Embed Size (px)
Citation preview

Question 1: Name, PDB codes• Eric Martz• [email protected]• Keiichi Namba• Macromolecular visualization
I chose:• 3onz• HUMAN TETRAMERIC HEMOGLOBIN: PROXIMAL NITRITE LIGAND AT BETA• Function: Oxygen transport• Resolution: 2.09 Å• R = 0.218• Rfree = 0.280 “WORSE THAN AVERAGE at this resolution”
Minamino-san gave to me:• 1d66• DNA-binding domain of Gal4.• Transcriptional regulation.• Resolution: 2.70 Å• R = 0.230• Rfree not published

Question 2: Number of chains
Protein: 2 chains (2 distinct)DNA: 0 chainsRNA: 0 chains
Chain A:HEMOGLOBIN SUBUNIT ALPHA141 residues (1 missing) of Protein.Source: Homo sapiens (Human).Other_details: Blood.
Chain B:HEMOGLOBIN SUBUNIT BETA146 residues (12 missing) of Protein.Source: Homo sapiens (Human).Other_details: Blood.

3orn:2 HEM: Protoporphyrin ix containing Fe2 NO2: Nitrite ion4 MBN: Toluene
1d66:4 CD : Cadmium ion
Question 3: Ligands and non-standard residues

Chain A:Crystal: 146 amino acidsFull length: 147 amino acids
Chain B:Crystal: 141 amino acidsFull-length: 142 amino acids
Question 4: Full length vs. crystallized sequence

This structure is all alpha helices (red) and “coil” (white). It has no beta strands.
80.7% alpha helices0% beta strands19.3% neither
Question 5: Secondary structure

I found no patches of all positive or all negative charges.
For 3onz chain A:pI = 8.69.Charges at pH• 4.0: +20.7• 7.0: +4.3• 10.0: -7.0
Question 6: Charge distribution
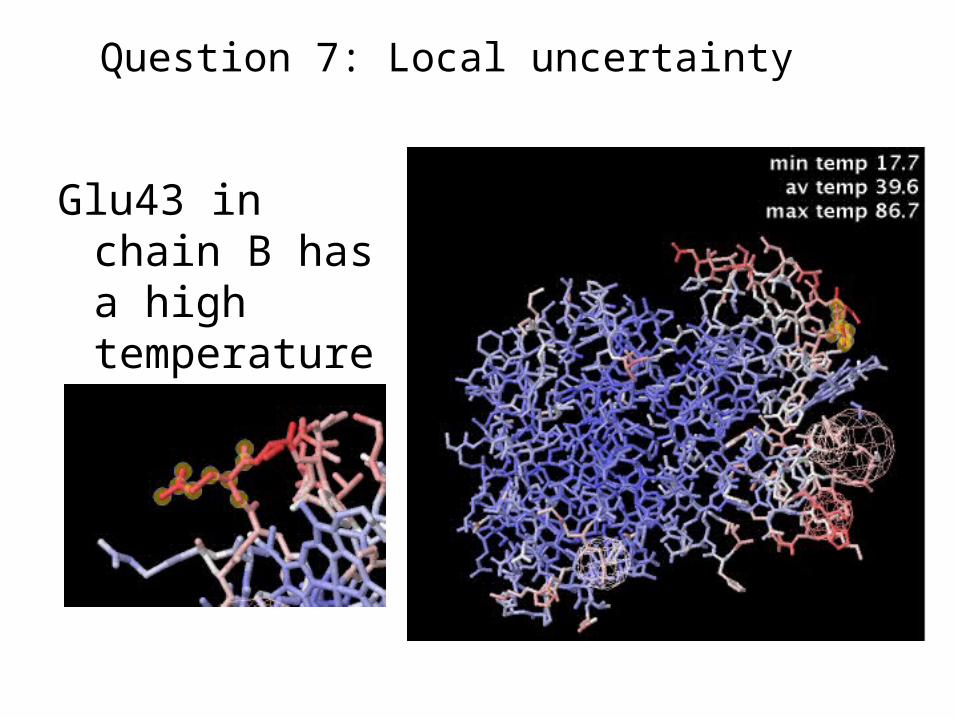
Glu43 in chain B has a high temperature.
