Embed Size (px)
Citation preview

Feature ReviewSkin Immunity to CandidaalbicansSakeen W. Kashem1 and Daniel H. Kaplan2,*
Candida albicans is a dimorphic commensal fungus that colonizes healthyhuman skin, mucosa, and the reproductive tract. C. albicans is also a predomi-nantly opportunistic fungal pathogen, leading to diseasemanifestations such asdisseminated candidiasis and chronic mucocutaneous candidiasis (CMC). Thediffering host susceptibilities for the sites of C. albicans infection have revealedtissue compartmentalization with tailoring of immune responses based on thesite of infection. Furthermore, extensive studies of host genetics in rare cases ofCMC have identified conserved genetic pathways involved in immune recogni-tion and the response to the extracellular pathogen. We focus here on humanand mouse skin as a site ofC. albicans infection, and we review established andnewly discovered insights into the cellular pathways that promote cutaneousantifungal immunity.
Compartmentalization of Immunity Against C. albicans Skin InfectionC. albicans is the most common and well-studied of the disease-causing Candida spp., andnaturally colonizes the skin, genital, and/or intestinalmucosa in up to 70%of healthy individuals [1].Under normal circumstances, the fungus does not cause disease but the absence of appropriateimmune recognition and response mechanisms can lead to the inability to control C. albicanscolonization and invasion. CMC is a rare non-life threatening condition that occurs in the setting ofprimary and acquired immunodeficiencies resulting in oropharyngeal candidiasis (OPC) or super-ficial mucosal and cutaneous lesions with thickening, hyperkeratosis, and erythema of the skin orthe nailbeds. The genomic sequencing of HIV-negative CMC patients has identified many genesthat are crucial for host defense against Candida albicans. Subsequent mechanistic studies havefurther defined the importance of specific pattern-recognition receptors, dendritic cells, cytokines,and T cell signaling events in immunity against C. albicans with the common theme of defects ininnate and/or adaptive interleukin-17 (IL-17) pathways (referred to as type 3 immunity) (Table 1).
In addition to infections at barrier surfaces, Candida albicans is also the leading cause of fatalfungal bloodstream infections. The increased rate is thought to result from increased use ofimmunosuppressive agents, anticancer treatments, increased antimicrobial resistance, and thefrequency of invasive surgeries [1]. The genetic susceptibilities associated with systemic candi-diasis as well as infections of gastrointestinal and female reproductive organs differ significantlyfrom those associated with CMC [2]. Unlike CMC patients, who have defects in type 3 immunity,genetic defects upstream or downstream of IL-17 have not been implicated in patients withdisseminated candidiasis [3]. Instead, type I interferons (IFNs) have been demonstrated to playan important role in patients with systemic infections. In mice, IL-17 plays a role in systemiccandidiasis, but IFN-g from type 1 T helper (Th1) and natural killer (NK) cells have been recentlyappreciated as key players in the host response [4–7]. Susceptibility to gastrointestinal andvulvovaginal candidiasis is largely dictated bymicroenvironmental factors such as local nutrients,pH, bile acids, and local commensal flora [8,9]. The host response to C. albicans infection intissues other than the skin has been recently reviewed in depth [8–10].
TrendsPrimary immunodeficiencies revealconserved IL-17 stimulating/signalingpathways involved in mucocutaneousdefense against C. albicans.
Mouse models have demonstratedboth innate and adaptive sources ofIL-17 in response to C. albicans.
Immunity against C. albicans is com-partmentalized towards sites ofinfection.
Non-hematopoietic cells provide anti-fungal immunity in the skin.
1Department of Dermatology, Centerfor Immunology, University ofMinnesota, Minneapolis, MN 55455,USA2Departments of Dermatology andImmunology, University of Pittsburgh,Pittsburgh, PA 15216, USA
*Correspondence: [email protected](D.H. Kaplan).
TREIMM 1284 No. of Pages 11
Trends in Immunology, Month Year, Vol. xx, No. yy http://dx.doi.org/10.1016/j.it.2016.04.007 1© 2016 Elsevier Ltd. All rights reserved.
TREIMM 1284 No. of Pages 11
Mouse models of OPC and cutaneous candidiasis demonstrate high fidelity to the humandisease. Mice with genetic defects as those found in patients with CMC have greatly increasedsusceptibility to skin and mucosal C. albicans infections [10]. Unlike humans, C. albicans is not acommensal fungus of mice [11]. Thus, mice allow the examination of primary innate and adaptiveimmune responses againstC. albicans. Cutaneous candidiasis models include either applicationof C. albicans onto stratum corneum-stripped epidermis or direct intradermal inoculation[12,13]. The well-characterized nature of the skin-resident and circulating leukocytes of theskin make the murine skin an ideal site to study both innate and adaptive antifungal immuneresponses. Specifically, the IL-17 cytokine family has been identified to be essential against hostdefense, driving neutrophil recruitment and antimicrobial peptide production [10]. It has beenappreciated that IL-17 can be produced by leukocytes from both the innate [e.g., innatelymphoid cells (ILCs), gd T cells] and adaptive immune systems (e.g., CD4+ [2_TD$DIFF] T cells) [5,14]. Thisreview focuses on recent advances in understanding the mechanisms by which the primaryinnate and the secondary adaptive type 3 immune response develop during C. albicansinfection. Better understanding these mechanisms can assist in the development of vaccinesagainst C. albicans and other extracellular pathogens as well as provide insight into type 3autoimmune diseases of the skin.
Innate Immune Response to C. albicansPattern RecognitionC-Type LectinsC-type lectins encompass a large family of receptors that bind to glycans through theirextracellular carbohydrate recognition domain and mediate intracellular signaling via variouscytoplasmic domains. Dectin-1 is one of the most commonly studied and reviewed C-typelectins [2,15]. Dectin-1 recognizes b-glucan on the cell wall of most species of fungi including C.albicans [16]. Signaling by Dectin-1 activates cells by a Syk-dependent pathway, resulting information of the CARD9–BCL10–MALT1 trimer and activation of NF-kB, leading to transcriptionof proinflammatory cytokines such as IL-1b, IL-6, and IL-12 [15].
Humans with SNP variants of Dectin-1 and mutations of Card9 develop CMC [17,18]. Thesepatients demonstrate impaired IL-1b, IL-6, IL-22, and IL-17 production alongside impaired
Table 1. Primary Immunodeficiencies Leading to CMC
Gene Phenotype Refs
IL2RG, RAG1, RAG2, ADA Loss of T and/or B cells [30]
UNC119, MAGT1, RAG1 Idiopathic CD4 lymphopenia [99,100]
STAT3 HIES due to defective IL-23R and IL-6R signaling anddecreased Th17
[101,102]
DOCK8 HIES due to defective T cell synapse formationand dysregulated DC migration
[103]
IkBA Impaired TCR and NF-kB signaling [29]
IL-17F/IL-17RA/IL-17RC/Act1 Defective IL-17A/F signaling [104–106]
RORC Loss of IL-17A/F production, lymph nodes, and impairedIFN-g response
[107]
Aire Autoantibodies to IL-17A/F/IL-22 [108]
STAT1 Gain of function mutation leading to excessive type Iand II IFN responses leading to decreased Th17
[104,109]
IL-12RB1 Abolished response to IL-23 and IL-12 [66]
Clec7A/Card9 Defective phagocytosis, IL-1b, and IL-6 productionby PBMCs and IL-17-producing T cells
[17,18]
2 Trends in Immunology, Month Year, Vol. xx, No. yy

TREIMM 1284 No. of Pages 11
phagocytosis of C. albicans yeasts [18]. Work in mouse models has revealed that the role ofDectin-1 is more nuanced. Recognition by Dectin-1 varies by species, strain, and life form (i.e.,yeast vs filamentous). Dectin-1 signaling has been shown to be required for protection fromintravenous and oral candidiasis in vivo, but has also been shown to be redundant [19,20]. Theseinitially discordant results resulted from the use of different strains ofC. albicans. Some strains ofC. albicans have increased cell wall chitin resulting in lower availability of cell wall b-glucans forrecognition by Dectin-1 [21]. In addition, Dectin-1 specifically recognizes C. albicans yeast andnot filamentous forms owing to a difference in b-glucan availability (covered below) [22].Recognition of fungi by other C-type lectin receptors, including the important concept of trainedimmunity, has been reviewed in depth elsewhere [2,15].
