Embed Size (px)
Citation preview

Research Collection
Doctoral Thesis
Reaktionskinetische Untersuchungen der Hydratisierung vonAcetylen zu Acetaldehyd an modifizierten Molekularsieben
Author(s): Aufdereggen, Klaus
Publication Date: 1968
Permanent Link: https://doi.org/10.3929/ethz-a-000093160
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

Diss. Nr. 4053
Reaktionskinetische Unter¬
suchungen der HydratisierungvonAcetylenzuAcetaldehyd anmodifizierten Molekularsieben
Abhandlungzur Erlangung der Würde eines
Doktors der technischen Wissenschaften
der
EIDGENÖSSISCHEN TECHNISCHEN HOCHSCHULE
ZÜRICH
vorgelegt von
Klaus Aufdereggendipl. Ing.-Chem. ETH
geboren am 2. April 1938
von Obergestein, Kanton Wallis
Angenommen auf Antrag von
Herrn Prof. Dr. A. Guyer, Referent
Herrn P.D. Dr. G. Gut, Korreferent
1968 Zürich
Offsetdruck P. Schmidberger

Meinen lieben Eltern und
meiner lieben Frau
gewidmet

Herrn Prof. Dr.A. Guyer, unter de s s en Leitung die
vorliegende Arbeit ausgeführt wurde, mochte ich fur sein
mir entgegengebrachtes Wohlwollen meinen herzlichsten
Dank aussprechen.
Besonders mochte ich auch Herrn PD Dr. G . Gut fur
seme wertvollen Anregungen und seine Hilfsbereitschaft,
die zum Gelingen dieser Arbeit beigetragen haben, herz¬
lich danken.
Aufrichtigen Dank schulde ich auch der Firma LONZA
AG, die die Durchfuhrung dieser Arbeit finanziell unter¬
stutzte.

INHALTSVERZEICHNIS
1. EINLEITUNG
2. LITERATURTEIL
2.1. Einfuhrung
2.2. Die Hydratisierung von Acetylen zu Acetaldehyd
2.2.1. Die Flussigphasenhydratisierung
2.2.1.1. Quecksilberkatalysatoren
2.2.1.2. Zink- und Cadmiumkatalysatoren
2.2.1.3. Kupfer- und Silberkatalysatoren
2.2.2. Die Gasphasenhydratisierung
2.3. Prinzipien der Katalysatorauswahl
2.3.1. Prinzipien der Katalysatorauswahl fur die Flussigphasenhydratisierung
2.3.2. Prinzipien der Katalysatorauswahl fur die Gasphasenhydratisierung
2.4. Reaktionsmechanismen und Kinetik
2.4.1. Reaktionsmechanismen und Kinetik der Flussigphasenhydratisierung
2.4.2. Reaktionsmechanismen und Kinetik der Gasphasenhydratisierung
3. EXPERIMENTELLER TEIL
3.1. Einfuhrung
3.2. Allgemeine Arbeltsgrundlagen
3.2.1. Apparatur
3.2.2. Versuchsschema
3.2.3. Analytik
3.2.4. Katalysatoren
3.2.4.1. Chemische Zusammensetzung und Textur des Molekularsiebs 13X
3.2.4.2. Herstellung von abgeänderten Molekularsieben 13X
3.3. Stofftransporteinflusse
3.3.1. Kornexterner Stofftransport
3.3.2. Porendiffusion

3.4. Katalysatorvergiftung
3.5. Kinetik
3.5.1. Zink-13X-Katalysatoren
3.5.2. Cadmium-13X-Katalysatoren
3.5.3. Quecksilber-13X-Katalysatoren
3.5.4. Kupfer-13X-Katalysatoren
3.5.5. Silber-13X-Katalysatoren
4. ZUSAMMENFASSUNG

1
1. EINLEITUNG
Acetaldehyd konnte 1881 von Kutscher off (1) erstmals aus Acetylen synthetisiert
werden. 1910 patentierte die Chemische Fabrik Griesheim (2) m Deutsch¬
land das Kutscheroff-Verfahren. 1916 wurde nach diesem Patent die Acetaldehydfabri-
kation in flussiger Phase mit Quecksilberkatalysatoren aufgenommen. In den folgenden
Jahren bis m die neueste Zeit versuchte man, dieses Verfahren zu verbessern, und das
sehr giftige Quecksilber durch andere geeignete Katalysatoren zu ersetzen. Wohl konn¬
ten im Lauf der Jahre technologische Fortschritte erzielt werden, im wesentlichen ist
das Verfahren aber gleich geblieben.
Von 1915 an gelang die Acetylenhydratisierung auch in der Gasphase (3). Diese Verfah¬
ren wurden aber nie zur Industriereife weiterentwickelt. Der Grund hiefur durfte dann
liegen, dass im Gegensatz zur Flussigphasenhydratisierung nur eine kleine Zahl grund¬
legende Arbeiten ausgeführt wurden, und dass die erzielten Raum-Zeit-Ausbeuten re¬
lativ gering waren.
Acetaldehyd ist ein chemisches Basisprodukt (4) und dient als Grundkorper zur Herstel¬
lung von Essigsaure, Butadien, n-Butanol, Buttersaure u.a.m.. Das grosse Interesse an
einer wirtschaftlichen Acetaldehydsynthese geht aus der umfangreichen Patentliteratur
hervor, sind doch bis auf den heutigen Tag mehr als 400 Patente veröffentlicht worden.
Obwohl Acetaldehyd auch durch Oxydation von Aethylalkohol (5), n-Butan (6) und in neu¬
ester Zeit von Aethylen (7-10) gewonnen wird, ist doch die Acetylenhydratisierung heu¬
te noch wichtig, weswegen diese Arbeit unternommen wurde.
Das Ziel der vorliegenden Arbeit bestand nun darin, die massgebenden Veröffentlichun¬
gen über die Entwicklung der Flussigphasen - wie auch der Gasphasenhydratisierung von
Acetylen zu Acetaldehyd zu besprechen, und die sich aus der Literatur ergebenden ge¬
nerellen Grundlagen abzuleiten, die fur eine Auswahl geeigneter Katalysatoren bedeu¬
tungsvoll sein können. Im weiteren sollen Arbeiten über Reaktionsmechanismen und
Kinetik miteinbezogen werden, um ein möglichst klares Bild über die Wirkungsweise
und Aktivität der Katalysatorsysteme zu erhalten.
Anschliessend soll der makrokinetische Ablauf der heterogenen Gasphasenhydratisie-
rung von Acetylen mit Wasserdampf in einem Differentialreaktor an den gebräuchli¬
chen, katalytisch aktiven Ionen zu Acetaldehyd durchgeführt werden. Basierend auf den

Versuchsergebnissen und den theoretischen Berechnungen soll alsdann das beobach¬
tete Verhalten durch einzelne Teilschritte zu erklaren versucht werden.

3
2. LITERATURTEIL
2.1. Einfuhrung
Im vorliegenden Abschnitt sollen die massgebenden Veröffentlichungen über die Ent¬
wicklung der Flussigphasen- wie auch der Gasphasenhydratisierung besprochen, und
die aus der Literatur sich ergebenden generellen Grundlagen abgeleitet werden, die
fur eine Auswahl geeigneter Katalysatoren bedeutungsvoll sind. Im weiteren sollen Ar¬
beiten über Reaktionsmechanismen und Kinetik miteinbezogen werden.
2.2. Die Hydratisierung von Acetylen zu Acetaldehyd
Es existiert heute eine sehr umfangreiche Literatur über die Hydratisierung von Ace¬
tylen zu Acetaldehyd. Da die wichtigsten Arbeiten bis ms Jahr 1950 im Beilstein (11),
Kirk - Othmer (12) und Ullmann (13) bzw. m zusammenfassenden Veröffentlichun¬
gen von Niewland et al. (14), Schwabe (15), Miocque et al. (16) und Kotlya-
revskn (17) beschrieben sind, soll auf diese Literatur nur insofern eingegangen wer¬
den, als sie fur das Verständnis des Reaktionsablaufes von Bedeutung ist.
Vorerst sollen die Katalysatorsysteme der Flussigphasenhydratisierung, wie auch der
Gasphasenhydratisierung zusammenfassend besprochen werden, um alsdann etwas na¬
her auf die in der Literatur bekannten optimalen Hydratisierungsbedingungen einzuge¬
hen.
2.2.1. Die Flussigphasenhydratisierung
Aus der Literatur geht hervor, dass vor allem Quecksilber-, Cadmium-, Zink-, Kupfer-
und Silbersalze die Wasseranlagerung an Acetylen katalysieren.
2.2.1.1. Quecksilberkatalysatoren
Wird Acetylen durch eine wasserige Quecksilber-(I)- oder Quecksilber-(II)-salzlosung
geleitet, bildet sich in geringen Mengen Acetaldehyd (11-37). Durch Zugabe von starken
organischen Sauren oder von Mineralsauren wird die katalytische Aktivität der Queck-
silbersalze wesentlich erhöht.
Als Aktivatoren gelangen vor allem Schwefelsaure (11-17, 25, 28, 31, 34-37), Salzsaure
(11-17), Phosphorsaure (11-17), Essigsaure (11-17, 31, 32) sowie organische Sulfonsau-

4
ren (11-17) zur Anwendung. Die gunstigsten Temperaturen liegen dabei zwischen 50
und 100°C (11-37).
Der gebildete Aldehyd muss rasch aus dem Reaktionsmedium entfernt werden, weil
er sonst zu Crotonaldehyd oder höheren Polymerisaten umgesetzt wird. Es wurde des¬
halb versucht, durch hohe Acetylendurchsatzgeschwindigkeiten (11, 12), Ansetzen von
Vakuum (11-17) oder durch Zugabe organischer Losungsmittel wie z.B. Benzol (27),
Alkohol (11-16), Phenol (11-16) oder Ketonen (11-18) den Acetaldehyd möglichst rasch
aus der Reaktionszone zu entfernen. Technische Anwendungen erlangten aber nur Ver¬
fahren mit hohen AcetylendurchSatzgeschwindigkeiten (Höchster Nassoxydationsver-
fahren) (11, 12, 13), sowie ein solches mit einer Kombination von Vakuum- und Druck¬
operationen (38) (Chisso Prozess).
Die Kontaktflussigkeit muss periodisch regeneriert werden, weil der Acetaldehyd mit
fortschreitender Reaktionsdauer die Quecksilber-(II)-ionen zu Quecksilber-(I)-ionen
und letztere bis zu metallischem Quecksilber reduziert. Als Regenerationsmittel wer¬
den Salpetersaure (11-16), Ozon (21), Chlor (11-16), reiner Sauerstoff (11-16) oder die
anodische Elektrolyse (11-16) vorgeschlagen. Um die Reduktionen zu verzögern, wur¬
den aber auch verschiedene Oxydationsmittel wie Eisen-(III)- (11-16, 19, 25, 28, 29,
30, 34, 35), Cer-(IV)- (11-16), Cer-(IH)- (19), Mangan-(IV)- (11-16, 19), Titan-(IV)-
(19), Blei-(IV)- (11-16), Vanadin-(IV)- (11-16) und Wolfram-(IV)-verbindungen (34) zur
Kontaktlosung zugegeben.
Acetylen kann auch mit Stickstoff (37), Wasserstoff, Methan und ungesättigten Kohlenwas¬
serstoffen (11-16, 21) verdünnt werden, ohne dass diese Zusätze den Reaktionsablauf stö¬
ren wurden.
Die technisch realisierten Verfahren arbeiten bei Normaldruck oder germgem Ueber-
druck. Es wurden zwar auch Versuche unternommen, den Prozess bei erhöhtem Druck
zu fahren, wobei man aber keine wesentlichen Vorteile erzielte (28, 33).
Die Apparatur muss gegen starke Sauren wie auch gegen Oxydationsmittel bestandig sein.
In der Literatur werden daher Blei, keramisches Material, Eisen-Siliciumlegierungen
und V^A-Stahl als Werkstoffe vorgeschlagen (11-16).
Die Anwendung von synthetischen Kationenaustauschern wie Dowex 50 (39, 43), KU-2
(sulfoniertes Polystyrolharz) (40, 42) und CS-1 (Phenol-Formaldehyd-Harz) (41), in de¬
nen die Wasserstoffionen teilweise oder total durch Quecksilber-(II)-ionen ersetzt wur-

5
den, zeigt keine grossere Stabilität und höhere Selektivität gegenüber dem Standard¬
katalysator (12). Die gunstigsten Temperaturen liegen auch hier zwischen 50 und 100 C
(44).
Aus der Literatur (11-44) lassen sich als optimale Hydratisierungsbedingungen die
nachfolgenden ableiten : Kontaktflussigkeit, bestehend aus 0,1-2 Gew. % Quecksilber-
(Il)-sulfat, 8-25 % Schwefelsaure, 5-20 % Eisen-(III)-sulfat und 86,9-53 % Wasser;
Temperatur der Kontaktlosung: 80-95°C; Acetaldehydausbeute, bezogen auf umgesetz¬
tes Acetylen: 92-93 % ; Crotonaldehyd- bzw. Essigsaureausbeute: 0,5-1 % des umge¬
setzten Acetaldehyds.
Die Acetylendurchsatzgeschwindigkeit wird so gewählt, dass sich nur etwa 50-70 % um¬
setzt. Nicht reagiertes Acetylen wird rezirkuliert. Als Regenerationsmittel dient mei¬
stens Salpetersaure.
2.2.1.2. Zink- und Cadmiumkataly s atoren
Kutscheroff (45) entdeckte 1909 die katalytische Wirksamkeit von Zink- und Cad-
miumsalzen, aber erst in neuester Zeit wurden eingehendere Studien über deren Wir¬
kungsweise ausgeführt (46-52).
Hoch konzentrierte wasserige Losungen der Zink-(II)- und Cadmium-(II)-salze zwischen
100 und 170 C zeigten eine mittlere katalytische Aktivität (11-16, 46-52), die aber durch
Druckerhohung verbessert wird (48, 49). Als Nebenprodukte werden Crotonaldehyd und
Harzprodukte gebildet.
Wasserige Aufschlemmungen verschiedener Kationenaustauscher wie CS-1, KU-2 und
IRC 50, deren Wasserstoffionen teilweise oder ganz durch Zink- und Cadmiumionen er¬
setzt worden waren, brachten kerne höheren Aktivitäten und Selektivitäten. Als Neben¬
wirkung trat vielmehr meist eine erhöhte Tendenz zu Polymerisation zu Vinyl- wie auch
zu Divinylacetylen m Erscheinung (52).
In der Literatur (47) wird ein Beispiel mit folgenden optimalen Bedingungen angegeben:
Kontaktlosung: 12-13 Gew. % Zinkchlond, 88-87 % Wasser; Temperatur: 160-17O°C;
Druck: 31 atm; Acetaldehydausbeute, bezogen auf umgesetztes Acetylen: 95 % ; Acety-
lenumsatz: 45-65 %; Reaktionsgeschwindigkeit: 2-4 Mole Acetaldehyd / Liter Kontakt¬
losung und Stunde.
Das vorliegende Verfahren wurde aber nur bis zur Pilot-Stufe entwickelt (51).

6
2.2.1.3. Kupfer- und Silberkatalysatoren
In sauren, wasserigen Medien zeigen Kupfer-(I)- und teilweise Kupfer-(II)-salze hohe
katalytische Wirksamkeit, wie auch lange Lebensdauer (11-16, 53-64). Die Kontakt¬
losungen können dabei aus folgenden Systemen bestehen: Kupfer-(l)-chlond - Ammo-
mumchlorid - Salzsaure - Wasser (11-16, 58-60); Kupfer-(I)-sulfat -Ammoniumsul¬
fat -Schwefelsaure - Wasser (64); Kupfer-(I)-chlorid - Ammomumchlond - Phosphor¬
saure - Wasser (54); Kupfer-(I)-chlorid - Ammomumchlond - Schwefelsaure - Wasser
(56-58, 62). Die gunstigsten Temperaturen sind 50-100°C (11-16, 56, 57, 59, 60). Zu¬
sätze wie Leucm und Tyrosin (60) oder anorganische Sulfide (56, 57, 61, 62) und Thio-
cyanate (61) erhohen die katalytische Lebensdauer. Als Nebenprodukte bilden sich Cro-
tonaldehyd (56, 57), Vinyl- und Divinylacetylen (56-58) nebst höheren Polymerisaten.
In Losungen, die Chlorionen enthalten, wird Acetylen auch zu Vinylchlorid umgesetzt
(58, 60).
Die Anwendung von Kationenaustauschern (CS-1, KU-2 und IRC 50) mit emgelagerten
Kupfer-(I)- und Kupfer-(II)-ionen erwies sich mit einer Ausnahme als ungunstig. CS-
1-Harze mit Kupfer-(I)-ionen zeigen dagegen eine relativ hohe Aktivität und lange Le¬
bensdauer (52).
In einer Versuchsanlage wurden folgende optimale Bedingungen gefunden : Kontaktflus-
sigkeit: 36-80 Gew. % Kupfer-(l)-chlond, 19-20 % Ammomumchlond, 10-15 % Schwe¬
felsaure, 35-27 % Wasser und 0,1-0,5 % Sulfide oder 37-39 % Kupfer-(I)-Chlorid, 19-
21 % Ammomumchlond, 2-3 % Salzsaure und 42-37% Wasser; Durchsatzgeschwmdig-
keit: 9-10 m Acetylen und 4-5 Liter Schwefelwasserstoff pro Stunde und 170 kg Losung;
Acetylenumsatz: 10-12 %; Acetaldehydausbeute, bezogen auf umgesetztes Acetylen: 87 %;
Nebenprodukte: Vinylchlorid, Vinylacetylen und Spuren von Crotonaldehyd (58).
Der Vorteil der oben aufgeführten Kontaktsysteme liegt dann, dass durch Variation der
Parameter entweder Acetaldehyd oder aber Vinylchlorid hergestellt werden kann.
Kupfer-(I)- und Kupfer-(II)-salze werden auch als Aktivatoren zu Quecksilber- (11-16)
und Zinksalzen (53) zugegeben. Vor allem wurde das System Kupfer-(I)-chlond - Zmk-
(H)-chlond - Wasser bei 170°C genauer untersucht. Die katalytische Wirkung dieses
Systems lasst sich mit derjenigen der Quecksilbersalze vergleichen. Der Umsatz wird
mit 38-44 % und die Ausbeute mit 92-97 % angegeben.
Silber-(I)-Salzlösungen smd ungeeignete Katalysatoren, weil die Raum-Zeit-Ausbeuten
sehr gering smd (65, 66).

7
2.2.2. Die Gasphasenhydratisierung
Schon relativ früh wurde versucht, Acetylen und Wasserdampf an aktiven Kontakten zu
Acetaldehyd umzusetzen (3, 11-17). Obwohl eine grosse Zahl Patente alteren Datums
existieren (11-17), ist es erst in neuester Zeit gelungen, etwas Klarheit über die kom¬
plexen Katalysatorsysteme zu erhalten.
Aus der Literatur geht hervor, dass vor allem Zinksalze der Phosphorsaure (68, 71-77),
Chromsaure (78), Molybdansaure (79, 80), Wolframsaure (81), Kieselsaure (82), Zirkon-
saure (83) und Wasser (67-70), sowie auch Cadmiumsalze der Phosphorsaure (72-74, 77,
84, 85), Pyrophosphorsaure (82, 84), Salpetersaure (86), Kieselsaure (82), Molybdansau¬
re (79), Chromsaure (78, 87, 88), als auch Kupfer-(II)-salze der Phosphorsaure (76, 89)
und Essigsaure (90), nebst einer grossen Anzahl Salzen mit gleichen oder unterschied¬
lichen Kationen wie Quecksilber-(II)- und Silber-(I)-salze (11-17) die Wasseranlage¬
rung an Acetylen katalysieren. Die angegebenen Temperaturen schwanken zwischen 100
und 500 C. Silber- und vor allem Kupfersalze smd besonders gute Aktivatoren fur Zmk-
und Cadmiumkontakte (72, 75, 76, 82, 87, 88, 91).
Die katalytische Wirkung der obigen Systeme kann durch Zumischen von Erdalkalime-
tallverbindungen (74, 84, 86) und / oder Sauren erhöht werden. Phosphorsaure scheint
ihres hohen Siedepunktes wegen besonders geeignet zu sein (73, 75-77, 82, 89, 90, 92-
94).
Phosphorsaure allein kann ebenfalls katalytisch wirksam sein (93, 95, 96). Auch Schwer-
metallverbindungen, wie Antimonpentoxid (94), Nickelphosphat (97), Vanadium- oder
Chromoxid (97), Mangan- oder Kobaltmolybdat (79), Nickel-, Zinn- und Wismuthmolyb-
dat (98) nebst einer Vielzahl anderer Verbindungen (11-17) sind ebenfalls, wenn auch
in sehr geringem Ausmass katalytisch wirksam.
Als Katalysatortrager kommen Holzkohle (71), Aktivkohle (75, 93), Kieselgel (93), Quarz
(72, 91), Glaskörper (72, 91), Cupren (99), Bimsstein und Zeohthe (11-17) in frage.
Alle diese oben genannten Katalysatoren verlieren ihre Aktivität aber oft sehr schnell.
Fur die praktische Anwendung ist die Möglichkeit der Regenerierung entscheidend. Mit
Luft oder Luft-Wasserdampf (72, 85, 87, 88, 100) gelingt es oft, eine teilweise oder to¬
tale Regenerierung zu erzielen. Katalysatoren auf brennbaren Tragern können dagegen
nicht regeneriert werden.
Bei Zink- und Cadmiumkatalysatoren wird zur Hauptsache Acetaldehyd gebildet, neben

8
kleinen Mengen von Crotonaldehyd, Essigsaure, Wasserstoff (69, 70, 101), Kohlensau¬
re (69, 70, 101), Kohlenmonoxyd (101), Methan (69, 70, 101), 1- und 2-Penten (101), Cup¬
ren (102) und öligen Produkten (101), Bei andern Katalysatoren ist es möglich, dass bis
zu 100 % Essigsaure oder Aceton gebildet wird, je nachdem, wie die Hydratisierungs-
bedingungen gewählt werden (103, 104).
Die Druckanwendung beschleunigt die Polymerisation von Acetylen und verkürzt da¬
durch die Lebensdauer des Katalysators (11-17, 100).
Gönn et al. (99, 101, 105-107) untersuchten einige fur die Industrie anwendbare Ka¬
talysatorsysteme und stellten fest, dass Katalysatoren, welche freie Phosphorsaure mit
Zink- oder Kupfersalzen auf Cupren oder Aktivkohle aufgetragen enthalten, nicht die Sta¬
bilität besitzen, welche fur industrielle Anwendung unerlasslich ist. Auch freie Phosphor¬
saure allein ist ungenügend. Zudem erlauben die brennbaren Trager eine Regenerierung
mit Luft oder Luft- Wasserdampf nicht. Der Grund der Desaktivierung ist das Absetzen
der Acetylen- und Acetaldehydpolymerisationsprodukte auf dem Katalysator (99).
Zinkphosphat allem oder solches mit 0,4 Gew. % Kupfer-(U)-phosphat besitzt eine be¬
friedigende Aktivität, Selektivität und Stabilität. Eine Regenerierung mit Luft und Was¬
serdampf ist möglich. Dem industriellen Einsatz stehen aber die kurzen Kontaktzyklen
im Wege. Als Maximalleistung erhalt man 150 g Acetaldehyd pro Liter Katalysator und
Stunde (99).
Die Zugabe von 0,1 - 0,3 Gew. % Kupfer-(II)-phosphat zu Calciumphosphat gibt einen
hoch aktiven und selektiven Katalysator (99, 106), wobei mit Kontaktzyklen von 40 - 100
Stunden gearbeitet werden kann. Die optimale Kupferphosphatkonzentration liegt bei 0,3
Gew. %. Bei höheren Konzentrationen bilden sich unerwünschte Nebenprodukte und die
Ausbeute smkt sehr rasch ab. Bezüglich des Anions ist die angenäherte Neutralorthophos-
phatzusammensetzung am gunstigsten. Die Aktivität von metallischem Kupfer oder Kupfer-
(I)-chlorid ist gering. Die nach dem besprochenen Prinzip hergestellten Katalysatoren kön¬
nen der Regenerierung mit Luft und Wasserdampf unterworfen werden. Die Stabilität ist
jedoch unbefriedigend und betragt im Maximum 300 - 600 Arbeitsstunden; es kann aber
mit emem Maximalertrag von 400 - 600 g Acetaldehyd pro Liter Katalysator und Stun¬
de gerechnet werden.
Nach Literaturangaben schemt ein Cadmium-Calciumphosphat-Katalysator (99, 107-109),
mit einer Zusammensetzung von 30 - 50 % Cadmiumphosphat und 70 - 50 % Calciumphos¬
phat sehr vielverspreche id zu sein.

9
In einer Pilot-Anlage wurden folgende optimale Bedingungen gefunden : Anfangstempe¬
ratur: 340 - 350°C; Endtemperatur: 400 - 410°C; Acetylen-Wasserverhaltnis: 1:7 bis
1:10 Vol. %; Acetylendurchsatzgeschwindigkeit: 150 - 250 Liter/Stunde; Umsatz: 45 -
50 %; Acetaldehydausbeute, bezogen auf umgesetztes Acetylen: 88 - 90 %; Crotonal-
dehydausbeute, bezogen auf umgesetztes Acetylen: 4 - 6 %; Regenerierung im Luft-Was¬
serdampfstrom; Kontaktzyklen: 70 - 100 Stunden; Ertrag: 140 - 215 g Acetaldehyd pro
Liter Katalysator und Stunde; totale Stabilität: 2600 Arbeitsstunden. Als Apparatur dien¬
te eine zylindrische Kolonne mit 10 Siebboden, auf denen der Katalysator ausgebreitet
lag, wodurch eine maximale Warmeabfuhr gewährleistet war.
Das oben angeführte System entspricht ungefähr den Anforderungen, die an Industrie¬
katalysatoren gestellt werden (105).
2.3. Prinzipien der Katalysatorauswahl
Bei der Analyse von Literaturdaten (66, 110, 111) wird festgestellt, dass einige Ionen,
welche eine komplette d-Schale haben, die Addition von Wasser an Acetylen katalysie¬
ren. Es sind dies die Metallionen der ersten bis dritten Nebengruppe im Periodensystem:
Cu+ (Cu2+) Zn2+ Ga*
Ag+ Cd2+ In*
(Au+) Hg2+ T13+
Diese Ionen aktivieren sowohl die Wasser- als auch die Acetylenmolekule. Als Ueber-
gangszustand bildet sich ein Komplex der folgenden Form:
m . C2H2 + n . H20 + [K] « {[Kl . (H20)n . (0^)1 ^
[II
{[K] . (H20)n_1 . (C2H2)m_A + CHgCHO + 36 kcal /Mol
[Kl : Metalhon
Die Kräfte, die zwischen dem aktiven Ion und dem Wasser einerseits und dem Acetylen-
molekul anderseits wirken, sind verschiedener Natur. Die Wechselwirkung des Kations
mit dem Wasser besteht aus einem Ion-Dipol-Typ. Die Wechselwirkung mit dem Acety-
lenmolekul besteht in einem ff- und einem "jf-Bindungsanteil, wobei der erstere durch

10
ein Ueberlappen von bindendenT-Orbitalen des Acetylens mit freien Orbitalen des
Zentralatoms und der zweite durch Kombination von besetzten d-Zustanden des Metall-
10ns mit antibindenden Orbitalen des Acetylens gedacht werden kann. Die zweite Art
der Wechselwirkung wird "dative"-Bindung genannt.
Zwischen den Parametern, welche einen wichtigen Einfluss auf die Natur der Bindungen
und die Charakteristik des Uebergangskomplexes haben, werden folgende erwähnt: Io¬
nenradius in Lösung, Hydratisierungswarme des Metallions, Ionisationspotential und
Standard-Elektrodenpotential zu einem weniger stabilen tieferen Oxydationszustand,so¬
wie die Wasserstoffionenkonzentration in wasserigen Losungen.
Das Elektrodenpotential gibt eine qualitative Beziehung der Donator-Acceptor-Wech-
selwirkung. Diese Tatsache ist durch eine Reihe von Experimenten bestätigt worden
(112-119), besteht doch eine lineare Beziehung zwischen dem Logarithmus der Reak¬
tionsgeschwindigkeitskonstanten und dem Elektrodenpotential in der Kontaktlosung (117).
Das lonisationspotential, der Ionenradius und die Hydratisierungswarme bilden in erster
Näherung em Mass fur die Fähigkeit der Ionen, "dative"-Bmdungen mit Acetylen einzu¬
gehen. Das lonisationspotential wachst von links nach rechts im Periodensystem und von
oben nach unten in der Gruppe. Der lonenradius fallt von links nach rechts und wachst
von oben nach unten.
Die Fähigkeit der genannten Metallionen, mit Acetylen eme "dative"-Bindung einzugehen,
fallt mit zunehmendem lonisationspotential und abfallendem lonenradius. Die Affinitat
des Kations zu Wasser wachst mit höherer Ionenladung und reduziertem Radius. Daraus
folgt, dass die Fähigkeit dieser Kationen mit Acetylen einen Komplex zu bilden, von
links nach rechts im Periodensystem abfallt und von oben nach unten in der Gruppe zu¬
nimmt. Die hydrophile Natur der Kationen lauft genau umgekehrt.
ICu+ Zn2+ Ca3*"
1»+ 2+
T*+
• Ag Cd In
^Au Hg Tl
Zunahme der Komplexbildung mit Acetylen
Zunahme der Ion-Dipol-Bindung mit Wasser
Aus dieser Darstellung geht hervor, dass die Bedingungen fur die Bildung des aktiven
Zwischenzustandes fur die einzelnen Kationen verschieden sind.

11
2.3.1. Prinzipien der Katalysatorauswahl fur die Flus sigphasen-
hydrati sie rung
Kupfergruppe ( Cu ,Cu , Ag ,
Au )
Die Hauptprodukte bei der Einwirkung von Acetylen auf Silber- und Kupfersalze sind
Acetylide, Halbacetylide sowie lösliche Metallion-Acetylenkomplexe. Acetylen wird al¬
so von den Kationen so stark gebunden, dass es zu einer Schwächung der C-H-Bindung
kommt, woraus alsdann eme Protonenabspaltung resultiert (120). Um die Acetylid- und
die Halbacetylidbildung zu unterdrucken, ist eine hohe Wasserstoffkonzentration not¬
wendig. Der gleiche Effekt kann aber auch durch Wahl eines geeigneten Anions oder
durch Einfuhrung eines Salzes erzielt werden, insofern das Elektrodenpotential in der
Kontaktlosung herabgesetzt wird.
Goldsalze lassen sich nicht m das angegebene Schema einordnen und sind fur die
Acetylenhydratisierung ungeeignet (66).
Silber salze bilden sehr bestandige Acetylide (AG9q„ von Ag„C, : -7,9 kcal/Mol ),
zeigen demgegenüber aber nur eine geringe Tendenz zur Hydratbildung und die Acidi-
tat, die zur Unterdrückung der Acetylidbildung notwendig ist, verrmgert die Tendenz
zur Hydratbildung noch mehr. Bei Sübersalzen gelingt es daher nicht, geeignete Be¬
dingungen zu schaffen, um eine annehmbare katalytische Wirkung fur die Hydratisie¬
rung zu erzielen.
Anders sind die Verhaltnisse bei Kupfer s alzen. Diese bilden weniger stabile Kupfer-
acetyhde ( AG„g„ = -4,72 kcal/Mol ) und zeigen eine grössere Tendenz zur Hydratbil¬
dung. Bei Kupfersalzen gelingt es daher, Bedingungen zu schaffen, bei denen ein optimal
aktiver Zwischenzustand gewahrleistet ist. Diese Tatsache kann durch folgendes Reak¬
tionsschema erläutert werden (120) :
C2H2 (aq) + n.Cu2Cl2 (aq) ,PH<0 > C2H2.n.Cu2Cl2 (aq) ,>
° < pH <*'*a
[2]
H-C = C-Cu (n-l)-Cu2Cl2 ,
pH >1,5>Cu2C2(n-2)Cu2Cl2 i
pH = 7pCu2C2
Die angegebenen Zwischenprodukte sind nicht stabil und können, wie aus Tabelle 1 er¬
sichtlich, zu Vinylacetylen, Acetaldehyd oder Vinylchlond umgesetzt werden.

12
Tabelle 1 Einfluss der Wasserstoffionenkonzentration auf die
Bildung von Produkten
pH Molverhaltms HCl : Cu„Cl2 Produkte
1,5
<0
«0
0,01-0,03
0,3-0,5
1-10
Vinylacetylen
Acetaldehyd
Vinylchlorid
Das Reaktionsschema [2] gibt die Reaktionsverhaltmsse aber in sehr vereinfachter
Form wieder. Es muss immerhin beachtet werden, dass in sehr konzentrierten Lösun¬
gen (siehe unter 2.2.1.3.) praktisch keine freien Kupferionen vorkommen. In der Losung
bilden sich vor allem Komplexe der allgemeinen Form : Cu(H,0) .(Cl) . In der Li-
teratur werden als Beispiele Cu(H„0) Cl, und Cu(H,0) Cl," angegeben (66). Diese Komp-^ 111 £• £• 111 O
lexe sind aber imstande, zusatzlich noch Acetylen anzulagern.
Aus diesen Darlegungen geht hervor, dass sehr komplizierte Komplexe gebildet wer¬
den, zudem sind die Komplexbildungskonstanten sehr schwierig zu erfassen, weil die
erwähnten Komplexe nicht stabil sind.
Zinkgruppe ( Zn2+, Cd2+, Hg2+ )
Die Donator-Acceptor-Eigenschaften von Zink- und Cadmium s alzen sind nicht
sehr ausgeprägt. Aus der Literatur sind auch keine definierten Zink- und Cadmiumace-
tylenkomplexe bekannt, und es sind daher hohe Eduktkonzentrationen, wie auch relativ
hohe Temperaturen erforderlich, um eine katalytische Wirkung zu erzielen. Die aktiv¬
sten Katalysatoren sind Quecksilbersalze. Sie bilden in saurem Medium sowohl
mit Wasser wie mit Acetylen Komplexe. Die Hydratbildung gegenüber Kupfer- und Sil-
benonen ist ungefähr gleich ; die Acetylidbildung dagegen ist aber viel geringer und
Quecksilberacetylide bilden sich erst in alkalischem Medium.
Galliumgruppe ( Ga3*, In3*, Tl31" )
Die Kationen der Galliumgruppe zeigen nur eine äusserst geringe Tendenz zur Komplex¬
bildung mit Acetylen. Zudem verlieren diese Metallionen infolge Reduktion zum Metall
ihre Aktivität sehr schnell. Ganz allgemein kann gesagt werden, dass eine starke Do-
nator-Acceptor-Wirkung die Reduktion der Metallionen verhindert. Diese Wirkung ist
aber hier nicht vorhanden. Die Kationen dieser Gruppe haben daher keine industrielle
Bedeutung (66).

