Embed Size (px)
Citation preview

Structure of MsbA from E. coli:A Homolog of the Multidrug
Resistance ATP Binding Cassette(ABC) Transporters
Geoffrey Chang* and Christopher B. Roth
Multidrug resistance (MDR) is a serious medical problem and presents a majorchallenge to the treatment of disease and the development of novel thera-peutics. ABC transporters that are associated with multidrug resistance (MDR-ABC transporters) translocate hydrophobic drugs and lipids from the inner tothe outer leaflet of the cell membrane. To better elucidate the structural basisfor the “flip-flop” mechanism of substrate movement across the lipid bilayer,we have determined the structure of the lipid flippase MsbA from Escherichiacoli by x-ray crystallography to a resolution of 4.5 angstroms. MsbA is organizedas a homodimer with each subunit containing six transmembrane a-helices anda nucleotide-binding domain. The asymmetric distribution of charged residueslining a central chamber suggests a general mechanism for the translocation ofsubstrate by MsbA and other MDR-ABC transporters. The structure of MsbA canserve as a model for the MDR-ABC transporters that confer multidrug resistanceto cancer cells and infectious microorganisms.
The increasing incidence of multidrug re-sistance is a significant health problem thathas profoundly impacted the treatment ofinfectious diseases and cancer. The WorldHealth Organization has recently reportedthat multidrug-resistant bacteria can ac-count for up to 60% of all hospital-acquiredinfections globally (1). Multidrug resis-tance in the treatment of cancer is respon-sible for tens of thousands of deaths peryear. Multidrug resistance can be conferredby a number of transporters that pumpdrugs out of cells. Certain multidrug resis-tance transporters such as the human P-glycoprotein, MDR1, can transport a di-verse class of amphipathic drug molecules(2). There is evidence that some of thesedrug transporters may act as phospholipidflippases and it has been proposed thatmultidrug transporters may function asdrug flippases, translocating drugs from theinner to the outer leaflet of the lipid bilayer.In an effort to understand the structuralbasis of multidrug resistance, we have de-termined the crystal structure of the multi-drug resistance transporter homolog,MsbA, from Escherichia coli (Eco-msbA).The msbA gene product belongs to a super-family of transporters that contain an aden-osine triphosphate (ATP) binding cassette(ABC), which is also called a nucleotide-binding domain (NBD) (3, 4 ). ABC trans-porters translocate a wide variety of sub-
strates, including amino acids, peptides,ions, sugars, toxins, lipids, and drugs andare implicated in a number of serious hu-man diseases, including cystic fibrosis andseveral disorders of the immune system(5–7 ).
MsbA is a member of the MDR-ABCtransporter group by sequence homology andis more closely related to the mammalianP-glycoproteins than any other bacterial ABCtransporter (8, 9). Although LmrA from Lac-tococcus lactis is functionally more similar tothe P-glycoproteins, MsbA is even more con-served by sequence homology (10, 11).MDR-ABC transporters have been proposedto act as “hydrophobic vacuum cleaners” be-cause of their ability to remove lipids anddrugs from the inner membrane leaflet (12).MsbA transports lipid A, a major componentof the bacterial outer cell membrane, and isthe only bacterial ABC transporter that isessential for cell viability (13). Loss of MsbAfrom the cell membrane or a mutation thatdisrupts transport results in a lethal accumu-lation of lipid A in the cytoplasmic leaflet(14, 15). Several bacterial homologs of msbAthat include the flippase ValA from Fran-cisella novicida and LmrA, have been report-ed in over 30 divergent prokaryotic species(16).
The overall organization of MsbA is consis-tent with most bacterial MDR-ABC transport-ers and its amino acid sequence is remarkablysimilar to several mammalian P-glycoproteinsinvolved in multidrug resistance. All knownABC transporters are composed of four mod-ules, including two membrane spanning regionsand two NBDs. Unlike the mammalian P-gly-
coproteins, which have these components fusedinto a single polypeptide, the msbA gene en-codes a half transporter that contains a singlemembrane spanning region fused with a NBD.MsbA is assembled as a homodimer with a totalmolecular mass of 129.2 kD. Hydropathy anal-ysis indicates six membrane spanning regionswith the NBD located on the cytoplasmic sideof the cell membrane (17). The primary role ofthe transmembrane domain is to recognize andtransport substrates across the lipid bilayer. TheABC, which is the hallmark of the MDR-ABCtransporter family and is located in the NBD,couples the energy of ATP hydrolysis to sub-strate translocation. Although the NBD struc-tures of the histidine transporter (HisP), themaltose transporter (MalK), the DNA repairenzyme (Rad50), and the branched-chain ami-no acid transporter from Methanococcus jann-aschii (MJ1267) have been determined, thestructural basis for substrate translocationthrough the cell membrane is not clear (18–21).
The structure of MsbA establishes thegeneral architecture of the MDR-ABCtransporter family, and facilitates our un-derstanding of the fundamental flippingmechanism that moves hydrophobic sub-strates from the inner to the outer mem-brane leaflet. The protein sequence of Eco-msbA is 36 and 32% identical to the NH2-terminal and COOH-terminal halves of hu-man MDR1, respectively (Fig. 1) (22).Human MDR3, which is a phosphatidyl-choline flippase and is 73% identical inprotein sequence to human MDR1, is 31%identical in protein sequence to Eco-msbA(23). The similarity in protein sequence andfunction between MsbA and human MDR1/MDR3 suggests a common evolutionaryorigin and, therefore, they may have com-mon mechanisms by which they catalyzethe flipping of substrates. The crystal struc-ture of Eco-msbA determined to 4.5 Å inresolution provides a framework for deci-phering P-glycoproteins and suggests ageneral mechanism for the transport of sub-strate across the lipid bilayer.
