Embed Size (px)
Citation preview

1

2

3
Sommaire de la ronéo n°2 du 2er trimestre
Semaine 3 du 15 novembre au 21 janvier
UE 6 : Le système digestif
Physiologie :
Cours 1 : Sécrétions digestives ……………………………………………….………………….…..p5
Cours 7 : Physiologie digestive : digestion et absorption………………………………………..p89
Cours 9 : Transport intestinal d’eau et d’électrolytes, en situation normale ou anormale..p117
Sémiologie :
Cours 2 : Sémiologie et exploration de l’intestin grêle ………………………..………………..p25
Cours 8 : Sémiologie et exploration de l’intestin grêle, du côlon et du rectum ………..…..p107
Anatomopathologie :
Cours 3: Dépistage et pathogénie du cancer colique ……………………………………….……p41
Anatomie :
Cours 4: Anatomie du rectum et du canal anal …………………………………….………………p55
Histologie :
Cours 5: Histologie du tube digestif (2ème partie)……………………..…………….….……….p73
Cours 6: Histologie du tube digestif (3ème partie) et embryologie du tube digestif ……..….p81
Pharmacologie
Cours 10 : Hépatites médicamenteuses …………………………………………………..………p135

4

5
UE6 – SD – PHYSIOLOGIE – n°2 15/01/2018
Pascal Houillier
RT : Ketty Christophe RL : Quynh-Anh Phung
Sécrétions digestives
I. Sécrétions gastriques A. Structure d’une « glande » gastrique B. Composition électrolytique du liquide gastrique C. Les cellules sécrétrices de l’épithélium gastrique D. Transports électrolytiques de la cellule pariétale E. Régulation de la sécrétion d’acide de la cellule pariétale F. Evolution temporelle de la sécrétion d’acide et du pH luminal
après un repas G. La sécrétion d’acide gastrique H. Les autres sécrétions gastriques
i. Sécrétion du facteur intrinsèque ii. Sécrétion du pepsinogène
iii. Sécrétion du mucus
II. Sécrétions pancréatiques A. Fonction exocrine du pancréas B. Le liquide pancréatique C. Régulation de la sécrétion pancréatique
III. Sécrétions biliaires A. La bile et les acides biliaires B. Cycle entéro-hépatique des acides biliaires C. Régulation des sécrétions biliaires D. Lithiase biliaire
IV. Points-clé
Abréviations : Ach : Acétylcholine – AINS : Anti Inflammatoires non Stéroïdiens - CCK :Cholécystokinine – ECL : Enterochromaffin Like Cells – HCL : Acide chlorhydrique – NaCl : chlorure de sodium – SN et P :Système Nerveux sympathique et parasympathique
Mot du RT : Le cours est assez long. J’ai essayé de mettre en évidence les éléments importants et de le rendre le plus explicite possible. Bonne lecture !!

6
Objectifs : Connaître et comprendre les mécanismes, les déterminants et les rôles des
principales sécrétions dans le tube digestif
Avant propos Les sécrétions salivaires ne seront pas abordées par faute de temps, bien qu’elles appartiennent aux sécrétions digestives. Il y a deux éléments importants sur les sécrétions salivaires. D’une part, elles sont stimulées par l’alimentation et leur débit est différent selon que l’on est en période de jeûne ou postprandiale. D’autre part, la ptyaline (amylase salivaire digérant l’amidon) est pratiquement la seule enzyme que l’on retrouve. Toutes les sécrétions sont contrôlées et varient au cours du temps en fonction des besoins. Petit rappel sur les modes de communication cellulaire observables à la paroi du tube digestif : Mode endocrine : la cellule répond à
l’événement en sécrétant une hormone, dont la cible est à distance et atteignable par voie sanguine.
Mode neurocrine : un neurone sensitif perçoit
l’information et se connecte à un réseau d’inter-neurones intégrateurs. Ils permettent d’activer un neurone effecteur sécrétomoteur (cellule neuroendocrine) qui libérera un neuromédiateur.
Mode paracrine : le médiateur est sécrété dans
le milieu et n’emprunte pas la circulation sanguine.
D’autres types de communication existent comme le mode autocrine, où la cellule détectrice et la cellule cible correspond à la même cellule.

7
I. Sécrétions gastriques D’un point du fonctionnel, on divise l’estomac en trois régions :
Région cardiale ou fundique (partie supérieure de l’estomac) : <5% de la surface : sécrétion de mucus, pepsinogène et ghréline (l’hormone qui donne faim)
Région oxyntique (corps de l’estomac) : 75% : sécrétion de mucus, pepsinogène, ghréline, HCL, facteur intrinsèque et lipase gastrique.
Région pylorique (antre gastrique) : 20-25% : sécrétion de gastrine, de mucus et pepsinogène
Ces sécrétions sont assurées par l’épithélium gastrique. On peut remarquer que le mucus et le pepsinogène sont sécrétés à tous les niveaux de l’estomac.
A. Structure d’une « glande » gastrique (région oxyntique) Les invaginations formées par l’épithélium constituent les glandes gastriques. Elles sont organisées en grande partie dans la profondeur de l’épithélium. Elles sont dotées d’une grande hétérogénéité cellulaire. De la surface à la profondeur, on distingue les :
Cellules épithéliales de surface Cellules muqueuse du collet Cellules souches Cellules pariétales (fonction principale : sécrétion d’HCL, facteur intrinsèque) Cellules principales (fonction principale : sécrétion de pepsinogène) Cellules endocrines
B. Composition électrolytique du liquide gastrique
La composition du liquide gastrique varie de manière qualitative et quantitative selon les situations En situation de jeûne, la sécrétion est à petit débit et le liquide gastrique correspond à une solution de NaCl, où il y a des concentrations faibles en potassium et ions H+. En situation postprandiale, le liquide gastrique contient surtout de l’acide chlorhydrique et le débit de sécrétion est augmenté (il passe de moins de 1mL/min à 3mL/min). Il y a un enrichissement modéré de la cellule en ions chlorure et une augmentation significative de la concentration en ions H+. En parallèle, la concentration de sodium chute considérablement.

8
C. Les cellules sécrétrices de l’épithélium gastrique La sécrétion d’électrolytes est un phénomène possible grâce à 2 types de cellules, dispersées dans l’épithélium :
Les cellules épithéliales de surface sécrètent des bicarbonates de sodium en situation basale et postprandiale
Les cellules pariétales, en situation postprandiale, sécrètent de l’acide chlorydrique à leur membrane apicale grâce à la pompe H/K ATPase (sortie de H+ et entrée de K+) (même famille que la Na/K ATPase) et à deux canaux, un canal potassique (sortie de K+) et un canal chlore (sortie de Cl- permettant la formation rapide de HCl).
L’activation de ces systèmes de sécrétion avec notamment l’entrée et la sortie de potassium, met en évidence le recyclage du potassium à travers la membrane apicale.
Les cellules pariétales subissent des modifications structurales et fonctionnelles rapides.
Au repos c’est-à-dire à jeun, on parle de cellules pariétales quiescentes (image A) : la surface de la membrane apicale (en contact avec la lumière gastrique) est réduite et les membranes tubulo-vésiculaires (structures membranaires) sont repliées sur elle-même à l’intérieur de la cellule. Il n’y a pas de contact avec la lumière. La cellule possède un nombre déjà relativement élevé de mitochondries et des canalicules cellulaires, qui eux non plus ne communiquent pas avec la lumière.
En situation postprandiale, les cellules pariétales sont activées et deviennent sécrétantes (image B) : la cellule s’enrichit fortement en mitochondries et il y a fusion des structures membranaires avec la membrane apicale. Cette fusion augmente considérablement la surface de la membrane apicale et le débit de sécrétion ionique. Le grand nombre de mitochondries permet de fournir suffisamment d’énergie à la H/K-ATPase, véritable moteur de la sécrétion gastrique.
D. Transports électrolytiques par la cellule pariétale D’un point de vue fonctionnel, en situation d’activation, la membrane apicale ainsi
développée présente des H/K-ATPase de type 1= gastrique, des canaux K+ et Cl-. Les cellules pariétales ont pour fonction la sécrétion d’acide chlorhydrique (HCl). Or, d’où viennent les ions H+ ? Ils proviennent de la dissociation de l’acide carbonique CO2, produisant un ion H+ et un bicarbonate. Ainsi, le bicarbonate généré sort de la cellule par un canal basolatéral. Cette sortie du bicarbonate, possible grâce à l’échangeur chlore/bicarbonate, est nécessaire pour maintenir le pH intracellulaire. Sinon, la cellule s’alcalinisera et produira moins d’acide. Lorsque l’estomac

9
travaille, on observe une augmentation transitoire dans les 10 minutes de la concentration de bicarbonates dans le plasma. On remarque également que le chlore entrant grâce à l’échangeur chlore/bicarbonate ressortira par un canal chlore de la membrane apicale.
E. Régulation de la sécrétion d’acide de la cellule pariétale L’activité sécrétoire des cellules pariétales est contrôlée par différents récepteurs. Certains activent la sécrétion et d’autres l’inhibent. Récepteurs activateurs :
CCKB = Récepteur de la gastrine : la liaison de la gastrine active la voie d’hydrolyse des phosphoinositides de membrane (via la protéine Gq) aboutissant à l’activation de la H/K-ATPase.
Récepteur muscarinique M3 = Récepteur de l’acétylcholine : même voie d’activation que CCKB (voir schéma)
Récepteur H2 = Récepteur de l’histamine : voie de signalisation différente car dépendante de la production d’AMPcyclique. L’histamine couplée à son récepteur entraîne l’activation de l’adénylate cyclase via la protéine GS, l’AMPC active ensuite la PKA qui active finalement la H/K-ATPase.
Ainsi, les 3 déterminants positifs majeurs sont : la gastrine + l’acétylcholine +l’histamine Par ailleurs, tout ce qui augmente l’activité de la PKA et de la PKC régule positivement la sécrétion d’acide par la cellule pariétale et inversement. Récepteurs inhibiteurs :
Récepteur à la somatostatine : couplage négatif à l’adénylate cyclase via Gi Récepteur des prostaglandines : même voie d’inhibition de la H/K-ATPase
Toutes les voies de signalisations ci-dessus sont décrites dans le schéma suivant.

10
F. Évolution temporelle de la sécrétion gastrique acide et du pH luminal après un repas
A l’état basal, la sécrétion d’HCl est très faible. Après un repas, on observe une augmentation extrêmement précoce et ample de l’activité de sécrétion acide. Elle atteint un maximum entre 1-2h après le repas. Puis, il y a une diminution progressive d’acide pour revenir à l’état basal. En terme de pH, il se situe entre 1 et 2 à jeun. Après un repas, on observe une élévation modérée du pH, parce qu’un phénomène de tamponnement et de dilution des ions H+ se produit. Puis, il y a une diminution progressive du pH jusqu’à atteindre une valeur entre 1 et 2, qui s’explique par la production massive d’HCL. Il s’agit des valeurs les plus acides que l’on puisse observer dans les liquides biologiques.
G. La sécrétion d’acide gastrique
On distingue 3 phases pour la régulation de la sécrétion gastrique : la phase céphalique, la phase gastrique et la phase intestinale. L’addition des ces 3 phases rend compte de l’intégralité de la sécrétion d’acide (minimale au repos et maximale 1-2h après le repas). La contribution de la phase céphalique et intestinale est minoritaire. En effet, la majorité de l’acide est sécrétée pendant la phase gastrique.

11
i. La Phase Céphalique
Correspond à la sécrétion d’acide débutant avant même que les aliments aient été ingérés. Elle est déclenchée par la mastication, le goût, l’odorat, la vue ou bien la pensée.
Cette phase repose sur la libération d’Ach par le nerf vague. L’Ach produira un effet direct
sur les cellules pariétales, ainsi qu’un effet indirect sur les cellules G pyloriques (sécrétrices de gastrine) et les cellules entérochromaffines = ECL (sécrétrices d’histamine). Les 3 médiateurs positifs (Gastrine+ Histamine + Ach) stimulent la sécrétion d’HCl (voir schéma).
ii. La Phase Gastrique
Constitue l’essentiel de la sécrétion d’acide. Elle est déclenchée par la distension gastrique, qui active le nerf vague +++. Ainsi, l’Ach produira les mêmes effets que ceux décrits précédemment.
De plus, certains peptides renforceront la production de gastrine par les cellules G pyloriques. On observe également un début de rétrocontrôle par la somatostatine (fabriquée par les cellules D présentent en partie dans la région pylorique). Or, les cellules D sont sensibles à la diminution du pH. Ainsi, lorsque le pH diminue, les cellules D vont sécréter la somatostatine qui

12
inhibera indirectement les cellules G (gastrine), les cellules ECL (histamine) et directement les cellules pariétales (HCl). On notera que l’effet de la somatostatine est inférieur aux déterminants positifs, qui l’emportent.
iii. La Phase intestinale
Débute lorsque l’estomac commence à se vider dans le duodénum, qui se remplit. Dès lors, la distension mécanique du duodénum active des récepteurs neuronaux entériques (appartenant au SN autonome) et des cellules endocrines du duodénum afin de stimuler la sécrétion gastrique d’acide positivement ou négativement.
Lors de la diminution du pH dans le duodénum, la présence d’acides gras et d’un liquide
hyper osmotique constitue un déterminant négatif de la sécrétion d’acide. Les voies inhibitrices d’acide sont neuronales et endocrines. Dans le dernier cas, elles mettent en jeu des entérogastrones (hormones provenant de l’intestin et agissant sur l’estomac) comme la sécrétine, la bulbogastrone, CCK (cholécystokinine) et GIP (peptide inhibiteur gastrique). Ainsi, la sécrétion d’acide par les cellules pariétales et les cellules G sera inhibée.
Au fur et à mesure, que l’on s’éloigne du repas, les déterminants négatifs de la sécrétion
acide prennent le pas sur les positifs afin de retourner à la situation de repos.

