Embed Size (px)
Citation preview

intl.elsevierhealth.com/journals/thre
Thrombosis Research (2008) 122, 191–202
REGULAR ARTICLE
Simvastatin induces apoptosis in human breastcancer cells in a NFκB-dependent manner andabolishes the anti-apoptotic signaling of TF/FVIIaand TF/FVIIa/FXa
Mikael Åberg, Malin Wickström, Agneta Siegbahn ⁎
Department of Medical Sciences, Clinical Chemistry and Pharmacology, Akademiska Hospital, S-751 85 Uppsala, Sweden
Received 16 May 2007; received in revised form 17 August 2007; accepted 18 September 2007Available online 26 November 2007
⁎ Corresponding author. Tel.: +46 18E-mail address: agneta.siegbahn@a
0049-3848/$ - see front matter © 200doi:10.1016/j.thromres.2007.09.017
Abstract
Introduction: Statins have benefits independent of the plasma cholesterol propertiesamong cancer patients and tissue factor (TF)/FVIIa induce PI3-kinase/AKT dependentanti-apoptosis during serum starvation. We analyzed how simvastatin inducesapoptosis in human breast cancer cells and the influence of FVIIa and/or FXa onthe proposed apoptosis.Materials and methods: MDA-MB-231 cells were serum starved or treated with 5 μMsimvastatin and incubated with 10 and 100 nM FVIIa or 5/130 nM FVIIa/FX. RhoA wasanalyzed by confocal microscopy and caspase-3, nuclear fragmentation, and NFκBtranslocation were measured using the ArrayScan microscope. mRNA for BCL-2, AKT1and TF were analyzed with RT-PCR or TaqMan. Protein levels and phosphorylation ofPKB/AKTwere determined by western blotting.Results and conclusions: Simvastatin-induced apoptosis was recorded at 48 h in theMDA-MB-231 cells. Addition of FVIIa to the cells induced PKB/AKT phosphorylation at24 h and rescued serum-deprived cells from apoptosis. However, in the presence ofsimvastatin we were unable to report any phosphorylation of PKB/AKT or anti-apoptotic effect mediated by the TF/FVIIa or TF/FVIIa/FXa complexes. This was due toa RhoA-dependent retention of NFκB to the cytosol at 12 h which led to atranscriptional down-regulation of the anti-apoptotic protein BCL-2 as well as reducedAKT1 mRNA production at 24 h and thus diminished levels of PKB/AKT protein. Atranscriptional down-regulation of TF at 12 h possibly also contributed to the absentanti-apoptotic signaling. These results thereby support a role for simvastatin in cancertreatment and emphasize the importance of PKB/AKT in TF-signaling.© 2007 Elsevier Ltd. All rights reserved.
KEYWORDSStatins;Apoptosis;NFκB;Tissue factor;FVIIa;Cell signaling
611 4251; fax: +46 18 552562.kademiska.se (A. Siegbahn).
7 Elsevier Ltd. All rights reserv
ed.
192 M. Åberg et al.
Statins, or HMG-CoA reductase inhibitors, areused for treating hypercholesterolemia by blockingthe conversion of HMG-CoA to mevalonic acid in thebiosynthesis of cholesterol [1]. Statins have alsoreceived attention regarding unpredictable bene-fits regarding the development of cancer and seemto reduce the overall cancer incidence by 14–28%[2]. The effects by statins may be both dependentand independent of the HMG-CoA reductase inhibi-tion. The inhibition of the integrin LFA1 resulting inimpaired leukocyte migration and T-cell activationis, for instance, dependent on the direct binding ofstatins to the protein [3] whereas the evidence for apro-apoptotic effect of statin treatment in cancercells seems to depend on a total blockage of themevalonate pathway [4,5]. The exact mechanismsbehind the pro-apoptotic effects are, however, stillunknown but the inhibition of the synthesis of theisoprenoid intermediate geranyl–geranyl pyro-phosphate (GGPP) in the mevalonate pathway isof importance [6]. The isoprenoids are fatty mole-cules which attach to, i.e. prenylate, and anchormany proteins involved in signal transduction to cellmembranes [7]. This includes the Rho GTPaseswhich are highly involved in several intracellularkey processes such as regulation of transcription,cell size, controlled cell death, proliferation,immune and inflammatory responses, and woundhealing [8]. Furthermore, the RhoA GTPases areover expressed in breast cancer cells and theexpression seems to correlate to the histologicalgrade and proliferation index in tumor samples [9].
Tissue factor (TF), the main initiator of coagula-tion is a structural relative to type II cytokine recep-tors and the 21 amino acids long cytoplasmic tail isendowed with three serines as potential phosphory-lation sites [10]. TF is present on a variety of celltypes and some malignant cells produce TF up-regulating cytokines while others have a constitutiveexpression of TF [11–13]. Apart from thrombosisbeing a frequent complication in cancer and thesecond leading cause of death in cancer patients [14],analyses of tumors have revealed TF as a significantand independent risk factor for metastases in colo-rectal cancer and a prognostic marker for overallsurvival in breast and pancreatic cancer [15–17]. TFalso correlates with the histologic grade of malignan-cy in glioma [18]. Binding of FVIIa to TFmediates bothproteolytic activity and intracellular signaling [10]and the TF/FVIIa complex is reported to increasesurvival of serum-starved TF-expressing baby ham-ster kidney-cells, chinese hamster ovary-cells, andAdr-MCF cells in a PI3-kinase/AKT-dependent manner[19–21]. Themechanisms bywhich TF/FVIIa preventsapoptosis are today not clear. In addition, data on TF/VIIa-mediated effects on apoptosis induced by other
mechanisms than serum starvation are completelylacking.
In this study we treated TF-expressing humanbreast cancer cells with simvastatin in the presenceor absence of FVIIa and/or FXa. Our primary aim wasto determine whether simvastatin induces apoptosisin MDA-MD-231 cells and also to further elucidate thetime lapse and the pro-apoptotic signaling pathwaysinvolved. Secondly, we examined whether TF/FVIIa,TF/FVIIa/FXa, or FXa would influence the proposedapoptotic process. Treatment with FVIIa-inducedPKB/AKT phosphorylation and significantly increasedthe survival of serum-starved MDA-MB-231 cells at24 h. On the other hand, treatment with simvastatinresulted in apoptosis at 48 h regardless of thepresence of FVIIa and/or FXa. At 12 h simvastatinwas found to induce a RhoA-dependent retention ofthe transcription factor NFκB from the nucleus to thecytosol. This led to a transcriptional down-regulationof the anti-apoptotic protein BCL-2 and also reducedproduction of AKT1 and TFmRNAwhichmight explainthe lack of anti-apoptotic signaling by TF/FVIIa. Theresults presented here thereby support a role forsimvastatin in cancer treatment and emphasize theimportance of PKB/AKT in TF-signaling.
Materials and methods
Reagents
Simvastatin, geranyl–geranyl pyrophosphate (GGPP) and staur-osporine were obtained from Sigma-Aldrich Sweden AB (Stock-holm, Sweden). Simvastatin was activated prior to theexperiments by alkaline hydrolysis of the lactone ring accordingto the manufacturer's protocol (MSD, Harleem, Netherlands). Theused concentration of simvastatin was based on previousexperiments [22]. Recombinant human FVIIa and active siteinhibited human FVIIa (FVIIai) were a kind gift from prof. LCPetersen, NovoNordisk A/S (Maaloev, Denmark). Human FX andFXa were obtained from Enzyme Research lab (South Bend, IN,USA).