13
2.3.2. Prinzipien der Katalysatorauswahl fur die Gasphasen¬
hydratisierung
Generell können alle Metallionen, die die Hydratisierung von Acetylen in Gussiger Pha¬
se katalysieren, auch in der Gasphase gebraucht werden, dabei sind folgende Faktoren
fur die Katalyse von Bedeutung: Acceptor-Donator-Eigenschaften der Kationen, Wahl
der Amonen, der Tragersubstanzen, der Temperatur und des Wasser-Acetylen-Verhalt-
msses sowie der Einfluss der Wasserstoffionenkonzentration.
a) Acceptor-Donator-Eigenschaften der Kationen
Gönn et al. (102) untersuchten die Aktivität von Kupfer-(II)-, Silber-(I)-, Zink-(II)-,
Cadmium-(II)- und Quecksilber-(II)-orthophosphaten unter gleichen Bedingungen und
stellten dabei fest, das s Kupferorthophosphat schon bei tiefen Temperaturen sehr ak¬
tiv, die Selektivität hingegen sehr gering ist. Cadmiumphosphat ist weniger aktiv und
verlangt Temperaturen zwischen 300 und 350 C, wirkt dagegen selektiver und ist sta¬
biler. Zinkphosphat stellt der inaktivste der drei Katalysatoren dar, wahrend Quecksil¬
berphosphat sehr schnell zu metallischem Quecksilber reduziert wird und deshalb fur
die Gasphasenhydratisierung kaum gebraucht werden kann. Demgegenüber kommt bei
Silberphosphat die Reaktion erst oberhalb 285 C in Gang. Praktische Bedeutung fur die
Gasphasenhydratisierung von Acetylen zu Acetaldehyd haben daher nur Zink-, Cadmium-
und Kupfersalze. Zum gleichen Resultat kamen auch Marton et al. (103).
b) Einfluss der Amonen
Der verschiedene Ionencharakter der Bindung zwischen Anion und Kation fuhrt zu einem
unterschiedlichen Grad der Ionisation, wodurch die Acceptor- und Donator-Eigenschaf¬
ten beeinflusst werden. Je grösser der Anteil der ionischen Bindung ist, umso geeigneter
ist der Katalysator. Bei der Untersuchung einiger Cadmiumsalze der Phosphorsaure, Py-
rophosphorsaure, Molybdansaure, Vanadiumsaure, Kieselsaure und des Wassers wurde
festgestellt, das s vor allem Cadmiumorthophosphate und -wolframate aktive Katalysa¬
toren darstellen. Die Aktivität von Cadmiumhydroxyd ist nur gering (104). In einer an¬
dern Veröffentlichung wurde gezeigt, dass von den untersuchten Zink- und Cadmiumsal-
zen der Phosphorsaure, Metaphosphorsaure, Pyrophosphorsaure, Arsensaure, Wolfram¬
saure und Vanadiumsaure die Ortho- und Pyrophosphate grosse katalytische Aktivität
besitzen (103).
Weiter kann die Selektivität der Hydratisierung durch die Auswahl der Amonen beträcht¬
lich beeinflusst werden (103, 104). Bei den Zink- und Cadmiumorthophosphaten und -pyro-

14
Phosphaten wird neben Acetaldehyd in geringen Mengen auch Crotonaldehyd gebildet.
Bei Cadmiumvanadaten setzt sich Acetylen und Wasserdampf nur zu Essigsaure und
Aceton um, bei Cadmiumhydroxiden ausschliesslich zu Essigsaure.
c) Einfluss der Träger Substanzen
Elektrisch neutrale Trager sind ungunstig. Eine polarisierende Wirkung der Trager¬
stoffe ist vorteilhaft und erhöht die katalytische Aktivität des Katalysatorsystems. Zu¬
dem müssen die Trägerstoffe unbrennbar sein, weil das Abbrennen wegen dem durch
Absetzen von Polymerisationsprodukten bedingten AkUvitatsverlust fur industrielle
Verhaltnisse unerlàsslich ist. Aus der Literatur ist bekannt, dass vor allem Erdalka¬
liphosphate gute Trager sind (121).
d) Einfluss der Temperatur
Obwohl die katalytische Aktivität bei Erhöhung der Temperatur zunimmt, darf die Re¬
aktionstemperatur eine gewisse Höhe nicht übersteigen, denn bei erhöhter Temperatur
neigen sowohl Acetylen, als auch Acetaldehyd zur Polymerisation und der Katalysator
wird vergiftet (76). Daneben tritt die Bildung von Essigsaure gegenüber Acetaldehyd in
den Vordergrund (79).
e) Einfluss des Wa s ser-Acetylen-Verhaltni s s e s
Je grösser das Wasser-Acetylen-Verhaltms ist, umso langer ist der Katalysator aktiv,
weil ein grosser Wasserdampfuberschuss die irreversible und unerwünschte Polymeri¬
sation von Acetylen und Acetaldehyd verringert (75, 103, 122). In der Literatur werden
Wasser-Acetylen-Verhaltms se von 5:1 bis 20:1 angegeben (76, 79, 103, 122).
f) Einfluss der Was ser Stoff îonenkonzentration
Es ist bekannt, dass auch Sàuren fur sich allein katalytisch wirksam sind (75, 93, 103,
104, 123). Ihre Aktivität ist aber sehr gering. Auch die Zugabe von freien Sauren zu ak¬
tiven Katalysatorsystemen zeigt kerne wesentliche Erhöhung der Aktivität, Selektivität
und Stabilität (75, 103, 107, 108).
2.4. Reaktionsmechanismen und Kinetik
Im folgenden Abschnitt sollen Reaktionsmechanismen und Kinetik der Flussigphasen-
und Gasphasenhydratisierung der Quecksilber-(U)-, Zink-(II)-, Cadmium-(II)-, Kupfer-
(I)-, und Silber-(l)-ionen miteinander verglichen werden.

15
2.4.1. Reaktionsmechanismen und Kinetik der Flussigphasen-
hydratisierung
a) Quecksilber-(II) -Ionen
F rieman et al. (124) untersuchten als erste den Reaktionsmechanismus und die Ki¬
netik der Acetylenhydratisierung mit Quecksilber-(II)-sulfat als Katalysator in schwe¬
felsaurer Losung. Sie postulierten, dass die Anfangsgeschwindigkeit bezüglich Acety¬
len erster Ordnung ist, bezuglich Quecksüber-(Il)-sulfat aber zweiter Ordnung. Der an¬
gegebene Reaktionsmechanismus scheint unwahrscheinlich zu sein und wurde durch neu¬
ere Untersuchungen widerlegt (115-119, 125-128).
Schwabe et al. (125, 126) verfolgten durch polarographische Acetaldehydbestimmun-
gen den zeitlichen Verlauf der Hydratisierung unter gleichen Bedingungen (124). Die er¬
ste Ordnung bezüglich Acetylen wurde auch von diesen Autoren gefunden. Zusätzlich wur¬
de aber festgestellt, dass die Geschwindigkeit dem Quadrat der Saurekonzentration pro¬
portional ist. Ein als weisser Niederschlag auftretendes Nebenprodukt wurde als Kata¬
lysator angenommen. Die Untersuchungen ergaben eine Aktivierungsenergie von 13,5
kcal/Mol.
Kalf u s et al. (115, 116) bestimmten die Beständigkeit der Quecksliberkomplexe mit
Hilfe der potentiometrischen Methode in einer modifizierten Kontaktlosung: Schwefel¬
saure, A]umimum-(III)-sulfat anstatt Eisen-(III)-sulfat und Magnesiumfulfat anstatt Ei-
sen-(II)-sulfat. Es wurden Beweise erbracht, dass zwei verschiedene Quecksilber-(n)-
Acetylenkomplexe gebildet werden, die hinsichtlich ihrer Beständigkeit unterscheidbar
sind. Die Konzentration dieser Komplexe steht nicht in direkter Abhängigkeit von der
totalen Quecksüber-(II)-konzentration. Es wurde gezeigt, dass als Katalysator ein Di-
sulfato-Quecksilber-(II)-anion auftritt (129). Die Reaktion ist bezuglich dieses Kom¬
plexes erster Ordnung und nicht wie furher angenommen wurde, zweiter Ordnung hin¬
sichtlich der totalen Quecksilber-(II)-konzentration. Auf Grund experimenteller Daten
wurde folgender Reaktionsmechanismus abgeleitet :
Hg(S04)2.n H202"
+ C2H2«3 Hg(S04)2.n H2O.C2H22~
*
13]
Hg(S04)2.(n-l)H202" + CHgCHO.
Der Mechanismus erklart auch die zweite ReaktionsOrdnung bezüglich Schwefelsàure-
konzentration. Dieser Befund lag zwar schon früher vor (125, 126), es konnte dafür aber

16
keine theoretische Erklärung gefunden werden. Zudem wurde bewiesen, dass der schon
früher beobachtete weisse Niederschlag nur ein Nebenprodukt darstellt und fur die Ace-
tylenhydratisierung inaktiv ist. Die Komplexbildungskonstate von Hg(SO.L wurde ge¬
messen und betragt in 1,17 M Magnesiumsulfat-, 0,14 M Aluminium-(HI)-sulfat- und
1,125 M Schwefelsaurelösung bei 25°C 64,4 Liter/Mol.
In neuester Zeit wurde am Lomosovinstitut m Moskau das Studium der Kinetik und des
Mechanismus der Acetylenhydratisierung neu aufgenommen (117-119, 129, 130). In ei¬
ner ersten Veröffentlichung (129) konnte bewiesen werden, dass Quecksilber-(II)-sulfat
in schwefelsaurer Losung praktisch vollständig in Form des Komplexamons Hg(SO.)„
auftritt, was bereits von Kalf u s (115, 116) postuliert worden war.
In einer weiteren Arbeit wurden die EMK-Werte der folgenden Reaktionen bestimmt
(131):
3+. „ „ „,, „
2+2 Fe
2 Fe2+
Hg'2+
2 Hg;
2Hg:
Hg
"2 Fe
2+r2Fe
3+
u2+
Hg2
Hg2
u2+
Hg2
[4]
[51
[6]
Die freien Reaktionsenthalpien dieser Redoxsysteme sind in Tabelle 2 zusammenge¬
stellt.
Tabelle 2 : Freie Reaktionsenthalpien der Reaktionen [4], [5] und [6]
Reaktion
[Nr.]"AG298[kcal/Moll
-AG363[kcal/Moll
[4]
[5]
[6]
3,98
0,37
4,10
6,50
1,45
5,15
Katalytisch wirksam ist aber nur das Quecksilber-(H)-ion. Aus diesen Redoxsystemen
ist eindeutig ersichtlich, dass die Zugabe von Oxydationsmitteln zur Kontaktsaure die
Lebensdauer des Katalysators verlängert, denn eine Verhinderung der InaJctivierung ist
wegen der reduzierenden Eigenschaften von Acetaldehyd nicht möglich.

17
Untersuchungen über die Kinetik und den Mechanismus unter industriellen Bedingungen
bei 97 C ergaben eine Bestätigung der ersten Ordnung bezüglich Acetylen (117). Aus¬
serdem wurde gefunden, das s in allen Fallen und unabhängig von der Zusammensetzung
der Kontaktlosung folgende Beziehungen gelten:
In k = A + B.Ej [7] bzw. In k = C + D.fEj + E2) [8]
k
A, B, C, D
E,
Jl
Reaktionsgeschwindigkeitskonstante
Konstanten
2+ +
Redoxpotential der Reaktion: Hg >Hg + le
Redoxpotential der Reaktion: Hg »Hg + le
In den Versuchen, bei denen die Kontaktlosung nur Quecksilber-(II)-salze und Sauren
enthielt, blieb die Lmeantat zwischen In k und E. sowie zwischen In k und Ei+E, un~
abhangig von der Natur des Anions und der Saure erhalten. In den Versuchen, bei de¬
nen die Kontaktlosung ausser Quecksilber-(Il)-salzen und Sauren noch weitere Salze ent¬
hielt wie Kaliumchlorid, Eisen-(III)-sulfat oder Ammomumsulfat, blieb diese lineare Ab¬
hängigkeit lediglich zwischen In k und E. bestehen, dagegen wurde die Konstante C eine
Funktion der Anionen. Im Lichte dieser Tatsache können auch die Resultate von Tr a-
vagli (132-134) erklart werden. Die katalytische Wirkung der Quecksilbersalze in sau¬
rer Losung fallen in folgender Reihenfolge ab: Perchlorat > Sulfat > Nitrat > Phosphat
Chlorid > Bromid > Jodid. Weiter konnte bewiesen werden, dass zwischen dem Redox¬
potential der Kontaktlosung und der Aktivierungsénergie ein definierter Zusammenhang
besteht. Bei extremen Bedingungen erhielt man Aktivierungsenergien in der Grossenord-
nüng von nur 3,1 kcal/Mol. Als geschwmdigkeitsbestimmender Schritt wurde die Bildung
irgend welcher intermediärer Komplexe zwischen Acetylen und dem hydratisierten, komp-2-
lexen Quecksilberberanion Hg(SO.)2 angenommen.
Dagegen steht die Behauptung von Dorfmann et al. (127, 128), dass die Saure am li¬
mitierenden Schritt teilnimmt.
Bei Untersuchungen mit deutenertem Wasser und Acetylen bzw. Wasser mit deutener-
tem Acetylen zwischen 75 und 80 C wurde beobachtet, dass schweres Wasser langsamer
mit Acetylen reagiert als leichtes Wasser. Aus dieser Tatsache wurde gefolgert, dass
der geschwindigkeitsbestimmende Schritt im Zerfall des Quecksilber-(II)-Acetylen-Was-
ser-Komplexes besteht (135).

18
Flid et al. (118, 119) befassten sich mit der Thermodynamik der Komplexbildungs -
reaktionen von Acetylen in schwefelsaurer Quecksilber-(II)-sulfatlosung mit Hilfe der
potentiometrisehen Methode. Es konnte gezeigt werden, das s folgende Komplexe ge¬
bildet werden :
Hg2+ + SQ42' ^==1 -HgSQ4 [9]
Hg2+ + 2S042"i==K2=>Hg(S04)22" [10]
Hg2+ + C2H2 ; -K3 =* Hg.C2H22+ [ll]
HgS04 + C2H2 ^ ^4 =sHgSQ4.C2H2 [12]
Hg(S04)2" + C2H2 :i=K5=~ Hg(S04)2.C2H22" [13]
Im Gegensatz zu früheren Ansichten (115-117, 129) wurde postuliert, dass im Konzen-
2+trationsintervall von 1,5 bis 3 M Schwefelsaure zur Hauptsache Hg.C2H2 und nicht
Hg(S04)„.C9H9 gebildet wird. Die Umwandlung diesesT-Komplexes mit Wasser in
Acetaldehyd ist geschwindigkeitsbestimmend.
Hg.C2H22+ + H20 «
k=£. Hg2+ + CH3CHO [14]
Die beobachtete Reaktionsgeschwindigkeitskonstante k kann wie folgt ausgedruckt wer¬
den:
kx = k. K3 . [Hg2+] [15]
Geschwindigkeitskonstante der Reaktion [14]
Gleichgewichtskonstante der Reaktion [11]
Gleichgewichtskonzentration von Quecksilber-(II)-sulfat
In all den besprochenen Arbeiten wurde angenommen, dass Quecksilber-Acetylen-Kom-
plexe 1:1 gebildet werden. In neuesten Untersuchungen ist es aber gelungen, Diacetylen-
Quecksilber-(II)-Komplexe in Quecksilber-(II)-perchlorat-Perchlorsaure-Losungen
nachzuweisen (136, 137). Nach den grundlegenden früheren Untersuchungen muss aber
trotzdem angenommen werden, dass der Acetaldehyd über den Quecksilber-Acetylen-
Komplex 1:1 gebildet wird, sonst hatte man eine zweite Ordnung bezüglich Acetylen fin¬
den müssen, was jedoch nicht der Fall war.
In Tabelle 3 sind die Gleichgewichtskonstanten K und K, aufgeführt.
32+
[Hg2+I

19
K = K3 + K4.Kr[S042-] + K5.K2. [Su/"] [16]
Tabelle 3 : Gleichgewichtskonstanten K und K, in 8,45.10 M Schwefelsaureà
(118, 119)
[H2S04] T [C2H2].102 (141-143) K K3
[Mol/Liter J [°c] [Mol/1000g H20 ] [Liter/Mol] [Liter/Mol]
1,44 40 2,90 5.740.105 7.75.105
0,288 60 2,42 1980.104 1,85.105
1,44 60 2,38 5.6.104 -
0,288 90 0,977 442.103 1.5.103
1,44 90 1,17 0.536.103 -
3,07 90 1,59 0.056.103 -
Die Geschwindigkeitskonstanten k als Funktion der Quecksilber-(II)-sulfatkonzentra-
tion sind m Tabelle 4 aufgeführt.
Tabelle 4 : Abhängigkeit der Geschwindigkeitskonstante k von der Quecksil-
ber-(II)-sulfatkonzentration m 10 Gew. %iger Schwefelsaure bei
90°C (118, 119)
[HgS04] CHgCHO k.103l
[Gew.%] [Ausbeute, %] [sek"XI
0,025 10 2,315
0,050 15 3,66
0,075 24 6,11
0,10 39 10,9
b) Zink-(II)- und Cadmium - (II)-îonen
Ban et al. (46, 47, 50) studierten die Kinetik der Acetylenhydratisierung in konzentrier¬
ten, wasserigen Zink- und Cadmiumsalzlosungen bei normalem und erhöhtem Druck. In
jedem Fall konnte die Geschwindigkeit der Acetaldehydbildung nach folgendem Gesetz
formuliert werden :
r = k. [Salz] . [C2H2] [17]

20
Zudem konnte gezeigt werden, das s die Acetaldehydausbeute von dem Ausmas s der Al-
dolkondensation zu Crotonaldehyd und höheren Polymerisaten abhangig ist. Die Geschwin¬
digkeit der Aldolkondensation ist dem Quadrat der Acetylenkonzentrauon proportional.
Potentiometrische Messungen an Zink- und Cadimumsalzlos jngen wurden keine veröf¬
fentlicht. Dagegen gelang es aber, eine lineare Abhängigkeit zwischen dem Logarith¬
mus der Reaktionsgeschwindigkeitskonstanten und dem Elektrodenpotential von Kupfer-
(I)-ionen im System Zink-chlorid-Kupfer-(I)-chlorid-Wasser bei 170 C nachzuweisen
(114). Aus dieser Tatsache wurde geschlossen, dass die Kupfer-(I)-ionen Acetylen ak¬
tivieren, die Zinkionen dagegen Wasser. Die erste Ordnung bezuglich Acetylen konnte
auch hier experimentell bewiesen werden. Es wurde eine Reaktionsgeschwindigkeits--2 -1
konstante von 7,4.10 sek bei einer Zusammensetzung der Kontaktlosung von 75 Gew.
% Zinkchlorid, 7,3 % Kupfer-(I)-chlorid und 17,7 % Wasser gemessen.
c) Kupfer-(I)-ionen
Mit Hilfe potentiometrischer Messungen gelang es, die Kinetik der Acetylenhydratasie-
rung in den Systemen Kupfer-(I)-sulfat-Schwefelsaure-Wasser und Kupfer-(I)-chlond-
Ammoniumchlorid-Salzsaure-Wasser zu bestimmen (138, 139). Beim Einleiten von Ace¬
tylen in die Kupfer-(I)- Salzlösungen wurde ein irreversibler und ein reversibler Poten-
tialabfall festgestellt. Der irreversible Potentialabfall ist auf die Bildung von unlöslichen
Acetyhden zurückzuführen, der reversible auf die Bildung von löslichen Komplexver¬
bindungen. Das folgende Reaktionsschema gibt einige lösliche Komplexe wieder, wobei
X ein Ligandanion bezeichnet (140):
[Cu+IX] + C2H2 «Kl=» [Cu+IX] .c2H2 [18]
[Cu+IX] + C2H2 „K2=£ [Cu+IX] .c2H" + H+ [19]
[Cu+IX] .C2H2 ^3==;: [Cu+IX] .C2H" + H+ [20]
Die Acetylidbildung in 0,096 M Schwefelsaure ist vernachlas sigbar klein. Die Gleich-
gewichtskonstanten der Reaktionen [18], [19] und [20] andern sich mit der Temperatur
und der Schwefelsaurekonzentration sehr stark (Tabellen 5 und 6).
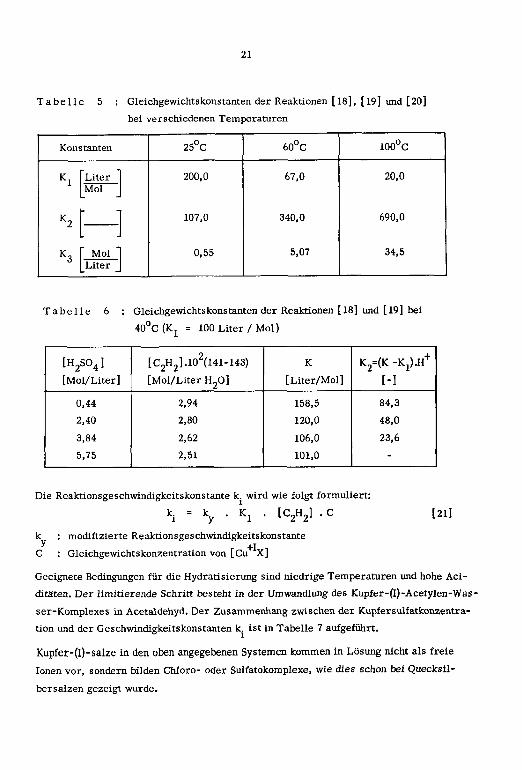
21
Tabelle 5 : Gleichgewichtskonstanten der Reaktionen [ 18], [19] und [20]
bei verschiedenen Temperaturen
Konstanten 25°C 60°C 100°C
Kl
*2
K3
Liter 200,0
107,0
0,55
67,0
340,0
5,07
20,0
690,0
34,5
Mol
r i
L JMol
"
Liter_
Tabelle 6 : Gleichgewichtskonstanten der Reaktionen [18] und [19] bei
40°C (K = 100 Liter /Mol)
[H2S04 ] [C2H2].102(141-143) K K2=(K -Kj).H+[Mol/Liter] [Mol/Liter H20] [Liter/Mol] [-]
0,44 2,94 158,5 84,3
2,40 2,80 120,0 48,0
3,84 2,62 106,0 23,6
5,75 2,51 101,0 -
Die Reaktionsgeschwindigkeitskonstante k wird wie folgt formuliert:
\ = ky KA • [C2H2] . C [21]
k : modifizierte Reaktionsgeschwindigkeitskonstante+1
C : Gleichgewichtskonzentration von [Cu X]
Geeignete Bedingungen fur die Hydratisierung sind niedrige Temperaturen und hohe Aci-
ditaten. Der limitierende Schritt besteht in der Umwandlung des Kupfer-(I)-Acetylen-Was-
ser-Komplexes in Acetaldehyd. Der Zusammenhang zwischen der Kupfersulfatkonzentra¬
tion und der Geschwindigkeitskonstanten k ist in Tabelle 7 aufgeführt.
Kupfer-(I)-salze in den oben angegebenen Systemen kommen in Losung nicht als freie
Ionen vor, sondern bilden Chloro- oder Sulfatokomplexe, wie dies schon bei Quecksil-
bersalzen gezeigt wurde.
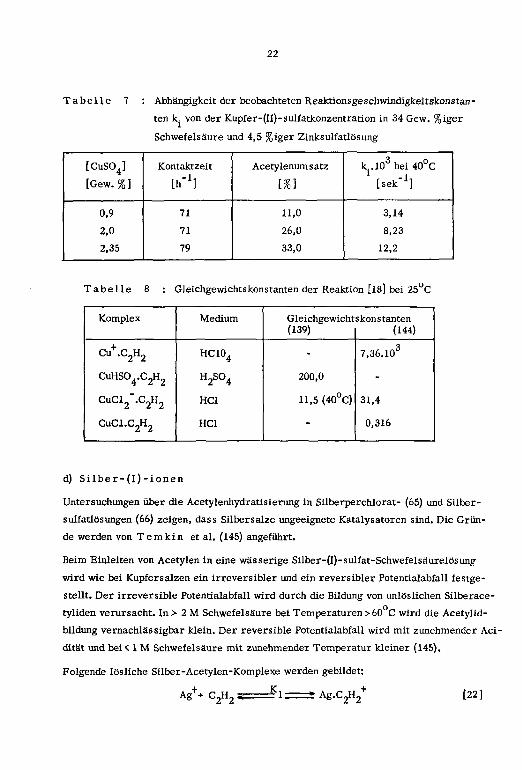
22
Tabelle 7 : Abhängigkeit der beobachteten Reaküonsgeschwindigkeitskonstan
ten k von der Kupfer-(II)-sulfatkonzentration in 34 Gew. %iger
Schwefelsaure und 4,5 %iger Zinksulfatlosung
[CuS04][Gew. %]
Kontaktzeit
[h"1]Acetylenumsatz
[%]
k .103 bei 40°C
[sek"1]
0,9
2,0
2,35
71
71
79
11,0
26,0
33,0
3,14
8,23
12,2
Tabelle 8 : Gleichgewichtskonstanten der Reaktion [18] bei 25°C
Komplex Medium Gleichgewicht(139)
skonstanten
(144)
Cu+.C2H2 HC10.4
- 7.36.103
CuHSO..C,H,4 2 2 H2S04 200,0 -
CuCl2-.C2H2 HCl 11,5 (40°C) 31,4
CuCl.C2H2 HCl - 0,316
d) Silber-(I)-lonen
Untersuchungen über die Acetylenhydratisierung in Silberperchlorat- (65) und Silber-
sulfatlösungen (66) zeigen, dass Silbersalze ungeeignete Katalysatoren sind. Die Grun¬
de werden von Temkin et al. (145) angeführt.
Beim Einleiten von Acetylen m erne wasserige Silber-(I)-sulfat-Schwefelsaurelösun^
wird wie bei Kupfersalzen ein irreversibler und ein reversibler Potentialabfall festge¬
stellt. Der irreversible Potentialabfall wird durch die Bildung von unlöslichen Silberace-
tyliden verursacht. In > 2 M Schwefelsaure bei Temperaturen > 60 C wird die Acetyhd-
büdung vemachlàssigbar klein. Der reversible Potentialabfall wird mit zunehmender Aci-
ditat und bei< 1 M Schwefelsaure mit zunehmender Temperatur kleiner (145).
Folgende losliche Silber-Acetylen-Komplexe werden gebildet:
Ag++ C2H2< gl: Ag.C2H2+ [22]

23
Ag+- C2H2 „K2 : ^ Ag.C2H + H+ 23
Tabelle 9 : Gleichgewichtskonstanten der Reaktionen [22] und [23] (145)
[H2S04] [C2H2] .102 T K Kl K^K-Kj). H+
[Mol/Liter] [Mol/lOOOg H20] [uc] [Liter/Mol] [Liter/Mol] [-]
0,288 3,06 40 79,1 16,0 43,8
0,288 2,42 60 103,3 4,4 70,6
0,274 0,121 100 1008,0 - 196,0
0,98 0,218 100 79,8 - 79,8
3,20 0,827 100 6,7 - 23,9
Eine Geschwindigkeitsgleichung wurde nicht angegeben. Die Hydraüsierung erfolgt aber
ebenfalls über die Bildung und Umsetzung des Silber-Acetylen-Komplexes mit Wasser
zu Acetaldehyd.
e) Vergleich der katalyti s chen Aktivitäten von Silber-(I)-, Kupfer-
(II)-, Zink-(II)- und Quecks llber-(II) - salzen
Die Anwendung der potentiometrischen Methoden erlaubt das Bestimmen der spezifi¬
schen katalytischen Aktivität von Silber-(I)-, Kupfer-(I)-, Zink-(II)- und Quecksilber-
(Il)-salzen. Beim Studium des Mechanismus dieser Reaktionen konnte gezeigt werden,
dass die Geschwindigkeit der Acetylenhydratisierung durch ein und dieselbe kinetische
Gleichung beschrieben werden kann:
k = k=
l 5
n+7
g K1 . [M"T] [24]
beobachtete Reaktionsgeschwindigkeitskonstante
Geschwindigkeitskonstante der Reaktion:
n+
Mn+.C2H2 + H20 -»•M + CHgCHO
Gleichgewtchtskonstante der Reaktion:
*n+M"T+C,H, (aq)*
2 2= Mn+.C2H2
Gleichgewichtskonstante der Reaktion:
C2H2.(g)^C2H2 (aq)
Wird Gleichung [24] durch die Metallionenkonzentration dividiert, erhalt man Gleichung

24
[25]:
ky= y[MI1+] = k5 . Kg . K2 L25j
Aus der Analyse dieser Funktion folgt, dass sich k mit K. und k„ ändert, d.h. dass
sich fur eine gegebene Reihe von Ionen die Geschwindigkeitsionstanten der Umwand¬
lung der entsprechenden IT-Komplexe mit den Gleichgewichtskonstanten der Komplex¬
bildung andern (Tabelle 10).
Tabelle 10 : Abhängigkeit der spezifischen Geschwindigkeitskonstanten
k der Acetylenhydratisierung von den Gleichgewichtskon- •
stanten der entsprechenden TT-Komplexe bei 100 C (146)
T-Komplexe Inky
In K373 -AH298[kcal/Mol]
Hg2+.C2H2 6,66 3,55 25,9
Cu+.C2H2 0,44 1,33 5,8
Ag+.C2H2 -0,10 -0,27 13,2
2+Zn .C2H2 -4,2 -2,22 -
2.4.2. Reaktions mechanismen und Kinetik der Gasphasenhydra¬
tisierung
T s y bin a et al. (123) untersuchten die Kinetik der Acetylenhydratisierung mit Phosphor¬
saure auf Aktivkohle zwischen 261 und 302 C in einem Differentialreaktor. Die Reak¬
tionsgeschwindigkeit ist der Acetylenkonzentration proportional und kann mit folgendem
mathematischen Gesetz formuliert werden:
f
r = k . K . h . C [26]o f*
k : Geschwindigkeitskonstante des limitierenden Schrittes
K : Gleichgewichtskonstante des IT-Komplexes
h : Konstanteo
f, f* : Aktivitätskoeffizienten desTT-Komplexes bzw. der Protonensaure im
Uebergangszustand
C : Saurekonzentration

25
Der Reaktionsmechanismus kann folgendermassen formuliert werden:
HC = CH + H+ï==S HC=î=CH «=i H„C =CH
hT2
H2C = CH + H20^=3,H2C=CH-èH2^=-H2C=CH2-œ + H+ [27]
H2C=CH-OH + H+—-»CHg-CH-OH—»CHgCHO + H+
Die Aktivierungsenergie betragt bei einem Phosphorpentoxidgehalt des Katalysators von
75 Gew. % 240 kcal/Mol bzw. 38,4 kcal/Mol bei 76 %.
Dieselben Autoren untersuchten auch die Kinetik der Acetylenhydratisierung mit neu¬
tralem Zinkphosphat auf Aktivkohle bei 278 - 351 C. Es wurde festgestellt, dass die
Aktivität mit zunehmender Reaktionsdauer verloren geht. Die Menge an höheren Alde¬
hyden betragt maximal 5 %. Der Nebenproduktanfall wachst mit zunehmendem Acety-
len-Wasser-Verhaltnis und mit höherer Temperatur. Die Reaktionsgeschwindigkeit in
3 2den untersuchten Grenzen betragt 0,49 - 7,5 Ncm / m Oberflache und Sekunde (Ober-
2flache des Katalysators : 6 - 6,5 m /g).
Die Reaktionsgeschwindigkeit ist dem Acetylenpartialdruck proporüonal (210) :
r = k.pAc [28]
r : Reaktionsgeschwindigkeit
k : Reaktionsgeschwindigkeitskonstante
p. : Acetylenpartialdruck
Die nullte Ordnung fur Wasser und die erste Ordnung fur Acetylen lasst sich durch das
angegebene Reakaons schema gut erklaren, wobei als geschwindigkeitsbestimmender
Schritt die Bildung des Carboniumions angenommen wird:
Zn2+ + HC = CH -j*HC =CH-Zn+ [2o]
HC=CH-Zn+ + H205=*-H20"-HC=CH-Zn+—*CHg-CHO + Zn
Als Aktivierungsenergie wurde 23 kcal/Mol gefunden.
Szonyi et al. (148, 149) bestimmten die Kinetik der Acetylenhydratisierung in der
Gasphase mit einem Zinkphosphat-Phosphorsaure-Aktivkohle-Katalysator in einem Dif¬
ferentialreaktor. Die katalytische Aktivität verminderte sich dabei mit zunehmender
Reaktionsdauer (innerhalb von 60 Stunden um 50 %). Neben Acetaldehyd konnten noch

26
Crotonaldehyd und Spuren von Essigsäure identifiziert werden. Die Geschwindigkeit
war eine Funktion der Acetylen- und Wasserkonzentration, der Temperatur und der
Reaktionsdauer.
7- rar + vè- m
la 2 w
k. : Geschwindigkeitskonstante der Wasseranlagerung an Acetylen
k„ : Geschwindigkeitskonstante der Wasseradsorption
C, C : Acetylen- bzw. Wasserkonzentration
a w'
Die Aktivierungsenergien betrugen für k, 13,6 kcal/Mol und für k. 20,4 kcal/Mol.

27
3. EXPERIMENTELLER TEIL
3.1. Einfuhrung
Als erstes wird der makrokinetische Ablauf der heterogenen Gasphasenhydratisierung
von Acetylen mit Wasserdampf in einem Festbettreaktor untersucht. Basierend auf den
Versuchsergebmssen und einigen theoretischen Berechnungen wird alsdann das beobach¬
tete Verhalten durch einzelne Teilschritte zu erklaren versucht.
In diesem Zusammenhang schien es angezeigt, zuvor eventuell vorhandene Stofftrans -
porteinflusse im Hauptstrom der Reaktionsgase zu überprüfen. Zusatzlich war zu be¬
rücksichtigen, das s neben der Struktur auch die Textur der verwendeten Kontakte eine
Rolle spielen kann.
3.2. Allgemeine Arbeitsgrundlagen
3.2.1. Apparatur
Die in Figur 1 wiedergegebene Apparatur bestand im wesentlichen aus Dosiervorrich¬
tung, Reaktor und Abscheider.
a) Dosiervorrichtung
Das verwendete Acetylen gelangte durch Waschflasche (1), Trockenturm (2) über Re¬
duzierventil (3) durch Rotameter (4) in das Vorwarmrohr (8/1). Waschfalsche (1) war
mit Chromschwefelsaure (15g Kaliumdichromat in 500 ml 96 %iger chemisch reiner
Schwefelsaure) und Trockenturm (2) mit fester Natronlauge gefüllt, um Acetylenverun-
reinigungen zu entfernen.
Wasser wurde aus dem Vorratsgefàss (6) mit der Mikrokolbenpumpe (7) dem Stickstoff¬
strom zugespiesen. Der Stickstoff gelangte über Reduzierventil (3) durch Rotameter (5)
in das Vorwarmrohr (8/II). Acetylen, Wasserdampf und Stickstoff wurden im ReakUons-
rohr (9/1) zusammengeführt. Der totale Gasdruck wurde am Quecksilbermanometer (19)
abgelesen.
b) Reaktor
Der Reaktionsofen bestand aus emem isolierten, 110 cm langen rostfreien Stahlrohr mit

28
einem Innendurchmesser von 20 cm. In diesem befanden sich vier vertikale Rohre (9/1-
IV) aus rostfreiem Stahl mit 1,5 cm innerem Durchmesser, die miteinander verbunden
waren. Alle vier Rohre wiesen oben Einfullstutzen auf. Das Reaktionsrohr (9/III) trug
unten einen Stutzen mit befestigtem Siebchen, auf dem der Katalysator lag. Weiter konn¬
te ein Thermoelement (14) in die Schuttschicht eingeführt werden. Als Warmeubertra-
gungsmittel zirkulierte eine 1:1 Mischung von Natrium- und Kaliummtrat (150). In der
Mischung befanden sich zwei regelbare elektrische Heizspiralen von je 1600 Watt (17)
und ein Propellerruhrer (18). Das eintretende Acetylen-Wasser-Stickstoff-Gemisch er¬
wärmte sich m den Vorwarmrohren (8/1 -II) und den Reaktionsrohren (9/I-II) und durch¬
strömte von oben her das mit Katalysator beschickte Rohr (9/III).
c) Abs cheider
Das Reaktionsgemisch gelangte über Reaktionsrohr (9/IV) durch Kuhler (11) m den Ab¬
scheider (12). Der Kuhler (11) wurde mit Hilfe eines Kryomaten konstant bei 2 C ge¬
halten. Die nicht abgeschiedenen Produkte konnten entweder direkt oder durch die Frit-
tenwaschflasche (15) und durch Gasuhr (16) m die Kapelle abgelassen werden.
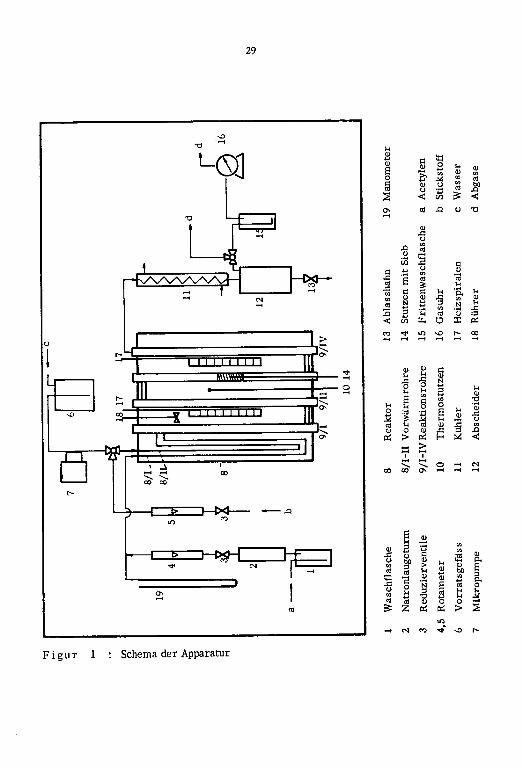
Abgase
dRuhrer
18
Wasser
cHeizspiralen
17
Stickstoff
bGasuhr
16
Acetylen
aFrittenwaschflasche
15
Sieb
mit
Stutzen
14
Manometer
19
Ablasshahn
13
Abscheider
12
Kühler
11
Thermostutzen
10
Reaküonsrohre
9/1-IV
Vorwarmrohre
8/I-II
Reaktor
8
Mikropumpe
7
Vorratsgefass
6
Rotameter
4,5
Reduzierventile
3
Natronlaugeturm
2
Waschflasche
1
0"
14
10
9/IV
13>
i:
rW
12
d—
11
9/II
9/1
V.
"
JT
tto
8/11.
8/1-
.1
4<¥
aX
À
-iffI
19
£TO
30
3.2.2. Ver suchs schema
Nach dem Einfüllen des Katalysators wurde die Apparatur wahrend einer halben Stun¬
de mit Stickstoff gespult, um Acetylen-Sauerstoffexplosionen zu verhindern. Anschlies¬
send leitete man Acetylen und Wasser, verdünnt mit Stickstoff, über den Katalysator.
Die kondensierbaren Produkte konnten im Abscheider (12) aufgefangen werden. Die Rest¬
gase gelangten in den Abzug. In gewissen Zeitabstanden wurden Proben entnommen. Vor
jeder Probenahme wurde der Abschieder (12) geleert. Wahrend einer Stunde konnten
die Produkte im Behalter {12) gesammelt und anschliessend analysiert werden. Die nicht
kondensierten Gase leitete man durch die Frittenwaschflasche (15), die, um den Ace-
taldehyd vollständig zu absorbieren, mit alkoholischer Hydroxylaminhydrochlondlö-
sung gefüllt worden war.
3.2.3. Analytik
Wie gaschromatografische Analysen eimger Versuche zeigten, sind Acetylen, Wasser,
Acetaldehyd, Crotonaldehyd und Spuren von Essigsaure in den Reaktionsprodukten ent¬
halten.
Tabelle 11 : Physikalische Daten der Edukte und Produkte (151)
Stoff Mol-Gewicht Schmelzpunkt Siedepunkt
[g/MolI [°C] [°CI
Acetylen 26,04 -81,8 -83,6
Wasser 18,016 0,00 100,00
Stickstoff 14,008 -210,0 -195,8
Acetaldehyd 44,05 -123,5 20,2
Crotonaldehyd 70,09 -69,0 102,2
Essigsaure 60,05 16,6 118,1
Bei den verwendeten Quecksilber-(II)-, Cadmium-(H)-, Zmk-(II)-, Kupfer-(II)- und Sü-
ber-(I)-13X-Katalysatoren wurden die Produkte fur je einen Versuch qualitativ mit dem
Gaschromatografen untersucht.