13
H. Les autres sécrétions gastriques
i. Sécrétion du facteur intrinsèque
La deuxième fonction de la cellule pariétale est la production du facteur intrinsèque (glycoprotéine de 55 kDa), essentielle à l’absorption de la vitamine B12. Les déterminants de cette sécrétion sont les mêmes que ceux de la sécrétion acide. En effet, elle n’est pas permanente mais phasique puisqu’elle suit les périodes d’alimentation et de jeûne. La sécrétion du facteur intrinsèque est l’unique fonction gastrique indispensable à la vie.
Lors d’une gastrectomie, on conserve l’absorption de la vitamine B12 par des injections intramusculaires at vitam.
ii. Sécrétion du pepsinogène
Les cellules principales synthétisent et sécrètent des pepsinogènes. Ceux sont des pro-
enzymes activées si le pH ≤ 3 : les pepsinogènes deviennent des pepsines. Un sujet normal dispose de ce pH acide en situation postprandiale, mais on n’observera
plus ces conditions de pH dès qu’un traitement inhibera la H/K-ATPase. L’Oméprasole (chef de file des antagonistes de la H/K-ATPase) diminue la sécrétion d’acide. Or, le blocage des ions H+ dans la cellule pariétale modifie le pH gastrique. Donc, l’activation des pepsinogènes n’aura plus lieu et les protéines ne seront pas digérées dans l’estomac. Ceci n’a pas d’importance en terme d’efficacité globale de la digestion. Les protéines seront digérées dans l’intestin grêle grâce aux sécrétions pancréatiques amplement suffisantes. L’ensemble de la digestion est ainsi déporté dans l’intestin grêle tandis qu’en temps normal 15 à 20% de protéines sont digérées dans l’estomac. On distingue 2 groupes de cellules principales : (qui ne sont pas à retenir)
Groupe I : sécrétion de pepsinogènes par la muqueuse oxyntique Groupe II : sécrétion de pepsinogènes par l’ensemble de l ’estomac et par les glandes de
Brunner duodénales En ce qui concerne les déterminants de la sécrétion de pepsinogènes :
- Les cellules principales répondent à une stimulation beta adrénergique, à la sécrétine et au VIP en induisant une exocytose des pepsinogènes AMPc-dépendante.
- Elles répondent à une stimulation par l’Ach, la CCK et la Gastrine en activant une exocytose Ca++-dépendante
iii. Sécrétion de mucus
Le mucus est fabriqué par les cellules muqueuses, il a un rôle protecteur sur l’épithélium gastrique et l’intégrité de la paroi face à un environnement chimique et mécanique agressif.
Il est composé de glycoprotéines de haut poids moléculaire (500kDa), dont les chaines protéiques, largement recouvertes d’hydrates de carbone (85%), les protègent de la dégradation protéolytique. Ces monomères de glycoprotéines se tétramérisent grâce à des liaisons disulfures (2 millions de Da) afin de rendre le mucus visqueux pour former un gel. Au niveau de la partie non glycosylée de ces tétramères, c’est-à-dire non protégée par les hydrates de carbones, les liaisons vont être digérées par les pepsines (protéases). Les monomères vont donc être libérés. C’est pourquoi, il doit y avoir une production permanente de mucus, pour compenser ce qui est détruit par les pepsines. La concentration des glycoprotéines du mucus est élevée et de 50g/L.

14
Le liquide dans lequel baigne le mucus contient aussi des alcalins, puisque les cellules muqueuses de surface sécrètent du bicarbonate. Ainsi, cette couche protectrice visqueuse et alcaline d’1mm d’épaisseur est très adaptée à l’environnement gastrique et protège efficacement la paroi gastrique.
De plus, le mucus est aussi en contact avec un liquide extrêmement acide, le bicarbonate va donc être consommé par l’acide présent dans la lumière de l’estomac. Pour maintenir la couche alcaline il faut donc sécréter du bicarbonate en permanence. La sécrétion de mucus est :
- stimulée par : distension mécanique + prostaglandines + SN et P - inhibée par : Anti Inflammatoires Non Stéroïdiens
Or, les AINS abolissent les défenses locales de l’estomac en diminuant la production des prostaglandines. D’où la vulnérabilité de l’estomac face aux agressions chimiques.
I. Régulation de la satiété L’estomac est une glande endocrine capable de sécréter des hormones comme la grhéline.
La prise alimentaire est dépendante de la perception de satiété, qui est sous contrôle nerveux et endocrine. Or, quand la satiété est atteinte et que la prise alimentaire s’interrompe, les déterminants de la satiété vont progressivement diminuer et parallèlement le principal déterminant du déclenchement de la prise alimentaire (la ghréline) va augmenter.
La ghréline est produite lorsque l’estomac est vide depuis longtemps et sa sécrétion s’arrête lorsque l’estomac se remplit. Cette hormone agit sur l’Hypothalamus et déclenche la sécrétion de neuromédiateurs comme NPY (neuropeptide Y), AGRP, ainsi que la sécrétion de GHRH (hormone hypothalamique entraînant la sécrétion d’hormone de croissance GH par l’hypophyse). Il y aura une augmentation d’hormones de croissance parallèle à l’augmentation de ghréline en période de jeun.
Ces hormones vont augmenter la sensation de faim, donc déclencher la prise alimentaire,
augmenter le poids (en cas de prise alimentaire non contrôlée), de diminuer l’utilisation des lipides et d’aboutir au développement du tissu adipeux. En situation normale, quand la quantité d’hormones appropriée est sécrétée, on peut maintenir un poids stable.

15
II. Sécrétions pancréatiques Le pancréas est un organe endocrine (insuline et glucagon) et exocrine mais on ne parlera dans ce cours que du pancréas exocrine
A. Fonction exocrine du pancréas
Le pancréas est une petite glande exocrine de 100g chez l’adulte. Ses fonctions exocrines sont essentielles à l’absorption et la digestion des aliments. Il est composé de lobules, dans lesquels se trouvent des cellules épithéliales branchées sur une circulation ramifiée et confluente avec des canalicules intra-lobulaires. Ils deviendront ensuite extra-lobulaires, puis formeront les 2 canaux pancréatiques principaux.
Les sécrétions pancréatiques sont abondantes : au moins 1L/j (elles peuvent atteindre 2L/j chez certaines personnes).
La sécrétion exocrine est un mélange d’eau, d’électrolytes et d’enzymes sécrétées le plus
souvent sous forme de pro-enzymes. Un pancréas ne fonctionne pas à son activité maximale en permanence. Son activité est
contrôlée : elle augmente en période postprandiale et se calme en période de jeûne. Deux hormones jouent un rôle majeur et non exclusif : la sécrétine et la CCK. La sécrétine augmente la phase électrolytique de la sécrétion, tandis que la CCK augmente la phase enzymatique de la sécrétion pancréatique.
B. Le liquide pancréatique
i. Composition du liquide pancréatique excrété
En période de jeun : liquide sécrété à faible débit, riche en sodium et en chlore. Il a une composition proche du liquide extracellulaire. En période postprandiale : liquide alcalin (bicarbonate de sodium), sécrété à débit élevé. Il sera sécrété dans le duodénum pour neutraliser l’acide sortant de l’estomac et pour ramener des conditions de pH compatibles avec l’activité des enzymes pancréatiques.
ii. La composante hydro-électrolytique
Le liquide pancréatique provient des cellules épithéliales en contact direct avec les canaux pancréatiques. Il est proche du liquide extracellulaire en période de jeûne.
En période postprandiale, une activité de transport ionique apparaît. Les cellules épithéliales possèdent un échangeur chlore/bicarbonate et un canal chlore CFTR (mucoviscidose). Ainsi, les cellules activées sécrètent du bicarbonate et absorbent du chlore, qui fera une navette grâce au canal CFTR. La grande quantité de bicarbonates augmentera le pH du liquide pancréatique.
La sécrétion de bicarbonates dépend de la sécrétine (AMPC-dépendante), qui circulera à haute concentration en situation postprandiale.

16
iii. La composante enzymatique
Les enzymes sont produites par les cellules acinaires, qui les stockent dans des granules intracellulaires. Leur sécrétion s’obtient par la fusion de la membrane des vésicules avec la membrane plasmique apicale. L’activité de sécrétion enzymatique est contrôlée par des déterminants, qui utilisent soit la voie de l’AMPcyclique, soit la voie du calcium cytosolique en aval de la Phospholipase C : - VIP et sécrétine agissent via la voie de l’AMPcyclique - GRP, Ach (récepteur M3) et CCK (via le récepteur CCK-1) agissent par la voie calcium
dépendante Les protéases sont nombreuses et fabriquées sous forme de proenzymes inactives pour éviter que le pancréas ne se digère lui même. Parmi lesquelles, on retrouve : le Trypsinogène (1/3 des enzymes), le Chymotrypsinogène , les Procarboxypeptidases, les Proélastase et les Ribonucléases Ces pro-enzymes sont activées lorsqu’elles entrent en contact avec les aliments dans le duodénum. L’Entéropeptidase (aussi appelée entérokinase), produite uniquement dans le duodénum, est capable d’activer les protéases. Par exemple, de transformer le trypsinogène en trypsine. La trypsine est quant à elle capable de s’auto activer mais aussi d’activer les autres pro-enzymes. Il y a donc une compartimentalisation des fonctions qui préserve l’intégrité du pancréas. L’amylase est une enzyme glycolytique qui digère l’amidon. On compte 3 lipases principales : la Triacylglycérol hydrolase qui digère les tiglycérides, (c’est la plus abondante) la Cholestérol ester hydrolase qui digère les esters de cholestérol la Phospholipase A2 qui digère les phospholipides Ces 3 enzymes sont sécrétées en période postprandiale directement sous formes actives.
C. Régulation de la sécrétion pancréatique
iv. La phase céphalique Débute avant même que les aliments aient été ingérés. Représente 25 % de la sécrétion pancréatique totale. Ses initiateurs sont la vue, l’odeur et le goût. Son médiateur principal est le nerf vague. Durant cette phase, c’est principalement la sécrétion hydroélectrolytique qui augmente.
v. Phase gastrique Débute quand les aliments arrivent dans l’estomac. Représente 10 % de la sécrétion pancréatique totale. Elle est initiée par la distension gastrique. Ses médiateurs sont le nerf vague et le système nerveux entérique (par relais).
vi. Phase intestinale Débute quand le duodénum commence à se remplir avec ce qui provient de l’estomac. Représente l’essentiel de la sécrétion (65 %).

17
Ses initiateurs sont mécaniques avec la distension du duodénum ; et chimiques avec la baisse du pH duodénal (<4.5), la présence de peptides, d’acides aminés (phénylalanine, valine, méthionine), d’acides gras et de monoglycérides dans le duodénum. Les principaux médiateurs sont la sécrétine, la CCK et le nerf vague. Ces médiateurs déclencheront la sécrétion enzymatique des cellules acinaires du pancréas.
vii. Inhibition de la sécrétion pancréatique Les initiateurs de l’inhibition sont l’élévation de la glycémie et de la concentration sanguine d’acides aminés. En effet, une augmentation de la concentration des acides aminés dans le sang montrent que la digestion a été efficace. Ainsi, on peut progressive arrêter la digestion. Les 2 médiateurs de l’inhibition sont le glucagon et la somatostatine.
III. Sécrétions biliaires
La sécrétion de la bile est une des nombreuses fonctions du foie. C’est la seule qui nous
intéresse ici pour la digestion. Les hépatocytes sont organisés à l’intérieur de lobules. Chaque hépatocyte est une cellule
épithéliale polarisée en contact avec la circulation sanguine (veine porte) et les voies biliaires. On observe des transports du compartiment sanguin vers le compartiment biliaire et peu dans l’autre sens.
Les voies biliaires confluent pour se réunir dans une voie biliaire principale. Cette dernière trouvera un appendice de stockage, la vésicule biliaire, et s’abouchera dans le duodénum au même endroit que les canaux pancréatiques. La communication entre les voies biliaires, pancréatiques et le duodénum est permise par le sphincter d’Oddi.
A. La bile et les acides biliaires La bile est un liquide hétérogène essentiellement composé d’acides biliaires (68%) quand on regarde son poids sec (on retire l’eau). Il contient aussi un peu de phospholipides (23%), des protéines (5%), du cholestérol (4%) et de la bilirubine (1%). La couleur jaune verdâtre caractéristique de la bile provient de la bilirubine. La phase aqueuse est constituée d’électrolytes, dont la composition est proche de celle du plasma (riche en sodium et chlore, une concentration modérée en bicarbonates et faible en potassium). Cette bile primitive s’écoule dans les voies biliaires et la composition en termes d’eau et d’électrolytes sera modifié en situation postprandiale. Comme pour le liquide pancréatique, on observera un enrichissement en alcalins avant sa sécrétion. La Sécrétine favorise cet enrichissement en bicarbonates. Les acides biliaires dérivent du cholestérol. 1 Les hépatocytes transforment le cholestérol en 2 acides biliaires primaires (acide cholique et
l’acide chénodéoxycholique). Ils seront dans la bile primitive qui arrive dans l’intestin grêle. 2 Dans le l’intestin grêle, les bactéries modifient la structure des acides biliaires primaires en
retirant des radicaux hydroxyles. Ils deviennent alors des acides biliaires secondaires (acide deoxycholique pour l’acide cholique, acide ursodoxycholique ou acide lithocholique pour l’acide chénodéoxycholique ). Les noms ne sont pas à retenir. Les acides biliaires secondaires ne peuvent pas être directement produits par les hépatocytes.

