete example of KLK-dependent signal-ing can be seen forKLK1. Like plasmakallikrein (above),one of the KLK family members, namely KLK1, can pro-duce bradykinin receptor-stimulating peptides from akininogen precursor. Thus KLK1, also formerly knownas tissue, urinary, pancreatic, or renal kallikrein, unlikethe other KLKs, possesses kininogenase activity. Thisactivity is responsible for the cleavage of a low molecularweight precursor kininogen, produced by the liver, torelease lysyl-bradykinin (Fig. 5) (Moreau et al., 2005).Lysyl-bradykinin in turn activates the bradykinin B1receptor (Leeb-Lundberg et al., 2005; Moreau et al.,2005). KLK1, via its production of lysyl-bradykinin fromkininogen has been implicated in various physiologicprocesses, including the control of blood pressure andelectrolyte balance, aswell as in inflammation (Clements,1989; Margolius, 1998a,b).
6. Interplay between Kallikrein-Related Peptidases,Coagulation Proteinases, and the Complement Cascade.The coagulation cascade is integrally linked to thecomplement and kallikrein-kinin cascades. Observa-tions in the 1980s pointed to the existence of signifi-cantly higher levels of complement activation productsin human serum compared with anticoagulated blood,strongly suggesting the development of complementactivation during blood clotting (Mollnes et al., 1988).It is now clear that there is crosstalk between thecomplement and coagulation cascades, either at thelevel of anaphylatoxin generation (Wiggins et al., 1981;DiScipio, 1982; Hiemstra, 2002; Amara et al., 2010),upstream in the complement pathways (Ghebrehiwetet al., 1981, 1983), or via the generation of activatedthrombin (Krarup et al., 2007; Gulla et al., 2010).Complement effectors can also facilitate biochemicaland morphologic changes that can indirectly affectcoagulation (Oikonomopoulou et al., 2012). For exam-ple, the anaphylatoxin C3a and its derivative C3adesArg
can induce platelet activation and aggregation directly(Polley and Nachman, 1983).
The KLKs are also known to interconnect with thecoagulation proteinases, mainly by cross-activating/deactivating proteinases of each other’s cascade or byaffecting receptor signaling, e.g., via the proteinase-activated receptor family (PARs) and the urinaryplasminogen activator receptor uPAR (Blaber et al.,2010). Furthermore, an impact of the KLKs on the com-plement cascade is also now known (Oikonomopoulouet al., 2013). Thus KLKs are able to generate theanaphylatoxin, C3a peptide, by acting on C3 outside ofthe “regular” complement activation cascade. In thecase of KLK14, the anaphylatoxin was shown to pos-sess significant biologic functions (Oikonomopoulouet al., 2013). In summary, four key proteolytic cascades,involving enzymes of the coagulation pathway, the com-plement system, plasma kallikrein, and the kallikrein-related peptidase/KLK family play important roles inthe innate inflammatory response by generating activepeptides that signal to cells via their specific receptors.This “indirect mode” of regulating cell signaling via thegeneration of peptide agonists for receptors is paralleledby the direct action of some of these enzymes to regulatea number of cell surface receptors, including growthfactor receptors like the one for insulin, G protein-activatedreceptors, and ion channels, as outlined in the followingsections
C. Hormone-like Signaling by Proteinases: Mimickingthe Action of Insulin and Mitogenic Polypeptides
In the mid-1960s, it was discovered that enzymes liketrypsin and chymotrypsin can cause hormone-likemetabolic-anabolic effects in tissues and cells akin tothe actions of insulin (Rieser and Rieser, 1964; Rieser,1967; Kono and Barham, 1971). This “insulin-like”action of trypsin on fat cells and rat diaphragm tissue
Proteinase-Mediated Signaling and Inflammation 1117

is now known to result from the cleavage of theextracellular alpha-subunit of the insulin receptor.That cleavage removes a negative regulatory domainfrom the extracellular insulin receptor alpha-subunitto release its inhibitory control of receptor functionand to enable receptor autophosphorylation-activation(Shoelson et al., 1988). At higher trypsin concentra-tions, the insulin binding site on the receptor is re-moved, thus “disarming” the receptor and preventinginsulin-triggered signal transduction (Cuatrecasas,1969, 1971). These data obtained for signaling by theinsulin receptor very likely also apply to signaling bythe insulin-like growth factor receptors. The dataobtained for the effect of trypsin on insulin signalingestablished the principle that tissue proteinases re-leased at sites of inflammation can have a “bidirec-tional” direct impact on receptors both to activate andsilence signaling. These anabolic actions of trypsin andother proteinases mimic the mitogenic receptor-mediated effects of polypeptide growth factors such asinsulin and epidermal growth factor (Burger, 1970;Sefton and Rubin, 1970; Chen and Buchanan, 1975;Carney and Cunningham, 1977, 1978). To date, thiseffect of tissue proteinases on insulin receptor signaling,and by extension, signaling via the insulin-like growthfactor-1 receptor, has gone largely unrecognized andhas not yet been explored in any depth. In principle, thisproteolytic mode of activation of the insulin receptorcould play a role in inflammatory disease (Hyun et al.,2010). The impact of proteinases on the regulation ofother “growth factor” receptors is a topic of interest forfurther study. The action of trypsin to signal via theinsulin receptor heralded the discovery of its ability toregulate cell function by activating a novel family of Gprotein-coupled receptors, the “PARs.” The theme thatlinks the proteinase-mediated activation of a growthfactor receptor and a G protein-coupled receptor is thatthere is a commonality in the downstream signalpathways that are ultimately triggered by both kindsof receptors.
D. Proteinase-Activated G Protein-Coupled Receptors:A New Paradigm for G Protein-CoupledReceptor Signaling
At the time the Riesers discovered the anabolicinsulin-like actions of trypsin and other proteinases instriated muscle (Rieser and Rieser, 1964; Rieser, 1967),it also became clear that the catalytic activity ofthrombin and other serine proteinases can regulateplatelet function (Davey and Luscher, 1967; Ganguly,1974; Martin et al., 1975). However, the mechanismswhereby the proteolytic enzymes activated plateletsand stimulated fibroblast mitogenesis were not known.It was not until the early 1990s that a receptormechanism for these “hormone-like” actions of proteo-lytic enzymes was discovered. That discovery resultedfrom a search that spanned over 30 years (time frame
shown in Table 2) to identify the “receptors” responsiblefor the action of thrombin 1) to stimulate humanplatelet function and 2) to trigger mitogenesis incultured cells. To the surprise of many, it turned outthat thrombin causes these effects by stimulating aproteolytically activated receptor (PAR) that is a mem-ber of the G protein-coupled receptor (GPCR) super-family (Rasmussen et al., 1991; Vu et al., 1991; Adamset al., 2011). This receptor mechanism whereby protein-ases can signal to cells is shown in Fig. 6. One mainfocus of this review is on these proteolytically triggeredproteinase-activated receptors or PARs, for which theunique mechanism of proteolytic activation was discov-ered by the Coughlin laboratory (Vu et al., 1991). It isimportant to recognize that although thrombin was theindex proteinase leading to the discovery of the PARs,enzymes other than those of the coagulation cascade,like trypsin, the KLKs, and neutrophil-secreted en-zymes can also signal via these unique receptors.Furthermore, after the discovery of PAR1 (known onlyas “the thrombin receptor” at the time), additionalpharmacological (Hollenberg et al., 1993; Kinlough-Rathbone et al., 1993) and gene knockout studies veryrapidly pointed to the existence of additional membersof this proteolytically activated family of receptors. Asoutlined in Tables 2 and 3, continued work then led tothe discovery of PAR2 (Nystedt et al., 1994; Bohm et al.,1996b), PAR3 (Ishihara et al., 1997), and finally PAR4(Kahn et al., 1998; Xu et al., 1998). The PAR-triggeredsignaling mechanism, representing a paradigm shift forunderstanding proteinase function, can be seen to becomplementary to the other mechanisms illustrated inFig. 2, whereby circulating and tissue-derived protein-ases can play a role in inflammation.
1. “Tethered Ligand” Receptor Domains Responsiblefor Proteinase-Activated Receptor Activation. In addi-tion to cloning the human PAR1 thrombin receptor, theCoughlin group showed that the mechanism wherebythrombin activates PAR1 involves the proteolyticunmasking of a cryptic N-terminal receptor ligand, theso called tethered ligand (TL), that remaining attached,activates the receptor as shown in Fig. 6A, middle. Themechanism identified for PAR1 holds true for PAR2 andPAR4 as well. PAR2 is preferentially activated bytrypsin, although at relatively high concentrationsthrombin can also activate PAR2 (Mihara et al., 2016).Moreover, both trypsin and thrombin can activate PAR4with comparable potencies. The revealed tetheredligand of the PARs is thought to interact with extracel-lular loop-2 of the receptor (Lerner et al., 1996; Al-Aniet al., 1999). In the case of PAR3, thrombin cleavage canalso expose a tethered ligand sequence, but the abilityof this sequence to cause PAR3 activation to signal onits own is still unclear, despite some evidence in favorof independent signaling due to PAR3 activation(Ostrowska and Reiser, 2008). Instead, PAR3 appearsto act as a cofactor for thrombin-mediated activation of
1118 Ramachandran et al.

PAR4 (Nakanishi-Matsui et al., 2000). PAR3 may alsosignal as part of a heterodimer with PAR1, wherein theunmasked TL of PAR3 “reaches over” to activate PAR1(McLaughlin et al., 2007). Part of the confusion aboutthe signaling role of PAR3 stems from the fact thatsynthetic peptides based on its tethered ligand se-quence unmasked by thrombin efficiently activatePARs 1 and 2 (Hansen et al., 2004). Thus, some of theeffects attributed to PAR3 due to the actions of thePAR3-derived tethered ligand peptides are very likelydue to PARs 1 and 2 and not to PAR3 as suggested bysome publications.2. Synthetic Receptor-activating Peptides Based on
the Tethered Ligands. Along with the discovery thatthe thrombin PAR1 receptor is activated by a cryptictethered ligand peptide sequence, Coughlin and col-leagues found that short synthetic peptides based on thetethered ligand sequence could also trigger activation ofPAR1 without the need for receptor proteolysis (Fig. 6B,right) (Vu et al., 1991). In a similar way, syntheticpeptides based on the cryptic tethered ligand sequencesunmasked by proteolysis in PARs 2 and 4 have beendeveloped for the selective activation of PARs 2 and4 (Ramachandran et al., 2012b). Although these PAR-activating peptides are quite selective for activating thePARs (e.g., TFLLR-amide for PAR1, 2-furoyl-LIGRLO-amide for PAR2; AYPGKF-amide for PAR4), there canbe some off-target actions of the peptides. For instance,
the PAR2-activating peptides can cause effects in PAR2-null mice (McGuire et al., 2002), possibly by activatingthe Mas-related G protein-coupled receptor, MrgprC11(Liu et al., 2011). These off-target effects of the PAR-activating peptides canbe resolved by obtaining structure-activity profiles of the peptide-generated responses (Liuet al., 2011).
3. “Noncanonical” Proteinase-Activated ReceptorTethered Ligands Revealed by Proteinases other thanThrombin and Trypsin. In the studies resulting inthe cloning of PARs 1, 2, and 4, the key cleavage sitefor both thrombin (PARs 1 and 4) and trypsin (PAR2)(here designated by: //) was found to be at the serineproteinase-targeted N-terminal arginine in the humanPARs: R//SFLLRN—, for PAR1; R//SLIGKV—, forPAR2; and R//GYPGQV—for PAR4. However, it sub-sequently became apparent that, like trypsin andthrombin, other serine proteinases (for instance, theKLK peptidases), could also unmask the same tetheredligands revealed by thrombin and trypsin. Nonetheless,it was soon found that quite different proteinases[e.g., matrix metalloproteinase-1 (MMP-1), activatedprotein-C (APC), neutrophil elastase, proteinase-3, andcathepsin-S could also cause PAR activation/signalingby cleaving at distinct N-terminal residues to unmaskdifferent “noncanonical” receptor-activating tetheredligand’ sequences (Boire et al., 2005; Ramachandranet al., 2011; Mosnier et al., 2012; Mihara et al., 2013;
TABLE 2Time frame for discovery of the proteinase activated receptor family and its mechanism of activation
Year Milestone Observations Comment References
1967 Thrombin activates platelets bydi-isoflluoro-phosphate-sensitive serineproteinase action
Platelet activation by thrombin requiresits proteolytic activity
Davey and Luscher, 1967
1975 The thrombin catalytic step is followed byan irreversible, thrombin-independentplatelet process leading to secretion
Thrombin cleavage is seen to precede asecond independent platelet activationevent
Martin et al., 1975
1980 Thrombin platelet binding observed to beindependent of a subsequentchymotrypsin-sensitive plateletsignaling event
Thrombin activation is independent of adownstream chymotrypsin-sensitivereceptor activation process
Tam et al., 1980
1991 The thrombin “receptor” is identified byexpression cloning of its target fromhuman megakaryocytes and hamsterfibroblasts
The thrombin receptor is discovered to bea G protein-coupled receptor
Rasmussen et al., 1991;Vu et al., 1991
1991 Thrombin proteolysis unmasks anN-terminal receptor-activatingtethered ligand; peptides based on therevealed sequence activate the receptordirectly: Proteolytically activatedGPCRs (PARs) are identified.
The tethered ligand mechanismdistinguishes PARs from other GPCRs
Vu et al., 1991
1993 Bioassays provide evidence for multiplemembers of the PAR family
PAR-activating peptides stimulatehuman but not rat and rabbit platelets;distinct structure-activity profiles areobserved for PAR-activating peptides indifferent tissues
Hollenberg et al., 1993;Kinlough-Rathboneet al., 1993
1994-1998 Cloning of PARs 2, 3, and 4. All PARs have a target serine proteinasearginine cleavage-activation site incommon
Nystedt et al., 1994; Bohmet al., 1996b; Ishiharaet al., 1997; Kahn et al.,1998; Xu et al., 1998
2014 Adhesion G protein-coupled receptors(ADGRs) are found to be activated via atethered ligand mechanism in commonwith the PARs
The tethered ligand mechanism may notapply to all of the ADGR familymembers
Hamann et al.,. 2015;Liebscher et al., 2014b
Proteinase-Mediated Signaling and Inflammation 1119

Schuepbach et al., 2012; Zhao et al., 2014a,b). Unmask-ing of these distinct noncanonical tethered ligandsequences triggers so-called functional selectivity orbiased PAR signaling (Kenakin, 2011, 2013; Kenakinand Miller, 2010; Kenakin and Christopoulos, 2013)that specifically activates unique downstream signalingpathways that differ from those triggered by eitherthrombin (for PAR1) or trypsin (for PAR2). Moreover,synthetic peptides based on these noncanonical teth-ered ligand sequences can also behave as biasedagonists for the PARs (Mihara et al., 2013). Themultiple sites at which PARs 1 and 2 can be cleaved tounmask either nonbiased or biased tethered activatingligands is illustrated by the sequences at Fig. 6, bottom.It is also possible that proteinase cleavage of theextracellular domains or a PAR can destabilize receptorconformation to stimulate effector interactions andsignaling via a process that does not involve docking ofa PAR tethered ligand. This diversity of tetheredligands that are sequestered in the N-terminal se-quences of the PARs and the possible activation ofPARs via a tethered ligand-independent mechanismleads to a substantial flexibility with which microenvi-ronment inflammation-related proteinases can regulate
signaling via the PARs. Thus in principle, three outcomesare possible for the impact of proteinases on PARsignaling: 1) canonical unbiased signaling via cleavageat the serine proteinase sites singled out by thrombin ortrypsin; 2) biased signaling, triggered by cleavage at thenoncanonical sites targeted bymatrixmetalloproteinases-1 and -13 (MMP1; MMP13), activated protein-C (APC),neutrophil proteinases (elastase and proteinase-3),cathepsin-S, and by the other enzymes shown in Fig. 6,bottom; 3) silencing of signaling caused by cleavage atsite(s) that remove any potential tethered ligand se-quences to prevent signaling (i.e., disarming by remov-ing the thrombin-unmasked tethered ligand sequenceshown in Fig. 6A). This disarming can be caused bythe kallikrein-related peptidases, KLKs 8 and 14(Oikonomopoulou et al., 2006; Ramachandran et al.,2012a). As summarized in Table 1, substantial prog-ress has been made in identifying proteinases that canregulate the PARs and in developing agonists and antag-onists for PARs 1, 2, and 4.
4. Regulation of Proteinase-Activated Receptor Sig-naling: Desensitization, Internalization, and Intracel-lular Targeting. Once unmasked by a PAR-activatingproteinase, the tethered ligand remains attached to the
Fig. 6. Signaling by proteinase -activated receptors. (A) Proteinase-activated receptors are a family of GPCRs activated by proteolytic processing of thereceptor N terminus to unmask a cryptic receptor-activating tethered ligand. The middle panel (A) illustrates this activation process, with theproteolytically revealed tethered ligand (SFLLRN: gray sequence) shown as nestled into the receptor. The receptor can also be “disarmed” byproteinases that cleave downstream of the tethered ligand sequences thereby preventing receptor activation by “canonical” activators like thrombin(PARs 1 and 4) or trypsin (PAR2). (B) Receptor activation can be triggered in the absence of proteolysis by the exogenous addition of synthetic peptidesthat mimic the proteolytically revealed ligand (TFLLR: pink sequence). As outlined in the text, cleavage at PAR1 sites other than the one targeted bythrombin can also cause receptor activation by unmasking a noncanonical tethered ligand that generates biased signaling. The possible activation ofPAR signaling via a tethered ligand-independent process is not shown. (C) The lower panel shows the sequence of PAR1 and its sites of cleavage byenzymes that stimulate either biased or unbiased signaling. This tethered-ligand mechanism resulting from receptor proteolysis is in common for all ofthe PARs. APC, activated protein C; CG, cathepsin-G; MMP-1,13, matrix metalloproteinases 1 and 13; NE, neutrophil elastase; PR3, proteinase-3;VIIa/Xa, coagulation factor VIIa/Xa complex.
1120 Ramachandran et al.

TABLE
3PARs:
Nom
enclature,receptor-activatingor
disa
rmingpr
oteina
ses,
agon
ists/antag
onists
andsign
alingpa
thway
sThetableliststhefourPARsaccord
ingto
theirge
neno
men
clatur
e,alon
gwiththePAR-reg
ulating
enzy
mes,PAR
agon
ists,PAR
antago
nists,r
adioliga
ndbindingpr
obes,an
dtheG
proteinsthat
thePARsactiva
te.
Recep
tor
Gen
eAgo
nistProteinas
esAgo
nistPep
tide
Pep
tido
-Mim
etic
Ago
nist
Antago
nistor
Inhibiting
Proteinas
esRad
ioliga
nd
GProtein
PAR1
F2R
(Hs),
F2r(M
m),
F2r(R
n)
Thrombin(V
uet
al.,19
91)
TFLLR-N
H2
Non
eVorap
axar
(Cha
ckalam
annilet
al.,
2005
,20
08),Atopa
xar
(Kog
ushi
etal.,20
11),
RWJ5
6110
(Andr
ade-
Gordo
net
al.,19
99)
KLK4po
ssible
disa
rming
(Ram
sayet
al.,20
08)
[3H]haT
RAP
(Ahnet
al.,
1997
)Gq/11
,Gi/o
,G12
/13
Riewaldet
al.,20
01;Riewaldan
dRuf,
2001
KLK8disa
rming
(Ram
acha
ndranet
al.,
2012
a)Activated
proteinC
(Riewaldet
al.,
2002
,20
03),Plasm
in(K
imura
etal.,
1996
;Kuliop
ulos
etal.,19
99)
KLK14
disa
rming
(Oikon
omop
oulouet
al.,
2006
a,b)
Gingipa
ins-R
(Rgp
Ban
dHrgpA
;arginine-sp
ecific
cysteine
proteina
ses)
(Lou
rbak
oset
al.,
2001
a,b),MMP1(B
oire
etal.,20
05;
Trive
diet
al.,20
09),MMP13
(Jaffre
etal.,20
12),Neu
trop
hilElastas
e(M
iharaet
al.,20
13),Cathep
sin-G
(Molinoet
al.,19
95)
KLK4(R
amsa
yet
al.,20
08;M
izeet
al.,
2008
;Wan
get
al.,20
10)
KLK6(V
ande
llet
al.,20
08)
KLK14
(Oikon
omop
oulouet
al.,20
06a,b)
PAR2
F2R
L1(H
s),
F2rl1(M
m),
F2rl1(R
n)
Tryps
in(N
ystedt
etal.,19
94,19
95a,b;
Boh
met
al.,19
96a;
Saifedd
ine
etal.,19
96)
2f-LIG
RLO-N
H2(M
cGuire
etal.,20
04),SLIG
RL-
NH
2,S
LIG
KV-N
H2
GB11
0(B
arry
etal.,20
10),
AC-555
41an
dAC-264
613;
(Garde
llet
al.,
2008
)
GB88
(Sue
net
al.,20
12);
P2p
al18
Spe
pducin
(Sev
igny
etal.,20
11)
2-furoyl-LIG
RL[N
[3H]propion
yl]-O-N
H2
(Hollenb
erget
al.,20
08),
[3H]2-fur
oyl-LIG
RL-N
H2
(Kan
keet
al.,20
05)
Gq/11
,Gi/o
,G12
/13
Tryps
inIV
(Cottrellet
al.,20
04),
Tryptas
e(M
irza
etal.,19
97;Molino
etal.,19
97)
Tissu
efactor-FactorVIIa-Xa(C
amerer
etal.,20
00),Mem
bran
e-type
serine
protea
se1(Tak
euch
iet
al.,20
00)
Acrosin
(Smithet
al.,20
00)
Dus
tmitepr
oteina
sesDer
P3an
dDer
P9(Sunet
al.,20
01)
Gingipa
ins-R
(Rgp
Ban
dHrgpA
;arginine-sp
ecific
cysteine
proteina
ses
(Lou
rbak
oset
al.,19
98)
Cockroa
chprotea
ses(Arizm
endi
etal.,
2011
;Pag
eet
al.,20
10),Neu
trop
hil
Elastase(Ram
acha
ndranet
al.,20
11)
KLK2(M
izeet
al.,20
08)
KLK4(R
amsa
yet
al.,20
08;Mize
etal.,20
08)
KLK5(O
ikon
omop
oulouet
al.,20
06a,b;
Stefansson
etal.,20
08;Briot
etal.,20
09)
KLK6(O
ikon
omop
oulouet
al.,20
06a,b;
Van
dellet
al.,20
08)
(con
tinued
)
Proteinase-Mediated Signaling and Inflammation 1121