Structure determination. Membraneprotein x-ray crystallography of transportersand ion channels presents new challengesowing to the disorder caused by detergent andthe inherent movement of transmembranea-helices. We have, therefore, adopted astrategy of rapidly exploring crystallizationspace by cloning, overexpressing, and puri-fying more than 20 full-length bacterial trans-porters and their homologs derived from sev-eral MDR-ABC transporter families and 12bacterial species (24). Our expectation wasthat one or more of these natural variantswould be more optimal for protein expres-sion, purification, and crystal formation.Each full-length MDR-ABC transporter wascloned and recombinantly expressed in aBL21 strain of E. coli (25). Although func-
Department of Molecular Biology, MB-9, The ScrippsResearch Institute, La Jolla, CA 92037, USA.
*To whom correspondence should be addressed. E-mail: [email protected]
R E S E A R C H A R T I C L E
www.sciencemag.org SCIENCE VOL 293 7 SEPTEMBER 2001 1793

tional activity of the purified material was notassayed, the integrity of each transporterdimer was confirmed by gel filtration. Afterscreening and refining approximately 96,000crystallization conditions for several MDR-ABC transporters and their homologs using;20 detergents, we obtained more than 35distinct membrane protein crystal forms.The MsbA MDR-ABC transporter from E.coli yielded crystals of good diffraction
quality that were used for x-ray structuredetermination.
Eco-msbA crystallized in space group P1(a 5 107.8 Å, b 5 126.1 Å, c 5 206.6 Å, a5 83.5°, b 5 76.3°, g 5 84.1°) using dode-cyl-a-D-maltoside (a-DDM) (Table 1) (26).The native crystals diffracted to a resolutionof ;6.2 Å using synchotron radiation butdata was fairly anisotropic. In an effort tostrengthen protein lattice contacts and de-
crease the disorder within these crystals, weapplied a crystal refinement strategy that in-cluded the screening of an extensive matrixof detergents, detergent concentrations, salts,temperatures, organics, additives, deuteriumoxide, and heavy metals. One compound,OsCl3, significantly improved the diffrac-tion quality to a limiting resolution of 4.5Å. This compound was later found to bindat crystal lattice contacts between NBDs of
Fig. 1. Amino acid sequence alignment (56). The inferred amino acid sequenceof Eco-msbA is shown with MsbA from H. influenzae (H.Inf-msbA), the NH2-terminal and COOH-terminal half of human MDR1, LmrA, and HisP. Identicaland similar residues are shaded blue and gray, respectively. Black asterisksindicate functional mutations of Eco-msbA. Transmembrane regions and a-he-lical segments are indicated by gray and red helices, respectively. Blue arrowslabel b-sheets. Extracellular loops are labeled “EC,” and gray bars denotedisordered regions of the structure. The Walker A/B motifs and the ABC
signature motif regions are boxed and labeled by A, B, and C, respectively. Alignment was done with the program ClustalW (50).
R E S E A R C H A R T I C L E
7 SEPTEMBER 2001 VOL 293 SCIENCE www.sciencemag.org1794

one transporter pair (Fig. 2). In view of theimproved diffraction quality, data collectedfrom this soaked crystal was used as the“native” data set for x-ray structure deter-mination. Protein phases were determinedby the method of single isomorphous re-placement and single anomalous scattering(SIR/SAS) using two evenly split frag-ments from a single crystal and also byphase combination with a two-wavelengthmultiple anomalous dispersion (MAD) us-ing the computer package PHASES (27,28). Iterative eightfold noncrystallographicsymmetry averaging, solvent flattening/flipping, phase extension, and amplitudesharpening using in-house programs yield-ed electron density maps of excellent qual-ity for tracing a polypeptide chain (Fig. 3)(29). A chemical model was built using theprogram CHAIN (30), and the protein se-quence registration was established by thepresence of electron density for bulky aro-matic groups in the transmembrane helices(Fig. 3C). Regions of the HisP model wereuseful as a guide for building the NBD.
The structure refinement of Eco-msbAwas complicated by a rapid decrease in theintensity of the diffraction pattern as afunction of resolution, corresponding to anoverall temperature factor of ;150 Å2.Similar temperature factors were reportedfor the K1 ion channel from Streptomyceslividans (KcsA) (31) as well as for themechanosensitive ion channel from Myco-bacterium tuberculosis (Tb-mscL) (32).The experimentally phased electron densitymaps, however, appeared to be of muchhigher quality than one would expect forthis temperature factor (33). This suggestedthat there were predominant orientationsfor the transporters in the crystal that werelikely stabilized by heavy-atom bindingwith additional orientations that introduceda degree of positional disorder. As a con-sequence, whereas the diffraction patternremained relatively strong at lower resolu-tion, the scattering contributions from thisensemble of transporters and associated de-tergent interfered at higher resolution, re-sulting in a rapid decrease in diffractionintensities to a higher angle of diffractionresolution. Standard crystallographic re-finement protocols are generally inade-quate for modeling this type of positionaldisorder and, as a result, we were unable torefine a single model of Eco-msbA to val-ues of R and Rfree below ;38 and ;45%,respectively. In an effort to better simulatethe data, 16 copies of the asymmetric unit(eight molecules per asymmetric unit) weresimultaneously refined against the OsCl3data (Set 2) with very strict eightfold non-crystallographic harmonic constraints(2000 kcal/mol) between the monomers us-ing the program XPLOR (34–38). After
Fig. 2. Crystal packing diagram of Eco-msbA transporters (chain trace in green). This orientationshows the pseudo-222 noncrystallographic symmetry. White arrows indicate the direction of thepseudo twofold operators relating monomers within a MsbA transporter. The space group is P1 andthe unit cell is superimposed onto the crystal lattice. Yellow spheres indicate OsCl3 bound atcontacts between two Eco-msbA transporters, which improved diffraction. The image was gener-ated with the program INSIGHTII (MSI, San Diego, California).