31
Dabei erwiesen sich folgende Bedingungen als günstig:
Apparat
Chromatografiesaule
Saulenmatenal
Fractovap Modell B der Firma Carlo Erba mit
Thermistor-Detektor
5 mm Durchmesser, 2 m Lange
10 % Ethofat auf Fluoropak der Firma Fluorocarbon
Co.
Saulentemperatur 110°C
Detektor ström 20 mA
Tragergas Helium
Gasgeschwindigkeit 1,8 Liter/Stunde
Gasdruck 1,5 atu
Bei den qualitativen Analysen wurde festgestellt, dass im Abscheider (12) Essigsaure
und Crotonaldehyd quantitativ, Wasser und Acetaldehyd aber nur teilweise abgeschieden
werden. Um den Acetaldehyd quantitativ zu erfassen, wurden die Gase anschliessend
durch die Frittenwaschflasche (15), die mit 200 ml 0,5 M alkoholischer Hydroxylamin-
hydrochloridlosung beschickt worden war, geleitet. Diese Losung genügten in allen Fal¬
len zur vollständigen Acetaldehyd-Absorption.
Wegen des grossen Wasseruberschusses (5-20fach) und den geringen Acetylenumsatzen
(durchschnittlich 1-7 %) genügte eine quantitative gaschromatografische Analyse nicht.
Deshalb wurden die Produkte wie folgt analysiert :
a) Essigsaure
Zur Bestimmung der Essigsaure wurden Proben mit 0,1 N Natronlauge gegen Phenol¬
phthalein titriert.
b) Crotonaldehyd
In einem aliquoten Teil konnte Crotonaldehyd kolorimetnsch bestimmt werden (152).
Bei Zugabe einer alkoholischen m-Phenylendiamin-dihydrochlond-Lösung bildete sich
ein farbiger Komplex mit Crotonaldehyd, dessen Extinktion bei 420 mp mit einem Lu-
metron-Kolorimeter (Modell 402 E) der Fotovolt Co. gemessen wurde. Acetaldehyd und
Spuren von Essigsaure stören die Messungen nicht.

32
c) Acetaldehyd
Ein aliquoter Teil der anfallenden Produktlosung wurde mit einem 3-10fachen Ueber-
schuss an Hydroxylaminhydrochloridlosung zusammen gegeben und mit 0,1 N Natron¬
lauge titriert. Der Bromphenolblauindikatorumschlag war zu ungenau und deshalb wur¬
de mittels pH-Messgerat der Endpunkt der Titration bestimmt. Wie Blmdversuche zeig¬
ten, wird Crotonaldehyd ebenfalls miterfasst, nicht aber Essigsaure.
3.2.4. Katalysatoren
Als Katalysatoren wurden modifizierte Molekularsiebe eingesetzt. Molekularsiebe sind
kristalline Metall- Aluminium-Silikate (Zeolithe) mit einheitlichen, molekularen Po-
rendimensionen (153-158). Durch Ionenaustausch können diese Siebe in Katalysatoren
umgewandelt werden, die sich von den herkömmlichen Kontakten in einigen wichtigen
Beziehungen unterscheiden. Die im Kristall vorhandenen Ionen zeigen eine ungewöhnli¬
che Fähigkeit zur Bildung von Carboniumionen. Die austauschbaren Natrium- oder Cal-
ciumionen erleichtern das Einfuhren von katalytisch wirksamen Metallionen in feinster
Verteilung. Die kauflichen, synthetischen 4A- und 13X-Typen haben sich fur grosstech¬
nische Katalysen am besten bewahrt. Infolge der grosseren Porendurchmesser des Mo-
lekularsiebs 13X von 10 Â gegenüber dem Molekularsieb 4A von 4 A wurde in der vor¬
liegenden Arbeit ersteres gewählt.
3.2.4.1. Chemische Zusammensetzung und Textur des Molekular¬
siebs 13X
Die Bezeichnung 13 bezieht sich auf den mittleren Porendurchmesser von 13 A und X
deutet die Struktur an (Faujasitstruktur; Faujasit ist ein natürlicher Zeolith). Der drei¬
dimensionale Aufbau besteht aus Kubooktaeder-Emheiten, d.h. Polyedern aus 24 SiO .-
und A10 .-Tetraedern, die bei vierfacher Koordination den X-Typ ergeben. Die Gitter¬
struktur besitzt zwei verschiedene miteinander verbundene Poren- und Hohlraumsy¬
steme. Die Hohlräume innerhalb der einzelnen Kubooktaeder haben einen Durchmesser
von ca. 11,4 Ä. Sie smd durch 8 hexaedrische und 6 viereckige Ringe oder Poren nach
aussen verbunden. Die Hohlräume zwischen den einzelnen Kubooktaedern besitzen ei¬
nen Durchmesser von 12 A. Diese Hohlräume sind durch 12-gliednge Porenöffnungen
mit einem freien Durchmesser von 8-9 A miteinander verbunden. Die Hohlräume kön¬
nen bis zu 32 Wassermolekule enthalten (154, 159). Die Dimensionen der Kationen, sie
neutralisieren die negative Ladung des Amonengerustes, und vor allem ihre Lage in
der Geruststruktur sind massgebend fur den effektiven Porendurchmesser.

33
Das Molekularsieb 13X hat folgende physikalische und chemische Eigenschaften (159-
167) :
Grundtyp
Chemische Formel
Bindemittel
Schuttdichte
Nominaler Porendurchmesser
Max. Adsorptionswarme
Wasserkapazitat
Regenerierung
pH wasseriger Aufschlammungen
pH-Be standigkeit
H-Ionenaustausch
Kationenaustausch
13X ( 1/16"-Stabchen)
Na86(A102W',lO2>106-276H2Omeistens Kaolin oder Ton bis 20 Gew. %
0,640 g/cm310 K
1000 kcal /kg Wasser
28,5 Gew. %
erhitzen auf 200-300°C
9-12
5-12
30 %, sonst geht die Knstallimtat ver¬
loren und das Molekularsieb löst sich auf
: allgemein nicht mehr als 50-60 %, sonst
beträchtlicher Verlust der Knstallimtat
In Tabelle 12 ist die Textur des Molekularsiebs 13X fur die in den Versuchen verwen¬
deten Zylinder (Durchmesser : 1,59 mm; durchschnittliche Lange : 3 mm) angegeben.
Die spezifischen Oberflachen wurden nach der BET-Methode bestimmt.
Tabelle 12 : Textur des Molekularsiebs 13X (l/16"-Stabchen)
Schuttdichte im 1,5 cm Rohr
Wahre Dichte (mit He bestimmt)
Scheinbare Dichte (Hg-Pyknometer)
9b= 0,628 g/cm3
Sw= 2,610 g/cm3
9p= 1,035 g/cm3
Leervolumenanteil
Porosität
•^b
£ = 1- -2- = 0,394
?P
0 = 1- -E- = 0,604
Spez. Porenvolumen (BET)(fur Poren <75 Â) Vg (BET) = 0,282 cm3/g
Spez. Oberflache (BET) Sg (BET) = 424m2/g
Aequivalenter Porenradius
(fur Poren < 75 K)
2V
re= S"5 = 13'3Â
g

34
3.2.4.2. Her Stellung von abgeänderten Molekular sieben 13X
Durch lonenaustausch wurde das Molekularsieb 13X so modifiziert, dass es als Hydra-
usierungskatalysator gebraucht werden konnte (168-170). Auf diese Art konnte eine fei¬
ne Verteilung der aktiven Quecksilber-(II)-, Cadmium-(II)-, Zink-(II)-, Kupfer-(II)-
und Silber-(I)-ionen erreicht werden. Um die maximale Aufnahmekapazitat und die Ab¬
hängigkeit der Hydratisierungsgeschwindigkeit vom Austauschgrad der Ionen zu er¬
mitteln, wurden die Gleichgewichtskurven der genannten Ionen aufgenommen.
a)Bestimmung der Gleichgewichtskurven nach der Progressive-
batch-Methode (171,172)+ 2+
Die Methode soll am Beispiel Na - Zn erläutert werden. Eine bestimmte Menge des
wasserfreien Molekularsiebs 13X wurde mit einer Losung von bekanntem Zinkgehalt bei
Raumtemperatur ins Gleichgewicht gebracht. Nach der Gleichgewichtseinstellung ent¬
nahm man einen aliquoten Teil der Losung, analysierte ihn auf den Zinkgehalt und rech¬
nete auf die gesamte Losungsmenge um. Die Differenz zwischem dem Zinkgehalt vor
und nach der Gleichgewichtseinstellung entsprach dem Zmkgehalt des Molekularsiebs
13X nach der Gleichgewichtseinstellung. Zur restlichen Losung, in welcher sich das Mo¬
lekularsieb befand, wurde eine neue Menge reiner Zinkldsung hinzugegeben und nach
der erneuten Gleichgewichtseinstellung wieder ein Teil analysiert. Durch mehrmaliges
Wiederholen dieser Schritte konnte das Konzentrattonsverhaltnis der Ionen in der Lo¬
sung und damit auch das Beladungsverhàltnls auf dem Molekularsieb 13X kontinuierlich
verändert werden.
Ausfuhrung: In ein Gefass von 1,5 Liter Inhalt mit KPG-Ruhrer wurden 10 g wasserfrei¬
es Molekularsieb 13X (1/16"-Stabchen) und 300 ml 0,2088 N Zinknitratlösung gefüllt,
verschlossen und wahrend ca. 12 Stunden bei Raumtemperatur gerührt. Die nach dieser
Einstellzeit analysierte Lösungsprobe ergab die Zinkaufnahme des Molekularsiebs. Da¬
rauf wurde frische Lösung zur Restlosung gegeben und wieder gleich verfahren. Dieses
Verfahren wurde so lange wiederholt, bis der Zmkgehalt vor und nach der Gleichgewichts¬
einstellung annähernd gleich war. Die Addition der bei jedem Schritt aufgenommenen Zink¬
menge ergab die Totalkapazitàt des Molekularsiebs, mit der die Aequivalentbruche der
Zinkionen im Molekularsieb nach jeder Gleichgewichtseinstellung berechnet und gegen
die entsprechenden Aequivalentbruche der Zinkionen in der Losung aufgetragen werden
konnten.
Die Bestimmungen der übrigen Gleichgewichtskurven erfolgte in analoger Weise. Mit
35
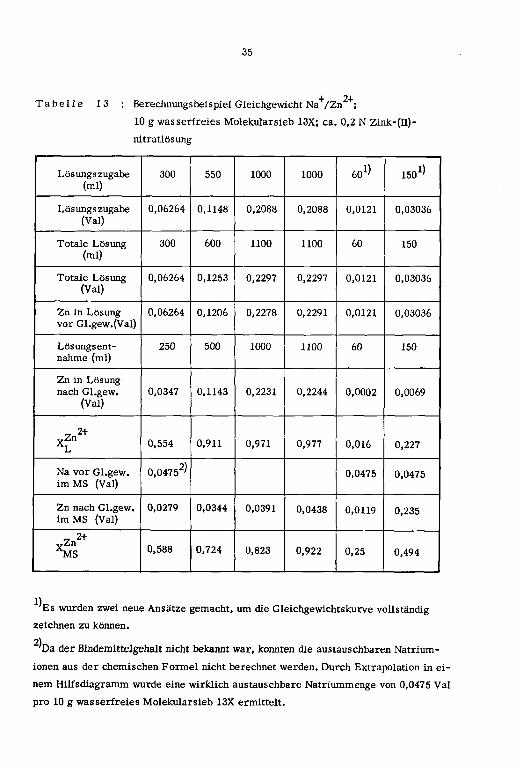
+ 2+Tabelle 13 : Berechnungsbeispiel Gleichgewicht Na /Zn ;
10 g wasserfreies Molekularsieb 13X; ca. 0,2 N Zink-(n)-
nitratlösung
Losungszugäbe(ml)
300 550 1000 1000 601* 1501*
Losungszugabe
(Val)0,06264 0,1148 0,2088 0,2088 0,0121 0,03036
Totale Losung(ml)
300 600 1100 1100 60 150
Totale Losung(Val)
0,06264 0,1253 0,2297 0,2297 0,0121 0,03036
Zn in Losungvor Gl.gew.(Val)
0,06264 0,1206 0,2278 0,2291 0,0121 0,03036
Losungsent¬nähme (ml)
250 500 1000 1100 60 150
Zn in Losungnach Gl.gew.
(Val)0,0347 0,1143 0,2231 0,2244 0,0002 0,0069
xZn2+XL 0,554 0,911 0,971 0,977 0,016 0,227
Na vor Gl.gew.im MS (Val)
0.04752) 0,0475 0,0475
Zn nach Gl.gew.im MS (Val)
0,0279 0,0344 0,0391 0,0438 0,0119 0,235
„Zn2+*MS 0,588 0,724 0,823 0,922 0,25 0,494
Es wurden zwei neue Ansätze gemacht, um die Gleichgewichtskurve vollständig
zeichnen zu können.
2)
'Da der Bindemittelgehalt nicht bekannt war, konnten die austauschbaren Natrium¬
ionen aus der chemischen Formel nicht berechnet werden. Durch Extrapolation in ei¬
nem Hüfsdiagramm wurde eine wirklich austauschbare Natriummenge von 0,0475 Val
pro 10 g wasserfreies Molekularsieb 13X ermittelt.
36
Quecksilbersalzlosungen gelang es jedoch mcht, die Gleichgewichtskurve aufzunehmen,
denn bei der Zugabe von Quecksilbernitratlosung zum Molekularsieb 13X fiel Quecksil-
berhydroxid aus. Um dies zu verhindern, wurde Essigsaure oder ein Natriumacetat-
Essigsaure-Puffer 1:1 zugegeben. Die Gleichgewichtskurve konnte alsdann aber auch
nicht bestimmt werden, weil sich das Molekularsieb aufzulösen begann.
Alle Titrationen wurden komplexometrisch durchgeführt (173). Ein Berechnungsbeispiel
ist in Tabelle 13 aufgeführt. Die Gleichgewichtskurven sind in Figur 2 gegeben.
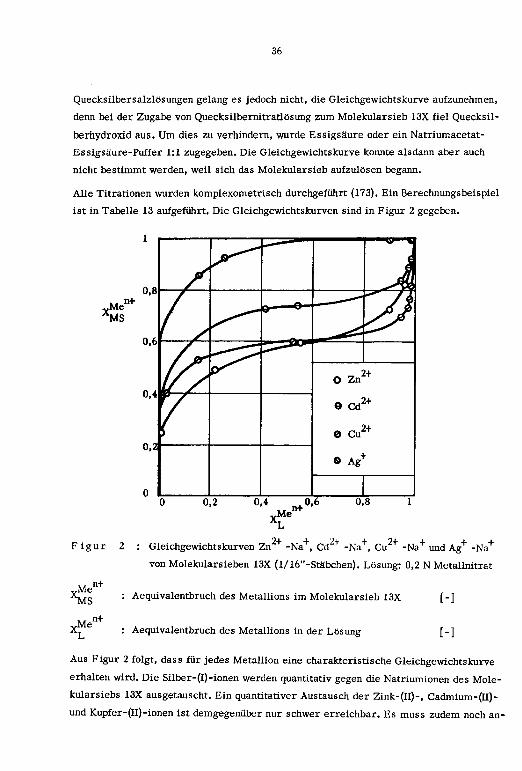
Figur 2 : Gleichgewichtskurven Zn+ -Na+, Cd
+ -Na+, Cu+ -Na+ und Ag+ -Na+
von Molekularsieben 13X (1/16"-Stabchen). Losung: 0,2 N Metallnitrat
YMe*MS
n+
„Men+
Aequivalentbruch des Metaliions im Molekularsieb 13X [-J
Aequivalentbruch des Metallions in der Losung [-]
Aus Figur 2 folgt, dass fur jedes Metalhon eine charakteristische Gleichgewichtskurve
erhalten wird. Die Silber-(I)-ionen werden quantitativ gegen die Natriumionen des Mole¬
kularsiebs 13X ausgetauscht. Ein quantitativer Austausch der Zink-(II)-, Cadmium-(II)-
und Kupfer-(II)-îonen ist demgegenüber nur schwer erreichbar. Es muss zudem noch an-

37
genommen werden, dass beim totalen Austausch der zweiwertigen Metallionen die Kri-
stallinitat des Molekularsiebs sich verändert oder sogar verloren geht, welche Tatsa¬
che mit Hilfe röntgenographischer Messungen bestätigt wurde (159-167).
b)Textur des Molekularsiebs Zn-13X
Da zu erwarten war, dass durch den Austausch der Natriumionen durch Zink-(n)-, Cad-
mium-(II)-, Kupfer-(n)- und Silber-(I)-ionen die physikalischen Eigenschaften des Mo¬
lekularsiebs 13X geändert werden, wurde die Textur des Molekularsiebs Zn-13X in Ab¬
hängigkeit des Austauschgrads gemessen (Tabelle 14).
Tabelle 14 : Textur des Molekularsiebs Zn-13X (1/16"-Stabchen) in Abhängig¬
keit des Zinkaustauschgrads
Zinkaustauschgrad a [%] 49,12 68,00 92,29
Schuttdichte im 1,5 cm Rohr ?,b
Wahre Dichte (mit He bestimmt) 3
Scheinbare Dichte (Hg-Pyknometer) 5
lg/cm ]
[g/cm I
[g/cm I
0,637 0,642
2,430
1,205
0,645
Leervolumenanteil £
Porosität 0
[-1
[-1
- 0,467
0,505 .
Spez. Porenvolumen V (BET)
(Radien < 75 R)
Spez. Porenvolumen V (Drupo)
(75 R < r < 75000 R)
[cm /gl
[cm /g]
0,316 0,257
0,340
0,279
Spez. Oberflache S (BET)
Spez. Oberflache S (Drupo)
[m2/gl[m2/g]
436 304
10
362
Aequivalenter Porenradius r (BET)
Aequivalenter Porenradius r (Drupo)
[R]
[R]
14,5 16,9
680
15,4
Aus Tabelle 14 geht hervor, dass das Molekularsieb Zn-13X zwei verschiedene Hohlraum¬
systeme besitzt, die sich hinsichtlich des Volumens, der Porenradien und der Oberflache
unterscheiden. Das Volumen fur Poren kleiner 75 A betragt 0,257 cm /g, dasjenige fur Po¬
ren zwischen 75 und 75000 R 0,340 cm /g bei einem Zinkaustauschgrad von 68 %. Die ent-

38
2 2
sprechende Oberfläche für kleine Poren ist 304 m /g, diejenige für grosse 10 m /g.
Die aequivalenten Radien betragen 16,9 bzw. 680 R..
Zudem ist aus der Tabelle ersichtlich, dass mit zunehmendem Zinkaustauschgrad das
Volumen und die Oberfläche der kleinen Poren abnehmende Tendenzen zeigen, was da¬
durch erklärt werden kann, dass die Zinkionen gegenüber den Natriumionen mehr Raum
beanspruchen. Dagegen zeigen die aequivalenten Porenradien eine zunehmende Tendenz,
weil sich sehr wahrscheinlich die Gitterstruktur ändert.
3.3. Stofftransporteinflüsse
Bei heterogenen Gaskatalysen können oft Stofftransporte die eigentliche chemische Re¬
aktion hemmen. Es sind dies:
- Film- oder externe Diffusion der Edukte zum Katalysator oder der Produkte
zurück in den Gasstrom.
- Diffusion der Edukte in die Poren oder der Produkte aus der unmittelbaren Nä¬
he der innern Kontaktoberfläche (Porendiffusion).
- Oberflächenreaktion (Adsorption, chemische Reaktion).
Aus diesen Tatsachen resultieren auch die komplexen Temperaturabhängigkeiten der
"Bruttoreakäonsgeschwindigkeitskonstanten", wie sie in Figur 3 wiedergegeben sind.
Ausführlichere Zusammenstellungen dieser Teilschritte finden sich u.a. in den Litera¬
turstellen (181-184).
Es schien nun angezeigt, zuvor diese Stofftransporteinflüsse festzustellen, um dann in
einer zweiten Phase die eigentlichen Reaktionsgeschwindigkeitskonstanten der Acetylen-
hydratisierung an den Zink-(H)-, Cadmium-(H)-, Kupfer-(II)-, Silber-(I)- und Quecksil-
ber-(II)-13X-Katalysatoren bestimmen zu können.
^r V\ c
\t
\Ta
l/T [V1)
Figur 3 : Arrhenius-Darstellung
der Bruttoreaktionsgeschwindigkeitskon-
stante von heterogenen Gasphasenreak¬
tionen (E: Aktivierungsenergie der Kata¬
lyse)

39
a: Adsorption, chemische Reaktion (Neigung = -E/R)
b: Porendiffusion (Neigung = -E/2R)
c: Filmdiffusion (Neigung = (l-2)/R)
d: homogene Reaktion
3.3.1. Einfluss des kornexternen Stoff trans portes
Der externe Stofftransport kann sich sowohl in Form der Fimdiffusion als auch in Form
der Ruckmischung auf den Ablauf der eigentlichen chemischen Umsetzung auswirken.
a) Filmdiffusion
Um den Einfluss der externen Diffusion der Acetylenhydratisierung bestimmen zu kön¬
nen, wurden bei 280 C und konstantem Zeitfaktor (t.) der Anfangsacetylenumsatz als
Funktion der modifizierten Reynoldszahl (Re') fur zwei verschiedene Zn-13X-KataIy-
satoren gemessen. Zudem wurde mit Hilfe theoretischer Berechnungen der Einfluss der
Filmdiffusion abgeschätzt.
An Stelle der bei homogenen Katalysen üblichen Kontaktzeit wurde der in heterogenen
Systemen geeignetere Zeitfaktor (t.) eingeführt:
Ac,o
tf : Zeitfaktor, mit [E ] abgekürzt [(l-Schuttvol.)h/Mol Ac.ol3
V„. : Schuttvolumen [dm ]
m\ : Anfangsmolendurchsatz von Acetylen [Mol/h]
Die modifizierte Reynoldszahl (Re') ist ein Mass fur die im Katalysatorbett herrschen¬
den Stromungsbedingungen.S.v.d
Re'=—-£ [32] (174)Mm
Re' : modifizierte Reynoldszahl [— ]
s : Dichte des Reaktionsgemisches [kg/m ]
v : Geschwindigkeit im leeren Rohr [m/s]
d : mittlerer Durchmesser der Katalysatorkorner [m]
u : Viskosität der Gasmischung [kg/m.s]
Der aequivalente Kugeldurchmesser (d ) fur zylindrische Korner berechnet sich nach
Formel [33]:

40
(d~.x +T2/2)1/2 [33] (180)
mittlere Lange der Zylinder
Durchmesser der Zylinder
aequivalenter Kugeldurchmesser
[m]
[m]
[m]
Die Viskosität (u ) des Acetylen-Wasser-Stickstoff-Gemisches kann nach Beziehung
[34] ermittelt werden.
M
= ^ [y .u . (M )0,5] / [ ^ ym 1=1 î î î 1=1 ^i
Molanteil der Komponente i
Viskosität der Komponente i
Molekulargewicht der Komponente l
(m/'5] [34] (175, 176)
(177, 178)
Gelingt es nun, die Acetylenumsatze als Funktion der modifizierten Reynoldszahl (Re')
zu bestimmen, kann der Einfluss der Filmdiffusion abgeschätzt werden.
In Vorversuchen wurde festgestellt, dass die Aktivität als Funktion der Reaktionsdauer
abnimmt. Deshalb mussten bei jedem Versuch mehrere Proben entnommen und analy¬
siert werden. Beim Auftragen der reziproken Acetylenhydratisierungsgeschwindigkei-
ten als Funktion der Reaktionsdauer wurden Geraden erhalten (siehe Kapitel 3.4.). Durch
graphische Extrapolation konnten die Anfangsgeschwindigkeiten und daraus die Anfangs-
acetylenumsatze ermittelt werden.
Die Reaktionsvariablen sowie die zeitliche Abnahme der Reaktionsprodukte und der Hy¬
dratisierungsgeschwindigkeit in Abhängigkeit des Zeitfaktors (t.) sind in den Tabellen
15 und 16 zusammengestellt.
Tabelle 15 : Reaktionsvariablen; Zn-13X-Katalysatoren
Versuch fK V Re' m*Ac,o W,o mt
1cm ]
a' Pt tp
[Nr.] L°C] Lm/s] L-] [Mol/h] [Mol/h] [Mol/h] [%} [arm] [h]
1 280 0,485 25,6 0,67 4,34 6,52 10,6 63 0,967 12 280 0,97 51,2 1,34 8,68 13,1 21,2 63 0,974 1/23 280 0,485 25,2 0,435 4,34 6,52 10,6 70 0,967 14 280 0,97 50,4 0,87 8,68 13,1 21,2 70 0,974 1/2
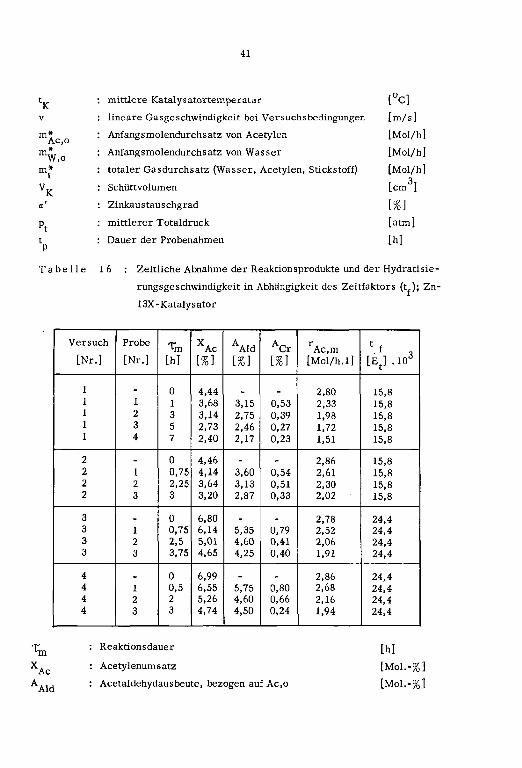
41
Ac,o
W,o
t
P
Tabel
: mittlere Katalysatortemperatur [ C]
: lineare Gasgeschwindigkeit bei Versuchsbedingungen [m/s]
: Anfangsmolendurchsatz von Acetylen [Mol/h]
: Anfangsmolendurchsatz von Wasser [Mol/h]
: totaler Gasdurchsatz (Wasser, Acetylen, Stickstoff) [Mol/h]r 3,
: Schuttvolumen Lern J
: Zinkaustauschgrad [%]
: mittlerer Totaldruck [atm]
: Dauer der Probenahmen [h]
le 16 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisie¬
rungsgeschwindigkeit in Abhängigkeit des Zeitfaktors (t ); Zn-
13X-Katalysator
Versuch ProbeTm
x. A A^, r. t„Ac Aid Cr Ac,m f
3
[Et] .103[Nr.] [Nr.] [h] [%] [%] [%I [Mol/h.1]
. 0 4,44 _ _ 2.80 15,81 1 3,68 3,15 0,53 2,33 15,82 3 3,14 2,75 0,39 1,98 15,83 5 2,73 2,46 0,27 1,72 15,84 7 2,40 2,17 0,23 1,51 15,8
2 - 0 4,46 - - 2,86 15,82 1 0,75 4,14 3,60 0,54 2,61 15,82 2 2,25 3,64 3,13 0,51 2,30 15,82 3 3 3,20 2,87 0,33 2,02 15,8
3 - 0 6,80 - - 2,78 24,43 1 0,75 6,14 5,35 0,79 2,52 24,43 2 2,5 5,01 4,60 0,41 2,06 24,43 3 3,75 4,65 4,25 0,40 1,91 24,4
4 » 0 6,99 - . 2,86 24,44 1 0,5 6,55 5,75 0,80 2,68 24,44 2 2 5,26 4,60 0,66 2,16 24,44 3 3 4,74 4,50 0,24 1,94 24,4
T
Ac
^Ald
: Reaktionsdauer
: Acetylenumsatz
: Acetaldehydausbeute, bezogen auf Ac,o
[hl
[Mol.-%]
[Mol.-%]

42
"Cr
r.Ac,m
: Crotonaldehydausbeute, bezogen auf Ac,o
: mittlere Hydratisierungsgeschwindigkeit
[Mol.-%I
[Mol/h(l-Schuttvol.)l
In Figur 3a ist der Anfangsacetylenumsatz (X, ) als Funktion der modifizierten Rey¬
noldszahl (Re') aufgetragen.
8
Umsatz
[Mol.-%]
"V
"V•^
a" : 70 %O
tf : 24,4 [Et]
0o' : 63 %
9
tf : 15,8 [E(]
10 20 30
Re'
40
5±!
50 60
Figur 3a : Anfangsumsatz des Acetylens als Funktion der modifizierten Reynolds¬
zahl bei konstantem Zeitfaktor (Kontrolle des Filmdiffusionseinflusses)
Wie zu erwarten war, ist der Umsatz im turbulenten Gebiet ( Re'>10 ) (179) unabhängig
von der Gasgeschwindigkeit. Alle folgenden Versuche wurden deshalb bei Re'>20 durch¬
geführt, um sicher ausserhalb des Filmdiffusionseinflusses zu sem.
Im weiteren gibt es auch rechnerische Methoden, um abzuschätzen, ob die Filmdiffusion
die Gesamtreaktion beeinflusst. Nachstehend sei ein solches Kriterium näher erörtert.
Das Ziel dieser Methode besteht darin, die Geschwindigkeit des Stofftransportes mit der
Geschwindigkeit der chemischen Reaktion zu vergleichen.
Der StoffÜbergangskoeffizient (kr) ist eine Funküon der modifizierten Reynoldszahl (Re'):
kG.p{,,M.
( ^ )\ 3-DAc.m I
2/3
f(Re')=JD [35] (180)
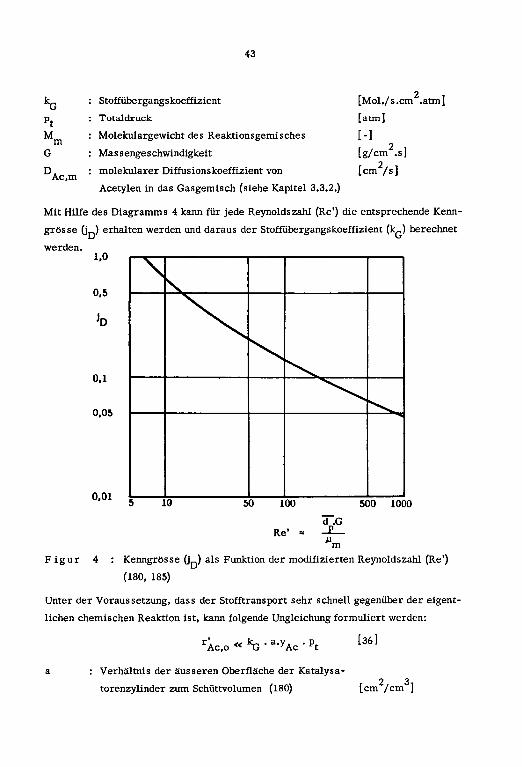
43
M
Ac,m
Stoffubergangskoeffizient
Totaldruck
Molekulargewicht des Reaktionsgemisches
Massengeschwindigkeit
molekularer Diffusionskoeffizient von
Acetylen in das Gasgemisch (siehe Kapitel 3.3.2.)
[Mol./s.cm .atm]
[atm]
t-I
[g/cm .s]
[cm /s]
Mit Hilfe des Diagramms 4 kann fur jede Reynoldszahl (Re') die entsprechende Kenn-
kG>
1,0
grosse (j_) erhalten werden und daraus der Stoffubergangskoeffizient (kn) berechnet
werden.
0,5
0,1
0,05
0,01500 1000
d .G
Re' =
Figur 4 : Kenngrosse (j_) als Funktion der modifizierten Reynoldszahl (Re')
(180, 185)
Unter der Voraussetzung, dass der Stofftransport sehr schnell gegenüber der eigent¬
lichen chemischen Reaktion ist, kann folgende Ungleichung formuliert werden:
rÄc,o « ka • a-yAc • pt [36]
Verhältnis der äusseren Oberflache der Katalysa¬
torenzylinder zum Schuttvolumen (180) [cm /cm ]

44
Ac,o: Acetylenhydratisierungsgeschwindigkeit [Mol/cm Schuttvol.s]
In Tabelle 17 sind die Anfangsacetylenhydratisierungsgeschwindigkeiten und die Ge¬
schwindigkeit des Acetylentransportes zum Katalysatorkorn fur die Versuche 1 und 3
aufgeführt.
Tabelle 17 : Geschwindigkeit der Acetylenhydratisierung und des Acetylen-
transportes zum Katalysatorkorn
Versuch
[Nr.]
Re' Sc
[-]
JDH [Mol/s.cm .atm]
r'.Ac,o „
[Mol/cm .s]V^Aç/t[Mol/cm .s]
1
3
25,6
25,2
0,650
0,6551
0,3
0,3
6,42.10"46.38.10"4
0,779.10"60.772.10"6
0.900.10"30.576.10"3
Der Wert fur a wurde aus einer Tabelle in (180) entnommen und betragt ca. 14 cm /
Aus den berechneten Werten der Tabelle 17 geht eindeutig hervor, dass die chemische
Reaktion ca. lOOOmal langsamer ablauft als der Acetylentransport zum Katalysatorkorn.
Es ware somit durchaus möglich, bei noch kleineren Geschwindigkeiten zu arbeiten, oh¬
ne dass sich die Filmdiffusion auf die Gesamtreaktion hemmend auswirken wurde.
b) Ruckmischung
Zusatzlich kann sich der Emfluss des externen Stofftransportes auch m Form der Ruck¬
mischung auf die Gesamtreaktion auswirken.
Bov.L
[37] Bo = Bo'
*P138]
L
D
lineare Gasgeschwindigkeit
charakteristische Lange
axialer Diffusionskoeffizient
[m/s]
[mj
[m2/s]
Fur turbulent stromende Gase (Re' > 10) ist Bo' = 2 (179). Zudem gilt, dass sich der
Einfluss der Ruckmischung bei Bo > 50 vernachlässigen lasst, sofern der Umsatz einer
Reaktion nicht genauer als 1 % bestimmt werden kann. Bei den eigenen Versuchen konn¬
te der Umsatz nicht genauer als - 5% bestimmt werden und die modifizierten Reynolds -
zahlen waren stets > 10. Fur einen mittleren Korndurchmesser d =2,64 mm war des-P