Toll-Like Receptors (TLRs)Several TLRs recognizeC. albicans cell-wall polysaccharides including TLR-2, which recognizesphospholipomannans, and TLR-4, which recognizes O-linked mannans [23]. Activation of TLRsby their ligands leads to triggering of intracellular signaling pathways, such as MAPK (mitogen-activated protein kinase) and NF-kB (nuclear factor k light-chain enhancer of activated B cells)pathways, leading to transcription and secretion of TNF-/, IL-6, and/or type I IFNs [24]. TLRsare expressed differentially on numerous cell types including KCs, melanocytes, dendritic cells,and macrophages and can also cooperate with C-type lectins or the inflammasome to drive IL-1b, TNF-/, and IL-12 production [25,26].
Deficiency for TLRs or for the adapter protein Myd88 significantly alters the survival of mice tointravenous C. albicans infection in vivo, and Myd88 deficiency in Langerhans cells (LCs) rendersmice unable tomount a Th17 response toC. albicans skin infection [27,28]. However, deficienciesfor IRAK-4 orMyd88 lead to recurrent pyogenic bacterial infections andcold abscessesbut do notlead to CMC in humans [29,30]. Mutations of IkBa, which acts downstream of TLRs and of the Tcell receptor (TCR), lead to CMC. Given the disparity of CMC susceptibility in IkBa variant patientscompared to those with Myd88/IRAK4 deficiency, it is likely that the role of T cells trumps that ofpattern recognition through TLRs in human resistance against CMC [29].
Nod-Like Receptors (NLRs)NLRs generally interact with ASC (apoptosis-associated speck-like protein containing a CARD)and procaspase-1 to form the inflammasome to convert procaspase-1 into active caspase-1,which in turn converts pro-IL-1b and pro-IL-18 into mature IL-1b and IL-18 [31]. In humans, NLRmutations and polymorphisms have not been identified with CMC but a defective NLRP3activation increases C. albicans colonization of the gut and NLRP3 polymorphism predisposespatients to recurrent vulvovaginal candidiasis (RVVC) [32,33]. In mice, macrophage-derivedNlrp3 recognizes filamentous forms of C. albicans, and deficiencies for Asc, Nlrp3, or IL-1R1lead to decreased C. albicans resistance in intravenous and oral routes of C. albicans infection[34,35]. Nlrp10 has also been implicated in host defense against C. albicans by the induction ofdendritic cell migration and differentiation of T helper cells [36,37]. However, subsequent studiesdemonstrated that Nlrp10-deficient mice also possessed functional mutations of Dock8. Dock8,but not Nlrp10, was found to be responsible for DC migration [38]. This supports the clinicalassociation between hyper IgE syndrome (HIES) caused by Dock8 mutations and CMC [39].Finally, stromal cell-derived NLRC4 has also been demonstrated to the crucial for mediatingimmunity against oral candidiasis [40]. While there has been some preliminary characterization ofNLRs in immunity against C. albicans disseminated and oral infections, the role of the inflam-masome in mediating immunity against C. albicans skin infection is currently unknown.
Stromal CellsThe uppermost layer of the avascular epidermis is the cornefied envelope that consists of deadkeratinocytes (KCs), keratin, and various hydrophobic lipids; this provides a physical barrier
Trends in Immunology, Month Year, Vol. xx, No. yy 3

TREIMM 1284 No. of Pages 11
against the environment and potential pathogens. In addition, the cornefied envelope containsantimicrobial and anti-Candida peptides, such as b-defensins and cathelicidins, which areproduced by KCs in response to infection or colonization [41]. Notably, mice deficient for akey component of the corneal layer – ceramide synthase 3 – are susceptible to C. albicans skininfections [42].
Beneath the cornefied envelope are the granular, spinous, and basal layers that are composed ofKCs that express PRRs. These KCs initiate the early cutaneous immune responses. C. albicanshas been shown to adhere to human KCs and induce proinflammatory cytokine secretion in vitrobut their role in directly activating KCs in vivo is unknown [43]. KCs constitutively expressreceptors for TNF-/, IL-17A, and IL-22. TNF-/ and IL-17 act on KCs and epithelial cells to driveproduction of antimicrobial peptides, such as b-defensins and S100 proteins, and chemokinesto drive the recruitment of neutrophils and inflammatory cells [44]. IL-22 acts on KCs to drive theirproliferation by downregulating genes involved in terminal differentiation [45]. Recent studieshave also identified the role of KCs in initiating immune responses through control of dendritic cellmigration and recruitment of memory T cells into the skin [46–48]. Thus, the interaction betweenC. albicans and epithelial cells of tissues is a crucial topic of investigation going forward.
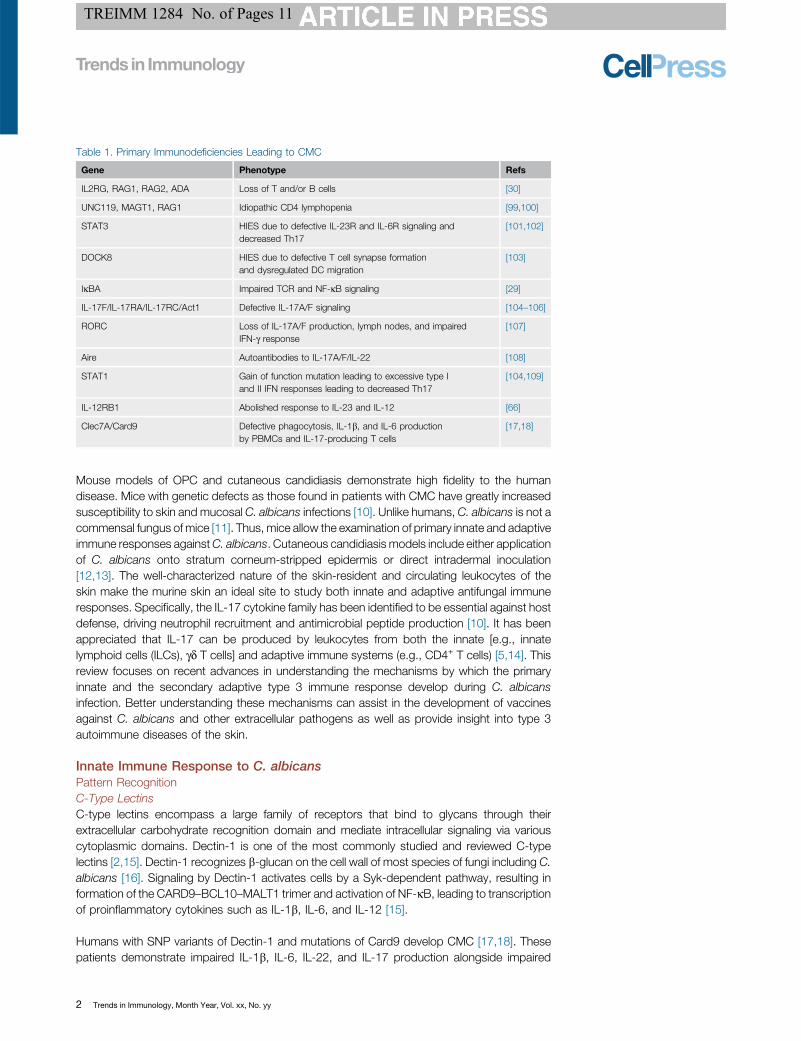
In addition to KCs, melanocytes can also respond to C. albicans. Melanocytes are located in thebasal layer of the epidermis and synthesize melanin to provide skin pigmentation. In invertebrates,melanocytesmodulatemelanin production during infection, and inflammation can lead to hypo- orhyperpigmentation in humans [49]. Melanin has also been demonstrated to have antimicrobialproperties [50]. While early studies demonstrated that C. albicans negatively regulates thetranscription of melanogenesis genes, more recent studies have shown that melanocytes mayrecognize C. albicans via TLR4 to increase melanization and inhibit infection (Figure 1) [51,52].