Question 7: Local uncertainty

Question 8 - Hydrophobic cores
Yes. Each of the two chains in 3onz has a hydrophobic core (circled in red).
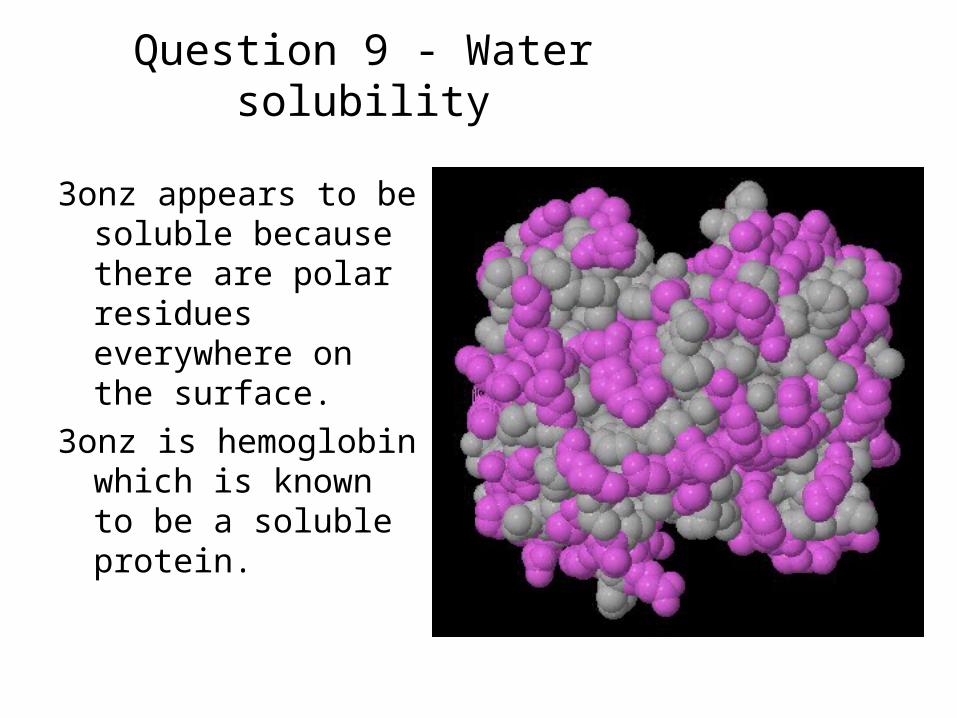
Question 9 - Water solubility
3onz appears to be soluble because there are polar residues everywhere on the surface.
3onz is hemoglobin which is known to be a soluble protein.

Question 10 - Disulfide bonds

Question 11 - Missing residues
13 Missing Residues including 2-, 3+ charged amino acids!
All sidechains are complete.
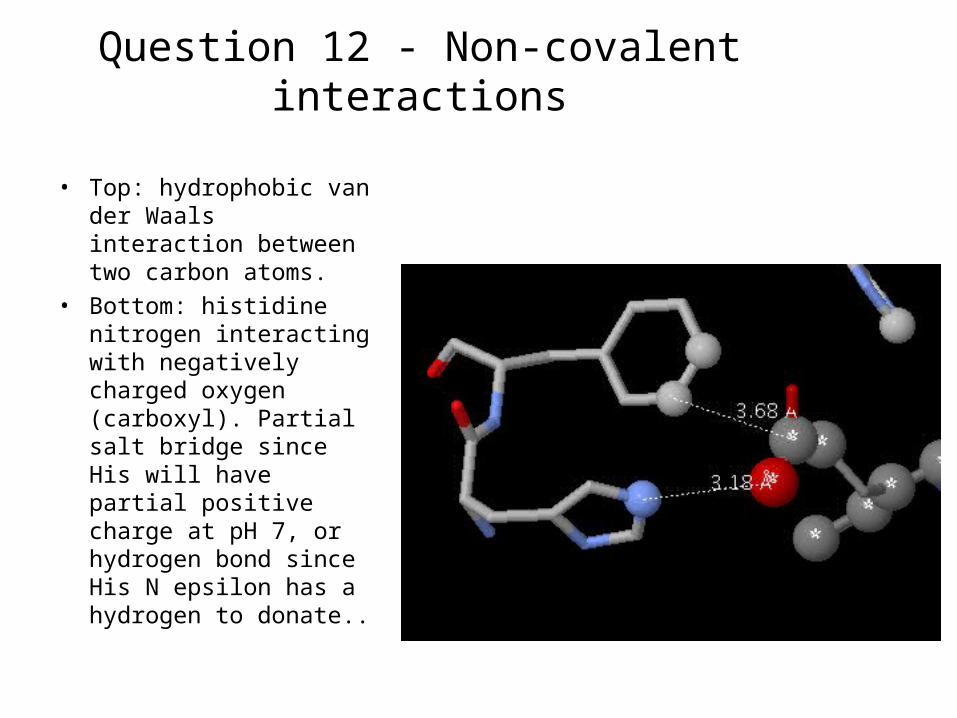
Question 12 - Non-covalent interactions
• Top: hydrophobic van der Waals interaction between two carbon atoms.
