Embed Size (px)
Citation preview

107
CHAPTER 5
Reactions of β-Chloroenones with Cyanomethylene
Compounds: Synthesis of 2-Pyridones and
2-Hydroxypyridines
5.1. Introduction
Heterocycles are among the most important structural classes of chemical
substances and are particularly well-represented among natural products and
pharmaceuticals. One striking structural feature inherent to heterocycles, which
continues to be exploited to great advantage by the drug industry, lies in their
ability to manifest substituents around a core scaffold in a defined three-
dimensional representation, thereby allowing for far less degrees of conformational
freedom than the corresponding conceivable acyclic structures. In addition, as a
result of the presence of heteroatoms such as O, N, and S, heterocycles often
exhibit altered absorption, distribution, metabolism, and excretion properties.
Among them pyridine and its derivatives are pharmacologically important
molecules. They are extensively used in medicines as drugs for the treatment of
various diseases.1 They also find applications as agrochemicals and anticancer
agents.2 Extensive studies have been carried out on the synthesis of these valuable
compounds owing to their importance as drugs and biologically active natural
products.3 A recent pharmacological evaluation revealed the importance of various
functionalized 2-pyridones as cardiotonic agents similar to milrinone which is the
most effective nonglycosidic cardiotonic agent clinically used for the treatment of
severe heart failure.4 An important analogue of milrinone, 5-aroyl-2-oxo-3-
pyridinecarbonitrile was synthesized in our laboratory from aroylformylketene
dithioacetal following a two step reaction strategy involving the condensation of
2-aroyl-3,3-bis(alkylsulfanyl)acrylaldehydes with malononitrile followed by
cyclization with conc. HCl or Br2 or reaction of aroylformylketene dithioacetal

108
with ethylcyanoacetamide in the presence of ammonium acetate and acetic acid.
Further, -chloroenones, being another valuable 1,3-bielectrophile, were
investigated for their reactions with cyanomethylene compounds, which is the
subject matter of the present Chapter.
NH
N
NC
H3C O
Milrinone
5.2. General Methods of synthesis of pyridones and its derivatives
2-Pyridones are generally prepared from N-substituted pyridinium salts
which are obtained from functionalized pyridines by alkylation or oxidation..5
Extensive use of this methodology has been made for the synthesis of biologically
active aroylpyridones and naturally occurring alkaloids. One of the general
methods for the synthesis of 2-pyridone derivatives involve cross condensation of
cyanoacetamides with -dicarbonyl compounds accompanied by cyclization yielding
2-pyridones. For example the reaction of substituted acetylacetones 1 with
cyanoacetamides resulted in the formation of 4,5,6-trisubstituted-2-oxo-1, 2-dihydro-
3-pyridinecarbonitriles 3 (Scheme 1).6
Me Me
O O
R1
CN
NH2
ON
Me
R1
Me
CN
OH
1 2 3
Scheme 1
Recently N-substituted cyanoacetamides were treated with 1,3-dicarbonyl
compounds 4 under microwave irradiation to prepare 2-hydroxypyridine
derivatives 6 (Scheme 2).7

109
Me Me
O O CNHN
ON
Me
Me
CN
OH
4 5 6
R
Piperidine
Microwave
R
Scheme 2
Regioselective synthesis of bicyclic 3-cyanopyridin-2(1H)-ones 8 have
been carried out by using cyclic 1,3-diketones 7 as substrates with
cyanoacetamides 2 (Scheme 3).8
CN
NH2
O
7 2 8
CH2
R
OO
NH
H2C
CN
O( )n
( ) n
Scheme 3
Unsymmetrical 1,3-carbonyl compounds 9 when condensed with
cyanoacetamide 2 led to the formation of two isomeric products 10 and 11
(Scheme 4).9
Ar CH3
O O CN
NH2
O NH
Ar
H3C
CN
O NH
CH3
Ar
CN
O
+
9
210 11
Scheme 4
Reactions of 1,3-diketones 4 with ethyl cyanoacetate or malononitrile
afforded 4,6-disubstituted-2-oxopyridine-3-carboxylates or nicotinonitrile
derivative 12 (Scheme 5).10
Me Me
O O
NH
O
X
Me
MeEt2NH
CNC H2X
X = CN or COOEt4
12
Scheme 5

110
Versatile synthetic intermediates such as enaminones along with alkyl
cyanoesters are used for the synthesis of 2-pyridone derivatives. Synthesis of 1,2-
dihydro-2-oxo-3-pyridinecarbonitriles 15 have been carried out recently by Mosti
et al from enaminoketones 13 by treating them with ethyl cyanoacetate 14 in acetic
acid followed by hydrolysis with KOH in ethanol and the products were obtained
in good yields (Scheme 6).11
R R
O O
NH
O
COOEt
R
14 15
N
CH3
H3C
NCOEt
O
R
O
13
1. CH3COOH
2. KOH / EtOH
Scheme 6
The reaction of chalcones and acetamides is a popular method for decades
in the preparation of 2-pyridones. Synthesis of 3-cyano-2-pyridones 18 have been
carried out from ,-unsaturated carbonyl compounds 16 by treating them with
cyanoacetamides 17 by Ciufolini et al (Scheme 7).12
This methodology had also
been later applied for the synthesis of natural product Nothapodytine B.
R1
O
R2
H2N
O CN NH
CN
O
R2
R1
+t-BuOK
DMSO, O 2R1 = alkyl, arylR2 = aryl
16 17 18
Scheme 7
A simple synthesis of novel 2-pyridones from chalcones was put forward
by Mohammad Al-Arab.13
Recently, Rong et al. have put forward a facile method
for the synthesis of 1,2-dihydro-2-oxo-4,6-diarylpyridine-3-carbonitrile from
cyanoacetamides and enecarbonyl compounds under solvent free conditions.14, 15
Such compounds have found applications in the synthesis of some natural products
like Nothapodytine B.16
2-Pyridones 20 have also been synthesized from
enaminoketones 19 and cyanoacetamide in the presence of strong basic catalysts
such as sodium hydride or alkoxides by Keshk et al (Scheme 8).17

111
R1 =
O OMe
OMe
R1
O
NMe 2
H2N
O CN NH
CN
OR1
+
19 17 20
strong base
Scheme 8
Dong and coworkers have reported a facile and efficient synthesis of
halogenated pyridine-2(1H)-ones 22 from a series of readily available enaminones
21 under Vilsmeier-Haack reaction condtion.11
The report has shown a series of
halogenation, formylation and intramolecular nucleophilic cyclization under
Vilsmeier-Haack reaction condition shows the importance of the reaction in the
synthesis functionalized heterocycles (Scheme 9).
O O
NHAr
NMe 2H
N
O
X
OHC Ar
X = Cl or Br
POCl3 or PBr3 / DMF
( 4 equivalent ), 125o
C
21 22
Scheme 9
A readily available starting material like 1-acetyl,1-
carbamoylcyclopropanes 23 were efficiently converted to pyridin-2(1H)-ones 24
and 25 by Vilsmeier-Haack reaction. The mechanism of the formation of the
pyridine derivatives shows sequential ring opening, haloformylation and
intramolecular cyclization.18
A slight change in the reaction condition gave
different product from the same substrate, indicates that Vilsmeier-Haack reaction
can be effectively used for the synthesis of highly functionalized heterocycles
(Scheme 10).