Cutaneous NervesThe skin is also a sensory organ that is highly innervated by terminal nerve fibers. Neuro-immuneinteractions have been appreciated in human and in mouse inflammatory and infectious diseasemodels [53]. Myeloid cells in the dermis and epidermis have been shown to localize closely withcutaneous nerve fibers, specifically fibers expressing calcitonin gene related peptide (CGRP)[54,55]. CGRP is a neuropeptide produced and released by sensory neurons that mediate painsignaling, as well as other functions. In the context of C. albicans vulvovaginal infection, C.albicans has been shown to stimulate pain, allodynia, and the development of CGRP+ nervefibers [56]. In addition, zymosan, a derivative of yeast cell walls, has been used as an experi-mental trigger of pain in rodent models [57]. Recently, pathogens such as C. albicans and S.aureus have been shown to directly activate sensory neurons taken from murine dorsal rootganglions [14,58]. Zymosan can activate sensory neurons directly, while Staphylococcus aureuscan activate neurons both directly and by causing membrane permeability in nerves through itssecreted toxins [14,58]. Thus, is possible that neurons may be activated by recognition of cell-wall products through neuronal PRRs as well as through secreted microbial products. C.albicans can also metabolize host arachnoid acid to byproducts that can directly activatenociceptor transient receptor potential cation channel subfamily V member 1 (TRPV1), suggest-ing that multiple pathways of activation may exist [59].
The activation of neurons by C. albicans leads to increased secretion of the neuropeptideCGRP [14]. CGRP has been demonstrated to have antifungal properties in vitro, to act on KCsto drive proliferation in vitro, to skew LCs towards type 2 responses in vitro, and to induce IL-23secretion by dermal DCs in vivo [14,60–62]. In the mouse model of C. albicans epicutaneousinfection, mechanical ablation of all cutaneous nerves or chemical denervation of TRPV1+
nociceptors (i.e., pain sensing nerves) with resineferatoxin (RTX) rendered mice less able toresist C. albicans skin infection as a result of diminished IL-23/IL-17 production (Figure 1).
4 Trends in Immunology, Month Year, Vol. xx, No. yy
TREIMM 1284 No. of Pages 11
Addition of CGRP in RTX-treated mice reversed the increased fungal burden [14]. Thus, painsensation via TRPV1 channels and CGRP secretion by sensory nerves is crucial for cutaneoushost defense against C. albicans.
IL-23 and Antigen-Presenting CellsIL-23 is a heterodimeric protein composed of IL-12p40 and IL-23p19 subunits that signalsthrough heterodimeric IL-12Rb1 and IL-23R [63]. IL-23 has crucial roles in protection frompathogens and in the pathogenesis of autoimmunity by inducing a unique inflammatory genesignature that includes Il17a, Il17f, Csf2, Tnfa, and others [64]. Mutation of IL-12Rb1 or ofdownstream STAT3 leads to reduced numbers of Th17 cells and CMC in humans [65,66]. Inmice, IL-23, but not IL-12, deficiency leads to increases susceptibility to intravenous, oral,epidermal, and intradermal primary and secondary infection with C. albicans due to decreasedIL-17, as well as reduced neutrophil infiltrates and epithelial hyperplasia [13,14,67,68].
IL-23 has been demonstrated to be produced by dendritic cells afterC. albicans infection in miceand humans [14,69]. The skin harbors three major subtypes of dendritic cells. LCs are the onlyMHC-II positive cells in the epidermis, while CD11b+ dermal DCs (dDCs) and CD103+ dDCsmake up the majority and minority of the DC subsets in the dermis, respectively [70]. Therequirement of skin DC subsets for imiquimod-induced psoriatic inflammation is unclear, withreports showing both LCs and CD11b+ dDCs are nonredundant producers of IL-23 in vivo[71,72]. In response to C. albicans skin infection, LCs are not required for IL-23 production or
C. albicans
CGRP IL-17
TRPV1+ NerveTLR-4
CD301b+
dDC
AMPs
PMN
Melanocyte Kera�nocyte
LCMelanin
γδT cell
NKcell
IL-23
Figure 1. Innate Immunity Against C. albicans Skin Infection. The skin has a layered innate immune system. C.albicans directly activates cutaneous sensory nerves to induce the release of calcitonin gene related peptide (CGRP). CGRPacts on CD301b+ dermal dendritic cells (dDC), which subsequently release IL-23. IL-23 acts on dermal gd T cells to drive IL-17 production in the skin, leading to anti-C. albicans resistance through the presumed activation of neutrophils andantimicrobial peptides (AMPs) such as b-defensins [14]. In addition, melanocytes in the basal epidermis can also recognizeC. albicans via TLR-4 to drive production and release of melanin granules, which are antimicrobial in nature [52]. Finally,Langerhans cells (LC) of the epidermis can suppress liver-derived CD49a+ natural killer (NK) cells in response to C. albicansthrough unknown mechanisms [73]. Abbreviation: PMN, polymorphonuclear leukocyte.
Trends in Immunology, Month Year, Vol. xx, No. yy 5

TREIMM 1284 No. of Pages 11
innate immune resistance against the fungus. Mice deficient for LCs, however, have exagger-ated NK cell-mediated inflammation in response to heat-killed C. albicans application to thefootpad (Figure 1) [14,73]. In addition, ablation of LCs in mice induces exaggerated contacthypersensitivity (CHS) and delayed-type hypersensitivity (DTH) responses, suggesting that LCsare immune-suppressive in these contexts [74–76].
Furthermore, mice deficient for both LCs and CD103+ dDCs have intact immunity against C.albicans. Mgl2-DTR mice that can be depleted of CD11b+ dDCs and tissue-resident macro-phages have decreased IL-23 transcription, IL-17 production, and increased fungal burden inskin after infection with C. albicans [14]. Mixed bone-marrow chimeric mice in which CD11b+
dDCs lack IL-23 production have a defective immune response to primary C. albicans skininfection, particularly owing to decreased IL-17 production by dermal gd T cells (see below) [14].Thus, CD11b+ dDCs are both necessary and sufficient for IL-23-driven anti-C. albicansresponses. Interestingly, CD11b+ dDC reside alongside dermal nerve fibers and also expressthe receptor for CGRP [14,55]. CGRP released by sensory nerves afterC. albicans infection actson the CD11b+ dermal DCs to stimulate IL-23 production. Thus, the sensory nervous systemand CD11b+ dermal DCs participate in a crucial cutaneous inflammatory circuit that drives hostresistance to C. albicans.
