Embed Size (px)
Citation preview

Solid-state NMR of proteins sedimentedby ultracentrifugationIvano Bertinia,1, Claudio Luchinata,1, Giacomo Parigia, Enrico Raveraa, Bernd Reifb, and Paola Turanoa
aMagnetic Resonance Center (CERM) and Department of Chemistry, University of Florence, Via L. Sacconi 6, 50019 Sesto Fiorentino, Italy; andbDepartment Chemie, Technische Universitaet Muenchen, Lichtenbergstrasse 4, D85747 Garching, Germany
Edited* by Alexander Pines, University of California Berkeley and Lawrence Berkeley National Laboratory, Berkeley, CA, and approved May 13, 2011(received for review March 11, 2011)
Relatively large proteins in solution, spun in NMR rotors for solidsamples at typical ultracentrifugation speeds, sediment at the rotorwall. The sedimented proteins provide high-quality solid-state-likeNMR spectra suitable for structural investigation. The proteinsfully revert to the native solution state when spinning is stopped,allowing one to study them in both conditions. Transiently sedi-mented proteins can be considered a novel phase as far as NMRis concerned. NMR of transiently sedimented molecules under fastmagic angle spinning has the advantage of overcoming proteinsize limitations of solution NMR without the need of sample crys-tallization/precipitation required by solid-state NMR.
ferritin ∣ magic angle spinning NMR ∣ sedimentation ∣ gravity ∣high molecular weight
“We built a new ultracentrifuge which permitted the study ofsolutions in fields of force of up to about 100,000 times
gravity…it is possible to cover the entire sector of the colloids downto the smallest particle sizes, and even…reach…the substances ofhigh molecular weight, such as haemoglobin, protein, starch, etc.The limit of the possible has not yet been reached” (from Nobellecture “The Ultracentrifuge” by Theodore Svedberg in 1927).
Today ultracentrifuges reach above 1 million g, and virtuallyevery protein can be ultracentrifuged. Ultracentrifuged proteinsare usually well behaved. The sediment resolubilizes as soon asthe centrifugal force is removed, and the protein generallymaintains its native state throughout (1–3). Therefore, the ultra-centrifuged, or sedimented, “state” of proteins, or of other bio-molecules, is at least as appealing for structural studies as thesolution or the crystalline state.
NMR as a structural method can be applied to both solutionsand solids. NMR on solid samples needs to be performed by ro-tating the sample about an axis tilted by the so-called magic angle(54.73°) with respect to the external magnetic field (magic anglespinning, MAS), and fast enough to average all chemical shift an-isotropies and dipolar interactions (4–6). The solid sample isplaced in a cylindrical rotor the internal diameter of which mayrange between a few millimeters and a fraction of a millimeter.Typical angular speeds range from a few kilohertz to about70 kHz. NMR rotors are thus ultracentrifuges, creating a field offorce of up to a few million g at their maximum speed (Table 1).Therefore, a protein in solution spun in a “solid-state” NMRrotor should sediment at the internal walls of the cylinder, andits NMR signals should be observable and studied as if the proteinwere in the solid state. The purpose of this work is to provide theproof of principle of this previously undescribed way of perform-ing NMR spectroscopy.
NMR of proteins in solution is a powerful structural methodthat, however, becomes increasingly difficult with increasingmolecular weight (7–20). The larger the protein, the lower thespectral resolution: Besides the increase in the number of signals,the linewidths increase linearly with molecular weight. Conver-sely, in the solid state the linewidth is independent of the mole-cular weight (21), and larger systems can be afforded provided thenumber of signals is not too large. For example, the iron-storage
protein ferritin—a 24-mer of 480-kDa molecular weight in theapo form (i.e., the form devoid of the iron oxide core)—couldhardly be studied in solution, and only in the perdeuterated form,because of its size, despite the fact that the number of signalsis only that of a 20-kDa protein. Luckily, apoferritin can beobtained in the microcrystalline state.
Apoferritin is an ideal case to test our hypothesis: It yields verywell-resolved solid-state NMR spectra (22), and its molecularweight ensures that it should sediment in an NMR rotor evenat nonextreme speeds.