Cell culture
The estrogen receptor-negative, p53-mutated breast cancer cell lineMDA-MB-231 was grown in RPMI1640 (Invitrogen AB, Stockholm,Sweden) supplemented with 10% foetal bovine serum (FBS), 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in ahumified chamber at 37°C in the presence of 5% CO2 and wereobtained from ATCC. The cells have high expression of TF combinedwith PAR-1/-2 receptors [23–26] and are characterized by aconstitutive activation of RAS [27] and NFκB [28] as well as over-expression of RhoA [29].
Apoptosis assay
Confluent MDA-MB-231 cells were washed once with sterile PBS,detached with Accutase® (Sigma-Aldrich Sweden AB, Stockholm,Sweden) and seeded into flat-bottomed 96-well plates (PerkinElmerInc., Wellesley, MA, USA) at a concentration of 4×104 cells/ml in
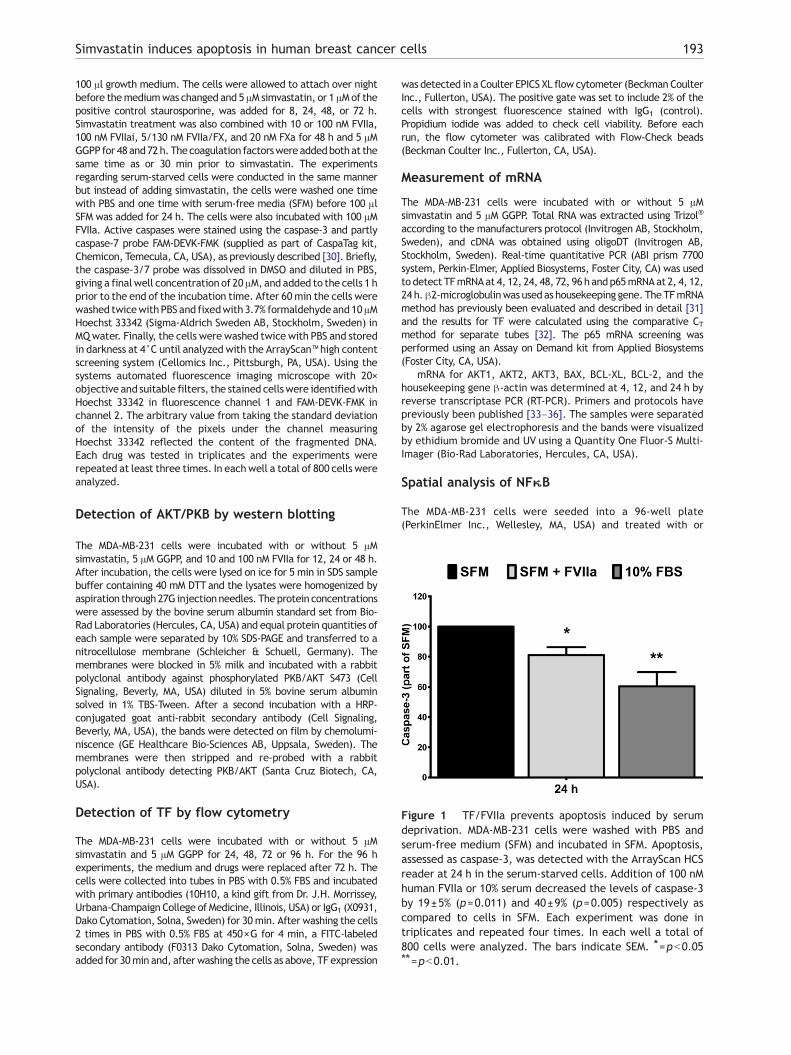
Figure 1 TF/FVIIa prevents apoptosis induced by serumdeprivation. MDA-MB-231 cells were washed with PBS andserum-free medium (SFM) and incubated in SFM. Apoptosis,assessed as caspase-3, was detected with the ArrayScan HCSreader at 24 h in the serum-starved cells. Addition of 100 nMhuman FVIIa or 10% serum decreased the levels of caspase-3by 19±5% (p=0.011) and 40±9% (p=0.005) respectively ascompared to cells in SFM. Each experiment was done intriplicates and repeated four times. In each well a total of800 cells were analyzed. The bars indicate SEM. ⁎=pb0.05⁎⁎=pb0.01.
193Simvastatin induces apoptosis in human breast cancer cells
100 μl growth medium. The cells were allowed to attach over nightbefore themediumwas changed and 5 μM simvastatin, or 1 μMof thepositive control staurosporine, was added for 8, 24, 48, or 72 h.Simvastatin treatment was also combined with 10 or 100 nM FVIIa,100 nM FVIIai, 5/130 nM FVIIa/FX, and 20 nM FXa for 48 h and 5 μMGGPP for 48and72h. Thecoagulation factorswere addedbothat thesame time as or 30 min prior to simvastatin. The experimentsregarding serum-starved cells were conducted in the same mannerbut instead of adding simvastatin, the cells were washed one timewith PBS and one time with serum-free media (SFM) before 100 μlSFM was added for 24 h. The cells were also incubated with 100 μMFVIIa. Active caspases were stained using the caspase-3 and partlycaspase-7 probe FAM-DEVK-FMK (supplied as part of CaspaTag kit,Chemicon, Temecula, CA, USA), as previously described [30]. Briefly,the caspase-3/7 probe was dissolved in DMSO and diluted in PBS,giving a finalwell concentration of 20μM, and added to the cells 1 hprior to the end of the incubation time. After 60min the cells werewashed twicewithPBS and fixedwith 3.7% formaldehydeand10μMHoechst 33342 (Sigma-Aldrich Sweden AB, Stockholm, Sweden) inMQwater. Finally, the cells were washed twice with PBS and storedin darkness at 4°C until analyzed with the ArrayScan™ high contentscreening system (Cellomics Inc., Pittsburgh, PA, USA). Using thesystems automated fluorescence imaging microscope with 20×objective and suitable filters, the stained cells were identifiedwithHoechst 33342 in fluorescence channel 1 and FAM-DEVK-FMK inchannel 2. The arbitrary value from taking the standard deviationof the intensity of the pixels under the channel measuringHoechst 33342 reflected the content of the fragmented DNA.Each drug was tested in triplicates and the experiments wererepeated at least three times. In eachwell a total of 800 cells wereanalyzed.
Detection of AKT/PKB by western blotting
The MDA-MB-231 cells were incubated with or without 5 μMsimvastatin, 5 μM GGPP, and 10 and 100 nM FVIIa for 12, 24 or 48 h.After incubation, the cells were lysed on ice for 5 min in SDS samplebuffer containing 40 mM DTTand the lysates were homogenized byaspiration through27G injectionneedles. Theprotein concentrationswere assessed by the bovine serum albumin standard set from Bio-Rad Laboratories (Hercules, CA, USA) and equal protein quantities ofeach sample were separated by 10% SDS-PAGE and transferred to anitrocellulose membrane (Schleicher & Schuell, Germany). Themembranes were blocked in 5% milk and incubated with a rabbitpolyclonal antibody against phosphorylated PKB/AKT S473 (CellSignaling, Beverly, MA, USA) diluted in 5% bovine serum albuminsolved in 1% TBS-Tween. After a second incubation with a HRP-conjugated goat anti-rabbit secondary antibody (Cell Signaling,Beverly, MA, USA), the bands were detected on film by chemolumi-niscence (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Themembranes were then stripped and re-probed with a rabbitpolyclonal antibody detecting PKB/AKT (Santa Cruz Biotech, CA,USA).