IL-17 and gd T cellsThe IL-17 family consists of six cytokines (IL-17A–IL-17F) that signal through five receptors (IL-17RA–IL-17RE). IL-17A and IL-17F form homo- and heterodimers and signal through a dimer ofIL-17RA and IL-17RC. Mutations in IL-17F, IL-17RA, IL-17RC, and a downstream signalingmolecule Act1 all lead to CMC in humans [10,64]. IL-17RA expression is found in bothhematopoietic and non-hematopoietic cells. In hematopoietic cells, IL-17 can signal on neu-trophils and NK cells to drive antifungal immunity [6,77]. In non-hematopoietic cells such as KCs,IL-17 induces the transcription of proinflammatory cytokines such as IL-6, G-CSF, chemokinessuch as CCL20, and the antimicrobial peptides b-defensins and S100A proteins [44,78]. TheseKC signals are important for neutrophil trafficking and function, as well as for host defenseagainst C. albicans and other extracellular pathogens. Unlike IL-17A/F, IL-17C is produced byepithelial KCs, but not by hematopoietic cells, and signals through IL-17RA and IL-17RE [64]. IL-17C induces a gene profile strikingly similar to that induced by IL-17A, and plays an importantrole in psoriasis pathogenesis, but plays no detectable role in protection against oral, dermal, ordisseminated candidiasis in vivo [79,80]. Similarly, IL-17RE was dispensable for protectionagainst candidiasis, further demonstrating the specific necessity for IL-17A/F signaling [80].
Th17 cells have been long considered the predominant source of IL-17 during C. albicansmucocutaneous infections (see below). Recently, tissue-resident type 3 innate lymphoid cells(ILC3s) and gd T cells have been appreciated as primary producers of effector cytokines at thesite of C. albicans infection in mouse models [81,82]. In humans, loss of function STAT3mutations lead to decreased number and function of IL-17-producing unconventionalmucosa-associated invariant T cells [83]. Unlike CD4+ T cells, which are primed by DC antigenpresentation and signals in secondary lymphoid organs to become IL-17 producers, innatelymphoid cells and gd T cells are pre-programmed into the IL-17-secreting lineage in the bonemarrow and thymus, respectively [84]. In humans, gd T cells have been shown to make IL-17 inresponse to IL-23 produced by DCs after C. albicans stimulation [69]. Thus, these cells mayserve as primary responders in patients who have not yet developedC. albicans-specific effectoror memory T cells and may augment early immunity. In mouse models of OPC, some groupshave found that gd T cells and innate-like CD4+ T cells in tissues are both crucial for resistancewhile other groups have demonstrated the need of ILCs for protection [81,82]. In the skin, dermalgd T cells are the obligate source of IL-17 after epicutaneous C. albicans [14]. Most IL-17-secreting dermal gd T cells have a Vg4 TCR [85]. These embryonically derived gd T cells with
6 Trends in Immunology, Month Year, Vol. xx, No. yy

TREIMM 1284 No. of Pages 11
constitutive expression of IL-23R can produce IL-17 and proliferate rapidly in response to IL-23from CD301b+ dermal DC [14,55]. Thus, IL-17 production is not restricted to CD4+ T cells afterinfection, and tissue-resident cells are important mediators of antifungal immunity.
NeutrophilsIt has long been appreciated that IL-17 is a crucial cytokine that drives the recruitment andactivation of neutrophils [86]. Neutrophils have been demonstrated to be required for protectionagainst mucosal and systemic C. albicans infections [77]. Recently, neutrophils have also beenshown to constitutively express RORgt and to produce and respond to IL-17 in a mouse modelof Aspergillus fumigatus [86]. Neutrophils are also crucial for phagocytosis of C. albicansbecause they sense pathogen size via Dectin-1 and release neutrophil extracellular traps inresponse to Candida albicans filaments but not to yeast [87]. In addition, adoptive transfer ofneutrophilic myeloid-derived suppressor cells have been shown to improve mice survivalfollowing invasive C. albicans infection [88]. Neutrophils in response to C. albicans infectionhave been studied extensively and are reviewed elsewhere, but their role in cutaneous hostdefense against C. albicans skin infections remains an open area of investigation [2,23,89].
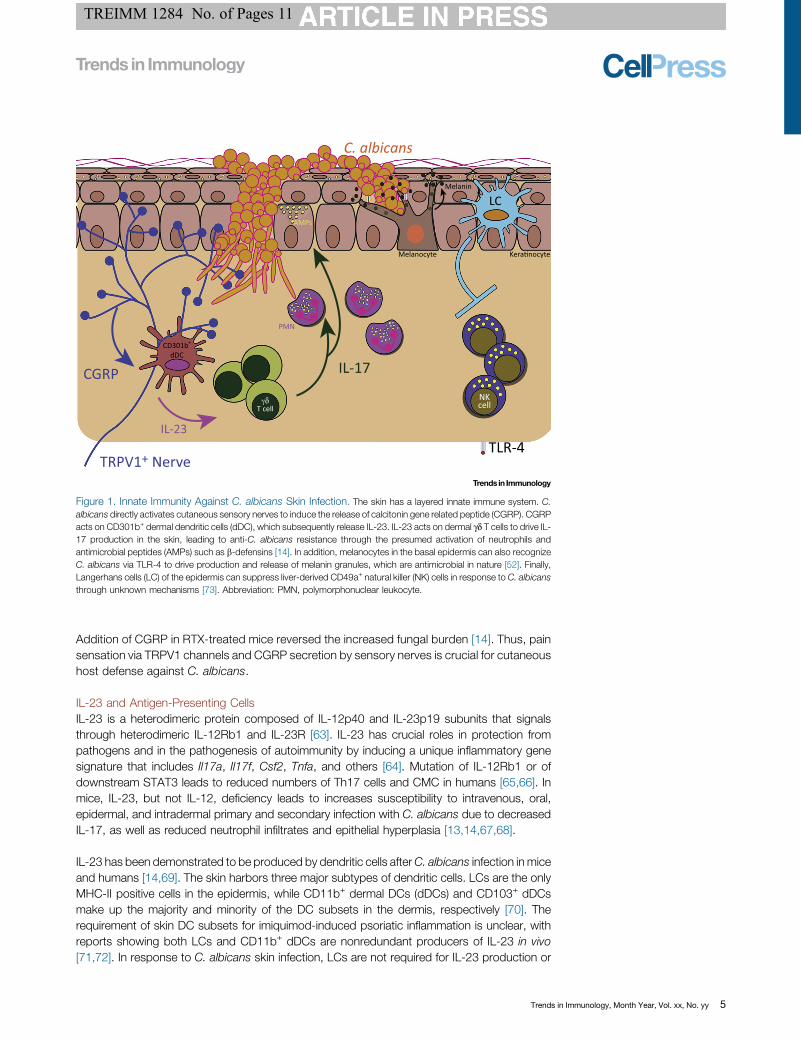
Adaptive Immunity Against C. albicans Skin InfectionThe importance of adaptive IL-17 producing CD4+ T cells in protection against C. albicans hasbeen demonstrated in mice and humans. In humans, Th17 cells are the crucial mediators ofantifungal barrier immunity. Patients with Th17 deficiencies in the context of STAT3-deficiency/HIES have increased susceptibility to mucocutaneous candidiasis [65]. Stimulation of naïvehuman CD4+ T cells with C. albicans can induce the expansion of T cell clones and induce theproduction of IL-17 and IFN-g that depended on IL-1b [90]. Interestingly, memory T cellresponses to C. albicans demonstrated functional heterogeneity, with distinct Th1/Th2/Th17cell subsets sharing the same T cell clone, suggesting that polarized T cell responses mightresult from preferential expansion rather than T cell priming [91]. However, in mouse models ofcutaneous candidiasis, both fungal morphology and DC subsets have been demonstrated to beimportant for the differentiation of specific T helper subsets that lead to a compartmentalized,tissue-specific response against secondary exposure of C. albicans (Figure 2) [5].