45
halb nach Gleichung [38] nur bei Versuchen mit Katalysatorschichten wesentlich unter
6 cm eine feststellbare Beeinflussung des Umsatzes durch die Ruckmischung; zu erwar¬
ten. Bei den eigenen Versuchen war die Betthohe 6 bzw. 12 cm. Demzufolge ist der Ern-
fluss der Ruckmischung vernachlassigbar klein.
Zusammenfassend sei festgehalten, dass bei den folgenden Untersuchungen weder Film-
diffusion noch Ruckmischung den Ablauf der Acetylenhydratisierung zu beeinflussen ver¬
mögen, wenn die modifizierte Reynoldszahl Re'> 10 und die Betthohe > 6 cm sind, was
auch immer zutraf.
3.3.2. Einfluss der Porendiffusion
Da bei katalytischen Reaktionen vor allem bei Katalysatorkornern mit kleinen Poren mit
einer Beeinflussung der Porendiffusion auf die Gesamtreaktion zu rechnen ist, sollen ne¬
ben den Grundlagen zur Beurteilung des Porendiffusionseinflusses auch die Auswirkungen
der korninternen Temperaturgradienten und der Adsorption auf die Porendiffusion selbst
naher untersucht werden. Das vorausberechnete Verhalten soll an Hand einiger Experi¬
mente verifiziert werden.
Der Einfluss der Porendiffusion kann sich auf drei Arten auswirken
a) gewöhnliche Diffusion
b) Knudsen-Diffusion
c) Oberflachendiffusion
Um beurteilen zu können, ob Knudsen- oder gewohnliche Diffusion vorherrscht, wurden
die betreffenden Diffusionskoeffizienten miteinander verglichen.
Der gewöhnliche Diffusionskoeffizient von Acetylen in ein Wasserdampf-Stickstoffge-
misch kann nach Gleichung [39] berechnet werden (180) :
Dlm - 0-yi) / g ^ [39]
D. : Diffusionskoeffizient von Acetylen in das Stickstoff-Wasser¬
dampfgemisch [cm /s]
D. : Diffusionskoeffizient von Acetylen in Stickstoff bzw. Wasser¬
dampf [cm /s]
y : Molenbruch [-]

46
0,001858.T3/2 [(M.+M )/M,.M ]I/2D =
2t401
T, p : Absoluttemperatur und -druck [ K, atm]
G" : Konstante aus der Leonard-Jones-Gleichung (180)
&D : Stos s integral (180)
M : Molekulargewichte
Der Knudsendiffusionskoeffizient ist eine Funktion des aequivalenten Porenradius, der
Temperatur und des Molekulargewichtes.
DK = 9700.re (T/M)1/2[41]
r : aequivalenter Porenradius des Katalysators; streng nur fur zylindrische Po¬
ren und em enges Porenspektrum gültig [cm]
Die jeweiligen Betrage fur den totalen effektiven Diffusionskoeffizienten ergeben sich
aus der Gleichung [42] .wobei sich die effektiven Diffusionskoeffizienten D.„und
K1,eff
l-U+N.j/NpyjD«
D,.,
D„«
eff lm.eff K.,eff
[42]
N2/N. ; Verhältnis des Diffusionsstromes aus den Poren zum [-]
Strom m die Poren hinein nach dem 1. Fick'schen Gesetz
y. : Anfangsmolenbruch des Acetylens [-]
Deff = D f M
0 : Porosität des Katalysatorkorns [-1
T : Tortuositatsfaktor (berücksichtigt, das s die Poren nicht
geradlinig verlaufen) [-]
In Tabelle 18 und Figur 5 sind die gewöhnlichen (D, ), Knudsen-, (D„ ) und totalen ef-lm K,
fektiven Diffusionskoeffizienten (D „) von Acetylen in ein Wasserdampt-Stickstoff-Ge-misch in Abhängigkeit der Temperatur aufgeführt. Die Werte fur D beziehen sich auf
Anfangsmolenbruche von 0,103 fur Acetylen, 0,232 fur Stickstoff und 0,665 fur Wasser.
D„ bezieht sich auf einen Druck von 0,967 atm, einen aequivalenten Porenradius von
Kl

47
16,9 bzw. 680 Ä und eine Porosität von 0,505. Alle diese verwendeten Daten entsprechen
dem im Kapitel 3.2.4.2. aufgeführten Zn-13X-Katalysator mit einem Austauschgrad von
68%. Das Verhältnis des Diffusionsstromes war wegen des grossen Wasseruberschus-
ses immer ca. eins. Die Genauigkeit des effektiven Diffusionskoeffizienten wird durch
den schwer abzuschätzenden Tortuositatsfaktor (T) beeinträchtigt. Nach Angaben von
Satterfield et al. (180) und Weis z et al. (186) wurde furf=2 eingesetzt.
Tabelle 18 : Temperaturabhangigkeit des gewöhnlichen (Dt ), Knudsen-
(D, ) und des totalen effektiven Diffusionskoeffizienten (D ,_)k. eti
von Acetylen in ein Wasserdampf-Stickstoff-Gemisch; D„ in
1Bezug auf den Zn-13X-Katalysator mit einem Austauschgrad
von 68 %
T Dlm DK> bezog
r =16,9 Ke
en auf
re=680 ADeff, bez
rg= 16,9 X
ogen auf
r =680 Ke
T
[°K] [cm /s] [cm2/s].103 [cm /s] [cm2/s].103 [cm /s] [°c]
473 0,532 6,96 0,280 1,73 0,0498 200
523 0,644 7,34 0,295 1,83 0,0545 250
573 0,756 7,69 0,309 1,92 0,0586 300
623 0,88 8,03 0,323 2,02 0,0629 350
673 1,00 8,30 0,334 2,08 0,0665 400
Wie schon im Kapitel 3.2.4.2. besprochen wurde, besitzt das Molekularsieb Zn-13X
zwei verschiedene Porensysteme. Demzufolge muss auch die Diffusionsgeschwmdigkeit
der Acetylenmolekule in diese Hohlraumsysteme verschieden gross sein. Aus Tabelle
18 geht hervor, dass der totale effektive Diffusionskoeffizient von Acetylen in die klei¬
nen Poren hinein im Temperaturintervall von 200-400°C 1,73 bis 2,08.10 cm/s, der
2entsprechende Diffusionskoeffizient in die grossen Poren aber 0,0498 bis 0,0665 cm /s
betragt. Der totale effektive Diffusionskoeffizient in die kleinen Poren hangt demnach
allein vom Knudsendiffusionskoeffizient ab, derjenige in die grossen Poren sowohl vom
Knudsen- als auch vom gewöhnlichen Diffusionskoeffizienten.
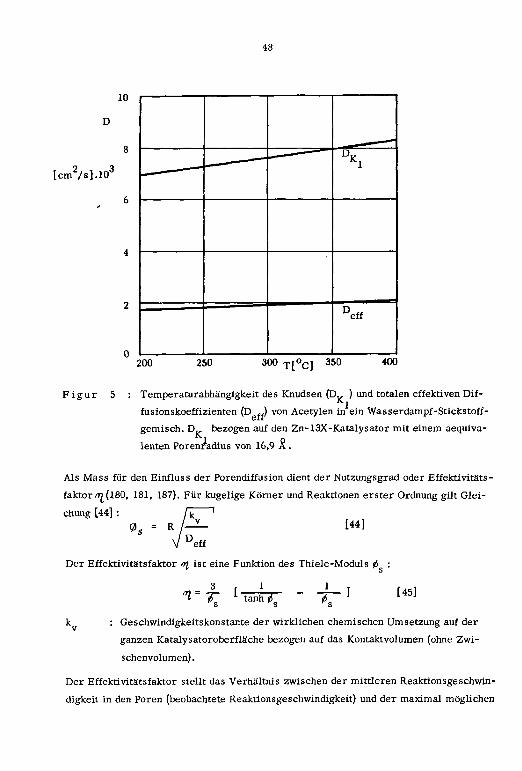
48
10
[cm2/s].103
DK
Kl
Deff
200 250 300 T[°C] 350 400
Figur 5 : Temperaturabhangigkeit des Knudsen (D„ ) und totalen effektiven Dif-
1fusionskoeffizienten (D .,) von Acetylen in ein Wasserdampf-Stickstoff-
gemisch. D„ bezogen auf den Zn-13X-Katalysator mit einem aequiva-
1 n
lenten Porenradius von 16,9 A.
Als Mass fur den Einfluss der Porendiffusion dient der Nutzungsgrad oder Effektivitats -
faktor it? (180, 181, 187). Fur kugelige Korner und Reaktionen erster Ordnung gilt Glei¬
chung [44] :
0, = R /— [44]
[45]
Der Effektivitatsfaktor *l ist eine Funktion des Thiele-Moduls 0
T± [_i_i>
ltanh 0
1I
k : Geschwindigkeitskonstante der wirklichen chemischen Umsetzung auf der
ganzen Katalysatoroberflache bezogen auf das Kontaktvolumen (ohne Zwi¬
schenvolumen).
Der Effektivitatsfaktor stellt das Verhältnis zwischen der mittleren Reaktionsgeschwin¬
digkeit in den Poren (beobachtete Reaktionsgeschwindigkeit) und der maximal möglichen

49
Geschwindigkeit dar, welche sich beobachten liesse, wenn über der ganzen inneren Ka¬
talysatoroberflache dieselbe Konzentration und Temperatur herrschten wie an der Teil-
chenau s s enflache.
Da k meist unbekannt ist, wird nj leichter aus graphischen Darstellungen der Hilfs-
funktion [46] (Figur 6) erhalten, welche nur experimentell ermittelbare Grossen ent¬
halt.
T R r l dnl 1 R,2
[46]
3V : totales Kornvolumen (Feststoff) [cm ]
dn : beobachtete Abnahmegeschwindigkeit des Eduktes [Mol/s]dt
c : Edukt-Konzentration an der Kornaussenflache; ists
die externe Diffusion vernachlas sigbar, wird c lden-S
3tisch mit der Konzentration c im Hauptstrom. [Mol/cm ]
Wird Gleichung [46] an die eigenen Verhaltnisse angepasst, erhalt man die Beziehung
M'-T R2 kE"VVM* =
frr d-WA -3600w
eff "Ac.o
kE : Reaktionsgeschwindigkeitskonstante l(g-Katalysator)h ^
3
j, : Schuttdichte der Katalysatorschicht [g-Kat./cm Schuttvol.]
£ : Leervolumenanteil in der Schuttschicht [--]3
V„ : Molvolumen [cm /g-Mol]
R : Radius der kugelförmigen Korner [cm]2
D,, : totaler effektiver Diffusionskoeffizient [cm /s]
Zusammen mit Diagramm 6 gestattet Gleichung [47] aus experimentellen Reaktionsge¬
schwindigkeitskonstanten erster Ordnung den Einfluss der Porendiffusion in kugelförmi¬
gen Katalysatorkornern abzuschätzen.
Die bis dahin dargelegten Beziehungen zum Ermitteln von Effektivitätsfaktoren gelten
einzig unter den vereinfachenden Voraussetzungen, dass die betrachtete Reaktion in¬
nerhalb eines Katalysatorkornes keinen Temperaturgradienten erzeugt und keinen Ad-
sorptionseinfluss zeigt.
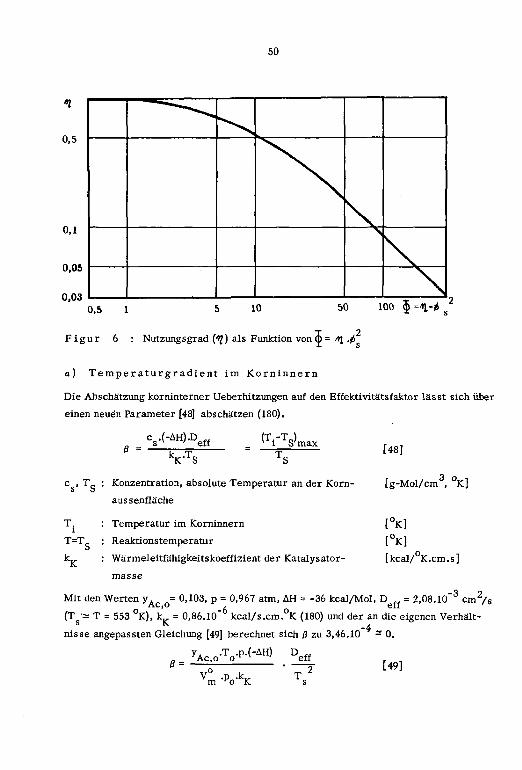
50
1
0,5
0,1
0,05
0,03 _
0,5 1 5 10 50 100 $=<V*S
Figur 6 : Nutzungsgrad (^) als Funktion von q> = />I .i>
a) Temperaturgradient im Korninnern
Die Abschätzung korninterner Ueberhitzungen auf den Effektivitatsfaktor lasst sich über
einen neuen Parameter [48] abschätzen (180).
c .(-AH).D„
s' '
eff
k TKK-1S
(T -Tc)v
l S'max
VTs
i
T=T„
Konzentration, absolute Temperatur an der Korn¬
aussenflache
Temperatur im Korninnern
Reaktionstemperatur
Warmeleitfahigkeitskoeffizient der Katalysator¬
masse
[48]
[g-Mol/cm3, °K]
[°K]
[°K]
[kcal/ K.cm.s]
Mit den Werten y = 0,103, p = 0,967 atm, AH = -36 kcal/Mol, D = 2,08.10" cni /sAC|0 ., 611
(T = T = 553 °K), k^ = 0,86.10 kcal/s.cm.°K (180) und der an die eigenen Verhalt-s K
-4msse angepassten Gleichung [49] berechnet sich 0 zu 3,46.10 - 0.
„yAc,o-To-P^AH) Deff
f ,
m *o K

51
Im Innern der Katalysatorkorner tritt somit unter den gegebenen Bedingungen eine
maximale Temperaturdifferenz von nur 0,2 K auf, die zur Berechnung des Effekti-
vitatsfaktors vernachlässigt werden darf.
ß) Einfluss der Adsorption
Bei der Acetylenhydratisierung herrscht nicht die physikalische, sondern die "akti¬
vierte" Adsorption oder Chemisorption vor. Da Adsorption und Desorption meist sehr
schnell verlaufen und analog zu den chemischen Umsetzungen exponentiell von der Tem¬
peratur abhangen, wirken sie allein durch die Lage des Gleichgewichtes und nicht durch
ihre Geschwindigkeit auf das kinetische System ein. Diese Vorgange bestimmen somit
die Konzentration der Reaktanden auf der Katalysatoroberflache, kontrollieren die Re¬
aktion aber nicht.
Der Einfluss der Chemisorption lasst sich nur schwer vorausberechnen, denn die da¬
zu notwendigen Adsorptionsgleichgewichtskonstanten der Edukte und Produkte sind nur
bei Bedingungen zuganglich, unter welchen zugleich die chemische Reaktion ablauft (188).
Acetylen und eventuell auch Wasser werden an den aktiven Zentren chemisorbiert und
wahrscheinlich bedeckt auch Acetaldehyd den Katalysator.
Fuhren also Edukte und Produkte zu Adsorptionsgleichgewichten, muss der Thiele-Mo¬
dul 0 durch die modifizierte Form i> [50] ersetzt werden (189).
lc* R T
^m = L-üfr [50]A.eff
k* : modifizierte Reaktionsgeschwindigkeitskonstante, [Mol/arm.cm ,s]
schliesst Adsorptionstherme ein
L : Dicke der Katalysatorplatte [cm]
''A.eff2
D, .. : effektiver Diffusionskoeffizient des Eduktes [cm Is\
Auch hier exmpfiehlt es sich, eine Variable (j). einzufuhren, welche nur experimentell
erfassbare Grossen enthalt (Figur 7).
K[atm ] berücksichtigt den Adsorptionseinfluss; p, entspricht dem Acetylenpartial-
druck an der Katalysatoraus senflache. Der Parameter (K.p4 ) stellt somit ein Mass furAC
die Starke und Art der Adsorption dar (siehe Figur 7). Zwischen der Dicke L der Kata¬
lysatorplatte und dem Radius R kugeliger Katalysatorkorner bei einer Reaktion erster
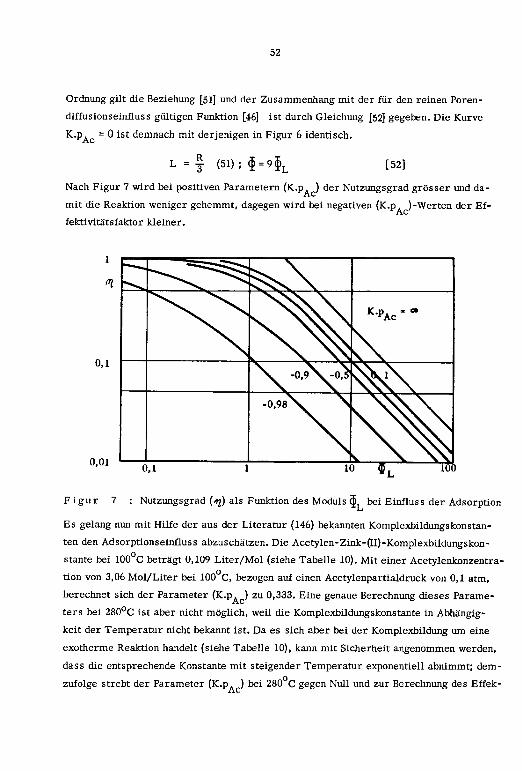
52
Ordnung gilt die Beziehung [51] und der Zusammenhang mit der fur den reinen Poren¬
diffusionseinflus s gültigen Funktion [46] ist durch Gleichung [52] gegeben. Die Kurve
K.p. = 0 ist demnach mit derjenigen in Figur 6 identisch.
RL =
f (51); (j> = 9(j>L [52]
Nach Figur 7 wird bei positiven Parametern (K.p4 ) der Nutzungsgrad grosser und da-AC
mit die Reaktion weniger gehemmt, dagegen wird bei negativen (K.p. )-Werten der Ef-
fektivitatsfaktor kleiner.
0,1
0,01
K-PAc=-
\. -0,9\-0,5 xvs/x
-0.98N. \
0 .1 1 1 o $L 100
Figur 7 : Nutzungsgrad (/rç) als Funktion des Moduls (]j bei Einflus s der Adsorption
Es gelang nun mit Hilfe der aus der Literatur (146) bekannten Komplexbildungskonstan¬
ten den Adsorptionseinfluss abzuschätzen. Die Acetylen-Zink-(II)-Komplexbildungskon-
stante bei 100 C betragt 0,109 Liter/Mol (siehe Tabelle 10). Mit einer Acetylenkonzentra-
tion von 3,06 Mol/Liter bei 100 C, bezogen auf einen Acetylenpartialdruck von 0,1 atm,
berechnet sich der Parameter (K.p, ) zu 0,333. Eine genaue Berechnung dieses Parame¬
ters bei 280°C ist aber nicht möglich, weil die Komplexbildungskonstante m Abhängig¬
keit der Temperatur nicht bekannt ist. Da es sich aber bei der Komplexbildung um eine
exotherme Reaktion handelt (siehe Tabelle 10), kann mit Sicherheit angenommen werden,
das s die entsprechende Konstante mit steigender Temperatur exponentiell abnimmt; dem¬
zufolge strebt der Parameter (K.p, ) bei 280 C gegen Null und zur Berechnung des Effek-

53
tivitatsfaktors kann Diagramm 6 und Gleichung [46] bzw. [47] angewandt werden.
Mit den Werten R = 0,132 cm, k„ = 3,64.10"3Mol(g-Kat.)h (Versuche 5-8), 9= 0,642
g/cm3, VM = 47.JO3 cm3/Mol, D* = 1,9.10"3 cm2/s, E= 0,467 und yAc= 0,103 berech¬
net sich $ zu 5 und />j= 0,7 (j> = 3, fi - 1). Dieser Wert gilt fur Poren mit einem aequi-
valenten Radius von 16,9 A. Das Katalysatorkorn besitzt aber noch Poren mit einem mitt¬
leren Radius von 680 A. Fur diese rund 40mal grosseren Poren wird <| = 0,18 und /?£ = 1.
In den grossen Poren herrscht somit überall die gleiche Konzentration und da die exter¬
ne Diffusion nicht den geschwindigkeitsbestimmenden Schritt darstellt, wird die Konzen¬
tration in diesen Poren gleich derjenigen im Gasstrom.
Die totale Oberflache des Zn-13X-Katalysators mit einem Austauschgrad von 68 % (sie¬
he Tabelle 14) betragt 304 m /g, davon entfallen auf die Poren über 75 Ä 10 m /g und
-3 2auf die äussere Oberflache ungefähr 2,2.10 m /g (Partikeldurchmesser: 2,64 mm).
Wird nun durch Zerkleinern der Katalysatorkorner die äussere Oberflache vergrossert
-3 -3 2auf ca. 6,0.10 bzw. 15.10 m /g entsprechend dem Partikeldurchmesser von 0,8 bzw.
0,6 mm, bleibt trotzdem die innere Oberflache der grossen und folglich auch der klei¬
nen Poren vollumfanglich erhalten. Theoretisch sollte demnach ein Zerkleinern der Ka¬
talysatorkorner von 2,64 auf 0,60 mm Durchmesser kernen Einfluss auf die Gesamtreak¬
tion haben.
Diese Tatsache konnte durch die in Tabelle 19 und Figur 8 zusammengestellten Versu¬
che bestätigt werden. Die Acetylenhydratisierungsgeschwindigkeit wurde als Funktion
der Reaktionsdauer fur Katalysatorkörner von 2,64 bis 0,424 mm Durchmesser gemes¬
sen. Die Reaktionsgeschwindigkeiten sind in den genannten Grenzen unabhängig von der
Partikelgrosse.
Aus den Berechnungen und aus den Resultaten der Tabelle 19 lasst sich zusammenfas¬
send festhalten, dass die Acetylendiffusion in Poren unter 75 A die Gesamtreaktion zwar
hemmt, experimentell aber nicht feststellbar ist, wie dies aus theoretischen Erörterun¬
gen auch vorausgesehen werden konnte. Dagegen herrscht in Poren über 75 A eine gleiche
Acetylenkonzentration wie im Hauptstrom und die Diffusion in diese Poren hinein ist nicht
ge schwindigkeits be stimmend.
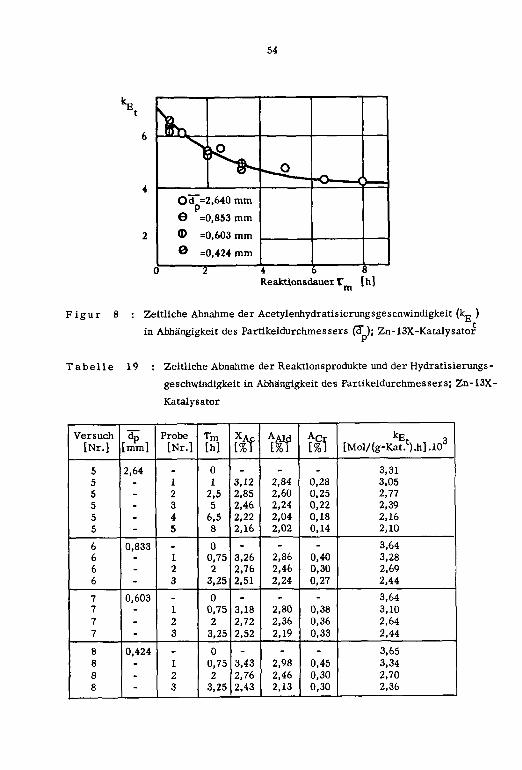
54
6
4
2
i,0
""-O-—
Od =2,640 mmP
0 =0,853 mm
© =0,603 mm
0 =0,424 mm
C 2 4 6 3
Reaktlonsdauer V [ h]
Figur 8 : Zeitliche Abnahme der Acetylenhydratisierungsgescnwindigkeit (kp )
in Abhängigkeit des Partikeldurchmessers (3" ); Zn-13X-Katalysator
Tabelle 19 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisierungs¬
geschwindigkeit in Abhängigkeit des Partikeldurchmessers; Zn-13X-
Katalysator
Versuch
[Nr.]dp[mm]
Probe
[Nr.]Tm[h]
xAc[561 ttff ACr
[Mol/(g-Kat. ).h] 103
5 2,64 . 0 . - . 3,31
5 - 1 1 3,12 2,84 0,28 3,05
5 - 2 2,5 2,85 2,60 0,25 2,77
5 - 3 5 2,46 2,24 0,22 2,39
5 - 4 6,5 2,22 2,04 0,18 2,16
5 - 5 8 2,16 2,02 0,14 2,10
6 0,833 . 0 - - - 3,64
6 - 1 0,75 3,26 2,86 0,40 3,28
6 - 2 2 2,76 2,46 0,30 2,69
6 - 3 3,25 2,51 2,24 0,27 2,44
7 0,603 - 0 - - - 3,64
7 - 1 0,75 3,18 2,80 0,38 3,10
7 - 2 2 2,72 2,36 0,36 2,64
7 - 3 3,25 2,52 2,19 0,33 2,44
8 0,424 - 0 - - - 3,65
8 - 1 0,75 3,43 2,98 0,45 3,34
8 - 2 2 2,76 2,46 0,30 2,70
8 - 3 3,25 2,43 2,13 0,30 2,36

55
Mittlere Katalysatortemperatur 280°C
Katalysatormenge 6,83 g
Zmkaustauschgrad 68%
Totaler Gasdurchsatz 6,52 Mol/h
Anfangsmolendurchsatz von Acetylen 0,67 Mol/h
Anfangsmolendurchsatz von Wasser 4,34 Mol/h
Totaldruck 0,967 atm
Dauer der Probenahmen lh
3.4. Katalysatorvergiftung
Da die Aktivität mit zunehmender Reaktionsdauer abnimmt (siehe Figur 8), wurde ver¬
sucht, die Katalysatorvergiftung mathematisch zu formulieren, das Gift zu identifizie¬
ren und die Möglichkeit einer Regenerierung zu untersuchen.
Bei der Katalysatorvergiftung durch einen Fremdstoff können zwei verschiedene Falle
unterschieden werden. Im emen Fall wird das Gift nur schwach adsorbiert und gleich-
massig in allen Poren verteilt, Ist die totale Katalysatoroberflache fur die Reaktion zu¬
gänglich (tg-el), dann ist die katalytische Aktivität direkt proportional der unvergifteten
Oberflache (Figur 9, Kurve B). Ist hingegen die Oberflache nur teilweise zuganglich (/?j
*0, ), fallt die Aktivität langsamer als linear mit der Giftansammlung ab (Kurve A). Im
andern Fall wird das Gift so stark adsorbiert, das s die Poreneingànge verstopfen, bevor
das Poreninnere vergiftet wird (Kurve C).
Wheeler (187) hat diese Vergiftungserscheinungen fur isotherme Reaktionen erster
Ordnung in zylindrischen Poren mathematisch formuliert. Das Verhaltms der Geschwin¬
digkeit des vergifteten zur Geschwindigkeit des nicht vergifteten Katalysators kann durch
die Gleichung [53] mit Hilfe der Beziehung [54] ausgedruckt werden.
F =
^./l-a.tanh itUo.jï=i)*L,o- tanML,o
Ac,m
Ac,o
3.0L,o
*.s.o
[53]
[54]
Verhältnis der Aktivität des vergifteten zur Aktivität
des mcht vergifteten Katalysators
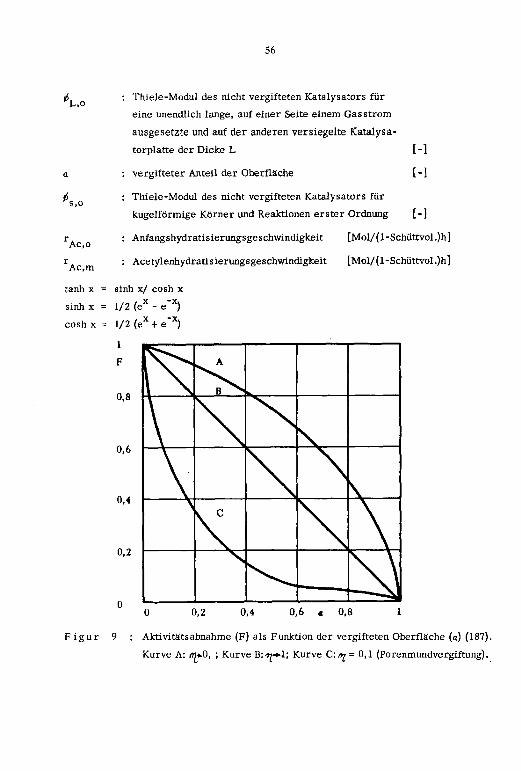
56
"L.o
Ac,o
r,Ac,m
tanh x
sinh x
cosh x
: Thiele-Modul des nicht vergifteten Katalysators fur
eine unendlich lange, auf einer Seite einem Gasstrom
ausgesetzte und auf der anderen versiegelte Katalysa¬
torplatte der Dicke L
: vergifteter Anteil der Oberfläche
: Thiele-Modul des nicht vergifteten Katalysators fur
kugelförmige Korner und Reaktionen erster Ordnung
: Anfangshydratisierungsgeschwindigkeit
: Acetylenhydratisierungsgeschwindigkeit
smb. x/ cosh x
1/2 (ex - e"x)
1/2 (eX + e"x)
1
F
0,8
[-]
[-]
[-]
[Mol/(l-Schuttvol.)h]
[Mol/(l-Schuttvol.)h]
0,6
0,4
0,2
A
V C
0,2 0,4 0,6 0,8
Figur 9 : Aktivitatsabnahme (F) als Funktion der vergifteten Oberflache (a) (187).
Kurve A: /)j*0, ; Kurve B:^-»l; Kurve C-./y = 0,1 (Porenmundvergiftung).
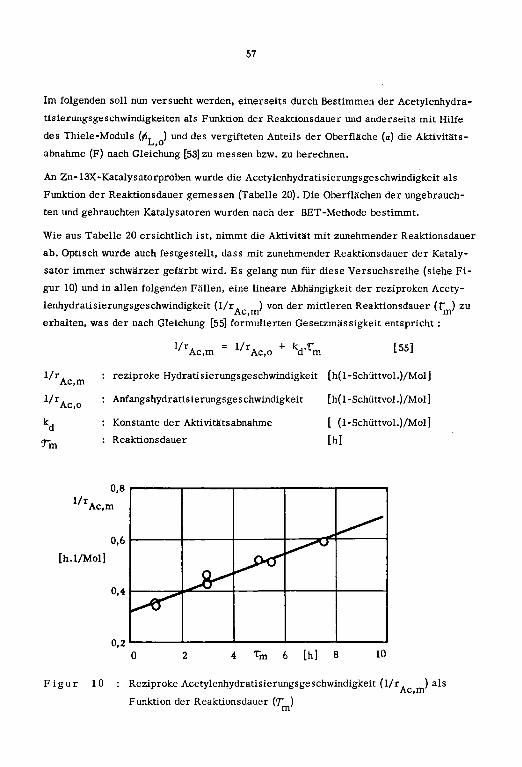
57
Im folgenden soll nun versucht werden, einerseits durch Bestimmen der Acetylenhydra -
tisierungsgeschwindigkeiten als Funktion der Reaktionsdauer und anderseits mit Hilfe
des Thiele-Moduls (>S ) und des vergifteten Anteils der Oberflache (a) die Aktivitats-
abnahme (F) nach Gleichung [53] zu messen bzw. zu berechnen.
An Zn-13X-Katalysatorproben wurde die Acetylenhydratisierungsgeschwindigkeit als
Funktion der Reaktionsdauer gemessen (Tabelle 20). Die Oberflachen der ungebrauch¬
ten und gebrauchten Katalysatoren wurden nach der BET-Methode bestimmt.
Wie aus Tabelle 20 ersichtlich ist, nimmt die Aktivität mit zunehmender Reaktionsdauer
ab. Optisch wurde auch festgestellt, dass mit zunehmender Reaktionsdauer der Kataly¬
sator immer schwarzer gefärbt wird. Es gelang nun fur diese Versuchsreihe (siehe Fi¬
gur 10) und in allen folgenden Fallen, eine lineare Abhängigkeit der reziproken Acety-
lenhydratisierungsgeschwindigkeit (1/r. ) von der mittleren Reaktionsdauer (T ) zu
erhalten, was der nach Gleichung [55] formulierten Gesetzmassigkeit entspricht :
1/rAc,m = 1/rAc,o + kd-rm I»!
reziproke Hydratisierungsgeschwindigkeit [h(l-Schuttvol.)/Mol]
Anfangshydratisierungsgeschwindigkeit [h(l-Schuttvol.)/Mol]
Konstante der Aktivitatsabnahme [ (l-Schuttvol.)/Mol]
1/r
1/r
Ac,m
Ac,o
d
rmReaktionsdauer [h]
1/rAc,m
[h.l/Mol]
4 Tm 6 [h] 8
Figur 10 : Reziproke Acetylenhydratisierungsgeschwindigkeit (1/r ) als
Funktion der Reakuonsdauer (T )nr

58
Tabelle 20 : Reaktionsprodukte und Hydratisierungsgeschwindigkeit als Funk¬
tion der Reaktionsdauer; Zn-13X-Katalysatoren
Mittlere Katalysatortemperatur 335°C
Schuttvolumen 10,6 cm3
Zinkaustauschgrad 60,8%
Totaler Gasdurchsatz 6,52 Mol/h
Anfangsmolendurchsatz von Acetylen 0,67 Mol/h
Anfangsmolendurchsatz von Wasser 4,34 Mol/h
Totaldruck 0,967 atm
Dauer der Probenahmen lh
Versuch
[Nr.]
Probe
[Nr.] [h]
XAc[%]
AAld ACr[SB!
Ac,m
[Mol/h.l]
14 1 1 4,43 3,56 0,87 2,80
14 2 3 3,72 3,20 0,52 2,35
15 1 1 4,35 3,70 0,65 2,75
15 2 3 3,40 2,90 0,50 2,14
15 3 5 3,03 2,66 0,37 1,91
16 1 1 4,58 3,93 0,65 2,89
16 2 3 3,66 3,14 0,52 2,31
16 3 5,5 3,12 2,80 0,32 1,97
16 4 7,5 2,66 2,43 0,23 1,68
Mit Hilfe der Gesetzmassigkeit [55] war es nun möglich, fur jede Reaktionsdauer (f*)
die Acetylenhydratisierungsgeschwindigkeit (r, ) zu berechnen. Daraus konnte die
Aküvitatsabnahme (F ) nach Formel [56] ermittelt werden.
[56]Acm
gAc,o
Da die Molekularsiebe nur eine monomolekulare Giftschicht anlagern können, ist die
Oberflachenmessung nach BET gemäss Formel [57] repräsentativ fur den vergifteten
Anteil der Oberflache.