receptor and is thus not able to diffuse away, as is thecase for conventional G protein-coupled receptor ago-nists. Thus the mechanisms that regulate PAR signal-ing would be expected to be in many respects distinctfrom those that affect other agonist-activated GPCRs,e.g., in terms of internalization and recycling to the cellsurface after activation. A number of studies have beendone to understand this aspect of the molecular ma-chinery involved in the trafficking of PAR1 and PAR2.However, in contrast to PARs 1 and 2, limited insightsare available for understanding the dynamics of PAR4signaling and the potential role(s) that PAR3 mobilityand internalization might play in terms of signal trans-duction (Marchese et al., 2008; Adams et al., 2011).What is clear is that the control of PAR signaling occurson several levels, in terms of both 1) the time frame ofsignaling desensitization and 2) the process of receptorinternalization, turnover, and trafficking of new recep-tors to the cell surface. Thesemechanisms are discussedhere because they may well be subject to regulation byextracellular proteinases such as chymotrypsin, whichhas been observed to target a cell surface event sub-sequent to platelet activation by thrombin, but beforethe PAR-stimulated platelet response (Tam et al., 1980).Of note, PAR1 is reported to internalize through bothconstitutive and agonist-triggered internalization path-ways (Shapiro et al., 1996; Shapiro and Coughlin, 1998).This process of constitutive internalization has not yetbeen evaluated in depth for either PAR2or the otherPARs.Whether extracellular membrane constituents involvedin receptor trafficking and internalization after PARactivation might be regulated by proteolysis is an issuethat merits attention.
5. Proteinase-Activated Receptor 1. PAR1 internal-ization is dependent on dynamin and clathrin-coatedpits and involves an activation-dependent phosphory-lation of the receptor via the receptor-targeted kinasesGRK3 and GRK5 (Ishii et al., 1994; Hammes et al.,1999; Trejo et al., 2000). The requirement for arrestinsfor the internalization of PAR1 is unclear (Chen et al.,2004). It is suggested that PAR1 can interact directlywith the clathrin adaptor AP2 complex through theYXXL motif (Paing et al., 2004; Dores et al., 2012). Asecond tyrosine motif proximal to the transmembranedomain is also present in the PAR1 C terminus andinteracts with AP3 (Canto and Trejo, 2013). Constitu-tive internalization of unactivated PAR1 is AP2 de-pendent and negatively regulated by ubiquitination(Wolfe et al., 2007; Chen et al., 2011), whereas thetrafficking of internalized receptor from endosomes tolysosomes isAP3dependent (Canto andTrejo, 2013). Afteractivation, basally ubiquitinated PAR1 is deubiquitinatedby unknown mechanisms and is then phosphorylatedand internalized in a bicaudal D1-dependent mecha-nism to early endosomes (Swift et al., 2010). ActivatedPAR1 is then sorted through late endosomesand targetedto lysosomes for degradation in a process dependent
TABLE
3—Con
tinued
Recep
tor
Gen
eAgo
nistProteinas
esAgo
nistPep
tide
Pep
tido
-Mim
etic
Ago
nist
Antago
nistor
Inhibiting
Proteinas
esRad
ioliga
ndG
Protein
KLK14
(Oikon
omop
oulouet
al.,20
06a,b;
Stefansson
etal.,20
08;Gratioet
al.,
2011
;Chunget
al.,20
12;
Ram
achan
dran
etal.,20
12a)
PAR3
F2R
L2(H
s),
F2rl2
(Mm),
F2rl2
(Rn)
Thrombin(Ish
iharaet
al.,19
97)(PAR3
canbe
clea
vedby
thrombin,h
owev
ertheab
ilityof
this
receptor
tosign
alis
unclea
r).
TFRGAP
(Bretsch
neide
ret
al.,20
03)(Thisis
the
pred
ictedTL
derive
dsequ
ence
forthrombin
clea
vedPAR3.
How
ever
thesign
alingpr
operties
ofthis
peptideare
unclea
r)(H
ansenet
al.,
2004
).
Non
eNon
e
PAR4
F2R
L3(H
s),
F2rl3
(Mm),
F2rl3
(Rn)
Thrombin(K
ahnet
al.,19
98;Xu
etal.,19
98)
AYPGKF-N
H2(Faruqi
etal.,20
00;Hollenb
erg
andSaifedd
ine,
2001
).
Non
eYD-3
(Wuet
al.,20
02);
ML35
4(Y
oung
etal.,
2013
;Wen
etal.,20
14)
Gq/11
,Gi/o
Tryps
ins(X
uet
al.,19
98)
Tryps
inIV
(Cottrellet
al.,20
04)
Cathe
psin
G(Sam
bran
oet
al.,20
00)
Plasm
in(Q
uintonet
al.,20
04)
Gingipa
ins-R
(Rgp
Ban
dHrgpA
;arginine
-spe
cificcysteine
proteina
ses)
(Lou
rbak
oset
al.,20
01b)
KLK14
(Oikon
omopou
louet
al.,20
06a,b)
PAR,p
roteinas
e-activa
tedreceptor.
1122 Ramachandran et al.

on sorting nexin 1 (SNX1), mammalian homologs ofthe yeast Vps5p retromer complex that functions inendosome-to-Golgi trafficking (Trejo et al., 1998; Wanget al., 2002). Although this internalization-sortingprocess has been documented for the activation ofPAR1 via its canonical thrombin-exposed tetheredligand and for activation triggered by nonbiasedPAR1-activating peptides, the situation for the acti-vation of the receptor via a noncanonical tetheredligand or via a biased peptide agonist has yet to beevaluated. Thus neutrophil-elastase-activated PAR1remains at the cell membrane (Mihara et al., 2013),and the mechanisms of its activation-dependent de-sensitization and turnover remain a most interestingtopic for further study.6. Proteinase-Activated Receptor 2. PAR2 internal-
izes solely through agonist-triggered mechanisms(Bohm et al., 1996a). In contrast with PAR1, activatedPAR2 is phosphorylated and binds b-arrestin 1 andb-arrestin 2 (Dery et al., 1999). PAR2 internalizes to theearly endosomes in an ubiquitination-dependent pro-cess also requiring the GTPase Rab5a (Roostermanet al., 2003; Jacob et al., 2005). Continued sorting ofPAR2 requires hepatocyte growth factor-regulated ty-rosine kinase substrate (Hasdemir et al., 2007). In thelate endosomes, PAR2 is deubiqutinated before lyso-somal degradation. PAR2 resensitization at the cellsurface with de novo Golgi synthesized receptors occursin a Rab11a-dependent process (Roosterman et al.,2003). As for the noncanonical activation of PAR1 viaenzymes like neutrophil elastase, the activation ofPAR2 via neutrophil elastase does not trigger eitherPAR2 internalization or the activation of Gq-coupledcalcium signaling (Ramachandran et al., 2009, 2011).Thus, for both PARs 1 and 2, the dynamics and locationof receptor activation (i.e., cell surface versus internal-ized scaffolds) plays a key role in the activation ofdistinct downstream signaling pathways.7. Proteinase-Activated Receptors 3 and 4.
Mechanisms contributing to PAR3 and PAR4 internal-ization and trafficking remain largely unknown. In thecase of PAR4, it is reported that the kinetics of signaltermination after agonist occupancy is slower comparedwith PAR1 and PAR2 (Shapiro et al., 2000); however,the underlying mechanisms are unknown. Further-more, the impact of proteolytic activation of PAR4 viaa noncanonical tethered ligand on receptor traffickinghas yet to be evaluated8. Effectors that Mediate Proteinase-Activated Re-
ceptor Signaling. In keeping with the “mobile” or“floating” model of receptor function (de Haen, 1976;Jacobs and Cuatrecasas, 1976), it is not surprising thatPARs, like other receptors, can regulate multiple effec-tors to result in biased signaling via processes describedin detail by Kenakin and coworkers (Kenakin andMiller, 2010; Kenakin, 2013; Kenakin and Christopou-los, 2013; Christopoulos et al., 2014). Of importance, we
also know now that, like other GPCRs, the PARs cansignal as either homo- or heterodimers as reviewedelsewhere (Gieseler et al., 2013). However, this areais not sufficiently clarified for its impact on signalingand will not be dealt with here. Nonetheless, either asa monomer or dimer, PAR2 can signal via multipleeffectors.
Although G protein interactions are without doubt akey for PAR signal transduction (e.g., Gq, Gi, G12/13),PAR signaling can in principle involve a variety of othersignal effectors. For instance, PAR2 signaling that doesnot depend on Gq-alpha is driven by an internalizedPAR2-beta-arrestin scaffold that leads to MAPKinasesignaling (Defea, 2008). Furthermore, the interactionsof PARs with selected G proteins can involve multipleadditional effectors. For instance, the disheveled pro-tein, an upstream Wnt signaling protein, can be selec-tively bound to the PAR1-activated G13 alpha-subunitto affect beta-catenin signaling (Turm et al., 2010). TheC-terminal domains of the PARs are also involved insuch receptor-effector interactions, possibly involvingPAR-PAR homo- or heterodimer-effector interactions asalluded to above (Gieseler et al., 2013). In particular,C-terminal sequences in PAR2 have been found toregulate calcium and MAPKinase signaling and tomodulate Akt activation via a plekstrin homology-domain binding motif (Seatter et al., 2004; Kancharlaet al., 2015).
In summary, the PARs are able to generate signals byinteracting with multiple effectors that, depending onthe cell host, may drive quite distinct signals. Thedifferential signaling triggered by PARs either viadistinct effector interactions in different cell environ-ments or via biased signaling stimulated by signal-selective enzymes or synthetic agonists remains afruitful area for further study.
9. Posttranslational Modifications of Proteinase-Activated Receptors and Proteinase-Activated ReceptorFunction. Soon after the cloning of PAR1 it wasrecognized that the PARs could be regulated by post-translational modifications. Using an antibody thattargeted the N terminus of PAR1, Brass et al. (1992)showed that PAR1 migrated as a 66-kDa protein onWestern blots, as opposed to the mass of the receptorpredicted from its amino acid sequence of around35 kDa. Subsequent work has shown that PAR1glycosylation is important for plasma membrane ex-pression of the receptor (Xiao et al., 2011) as well as forligand-induced receptor activation and internalization(Soto and Trejo, 2010). Similarly, our own work showedthat PAR2 is also extensively glycosylated and that lossof glycosylation sequons negatively affects cell surfacereceptor expression (Compton et al., 2002). In the caseof PAR2, it was demonstrated further that glycosyla-tion of the receptor also serves to regulate activationof the receptor by endogenous enzymes, with mastcell-derived tryptase being unable to cleave a fully
Proteinase-Mediated Signaling and Inflammation 1123

glycosylated cell surface-expressedPAR2receptor,whereasbeing able efficiently to cleave synthetic peptidesrepresenting the nonglycosylated PAR2 N-terminal se-quence or to cleave deglycosylated PAR2 expressed incells (Compton et al., 2001).Another important posttranslational modification
that appears to regulate PAR function is palmitoylationof the receptor, as is reported for a number of otherGPCRs (Qanbar and Bouvier, 2003). The palmitoylationof PAR1 at two C-terminal cysteine residues is impor-tant for the interaction of the activated receptor withthe clathrin adaptor protein complex so as to affectreceptor trafficking. That said, mutated PAR1 (CC/AA),lacking the cysteine palmitoylation sites, is nonethelessfully competent for signaling, as determined by an[3H]inositol phosphate accumulation assay (Cantoand Trejo, 2013). By comparing the wild-type andpalmitoylation-deficient CC/AA-mutated PAR1 con-structs, it was determined that an enhanced constitu-tive internalization and lysosomal degradation of thereceptor occurs in the absence of receptor palmitoyla-tion. In the case of PAR2, however, the palmitoylation ofthe receptor occurs primarily at the C-terminal cysteine361 and a palmitoylation-deficient C/A mutant receptorhas greater cell-surface expression but a decreasedability to cause intracellular calcium signaling. How-ever, the palmitoylation-deficient PAR2-expressingcells triggered greater and more prolonged ERK/MAP-Kinase phosphorylation compared with the wild-typereceptor in a Gai-dependent manner (Botham et al.,2011). Given the ability of PARs to trigger biased signal-ing as described below, it will be of importance to assessthe roles for posttranslational modifications in regulat-ing PAR signaling when activated by different protein-ases. For instance, it is not yet known if the receptordynamics triggered by neutrophil or pathogen-derivedproteinases that can stimulate biased signaling are thesame as the regulation of PAR mobility and internali-zation triggered either by the canonical PAR activators,thrombin and trypsin, or by the tethered ligand-derivedreceptor-activating peptides.
E. Endogenous Proteolytic Regulators of Proteinase-Activated Receptor Function and Proteinase-ActivatedReceptor Signaling
The activation of PARs by enzymes in the tissue micro-environment can play key roles in inflammation andcancer (Fig. 1). These enzymes may also play a rolein regulating the function of integrin and adhesionADGR receptors (below). After the initial discovery ofPAR1, PAR3, and PAR4 as thrombin-cleaved receptorsand PAR2 as a trypsin-cleaved receptor, a key questionwas: what are the endogenous proteinases that regulatePAR activity in vivo? Understandably, thrombin can beseen as an important “physiological” blood stream-localized regulator for PARs 1 and 4; but what aboutthe other coagulation proteinases? Furthermore, are
there localized sites of synthesis of the coagulationproteinases, including thrombin, that release the en-zymes only in the tissue microenvironment and not intothe circulation? As opposed to thrombin, trypsin isthought of primarily as an intestinal digestive enzyme.Thus, which trypsin-like enzymes outside the intesti-nal lumen might be responsible for regulating PAR2?Clearly, the many diverse sites of expression of PARsin the body necessitate the existence of multiple endog-enous proteinases to regulate PAR function in vivo.Thus, once the PARs were cloned and their mechanismof activation understood, studies were focused not onlyon the molecular pharmacology and pathophysiology ofPAR signaling but also on identifying the enzymes thatare responsible for PAR regulation in vivo in a tissue-and disease-specific context.
1. Coagulation Cascade Enzymes. After the discov-ery of PAR1 as a thrombin-activated receptor, addi-tional enzymes in the coagulation cascade (Fig. 3, right)were evaluated for their ability to regulate the PARs. Inparticular, much evidence points to an important rolefor the factor VIIa/Xa complex as a regulator of PARsignaling. The activation of PAR2 by factor VIIa re-quires the presence of its cellular cofactor, tissue factor(Camerer et al., 2000). In addition to activating PAR2directly, the tissue factor (TF)-VIIa complex convertsthe inactive zymogen factor X to the activate factor Xa(Fig. 3), which also has the ability to activate PAR2signaling (Camerer et al., 2000; Riewald and Ruf, 2001).It is unclear as to what role this response would play inplatelet-regulated hemostasis, because PAR2 is notexpressed on either human or rodent platelets. However,the activation of PAR2 by the TF-VIIa-Xa complex pre-sumably has important roles in regulating vascularendothelial function and in tumor biology. PAR2 activa-tion by the TF-VIIa-Xa complex drives breast cancermigration as assessed both in the Adr-MCF7 cell line(Jiang et al., 2004) as well as in the MDA-MB-231 andBT549 breast cancer cell lines (Morris et al., 2006).
PAR2 is expressed in both vascular endothelial andsmooth muscle cells. Thus factor Xa-dependent acti-vation of PAR2 can potentially trigger responses notonly in intact vessels (Al-Ani et al., 1995; El-Dalyet al., 2014) but also in coronary artery smooth musclecells, aortic smooth muscle cells, adventitial fibro-blasts, and aortic endothelial cells (McLean et al.,2001). Furthermore, although PAR2 has not beenthought of as a target for thrombin activation, recentdata show that at concentrations of thrombin that canbe achieved in the setting of acute injury or tumorinvasion (e.g., 10 to 25 U/ml), PAR2 can be activateddirectly both in cultured cells and in the endotheliumof intact vessels to cause vasorelaxation (Mihara et al.,2016).
An additional regulatory mechanism for the TissueFactor-VIIa-Xa complex activation of PARs involves theendothelial cell protein C receptor (EPCR). EPCR can
1124 Ramachandran et al.

scaffold the interaction of factor X with TF-VIIa com-plexes (Disse et al., 2011) and also acts as a dockingsite for factor VIIa and activated protein C on endo-thelial cells (Ghosh et al., 2007; Sen et al., 2011). SincePAR2 and Tissue Factor-VIIa-Xa are co-localized inlipid rafts and caveolae, their proximity may act tosegregate signaling spatially through this pathway(Awasthi et al., 2007). The endothelial protein Creceptor (EPCR) is also reported to support endothe-lial PAR1 activation by factor Xa (Feistritzer et al.,2005; Schuepbach and Riewald, 2010). However, it isnot clear whether this endothelium-localized activa-tion of PAR1 by Factor Xa would complement theactivation of PAR1 by thrombin which is also gener-ated in step with Xa activation.Once hemostasis has been restored, the fibrinolytic
cascade is engaged to remove fibrin and disassemble theclot (Nesheim, 2003). In addition, upon binding throm-bomodulin, thrombin causes the activation of protein Cand the thrombin-activatable fibrinolysis inhibitor.This process leads to the generation of the anticoagu-lant, activated protein C (APC), and the inhibition offibrin generation (Esmon, 2003, 2014; Ito andMaruyama,2011). Activated protein-C is also an important regula-tor of PAR1 signaling but does not activate PAR2. Theactivation of PAR1 by the protein C pathway was firstreported by Ruf and colleagues (Riewald et al., 2002)who found that APC activation of PAR1 can triggerprotective anti-inflammatory, antiapoptotic signalingin endothelial cells (Riewald et al., 2003). Given thatPAR1 signaling by thrombin itself in endothelial cells isreported to trigger proinflammatory responses that arethe opposite of those stimulated by APC, Ruf andcolleagues went on to explore these different PAR1responses caused by activation of the same receptor bytwo different enzymes. Large-scale gene expressionprofiling studies showed a significant divergence inendothelial genes that are up- and downregulated bythrombin and APC (Riewald and Ruf, 2005). Thesestudies showed that although thrombin generation isongoing in the context of an injury, the concurrentactivation of PARs by other coagulation cascade en-zymes can signal in a different way via the samereceptor (Mohan Rao et al., 2014; Mosnier et al., 2012;Schuepbach et al., 2012).This kind of functional selectivity or biased signaling,
well characterized for other G protein-coupled recep-tors (Kenakin and Miller, 2010; Kenakin, 2011, 2013;Kenakin and Christopoulos, 2013), is now known toapply to the activation of PARs by different proteinases(Hollenberg et al., 2014; Zhao et al., 2014a,b). Further-more, as described above for the activation of PAR2 bythe TT-Factor VIIa-Xa complex, membrane lipid raftmicrodomains are important orchestrators of the acti-vated protein C/PAR1 signaling axis. Thus the specialmembrane locale where all components are broughttogether to enable APC-mediated activation of PAR1
can lead to site-selective biased signaling (Bae et al.,2007, 2008). In sum, several enzymes of the coagulationcascade including thrombin itself can activate PARsignaling. This activation can exhibit functional selec-tivity, depending on the proteinase and its microenvi-ronment wherein PARs are cleaved.
2. Kallikrein-Related Peptidases and Proteinase-Activated Receptor Signaling. When PAR2 was firstcloned, its expression was observed in unanticipatedsettings like the retina and prostate gland (Bohm et al.,1996b; Nystedt et al., 1994, 1995a). Because we werealready aware of the expression of the prostate-specificantigen/KLK3 family of serine proteinases in the pros-tate, we suggested that the KLKs might take part in anautocrine signaling loop in this locale, where thelocalized production of active KLK proteinases mightin turn signal to the coexpressed PARs. In support ofthis hypothesis, we were able to show that KLKs areindeed able to generate cell signaling by cleaving andactivating the PARs (Oikonomopoulou et al., 2006).It was later shown that the KLKs exhibit variablepharmacological potencies for cleaving at the sametethered ligand site required for PAR activation andcan trigger distinct G protein-coupled mechanismsdepending on 1) which PAR member is the target(e.g., PAR1 versus PAR2 versus PAR4), 2) the locali-zation of the KLKs or their upregulation in diseasesettings, and 3) their local concentration (Ramsayet al., 2008; Stefansson et al., 2008; Gratio et al.,2010; Oikonomopoulou et al., 2010a; Ramachandranet al., 2012a). For example, KLK7 (Stefansson et al.,2008) andKLK8 (Stefansson et al., 2008; Ramachandranet al., 2012a) are not able to cause PAR2 calcium signal-ing, whereas KLKs 5, 6, and 14 can induce prominentPAR2 signals. Furthermore, KLK8 and KLK14 are nowknown to disarm PAR1, shutting down its signalingpotential by other proteinases (Oikonomopoulou et al.,2006; Ramachandran et al., 2012a). In contrast, KLK4has been shown to induce signals via both PARs 1 and2 (Ramsay et al., 2008). KLK-triggered PAR signal-ing has been found to play roles in many settings,including skin inflammation, as well as in neuronalpathologies and cancer (Oikonomopoulou et al., 2010b).Specifically in inflammatory skin settings, a role hasbeen established clinically for KLKs in the pathobiologyof Netherton syndrome (Chavanas et al., 2000; Briotet al., 2009, 2010; Hovnanian, 2013). As reviewedelsewhere (de Veer et al., 2014), a signaling pathwayhas been discovered whereby KLK5 activates skin PAR2to increase expression of proinflammatory cytokines andchemokines, such as tumor necrosis factor alpha andinterleukin-8 in human keratinocytes. These data in-dicated that active KLK5 in the epidermis of Nethertonsyndrome patients triggers inflammation via PAR2,independently of the environmental stimuli and theadaptive immune system (Briot et al., 2009; Hovnanian,2013; de Veer et al., 2014).
Proteinase-Mediated Signaling and Inflammation 1125