Detection of TF by flow cytometry
The MDA-MB-231 cells were incubated with or without 5 μMsimvastatin and 5 μM GGPP for 24, 48, 72 or 96 h. For the 96 hexperiments, the medium and drugs were replaced after 72 h. Thecells were collected into tubes in PBS with 0.5% FBS and incubatedwith primary antibodies (10H10, a kind gift from Dr. J.H. Morrissey,Urbana-Champaign College of Medicine, Illinois, USA) or IgG1 (X0931,Dako Cytomation, Solna, Sweden) for 30min. After washing the cells2 times in PBS with 0.5% FBS at 450×G for 4 min, a FITC-labeledsecondary antibody (F0313 Dako Cytomation, Solna, Sweden) wasadded for 30min and, afterwashing the cells as above, TF expression
was detected in a Coulter EPICS XL flow cytometer (BeckmanCoulterInc., Fullerton, USA). The positive gate was set to include 2% of thecells with strongest fluorescence stained with IgG1 (control).Propidium iodide was added to check cell viability. Before eachrun, the flow cytometer was calibrated with Flow-Check beads(Beckman Coulter Inc., Fullerton, CA, USA).
Measurement of mRNA
The MDA-MB-231 cells were incubated with or without 5 μMsimvastatin and 5 μM GGPP. Total RNA was extracted using Trizol®
according to the manufacturers protocol (Invitrogen AB, Stockholm,Sweden), and cDNA was obtained using oligoDT (Invitrogen AB,Stockholm, Sweden). Real-time quantitative PCR (ABI prism 7700system, Perkin-Elmer, Applied Biosystems, Foster City, CA) was usedto detect TFmRNAat 4, 12, 24, 48, 72, 96h andp65mRNAat 2, 4, 12,24 h.β2-microglobulinwas used as housekeeping gene. TheTFmRNAmethod has previously been evaluated and described in detail [31]and the results for TF were calculated using the comparative CTmethod for separate tubes [32]. The p65 mRNA screening wasperformed using an Assay on Demand kit from Applied Biosystems(Foster City, CA, USA).
mRNA for AKT1, AKT2, AKT3, BAX, BCL-XL, BCL-2, and thehousekeeping gene β-actin was determined at 4, 12, and 24 h byreverse transcriptase PCR (RT-PCR). Primers and protocols havepreviously been published [33–36]. The samples were separatedby 2% agarose gel electrophoresis and the bands were visualizedby ethidium bromide and UV using a Quantity One Fluor-S Multi-Imager (Bio-Rad Laboratories, Hercules, CA, USA).
Spatial analysis of NFκB
The MDA-MB-231 cells were seeded into a 96-well plate(PerkinElmer Inc., Wellesley, MA, USA) and treated with or

Figure 2 Simvastatin activated caspase-3. (A) MDA-MB-231 cells were incubated with or without 5 μM simvastatin and 5 μMGGPP for the indicated times and cleaved caspase-3 was measured with the ArrayScan HCS reader. One μM staurosporine wasused as a positive control for the method and induced apoptosis at every time point. Simvastatin-induced apoptosis wasdetected at 48 and 72 h. Addition of GGPP completely blocked the pro-apoptotic effect of simvastatin. (B) MDA-MB-231 cellswere incubated with or without simvastatin for 48 h. Addition of 100 nM recombinant human FVIIa, 100 nM recombinant humanFVIIai, 5/130 nM human FVIIa/FX, or 20 nM human FXa at the same time as simvastatin had no effects on the induced apoptosis.The results are shown as percentage of the untreated control for each individual experiment and statistically compared to thecontrol value of 100%. Each experiment was done in triplicates and repeated three times. In each well a total of 800 cells wereanalyzed. The bars indicate SEM. ⁎⁎=pb0.01 ⁎⁎⁎=pb0.001.
194 M. Åberg et al.
without 5 μM simvastatin and 5 μM GGPP for 2, 4, 12 and 24 h. Fordetection of NFκB, the NFκB Activation HitKit™ was usedaccording to the manufacturer's protocol (Cellomics Inc.,
Figure 3 Simvastatin-induced DNA-fragmentation. (A) MDA-MBand 5 μM GGPP for the indicated times and fragmented DNA was mThe arbitrary value from taking the standard deviation of the intenwas recorded. Simvastatin-induced apoptosis was detected at 4apoptotic effect of simvastatin (o). (B) MDA-MB-231 cells were100 nM recombinant human FVIIa, 100 nM recombinant human FVIItime as simvastatin had no effects on the induced apoptosis. The reeach individual experiment and statistically compared to the contrepeated three times. In each well a total of 800 cells were anal
Pittsburgh, PA, USA). Briefly, the cells were fixed with 3.7%formaldehyde, permeabilized with permeabilization buffer fromthe kit and incubated with an antibody against NFκB. The primary
-231 cells were incubated with or without 5 μM simvastatineasured with an output feature in the ArrayScan HCS reader.sity of the pixels under the channel measuring Hoechst 333428 and 72 h. Addition of GGPP completely blocked the pro-incubated with or without simvastatin for 48 h. Addition ofai, 5/130 nM human FVIIa/FX, or 20 nM human FXa at the samesults are displayed as percentage of the untreated control forrol value of 100%. Each experiment was done in triplicates andyzed. The bars indicate SEM. ⁎⁎=pb0.01 ⁎⁎⁎=pb0.001.

Figure 4 Simvastatin down-regulated PKB/AKT. MDA-MB-231 cells were incubated with or without 5 μM simvastatin and100 nM recombinant human FVIIa in the presence of 10% serumfor 12, 24, or 48 h. The levels of PKB/AKTand phosphorylatedPKB/AKT (pAKT) were determined by western blotting. At 24and 48 h, addition of FVIIa induced a 1.5- to 2-fold increase inthe quote of pAKT and PKB/AKT as compared to untreatedcells. Simvastatin treatment, on the other hand, decreasedthe levels of PKB/AKT and pAKT at 48 h. Co-incubation ofsimvastatin with of 100 nM FVIIa did not change the PKB/AKTlevels or pAKT at 48 h as compared to simvastatin treatmentalone. The data represent one representative experiment outof four.
195Simvastatin induces apoptosis in human breast cancer cells
antibody was detected with an Alexa-Fluor 488-conjugatedsecondary antibody (A11029, Molecular Probes, Eugene, OR,USA) and the nucleus was identified by using Hoechst 33342. Themean nucleus-cytoplasm intensity difference was determinedusing the ArrayScan HCS system in combination with thecytoplasm-to-nucleus translocation application software. Eachtreatment was tested in triplicates and repeated three times. Ineach well 1000 cells were analyzed.