112
O O
NH
ArN
Ar
O
Cl
Cl
OHC
N
Ar
O
Cl
Cl
POCl3 / DMF(5 equiv)
100 o
C
POCl3 / DMF(5 equiv)
120 oC
24 23 25
Scheme 10
Kohler et al have discussed the reactions of δ-ketonitriles and their further
conversion to 2-pyridones under basic conditions in 1922 itself (Scheme 11).19
R1
O
R2 CN NH
CN
O
R2
R1
+t-BuOK
DMSO, O 2
R1 = alkyl, arylR2 = aryl
16
26
18
CN
R1
R2
CN
H3O
27
CN
Scheme 11
Synthetic potential of -substituted vinamidinium hexafluorophosphate
salts 29 have been effectively utilized by Marcoux et al for the preparation of 3,5-
substituted 2-pyridone 30 (Scheme 12),20
-substituted vinamidinium
hexafluorophosphate salts 29 in turn have been prepared by the Vilsmeier-Haack
reaction of acetic or substituted acetic acids or their acid chlorides.
EtO
R2
O
NH3C CH3
R3
NCH3
CH3
NH
R2
R3
O
PF6
1. t-BuOK/THF DABCO
2. NH4OAc, heat
28
29
30
Scheme 12
Ring expansion reactions can also be used to synthesize 2-pyridone
derivatives. This technique is used in the synthesis of 2-pyridone derivatives 32
and 34 from substituted oxazoles 31 and 33 (Scheme 13 & 14) respectively.21

113
N
O
Ph
R1
O
PhN
R2
OHR1
Ph
Ph
O
31 32
R2CHN 2
Scheme 13
N
OCl
HOOC
Cl
H3COOCNH
Cl
Cl
OH3COOC
HOOCCH3OH
HCl
33 34
Scheme 14
Reactions of various alkenes or alkynes with carbonyl compounds afford 2-
pyridone derivatives. Duchene et al have shown that (Z)-3-substituted-3-iodoprop-
2-enamides 36 can be synthesized from (Z)-iodovinylic acids 35 when cross-
coupled with tributylstannylallenes, which resulted in a one-pot synthesis of 4,6-
disubstituted-2-pyridones 37 (Scheme 15).22
R1
OH
I O
R1
HNR'
I O
SnBu3
R2 N
R1
OR
2
R'
(COCl) 2
Pd(OAc) 2, PPh 3
R'NH2
K2CO3, n-Bu4NBr
MeCN, 80 oC, 3h
3536
37
Scheme 15
Libraries of 3,5,6-trisubstituted-2-pyridone derivatives are generated by
rapid microwave assisted solution phase methods using a one-pot, two-step
protocol.23
The three-component condensation of CH–acidic carbonyl compounds
38, N,N-dimethylformamide dimethylacetal 39 and methylene active nitriles, leads
to the formation of 2-pyridones 41 and fused analogues in moderate to good yields
and high purities (Scheme 16).

114
R2
R1 O O
O
N
R2
R1
N
O
CN
R3
+
NH
OR1
R2
R3
38 39 4041
Scheme 16
Very recently Rao et al have described a facile one pot method for the
synthesis of novel 3,5,6-trisubstituted-2-pyridones 44 from the acetylated Baylis–
Hillman esters 43 and β-enamino esters or β-enaminonitriles 42 (Scheme 17).24
R
NH2AcO
R1
COOR2
NH
O
R1R
NaH (3 eq)THF, rt
4-6 hrs
+
42 43 44
Scheme 17
Reaction of amines with an allenic precursor 45 afforded aminoarylpyridones
46 in good yields (Scheme 19).25
These syntheses can be performed in two distinct
steps, allowing the possibility to introduce different substituents in the positions 1
and 4 (Scheme 18).
N
NHAr
Ar
O
Ar-NH2CO2MeO O
45
46
Scheme 18
Trimethylsilylketene 48 reacts with acyl isocyanates 47 to give
4-trimethylsiloxy-1,3-oxazin-6-ones 49 which smoothly undergo the Diels-Alder
reaction with dimethyl acetylenedicarboxylate 50 or methyl propiolate to furnish 2-
pyridones 51 (Scheme 19).26

115
R C
O
N C ON
O
O
RMe3SiO
MeO 2C CO2Me
NH
O
R
CO2Me
MeO 2CMe3SiCH=C=O
47
48
49
50
51
Scheme 19
4-Trimethylsiloxy-1,3-oxazin-6-ones 49 smoothly react with enamines 52
of cycloalkanones to give bicyclic 2-pyridones 53 (Scheme 20).27
N
O
O
RMe3SiO
N(CH2) n
NH
O
n(H2C)R
49
52
53
Scheme 20
Allavi and coworkers have described a new modular synthesis of
deoxypyridinoline in which 4,5-disubstituted 3-hydroxypyridine as the key
intermediate. In this synthesis the pyridine ring was constructed by a series of
reactions. The allylamine 54 was allowed to react with 2 equivalents of
bromoketone 55 into a pyridinium derivative 57, which was deallylated using
titanium II compound, generated from titanium isopropoxide and
propylmagnesium bromide for the intermediate substituted 3-hydroxypyridine 58
(Scheme 21).3

116
Br
ZHN CO2But
O
NHZ
+
K2CO3
CH3CN, rt NO
CO2ButZHN
O
NHZ
CO2But
N
CO2But
ZHN
NHZ
CO2But
ODBU / THF
Rt
N
CO2But
ZHN
NHZ
CO2But
HON
CO2
H3N
NH3
CO2
O
H3N CO2
Ti(OiPr)4 iPrMgBr
deoxypyridinoline
542 equivalents
5556
57
58
59
Scheme 21
A new synthetic route for the biologically important imidazo[4,5-b]pyridine
ring 62 system is described by Tenant etal.4 Readily available 5-chloro-4-
nitroimidazole was converted into its acid chloride derivatives 60 by known
chemistry. It was then treated with a variety of β-keto esters. The resulting
compounds 61 were subjected to reductive cyclizations to imidazo[4,5-
b]pyridinones 62 by catalytic hydrogenation in ethanol over palladium on charcoal
or by treatment of alkaline sodium borohydride in the presence of palladium.
Highly oxygenated derivatives of 1-deazapurines are thus readily available by this
method (Scheme 22).
N
N NO2
R1
R2
O
ClN
N NO2
R1
R2
HO O
EtO
O
R3
N
N
R1
R2
N
O O
OEt
OH
R3
O O
OEtR3
Mg(OEt)2
ether, reflux
H2, Pd-C
EtOH, Rt
60 61 62
Scheme 22

117
For the preaparation of a pyridone derivative, an intramolecular Michael-
type addition followed by retro-Michael elimination was reported during the total
synthesis of a naturally occurring alkaloid (-)-Myrtine. (S,S,R)- (+)-N-Sulfinyl-α-
amino-α-ketophosphonate 63, was treated with dimethylformamide dimethylacetal
at room temperature for 12 h. The resulting enaminone 64 was treated with 4N
HCl and isolated the pyridone derivative 65 in excellent yields (Scheme 23).9
O
Sp-Tolyl NH
Ph
O
P(OMe) 2
O Me2NCH(OMe) 2
12 h
O
Sp-Tolyl NH
Ph
O
P(OMe) 2
O
N NH
Ph
O (OMe) 2P
O4N HCl
63 64 65
Scheme 23
Alkylsulfanyl-substituted pyridones have been obtained from ketene
dithioacetals in high yields. -Oxoketene dithioacetals 66 when treated with
cyanoacetamide in the presence of sodium isopropoxide afforded 2-pyridones 67 in
good yields (Scheme 24).28
An inseparable 4-ethoxy and 4-methylthiopyridones
were obtained in the same reaction when sodium ethoxide was used instead of
sodium isopropoxide.
Ar SCH 3
O SCH 3
NH
CN
OAr
NCCH 2CONH 2
SCH 3
NaOPri/Pr
iOH
66 67
Scheme 24
Fused pyridones 69 have been obtained in a double cyclization process
involving substitution of 4-methylthio substituent of the pyridone ring by a second
molecule of the cyanoacetamide during the reaction of 68 with secondary α-
cyanoacetamides (Scheme 25).29
With α-oxoketene-N,S-acetals, the reaction
produces 3-cyano-4-aminopyridones in moderate to good yield.