• Bottom: histidine nitrogen interacting with negatively charged oxygen (carboxyl). Partial salt bridge since His will have partial positive charge at pH 7, or hydrogen bond since His N epsilon has a hydrogen to donate..
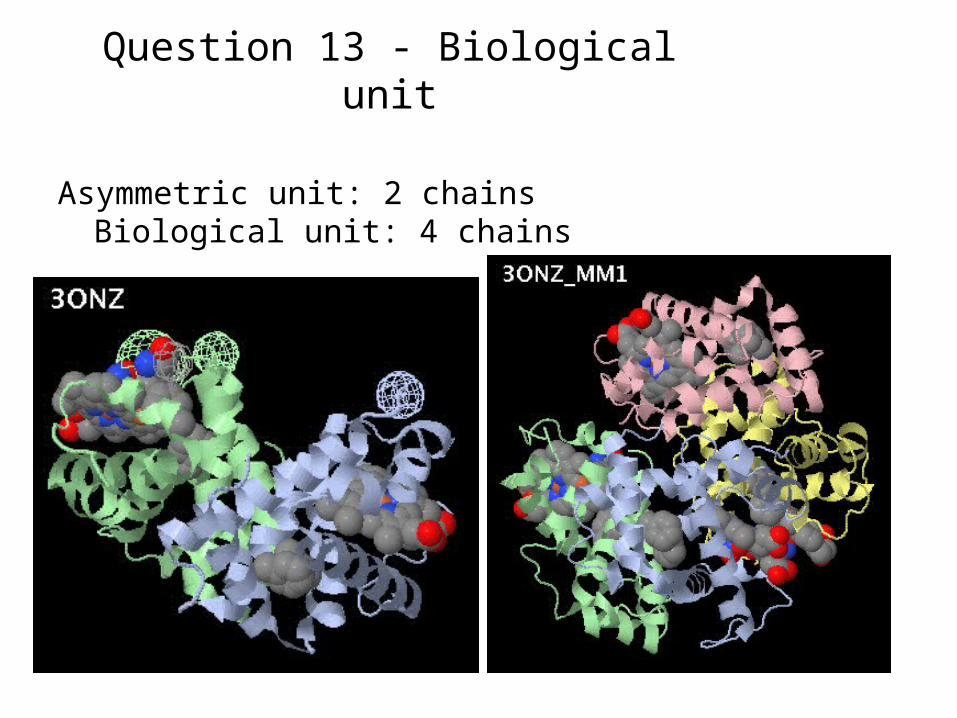
Question 13 - Biological unit
Asymmetric unit: 2 chains Biological unit: 4 chains