Immunofluorescence microscopy of RhoA
Figure 5 Simvastatin down-regulated AKT1 mRNA. (A) MDA-MB-231 cells were incubated with or without 5 μM simvastatinand 5 μM GGPP and the mRNA levels of AKT1, AKT2, AKT3, andβ-actin were determined by RT-PCR. The quote of AKT2 andAKT3mRNAand the housekeeping geneβ-actinwas not altered
The MDA-MB-231 cells were cultured in 8-well chamber slides(Lab-Tek™) for 4 and 24 h with or without 5 μM simvastatin and5 μM GGPP. The cells were then fixed and labeled with RhoAantibody according to previously published tricloroacetic acid(TCA)-fixation methodology [37,38]. Briefly, adherent cells werewashed with PBS containing 30 mM glycine (G-PBS) and fixed for15 min in freshly prepared ice cold TCA. The fixed cells weretreated with 0.2% Triton X-100 in G-PBS for 15 min and washedthree times with G-PBS. The samples were blocked for 1 h with G-PBS containing 10% FBS and 1% bovine serum albumin andincubated with primary RhoA antibody (26C4, Santa CruzBiotech, CA, USA). The slides were then washed three timeswith G-PBS and incubated with Alexa-Fluor® 488-conjugated goatanti-mouse IgG secondary antibody (A11029, Molecular Probes,Eugene, OR, USA) for 30 min. Finally, samples were washed threetimes and mounted in VectaShield® mounting medium (VectorLaboratories, Burlingame, CA, USA). The fluorescence wasvisualized using the LSM 510 confocal microscope (Carl ZeissAB, Stockholm, Sweden).
over time. (B) The quote of AKT1 mRNA and the housekeepinggene β-actin was decreased by 54±21% (p=0.042) after 24 h of
Statistics simvastatin treatment as compared to untreated controls.Addition of GGPP blocked the reduction of AKT1 mRNA bysimvastatin and seemed to increase the production of AKT3.The bars indicate SEM. The data are based on three experi-ments. ⁎=pb0.05.After development, WB and RT-PCR gels were analyzed inPhotoshop CS2 (Adobe Systems Inc., San Jose, CA, USA) andImageJ. The statistical work was done in Graph Pad Prism 5.0
(GraphPad software, Inc., San Diego, CA, USA) by using anunpaired t-test. When possible, results are expressed as mean±SEM and p-values (2-tailed) below 0.05 are considered statisti-cally significant.
Results
TF/FVIIa prevents apoptosis induced by serumdeprivation in MDA-MB-231 cells
To demonstrate a direct effect by TF/FVIIa in the MDA-MB-231cells and to confirm previously published data concerningthe anti-apoptotic effect by TF/FVIIa during serum deprivation,the MDA-MB-231 cells were grown with or without 10% FBS.Cleaved/activated caspase-3 was used as a marker for

Figure 6 Effects of simvastatin on tissue factor. MDA-MB-231 cells were incubated with or without 5 μM simvastatin and 5 μMGGPP for the indicated times and tissue factor (TF) protein and mRNA were determined by flow cytometry and real-timequantitative PCR respectively. (A) Simvastatin decreased the levels of TF surface protein in a time dependentmanner from 96.7±0.2 to 58.4±14% at 24 h and to 29.0±11% at 48 h. (B) The levels of TFmRNAwere not affected at 4 h of treatment, but at 12 h, thereduction was about 58%. GGPP blocked the effects of simvastatin both regarding the TF surface protein and the mRNAproduction. The results were statistically compared to the value of the untreated control cells at every time point. Eachexperiment was repeated at least three times and the bars indicate SEM. ⁎⁎=pb0.01 ⁎⁎⁎=pb0.001.
196 M. Åberg et al.
apoptosis and was analyzed with the ArrayScan HCS reader at24 h (Fig. 1). Addition of 100 nM FVIIa significantly reduced thelevels of caspase-3 in the serum-starved cells as did the pres-ence of serum.
Simvastatin induces apoptosis in breast cancercells regardless of the presence of TF/FVIIa orTF/FVIIa/FXa
Figure 7 Simvastatin induce retention of NFκB to thecytosol. MDA-MB-231 cells were incubated with or without5 μM simvastatin and 5 μM GGPP for the indicated times. Thetranslocation of NFκB from the cytosol to the nucleus wasmeasured with the ArrayScan HCS reader. Addition ofsimvastatin had no effect at 4 h but at 12 h (45±10%;p=0.009) and 24 h (39±3%; p=0.0002) the translocation wasreduced as compared to untreated controls (100). Addition ofGGPP blocked the effects of simvastatin. Each experimentwas done in triplicates and repeated at least three times andin each well 1000 cells were analyzed. The bars indicate SEM.⁎⁎=pb0.01 ⁎⁎⁎=pb0.001.
Increased levels of cleaved caspase-3 in the MDA-MB-231 cellswere detected after 48 h (387.1±88%) and 72 h (589.1±125%)of simvastatin treatment as compared to untreated controls(100%) (Fig. 2A). There was no increase in the cleavedcaspase-3 levels at 24 h (125±20%). Thus, the onset ofsimvastatin-induced apoptosis occurs between 24 and 48 hafter addition of simvastatin.
When 10 nM FVIIa was added together with 5 μMsimvastatin, no differences in simvastatins ability to induceapoptosis were recorded at 48 or 72 h (data not shown). Inorder to rule out the possibility of a dose-related response,the cells were also incubated with 5 μM simvastatin and100 nM FVIIa for 48 h. No effects were observed at this highconcentration of FVIIa or by the addition of FVIIai (Fig. 2B). FXbinds the binary complex of TF/FVIIa and thereby makes upthe ternary complex TF/FVIIa/FXa which was reportedsuperior in preventing apoptosis compared to the binarycomplex [20]. Addition of 5/130 nM FVIIa/FX was, however,equally ineffective in blocking the pro-apoptotic effect ofsimvastatin as TF/FVIIa and so was the treatment with 20 nMFXa (Fig. 2B). These results were the same regardless whetherthe coagulation factors were added at the same time as or30 min prior to (data not shown) simvastatin.
The cells were treated identically as for the caspase-3 assayand fragmented DNA, another marker for apoptosis, was mea-
sured with an output feature in the ArrayScan HCS reader. Theobtained data correlated with the data on caspase-3 activationin every aspect (Fig. 3).

Figure 8 Simvastatin down-regulatedBCL-2mRNA. (A)MDA-MB-231 cells were incubated with or without 5 μM simvastatinand 5 μMGGPPand themRNA levels of BCL-2, BCL-XL, BAX, andthe housekeeping geneβ-actinwas determined by RT-PCR. Thequote of BCL-2 mRNA and β-actin was decreased at 12 and 24 hsimvastatin treatment but not at 4 h. The transcription ofBCL-XL and BAX was not altered by simvastatin. (B) Ascompared to untreated controls, the quote of BCL-2 mRNAand the housekeeping gene β-actin was decreased by 31±3%(p=0.011) at 12 h of simvastatin treatment and by 64±11%(p=0.0009) at 24 h. Addition of GGPP blocked the reductionof BCL-2 mRNA by simvastatin and seemed to increase theexpression of BCL-XL. The data are based on four experi-ments. The bars indicate SEM. ⁎=pb0.05 ⁎⁎⁎=pb0.001.