KLKs, and more specifically KLK14, were also shownto induce receptor inflammatory signals indirectly viathe complement receptor for the anaphylatoxin C3a(known as C3AR) (Oikonomopoulou et al., 2013). Thisstudy pointed to a potential role for the KLK-C3ARreceptor axis in subcutaneous inflammation using anin vivo model of paw inflammation. Interestingly,trypsin-responsive PAR2 signals in the same modelare also known to trigger paw swelling, indicating apotential crosstalk of the two receptor systems in thismodel, namely PAR2 and the complement C3a receptor.The impact of this crosstalk in settings of pathologyremains to be explored. Taken together, the four pro-teolytic cascades (coagulation, complement, kininogen,and KLK) can be seen to integrate signaling along withthe PARs. Thus these proteolytic cascades can beconsidered as regulators of inflammation along withthe PARs.3. Neutrophil Proteinases as Regulators of Proteinase-
Activated Receptor Function. In the setting of acute in-flammation caused by traumatic or infectious events,the initial activation of the coagulation and comple-ment cascades is followed rapidly by the influx ofneutrophils. We have therefore been particularlyinterested in the potential roles of neutrophil protein-ases in driving the acute inflammatory response.Locally secreted neutrophil proteinases such as neu-trophil elastase, proteinase-3, and cathepsin-G canhave a dramatic impact on tissue function in partby regulating PAR activity (Ramachandran et al.,2011; Mihara et al., 2013). As already mentioned,these enzymes can cause either unbiased or biasedPAR signaling and can also disarm PARs by removingthe N-terminal sequences that harbor the cryptictethered ligand sequences. Although the neutrophilenzymes target PARs 1, 2, and 4, which are known tosignal autonomously, they can also cleave the N-terminaldomain of PAR3. However, the biologic impact of thiscleavage of PAR3 is difficult to interpret.One of the first studies that implicated neutrophil-
derived enzymes as regulators of PARs hypothesizedthat in the setting of an acute lung inflammation, thelarge neutrophil influx and release of serine proteinasessuch as elastase and cathepsin-Gmight act on epithelialcell expressed PAR2 (Dulon et al., 2003). Interestingly,in cultured A549 or 16HBE lung epithelial cells,elastase and cathepsin-G failed to trigger increases incalcium signaling but rather made the receptor re-fractory to subsequent activation by trypsin. Laterstudies also demonstrated that an elastolytic metal-loproteinase from Pseudomonas aeruginosa similarlycleaved and disarmed PAR2, thereby preventing itssubsequent activation by trypsin (Dulon et al., 2005).Based on molecular pharmacological studies with aPAR2 receptor that upon trypsin activation revealedmutated tethered ligand motifs (Al-Ani et al., 2004),we hypothesized that receptor cleavage, even in the
absence of an intact canonical tethered ligand, mightactivate some signaling downstream of the receptor(Ramachandran et al., 2009). Indeed we found that theintact trypsin-revealed tethered ligand was necessaryfor coupling to the calcium signaling response but wasnot a requirement for triggering MAPK responses fromPAR2 (Ramachandran et al., 2009). Extending this lineof thought we investigated whether an enzyme such asneutrophil elastase, which cleaves downstream of theclassic tethered ligand revealing site, might also triggersome but not all signaling downstream of PAR2. Wewere able to demonstrate that neutrophil elastase is abiased agonist for PAR2 and stimulates p42/44 MAPKactivation while disarming the calcium signaling thatwould otherwise be caused by trypsin activation(Ramachandran et al., 2011). In subsequent work weshowed that neutrophil elastase can also trigger com-parable biased signaling responses via PAR1 (Miharaet al., 2013). The functional consequences of the neu-trophil elastase-triggered biased signaling of PAR2 arestill being worked out, but it can be pointed out thatneutrophil elastase induces mucin production throughPAR2 activation (Tull et al., 2012) and neutrophil-mediated activation of epithelial proteinase-activatedreceptors 1 and 2 regulates barrier function and trans-epithelial migration (Chin et al., 2008). More recentlyit was shown that PAR2 activation causes cross-sensitization of the transient receptor potential familychannel TRPV4 to regulate a number of differentcellular responses (Grant et al., 2007; Poole et al.,2013; Saifeddine et al., 2015). Moreover, neutrophilelastase activation of PAR2 is able to support suchcrosstalk to affect cell function (Sostegni et al., 2015).
Although more is known about neutrophil elastase asa regulator of PARs, proteinase-3 can also cleave PAR1(Tull et al., 2012; Mihara et al., 2013). In contrast toneutrophil elastase, which cleaves PAR1 downstream ofthe thrombin cleavage-activation site, proteinase-3cleavage occurs upstream of the thrombin site andappears to trigger signaling that is distinct from theelastase-triggered response. As discussed above, EPCRacts as a cofactor to enable APC cleavage of PAR1.Proteinase-3 is shown to cleave EPCR, and this cleav-age in principle can regulate APC signaling via PAR1,although this possibility has not yet been directlydemonstrated (Villegas-Mendez et al., 2007).
In addition to the activation of PAR2 described above(Dulon et al., 2003), cathepsin-G can also activate PAR4on platelets (Sambrano et al., 2000) and on colonicepithelial cells (Dabek et al., 2009). It is not yet known ifthe activation of PAR4 by cathepsin-G triggers signal-ing that is identical to activation by thrombin. However,in contrast with the activation of PAR4 on humanplatelets, activation of PAR4 by cathepsin-G on murineplatelets does not support aggregation (Cumashi et al.,2001). Whether the observed disabling of murine plate-let PAR3 by cathepsin-G contributes to this observed
1126 Ramachandran et al.

difference is unclear. It can be noted that murine PAR3,which is disabled by cathepsin G, acts as a cofactor forPAR4 to facilitate PAR4 cleavage at low thrombinconcentrations.4. Matrix Metalloproteinases as Proteinase-Activated
Receptor Regulators. In addition to neutrophil elas-tase, proteinase-3, and cathepsin-G, neutrophil-derivedmatrix metalloproteinases (MMPs) are also thought toplay an important role in inflammation in localesranging from tumor microenvironments to the centralnervous system (Ardi et al., 2007; Deryugina et al.,2014; Han et al., 2014; Lee et al., 2014). MMPs 3, 8, and9 have been singled out for attention in addition toMMP1, which can be released by platelets along withthe other MMPs in an inflammatory situation. PARshave also been found to be targets of the MMPs. Inparticular, PAR1 activation by stromal MMP-1 pro-motes tumorigenesis in breast cancer (Boire et al., 2005)and promotes melanoma invasion and metastasis(Blackburn et al., 2009). In addition to acting directlyon tumor cell PAR1, activation of human platelet PAR1by MMP-1 also contributes to platelet activation andthrombogenesis (Trivedi et al., 2009). This MMP-1-stimulated PAR1 response can also be involved in theregulation of vascular integrity (Tressel et al., 2011).Thus the MMP-PAR1 signaling axis is of relevance tovascular regulation and angiogenesis in the setting of atumor.PAR1 activation by MMPs and thrombin is also
implicated in stroke-induced neurotoxicity (Xue et al.,2006, 2009). Another MMP-PAR1 interaction has beenfound in cardiac myocytes and fibroblasts wherein beta-adrenergic stimulation causes an MMP13-mediatedautocrine-paracrine activation of PAR1 (Jaffre et al.,2012). This cleavage unmasks a novel tethered ligandsequence DPRS(42)↓(43)FLLRN that differs from theone revealed by MMP1. MMP-1 specifically activatesPAR1 by cleaving at LD(39)↓P(40)RSFL rather than atthe LDPR4(41)↓S(42)FL thrombin cleavage site anddoes not cleave the other PARs. Of note, MMP1, alongwithMMP13, are biased agonists for PAR1, stimulatingdistinct downstream signaling in away that differs fromthe activation of PAR1 by thrombin (Austin et al., 2013).Indeed, given the high expression of PAR1 in cardiacfibroblasts (Snead and Insel, 2012) and the potentialimpact of PAR regulation on cardiac pathophysiology(Fukunaga et al., 2006; Pawlinski et al., 2007; Soninet al., 2013; Bode andMackman, 2015), theMMPs alongwith the PARs can be thought of as joint therapeutictargets for PAR-associated cardiomyopathies. Thus, inan inflammatory situation, activation of PAR1 by boththrombin and MMPs can be complementary, stimulat-ing PAR1 responses that are temporally and mechanis-tically distinct (Blackburn and Brinckerhoff, 2008). Todate, the ability of the other neutrophil-derived MMPsthat can play a part in the process of inflammation(MMPs 3, 8, and 9) to affect PAR signaling have not been
explored directly, for example, in terms of their abilitiesto “disarm” PAR1 activation. In this inflammatorysetting, the MMP inhibitors will also play a key role toregulate signaling. The upregulation of the TIMPSswould not only attenuate MMP-stimulated PAR signal-ing but would prevent remodeling of the extracellularmatrix that can sequester cell-regulating polypeptidesthat in turn can act along with the PARs to stimulatefibrosis (Chambers and Scotton, 2012; Arpino et al.,2015). Thus both the MMPs and their TIMP inhibitorsare of importance in regulating PAR signaling.
5. Cathepsins, Proteinase-Activated Receptor 2 Acti-vation, and Inflammatory Pain. Added to the rapidinflux of neutrophils at a site of inflammation, macro-phages soon migrate into the area to augment theinnate immune response and to contribute their se-creted proteinases to the microenvironment. Inflamma-tory mediators can trigger the secretion of proteinasesfrom macrophages (and microglial cells in the centralnervous system), including the cysteine proteinasecathepsin-S, which has been detected in a number ofinflammatory settings. Like other microenvironmentproteinases that we have discussed so far, cathepsin-Scan also signal via the processes outlined in Fig. 2,including mechanisms 4 (ion channel regulation),5 (PAR activation), and 6 (release of a membrane-tethered agonist). In this regard, cathepsin-S has beendocumented to activate the epithelial sodium channel(ENaC) as discussed below (Haerteis et al., 2012a) andmaywell affect other ion channels. Further, cathepsin-Scan release the membrane-tethered cytokine fractal-kine from neurons, thereby contributing to pain sensi-tization (Clark and Malcangio, 2012). Of relevance toPAR2 signaling, it was recently found that, like neutro-phil elastase, cathepsin-S can cleave and activate PAR2at an N-terminal sequence site downstream fromthe canonical “R//SLIGLV–” trypsin activation site.Cathepsin-S cleaves PAR2 at E(56)↓T(57) to revealyet another novel noncanonical tethered ligand se-quence, “TVFSVDEFSA—” (Zhao et al., 2014a,b). Whenunmasked by cathepsin-S, this newly discovered PAR2tethered ligand stimulates signaling in a completelydifferent way than that caused either by trypsin or byneutrophil elastase activation of PAR2. Thus, like theneutrophil elastase-revealed noncanonical tethered li-gand, the one unmasked by cathepsin-S does notstimulate either calcium signaling or an interactionwith beta-arrestin and does not cause receptor inter-nalization. However, unlike the elastase-stimulatedPAR2 response, cathepsin-S does not activate MAPKi-nase but rather stimulates adenylyl cyclase and proteinkinase A-dependentmechanisms to cause inflammationand hyperalgesia (Zhao et al., 2014a,b). This hyper-algesia results from a link between PAR2 activation andthe enhancement of neuronal calcium flux throughneuronal TRPV4 channels, as outlined above (Grantet al., 2007). The ability of this enzyme to regulate PAR1
Proteinase-Mediated Signaling and Inflammation 1127

signaling is very likely but has yet to be studied in anydepth. Thus, overall, cathepsin-S can be seen to join anumber of othermicroenvironment proteinases that canregulate inflammation and pain by the hormone-likemechanisms outlined in this chapter. What is evidentfrom the sections above is that enzymes of the majorityof the families outlined in Fig. 2 (metallo-, serine,threonine, and cysteine-proteinases), with very differ-ent mechanisms of catalysis and distinct target sub-strates, are all capable of triggering hormone-likesignals. No doubt many other proteinases secreted inthe course of an inflammatory response will have thesekinds of actions.
F. Proteinase-Activated Receptor Regulation byPathogen and Allergen-Derived Proteinases: Enzymesfrom Cockroaches, Molds, Dust Mites,and Trypanosomes
In addition to endogenously generated enzymes, wenow know that pathogen-derived proteinases to whichthe lung, skin, and other tissues are exposed are alsoable to regulate PAR activity. The search for theantibody-generating allergens detected by sera fromdust mite-allergic patients resulted in the identificationof highly allergenic proteinases from the Dermatopha-goides pteronyssinus (Der p) organism with tryptic,chymotryptic, and cysteine proteinase activities (Chuaet al., 1988). A number of other diverse environmentalallergens have since been found to contain proteolyticactivity, including several species of molds (Shin et al.,2006), pollens (Gunawan et al., 2008), and the Germancockroach (Wongtim et al., 1993). Recent work suggeststhat, similar to the house dust mite allergen, theproteinases in these many allergen sources may con-tribute to their allergenicity. Initially, the pathogenicproperties of these enzymes were thought to be due totheir ability to interact with IgE. No doubt, that in-teraction can explain some of the acute reaction ofsensitized individuals to the allergens. However, wenow believe that this pathogen-host interaction, whichgenerates an inflammatory allergenic response, is duein large part to the activation of proteinase-mediatedsignaling by one or more of the mechanisms outlined inFig. 2. Work by us and by others has focused onidentifying the serine proteinases present in suchallergens and evaluating their ability to signal bycleaving and activating PARs (Adam et al., 2006;Arizmendi et al., 2011; Boitano et al., 2011). We suggestthat this mechanism whereby pathogen-derived pro-teinases can enhance their inflammatory impact by themechanisms shown in Fig. 2, including PAR regulationmay be widespread. As an example, the sleepingsickness caused by the African trypanosome involves aPAR2-mediated mechanism, whereby the trypanoso-mal cysteine proteinase enables its passage into thecentral nervous system (Grab et al., 2009; Moxon et al.,2013). Thus pathogen-derived proteinases from organisms
ranging from bacteria to insect allergens and tissue-invading parasites can use the mechanisms that triggerinflammatory signaling to their advantage. Therefore,for these pathologies, the proteinases themselves aswell as their substrates like the PARs offer novel targetsto consider for therapy.
IV. Adhesion G Protein-Coupled Receptors: AProteinase-Activated Receptor-like Tethered
Ligand Signaling Mechanism Driven byReceptors with Intrinsic Proteinase Activity
In Fig. 2, cell-matrix interactions, which regulateintegrin “outside-in” and “inside-out” signaling, aresingled out as one potential proteinase signaling target(mechanism 7). However, signaling via cell adhesioninteractions is also now known to be regulated by a largesuperfamily of adhesion G protein-coupled receptors(aGPCRs) or ADGRs [recommended IUPHAR nomen-clature (Hamann et al., 2015)], which are described indetail elsewhere (Yona et al., 2008; Arac et al., 2012b;Paavola and Hall, 2012; Langenhan et al., 2013;Liebscher et al., 2014a; Hamann et al., 2015). Surpris-ingly, ADGRs and the PARs share the tethered ligandparadigm of activation but via a very different ADGRautoproteolytic process. There is thus a common signal-ing theme between the PARs and the adhesion-activated receptors, namely, receptor cleavage thatexposes a signal-generating tethered ligand. Further-more, it is possible that in addition to the intrin-sic proteolytic mechanism of ADGR activation,extracellular proteinases may also affect signaling bythis receptor superfamily. Thus ADGRs like PARsmay be targets for regulation by microenvironmentproteinases.
The large family of ADGRs, including 33 humanhomologs, is the second largest family of GPCRs, re-lated evolutionarily to the secretin family of GPCRsand expressed not only in all vertebrates but also inprimitive animals and unicellular metazoans (Hamannet al., 2015). These receptors have been found to playkey roles in diverse settings ranging from organ devel-opment to myelination and the innate inflammatoryresponse, as detailed in the above-referenced reviewarticles. The large extracellular domain that distin-guishes these aGPCRs/ADGRs from other receptorscomprises a unique heterodimeric structure generatedby an autocatalytic cleavage that occurs during receptorbiosynthesis andmaturation. The surprising outcome ofwork on these intriguing receptors, for which fewendogenous ligands have yet been identified, is thattheir mechanism of activation, as alluded to above,echoes the process used by the PARs, in terms of theunmasking of a tethered ligand (Fig. 7). The exposedtethered ligand of an adhesion-GPCR, termed the“Stachel” (German word for stinger) domain byLiebscher and colleagues, can drive receptor-G protein
1128 Ramachandran et al.

coupling (Liebscher et al., 2014a,b; Hamann et al.,2015). Whether this mechanism applies to all of theADGRs found in the human genome, apart from GPRs126 (ADGRG6) and 133 (ADGRD1), for which thetethered ligand mechanism has been validated, re-mains to be determined.The key to the activation of ADGRs lies in a common
GPCR-Autoproteolysis-INducing domain (acronym:GAIN domain; Fig. 7), that triggers receptor cleavageduring the process of endosomal maturation and trans-port to the cell surface (Arac et al., 2012a,b; Promelet al., 2013; Liebscher et al., 2014a; Hamann et al.,2015). Thus these receptors possess intrinsic proteolyticactivity that is integral to their signaling mechanism.The GAIN domain is found in common with all of thehuman ADGRs and functions as an autoproteolytic foldincluding a short proteolysis site (termed the G proteinproteolysis site, or GPS). The proteolytic cleavagemechanism at the GPS site echoes that of serine pro-teinases in which a basic histidine (or arginine in someADGRs) participates with the hydroxyl of a serine orthreonine (or a sulfhydryl of cysteine) to trigger anucleophillic attack on a leucine or other amino acidcarbonyl that results in peptide bond cleavage (Aracet al., 2012b). This cleavage leaves an N-terminal ADGRfragment tightly associated noncovalently with aC-terminal receptor domain as a heterodimer. Uponrelease of the N-terminal fragment from a docking siteor upon binding its cognate ligand [e.g., binding ofcollagen III by GPR56/ADGRG1 (Luo et al., 2014)], theheterodimer is believed to dissociate, revealing the cell-attached receptor portion, representing the C-terminalcleaved fragment of the GPS domain. It is theN-terminus of this exposed cell-attached extracellularreceptor sequence that can act as a tethered ligand todrive interactions of the receptor with its cognate Gproteins (Fig. 7) (Liebscher et al., 2014b). Evidently, thereleasedN-terminal sequence of anADGR can also havea “life of its own” to affect signaling via a G protein-independent process (Patra et al., 2014). As a parallelwith the PARs, the proteolytically released N-terminalsequence of PAR1 is also thought to be biologicallyactive (Zania et al., 2009).To sum up, the adhesion GPCRs, representing the
second largest GPCR superfamily, have a number ofmechanisms in common with the PARs in terms of theirtethered ligand mode of activation, their ability tocouple to multiple G proteins, and the potential activityof the “released” receptor segment that before pro-teolysis masks the tethered ligand. One issue not yetconsidered for the ADGRs is that their proteolysis byproteinases in the tissue microenvironment or bypathogen-derived proteinases may regulate their sig-naling via a noncanonical mechanism, as has beenobserved for the PARs. Thus, direct cleavage upstreamof the N-terminal GAIN domain of an adhesion GPCRheterodimer by microenvironment proteinases might
result in signaling, in addition to and quite apart fromany adhesion-mediated signaling process. Furthermore,secondary cleavage downstream of the ADGR tetheredligand sequences could in principle either silence thesereceptors or, as for the PARs, reveal alternate tetheredligand sequences that might confer biased ADGR signal-ing. These topics merit attention in terms of our hypoth-esis that proteinases released into the microenvironmentcan in principle disrupt integrin or ADGR-matrix inter-actions and thereby regulate tissue signaling. This kind ofprocess may be of particular importance in cancer tis-sues for the processes of oncogenesis and metastasis inwhich the ADGRs appear to play key roles. Clearly, theadhesion GPCR family represents a rapidly movingresearch target thatmerits close attention in the future.As for the PARs, the ADGR tethered ligand sequencesand their receptor docking sites may offer clues for thedevelopment of therapeutic agents. However, as out-lined above, unlike the ADGRs, the PARs depend onextracellular proteinases to unmask their tetheredligands and can therefore have distinct tethered ligandsunmasked by different proteinases
V. Proteolytic Regulation of Ion Channels
In addition to regulating PARs and the insulin recep-tor, proteolytic cleavage is also known to generate signalsby cleaving ion channels. Proteinases are central modu-lators of ion channels in epithelial, endothelial, andneural tissues. Moreover, we now know that proteolyti-cally mediated PAR activation can also regulate thefunction of members of the transient receptor potentialvanilloid family members, TRPV1 and TRPV4. Thusproteolytic activation of PARs and ion channels bymicroenvironment proteinases can be envisioned as acoordinated process. Proteolytic activation of ion chan-nels is particularly important as an inflammatory sen-sory mechanism that constitutes an “early warningsystem” for the organism. This biologic process under-lies the transduction of noxious and damaging signalsduring injury/trauma or infection and triggers pro-tective cellular responses that initiate defense mecha-nisms to counterregulate inflammation in the tissueand promote resolution. In addition, the modulation ofion channels by proteinases was recently reported as acritically important signaling mechanism in the con-text of diseases such as cancer (Szabo and Bugge,2011). We will not cover the indirect regulation of ionchannels by intracellular proteinases activated in thecourse of GPCR signaling (for review, see Veldhuis et al.,2015), but will deal with the direct activation of thesechannels by membrane-associated intracellular and se-creted proteinases.
A. Proteolytic Activation of L-type Calcium Channels
The voltage-gated L-type calcium channels controlthe influx of calcium in cardiac, vascular, and neuronal
Proteinase-Mediated Signaling and Inflammation 1129
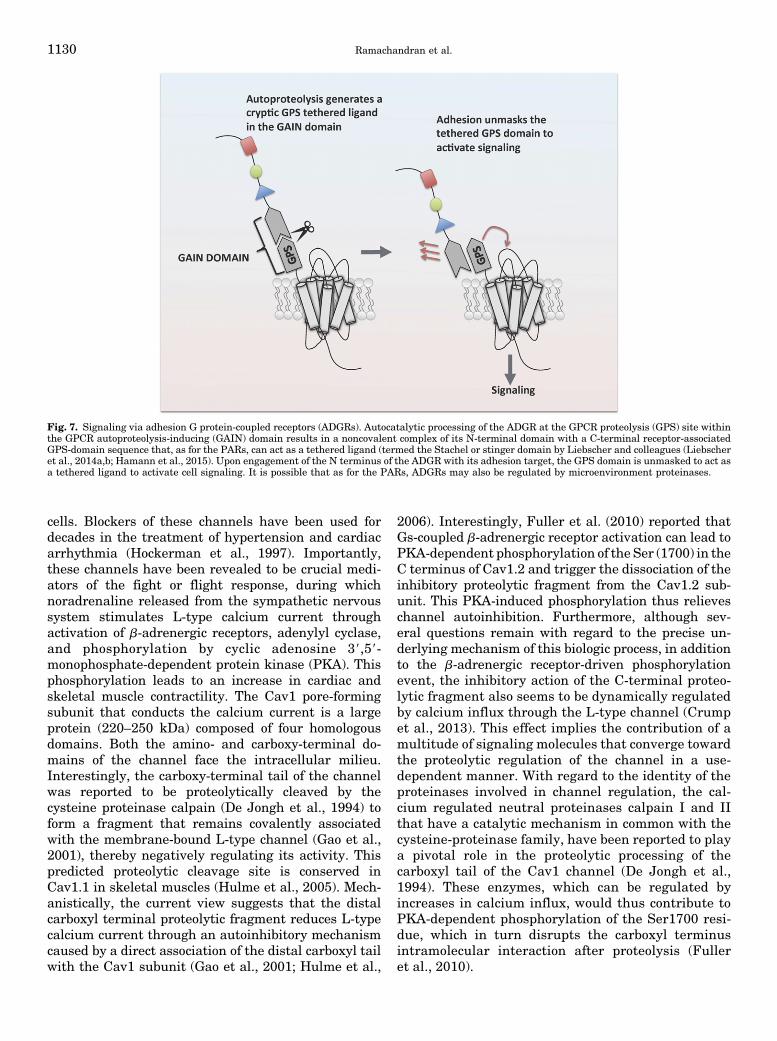
cells. Blockers of these channels have been used fordecades in the treatment of hypertension and cardiacarrhythmia (Hockerman et al., 1997). Importantly,these channels have been revealed to be crucial medi-ators of the fight or flight response, during whichnoradrenaline released from the sympathetic nervoussystem stimulates L-type calcium current throughactivation of b-adrenergic receptors, adenylyl cyclase,and phosphorylation by cyclic adenosine 39,59-monophosphate-dependent protein kinase (PKA). Thisphosphorylation leads to an increase in cardiac andskeletal muscle contractility. The Cav1 pore-formingsubunit that conducts the calcium current is a largeprotein (220–250 kDa) composed of four homologousdomains. Both the amino- and carboxy-terminal do-mains of the channel face the intracellular milieu.Interestingly, the carboxy-terminal tail of the channelwas reported to be proteolytically cleaved by thecysteine proteinase calpain (De Jongh et al., 1994) toform a fragment that remains covalently associatedwith the membrane-bound L-type channel (Gao et al.,2001), thereby negatively regulating its activity. Thispredicted proteolytic cleavage site is conserved inCav1.1 in skeletal muscles (Hulme et al., 2005). Mech-anistically, the current view suggests that the distalcarboxyl terminal proteolytic fragment reduces L-typecalcium current through an autoinhibitory mechanismcaused by a direct association of the distal carboxyl tailwith the Cav1 subunit (Gao et al., 2001; Hulme et al.,
2006). Interestingly, Fuller et al. (2010) reported thatGs-coupled b-adrenergic receptor activation can lead toPKA-dependent phosphorylation of the Ser (1700) in theC terminus of Cav1.2 and trigger the dissociation of theinhibitory proteolytic fragment from the Cav1.2 sub-unit. This PKA-induced phosphorylation thus relieveschannel autoinhibition. Furthermore, although sev-eral questions remain with regard to the precise un-derlying mechanism of this biologic process, in additionto the b-adrenergic receptor-driven phosphorylationevent, the inhibitory action of the C-terminal proteo-lytic fragment also seems to be dynamically regulatedby calcium influx through the L-type channel (Crumpet al., 2013). This effect implies the contribution of amultitude of signaling molecules that converge towardthe proteolytic regulation of the channel in a use-dependent manner. With regard to the identity of theproteinases involved in channel regulation, the cal-cium regulated neutral proteinases calpain I and IIthat have a catalytic mechanism in common with thecysteine-proteinase family, have been reported to playa pivotal role in the proteolytic processing of thecarboxyl tail of the Cav1 channel (De Jongh et al.,1994). These enzymes, which can be regulated byincreases in calcium influx, would thus contribute toPKA-dependent phosphorylation of the Ser1700 resi-due, which in turn disrupts the carboxyl terminusintramolecular interaction after proteolysis (Fulleret al., 2010).
Fig. 7. Signaling via adhesion G protein-coupled receptors (ADGRs). Autocatalytic processing of the ADGR at the GPCR proteolysis (GPS) site withinthe GPCR autoproteolysis-inducing (GAIN) domain results in a noncovalent complex of its N-terminal domain with a C-terminal receptor-associatedGPS-domain sequence that, as for the PARs, can act as a tethered ligand (termed the Stachel or stinger domain by Liebscher and colleagues (Liebscheret al., 2014a,b; Hamann et al., 2015). Upon engagement of the N terminus of the ADGR with its adhesion target, the GPS domain is unmasked to act asa tethered ligand to activate cell signaling. It is possible that as for the PARs, ADGRs may also be regulated by microenvironment proteinases.
1130 Ramachandran et al.