18
3 Par nature, les acides biliaires sont liposolubles. C’est pourquoi, ils sont conjugués à des acides aminés, comme la glycine et la taurine, pour les rendre plus hydrophiles. Cette conjugaison est aussi le rôle des hépatocytes, alors que les bactéries intestinales favorisent la dé-conjugaison. Ces molécules s’organisent alors en structures micellaires de manière à présenter, à l’extérieur leur face hydrophile et à l’intérieur leur face lipophile. Les micelles permettent aux lipides digérés d’être maintenus en suspension dans un environnent aqueux et joue un rôle essentiel dans l’élimination et le stockage du cholestérol.
Cette organisation en micelles nécessite que la concentration en acides biliaires soit supérieur à la concentration micellaire critique. C’est toujours le cas en situation normale.
B. Cycle entéro-hépatique des acides biliaires
Les hépatocytes synthétisent et conjuguent les acides biliaires primaires. Ils sont sécrétés dans les voies biliaires. Entre les repas, ils peuvent être stocké dans la vésicule biliaire. Lorsqu’ils arrivent dans le duodénum, ils permettent la digestion et l’absorption des graisses. Dans l’iléon terminal tous les processus de digestion ont dû avoir lieu mais ils restent toujours les acides biliaires. Quand ils ne sont plus utiles, ils vont être réabsorbés pour éviter de les gâcher, par des transporteurs selon une absorption active ou une diffusion passive. - Transport actif : acides biliaires primaires conjugués - Transport passif : acides biliaires secondaires et dé-conjugués.
Ces molécules entrent dans la circulation porte, ils « retournent à la maison ». Les acides biliaires retournent au foie par le pôle sanguin et sont récupérés par hépatocytes quelque soit leur état. Ils seront re-conjugués et re-hydroxylés si besoin.
Autrement dit, il existe un cycle entéro-hépatique des acides biliaires efficace. A l’âge adulte, on a besoin de sécréter 5g acide biliaire lors d’un repas normal. Donc on a besoin de 15g tous les jours (3 repas). Or, nous ne produisons de novo que 0,5g/jour, le reste provient du recyclage des acides biliaires. Ces 0,5g n’ont pas été réabsorbé par l’iléon car l’absorption ne peut pas être totale. Ainsi, la suppression de l’iléon ou la prise de médicaments empêchant la réabsorption des acides biliaires, imposera une augmentation de la novo synthèse d’acide biliaire par les hépatocytes. Ils sont tout à fait capables d’en fabriquer 15g de novo/jour sans problèmes.

19
C. Régulation des sécrétions biliaires
Ce phénomène de recyclage et de transports des acides biliaires repose sur l’expression de transporteurs spécifiques à la membrane des hépatocytes. Le long des canaux biliaires, comme le liquide pancréatique, le liquide biliaire peut devenir alcalin s’il y a une sécrétion de bicarbonates par les cellules épithéliales. Cette sécrétion de bicarbonates dépend de l’échangeur chlore/bicarbonate qui fonctionne en parallèle avec 2 canaux chlore dont un CFTR. Ainsi, les défauts de CFTR sont bien plus marqués sur le fonctionnement pancréatique que le biliaire. L’insuffisance pancréatique est une des complications évolutives de la mucoviscidose tandis que l’insuffisance biliaire ne l’est pas.
La vésicule biliaire est une zone de stockage appendue à la voie biliaire, dont les 3 principaux déterminants sont : - L’activation du nerf vague (Ach) - La CCK - La sécrétine
La contraction de la vésicule biliaire se fait via la CCK et l’Ach. A la suite d’un repas, le SNP est activé et les cellules le duodénum sécrètent de la CCK et de
la sécrétine. La relaxation du sphincter Oddi doit se relâcher pour que les sécrétions biliaires et pancréatiques atteignent la lumière du duodénum. Sa relaxation dépend de fibres P non cholinergique utilisant comme médiateurs le mononoxyde d’azote NO et le VIP.
Voici un tableau récapitulatif de la sécrétion biliaire :

20
i. Les 3 phases de la sécrétion Comme la sécrétion pancréatique, la sécrétion biliaire peut être divisée en 3 phases. 1- La phase céphalique est déclenchée par l’odeur et le goût. Son principal médiateur est le nerf vague, qui déclenche la vidange vésiculaire 2- La phase gastrique, déclenchée par la distension gastrique, renforce l’action du nerf vague et poursuit la vidange vésiculaire. 3- Pour la phase intestinale, la distension du duodénum stimule le nerf vague. La présence de lipides dans le duodénum induit la sécrétion de CCK, qui déclenche la sécrétion des acides biliaires et termine la vidange vésiculaire. En réponse à la baisse du pH duodénal, la sécrétine entraîne la sécrétion d’un liquide alcalin par les cellules canalaires des voies biliaires. Tous ces phénomènes permettent d’obtenir un liquide alcalin et d’augmenter rapidement la concentration en acides biliaires dans le duodénum. Dès lors, les actions de digestion et d’absorption des lipides peuvent s’établir. Dans l’iléon, les acides biliaires seront majoritairement réabsorbés. La concentration élevée dans le sang porte des acides biliaires va être détecté par les hépatocytes. Ceci renforcera la sécrétion des acides biliaires par les hépatocytes alors qu’en même temps leur synthèse serra diminuée. Un hépatocyte ne sécrète pas en même temps qu’il fait de la synthèse. En fait, il synthétise pendant les périodes inter-prandiales. Ainsi, au fur et à mesure que la digestion se termine : le pH du duodénum s’élève, il y a de moins en moins de lipides et on a de moins en moins besoin des acides biliaires. On revient en période inter-digestive avec un faible débit de bile, une faible concentration d’acides biliaires dans le sang porte. C’est à ce moment que la synthèse des acides biliaires augmente, tandis que la sécrétion biliaire est minimale.
ii. La bilirubine : un pigment biliaire
C’est la bilirubine qui colore la bile et non les acides biliaires. Elle est considérée comme un métabolite qu’il est nécessaire d’éliminer, parce qu’elle est produite suite à la dégradation des hématies. On dégrade environ 1% de nos hématies tous les jours. Cette dégradation libère 6g d’Hémoglobine par jour qui sera métabolisé en bilirubine. Afin de la rendre soluble et de pouvoir l’éliminer à forte concentration dans un volume réduit, la bilirubine doit être conjuguée par les hépatocytes. En effet, la bilirubine libre circulant dans le sang à faible concentration est peu soluble. La glycuronyl transférase est l’enzyme transformant la bilirubine libre en conjuguée. Sécrétée dans la bile, la bilirubine conjuguée sera éliminée dans les selles (principalement) ou l’urine (si elle est réabsorbée par l’épithélium intestinal). La capacité à conjuguer la bilirubine est absolument essentielle pour son élimination. Il existe 2 maladies au cours desquelles cette conjugaison est défaillante : - La maladie de Gilbert est une maladie autosomique dominante. On observe un déficit
monoallélique de la Glycuronyl transférase. Donc, ces personnes ont une activité glycuronyl transférase moindre, qui leur permet toutefois d’éliminer correctement la bilirubine. C’est pourquoi la maladie de Gilbert est plus considérée comme une curiosité puisqu’il n’y a pas vraiment de conséquences majeures. Face à la fatigue et un rhume, ils deviennent vaguement ictériques (blanc des yeux jaune par exemple).
- La forme biallélique de cette perte de fonction est tragique. Les nouveaux nés n’ont aucune possibilité de conjuguer la bilirubine et s’intoxiquent avec la bilirubine libre. Sa toxicité neuronale les rend infirmes.

21
D. Lithiase biliaire
La lithiase biliaire est une maladie fréquente se traduisant par des calculs dans les voies biliaires. En effet, 10-20% des adultes ont des calculs principalement dans la vésicule biliaire et sont asymptomatiques. Seul dans 2-4% des cas, il y a une symptomatologie associée qu’il faut traiter.
Dans 85% des cas, il s’agit de calculs de cholestérol. Ils sont radiotransparents donc on ne les voit pas. Ils se développent sous l’influence d’une concentration élevée de cholestérol dans la bile ( qui est un bon moyen pour l’éliminer). Or, le risque est que la concentration en cholestérol soit supérieure à sa solubilité dans la bile. Toutes les nuits, on se retrouve en situation de sursaturation de cholestérol dans la bile. Donc on a tous la possibilité de développer des calculs. S’ils s’agglomèrent suffisamment, ils entraîneront des calculs.
Dans 15% des cas, ceux sont des calculs de bilirubinate de calcium. Ils sont radio-opaques et s’expliquent par l’insolubilité de la bilirubine libre.
IV. Points-clé Traduction de 2 diapos en anglais données par le professeur.
• Les principales fonctions de l’estomac sont le stockage et l’initiation de la digestion des protéines
• La régulation des fonctions gastriques dépend des voies nerveuses intrinsèques et extrinsèques, ainsi que de facteurs humoraux (gastrine) et paracrines (histamine)
• Les sécrétions clé de l’estomac sont les sécrétions d’acide et de pepsinogènes, qui permettent de commencer la digestion des protéines
• Les ions H+ sont sécrétés à travers la membrane plasmique apicale des cellules pariétales via la H/K-ATPase, une pompe à protons
• La seule sécrétion gastrique nécessaire, est la sécrétion du facteur intrinsèque impliquée dans l’absorption de la vitamine B12.
• L’épithélium gastrique sécrète des bicarbonates HCO3- et du mucus pour former un gel, constituant une barrière protectrice contre les agressions acide et chimique.
• Les principales fonctions exocrines du pancréas sont les sécrétions alcalines et enzymatiques
• La régulation de la fonction pancréatique exocrine dépend de voies nerveuses intrinsèques et extrinsèques, ainsi que de médiateurs locaux (CCK et sécrétine)
• La fonction clé du foie vis à vis de la sécrétion digestive est la sécrétion des sels biliaires • La régulation de la sécrétion des sels biliaires dépend de facteurs neuronaux et de facteurs
humoraux (CCK et sécrétine) Voici les QCM vus en fin de cours
La sécrétion acide de l’estomac est assurée par A- les cellules principales B- les cellules pariétales C- les cellules G sécrétant la gastrine D- les cellules D E- les cellules à mucus La sécrétion acide de l’estomac est activée par A- la gastrine

22
B- l’acétylcholine C- l’histamine D- la somatostatine E- la cholecystokinine (CCK) Le facteur intrinsèque A- est sécrété par les cellules épithéliales du jéjunum B- est une glycoprotéine C- est nécessaire à l’absorption de la vitamine B12 D- permet la digestion de l’amidon E- permet la digestion des triglycérides La sécrétion pancréatique exocrine A- augmente après un repas B- est riche en trypsinogène C- est riche en lactase D- est activée par la cholécystokinine E- est activée par la somatostatine Les acides biliaires A- sont des métabolites du cholestérol B- constituent une part minoritaire de la bile C- sont nécessaires à la digestion des molécules liposolubles D- sont sécrétés à débit constant au cours de la journée E- colorent la bile Réponses : Q1-B Q2-ABC Q3-BC Q4-ABD Q5-AC

23
Fiche récapitulative : Sécrétions digestives I)Sécrétions gastriques - L’estomac est composé de 3 régions : cardiale (<5%), oxyntique (75%) et pylorique (20-25%).
- Les sécrétions sont assurées par les glandes gastriques qui sont des invaginations de l’épithélium gastrique. On observe une grande hétérogénéité cellulaire avec de la surface à la profondeur : les c. épithéliales de surface ($ bicarbonates de sodium), les c. muqueuses du collet, les c. souches, les c. pariétales ($ HCl), les c. principales et les c. endocrines.
- La composition du liq. gastrique varie selon les situations. En période de jeun, la sécrétion est à petit débit et la composition du liq. gastrique est proche de celle du liq. extracellulaire, alors qu’en situation postprandiale le débit de sécrétion est augmenté avec une élévation de la concentration en Cl- et H+ (acide chlorhydrique ++).
- Cette sécrétion d’acide gastrique est régulée et on distingue 3 phases : 1) phase céphalique : avant que les aliments aient été ingérés. Le nerf vague stimule la libération d’ACh. L’ACh stimule à son tour la sécrétion d’HCl : directement via les c. pariétales et indirectement via les c. G pyloriques (sécrétion de gastrine) et les c. entérochromaffines (=ECL) (sécrétion d’histamine). 2) phase gastrique (majorité de la sécrétion) : lorsque les aliments sont ingérés : la distension gastrique active le nerf vague (++). L’ACh produit les mêmes effets que pour la phase céphalique. Début de rétrocontrôle par la somatostatine. 3) phase intestinale : lorsque l’estomac se vide dans le duodénum. La présence d’acides gras et d’un liquide hyper osmotique dans le duodénum est un déterminant négatif de la sécrétion d’acide. Les voies inhibitrices d’acide sont neuronales et endocrines (entérogastrone).
- c. pariétale : quiescente à jeun avec une membrane apicale de surface réduite. Sécrétante en post-prandiale avec enrichissement en mitochondries et fusion des structures membranaires avec la mb apicale. 1ère fonction est de sécréter du HCl, elle possède des pompes H/K-ATPase à sa mb apicale. Rcpt activateurs de la sécrétion d’acide : CCKB, M3, H2 / Rcpt inhibiteurs : rcpt à la somatostatine et à la prostaglandine. 2ème fonction est la production du facteur intrinsèque qui est indispensable pour l’absorption de la vitamine B12.
- c. principale : sécrète des pepsinogènes = pro-enzymes activées si le pH ≤ 3
- c. muqueuse : production de mucus à rôle protecteur de l’épithélium gastrique. Le mucus est composé de glycoprotéines de haut poids moléculaire qui se tétramérisent pour donner la viscosité du mucus et forme une couche protectrice d’1mm d’épaisseur. Cette sécrétion est stimulée par la distension mécanique, les prostaglandines et le SN et P, inhibée par les AINS.
- la ghréline est une hormone qui augmente la sensation de faim. Elle est produite par l’estomac lorsqu’il est vide et agit sur l’hypothalamus. On note une augmentation parallèle d’hormones de croissance à celle de la ghréline en période de jeun. II) Sécrétions pancréatiques - les sécrétions pancréatiques sont abondantes (1L/j). La sécrétion d’électrolytes est augmentée par la sécrétine alors que celle d’enzymes est stimulée par la CCK.
- Le liquide pancréatique a une composition différente selon les situations. En période de jeun il a une composition proche du liquide cellulaire et est sécrété à petit débit alors qu’en période