Results and DiscussionThe theoretical treatment of equilibrium ultracentrifugation (23)can be adapted to the geometry of the NMR rotor to quantita-tively predict the behavior of biological samples in solution (seeSI Text). For a 0.125-mM apoferritin solution (3-mM monomer)in a 4-mm rotor the predicted behavior is shown in Fig. 1. Theconcentration of the protein as a function of the distance fromthe axis of the cylinder significantly increases with increasingspeed. Already at 3 kHz the protein concentration at the rotorwall approaches its maximum, which can be approximated byits density (1;340 kg∕m3, as determined by ultracentrifugationin 1944 (24), times 0.74 that corresponds to the maximum packingfactor for rigid spheres) divided by its molecular weight, i.e., closeto 3-mM 24-mer concentration. Beyond this point, the solvent islargely excluded, each protein molecule is in close contact with
Table 1. Maximum acceleration reached in commercial solid-stateMAS rotors at maximum speed, and their predicted proteinsedimentation capability
Externaldiameter,mm
Internalradius,mm
Maximumangular
speed, kHz
Maximumacceleration
relativeto Earth
gravity, a∕g
Minimumprotein MW,Da, for >90%sedimentationat maximum
speed*
7 2.8 8 7.2 × 105 141,0004 1.5 18 2.0 × 106 97,0003.2 1.2 23 2.6 × 106 93,0002.5 0.9 35 4.4 × 106 71,0001.3 0.35 70 6.9 × 106 118,000
Conditions: protein concentration 60 mg∕mL, T ¼ 290 K.*The minimum protein MW is inversely proportional to the squareof the product between the internal radius and the maximum angularspeed, as shown in the SI Text.
Author contributions: I.B., C.L., E.R., and B.R. designed research; E.R. and P.T. performedresearch; C.L., G.P., E.R., and P.T. analyzed data; and I.B., C.L., G.P., and P.T. wrote the paper.
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.1To whom correspondence may be addressed. E-mail: [email protected] [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1103854108/-/DCSupplemental.
10396–10399 ∣ PNAS ∣ June 28, 2011 ∣ vol. 108 ∣ no. 26 www.pnas.org/cgi/doi/10.1073/pnas.1103854108
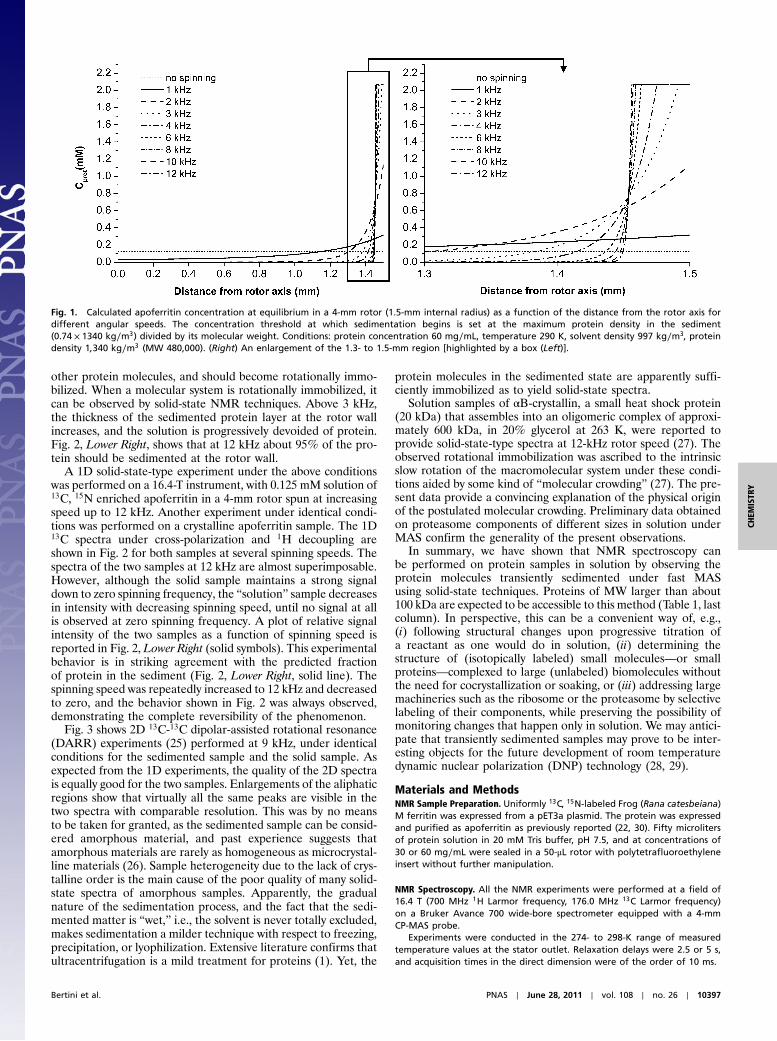
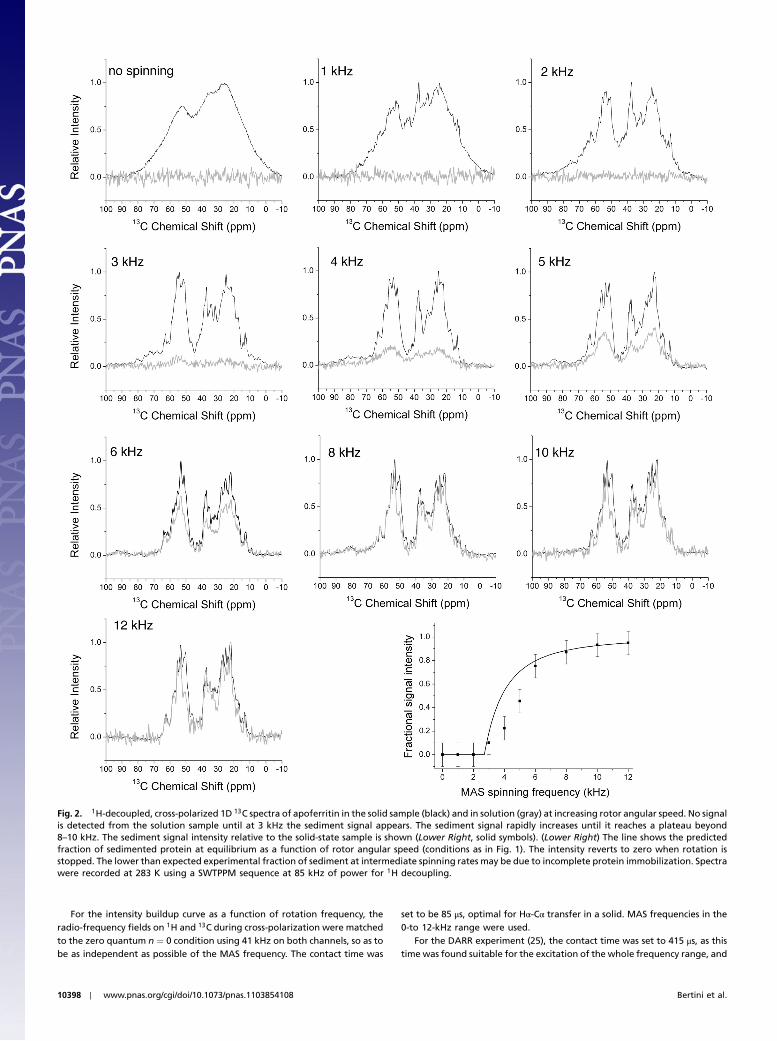
other protein molecules, and should become rotationally immo-bilized. When a molecular system is rotationally immobilized, itcan be observed by solid-state NMR techniques. Above 3 kHz,the thickness of the sedimented protein layer at the rotor wallincreases, and the solution is progressively devoided of protein.Fig. 2, Lower Right, shows that at 12 kHz about 95% of the pro-tein should be sedimented at the rotor wall.