Table 1. Data collection and crystallographic analysis. Diffraction data used in the structural analysiswere collected from single crystals at beamlines 11-1 (l 5 0.98 Å) and 9-2 for the multiwavelengthexperiments at the Stanford Synchotron Radiation Laboratory (SSRL). Additional data sets were collectedat beamline 5.0.2 at the Advanced Light Source (ALS) and on laboratory x-ray sources (MAR300/MAR345). Crystals were translated periodically during the data collection to adjust for the rapid decaydue to radiation damage. All data sets were collected at –165°C and processed using the programsHKL2000 (HKL Research) and MOSFLM/SCALA (49). The concentration of PEG 300 was graduallyincreased to 29% by vapor diffusion for optimal cryo-protection for flash-cooling in liquid nitrogen orpropane. The concentration of a-DDM and OsCl3 was critical for maintaining the integrity and diffractionquality of the crystals.
Parameter NativeOsCl3
crystal 1(l50.980 Å)
OsCl3crystal 1
(l51.139 Å)
OsCl3crystal 1
(l51.140 Å)
OsCl3crystal 2
(l50.980 Å)
Diffraction dataBragg spacing limits (Å) 80-6.2 80-5.75 80-5.4 80-6.2 80-4.5Total observations 197,870 150,453 192,102 185,241 384,156Unique observations 25,367 29,009 30,157 26,145 69,609Rsym (%) 4.7% 4.1% 6.7% 6.2% 4.6%Completeness 90% 99% 96% 98% 90%
Generation of experimental electron density
Phasing power (OsCl3) 2.4Density correlation coefficient between pairs of 36% lowest
monomers in asymmetric unit 64% highestOverall figure of merit 0.7
Refinement statistics Model geometry
Single model Bond length deviation 0.009 ÅR factor (80.0–4.5 Å) 38% Bond angle deviation 1.8°Rfree (80.0–4.5 Å) 45%
Sixteen modelsR factor (80.0–4.5 Å) 27%Rfree (80.0–4.5 Å) 38%
Overall B factor of the model 90 Å2
R E S E A R C H A R T I C L E
www.sciencemag.org SCIENCE VOL 293 7 SEPTEMBER 2001 1795

molecular dynamics refinement, an ensem-ble of very similar models (average rootmean square deviation of Ca atoms amongmodels of ,1.4 Å) was achieved with acrystallographic R value of 27% and anRfree of 38%. Residue positions in helicalregions were well defined in these models,whereas the loop regions were less ordered.An averaged model with good stereochem-istry was computed and used for structuralanalysis.
Structural organization of Eco-MsbA.The crystal structure of Eco-msbA trans-porter is consistent with the molecule beinga homodimer and each subunit is composedof two domains (Fig. 4). We have identifieda third domain bridging the transmembraneand nucleotide-binding domains. Eco-msbA is approximately 120 Å in length,with the transmembrane domain, includingthe membrane spanning region, accountingfor ;52 Å. All the transmembrane a-heli-ces are tilted between 30° and 40° from thenormal of the membrane, forming a coneshaped structure with two substantial open-ings on either side facing the lipid bilayer.These openings are ;25 Å wide in thelongest dimension and lead into a largecone-shaped chamber in the interior of themolecule’s transmembrane domain. Theouter membrane leaflet half of the trans-membrane domain forms the intermolecu-lar contacts holding the two monomers ofthe transporter together. The dimer inter-face, which is mostly contributed by thetransmembrane helices, buries approxi-mately 850 Å2 of solvent accessible surfacearea. The base of the chamber facing thecytoplasm is ;45 Å in the widest dimen-sion and the volume of the chamber caneasily accommodate a lipid A molecule.The resolved regions of the NBDs share nointermolecular contact and are separated by;50 Å in the closest dimension.
Transmembrane domain structure.Eco-msbA begins with the NH2-terminuson the cytoplasmic side as a helix (residues10 to 21) that is parallel with the lipidbilayer (Fig. 4). Residues Trp10, Phe13, andTrp17 would intercalate the inner leafletside of the cell membrane. The polypeptidechain continues into the first transmem-brane helix (TM1, residues 22 to 52) and
Fig. 3. Stereoviews of Eco-msbA experimental elec-tron density at 4.5 Å resolution. The Ca polypeptidetrace is superimposed on the electron density andcalculated at 1.5 s. View looking (A) into and (B)perpendicular to the chamber opening. (B) Showssharpened electron density of the transmembranedomain, intracellular domain, and the NBD. (C)Shows a close up of sharpened electron densitycontaining modeled bulky side chains revealinga-helical structural features and registration of thepolypeptide backbone. The densities were renderedwith the program CHAIN.