59
BET<r'
'bet= 1-a [57]
Die berechnete Aktivitatsabnahme (F, ) wurde nach der schon früher formulierten Ge¬
setzmassigkeit [47] und nach der von Wheeler angegebenen Formel [53] ermittelt,
wobei nachfolgende Werte eingesetzt wurden: R = 0,132 cm, r, = 3,085.10 Mol/
(cm3 Schuttvol.)h, V., = 51.6.103 cm3/Mol, D„
= 2.10"3 --'- '
' M eflcm /s, £ = 0,467 und y. =
0,103. Nach Gleichung [47] erhalt man fur $ = 7 und />£ = 0,6 (tf =4,2; *S = 1,4).
Die Acetylenhydratisierungsgeschwindigkeiten (r, ), die Oberflachen (OßE_) und die
gemessene (F J und berechnete (Fj Aktivitatsabnahme als Funktion der Reaktionsdauer
(T") bzw. des vergifteten Anteils der Oberflache (o) sind in Tabelle 21 und in Figur 11
ersichtlich.
Tabelle 21 Hydratisierungsgeschwindigkeit (r. ), Oberflache (OgET), ge¬
messene (FJ und berechnete (F.) Aktivitatsabnahme als Funktion
der Reaktionsdauer (y*) bzw. des vergifteten Anteils der Oberfla¬
che (a)
Versuch
[Nr.]
r
[h]
rAAc,m
[Mol/h.l]0BETf
[m76,83g]
Fg
[-]
Fb[-]
a
r-]
10 0 3,085 2609 1,00 1,000 0
11 0,5 2,94 2215 0,952 0,895 0,151
12 1 2,78 2097 0,90 0,858 0,196
13 2 2,50 1785 0,81 0,768 0,316
14 4,62 2,04 1403 0,66 0,640 0,462
15 5,5 1,90 1140 0,615 0,545 0,563
16 8,5 1,56 778 0,505 0,398 0,702
Sowohl die Acetylenhydratisierungsgeschwindigkeit als auch die Oberflache nehmen mit
zunehmendem Grad der Vergiftung ab. Beim graphischen Auftragen der gemessenen (F J
und berechneten (F.) Aktivitatsabnahmen in Abhängigkeit des vergifteten Anteils der Ober¬
flache erhalt man relaüv gute Ueberemstimmung, wie in Figur 11 ersichtlich ist. Aus die¬
ser Tatsache lasst sich folgern, dass die Modellvorstellungen der Vergiftung recht über-
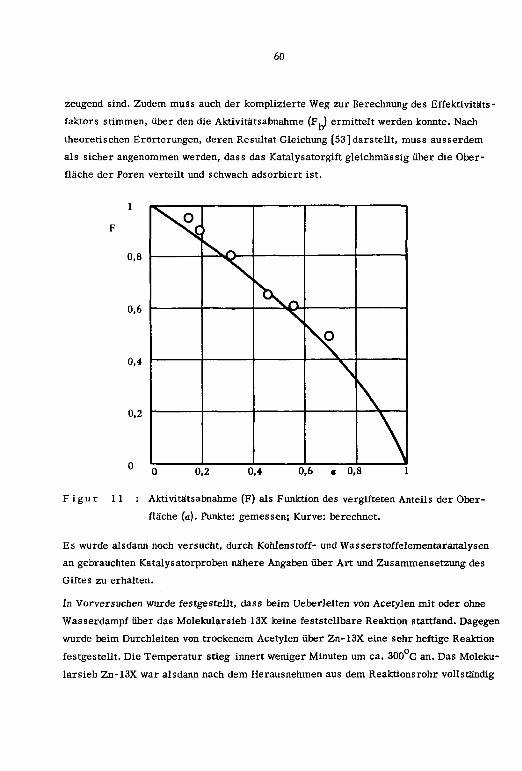
60
zeugend sind. Zudem muss auch der komplizierte Weg zur Berechnung des Effektivitats
faktors stimmen, über den die Aktivitätsabnahme (Fj ermittelt werden konnte. Nach
theoretischen Erörterungen, deren Resultat Gleichung [53] darstellt, muss ausserdem
als sicher angenommen werden, das s das Katalysatorgift gleichmassig über die Ober¬
flache der Poren verteilt und schwach adsorbiert ist.
1
F
0,8
0,6
0,4
0,2
"
0 0,2 0,4 0,6 a 0,8 1
Figur 11 : Aktivitatsabnahme (F) als Funktion des vergifteten Anteils der Ober¬
flache (a). Punkte: gemessen; Kurve: berechnet.
Es wurde alsdann noch versucht, durch Kohlenstoff- und Wasserstoffelementaranalysen
an gebrauchten Katalysatorproben nähere Angaben über Art und Zusammensetzung des
Giftes zu erhalten.
In Vorversuchen wurde festgestellt, dass beim Ueberleiten von Acetylen mit oder ohne
Wasserdampf über das Molekularsieb 13X keine feststellbare Reaktion stattfand. Dagegen
wurde beim Durchleiten von trockenem Acetylen über Zn-13X eine sehr heftige Reaktion
festgestellt. Die Temperatur stieg innert weniger Minuten um ca. 300 C an. Das Moleku¬
larsieb Zn-13X war alsdann nach dem Herausnehmen aus dem Reaktionsrohr vollständig

61
schwarz gefärbt. Demgegenüber vergiftete Acetaldehyd zusammen mit Wasserdampf
bereits das unbehandelte Molekularsieb 13X.
Aus diesen Vorversuchen und aus Literaturdaten muss angenommen werden, dass ober¬
halb ca. 200 C sowohl Acetaldehyd als auch Acetylen polymerisieren und homogen an der
Katalysatoroberflache haften bleiben.
Mit Hilfe von Kohlenstoff- und WasserStoffanalysen der gebrauchten Katalysatoren wur¬
de versucht, die Zusammensetzung des Giftes zu erfassen.
Die Kohlenstoff- bzw. Wasserstoffmenge der gebrauchten Zn-13X-Katalysatoren als Funk¬
tion der Reaktionsdauer sind in Tabelle 22 wiedergegeben. Die Aktivitäts abnähme in Ab¬
hängigkeit der Kohlenstoff- bzw. Wasserstoffmenge ist in Figur 12 dargestellt.
Tabelle 22 : Kohlenstoff-bzw. Wasserstoffmenge der Zn-I3X-Katalysatoren in
Abhängigkeit der Reaktionsdauer
Versuch
[Nr.]
T*
[h]
r.
Ac,m
[Mol/h.l]mc,t
[g/6,83g]mH,t
fe/6,83g]mC,t :mH,t[-]
10 0 3,085 0 0 ...
11 0,5 2,94 0,0824 0,0108 7,62
12 1 2,78 0,1605 0,0473 3,39
13 2 2,50 0,259 0,0274 9,44
14 4,62 2,04 0,391 0,034 11,5
15 5,5 1,90 0,409 0,0426 9,6
16 8,5 1,56 0,475 0,045 10,5
Wie zu erwarten war, fallt die Reaktionsgeschwindigkeit langsamer als linear mit der
Kohlenstoff bzw. Wasserstoffzunahme ab, wie in Figur 12 erkenntlich ist. Diese Tatsache
stimmt mit dem Befund der Figur II uberem. Aus Tabelle 22 geht zudem hervor, dass das
Gift eine C-H-Verbmdung ist, und nicht reiner Kohlenstoff allein, wie dies in der Litera¬
tur oft behauptet wird. Bei der Annahme, dass das Gift ein Polymerisationsprodukt von Ace¬
tylen oder Acetaldehyd oder eine Mischung derselben ist, wurde bei genügendem Polymen-
sationsgrad sowohl von Acetylen als auch von Acetaldehyd ein Gewichtsverhaltnis von Koh¬
lenstoff zu Wasserstoff von 12:1 erhalten. Das aus den Experimenten berechnete mittlere
Gewichtsverhaltnis von 9,7:1 deutet daraus hin, dass es sich höchst wahrscheinlich um ein
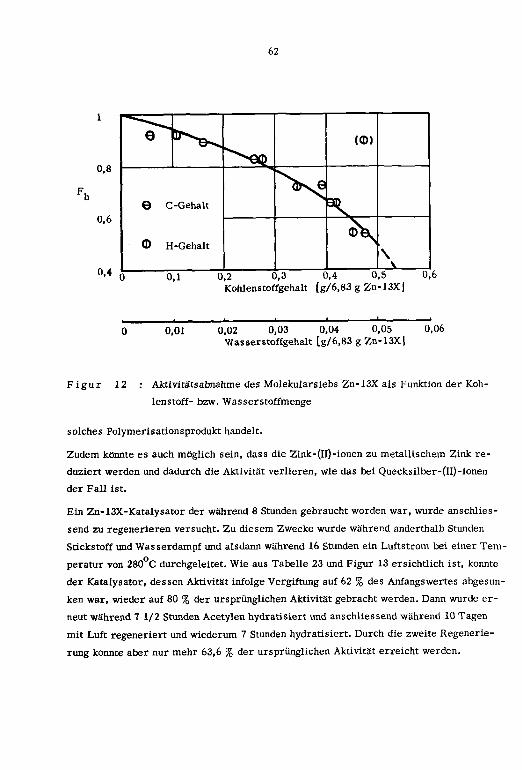
62
e «D>
Q C-Gehalt
JN6
O H-Gehalt
©"K
\
0 0,1 0,2 0,3 0,4 0,5 0,6
Kohlenstoffgehalt [g/6,83 g Zn-13X]
0 0,01 0,02 0,03 0,04 0,05 0,06
Wasserstoffgehalt [g/6,83 g Zn-13X]
Figur 12 : Aktivitatsabnahme des Molekularsiebs Zn-13X als Funktion der Koh¬
lenstoff- bzw. Wasserstoffmenge
solches Polymerisationsprodukt handelt.
Zudem könnte es auch möglich sein, das s die Zink-(II)-ionen zu metallischem Zink re¬
duziert werden und dadurch die Aktivität verlieren, wie das bei Quecksilber-(II)-ionen
der Fall ist.
Ein Zn-13X-Katalysator der wahrend 8 Stunden gebraucht worden war, wurde anschlies¬
send zu regenerieren versucht. Zu diesem Zwecke wurde wahrend anderthalb Stunden
Stickstoff und Wasserdampf und alsdann wahrend 16 Stunden ein Luftstrom bei einer Tem¬
peratur von 280 C durchgeleitet. Wie aus Tabelle 23 und Figur 13 ersichtlich ist, konnte
der Katalysator, dessen Aktivität infolge Vergiftung auf 62 % des Anfangswertes abgesun¬
ken war, wieder auf 80 % der ursprünglichen Aktivität gebracht werden. Dann wurde er¬
neut wahrend 7 1/2 Stunden Acetylen hydratisiert und anschliessend wahrend 10 Tagen
mit Luft regeneriert und wiederum 7 Stunden hydratisiert. Durch die zweite Regenerie¬
rung konnte aber nur mehr 63,6 % der ursprunglichen Aktivität erreicht werden.

63
Tabelle 23 : Reaktionsprodukte und Hydratisierungsgeschwindigkeit in Abhängig¬
keit der Reaktionsdauer; Zn-13X-Katalysator
Mittlere Katalysatortemperatur 280°C
Schuttvolumen 10,6 cm3
Zinkaustauschgrad 60,8%
Totaler Gasdurchsatz 6,52 Mol/h
Anfangsmolendurchsatz von Acetylen 0,67 Mol/h
Anfangsmolendurchsatz von Wasser 4,34 Mol/h
Totaldruck 0,967 arm
Versuch
[Nr.]
Probe
[Nr.] [h]Ac
[SEI
AAld[%]
ACr[%]
r.Ac.m
[Mol/h.l]
17a 1 1 3,69 3,23 0,46 2,33
17a 2 7 2,56 2,26 0,30 1,62
17b 1 1 2,94 2,56 0,38 1,84
17b 2 4,5 2,44 2,14 0,30 1,54
17b 3 7 2,22 2,02 0,20 1,40
17c I 1 2,42 2,12 0,30 1,53
17c 2 2,5 2,41 2,11 0,30 1,52
17c 3 4,5 2,25 1,99 0,26 1.42
17c 4 6,5 2,13 1,94 0,19 1,34
Aus den Regenerierungsversuchen kann geschlossen werden, dass der Katalysator bei
280 C mit Luft nur unvollständig regenerierbar ist. Die Aktivitatsabnahmen vor und nach
der ersten Regenerierung blieben ungefähr gleich. Auch bei einer Regenerierung von meh¬
reren Tagen konnte die ursprungliche Aktivität nicht mehr erreicht werden.
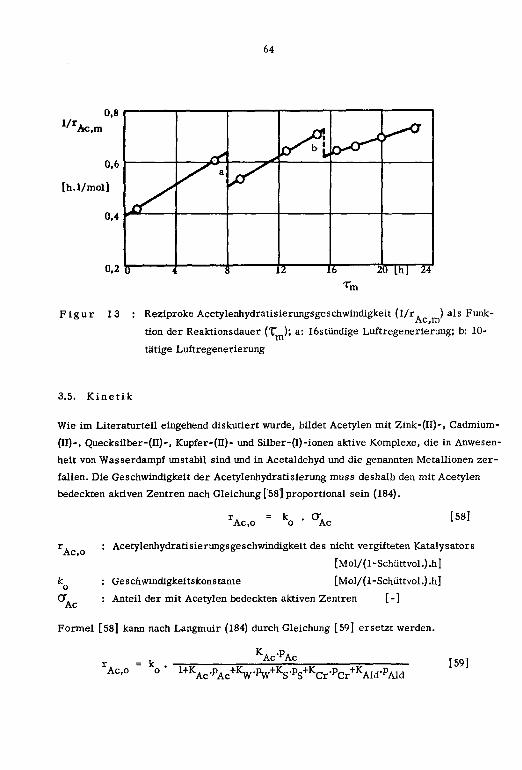
[59]l+KAc.PAc+Kw.pw+Ks.ps+KCr.Pcr+KAId.pAld
•
oAc,o
Ac'vAc.k
werden.ersetzt[59]Gleichungdurch(184)Langmuirnachkann[581Formel
[-]ZentrenaktivenbedecktenAcetylenmitderAnteil:CT
[Mol/(l-Schuttvol.).h]Geschwindigkeitskonstante:
[Mol/(1-Schuttvol.).h]
KatalysatorsvergiftetennichtdesAcetylenhydratisierungsgeschwindigkeit:r.
°Ack=
Ac,o[58]
(184).seinproportional[58]GleichungnachZentrenaktivenbedeckten
AcetylenmitdendeshalbmussAcetylenhydratisierungderGeschwindigkeitDiefallen.
zer¬MetallionengenanntendieundAcetaldehydinundsindunstabilWasserdampfvonheit
Anwesen¬indieKomplexe,aktiveSilber-(I)-ionenundKupfer-(II)-Quecksilber-(II)-,(II)-,
Cadmium-Zmk-(II)-,mitAcetylenbildetwurde,diskutierteingehendLiteraturteilimWie
Kinetik3.5.
Luftregenerierungtatige
10-b:Luftregenerierung;16stundigea:);(TReaktionsdauerdertion
Funk¬als)(l/r,AcetylenhydratisierungsgeschwindigkeitReziproke13Figur
24'
Ih|20TSTJ0,2
Ac,m
0,8
64

65
K : Adsorptionskonstanten von Acetylen (Ac), Wasser (W), Stickstoff (S), Cro-
tonaldehyd (Cr) und Acetaldehyd (Aid) an den aktiven Ionen [atm" ]
p : Partialdrucke der Edukte und Produkte [atm]
Sofern K^.p^+K^^+Kg.pg+K^.p^+K^.p^ <<; j [6Q] ^ erhal( m£m ^
Gesamtmässigkeit [61] bzw. [62] :
rAc,o= ko-KAc-PAc ^
rAc,o= ko-?Ac ^
k' : wahre Brutto-Reaktionsgeschwindigkeitskonstante, schliesst Acsorptions-
therme ein [Mol/(l-Schuttvol.)h.atm]
Hemmt zudem die Porendiffusion die chemische Reaktion (siehe Kapitel 3.3.2.), wird
nicht die wahre Reaktionsgeschwindigkeit gemessen.und die Geschwindigkeitsgesetze
bedürfen einer gewissen Korrektur, bzw. Formel [62] muss zu Gleichung [63] umge¬
formt werden.
rAc,o= kö-pAc-^ = lco-pAc [63]
k* : gemessene Reaktionsgeschwindigkeitskonstante [Mol/(l-Schuttvol.).h.atm]
Das Ziel der weiteren Untersuchungen bestand nun darin, die Richtigkeit dieser kineti¬
schen Gesetze fur die Acetylenhydratisierung an Zink-(II)-, Cadmium-(II)-, Quecksil-
ber-(II)-, Kupfer-(II)- und Silber-(I)-13X-Katalysatoren und damit indirekt auch den im
Literaturteil angegebenen Reaktionsmechanismus zu beweisen (210).
3.5.1. Zn-13X- Katalysatoren
Sofern Ungleichung [60] gilt, muss nach Formel [63] die Reaktionsgeschwindigkeit dem
Partialdruck direkt proportional sein. Um die Richtigkeit dieser Gesetzmassigkeit zu
beweisen, wurden die Acetylenhydratisierungsgeschwindigkeiten in Abhängigkeit der Ace-
tylenpartialdrucke gemessen. Die zeitliche Abnahme der Produkte und der Geschwindig¬
keit in Abhängigkeit des Acetylenpartialdruckes sind in Tabelle 24 aufgeführt und in Fi¬
gur 14 ersichtlich.
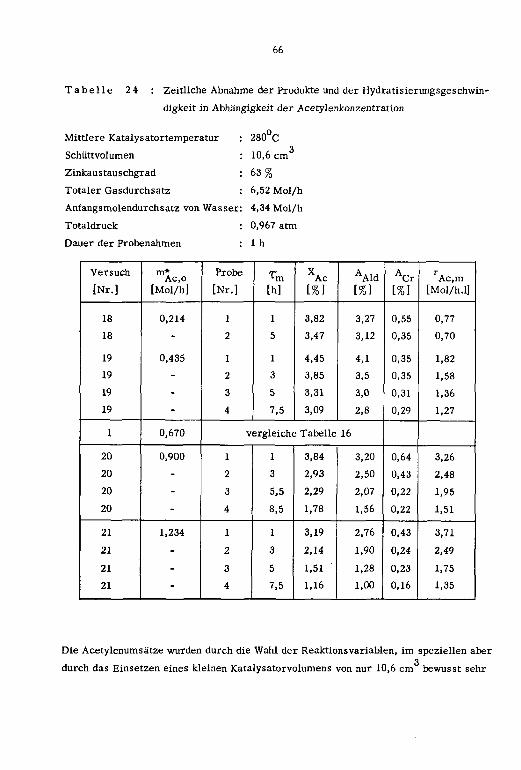
66
Tabelle 24 : Zeitliche Abnahme der Produkte und der Hydratisierungsgeschwin¬
digkeit in Abhängigkeit der Acetylenkonzentration
Mittlere Katalysatortemperatur 280°C
Schuttvolumen 10,6 cm3
Zinkaustauschgrad 63%
Totaler Gasdurchsatz 6,52 Mol/h
Anfangsmolendurchsatz von Wasser 4,34 Mol/h
Totaldruck 0,967 atm
Dauer der Probenahmen 1 h
Versuch
[Nr.]
m*Ac,o
[Mol/h]
Probe
[Nr.] [h]
XAc[%]
AAld[%]
ACr Ac,m
[Mol/h.l]
18
18
19
19
19
19
0,214
0,435
1
2
1
2
3
4
1
5
1
3
5
7,5
3,82
3,47
4,45
3,85
3,31
3,09
3,27
3,12
4,1
3,5
3,0
2,8
0,55
0,35
0,35
0,35
0,31
0,29
0,77
0,70
1,82
1,58
1,36
1,27
1 0,670 vergleiche Tabelle 16
20
20
20
20
0,900 1
2
3
4
1
3
5,5
8,5
3,84
2,93
2,29
1,78
3,20
2,50
2,07
1,56
0,64
0,43
0,22
0,22
3,26
2,48
1,95
1,51
21
21
21
21
1,234 1
2
3
4
1
3
5
7,5
3,19
2,14
1,51
1,16
2,76
1,90
1,28
1,00
0,43
0,24
0,23
0,16
3,71
2,49
1,75
1,35
Die Acetylenumsatze wurden durch die Wahl der Reaktionsvariablen, im speziellen aber
durch das Einsetzen eines kleinen Katalysatorvolumens von nur 10,6 cm bewusst sehr
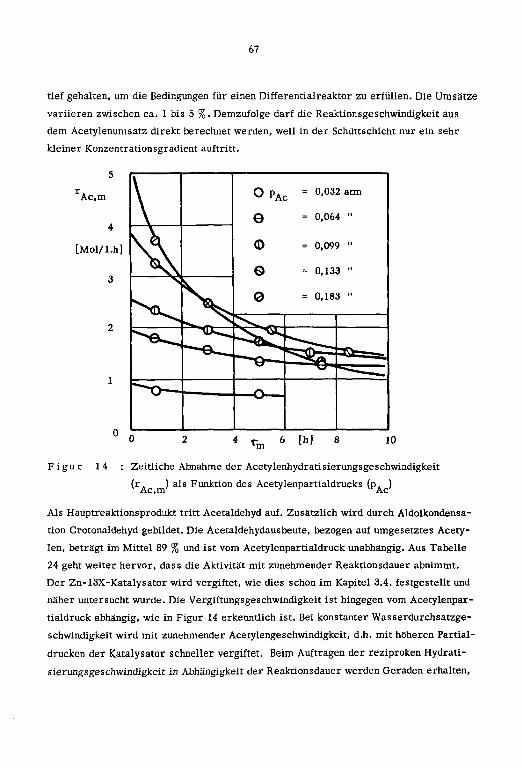
67
tief gehalten, um die Bedingungen fur einen Differentialreaktor zu erfüllen. Die Umsätze
variieren zwischen ca. 1 bis 5 %. Demzufolge darf die Reaktionsgeschwindigkeit aus
dem Acetylenumsatz direkt berechnet werden, weil in der Schuttschicht nur ein sehr
kleiner Konzentrationsgradient auftritt.
Figur 14 : Zeitliche Abnahme der Acetylenhydratisierungsgeschwindigkeit
(r ) als Funktion des Acetylenpartialdrucks (p )
Als Hauptreaktionsprodukt tritt Acetaldehyd auf. Zusatzlich wird durch Aldolkondensa-
tion Crotonaldehyd gebildet. Die Acetaldehydausbeute, bezogen auf umgesetztes Acety-
len, betragt im Mittel 89 % und ist vom Acetylenpartialdruck unabhängig. Aus Tabelle
24 geht weiter hervor, das s die Aktivität mit zunehmender Reaktionsdauer abnimmt.
Der Zn-13X-Katalysator wird vergiftet, wie dies schon im Kapitel 3.4. festgestellt und
naher untersucht wurde. Die Vergiftungsgeschwindigkeit ist hingegen vom Acetylenpar¬
tialdruck abhangig, wie in Figur 14 erkenntlich ist. Bei konstanter Wasserdurchsatzge¬
schwindigkeit wird mit zunehmender Acetylengeschwindigkeit, d.h. mit höheren Partial-
drucken der Katalysator schneller vergiftet. Beim Auftragen der reziproken Hydrati¬
sierungsgeschwindigkeit in Abhängigkeit der Reaktionsdauer werden Geraden erhalten,
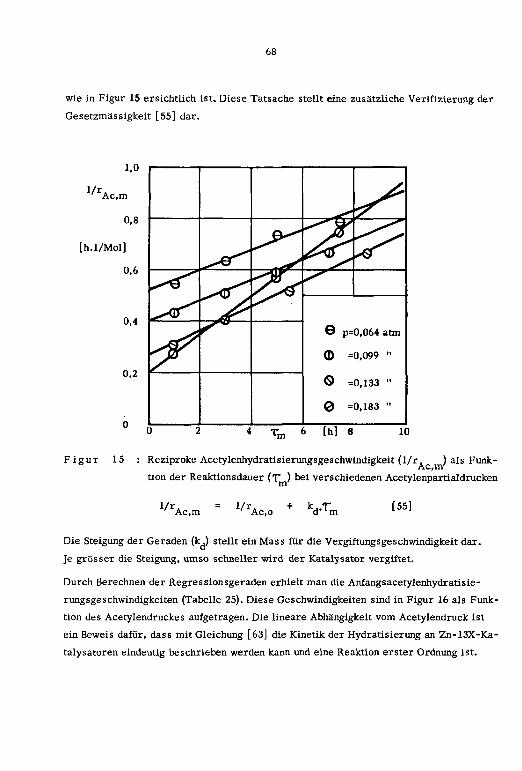
68
wie in Figur 15 ersichtlich ist. Diese Tatsache stellt eine zusätzliche Verifizierung der
Gesetzmassigkeit [55] dar.
Figur 15 : Reziproke Acetylenhydratisierungsgeschwindigkeit (1/r, ) als Funk-Ac,m
tion der Reaktionsdauer (T ) bei verschiedenen Acetylenpartialdrucken
1/rAc,m
1/r. + k..T
Ac,o dm[55]
Die Steigung der Geraden (k,) stellt em Mass fur die Vergiftungsgeschwindigkeit dar.
Je grosser die Steigung, umso schneller wird der Katalysator vergiftet.
Durch Berechnen der Regressionsgeraden erhielt man die Anfangsacetylenhydratisie-
rungsgeschwindigkeiten (Tabelle 25). Diese Geschwindigkeiten sind in Figur 16 als Funk¬
tion des Acetylendruckes aufgetragen. Die lineare Abhängigkeit vom Acetylendruck ist
ein Beweis dafür, dass mit Gleichung [63] die Kinetik der Hydratisierung an Zn-13X-Ka-
talysatoren eindeutig beschrieben werden kann und eine Reaktion erster Ordnung ist.
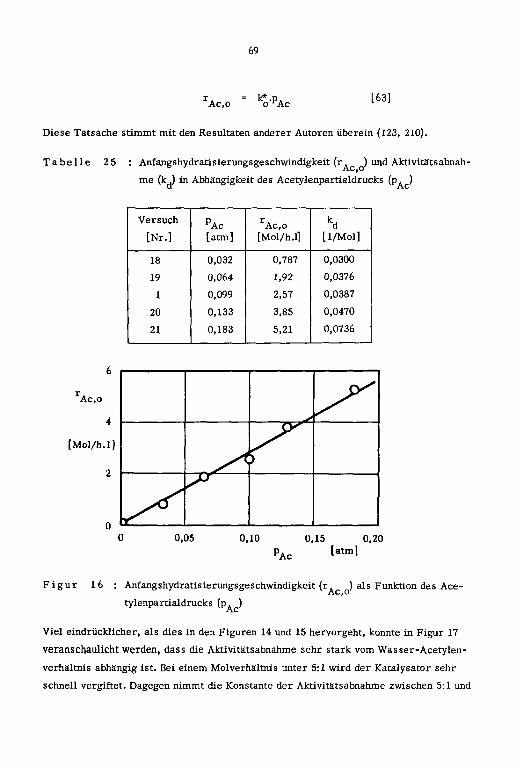
69
r. = k* .p,Ac.o o rAc
[63]
Diese Tatsache summt mit den Resultaten anderer Autoren uberein (123, 210).
Tabelle 25 : Anfangshydratisierungsgeschwindigkeit (r, ) und Aktivitatsabnah¬
me (k,) in Abhängigkeit des Acetylenpartialdrucks (p, )
Versuch
[Nr.]
PAc[atm]
Ac,o
[Mol/h.l]
kd[1/Mol]
18 0,032 0,787 0,0300
19 0,064 1,92 0,0376
1 0,099 2,57 0,0387
20 0,133 3,85 0,0470
21 0,183 5,21 0,0736
Ac,o
0,05 0,10
^Ac
0,15 0,20
[atm]
Figur 16 : Anfangshydratisierungsgeschwindigkeit (r ) als Funktion des Ace¬
tylenpartialdrucks (p, )
Viel eindrucklicher, als dies in den Figuren 14 und 15 hervorgeht, konnte in Figur 17
veranschaulicht werden, dass die Aktivitatsabnahme sehr stark vom Wasser-Acetylen-
verhaltnis abhangig ist. Bei einem Molverhaltnis unter 5:1 wird der Katalysator sehr
schnell vergiftet. Dagegen nimmt die Konstante der Aktivitatsabnahme zwischen 5:1 und

70
20:1 nur wenig ab. Der Grund dieses Verhaltens kann dadurch erklart werden, dass
ein grosser Wasserdampfuberschuss die irreversible und schädliche Polymerisation
von Acetylen oder Acetaldehyd zurückdrängt. Es ist somit nicht möglich, mit der stö-
chiometrischen Wasser-Acetylenzusammensetzung von 1:1 Acetaldehyd herzustellen.
0,09 I 1 1 1 1 1
0,07 2
[1/Mol] \
0,05 \r
0,03"~
<>=-
0 4 8 12 16 20
Molverhältnis von Wasser zu Acetylen [-]
Figur 17 : Aktivitatsabnahme (k) als Funktion des Wasser-Acetylenverhaltnis-
ses
Um eine mögliche Abhängigkeit der Acetylenhydratisierungsgeschwmdigkeit vom Zink-
austauschgrad festzustellen, wurden eine Anzahl Katalysatoren mit variablem Austausch¬
grad unter sonst konstanten Bedingungen getestet. Die Reaktionsprodukte und die Hydra¬
tisierung sgeschwindigkeit in Abhängigkeit des Zinkaustauschgrades sind In Tabelle 26
zusammengestellt.
Der Acetylenumsatz variiert zwischen 0,5 und 6 %. Die Acetaldehydausbeute betragt
im Mittel 89 % und ist vom Zinkaustauschgrad unabhängig. Die restlichen 11 % sind
Crotonaldehyd.
Durch Extrapolation der reziproken Hydratisierungsgeschwindigkeiten konnten die in Ta¬
belle 27 zusammengestellten Anfangsgeschwindigkeiten und die Konstanten der Aktivi-
tatsabnahmen in Abhängigkeit des Zinkaustauschgrades ermittelt werden. Die tabellari¬
schen Werte sind in Figur 18 graphisch dargestellt.

71
Aus den Untersuchungen lassen sich nachfolgende Gesetzmassigkeiten formulieren. Wie
zu erwarten war, nimmt mit zunehmender Zink-(Il)-ionenkonzentrationen die beobachte¬
te Reaktionsgeschwindigkeit von Null bis ca. 47 Mole pro Liter Schuttvolumen, Stunde
und Atmosphäre zu. Interessanterweise ist diese Zunahme nicht linear, wie in Figur 18
ersichtlich ist. Der sprunghafte Anstieg der Reaktionsgeschwindigkeit zwischen einem
Zinkaustauschgrad von 60 bis 65 % lasst sich wahrscheinlich dadurch erklaren, dass
sich der aequivalente Radius fur Poren unter 75 A in diesem Bereich stark ändert (sie¬
he Tabelle 13), und dass als Folge davon der hemmende Einfluss der Knudsendiffusion
etwas abnimmt. Ein ganz analoges Verhalten zeigt auch die Konstanten der Aktivitäts -
abnähme in Abhängigkeit des Zinkaustauschgrades. Die Grosse der Konstante variiert
von Null bis 0,065 Liter Schuttvolumen pro Mol umgesetztes Acetylen.
Im weiteren soll noch abgeklärt werden, inwiefern die Reaktionstemperatur und die An-
fangshydratisierungsgeschwindigkeit die Katalysatorvergiftung und die Zusammensetzung
der Produkte beeinflusst.
Da bei heterogenen Gaskatalysen oft komplexe Temperaturabhangigkeiten der "Brutto-
reaktionsgeschwindigkeitskonstanten" erhalten werden, wie sie in Figur 3 (siehe Kapi¬
tel 3.3.) wiedergegeben sind, schien es angezeigt, zuvor die an die eigenen Verhaltnisse
angepassten Gesetze zu formulieren, um sie dann zur Charakterisierung der erhaltenen
Resultate anzuwenden.
Wie aus Figur 3 ersichtlich ist, wird die beobachtete Anfangsreaktionsgeschwindigkeits-
konstante (k ) im Gebiet des hemmenden Einflusses der Porendiffusion linear von der
Temperatur abhangig (180, 189).
Ink* = In A* - E* / R.T [64]o g'
k* : beobachtete Anfangsreaktionsgeschwindigkeitskonstante, schliesst hemmenden
Einfluss der Porendiffusion ein [Mol/(l-Schuttvol.).h.atm]
A* : praexponentieller Faktor [Mol/(l-Schuttvol.).h.atmI
E* : beobachtete Aktivierungsénergie [kcal/Mol]
Ist die gesamte Oberflache fur die chemische Umsetzung zugänglich, wird nach der Arr-
henius'schen Gesetzmassigkeit [65] ebenfalls eine einfache Temperaturabhangigkeit er¬
halten.
Ink' = In A' - E' /R.T [65]° g
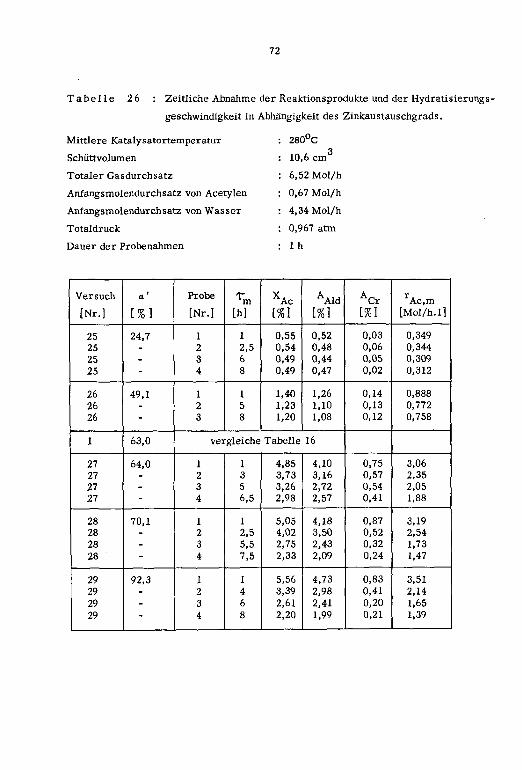
72
Tabelle 26 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisierungs¬
geschwindigkeit in Abhängigkeit des Zinkaustauschgrads.
Mittlere Katalysatortemperatur 280°C
Schuttvolumen 10,6 cm
Totaler Gasdurchsatz 6,52 Mol/h
Anfangsmolendurchsatz von Acetylen 0,67 Mol/h
Anfangsmolendurchsatz von Wasser 4,34 Mol/h
Totaldruck 0,967 atm
Dauer der Probenahmen lh
Versuch
[Nr.]
a'
[561
Probe
[Nr.]
rmlh]
XAc[561
AAld[%1
ACr[561
r.Ac,m
[Mol/h. 1]
25
25
25
25
24,7 1
2
3
4
1
2,56
8
0,55
0,54
0,49
0,49
0,52
0,48
0,44
0,47
0,03
0,06
0,05
0,02
0,349
0,344
0,309
0,312
26
26
26
49,1 1
2
3
1
5
8
1,40
1,23
1,20
1,261,10
1,08
0,14
0,13
0,12
0,888
0,772
0,758
1 63,0 vergleiche Tabelle 16
27
27
27
27
64,0 1
2
3
4
1
3
5
6,5
4,85
3,73
3,262,98
4,103,16
2,72
2,57
0,75
0,57
0,54
0,41
3,062,35
2,05
1,88
28
28
28
28
70,1 1
2
3
4
1
2,5
5,5
7,5
5,054,02
2,75
2,33
4,18
3,50
2,43
2,09
0,87
0,52
0,32
0,24
3,19
2,54
1,73
1,47
29
29
29
29
92,3 1
2
3
4
1
4
6
8
5,56
3,39
2,61
2,20
4,73
2,98
2,41
1,99
0,83
0,41
0,20
0,21
3,51
2,14
1,65
1,39
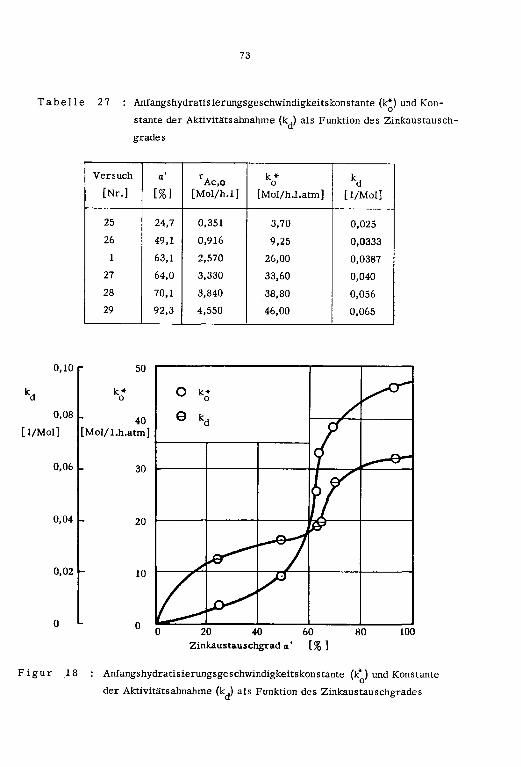
73
Tabelle 27 : Anfangshydratisierungsgeschwindigkeitskonstante (k*) und Kon¬
stante der Aktivitatsabnahme (k,) als Funktion des Zmkaustausch-
grades
Versuch
[Nr.]
a
[561
r
Ac,o
[Mol/h.l]
k*0
[Mol/h.l.atm]kd
[1/MolJ
25 24,7 0,351 3,70 0,025
26 49,1 0,916 9,25 0,0333
1 63,1 2,570 26,00 0,0387
27 64,0 3,330 33,60 0,040
28 70,1 3,840 38,80 0,056
29 92,3 4,550 46,00 0,065
0,10
kd
0,08
[1/Mol]
0,06
0,04
0,02
50
k*o
40
[Mol/l.h.atm]
30
20
10
O k(ô
k
*
°Kd
20 40 60
Zinkaustauschgrad a' [%]
80 100
Figur 18 : Anfangshydratisierungsgeschwindigkeitskonstante (k*) und Konstante
der Aktivitatsabnahme (k.) als Funktion des Zinkaustauschgrades