Th17 Cell DifferentiationDuring infection or inflammation, DCs migrate to the lymph node, upregulate co-stimulatorymolecules, and secrete cytokines that induce the proliferation and differentiation of effector andcytotoxic T cells [70]. Dock8 mutations in humans lead to defective T cell responses anddendritic cell migration that cause HIES and CMC [39,92]. In mice, skin DCs prime distinct Thelper responses that have differing functions in protective immunity against subsequent C.albicans infections [12]. LCs migrate to the lymph node 3–4 days after infection, where theyexpress high amounts of the Th17-differentiating cytokines IL-1b, IL-6, and TGF-b. Micedeficient for LCs have intact C. albicans-specific T cell expansion but have significantlydecreased Th17 cells [12]. In the epidermis, C. albicans colonizes as budding yeasts. C.albicans strains that are genetically locked into yeast, but not filaments, are capable of inducingTh17 differentiation through LCs [5]. The yeast morphology of C. albicans provides accessibilityligands for the b-glucan receptor Dectin-1 in its bud scars [22]. In mice, LCs express the patternrecognition receptor Dectin-1 [5]. Humans with Dectin-1 polymorphisms andmice with Dectin-1deficiency display a decreased C. albicans-specific Th17 response [18]. Binding of Dectin-1 byC. albicans induces the secretion of the proinflammatory cytokine IL-6 by human peripheralbloodmononuclear cells (PBMC) andmouse LCs [5,18]. Production of IL-6, but not IL-1b, IL-23,or TGF-b, from LC is necessary for Th17 cell differentiation, while IL-6 from other sources wasdispensable. Finally, LCs deficient for Myd88 have defective Th17 cell generation while LCmigration is unaffected [28]. Thus, pattern recognition of C. albicans via Dectin-1 and TLRs byLCs allows the elaboration of IL-6 that is required for Th17 cell differentiation.
Trends in Immunology, Month Year, Vol. xx, No. yy 7
TREIMM 1284 No. of Pages 11
As discussed previously, CD11b+ dermal DC are required for innate immunity against C.albicans and for Th17 generation against bacterial and fungal pathogens in other tissues[93]. CD11b+ dermal DCs also express Dectin-1 but are not required for Th17 generationafter C. albicans infection because of the inaccessibility of pseudohyphal Dectin-1 ligands inthe dermis [5,12]. Unlike LCs and CD11b+ dermal DCs, CD103+ dermal DCs do notexpress Dectin-1 and suppress Th17 cell differentiation presumably through anti-Th17cytokines such as IL-27 and IL-12 [5]. While CD103+ dDCs and CD11b+ dDCs are notrequired for Th17 differentiation, they do play a role in activating and differentiatingbystander IL-17-secreting CD8+ T cell responses that can protect against C. albicans[12,94].
Th1 Cell Differentiation and Compartmentalization of the Effector ResponseUnlike the yeast form of C. albicans, filamentous C. albicans invade past the stratum corneumand the dermis and disseminate to systemic tissues. Epicutaneous and intravenous infectionwith a strain of C. albicans that expresses model antigens under a filament-specific promoterinduces Th1 but not Th17 responses [5]. Given that Batf3-deficient mice have defective Th1 celldifferentiation in response to epicutaneous and disseminated C. albicans infection, CD103+
dermal DC likely induces Th1 cells through the secretion of IL-12 or IL-27 [12]. In the oralmucosa, CD103+ and CD11b+ migratory DCs collaborate to induce C. albicans-specific T cellexpansion and Th17 cell differentiation, while oral LCs were dispensable [95]. Thus, differentDCs in distinct tissues appear to play diverse roles. Interestingly, a patient with an IRF8 mutationand lacking both conventional and plasmacytoid dendritic cells was recently found to be infectedwith oral candidiasis [96].
C. albicans
LC
Dec�n-1TLR-2
Lymph node
NaiveCD4+
T cell
IL-12
IL-6
Skin Systemic IFN-γIL-17
Th17Th17 Th1
Th1
Th1
Th1
Th1Th17Th17
Th17
CD103+
dDC
Figure 2. Adaptive Immunity AgainstC. albicans Skin Infection. The skingenerates heterogeneous T helperresponse that provides compartmenta-lized immunity against C. albicans. Inthe epidermis, C. albicans exists asyeasts. C. albicans yeasts are recognizedby Dectin-1 on Langerhans cells (LCs).Dectin-1 engagement on LCs leads toproduction of IL-6 in the secondary lym-phoid organs that differentiate naïve CD4+
T cells to the type 17 T helper (Th17) celllineage. These Th17 cells provide protec-tion against secondary cutaneous infec-tions but not against secondary systemicinfections. Conversely, C. albicansinvades as filamentous pseudohyphae inthe dermis. Recognition of C. albicansfilaments by CD103+ dermal dendriticcells (dDCs), presumably through TLR-2, leads to Th1 cell differentiation in thesecondary lymphoid organs. These Th1cells provide protection against second-ary systemic infections but not againstsecondary skin infections [5,12]. Thus,recognition of distinct morphology of C.albicans via different DC subsets leads toa tailored immune response that providesprotection against specific subsequentroutes of infection.
8 Trends in Immunology, Month Year, Vol. xx, No. yy

TREIMM 1284 No. of Pages 11
Similarly to humans, mice infected withC. albicans demonstrate heterogeneous T helper profilesthat include both Th17 and Th1 cells. Mice lacking Th17 cells have impaired protection againstsecondary cutaneous, but not systemic,C. albicans infection, while mice with exaggerated Th17cells have greater protection against cutaneous, but not systemic, reinfection [5]. In addition,adoptive transfer of Th17 cells from C. albicans-primed mice into naïve animal afforded hostprotection against oral and cutaneous, but not systemic,C. albicans infection [5,97]. Conversely,mice lacking Th1 cells have impaired protection against secondary systemic, but not cutaneous,C. albicans infection, while mice with exaggerated Th1 cells have increased resistance againstsystemic but not cutaneous reinfection. Finally, adoptive transfer of Th1 cells from C. albicans-primed mice into naïve animals afforded host protection against systemic but not cutaneous C.albicans infection [5]. Thus, specific T helper subsets have compartmentalized immunity againstdistinct routes of C. albicans reinfection. While differential priming of T helper subsets to distinctroutes of infection has been appreciated in other models, the mechanism of compartmentalizedprotection is still unknown [98]. It remains to be determined whether distinct T helper subsetshave different tissue homing, persistence, and/or function.
