Embed Size (px)
Citation preview

Qualification and Validation
An Overview

Contents• Training Objectives• References• Qualification and Validation Definitions• Order of Q and V Activities• Validation Responsibilities• Important Validation Considerations• Basic Checking of Validation
Documentation• Revalidation• Typical Validation Errors• Conclusion

Training Objectives
• To provide a high level introduction to the principles of
Qualification and Validation for people not working directly
with this area
• To cover the main aspects associated with each element of
Qualification and Validation and how they inter-relate
• To provide an indication of things to look for when reviewing
validation documentation
• To provide further reference for people who are interested
to learn more about the topic.

References (1)
• References EU GMP volume 4 Annexe 15 “Qualification & Validation”
• Qualification: Action of proving that any EQUIPMENT works correctly and actually leads to the expected results. The word Validation is sometimes widened to incorporate the concept of qualification
• Validation: Action of proving, in accordance with the principles of GMPs, that any PROCEDURE, PROCESS, MATERIAL, ACTIVITY or SYSTEM actually leads to the expected results

References (2)
• References EU GMP volume 4 (1997)Chapter 5 Production, Validation : 5.22 – 5.24
“When any new manufacturing formula or method of preparation is adopted (after validation), steps should be taken to demonstrate its suitability for routine processing. The defined process, using the materials and equipment specified, should be shown to yield a product consistently of the required quality”
“Significant amendments to the manufacturing process, including any change in equipment or materials, which may affect product quality and/or the reproducibility of the process should be validated”
“Processes and procedures should undergo periodic critical revalidation to ensure that they remain capable of achieving the intended results.”

Qualification and Validation definitions (1)
• Qualification
Identification of particular attributes of equipment, utilities or processes related to the performance of a particular function, such as design (DQ), Installation (IQ) or Operation (OQ) and the allocation of certain limits or restrictions to those attributes and finally, the measurement of those attributes in those ranges for those functions, such as performance (PQ)
• Validation
Documented evidence that the item under consideration does what it purports to do. Validation includes but is not limited to: Manufacturing processes, cleaning procedures, Computerized Systems, facilities, utilities and analytical methods.

Qualification and Validation definitions (2)
• So let us simplify these definitions by the following practical facts:
• The term Qualification tends to get used when discussing personnel , equipment and utilities (e.g. filling machines, laboratory equipment, nitrogen systems)
• The term Validation tends to get used when discussing methods and processes (e.g. manufacturing, packaging, cleaning, sterilization and computer systems), and can sometimes include Qualification activities
• For example, a purified water system is considered “validated” when all phases of the IQ, OQ, and PQ are completed.

Qualification and Validation definitions (3)
• There are a few important process validation terms that can be explained as follows:
• Prospective Validation required to be completed before a process is
used, or before a Product is released
• Concurrent A Validation exercise performed at the same time as
ongoing commercial production
• Retrospective (not recommended) Validation of a Process already in use based upon
accumulated historical data’s conformance to predetermined acceptance criteria. Use of Retrospective Validation is restricted to APIs under limited circumstances without significant changes.

Order of Q and V Activities (1)
• There are distinct phases that relate to Qualification and these take place in the following order - using equipment as an example:
• Design Qualification (DQ) ensures the critical design requirements have been met during equipment construction (e.g. grade of stainless steel specified)
• Installation Qualification (IQ) ensures that the equipment has been installed correctly (e.g. correctly electrical wiring, piping etc versus approved drawings)
• Operational Qualification (OQ) ensures that the equipment can perform the basic operations across its operating range (e.g. motors operate across defined speed range)
• Performance Qualification (PQ) ensures that an operation, when carried out versus a defined protocol, will consistently perform its intended function to meet pre-determined acceptance criteria.

Order of Q and V Activities (2)
• The following Q and V documentation is normally generated
Qualification/Validation Plan – that sets out the overall plan of the qualification or validation, and includes responsibilities, number of batches etc
Qualification/Validation Protocol – that details the work that must be performed for each batch/run, the tests to be performed, and clearly shows the acceptance criteria
Qualification/Validation Report – that documents the obtained results, and assesses them versus the acceptance criteria
Qualification/Validation Summary Report – this document summarises the overall validation, and provides a conclusion about the validation status.