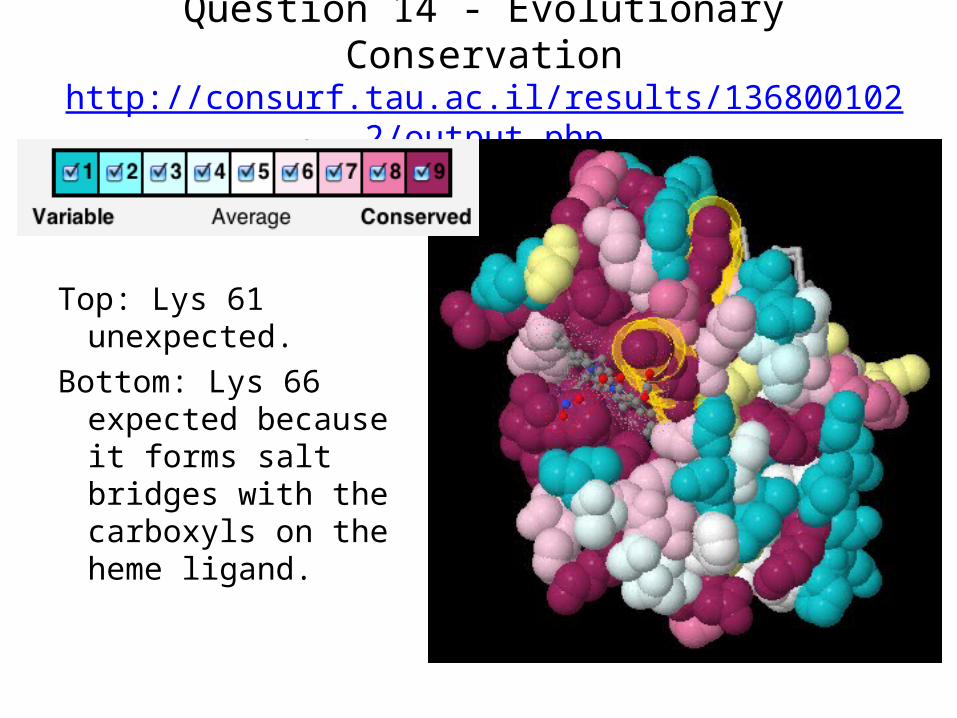
Question 14 - Evolutionary Conservationhttp://consurf.tau.ac.il/results/1368001022/output.php
Top: Lys 61 unexpected.
Bottom: Lys 66 expected because it forms salt bridges with the carboxyls on the heme ligand.
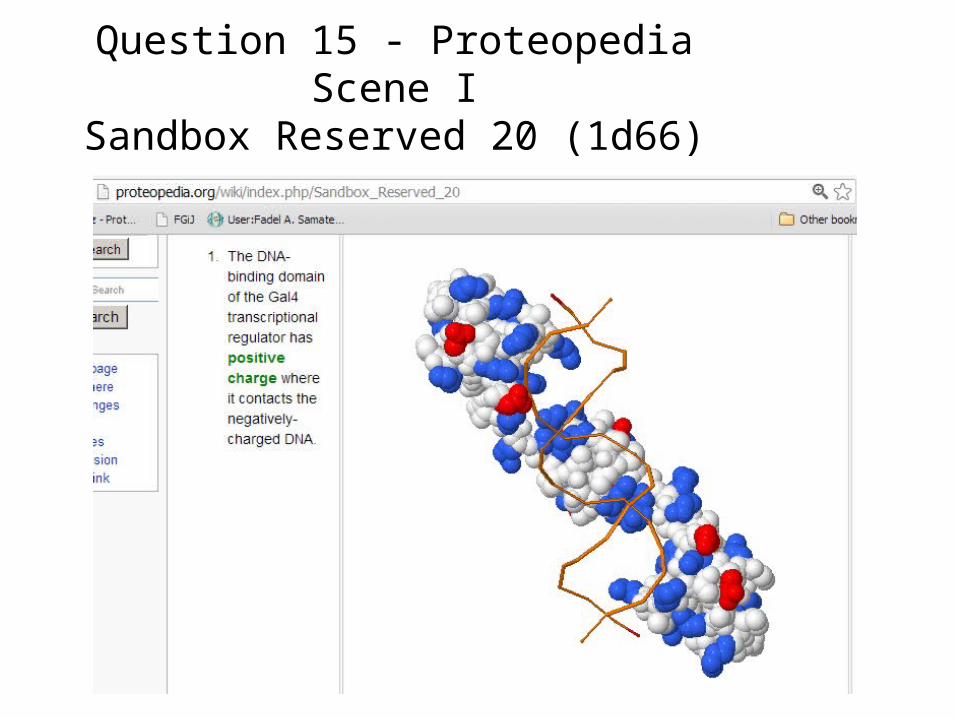