197Simvastatin induces apoptosis in human breast cancer cells
Simvastatin down-regulated TF/FVIIa-inducedphosphorylation of PKB/AKT
The TF/FVIIa-initiated signaling leading to anti-apoptosis has beenclosely linked to the phosphorylation of the serine/threonine kinasePKB/AKT [19,20]. Therefore, the levels of PKB/AKTwere assessed bywestern blotting in the untreated and treated MDA-MB-231 cells.Incubation with 10 nM (data not shown) and 100 nM FVIIa in thepresence of 10% FBS during 24 and 48 h resulted in a 1.5- to 2-foldincrease of the pAKT/AKT quote when compared to the untreatedcontrols (Fig. 4). Simvastatin, with or without 100 nM FVIIa,decreased the levels of pAKTas well as those of PKB/AKTafter 48 hof treatment as compared to the untreated cells.
The pAKTand PKB/AKTantibodies recognize all three isoformsof the protein. In order to analyze whether simvastatin actedselectively and on a transcriptional level, themRNAof AKT1, AKT2,and AKT3 were investigated with RT-PCR (Fig. 5A). The quote ofAKT1 mRNA and the housekeeping gene β-actin was not altered at12 h but decreased with 54±21% (p=0.042) at 24 h as compared tountreated controls (Fig. 5B). No changes in themRNA levels of AKT2and AKT3 were recorded.
Simvastatin down-regulated TF mRNA
The binding of FVIIa toTF and the subsequent intracellular signalingis of crucial importance for the anti-apoptotic effect [19]. It haspreviously been reported that statins down-regulate the expressionof TF in different cell types [6,22]. Before incubation withsimvastatin, 96.7±0.2% of the MDA-MB-231 cell populationexpressed TF (Fig. 6A). After 24 h of simvastatin treatment theexpression of TF surface levels was decreased to 58.4±14%, andafter 48 h to 29.0±11% of the initial levels. Therefore, at the onsetof simvastatin-induced apoptosis, i.e. between 24 h and 48 h ofstatin incubation, about 60% to 30% of the cell population stillexpressed TF.
In order to investigate whether simvastatin acted on thetranscription of TF, TF mRNA was measured with real-timequantitative PCR (Fig. 6B). The mRNA production, assessed as thequote of TF and the housekeeping gene β2-microgobulin, was notaffected at all at 4 h, itwas reduced by approximately 58% after 12 h,and was undetectable after 48 h of simvastatin treatment.
Simvastatin reduced the translocation of NFκB
To further clarify the mechanism behind the simvastatin-inducedapoptosis and the absent anti-apoptotic effect by TF/FVIIa, wefocused on the altered levels of PKB/AKT. Recent work suggeststhat the expression and regulation of PKB/AKTmight be controlledby the transcription factor NFκB [39,40]. Since simvastatindecreased the transcription of AKT1 mRNA (Fig. 5), the transloca-tion of NFκB from the cytosol to the nucleus was measured in theMDA-MB-231 cells in the presence or absence of 5 μM simvastatin.Statin treatment reduced the NFκB translocation with 45±10%(p=0.009) at 12 h. (Fig. 7). The cytosolic retention was not seen at4 h but was consistent at 24 h (39±3%; p=0.0002).
Simvastatin down-regulated BCL-2 mRNA
After IκB degradation and activation of NFκB, NFκB-dimers mayinduce the expression of a number of different genes in thenuclei. One of those is the BCL-2 family of apoptosis-regulatinggenes [41]. The mRNA levels of the pro-apoptotic BAX and theanti-apoptotic proteins BCL-2 and BCL-XL were thereforedetermined by RT-PCR at 4, 12, and 24 h of simvastatin
incubation (Fig. 8). The housekeeping gene β-actin was usedfor mRNA integrity control.
The quote of BCL-2 mRNA and β-actin was unaltered at 4 h ofsimvastatin treatment whereas it was diminished by 31±3%(p=0.011) at 12 h and by 64±11% (p=0.0009) at 24 h (Fig. 8B).There were no changes in the BAX and BCL-XL expression at anytime point.
Simvastatin delocalized RhoA from the cellmembrane
Statins have been shown to act by blocking GGPP, an intermediatein the cholesterol biosynthesis pathway responsible for

Figure 9 Simvastatin delocalized RhoA to the cytosol. MDA-MB-231 cells were incubated with or without 5 μM simvastatin and5 μM GGPP for 4 and 24 h. The cells were then stained for RhoA and visualized with confocal microscopy. Membrane localized/active RhoA is observed as thin, glowing edges lining the cells. (A) The untreated cells had a high membrane localization of RhoAwhereas RhoA was delocalized in the simvastatin-treated cells at (B) 4 and (C) 24 h. (D) GGPP blocked the effect of simvastatinseen at 24 h but (E) did not mediate an effect on its own. The data are representative for one experiment out of two.
198 M. Åberg et al.
prenylation/activation of proteins in the Rho-family of GTPases[7] which in turn play an important role in the development ofbreast cancer [9]. Therefore, the impact of simvastatin on themembrane localization of RhoA, a pre-requisite for RhoAsignaling, was examined by confocal microscopy. As previouslydescribed, the MDA-MB-231 cells expressed activated/membranelocalized RhoA constitutively (Fig. 9A and [29]). Treatment withsimvastatin deactivated/delocalized RhoA to the cytosol. Thiseffect was recorded already at 4 h of treatment (Fig. 9B) and waseven more pronounced at 24 h (Fig. 9C).
Addition ofGGPPblocked the effects of simvastatin
The MDA-MB-231 cells were incubated with simvastatin and 5 μMGGPP in order to investigate whether the observed effect ofstatins could be further linked to RhoA. Both the pro-apoptoticeffect of simvastatin (Figs. 2A, 3A), and the effects recorded onAKT1 (Fig. 5), TF (Fig. 6), NFκB (Fig. 7A), and BCL-2 (Fig. 8) werecompletely blocked by GGPP while RhoA's membrane localizationwas preserved (Fig. 9D). Incubation of the cells with 5 μM GGPPalone did not seem to increase the RhoA membrane localizationas compared to the control cells (Fig. 9E).
Discussion
Accumulating data have clearly designated statins tohave benefits beyond lipid lowering independent ofthe plasma cholesterol properties. These effectsconcern endothelial function, anti-inflammatoryreactions, anti-thrombotic and anti-angiogenic reg-ulations, inhibition of tumor proliferation [2], andability to induce apoptosis [4,5]. The present studywas designed to investigate whether simvastatininduces apoptosis in human breast cancer cells andto furthermore clarify the time lapse and the pro-apoptotic signaling pathways involved. As a second-ary aim we examined the impact of the binary andthe ternary TF-signaling complexes on the inducedapoptosis.
For the experiments, MDA-MB-231 breast cancercells were used. These cells have the pre-requisite forTF/FVIIa signaling due to high TF expression combinedwith PAR1 and PAR2 receptors [23–26]. In ourexperimental system, addition of FVIIa significantly
decreased the levels of caspase-3 in serum-deprivedcells and increased the phosphorylation of PKB/AKT.This is in accordancewith previous studies with serum-starved cells, in which TF/FVIIa-induced activation ofPI3-kinase/AKT was mandatory to prevent apoptosis[19–21].