118
R SCH 3
O SCH 3
NR2
OR
NCCH 2CONHR 2
NaOPri/Pr
iOH
Reflux
NR2
O
NC
NH2
R = alkyl, aryl
68 69
Scheme 25
N-Substituted pyridones 71 were obtained when secondary α-
cyanoacetamide derivatives were reacted with 2-cyano-3,3-bis(methylthio)
acrylonitrile 70 in DMF or in acetonitrile containing K2CO3 (Scheme 26).30
NC
SCH 3H3CS N
NC CN
OH2N
NCCH 2CONHR
K2CO3 / DMF
CN
SMe
RO CHOR =
70 71
Scheme 26
N-Amino-2-pyridones 73 were obtained when ketene dithioacetal 72
reacted with cyanoacetohydrazide at room temperature in the presence of
potassium hydroxide in 1,4-dioxane (Scheme 27).31
NC
SCH 3H3CS N
NC CN
OH2N
NCCH 2CONHNHRCN
SMe
NHR
KOH / dioxane
R = H, COC6H5
72 73
Scheme 27
Junjappa et al have shown that polarized dienes 75, obtained by the 1,2-
addition of Reformatsky reagent to -oxoketene dithioacetals 74, when heated
with ammonium acetate/acetic acid buffer afford substituted 2-pyridones 76
(Scheme 28).32

119
R1
SCH3
O
H3CS
OEt
OZnBr
O
SCH 3H3CS
OEt
NH
O
SCH 3R1
R1I2/ C6H6/ heat
NH4OAc / AcOH
heat
74 75 76
Scheme 28
Similarly, 2-pyridone derivatives 78 were afforded in good yields by the acid
hydrolysis of ketene dithioacetals of alkylidenemalononitrile 77 (Scheme 29).33
NC
CN
SCH3
SCH3 NH
OH3CS
CN(H3O)+
77 78
Scheme 29
-Oxoketene-N,S-acetals 79 react with malonyl chloride 80 to afford 4-hydroxy-
6-(methylsulfanyl)-2(1H)-pyridones 81 (Scheme 30) in good yields.34
R1
NHR2
O SCH 3Cl
Cl
O
O N
R1
O OH
OH3CS
R2
Et3N/C6H6
71-89%+
79 80 81
Scheme 30
A one-pot synthesis of 6-methylthio-N-aryl-2-pyridone 84 and its
deazapurine analogues by the reaction of ketene dithioacetal 82 with substituted
acetanilides 83, has been reported by Elgemee et al (Scheme 31).35
O
H3CS NH
OO SCH 3
SCH 3
N
O
+MeOH
82 83 84
Scheme 31
Synthesis of 2-pyridones 87 can also be carried out using ketene-N,N-
aminals 85 on heating with methyl propiolate 86 in methanol (Scheme 32).36
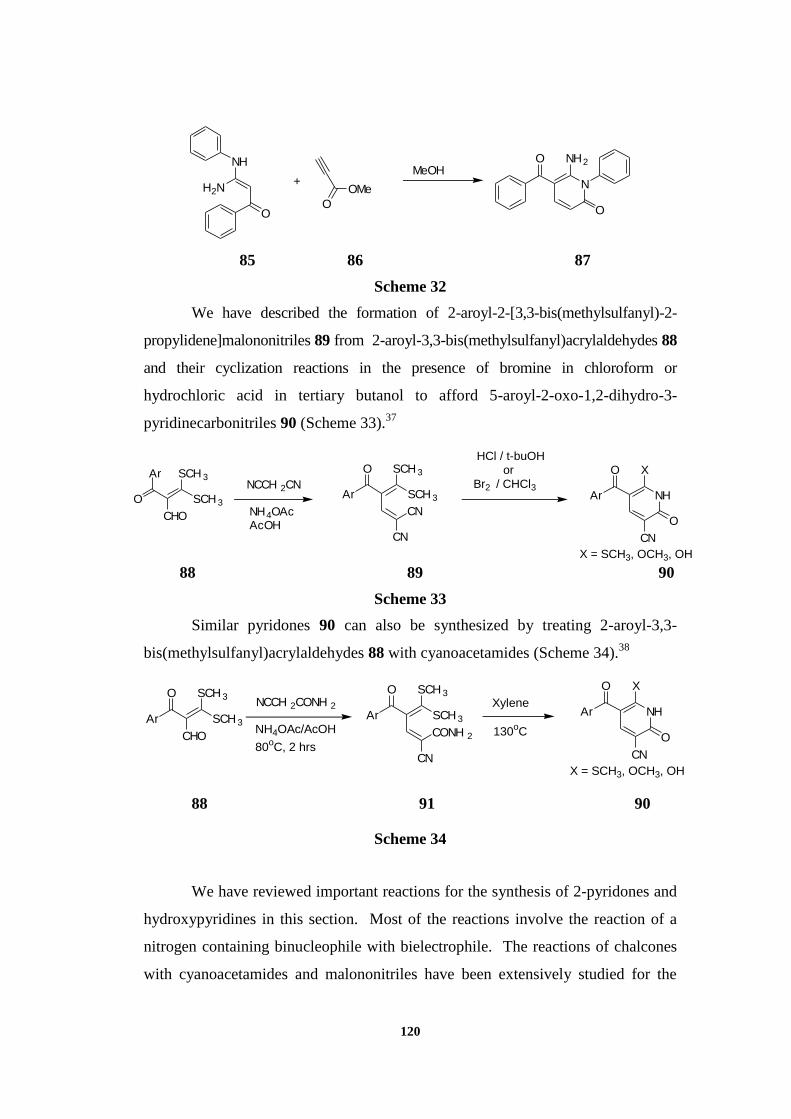
120
O
H2N OMe
O
O NH2NH
N
O
+MeOH
85 86 87
Scheme 32
We have described the formation of 2-aroyl-2-[3,3-bis(methylsulfanyl)-2-
propylidene]malononitriles 89 from 2-aroyl-3,3-bis(methylsulfanyl)acrylaldehydes 88
and their cyclization reactions in the presence of bromine in chloroform or
hydrochloric acid in tertiary butanol to afford 5-aroyl-2-oxo-1,2-dihydro-3-
pyridinecarbonitriles 90 (Scheme 33).37
Ar
SCH 3O
SCH 3
CHO
Ar SCH 3
O SCH 3
CN
CN
Ar NH
O X
CN
O
X = SCH3, OCH3, OH
NCCH 2CN
NH4OAcAcOH
HCl / t-buOH
or
Br2 / CHCl3
88 89 90
Scheme 33
Similar pyridones 90 can also be synthesized by treating 2-aroyl-3,3-
bis(methylsulfanyl)acrylaldehydes 88 with cyanoacetamides (Scheme 34).38
Ar SCH 3
O SCH 3
CHO
Ar SCH 3
O SCH 3
CONH 2
CN
Ar NH
O X
CN
O
X = SCH3, OCH3, OH
NCCH 2CONH 2
NH4OAc/AcOH
80oC, 2 hrs
Xylene
130oC
88 91 90
Scheme 34
We have reviewed important reactions for the synthesis of 2-pyridones and
hydroxypyridines in this section. Most of the reactions involve the reaction of a
nitrogen containing binucleophile with bielectrophile. The reactions of chalcones
with cyanoacetamides and malononitriles have been extensively studied for the

121
preparation of 2-pyridones.39
Still the synthesis of -chloroenones and their further
transformation to 2-oxo/2-hydroxy-5,6-bisarylnicotinonitriles deserve much
attention, as the main challenge to synthetic chemist is the synthesis of molecules
in good yields with appropriate functional moieties at suitable position of the
carbon skeleton.
5.3. Results and discussions
5.3.1. Reactions of 3-chloro-1,2-diaryl-2-propen-1-ones with
malononitrile: synthesis of 2-hydroxy-5,6-diarylnicotinonitriles
Initially we investigated the reaction of 3-chloro-1,2-diphenyl-2-propen-1-
one 92a with malononitrile in ammonium acetate/acetic acid buffer. The
optimization studies are concluded in Table 1. All the reactions were monitored by
TLC, by a 7 GF silica gel stationary phase, for every half an hour.
Table 1 Optimization studies for the synthesis of 2-hydroxy-5,6-
diphenylnicotinonitrile 93a
Sl No Temperature-Time Yield %
1 room temperature-10hrs 0
2 40C-10hrs 0
3 60C-10hrs 0
4 70C-15hrs 22
5 72C-15hrs 35
6 74C-15hrs 50
7 76C-12hrs 65
8 78C-8hrs 85
6 80C-8hrs 84
7 90 C-3h Complex reaction
mixture
----------------------------------------------------------------------------------------------------