24
postprandiale c’est un liquide alcalin sécrété à débit élevé. Il est sécrété dans le duodénum pour neutraliser l’acide sortant de l’estomac et permettre des conditions de pH compatibles avec l’activité des enzymes pancréatiques.
- En période postprandiale on a donc une activité de transport ionique qui apparaît : les c. épithéliales sécrètent du bicarbonate et absorbent du chlore grâce à un échangeur chlore/bicarbonate et un canal chlore CFTR.
- Les enz. pancréatiques sont produites par les c. acinaires. Elles sont stockées dans des granules intracellulaires, puis libérées lors de la fusion de la mb des vésicules avec la mb plasmique apicale. Les protéases sont nombreuses et produites sous forme de pro-enzymes inactives (trypsinogène, chymotrypsinogène,..). Elles sont activées lorsqu’elles entrent en contact avec les aliments dans le duodénum ou par l’entéropeptidase, uniquement produite dans le duodénum. L’amylase est une enzyme glycolitique qui digère l’amidon. Les lipases sont sécrétées en période postprandiale directement sous formes actives (triacylglycérol hydrolase, cholestérol ester hydrolase, phospholipase A2).
- La sécrétion pancréatique est régulée par 3 phases : 1) phase céphalique (25%) : Le médiateur principal est le nerf vague. Principalement la sécrétion hydro-électrolytique qui augmente. 2) phase gastrique (10%) : les médiateurs sont le nerf vague et le système nerveux entérique. 3) phase intestinale (65%) : initiateurs sont mécaniques (distension du duodéum) et chimiques (baisse du pH). Principaux médiateurs sont la sécrétine, la CCK et le nerf vague.
- La sécrétion pancréatique est inhibée par l’élévation de la glycémie et de la concentration sanguine d’acides aminés. III) Sécrétions biliaires - La bile est sécrétée par le foie. C’est un liq. hétérogène composé d’acides biliaires (68%), de phospholipides (23%), protéines (5%), de bilirubine (1%) et d’une phase aqueuse de composition proche de celle du plasma. De plus, lors de son passage dans les voies biliaires, la bile s’enrichit en bicarbonates (favorisé par la sécrétine). - Les hépatocytes transforment le cholestérol en 2 acides biliaires primaires (acide cholique et l’acide chénodéoxycholique). Dans l’intestin grêle, les bactéries transforment les acides biliaires primaires en secondaires. Les acides biliaires sont conjugués à des AA pour les rendre plus solubles. La conjugaison est assurée par les hépatocytes alors que les bactéries intestinales favorisent la dé-conjugaison. Ces molécules s’organisent alors en micelles. Les a. biliaires sont réabsorbés au niveau de l’iléon. - Au cours de la digestion, via le nerf vague pendant la phase céphalique et gastrique, la vésicule biliaire initie sa vidange, puis pendant la phase intestinale, la sécrétion de CCK induit la vidange de la vésicule biliaire dans le duodénum. - Les lithiases biliaires sont des maladies fréquentes, des calculs se forment dans la vésicule biliaire.

25
UE 6 – SD – Sémiologie - n°3
15/01/2018
Pr. Georgia Malamut [email protected]
RT : Amaury CINQUIN
RL : Leria PIETRI
Sémiologie et exploration de l’intestin grêle
Plan :
I. Rappels anatomo-physiologiques A. Rappels anatomiques B. Rappels histologiques C. Rappels physiologiques D. Pathologies et sémiologie de l’intestin grêle
II. La diarrhée
A. La diarrhée aiguë B. 5 grands mécanismes
i. Diarrhées osmotiques ii. Diarrhées sécrétoires iii. Diarrhées par trouble de la motricité intestinale iv. Diarrhées par malabsorption v. Diarrhées exsudatives
III. Le syndrome de malabsorption
A. Signes cliniques B. Signes biologiques C. Etiologies
i. Malabsorptions d’origine pariétale intestinale ii. Malabsorptions d’origine pancréatique
IV. Explorations de l’intestin grêle
A. Explorations biologiques B. Explorations fonctionnelles C. Explorations endoscopiques classiques D. Explorations morphologiques

26
i. Transit du grêle ii. Entéroscanner iii. Entéro-IRM
E. Explorations endoscopiques spécifiques i. Vidéocapsules endoscopiques ii. Entéroscopie
V. Hémorragies d’origine grêlique

27
I. Rappels anatomo-physiologiques A. Rappels anatomiques L’IG a une longueur de 4 à 7m. Il est composé de deux parties : le jéjunum qui représente les 2/5 de la longueur totale, et l’iléon, les 3/5 restants. La surface de contact avec le suc alimentaire est considérable car elle représente de 250 à 400 m2 si on dépliait totalement l’IG.
B. Rappels histologiques La cellule fonctionnelle principale de l’IG est l’entérocyte, une cellule épithéliale. L’IG est vascularisé par l’artère mésentérique supérieure. Ce qui donne à l’IG cette très grande surface, ce sont ses villosités intestinales qui permettent d’absorber les nutriments, et qui sont bordées d’un épithélium composé d’entérocytes. Ces villosités sont vascularisées par le système veineux, artériel et lymphatique.
C. Rappels physiologiques L’IG est le site le plus important d’absorption des nutriments, en particulier de l’eau et des électrolytes. Pour pouvoir être absorbés, les aliments doivent d’abord être digérés grâce aux sucs gastriques, à l’action mécanique et aux enzymes pancréatiques. Les nutriments sont ensuite au contact de la lumière de l’IG et sont absorbés au travers des entérocytes. Au niveau du duodénum et du jéjunum, se fait l’absorption du fer, du calcium, de vitamines liposolubles (A, D, E, et K), d’albumine… Au niveau de l’iléon se fait l’absorption de la vitamine B12 qui est couplée au facteur intrinsèque (sécrété dans l’estomac), et c’est également dans l’iléon qu’il y a l’absorption des sels biliaires.
D. Pathologies et sémiologie de l’intestin grêle Plusieurs éléments sémiologiques permettent de s’orienter vers une pathologie : - Le syndrome de malabsorption : se caractérise généralement par une diarrhée chronique, des carences et une altération de l’état général (AEG). Il y a plusieurs causes possibles : soit une atrophie villositaire (villosités intestinales toutes aplaties comme au

28
cours de la maladie cœliaque à cause d’une destruction des cellules épithéliales de façon auto-immune), une atteinte inflammatoire (ex : maladie de Crohn, en particulier au niveau de l’IG terminal, et en cas de cas ancien ou étendu au niveau de l’IG terminal, il y a fréquemment une malabsorption de la vitamine B12 et des sels biliaires) ou à un trouble moteur (ralentissement ou stagnation du péristaltisme qui favorise la pullulation microbienne entraînant un syndrome de malabsorption, surtout des graisses). - Le saignement chronique ou aigu : peut se manifester cliniquement par une anemie ( < 13g/dL chez la femme et < 12g/dL chez l’homme), et peut s’extérioriser par un méléna (sang foncé et malodorant) ou par des rectorragies (sang rouge). Il faudra alors évoquer une tumeur, une maladie inflammatoire chronique de l’intestin (MICI) ou une angiodysplasie (petits vaisseaux pouvant saigner d’autant plus si le patient prend des anticoagulants). - Obstruction (occlusion) : arre t du transit intestinal pouvant e tre cause par une tumeur, une sténose (rétrécissement inflammatoire (MICI, maladie de Crohn)), une bride (= bande de tissu conjonctif fibreux réunissant anormalement deux organes. Lorsque les patients ont déjà été opérés de l’abdomen, l’IG peut s’incarcérer au niveau de ces brides), ou encore la prise importante et chronique d’AINS (favorisent les ulcérations qui peuvent évoluer en sténose). - Inflammation (duodenite, jejunite, iléite) : peut e tre liee a une infection, une maladie inflammatoire (MICI) ou bien à une immunodépression.
II. La diarrhée Définition : Emissions quotidiennes trop fréquentes de selles trop abondantes, liquides ou très molles avec un poids > 300g/j. Définition de l’OMS : au moins 3 selles molles à liquides par jour. - Diarrhee aigue : < 2 semaines - Diarrhee prolongee : 2 a 4 semaines - Diarrhée chronique : > 4 semaines
A. La Diarrhée aiguë La diarrhee aigue a un debut soudain/brutal sur un transit normal et est inferieure a 15 jours (il y a des exceptions…). Elle ne récidive pas à court terme et régresse le plus souvent spontanément ou sous traitement symptomatique (notamment pour la gastroentérite virale : on traite les conséquences, la déshydratation principalement, et non la cause). L’étiologie principale est infectieuse ++, virale ou bactérienne. Dans les pays en développement, c’est une cause majeure de mortalité mais elle est beaucoup plus bénigne dans les pays industrialisés, grâce à de meilleurs moyens de prise en charge et de prévention. La diarrhée infectieuse se caractérise par des douleurs abdominales, des vomissements. Lorsqu’il y a de la fièvre et des saignements (rectorragies), il faut penser à une diarrhée causée par des germes entéro-invasifs qui vont créer des lésions au niveau de la muqueuse intestinale.
B. 5 grands mécanismes Lorsqu’un patient se pointe avec une diarrhée (en particulier chronique) et qu’on veut aborder le mécanisme, il faut évoquer :
i. La diarrhée osmotique :

29
Caractérisée par des selles abondantes, jaunâtres et mousseuses, elle est typiquement liée à une substance osmotiquement active (soit des laxatifs osmotiques – pour préparer par exemple un patient pour une coloscopie, on lui donne du PEG – soit certains bonbons), qui entraîne un appel d’eau dans la lumière intestinale (intraluminale). On peut, au cours du fécalogramme, voir qu’il y a une augmentation du « trou osmotique » : (osmolarité normale des selles = 290 mOsm) – (osmolarité du Na+ + celle du K+) Le trou osmotique devient alors > 125 mOsm (dans le cas d’une diarrhée osmotique). Les substances osmotiques font baisser l’osmolarité du Na+ et du K+ des selles.
ii. La diarrhée sécrétoire : Très aqueuse (ex : entérite cholérique), elle peut être liée à des laxatifs ou des prostaglandines. Elle augmente la sécrétion d’eau et d’électrolytes (Na+ et K+) par l’entérocyte. L’osmolarité des électrolytes augmente, donc pas de trou osmotique élevé ( < 50 mOsm en général).
iii. Les diarrhées par trouble de la motricité intestinale : Postprandiales et impérieuses, elles peuvent être secondaires soit à une hypermotricité avec une accélération du transit intestinal (comme au cours de l’hyperthyroïdie) soit au contraire à une inhibition de la motricité qui va ralentir le transit et ainsi favoriser une pullulation microbienne (qui peut dans un second temps favoriser une malabsorption).
iv. Les diarrhées par malabsorption : Deux causes principales : - en amont de l’IG : la maldigestion (défaut de sécrétion gastrique, biliaire, et pancréatique) ; - un défaut vrai d’absorption par l’entérocyte (comme au cours de la maladie cœliaque, ou d’une destruction inflammatoire dans la maladie de Crohn).
v. Les diarrhées exsudatives : Exsudation éventuellement de lymphe (exsudations des lymphangiectasies secondaires lorsqu’il y a un blocage sur le canal thoracique lymphatique => hyperpression dans les capillaires lymphatiques, donc les villosités vont perler de la lymphe – villosités blanches sur endoscopie) qui contient des protéines et des lymphocytes (surtout CD4). Dans le cas des lymphangiectasies primitives (on ne connait pas le mécanisme), c’est la maladie de Waldmann, il y a aussi une exsudation de lymphe qui perle des villosités intestinales. Dans les MICI, on a des ulcérations avec exsudation au travers de ces ulcérations avec pertes protéiques aussi. En conséquence des diarrhées exsudatives, on a une baisse de l’albuminémie, une perte de protéines dans le sérum, donc on peut voir des œdèmes (des membres inférieurs notamment), des épanchements pleuraux, de l’acide dans le ventre…

30
III. Le syndrome de malabsorption
A. Signes cliniques
● Diarrhée graisseuse (stéatorrhée) ou hydrique
● Douleurs abdominales inconstantes
● AEG : amaigrissement, anorexie (perte de l’appétit), asthénie (fatigue extrême)
● Œdèmes des membres inférieurs, anasarque (hypoalbuminémie)
● Anémie : carence en fer et folates (ex : maladie cœliaque car l’atrophie n’atteint pas
l’IG distal dans la plupart des cas, l’IG proximal - duodénum et jéjunum - est le plus
touché et c’est le site privilégié d’absorption du fer et des folates), ou en vitamine
B12 (ex : maladie de Crohn car elle touche plutôt l’iléon terminal, site privilégié
d’absorption de la VB12, et le côlon)
● Ostéoporose, ostéomalacie (carence en vitamine D, calcium)
● Tétanie (carence en vitamine D, calcium, magnésium)
● Hémorragie/hématome (carence en facteurs liposolubles A, D, E, et notamment
vitamine K et donc en facteurs de la coagulation)
● Lésions de la peau et des phanères (du fait de la carence vitaminique, Fer, Ca2+)

31
B. Signes biologiques
● Baisse de l’albumine et des protides
● Baisse du calcium, du phosphore et du magnésium,
● Augmentation des phosphatases alcalines osseuses liée à l’ostéomalacie
● Anémie micro ou macrocytaire si carence en fer (surtout), folates, vitamine B12, et
baisse de la ferritinémie
● Baisse des facteurs de coagulation vitamine K - dépendants
C. Etiologies Une maladie peut se présenter par un overlap de deux syndromes. Par exemple, la maladie de Crohn peut se manifester à la fois par un syndrome de malabsorption en raison de l’atteinte de l’IG, mais aussi par un syndrome exsudatif en raison des ulcérations et de la perte de protéines. i. Malabsorptions d’origine pariétale intestinale : C’est l’atteinte de l’entérocyte. On distingue : - la principale cause : les atrophies villositaires (maladie cœliaque) ; - les ulcérations (maladie de Crohn) ; - les parasitoses et viroses chroniques (ex : déficit immunitaire) ; - les résections chirurgicales étendues du grêle : syndrome du grêle court (on réduit la surface d’absorption des nutriments – auparavant lorsqu’il n’y avait pas encore de traitement pour la maladie de Crohn –, 1m d’IG n’est généralement pas suffisant pour être autonome, il faut un cathéter central pour la nutrition) ; - une pullulation bactérienne chronique du grêle par malabsorption des graisses ; - une insuffisance artérielle mésentérique chronique.
ii. Malabsorptions d’origine pancréatique (maldigestion) : Elles sont liées à une insuffisance pancréatique exocrine (défaut d’enzymes pancréatiques), il y a un problème avec la fonction exocrine pancréatique comme au cours de la pancréatite chronique. C’est beaucoup moins problématique que les malabsorptions d’origine pariétale intestinale puisqu’on peut aisément donner des extraits pancréatiques pour remplacer les enzymes déficientes.
IV. Exploration de l’intestin grêle
A. Biologie
● Bilan sanguin : NFS, plaquettes, taux de prothrombine (TP)
● Ionogramme, créatinine, taux de CRP (protéine C réactive)
● Fer, ferritine, folates, vitamine B12, vitamine B1, B6
● Ca2+, phosphore, vitamine D, Mg2+, albumine
● Phosphatases alcalines osseuses (sont-elles augmentées ?)