Order of Q and V Activities (3)
• It is essential that Qualification and Validation are performed in the correct order within the site otherwise the work will be invalid
• If we consider a new product as an example, we need to ensure that key equipment, utilities, computer systems are qualified/validated prior to trying to validate our product
• In addition, there is no point in QC testing the validation samples of the new product if they have not qualified their laboratory equipment nor validated their QC testing method
• This is shown diagrammatically on the following slide, indicating the activities that are pre-requisites to process validation.
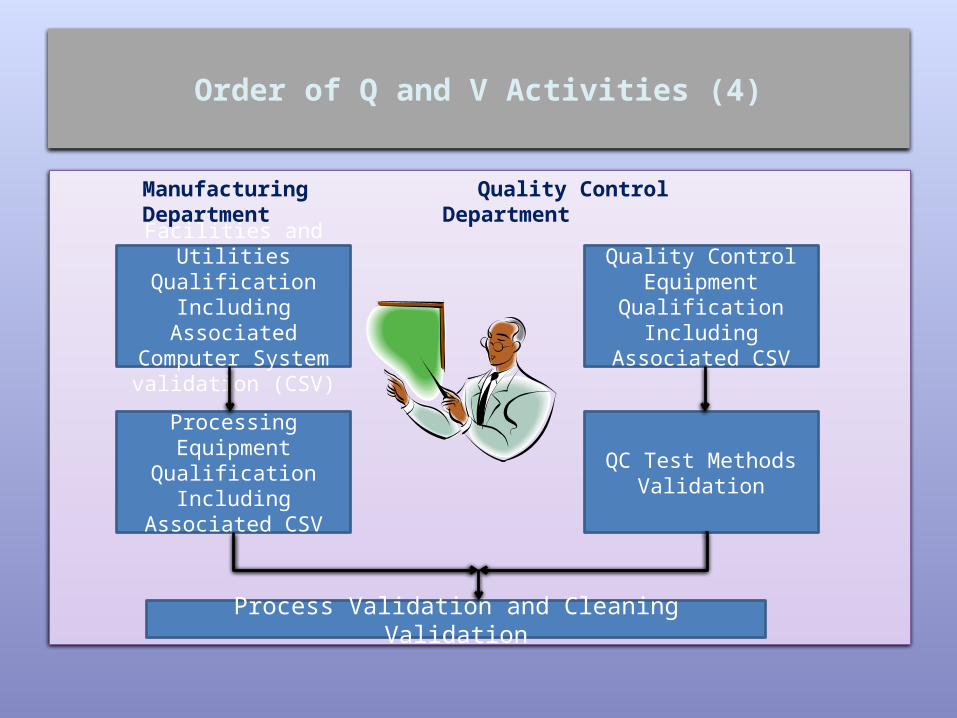
Order of Q and V Activities (4)
Manufacturing Quality ControlDepartment Department
Facilities and Utilities
Qualification Including
Associated Computer System validation (CSV)
Quality Control Equipment
Qualification Including
Associated CSV
Processing Equipment
Qualification Including
Associated CSV
QC Test Methods Validation
Process Validation and Cleaning Validation
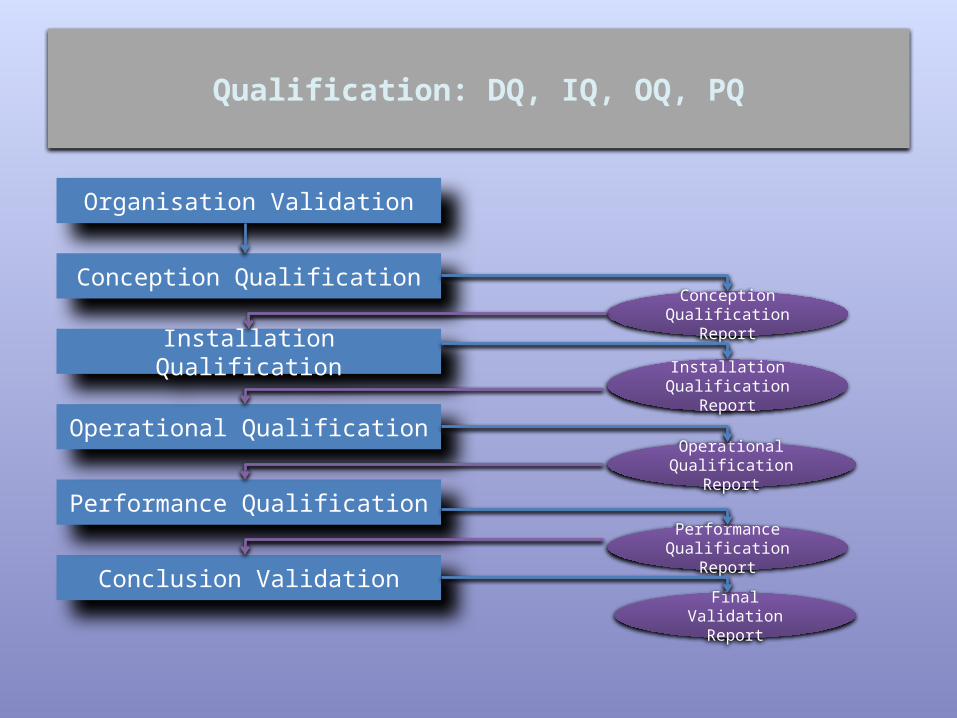
Qualification: DQ, IQ, OQ, PQ
Organisation Validation
Conception Qualification
Conclusion Validation
Performance Qualification
Operational Qualification
Installation Qualification
Final Validation Report
Performance Qualification
Report
Operational Qualification
Report
Installation Qualification
Report
Conception Qualification
Report