122
From the table it is clear that 3-chloro-1,2-diphenyl-2-propen-1-one 92a is
unreactive with malononitrile at room temperature or at a temperature below 70C.
The 2-hydroxy-5,6-diphenylnicotinonitrile 93a, having melting point 182-184C,
was obtained in 85 % yield when the reaction was conducted at 78 C for 8h
(Scheme 35).
O
ClH
NC CN
NH4OAc / CH3COOH
N OH
CN
HN
CN
O
78 oC-8h
26
92a 93a
Scheme 35
2-Hydroxy-5,6-diphenylnicotinonitrile 93a was characterized on the basis
of conventional spectroscopic methods and Single crystal X-ray analysis. In the IR
spectrum, the compound 93a showed characteristic OH and NH stretching at 3463
cm-1
, 3433 cm-1
and 3352 cm-1
due to the pyridine-pyridone equilibrium. Aromatic
C-H stretching was observed at 3178 cm-1
. Characteristic cyano peak observed at
2216 cm-1
and the carbonyl stretching at 1645 cm-1
. The C=N and C-N stretching
was observed at 1591 cm-1
and 1463 cm-1
. The C=C stretching was observed at
1627, 1591, 1544 cm-1
of both aromatic and pyridine rings respectively. The
GCMS of the compound showed the molecular ion peak at m/z 272 and the base
peaks at m/z 270 and 271 respectively. The 1H NMR spectrum of the compound
showed two aromatic proton singlets at 7.75 and 7.65 respectively
corresponding to the pyridinyl protons of the two tautomers. Two multiplets were
appeared at 6.92-6.97 and 7.05-7.1 in which each peak corresponds to two
hydrogens atoms in the phenyl rings. Other aromatic protons appeared are 7.11-
7.19 as a 5 hydrogen multiplet and 7.21-7.34 as an 11 hydrogen multiplet
respectively. The hydroxyl and amino protons found to be merged at 5.25 as a
two proton broad signal. The 13
C NMR spectrum of the compound has shown a
carbonyl carbon peak at 160.37 due to the pyridone ring carbonyl carbon and the
two cyano group signals at 115.79 and 115.49 due to the hydroxypyridine-pyridone
equilibrium. The other aromatic and heterocyclic carbons were appeared at

123
157.71, 150.77, 149.38, 143.52, 138.87, 138.3, 138.03, 136.41, 131.94, 129.64,
129.55, 129.43, 128.84, 128.77, 128.42, 128.31, 128.15, 127.95, 127.22, 126.84,
116.61, 99.11, 96.41, 89.87, 77.32, 77, 76.68 and 74.15 ppm. The CHN analysis
of the compound 93a gave the percentage of various elements presented in the
molecule as Carbon = 79.385, Hydrogen = 4.33, Nitrogen = 10.295, which are
found to be agreeing with the calculated data as Carbon-79.39, Hydrogen-4.44,
Nitrogen-10.29 and confirmed the molecular formula as C18H12N2O. Literature
review supports the above spectral data as 2-pyridones usually exist in
2-pyridone/2-hydroxy pyridine equilibrium.40
Figure 1 IR spectrum of 2-hydroxy-5,6-diphenylnicotinonitrile 93a

124
Figure 2 GCMS spectrum of 2-hydroxy-5,6-diphenylnicotinonitrile 93a
Figure 3 1HNMR spectrum of spectrum of 2-hydroxy-5,6-diphenylnicotinonitrile
93a

125
Figure 4 13
C NMR spectrum of 2-hydroxy-5,6-diphenylnicotinonitrile 93a
The proposed mechanism of the reaction is as follows: Initially the
-chloroenone underwent Knoevenagel reaction with malononitrile in the presence
of ammonium acetate/acetic acid mixture to get an adduct 94. The
alkylidenemalononitrile formed then, underwent an intramolecular addition-
elimination reaction followed by hydroxylation to get 2-hydroxypyridine. Due to
the comparable stability of the 2-pyridone ring systems in its crystal lattice,
2-hydroxypyridine exists as 2-hydroxypyridine/2-pyridone tautomeric mixture
(Scheme 36).
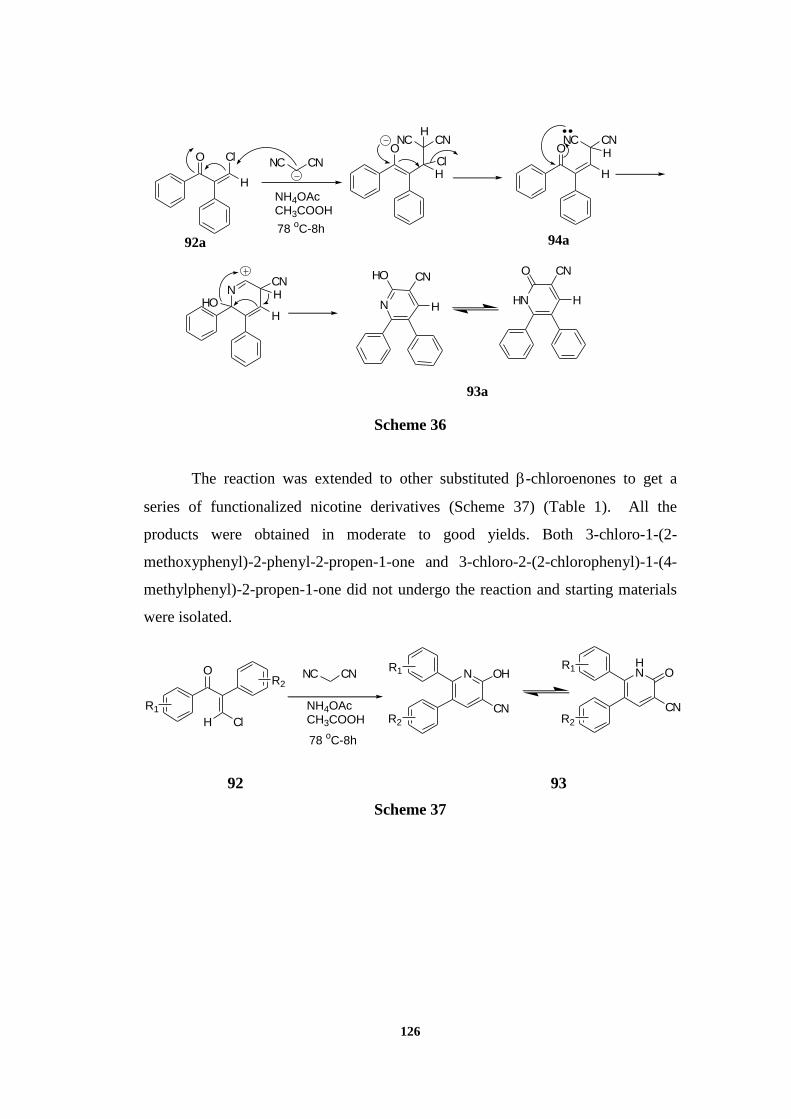
126
O Cl
H
NC CN
NH4OAc CH3COOH
HN
CNO
78 oC-8h
H
ONC CN
ClH
ONC CN
H
NCN
HOH
H
H
HN
CNHO
H
92a 94a
93a
Scheme 36
The reaction was extended to other substituted -chloroenones to get a
series of functionalized nicotine derivatives (Scheme 37) (Table 1). All the
products were obtained in moderate to good yields. Both 3-chloro-1-(2-
methoxyphenyl)-2-phenyl-2-propen-1-one and 3-chloro-2-(2-chlorophenyl)-1-(4-
methylphenyl)-2-propen-1-one did not undergo the reaction and starting materials
were isolated.
O
ClH
NC CN
NH4OAcCH3COOH
N OH
CN
HN
CN
O
78 oC-8h
R1
R2
R1
R2
R1
R2
92 93
Scheme 37