32
● Anticorps anti-transglutaminase (en première intention si on recherche une
maladie cœliaque), dosage des immunoglobulines (pour vérifier qu’il n’y ait pas de
déficit immunitaire)
B. Explorations fonctionnelles
● Le fécalogramme étudie la malabsorption. On recueille sur trois jours les selles (et
uniquement les selles, si en plus on recueille l’urine, les résultats sont totalement
faussés) des 24h du patient et on les pèse chaque jour. Si les selles ont une masse
de graisses supérieure à 7g/j, on parle de stéatorrhée, càd une quantité trop
importante de graisses dans les selles due à une MALABSORPTION des graisses.
● Le test au D-Xylose pour explorer le grêle proximal en faisant ingérer une dose
initiale de 25g d’un xylose (un sucre…) marqué qui n’est pas métabolisé et est
excrété dans les urines afin de voir s’il y a un défaut d’excrétion. Deux heures après
l’ingestion, on mesure le taux sanguin de D-Xylose (normale > 1,66 mmol/L), et on
mesure le taux urinaire pendant 5 heures après l’ingestion.
● La clairance de l’alpha1-antitrypsine : ça nécessite d’une part le recueil de selles,
mais il ne faut pas oublier la prise de sang le dernier jour sinon il n’y aura pas de
clairance. Elle permet de voir s’il y a une exsudation protéique (entéropathies
exsudatives). L’alpha1-antitrypsine est une protéine plasmatique très peu
éliminée par voie digestive, donc s’il y en a beaucoup, ça veut dire qu’il y a une
exsudation des protéines du plasma liée à une lymphangiectasie ou à une
ulcération. Donc la clairance est très élevée dans les lymphangiectasies primitives
(maladie de Waldmann) ou secondaires. La perte fécale est proportionnelle à
l’exsudation plasmatique. La normale est < 20 mL/24h.
● Le test respiratoire à l’hydrogène pour voir s’il y a une pullulation microbienne :
on fait ingérer 50g de glucose pendant 4h et on mesure le dihydrogène expiré. Si
celui-ci est augmenté, alors cela indique que le glucose est d’avantage transformé
et donc que ça pullule (ex : colonisations bactériennes chroniques).
● Le test au rouge carmin permet d’étudier la durée du transit intestinal : on fait
avaler des pastilles rouge carmin, et on note le temps à partir duquel elles sont
évacuées dans les selles. Si ce temps est < 6h, alors on a une accélération du transit.
On peut également utiliser des marqueurs radio-opaques, mais c’est plutôt pour
les études de constipation.
C. Explorations endoscopiques classiques Les explorations fonctionnelles se faisaient auparavant beaucoup plus en première intention. Mais aujourd’hui, dans un bilan de diarrhée chronique, on a davantage recours aux endoscopies. On fait des tests fonctionnels plutôt en seconde intention si on a des doutes diagnostiques. Les examens endoscopiques classiques sont :
● La fibroscopie œsogastroduodénale, sans oublier les biopsies duodénales qui nous
renseignent sur l’état de la muqueuse : juste après l’estomac c’est le bulbe, le D1

33
puis le D2 et c’est là (au niveau du D2) qu’il faut faire des biopsies pour voir s’il n’y
a pas d’atrophies villositaires, afin de faire le diagnostic d’une maladie cœliaque.
On peut également faire des biopsies dans l’estomac, notamment dans le fundus
pour éliminer l’hypothèse de la maladie de Biermer. Cette dernière correspond à
une atteinte auto-immune des cellules pariétales gastriques qui ne vont plus
sécréter de facteur intrinsèque (normalement chargé de l’absorption de la
vitamine B12), donc on a une malabsorption de la vitamine B12 au niveau de l’iléon
terminal.
● Au cours de la même anesthésie générale, on peut réaliser une iléocoloscopie où
l’on observe tout le cadre colique jusqu’à la jonction avec l’IG. On fait des biopsies
iléales et coliques étagées pour éliminer l’hypothèse d’une colite microscopique
qu’on détecte sur les biopsies. Une colite microscopique lymphocytaire au
collagène est pourvoyeuse d’une diarrhée sécrétoire. Donc on peut avoir des
diarrhées avec grande hypokaliémie puisqu’il y a une forte sécrétion de potassium
au moment des poussées. Il faut aller voir jusque dans l’IG terminal (franchir la
valvule iléo-caecale) pour voir s’il n’y a pas de maladies de Crohn purement
grêliques.
Sur la photo où il y a la maladie cœliaque avec l’atrophie villositaire, on remarque une infiltration par des lymphocytes intra-épithéliaux au niveau des entérocytes détruits. Lorsque le traitement repose sur un régime sans gluten, les villosités repoussent totalement au bout d’1 à 2 années. En effet, c’est le gluten qui déclenche la réaction auto-immune et la destruction des entérocytes.
IG normal
avec de
belles
villosités
Aspect macroscopique de
fibroscopie œsogastroduodénale :
aspect fissuraire pavimenteux
d’atrophie villositaire, effacement
des plis duodénaux normalement
réguliers.
Atrophie
villositaire, la
muqueuse est
plate, mais
hyperplasie
des cryptes
compensatric
es

34
D. Explorations morphologiques Elles sont réalisées grâce à des examens d’imagerie. i. Le transit du grêle : Cet examen a une faible rentabilité diagnostique (0-10%) donc on ne le fait plus. De plus, il est le plus irradiant et est peu informatif. ii. L’entéroscanner : Moins irradiant et bien plus pratiqué que le précédent, l’entéroscanner permet de diagnostiquer des maladies de Crohn, des sténoses, des occlusions, des complications (abcès...). Il permet aussi de localiser les polypes et les tumeurs (ainsi que de mesurer leur extension). Cet examen reste quand même très irradiant, il ne faut donc pas le répéter, d’autant plus qu’il est inconfortable. iii. L’entéro-IRM : Chez les patients qui ont une maladie de Crohn et qu’on doit surveiller régulièrement par une imagerie de l’IG, on pratique plutôt l’entéro-IRM car il n’y a pas d’irradiation. Il permet de voir l’extension des lésions et donc de détecter la maladie de Crohn, une sténose inflammatoire ou fibreuse, voire une tumeur ou un polype.
E. Explorations endoscopiques spécifiques Même si on peut voir le duodénum lors d’une fibroscopie œsogastroduodénale, ou encore l’iléon terminal lors d’une iléocoloscopie, la majorité de l’IG n’est pas explorée par ces examens. Il y a donc eu le développement de vidéocapsules endoscopiques et d’entéroscopies. i. Les vidéocapsules endoscopiques : C’est comme un gros comprimé qu’on avale, sauf qu’il comporte une caméra. Le patient porte un boîtier avec des électrodes, et il va recueillir les images émises par la capsule. Cette dernière parcourt tout le transit, et sa circulation est favorisée par la mobilité du patient. La capsule est évacuée dans les selles. Cette technique nécessite une préparation

35
moins lourde que pour la coloscopie : il faut boire un litre et demi de PEG. On regarde ensuite le film de la capsule sur ordinateur et on peut voir des trucs comme :
Quelques petites images pour ceux qui ont un petit creux… Remarque : Une angiodysplasie est une petite lésion vasculaire qui est fréquente notamment chez les patients qui sont sous dialyse, insuffisants rénaux ou sous anticoagulants. On peut les coaguler au cours d’une entéroscopie (cf ii.) avec du plasma argon.

36
ii. L’entéroscopie : Une fois qu’on a dépisté la lésion en vidéocapsule, on sait en fonction du temps auquel l’image a été prise, à peu près à quel niveau la lésion se situe. On peut ensuite aller rechercher la lésion à l’aide d’un tube beaucoup plus long que le fibroscope au cours d’une entéroscopie digestive haute (si on passe par voie haute – on voit alors 2,5 à 3 m d’IG) ou basse (si on passe par voie basse – on voit alors 1,5 à 2 m d’IG).
● Avant il y avait l’entéroscopie poussée qui permettait une exploration limitée
(1,2m jéjunum) avec une faible rentabilité.
● Maintenant on pratique l’entéroscopie double ballon qui permet de retrousser l’IG
sur l’entéroscope et ainsi de progresser dedans. Grâce à cette technique, on
bénéficie d’une exploration étendue de l’IG (2 à 3 m), d’une rentabilité
diagnostique plus élevée (60-70%). Elle permet de biopsier les lésions (lorsqu’on
a une ulcération et qu’on pense que c’est un Crohn, qu’il n’y avait rien sur la
gastroscopie, ni sur la coloscopie, ou encore lorsqu’on suspecte une tumeur
neuroendocrine) et de les traiter en coagulant au plasma argon ou au BICAP une
angiodysplasie par exemple, ou en faisant une polypectomie.
Si on ne voit pas de
lésion au cours de l’entéroscopie, on peut tatouer l’IG et y retourner en per opératoire. La dernière génération d’entéroscopies est l’entéroscopie spiralée qu’on n’utilise moins…

37
V. Hémorragies d’origine grêlique Hématémèse : émission de sang par la bouche. Même si l’origine grêlique n’est pas la plus fréquente, toute lésion du duodénum (ulcère par exemple) peut se manifester par une hématémèse puisque cette dernière concerne les saignements qui se situent au-dessus de l’angle de Treitz (angle duodénojéjunal). De façon beaucoup plus fréquente, une des manifestations d’hémorragie est le méléna : un sang noir, pâteux et nauséabond mélangé ou non aux selles puisqu’il est partiellement digéré après avoir transité un minimum. Son origine est donc relativement haute : si le saignement provient de l’IG ou du côlon ascendant, on verra probablement du méléna, alors qu’à partir du côlon transverse, il se manifestera plutôt par une rectorragie. Le sang d’une rectorragie est rouge vif et frais mêlé aux selles. Il provient généralement d’une atteinte basse (recto-sigmoïdienne, du côlon gauche, ou du canal anal – les hémorroïdes en sont une cause fréquente) s’il est en faible abondance. Par contre, si l’hémorragie est importante, on ne peut pas préjuger de son origine ; en effet, toute hémorragie digestive très abondante peut se manifester par une rectorragie (la quantité importante de sang fait qu’il s’écoule rapidement et qu’il n’a pas le temps d’être digéré), le saignement peut provenir de l’IG par exemple… Si un patient a des rectorragies avec une baisse importante de l’hémoglobine (anémie), il faudra donc vérifier si l’hypothèse d’une origine digestive haute est possible ou pas. Dans certaines circonstances, il y a une anémie, mais pas d’hémorragie extériorisée. On peut alors avoir une anémie ferriprive d’origine inconnue ou occulte, avec une baisse de l’hémoglobine et de la ferritinémie ainsi qu’une microcytose. L’origine grêlique ne représente que 5-10% des cas. Les étiologies d’hémorragies digestives d’origine inconnue sont souvent dues à des lésions vasculaires, beaucoup plus qu’à des polypes, des tumeurs, des ulcères ou des sténoses. Abréviations : IG = intestin grêle AEG = altération de l’état général MICI = maladie inflammatoire chronique de l’intestin AINS = anti-inflammatoire non stéroïdien (au cas où…) NFS = Numération formule sanguine VB12 = Vitamine B12
Mot du RT : La prof a dit qu’il fallait retenir : - la diarrhée chronique dure plus de 4 semaines ; - les mécanismes suivants : osmotiques, sécrétoires, de trouble de la motricité, le syndrome de malabsorption et les entéropathies exsudatives (avec notamment les lymphangiectasies) ; - dans le syndrome de malabsorption il y a à la fois des diarrhées chroniques, une AEG, et des carences ; - pour explorer un syndrome de malabsorption on a : la biologie, les explorations fonctionnelles, les endoscopies standard avec les biopsies étagées, les explorations morphologiques, et endoscopiques de l’IG pour avoir une preuve histologique (biopsies).