A 1D solid-state-type experiment under the above conditionswas performed on a 16.4-T instrument, with 0.125 mM solution of13C, 15N enriched apoferritin in a 4-mm rotor spun at increasingspeed up to 12 kHz. Another experiment under identical condi-tions was performed on a crystalline apoferritin sample. The 1D13C spectra under cross-polarization and 1H decoupling areshown in Fig. 2 for both samples at several spinning speeds. Thespectra of the two samples at 12 kHz are almost superimposable.However, although the solid sample maintains a strong signaldown to zero spinning frequency, the “solution” sample decreasesin intensity with decreasing spinning speed, until no signal at allis observed at zero spinning frequency. A plot of relative signalintensity of the two samples as a function of spinning speed isreported in Fig. 2, Lower Right (solid symbols). This experimentalbehavior is in striking agreement with the predicted fractionof protein in the sediment (Fig. 2, Lower Right, solid line). Thespinning speed was repeatedly increased to 12 kHz and decreasedto zero, and the behavior shown in Fig. 2 was always observed,demonstrating the complete reversibility of the phenomenon.
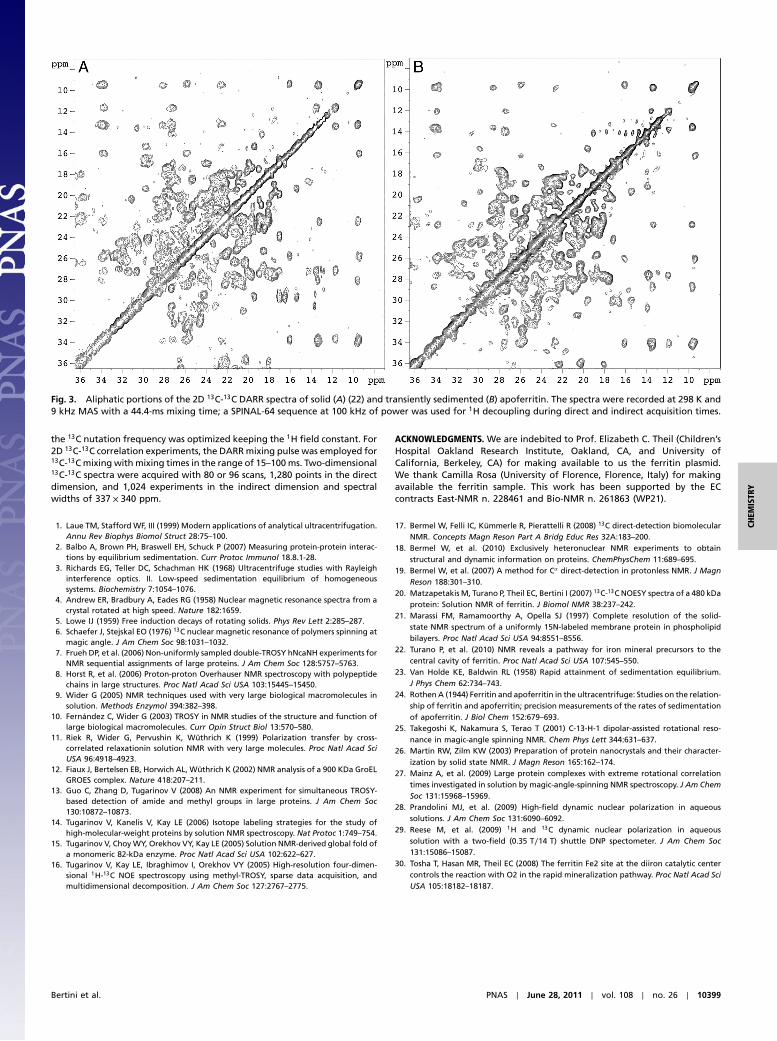
Fig. 3 shows 2D 13C-13C dipolar-assisted rotational resonance(DARR) experiments (25) performed at 9 kHz, under identicalconditions for the sedimented sample and the solid sample. Asexpected from the 1D experiments, the quality of the 2D spectrais equally good for the two samples. Enlargements of the aliphaticregions show that virtually all the same peaks are visible in thetwo spectra with comparable resolution. This was by no meansto be taken for granted, as the sedimented sample can be consid-ered amorphous material, and past experience suggests thatamorphous materials are rarely as homogeneous as microcrystal-line materials (26). Sample heterogeneity due to the lack of crys-talline order is the main cause of the poor quality of many solid-state spectra of amorphous samples. Apparently, the gradualnature of the sedimentation process, and the fact that the sedi-mented matter is “wet,” i.e., the solvent is never totally excluded,makes sedimentation a milder technique with respect to freezing,precipitation, or lyophilization. Extensive literature confirms thatultracentrifugation is a mild treatment for proteins (1). Yet, the
protein molecules in the sedimented state are apparently suffi-ciently immobilized as to yield solid-state spectra.