127
Table 1 Synthesis of 5,6-diaryl-2-hydroxynicotinonitriles 93a-k
93 R1 R2 Yields %
a H H 85
b Br H 75
c 4-OCH3 H 82
d 4-Cl H 88
e 4-CH3 H 78
f 4-Cl 2-Cl 82
g H 2-Cl 77
h 4-CH3 4-OCH3 71
i 4-CH3 2-Cl 0
j 2-OCH3 H 0
k H 4-OCH3 90
5.3.2 Crystal structure and structure refinement of 2-hydroxy-5-(4-
methoxyphenyl)-6-phenylnicotinonitrile 93k
The structure refinement on 93k showed that the compound exits as 2-
hydroxy-5-(4-methoxyphenyl)-6-phenylnicotinonitrile (molecular formula: C19H12N2O)
in its crystal lattice (Figure 5, 6 &7). The crystal parameters are as follows: Space
group, P 2 1/c; a, 12.0731 (14); b, 14.1000 (17); c, 9.8157 (12); α, 90.00; β,
108.797 (7); γ, 90.00; V, 1581.82, Z, Z’= Z: 4, Z’: 0, R-Factor [%] = 5.91 and
CCDC 791271. Two neighboring molecules are bound together with intermolecular
hydrogen bonding (N-H-O) between adjacent hydroxyl pyridine moieties (Figure
7). Another important hydrogen bonding was observed between the nitrogen atom
of the cyano group and the hydrogen atom presented at the para position of the
nicotinonitrile. The molecule is arranged in the three dimensional crystal lattice
due to the aromatic stacking interactions and weak polar interactions between
methoxy groups and aromatic stacking interactions (Figure 5, 6 & 7). Various

128
bond lengths obtained by the analysis according to the atoms shown in Figure 5 is
displayed in Table 2 and the bond lengths obtained are in mutual agreement with
earlier reported values.
Figure 5 Ortep diagram of 2-hydroxy-5-(4-methoxyphenyl)-6-
phenylnicotinonitrile 93k with atom label
Figure 6 Ortep diagram of 2-hydroxy-5-(4-methoxyphenyl)-6-
phenylnicotinonitrile 93k

129
Figure 7 Three dimensional arrays of 2-hydroxy-5-(4-methoxyphenyl)-6-
phenylnicotinonitrile in its crystal lattice 93k
Table 2: Bond lengths of various bonds presents in 2-hydroxy-5-(4-
methoxyphenyl)-6-phenylnicotinonitrile 93k obtained from Single
Crystal Analysis
No. Atom 1 Atom 2 Bond length (Ao unit)
1. N2 C13 1.351
2. N2 C12 1.344
3. C8 C13 1.399
4. C8 C9 1.394
5. C8 C5 1.489
6. C10 C12 1.414
7. C10 C9 1.389
8. C10 C11 1.425
9. C13 C14 1.482
10. C12 O2 1.375
11. C9 H9 0.930
12. C14 C15 1.388
13. C14 C19 1.397
14. C5 C6 1.392

130
15. C5 C4 1.389
16. C2 C7 1.389
17. C2 C3 1.368
18. C2 O1 1.367
19. C7 H7 0.930
20. C7 C6 1.373
21. C11 N1 1.154
22. C15 H15 0.930
23. C15 C16 1.381
24. C6 H6 0.930
25. C3 H3 0.930
26. C3 C4 1.395
27. C19 H19 0.931
28. C19 C18 1.386
29. C18 H18 0.930
30. C18 C17 1.368
31. C4 H4 0.929
32. C16 H16 0.930
33. C16 C17 1.357
34. C17 H17 0.930
35. C1 H1a 0.961
36. C1 H1c 0.960
37. C1 H1b 0.960
38. C1 O1 1.414
39. O2 H20 0.820
5.3.3. Reactions of 3-chloro-1,2-diaryl-2-propen-1-ones with
cyanoacetamide
As a continuation to earlier studies in our laboratory, 3-chloro-2,3-
diarylpropan-1-ones were treated with cyanoacetamide in the presence of
ammonium acetate/acetic acid mixture at 78C. The reaction afforded a complex

131
mixture of products and found difficulty in exploring the reaction for the synthesis
of pyridine derivatives.
5.4 Conclusion
The synthesis of functionalized 2-hydroxy-5,6-diarylnicotinonitriles from
3-chloro-1,2-diaryl-2-propen-1-ones is found to be very efficient and it can be
considered as a general method for the synthesis of pyridine derivatives.
5.5. Experimental
Melting points were determined on a Buchi 530 melting point apparatus
and were uncorrected. The IR spectra were recorded by KBr pellet method on a
Shimadzu FTIR 470 spectrometer and the frequencies are reported in cm-1
. Mass
spectra were recorded on a GCMS Shimadzu 5050 model instrument. 1H NMR
spectra were recorded on a Bruker EM 400 MHz spectrometer using CDCl3 as the
solvent. 13
C NMR spectra were recorded on a Bruker EM 400 MHz spectrometer
using CDCl3 as the solvent. Both 13
C NMR and
1H NMR values are expressed in δ
(ppm). Elemental analyses were done on a Shimadzu CHN analyzer.
All reagents were commercially available and were purified before use. All
the solvents used were dried and distilled under reduced pressure. 3-Chloro-1,2-
diaryl-2-propen-1-ones were prepared according to the procedure explained in
Chapter 3 of this thesis. All the purified compounds gave single spot upon TLC
analyses on silica gel 7GF using ethyl acetate/hexane as eluent. Iodine vapors or
KMnO4 in water was used as the developing agent for TLC.
5.5.1. General procedure for the synthesis of 5,6-diaryl-2-hydroxy-
nicotinonitriles
In a 250 ml R. B. flask fitted with a guard tube and a bar magnet inside, a
mixture of malononitrile (0.950 g, 14.4 mmol), ammonium acetate (2.96 g, 38.4
mmol) and acetic acid (9.6 ml) were taken and refluxed with stirring for 10 min at 70
0C. To this 3-chloro-1,2-diaryl-2-propen-1-one (9.6 mmol) was added and stirring
was continued for 8 hrs. The reaction was monitored by TLC and after complete
consumption of the starting material; it was allowed to cool up to room temperature
and poured into ice cold water. Extracted with chloroform (25 x 3), washed with

132
water (25 x 3), dried over anhydrous sodium sulfate and the chloroform layer was
evaporated to afford the crude product. The crude product was subjected to column
chromatography over 60-120 mesh silica gel using ethyl acetate : hexane (1:9) as
eluent and the products were isolated in pure and better yields.
N OH
CN
C18H12N2O
Mol. Wt.: 272.30
2-Hydroxy-5,6-diphenylnicotinonitrile 93a was obtained
by the reaction of 3-chloro-1,2-diphenyl-2-propen-1-one 92a
(2.32 g, 9.6 mmol) with malononitrile (0.950 g, 14.4 mmol)
in the presence of ammonium acetate (2.96 g, 38.4 mmol)
and acetic acid (9.6ml) at 780C as a pale yellow powder; mp,
182-184C; yield, 2.22 g (85 %); IR (KBr, ν max) = 3463
(OH), 3284 (NH), 2216 (CN), 1645 (CO), 1627 (C=C) cm-1
;
GCMS (m/z) = 272 (M)+, 271 (base peak), 270, 253, 226,
135.2, 140, 121.5, 94.5, 77; 1H NMR (400 MHz, CDCl3)
δ = 5.25 (2H, broad, NH and OH), 6.92-6.97 (2H, m, ArH),
7.05-7.1 (2H, M, ArH), 7.11-7.19 (5H, m, ArH), 7.21-7.34
(11H, m, ArH), 7.65 (1H, s, pyridine ring hydrogen), 7.75
(1H, s, pyridine ring hydrogen); 13
C NMR (400 MHz,
CDCl3) δ = 160.37 (CO), 157.71, 150.77, 149.38, 143.52,
138.87, 138.03, 136.41, 131.94, 129.64, 129.55, 129.43,
128.84, 128.77, 128.42, 128.31, 128.15, 127.95, 127.22,
126.84, 116.61, 115.79 (CN), 115.49 (CN), 99.11, 96.41,
89.87; Anal Calcd. for C18H12N2O: Carbon-79.39;
Hydrogen-4.44; Nitrogen-10.29. Found: Carbon = 79.385;
Hydrogen = 4.33; Nitrogen = 10.295.
N OH
CN
Br
C18H11BrN2O
Mol. Wt.: 351.20
6-(4-Bromophenyl)-2-hydroxy-5-phenylnicotinonitrile
93b was obtained by the reaction between 1-(4-
bromophenyl)-3-chloro-2-phenyl-2-propen-1-one 92b (3.08
g, 9.6 mmol) with 1.5 equivalent malononitrile (0.950 g, 14.4
mmol) in ammonium acetate (2.96 g, 38.4 mmol) and acetic