38
Fiche récapitulative
Rappels anatomo-physiologiques IG = jéjunum (2/5) + iléon (3/5) Surface de contact considérable (250-400m²) → villosités intestinales Entérocyte = cellule fonctionnelle principale Vascularisation : artère mésentérique supérieure Site quasi exclusif d’absorption des nutriments (eau et électrolytes ++) 2 mécanismes : digestion + absorption Jéjunum : fer, Ca, vitamines liposolubles, albumine... Iléon : VB12 + sels biliaires Pathologies : -malabsorption : diarrhée chronique, carences et AEG -saignement chronique ou aigu (anémie, méléna et rectorragies) -obstruction/occlusion -inflammation La diarrhée Définition : émissions quotidiennes trop fréquentes de selles trop abondantes, liquides ou très molles (poids > 300g/j) - aiguë < 2 semaines (nature infectieuse ++ avec douleurs abdominales, vomissements, fièvre, rectorragies) - chronique > 4 semaines 5 grands mécanismes : - diarrhée osmotique : substance osmotiquement active → appel d'eau intraluminal → trou osmotique - diarrhée sécrétoire : augmentation sécrétion d'eau et d'électrolytes par l'entérocyte - diarrhée par trouble de la motricité intestinale : secondaire à inhibition de motricité (pullulation microbienne) ou hypermotricité - diarrhée par malabsorption : maldigestion par défaut de sécrétion ou défaut vrai d'absorption - diarrhée exsudative : inflammation/ulcération de la muqueuse avec perte de plasma/sang Le syndrome de malabsorption Signes cliniques : - stéatorrhée ou diarrhée hydrique - douleurs abdo inconstantes - AEG (amaigrissement, anorexie, asthénie) - œdèmes des membres inférieurs, anasarque - anémie - ostéoporose, ostéomalacie - tétanie - hémorragie

39
- lésions de la peau et des phanères Signes biologiques : - baisse albumine, protides, Ca, phosphore, Mg - anémie micro/macrocytaire, baisse ferritinémie - baisse facteurs de coagulation VK-dépendants - augmentation phosphatases alcalines Malabsorption d'origine pariétale intestinale : atrophies villositaires, ulcérations, pullulations bactériennes... Malabsorption d'origine pancréatique : maldigestion, insuffisance pancréatique exocrine… Explorations de l’intestin grêle Bilan biologique : NFS, vérifier carences, phosphatases alcalines, dosage Ig... Explorations fonctionnelles : - fécalogramme (étude malabsorption +++) - test au D-Xylose (grêle proximal) - clairance de l'alpha1-antitrypsine (altérée si entéropathies exsudatives) - test respiratoire à l'hydrogène (altéré si colonisations bactériennes chroniques) - test au rouge carmin (durée du transit intestinal) Explorations morphologiques : - transit du grêle (rentabilité -- et irradiations ++) - entéroscanner (irradiant) - entéro-IRM (non irradiant, suivi des maladies de Crohn) Endoscopies standard : - gastroscopie ou fibroscopie œsogastroduodénale (FOGD) avec biopsies duodénales (état de la muqueuse dans diarrhée chronique inexpliquée et malabsorption intestinale ou carence) - iléoscopie avec biopsies iléales et coliques étagées Explorations endoscopiques : - vidéocapsule endoscopique - entéroscopie poussée, double ballon (++), spiralée, per opératoire Hémorragies d’origine grêlique - Hématémèse : émission de sang par la bouche, origine au-dessus de l'angle de Treitz - Méléna : sang noir, pâteux et nauséabond (digestion partielle), origine relativement haute - Rectorragie : sang rouge vif et frais, atteinte basse en général /!\ si hémorragie importante, on ne peut préjuger de son origine - Anémie ferriprive d'origine inconnue ou occulte : pas d'extériorisation (généralement lésions vasculaires)

40
UE 6 – Système Digestif (SD) –
Anatomopathologie – Cours n° 2
15/01/2018
Benoit TERRIS
RT : Andrea CLEMENT DE GIVRY
RL : Xavier PINUS
Dépistage et pathogénie du cancer colique
I. Généralités
A. Epidémiologie
B. Causes du cancer colorectal C. Le cancer D. Adénome et adénocarcinome
II.Le cancer colorectal
A. Histologie de la paroi colique
B. La classification TNM
C. Les adénomes coliques
III.Cancérogénèse
A. La population générale : les cas sporadiques B. Les populations à haut risques : prédisposition génétique C. Le dépistage du CCR

41
I. Généralités
A. Epidémiologie En France, le cancer colorectal (CCR) est un véritable problème de santé publique. On
recense 36 OOO (voire 40 000) nouveaux cas par an, ce qui lui donne le titre de 1er cancer
digestif. Chez la femme, c’est le deuxième cancer le plus commun après le cancer du sein et chez
l’homme le troisième après le cancer de la prostate et du poumon.
C’est un cancer relativement grave en raison de sa mortalité importante (16 OOO décès
par an) ce qui représente environ un décès toutes les deux heures ou encore 4 fois le nombre de
morts sur la route.
Entre 1980 et 2000, le nombre de nouveaux cas de cancer colique augmente de 50%,
survenant surtout dans la région du colon droit. En conséquence, le nombre de personnes
vivantes atteintes d’un CCR ou avec des antécédents de CCR est aujourd’hui de 200 000.
Le CCR est un cancer qui se développe plutôt à un âge avancé, à partir de 70 ans environ.
Pourtant, il existe des cas précoces (avant 50 ans) dans 6% des cas.
B. Causes du cancer colorectal De manière générale, les causes du CCR sont mal connues. On attribue néanmoins un rôle
important à l’alimentation et aux lésions précancéreuses.
• Facteurs Alimentaires • Lésions précancéreuses
- Régime riche en graisses
- Régime pauvre en fibres alimentaires
• Conséquence : Inégale répartition du CCR
dans le monde car les pays en voie de
développement ont plutôt tendance à avoir un
régime pauvre en graisse et riche en fibre à
l’inverse des pays développés.
- Anomalies histopathologiques qui, si
elles persistent suffisamment
longtemps, peuvent aboutir à
l’apparition d’un cancer.
• (gastrites chroniques, ulcère
gastrique…)
- Au niveau de colon :
Polypes (adénomes)
Lésions inflammatoires
chroniques de l’intestin
• (ex. Maladie de Crohn)
C. Le cancer Le cancer est un processus cellulaire, constitué par une prolifération de cellules et possédant des anomalies fonctionnelles et morphologiques. Il envahit et infiltre les tissus aux alentours (phase locale), avant d’être à l’origine d’une extension à distance sous la forme de métastases (phase générale).

42
Les signes cliniques du cancer colorectal :
- Rectorragies : sang dans les selles
- Trouble du transit : souvent apparition d’une constipation chronique
- Altération de l’état général
Le diagnostic du CCR se fait grâce à une exploration par coloscopie qui consiste à
observer le rectum et la totalité du cadre colique (jusqu’à la dernière anse du grêle au niveau de
la jonction iléo-sécale) grâce à une sonde optique.
D. Adénome et adénocarcinome Un adénome est une lésion précurseur d’un cancer. C’est une tumeur bénigne
constituée par la prolifération d’un épithélium glandulaire dans une muqueuse ou une glande
exocrine ou endocrine. L’adénome a un potentiel malin.
Un adénocarcinome est une lésion cancéreuse établie. C’est une tumeur maligne dont
la structure reproduit celle d’un épithélium glandulaire, que son développement soit à partir
d’une muqueuse glandulaire ou à partir d’une glande endocrine ou exocrine. Dans le colon, les
adénocarcinomes reproduisent souvent l’épithélium qui leur à donner naissance.
- Il peut être typique ou bien différencié, ou atypique ou peu différencié.
- Il peut avoir plusieurs aspects : canalaire ou tubulaire, trabéculaire, papillaire, à cellules indépendantes.
II. Le cancer colorectal
A. Histologie de la paroi colique Au plan microscopique, la paroi colique normale à une structure en couches distinctes (de la
lumière vers l’intérieur) :
- Muqueuse : épithélium de surface avec des glandes, limitée en profondeur par sa
musculaire-muqueuse (à ne pas confondre avec la musculeuse)
- Sous-muqueuse
- Musculeuse
- Sous-séreuse tapissée par le péritoine

43
- Séreuse (péritoine)
• Quand la paroi colique est coupée
transversalement, on peut observer un aspect en
champs de marguerite qui correspond aux glandes
coliques. Ces glandes sont tapissées par des cellules
caliciformes dont le noyau est repoussé en position
basale et dont le cytoplasme contient une
volumineuse vacuole de mucus.
•
Sur le plan histopathologique, dans le cadre d’un cancer colique la paroi se désorganise et
les cellules changent d’aspect. Les glandes, normalement bien définies, se confondent et leur
muco-sécrétion diminue.
Macroscopiquement, la muqueuse colique normale est interrompue par la tumeur qui peut
occuper toute la circonférence du colon ou pas, et bourgeonner dans la lumière du tube
entrainant ainsi une sténose (source de potentielle constipation). Cette tumeur va aussi saigner
expliquant la rectorragie associée au CCR. Dans la photo ci-dessous, on observe une tumeur
ulcéro-bourgeonnante circonférentielle typique.
Tumeur ulcéro-bourgeonnante circonférentielle

44
B. Classification TNM La classification TNM permet de décrire l’évolution d’un cancer selon son extension
anatomique.
- T : Degré d’extension de la tumeur dans un tissu
- N : Extension métastatique ganglionnaire (lymph nodes en anglais)
- M : Présence de métastase
NB : Chaque lettre se divise en plusieurs catégories
La gradation des tumeurs grâce à la lettre T va de 1 à 4 :
- T1 : infiltre la muqueuse et la sous-muqueuse
sans atteindre la musculeuse.
- T2 : va jusqu’à la musculeuse.
- T3 : va au-delà la musculeuse jusqu’à la sous-
séreuse.
- T4 : est présente dans toutes les couches de la
paroi colique et perfore la séreuse. Elle est à
l’origine d’une carcinose péritonéale.
NB : Plus la tumeur est évoluée plus le pronostic est
mauvais
Exemple de classification en lien avec le pronostic :
- Tumeur de stade 1 (T1 ou T2) qui ne s’accompagne pas de métastases ganglionnaires ou de
métastases autres (en particulier hépatique)
Le pronostic est bon : plus de 90% des patients seront vivants à 5 ans.
- Tumeur de stades 3 ou 4 – sachant que le stade 4 est défini par la présence d’une métastase
hépatique.
Le pronostic chute avec moins de 10% des patients vivants à 5 ans.
C. Les adénomes coliques 60 à 80% des CCR naissent à partir de tumeurs bénignes, les adénomes. En cas d’exérèse
chirurgicale des adénomes, l’incidence et la mortalité sont fortement diminuées.
Les adénomes sont très fréquents (présent chez 1 sujet sur 3 de plus de 65 ans) mais ils
n’évoluent pas tous vers la malignité. En effet, seulement 10% de ces adénomes augmentent de
taille (notamment au-delà d’1cm) et peuvent évoluer vers un cancer.
Tous les adénomes s’accompagnent d’une dysplasie, des lésions acquises dues à une
anomalie de maturation d’un tissu à renouvellement rapide.
L’évolution de l’adénome à l’adénocarcinome dure en moyenne 10 ans, elle est donc lente. Le
dépistage est ainsi un facteur essentiel de la prévention de l’apparition d’un cancer.
Un adénome se présente le plus souvent la forme d’un polype, soit une formation
circonscrite, sessile ou pédiculée, saillant dans la lumière intestinale. La présence d’un polype un
signe macroscopique recherché grâce à la coloscopie. Sa présence ne préjuge en rien d’une
nature précancéreuse, il faut donc compléter l’exérèse par une analyse histologique.

45
NB : Un polype peut aussi être de nature non épithélial (léiomyome – donc pas un adénome), ou de
nature épithéliale dans le cas des polypes hyperplasique qui sont des glandes modifiées ne
conduisant pas à un cancer.
Le terme de « polype malin » est aussi utilisé pour décrire un adénome dégénéré en
cancer (adénocarcinome).
Un adénome colique peut avoir plusieurs aspects :
- Pédiculé ou sessile (pour décrire comment il se rattache à la paroi)
- Tubuleux, villeux ou tubulo-villeux (pour décrire sa morphologique)
• Moyen
de rattachement
à la paroi
• Pédiculé
- Polype rattaché à la paroi colique par un pédicule
à base d’implantation fine
- Exérèse chirurgicale grâce à un lasso coupant
l’excroissance au pied.
• Sessile
- Polype rattaché à la paroi colique par une large
base d’implantation.
- Exérèse chirurgicale au lasso, suite à une injection
permettant de lui donner une structure semi-
pédiculée en le surélevant.
• Aspect
morphologique
• Tubuleux
- Prolifération des glandes identiques aux glandes
de Lieberkühn – structure en champ de
marguerite (cf. Histologie)
- Mucosécrétion diminuée (dédifférenciation)
• Villeux
- Masse molle et sessile
- Axes conjonctifs grêles recouverts d’une couche
de cellules épithéliales atypiques muco-
sécrétantes (entrainant parfois des diarrhées
muco-sécrétantes)
- Souvent au niveau du rectum
• Tubulo-
villeux - Un mélange des deux précédents

46
•
•
Critère de définition d’un « adénome avancé » (avec le plus de risque d’évoluer en cancer) :
- Taille (>1cm)
- Villeux
- Dysplasie de haut grade (DHG)
En fonction du stade de l’infiltration tumorale dans la paroi digestive, les traitements
possibles ne seront pas les mêmes. Pour une tumeur allant de la muqueuse à la sous-muqueuse
un traitement endoscopique suffit alors que pour une tumeur infiltrant profondément dans la
paroi colique, une exérèse chirurgicale est le seul moyen de l’enlever.
Plus une tumeur progresse en profondeur plus son risque d’infiltrer les vaisseaux
lymphatiques ou vasculaires est augmenté, d’autant plus que le front de progression d’une
tumeur peut être constitué de cellules indépendantes. Le drainage de l’intestin étant effectué par
la veine porte, la plupart des métastases du CCR se retrouvent dans le foie.
De plus, en règle générale, on remarque que les lésions pré-néoplasiques peuvent coexister
avec des lésions cancéreuses.
III. Cancérogénèse Les anomalies génétiques sont mises en cause dans le développement du CCR. Les connaitre
va permettre une meilleure compréhension de sa cancérogénèse mais aussi contribuer à
l’amélioration de son dépistage.
De plus, il existe des populations à haut risque de développer un CCR à cause de leurs
prépositions héréditaires notamment dans les pathologies suivantes : la polypose adénomateuse
familiale (PAF) et le syndrome de Lynch. Ces cancers héréditaires constituent 5% des cancers
coliques.
Polype pédiculé Polype sessile Polype villeux, tubuleux, tubulo-villeux