Qualification: IQ, OQ, PQ (1)
• IQ - Each element of the system is: adequately installed, in conformity with user needs, supplier technical
specifications and the regulatory requirements
• Protocol
description of the system to be validated Technical verification tests (individual component, critical
parameters, control and functioning systems, system environment, power, and utilities, accessibility / material flow, safety)
documentation checking.

Qualification: IQ, OQ, PQ (2)
• OQ - Each element of the system works within the predefined limits
• Protocol:
description of the system to be validated technical verification tests and acceptance criterion
(individual functions, calibration of the critical measurement systems, working of the whole system, control and working systems, system environment, power and utilities, safety and alarms)
documentation checking (operational documents).

Qualification: IQ, OQ, PQ (3)
• PQ - The system or the product process will regularly produce the expected results within the predefined limits
• Protocol :
verification of the documents of validation achieved and approved
verification of the presence of all organisational procedures, operational documents and training documents
verification tests for the general process during a determined period or on defined batches and acceptance criteria.

Validation Responsibility (1)
• Site validation activities need to be well coordinated to ensure they are performed in a suitable and timely manner
• The validation work needs to include the appropriate cross-functional expertise from the planning stage
• A cross-functional Site Validation Forum may well exist, To publish Site Validation Master Plan To generate validation SOP’s and templates To agree validation priorities and timings To review completed validation documents To plan re-validation activities
• Process owners are generally responsible for performing and documenting their validation work within their area using the approved site systems, SOP’s, and templates
• QA are responsible for final approval of the validation plans, protocols, and reports.

Validation Responsibility (2)
• Each site must develop, and keep current, a Site Qualification and Validation Master Plan that includes, but is not limited to:Site descriptionSite or global project description as applicableList of systems and equipments requiring qualification
and validation (e.g. inventory list)Qualification and validation status and history of each
system or equipment unitSite Qualification and Validation Plan (See next slide)