Of note, proteolytic cleavage of L-type channels hasbeen found to differ with aging. Indeed, Michailidiset al. (2014) reported that age-dependent proteolyticprocessing of neuronal L-type channels by the calcium-dependent proteinase, calpain, can generate midchan-nels” that remain functional at the plasma membranebut that exhibit biophysical properties differing fromthe full-length “uncleaved” channel. This difference inproteolytic processing by calpain may alter L-typechannel-mediated neuronal functions such as synapticplasticity and neurotransmission. Therefore, this pro-cess provides newly identified roles for proteolysis-dependent regulation of ion channels in physiologicstates like aging.
B. Proteolytic Cleavage of the Amiloride-sensitiveEpithelial Na+ Channel (ENaC/Degenerin Family):Epithelial Na+ Channel Activation byProteolytic Enzymes
Among mechanisms that control sodium ion homeo-stasis, the absorption of sodium by epithelial tissue ofthe colon, the airways or the kidney must be finelyregulated. The epithelial sodium channel (ENaC) is acritically important regulator of epithelial sodiumtransport. ENaC channels belong to the ENaC/dege-nerin family of non-voltage-gated ion channels thatconsists of ENaC, the neuronal mechanosensitivedegenerins of nematodes (UNC, MEC, DEG, DEL),and the neuronal acid-sensitive ion channels (ASICs)(Gamper and Shapiro, 2007). Dysfunction and/or dysreg-ulation of ENaC channels leads to numerous disorderssuch as Liddle syndrome, which is associated withexcess reabsorption of Na+ and thus severe hyperten-sion (Kellenberger and Schild, 2002, 2015). At themolecular level, ENaC is a heteromeric channel formedby assembly of structurally related a, b, and g subunits(Canessa et al., 1994). Each subunit has two trans-membrane domains (M1 and M2) and a pore regionadjacent to the M2 domains of the three subunits (Fig.8). Proteolytic cleavage of ENaC channels has beenfound to be the main mechanism of channel activationby increasing the open probability of the channel(Kleyman et al., 2009; Rossier and Stutts, 2009). Recentadvances in the understanding of the mechanisms ofENaC channel activation have provided key informa-tion with regards to the site(s) of proteolytic cleavagewithin ENaC subunits and the identification of theproteinases that mediate channel activation (Rossierand Stutts, 2009).Previous work using the trypsin inhibitor aprotinin
suggested that the ENaC channel can be activated byserine proteinases (Chraibi et al., 1998; Vuagniauxet al., 2002). Biochemical studies have confirmed thatthe a and g subunits of ENaC are processed byproteinases. Interestingly, proteolytic channel frag-ments can be detected in both cytosolic and plasmamembrane fractions, indicating that proteolytic
processing of the channel can result from intracellularproteinases, membrane-anchored proteinases, extracel-lular matrix-bound proteinases (metalloproteinases),and proteinases released into the extracellular milieu(Hughey et al., 2004a,b). A role for pathogen-derivedinflammatory proteinases for ENaC regulation has yetto be evaluated in any depth. The first membrane-bound serine proteinase described to mediate ENaCchannel activation was channel-activating proteinase-1, an ortholog of human prostasin. This proteinasebelongs to the subgroup of S1 proteinases that arebound to the plasma membranes either by a C-terminaltransmembrane domain (type I), an N-terminal proxi-mal transmembrane domain (type II, or TTSP), or aglycosylphosphatidylinositol-anchored motif (Netzel-Arnett et al., 2003). In recent years, considerableadvances have been made in understanding the molec-ular mechanisms of proteolytic activation of ENaC andin identifying the nature of the proteinases involved.The extracellular domains of the ENaC a and g
Fig. 8. Regulation of epithelial sodium channel (ENaC) by extracellularproteinases. The epithelial cell sodium channel (ENaC) represents aprototype channel that is regulated by proteolysis. ENaC is a heteromericchannel formed by assembly of structurally related alpha, beta, andgamma subunits. Proteolytic processing of the channel can result fromthe activation of intracellular (furin), membrane-anchored (metallopro-teinases), and soluble proteases. Although furin has been reported tofacilitate the trafficking of ENaC and to remove the autoinhibition of thechannel through cleavage of the a subunit, a membrane-anchoredproteinase (i.e., prostasin) cleaves the gamma subunit to activate thechannel fully. Microenvironment extracellular proteinases such astrypsins I and IV and cathepsin-S have also been shown to be keymediators of proteolysis-induced activation of apical ENaC.
Proteinase-Mediated Signaling and Inflammation 1131

subunits each contain two consensus recognition andcleavage sequences for furin and furin-like convertases.It has been found that proteolytic cleavage of thesesubunits occurs in intracellular compartments beforechannel anchoring at the plasma membrane. Thesefindings suggested that proteolytic processing may beessential for maturation of the channel a and g subunitsen route to the cell surface (Hughey et al., 2004a).Therefore, the current view is that the unprocessedENaC channel has a low open probability, thus trigger-ing small sodium currents. This autoinhibitory mecha-nism is mediated by a disulfide-bonded loop in the asubunit. The serine proteinase furin cleaves the asubunit and thus removes autoinhibition during ENaCtrafficking to the plasma membrane. This cleavageleads to an increase in channel open probability.Additionally, a second disulfide-bonded loop in the gsubunit can also be cleaved by furin to elicit partialactivation of ENaC. Complete removal of the g subunitautoinhibitory loop can be achieved by membrane-anchored prostasin at the cell surface, thus mediatingfull activation of the channel (Szabo and Bugge, 2011).It is likely that other members of membrane-
anchored and soluble serine proteinases present in theextracellular milieu may also activate furin-processedENaC in a similar manner, because putative cleavagesites in the g subunit of ENaC have been postulated forplasmin, elastase, tissue kallikrein (KLK1), and chy-motrypsin (Adebamiro et al., 2007; Bruns et al., 2007;Passero et al., 2008; Haerteis et al., 2012b; Patel et al.,2012). Interestingly, proteinase-specific mechanisms ofENaC activation may provide differential degrees andmodalities of channel activation in a tissue-dependentmanner. For instance, Haerteis et al. (2014) identifiedtwo distinct cleavage sites for trypsin IV and trypsin I ing ENaC. They showed that trypsin IV cleaves the gsubunit at Lys189, whereas trypsin I cleaves thechannel at multiple sites. Given that trypsin IV isabundantly expressed in human epithelial cells of thekidney, the trypsin IV-mediated activation of ENaCmay provide a unique mechanism of channel activationthat could vary in pathophysiological states where tryp-sin IV expression/secretion is upregulated.It should be noted that other groups of proteinases
such as the ones that belong to the cysteine proteinasefamily have also been found to stimulate apical ENaCchannels (Haerteis et al., 2012a). For instance, Haerteisand coworkers reported that purified cathepsin-S is ableto cleave g ENaC in a heterologous expression system.This work identified two valine residues (V182 andV193) that are essential for mediating cathepsin-S-dependent activation of the channel. Of importance, asalready pointed out above, cathepsin-S can also cleave/activate PAR2 to cause biased signaling via adenylylcyclase activation. Thus ENaC ion channel and PAR2signaling can be coregulated by microenvironmentproteinases like cathepsin-S. In summary, the ENaC
channel can be taken to represent a “prototype” ionchannel regulated in a complex way by both intracellu-lar (e.g., furins) and extracellular proteinases (e.g.,trypsin IV). Multiple extracellular enzymes appear tobe able to regulate channel activity by cleaving theg-subunit at distinct sites. This process may be ofrelevance not only for normal physiologic function butalso for the effects of microenvironment proteinasesproduced by tumor cells and for the untoward impact ofpathogen-derived proteinases.
C. Proteolytic Cleavage of the Acid-SensingIon Channels.
The acid-sensing ion channel-1 (ASIC1) is a hydrogenion-gated channel that belongs to the ENaC/degenerinfamily. ASIC channels consists of a two transmembranesubunit that likely form trimers to generate a functionalchannel. ASIC1 is the first of four ASIC genes expressedin sensory neurons of the peripheral nervous system(Kellenberger and Schild, 2002, 2015). Regulation ofASIC1 channels by the serine proteinases trypsin,chymotrypsin, and proteinase K has been measured inheterologous expression systems where proteolytic pro-cessing inhibits acid-induced currents and lowers thepH of channel activation (Poirot et al., 2004; Vukicevicet al., 2006). More recently, the epithelial transmem-brane serine proteinase, matriptase, has also beenimplicated in modulating ASIC1 channel, specificallythrough three cleavage sites (Arg145, Lys185, andLys384) located in the extracellular loop of the channel(Clark et al., 2010). In pathophysiological settings,ASIC channels are critically important for the responseto ischemia and acidosis that induce endoplasmic re-ticulum stress and cell death. Along these lines, Su et al.(2011) documented a protective role for tissue kallikrein(very likely, KLK1) during ischemia/acidosis-inducedinjury. This protection may be mediated by the specificproteolytic regulation of the ASIC1a channel. Collec-tively, although serine proteinases modulate the ENaC/degenerin family, they exhibit opposite regulation ofthe different members of this family. Hence, it remainsto be fully understood how proteolytic cleavage of ENaCsubunits at some sites promotes an increase in channelopen probability, leading to larger sodium current,whereas cleavage of other distinct sites inhibits theASIC current.
D. Proteolytic Regulation of Voltage-Gated IonChannels by b-Secretase and ɣ-Secretase
Voltage-gated sodium and potassium channels arecritically important in cellular excitability by initiatingand propagating action potentials along the axon ofnerve cells. Early electrophysiological work studyingvoltage-clamped squid axons showed that pronaseapplication removes the voltage-dependent sodiumchannel inactivation, probably through cleavage of theintracellular inactivation gate intrinsic to the channel
1132 Ramachandran et al.

(Armstrong et al., 1973). Similar effects were found forthe Shaker potassium channel. These studies providedthe key basis to establish the “ball and chain” mecha-nism of mammalian Kv potassium channel inactivation(Hoshi et al., 1990). This mechanism involves theamino-terminal domain of the channel that forms theinactivation gate (ball) that is tethered to the channelpore upon depolarization, thus preventing the flow ofpotassium through the channel (Murrell-Lagnado andAldrich, 1993). Proteolytic removal of the amino-terminal tail thus blocks Kv channel inactivation. Morerecently, voltage-dependent sodium channels have beenshown to be proteolytically cleaved by the secretasefamily of proteinases, a multisubunit complex thatcleaves transmembrane proteins. The a- and b-secretase(b-site of amyloid precursor protein-cleaving enzyme 1;BACE1) enzymes have been recognized as centralproteinases implicated in the cleavage of amyloid pre-cursor protein and the production of amyloid b (Ab)peptides found in senile plaques in Alzheimer’s disease(Hardy and Selkoe, 2002). Interestingly, new evidencesuggests that the aspartyl proteinases, b-secretase(BACE1) and g-secretase can mediate proteolysis ofvoltage-gated potassium and sodium channels, respec-tively. The transmembrane topology of voltage-gatedsodium channels is very similar to that of voltage-gatedcalcium channels (see above). Like most of the voltage-gated ion channels, their function and cellular traffick-ing is regulated by auxiliary subunits that associatedirectly with the main pore-forming a subunit (Savio-Galimberti et al., 2012; Catterall, 2014). Among theseauxiliary subunits, the sodium channel-associated b2subunit has been shown to be a substrate of BACE1, adisintegrin and metalloproteinase domain-containingprotein 10 (ADAM10), and g-secretase (Kim et al., 2005;Wong et al., 2005). Consequently, proteolytic modula-tion of b2 by BACE1 and g-secretase activities promotesan increase in channel expression and thus neuronalmembrane excitability (Kim et al., 2007; Hu et al., 2010;Huth et al., 2011). Other ion channels such as thecardiac hyperpolarization-activated cation channel areblocked by trypsin-induced proteolysis (Budde et al.,1994), and this inhibition may represent an importantregulatory factor during heart failure (Chapman andSpinale, 2004). In fact, growing evidence has pointed toa role of matrix metalloproteinases (MMPs) as keyplayers of left ventricle remodeling in patients withpostmyocardial infarction (Sutton and Sharpe, 2000).Modulation of cardiac ion channels by MMPs in in-farcted tissuemay lead to functional cardiac remodelingand hence targeting MMP action on cardiac ion chan-nels could represent an innovative and effective thera-peutic approach.In conclusion, the entire repertoire of ion channels
modulated by proteolytic cleavage and importantly, theproteinases involved in this biologic process remainsto be established. Attention is rarely given to the
likelihood that the extracellular domains of many ionchannels can be targeted by microenvironment protein-ases in a way that may alter channel function. This areamerits close attention in the future. Given the manyroles that extracellular proteinases can play in cellularand tissue homeostasis, it will be crucial to understandhow the proteolytic regulation and dysregulation of ionchannels may participate in health and disease re-spectively. In addition to their regulation by endoge-nous proteinases, accumulating evidence indicates thation channels may also be subject to regulation bypathogen-derived proteinases in the course of infec-tious processes. These mechanisms have begun to bedescribed, for instance, in the context of enteritis ormyocarditis after virus infections. The identification ofthe proteinases and ion channels substrates that par-ticipate in host-pathogen interactions may identify newtargets for the development of clinically relevant ther-apeutic agents.
VI. Therapeutic Targeting of the ProteolyticSignaling System
Major roles for the proteinase-mediated signalingmechanisms outlined in Fig. 2 are to be found in thesettings of acute and chronic inflammatory disorders,like tissue trauma, arthritis, colitis, asthma, cardiomyo-pathies, and neurodegenerative diseases as well asin cancer cell growth, invasion, and metastasis, asreviewed by us inmore detail elsewhere (Ramachandranand Hollenberg, 2008; Ramachandran et al., 2012b).Thus the proteinases themselves along with theirsignal-generating substrates, including the substrate-triggered signal transduction pathways, can be consid-ered as fruitful therapeutic targets. For instance,angiotensin converting enzyme (ACE) inhibitors andangiotensin AT1 receptor blockers have proved ofenormous clinical benefit for the treatment of hyper-tension. This principle, targeting both proteolytic ago-nist production and the activated receptor, has alsobeen used very successfully to regulate signaling stim-ulated by the coagulation pathway. For that pur-pose, coagulation inhibitors have been developed thateither prevent the post-translational carboxylation-capacitation of prothrombin and factorVII (i.e., warfarin)or inhibit the catalytic activity of the enzymes thatregulate signaling (e.g., dabigatran, a peptidomimeticthrombin inhibitor). Furthermore, inhibitors that blocksignaling by the PAR1 target of thrombin have also beendeveloped (e.g., Vorapaxar). The PAR1 inhibitors haveproved both of clinical utility and of value for assessingthe role(s) of PAR1 in animal models of inflammatorydisease. That said, the clinical use of Vorapaxar hasbeen limited by its unexpected association with in-creased cerebral hemorrhage in a subset of treatedpatients (O’Donoghue et al., 2011; Wiviott et al., 2011;Goto and Tomita, 2014). Similarly, PAR2 antagonists
Proteinase-Mediated Signaling and Inflammation 1133

that diminish inflammation in animal models are nowavailable (Ramachandran et al., 2012b; Yau et al.,2013), but their clinical utility in humans has yet tobe established. The challenge of considering the PARsor their activating proteinases for therapeutic pur-poses has been summarized in some depth elsewhere(Ramachandran, 2012; Ramachandran et al., 2012b).The “take-home” messages from the literature deal-
ing with proteinase signaling systems as therapeutictargets, as summarized in the above-quoted articles, arethat 1) the proteinases clearly play key roles in a varietyof inflammatory settings, but 2) targeting either theenzymes themselves or their signal-generating systemsrepresents a substantial challenge. The therapeuticapproach for proteinase-mediated signaling thus differsin complexity from the more straightforward develop-ment of conventional receptor antagonists. This chal-lenge is due firstly to the common catalytic mechanismsshared between proteinases of a given class (e.g., serineproteinases with different substrate sequence targets)that render enzyme selectivity and potency difficult,and secondly, to the multiple proteinases of differentclasses that may cleave the same signal-generatingsubstrate (but at slightly different sites) to yield acommon inflammatory signal. Furthermore, even withvery successful proteinase antagonists, like the angio-tensin converting enzyme (ACE) inhibitors, which blockthe production of the hypertensive agonist (i.e., angio-tensin II), there can be unanticipated side effects. Thus,with the ACE inhibitors, blocking angiotensin produc-tion comes hand-in-hand with the preservation of theactivity of inflammatory peptides, i.e., the kinins.This issue has surfaced with an appreciation of the
angioedema encountered either idiosyncratically or ingenetically susceptible individuals treated with ACEinhibitors (Roberts et al., 2012; Bas et al., 2015). Thelife-threatening angioedema is due to the lack of me-tabolism of plasma kallikrein-generated bradykinin, inkeeping with the mechanism outlined in Fig. 5. None-theless, treatment of the hereditary form of thisangioedema disorder has been developed from the useof both 1) a proteinase inhibitor that blocks the genera-tion of kinins by plasma kallikrein (Kalbitor/Ecallantide,a Kunitz domain plasma kallikrein inhibitor) and 2) aGPCR inhibitor, Icatibant/Firazyr, that blocks thebradykinin B2 receptor (Bork, 2014). Thus the thera-peutic strategy for ACE-induced angioedema deals ontwo levels with the mechanism illustrated in Fig. 5: 1)blocking the enzyme and 2) blocking the receptor. Ofnote, the successful enzyme inhibitor is not one thattargets the enzyme catalytic site but rather an enzyme-selective Kunitz-domain inhibitor. This approach usingendogenous enzyme-selective allosteric site inhibitorsmay prove to be more successful in the selective block-ing of proteinase signaling than the design of catalyticsite inhibitors. In sum, in keeping with mechanism 2in Fig. 2, it has been possible to develop successful
therapeutic agents for treating hypertension and he-reditary angioedema. In a similar vein it has beenpossible to generate successful therapeutic agents forcoagulation disorders by targeting both the enzyme(thrombin) and its target receptor (PAR1). For thecomplement system, recent work shows promise of de-veloping effective C3a receptor antagonists that inprinciple can be coupled with inhibitors of the comple-ment proteinase cascade. One can be optimistic that acomparable approach will succeed in dealing with otherproteinase mediators of inflammation that may work byone or more of the mechanisms outlined in Fig. 2.
To place these successes in context, it is important toillustrate failures in targeting proteinases for thera-peutic purposes. This issue is well illustrated by thesearch for potent matrix metalloproteinase inhibitorsthat can block MMP-induced cell migration and tissueinvasion in the setting of cancer. Given the pathologicroles of MMPs in cancer and other diseases and thesuitability of the enzymes as therapeutic targets, over50 MMP inhibitors, were developed that blocked can-cer progression in murine tumor models. However,although successful in the animal models of cancer,MMP inhibitors failed in clinical trials in humans. Thefailures were due to the as yet unexplained differencesbetween humans and rodents in terms of the innateinflammatory responses that MMPs drive and becauseof insufficient knowledge about the distinct biologicroles played by the differentMMPs. That said, there is arenewed understanding of MMP biology and there isindeed new hope that the suitably selective enzymeinhibitors may find a place in the therapeutic arma-mentarium (Vandenbroucke and Libert, 2014). Forinstance, it is now known that in addition to causingthe degradation of extracellularmatrix to enable cells toexit and metastasize from a tumor site, distinct MMPscan now signal to cells by activating PAR1 in quitedifferent ways (Boire et al., 2005; Jaffre et al., 2012).Thus, effective therapymay depend on targeting both theenzyme and the effector signaling system that is acti-vated enzymatically. One keywill be to identify themodelsystems that best reflect the human pathology environ-ment. In this regard, it is important to point out thatacross species, there is considerable variation in theabundances and types of proteinases in any given tissue.This point may be of importance for elements like mastcells, which can have a quite different complement ofenzymes from one species to another. A further issuerelates to the correlation (or not) of inflammatory re-sponse genes upregulated in rodents, compared withhumans (Seok et al., 2013; Takao and Miyakawa, 2015).Thus animal models alone must be complemented byother approaches involving human-derived systems forpredicting efficacy in human pathologies. This challengeis being met by the development of human tissue-derived“organoid” culture systems that retain some of thephenotypes of the parent intact organs.
1134 Ramachandran et al.