R E S E A R C H A R T I C L E
7 SEPTEMBER 2001 VOL 293 SCIENCE www.sciencemag.org1796
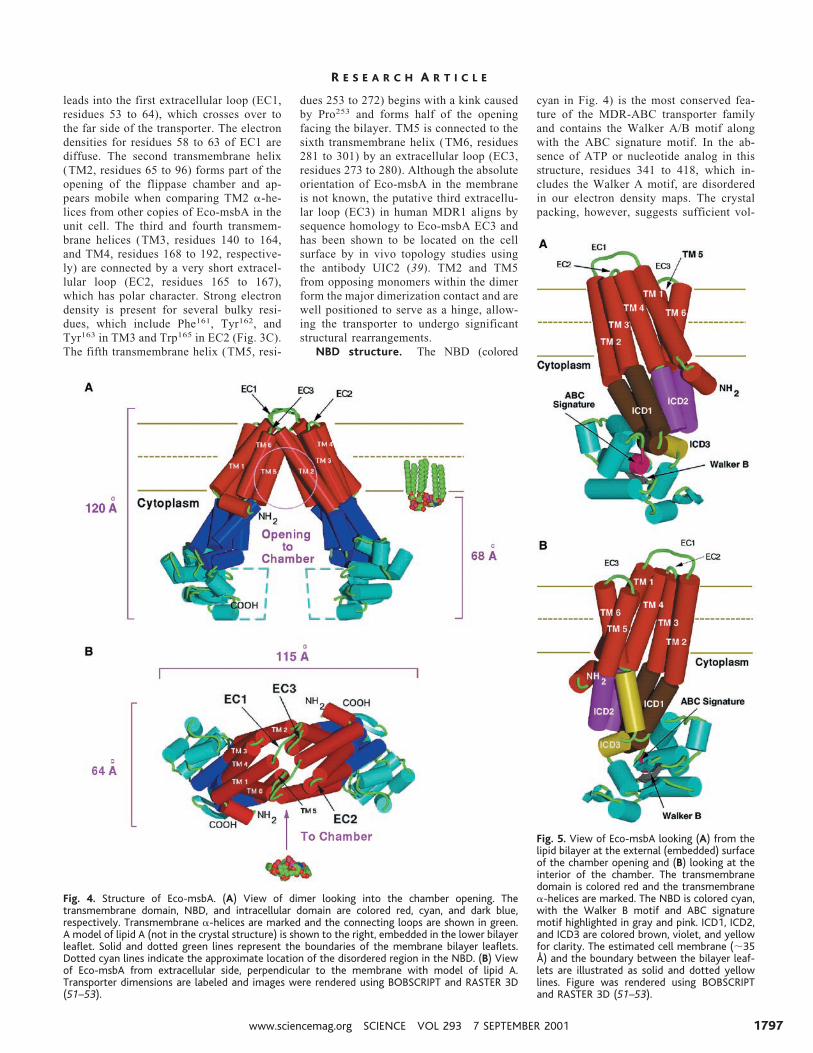
leads into the first extracellular loop (EC1,residues 53 to 64), which crosses over tothe far side of the transporter. The electrondensities for residues 58 to 63 of EC1 arediffuse. The second transmembrane helix(TM2, residues 65 to 96) forms part of theopening of the flippase chamber and ap-pears mobile when comparing TM2 a-he-lices from other copies of Eco-msbA in theunit cell. The third and fourth transmem-brane helices (TM3, residues 140 to 164,and TM4, residues 168 to 192, respective-ly) are connected by a very short extracel-lular loop (EC2, residues 165 to 167),which has polar character. Strong electrondensity is present for several bulky resi-dues, which include Phe161, Tyr162, andTyr163 in TM3 and Trp165 in EC2 (Fig. 3C).The fifth transmembrane helix (TM5, resi-
dues 253 to 272) begins with a kink causedby Pro253 and forms half of the openingfacing the bilayer. TM5 is connected to thesixth transmembrane helix (TM6, residues281 to 301) by an extracellular loop (EC3,residues 273 to 280). Although the absoluteorientation of Eco-msbA in the membraneis not known, the putative third extracellu-lar loop (EC3) in human MDR1 aligns bysequence homology to Eco-msbA EC3 andhas been shown to be located on the cellsurface by in vivo topology studies usingthe antibody UIC2 (39). TM2 and TM5from opposing monomers within the dimerform the major dimerization contact and arewell positioned to serve as a hinge, allow-ing the transporter to undergo significantstructural rearrangements.
NBD structure. The NBD (colored
cyan in Fig. 4) is the most conserved fea-ture of the MDR-ABC transporter familyand contains the Walker A/B motif alongwith the ABC signature motif. In the ab-sence of ATP or nucleotide analog in thisstructure, residues 341 to 418, which in-cludes the Walker A motif, are disorderedin our electron density maps. The crystalpacking, however, suggests sufficient vol-
Fig. 4. Structure of Eco-msbA. (A) View of dimer looking into the chamber opening. Thetransmembrane domain, NBD, and intracellular domain are colored red, cyan, and dark blue,respectively. Transmembrane a-helices are marked and the connecting loops are shown in green.A model of lipid A (not in the crystal structure) is shown to the right, embedded in the lower bilayerleaflet. Solid and dotted green lines represent the boundaries of the membrane bilayer leaflets.Dotted cyan lines indicate the approximate location of the disordered region in the NBD. (B) Viewof Eco-msbA from extracellular side, perpendicular to the membrane with model of lipid A.Transporter dimensions are labeled and images were rendered using BOBSCRIPT and RASTER 3D(51–53).
Fig. 5. View of Eco-msbA looking (A) from thelipid bilayer at the external (embedded) surfaceof the chamber opening and (B) looking at theinterior of the chamber. The transmembranedomain is colored red and the transmembranea-helices are marked. The NBD is colored cyan,with the Walker B motif and ABC signaturemotif highlighted in gray and pink. ICD1, ICD2,and ICD3 are colored brown, violet, and yellowfor clarity. The estimated cell membrane (;35Å) and the boundary between the bilayer leaf-lets are illustrated as solid and dotted yellowlines. Figure was rendered using BOBSCRIPTand RASTER 3D (51–53).
R E S E A R C H A R T I C L E
www.sciencemag.org SCIENCE VOL 293 7 SEPTEMBER 2001 1797

ume to accommodate the mass of the Walk-er A region. The remaining portion of theNBD is well resolved in our structure andincludes an a-helix (residues 331 to 340)and residues 418 to 564, which contains theABC signature motif (colored pink in Fig.5) and the Walker B region (colored gray inFig. 5). The NBD of Eco-msbA has signif-icant similarity in sequence and structure tothe corresponding regions found in HisPwith an rms deviation of ;1.5 Å (Ca) forresidues 445 to 528. NBD residues that arein direct contact with the intracellular do-main (residues 420 to 448, 500 to 508, and531 to 556) are among some of the mosthighly conserved regions among all mem-bers of the MDR-ABC transporter family.The COOH-terminus end of Eco-msbA(residues 565 to 582) is not resolved.