Question 15 - Proteopedia Scene ISandbox Reserved 20 (1d66)
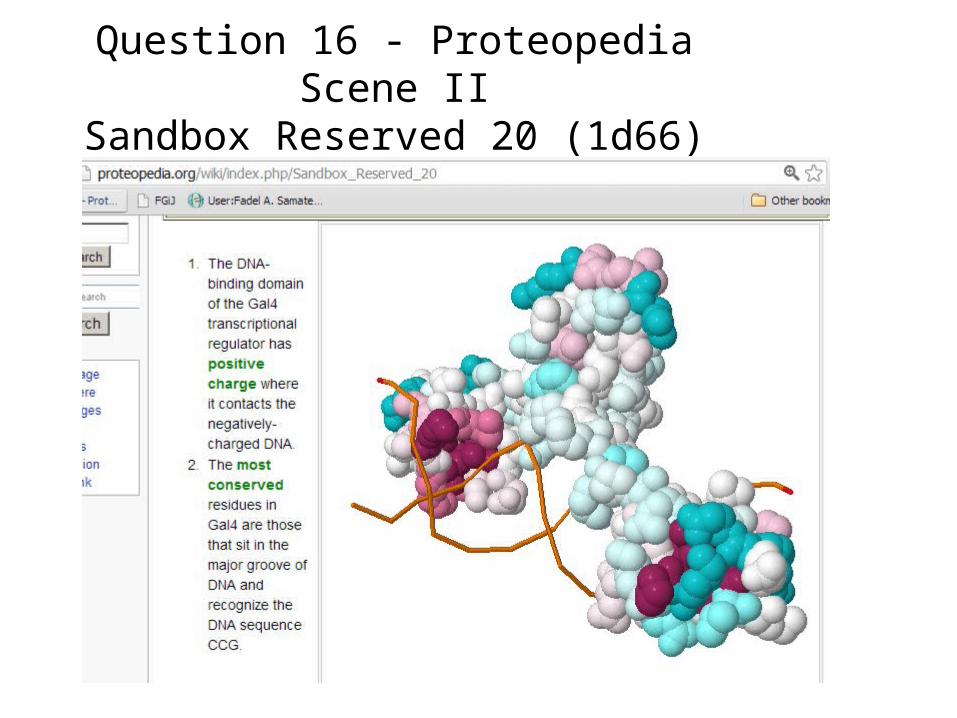
Question 16 - Proteopedia Scene IISandbox Reserved 20 (1d66)

Question 17a - Cation-pi interaction
There are no significant cation-pi interactions in 3onz.1d66 contains 3 significant cation-pi interactions. One is Lys25 with Tyr40 in