VII. Future Perspectives
Given the complexities of signaling by proteinasesoutlined in Fig. 2 and as discussed in this review, it isimportant to ask: what does the future hold for target-ing proteinases and their signal-generating substratesfor therapeutic purposes? The answer in part is outlinedin the previous section summarizing some of the suc-cesses and failures of the approach. That said, mightthere be some guiding principles based on the experi-ence to date?
A. Targeting the Receptor
It can be suggested that, if the signaling processinvolves the generation of a peptide agonist that singlesout a principle receptor as for the renin-angiotensinsystem, it makes good sense to target the relevantreceptor. Thus, despite the unequivocal success of theangiotensin converting enzyme inhibitors for treatinghypertension, the angiotensin AT1 receptor antagonistsappear to us to be a pharmacologically more attractiveand direct solution to the problem. Fortunately, unlikethe production of prostaglandins that can cross over atmultiple EP/FP/TP receptors, the proteolytically gener-ated agonist peptides in general are more restricted intheir receptor targets. Thus, if a single receptor systemcan be identified as responsible for the pathology, areceptor antagonist might be the best target. Thatavenue is one suggested choice for a therapeutic answerto the above question.
B. Targeting the Proteinase
In the complex setting of tissue inflammation orinflammation-related tumor growth and metastasis,multiple receptors triggered by proteolytic mechanismsare undoubtedly involved. As a well-known exampleof common agonists generating diverse signals,inflammation-generated prostaglandins can in princi-ple be seen to act on multiple target receptors (e.g.,EP/TP/FP). Thus, for proteinase-mediated signalingwhere multiple signaling targets can be affected, itmay provemore efficient to target the ligand-generatingenzyme rather than the ligands themselves or theirreceptors.With proteinases like those of the coagulationcascade, which are relatively substrate-selective, tar-geting the enzyme catalytic activity can clearly work, asevidenced by the therapeutic success of thrombininhibitors and agents that block the posttranslationalactivation of the coagulation pathway enzymes. How-ever, many other members of the serine proteinase andmatrix metalloproteinase families have broad substratespecificities and have common catalytic sites thatrender substrate-selective enzyme inhibitors very chal-lenging to develop. One fruitful alternative is to seekcompounds that interact with unique allosteric enzymesites that inhibit catalytic activity in an enzyme selec-tive manner. However, as for the kallikrein-related
peptidase family, these inflammation-involved protein-ases can often work as “cascades” in keeping with thecoagulation and complement cascades. Thus, inhibitinga single enzyme in an inflammatory setting might notprove therapeutically effective, because multiple en-zymes in an inflammatory tissue microenvironmentmight provide a redundant/alternative pathway. Allthis is to say that dealing with a complex inflammatorysetting may necessitate the inhibition of more than asingle enzyme, for instance in the case of the multipleproteinases released by mast cells during an inflamma-tory response (Douaiher et al., 2014; Reber et al., 2014).Therefore, if mast cell stabilizers are not able to blockthe release of multiple proteinases into the microenvi-ronment, a single proteinase inhibitor cannot possiblystop the signaling process. An approach to this situationis outlined in the following paragraph.
C. Potential Use of Broad-Spectrum EnzymeInhibitors in a Restricted Setting over a Limited TimeFrame: A Heretical Approach to Consider
Although an inflammatory setting may result in theactivation of multiple proteinase families, the basiccatalytic mechanisms of the inflammatory enzymesare relatively few in number (Fig. 2). Thus a majorityof the proteinases secreted into the inflammatorymicroenvironment act via serine, cysteine, or metal-loproteinase mechanisms. Therefore, a “heretical ap-proach” might be to consider the localized and timeframe-restricted administration of broad-spectrum in-hibitors that target multiple classes and multipleenzymes of the same class. As an example, one canpoint to the limited, but clinically successful, use of anintranasal proteinase inhibitor for alleviating allergicrhinitis (Erin et al., 2006). Of note, the inhibitor, RWJ-58643, was able to target not only b-tryptase, but alsotrypsin and presumably many other trypsin familymembers released in the nasal cavity. This approachillustrates three important principles: first, the evalu-ation was done in humans and not other animal species,where different enzymes would very likely be active inthe inflammatory setting; second, that multiple en-zymes with a common catalytic mechanism can beblocked simultaneously and importantly; third, theinhibitors can be administered in a localized settingover a limited time frame. Clearly, the approach wouldnot be appropriate for a systemic dosing of the inhibi-tors, which would certainly cause many “off-target:complications, and the approach takes advantage ofthe “fourth dimension” of therapeutics, namely the timefactor. Thus, by limiting the time frame of enzymeinhibition to an acute exposure setting, it is possible toattenuate some of the rapid inflammatory signal path-ways, leaving the signal pathways required for theresolution of inflammation at later time points un-affected. Thus the same signal stimuli that can bedamaging early on may be required at a later stage for
Proteinase-Mediated Signaling and Inflammation 1135

the healing process. Thinking as a pharmacologist, onecan use these principles for selected sites of drugdelivery such as the skin, airway, gastrointestinal tract(oral cavity and intestinal lumen from esophagus torectum), joint space, bladder, ear canal, and conjuncti-val sac, among others. Optimally, the agents usedshould be poorly absorbed, so as not to be systemicallyactive, where they would cause untoward side effects.We suggest that by examining in detail the molecularpharmacology of the mechanisms of signal generationand by designing novel approaches to inhibit theenzymes either selectively or nonselectively as a family,it should be possible to design novel agents that cancontrol the inflammatory process. To that end, it ishoped that the principles outlined in this review willlead to new therapeutic modalities to treat inflamma-tory disease in the future.
Acknowledgments
The authors are indebted to the editor and reviewers for theirconstructive comments that have led to a much improved quality ofour text.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Ramachandran,Altier, Oikonomopoulou, Hollenberg.
ReferencesAdam E, Hansen KK, Astudillo Fernandez O, Coulon L, Bex F, Duhant X, JaumotteE, Hollenberg MD, and Jacquet A (2006) The house dust mite allergen Der p 1,unlike Der p 3, stimulates the expression of interleukin-8 in human airway epi-thelial cells via a proteinase-activated receptor-2-independent mechanism. J BiolChem 281:6910–6923.
Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD,and Hooper JD (2011) Structure, function and pathophysiology of protease acti-vated receptors. Pharmacol Ther 130:248–282.
Adebamiro A, Cheng Y, Rao US, Danahay H, and Bridges RJ (2007) A segment ofgamma ENaC mediates elastase activation of Na+ transport. J Gen Physiol 130:611–629.
Ahn HS, Foster C, Boykow G, Arik L, Smith-Torhan A, Hesk D, and Chatterjee M(1997) Binding of a thrombin receptor tethered ligand analogue to human plateletthrombin receptor. Mol Pharmacol 51:350–356.
Al-Ani B, Hansen KK, and Hollenberg MD (2004) Proteinase-activated receptor-2:key role of amino-terminal dipeptide residues of the tethered ligand for receptoractivation. Mol Pharmacol 65:149–156.
al-Ani B, Saifeddine M, and Hollenberg MD (1995) Detection of functional re-ceptors for the proteinase-activated-receptor-2-activating polypeptide, SLIGRL-NH2, in rat vascular and gastric smooth muscle. Can J Physiol Pharmacol 73:1203–1207.
Al-Ani B, Saifeddine M, Kawabata A, and Hollenberg MD (1999) Proteinase activatedreceptor 2: Role of extracellular loop 2 for ligand-mediated activation. Br J Phar-macol 128:1105–1113.
Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M,Peters JA, Harmar AJ, and Collaborators C; CGTP Collaborators (2013) TheConcise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br JPharmacol 170:1459–1581.
Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, Brückner UB, Nilsson B,Gebhard F, Lambris JD, et al. (2010) Molecular intercommunication between thecomplement and coagulation systems. J Immunol 185:5628–5636.
Andrade-Gordon P, Maryanoff BE, Derian CK, Zhang HC, Addo MF, Darrow AL,Eckardt AJ, Hoekstra WJ, McComsey DF, Oksenberg D, et al. (1999) Design,synthesis, and biological characterization of a peptide-mimetic antagonist for atethered-ligand receptor. Proc Natl Acad Sci USA 96:12257–12262.
Araç D, Aust G, Calebiro D, Engel FB, Formstone C, Goffinet A, Hamann J, Kittel RJ,Liebscher I, Lin HH, et al. (2012a) Dissecting signaling and functions of adhesion Gprotein-coupled receptors. Ann N Y Acad Sci 1276:1–25.
Araç D, Boucard AA, Bolliger MF, Nguyen J, Soltis SM, Südhof TC, and Brunger AT(2012b) A novel evolutionarily conserved domain of cell-adhesion GPCRs mediatesautoproteolysis. EMBO J 31:1364–1378.
Ardi VC, Kupriyanova TA, Deryugina EI, and Quigley JP (2007) Human neutrophilsuniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator ofangiogenesis. Proc Natl Acad Sci USA 104:20262–20267.
Arizmendi NG, Abel M, Mihara K, Davidson C, Polley D, Nadeem A, El Mays T,Gilmore BF, Walker B, Gordon JR, et al. (2011) Mucosal allergic sensitization tocockroach allergens is dependent on proteinase activity and proteinase-activatedreceptor-2 activation. J Immunol 186:3164–3172.
Armstrong CM, Bezanilla F, and Rojas E (1973) Destruction of sodium conductanceinactivation in squid axons perfused with pronase. J Gen Physiol 62:375–391.
Arpino V, Brock M, and Gill SE (2015) The role of TIMPs in regulation of extracel-lular matrix proteolysis. Matrix Biol 44-46:247–254.
Austin KM, Covic L, and Kuliopulos A (2013) Matrix metalloproteases and PAR1activation. Blood 121:431–439.
Awasthi V, Mandal SK, Papanna V, Rao LV, and Pendurthi UR (2007) Modulation oftissue factor-factor VIIa signaling by lipid rafts and caveolae. Arterioscler ThrombVasc Biol 27:1447–1455.
Bae JS, Yang L, and Rezaie AR (2007) Receptors of the protein C activation andactivated protein C signaling pathways are colocalized in lipid rafts of endothelialcells. Proc Natl Acad Sci USA 104:2867–2872.
Bae JS, Yang L, and Rezaie AR (2008) Lipid raft localization regulates the cleavagespecificity of protease activated receptor 1 in endothelial cells. J Thromb Haemost6:954–961.
Ban Y, Wang MC, Watt KW, Loor R, and Chu TM (1984) The proteolytic activity ofhuman prostate-specific antigen. Biochem Biophys Res Commun 123:482–488.
Barrett AJ (1981) Alpha 2-macroglobulin. Methods Enzymol 80:737–754.Barrett AJ and Starkey PM (1973) The interaction of alpha 2-macroglobulin withproteinases. Characteristics and specificity of the reaction, and a hypothesis con-cerning its molecular mechanism. Biochem J 133:709–724.
Barry GD, Suen JY, Le GT, Cotterell A, Reid RC, and Fairlie DP (2010) Novelagonists and antagonists for human protease activated receptor 2. J Med Chem 53:7428–7440.
Bas M, Greve J, Stelter K, Havel M, Strassen U, Rotter N, Veit J, Schossow B,Hapfelmeier A, Kehl V, et al. (2015) A randomized trial of icatibant in ACE-inhibitor-induced angioedema. N Engl J Med 372:418–425.
Bhoola KD, Figueroa CD, and Worthy K (1992) Bioregulation of kinins: kallikreins,kininogens, and kininases. Pharmacol Rev 44:1–80.
Blaber M, Yoon H, Juliano MA, Scarisbrick IA, and Blaber SI (2010) Functionalintersection of the kallikrein-related peptidases (KLKs) and thrombostasis axis.Biol Chem 391:311–320.
Blackburn JS and Brinckerhoff CE (2008) Matrix metalloproteinase-1 and thrombindifferentially activate gene expression in endothelial cells via PAR-1 and promoteangiogenesis. Am J Pathol 173:1736–1746.
Blackburn JS, Liu I, Coon CI, and Brinckerhoff CE (2009) A matrix metal-loproteinase-1/protease activated receptor-1 signaling axis promotes melanomainvasion and metastasis. Oncogene 28:4237–4248.
Bode MF and Mackman N (2015) Protective and pathological roles of tissue factor inthe heart. Hamostaseologie 35:37–46.
Böhm SK, Khitin LM, Grady EF, Aponte G, Payan DG, and Bunnett NW (1996a)Mechanisms of desensitization and resensitization of proteinase-activated re-ceptor-2. J Biol Chem 271:22003–22016.
Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, Kahn M,Nelken NA, Coughlin SR, Payan DG, et al. (1996b) Molecular cloning, expressionand potential functions of the human proteinase-activated receptor-2. Biochem J314:1009–1016.
Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, and Kuliopulos A (2005) PAR1 is amatrix metalloprotease-1 receptor that promotes invasion and tumorigenesis ofbreast cancer cells. Cell 120:303–313.
Boitano S, Flynn AN, Sherwood CL, Schulz SM, Hoffman J, Gruzinova I, and Daines MO(2011) Alternaria alternata serine proteases induce lung inflammation and airway epi-thelial cell activation via PAR2. Am J Physiol Lung Cell Mol Physiol 300:L605–L614.
Bond M, Chase AJ, Baker AH, and Newby AC (2001) Inhibition of transcriptionfactor NF-kappaB reduces matrix metalloproteinase-1, -3 and -9 production byvascular smooth muscle cells. Cardiovasc Res 50:556–565.
Bond M, Fabunmi RP, Baker AH, and Newby AC (1998) Synergistic upregulation ofmetalloproteinase-9 by growth factors and inflammatory cytokines: an absoluterequirement for transcription factor NF-kappa B. FEBS Lett 435:29–34.
Borgoño CA and Diamandis EP (2004) The emerging roles of human tissue kalli-kreins in cancer. Nat Rev Cancer 4:876–890.
Bork K (2014) An evidence based therapeutic approach to hereditary and acquiredangioedema. Curr Opin Allergy Clin Immunol 14:354–362.
Botham A, Guo X, Xiao YP, Morice AH, Compton SJ, and Sadofsky LR (2011) Pal-mitoylation of human proteinase-activated receptor-2 differentially regulatesreceptor-triggered ERK1/2 activation, calcium signalling and endocytosis. Biochem J438:359–367.
Brass LF, Vassallo RR Jr, Belmonte E, Ahuja M, Cichowski K, and Hoxie JA (1992)Structure and function of the human platelet thrombin receptor. Studies usingmonoclonal antibodies directed against a defined domain within the receptor Nterminus. J Biol Chem 267:13795–13798.
Bretschneider E, Spanbroek R, Lötzer K, Habenicht AJ, and Schrör K (2003) Evi-dence for functionally active protease-activated receptor-3 (PAR-3) in human vas-cular smooth muscle cells. Thromb Haemost 90:704–709.
Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, Dubus P,and Hovnanian A (2009) Kallikrein 5 induces atopic dermatitis-like lesionsthrough PAR2-mediated thymic stromal lymphopoietin expression in Nethertonsyndrome. J Exp Med 206:1135–1147.
Briot A, Lacroix M, Robin A, Steinhoff M, Deraison C, and Hovnanian A (2010) Par2inactivation inhibits early production of TSLP, but not cutaneous inflammation, inNetherton syndrome adult mouse model. J Invest Dermatol 130:2736–2742.
Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, Hughey RP,and Kleyman TR (2007) Epithelial Na+ channels are fully activated by furin- andprostasin-dependent release of an inhibitory peptide from the gamma-subunit. JBiol Chem 282:6153–6160.
Bryant RJ and Lilja H (2014) Emerging PSA-based tests to improve screening. UrolClin North Am 41:267–276.
Budde T, White JA, and Kay AR (1994) Hyperpolarization-activated Na(+)-K+ cur-rent (Ih) in neocortical neurons is blocked by external proteolysis and internalTEA. J Neurophysiol 72:2737–2742.
1136 Ramachandran et al.