Intracellular domain (ICD) structure.A distinctive feature of the MDR-ABCtransporter family is two extensive intracel-lular regions called ICD1 (residues 97 to139) and ICD2 (residues 193 to 252). Wehave identified a third intracellular subdo-main that connects TM6 with the NBD thatwe label ICD3 (residues 302 to 327). Wecollectively designate the region locatedbetween the transmembrane domain and theNBD as the intracellular domain (coloreddark blue in Fig. 4). Most of the residuesthat correspond to structural elements inICD1 that are in contact with either thetransmembrane domain or the NBD arehighly conserved throughout the MDR-ABC transporter family. ICD1 (coloredbrown in Fig. 5), which is sandwiched be-tween ICD2 (colored violet in Fig. 5) andthe NBD, is composed of three a-helicesconnected by short loops to form a “U”-likestructure. The second a-helix of ICD1 (res-idues 111 to 121) is highly conserved and isnestled against residues 420 to 430 of theNBD. The well-ordered portions of ICD2 inour electron density maps are mostly a-he-
lical (residues 193 to 207 and 237 to 252).The electron densities for residues 208 to236, however, are diffuse within the crys-tal. ICD3 links TM6 and the NBD andforms two a-helices connected by shortloops (colored yellow in Fig. 5B). Thea-helix just preceding the NBD domain(residues 318 to 327) is conserved amongMDR-ABC transporters and is in directcontact with both ICD1 and ICD2.
Chamber structure. The chamber is asymmetric structure that is formed fromtwo Eco-msbA transmembrane domainsand extends along the pseudo twofold axisperpendicular to the cell membrane (Fig.6A). The chamber has an opening (;25 Å)on either side facing the bilayer providingfree access of substrate from the cytoplas-
mic leaflet of the lipid bilayer while ex-cluding molecules from the outer leaflet.The openings to the chamber are defined byintermolecular interactions between TM2of one monomer and TM5 of another (Fig.4A). Residues 268 to 273 of TM2 lining theopening of the chamber are partially shield-ed from the bilayer by TM5. The residueslining the chamber are contributed by all 12transmembrane a-helices and could behighly solvated. The inner membrane leaf-let side of the chamber contains a cluster ofpositively-charged residues (Arg148,Arg183, Lys187, Arg190, Lys194, and Arg296)(Fig. 6B), which contrasts with the signif-icantly less charged and more hydrophobicenvironment within the outer membraneleaflet side.
Fig. 6. Molecular surface render-ing of Eco-msbA with electro-static potentials. This figure wasgenerated with the programGRASP (54), assuming an ionicstrength of 100 mM NaCl anddielectric constant of 2 and 80for protein and solvent, respec-tively. The surface potential var-ies continuously from blue (pos-itive) to red (negative). The cellmembrane, which is ;35 Åwide, is represented with yellowlines. The transmembrane do-main (TMD), intracellular do-main (ICD), and NBD are indi-cated. (A) Side view of dimerlooking into the chamber. Theopening of the chamber spansthe lower bilayer leaflet. (B) View of Eco-msbA monomer looking at the inner surface of the chamber. The surface potential within the lower half ofthe chamber is highly positive due to a clustering of lysine and arginine residues.
Fig. 7. Potential model forlipid A transport by Eco-msbA. Stages 1 to 3 beginat top and proceedsclockwise. See text for de-tails. (1) Lipid A binding,triggering of ATP hydroly-sis, and recruitment ofsubstrate to chamber. (2)Closure of the chamberand translocation of lipidA. Interaction betweenthe two NBDs is possible.(3) Opening of the cham-ber, movement of TM2/TM5, release of lipid A tothe outer bilayer leaflet,and nucleotide exchange.A small yellow rectangleand a green circle denotethe hydrophobic tails andsugar head groups of lipidA, respectively. The trans-membrane domain (TM),intracellular domain (ICD), and nucleotide-binding domain (NBD) are labeled. The cell membraneis represented as a set of two horizontal lines separated by a dash to indicate the separation ofbilayer leaflets. Blue regions indicate positive charge lining the chamber, and purple regionsrepresent the intracellular domain. The gray region on the outer membrane side of the chamber ishydrophobic. Red and black arrows show the movement of substrate and the structural changes ofMsbA, respectively.
R E S E A R C H A R T I C L E
7 SEPTEMBER 2001 VOL 293 SCIENCE www.sciencemag.org1798

Possible flipping mechanism. Thestructure of MsbA suggests a general mech-anism for hydrophobic substrate transloca-tion by members of the MDR-ABC trans-porter group (Fig. 7). Studies of P-glyco-protein function indicate that residues lin-ing the proposed chamber opening (TM2,TM5, and TM6) play an important role insubstrate recognition (40). There is signif-icant evidence indicating a cooperative in-teraction between the two opposing NBDs(41). The extensive contact of the intracel-lular domain (colored purple in Fig. 7) withboth the transmembrane domain and theNBD allow it to function as a conduit cou-pling the triggering of ATP hydrolysis bythe NBD to tertiary arrangements of thetransmembrane a-helices. Upon binding ofa lipid A molecule, conformational changesrelayed from the transmembrane domain tothe intracellular domain initiate nucleotidehydrolysis by the NBD (step 1 in Fig. 7).Movement of TM2, TM5, TM6, and thetwo nucleotide-binding domains serves torecruit the substrate and close the chamber(step 2 in Fig. 7). Hydrolysis of ATP by theNBD may result in a conformational shiftthat promotes the interaction of the adja-cent NBDs. The cluster of charges liningthe chamber on the inner membrane leafletside creates an energetically unfavorablemicroenvironment for a hydrophobic sub-strate. The asymmetric charge distributionin the interior of the chamber is consistentwith the vectored transport of substratefrom the inner to the outer membrane leaf-let. Faced with both the charge and thehighly polar contribution of potentiallybound solvent, the lipid A molecule “flips”into an energetically more favorable posi-tion within the outer membrane leaflet sideof the chamber (colored gray in Fig. 7)where it can form hydrophobic interactions.The substrate is now properly orientated toenter the outer bilayer leaflet. The flippingof substrate signals the chamber to undergostructural rearrangements that include theseparation of the NBDs and repositioningof TM2 and TM5, enabling the expulsion ofthe substrate to the outer membrane leaflet(step 3 in Fig. 7). Alternatively, it has beenproposed that some drug transporters canextrude their substrates directly into theextracellular medium from the inner leafletof the bilayer (42). Substrate release signalsthe NBD to exchange ATP for ADP, reset-ting the system. This entropically driven“flip-flop” mechanism could account forthe unusually broad range of hydrophobicdrugs and lipids transported by members ofthe MDR-ABC transporter family. Severalother variations of this mechanism areplausible and will be refined using bio-chemical and structural information de-rived from additional MsbA studies.