133
acid (9.6ml) mixture at 78C as a pale yellow powder,
melting point = 204-206C. Yield = 2.53 g (75 %); IR
(KBr, ν max) = 3402 (OH), 3342 (NH), 3150 (aromatic C-
H), 3050 (aromatic C-H), 2221 (CN), 1656 (C=O), 1587
(C=N), 1544 (C=N), 1469 (C=C), 1390 (C-C) cm-1
; GCMS
(m/z) = 353(M+2)+, 352 (M+2)
+, 351 (M+1)
+, 350 (M)
+,
288, 286, 270 (base peak), 255, 242, 227, 214, 184, 156, 140,
75; 1H NMR (400 MHz, CDCl3) δ = 7.739 (1H, s, pyridine
ring hydrogen), 7.64 (1H, s, pyridine ring hydrogen), 7.43
(4H, d, J = 10 Hz, ArH), 7.16-7.23 (5H, m, ArH), 7.025 (4H,
d, J = 10 Hz, ArH), 6.91-6.97 (3H, m, ArH), 5.83 (2H,
Broad, NH and OH); 13
C NMR (400 MHz, CDCl3) δ = 207,
157.67, 150.85, 147.9, 143.72, 138.24, 137.94, 137.55,
135.26, 131.71, 131.62, 131.33, 131.18, 129.39, 129.31,
128.62, 128.35, 127.43, 126.69, 123.37, 116.44, 115.63
(CN), 115.35 (CN), 98.68, 96.7, 90.14.
N OH
CN
MeO
C19H14N2O2
Mol. Wt.: 302.33
2-Hydroxy-6-(4-methoxyphenyl)-5-phenylnicotinonitrile
93c was obtained by the reaction between 3-chloro-1-(4-
methoxyphenyl)-2-phenyl-2-propen-1-one 92c (2.61 g, 9.6
mmol) with 1.5 equivalent malononitrile (0.950 g, 14.4
mmol) in ammonium acetate (2.96 g, 38.4 mmol) and acetic
acid (9.6ml) mixture at 780C as a pale yellow powder,
melting point = 178-1800C; Yield = 2.38 g (82 %); IR
(KBr, ν max) = 3413 (OH), 3348 (NH), 3249 (aromatic C-
H), 3060 (aromatic C-H), 3028 (aromatic C-H), 3002
(aromatic C-H), 2217 (CN), 1660 (C=O), 1602 (C=N), 1512,
1492, 1452, 1425 cm-1
; GCMS (m/z) = 325 (M+Na+, base
peak)+, 309, 294, 281, 265, 227, 155, 141, 127, 114, 100;
1H
NMR (400 MHz, CDCl3) δ = 8.01 (1H, s, pyridine ring
hydrogen), 7.63 (1H, s, pyridine ring hydrogen), 7.194 (2H,
t, ArH), 7.069 (2H, dd, ArH), 6.954 (8H, d, j = 8.8 Hz, ArH),

134
6.81 (2H, dd, ArH), 3.885 (3H, 3, OMe), 3.793 (3H, s,
OMe); 13
C NMR (400 MHz, CDCl3) δ = 171.5, 164, 132.36,
131.05, 129.45, 128.24, 121.6, 113,78 (CN), 113.74 (CN),
55.49.
N OH
CN
Cl
C18H11ClN2O
Mol. Wt.: 306.75
6-(4-Chlorophenyl)-2-hydroxy-5-phenylnicotinonitrile
93d was obtained by the reaction between 3-chloro-1-(4-
chlorophenyl)-2-phenyl-2-propen-1-one 92d (2.65 g, 9.6
mmol) with 1.5 equivalent malononitrile (0.950 g, 14.4
mmol) in ammonium acetate (2.96 g, 38.4 mmol) and acetic
acid (9.6ml) mixture at 780C as a pale yellow powder,
melting point = 200-2020C. Yield = 2.59 g (88 %); IR
(KBr, ν max) = 3407(OH), 3342(NH), 3232 (aromatic C-H),
2219 (CN), 1649 (C=O), 1591 (C=C) cm-1
; GCMS (m/z) =
308 (m+2), 307 (m+1), 306 (m+), 305 (base peak)), 304,
269, 253, 252, 226, 214, 134, 121, 107; 1H NMR (400 MHz,
CDCl3) δ = 7.71 (1H, s, pyridine ring hydrogen), 7.65 (1H, s,
pyridine ring hydrogen), 7.25-7.29 (4H, m, ArH), 7.17-7.22
(6H, m, ArH), 7.05-7.11 (4H, m, ArH), 6.91-6.96 (3H, m,
ArH), 5.3 (2H, broad, OH and NH); 13
C NMR (400 MHz,
CDCl3) δ = 158.88, 157.66, 150.82, 147.94, 143.71, 138.22,
137.99, 137.59, 137.24, 135.11, 134.97, 134.79, 131.84,
131.09, 130.95, 129.4, 129.33, 128.85, 128.7, 128.63,
128.36, 128.2, 127.44, 126.76, 116.46, 115.64 (CN), 115.36
(CN), 98.81, 96.72, 90.14.
N OH
CN
H3C
C19H14N2O
Mol. Wt.: 286.33
2-Hydroxy-6-(4-methylphenyl)-5-phenylnicotinonitrile
93e was obtained by the reaction between 3-chloro-1-(4-
methylphenyl)-2-phenyl-2-propen-1-one 92e (2.46 g, 9.6
mmol) with 1.5 equivalent malononitrile (0.950 g, 14.4
mmol) in ammonium acetate (2.96 g, 38.4 mmol) and acetic
acid (9.6ml) mixture at 780C as a pale yellow powder,