Treatment with 5 μM simvastatin affected theMDA-MB-231 cells in several ways as summarized inFig. 10. Apoptosis was recorded at 48 h of simvas-tatin incubation and neither the binary TF/FVIIa- northe ternary TF/FVIIa/FXa complexes were able toprevent this. Simvastatin reduced the surface levelsof TF due to diminished mRNA production but at theonset of apoptosis about 60% to 30% of the cellpopulation still expressed TF and would thus be ableto bind FVIIa and signal. Therefore we propose thetranscriptional down-regulation of AKT1 mRNA at24 h by the statin as a strong contributor for theabolished TF/FVIIa mediated phosphorylation ofPKB/AKT at 48 h. This assumption is strengthen bythe fact that the housekeeping genes β2-mikroglo-bulin and β-actin were unaffected by simvastatinand accordingly the decreases in steady state mRNAlevels solely reflect transcription and not a generaldecrease in mRNA stability due to simvastatintreatment.
Previous work using both mouse and human cells,have demonstrated NFκB activation by PKB/AKT viaphosphorylation of IKKα and IKKβ [42,43]. On thecontrary, work by Meng and others [39,40] indicatethat the expression and regulation of PKB/AKT iscontrolled by NFκB. Events necessary for NFκBactivation occur prior to increase in PKB/AKT phos-phorylation and addition of NFκB inhibitors block thePKB/AKTactivation. Moreover, over-expression of p65leads to increased PKB/AKT phosphorylation, whereasa similar over-expression of IκB inhibits the phosphor-ylation [39,40].
We measured the spatial translocation of NFκBbetween the cell cytoplasm and the nucleus after 12-hour incubation with simvastatin and 50% retention ofNFκB in the cytoplasm was recorded. Since no effect

Figure 10 Effects of simvastatin and FVIIa in MDA-MB-231 cells over time. The figure summarizes the experiments presentedin Figs. 1–8. MDA-MB-231 breast cancer cells were incubated with or without 5 μM simvastatin or 100 nM FVIIa in the presence of10% serum for the indicated times. Co-incubation of simvastatin and FVIIa was equivalent to incubation with simvastatin alone.At 4 h simvastatin treatment delocalized/deactivated RhoA from the cell membrane. This led to NFκB retention to the cytosoland reduction of NFκB-dependent gene expression of TF and BCL-2. At 24 h, AKT1 mRNA was reduced as well as the surfacebound TF. At 48 h, PKB/AKT protein levels were decreased and the phosphorylated form of the protein (pAKT) was notdetectable by WB. Caspase-3 and fragmentation of the DNA were also evident at this time. When the MDA-MB-231 cells weretreated with FVIIa alone, PKB/AKTwas phosphorylated at 24 and 48 h and apoptosis induced by serum deprivation was reducedat 24 h. No experiments regarding PKB/AKTwas performed beyond 48 h.
199Simvastatin induces apoptosis in human breast cancer cells
was observed at 4 h and extended treatment withsimvastatin (24 h) had no further effect, the suppres-sion by simvastatin regarding NFκB translocationseems to reach its maximum between 4 and 12 h.PKB/AKTwas unaffected by simvastatin at these timepoints on both the protein and the mRNA levels.Therefore it is likely that PKB/AKTis regulatedbyNFκBin the simvastatin-treated MDA-MB-231 cells in thiscontext.
In our experimental set-ups, NFκB was still de-tected inside the nucleus at 12 and 24 h of treatment.Thus, simvastatin does only act on the translocation ofsome of the proteins in the Rel-family. Both TF and theanti-apoptotic protein BCL-2 have regulatory promot-er regions to which combinations of transcriptionfactor complexes containing p65 may bind [41,44] andthe activity of the AKT1 promoter is increased 20-foldby p65 over-expression in NIH3T3 cells [40]. The down-regulation of mRNA for these proteins was, however,not explained by a simvastatin-dependent decrease inthe transcription of p65 at 2, 4, 12 or 24 h (data notshown). InMDA-MB-231 cells simvastatin (5 μM) did notsuppress themRNA levels of the anti-apoptotic proteinBCL-XL. In contrast, it was recently demonstrated thatsimvastatin at a ten-fold higher concentration canmodulate expression of this anti-apoptotic geneproduct induced by TNF-α in human myeloid KBM-5cells [45]. The contradictory results may be explainedby different cellular systems and the much lowerconcentration of simvastatin used in our experiments.
Increase in the amount of RhoGTPases, in particularRhoA, appears tobea frequent event in different typesof human tumors and so also in MDA-MB-231 cells[9,29]. In spite of this, RhoA was deactivated bysimvastatin already after 4 h of incubation. RhoA has
been implicated in the regulation of NFκB in HUVECcells by IKKβ-dependent degradation of IκBα [46] andin vascular smooth muscle cells, statin treatmentprevented NFκB activation induced by oxidative stressby about 50% due to inhibition of IKKα/IKKβ andsubsequently IκBα [47]. Similar results were observedin human blood monocytes and vascular smoothmuscle cells [48,49]. It is thus possible that the effecton NFκB observed in the present study was due to aRhoA-dependent inhibition of IKKα/IKKβ.
Although TF was still expressed at the time for theonset of apoptosis and thereby most likely still signals,the total down-regulation of TF transcription bysimvastatin is significant. TF is linked to colorectalcancer, pancreatic cancer, breast cancer and glioma inpatients [15–18] and there are substantial evidence inthe literature describing the importance of TF inmalignancy, either by clotting-dependentmechanismsinvolving FXa and thrombin signaling through PAR-receptors and cross-linked fibrin building, or byclotting-independent mechanism mediated throughTF/FVIIa intracellular signaling [50]. TF expression hasbeen associated to tumor angiogenesis by up-regula-tion of VEGF and other angiogenesis-related genes andby offering a proangiogenic environment via clotformation [51,52]. Furthermore, the metastaticpotential of cancer cells is closely correlated to theTFexpression [53] and so is cellmigration [54] and thepresence of metastases increases the risk for venousthromboembolism from 7 to 58-fold as compared to acontrol group [55]. There is also evidence for aconnection between TF and the up-regulation of theurokinase-type plasminogen receptor (uPAR) oncancer cells [56]. Previous studies have revealedthat uPAR and the plasminogen activation pathway

200 M. Åberg et al.
play a central role in the promotion of tumor cellinvasion and metastasis [57]. Therefore, the dimin-ished transcription of TF induced by simvastatin inbreast cancer cells is an important finding in thiscontext.
A μM dose of statins is rather high. The relevancefor statins pleiotropic effects in vivo has beenquestioned [58]. Though some non-lipid statineffects shown in vitro were achieved using nano-molar drug concentrations, a number of in vitroexperiments, including this study, have used statinconcentrations considerably higher. However, in arecent study by van der Spek et al. [59] it wasclearly demonstrated that simvastatin could safelybe administered, with acceptable toxicity, topatients at a dose of 15 mg/kg/day for 7 days andat this high dose (normal dosage range 5–80 mg/day) in vivo down-regulation of proteins andinhibition of prenylation were indeed achieved.
The effects by statins may be both dependentand independent of the HMG-CoA reductase inhibi-tion ([3–5] and this study). Although we cannot ruleout the possibility that simvastatin-induced apo-ptosis is also associated with other, substantialsignaling pathways in the MDA-MB-231 cells, such asthe JNK as suggested by Koyuturk et al. [60], wepropose simvastatin to exert its pro-apoptoticeffects, at least in part, through a RhoA-dependentretention of NFκB to the cytosol due to inhibition ofHMG-CoA reductase (Fig. 10). This leads to apopto-sis by transcriptional down-regulation of BCl-2 andabolishes signaling by TF/FVIIa and TF/FVIIa/FXadue to reduced levels of AKT1 mRNA and possiblyTF. The results presented here thus contribute to awider understanding of the pleiotropic propertiesof simvastatin and emphasize the importance ofPKB/AKT in TF-signaling.