Concluding RemarksIn this review we highlight the mechanisms underlying innate and adaptive immunity in responseto C. albicans skin infections. Innate immunity against C. albicans skin infections is driven byrecognition of the pathogen by the cutaneous stromal and nervous system that alarm DCs toactivate tissue-resident IL-17 secreting gd T cells. Adaptive immunity is dependent on recogni-tion of specific morphologies of C. albicans by distinct DC subsets, leading to compartmental-ized T helper responses. In conclusion, C. albicans skin and mucosal infections have been afruitful model of investigation of how IL-17 responses are developed. Findings addressed in thisreview provide a mechanistic insight into skin immunity that may have implications for bothvaccination strategies and treatment autoimmunity. Investigation into fungal recognition, stro-mal–immune interaction, and the development of the memory CD4+ response to C. albicans areimportant areas for future research.
AcknowledgmentsWe would like to thank the members of the laboratory of D.H.K. and the Center for Immunology for fruitful discussions. This
work was supported by grants from the National Institutes of Health (NIH) (AR056632 and AR060744) to D.H.K. S.W.K. was
supported by the University of Minnesota NIH MSTP grant T32 GM008244, Immunology Training Grant T32 AI007313, and
University of Minnesota CTSI Translational Research Development Program Grant UL1TR000114. The authors have no
conflicting financial interests. Finally, we would like to apologize for any omissions in referencing owing to space restrictions.
References1. Perlroth, J. et al. (2007) Nosocomial fungal infections: epidemi-
ology, diagnosis, and treatment. Med. Mycol. 45, 321–346
2. Underhill, D.M. and Pearlman, E. (2015) Immune interactions withpathogenic and commensal fungi: a two-way street. Immunity43, 845–858
3. Milner, J.D. and Holland, S.M. (2013) The cup runneth over:lessons from the ever-expanding pool of primary immunodefi-ciency diseases. Nat. Rev. Immunol. 13, 635–648
4. Smeekens, S.P. et al. (2013) Functional genomics identifies type Iinterferon pathway as central for host defense against Candidaalbicans. Nat. Commun. 4, 1342
5. Kashem, S.W. et al. (2015) Candida albicans morphology anddendritic cell subsets determine T helper cell differentiation.Immunity 42, 356–366
6. Bar, E. et al. (2014) IL-17 regulates systemic fungal immunity bycontrolling the functional competence of NK cells. Immunity 40,117–127
7. LeibundGut-Landmann, S. et al. (2007) Syk- and CARD9-dependent coupling of innate immunity to the induction of Thelper cells that produce interleukin 17. Nat. Immunol. 8, 630–638
8. Kumamoto, C.A. (2011) Inflammation and gastrointestinal Can-dida colonization. Curr. Opin. Microbiol. 14, 386–391
9. Cassone, A. (2015) Vulvovaginal Candida albicans infections:pathogenesis, immunity and vaccine prospects. BJOG 122,785–794
10. Conti, H.R. and Gaffen, S.L. (2015) IL-17-mediated immunity tothe opportunistic fungal pathogenCandida albicans. J. Immunol.195, 780–788
11. Iliev, I.D. et al. (2012) Interactions between commensal fungi andthe C-type lectin receptor Dectin-1 influence colitis. Science 336,1314–1317
12. Igyártó, B.Z. et al. (2011) Skin-resident murine dendritic cellsubsets promote distinct and opposing antigen-specific T helpercell responses. Immunity 35, 260–272
13. Kagami, S. et al. (2010) IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candidaalbicans. J. Immunol. 185, 5453–5462
14. Kashem, S.W. et al. (2015) Nociceptive sensory fibers driveinterleukin-23 production from CD301b+ dermal dendritic cellsand drive protective cutaneous immunity. Immunity 43, 515–526
Outstanding QuestionsMajor disease-causing Candida spe-cies include C. dubliniensis, C. para-psilosis, C. glabrata, C. tropicalis, andC. krusei. How do the immuneresponses to these species of Candidadiffer from skin immunity against C.albicans?
How is C. albicans recognized by sen-sory neurons? Understanding whetherneurons express pattern-recognitionreceptors, and what specific C. albi-cans element is being sensed, will becrucial for deciphering the interactionbetween the pathogen and the nervoussystem in the skin innate immuneresponse.
What are the roles of innate lymphoidcells, neutrophils, and recruited mye-loid cells in C. albicans skin infection?While much is known about the func-tion of these leukocytes in other sys-tems of C. albicans infection or in vivo,very little is known about their specificcontributions to immunity in the contextof C. albicans infection.
Why and how is T cell immunity againstC. albicans compartmentalized to themorphology of the pathogen? Do dis-tinct T helper subsets have divergenttissue migration? What is the role ofIFN-g in protective immunity againstskin and invasive C. albicans infec-tions? How is T cell memory generatedand maintained in the skin in responseto C. albicans? Are there C. albicans-specific tissue-resident Th17 cells inthe skin? What stromal signals, cyto-kines, and antigen-presenting cellscontribute to the maintenance of thesecells?
How does the immune response toother common skin pathogens suchas Staphylococcus aureus and Strep-tococcus pyogenes differ from skinimmunity against C. albicans?
Trends in Immunology, Month Year, Vol. xx, No. yy 9

TREIMM 1284 No. of Pages 11
15. Dambuza, I.M. and Brown, G.D. (2015) C-type lectins in immu-nity: recent developments. Curr. Opin. Immunol. 32, 21–27
16. Brown, G.D. and Gordon, S. (2001) A new receptor for beta-glucans. Nature 413, 36–37
17. Glocker, E.-O. et al. (2009) A homozygous CARD9 mutation in afamily with susceptibility to fungal infections. N. Engl. J. Med.361, 1727–1735
18. Ferwerda, B. et al. (2009) Human dectin-1 deficiency and muco-cutaneous fungal infections. N. Engl. J. Med. 361, 1760–1767
19. Taylor, P.R. et al. (2007) Dectin-1 is required for beta-glucanrecognition and control of fungal infection. Nat. Immunol. 8, 31–38
20. Saijo, S. et al. (2007) Dectin-1 is required for host defense againstPneumocystis carinii but not against Candida albicans. Nat.Immunol. 8, 39–46
21. Marakalala, M.J. et al. (2013) Differential adaptation of Candidaalbicans in vivomodulates immune recognition by dectin-1.PLoSPathog. 9, e1003315
22. Gantner, B.N. et al. (2005) Dectin-1 mediates macrophage rec-ognition of Candida albicans yeast but not filaments. EMBO J.24, 1277–1286
23. Gow, N.A.R. et al. (2012) Candida albicans morphogenesis andhost defence: discriminating invasion from colonization.Nat. Rev.Microbiol. 10, 112–122
24. Takeda, K. et al. (2003) Toll-like receptors. Annu. Rev. Immunol.21, 335–376