Although we believe that MsbA may notbe able to serve as a close structural repre-sentative of the ABC transporters that trans-locate hydrophilic substrates, there is signif-icant evidence that suggests that humanMDR1 and other MDR-ABC transportersshare transport mechanisms and structuralcomponents similar to Eco-msbA (43). Firstand foremost, human MDR1, mouse MDR3,and LmrA are lipid flippases like Eco-msbAand transport short-chain lipids (44), phos-phatidylcholine (45), and phophatidylethano-lamine (PE) (46), respectively. Secondly, thesize and shape of the chamber of Eco-msbAcould accommodate and transport a wide va-riety of amphipathic molecules, which ex-plains why human MDR1 and mouse MDR3can extrude an unusually broad range of sub-strates. Finally, the transport kinetics of P-glycoproteins reconstituted in lipid vesiclesdeviate significantly from those made for“pore-like” transport systems (47, 48). Theexplanation is now apparent. The crystalstructure reveals that Eco-msbA is not a porethrough the cell membrane but is a molecularmachine scanning the lower bilayer leaflet forsubstrates, accepting them laterally, and flip-ping them to the outer membrane leaflet. Thex-ray structure of Eco-msbA provides a foun-dation for understanding the bioenergetics oflipid/drug “flip-flop” for the entire MDR-ABC transporter family. The structure canalso help elucidate the mechanism underlyingthe multidrug resistance phenotype, whichcould have a profound impact on the devel-opment of novel therapeutics used to treatcancer and infectious disease.
References and Notes1. “Drug Resistance Threatens to Reverse Medical
Progress,” WHO Press Release 41 ( World HealthOrganization, Geneva, Switzerland, 2000).
2. C. Chen et al., Cell 47, 3 (1986).3. B. Sarkadi, E. M. Price, R. C. Boucher, U. A. Germann,
G. A. Scarborough, J. Biol. Chem. 267, 7 (1992).4. S. V. Ambudkar et al., Proc. Natl. Acad. Sci. U.S.A. 89,
18 (1992).5. I. B. Holland, M. A. Blight, J. Mol. Biol. 293, 2 (1999).6. C. M. Fuller, D. J. Benos, Am. J. Physiol. 263 (no. 2,
part 1), C267 (1992).7. J. Trowsdale et al., Nature 348, 6303 (1990).8. K. Tomii, M. Kanehisa, Genome Res. 8, 1048 (1998).9. A. L. Hughes, Mol. Biol. Evol. 11, 6 (1994).
10. H. W. van Veen et al., Nature 391, 6664 (1998).11. For the purpose of this work, which focuses on
using MsbA as a structural model, we use sequencehomology as a basis for grouping MsbA with theMDR-ABC transporters. By sequence identity cal-culated with the program BLAST, MsbA is 32% andLmrA is 30% identical to the NH2-terminal half ofhuman MDR1.
12. Y. Raviv, H. Pollard, E. P. Bruggermann, I. Pastan, M. M.Gottesman, J. Biol. Chem. 265, 7 (1990).
13. M. Karow, C. Georgopoulos, Mol. Microbiol. 7, 1(1993).
14. Z. Zhou, K. A. White, A. Polissi, C. Georgopoulos, C. R.Raetz, J. Biol. Chem. 273, 20 (1998).
15. W. T. Doerrler, M. C. Reedy, C. R. Raetz, J. Biol. Chem.276, 15 (2001).
16. M. K. McDonald, S. C. Cowley, F. E. Nano, J. Bacteriol.179, 24 (1997).
17. B. Rost, P. Fariselli, R. Casadio, Protein Sci. 5, 8 (1996).18. L. W. Hung et al., Nature 396, 6712 (1998).
19. J. Diez et al., J. Mol. Biol. 305, 4 (2001).20. K. P. Hopfner et al., Cell 101, 7 (2000).21. N. Karpowich et al., Structure 9, 7 (2001).22. S. F. Altschul, W. Gish, W. Miller, E. W. Myers, D. J.
Lipman, J. Mol. Biol. 215, 3 (1990).23. J. J. Smit et al., Cell 75, 3 (1993).24. The nucleotide sequence of E. coli msbA used for clon-
ing was obtained from the published genomic sequenceof E. coli K-12, locus AE000193.1:9950..11698 as de-fined by the NCBI Entrez database. The predicted trans-lation of Eco-msbA produces a 582–amino acidpolypeptide with a molecular weight of 64,461 and wasconfirmed to within 0.07% by mass spectrometry.MsbA homologs and other putative MDR-ABC trans-porters were identified using a BLAST search of theNCBI microbial genomes database, using the amino acidsequence of msbA predicted from the E. coli gene.Clones were isolated by polymerase chain reaction frompurified genomic DNA of several bacterial species in-cluding E. coli, Bacillus subtilis, Vibrio cholerae, Acti-nobacillus actinomycetemcomitans, Haemophilus influ-enzae, Helicobacter pylori, Klebsiella pneumoniae,Streptococcus pneumoniae, Pseudomonas aeruginosa,Enterococcus faecalis, Pasteurella multocida, Salmonellatyphimurium, S. paratyphi, and Chlorobium tepidum.