chain B. See next slide for snapshot.Here is the report from CaPTURE:
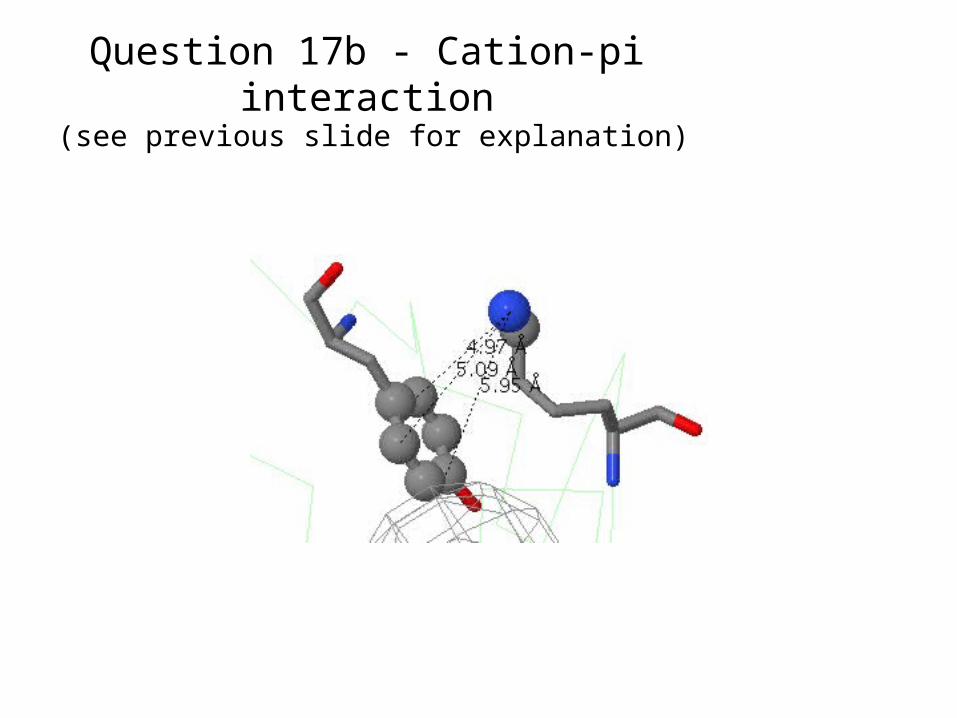
(see previous slide for explanation)
Question 17b - Cation-pi interaction
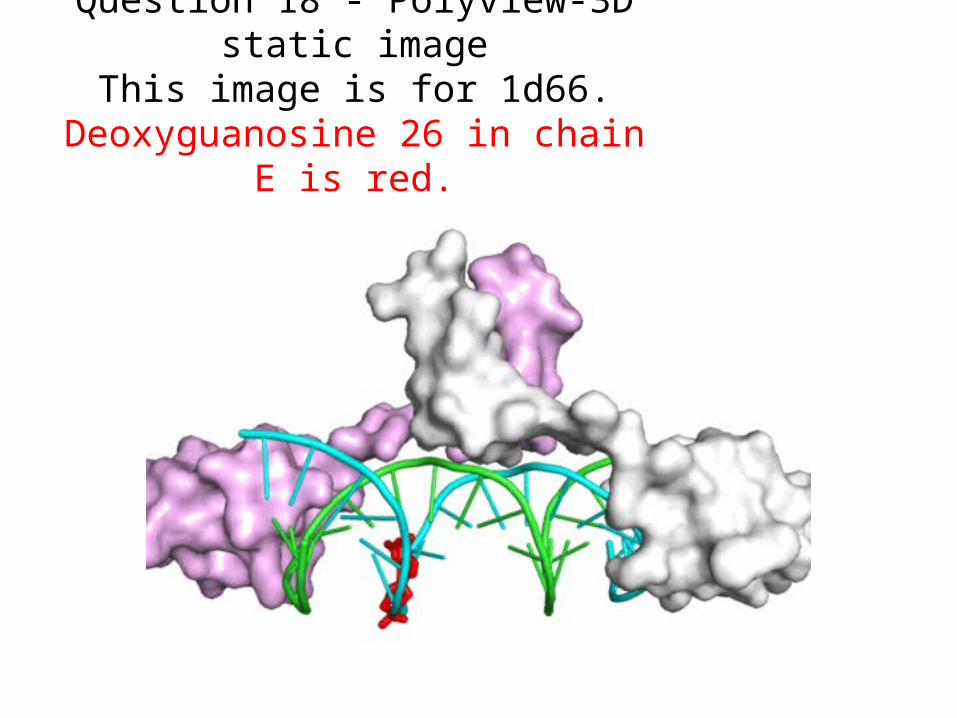
Question 18 - Polyview-3D static imageThis image is for 1d66.
Deoxyguanosine 26 in chain E is red.