Solution samples of αB-crystallin, a small heat shock protein(20 kDa) that assembles into an oligomeric complex of approxi-mately 600 kDa, in 20% glycerol at 263 K, were reported toprovide solid-state-type spectra at 12-kHz rotor speed (27). Theobserved rotational immobilization was ascribed to the intrinsicslow rotation of the macromolecular system under these condi-tions aided by some kind of “molecular crowding” (27). The pre-sent data provide a convincing explanation of the physical originof the postulated molecular crowding. Preliminary data obtainedon proteasome components of different sizes in solution underMAS confirm the generality of the present observations.
In summary, we have shown that NMR spectroscopy canbe performed on protein samples in solution by observing theprotein molecules transiently sedimented under fast MASusing solid-state techniques. Proteins of MW larger than about100 kDa are expected to be accessible to this method (Table 1, lastcolumn). In perspective, this can be a convenient way of, e.g.,(i) following structural changes upon progressive titration ofa reactant as one would do in solution, (ii) determining thestructure of (isotopically labeled) small molecules—or smallproteins—complexed to large (unlabeled) biomolecules withoutthe need for cocrystallization or soaking, or (iii) addressing largemachineries such as the ribosome or the proteasome by selectivelabeling of their components, while preserving the possibility ofmonitoring changes that happen only in solution. We may antici-pate that transiently sedimented samples may prove to be inter-esting objects for the future development of room temperaturedynamic nuclear polarization (DNP) technology (28, 29).
Materials and MethodsNMR Sample Preparation. Uniformly 13C, 15N-labeled Frog (Rana catesbeiana)M ferritin was expressed from a pET3a plasmid. The protein was expressedand purified as apoferritin as previously reported (22, 30). Fifty microlitersof protein solution in 20 mM Tris buffer, pH 7.5, and at concentrations of30 or 60 mg∕mL were sealed in a 50-μL rotor with polytetrafluoroethyleneinsert without further manipulation.
NMR Spectroscopy. All the NMR experiments were performed at a field of16.4 T (700 MHz 1H Larmor frequency, 176.0 MHz 13C Larmor frequency)on a Bruker Avance 700 wide-bore spectrometer equipped with a 4-mmCP-MAS probe.
Experiments were conducted in the 274- to 298-K range of measuredtemperature values at the stator outlet. Relaxation delays were 2.5 or 5 s,and acquisition times in the direct dimension were of the order of 10 ms.
Fig. 1. Calculated apoferritin concentration at equilibrium in a 4-mm rotor (1.5-mm internal radius) as a function of the distance from the rotor axis fordifferent angular speeds. The concentration threshold at which sedimentation begins is set at the maximum protein density in the sediment(0.74 × 1340 kg∕m3) divided by its molecular weight. Conditions: protein concentration 60 mg∕mL, temperature 290 K, solvent density 997 kg∕m3, proteindensity 1;340 kg∕m3 (MW 480,000). (Right) An enlargement of the 1.3- to 1.5-mm region [highlighted by a box (Left)].
Bertini et al. PNAS ∣ June 28, 2011 ∣ vol. 108 ∣ no. 26 ∣ 10397
CHEM
ISTR
Y
For the intensity buildup curve as a function of rotation frequency, theradio-frequency fields on 1H and 13C during cross-polarization were matchedto the zero quantum n ¼ 0 condition using 41 kHz on both channels, so as tobe as independent as possible of the MAS frequency. The contact time was
set to be 85 μs, optimal for Hα-Cα transfer in a solid. MAS frequencies in the0-to 12-kHz range were used.