47
La population générale et la population avec des facteurs de prédisposition sont donc à
distinguer.

48
A. La population générale : les cas sporadiques
Les CCR sporadiques sont les plus communs. On observe qu’ils ont un aspect homogène et qu’ils
se développent selon deux mécanismes moléculaires :
- Instabilité chromosomique (80% à 85%)
- Instabilité génétique, ou microsatellite (15%)
- Dans chacun de ces mécanismes les voies de signalisation impliquées sont identiques.
NB : L’un exclu l’autre c’est-à-dire qu’un patient avec une instabilité chromosomique sera
génétiquement stable et vice versa.
• CCR à
instabilité
chromosomique
- Surviennent surtout aux alentours de 60 à 70 ans
• CCR à
instabilité
microsatellite
- Surviennent davantage chez la femme au niveau du colon droit
(méthylation du promoteur MLH1)
i. Instabilité chromosomique (MSS) Ce sont des tumeurs caractérisées par des pertes alléliques ou des pertes d’hétérozygoties :
- D’abord il y a la perte d’un chromosome par anomalie de ségrégation (ex. Non
disjonction méiotique) ou par un autre mécanisme plus complexe.
- Puis, le dernier allèle restant est perdu.
- Les parties chromosomiques les plus souvent perdus sont : 17p (p53), 8p, 18q,
5q (APC), 22q
Ces tumeurs sont dites LOH+ (loss of heterozygosity) et ont des mutations fréquentes des
gènes suppresseurs de tumeurs comme :
p53 gène suppresseur de tumeur
APC : anomalies de ségrégation chromosomique
ii. Instabilité génétique ou microsatellite instable (MSI) Les tumeurs génétiquement instables sont dues à une instabilité des locus microsatellites :
- Les microsatellites sont des séquences mono- à tétra-nucléotidiques répétées du génome
humain. Elles sont propres aux individus (utilisées en médecine légale).
- Lors de la réplication, ces régions hautement répétées sont sujettes soit à des erreurs
d’appariement de nucléotides donc à des mésappariements de l’ADN.
Normalement, les erreurs de réplication nécessitent l’intervention de protéines réparatrices
de l’ADN. Or, on remarque que ces tumeurs sont souvent déficitaires en protéines du système de
réparation des mésappariements de l’ADN. Ce sont des tumeurs dites MSI+ :
Cette déficience peut être liée à la méthylation du promoteur d’une protéine de réparation
comme la protéine MLH1. Ceci va aboutir à une accumulation de mutations secondaires au
niveau des séquences répétées avec un effet délétère dans régions codantes. Enfin, par cette
accumulation de mutation, il y aura une inactivation des gènes impliqués dans le cycle cellulaire
et l’apoptose.

49
Pour identifier ce type de tumeur on cherche avec des anticorps ces protéines réparatrices
de l’ADN grâce à des lames de biopsies. L’ADN de la tumeur peut aussi être utilisé et par
comparaison de longueur déterminer si les régions satellites ont été altérées.
B. Les populations à haut risques : prédisposition génétique Les CCR faisant suite à une prédisposition héréditaire surviennent de manière beaucoup
plus précoce que les cancers sporadiques même s’ils suivent les mêmes modes de
développement que les cancers sporadiques. Ils représentent 5% des cancers coliques.
Ils sont transmis selon un mode autosomal dominant (dans 1% à 5%) par mutation
germinale chez un des parents. Il suffit d’un seul évènement pour aboutir à l’altération de l’allèle
non atteint ce qui explique l’âge plus précoce auquel se développe les CCR héréditaires. Ceci
s’oppose aux mutations somatiques qui surviennent dans les cas sporadiques et qui ne sont pas
transmissibles à la descendance. Les mutations somatiques doivent accumuler des mutations sur
les deux allèles, donc les CCR sporadiques se développent à un âge plus avancé.
Mécanismes de cancérogénèse (bilan)

50
Il est important de connaitre le profil héréditaire d’un individu pour adapter le dépistage,
notamment le commencer plus jeune, et pour avoir une meilleure compréhension de la
carcinogénèse.
Les prédispositions familiales au cancer peuvent être d’ordre environnemental ou génétique.
Dans ce cas, il fau rechercher des anomalies constitutionnelles (germinales) et connaitre les
antécédents familiaux (précocité et multiplicité des tumeurs).
Polypose adénomateuse familiale (PAF) :
La PAF est liée à la mutation germinale d’APC (mécanisme autosomique dominante). Elle
concerne environ 1 naissance pour 8000
- Polypose colique et rectale se caractérise par la présence de nombreux adénomes (>100)
dégénérant inéluctablement en cancer avant l’âge de 60 ans.
- Le traitement principal est la colectomie prophylactique (enlever le colon avant 20 ans).
- Il existe aussi des manifestations extra-coliques de la PAF qui sont plutôt inconstantes : adénomes duodénaux évoluant potentiellement vers des adénocarcinomes. (polypose fundique glandulokystique, hypertrophie de l'épithélium pigmentaire rétinien (CHRPE),
ostéomes, tumeurs desmoïdes)
Syndrome de Lynch (HNPCC) :
Cette maladie est liée à la mutation constitutionnelle d’un gène de réparation des
mésappariements de l’ADN (hMLH1, hMSH2, hMSH6). Elle est donc microsatellite instable
(MSI+).
C’est une maladie fréquente : Syndrome HNPCC (Lynch) :
- 3 sujets atteints dans une famille avec des parents au 1° degré, sur 2 générations atteints ou 1 cancer diagnostiqué < 50 ans
Les patients souffrant de ce syndrome sont touchés par plusieurs pathologies :
- Cancer du colon (90%) : colon droit, mucineux, inflammatoire
- Polypes adénomateux et/ou hyperplasiques
- Cancer de l’endomètre : 40%
- Autres cancers (estomac (9%), intestin grêle (1%), voies biliaires (<10%), voies excrétrices
urinaires (8%))
Incidence du CCR en fonction de l’âge et de la pathologie sous-jacente
PAF : entre 20 et 40 ans
Syndrome de lynch : 40 et 50
ans

51
C. Le dépistage du CCR Il y a une forte filiation entre polype – cancer (ou du moins adénome et cancer). Si on faisait
une coloscopie totale à tous les patients de plus de 40 ans tous les 1 à 5 ans, en fonction des
résultats, on pourrait faire disparaître le CCR. Or, ceci n’est pas réalisable car trop coûteux et
anxiogène par rapport au avantages engendrés (balance bénéfice-risque).
Le dépistage concerne aujourd’hui deux types de populations :
- Les groupes à risque (risque élevé ou très élevé) :
Personnes présentant des symptômes d’alerte et/ou ayant des antécédents
familiaux de CCR
Personnes ayant des prédispositions constitutionnelles (PAF, Lynch)
Personnes souffrant de colites inflammatoires chroniques
- Les groupes à risque moyen :
Recherche de saignements occultes dans les selles grâce à un test immunologique
permettant de déceler une présence de sang humain dans les selles.
Ce test est un test de sélection des personnes éligibles à la coloscopie en fonction
des résultats et n’a pas une valeur de diagnostic.
Il est proposé tous les 2 ans aux personnes de 50 à 74 ans mais peu de gens le
font vraiment ce qui pose la question de son efficacité.
Il a permis une diminution de 25% de la mortalité par CCR dans la population
soumise au dépistage.
CONCLUSION :
- CCR est un cancer fréquent et de plus en plus à cause du vieillissement de la population
- Les lésions précancéreuses sont des adénomes coliques (souvent des polypes)
- Les campagnes de dépistage pour les saignements occultes sont très importantes et il
faut encourager le plus de monde possible à le faire.
- Les CCR, comme d’autres cancers se développent suite à une cascade d’évènements
génétiques.
- 5% des CCR surviennent dans le cadre de 2 prédispositions génétiques (PAF et HNPCC).
Abréviations :
CCR – Cancer colorectal

52
Fiche récapitulative : Dépistage et pathogénie du cancer
colique
I. Généralités :
- Cancer colorectal (CCR) = vrai problème de santé publique : 36000 nouveaux cas/an, 1er
cancer digestif, deuxième cancer chez la femme, troisième chez l’homme
Cancer avec une mortalité importante : 16000 décès/an
Son incidence augmente (+50% en 20 ans) avec 200000 personnes vivantes atteintes ou avec
des antécédents de CCR
Cancer qui se développe très majoritairement à un âge avancé
- Causes du CCR : mal connues mais rôle important de l’alimentation (régime riche en graisses et
pauvre en fibres alimentaires) et des lésions précancéreuses (lésions pouvant aboutir à un
cancer, sous forme de polypes au niveau du colon)
- Le cancer : processus cellulaire constitué par une prolifération de cellules et possédant des
anomalies fonctionnelles et morphologiques, avec une phase locale puis générale
Signes cliniques du CCR : rectorragies, constipation chronique, altération de l’état général
Diagnostic grâce à la coloscopie
Adénome : tumeur bénigne, lésion précurseur d’un cancer
Adénocarcinome : lésion cancéreuse établie, tumeur maligne
II. Le cancer colorectal :
- Paroi colique : constituée de la lumière vers l’intérieur : muqueuse (limitée en profondeur par
une musculaire muqueuse), sous-muqueuse, musculeuse, sous-séreuse, séreuse
Aspect histologique en champs de marguerite : glandes coliques tapissées de cellules
caliciformes
Si cancer : glandes qui se confondent avec diminution de la muco-sécrétion
Macroscopiquement : tumeur qui bourgeonne dans la lumière du tube, interrompant la
muqueuse colique normale
- Classification TNM : gradation des tumeurs de T1 à T4 (T décrivant l’extension de la tumeur
dans un tissu) : T1 atteint la muqueuse et la sous-muqueuse, T2 atteint la musculeuse, T3 atteint
sous-séreuse, T4 perfore la séreuse (carcinose péritonéale)
- Adénomes coliques : tumeurs bénignes donnant naissance à 60 à 80% des CCR
Très fréquents (1/3 des sujets de plus de 65 ans), mais seuls 10% augmentent de taille
Evolution en adénocarcinome très lente (10 ans en moyenne) d’où l’importance du dépistage
Adénome souvent sous la forme de polype (formation circonscrite, sessile ou pédiculée, saillant
dans la lumière intestinale
Polype malin = adénome dégénéré en cancer

53
Un adénome peut être pédiculé ou sessile (manière dont il se rattache à la paroi) et tubuleux,
villeux ou tubulo-villeux (aspect morphologique)
Adénome avancé à risque : taille>1cm, villeux, dysplasie de haut grade(DHG)
Traitements différents selon l’infiltration tumorale : peu infiltrée traitement endoscopique,
sinon exérèse chirurgicale nécessaire
Les CCR donnent des métastases au niveau du foie car drainage de l’intestin par la veine porte
III. Cancérogénèse :
- Il est nécessaire de distinguer la population générale (cas sporadiques), des populations à haut
risque à cause de prédispositions génétiques
- Cas sporadiques : se développent selon deux mécanismes : instabilité chromosomique (80 à
85%) et instabilité génétique ou microsatellite (15%)
- Instabilité chromosomique : due à des pertes alléliques ou d’hétérozygoties, gènes souvent
touchés : P53 et APC (gènes suppresseurs de tumeurs)
- Instabilité génétique ou microsatellite : due à l’instabilité des locus microsatellites (séquences
hautement répétées), avec souvent un déficit en protéines du système de réparation des
mésappariements de l’ADN comme MLH1.
- Cas héréditaires : ils surviennent de manière beaucoup plus précoce mais suivent les mêmes
modes de développement que les cas sporadiques ; ne représentent que 5% des CCR.
Du à la présence d’une mutation germinale chez un des parents, il suffit alors d’un seul
évènement pour altérer l’allèle non atteint, ce qui explique leur précocité
- Polypose adénomateuse familiale (PAF) : mutation germinale d’APC, 1 naissance sur 8000
Présence de très nombreux adénomes (plus de 100), seul traitement : colectomie prophylactique
(enlever le colon avant 20 ans) car un des adénomes dégénèrera forcément en adénocarcinome
Mais il existe aussi des manifestations extra-coliques inconstantes (adénomes duodénaux,
polypose fundique glandulokystique)
- Syndrome de Lynch : mutation germinale d’un gêne de réparation des mésappariements de
l’ADN, à l’origine d’un CCR de type microsatellite instable
Maladie fréquente, suspectée si dans une famille on a 3 sujets atteints parents au 1er degré sur 2
générations, et 1 cancer diagnostiqué avant 50 ans
Plusieurs pathologies chez les sujets atteints : CCR (90%), cancer de l’endomètre (40%), autres
cancers (estomac, intestin grêle, voies biliaires, voies excrétrices urinaires) de façon plus rare
- Dépistage du CCR : concerne deux types de population : les groupes à risque (individus avec
antécédents, prédispositions constitutionnelles, colites inflammatoires chroniques) et les
groupes à risque moyen (chez qui on recherche des saignements occultes dans les selles puis
une éventuelle coloscopie)
Test proposé tous les 2 ans chez les individus de 50 à 74 ans

54
UE6 – SD – Anatomie – n°4
16 janvier 2018
Pr Richard Douard
RT : Marianne Clemot
RL : Marie Payer
Anatomie du rectum et du canal anal
Plan :
I. Introduction
II. Le rectum
A. Les « trois étages » du rectum
B. La loge rectale
C. Vascularisation
i. Les artères
ii. Les lymphatiques
iii. Les veines
III. Le canal anal
IV. L’appareil sphinctérien externe
V. Physiologie de la défécation
VI. Les pathologies
A. Les hémorroïdes
B. Les fistules
VII. Le muscle élévateur de l’anus
VIII. Conclusion
Mot du RT : Le professeur a parlé d’un référentiel sur le système digestif disponible sur le Moodle. J’ai mis en italique les apartés faits par le professeur, notamment historiques.