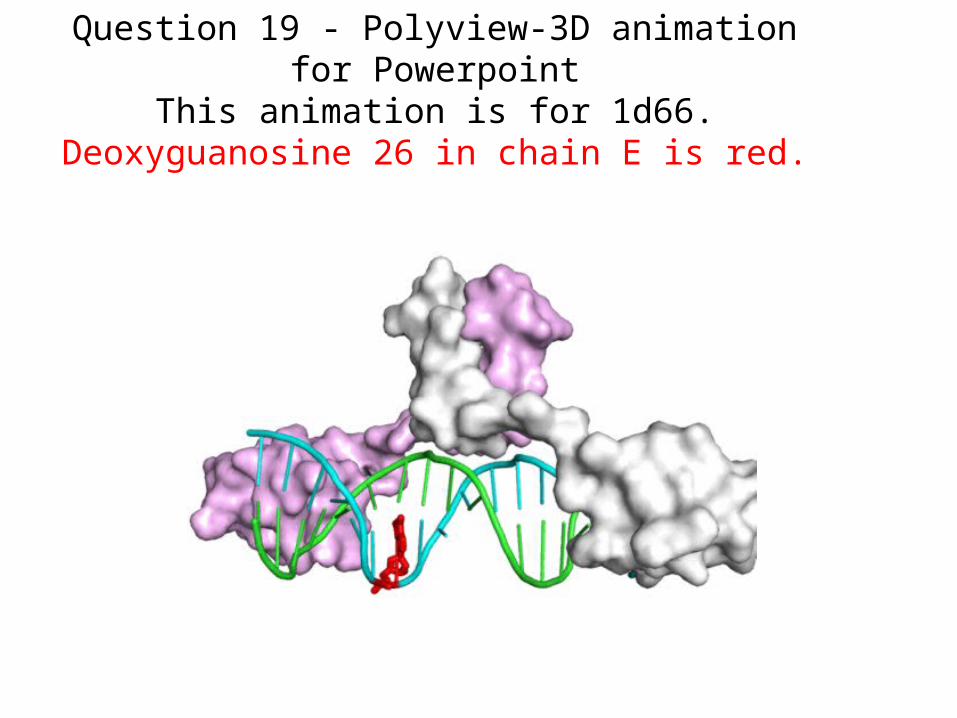
Question 19 - Polyview-3D animation for PowerpointThis animation is for 1d66.
Deoxyguanosine 26 in chain E is red.
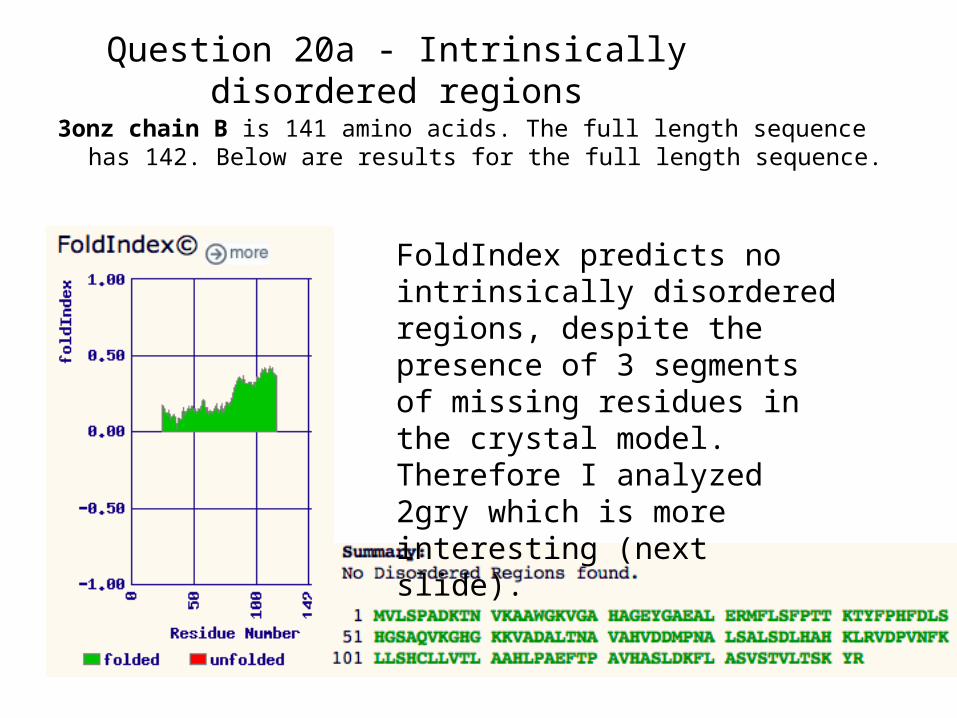
Question 20a - Intrinsically disordered regions
3onz chain B is 141 amino acids. The full length sequence has 142. Below are results for the full length sequence.