74
k' : beobachtbare Anfangsreaktionsgeschwindigkeitskonstante ohne hemmenden
Einfluss der Porendiffusion [Mol/(l-Schuttvol.).h.atm]
E' : beobachtbare Aktivierungsenergie [kcal/Mol]
Der Unterschied der Gleichungen [64] und [65] besteht darin, dass die beobachtbare
Aktivierungsenergie (EL) doppelt so gross ist als die beobachtete Aktivierungsenergie
Die wahre Aktivierungsenergie (E ) der chemischen Umsetzung setzt sich zusammen
aus der beobachtbaren Aktivierungsenergie (E') und der Adsorptionsenergie (AH ):
E = E' + AH [66]w g a
E : wahre Aktivierungsenergie der chemischen Umsetzung [kcal/Mol]
AH : Adsorptionsenergie der Edukte [kcal/Mol]
Die Energiemveaux der Edukte und Produkte, und die entsprechenden Aktivierungsener¬
gien sind in Figur 19 schematisch dargestellt.
Um nun den Temperaturemfluss auf die Acetylenhydratisierungsgeschwindigkeit, die
Katalysatorvergiftung und die Zusammensetzung der Reaktionsprodukte festzustellen,
wurde die zeitliche Abnahme der Produkte und der daraus berechneten Geschwindigkeit
im Temperaturbereich von 240 bis 424°C gemessen. Die Versuchsdaten und Ergebnisse
sind in Tabelle 28 zusammengestellt. Die in Tabelle 29 aufgeführten gemessenen (k*)
und wahren (k') Anfangsgeschwindigkeitskonstanten und die Konstanten der Aktivitäts-
abnahme (k,) sind in Figur 20 graphisch dargestellt.
Aus den Resultaten geht hervor, dass die Acetaldehydausbeute, bezogen auf umgesetztes
Acetylen von 90 % bei 240 C auf 84 % bei 424 C abfallt. Entsprechend nimmt die Croto-
naldehydausbeute zu. Ausserdem nehmen mit steigender Temperatur die extrapolierten
Anfangsgeschwindigkeitskonstanten von 14,34 bis 92,2 Mole pro Liter Schuttvolumen, Stun¬
de und Atmosphäre zu. Die Konstanten smd nach Arrheruus linear von der Temperatur
abhangig. Die aus der Steigung berechnete Aktivierungsenergie betragt nur 6,1 kcal/Mol,
was ein zusätzlicher Beweis ist, dass die Knudsendiffusion die eigentliche chemische Re¬
aktion hemmen muss. Mit Hilfe der im Abschnitt 3.3.2. angegebenen Methode zur Berech¬
nung des Porendiffusionseinflusses wurde versucht, die wahren Reaktionsgeschwindigkeits¬
konstanten zu ermitteln. Die Resultate der Berechnungen zeigen, dass der Nutzungsgrad
der Reaktion bezogen auf einen aequivalenten Porenradius von 15 A m den untersuchten
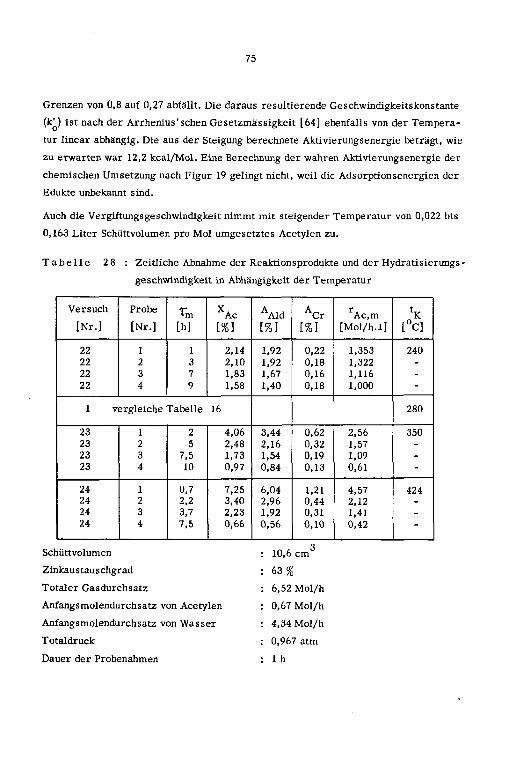
75
Grenzen von 0,8 auf 0,27 abfallt. Die daraus resultierende Geschwindigkeitskonstante
(k^) ist nach der Arrhemus'sehen Gesetzmassigkeit [64] ebenfalls von der Tempera¬
tur linear abhangig. Die aus der Steigung berechnete Aktivierungsenergie betragt, wie
zu erwarten war 12,2 kcal/Mol. Eme Berechnung der wahren Aktivierungsenergie der
chemischen Umsetzung nach Figur 19 gelingt nicht, weil die Adsorptionsenergien der
Edukte unbekannt sind.
Auch die Vergiftungsgeschwindigkeit nimmt mit steigender Temperatur von 0,022 bis
0,163 Liter Schuttvolumen pro Mol umgesetztes Acetylen zu.
Tabelle 28 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisierungs¬
geschwindigkeit in Abhängigkeit der Temperatur
Versuch
[Nr.]
Probe
[Nr.]
rm
[h]
XAc[SB]
AAld[%I
ACr[%]
Ac,m
[Mol/h.1] [°C]
22
22
22
22
1
2
3
4
1
3
7
9
2,14
2,10
1,83
1,58
1,92
1,92
1,67
1,40
0,22
0,18
0,16
0,18
1,353
1,322
1,116
1,000
240
1 vergleiche Tabelle 16 280
23
23
23
23
1
2
3
4
2
5
7,510
4,06
2,48
1,73
0,97
3,44
2,16
1,54
0,84
0,62
0,32
0,19
0,13
2,56
1,57
1,09
0,61
350
24
24
24
24
1
2
3
4
0,7
2,2
3,7
7,5
7,25
3,40
2,23
0,66
6,042,96
1,92
0,56
1,21
0,44
0,31
0,10
4,57
2,12
1,41
0,42
424
Schuttvolumen 10,6 cm3
Zmkaustauschgrad 63%
Totaler Gasdurchsatz 6,52 Mol/h
Anfangsmolendurchsatz von Acetylen 0,67 Mol/h
Anfangsmolendurchsatz von Wasser 4,34 Mol/h
Totaldruck 0,967 atm
Dauer der Probenahmen 1 h
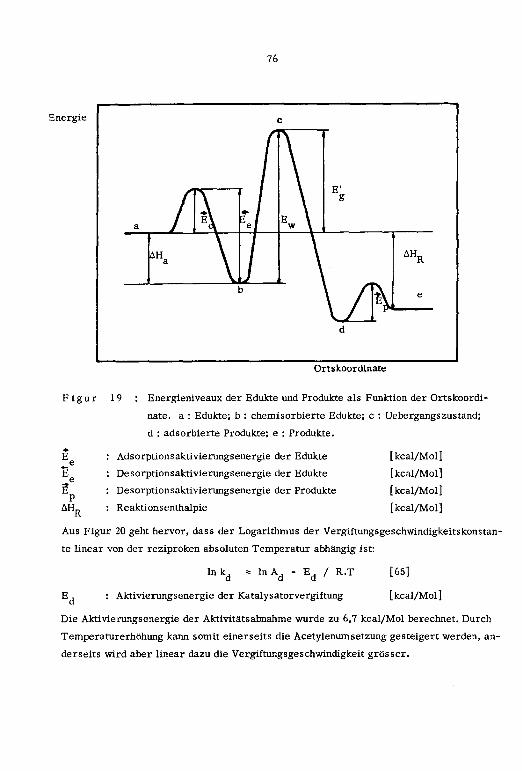
76
Energie
JX-,/ E \w \
E'g
\AH \
V
\
AHR
eb
V3d
Ortskoordinate
Figur 19 : Energiemveaux der Edukte und Produkte als Funktion der Ortskoordi¬
nate, a : Edukte; b : chemisorbierte Edukte; c : Uebergangszustand;
d : adsorbierte Produkte; e : Produkte.
E : Adsorptionsaktivierungsenergie der Edukte [kcal/Mol]
E : Desorptionsaktivierungsenergie der Edukte [kcal/Mol]
En : Desorptionsaktivierungsenergie der Produkte [kcal/Mol]
Reaküonsenthalpie [kcal/Mol]AH,
'R
Aus Figur 20 geht hervor, dass der Logarithmus der Vergiftungsgeschwindigkeitskonstan-
te linear von der reziproken absoluten Temperatur abhangig ist:
Ink = In A - E / R.T [65]
Aktivierungsenergie der Katalysatorvergiftung [kcal/Mol]
Die Aktivierungsenergie der Aktivitatsabnahme wurde zu 6,7 kcal/Mol berechnet. Durch
Temperaturerhöhung kann somit einerseits die Acetylenumsetzung gesteigert werden, an¬
derseits wird aber linear dazu die Vergiftungsgeschwindigkeit grosser.
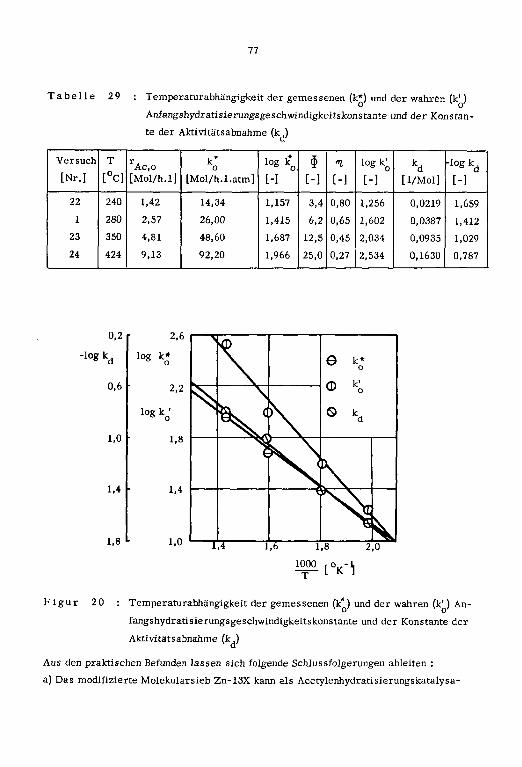
77
Tabelle 29 Temperaturabhangigkeit der gemessenen (k*) und der wahren (k1 )
Anfangshydratisierungsgeschwindigkeitskonstante und der Konstan¬
te der Aktivitatsabnahme (k,)
Versuch T r.Ac,o
k*0 logk; $ 1 logk; kd -log kd
[Nr.] [uc] [Mol/h.l] [Mol/h.l.atm] [-] [-] [-] [-] [1/Mol] [-]
22 240 1,42 14,34 1,157 3,4 0,80 1,256 0,0219 1,659
1 280 2,57 26,00 1,415 6,2 0,65 1,602 0,0387 1,412
23 350 4,81 48,60 1,687 12,5 0,45 2,034 0,0935 1,029
24 424 9,13 92,20 1,966 25,0 0,27 2,534 0,1630 0,787
0,2
-log k.
0,6
1,0
1,8 L
2,6
log k*
2,2
logic,'
1,8
1.4 1,4
1,0
© k*
^k c >v Q kHa
1 4 1 ,6 1 ,8 2 ,0
i2 [V1!
Figur 20 : Temperaturabhangigkeit der gemessenen (k*) und der wahren (k') An-
fangshydratisierungsgeschwindigkeitskonstante und der Konstante der
Aktivitatsabnahme (fc )
Aus den praktischen Befunden lassen sich folgende Schlussfolgerungen ableiten :
a) Das modifizierte Molekularsieb Zn-13X kann als Acetylenhydratisierungskatalysa-

78
tor eingesetzt werden. Mit zunehmender Reaktionsdauer ( T ) wird aber der Katalysa¬
tor nach folgendem Gesetz vergiftet :
1/r. =l/r,„ + k,.r [55]'
Ac,m' Ac,o d lm
Die Vergiftungsgeschwindigkeitskonstante (k ,) ist vom Wasser-Acetylenverhaltms, vom
Zinkaustauschgrad und von der Temperatur abhängig. Em Wasser-Acetylen-Molverhalt-
nis von 5:1 sollte nicht unterschritten werden, sonst verliert der Katalysator seme Akti¬
vität sehr schnell. Die Vergiftungsgeschwindigkeit nimmt auch mit zunehmendem Zink-
austauschgrad und mit steigender Temperatur nach folgender Gesetzmassigkeit zu :
lnkd = lnAd-Ed/R.T [65]
Die aus Formel [65] berechnete De s aktivierungsénergie (E ,) betragt 6,7 kcal/Mol.
b) Die Hydratisierungsgeschwindigkeit des nicht vergifteten Katalysators ist vom Ace-
tylenpartialdruck, vom Zinkaustauschgrad und von der Temperatur abhangig.
Die Anfangsacetylenumsetzungsgeschwindigkeit (r ) ist dem Acetylenpartialdruck
(p, ) direkt proportional:^Ac
Ac,o o Ac
Analog der Vergiftungs- nimmt auch die Hydratisierungsgeschwindigkeit mit höherer
Zink-(II)-ionenkonzentration bei konstantem Katalysatorvolumen zu. Die beobachtete Re¬
aktionsgeschwindigkeitskonstante (k ) ist nach Formel [64] linear von der Temperatur
abhangig:
Ink* = In A - E* / R.T [64]
Die entsprechende Aktivierungsenergie (E*) betragt 6,1 kcal/Mol.
c) Als Hauptprodukt fallt Acetaldehyd an. Die Ausbeute, bezogen auf umgesetztes Acetylen,
ist vom Zinkaustauschgrad und vom Acetylenpartialdruck unabhängig und betragt durch¬
schnittlich 89 %. Die restlichen 11 % sind Crotonaldehyd. Dagegen fallt die Ausbeute mit
zunehmender Temperatur von 90 % bei 240°C auf 84 % bei 424 °C ab.
3.5.2. Cd-13X-Katalysatoren
Es soll nun abgeklärt werden, ob die fur Zink-(II)-13X-Katalysatoren gefundenen Gesetz-

79
massigkeiten auch fur die entsprechenden Cadmium-(II)-13X-Molekularsiebe gelten.
Zu diesem Zwecke wurde vorerst die zeitliche Abnahme der Acetylenumsatzgeschwin-
digkeit und der Produkte in Abhängigkeit des Acetylenpartialdruckes gemessen. Ver¬
suchsbedingungen und Ergebnisse sind m Tabelle 30 zusammengestellt.
Die Temperatur konnte im Gegensatz zu den Zinkkatalysatoren wahrend der ersten 15
Minuten nicht konstant gehalten werden. Sie stieg kurz nach dem Einfahren mit Acetylen
stark an, sank dann aber wieder schnell auf die Salzbadtemperatur ab. Der Grund des
Temperaturanstieges kann darin liegen, dass durch Adsorption der Edukte, vor allem
aber durch Komplexbildung von Acetylen an Cadmium-(II)-ionen eine momentane gros¬
se Wärmemenge frei wird, die nicht schnell genug abgeleitet wird.
Wie aus Tabelle 30 hervorgeht, variieren die Acetylenumsatze zwischen 0,5 und 25 %,
im Mittel übersteigen sie aber 7 % nicht, sodass die Bedingungen fur einen Differential -
reaktor trotzdem eingehalten sind. Als Hauptreaktionsprodukt fallt Acetaldehyd an. Wei¬
ter bilden sich noch geringe Mengen von Crotonaldehyd. Die Acetaldehydausbeute, bezo¬
gen auf umgesetztes Acetylen, betragt durchschmttlich 87,5 %. Eine eindeutige Abhängig¬
keit der Selektivität der Acetylenhydratisierung vom Acetylenpartialdruck lasst sich
nicht erkennen. Weiter ist aus dieser Tabelle ersichtlich, dass auch der Cadmium-13X-
Katalysator mit fortschreitender Reaktionsdauer vergiftet wird. Die gebrauchten Kataly-
satorkorner waren immer dunkel bis schwarz gefärbt. Es ist deshalb wahrscheinlich,
dass es sich im vorliegenden Fall um die gleiche Vergiftungsart wie bei den Zinkkata¬
lysatoren handelt. Diese Vermutung wird durch die Tatsache bestätigt, dass Gleichung
[55] vollumfanglich zur Beschreibung der Katalysatorvergiftung gilt. Somit konnten durch
Berechnung der Regressionsgeraden die Anfangshydratisierungsgeschwindigkeiten (r )
und aus den Steigungen der Geraden die Vergiftungskonstanten (k.),die m Tabelle 31 auf¬
geführt sind, ermittelt werden.
Die tabellarischen Werte sind in den Figuren 21 und 22 graphisch dargestellt. Wie aus
Figur 21 hervorgeht, ist die Reaktionsgeschwindigkeit des unvergifteten Katalysators
dem Acetylenpartialdruck direkt proportional, was ein Beweis dafür ist, dass Formel
[63] bei Cd-13X-Katalysatoren zur Beschreibung der Kinetik gilt. Die Geschwindigkeit
steigt linear von Null bis 25 Mole pro Liter Schuttvolumen und Stunde bei Erhöhung des
Druckes von Null auf 0,133 Atmosphären an. Entsprechend, aber nicht linear, nimmt die
Vergiftungsgeschwindigkeit von Null bis 0,24 Liter Schuttvolumen pro Mol umgesetz¬
tes Acetylen zu. Ausserdem ist die Konstante der Aktivitatsabnahme eine Funktion des
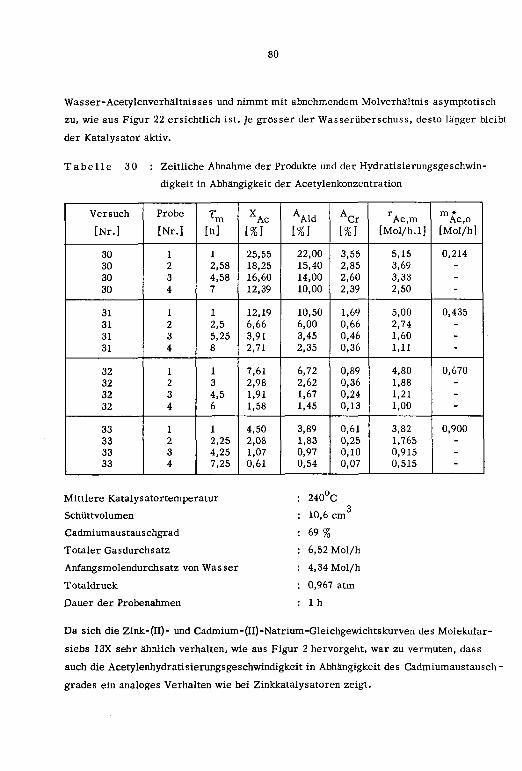
80
Wasser-Acetylenverhaltnisses und nimmt mit abnehmendem Molverhaltnis asymptotisch
zu, wie aus Figur 22 ersichtlich ist. Je grosser der Wasseruberschuss, desto langer bleibt
der Katalysator aktiv.
Tabelle 30 : Zeitliche Abnahme der Produkte und der Hydratisierungsgeschwin¬
digkeit m Abhängigkeit der Acetylenkonzentration
Versuch
[Nr.]
Probe
[Nr.]
Tm[h]
XAc[%]
AAld[%]
ACrtSBl
Ac,m
[Mol/h.1]
m *
Ac,o
[Mol/h]
30
30
30
30
1
2
3
4
1
2,58
4,587
25,55
18,25
16,60
12,39
22,00
15,40
14,00
10,00
3,55
2,85
2,60
2,39
5,15
3,69
3,33
2,50
0,214
31
31
31
31
1
2
3
4
1
2,5
5,25
8
12,19
6,66
3,91
2,71
10,50
6,00
3,45
2,35
1,69
0,660,46
0,36
5,00
2,74
1,60
1,11
0,435
32
32
32
32
1
2
3
4
1
3
4,56
7,61
2,98
1,91
1,58
6,72
2,62
1,67
1,45
0,89
0,360,24
0,13
4,80
1,88
1,21
1,00
0,670
33
33
33
33
1
2
3
4
1
2,25
4,25
7,25
4,50
2,08
1,07
0,61
3,89
1,83
0,97
0,54
0,61
0,25
0,10
0,07
3,82
1,765
0,915
0,515
0,900
Mittlere Katalysatortemperatur 240°C
Schuttvolumen 10,6 cm3
Cadmiumaustauschgrad 69%
Totaler Gasdurchsatz 6,52 Mol/h
Anfangsmolendurchsatz von Wasser 4,34 Mol/h
Totaldruck 0,967 atm
Dauer der Probenahmen 1 h
Da sich die Zink-(II)- und Cadmium-(II)-Natrium-Gleichgewichtskurven des Molekular-
siebs 13X sehr ähnlich verhalten, wie aus Figur 2 hervorgeht, war zu vermuten, dass
auch die Acetylenhydratisierungsgeschwindigkeit in Abhängigkeit des Cadmiumaustausch -
grades ein analoges Verhalten wie bei Zinkkatalysatoren zeigt.

81
Tabelle 31 : Anfangshydratisierungsgeschwindigkeit (r ) und Aktivitätsabnäh¬
me (k.) in Abhängigkeit des Acetylenpartialdruckes (p. )u AC
Versuch
[Nr.!Ac,m
[atm]
r.
Ac,o
[Mol/h.1]
kd[1/MolI
30 0,032 6,25 0,035
31 0,064 10 0,10
32 0,099 20 0,158
33 0,133 25 0,24
32
rA
Ac,o
24
[Mol/h.1]
16
o
o
0,05 0,1
^Ac
0,15
[atm]
Figur 21 : Anfangshydratisierungsgeschwindigkeit (r ) als Funktion des Ace¬
tylenpartialdrucke s (p )
Um diese Ansicht zu beweisen, wurde die zeitliche Abnahme der Reaktionsgeschwindig¬
keit und der anfallenden Produkte in Abhängigkeit der Cadmium-(Il)-ionenkonzentration
auf dem Molekularsieb 13X gemessen. Die Hydratisierungsgeschwindigkeit und die Zusam¬
mensetzung der Produkte sind in Tabelle 32 und die daraus extrapolierten Umsatz- und
Vergiftungsgeschwindigkeitskonstanten in Tabelle 33 aufgeführt. Die tabellarischen Wer-

82
te sind in Figur 23 graphisch dargestellt. Die erhaltenen Acetylenumsatze m Abhängig¬
keit des Cadmiumaustauschgrades variieren von 1 bis 8 %. Die Acetaldehydausbeute,
bezogen auf umgesetztes Acetylen betragt rund 87,5 %. Unter den in Tabelle 32 gege¬
benen Bedingungen steigt die Reaktionsgeschwindigkeitskonstante von Null bis ca. 340
Mole pro Liter Schuttvolumen, Stunde und Atmosphäre an, chejemge der Vergiftung von
Null bis rund 0,13 Liter Schuttvolumen pro Mol umgesetztes Acetylen. Tatsächlich tritt,
wie zu vermuten war und aus Figur 23 ersichtlich ist, ein sprunghafter Anstieg sowohl
der Acetylenumsatzgeschwmdigkeit als auch der Vergiftung m Abhängigkeit des Cadmium
austauschgrades zwischen 55 und 65 % auf. Dieses interessante Verhalten wurde schon
bei den Zn-13X-Katalysatoren festgestellt.
0,32
kd
0,24
[1/Mol]
0,16
0,08
"
0"
5 10 15 20
Molverhaltnis von Wasser zu Acetylen [-]
Figur 22 : Aktivitatsabnahme (k.) als Funktion des Wasser-Acetylenverhaltnis-
ses
Um den Einfluss der Temperatur festzustellen, wurde die Acetylenum satzgesehwindig -
keit und die Zusammensetzung der Produkte in Abhängigkeit der Temperatur gemessen.
Die Versuchsbedingungen und Resultate sind in Tabelle 34 zusammengestellt.
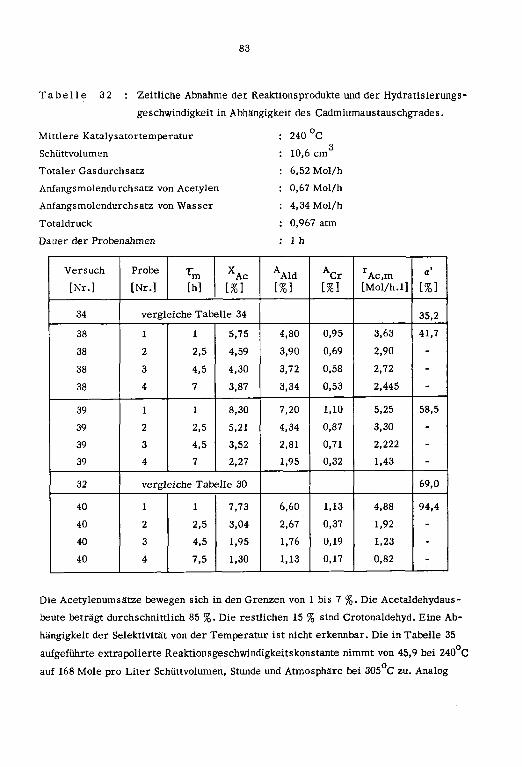
83
Tabelle 32 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisierungs¬
geschwindigkeit in Abhängigkeit des Cadmiumaustauschgrades.
Mittlere Katalysatortemperatur
Schuttvolumen
Totaler Gasdurchsatz
Anfangsmolendurchsatz von Acetylen
Anfangsmolendurchsatz von Wasser
Totaldruck
Dauer der Probenahmen
240 °C
10,6 cm3
6,52 Mol/h
0,67 Mol/h
4,34 Mol/h
0,967 atm
1 h
Versuch
[Nr.]
Probe
[Nr.] [h]
XAc[%]
AAld[%]
A~
Cr
[%1Ac,m
[Mol/h.1]
a'
[%]
34 vergleiche Tabelle 34 35,2
38 1 1 5,75 4,80 0,95 3,63 41,7
38 2 2,5 4,59 3,90 0,69 2,90 -
38 3 4,5 4,30 3,72 0,58 2,72 -
38 4 7 3,87 3,34 0,53 2,445 -
39 1 1 8,30 7,20 1,10 5,25 58,5
39 2 2,5 5,21 4,34 0,87 3,30 -
39 3 4,5 3,52 2,81 0,71 2,222 -
39 4 7 2,27 1,95 0,32 1,43 -
32 vergleiche Tabelle 30 69,0
40 1 1 7,73 6,60 1,13 4,88 94,4
40 2 2,5 3,04 2,67 0,37 1,92 -
40 3 4,5 1,95 1,76 0,19 1,23 -
40 4 7,5 1,30 1,13 0,17 0,82 -
Die Acetylenumsatze bewegen sich m den Grenzen von 1 bis 7 %. Die Acetaldehydaus-
beute betragt durchschnittlich 85 %. Die restlichen 15 % sind Crotonaldehyd. Eme Ab¬
hängigkeit der Selektivität von der Temperatur ist nicht erkennbar. Die in Tabelle 35
aufgeführte extrapolierte Reaktionsgeschwindigkeitskonstante nimmt von 45,9 bei 240 C
auf 168 Mole pro Liter Schuttvolumen, Stunde und Atmosphäre bei 305 C zu. Analog

84
steigt auch die Vergiftungsgeschwindigkeit von 0,034 bis 0,19 Liter Schuttvolumen pro
Mol umgesetztes Acetylen an.
Tabelle 33 Anfangshydratisierungsgeschwmdigkeitskonstante (k*) und Konstan¬
te der Aktivitatsabnahme (k,) als Funktion des Cadmiumaustausch¬
grades
Versuch
[Nr.]
a
[%]
r.
Ac,o
[Mol/h.l]
Vr.Ac,o
[h.l/Mol]
k*0
[Mol/h.l.atm]
kd[1/Mol]
34 35,2 4,55 0,22 45,9 0,034
38 41,7 4,55 0,22 45,9 0,034
39 58,5 10,00 0,10 101 0,084
32 69,0 20,00 0,05 202 0,158
40 94,4 25,00 0,04 252 0,170
0,20
kd
0,16
[ 1/Mol]
0,12
0,08
0,04
400
k*o
320
[Mol/l.h.atm]
240
160
80
O k«
e *d
20 40 60 80
Cadmiumaustau s chgrad a' [%]
100
Figur 2 3 : Anfangshydratisierungsgeschwmdigkeitskonstante (k*) und Konstante
der Aktivitatsabnahme (k,) als Funktion des Cadmiumaustauschgrades

85
Zur Charakterisierung der Temperaturabhangigkeit dieser Konstanten gelten Gleichun¬
gen [64] und [66],wie aus Figur 24 erkenntlich ist, mit den entsprechenden Aktivierungs¬
energien von 10,2 kcal/Mol fur die Hydratisierung und 15,7 kcal/Mol fur die Katalysa¬
torvergiftung.
In Tabelle 35 ist ausserdem der Nutzungsgrad der katalytisch wirksamen Oberflache
ersichtlich, der bezogen auf einen Porenradius von 15 A mit steigender Temperatur
von 0,49 auf 0,18 abnimmt. Die dazugehörige Aktivierungsenergie, die dann erhalten
wurde, wenn die totale Oberflache fur die katalytische Acetylenumsetzung zugänglich
ware, berechnet sich zu 20,4 kcal/Mol.
Zusammenfassend sei festgehalten, dass alle Gesetzmassigkeiten, die bei der Untersu¬
chung der Kinetik der Acetylenhydratisierung mit Zn-13X-Katalysatoren gefunden wur¬
den, auch bei den Cd-13X-Molekularsiebkatalysatoren gelten. Somit ist es möglich, die
Aktivitäten der beiden genannten Katalysatortypen z.B. unter folgenden Bedingungen mit¬
einander zu vergleichen: mittlere Katalysatortemperatur: 280 C; Acetylendurchsatzge-
schwindigke«: 0,67 Mol/ Stunde; Wasserdurchsatzgeschwindigkeit: 4,34 Mol/Stunde;
Stickstoffdurchsatzgeschwindigkeit: 1,51 Mol/Stunde; Zink- bzw. Cadmiumaustausch-
grad: 100 %; Schuttvolumen: 10,6 cm ; totaler Gasdruck: 0,967 atm.
Unter den angegebenen Bedingungen betragt die Reaktionsgeschwindigkeitskonstante des
unvergifteten Katalysators fur das Zn-13X-Molekularsieb 47 Mole pro Liter Schuttvolu¬
men, Stunde und Atmosphäre mit einer Vergiftungsgeschwmdigkeitskonstante von 0,06
Liter Schuttvolumen pro Mol umgesetztes Acetylen. Die entsprechenden Konstanten fur
das Cd-13X-Molekularsieb betragen 520 fur die Hydratisierung und 0,55 fur die Vergif¬
tung. Die Cadmiumionen setzen somit rund lOmal mehr Acetylen um, werden aber auch
ca. lOmal schneller vergiftet. Da die Geschwindigkeitskonstanten der Acetylenumsetzungund der Katalysatorvergiftung beim Cadmium starker von der Temperatur abhangig
sind als beim Zink, sollte es somit möglich sein, unter Anwendung von Cadmiumkon-
takten bei tieferen Temperaturen ein gunstigeres Verhältnis von Reaktionsgeschwindig¬
keit zu Katalysatorvergiftung zu erhalten. Eine grobe Abschätzung bestätigt dies auch.
Unter den gegebenen Bedingungen setzt der Cd-13X-Kontakt schon bei 170°C 47 Mole
Acetylen pro Liter Schuttvolumen, Stunde und Atmosphäre mit einer Vergiftungsgeschwin-
digkeitskonstante von nur rund 0,003 Liter Schuttvolumen pro Mol Acetylen um. Mit Cd-
13X-Molekularsiebkatalysatoren gelingt somit die Wasseranlagerung an Acetylen schon
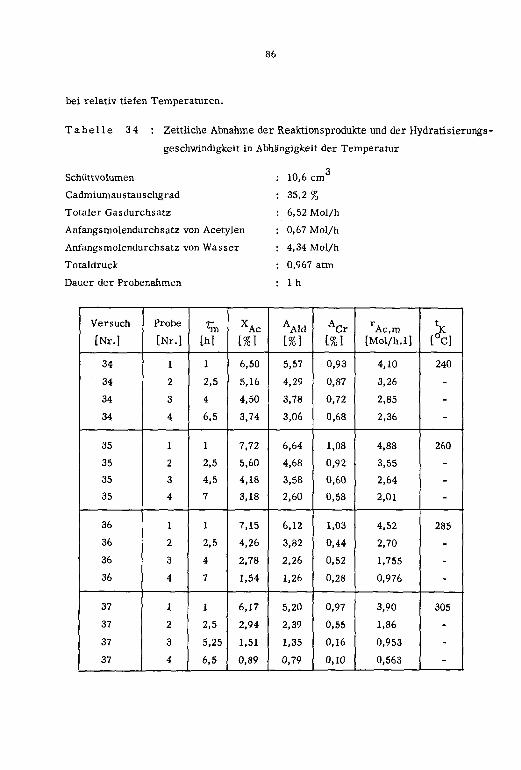
86
bei relativ tiefen Temperaturen.
Tabelle 34 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisierungs¬
geschwindigkeit in Abhängigkeit der Temperatur
Schuttvolumen 10,6 cm3
Cadmiumaustauschgrad 35,2%
Totaler Gasdurchsatz 6,52 Mol/h
Anfangsmolendurchsatz von Acetylen 0,67 Mol/h
Anfangsmolendurchsatz von Wasser 4,34 Mol/h
Totaldruck 0,967 atm
Dauer der Probenahmen lh
Versuch
[Nr.]
Probe
[Nr.l [hl
XAc[%]
AAld[%]
ACr[%1
r.
Ac,m
[Mol/h.1] [°C]
34
34
34
34
1
2
3
4
1
2,5
4
6,5
6,50
5,16
4,50
3,74
5,57
4,29
3,78
3,06
0,93
0,87
0,72
0,68
4,10
3,26
2,85
2,36
240
35
35
35
35
1
2
3
4
1
2,5
4,5
7
7,72
5,60
4,18
3,18
6,64
4,68
3,58
2,60
1,08
0,92
0,60
0,58
4,88
3,55
2,64
2,01
260
36
36
36
36
1
2
3
4
1
2,5
4
7
7,15
4,26
2,78
1,54
6,12
3,82
2,26
1,26
1,03
0,44
0,52
0,28
4,52
2,70
1,755
0,976
285
37
37
37
37
1
2
3
4
1
2,5
5,25
6,5
6,17
2,94
1,51
0,89
5,20
2,39
1,35
0,79
0,97
0,55
0,16
0,10
3,90
1,86
0,953
0,563
305
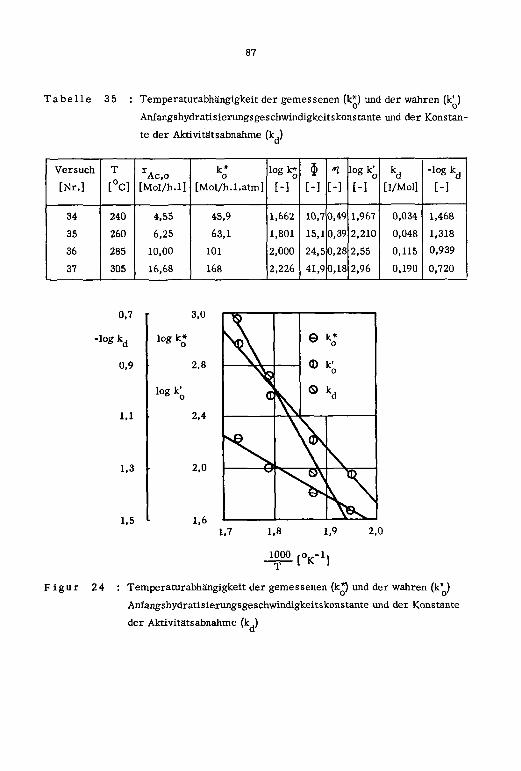
87
Tabelle 35 : Temperaturabhangigkeit der gemessenen (k*) und der wahren (k')
Anfangshydratisierungsgeschwindigkeitskonstante und der Konstan¬
te der Aktivitätsabnähme (k,)
Versuch TAc,o
k*o
logk* $ «Z logk; kd -log kd[Nr.] [°C] [Mol/h.l] [Mol/h.l.atm] [-] [-] [-1 [-1 [1/Mol] [-]
34 240 4,55 45,9 1,662 10,7 0,49 1,967 0,034 1,468
35 260 6,25 63,1 1,801 15,1 0,39 2,210 0,048 1,318
36 285 10,00 101 2,000 24,5 0,28 2,55 0,115 0,939
37 305 16,68 168 2,226 41,9 0,18 2,96 0,190 0,720
0,7
-log kd
0,9
1,1
1,3
1,5
logk*
1°S^
3,0
2,8
2,4
2,0
1,6
e k*0
w"o
o kd
"V.^\
1,7 1,8 1,9 2,0
ÄtV1]
Figur 24 Temperaturabhangigkeit der gemessenen (k*) und der wahren (k' )
Anfangshydratisierungsgeschwindigkeitskonstante und der Konstante
der Aktivitàtsabnahme (k.)