Validation Responsibility (3)
• A site qualification and validation master plan that includes, but is not limited to:responsibilities for Q&V and organizational
structure for validation activitiesdefinition of terms used in the site qualification and
validation master planmethodology and/or procedures that govern the
qualification and validation master planidentification of revalidation or requalification
requirements, the change control process and evidence that will maintain validation documentation in a current state of control.
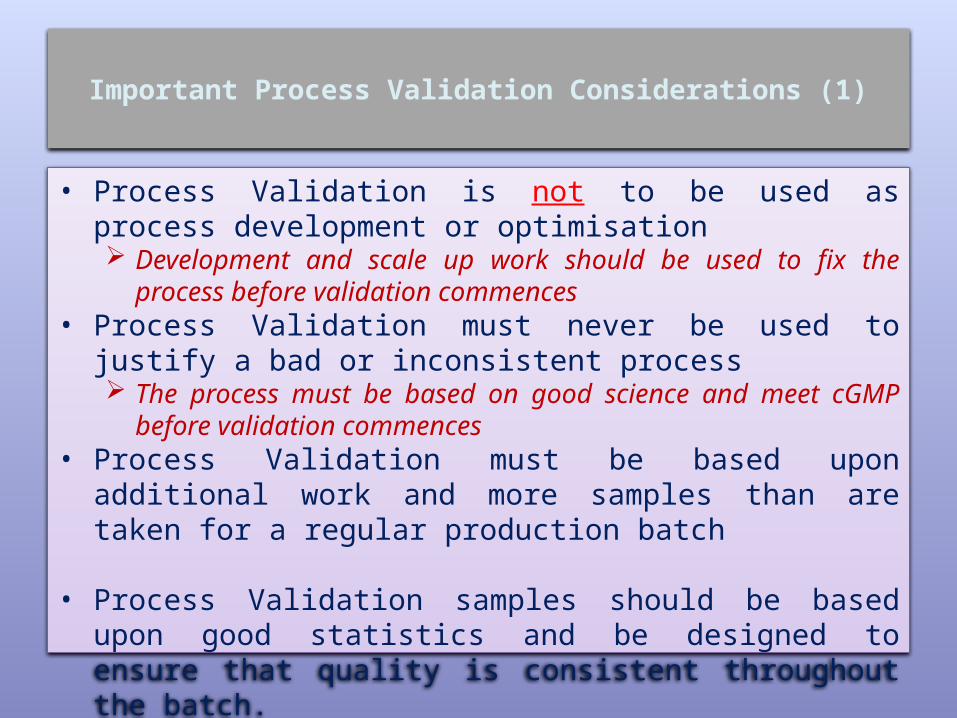
Important Process Validation Considerations (1)
• Process Validation is not to be used as process development or optimisation Development and scale up work should be used to fix
the process before validation commences• Process Validation must never be used to justify a bad
or inconsistent process The process must be based on good science and meet
cGMP before validation commences• Process Validation must be based upon additional
work and more samples than are taken for a regular production batch
• Process Validation samples should be based upon good statistics and be designed to ensure that quality is consistent throughout the batch.

Important Process Validation Considerations (2)
• A minimum of three consecutive validation batches are normally used to consider a process validated
• All deviations from the validation protocol need to be fully justified, and approved by Quality Assurance
• Process Validation batches are normally placed on the stability testing program.

Basic Checking of Validation Documentation (1)
• Validation documents are often reviewed by regulatory authorities either as part of product registration dossiers or during audits
• Any validation activity should be able to be read like a book A beginning (a clear and precise plan, requirements, and acceptance
criteria) A middle (documented results versus the acceptance criteria) An end (a summary and clear conclusion)
• The next few slides indicate basic checks that should be performed to ensure that the most commonly found validation documentation errors are not present - even if you are not an expert in that area or product.

Basic Checking of Validation Documentation (2)
• Look for examples of the following general document errors:
Was the Q and V work performed in the correct order ? Was one phase completed before the next commenced ? Is the report completed with results, signatures and
dates ? Do the results make common sense ? Do the dates make sense ? Is there a trace to the raw data, measuring equipment,
calibration certificates etc.

Basic Checking of Validation Documentation (3)
• Ensure that you start by reviewing the approved validation plans and protocolsIdentify precisely what was being planned:
– Does validation mimic production process?– How many batches/runs? (normally minimum 3 consecutive
batches)– How many samples will be taken, and from where?– How will samples be tested? (individual or pooled)– What specification will the samples be tested against?– Are critical process parameters defined? (e.g. mix time,
temperature)– What are the acceptance criteria? (e.g. 30 minutes, 25 –
30oC)– Are batches being put onto stability testing program?