25. Gantner, B.N. et al. (2003) Collaborative induction of inflamma-tory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med.197, 1107–1117
26. Ferwerda, G. et al. (2008) Dectin-1 synergizes with TLR2 andTLR4 for cytokine production in human primary monocytes andmacrophages. Cell. Microbiol. 10, 2058–2066
27. Bellocchio, S. et al. (2004) The contribution of the Toll-like/IL-1receptor superfamily to innate and adaptive immunity to fungalpathogens in vivo. J. Immunol. 172, 3059–3069
28. Haley, K. et al. (2012) Langerhans cells require MyD88-depen-dent signals for Candida albicans response but not for contacthypersensitivity or migration. J. Immunol. 188, 4334–4339
29. Picard, C. et al. (2011) Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkB/ deficiency. Clin. Microbiol. Rev. 24,490–497
30. Lanternier, F. et al. (2013) Primary immunodeficiencies underlyingfungal infections. Curr. Opin. Pediatr. 25, 736–747
31. Martinon, F. et al. (2009) The inflammasomes: guardians of thebody. Annu. Rev. Immunol. 27, 229–265
32. Rehaume, L.M. et al. (2010) Lessons from the inflammasome: amolecular sentry linking Candida and Crohn's disease. TrendsImmunol. 31, 171–175
33. Lev-Sagie, A. et al. (2009) Polymorphism in a gene coding for theinflammasome component NALP3 and recurrent vulvovaginalcandidiasis in women with vulvar vestibulitis syndrome. Am. J.Obstet. Gynecol. 200, 303.e1–6
34. Hise, A.G. et al. (2009) An essential role for the NLRP3 inflam-masome in host defense against the human fungal pathogenCandida albicans. Cell Host Microbe 5, 487–497
35. Joly, S. et al. (2009) Cutting edge: Candida albicans hyphaeformation triggers activation of the Nlrp3 inflammasome. J.Immunol. 183, 3578–3581
36. Joly, S. et al. (2012) Cutting edge: Nlrp10 is essential for protec-tive antifungal adaptive immunity against Candida albicans. J.Immunol. 189, 4713–4717
37. Eisenbarth, S.C. et al. (2012) NLRP10 is a NOD-like receptoressential to initiate adaptive immunity by dendritic cells. Nature484, 510–513
38. Krishnaswamy, J.K. et al. (2015) Coincidental loss of DOCK8function in NLRP10-deficient and C3H/HeJmice results in defec-tive dendritic cell migration. Proc. Natl. Acad. Sci. U.S.A. 112,3056–3061
39. Farmand, S. and Sundin, M. (2015) Hyper-IgE syndromes: recentadvances in pathogenesis, diagnostics and clinical care. Curr.Opin. Hematol. 22, 12–22
40. Tomalka, J. et al. (2011) A novel role for the NLRC4 inflamma-some in mucosal defenses against the fungal pathogen Candidaalbicans. PLoS Pathog. 7, e1002379
41. Ali, R.S. et al. (2001) Expression of the peptide antibiotics humanbeta defensin-1 and human beta defensin-2 in normal humanskin. J. Invest. Dermatol. 117, 106–111
42. Jennemann, R. et al. (2012) Loss of ceramide synthase 3 causeslethal skin barrier disruption. Hum. Mol. Genet. 21, 586–608
43. Lopez, C.M. et al. (2014) Candida albicans uses the surfaceprotein Gpm1 to attach to human endothelial cells and to ker-atinocytes via the adhesive protein vitronectin. PLoS ONE 9,e90796
44. Liang, S.C. et al. (2006) Interleukin (IL)-22 and IL-17 are coex-pressed by Th17 cells and cooperatively enhance expression ofantimicrobial peptides. J. Exp. Med. 203, 2271–2279
45. Nograles, K.E. et al. (2008) Th17 cytokines interleukin (IL)-17 andIL-22 modulate distinct inflammatory and keratinocyte-responsepathways. Br. J. Dermatol. 159, 1092–1102
46. Nagao, K. et al. (2012) Stress-induced production of chemokinesby hair follicles regulates the trafficking of dendritic cells in skin.Nat. Immunol. 13, 744–752
47. Mohammed, J. et al. (2016) Stromal cells control the epithelialresidence of DCs and memory T cells by regulated activation ofTGF-b. Nat. Immunol. 17, 414–421
48. Bobr, A. et al. (2012) Autocrine/paracrine TGF-b1 inhibits Lan-gerhans cell migration. Proc. Natl. Acad. Sci. U.S.A. 109, 10492–10497
49. Gasque, P. and Jaffar-Bandjee, M.C. (2015) The immunologyand inflammatory responses of human melanocytes in infectiousdiseases. J. Infect. 71, 413–421
50. Mackintosh, J.A. (2001) The antimicrobial properties of melano-cytes, melanosomes and melanin and the evolution of black skin.J. Theor. Biol. 211, 101–113
51. Kippenberger, S. et al. (1997) Candida albicans suppressestranscription of melanogenesis enzymes in cultured melano-cytes. Mycoses 40, 373–375
52. Tapia, C.V. et al. (2014)Melanocytes andmelanin represent a firstline of innate immunity againstCandida albicans.Med.Mycol. 52,445–454
53. Ordovas-Montanes, J. et al. (2015) The regulation of immuno-logical processes by peripheral neurons in homeostasis anddisease. Trends Immunol. 36, 578–604
54. Hosoi, J. et al. (1993) Regulation of Langerhans cell function bynerves containing calcitonin gene-related peptide. Nature 363,159–163
55. Riol-Blanco, L. et al. (2014) Nociceptive sensory neurons driveinterleukin-23-mediated psoriasiform skin inflammation. Nature510, 157–161
56. Farmer, M.A. et al. (2011) Repeated vulvovaginal fungal infectionscause persistent pain in amousemodel of vulvodynia.Sci. Transl.Med. 3, 101ra91
57. Ren, K. and Dubner, R. (1999) Inflammatory models of pain andhyperalgesia. ILAR J. 40, 111–118
58. Chiu, I.M. et al. (2013) Bacteria activate sensory neurons thatmodulate pain and inflammation. Nature 501, 52–57
59. De Petrocellis, L. et al. (2009) Chemical synthesis, pharmacolog-ical characterization, and possible formation in unicellular fungi of3-hydroxy-anandamide. J. Lipid Res. 50, 658–666
60. Karim and El, I.A. et al. (2008) Antimicrobial activity of neuro-peptides against a range of micro-organisms from skin, oral,respiratory and gastrointestinal tract sites. J. Neuroimmunol.200, 11–16
61. Roggenkamp, D. et al. (2013) Epidermal nerve fibers modulatekeratinocyte growth via neuropeptide signaling in an innervatedskin model. J. Invest. Dermatol. 133, 1620–1628
62. Ding, W. et al. (2008) Calcitonin gene-related peptide biasesLangerhans cells toward Th2-type immunity. J. Immunol. 181,6020–6026
63. Parham, C. et al. (2002) A receptor for the heterodimeric cytokineIL-23 is composed of IL-12Rbeta1 and a novel cytokine receptorsubunit, IL-23R. J. Immunol. 168, 5699–5708
10 Trends in Immunology, Month Year, Vol. xx, No. yy

TREIMM 1284 No. of Pages 11
64. Gaffen, S.L. et al. (2014) The IL-23–IL-17 immune axis: frommechanisms to therapeutic testing. Nat. Rev. Immunol. 14, 585–600
65. de Beaucoudrey, L. et al. (2008) Mutations in STAT3 andIL12RB1 impair the development of human IL-17-producing Tcells. J. Exp. Med. 205, 1543–1550
66. Ouederni, M. et al. (2014) Clinical features of candidiasis inpatients with inherited interleukin 12 receptor b1 deficiency. Clin.Infect. Dis. 58, 204–213