For the DARR experiment (25), the contact time was set to 415 μs, as thistime was found suitable for the excitation of the whole frequency range, and
Fig. 2. 1H-decoupled, cross-polarized 1D 13C spectra of apoferritin in the solid sample (black) and in solution (gray) at increasing rotor angular speed. No signalis detected from the solution sample until at 3 kHz the sediment signal appears. The sediment signal rapidly increases until it reaches a plateau beyond8–10 kHz. The sediment signal intensity relative to the solid-state sample is shown (Lower Right, solid symbols). (Lower Right) The line shows the predictedfraction of sedimented protein at equilibrium as a function of rotor angular speed (conditions as in Fig. 1). The intensity reverts to zero when rotation isstopped. The lower than expected experimental fraction of sediment at intermediate spinning rates may be due to incomplete protein immobilization. Spectrawere recorded at 283 K using a SWTPPM sequence at 85 kHz of power for 1H decoupling.
10398 ∣ www.pnas.org/cgi/doi/10.1073/pnas.1103854108 Bertini et al.
the 13C nutation frequency was optimized keeping the 1H field constant. For2D 13C-13C correlation experiments, the DARRmixing pulse was employed for13C-13Cmixing with mixing times in the range of 15–100ms. Two-dimensional13C-13C spectra were acquired with 80 or 96 scans, 1,280 points in the directdimension, and 1,024 experiments in the indirect dimension and spectralwidths of 337 × 340 ppm.
ACKNOWLEDGMENTS. We are indebited to Prof. Elizabeth C. Theil (Children’sHospital Oakland Research Institute, Oakland, CA, and University ofCalifornia, Berkeley, CA) for making available to us the ferritin plasmid.We thank Camilla Rosa (University of Florence, Florence, Italy) for makingavailable the ferritin sample. This work has been supported by the ECcontracts East-NMR n. 228461 and Bio-NMR n. 261863 (WP21).
1. Laue TM, Stafford WF, III (1999) Modern applications of analytical ultracentrifugation.Annu Rev Biophys Biomol Struct 28:75–100.
2. Balbo A, Brown PH, Braswell EH, Schuck P (2007) Measuring protein-protein interac-tions by equilibrium sedimentation. Curr Protoc Immunol 18.8.1-28.
3. Richards EG, Teller DC, Schachman HK (1968) Ultracentrifuge studies with Rayleighinterference optics. II. Low-speed sedimentation equilibrium of homogeneoussystems. Biochemistry 7:1054–1076.
4. Andrew ER, Bradbury A, Eades RG (1958) Nuclear magnetic resonance spectra from acrystal rotated at high speed. Nature 182:1659.
5. Lowe IJ (1959) Free induction decays of rotating solids. Phys Rev Lett 2:285–287.6. Schaefer J, Stejskal EO (1976) 13C nuclear magnetic resonance of polymers spinning at
magic angle. J Am Chem Soc 98:1031–1032.7. Frueh DP, et al. (2006) Non-uniformly sampled double-TROSY hNcaNH experiments for
NMR sequential assignments of large proteins. J Am Chem Soc 128:5757–5763.8. Horst R, et al. (2006) Proton-proton Overhauser NMR spectroscopy with polypeptide
chains in large structures. Proc Natl Acad Sci USA 103:15445–15450.9. Wider G (2005) NMR techniques used with very large biological macromolecules in
solution. Methods Enzymol 394:382–398.10. Fernández C, Wider G (2003) TROSY in NMR studies of the structure and function of
large biological macromolecules. Curr Opin Struct Biol 13:570–580.11. Riek R, Wider G, Pervushin K, Wüthrich K (1999) Polarization transfer by cross-
correlated relaxationin solution NMR with very large molecules. Proc Natl Acad SciUSA 96:4918–4923.
12. Fiaux J, Bertelsen EB, Horwich AL, Wüthrich K (2002) NMR analysis of a 900 KDa GroELGROES complex. Nature 418:207–211.