Basic Checking of Validation Documentation (4)
• Review the validation reports and summary report, and compare the actual data versus the plan:
Were there the required number of batches/runs? Were the batches/runs consecutive? Were the required number (and location) of samples taken? Were the samples tested individually or pooled? Was the correct test specification used? Were there records for all of the critical process parameters? Did all results meet the pre-determined acceptance criteria? Are all deviations from the protocol fully justified? Have process validation batches been put on stability program? Is there a clear summary/concluding statement on validation
status?

Basic Checking of Validation Documentation (5)
• A useful check for an established process is to check the actual process (reality) versus the validation documentation and the submitted registration dossier
• This will identify whether the actual process still matches the one that has been validated and registered
• For the chosen product, compare in detail the following three key documents:
The batch record from the most recently released batch The most recent process validation report/summary report The most recently submitted CMC part of the dossier
• Ideally, the reality should match theory. If not then review the change control documents to understand why there is a difference.
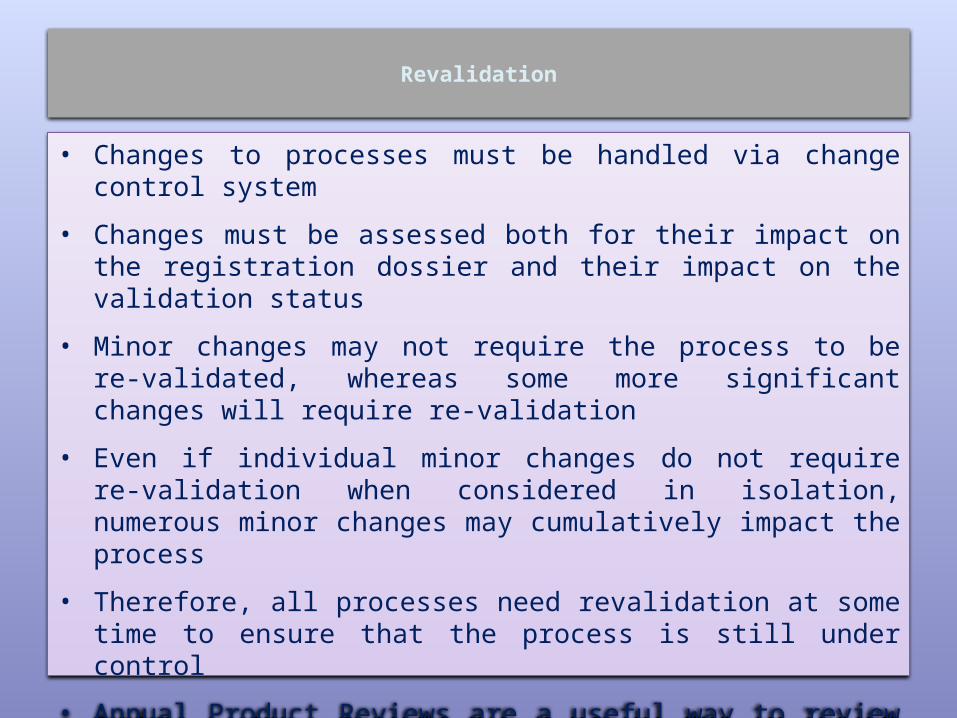
Revalidation
• Changes to processes must be handled via change control system
• Changes must be assessed both for their impact on the registration dossier and their impact on the validation status
• Minor changes may not require the process to be re-validated, whereas some more significant changes will require re-validation
• Even if individual minor changes do not require re-validation when considered in isolation, numerous minor changes may cumulatively impact the process
• Therefore, all processes need revalidation at some time to ensure that the process is still under control
• Annual Product Reviews are a useful way to review recent changes that may have an impact on the status of process validation.

Typical Validation Pitfalls
• The next few slides will cover some of the most frequent pitfalls noted during review of validation documentation
• We will consider the generalities of validation in the following distinct phasesValidation planningValidation protocolsSampling and TestingDocumentingReview and Sign offWhat happens after validation

Which of These Validation Statements is True?
• Validation is generally the final stage of a project and there is always a lot of pressure to just “make it happen”
• Generally, too little time is spent planning the validation activities
• The validation activities nearly always take longer and utilise more resource than originally thought
• Validation activities are therefore on many occasions under resourced
• Each validation does not have an identified owner to drive the process (organisation)
• Major reason : Lack of realistic validation planning?