Acknowledgements
This study was supported by grants from the SwedishResearch Council, the Swedish Heart and Lung Foun-dation, and Gustaf V and Queen Victoria Foundation.
References
[1] Pedersen TR. Randomised trial of cholesterol lowering in4444 patients with coronary heart disease: the ScandinavianSimvastatin Survival Study (4S). Lancet 1994;344:1383–9.
[2] Demierre MF, Higgins PD, Gruber SB, Hawk E, Lippman SM.Statins and cancer prevention. Nat Rev Cancer 2005;5:930–42.
[3] Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T,Kallen J, Bruns C, Cottens S, Takada Y, Hommel U. Statinsselectively inhibit leukocyte function antigen-1 by bindingto a novel regulatory integrin site. Nat Med 2001;7:687–92.
[4] Chapman-Shimshoni D, Yuklea M, Radnay J, Shapiro H,Lishner M. Simvastatin induces apoptosis of B-CLL cells byactivation of mitochondrial caspase 9. Exp Hematol 2003;31:779–83.
[5] Cafforio P, Dammacco F, Gernone A, Silvestris F. Statinsactivate the mitochondrial pathway of apoptosis in humanlymphoblasts and myeloma cells. Carcinogenesis 2005;26:883–91.
[6] Denoyelle C, Vasse M, Korner M, Mishal Z, Ganne F, Vannier JP,Soria J, Soria C. Cerivastatin, an inhibitor of HMG-CoAreductase, inhibits the signaling pathways involved in theinvasiveness andmetastatic properties of highly invasive breastcancer cell lines: an in vitro study. Carcinogenesis2001;22:1139–48.
[7] BrownJH,Del ReDP, SussmanMA.TheRacandRhohall of fame:a decade of hypertrophic signaling hits. Circ Res 2006;98:730–42.
[8] Benitah SA, Valeron PF, van AL, Marshall CJ, Lacal JC. RhoGTPases in human cancer: an unresolved link to upstreamand downstream transcriptional regulation. Biochim BiophysActa 2004;1705:121–32.
[9] Fritz G, Brachetti C, Bahlmann F, Schmidt M, Kaina B. RhoGTPases in human breast tumours: expression and mutationanalyses and correlation with clinical parameters. Br JCancer 2002;87:635–44.
[10] Siegbahn A. Cellular consequences upon factor VIIa bindingto tissue factor. Haemostasis 2000;30(Suppl 2):41–7.
[11] Mackman N, Fowler BJ, Edgington TS, Morrissey JH. Func-tional analysis of the human tissue factor promoter andinduction by serum. Proc Natl Acad Sci U S A 1990;87:2254–8.
[12] Mann KG. Thrombin formation. Chest 2003;124:4S–10S.[13] Lip GY, Chin BS, Blann AD. Cancer and the prothrombotic
state. Lancet Oncol 2002;3:27–34.[14] Prandoni P, Falanga A, Piccioli A. Cancer and venous
thromboembolism. Lancet Oncol 2005;6:401–10.[15] Shigemori C, Wada H, Matsumoto K, Shiku H, Nakamura S,
Suzuki H. Tissue factor expression and metastatic potentialof colorectal cancer. Thromb Haemost 1998;80:894–8.
[16] Ueno T, Toi M, Koike M, Nakamura S, Tominaga T. Tissuefactor expression in breast cancer tissues: its correlationwith prognosis and plasma concentration. Br J Cancer2000;83:164–70.
[17] Kakkar AK, Lemoine NR, Scully MF, Tebbutt S, Williamson RC.Tissue factor expression correlates with histological grade inhuman pancreatic cancer. Br J Surg 1995;82:1101–4.
[18] Hamada K, Kuratsu J, Saitoh Y, Takeshima H, Nishi T, Ushio Y.Expression of tissue factor correlates with grade ofmalignancy in human glioma. Cancer 1996;77:1877–83.
[19] Sorensen BB, Rao LV, Tornehave D, Gammeltoft S, PetersenLC. Antiapoptotic effect of coagulation factor VIIa. Blood2003;102:1708–15.
[20] Versteeg HH, Spek CA, Richel DJ, Peppelenbosch MP.Coagulation factors VIIa and Xa inhibit apoptosis andanoikis. Oncogene 2004;23:410–7.
[21] Jiang X, Guo YL, Bromberg ME. Formation of tissue factor–factor VIIa-factor Xa complex prevents apoptosis in humanbreast cancer cells. Thromb Haemost 2006;96:196–201.
[22] Lindmark E, Siegbahn A. Tissue factor regulation and cytokineexpression in monocyte-endothelial cell co-cultures: effects ofa statin, an ACE-inhibitor and a low-molecular-weight heparin.Thromb Res 2002;108:77–84.
[23] Ge L, Shenoy SK, Lefkowitz RJ, DeFea K. Constitutive protease-activated receptor-2-mediatedmigrationofMDAMB-231breastcancer cells requires both beta-arrestin-1 and -2. J Biol Chem2004;279:55419–24.
[24] Morris DR, Ding Y, Ricks TK, Gullapalli A, Wolfe BL, Trejo J.Protease-activated receptor-2 is essential for factor VIIa and

201Simvastatin induces apoptosis in human breast cancer cells
Xa-induced signaling, migration, and invasion of breast cancercells. Cancer Res 2006;66:307–14.
[25] Liu Y, Mueller BM. Protease-activated receptor-2 regu-lates vascular endothelial growth factor expression inMDA-MB-231 cells via MAPK pathways. Biochem BiophysRes Commun 2006;344:1263–70.
[26] Hjortoe GM, Petersen LC, Albrektsen T, Sorensen BB,Norby PL, Mandal SK, Pendurthi UR, Rao LV. Tissue factor-factor VIIa-specific up-regulation of IL-8 expression inMDA-MB-231 cells is mediated by PAR-2 and results inincreased cell migration. Blood 2004;103:3029–37.
[27] Kozma SC, Bogaard ME, Buser K, Saurer SM, Bos JL, Groner B,Hynes NE. The human c-Kirsten ras gene is activated by anovel mutation in codon 13 in the breast carcinoma cell lineMDA-MB231. Nucleic Acids Res 1987;15:5963–71.
[28] Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet Jr RJ,Sledge Jr GW. Constitutive activation of NF-kappaB duringprogression of breast cancer to hormone-independentgrowth. Mol Cell Biol 1997;17:3629–39.
[29] Fritz G, Just I, Kaina B. Rho GTPases are over-expressed inhuman tumors. Int J Cancer 1999;81:682–7.
[30] Lovborg H, Nygren P, Larsson R. Multiparametric evalua-tion of apoptosis: effects of standard cytotoxic agents andthe cyanoguanidine CHS 828. Mol Cancer Ther2004;3:521–6.
[31] Malarstig A, Tenno T, Jossan S, Aberg M, Siegbahn A.A quantitative real-time PCR method for tissue factormRNA. Thromb Res 2003;112:175–83.
[32] Perkin-Elmer Corp. ABI PRISM 7700 sequence detectionsystem. User Bulletin #2; 1997. p. 11–6.