Burger MM (1970) Proteolytic enzymes initiating cell division and escape from con-tact inhibition of growth. Nature 227:170–171.
Camerer E, Huang W, and Coughlin SR (2000) Tissue factor- and factor X-dependentactivation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci USA97:5255–5260.
Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, and RossierBC (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologoussubunits. Nature 367:463–467.
Canto I and Trejo J (2013) Palmitoylation of protease-activated receptor-1 regulatesadaptor protein complex-2 and -3 interaction with tyrosine-based motifs andendocytic sorting. J Biol Chem 288:15900–15912.
Carney DH and Cunningham DD (1977) Initiation of check cell division by trypsinaction at the cell surface. Nature 268:602–606.
Carney DH and Cunningham DD (1978) Transmembrane action of thrombin initiateschick cell division. J Supramol Struct 9:337–350.
Catterall WA (2014) Sodium channels, inherited epilepsy, and antiepileptic drugs.Annu Rev Pharmacol Toxicol 54:317–338.
Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y,Palamanda J, Agans-Fantuzzi J, et al. (2008) Discovery of a novel, orally activehimbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplateletactivity. J Med Chem 51:3061–3064.
Chackalamannil S, Xia Y, Greenlee WJ, Clasby M, Doller D, Tsai H, Asberom T,Czarniecki M, Ahn HS, Boykow G, et al. (2005) Discovery of potent orally activethrombin receptor (protease activated receptor 1) antagonists as novel antith-rombotic agents. J Med Chem 48:5884–5887.
Chambers RC and Scotton CJ (2012) Coagulation cascade proteinases in lung injuryand fibrosis. Proc Am Thorac Soc 9:96–101.
Chapman RE and Spinale FG (2004) Extracellular protease activation and unrav-eling of the myocardial interstitium: critical steps toward clinical applications. AmJ Physiol Heart Circ Physiol 286:H1–H10.
Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, Bonafé JL,Wilkinson J, Taïeb A, Barrandon Y, et al. (2000) Mutations in SPINK5, encoding aserine protease inhibitor, cause Netherton syndrome. Nat Genet 25:141–142.
Chen B, Dores MR, Grimsey N, Canto I, Barker BL, and Trejo J (2011) Adaptorprotein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 in-ternalization via phosphorylation- and ubiquitination-dependent sorting signals. JBiol Chem 286:40760–40770.
Chen CH, Paing MM, and Trejo J (2004) Termination of protease-activated receptor-1signaling by beta-arrestins is independent of receptor phosphorylation. J BiolChem 279:10020–10031.
Chen LB and Buchanan JM (1975) Mitogenic activity of blood components. I.Thrombin and prothrombin. Proc Natl Acad Sci USA 72:131–135.
Chin AC, Lee WY, Nusrat A, Vergnolle N, and Parkos CA (2008) Neutrophil-mediated activation of epithelial protease-activated receptors-1 and -2 regulatesbarrier function and transepithelial migration. J Immunol 181:5702–5710.
Chraïbi A, Vallet V, Firsov D, Hess SK, and Horisberger JD (1998) Protease modu-lation of the activity of the epithelial sodium channel expressed in Xenopus oocytes.J Gen Physiol 111:127–138.
Chrétien M (2012) My road to Damascus: how I converted to the prohormone theoryand the proprotein convertases. Biochem Cell Biol 90:750–768.
Christensson A, Laurell CB, and Lilja H (1990) Enzymatic activity of prostate-specific antigen and its reactions with extracellular serine proteinase inhibitors.Eur J Biochem 194:755–763.
Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA,Olsen RW, Peters JA, Neubig RR, Pin JP, et al. (2014) International Union of Basicand Clinical Pharmacology. XC. multisite pharmacology: recommendations for thenomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev 66:918–947.
Chua KY, Stewart GA, Thomas WR, Simpson RJ, Dilworth RJ, Plozza TM,and Turner KJ (1988) Sequence analysis of cDNA coding for a major house dustmite allergen, Der p 1. Homology with cysteine proteases. J Exp Med 167:175–182.
Chung DW, Fujikawa K, McMullen BA, and Davie EW (1986) Human plasma pre-kallikrein, a zymogen to a serine protease that contains four tandem repeats.Biochemistry 25:2410–2417.
Chung H, Hamza M, Oikonomopoulou K, Gratio V, Saifeddine M, Virca GD, DiamandisEP, Hollenberg MD, and Darmoul D (2012) Kallikrein-related peptidase signaling incolon carcinoma cells: targeting proteinase-activated receptors. Biol Chem 393:413–420.
Clark AK and Malcangio M (2012) Microglial signalling mechanisms: Cathepsin Sand Fractalkine. Exp Neurol 234:283–292.
Clark EB, Jovov B, Rooj AK, Fuller CM, and Benos DJ (2010) Proteolytic cleavage ofhuman acid-sensing ion channel 1 by the serine protease matriptase. J Biol Chem285:27130–27143.
Clark IM, Swingler TE, Sampieri CL, and Edwards DR (2008) The regulation ofmatrix metalloproteinases and their inhibitors. Int J Biochem Cell Biol 40:1362–1378.
Clements J, Hooper J, Dong Y, and Harvey T (2001) The expanded human kallikrein(KLK) gene family: genomic organisation, tissue-specific expression and potentialfunctions. Biol Chem 382:5–14.
Clements JA (1989) The glandular kallikrein family of enzymes: tissue-specific ex-pression and hormonal regulation. Endocr Rev 10:393–419.
Clements JA (2008) Reflections on the tissue kallikrein and kallikrein-related pep-tidase family - from mice to men - what have we learnt in the last two decades? BiolChem 389:1447–1454.
Compton SJ, Renaux B, Wijesuriya SJ, and Hollenberg MD (2001) Glycosylation andthe activation of proteinase-activated receptor 2 (PAR(2)) by human mast celltryptase. Br J Pharmacol 134:705–718.
Compton SJ, Sandhu S, Wijesuriya SJ, and Hollenberg MD (2002) Glycosylation ofhuman proteinase-activated receptor-2 (hPAR2): role in cell surface expression andsignalling. Biochem J 368:495–505.
Conus S and Simon HU (2008) Cathepsins: key modulators of cell death and in-flammatory responses. Biochem Pharmacol 76:1374–1382.
Cottrell GS, Amadesi S, Grady EF, and Bunnett NW (2004) Trypsin IV, a novelagonist of protease-activated receptors 2 and 4. J Biol Chem 279:13532–13539.
Crump SM, Andres DA, Sievert G, and Satin J (2013) The cardiac L-type calciumchannel distal carboxy terminus autoinhibition is regulated by calcium. Am JPhysiol Heart Circ Physiol 304:H455–H464.
Cuatrecasas P (1969) Interaction of insulin with the cell membrane: the primaryaction of insulin. Proc Natl Acad Sci USA 63:450–457.
Cuatrecasas P (1971) Properties of the insulin receptor of isolated fat cell mem-branes. J Biol Chem 246:7265–7274.
Cumashi A, Ansuini H, Celli N, De Blasi A, O’Brien PJ, Brass LF, and Molino M(2001) Neutrophil proteases can inactivate human PAR3 and abolish theco-receptor function of PAR3 on murine platelets. Thromb Haemost 85:533–538.
Dabek M, Ferrier L, Roka R, Gecse K, Annahazi A, Moreau J, Escourrou J, Cartier C,Chaumaz G, Leveque M, et al. (2009) Luminal cathepsin g and protease-activatedreceptor 4: a duet involved in alterations of the colonic epithelial barrier in ul-cerative colitis. Am J Pathol 175:207–214.
Davey MG and Lüscher EF (1967) Actions of thrombin and other coagulant andproteolytic enzymes on blood platelets. Nature 216:857–858.
Davie EW and Ratnoff OD (1964) Waterfall Sequence For Intrinsic Blood Clotting.Science 145:1310–1312.
de Haën C (1976) The non-stoichiometric floating receptor model for hormone sen-sitive adenylyl cyclase. J Theor Biol 58:383–400.
De Jongh KS, Colvin AA, Wang KK, and Catterall WA (1994) Differential proteolysisof the full-length form of the L-type calcium channel alpha 1 subunit by calpain. JNeurochem 63:1558–1564.
de Veer SJ, Furio L, Harris JM, and Hovnanian A (2014) Proteases: common culpritsin human skin disorders. Trends Mol Med 20:166–178.
Defea K (2008) Beta-arrestins and heterotrimeric G-proteins: collaborators andcompetitors in signal transduction. Br J Pharmacol 153 (Suppl 1):S298–S309.
Déry O, Thoma MS, Wong H, Grady EF, and Bunnett NW (1999) Trafficking ofproteinase-activated receptor-2 and beta-arrestin-1 tagged with green fluorescentprotein. beta-Arrestin-dependent endocytosis of a proteinase receptor. J Biol Chem274:18524–18535.
Deryugina EI, Zajac E, Juncker-Jensen A, Kupriyanova TA, Welter L, and Quigley JP(2014) Tissue-infiltrating neutrophils constitute the major in vivo source ofangiogenesis-inducing MMP-9 in the tumor microenvironment.Neoplasia 16:771–788.
DiScipio RG (1982) The activation of the alternative pathway C3 convertase by hu-man plasma kallikrein. Immunology 45:587–595.
Disse J, Petersen HH, Larsen KS, Persson E, Esmon N, Esmon CT, Teyton L,Petersen LC, and Ruf W (2011) The endothelial protein C receptor supports tissuefactor ternary coagulation initiation complex signaling through protease-activatedreceptors. J Biol Chem 286:5756–5767.
Dores MR, Chen B, Lin H, Soh UJ, Paing MM, Montagne WA, Meerloo T, and Trejo J(2012) ALIX binds a YPX(3)L motif of the GPCR PAR1 and mediates ubiquitin-independent ESCRT-III/MVB sorting. J Cell Biol 197:407–419.
Douaiher J, Succar J, Lancerotto L, Gurish MF, Orgill DP, Hamilton MJ, Krilis SA,and Stevens RL (2014) Development of mast cells and importance of their tryptaseand chymase serine proteases in inflammation and wound healing. Adv Immunol122:211–252.
Dubin G (2005) Proteinaceous cysteine protease inhibitors. Cell Mol Life Sci 62:653–669.
Dulon S, Candé C, Bunnett NW, Hollenberg MD, Chignard M, and Pidard D (2003)Proteinase-activated receptor-2 and human lung epithelial cells: disarming byneutrophil serine proteinases. Am J Respir Cell Mol Biol 28:339–346.
Dulon S, Leduc D, Cottrell GS, D’Alayer J, Hansen KK, Bunnett NW, HollenbergMD, Pidard D, and Chignard M (2005) Pseudomonas aeruginosa elastase disablesproteinase-activated receptor 2 in respiratory epithelial cells. Am J Respir Cell MolBiol 32:411–419.
Eggertsen G, Hellman U, Lundwall A, Folkersen J, and Sjöquist J (1985) Charac-terization of tryptic fragments of human complement factor C3. Mol Immunol 22:833–841.
El-Daly M, Saifeddine M, Mihara K, Ramachandran R, Triggle CR, and HollenbergMD (2014) Proteinase-activated receptors 1 and 2 and the regulation of porcinecoronary artery contractility: a role for distinct tyrosine kinase pathways. Br JPharmacol 171:2413–2425.
Erin EM, Leaker BR, Zacharasiewicz A, Higgins LA, Nicholson GC, Boyce MJ, deBoer P, Jones RC, Durham SR, Barnes PJ, et al. (2006) Effects of a reversible beta-tryptase and trypsin inhibitor (RWJ-58643) on nasal allergic responses. Clin ExpAllergy 36:458–464.
Esmon CT (2003) The protein C pathway. Chest 124(3, Suppl)26S–32S.Esmon CT (2014) Targeting factor Xa and thrombin: impact on coagulation andbeyond. Thromb Haemost 111:625–633.
Faruqi TR, Weiss EJ, ShapiroMJ, Huang W, and Coughlin SR (2000) Structure-functionanalysis of protease-activated receptor 4 tethered ligand peptides. Determinants ofspecificity and utility in assays of receptor function. J Biol Chem 275:19728–19734.
Feistritzer C, Lenta R, and Riewald M (2005) Protease-activated receptors-1 and -2can mediate endothelial barrier protection: role in factor Xa signaling. J ThrombHaemost 3:2798–2805.
Frenette G, Deperthes D, Tremblay RR, Lazure C, and Dubé JY (1997) Purification ofenzymatically active kallikrein hK2 from human seminal plasma. Biochim BiophysActa 1334:109–115.
Fukunaga R, Hirano K, Hirano M, Niiro N, Nishimura J, Maehara Y, and Kanaide H(2006) Upregulation of proteinase-activated receptors and hypercontractile re-sponses precede development of arterial lesions after balloon injury. Am J PhysiolHeart Circ Physiol 291:H2388–H2395.
Fukuoka Y, Xia HZ, Sanchez-Muñoz LB, Dellinger AL, Escribano L, and SchwartzLB (2008) Generation of anaphylatoxins by human beta-tryptase from C3, C4, andC5. J Immunol 180:6307–6316.
Proteinase-Mediated Signaling and Inflammation 1137

Fuller MD, Emrick MA, Sadilek M, Scheuer T, and Catterall WA (2010) Molecularmechanism of calcium channel regulation in the fight-or-flight response. Sci Signal3:ra70.
Gamper N and Shapiro MS (2007) Regulation of ion transport proteins by membranephosphoinositides. Nat Rev Neurosci 8:921–934.
Ganguly P (1974) Binding of thrombin to human platelets. Nature 247:306–307.Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, Eick RT,and Hosey MM (2001) C-terminal fragments of the alpha 1C (CaV1.2) subunitassociate with and regulate L-type calcium channels containing C-terminal-truncated alpha 1C subunits. J Biol Chem 276:21089–21097.
Gardell LR, Ma JN, Seitzberg JG, Knapp AE, Schiffer HH, Tabatabaei A, Davis CN,Owens M, Clemons B, Wong KK, et al. (2008) Identification and characterization ofnovel small-molecule protease-activated receptor 2 agonists. J Pharmacol ExpTher 327:799–808.
Gettins PG (2002) Serpin structure, mechanism, and function. Chem Rev 102:4751–4804.
Ghebrehiwet B, Randazzo BP, Dunn JT, Silverberg M, and Kaplan AP (1983)Mechanisms of activation of the classical pathway of complement by Hagemanfactor fragment. J Clin Invest 71:1450–1456.
Ghebrehiwet B, Silverberg M, and Kaplan AP (1981) Activation of the classicalpathway of complement by Hageman factor fragment. J Exp Med 153:665–676.
Ghosh S, Pendurthi UR, Steinoe A, Esmon CT, and Rao LV (2007) Endothelial cellprotein C receptor acts as a cellular receptor for factor VIIa on endothelium. J BiolChem 282:11849–11857.
Gieseler F, Ungefroren H, Settmacher U, Hollenberg MD, and Kaufmann R (2013)Proteinase-activated receptors (PARs) - focus on receptor-receptor-interactions andtheir physiological and pathophysiological impact. Cell Commun Signal 11:86.
Gomez DE, Alonso DF, Yoshiji H, and Thorgeirsson UP (1997) Tissue inhibitors ofmetalloproteinases: structure, regulation and biological functions. Eur J Cell Biol74:111–122.
Goto S and Tomita A (2014) New antithrombotics for secondary prevention of acutecoronary syndrome. Clin Cardiol 37:178–187.
Grab DJ, Garcia-Garcia JC, Nikolskaia OV, Kim YV, Brown A, Pardo CA, Zhang Y,Becker KG, Wilson BA, de A Lima AP, et al. (2009) Protease activated receptorsignaling is required for African trypanosome traversal of human brain micro-vascular endothelial cells. PLoS Negl Trop Dis 3:e479.
Grant AD, Cottrell GS, Amadesi S, Trevisani M, Nicoletti P, Materazzi S, Altier C,Cenac N, Zamponi GW, Bautista-Cruz F, et al. (2007) Protease-activated receptor2 sensitizes the transient receptor potential vanilloid 4 ion channel to cause me-chanical hyperalgesia in mice. J Physiol 578:715–733.
Gratio V, Beaufort N, Seiz L, Maier J, Virca GD, Debela M, Grebenchtchikov N,Magdolen V, and Darmoul D (2010) Kallikrein-related peptidase 4: a new activatorof the aberrantly expressed protease-activated receptor 1 in colon cancer cells. AmJ Pathol 176:1452–1461.
Gratio V, Loriot C, Virca GD, Oikonomopoulou K, Walker F, Diamandis EP, HollenbergMD, and Darmoul D (2011) Kallikrein-related peptidase 14 acts on proteinase-activated receptor 2 to induce signaling pathway in colon cancer cells. Am J Pathol179: 2625–2636.
Gulla KC, Gupta K, Krarup A, Gal P, Schwaeble WJ, Sim RB, O’Connor CD,and Hajela K (2010) Activation of mannan-binding lectin-associated serine prote-ases leads to generation of a fibrin clot. Immunology 129:482–495.
Gunawan H, Takai T, Ikeda S, Okumura K, and Ogawa H (2008) Protease activity ofallergenic pollen of cedar, cypress, juniper, birch and ragweed. Allergol Int 57:83–91.
Haerteis S, Krappitz A, Krappitz M, Murphy JE, Bertog M, Krueger B, Nacken R,Chung H, Hollenberg MD, Knecht W, et al. (2014) Proteolytic activation of thehuman epithelial sodium channel by trypsin IV and trypsin I involves distinctcleavage sites. J Biol Chem 289:19067–19078.
Haerteis S, Krappitz M, Bertog M, Krappitz A, Baraznenok V, Henderson I, LindströmE, Murphy JE, Bunnett NW, and Korbmacher C (2012a) Proteolytic activation of theepithelial sodium channel (ENaC) by the cysteine protease cathepsin-S. Pflugers Arch464:353–365.
Haerteis S, Krappitz M, Diakov A, Krappitz A, Rauh R, and Korbmacher C (2012b)Plasmin and chymotrypsin have distinct preferences for channel activatingcleavage sites in the g subunit of the human epithelial sodium channel. J GenPhysiol 140:375–389.
Hamann J, Aust G, Araç D, Engel FB, Formstone C, Fredriksson R, Hall RA, HartyBL, Kirchhoff C, Knapp B, et al. (2015) International Union of Basic and ClinicalPharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol Rev 67:338–367.
Hammes SR, Shapiro MJ, and Coughlin SR (1999) Shutoff and agonist-triggeredinternalization of protease-activated receptor 1 can be separated by mutation ofputative phosphorylation sites in the cytoplasmic tail. Biochemistry 38:9308–9316.
Han J, Bae SY, Oh SJ, Lee J, Lee JH, Lee HC, Lee SK, Kil WH, Kim SW, Nam SJ,et al. (2014) Zerumbone suppresses IL-1b-induced cell migration and invasion byinhibiting IL-8 and MMP-3 expression in human triple-negative breast cancercells. Phytother Res 28:1654–1660.
Hansen KK, Saifeddine M, and Hollenberg MD (2004) Tethered ligand-derivedpeptides of proteinase-activated receptor 3 (PAR3) activate PAR1 and PAR2 inJurkat T cells. Immunology 112:183–190.
Hardy J and Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease:progress and problems on the road to therapeutics. Science 297:353–356.
Hasdemir B, Bunnett NW, and Cottrell GS (2007) Hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) mediates post-endocytic trafficking ofprotease-activated receptor 2 and calcitonin receptor-like receptor. J Biol Chem282:29646–29657.
Hiemstra PS (2002) Novel roles of protease inhibitors in infection and inflammation.Biochem Soc Trans 30:116–120.
Hockerman GH, Peterson BZ, Johnson BD, and Catterall WA (1997) Molecular de-terminants of drug binding and action on L-type calcium channels. Annu RevPharmacol Toxicol 37:361–396.
Hollenberg MD, Laniyonu AA, Saifeddine M, and Moore GJ (1993) Role of the amino-and carboxyl-terminal domains of thrombin receptor-derived polypeptides in bio-logical activity in vascular endothelium and gastric smooth muscle: evidence forreceptor subtypes. Mol Pharmacol 43:921–930.
Hollenberg MD, Mihara K, Polley D, Suen JY, Han A, Fairlie DP, and RamachandranR (2014) Biased signaling and proteinase-activated receptors (PARs): Targetinginflammatory disease. Br J Pharmacol 171: 1180–1994.
Hollenberg MD, Renaux B, Hyun E, Houle S, Vergnolle N, Saifeddine M,and Ramachandran R (2008) Derivatized 2-furoyl-LIGRLO-amide, a versatile andselective probe for proteinase-activated receptor 2: binding and visualization. JPharmacol Exp Ther 326:453–462.
Hollenberg MD and Saifeddine M (2001) Proteinase-activated receptor 4 (PAR4):activation and inhibition of rat platelet aggregation by PAR4-derived peptides. CanJ Physiol Pharmacol 79:439–442.
Hoshi T, Zagotta WN, and Aldrich RW (1990) Biophysical and molecular mechanismsof Shaker potassium channel inactivation. Science 250:533–538.
Hovnanian A (2013) Netherton syndrome: skin inflammation and allergy by loss ofprotease inhibition. Cell Tissue Res 351:289–300.
Hu X, Zhou X, He W, Yang J, Xiong W, Wong P, Wilson CG, and Yan R (2010) BACE1deficiency causes altered neuronal activity and neurodegeneration. J Neurosci 30:8819–8829.
Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD,Johnson JP, Stockand JD, and Kleyman TR (2004a) Epithelial sodium channelsare activated by furin-dependent proteolysis. J Biol Chem 279:18111–18114.
Hughey RP, Bruns JB, Kinlough CL, and Kleyman TR (2004b) Distinct pools ofepithelial sodium channels are expressed at the plasma membrane. J Biol Chem279:48491–48494.
Hulme JT, Konoki K, Lin TW, Gritsenko MA, Camp DG 2nd, Bigelow DJ,and Catterall WA (2005) Sites of proteolytic processing and noncovalent associa-tion of the distal C-terminal domain of CaV1.1 channels in skeletal muscle. ProcNatl Acad Sci USA 102:5274–5279.
Hulme JT, Yarov-Yarovoy V, Lin TW, Scheuer T, and Catterall WA (2006) Auto-inhibitory control of the CaV1.2 channel by its proteolytically processed distalC-terminal domain. J Physiol 576:87–102.
Huth T, Rittger A, Saftig P, and Alzheimer C (2011) b-Site APP-cleaving enzyme1 (BACE1) cleaves cerebellar Na+ channel b4-subunit and promotes Purkinjecell firing by slowing the decay of resurgent Na+ current. Pflugers Arch 461:355–371.
Hyun E, Ramachandran R, Cenac N, Houle S, Rousset P, Saxena A, Liblau RS,Hollenberg MD, and Vergnolle N (2010) Insulin modulates protease-activated re-ceptor 2 signaling: implications for the innate immune response. J Immunol 184:2702–2709.
Isaacs WB and Coffey DS (1984) The predominant protein of canine seminal plasmais an enzyme. J Biol Chem 259:11520–11526.
Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T,and Coughlin SR (1997) Protease-activated receptor 3 is a second thrombin re-ceptor in humans. Nature 386:502–506.
Ishii K, Chen J, Ishii M, Koch WJ, Freedman NJ, Lefkowitz RJ, and Coughlin SR(1994) Inhibition of thrombin receptor signaling by a G-protein coupled receptorkinase. Functional specificity among G-protein coupled receptor kinases. J BiolChem 269:1125–1130.
Ito T and Maruyama I (2011) Thrombomodulin: protectorate God of the vasculaturein thrombosis and inflammation. J Thromb Haemost 9 (Suppl 1):168–173.
Jacob C, Cottrell GS, Gehringer D, Schmidlin F, Grady EF, and Bunnett NW (2005)c-Cbl mediates ubiquitination, degradation, and down-regulation of humanprotease-activated receptor 2. J Biol Chem 280:16076–16087.
Jacobs S and Cuatrecasas P (1976) The mobile receptor hypothesis and “coopera-tivity” of hormone binding. Application to insulin. Biochim Biophys Acta 433:482–495.
Jaffré F, Friedman AE, Hu Z, Mackman N, and Blaxall BC (2012) b-adrenergicreceptor stimulation transactivates protease-activated receptor 1 via matrixmetalloproteinase 13 in cardiac cells. Circulation 125:2993–3003.
Jiang X, Bailly MA, Panetti TS, Cappello M, Konigsberg WH, and Bromberg ME(2004) Formation of tissue factor-factor VIIa-factor Xa complex promotes cellularsignaling and migration of human breast cancer cells. J Thromb Haemost 2:93–101.
Joseph K and Kaplan AP (2005) Formation of bradykinin: a major contributor to theinnate inflammatory response. Adv Immunol 86:159–208.
Kahn ML, Zheng YW, Huang W, Bigornia V, Zeng D, Moff S, Farese RV Jr, Tam C,and Coughlin SR (1998) A dual thrombin receptor system for platelet activation.Nature 394:690–694.
Kancharla A, Maoz M, Jaber M, Agranovich D, Peretz T, Grisaru-Granovsky S,Uziely B, and Bar-Shavit R (2015) PH motifs in PAR1&2 endow breast cancergrowth. Nat Commun 6:8853.
Kanke T, Ishiwata H, Kabeya M, Saka M, Doi T, Hattori Y, Kawabata A, and PlevinR (2005) Binding of a highly potent protease-activated receptor-2 (PAR2) activatingpeptide, [(3)H]2-furoyl-LIGRL-NH(2), to human PAR2. Br J Pharmacol 145:255–263.
Kaplan AP and Ghebrehiwet B (2010) The plasma bradykinin-forming pathways andits interrelationships with complement. Mol Immunol 47:2161–2169.
Kellenberger S and Schild L (2002) Epithelial sodium channel/degenerin family of ionchannels: a variety of functions for a shared structure. Physiol Rev 82:735–767.
Kellenberger S and Schild L (2015) International Union of Basic and ClinicalPharmacology. XCI. structure, function, and pharmacology of acid-sensing ionchannels and the epithelial Na+ channel. Pharmacol Rev 67:1–35.
Kenakin T (2011) Functional selectivity and biased receptor signaling. J PharmacolExp Ther 336:296–302.
1138 Ramachandran et al.