Validation Planning (1)
• In many cases validation protocols:
do not embrace all of the validation requirements are too complicated for people to follow (too “wordy”) are not specific enough in terms of samples (e.g. size, location) Are not specific enough about QC testing (e.g. individual assays) do not specify clear responsibilities are not communicated to involved parties
• We can probably all think of examples where the technical aspects of a process are ok, but the validation failed or was severely delayed because:
The people took the wrong sample The samples were inadequately labelled and were mixed up Not all samples were taken as required The samples were inappropriately tested.

Validation Planning (2)
• There is sometimes no clear indication of all the identified critical process parameters mixing speed mixing time water temperature
• Operating conditions are not specified – sometimes because the process has not yet been fixed. We see examples as follows: Validation Batch A 15 minutes mixing – failed assay Validation Batch B 20 minutes mixing – failed assay Validation Batch C 25 minutes mixing – satisfactory assay Validation status : Passed !!!
• Do NOT use validation as Process Development• Acceptance Criteria are not clearly defined – so people do not know the
parameters to work within.

Validation Planning: Process validation Method
• The validation protocol should be prepared by the technical personnel responsible for carrying out the validation exercise. It should state the way in which the process is to be operated, identify the controls to be exerted, specify the variables to be monitored, state the samples to be taken for subsequent testing, specify the product performance characteristics/attributes to be monitored along with acceptable limits and refer to the test methods to be used
• Examples of process variables are:-Temperature - Time - Volume - Capacity - Appearance - Cooling/Heating rate - Heat distribution - Particles size - Chemical composition of the environment in contact with the product (e.g. oxygen, carbon dioxide, water vapour) – power consumption - pressure - humidity - vacuum - materials - speed.

Critical Process Parameters: Example process “Mixing of Solids”
• Equipment: Blade mixers, Free fall mixers
• Variables of Process: type, shape and position of mixing blades, sequence of addition; capacity; percent fill; time; blade/container rpm or peripheral speed
• Product properties affected by these variables: Homogeneity; particles size; drug release (wear of coats or coating with lubricant)
• Control of the Process variables or IPC of the process step: Time; rate/rpm; power consumption; temperature
• Control of the characteristics of the Product: Content uniformity; particles size analysis; disintegration time; dissolution rate.

Critical Process Parameters: Example process “Wet Granulation”
• Equipment: Fluidized bed granulator
• Variables of Process: Type; capacity; position; angle & bore of nozzle; spray cone; batch size; inlet/outlet temperature; airflow rate at inlet/outlet; spraying rate; material temperature; filter shaking period; drying time; filter bags; climatic conditions
• Product properties affected by these variables: particles size distribution; bulk/tapped density; flow properties; hardness; homogeneity; residual moisture
• Control of the Process variables or IPC of the process step: airflow meter; temp. of air at inlet & outlet; feeding rate; pressure of air; process timing; humidity of air at outlet
• Control of the characteristics of the Product: residual moisture; particle size analysis; bulk/tapped density; flow properties; granule friability; yield; disintegration time.

Critical Process Parameters: Example process “Tableting/Slugging”
• Equipment: tableting machines (reciprocating & rotating)• Variables of Process: Type of machine; shape & material of tools;
condition of tools; machine speed; filling depth; pre-compression pressure; compression pressure; percent hopper fill; feeding rate/speed of feeder
• Product properties affected by these variables: weight; weight distribution; thickness; hardness; friability; disintegration time; dissolution time
• Control of the Process variables or IPC of the process step: inspection of tools; machine speed control; volume fill/feeder speed control; compression pressure; ejection force; metal detection
• Control of the characteristics of the Product: hardness; thickness; weight; uniformity of weight & contents; appearance; yield; (moisture content); disintegration time; dissolution rate.