25. MsbA homologs were cloned into the pET19b ex-pression vector (Novagen, Madison, WI), whichcontains a 23-residue fusion leader containing anNH2-terminal deca-histidine tag. Clones were ex-pressed in E. coli BL21(DE3) (Novagen, Madison,WI) in a 50-liter batch fermentor at 37°C using 2mM IPTG (Anatrace, Maumie, OH) as an inducer.Eco-msbA protein was extracted from ;1 kg of E.coli by agitation in the presence of 1% dodecyl-a-D-maltoside (a-DDM). Extracted MsbA was puri-fied in the presence of 20 mM Tris-HCl ( pH 7.5), 20mM NaCl, and 0.05% a-DDM using nickel-chelateand ion-exchange chromatography. Purified Eco-msbA was assayed for purity by Coomassie bluestaining and mass spectrometry, and then concen-trated to 10 to 15 mg/ml protein with a finaldetergent concentration of 0.05% a-DDM.
26. Crystallization trials were performed using a multi-variate crystallization matrix of temperatures, deter-gents, precipitants, salts, and additives. The best Eco-msbA crystals were obtained using the sitting dropmethod at 5°C, by combining protein with precipi-tant at a ratio of 1 to 3 :1. The precipitant solutionconsisted of 20 mM tris-HCl (pH 7.5), 100 to 200mM citrate (pH 4.8 to 5.4), 15 to 20% PEG 300, 80to 120 mM Li2S04, and 0.05% a-DDM. Crystals ap-peared within 3 weeks and continued to grow for 2months to a full size of (0.4 mm by 0.8 mm by 0.3mm). To verify identity, crystals were washed anddissolved, and NH2-terminal amino acid sequencewas determined to five residues. Mass spectrometryanalysis confirmed the predicted molecular weight.
27. W. Furey, S. Swaminathan, Methods Enzymol. 277,590 (1997).
28. Initial electron density maps clearly revealed thatthe asymmetric unit contained four complete Eco-msbA transporters (eight monomers) in a pseudo-222 arrangement consistent with the operators inthe self-rotation function and corresponding to asolvent/detergent content of ;75%. Electron den-sity correlations on experimentally derived mapsindicated that there were some differences be-tween the transporters that did and did not bindosmium heavy atoms. Transporters that did notbind OsCl3 did not adhere to a perfect twofoldrelating monomers within a dimer. These differ-ences were accommodated in the averaging masksand density modifications.
29. The package PHASES was used for all phase calcu-lations with multiple isomorphous and all anoma-lous scattering data. The isomorphous and anom-alous difference Pattersons yielded strong peaks(.21 s) for the OsCl3 sites. The correct hand ofthe structure was established by observing thehand of the a-helices in the sharpened 4.5 Åelectron density map and also confirmed when
R E S E A R C H A R T I C L E
www.sciencemag.org SCIENCE VOL 293 7 SEPTEMBER 2001 1799

docking a fragment of the hisP to the NBD density.The initial experimentally phased electron densitymap revealed eight monomers of Eco-msbA withreal-space correlation coefficient ranging from 36to 64% between monomers. Eightfold noncrystal-lographic symmetry averaging, solvent flattening/flipping, phase extension, and amplitude sharpen-ing were accomplished using locally written soft-ware (G. Chang, unpublished data) and yieldedelectron density maps that were of excellent qual-ity for model building. The final inversion R valueof 38% reflects the relatively large solvent con-tent, the intensity distribution of the data, thedisorder due to the missing regions of the mole-cule, and the presence of detergent in the crystal.
30. J. S. Sacks, J. Mol. Graphics 6, 224 (1988).31. D. A. Doyle et al., Science 280, 69 (1998).32. G. Chang, R. H. Spencer, A. T. Lee, M. T. Barclay, D. C.
Rees, Science 282, 5397 (1998).33. Numerous rounds of vector refinement were per-
formed using the program XPLOR to best fit themodel into the sharpened electron density. Using thispreliminary model, anisotropic correction to the dif-fraction was applied using XPLOR. Wilson plots indi-cated a sharp drop of the mean diffraction intensitiesas a function of resolution with an overall B factor forthe data ;150 Å2.
34. M. Pellegrini, N. Gronbech-Jensen, J. A. Kelly, G. M.Pfluegl, T. O. Yeates, Proteins 29, 426 (1997).
35. P. Gros, W. F. van Gunsteren, W. G. J. Hol, Science235, 458 (1987).
36. J. Kuriyan et al., Proteins 10, 340 (1991).37. A. T. Brunger, J. Kuriyan, M. Karplus, Science 235, 458
(1987).38. Multicopy refinements were done using 5, 8, 10, or
16 copies of the asymmetric unit. These multicopyrefinements yielded similar crystallographic R fac-tors (27%), Rfree values (38%), and averaged modelstructures. The significant drop in the Rfree valueduring the multicopy refinements and the similar-ity of the independently averaged model structuressuggested a proper fit of the data. The choice ofusing 16 copies of the asymmetric unit provided anopportunity to more finely observe their spatialdistribution. Helical regions were more similarwhile loop regions were generally more disordered.B factors for all atoms were fixed at 90 Å2 duringthe multicopy refinement. An image of superim-posed models (Ca traces) generated from the mul-ticopy refinement can be found on Science Online(www.sciencemag.org/cgi/content/full/293/5536/1793/DC1).
39. Y. Zhou, M. M. Gottesman, I. Pastan, Arch. Biochem.Biophys. 367, 1 (1999).
40. S. V. Ambudkar et al., Annu. Rev. Pharmacol. Toxicol.39, 361 (1999).
41. A. Senior, S. Bhagat, Biochemistry 37, 3 (1998).42. H. Bolhuis et al., EMBO J. 15, 4239 (1996).43. C. F. Higgins, M. M.Gottesman, Trends Biosci. 17, 1
(1992).44. A. van Helvoort et al., Cell 87, 3 (1996).45. A. J. Smith et al., FEBS Lett. 354, 3 (1994).46. A. Margolles, M. Putman, H. W. van Veen, W. K.