135
melting point = 178-1800C. Yield = 2.14 g (78 %); IR
(KBr, ν max) = 3456 (OH), 3402 (NH), 3350 (NH), 3340
(OH), 3249 (aromatic C-H), 3056 (aromatic C-H), 3029
(aromatic C-H), 2223(CN), 1656 (CO), 1649 (C=N), 1589
(C=C), 1546 (C=C) cm-1
; GCMS (m/z) = 286 (M)+, 285, 284
(base peak), 269, 252, 240, 227, 207, 141, 128, 101; 1H
NMR (400 MHz, CDCl3) δ = 7.9 (4H, d, j = 10Hz, ArH),
7.72 (1H, s, pyridine ring hydrogen), 7.63 (1H, s, pyridine
ring hydrogen), 7.26-7.15 (5H, m, ArH), 7.02 (4H, d, j =
10Hz, ArH), 6.93-6.97 (3H, m, ArH), 5.24 (4H, broad, OH
and NH), 2.3185 (6H, s, CH3); 13
C NMR (400 MHz, CDCl3)
δ = 160, 157, 150.8, 149.55, 143.49, 138.83, 138.11, 138.02,
133.41, 131.91, 129.61, 129.44, 129.04, 128.67, 128.43,
128.15, 127.14, 115.88 (CN), 115.7 (CN), 99.14, 96.12,
89.1, 21.3.
N OH
CN
Cl
Cl
C18H10Cl2N2O
Mol. Wt.: 341.19
5-(2-Chlorophenyl)-6-(4-chlorophenyl)-2-
hydroxynicotinonitrile 93f was obtained by the reaction
between 3-chloro-2-(2-chlorophenyl)-1-(4-chlorophenyl)-2-
propen-1-one 92f (2.98 g, 9.6 mmol) with 1.5 equivalent
malononitrile (0.950 g, 14.4 mmol) in ammonium acetate
(2.96 g, 38.4 mmol) and acetic acid (9.6ml) mixture at 780C
as a pale yellow powder, melting point = 178-1800C. Yield
= 2.68 g (82 %); IR (KBr, ν max) = 3417(OH), 3342(NH),
3245 (aromatic C-H), 3184 (aromatic C-H), 2221 (CN), 1649
(C=O) cm-1
; GCMS (m/z) = 344(M+4)+, 343 (M+3)
+, 342
(M+2)+, 341 (M+1)
+, 340 (M)
+, 339 (base peak), 316, 325,
324, 306, 249, 230, 168, 112, 76; 1H NMR (400 Mhz,
CDCl3) δ = 7.34-7.28 (2H, m, ArH), 7.27-7.21 (3H, m, ArH),
7.21-7.16 (2H, m, ArH), 7.14-7.07 (5H, m, ArH), 6.95-6.91
(2H, m, ArH), 5.35 (2H, broad, OH and NH); 13
C NMR
(400 Mhz, CDCl3) δ = 162.1, 161.8, 152.09, 145.67, 140.66,

136
140.06, 139.5, 135.87, 134.76, 133.21, 132.55, 132.45, 132,
130.4, 130.04, 129.7, 128.99, 128.76, 128.53, 128.23, 125.4,
119.05, 116.07, 114.3 (CN), 114.8 (CN), 102.9, 96.78.
N OH
CN
Cl
C18H11ClN2O
Mol. Wt.: 306.75
5-(2-Chlorophenyl)-2-hydroxy-6-phenylnicotinonitrile
93g was obtained by the reaction between 3-chloro-2-(2-
chlorophenyl)-1-phenyl-2-propen-1-one 92g (2.65 g, 9.6
mmol) with 1.5 equivalent malononitrile (0.950 g, 14.4
mmol) in ammonium acetate (2.96 g, 38.4 mmol) and acetic
acid (9.6ml) mixture at 780C as a pale yellow powder,
melting point = 132-1340C. Yield = 2.26 g (77 %); IR (KBr,
ν max) = 3417(OH), 3340(NH), 3244 (aromatic C-H), 3056
(aromatic C-H), 2219 (CN), 1650 (C=O) cm-1
; GCMS (m/z)
= 308 (M+2)+, 307 (M+1)
+, 306 (M)
+, 305, 304, 281, 270,
253, 242, 226, 207, 191, 174, 147, 134, 121, 107, 94, 83
(base peak); 1H NMR (400 MHz, CDCl3) δ = 7.55 (1H, s,
pyridine ring hydrogen), 7.27-7.31 (2H, m, ArH), 7.13-7.18
(2H, m, ArH), 7.02-7.09 (10H, m, ArH), 6.9-6.94 (2H, m,
ArH), 5.3461 (2H, broad, OH and NH); 13
C NMR (400
MHz, CDCl3) δ = 151.31, 150.37, 138.84, 138.45, 136.82,
133.49, 133, 132.04, 129.42, 129.1, 128.96, 128.78, 126.41,
115.74 (CN), 115.56 (CN), 98.7, 95.69.
N OH
CN
MeO
H3C
C20H16N2O2
Mol. Wt.: 316.35
2-Hydroxy-5-(4-methoxyphenyl)-6-(4-
methylphenyl)nicotinonitrile 93h was obtained by the
reaction between 3-chloro-2-(4-methoxyphenyl)-1-(4-
methylphenyl)-2-propen-1-one 92h (2.75 g, 9.6 mmol) with
1.5 equivalent malononitrile (0.950 g, 14.4 mmol) in
ammonium acetate (2.96 g, 38.4 mmol) and acetic acid
(9.6ml) mixture at 780C as a deep yellow powder, melting
point = 188-1900C. Yield = 2.15 g (71 %); IR (KBr, ν max)
= 3481(OH), 3363(NH), 3234, 3014 (aromatic C-H), 2217

137
(CN), 1631 (C=O) cm-1
; GCMS (m/z) = 316 (M)+, 315 (base
peak), 314, 300, 285, 271, 257, 256, 239, 227, 207, 191, 170,
150, 135, 128, 101, 88; 1H NMR (400 MHz, CDCl3) δ = 7.6
(1H, s, pyridine ring hydrogen), 7.11 (4H, d, j = 10 Hz,
ArH), 7.03 (4H, d, j = 10 Hz, ArH), 6.86 (4H, d, j = 10 Hz,
ArH), 6.7 (4H, d, j = 10Hz, ArH), 5.22 (2H, broad, OH and
NH), 3.75 (6H, s, OMe), 2.33 (6H, s, CH3); 13
C NMR (400
MHz, CDCl3) δ = 170.47, 163.93, 155.74, 150.55, 149.43,
138.71, 137.94, 133.58, 132.28, 131.61, 130.51, 130.41,
130.19, 129.41, 129.23, 129.08, 127.89, 126.52, 122.75,
121.61, 117.52, 115.95, 115.75, 114.82 (CN), 113.71 (CN),
113.59, 99.14, 96.10.
N OH
CN
MeO
C19H14N2O2
Mol. Wt.: 302.33
2-Hydroxy-5-(4-methoxyphenyl)-6-phenylnicotinonitrile
93k was obtained by the reaction between 3-chloro-2-(4-
methoxyphenyl)-1-phenyl-2-propen-1-one 92k (2.61 g, 9.6
mmol) with 1.5 equivalent malononitrile (0.950 g, 14.4
mmol) in ammonium acetate (2.96g, 38.4 mmol) and acetic
acid (9.6ml) mixture at 780C as a pale yellow powder,
melting point = 204-2060C; Yield = 2.61 g (90 %); IR
(KBr, ν max) = 3479 (OH), 3296 (NH), 3186 (aromatic C-
H), 2212 (CN), 1620 (CO) cm-1
; GCMS (m/z) = 302 (m+),
301, 300, 287, 286, 285, 270, 214, 207, 143, 128, 89, 76; 1H
NMR (400 MHz, CDCl3) δ = 3.74 (3H, s, OMe), 3.79 (3H, s,
OMe), 5.2 (2H, broad, NH and OH), 6.7 (1H, s, dd, ArH),
6.78 (2H, m, ArH), 6.86 (1H, dd, ArH), 6.98 (2H, m, ArH),
7.14 (1H, dd, ArH), 7.1-7.34 (6H, m, ArH), 7.63 (1H, s,
pyridine ring hydrogen), 7.72 (1H, s, pyridine ring
hydrogen); 13
C NMR (400 MHz, CDCl3) δ = 160.23 (CO),
158.79, 157.52, 150.54, 149.23, 144.25, 143.38, 142.04,
139.04, 137.97, 136.6, 131.58, 130.51, 130.45, 130.22,
129.57, 129.51, 128.74, 128.65, 128.33, 127.96, 126.46,

138
116.67, 115.86, 115.54, 113.88 (CN), 113.6 (CN), 104.24,
99.08, 96.37, 89.84, 77.32, 77, 76.68, 67.57, 61.22, 55.19,
43.44, 39.31, 30.87; Single Crystal Data : CCDC = 791271,
molecular formula = C19H12N2O, space group = P 2 1/c, Cell
lengths = a 12.0731(14) b 14.1000(17) c 9.8157(12), Cell
angles = α90.00 β108.797(7) γ90.00, cell volume = 1581.82,
Z, Z’ = Z : 4, Z’ : 0, R-Factor[%] = 5.91.