Critical Process Parameters: Example process “Steam
Sterilization”• Equipment: Autoclave• Variables of Process: Type, size, temperature,
pressure, cycle time, load and arrangement of load, distribution and penetration of heat, cooling system
• Product properties affected by these variables: Sterility
• Control of the Process variables or IPC of the process step: temperature, pressure, cycle time, bio indicator
• Control of the characteristics of the Product: sterility, apyrogenicity (where applicable).

Critical Process Parameters: Example process
“Lyophilising”• Equipment: Freeze-drier• Variables of Process: Type; capacity; temperature of shelf /
product /condenser; distribution of temperature; time; vacuum; closing mechanism (for sterile products); air filter
• Product properties affected by these variables: uniformity of weight; residual solvent; microbial contamination
• Control of the Process variables or IPC of the process step: vacuum – temperature (shelf, product, condenser) – time profile
• Control of the characteristics of the Product: uniformity of weight and content; solvent residues; time of dissolution in the solvent for reconstitution; moisture content; head space pressure; sterility test (where applicable).

Validation Planning – Worse Case scenarios
• The earlier part of the Qualification process should help you to better understand your equipment and processes, which will help you to identify any potential weaknesses
• Early Qualification work does not always consider the potential worse case scenarios:
What happens if the power fails to the mixer? What happens if the water temperature goes above 30
degrees centigrade? What happens if the operator tries to modify data and save
it? What happens if a re-print is requested?
• Knowing the answers to key questions can help to save expensive product when there are problems when running live product.

Inappropriate Sampling and Testing: Some Real Life Examples
• Inadequate sampling: The quantity of validation samples are the same number as
routine production samples The samples are not statistically valid The sampling details are not specified in the validation
protocol (sample size, location, sample container, etc.)
• Inadequate testing: Validation samples are pooled although validation protocol
requires individual testing Cleaning validation samples are not tested for all active
materials and residual cleaning agents Test method used for testing of validation samples is not
developed / validated.

Failure to Record Key Information During Validation
• There is no documented evidence within the validation to support the validation parameters, for example:Actual machine speedActual mixer speedActual water temperature
• There is no direct audit trail to support the validation work, for example:No cross reference to measuring devicesMeasuring devices are not calibratedRaw data is missingA protocol or report is missing.

Inappropriate Review/Sign off (Some Real life Examples)
• The review of validation data is sometimes poor: Deviations from the validation protocol are not noticed Validation results do not meet acceptance criteria but
no comment is made
• Examples of Deviations from Validation Protocol: Machine running speed only 2/3 of speed required
within protocol, but signed off with no comments two validation batches instead of three that were
required in the protocol, but signed off with no comment
One acceptance criteria required the equipment to be visually clean. This was not met – but some trickery words used to justify why this was ok.

Inadequate Conclusion and Sign Off
• There is no clear statement or conclusion in the validation report to indicate the status - such as “the process for product X is considered validated”
• Work is not performed in sequence, or work is not signed off in a timely manner PQ commenced before IQ signed off Work performed but not signed off for months (or years !)
• Make sure the routine batch running parameters actually reflect your process validation parameters ! Validated rinse time for cleaning vessel = 30 minutes Routine Production rinse time 20 minutes !!.

What Happens After Validation?
• Do clean room operators change the clean room air pressure?– definitely not as they should not have access
• Do process operators change the granulation blending time?– highly unlikely because of:
Pre-set Programmable Logic Controller (PLC) Requirement to record actual value in batch documentation Operators generally understand the consequences of not mixing for
long enough
• Do operators/technicians change equipment parameters such as filling/Packaging line speed, washing machine temperature/time, heat sealing temperature/time?
– unfortunately they do !!!

Conclusion
• Validation is simple if planned properly
• Validation does not always require masses of documentation if performed correctly
• Validation plans/protocols need to be very clear to enable people to follow them precisely, and for reviewers to check the key pieces of data
• Once the validated conditions have been established – consider how the equipment controls can be “locked”
• Minor deviations from the validation protocol may happen occasionally, and need to be properly explained and justified
• The protocol sets the critical parameters and acceptance criteria. Failure to meet these acceptance criteria must be investigated and documented.

Thank You
Any Questions





