Konings, Biochemistry 38, 49 (1999).47. M. Horio, M. M.Gottesman, I. Pastan, Proc. Natl.
Acad. Sci. U.S.A. 85, 10 (1988).48. D. F. Cano-Gauci, F. S. Seibert, A. R. Safa, J. R. Riordan,
Biochem. Biophys. Res. Commun. 209, 2 (1995).49. S. Bailey, CCP4 Project, Acta Crystallogr. D 50, 760
(1994).
50. J. D. Thompson, D. G. Higgins, T. J. Gibson, NucleicAcids Res. 22, 22 (1994).
51. R. M. Esnouf, J. Mol. Graphics 15, 132 (1997).52. P. J. Kraulis, J. Appl. Crystallogr. 24, 946 (1991).53. E. A. Merritt, D. J. Bacon, Methods Enzymol. 277, 505
(1997).54. B. Honig, A. Nicholls, Science 268, 1144 (1995).55. We thank M. Elsliger for computer support in the
multicopy refinement and X. Dai for assisting inthe processing of the diffraction data. We thank H.Banie, C. McCarthy, and M. Hornsby for technicalassistance. We also thank the staff at the StanfordSynchrotron Radiation Laboratory (SSRL), the Ad-vanced Light Source (ALS), the Cornell High EnergySynchrotron Source (CHESS), and the AdvancedPhoton Source (APS) for their help in the datacollection and screening of a couple thousandMDR-ABC transporter crystals. We thank I. Wilson,J. Johnson, R. Milligan, W. Balch, P. Wright, and C.Higgins for thoughtful discussions. We thank P.Wright, J. Kelly, and R. Lerner for supporting mem-brane protein x-ray crystallography at The ScrippsResearch Institute ( TSRI). This project was sup-ported by start-up lab funds from TSRI, the NIH(GM61905-01), and the Presidential Early CareerAward for Scientists and Engineers (PECASE). Co-ordinates have been deposited with Protein DataBank (accession code 1JSQ).
56. Single-letter abbreviations for the amino acid resi-dues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F,Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn;P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; andY, Tyr.
6 July 2001; accepted 6 August 2001
R E P O R T S
Hydrogen 21-CentimeterEmission from a Galaxy at
Cosmological DistanceM. A. Zwaan,1* P. G. van Dokkum,2 M. A. W. Verheijen3,4
We have detected the neutral atomic hydrogen (HI) emission line at acosmologically significant distance [redshift (z) 5 0.18] in the rich galaxycluster Abell 2218 with the Westerbork Synthesis Radio Telescope. The HIemission originates in a spiral galaxy 2.0 h65
21 megaparsecs from the clustercore. No other significant detections have been made in the cluster, sug-gesting that the mechanisms that remove neutral gas from cluster galaxiesare efficient. We infer that fewer than three gas-rich galaxies were accretedby Abell 2218 over the past 109 years. This low accretion rate is qualitativelyconsistent with low-density cosmological models in which clusters arelargely assembled at z . 1.
Galaxies in clusters have evolved in thepast ;3 3 109 years. The number of bluegalaxies in clusters was higher in the past(the Butcher-Oemler effect) (1, 2) and spi-
ral galaxies were more prevalent (3–5). Ithas been argued that these effects arecaused by enhanced accretion of gas-richstar-forming galaxies from the surroundingfield (6–8). Detailed modeling suggests thatthe neutral gas disks of infalling galaxiescan be stripped by the hot x-ray gas thatenvelopes rich galaxy clusters (9–11). Be-cause the neutral gas provided the fuel forstar formation, the star formation rate dropsprecipitously after the cold gas has beenremoved. Hence, galaxies rapidly fade andredden after they have been accreted by a
rich cluster. The low neutral atomic hydro-gen (HI) content of galaxies in the cores ofthe nearby Coma (12) and Virgo (13) clus-ters is consistent with these models. How-ever, at higher redshift, at which the galaxyaccretion rate is predicted to be higher andspiral galaxies are more abundant in thecentral regions of rich clusters, these mod-els have not been tested by direct observa-tions of the neutral gas reservoir of infall-ing galaxies. Studies of HI at an emissionline of 21 cm wavelength have been limitedto the local universe (12–15), because radiosynthesis telescopes were not equipped tooperate at frequencies corresponding to theredshifted HI line or lacked the sensitivityto detect the HI line at higher redshifts.
We have initiated a program of deep HIimaging of galaxy clusters Abell 2218 andAbell 1689 at redshift (z) ; 0.2 to study thecontent and distribution of HI in cluster gal-axies at intermediate redshifts. Here, we re-port on observations of Abell 2218 at z 50.176 from the recently upgraded WesterborkSynthesis Radio Telescope (WSRT). Thecluster is extremely rich and massive (16,17), has a luminous and extended x-ray halo(18), and has become widely known for theHubble Space Telescope imaging that re-vealed a rich structure of strong gravitationalarcs (19).
Observations were performed with theWSRT during the commissioning of theupgraded system in the period from July to
1School of Physics, University of Melbourne, Victoria3010, Australia. 2California Institute of Technology,Mail Stop 105-24, Pasadena, CA 91125, USA. 3Depart-ment of Astronomy, University of Wisconsin, 475North Charter Street, Madison, WI 53706, USA. 4Na-tional Radio Astronomical Observatory, Post OfficeBox 0, Socorro, NM 87801, USA.
*To whom correspondence should be addressed. E-mail: [email protected]
R E S E A R C H A R T I C L E
7 SEPTEMBER 2001 VOL 293 SCIENCE www.sciencemag.org1800





