67. Whitney, P.G. et al. (2014) Syk signaling in dendritic cells orches-trates innate resistance to systemic fungal infection. PLoSPathog. 10, e1004276
68. Conti, H.R. et al. (2009) Th17 cells and IL-17 receptor signalingare essential for mucosal host defense against oral candidiasis. J.Exp. Med. 206, 299–311
69. Maher, C.O. et al. (2015) Candida albicans stimulates IL-23release by human dendritic cells and downstream IL-17 secretionby Vd1 T cells. J. Immunol. 194, 5953–5960
70. Kaplan, D.H. (2010) In vivo function of Langerhans cells anddermal dendritic cells. Trends Immunol. 31, 446–451
71. Yoshiki, R. et al. (2014) IL-23 from Langerhans cells is required forthe development of imiquimod-induced psoriasis-like dermatitisby induction of IL-17A-producing gd T cells. J. Invest. Dermatol.134, 1912–1921
72. Wohn, C. et al. (2013) Langerin(neg) conventional dendritic cellsproduce IL-23 to drive psoriatic plaque formation in mice. Proc.Natl. Acad. Sci. U.S.A. 110, 10723–10728
73. Scholz, F. et al. (2015) Langerhans cells suppress CD49a+ NKcell-mediated skin inflammation. J. Immunol. 195, 2335–2342
74. Kaplan, D.H. et al. (2005) Epidermal Langerhans cell-deficientmice develop enhanced contact hypersensitivity. Immunity 23,611–620
75. Bobr, A. et al. (2010) Acute ablation of Langerhans cells enhan-ces skin immune responses. J. Immunol. 185, 4724–4728
76. Igyártó, B.Z. et al. (2009) Langerhans cells suppress contacthypersensitivity responses via cognate CD4 interaction and Lan-gerhans cell-derived IL-10. J. Immunol. 183, 5085–5093
77. Huppler, A.R. et al. (2014) Role of neutrophils in IL-17-dependentimmunity to mucosal candidiasis. J. Immunol. 192, 1745–1752
78. Trautwein-Weidner, K. et al. (2015) IL-17-mediated antifungaldefense in the oral mucosa is independent of neutrophils.Muco-sal Immunol. 8, 221–231
79. Johnston, A. et al. (2013) Keratinocyte overexpression of IL-17Cpromotes psoriasiform skin inflammation. J. Immunol. 190,2252–2262
80. Conti, H.R. et al. (2015) Signaling through IL-17C/IL-17RE isdispensable for immunity to systemic, oral and cutaneous can-didiasis. PLoS ONE 10, e0122807
81. Gladiator, A. et al. (2013) Cutting edge: IL-17-secreting innatelymphoid cells are essential for host defense against fungalinfection. J. Immunol. 190, 521–525
82. Conti, H.R. et al. (2014) Oral-resident natural Th17 cells and gd Tcells control opportunistic Candida albicans infections. J. Exp.Med. 211, 2075–2084
83. Wilson, R.P. et al. (2015) STAT3 is a critical cell-intrinsic regulatorof human unconventional T cell numbers and function. J. Exp.Med. 212, 855–864
84. Cua, D.J. and Tato, C.M. (2010) Innate IL-17-producing cells: thesentinels of the immune system.Nat. Rev. Immunol. 10, 479–489
85. Gray, E.E. et al. (2013) Deficiency in IL-17-committed Vg4+ gd Tcells in a spontaneous Sox13-mutant CD45.1+ congenic mousesubstrain provides protection from dermatitis. Nat. Immunol. 14,584–592
86. Taylor, P.R. et al. (2014) Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated byIL-6, IL-23, RORgt and dectin-2. Nat. Immunol. 15, 143–151
87. Branzk, N. et al. (2014) Neutrophils sense microbe size andselectively release neutrophil extracellular traps in response tolarge pathogens. Nat. Immunol. 15, 1017–1025
88. Rieber, N. et al. (2015) Pathogenic fungi regulate immunity byinducing neutrophilic myeloid-derived suppressor cells. Cell HostMicrobe 17, 507–514
89. Netea, M.G. et al. (2015) Immune defence against Candidafungal infections. Nat. Rev. Immunol. 15, 630–642
90. Zielinski, C.E. et al. (2012) Pathogen-induced human TH17 cellsproduce IFN-g or IL-10 and are regulated by IL-1b. Nature 484,514–518
91. Becattini, S. et al. (2015) Functional heterogeneity of humanmemory CD4z T cell clones primed by pathogens or vaccines.Science 347, 400–406
92. Randall, K.L. et al. (2011) DOCK8 deficiency impairs CD8 T cellsurvival and function in humans and mice. J. Exp. Med. 208,2305–2320
93. Linehan, J.L. et al. (2015) Generation of Th17 cells in response tointranasal infection requires TGF-b1 from dendritic cells and IL-6from CD301b+ dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 112,12782–12787
94. Naik, S. et al. (2015) Commensal–dendritic-cell interaction speci-fies a unique protective skin immune signature.Nature 520, 104–108
95. Trautwein-Weidner, K. et al. (2015) Antigen-specific Th17 cellsare primed by distinct and complementary dendritic cell subsetsin oropharyngeal candidiasis. PLoS Pathog. 11, e1005164
96. Hambleton, S. et al. (2011) IRF8 mutations and human dendritic-cell immunodeficiency. N. Engl. J. Med. 365, 127–138
97. Hernández-Santos, N. et al. (2013) Th17 cells confer long-termadaptive immunity to oral mucosal Candida albicans infections.Mucosal Immunol. 6, 900–910
98. Pepper, M. et al. (2010) Different routes of bacterial infectioninduce long-lived TH1 memory cells and short-lived TH17 cells.Nat. Immunol. 11, 83–89
99. Ahmad, D.S. et al. (2013) Idiopathic CD4 lymphocytopenia:spectrum of opportunistic infections, malignancies, and autoim-mune diseases. Avicenna J. Med. 3, 37–47
100. Manchado Lopez, P. et al. (1999) Cutaneous infections by pap-illomavirus, herpes zoster and Candida albicans as the onlymanifestation of idiopathic CD4+ T lymphocytopenia. Int. J. Der-matol. 38, 119–121
101. Holland, S.M. et al. (2007) STAT3 mutations in the hyper-IgEsyndrome. N. Engl. J. Med. 357, 1608–1619
102. Milner, J.D. et al. (2008) Impaired T(H)17 cell differentiation insubjects with autosomal dominant hyper-IgE syndrome. Nature452, 773–776
103. Zhang, Q. et al. (2009) Combined immunodeficiency associatedwith DOCK8 mutations. N. Engl. J. Med. 361, 2046–2055
104. Puel, A. et al. (2011) Chronic mucocutaneous candidiasis inhumans with inborn errors of interleukin-17 immunity. Science332, 65–68
105. Ling, Y. et al. (2015) Inherited IL-17RC deficiency in patients withchronic mucocutaneous candidiasis. J. Exp. Med. 212, 619–631
106. Boisson, B. et al. (2013) An ACT1 mutation selectively abolishesinterleukin-17 responses in humans with chronic mucocutane-ous candidiasis. Immunity 39, 676–686
107. Okada, S. et al. (2015) Impairment of immunity to Candida andMycobacterium in humans with bi-allelic RORC mutations. Sci-ence 349, 606–613
108. Kisand, K. et al. (2010) Chronic mucocutaneous candidiasis inAPECED or thymoma patients correlates with autoimmunity toTh17-associated cytokines. J. Exp. Med. 207, 299–308
109. Liu, L. et al. (2011) Gain-of-function human STAT1 mutationsimpair IL-17 immunity and underlie chronic mucocutaneouscandidiasis. J. Exp. Med. 208, 1635–1648
Trends in Immunology, Month Year, Vol. xx, No. yy 11





