13. Guo C, Zhang D, Tugarinov V (2008) An NMR experiment for simultaneous TROSY-based detection of amide and methyl groups in large proteins. J Am Chem Soc130:10872–10873.
14. Tugarinov V, Kanelis V, Kay LE (2006) Isotope labeling strategies for the study ofhigh-molecular-weight proteins by solution NMR spectroscopy. Nat Protoc 1:749–754.
15. Tugarinov V, ChoyWY, Orekhov VY, Kay LE (2005) Solution NMR-derived global fold ofa monomeric 82-kDa enzyme. Proc Natl Acad Sci USA 102:622–627.
16. Tugarinov V, Kay LE, Ibraghimov I, Orekhov VY (2005) High-resolution four-dimen-sional 1H-13C NOE spectroscopy using methyl-TROSY, sparse data acquisition, andmultidimensional decomposition. J Am Chem Soc 127:2767–2775.
17. Bermel W, Felli IC, Kümmerle R, Pierattelli R (2008) 13C direct-detection biomolecularNMR. Concepts Magn Reson Part A Bridg Educ Res 32A:183–200.
18. Bermel W, et al. (2010) Exclusively heteronuclear NMR experiments to obtainstructural and dynamic information on proteins. ChemPhysChem 11:689–695.
19. Bermel W, et al. (2007) A method for Cα direct-detection in protonless NMR. J MagnReson 188:301–310.
20. Matzapetakis M, Turano P, Theil EC, Bertini I (2007) 13C-13C NOESY spectra of a 480 kDaprotein: Solution NMR of ferritin. J Biomol NMR 38:237–242.
21. Marassi FM, Ramamoorthy A, Opella SJ (1997) Complete resolution of the solid-state NMR spectrum of a uniformly 15N-labeled membrane protein in phospholipidbilayers. Proc Natl Acad Sci USA 94:8551–8556.
22. Turano P, et al. (2010) NMR reveals a pathway for iron mineral precursors to thecentral cavity of ferritin. Proc Natl Acad Sci USA 107:545–550.
23. Van Holde KE, Baldwin RL (1958) Rapid attainment of sedimentation equilibrium.J Phys Chem 62:734–743.
24. RothenA (1944) Ferritin and apoferritin in the ultracentrifuge: Studies on the relation-ship of ferritin and apoferritin; precision measurements of the rates of sedimentationof apoferritin. J Biol Chem 152:679–693.
25. Takegoshi K, Nakamura S, Terao T (2001) C-13-H-1 dipolar-assisted rotational reso-nance in magic-angle spinning NMR. Chem Phys Lett 344:631–637.
26. Martin RW, Zilm KW (2003) Preparation of protein nanocrystals and their character-ization by solid state NMR. J Magn Reson 165:162–174.
27. Mainz A, et al. (2009) Large protein complexes with extreme rotational correlationtimes investigated in solution by magic-angle-spinning NMR spectroscopy. J Am ChemSoc 131:15968–15969.
28. Prandolini MJ, et al. (2009) High-field dynamic nuclear polarization in aqueoussolutions. J Am Chem Soc 131:6090–6092.
29. Reese M, et al. (2009) 1H and 13C dynamic nuclear polarization in aqueoussolution with a two-field (0.35 T∕14 T) shuttle DNP spectometer. J Am Chem Soc131:15086–15087.
30. Tosha T, Hasan MR, Theil EC (2008) The ferritin Fe2 site at the diiron catalytic centercontrols the reaction with O2 in the rapid mineralization pathway. Proc Natl Acad SciUSA 105:18182–18187.
Fig. 3. Aliphatic portions of the 2D 13C-13C DARR spectra of solid (A) (22) and transiently sedimented (B) apoferritin. The spectra were recorded at 298 K and9 kHz MAS with a 44.4-ms mixing time; a SPINAL-64 sequence at 100 kHz of power was used for 1H decoupling during direct and indirect acquisition times.
Bertini et al. PNAS ∣ June 28, 2011 ∣ vol. 108 ∣ no. 26 ∣ 10399
CHEM
ISTR
Y





