88
3.5.3. Hg-13X-Katalysatoren
In einer analogen Apparatur, wie Figur 1 zeigt, wobei aber ein elektrisch beheiztes Re¬
aktionsrohr aus Glas von 1,5 cm innerem Durchmesser eingesetzt wurde, gelangten
Quecksilber-(Il)-13X-Katalysatoren zwischen 110 und 130 C zur Untersuchung. In al¬
len Fallen fiel die relativ grosse Anfangsgeschwindigkeit innert weniger Minuten auf
Null ab. Im Abscheider konnte elementares Quecksilber nachgewiesen werden. Infolge
der sehr schnellen Aktivitätsabnähme wurde auf eine kinetische Auswertung verzichtet.
Quecksilber-(II)-salze sind somit, wie bereits von einigen anderen Autoren (102, 103)
festgestellt wurde, fur die Acetylenhydratisierung in der Gasphase kaum geeignet.
3.5.4. Cu-13X-Katalysatoren
2+ 2+ 2+Aus der Literatur ist bekannt, das s ausser der Zinkgruppe (Zn , Cd , Hg ) auch die
+ 2+ +Metallionen der Kupfergruppe (Cu ,
Cu , Ag ) die Wasseranlagerung an Acetylen ka¬
talysieren.
Zu diesem Zwecke sollte nun abgeklärt werden, ob sich die ins Molekularsieb 13X ein¬
gelagerten Kupfer-(II)-îonen fur die Gasphasenhydratisierung von Acetylen zu Acetal-
dehyd eignen, und wie gross ihre Aktivität im Vergleich zu den Zink-(II)- und Cadmium-
(Il)-ionen ist.
Als erstes wurde die zeitliche Abnahme der Acetylenumsatzgeschwmdigkeit und der Zu¬
sammensetzung der Produkte in Abhängigkeit des Acetylenpartialdruckes gemessen. Ver¬
suchsbedingungen und Resultate sind in Tabelle 36 zusammengestellt.
Die Temperatur konnte wahrend der ersten halben Stunde nicht konstant gehalten werden,
sondern stieg beim Einfahren mit Acetylen sehr schnell und stark an, fiel dann aber wie¬
der langsam auf den Sollwert ab. Der Grund des Temperaturanstieges durfte darin be¬
stehen, dass im ersten Moment sehr stabile Acetylen-Kupfer-(II)-Komplexe, und Halb-
acetylide sowie Acetyhde gebildet werden. Bei der Entstehung dieser Verbindungen wird
eine grosse Wärmemenge freigesetzt, die nicht schnell genug abgeführt werden kann.
Die Enthalpien dieser Reaktionen zwischen 240 und 335 C sind zwar unbekannt, doch be¬
stätigen die Werte der Tabelle 10 diese Vermutung.
Aus Tabelle 36 geht hervor, dass die Acetylenumsatze in den untersuchten Grenzen von
0,2 bis 1 % variieren. Die Acetaldehydausbeute betrug durchschnittlich 92 %. Neben rund
7 % Crotonaldehyd wurden noch Spuren von Essigsäure identifiziert. Die Aküvitat des

89
Cu-13X-Katalysators sank innerhalb weniger Stunden praktisch auf Null ab. Das unge¬
brauchte Cu-13X-Molekularsieb war blau gefärbt, das gebrauchte hingegen braunschwarz,
dies deutet darauf hm, dass zu Beginn Acetylide gebildet werden, die ihrerseits, wie aus
der Literatur wohl bekannt ist (178), die Polymerisation von Acetylen katalysieren. Da¬
durch wird die Oberflache innert kürzester Zeit mit Acetylenpolymeren bedeckt, so dass
alle eventuell noch freien Kupfer-(II)-ionen, die als Hydratisierungskatalysatoren auf¬
treten können, dem Eduktstrom nicht mehr zugänglich sind. Die Acetylenpolymensa-
tion ist ebenfalls eine stark exotherme Reaktion, z.B. betragt die Reaktionsenthalpie von
Divinylacetylen aus zwei Molekülen Acetylen - 63 kcal/Mol bei 0 C, und kann somit ei¬
ne weitere Ursache fur den starken Temperaturanstieg sein.
Tabelle 36 : Zeitliche Abnahme der Produkte und der Hydratisierungsgeschwin¬
digkeit in Abhängigkeit der Acetylenkonzentration
Mittlere Katalysatortemperatur
Schuttvolumen
Kupferaustau s chgrad
Totaler Gasdurchsatz
Anfangsmolendurchsatz von Wasser
Totaler Gasdruck
Dauer der Probenahmen
280°C
10,6 cm3
66,6%
6,52 Mol/h
4,34 Mol/h
0,967 atm
lh
Versuch
[Nr.]
m.
Ac,o
[Mol/hl
Probe
[Nr.I [hl
XAcIS61
AAldm
ACr[561
rAAc,m
[Mol/h.l]
41 0,435 1 0,75 0,99 0,92 0,07 0,408
41 - 2 2 0,69 0,64 0,05 0,282
41 - 3 4 0,49 0,44 0,05 0,200
42 0,670 1 1 1,04 0,96 0,08 0,658
42 - 2 2,5 0,65 0,60 0,05 0,408
42 - 3 5 0,47 0,43 0,04 0,298
43 0,900 1 0,88 0,61 0,55 0,06 0,519
43 - 2 2 0,42 0,38 0,04 0,357
43 - 3 4,75 0,22 0,21 0,01 0,185
43 - 4 5,75 0,20 0,20 - 0,170

90
Aus diesen Ueberlegungen heraus, die durch die im Literaturteil aufgeführten experi¬
mentellen Befunde bestätigt werden, sind die fur den Cu-13X-Katalysator abgeleiteten
Reaktionsgeschwindigkeits- und Vergiftungsgeschwindigkeitskonstanten mit bedeutend
grosseren Fehlern behaftet als diejenigen fur die Zn-13X- und Cd-13X-Katalysatoren.
Diese Versuche wurden denn auch nur im Sinne emer experimentellen Bestätigung der
eingangs gemachten theoretischen Erörterungen durchgeführt, und sollten dadurch mit¬
helfen, die Kenntnisse über die Gasphasenhydratisierung von Acetylen zu Acetaldehyd
zu vervollständigen. Obwohl die Kupferionen kaum fur die Gasphasenhydratisierung ge¬
eignet sind, war es doch mteressant, zu untersuchen, ob die an Zn-13X- und Cd-13X-
Katalysatoren gefundenen Gesetzmassigkeiten auch fur die entsprechenden Cu-13X-Ka-
talysatoren gelten.
Durch Auftragen der reziproken Hydratisierungsgeschwindigkeiten in Abhängigkeit der
Reaktionsdauer wurden ebenfalls angenähert Geraden erhalten, aus denen sich die ent¬
sprechenden Konstanten abschätzen lies sen. Die extrapolierten Werte smd in Tabelle 37
aufgeführt. Die Anfangsgeschwindigkeit der Hydratisierung nimmt linear, wie aus Figur
25 erkenntlich, von Null bis 1 Mol pro Liter Schuttvolumen und Stunde zu, die entspre¬
chende Konstante der Aktivitatsabnahme, wie aus Tabelle 37 hervorgeht, von Null bis
0,9 Liter Schuttvolumen pro Mol umgesetztes Acetylen bei Aenderung des Acetylendruckes
von Null bis 0,133 Atmosphären. Die Vergrftungsgeschwindigkeit ist ausserdem eine Funk¬
tion des Wasser-Acetylenverhàltmsses, wie in Figur 26 zum Ausdruck kommt.
Ac,o
[Mol/h.l]
°'10[arm]
°'15
Figur 25 : Anfangshydratisierungsgeschwindigkeit (r, ) als Funktion des Acety-
lenpartialdrucks (p. )
Weiter wurde auch der Einfluss des Kupferaustauschgrades auf die Acetylenumsatzge-
schwindigkeit und auf die Zusammensetzung der Produkte nach der bereits beschriebe-
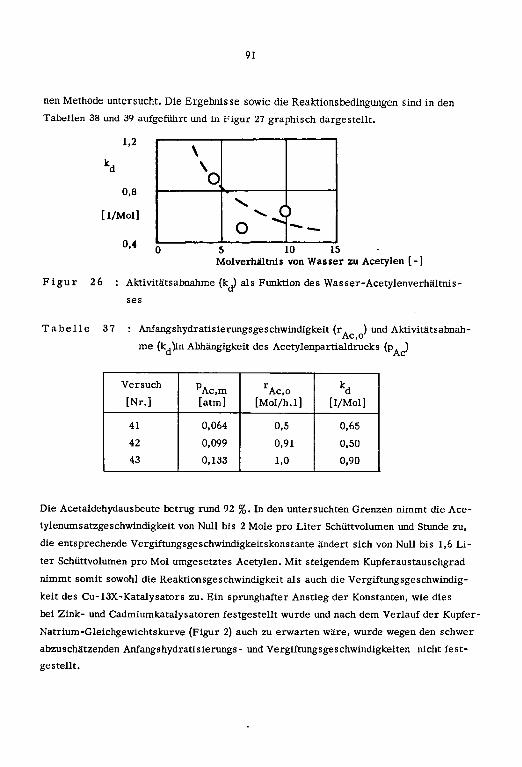
91
nen Methode untersucht. Die Ergebnisse sowie die Reaktionsbedingungen sind in den
Tabellen 38 und 39 aufgeführt und in Figur 27 graphisch dargestellt.
1,2
kd
0,8
[1/Mol]
0,40 5 10 15
Molverhaltnis von Wasser zu Acetylen [-]
Figur 26 : Aktivitatsabnahme (k.) als Funktion des Wasser-Acetylenverhaltnis-
\
\o
o ^~-
Tabelle 37 : Anfangshydratisierungsgeschwindigkeit (r ) und Aktivitatsabnah¬
me (k ,)in Abhängigkeit des Acetylenpartialdrucks (p )
Versuch
[Nr.]Ac,m[atm]
Ac,o
[Mol/h. 1]
kd[1/Mol]
41
42
43
0,064
0,099
0,133
0,5
0,91
1.0
0,65
0,50
0,90
Die Acetaldehydausbeute betrug rund 92 %. In den untersuchten Grenzen nimmt die Ace-
tylenumSatzgeschwindigkeit von Null bis 2 Mole pro Liter Schuttvolumen und Stunde zu,
die entsprechende Vergiftungsgeschwindigkeitskonstante ändert sich von Null bis 1,6 Li¬
ter Schuttvolumen pro Mol umgesetztes Acetylen. Mit steigendem Kupferaustauschgrad
nimmt somit sowohl die Reaktionsgeschwindigkeit als auch die Vergiftungsgeschwindig¬
keit des Cu-13X-Katalysators zu. Ein sprunghafter Anstieg der Konstanten, wie dies
bei Zink- und Cadmiumkatalysatoren festgestellt wurde und nach dem Verlauf der Kupfer-
Natnum-Gleichgewichtskurve (Figur 2) auch zu erwarten ware, wurde wegen den schwer
abzuschätzenden Anfangshydratisierungs- und Vergiftungsgeschwindigkeiten nicht fest¬
gestellt.
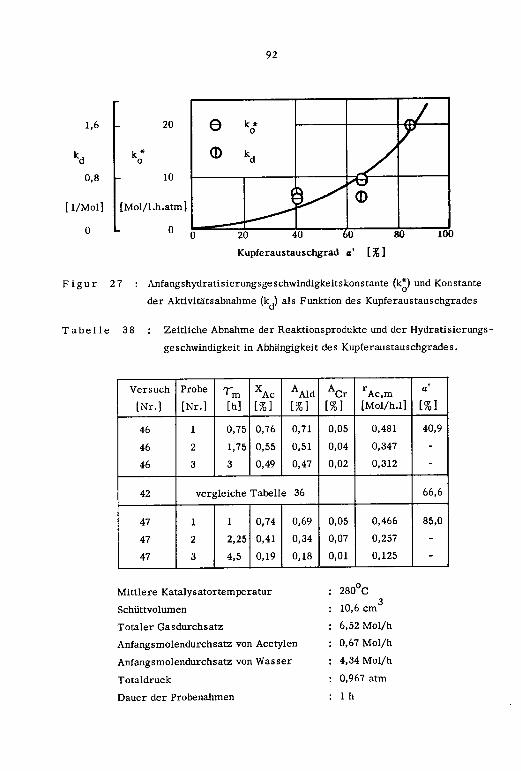
92
1,6
kd
0,8
[l/Uol]
0
20
10
[Mol/l.h.atm]
0
Ä It »
© kd
O
20 40 60
Kupferaustauschgrad a' [%]
80 100
Figur 2 7 : Anfangshydratisierungsgeschwindigkeitskonstante (k*) und Konstante
der Aktivitäts abnähme (k,) als Funktion des Kupferaustauschgrades
Tabelle 38 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisierungs¬
geschwindigkeit in Abhängigkeit des Kupferaustauschgrades.
Versuch
[Nr.]
Probe
[Nr.]
Tm
[h]
XAc[%]
AAld[%]
ACr[%I
r.Ac,m
[Mol/h.l]
a'
[%]
46 1 0,75 0,76 0,71 0,05 0,481 40,9
46 2 1,75 0,55 0,51 0,04 0,347 -
46 3 3 0,49 0,47 0,02 0,312 -
42 vergleiche Tabelle 36 66,6
47 1 1 0,74 0,69 0,05 0,466 85,0
47 2 2,25 0,41 0,34 0,07 0,257 -
47 3 4,5 0,19 0,18 0,01 0,125 -
Mittlere Katalysatortemperatur
Schuttvolumen
Totaler Gasdurchsatz
Anfangsmolendurchsatz von Acetylen
Anfangsmolendurchsatz von Wasser
Totaldruck
Dauer der Probenahmen
280°C
10,6 cm3
6,52 Mol/h
0,67 Mol/h
4,34 Mol/h
0,967 atm
1 h

93
Tabelle 39 : Anfangshydratisierungsgeschwindigkeitskonstante (k*) und Konstan¬
te der Aktivitatsabnahme (k ) als Funktion des Kupferaustauschgra¬
des
Versuch
[Nr.] [561
r.
Ac,o
[Mol/h.l]
k*0
[Mol/h.l.atm]
kd[1/Mol]
46
42
47
40,9
66,6
85,0
0,589
0,91
2,00
5,94
9,18
20,2
0,55
0,50
1,6
-log k.
0,2
0,4
0,6
0,8
log k
1,5
1,3
1,1
0,9
0,7
O k.
G kd
1,6 1,7 1,8 1.9 2,0
mo_ [Vij
Figur 2 8 : Temperaturabhangigkeit der gemessenen (k*) Anfangshydratisierungs-
geschwindigkeitskonstante und der Konstante der Aktivitätsabnahme (k,)
Um den Einfluss der Temperatur auf den Reaktionsablauf wie auch auf die Vergiftungs -
geschwindigkeit festzulegen, wurde die zeitliche Abnahme der Reaktionsprodukte in Ab¬
hängigkeit der Temperatur gemessen. Versuchsbedingungen und Resultate smd in den
Tabellen 40 und 41 wiedergegeben und in Figur 28 graphisch dargestellt.
Die mittlere Acetaldehydausbeute berechnet sich auch hier zu 92 %. Im Temperatur¬
intervall von 240 bis 335 C nimmt die beobachtete Reaktionsgeschwindigkeitskonstan-
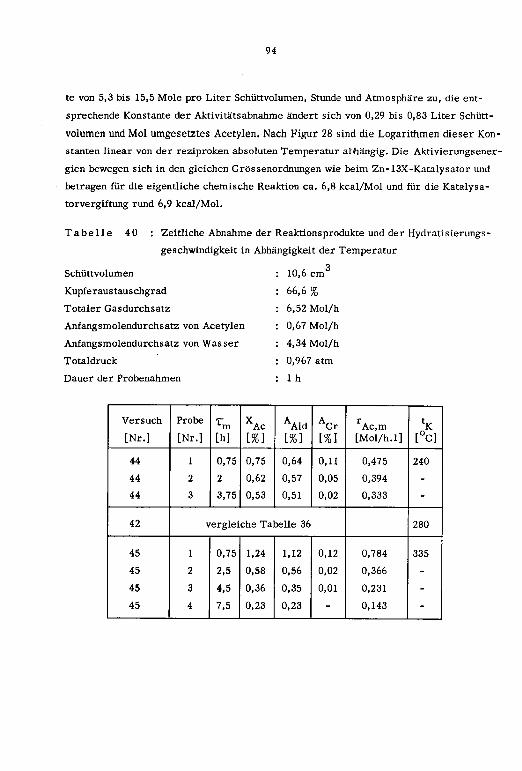
94
te von 5,3 bis 15,5 Mole pro Liter Schuttvolumen, Stunde und Atmosphäre zu, die ent¬
sprechende Konstante der Aktivitatsabnahme ändert sich von 0,29 bis 0,83 Liter Schutt¬
volumen und Mol umgesetztes Acetylen. Nach Figur 28 sind die Logarithmen dieser Kon¬
stanten linear von der reziproken absoluten Temperatur abhangig. Die Aktivierungsener¬
gien bewegen sich in den gleichen Grossenordnungen wie beim Zn-13X-Katalysator und
betragen fur die eigentliche chemische Reaktion ca. 6,8 kcal/Mol und fur die Katalysa¬
torvergiftung rund 6,9 kcal/Mol.
Tabelle 40 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisierungs¬
geschwindigkeit in Abhängigkeit der Temperatur
3Schuttvolumen
Kupferaustauschgrad
Totaler Gasdurchsatz
Anfangsmolendurchsatz von Acetylen
Anfangsmolendurchsatz von Wasser
Totaldruck
Dauer der Probenahmen
10,6 cm
66,6%
6,52 Mol/h
0,67 Mol/h
4,34 Mol/h
0,967 atm
lh
Versuch
[Nr.]
Probe
[Nr.]
rm[h]
Ac
[%]
AAld[561
ACr Ac,m
[Mol/h. 1]
lK[°C]
44 1 0,75 0,75 0,64 0,11 0,475 240
44 2 2 0,62 0,57 0,05 0,394 -
44 3 3,75 0,53 0,51 0,02 0,333 -
42 vergleiche Tabelle 36 280
45 1 0,75 1,24 1,12 0,12 0,784 335
45 2 2,5 0,58 0,56 0,02 0,366 -
45 3 4,5 0,36 0,35 0,01 0,231 -
45 4 7,5 0,23 0,23 - 0,143 -

95
Tabelle 41 : Temperaturabhangigkeit der gemessenen (k*) Anfangshydratisie-
rungsgeschwindigkeitskonstante und der Konstante der Aktivitäts
abnähme (k )
Versuch
[Nr.]
T
[°C]
1/T
[VWr.Ac,o
[Mol/h.l]
k*0
[Mol/h.l.atm]
log kQ*[-]
kd[1/Mol]
-log ka[-]
44 240 1,95 0,526 5,31 0,726 0,29 0,537
42 280 1,81 0,91 9,18 0,964 0,50 0,300
45 335 1,645 1,54 15,5 1,192 0,83 0,078
Zusammenfassend sei festgehalten :
a) Das modifizierte Molekularsleb Cu-13X kann wohl als Acetylenhydratisierungskata-
lysator eingesetzt werden, der grosse Nachteil gegenüber den Zn-13X- und Cd-13X-Ka-
talysatoren besteht darin, dass die Aktivität innert weniger Stunden praktisch auf Null
absinkt. Kupferionen bilden mit Acetylen in neutralem Gebiet Acetylide, die ihrerseits
die Polymerisation von Acetylen katalysieren. Als Folge tritt eine Bedeckung der kata-
lytisch wirksamen Oberflache mit Acetylenpolymeren auf. Die reziproke Acetylenhydra -
tisierungsgeschwindigkeit scheint auch m grober Näherung der Reaktionsdauer direkt
proportional zu sein, wie dies m Gleichung [55] zum Ausdruck kommt und fur Zn-13X-
Kontakte gefunden wurde. Die Vergiftungsgeschwindigkeit ist vom Wasser-Acetylen-
verhaltnis abhangig und nimmt mit abnehmendem Molverhaltnis zu. Ausserdem nimmt
die Konstante der Aktivitatsabnahme mit zunehmendem Kupferaustauschgrad und mit
steigender Temperatur zu. Die nach Formel [65] berechnete Aktivierungsenergie der
Katalysatorvergiftung betragt rund 6,9 kcal/Mol.
b) Die Hydratisierungsgeschwindigkeit des nicht vergifteten Katalysators ist vom Ace-
tylenpartialdruck, vom Zinkaustauschgrad und von der Temperatur abhangig. Die Anfangs-
acetylenumsetzung scheint zwar linear vom Acetylendruck abhangig zu sein, trotzdem
muss aber angenommen werden, dass Ungleichung [60] nicht mehr gilt, weil die Ace-
tylen-Kupfer-Komplexbildungskonstanten sehr gross sind, wie m den Kapiteln 2.3.1.
und 2.4. gezeigt wurde. Somit muss Gleichung [59] zur Beschreibung der Kinetik gel¬
ten. Da die Komplexbildungskonstanten in Abhängigkeit der Temperatur nicht bekannt
sind, konnten die eigentlichen Reaktionsgeschwindigkeitskonstanten (k*) nicht berech¬
net werden. Deshalb wurde eine Konstante (k*) angegeben, die alle Adsorptionstherme
einschliesst. Analog der Vergiftungs- nimmt auch die Hydratisierungsgeschwindigkeits-
konstante mit höherer Kupfer-(II)-ionenkonzentration bei konstantem Katalysatorvolu-

96
men und mit steigender Temperatur nach Gleichung [64] zu. Die entsprechende Akti¬
vierungsenergie betragt rund 6,8 kcal/Mol.
c) Als Hauptreaktionsprodukt fallt Acetaldehyd an. Die Ausbeute, bezogen auf umge¬
setztes Acetylen, betragt ca. 92 %. Die restlichen 8 % sind Crotonaldehyd und Spuren
von Essigsaure.
d) Beim Vergleich der Aktivität des Cu-13X-Katalysators mit derjenigen des Zinkkon¬
taktes unter den im Kapitel 3.5.2.angegebenen Bedingungen, erhalt man fur die Acetylen-
hydratisierungsgeschwindigkeit 30 Mole pro Liter Schuttvolumen, Stunde und Atmosphä¬
re und fur die Vergiftungsgeschwindigkeit rund 3,2 Liter Schuttvolumen pro Mol umge¬
setztes Acetylen. Bei praktisch identischer Anfangsaktivitat wird der Kupferkatalysator
aber rund 50mal schneller vergiftet. Kupfer-(II)-ionen sind somit fur die Gasphasen¬
hydratisierung von Acetylen zu Acetaldehyd ungeeignet.
3.5.5. Ag-1 3X-Katalysatoren
Um nun das im Kapitel 2.3. angegebene System zu vervollständigen und um zu unter¬
suchen, ob sich Ag-13X-Katalysatoren ebenfalls in das festgelegte Reaktionsschema ein¬
fugen lassen, wurde die zeitliche Abnahme der Hydratisierungsgeschwindigkeit und der
Reaktionsprodukte in Abhängigkeit des Acetylenpartialdruckes, des Silberaustauschgra-
des und der Temperatur gemessen.
Nach den Ergebnissen der durchgeführten Versuchsreihen, deren Bedingungen und Re¬
sultate in den Tabellen 42 bis 47 aufgeführt und in den Figuren 29 bis 32 graphisch dar¬
gestellt sind, ist eindeutig ersichtlich, dass sich die Silber-(I)-ionen als Hydratisie¬
rungskatalysatoren sehr ähnlich verhalten wie die Kupferionen. Diese Tatsache wurde
bereits im Literaturteil eingehend diskutiert und war vorauszusehen. Es soll deshalb
auf eine eingehende Besprechung der einzelnen Versuchsreihen verzichtet werden. Die
Ergebnisse sollen lediglich vergleichend zu den Kupferkatalysatoren summarisch zu-
sammengefasst werden.
Die Acetaldehydausbeute betrug, wie beim Kupferkontakt, durchs chmttlich 92 %. Die rest¬
lichen 8 % waren zur Hauptsache Crotonaldehyd neben Spuren von Essigsaure. Alle Ge¬
setzmassigkeiten, die zur Charakterisierung der Kupferkatalysatoren gefunden wurden,
waren auch im vorliegenden Fall gültig. Die beobachteten Acetylenumsatzgeschwindig-
keitskonstanten bewegen sich m den untersuchten Grenzen von Null bis rund 100 Mole
pro Liter Schuttvolumen, Stunde und Atmosphäre, die entsprechende Vergiftungsgeschwin-
digkeitskonstante von Null bis 1,5 Liter Schuttvolumen pro Mol umgesetztes Acetylen. Die-
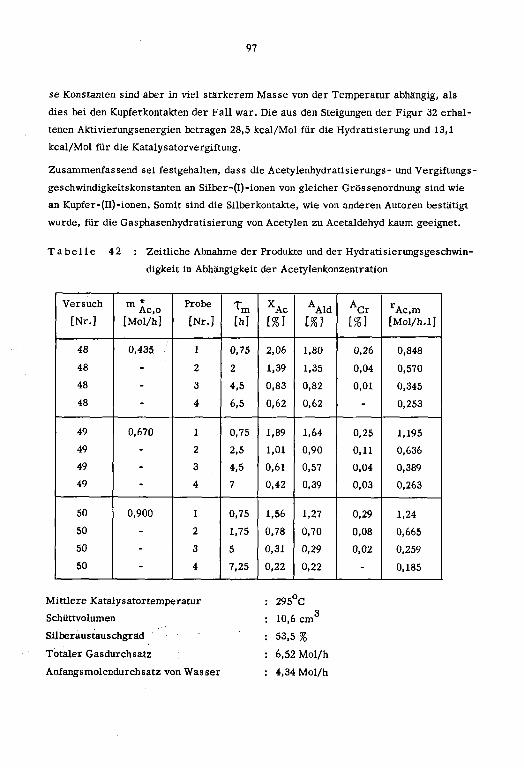
97
se Konstanten sind aber m viel stärkerem Masse von der Temperatur abhangig, als
dies bei den Kupferkontakten der Fall war. Die aus den Steigungen der Figur 32 erhal¬
tenen Aktivierungsenergien betragen 28,5 kcal/Mol fur die Hydratisierung und 13,1
kcal/Mol fur die Katalysatorvergiftung.
Zusammenfassend sei festgehalten, dass die Acetylenhydratisierungs- und Vergiftungs-
geschwmdigkeitskonstanten an Silber-(I)-ionen von gleicher Grossenordnung sind wie
an Kupfer-(II)-ionen. Somit sind die Silberkontakte, wie von anderen Autoren bestätigt
wurde, fur die Gasphasenhydratisierung von Acetylen zu Acetaldehyd kaum geeignet.
Tabelle 42 : Zeitliche Abnahme der Produkte und der Hydratisierungsgeschwin¬
digkeit in Abhängigkeit der Acetylenkonzentration
Versuch
[Nr.]Ac,o
[Mol/h]
Probe
[Nr.] [hl
XAc[561
AAld[SEI
Cr
[561rAc,m[Mol/h.l]
48 0,435 1 0,75 2,06 1,80 0,26 0,848
48 - 2 2 1,39 1,35 0,04 0,570
48 - 3 4,5 0,83 0,82 0,01 0,345
48 - 4 6,5 0,62 0,62 - 0,253
49 0,670 1 0,75 1,89 1,64 0,25 1,195
49 - 2 2,5 1,01 0,90 0,11 0,636
49 - 3 4,5 0,61 0,57 0,04 0,389
49 - 4 7 0,42 0,39 0,03 0,263
50 0,900 1 0,75 1,56 1,27 0,29 1,24
50 - 2 1,75 0,78 0,70 0,08 0,665
50 - 3 5 0,31 0,29 0,02 0,259
50 - 4 7,25 0,22 0,22 - 0,185
Mittlere Katalysatortemperatur 295°C
Schuttvolumen 10,6 cm3
Silberaustauschgrad 53,5%
Totaler Gasdurchsatz 6,52 Mol/h
Anfangsmolendurchsatz von Wasser 4,34 Mol/h
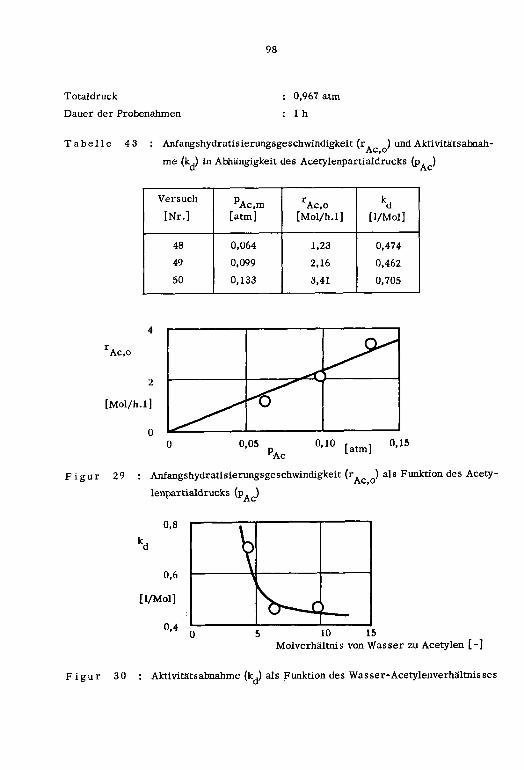
98
Totaldruck
Dauer der Probenahmen
0,967 atm
1h
Tabelle 43 : Anfangshydratisierungsgeschwindigkeit (r ) und Aktivitatsabnah¬
me (k,) in Abhängigkeit des Acetylenpartialdrucks (p )
Versuch
[Nr.]Ac,m
[atm]
r.Ac,o
[Mol/h.l]
kd[1/Mol]
48
49
50
0,064
0,099
0,133
1,23
2,16
3,41
0,474
0,462
0,705
0 0,05 0,10 r „ , 0,15PAc [ ]
Figur 29 : Anfangshydratisierungsgeschwindigkeit (r ) als Funktion des Acety¬
lenpartialdrucks (p, )
5 10 15
Molverhaltnis von Wasser zu Acetylen [-]
Figur 30 : Aktivitatsabnahme (k.) als Funktion des Wasser-Acetylenverhaltnisses

99
Tabelle 44 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisierungs-
geschwindigkeit in Abhängigkeit des Silberaustauschgrades.
Mittlere Katalysatortemperatur
Schuttvolumen
Totaler Gasdurchsatz
Anfangsmolendurchsatz von Acetylen
Anfangsmolendurchsatz von Wasser
Totaldruck
Dauer der Probenahmen
295"C
10,6 cm
6,52 Mol/h
0,67 Mol/h
4,34 Mol/h
0,967 atm
1h
Versuch
[Nr.] 1561
Probe
[Nr.] [h]
XAc[%]
AAld[%]
ACr[%]
r.
Ac,m
[Mol/h.l]
49 53,5 vergleiche Tabelle 42
54 85,9 1 0,75 1,33 1,24 0,09 0,87
54 - 2 3,25 0,39 0,39 - 0,245
54 - 3 5 0,24 0,24 - 0,151
54 - 4 7 0,18 0,18 - 0,111
55 100 1 0,75 1,22 1,07 0,15 0,769
55 - 2 2,75 0,37 0,37 - 0,232
55 - 3 4,75 0,21 0,21 - 0,130
Tabelle 45 : Anfangshydratisierungsgeschwindigkeitskonstante (k*) und Konstante
der Aktivitatsabnahme (k,) als Funktion des Silberaustauschgrades
Versuch
[Nr.]
a1
[SB]
r,Ac,o
[Mol/h.l]
k*0
[Mol/h.l.atm]
kd[l/Mol]
49
54
55
53,5
85,9
100
2,16
7,40
10
21,81
74,80
101
0,462
1,27
1,57
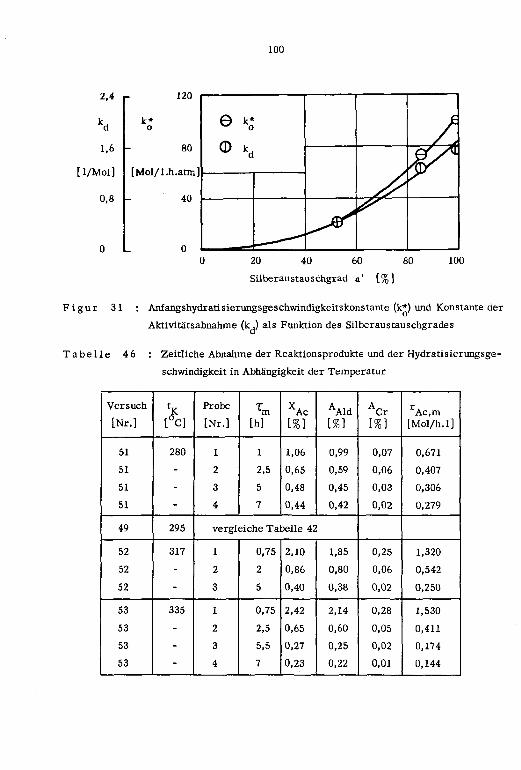
100
2,4
kd
1,6
[ 1/Mol]
0,8
0 L
120
k*o
80
[Mol/l.h.atm
40
\V k,a
20 40 60 80
Silberaustauschgrad a' [%]
100
Figur 31 : AnfangshydraQsierungsgeschwindigkeitskonstante (k*) und Konstante der
Aktivitatsabnahme (k.) als Funktion des Silberaustauschgrades
Tabelle 46 : Zeitliche Abnahme der Reaktionsprodukte und der Hydratisierungsge-
schwindigkeit in Abhängigkeit der Temperatur
Versuch
[Nr.] [°C]
Probe
[Nr.]
Tm[h]
Ac AAld[%]
ACr[%]
r,Ac,m
[Mol/h.1]
51 280 1 1 1,06 0,99 0,07 0,671
51 - 2 2,5 0,65 0,59 0,06 0,407
51 - 3 5 0,48 0,45 0,03 0,306
51 - 4 7 0,44 0,42 0,02 0,279
49 295 vergleiche Tabelle 42
52 317 1 0,75 2,10 1,85 0,25 1,320
52 - 2 2 0,86 0,80 0,06 0,542
52 - 3 5 0,40 0,38 0,02 0,250
53 335 1 0,75 2,42 2,14 0,28 1,530
53 - 2 2,5 0,65 0,60 0,05 0,411
53 - 3 5,5 0,27 0,25 0,02 0,174
53 - 4 7 0,23 0,22 0,01 0,144

101
Schuttvolumen
Silberaustauschgrad
Totaler Gasdurchsatz
Anfangsmolendurchsatz von Acetylen
Anfangsmolendurchsatz von Wasser
Totaldruck
Dauer der Probenahme
10,6 ein
53,5 %
6,52 Mol/h
0,67 Mol/h
4,34 Mol/h
0,967 atm
lh
Tabelle 47 : Temperaturabhangigkeit der gemessenen Anfangshydratisierungs-
geschwindigkeitskonstante (k*) und der Konstante der Aktivitatsab-
nahme (k )
Versuch
[Nr.]
T
[°C]
1/T
[Wr.
Ac,o
[Mol/h.l]
k*0
[Mol/h.l.atm]
logkQ[-]
kd[1/Mol]
-log kd[-]
51 280 1,81 0,725 7,32 0,865 0,340 0,468
49 295 1,76 2,16 21,81 1,339 0,462 0,335
52 317 1,695 4,35 43,9 1,642 0,754 0,123
53 335 1,645 10 101 2,0 0,998 0
Abschliessend sollen nochmals die Aktivitäten der Zink-(II)-, Cadmium-{II)-, Kupfer-(n)-
und Silber-(I)-13X-Katalysatoren miteinander verglichen werden. Zu diesem Zwecke wur¬
den die Acetylenumsatzgeschwindigkeitskonstanten (k») wie auch die Vergiftungsgeschwin-
digkeitskonstanten (k,) auf die in Tabelle 48 angegebenen Bedingungen extrapoliert.
Aus Tabelle 48 geht hervor, das s unter den angegebenen Bedingungen die Anfangsaktivi-
taten der Zink-(U)-, Kupfer-(ü)- und Süber-(I)-ionen ungefähr gleich gross sind, nur wer¬
den die Kupfer- und Süberkontakte rund 20 bis 50mal schneller vergiftet als die Zinkka¬
talysatoren, d.h. die Cu-13X- und Ag-13X-Molekularsiebe verlieren ihre Aktivität innert
weniger Stunden. Als Folge der schnellen Akavitatsabnahme sind diese Werte mit gros¬
sem Fehler behaftet und sind daher m Tabelle 48 in Klammern gesetzt.
Der Cadmiumkatalysator setzt im Vergleich zum Zinkkatalysator rund lOmal mehr Ace¬
tylen um, wird aber auch ca. lOmal schneller vergiftet. Da die Aktivierungsenergien der
Acetylenumsatz- und der Katalysatorvergiftungsgeschwindigkeit beim Cadmium starker
temperaturabhangig sind als beim Zink, ist die Möglichkeit gegeben, beispielsweise an

102
Cadmiumkontakten schon bei 170 C 47 Mole Acetylen pro Liter Schuttvolumen, Stunde
und Atmosphäre umzusetzen, wobei dann aber der Cd-13X-Katalysator im Vergleich zum
Zn-13X-Katalysator rund 20mal langsamer vergiftet wird. Es scheint, dass der Cadmium-
kontakt die grosste Aussicht auf industrielle Anwendung hat.
-0,2
logkd
0
0,2
0,4
0,6
0,8
logk'
2,8
2,4
2,0
1,6
1,2
0,8
O k*v-' 0
Q kü *d
1,6 1,7 1000 rOK-l, 1,8
Figur 32 : Temperaturabhangigkeit der gemessenen Anfangshydratisierungsgeschwin-
digkeitskonstante (k*) und der Konstante der Aktivitatsabnahme (k,)
Tabelle 48 : Zusammenstellung der Acetylenumsatzgeschwindigkeitskonstanten (k*)
und Vergiftungsgeschwindigkeitskonstanten (k,) und der beobachteten
Aktivierungsenergien der Acetylenhydratisierung (E*) und der Kataly¬
satorvergiftung (E,) fur Zink-(II)-, Cadmium-(II)-, Kupfer-(H)- und Sil-
ber-(I)-13X-Katalysatoren
Katalysator k*0
[Mol/l.h.atm]
kd[1/Mol]
E*g
[kcal/Mol]
Ed[kcal/Mol]
Zn-13X
Cd-13X
47
520
0,06
0,55
6,1
10,2
6,7
15,7
Cu-13X
Ag-13X
(30)(45)
(3,2)(1,2)
(6,8)(28,5)
(6,9)(13,1)

103
Mittlere Katalysatortemperatur 280°C
Molendurchsatz von Acetylen 0,67 Mol/h
Molendurchsatz von Wasser 4,34 Mol/h
Molendurchsatz von Stickstoff 1,51 Mol/h
Schüttvolumen 10,6 cm3
Metallionenaustauschgrad 100%
Totaldruck 0,967 atm

104
ZUSAMMENFASSUNG
Die sich aus der Literatur ergebenden generellen Grundlagen der Flussigphasen-
wie auch der Gasphasenhydratisierung von Acetylen zu Acetaldehyd wurden disku¬
tiert, wobei vor allem die fur eine Auswahl geeigneter Katalysatorsysteme wichti¬
gen Kriterien besondere Beachtung fanden. Bezüglich Katalysatorauswahl zeigte es
sich, dass vor allem Quecksilber-(II)-, Cadmium-(II)-, Zink-(II)-, Kupfer-(II)-,
Kupfer-(I)- und Silber-(I)-ionen die Wasseranlagerung an Acetylen katalysieren.
Kinetik und Aktivitäten dieser Metallionen wurden miteinander verglichen.
Durch Ionenaustausch wurden alsdann Quecksilber-(H)-, Cadmium-(II)-, Zink-(II)-,
Kupfer-(II)- und Silber-(I)-13X-Molekularsiebe hergestellt, an denen der makroki-
netische Verlauf der katalyüschen Gasphasenhydratisierung zwischen 110 und 424 C
in einem Differentialreaktor untersucht wurde.
Als Hauptprodukte traten Acetaldehyd und geringe Mengen von Crotonaldehyd auf.
Acetaldehyd wird über die als Zwischenprodukte auftretenden, unstabilen Acetylen-
Wasser-Metallionen-Komplexe gebildet. Crotonaldehyd entsteht durch Dimensie-
rung von Acetyldehyd mit anschliessender Wasserabspaltung. Essigsaure konnte in
Spuren nur bei Kupfer- und Silberkontakten nachgewiesen werden.
Alle Katalysatoren unterlagen mit zunehmender Reaktionsdauer einer gewissen Ak-
tivitatsabnahme. Die Geschwindigkeit der Vergiftung ist proportional zum Quadrat
der Hydratisierungsgeschwindigkeit. Auf Grund dieser Gesetzmassigkeit konnten
die Anfangshydratisierungsgeschwindigkeiten relativ genau extrapoliert werden. Die¬
se sind bezuglich der Acetylenkonzentration erster Ordnung.
Sowohl die Anfangsumsatzgeschwindigkeitskonstanten als auch die Vergiftungsge-
schwindigkeitskonstanten nehmen mit höherer Metalhonenkonzentration bei konstan¬
tem Katalysatorvolumen und mit steigender Temperatur nach Arrhemus zu. Die
entsprechenden Aktivierungsenergien der Hydratisierung und der Katalysatorver¬
giftung konnten ermittelt werden. Die Konstante der Aktivität sabnähme ist auch eine
Funktion des Wasser-Acetylen-Verhàltmsses und nimmt mit zunehmendem Molver-
hältms ab.