Kenakin T (2013) New concepts in pharmacological efficacy at 7TM receptors:IUPHAR review 2. Br J Pharmacol 168:554–575.
Kenakin T and Christopoulos A (2013) Signalling bias in new drug discovery: de-tection, quantification and therapeutic impact. Nat Rev Drug Discov 12:205–216.
Kenakin T and Miller LJ (2010) Seven transmembrane receptors as shapeshiftingproteins: the impact of allosteric modulation and functional selectivity on new drugdiscovery. Pharmacol Rev 62:265–304.
Khokha R, Murthy A, and Weiss A (2013) Metalloproteinases and their natural in-hibitors in inflammation and immunity. Nat Rev Immunol 13:649–665.
Kim DY, Carey BW, Wang H, Ingano LA, Binshtok AM, Wertz MH, Pettingell WH,He P, Lee VM, Woolf CJ, et al. (2007) BACE1 regulates voltage-gated sodiumchannels and neuronal activity. Nat Cell Biol 9:755–764.
Kim DY, Ingano LA, Carey BW, Pettingell WH, and Kovacs DM (2005) Pre-senilin/gamma-secretase-mediated cleavage of the voltage-gated sodium chan-nel beta2-subunit regulates cell adhesion and migration. J Biol Chem 280:23251–23261.
Kimura M, Andersen TT, Fenton JW 2nd, Bahou WF, and Aviv A (1996) Plasmin-platelet interaction involves cleavage of functional thrombin receptor. Am JPhysiol 271:C54–C60.
Kinlough-Rathbone RL, Rand ML, and Packham MA (1993) Rabbit and rat plateletsdo not respond to thrombin receptor peptides that activate human platelets. Blood82:103–106.
Kleyman TR, Carattino MD, and Hughey RP (2009) ENaC at the cutting edge: reg-ulation of epithelial sodium channels by proteases. J Biol Chem 284:20447–20451.
Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, and Köhl J (2009) The role of theanaphylatoxins in health and disease. Mol Immunol 46:2753–2766.
Klos A, Wende E, Wareham KJ, and Monk PN (2013) International Union of Basicand Clinical Pharmacology. [corrected]. LXXXVII. Complement peptide C5a, C4a,and C3a receptors. Pharmacol Rev 65:500–543.
Kogushi M, Matsuoka T, Kawata T, Kuramochi H, Kawaguchi S, Murakami K,Hiyoshi H, Suzuki S, Kawahara T, Kajiwara A, et al. (2011) The novel and orallyactive thrombin receptor antagonist E5555 (Atopaxar) inhibits arterial thrombosiswithout affecting bleeding time in guinea pigs. Eur J Pharmacol 657:131–137.
Komissarov AA, Mazar AP, Koenig K, Kurdowska AK, and Idell S (2009) Regulationof intrapleural fibrinolysis by urokinase-alpha-macroglobulin complexes intetracycline-induced pleural injury in rabbits. Am J Physiol Lung Cell Mol Physiol297:L568–L577.
Kono T and Barham FW (1971) Insulin-like effects of trypsin on fat cells. Localizationof the metabolic steps and the cellular site affected by the enzyme. J Biol Chem246:6204–6209.
Krarup A, Wallis R, Presanis JS, Gál P, and Sim RB (2007) Simultaneous activationof complement and coagulation by MBL-associated serine protease 2. PLoS One 2:e623.
Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, and Costello CE (1999)Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of trunca-tion, and implications for thrombolytic therapy. Biochemistry 38:4572–4585.
Kuriyama M, Wang MC, Papsidero LD, Killian CS, Shimano T, Valenzuela L,Nishiura T, Murphy GP, and Chu TM (1980) Quantitation of prostate-specificantigen in serum by a sensitive enzyme immunoassay. Cancer Res 40:4658–4662.
Langenhan T, Aust G, and Hamann J (2013) Sticky signaling–adhesion class Gprotein-coupled receptors take the stage. Sci Signal 6:re3.
Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, Rosado CJ,Langendorf CG, Pike RN, Bird PI, et al. (2006) An overview of the serpin super-family. Genome Biol 7:216.
Lawrence MG, Lai J, and Clements JA (2010) Kallikreins on steroids: structure,function, and hormonal regulation of prostate-specific antigen and the extendedkallikrein locus. Endocr Rev 31:407–446.
Le Friec G and Kemper C (2009) Complement: coming full circle. Arch Immunol TherExp (Warsz) 57:393–407.
Lee EJ, Han JE, Woo MS, Shin JA, Park EM, Kang JL, Moon PG, Baek MC, Son WS,Ko YT, et al. (2014) Matrix metalloproteinase-8 plays a pivotal role in neuro-inflammation by modulating TNF-a activation. J Immunol 193:2384–2393.
Leeb-Lundberg LM, Marceau F, Müller-Esterl W, Pettibone DJ, and Zuraw BL (2005)International union of pharmacology. XLV. Classification of the kinin receptorfamily: from molecular mechanisms to pathophysiological consequences. Pharma-col Rev 57:27–77.
Lerner DJ, Chen M, Tram T, and Coughlin SR (1996) Agonist recognition byproteinase-activated receptor 2 and thrombin receptor. Importance of extracellularloop interactions for receptor function. J Biol Chem 271:13943–13947.
Liebscher I, Ackley B, Araç D, Ariestanti DM, Aust G, Bae BI, Bista BR, BridgesJP, Duman JG, Engel FB, et al. (2014a) New functions and signaling mecha-nisms for the class of adhesion G protein-coupled receptors. Ann N Y Acad Sci1333:43–64.
Liebscher I, Schön J, Petersen SC, Fischer L, Auerbach N, Demberg LM, Mogha A,Cöster M, Simon KU, Rothemund S, et al. (2014b) A tethered agonist within theectodomain activates the adhesion G protein-coupled receptors GPR126 andGPR133. Cell Reports 9:2018–2026.
Liu Q, Weng HJ, Patel KN, Tang Z, Bai H, Steinhoff M, and Dong X (2011) Thedistinct roles of two GPCRs, MrgprC11 and PAR2, in itch and hyperalgesia. SciSignal 4:ra45.
López-Otín C and Bond JS (2008) Proteases: multifunctional enzymes in life anddisease. J Biol Chem 283:30433–30437.
Lourbakos A, Chinni C, Thompson P, Potempa J, Travis J, Mackie EJ, and PikeRN (1998) Cleavage and activation of proteinase-activated receptor-2 on humanneutrophils by gingipain-R from Porphyromonas gingivalis. FEBS Lett 435:45–48.
Lourbakos A, Potempa J, Travis J, D’Andrea MR, Andrade-Gordon P, Santulli R,Mackie EJ, and Pike RN (2001a) Arginine-specific protease from Porphyromonasgingivalis activates protease-activated receptors on human oral epithelial cells andinduces interleukin-6 secretion. Infect Immun 69:5121–5130.
Lourbakos A, Yuan YP, Jenkins AL, Travis J, Andrade-Gordon P, Santulli R,Potempa J, and Pike RN (2001b) Activation of protease-activated receptors bygingipains from Porphyromonas gingivalis leads to platelet aggregation: a newtrait in microbial pathogenicity. Blood 97:3790–3797.
Lundwall A (2013) Old genes and new genes: the evolution of the kallikrein locus.Thromb Haemost 110:469–475.
Lundwall A and Brattsand M (2008) Kallikrein-related peptidases. Cell Mol Life Sci65:2019–2038.
Luo R, Jeong SJ, Yang A, Wen M, Saslowsky DE, Lencer WI, Araç D, and Piao X(2014) Mechanism for adhesion G protein-coupled receptor GPR56-mediated RhoAactivation induced by collagen III stimulation. PLoS One 9:e100043.
MacFarlane RG (1964) An Enzyme Cascade In The Blood Clotting Mechanism, AndIts Function As A Biochemical Amplifier. Nature 202:498–499.
Magister S and Kos J (2013) Cystatins in immune system. J Cancer 4:45–56.Marceau F and Regoli D (2004) Bradykinin receptor ligands: therapeutic perspec-tives. Nat Rev Drug Discov 3:845–852.
Marchese A, Paing MM, Temple BR, and Trejo J (2008) G protein-coupled receptorsorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol 48:601–629.
Margolius HS (1998a) Kallikreins, kinins and cardiovascular diseases: a short re-view. Biol Res 31:135–141.
Margolius HS (1998b) Tissue kallikreins structure, regulation, and participation inmammalian physiology and disease. Clin Rev Allergy Immunol 16:337–349.
Martin BM, Feinman RD, and Detwiler TC (1975) Platelet stimulation by thrombinand other proteases. Biochemistry 14:1308–1314.
McGuire JJ, Dai J, Andrade-Gordon P, Triggle CR, and Hollenberg MD (2002)Proteinase-activated receptor-2 (PAR2): vascular effects of a PAR2-derived acti-vating peptide via a receptor different than PAR2. J Pharmacol Exp Ther 303:985–992.
McGuire JJ, Saifeddine M, Triggle CR, Sun K, and Hollenberg MD (2004) 2-furoyl-LIGRLO-amide: a potent and selective proteinase-activated receptor 2 agonist. JPharmacol Exp Ther 309:1124–1131.
McLaughlin JN, Patterson MM, and Malik AB (2007) Protease-activated receptor-3(PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad SciUSA 104:5662–5667.
McLean K, Schirm S, Johns A, Morser J, and Light DR (2001) FXa-induced responsesin vascular wall cells are PAR-mediated and inhibited by ZK-807834. Thromb Res103:281–297.
Michailidis IE, Abele-Henckels K, Zhang WK, Lin B, Yu Y, Geyman LS, Ehlers MD,Pnevmatikakis EA, and Yang J (2014) Age-related homeostatic midchannel pro-teolysis of neuronal L-type voltage-gated Ca²⁺ channels. Neuron 82:1045–1057.
Mihara K, Ramachandran R, Renaux B, Saifeddine M, and Hollenberg MD (2013)Neutrophil elastase and proteinase-3 trigger G protein-biased signaling throughproteinase-activated receptor-1 (PAR1). J Biol Chem 288:32979–32990.
Mihara K, Ramachandran R, Saifeddine M, Hansen KK, Renaux B, Polley D, GibsonS, Vanderboor C, and Hollenberg MD (2016) Thrombin-mediated direct activationof proteinase-activated receptor-2 (PAR2): another target for thrombin signaling.Mol Pharmacol 89:606–614.
Mize GJ, Wang W, and Takayama TK (2008) Prostate-specific kallikreins-2 and -4enhance the proliferation of DU-145 prostate cancer cells through protease-activatedreceptors-1 and -2. Mol Cancer Res 6:1043–1051.
Mirza H, Schmidt VA, Derian CK, Jesty J, and Bahou WF (1997) Mitogenic responsesmediated through the proteinase-activated receptor-2 are induced by expressedforms of mast cell alpha- or beta-tryptases. Blood 90:3914–3922.
Mohan Rao LV, Esmon CT, and Pendurthi UR (2014) Endothelial cell proteinC receptor: a multiliganded and multifunctional receptor. Blood 124:1553–1562.
Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, Hoxie JA,Schechter N, Woolkalis M, and Brass LF (1997) Interactions of mast cell tryptasewith thrombin receptors and PAR-2. J Biol Chem 272:4043–4049.
Molino M, Blanchard N, Belmonte E, Tarver AP, Abrams C, Hoxie JA, Cerletti C,and Brass LF (1995) Proteolysis of the human platelet and endothelial cellthrombin receptor by neutrophil-derived cathepsin G. J Biol Chem 270:11168–11175.
Mollnes TE, Garred P, and Bergseth G (1988) Effect of time, temperature and an-ticoagulants on in vitro complement activation: consequences for collection andpreservation of samples to be examined for complement activation. Clin ExpImmunol 73:484–488.
Moreau ME, Garbacki N, Molinaro G, Brown NJ, Marceau F, and Adam A (2005) Thekallikrein-kinin system: current and future pharmacological targets. J PharmacolSci 99:6–38.
Morris DR, Ding Y, Ricks TK, Gullapalli A, Wolfe BL, and Trejo J (2006) Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migra-tion, and invasion of breast cancer cells. Cancer Res 66:307–314.
Mosnier LO, Sinha RK, Burnier L, Bouwens EA, and Griffin JH (2012) Biased ago-nism of protease-activated receptor 1 by activated protein C caused by non-canonical cleavage at Arg46. Blood 120:5237–5246.
Moxon CA, Wassmer SC, Milner DA Jr, Chisala NV, Taylor TE, Seydel KB, MolyneuxME, Faragher B, Esmon CT, Downey C, et al. (2013) Loss of endothelial protein Creceptors links coagulation and inflammation to parasite sequestration in cerebralmalaria in African children. Blood 122:842–851.
Murrell-Lagnado RD and Aldrich RW (1993) Energetics of Shaker K channels blockby inactivation peptides. J Gen Physiol 102:977–1003.
Nakanishi-Matsui M, Zheng YW, Sulciner DJ, Weiss EJ, Ludeman MJ, and CoughlinSR (2000) PAR3 is a cofactor for PAR4 activation by thrombin. Nature 404:609–613.
Nesheim M (2003) Thrombin and fibrinolysis. Chest 124(3, Suppl)33S–39S.Netzel-Arnett S, Hooper JD, Szabo R, Madison EL, Quigley JP, Bugge TH,and Antalis TM (2003) Membrane anchored serine proteases: a rapidly expandinggroup of cell surface proteolytic enzymes with potential roles in cancer. CancerMetastasis Rev 22:237–258.
Proteinase-Mediated Signaling and Inflammation 1139

Nystedt S, Emilsson K, Larsson AK, Strömbeck B, and Sundelin J (1995a) Molecularcloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem 232:84–89.
Nystedt S, Emilsson K, Wahlestedt C, and Sundelin J (1994) Molecular cloning of apotential proteinase activated receptor. Proc Natl Acad Sci USA 91:9208–9212.
Nystedt S, Larsson AK, Aberg H, and Sundelin J (1995b) The mouse proteinase-activated receptor-2 cDNA and gene. Molecular cloning and functional expression.J Biol Chem 270:5950–5955.
O’Donoghue ML, Bhatt DL, Wiviott SD, Goodman SG, Fitzgerald DJ, Angiolillo DJ,Goto S, Montalescot G, Zeymer U, Aylward PE, et al.; LANCELOT-ACS Investi-gators (2011) Safety and tolerability of atopaxar in the treatment of patients withacute coronary syndromes: the lessons from antagonizing the cellular effects ofThrombin–Acute Coronary Syndromes Trial. Circulation 123:1843–1853.
Oikonomopoulou K, Batruch I, Smith CR, Soosaipillai A, Diamandis EP, and HollenbergMD (2010a) Functional proteomics of kallikrein-related peptidases in ovarian cancerascites fluid. Biol Chem 391:381–390.
Oikonomopoulou K, DeAngelis RA, Chen H, Diamandis EP, Hollenberg MD, RicklinD, and Lambris JD (2013) Induction of complement C3a receptor responses bykallikrein-related peptidase 14. J Immunol 191:3858–3866.
Oikonomopoulou K, Diamandis EP, and Hollenberg MD (2010b) Kallikrein-relatedpeptidases: proteolysis and signaling in cancer, the new frontier. Biol Chem 391:299–310.
Oikonomopoulou K, Hansen KK, Saifeddine M, Tea I, Blaber M, Blaber SI, ScarisbrickI, Andrade-Gordon P, Cottrell GS, Bunnett NW, et al. (2006a) Proteinase-activatedreceptors, targets for kallikrein signaling. J Biol Chem 281:32095–32112.
Oikonomopoulou K, Hansen KK, Saifeddine M, Vergnolle N, Tea I, Diamandis EP,and Hollenberg MD (2006b) Proteinase-mediated cell signalling: targetingproteinase-activated receptors (PARs) by kallikreins and more. Biol Chem 387:677–685.
Oikonomopoulou K, Ricklin D, Ward PA, and Lambris JD (2012) Interactions be-tween coagulation and complement–their role in inflammation. Semin Immuno-pathol 34:151–165.
Orr FW, Varani J, Kreutzer DL, Senior RM, and Ward PA (1979) Digestion of thefifth component of complement by leukocyte enzymes. Sequential generation ofchemotactic activities for leukocytes and for tumor cells. Am J Pathol 94:75–83.
Ostrowska E and Reiser G (2008) The protease-activated receptor-3 (PAR-3) cansignal autonomously to induce interleukin-8 release. Cell Mol Life Sci 65:970–981.
Paavola KJ and Hall RA (2012) Adhesion G protein-coupled receptors: signaling,pharmacology, and mechanisms of activation. Mol Pharmacol 82:777–783.
Page K, Ledford JR, Zhou P, Dienger K, and Wills-Karp M (2010) Mucosal sensiti-zation to German cockroach involves protease-activated receptor-2. Respir Res 11:62.
Paing MM, Temple BR, and Trejo J (2004) A tyrosine-based sorting signal regulatesintracellular trafficking of protease-activated receptor-1: multiple regulatorymechanisms for agonist-induced G protein-coupled receptor internalization. J BiolChem 279:21938–21947.
Passero CJ, Mueller GM, Rondon-Berrios H, Tofovic SP, Hughey RP, and KleymanTR (2008) Plasmin activates epithelial Na+ channels by cleaving the gammasubunit. J Biol Chem 283:36586–36591.
Patel AB, Chao J, and Palmer LG (2012) Tissue kallikrein activation of the epithelialNa channel. Am J Physiol Renal Physiol 303:F540–F550.
Patra C, Monk KR, and Engel FB (2014) The multiple signaling modalities of ad-hesion G protein-coupled receptor GPR126 in development. Receptors Clin Investig1:79.
Pawlinski R, Tencati M, Hampton CR, Shishido T, Bullard TA, Casey LM,Andrade-Gordon P, Kotzsch M, Spring D, Luther T, et al. (2007) Protease-activated receptor-1 contributes to cardiac remodeling and hypertrophy. Cir-culation 116:2298–2306.
Poirot O, Vukicevic M, Boesch A, and Kellenberger S (2004) Selective regulation ofacid-sensing ion channel 1 by serine proteases. J Biol Chem 279:38448–38457.
Polley MJ and Nachman RL (1983) Human platelet activation by C3a and C3a des-arg.J Exp Med 158:603–615.
Poole DP, Amadesi S, Veldhuis NA, Abogadie FC, Lieu T, Darby W, Liedtke W,Lew MJ, McIntyre P, and Bunnett NW (2013) Protease-activated receptor2 (PAR2) protein and transient receptor potential vanilloid 4 (TRPV4) proteincoupling is required for sustained inflammatory signaling. J Biol Chem 288:5790–5802.
Potempa J, Korzus E, and Travis J (1994) The serpin superfamily of proteinaseinhibitors: structure, function, and regulation. J Biol Chem 269:15957–15960.
Prömel S, Langenhan T, and Araç D (2013) Matching structure with function: theGAIN domain of adhesion-GPCR and PKD1-like proteins. Trends Pharmacol Sci34:470–478.
Puente XS, Sánchez LM, Gutiérrez-Fernández A, Velasco G, and López-Otín C (2005)A genomic view of the complexity of mammalian proteolytic systems. Biochem SocTrans 33:331–334.
Qanbar R and Bouvier M (2003) Role of palmitoylation/depalmitoylation reactions inG-protein-coupled receptor function. Pharmacol Ther 97:1–33.
Quinton TM, Kim S, Derian CK, Jin J, and Kunapuli SP (2004) Plasmin-mediatedactivation of platelets occurs by cleavage of protease-activated receptor 4. J BiolChem 279:18434–18439.
Ramachandran R (2012) Developing PAR1 antagonists: minding the endothelial gap.Discov Med 13:425–431.
Ramachandran R, Eissa A, Mihara K, Oikonomopoulou K, Saifeddine M, Renaux B,Diamandis E, and Hollenberg MD (2012a) Proteinase-activated receptors (PARs):differential signalling by kallikrein-related peptidases KLK8 and KLK14. BiolChem 393:421–427.
Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, DeFeaKA, Bouvier M, and Hollenberg MD (2011) Neutrophil elastase acts as a biasedagonist for proteinase-activated receptor-2 (PAR2). J Biol Chem 286:24638–24648.
Ramachandran R, Mihara K, Mathur M, Rochdi MD, Bouvier M, Defea K,and Hollenberg MD (2009) Agonist-biased signaling via proteinase activatedreceptor-2: differential activation of calcium and mitogen-activated protein kinasepathways. Mol Pharmacol 76:791–801.
Ramachandran R, Noorbakhsh F, Defea K, and Hollenberg MD (2012b) Targetingproteinase-activated receptors: therapeutic potential and challenges.Nat Rev DrugDiscov 11:69–86.
Ramsay AJ, Dong Y, Hunt ML, Linn M, Samaratunga H, Clements JA, and HooperJD (2008) Kallikrein-related peptidase 4 (KLK4) initiates intracellular signalingvia protease-activated receptors (PARs). KLK4 and PAR-2 are co-expressed duringprostate cancer progression. J Biol Chem 283:12293–12304.
Rasmussen UB, Vouret-Craviari V, Jallat S, Schlesinger Y, Pagès G, Pavirani A,Lecocq JP, Pouysségur J, and Van Obberghen-Schilling E (1991) cDNA cloning andexpression of a hamster alpha-thrombin receptor coupled to Ca2+ mobilization.FEBS Lett 288:123–128.
Rawlings ND, Tolle DP, and Barrett AJ (2004) Evolutionary families of peptidaseinhibitors. Biochem J 378:705–716.
Reber LL, Daubeuf F, Pejler G, Abrink M, and Frossard N (2014) Mast cellscontribute to bleomycin-induced lung inflammation and injury in mice througha chymase/mast cell protease 4-dependent mechanism. J Immunol 192:1847–1854.
Rehman AA, Ahsan H, and Khan FH (2013) a-2-Macroglobulin: a physiologicalguardian. J Cell Physiol 228:1665–1675.
Reid RC, Yau MK, Singh R, Hamidon JK, Lim J, Stoermer MJ, and Fairlie DP (2014)Potent heterocyclic ligands for human complement c3a receptor. J Med Chem 57:8459–8470.
Ricklin D, Hajishengallis G, Yang K, and Lambris JD (2010) Complement: a keysystem for immune surveillance and homeostasis. Nat Immunol 11:785–797.
Rieser P (1967) The insulin-like action of pepsin and pepsinogen. Acta Endocrinol(Copenh) 54:375–379.
Rieser P and Rieser CH (1964) Anabolic responses of diaphragm muscle to insulinand to other pancreatic proteins. Proc Soc Exp Biol Med 116:669–671.
Riewald M, Kravchenko VV, Petrovan RJ, O’Brien PJ, Brass LF, Ulevitch RJ,and Ruf W (2001) Gene induction by coagulation factor Xa is mediated by activa-tion of protease-activated receptor 1. Blood 97:3109–3116.
Riewald M, Petrovan RJ, Donner A, Mueller BM, and Ruf W (2002) Activation ofendothelial cell protease activated receptor 1 by the protein C pathway. Science296:1880–1882.
Riewald M, Petrovan RJ, Donner A, and Ruf W (2003) Activated protein C signalsthrough the thrombin receptor PAR1 in endothelial cells. J Endotoxin Res 9:317–321.
Riewald M and Ruf W (2001) Mechanistic coupling of protease signaling and initia-tion of coagulation by tissue factor. Proc Natl Acad Sci USA 98:7742–7747.
Riewald M and Ruf W (2005) Protease-activated receptor-1 signaling by activatedprotein C in cytokine-perturbed endothelial cells is distinct from thrombin sig-naling. J Biol Chem 280:19808–19814.
Roberts JR, Lee JJ, and Marthers DA (2012) Angiotensin-converting enzyme (ACE)inhibitor angioedema: the silent epidemic. Am J Cardiol 109:774–775.
Roosterman D, Schmidlin F, and Bunnett NW (2003) Rab5a and rab11a mediateagonist-induced trafficking of protease-activated receptor 2. Am J Physiol CellPhysiol 284:C1319–C1329.
Rossier BC and Stutts MJ (2009) Activation of the epithelial sodium channel (ENaC)by serine proteases. Annu Rev Physiol 71:361–379.
Saifeddine M, al-Ani B, Cheng CH, Wang L, and Hollenberg MD (1996) Ratproteinase-activated receptor-2 (PAR-2): cDNA sequence and activity ofreceptor-derived peptides in gastric and vascular tissue. Br J Pharmacol 118:521–530.
Saifeddine M, El-Daly M, Mihara K, Bunnett NW, McIntyre P, Altier C, HollenbergMD, and Ramachandran R (2015) GPCR-mediated EGF receptor transactivationregulates TRPV4 action in the vasculature. Br J Pharmacol 172:2493–2506.
Salvesen G, Farley D, Shuman J, Przybyla A, Reilly C, and Travis J (1987) Molecularcloning of human cathepsin G: structural similarity to mast cell and cytotoxicT lymphocyte proteinases. Biochemistry 26:2289–2293.
Sambrano GR, Huang W, Faruqi T, Mahrus S, Craik C, and Coughlin SR (2000)Cathepsin G activates protease-activated receptor-4 in human platelets. J BiolChem 275:6819–6823.
Savio-Galimberti E, Gollob MH, and Darbar D (2012) Voltage-gated sodium chan-nels: biophysics, pharmacology, and related channelopathies. Front Pharmacol 3:124.
Schuepbach RA, Madon J, Ender M, Galli P, and Riewald M (2012) Protease-activated receptor-1 cleaved at R46 mediates cytoprotective effects. J ThrombHaemost 10:1675–1684.
Schuepbach RA and Riewald M (2010) Coagulation factor Xa cleaves protease-activated receptor-1 and mediates signaling dependent on binding to the endo-thelial protein C receptor. J Thromb Haemost 8:379–388.
Sefton BM and Rubin H (1970) Release from density dependent growth inhibition byproteolytic enzymes. Nature 227:843–845.
Seatter MJ, Drummond R, Kanke T, Macfarlane SR, Hollenberg MD, and Plevin R(2004) The role of the C-terminal tail in protease-activated receptor-2-mediatedCa2+ signaling, proline-rich tyrosine kinase-2 activation, and mitogen-activatedprotein kinase activity. Cell Signal 16:21–29.
Sen P, Gopalakrishnan R, Kothari H, Keshava S, Clark CA, Esmon CT, PendurthiUR, and Rao LV (2011) Factor VIIa bound to endothelial cell protein C receptoractivates protease activated receptor-1 and mediates cell signaling and barrierprotection. Blood 117:3199–3208.
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR,McDonald-Smith GP, Gao H, Hennessy L, et al.; Inflammation and Host Responseto Injury, Large Scale Collaborative Research Program (2013) Genomic responsesin mouse models poorly mimic human inflammatory diseases. Proc Natl Acad SciUSA 110:3507–3512.
1140 Ramachandran et al.