[33] Floros KV, Thomadaki H, Katsaros N, Talieri M, Scorilas A.mRNA expression analysis of a variety of apoptosis-relatedgenes, including the novel gene of the BCL2-family, BCL2L12,in HL-60 leukemia cells after treatment with carboplatin anddoxorubicin. Biol Chem 2004;385:1099–103.
[34] Gajate C, Santos-Beneit AM, Macho A, Lazaro M, Hernan-dez-De RA, Modolell M, Munoz E, Mollinedo F. Involvementof mitochondria and caspase-3 in ET-18-OCH(3)-inducedapoptosis of human leukemic cells. Int J Cancer 2000;86:208–18.
[35] Nakatani K, Thompson DA, Barthel A, Sakaue H, Liu W,Weigel RJ, Roth RA. Up-regulation of Akt3 in estrogenreceptor-deficient breast cancers and androgen-indepen-dent prostate cancer lines. J Biol Chem 1999;274:21528–32.
[36] Zhu Z, Jia J, Lu R, Lu Y, Fu Z, Zhao L, Wang L, Jin M, Zhao L,Gao W, Yao Z. Expression of PTEN, p27, p21 and AKT mRNAand protein in human BEL-7402 hepatocarcinoma cells intransplanted tumors of nude mice treated with thetripeptide tyroservatide (YSV). Int J Cancer 2006;118:1539–44.
[37] Hayashi K, Yonemura S, Matsui T, Tsukita S. Immunofluo-rescence detection of ezrin/radixin/moesin (ERM) proteinswith their carboxyl-terminal threonine phosphorylated incultured cells and tissues. J Cell Sci 1999;112(Pt 8):1149–58.
[38] Yonemura S, Hirao-Minakuchi K, Nishimura Y. Rho localiza-tion in cells and tissues. Exp Cell Res 2004;295:300–14.
[39] Meng F, Liu L, Chin PC, D'Mello SR. Akt is a downstreamtarget of NF-kappa B. J Biol Chem 2002;277:29674–80.
[40] Meng F, D'Mello SR. NF-kappaB stimulates Akt phosphor-ylation and gene expression by distinct signaling mechan-isms. Biochim Biophys Acta 2003;1630:35–40.
[41] Catz SD, Johnson JL. Transcriptional regulation of bcl-2by nuclear factor kappa B and its significance in prostatecancer. Oncogene 2001;20:7342–51.
[42] Romashkova JA, Makarov SS. NF-kappaB is a target of AKT inanti-apoptotic PDGF signalling. Nature 1999;401:86–90.
[43] OzesON,MayoLD,Gustin JA,Pfeffer SR,Pfeffer LM,DonnerDB.NF-kappaB activation by tumour necrosis factor requires theAkt serine-threonine kinase. Nature 1999;401:82–5.
[44] Mackman N. Regulation of the tissue factor gene. FASEB J1995;9:883–9.
[45] Ahn KS, Sethi G, Aggarwal BB. Simvastatin potentiates TNF-{alpha}-induced apoptosis through the down-regulation of NF-{kappa}B-dependent antiapoptotic gene products: role of I{kappa}B{alpha} kinase and TGF-beta-activated kinase-1.J Immunol 2007;178:2507–16.
[46] Anwar KN, Fazal F, Malik AB, Rahman A. RhoA/Rho-associated kinase pathway selectively regulates throm-bin-induced intercellular adhesion molecule-1 expres-sion in endothelial cells via activation of I kappa B kinasebeta and phosphorylation of RelA/p65. J Immunol2004;173:6965–72.
[47] Ortego M, Gomez-Hernandez A, Vidal C, Sanchez-Galan E,Blanco-Colio LM, Martin-Ventura JL, Tunon J, Diaz C,Hernandez G, Egido J. HMG-CoA reductase inhibitorsreduce I kappa B kinase activity induced by oxidativestress in monocytes and vascular smooth muscle cells.J Cardiovasc Pharmacol 2005;45:468–75.
[48] Colli S, Eligini S, Lalli M, Camera M, Paoletti R, Tremoli E.Vastatins inhibit tissue factor in cultured human macro-phages. A novel mechanism of protection against athero-thrombosis. Arterioscler Thromb Vasc Biol 1997;17:265–72.
[49] Hilgendorff A, Muth H, Parviz B, Staubitz A, Haberbosch W,Tillmanns H, Holschermann H. Statins differ in their ability toblock NF-kappaB activation in human blood monocytes.Int J Clin Pharmacol Ther 2003;41:397–401.
[50] Versteeg HH, Spek CA, Peppelenbosch MP, Richel DJ. Tissuefactor and cancer metastasis: the role of intracellular andextracellular signaling pathways.Mol Med 2004;10:6–11.
[51] Abe K, Shoji M, Chen J, Bierhaus A, Danave I, Micko C, Casper K,Dillehay DL, Nawroth PP, Rickles FR. Regulation of vascularendothelial growth factor production and angiogenesis by thecytoplasmic tail of tissue factor. Proc Natl Acad Sci U S A1999;96:8663–8.
[52] Shoji M, Hancock WW, Abe K, Micko C, Casper KA, Baine RM,Wilcox JN, Danave I, Dillehay DL, Matthews E, Contrino J,Morrissey JH, Gordon S, Edgington TS, Kudryk B, Kreutzer DL,Rickles FR. Activation of coagulation and angiogenesis incancer: immunohistochemical localization in situ of clottingproteins and vascular endothelial growth factor in humancancer. Am J Pathol 1998;152:399–411.
[53] Bromberg ME, KonigsbergWH, Madison JF, Pawashe A, Garen A.Tissue factor promotes melanoma metastasis by a pathwayindependent of blood coagulation. Proc Natl Acad Sci U S A1995;92:8205–9.
[54] Siegbahn A, Johnell M, Rorsman C, Ezban M, Heldin CH,Ronnstrand L. Binding of factor VIIa to tissue factor onhuman fibroblasts leads to activation of phospholipase Cand enhanced PDGF-BB-stimulated chemotaxis. Blood2000;96:3452–8.
[55] Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies,prothrombotic mutations, and the risk of venous thrombosis.JAMA 2005;293:715–22.
[56] Taniguchi T, Kakkar AK, Tuddenham EG, Williamson RC,Lemoine NR. Enhanced expression of urokinase receptorinduced through the tissue factor-factor VIIa pathway inhuman pancreatic cancer. Cancer Res 1998;58:4461–7.
[57] Andreasen PA, Kjoller L, Christensen L, Duffy MJ. Theurokinase-type plasminogen activator system in cancermetastasis: a review. Int J Cancer 1997;72:1–22.
[58] Bonetti PO, Lerman LO, Napoli C, Lerman A. Statin effectsbeyond lipid lowering—are they clinically relevant?Eur Heart J 2003;24:225–48.

202 M. Åberg et al.
[59] van der Spek E, Bloem AC, van de Donk NW, Bogers LH, vander GR, Kramer MH, de WO, Wittebol S, Lokhorst HM. Dose-finding study of high-dose simvastatin combined withstandard chemotherapy in patients with relapsed orrefractory myeloma or lymphoma. Haematologica2006;91: 542–5.
[60] Koyuturk M, Ersoz M, Altiok N. Simvastatin induces apoptosisin human breast cancer cells: p53 and estrogen receptorindependent pathway requiring signalling through JNK.Cancer Lett 2007;250:220–8.