105
5. Zink- und Cadmiumkatalysatoren zeigen nur eine geringe, dagegen Kupfer- und
Silberkontakte eine grosse Vergiftungsgeschwindigkeit infolge Polymerisation von
Acetylen oder Acetaldehyd. Auch Quecksilberionen verlieren ihre Aktivität infolge
Reduktion zu metallischem Quecksilber schon bei 110 C ausserordentlich schnell.
6. Am Zink-13X-Molekularsieb wurde auf Grund der Kriterien von Satterfield und
Sherwood der Einfluss des kornexternen Stofftransportes und der Porendiffusion
berechnet. Das Fehlen eines hemmenden Einflusses des externen Stofftransportes
konnte sowohl rechnerisch wie auch experimentell bewiesen werden. Dagegen zeig¬
te es sich, das s die Diffusion in die Mikroporen hinein die chemische Reaktion
hemmt.
7. An Zinkkontakten wurde alsdann die Art und Zusammensetzung des Giftes studiert.
Da sich die Porendiffusion ruckwirkend auf die Geschwindigkeit der Aktivitatsabnah-
me auswirkt, konnten die praktischen Befunde auf Grund der von Wheeler angege¬
benen Kriterien auch rechnerisch bestätigt werden. Das Gift, welches homogen in
allen Poren verteilt ist, wird schwach adsorbiert. Die Möglichkeit der Katalysator¬
regenerierung wurde studiert.

106
NOMENKLATUR
^Ald
^Cr
Bo
c
D
D12
^Kd
_E-dc
E
G
AHa
Praexponentieller Faktor der Arrhenius-Gleichung
Acetaldehydausbeute, bezogen auf Ac,o
Crotonaldehydausbeute, bezogen auf Ac,o
Verhältnis der äusseren Katalysatoroberflache
zum Reaktorvolumen
Bodenstein'sehe Kenngrosse
Eduktkonzentration im Hauptstrom
Eduktkonzentration an der Kornaus senflache
Diffusionskoeffizient
gewöhnlicher Diffusionskoeffizient
Knudsendiffusionskoeffîzient
mittlerer Durchmesser der Katalysatorkörner
mittlere Lange der Katalysatorzylinder
Aktivierungsenergie
Verhältnis der Aktivität des vergifteten zur Akti¬
vität des nicht vergifteten Katalysators
spezifischer Massendurchsatz
Adsorptionsénergie
Reaktionsenthalpie
beobachtbare Reaktionsgeschwindigkeitskonstante
ohne Einfluss der Adsorption
wahre Reaktionsgeschwindigkeitskonstante, schliesst
Adsorptionstherme ein
gemessene Reaktionsgeschwindigkeitskonstante
beobachtete Reaktionsgeschwindigkeitskonstante
wirkliche Reaktionsgeschwindigkeitskonstante,
bezogen auf das Kontaktvolumen; 1. Ordnung
Stofftransportkoeffizient
Konstante der Aktivitätsabnahme
Komplexbildungskonstante
Wärmeleitzahl
Mol-%
Mol-%
2. 3cm /cm
Mol/cm
Mol/cm2/
cm /s2.
cm /s2,
cm /s
m
m
kcal/Mol
u /2
kg/m s
kcal/Mol
kcal/Mol
Mol/1.h
Mol/l.h.atm
Mol/l.h.atm
Mol/g-Kat..h
1/s2
Mol/s.cm .atm
l-Schuttvol./Mol
atm
kcal/h. K.m

107
L
M
Äc,o
W,o
m*
P
PiR
R
Re'
re
r,Ac,o
Ac,m
Sg
Se
T
T
t
P
M
YMe'
*MS„Me1
n+
n+
Ac
*i
Lange des Katalysatorbettes
Molekulargewicht der Komponente i
Anfangsmolendurchsatz des Acetylens
Anfangsmolendurchsatz des Wassers
totaler Gasdurchsatz
Absolutdruck
Partialdruck der Komponente 1
Gaskonstante
Kornradius
modifizierte Reynoldszahl
aequivalenter Porenradius
Anfangsacetylenhydratisierungsgeschwindigkeit
mittlere Acetylenhydratisierungsgeschwindigkeit
spezifische Oberflache des Katalysators
Schmidt' sehe Zahl
Absoluttemperatur
Temperatur der Kornaus senflache
mittlere Katalysatortemperatur
Zeitfaktor
Dauer der Probenahmen
Schuttvolumen
totales Kornvolumen
spezifisches Porenvolumen
Molvolumen
lineare Gasgeschwindigkeit
Aequivalentbruch des Metallions im MS 13X
Aequivalentbruch des Metallions in der Losung
Durchmesser der Katalysatorzylinder
Acetylenum satz
Molenbruch der Komponente 1
vergifteter Anteil der Oberflache
Austauschgrad des Metallions
Mol/h
Mol/h
Mol/h
atm
arm
kcal/°KJvlol
cm
cm oder A
Mol/(l-Schuttvol.).h
Mol/(1-Schuttvol.).h2.
m /g
(l-Schuttvol.)/h.Mol
h
3/cm /g
cm /Mol
m/s
m
Mol-%

108
ß maximale relative Temperaturdifferenz
6 Leervolumenanteil
1 Nutzungsgrad
e Porosität
»•mViskosität des Reaktionsgemisches kg/m. s
*D Stossintegral
S Dichte des Reaktionsgemisches kg/m
9b Schuttdichte /3
g/cm
g wahre Dichte /3
g/cm
9Pscheinbare Dichte /
3g/cm
Ö12 Konstante
ÖAcAnteil der mit Acetylen bedeckten Oberflache
T Tortuositatsfaktor
Tm mittlere Reaktionsdauer h
ts, jSL Thiele-Modul
$ Hilfsfunktion

109
LITERATURVERZEICHNIS
1) M. Kutscheroff, Ber. deutsch, ehem. Ges. 14, 1540 (1881)
2) Chemische Fabrik Griesheim, D.R.P. 250 356 (1910)
3) I. G. Farbenindustrie, D.R.P. 511 634 (1915)
4) S.A. Miller, Chem. Ind. (London) 1963, 4
5) W.L. Faith, D.B. Keyes, R.L. Clark, Industrial Chemicals, 2nd ed.,
John Wiley and Sons, Inc., New York, 1957,2
6) M . Sittig, Petrol. Refiner _41 (4), 157 (1962)
7) Consortium für Elektrochemische G.m.b.H., Can.P. 625 430 (1961)
8) Aldehyd G.m.B.H., Petrol. Refiner 40 (11), 206 (1961)
9) Farbwerke Hoechst, Petrol. Refiner 40 (11), 209 (1961)
10) Stone and Webster Engineering Co., Petrol. Refiner 40 (11), 213 (1961)
11) Beilstein, HI, 596(1918); EI 1, 322 (1928); Ell 1, 659 (1941); EIIIl, 2618
(1959) (je 4. Auflage)
12) Uli mann, Enzykl. der techn. Chem.J, 97 (1928); IV, 644 (1929); £I£,4 (1953)
(je 2. Auflage)
13) Kirk-Othmer, Encycl. of Chem. Techn. I, 42 (1947); 1, 77 (1963)
14) J. Nieuwland, R. Vogt, The Chemistry of Acetylene, New York 1945, 115
15) G.M. Schwabe, Katalyse in der org. Chemie 7 (2), 54 (1943)
16) M. Miocque, N. Manh Hung, Vo Quang Yen, Ann. Chim. 8, 157 (1963)
17) I.L. Kotlyarevskii, Trudy Vostochno-Sibir. Filiala, Akad. Nauk USSR, Ser.
Khim 1956 (4), 58 (Russisch)
cit. Chem. Abstr. 51, 12815 (1957)
18) E.V. Murphree, A.P. 2 183 148 (1940)
19) A.R. Globus, A.P. 2 402958 (1946)
20) F. Lieseberg, G. Wietzel, D.R.P. 717 951 (1942)
21) R.M. Isham, A.P. 2 303 279 (1943)
22) L. Mayer, D.R.P. 708 462(1941)
23) C. Toyama, Jap.P. 154 179 (1942)
cit. Chem. Abstr. 43, 3837 (1949)

110
24) M. Yamada, T. Kamioka, S. Kato, Jap.P. 176 406 (1948)
cit. Chem. Abstr. 45, 7137 (1951)
25) M. Randall, A.P. 2 511787 (1947)
26) T. Kamioka et al., Jap. P. 4154 (1952)
cit. Chem. Abstr. 48, 5208 (1954)
27) K. Oda, Y. Maeda, Jap. P. 1672 (1953)
cit. Chem. Abstr. 48, 3385 (1954)
28) T. Tokue, K. Shiga, Jap. P. 1620 (1958)
cit. Chem. Abstr. 53, 4136 (1959)
29) K.K. Showa Denko, Jap. P. 8220 (1960)
cit. Chem. Abstr. 55, 16857 (1961)
30) M.K. Kal'fus et al., USSR.P. 108 128 (1957)
cit. Chem. Abstr. 52, 6393 (1958)
31) R.W. Chapman, R.M. Dille, A.P. 3 116 333 (1963)
32) R.M. Dille, R.W. Chapmann et al., 3 095 453 (1963)
33) Consortium fur elektrochemische Industrie, F.P. 1 215 938 (1958);
Oe. P. 211 293 (1958)
34) D.V. Sokol'skii, S.K. Nurgozheva, Trudy Inst. Khim. Nauk, Akad. Nauk
Kaz. USSR 8, 163 (1962)
cit. Chem. Abstr. 58, 13782 (1963)
35) D.V. Sokols'kn et al., Trudy Inst. Khim. Nauk, Akad. Nauk Kaz. USSR
2, 158 (1958)
cit. Chem. Abstr. 53, 206 (1959)
36) V. Medonos, M. Kadlec, Chem. Prumysl^, 92 (1956) (Tschechisch)
cit. Chem. Abstr. 51, 1829 (1957)
37) V. Medonos, J. Brezina, Chem. Prumysl 8, 344 (1958) (Tschechisch)
cit. Chem. Abstr. 53, 1098 (1959)
38) D.F. Othmer, K. Kon, T. Igarashi, Ind. Eng. Chem. 48, 1258 (1956)
39) M.S. Newman, J. Am. Chem. Soc. 75, 4740 (1953)
40) S.A. Vartanyan, S.K. Pirenyan, N.G. Mana sy an, J. Gen.Chem.
(USSR) 31, 1176 (1961) (Engl. Transi.)
41) M. Fiti et al. Acad. Rep. Populäre Romine, Studii Cercetan Chim.
10, 243 (1962) (Rumänisch)
cit. Chem. Abstr. 58, 961 (1963)

Ill
42) A.A. Ponfilov, K.A. Grigoryan, V.l. Isagulayant s,Dokl. Akad.
Nauk. Arm. USSR 35, 63 (1962) (Russisch)
cit. Chem. Abstr. 58, 5500 (1963)
M.S. Newman, A.P. 2 853 520 (1953)
K. Yamamoto, Jap. P. 3873 (1952)
cit. Chem. Abstr. 48, 5208 (1954)
M. Kutscheroff, Ber. deutsch. Chem. Ges. 42, 2759 (1909)
M. Ban, K. Yamamoto, Kogyo Kagaku Zasshi 61, 708 (1958) (Japanisch)
cit. Chem. Abstr. £5, 12274 (1961)
M. Ban et al., Kogyo Kagaku Zasshi 61, 852 (1958) (Japanisch)
cit. Chem. Abstr. 57, 2892 (1962)
M. Ban et al., D.A.S. 1 043 313 (1955); E.P. 767 388 (1955);
A.P. 2 791 614 (1954)
M. Ban et al., Jap. P. 3674 (1955)
cit. Chem. Abstr. 51, 16516 (1957)
M. Ban, Kogyo Kagaku Zasshi 61, 1166 (1958) (Japanisch)
cit. Chem. Zentr. 1959, 13124
M. Ban et al., Kogyo Kagaku Zasshi 64, 945 (1961) (Japanisch)
eu. Chem. Abstr. 57, 4511 (1962)
M . Fiti, Akad. Rep. Populäre Romme, Rev. Phys. 8 (3), 239 (1963)
cit. Chem. Abstr. 60, 400 (1964) (Rumänisch)
R.M. Flid, O.N. Temkin, USSR.P. 117 494 (1958)
«t. Chem. Abstr. 53, 19882 (1959)
Nippon Carbide Ind. Co., Jap. P. 155 852 (1943)
cit. Chem. Abstr. 44, 2011 (1950)
A.L. Klebanski, V.D.Titov, Zh. Prikl. Khim. 20, 1005 (1947)
cit. Chem. Abstr. ^2, 4434 (1948) (Russisch)
S.A. Vartanyan, S.K. Pirenyan, N.G. Manas y an, Izv. Akad. Nauk
Arm. SSSR, Khim. Nauki 12, 345 (1959) (Russisch)
cit. Chem. Abstr. 54, 10841 (1960)
S.A. Vartanyan et al., J. Gen. Chem. (USSR) 31, 2273 (1961)
(Engl, transi.)
S.A. Vartanyan et al., Izv. Akad. Nauk Arm. SSSR, Khim Nauki 14,

112
565 (1961) (Russisch)
cit. Chem. Abstr. 58, 4412 (1963)
59) Japan. Celluloid Co., Jap. P. 158 670 (1943)
cit. Chem. Abstr. 43^7037 (1949)
60) Nippon Carpide Ind. Co., Jap. P. 156 649 (1943)
cit. Chem. Abstr._44, 1528 (1950)
61) American Cyanamid Co.,
A.P. 2 784 236 (1954)
62) S.A. Vartanyan, S.K. Pirenyan, USSR.P. 129 652 (1960)
cit. Chem. Abstr. 55, 4364 (1961)
63) Nippon Carbide Ind. Co., Jap. P. 153 300 (1942)
cit. Chem. Abstr._44, 1528 (1950)
64) I.K. Khnstich, USSR.P. 85 580 (1949)
cit. (66)
65) R. Vestin, Svensk Kern. Tidskr.J^ 66 (1954)
cit. (66)
66) R.M. Flid, O.N. Temkin, J. Phys. Chem. (USSR) 35, 219 (1961)
(Engl, transi.)
67) Tekko Society Inc., Jap. P. 155 905 (1943)
cit. Chem. Abstr. 44, 4023 (1950)
68) J. Marton, G. Zöllner et al., Ung.P. 145 057 (1956)
cit. Chem. Zentr. 1963, 9484
69) T. Sakaki, Kogyo Kagaku Zasshi_67, 117 (1946) (Japanisch)
cit. Chem. Abstr. 45 538 (1951)
70) T. Sakaki, Kogyo Kagaku Zasshi 69, 11 (1948) (Japanisch)
cit. Chem. Abstr. 46, 8490 (1952)
71) Stockholms Superfosfat Fabriks A .B.,
Schwed. P. 124 669 (1949)
cit. Chem. Abstr. 44, 1528 (1950)
72) Stockholms Superfosfat Fabriks A.B., Schwed.P. 127 558 (1950)
cit. Chem. Abstr. 46, 4562 (1952)
73) Stockholms Superfosfat Fabriks A.B., E.P. 654 792 (1948)
74) Hua-Hsueh Tung Pao, Chin.P. 1958, 655
cit. Chem. Abstr. 56^ 4607 (1962)
75) A.Y. Yakubovich et al., Zh. Prikl. Khim. 19, 973 (1946) (Russisch)
cit. Chem. Abstr. 41, 4367 (1947)

113
76) R.A. Melikyan, G.G. Badalyan, Sbormk Nauch. Truolov Erevan. Poli-
tekh. Inst. 1957, (16), 33 (Russisch)
cit. Chem. Abstr. 54, 8610 (1960)
77) R. Wendtlandt, G. Hoffmann, D.B.P. 963 240 (1948)
78) R. Nozu, Jap. P. 156 174(1943)
cit. Chem. Abstr. 44^ 3009 (1950)
79) A.N. Sorbaev, I.P. Kirillov, Izv. Vyss. Uchebn. Zavedenii, Khim. i
Khim. Tekhnol._7_(6), 948 (1964) (Russisch)
cit. Chem. Abstr. ^3, 2893 (1965)
80) A.N. Sorbaev et al., Izv. Vyss. Uchebn. Zavedenii, Khim. i. Khim.
Tekhnol. 8 (5), 799 (1965) (Russisch)
cit. Chem. Abstr. _64_, 12 541 (1966)
81) Chemische Werke Huis, E.P. 701 550 (1953)
82) Escambia Chemical Co., A.P. 2 810 764 (1953); A.P. 2 847 476 (1953)
83) Philipps Petroleum Co., A.P. 2 712 559 (1952)
84) Y.A. Gorin et al., USSR.P. 138 607(1960)
cit. Chem. Abstr. 56^ 8567 (1962)
85) Escambia Chemical Co.,
D.A.S. 1 064 493 (1959)
86) Nippon Chemical Ind. Co., Jap.P. 155 433 (1943)
cit. Chem. Abstr._44, 3008 (1950)
87) S. Kunichika, Bull. Inst. Chem. Research. Kyoto Univ. 20, 35 (1950);
21, 22 (1950)
cit. Chem. Abstr. 48, 3891 (1954)
88) S. Kunichika, Repts. Inst. Chem. Research, Kyoto Univ. 16, 37 (1947)
cit. Chem. Abstr. _46^ 425 (1952) (Japanisch)
89) K. Yoshino, Chem. High Polymers 1, 29 (1944) (Japanisch)
cit. Chem. Abstr. 4^ 5309 (1950)
90) Toyo Textile Mills Co., Jap. P. 158 604 (1943)
cit. Chem. Abstr. 44^ 3008 (1950)
91) J.A. Tebboth, R.L. Ladell, E.P. 842 798 (1958)
92) Stockholms Superfosfat Fabriks A .B.,
Schwed.P. 127 518 (1946)
93) A.L. Klebanski et al., Zh. Prikl. Khim. 20, 1005 (1947)
cit. Chem. Abstr. 42, 4434 (1948) (Russisch)

114
94) B. Kubota, T. Take shima, Rept. Sei, Research Inst. 24, 195 (1948)
cit. Chem. Abstr. ^5, 4201 (1951) (Japanisch)
95) Stockholms Superfosfat Fabriks A.B., Schwed.P. 124 737 (1949)
cit. Chem. Abstr. 44, 1524 (1950)
96) R.E. Schaad, V.N. Ipatieff, J. Am. Chem. Soc. 62, 178 (1940)
97) I.G. Farbenindustrie, D.B.P. 1 024 949 (1954)
98) I.P. Kirillov, A.N. Sorbaev, Izv. Vyss. Uchebn. Zavedenii, Khim. i
Khim. Tekhnol.2(4), 613 (1964) (Russisch)
cit. Chem. Abstr. 62, 2703 (1965)
99) Y.A. Gorin, Khim. Prom. 1959 (3), 194 (Russisch)
cit. Chem. Abstr. JS4, 7539 (1960)
100) I.L. Kotlyarevskii, V.G. Lipovich, Met. i Khim. Prom. Kaz., Nauchn.
Tekhn. Sb. 1961 (3), 74 (Russisch)
cit. Chem. Abstr. 58, 5500 (1963)
101) Y.A. Gorin, I.K. Gorn, Zh. Organ. Khim. j_(12), 2090 (1965); 2j4), 590
(1966) (Russisch)
cit. Chem. Abstr. 64, 11069 (1966)
102) Y.A. Gorin, I.K. Gorn, J. Gen. Chem. 28, 2366 (1958) (Engl, transi.)
(USSR)
103) J. Marton et al., Acta Chim. Acad. Sei. Hung. 21, 375 (1959) (Engl, transi.)
104) I.K. Gorn, Y.A. Gorin, J. Gen. Chem. (USSR) 29, 2090 (1959)
(Engl, transi.)
105) Y.A. Gorin et al., Khim. Prom. 1964, 265 (Russisch)
cit. Chem. Abstr. 6L_ 5505 (1964)
106) Y.A. Gorin et al., J. Gen. Chem. (USSR) 30, 3777 (1960) (Engl, transi.)
107) Y.A. Gorin et al., Zh. Organ. Khim._l (5) 852 (1965) (Russisch)
cit. Chem. Abstr. 63^ 6833 (1965)
108) I.P. Kirillov, A.A. Kalinin, Izv. Vyss. Uchebn. Zav., Khim. i Khim.
Tekhnol._7 (5), 801 (1964) (Russisch)
cit. Chem. Abstr. £2, 7627 (1965)
109) A.A. Kalinin et al., Izv. Vyss. Uchebn. Zav., Khim. i Khim. Tekhnol.
_8_(1), 88 (1965) (Russisch)
cit. Chem. Abstr. 63, 2427 (1965)
110) A.I. Gel'bstein et al., Kinetics Catalysis 5 (3), 402 (1964) (Engl, transi.)
(USSR)

115
111) R.M. Flid, Trudy Inst. Khim. Nauk, Akad. Nauk Kaz. SSR 5^81 (1959)
cit. Chem. Abstr. j>4, 10841 (1960) (Russisch)
112) O.N. Temkin, R.M. Flid, Kat. Reakt. v Zhid. Faze, Akad. Nauk Kaz.,
Kaz. Gos. Univ., Kaz. Resp. Prav. Mendel. Obshch, Tr. Vses. Konf. Alma Ata 1962,
347 (Russisch)
cit. Chem. Abstr. 60, 11412 (1964)
113) O.N. Temkin, R.M. Flid et al., Kmetika i Kataliz (USSR)_2, 205 (1961)
(Russisch)
cit. (66)
114) CS. Yang, R.M. Flid et al., Izv. Vyss. Uchebn. Zav., Khim. i Khim.
Tekhnol. 1961 (2), 4 (Russisch)
cit. Chem. Abstr. _55, 20577 (1961)
115) M.K. Kalfuss, D.V. Sokolsku, Nachr. Akad. Wiss. Kass. SSSR 13 (9),
74 (1957) (Russisch)
cit. Chem. Abstr. 52, 2510 (1958)
116) M.K. Kalfuss, Master's Thesis (Kaz. State Univ., Alma-Ata, 1957) (Russisch);
cit. (115)
117) R.M. Flid et al., Zh. Fig. Khim. 31, 904 (1957) (Russisch)
cit. Chem. Abstr. 52^ 59 (1958)
118) R.M. Flid, Doctoral Dissertation, Moscow Institute of Fine Chemical Tech¬
nology, Moscow (1959) (Russisch)
cit. (66)
119) O.N. Temkin, R.M. Flid et al., Kinetics Catalysis (USSR) 4, 233 (1963)
(Engl, transi.)
120) R.M. Flid, Zh. Fiz. Khim. 32, 2339 (1958) (Russisch)
cit. Chem. Zentr. 1961, 6452
121) Y.A. Gönn, I.K. Gorn, J. Gen. Chem. (USSR) 30, 3781 (1960)
(Engl, transi.)
122) G . Levai, Ung. Z. Chem. 66, 389 (1960) (Ungarisch)
cit. Chem. Zentr. 1961, 15195
123) E.N. Tsybina, A.I. Gel'bstein et al., Zh. Fiz. Khim. 32, 856 (1958)
(Russisch); cit. Chem. Abstr. 52j 19370 (1958)
124) R.H. Frieman, E.R. Kennedy, H.J. Lucas, J.Am. Chem. Soc. 59,
724 (1937)

116
125) K. Schabe, Abh. Sachs, Akad. Wiss. Leipzig, Math, naturw. Kl. 44, (6), 1
(1955)
126) K. Schwabe, J. Voigt, Z. Physik. Chem. A 203, 383 (1954)
127) Y.A. Dorfman, D.V. S okol ski l, Izv. Akad. Nauk. Kaz. SSSR, Ser. Khim.
16, (I), 42 (1966) (Russisch)
cit. Chem. Titles 1967
128) Y.A. Dorfman, D.V. S okol ski l, Izv. Akad. Nauk. Kaz. SSSR, Ser.
Khim._l6_(2), 50 (1966) (Russisch)
cit. Chem. Titles 1967
129) R.M. Flid, I.I. Moissev, J. Appl. Chem. 27, 1085 (1954) (USSR)
(Engl, transi.)
130) R.M. Flid, I.I. Moissev, J. Appl. Chem. 28, 677 (1955) (USSR)
(Engl, transi.)
131) D.V. Sokolskn et al., Trudy Inst. Khim. Nauk., Akad. Nauk. Kaz. SSSR 2^
173 (1958) (Russisch)
cit. (66)
132) G. Travagli, Ann. Univ. FerrarajS, Pt. 1, 25 (1948 - 1950)
133) G. Travagli, Gazz. chim. ital. 82, 528 (1952)
134) G. Travagli, Gazz. chrm. ital. 81, 64 (1951)
135) A.F. Rekasheva, I.P. Samchenko, Dokladay Akad. Nauk. SSSR 133,
1340 (1960); Proc. Acad. Sei. USSR, Chem. Sect. _133, 965 (1960) (Engl, transi.)
136) W.L. Budde, R.E. Dessy, J. Am. Chem. Soc. 85, 3964 (1964)
137) W.L. Budde, R.E. Dessy, Tetrahedron Letters 1963, 651
138) O.N. Temkin, E.D. German, R.M. Flid, J. Gen. Chem. (USSR) 30,
722 (1960) (Engl, transi.)
139) O.N. Temkin et al.. Kinetics Catalysis^ 190 (1961) (USSR) (Engl, transi.)
140) O.N. Temkin, E.D. German, R.M. Flid, Metallurg, l Khim. prom'st
Kaz. 2, 62 (1960) (Russisch)
cit. (66)
141) R.M. Flid, Y.F. Golynets, Izv. Vyss. Uchebn. Zav., Khim l Khim.
Tekhnol.J^ 173 (1959) (Russisch)
cit. Chem. Abstr..5JL_ 19525 (1959)
142) Y.F. Golynets, Master's Thesis, Moscow, 1954
cit. (141)

117
143) A. I. Gel'bstein et al., Dokladay Akad. Nauk SSSR 107, 108 (1956)
(Russisch)
cit. (139)
144) R. Vestin, A. Somersalo, R.Mueller, Acta, Chem. Scand. 7,
745 (1953)
145) O.N. Temkin, R.M. Flid, A.I. Molakhov, Kinetics Catalysis_3,
799 (1962) (USSR) (Engl. Transi.)
146) O.N. Temkin, R.M. Flid, Kat. Reakt. et al., Alma-Ata 1962, 347
cit. Chem. Abstr. 60, 11412 (1964)
147) E.N. Tsybina, A.I. Gel'bstein, M.I. Temkin, Zh. Fiz. Khim. 32,
995 (1958) (Russisch)
cit. Chem. Abstr. 52^ 19370 (1958)
148) A.J. Szonyi, W.F. Graydon, Can. J. Chem. Eng. 40, 183 (1962)
149) A.J. Szonyi, Ph. D. Thesis, Toronto, 1962
cit. (148)
150) Houben-Weyl, Meth. org. Chem. 1/2, 654 (1959), 4. Auflage
151) Chemiker Kalender, (1956)
152) R.B. Wearn, W.M. Murray, M.P. Ramsey, N. Chandler, Anal.
Chem. 20, 922 (1948)
153) M. Ineichen, Diss. ETH, Prom. Nr. 2727 (1957)
154) M. Graf, Diss. ETH, Prom. Nr. 3058 (1960)
155) R.M. Barrer, Quart. Reviews_3, 293 (1949)
156) T.B. Reed, D.W. Breck, J. Am. Chem. Soc. 78, 5973 (1956)
157) D.W. Breck, W.G. Eversole, R.M. Milton, T.L. Thomas, J.
Am. Chem. Soc. 78, 5963 (1956)
158) R.M. Barrer, W.M. Meier, Trans. Farad. Soc.£4, 1074 (1958)
159) R.M. Milton, AJ>. 2 882 243 (1959); A.P. 2 882 244 (1959)
160) Union Carbide, Mitteilung ICZ-G-1364
161) Union Carbide, Mitteilung ICZ-16E64-G
162) P. Jiru, O. Grubner, M. Ralek, Int. Chem. Eng. 3_(1), 76 (1963)
163) G. Bergerhoff, H. Koyama, W. Nowacki, Expenentia 12, 418 (1956)
164) R.M. Barrer, F.W. Baltitude, J.W. Shuterland, Trans. Farad.
Soc. 53, 1111 (1957)

118
165) R.M. Barrer, J.W. Baynham, F.W. Baltitutde, W.M. Meier,
J. Chem. Soc. 36, 195 (1959)
J.A. Rabo, A.P. 3 130 006 (1964)
Union Carbide, Mitteilung ICZ 1464
D.W. Breck, R.M. Milton, A.P. 3 013 982 (1961); A.P. 3 013 983 (1961);
A.P. 3 013 990 (1961); A.P. 3 013 985 (1961)
W. Bukata, CR. Ca stör, R .M. Milton, A.P. 3 013 988 (1961)
J.A. Rabo, P.E. Pickert, J.E. Boyle, A.P. 3 130 006 (1964)
J.D. Cosgrove, J.D. H. Strickland, J. Chem. Soc. 1950, 1845
M. Betschart, Diss. ETH, Prom. Nr. 2842 (1959)
G. Schwär zenbach, H. F la s chka, Die komplexometrische Titration (1965)
Perry, Chemical Engineer's Handbook, 1963, 4. Auflage
R.C. Reid, T.K. Shervood, The Properties of Gases an Liquide, 1966,
2. Auflage, McGraw-Hill
F. Herning, L. Zipperer, Gas und Wasserfach 79, 49 (1936)
Handbook of Chemistry and Physics, 1961/62
S.A. Miller, Acetylene, its Properties, Manufacture and Uses _1_, (1965)
H.J. Hofmann, H. Astheimer, Chem. Eng. Sei. _18,643 (1963)
C.N. Satterfield, T.K. Sherwood, The Role of Diffusion in Catalysis
(1963)
O. Levenspiel, Chemical Reaction Engineering (1962); Wiley International
Edition, N.Y., London
K.H. Yang, O.A. Hougen, Chem. Eng. Progr. 46, 146 (1950)
O.A. Hougen, K.M. Watson, Ind. Eng. Chem. 35, 529 (1943)
I.C. Jüngers, Cinétique Appliquée (1958), Paris
De Acetis, I.G. Th'odos, Ind. Eng. Chem. 52, 1003 (1960)
P.W. Weisz, A.B. Schwartz, J. CatalysisJ^ 399 (1962)
A. Wheeler, Advances in CataysisJ5, 249 (1951) und Catalysis_2, 105 (1955)
L.Y. Margolis, Acvances in Catalysis Hj 429 (1963)
G. Roberts, C.N. Satterfield, Ind. Eng. Chem. Fundamentals_4, 288
(1965)

LEBENSLAUF
Ich wurde am 2. April 1938 in Obergestein (Kt. Wallis) geboren.
Nach Beendigung der Primarschule in Obergestein besuchte ich
am Kollegium Brig die Mittelschule und bestand 1960 die Matu¬
ritätsprüfung (Typus A). Anschliessend immatrikulierte ich mich
an der Eidgenössischen Technischen Hochschule in Zurich, wo
ich im Frühjahr 1965 das Diplom eines Ingenieur-Chemikers er¬
warb. In der nachfolgenden Zeit widmete ich mich am technisch¬
chemischen Institut unter der Leitung von Herrn Prof. Dr. A. Guyer
der vorliegenden Promotionsarbeit.
Zurich, im Juli 1967
























![INAUGURAL - DISSERTATION · des 6H-Dibenzo[c,e][1,2]oxaphosphinin-6-oxids (DOPO), Diphenylphosphinoxids (DPhPO) und 5,5-Dimethyl-1,3,2-dioxaphosphinan-2-oxids (DDPO) synthetisiert](https://img.pdfslide.net/doc/110x75/5f48018442cb9a12a171df34/inaugural-dissertation-des-6h-dibenzoce12oxaphosphinin-6-oxids-dopo-diphenylphosphinoxids.jpg)
![Synonymaverzeichnis 6. Synonymaverzeichnis Acetaldehyd [Acetaldehyd] Acetan [Ethylen] Aceton [Aceton] Acetoxylsäure [Essigsäure] Acetylentrichlorid [Trichlorethylen] Acetylhydrür](https://img.pdfslide.net/doc/110x75/5e1d817c0b1a5723a07c9249/synonymaverzeichnis-6-synonymaverzeichnis-acetaldehyd-acetaldehyd-acetan-ethylen.jpg)