139
5.6. References
1 Ian Collins, Christopher Moyes, William B. Davey, Michael Rowley,
Frances A. Bromidge, Kathleen Quirk, John R. Atack, Ruth M. McKernan,
Sally-Ann Thompson, Keith Wafford, Gerard R. Dawson, Andrew Pike,
Bindi Sohal, Nancy N. Tsou, Richard G. Ball; Jose L. Castro; J. Med.
Chem.; 2002, 45, 1887-1900.
2 Balasubramanian, M.; Keay, J. G.; Comprehensive Heterocyclic Chemistry
II, ed. Katritzky, A. R.; Rees, C. W.; Scriven,;E. F. V.; Pergamon Press,
Oxford, 1996, 5, 245.
3 Jones, G.; Comprehensive Heterocyclic Chemistry II, ed. Katritzky, A. R.;
Rees C. W.; Scriven, E. F. V.; Pergamon Press, Oxford, 1996, 5, 167.
4 Fossa, P.; Menozzi, G.; Dorigo, P.; Floreani, M.; Mosti, L.; Bio Organic
and Medicinal Chemistry, 2003, 11, 4749.
5. Torres, M.; Gil, S.; Parra, M., Curr. Org. Chem., 2005, 17, 1757.
6. Mertel, H. E., “Pyridine and its derivatives”, part 2, Klingsberg, E. Ed.,
Interscience, New York., 1961, 326.
7. (a) Vieweg, H.; Wagner, G., Pharmazie, 1991, 46, 51; (b) Vieweg, H.;
Leistner, S., Pharmazie, 1986, 41, 827.
8. Yu, A. S.; Shestopalov, A. M.; Rodinovskaya, L. A.; Promonenkov, V. K.;
Litvinov, V. P., Zh. Org. Khim, 1984, 20, 2442.
9. (a) Vieweg, H.; Wagner, G., Pharmazie, 1991, 46, 51; (b) Vieweg, H.;
Leistner, S., Pharmazie, 1986, 41, 827.
10. (a) Chase, B. H.; Walker, J., J. Chem. Soc., 1953, 3518; (b) Chorvat, R. J.;
Radak, S. E., Tetrahedron Lett., 1980, 21, 421.
11. Fossa, P.; Menozzi, G.; Dorigo, P.; Floreani, M.; Mosti, L., Bioorg. Med.
Chem., 2003, 11, 4749.
12. Carles, L.; Narkunan, K.; Penlou, S.; Rousset, L.; Bouchu, D.; Ciufolini, M.
A., J. Org. Chem., 2002, 67, 4304.

140
13. Al-Arab, M. M., Journal of Heterocyclic Chemistry, 1990, 27(3), 523-5.
14. Rong, L.; Li, X.; Wang, H.; Shi, D.; Tu, S. Chem. Lett., 2006, 35, 1314.
15. Rong, L.; Wang, H.; Shi, J.; Yang, F.; Yao, H.; Tu, S.; Shi, D. J. Het.
Chem., 2007, 44, 1505.
16. Carles, L.; Narkunan, K.; Penlou, S.; Rousset, L.; Bouchu, D.; Ciufolini, M.
A. J. Org. Chem., 2002, 67, 4304.
17. Keshk, E. M., Heteroatom Chem., 2004, 15, 85.
18 Pan, W.; Dong, D.; Zhang, J.; Wu, R.; Xiang, D.; Liu, Q.; Org. Lett., 2007,
9, 2421-2423.
19. Kohler, E. P.; Souther, B. L. J. Am. Chem. Soc., 1922, 44, 2903.
20. Marcoux, J. F.; Marcotte, F. A.; Wu, J.; Dormer, P. G.; Davies, I. W.; Huges,
D.; Reider, P. J., J. Org. Chem., 2001, 66, 4194.
21. Andreani, A.; Bonazzi, D.; Rambaldi, M., Bull. Chim. Farm, 1976, 115, 732.
22. Cherry, K.; Abarbri, M.; Parrain, J.; Duchene, A., Tetrahedron. Lett., 2003, 44,
5791.
23. Gorobets, N. Y.; Yousefi, B. H.; Belaj, F.; and Kappe, C. O., Tetrahedron,
2004, 60, 39, 8633.
24. Ravinder, M.; Sadhu, P. S.; and Rao, V. J., Tet Lett., 2009, 50, 29, 4229.
25. Boisse, T.; Rigo, B.; Millet R.; and Hénichart, J. P., Tetrahedron, 2007, 63,
42, 10511.
26. Takaoka, K.; Aoyama, T.; and Shioiri, T., Tet Lett., 1996, 37, 28, 4973.
27. Takaoka, K.; Aoyama, T.; and Shioiri, T., Tet Lett., 1996, 37, 28, 4977.
28. Rastogi, R. R.; Ila, H.; Junjappa, H., J. Chem. Soc. Chem. Commun., 1975,
645.
29. Rastogi, R, R.; Kumar, A.; Ila, H.; Junjappa, H., J. Chem. Soc. Perkin Trans.1,
1978, 549.

141
30. Bartroli, R.; Diaz, M.; Quinococes, J.; Peseke, K., Sobre Deriv. Cana Azucar,
1990, 22, 44, CA112, 118601.
31. (a) Elgemeie, G. H.; Elgahndour, A. H.; Elzanate, A. M.; Abd Elaziz, G. W.,
Synth. Commun., 2003, 33, 253; (b) Elgemeie,G. H.; Elzanate, A. M., Synth.
Commun., 2003, 33, 2087.
32. (a) Datta, A.; Ila, H.; Junjappa, H., J. Org. Chem., 1990, 55, 5589; (b)
Asokan, C.V.; Ila, H.; Junjappa, H., Tetrahedron, 1990, 46, 16, 5423.
33. Rastogi, R. R.; Kumar, A.; Ila, H.; Junjappa, H., J. Chem. Soc., Perkin Trans.
1, 1978, 554.
34. Chakrasali, R. T.; Ila, H.; Junjappa, H., Synthesis, 1988, 87.
35. Elgemeie, G. H.; Ali, H. A.; Elghandour, A. H.; Abd Elaziz, G. W.,
Phosphorus, sulfur and silicon, 2000, 164, 189.
36. Hartmut, S.; Cristina, A.; Jordi, B.; Andreas, H. G.; Rolf, G.; Martin, M.;
Holger, P., J. Org. Chem., 2005, 70, 9463.
37. Anabha, E.R.; Asokan, C. V.; Synthesis, 2006, 1, 151.
38. Annie Mathews; Anabha, E. R.; Sasikala, K. A.; Lethesh, K. C.; Krishnaraj,
K. U.; Sreedevi, K. N.; Prasanth, M.;Devaky, K. S.; Asokan C. V.
Tetrahedron 2008, 64, 1671.
39 (a) Mont, N.; Teixido, J.; Borell, J. I.; Kappe, C. O.; Tetrahedron Letters,
2003, 44, 5385-5387; (b) Manna, F.; Chimenti, F.; Balasco, A.; Bizzarri,
B.; Filippelli, W.; Filippelli, A.; Gagliardi, L.; Eur. J. Med. Chem., 1999,
34, 245-254; (c) Ducker, J. W.; Gunter, M. J.; Aust. J. Chem., 1975, 28,
581-590.
40 (a) Ragaini, F.; Gallo, E.; Cenini, S.; Journal of Organometallic Chemistry,
2000, 593-594, 109-118; (b) Maris, A.; Ottaviani, P.; Caminati, W.;
Chemical Physics Letters, 2002, 360, 155-160; (c) Hong, S.; Li, Y.; Feng,
W.; Journal of Molecular Structure (Theochem), 2000, 530, 321-325;
(d) Ducker, J. W.; Gunter, M. J.; Aust. J. Chem., 1975, 28, 581-590.





