FoldIndex predicts no intrinsically disordered regions, despite the presence of 3 segments of missing residues in the crystal model. Therefore I analyzed 2gry which is more interesting (next slide).
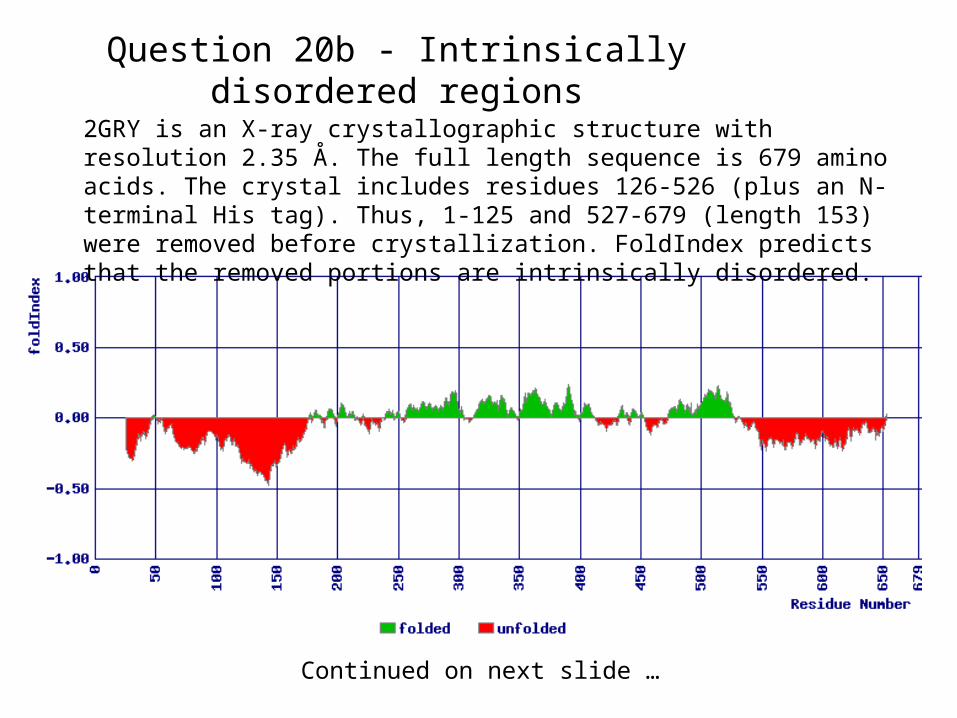
Question 20b - Intrinsically disordered regions
2GRY is an X-ray crystallographic structure with resolution 2.35 Å. The full length sequence is 679 amino acids. The crystal includes residues 126-526 (plus an N-terminal His tag). Thus, 1-125 and 527-679 (length 153) were removed before crystallization. FoldIndex predicts that the removed portions are intrinsically disordered.
Continued on next slide …

Question 20c - Intrinsically disordered regionsFoldIndex predicts that 5 segments of the crystallized sequence will be disordered.
Segments predicted to be disordered:
1. All but 7 of these 69 N terminal residues are missing in 2GRY.
2. The middle 4 of these 6 residues are missing in 2GRY.
3, 4, 5: None of these predictions overlap with the four additional segments of missing residues in 2GRY. Continued on next slide …

Question 20d - Intrinsically disordered regions
The two segments of amino acids missing in 2GRY are at the N terminus and near the N terminus. The flanking residues (marked with yellow halos) have mostly low temperatures, while other missing segments (not predicted by FoldIndex) have higher temperatures.
2GRY colored by temperature showing “empty baskets”, regions with missing residues. Yellow halos mark the alpha carbons flanking missing residues predicted to be disordered by FoldIndex.





