Sevigny LM, Zhang P, Bohm A, Lazarides K, Perides G, Covic L,and Kuliopulos A (2011) Interdicting protease-activated receptor-2-driveninflammation with cell-penetrating pepducins. Proc Natl Acad Sci USA 108:8491–8496.
Shahinian H, Loessner D, Biniossek ML, Kizhakkedathu JN, Clements JA, MagdolenV, and Schilling O (2014) Secretome and degradome profiling shows thatKallikrein-related peptidases 4, 5, 6, and 7 induce TGFb-1 signaling in ovariancancer cells. Mol Oncol 8:68–82.
Shapiro MJ and Coughlin SR (1998) Separate signals for agonist-independent andagonist-triggered trafficking of protease-activated receptor 1. J Biol Chem 273:29009–29014.
Shapiro MJ, Trejo J, Zeng D, and Coughlin SR (1996) Role of the thrombin receptor’scytoplasmic tail in intracellular trafficking. Distinct determinants for agonist-triggered versus tonic internalization and intracellular localization. J Biol Chem271:32874–32880.
Shapiro MJ, Weiss EJ, Faruqi TR, and Coughlin SR (2000) Protease-activated re-ceptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. JBiol Chem 275:25216–25221.
Shin SH, Lee YH, and Jeon CH (2006) Protease-dependent activation of nasal polypepithelial cells by airborne fungi leads to migration of eosinophils and neutrophils.Acta Otolaryngol 126:1286–1294.
Shoelson SE, White MF, and Kahn CR (1988) Tryptic activation of the insulin re-ceptor. Proteolytic truncation of the alpha-subunit releases the beta-subunit frominhibitory control. J Biol Chem 263:4852–4860.
Smith R, Jenkins A, Lourbakos A, Thompson P, Ramakrishnan V, Tomlinson J,Deshpande U, Johnson DA, Jones R, Mackie EJ, et al. (2000) Evidence for theactivation of PAR-2 by the sperm protease, acrosin: expression of the receptor onoocytes. FEBS Lett 484:285–290.
Snead AN and Insel PA (2012) Defining the cellular repertoire of GPCRs identifies aprofibrotic role for the most highly expressed receptor, protease-activated receptor1, in cardiac fibroblasts. FASEB J 26:4540–4547.
Sonin DL, Wakatsuki T, Routhu KV, Harmann LM, Petersen M, Meyer J,and Strande JL (2013) Protease-activated receptor 1 inhibition by SCH79797 at-tenuates left ventricular remodeling and profibrotic activities of cardiac fibroblasts.J Cardiovasc Pharmacol Ther 18:460–475.
Sostegni S, Diakov A, McIntyre P, Bunnett N, Korbmacher C, and Haerteis S (2015)Sensitisation of TRPV4 by PAR2 is independent of intracellular calcium signallingand can be mediated by the biased agonist neutrophil elastase. Pflugers Arch 467:687–701.
Sotiropoulou G and Pampalakis G (2010) Kallikrein-related peptidases: bridges be-tween immune functions and extracellular matrix degradation. Biol Chem 391:321–331.
Sotiropoulou G, Pampalakis G, and Diamandis EP (2009) Functional roles of humankallikrein-related peptidases. J Biol Chem 284:32989–32994.
Soto AG and Trejo J (2010) N-linked glycosylation of protease-activated receptor-1second extracellular loop: a critical determinant for ligand-induced receptor acti-vation and internalization. J Biol Chem 285:18781–18793.
Stefansson K, Brattsand M, Roosterman D, Kempkes C, Bocheva G, Steinhoff M,and Egelrud T (2008) Activation of proteinase-activated receptor-2 by humankallikrein-related peptidases. J Invest Dermatol 128:18–25.
Steiner DF (2011) On the discovery of precursor processing. Methods Mol Biol 768:3–11.
Steiner DF, Cunningham D, Spigelman L, and Aten B (1967) Insulin biosynthesis:evidence for a precursor. Science 157:697–700.
Stephan C, Jung K, Lein M, and Diamandis EP (2007) PSA and other tissue kalli-kreins for prostate cancer detection. Eur J Cancer 43:1918–1926.
Su J, Tang Y, Liu L, Zhou H, and Dong Q (2011) Regulation of acid-sensing ionchannel 1a function by tissue kallikrein may be through channel cleavage. Neu-rosci Lett 490:46–51.
Suen JY, Barry GD, Lohman RJ, Halili MA, Cotterell AJ, Le GT, and Fairlie DP(2012) Modulating human proteinase activated receptor 2 with a novel antagonist(GB88) and agonist (GB110). Br J Pharmacol 165:1413–1423.
Sun G, Stacey MA, Schmidt M, Mori L, and Mattoli S (2001) Interaction of miteallergens Der p3 and Der p9 with protease-activated receptor-2 expressed by lungepithelial cells. J Immunol 167:1014–1021.
Sutton MG and Sharpe N (2000) Left ventricular remodeling after myocardial in-farction: pathophysiology and therapy. Circulation 101:2981–2988.
Swift S, Xu J, Trivedi V, Austin KM, Tressel SL, Zhang L, Covic L, and Kuliopulos A(2010) A novel protease-activated receptor-1 interactor, Bicaudal D1, regulates Gprotein signaling and internalization. J Biol Chem 285:11402–11410.
Szabo R and Bugge TH (2011) Membrane-anchored serine proteases in vertebratecell and developmental biology. Annu Rev Cell Dev Biol 27:213–235.
Takao K and Miyakawa T (2015) Genomic responses in mouse models greatly mimichuman inflammatory diseases. Proc Natl Acad Sci USA 112:1167–1172.
Takayama TK, McMullen BA, Nelson PS, Matsumura M, and Fujikawa K (2001)Characterization of hK4 (prostase), a prostate-specific serine protease: activation ofthe precursor of prostate specific antigen (pro-PSA) and single-chain urokinase-type plasminogen activator and degradation of prostatic acid phosphatase. Bio-chemistry 40:15341–15348.
Takeuchi T, Harris JL, Huang W, Yan KW, Coughlin SR, and Craik CS (2000) Cel-lular localization of membrane-type serine protease 1 and identification ofprotease-activated receptor-2 and single-chain urokinase-type plasminogen acti-vator as substrates. J Biol Chem 275:26333–26342.
Tam SW, Fenton JW 2nd, and Detwiler TC (1980) Platelet thrombin receptors.Binding of alpha-thrombin is coupled to signal generation by a chymotrypsin-sensitive mechanism. J Biol Chem 255:6626–6632.
Trejo J, Altschuler Y, Fu HW, Mostov KE, and Coughlin SR (2000) Protease-activatedreceptor-1 down-regulation: a mutant HeLa cell line suggests novel requirementsfor PAR1 phosphorylation and recruitment to clathrin-coated pits. J Biol Chem275:31255–31265.
Trejo J, Hammes SR, and Coughlin SR (1998) Termination of signaling by protease-activated receptor-1 is linked to lysosomal sorting. Proc Natl Acad Sci USA 95:13698–13702.
Tressel SL, Kaneider NC, Kasuda S, Foley C, Koukos G, Austin K, Agarwal A, CovicL, Opal SM, and Kuliopulos A (2011) A matrix metalloprotease-PAR1 systemregulates vascular integrity, systemic inflammation and death in sepsis. EMBOMol Med 3:370–384.
Trivedi NR, Gilliland KL, Zhao W, Liu W, and Thiboutot DM (2006) Gene arrayexpression profiling in acne lesions reveals marked upregulation of genes involvedin inflammation and matrix remodeling. J Invest Dermatol 126:1071–1079.
Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O’Callaghan K, Covic L,and Kuliopulos A (2009) Platelet matrix metalloprotease-1 mediates thrombo-genesis by activating PAR1 at a cryptic ligand site. Cell 137:332–343.
Tull SP, Bevins A, Kuravi SJ, Satchell SC, Al-Ani B, Young SP, Harper L, WilliamsJM, Rainger GE, and Savage CO (2012) PR3 and elastase alter PAR1 signaling andtrigger vWF release via a calcium-independent mechanism from glomerular en-dothelial cells. PLoS One 7:e43916.
Turk B, Turk D, and Salvesen GS (2002) Regulating cysteine protease activity: es-sential role of protease inhibitors as guardians and regulators. Curr Pharm Des 8:1623–1637.
Turk B, Turk D, and Turk V (2012) Protease signalling: the cutting edge. EMBO J 31:1630–1643.
Turk V, Turk B, and Turk D (2001) Lysosomal cysteine proteases: facts and oppor-tunities. EMBO J 20:4629–4633.
Turm H, Grisaru-Granvosky S, Maoz M, Offermanns S, and Bar-Shavit R (2010) DVLas a scaffold protein capturing classical GPCRs. Commun Integr Biol 3:495–498.
Vandell AG, Larson N, Laxmikanthan G, Panos M, Blaber SI, Blaber M,and Scarisbrick IA (2008) Protease-activated receptor dependent and independentsignaling by kallikreins 1 and 6 in CNS neuron and astroglial cell lines. J Neurochem107:855–870.
Vandenbroucke RE and Libert C (2014) Is there new hope for therapeutic matrixmetalloproteinase inhibition? Nat Rev Drug Discov 13:904–927.
Veldhuis NA, Poole DP, Grace M, McIntyre P, and Bunnett NW (2015) The G protein-coupled receptor-transient receptor potential channel axis: molecular insights fortargeting disorders of sensation and inflammation. Pharmacol Rev 67:36–73.
Vicuña L, Strochlic DE, Latremoliere A, Bali KK, Simonetti M, Husainie D, ProkoschS, Riva P, Griffin RS, Njoo C, et al. (2015) The serine protease inhibitor SerpinA3Nattenuates neuropathic pain by inhibiting T cell-derived leukocyte elastase. NatMed 21:518–523.
Villegas-Mendez A, Montes R, Ambrose LR, Warrens AN, Laffan M, and Lane DA(2007) Proteolysis of the endothelial cell protein C receptor by neutrophil pro-teinase 3. J Thromb Haemost 5:980–988.
Vu TK, Hung DT, Wheaton VI, and Coughlin SR (1991) Molecular cloning of afunctional thrombin receptor reveals a novel proteolytic mechanism of receptoractivation. Cell 64:1057–1068.
Vuagniaux G, Vallet V, Jaeger NF, Hummler E, and Rossier BC (2002) Synergisticactivation of ENaC by three membrane-bound channel-activating serine proteases(mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase(Sgk1) in Xenopus Oocytes. J Gen Physiol 120:191–201.
Vukicevic M, Weder G, Boillat A, Boesch A, and Kellenberger S (2006) Trypsincleaves acid-sensing ion channel 1a in a domain that is critical for channel gating. JBiol Chem 281:714–722.
Wang W, Mize GJ, Zhang X, and Takayama TK (2010) Kal- likrein-related peptidase-4initiates tumor-stroma interactions in prostate cancer through protease-activatedreceptor-1. Int J Cancer 126:599–610.
Wang MC, Papsidero LD, Kuriyama M, Valenzuela LA, Murphy GP, and Chu TM(1981) Prostate antigen: a new potential marker for prostatic cancer. Prostate 2:89–96.
Wang MC, Valenzuela LA, Murphy GP, and Chu TM (1979) Purification of a humanprostate specific antigen. Invest Urol 17:159–163.
Wang Y, Zhou Y, Szabo K, Haft CR, and Trejo J (2002) Down-regulation ofprotease-activated receptor-1 is regulated by sorting nexin 1. Mol Biol Cell 13:1965–1976.
Wen W, Young SE, Duvernay MT, Schulte ML, Nance KD, Melancon BJ, Engers J,Locuson CW 2nd, Wood MR, Daniels JS, et al. (2014) Substituted indoles as se-lective protease activated receptor 4 (PAR-4) antagonists: Discovery and SAR ofML354. Bioorg Med Chem Lett 24: 4708–4713.
Wetsel RA and Kolb WP (1982) Complement-independent activation of the fifthcomponent (C5) of human complement: limited trypsin digestion resulting in theexpression of biological activity. J Immunol 128:2209–2216.
Whalley ET, Figueroa CD, Gera L, and Bhoola KD (2012) Discovery and therapeuticpotential of kinin receptor antagonists. Expert Opin Drug Discov 7:1129–1148.
Wiggins RC, Giclas PC, and Henson PM (1981) Chemotactic activity generated fromthe fifth component of complement by plasma kallikrein of the rabbit. J Exp Med153:1391–1404.
Wiviott SD, Flather MD, O’Donoghue ML, Goto S, Fitzgerald DJ, Cura F, Aylward P,Guetta V, Dudek D, Contant CF, et al.; LANCELOT-CAD Investigators (2011)Randomized trial of atopaxar in the treatment of patients with coronary arterydisease: the lessons from antagonizing the cellular effect of Thrombin–CoronaryArtery Disease Trial. Circulation 123:1854–1863.
Wolfe BL, Marchese A, and Trejo J (2007) Ubiquitination differentially regulatesclathrin-dependent internalization of protease-activated receptor-1. J Cell Biol177:905–916.
Wong HK, Sakurai T, Oyama F, Kaneko K, Wada K, Miyazaki H, Kurosawa M, DeStrooper B, Saftig P, and Nukina N (2005) beta Subunits of voltage-gated so-dium channels are novel substrates of beta-site amyloid precursor protein-cleaving enzyme (BACE1) and gamma-secretase. J Biol Chem 280:23009–23017.
Wongtim S, Lehrer SB, Salvaggio JE, and Horner WE (1993) Protease activity incockroach and basidiomycete allergens extracts. Allergy Proc 14:263–268.
Proteinase-Mediated Signaling and Inflammation 1141

Wu CC, Hwang TL, Liao CH, Kuo SC, Lee FY, Lee CY, and Teng CM (2002) Selectiveinhibition of protease-activated receptor 4-dependent platelet activation by YD-3.Thromb Haemost 87:1026–1033.
Xiao YP, Morice AH, Compton SJ, and Sadofsky L (2011) N-linked glycosylationregulates human proteinase-activated receptor-1 cell surface expression and dis-arming via neutrophil proteinases and thermolysin. J Biol Chem 286:22991–23002.
Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, DavieEW, and Foster DC (1998) Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci USA 95:6642–6646.
Xue M, Hollenberg MD, Demchuk A, and Yong VW (2009) Relative importance ofproteinase-activated receptor-1 versus matrix metalloproteinases in intracerebralhemorrhage-mediated neurotoxicity in mice. Stroke 40:2199–2204.
Xue M, Hollenberg MD, and Yong VW (2006) Combination of thrombin and matrixmetalloproteinase-9 exacerbates neurotoxicity in cell culture and intracerebralhemorrhage in mice. J Neurosci 26:10281–10291.
Yau MK, Liu L, and Fairlie DP (2013) Toward drugs for protease-activated receptor2 (PAR2). J Med Chem 56:7477–7497.
Yona S, Lin HH, Siu WO, Gordon S, and Stacey M (2008) Adhesion-GPCRs: emergingroles for novel receptors. Trends Biochem Sci 33:491–500.
Young SE, Duvernay MT, Schulte ML, Nance KD, Melancon BJ, Engers J, Wood MR,Hamm HE, and Lindsley CW (2013) A Novel and Selective PAR4 Antagonist:ML354, in Probe Reports from the NIH Molecular Libraries Program, NationalCenter for Biotechnology Information, Bethesda, MD.
Yoon H, Laxmikanthan G, Lee J, Blaber SI, Rodriguez A, Kogot JM, Scarisbrick IA,and Blaber M (2007) Activation profiles and regulatory cascades of the humankallikrein-related peptidases. J Biol Chem 282:31852–31864.
Yousef GM and Diamandis EP (2001) The new human tissue kallikrein gene family:structure, function, and association to disease. Endocr Rev 22:184–204.
Yousef GM and Diamandis EP (2009) The human kallikrein gene family: new bio-markers for ovarian cancer. Cancer Treat Res 149:165–187.
Zania P, Gourni D, Aplin AC, Nicosia RF, Flordellis CS, Maragoudakis ME,and Tsopanoglou NE (2009) Parstatin, the cleaved peptide on proteinase-activatedreceptor 1 activation, is a potent inhibitor of angiogenesis. J Pharmacol Exp Ther328:378–389.
Zhao P, Lieu T, Barlow N, Metcalf M, Veldhuis NA, Jensen DD, Kocan M, Sostegni S,Haerteis S, Baraznenok V, et al. (2014a) Cathepsin S causes inflammatory pain viabiased agonism of PAR2 and TRPV4. J Biol Chem 289:27215–27234.
Zhao P, Metcalf M, and Bunnett NW (2014b) Biased signaling of protease-activatedreceptors. Front Endocrinol (Lausanne) 5:67.
1142 Ramachandran et al.









![Review Extracellular vesicles in Inflammatory Skin Disorders ...MiRNAs Help discriminate between EV subpopulations [127] MiR-381-3p CD4+ T cells Induce Th1/Th17 polarization in psoriasis](https://img.pdfslide.net/doc/110x75/6001046ada2b32234b3be391/review-extracellular-vesicles-in-inflammatory-skin-disorders-mirnas-help-discriminate.jpg)



