55
I. Introduction
Il y a des problèmes de définitions, c’est toujours le problème en anatomie, savoir de quoi on parle,
et souvent c’est un peu l’histoire de la médecine aussi, il n’y a pas si longtemps, la nomenclature,
la façon de présenter les choses n’était pas la même, voire même si vous allez sur des sites
étrangers des pays francophones vous trouverez une autre façon de décrire. Alors évidemment le
rectum et l’anus sont les mêmes à l’étranger mais la façon de décrire peut être différente.
Autrefois (dans les années 70), on définissait le rectum comme la partie terminale du tube digestif,
séparé entre rectum pelvien et rectum périnéal. Cette distinction a vécu mais elle est encore
utilisée dans tous les pays francophones qui utilisent les anciennes façons de décrire, si on prend
les sites du Maghreb, on nous décrira le rectum pelvien et le rectum périnéal, alors que le rectum
périnéal correspond en fait au canal anal.
Or, la bonne façon de décrire, à la fois sur un plan embryologique et sur un plan pratique, est de
décrire deux organes différents : le rectum qui est dans la cavité pelvienne, et le canal anal qui est
périnéal.
C’est cette façon de décrire qui est utilisée actuellement, il faut donc faire attention et se méfier
des références que l’on utilise en fonction d’où on va les chercher, ou si on lit des livres anciens
sur l’anatomie.
II. Le rectum
A. Les « trois étages » du rectum

56
Sur cette coupe frontale il y a un problème c’est que l’anus et le rectum sont dans l’alignement, ce
qui est impossible puisque si c’était comme ça on aurait beaucoup de mal à retenir les matières,
elles pèseraient directement sur les sphincters. Il y a donc un angle entre l’anus et le rectum.
Cette coupe est schématisée.
On voit sur cette coupe les trois étages de ce rectum dans son ancienne acception, avec le rectum
dans la cavité pelvienne composé de deux parties, et le canal anal en dessous, dans le périnée.
Le rectum est donc composé de deux parties :
- Une partie péritonisée, c’est-à-dire recouverte de péritoine.
- Une partie sous péritonéale
La frontière entre le petit bassin et le périnée est formée par le muscle élévateur de l’anus.
Un des faisceaux de ce muscle va aller cravater la partie basse du rectum et donc créer l’angle
anorectal. Cet angle vaut 80°, il permet à l’anus et au rectum de ne pas être dans l’axe.
Le péritoine est une séreuse, comme la plèvre pour le poumon. Malgré ce qu’on peut voir sur
certaines coupes, il n’y a qu’une cavité péritonéale, comme il n’y a qu’une cavité péricardique, alors
qu’il y a deux cavités pleurales. Le péritoine quand il recouvre les organes fait partie
histologiquement de la paroi de l’organe. C’est une construction de description.
La charnière recto-sigmoïdienne est la fin de la racine primaire du sigmoïde (appelée ainsi parce
qu’elle ressemble à l’axe primitif du tube digestif embryonnaire, qui est sagittal, c’est un long tube
et il y a un péritoine qui est postérieur avec une racine verticale). Il y a donc deux racines du
sigmoïde : une secondaire, oblique vers le bas et la gauche, et une primaire qui est vers le bas
jusqu’à la jonction recto-sigmoïdienne (qui ressemble à la racine primitive, d’où son nom).
On va définir de part et d’autre du rectum péritonisé des espaces latéro-rectaux. Comme il n’y a
pas d’air dans le ventre (en principe), les six mètres d’intestin grêle tombent dans les fosses latéro-
rectales. On y trouve aussi l’appendice qui pend dans ces espaces si c’est un appendice pelvien.
Ceci explique pourquoi lorsqu’on fait de la radiothérapie pour les cancers du rectum, on le fait
pour les cancers sous-péritonisés du rectum, parce que si on le fait pour les cancers péritonisés,
on va irradier (même si les radiothérapeutes sont extrêmement précis) l’intestin grêle qui est de
part et d’autre, alors que l’objectif est d’irradier le rectum et pas le grêle.
B. La loge rectale La loge rectale est très importante en chirurgie du cancer puisque le traitement du cancer du
rectum, dans sa forme la plus fréquente est un traitement qui va être chirurgical et qui va
comporter l’exérèse, l’ablation, le retrait de la loge rectale. C’est ce qu’on apprendra sous le terme
d’exérèse totale du mésorectum (qui est un terme anglais qui ne fait que reprendre des notions
anatomiques parfaitement connues depuis les années 20).
Cela veut dire qu’entre la description par les Anglais de l’exérèse de la loge rectale et la description
de la loge rectale il y a 60 ans. 60 ans pour qu’on réalise que ce qu’il fallait faire en chirurgie c’est ce
qu’on connaissait en anatomie.
Ceci n’est pas spécifique : il y a parfois des avancées où l’anatomie va être en retard sur la chirurgie,
c’est même souvent le cas, ce sont les chirurgiens qui vont décrire des choses que les anatomistes vont
réutiliser.

57
On parle de loge rectale au niveau du rectum sous-péritonéal. En effet, on trouve au niveau du
rectum péritonisé un méso, tandis qu’au niveau sous péritonéal il n’y a plus de péritoine donc plus
de méso.
Heald, un anglais, a décrit le mésorectum juste à l’endroit où il n’y a pas de péritoine. Ce qui a posé
un problème aux anatomistes, c’est que les chirurgiens décrivaient un méso alors qu’il n’y a pas de
péritoine. Il aura fallu 20 ans pour que les chirurgiens et les anatomistes se mettent d’accord sur le
fait qu’en sous-péritonéal il faut enlever la loge rectale dans le cancer, et pour qu’on apprenne aux
chirurgiens que ce que les Anglais appelaient mésorectum n’était en fait pas un méso, mais la loge
rectale.
Les lames sacro-recto-génito-pubiennes sont des condensations de tissu conjonctif sous
péritonéales (ces condensations n’existent pas dans la cavité péritonéale). Dans ces condensations
se trouve le plexus hypogastrique ou pelvien latéral, c’est-à-dire que c’est dans ces condensations
que passent des nerfs sympathiques et parasympathiques qui vont innerver de façon végétative
le rectum, l’appareil génital et l’appareil urinaire. Ces nerfs végétatifs, qui sont aussi responsables
chez l’homme de l’érection, vont être la paroi latérale de la loge rectale.
Dans les années 80, on découvrit qu’on pouvait enlever un rectum sous-péritonéal sans
systématiquement compromettre l’innervation végétative des organes qu’on laissait.
En arrière du rectum, on trouve un fascia pré-sacral ou pré-sacré qui est une aponévrose
relativement solide qui définit en avant la loge rectale et en arrière l’espace pré-sacré. Dans
l’espace pré-sacré on trouve des veines, qui vont dans le sacrum, ainsi que l’artère sacrale médiane
ou sacrée moyenne (qui est une branche de l’aorte).
Longtemps quand on faisait l’exérèse du rectum on ne savait pas trop où passer, et souvent on allait
dans cet espace et on rencontrait les veines. Ces veines, quand on les rencontrait, se rétractaient dans
le sacrum provoquant un saignement en nappe qu’on n’arrivait pas à arrêter. Parfois, on était par
conséquent amenés à mettre des punaises en métal (dans les années 80) puisqu’on ne pouvait pas
suturer car elles se rétractaient dans l’os.
Le plan d’exérèse du sacrum est juste en avant du fascia pré-sacré.

58
Chez la femme, il y a trois loges : la loge rectale, la loge génitale et la loge urinaire. Entre la loge
rectale et le vagin, on trouve la cloison recto-vaginale, espace extrêmement fin, on y trouve très
peu de graisse péri-rectale (en blanc sur la coupe). On trouve beaucoup plus de graisse en arrière
du rectum qu’en avant. Si on fait, chez la femme, un toucher bimanuel, un doigt dans le vagin, un
doigt dans le rectum, les deux doigts se touchent, il n’y a quasiment pas d’espace, de la même façon
qu’on touche la prostate chez l’homme à travers la paroi du rectum. On fait des touchers bimanuels
afin notamment de savoir si une tumeur du bas rectum a envahi la paroi vaginale.
La loge rectale va être définie latéralement par les plexus pelviens latéraux au sein des lames
sacro-recto-génito-pubiennes, en arrière par le fascia pré-sacral, en avant, chez la femme, par la
cloison recto-vaginale.
/ !\ L’espace rétro-rectal est en avant du fascia pré-sacré alors que l’espace pré-sacré est en avant du sacrum et en arrière du fascia.
Les racines nerveuses sacrales vont donner des contingents végétatifs vers l’avant puis des
contingents somatiques vers l’arrière puisqu’en face du muscle piriforme on trouve le nerf
sciatique qui a des racines sacrales, somatiques (ce nerf permet de marcher). On trouve aussi le
muscle obturateur interne, l’ischion, le pubis.
La graisse péri-rectale dans cet espace rétro-rectal doit être retirée avec le rectum dans une
exérèse de la loge rectale à visée carcinologique. C’est ce qu’on appelle l’exérèse du mésorectum.
Mais il faut faire attention car on ne peut pas enlever le fascia pré-sacré, ni enlever les nerfs. Il y a
un autre feuillet qui est juste en dehors de la graisse et en dedans du fascia et des plexus, qui va
doubler ces parois. On l’appelle le fascia recti, que l’on va trouver lors de l’exérèse entre le fascia
et la graisse. Cet « emballage », ce feuillet va permettre de retirer la graisse du rectum qui y est
parfaitement emballée, il n’y a donc pas de besoin de gratter. Ceci suppose une dissection fine, qui
va avec les progrès de l’anesthésie et de la chirurgie, permettant de prendre plus de temps, de
mieux regarder.
Tel fut l’apport de Heald : il découvrit qu’il y a un fascia autour de la graisse qui évite de passer en
arrière du fascia pré-sacré où on trouve les veines. Rouvière en 1923 le décrivait déjà.
On voit sur cette coupe l’artère iliaque interne qui va donner une branche pour le rectum en sous-
péritonéal, l’artère rectale moyenne (ce qui sous-entend qu’il existe une artère rectale supérieure
et une inférieure). Il s’agit du tronc qui précède le bras d’artère légendé comme « aileron moyen
soulevé par l’artère rectale moyenne ».
À retenir ++ : la loge rectale est donc associée au rectum sous-péritonéal, il n’y a pas de
péritoine mais on parle quand même de mésorectum.
Chez l’homme, on retrouve trois étages : deux étages de rectum, un étage de canal anal : le rectum
péritonisé et le rectum sous péritonéal, puis ce qu’on appelait autrefois le rectum périnéal, c’est-
à-dire le canal anal. Sur cette coupe sagittale, le péritoine est en vert.
Entre les deux, on retrouve le cap anal, la courbure du rectum et de l’anus liée à l’empreinte du
muscle élévateur de l’anus qui explique que l’on fasse le toucher rectal en grande flexion, et
qu’autant on fait un toucher vaginal juste les jambes écartées, car le vagin est orienté vers le bas
et l’avant, alors que le rectum est orienté vers le bas et l’arrière, il faut donc le ramener dans l’axe,
soit en mettant le malade à quatre pattes, soit en le mettant sur le dos les jambes fléchies. C’est la
position logique, elle est expliquée par la clinique et par l’anatomie.

59
Chez l’homme, le péritoine recouvre la vessie qui est un organe sous-péritonéal, puis le relief des
vésicules séminales et, en arrière de ces vésicules, le fond du cul-de-sac génito-rectal ou cul-de-
sac de Douglas qui est plus bas que les vésicules séminales. Puis on trouve en avant l’aponévrose
de Denonvilliers (aponévrose prostato-péritonéale).
On comprend bien sur cette vue que le toucher rectal explore la prostate et qu’il n’y a pas d’espace
entre la prostate et le rectum (juste l’aponévrose), de la même façon qu’au niveau de la cloison
recto-vaginale.
Il y a deux courbures au rectum, la courbure sacrale (postérieure) et la courbure périnéale
(antérieure).
/!\ Rappel : on parle souvent d’un rectum pelvien, et d’un rectum périnéal, alors qu’il faut bien
distinguer deux organes : rectum et canal anal. (Le professeur insiste beaucoup sur ces définitions).
Sur la coupe sagittale chez la femme (ci-dessous), on retrouve la vessie avec un urètre court, le
vagin qui est orienté vers le bas et l’arrière (ce qui explique que le toucher vaginal se fasse
simplement, l’orientation est propice à une examination couchée sur le dos). L’utérus est
antéfléchi et antéversé (c’est-à-dire qu’il recouvre la vessie et est plié sur lui-même). Il a donc deux
orientations.

60
En arrière, on retrouve la cloison recto-vaginale qui descend vers le centre fibreux du périnée qui
est beaucoup plus faible chez la femme que chez l’homme, puisque ce qui tient la statique
pelvienne chez la femme est l’orientation opposée des organes (vagin vers arrière, rectum vers
l’arrière, tout se tient). Chez la femme, le diaphragme urogénital, les orifices sont plus larges, il y
a une fonction d’accouchement qui fait que cette forme d’encombrement stérique tient la statique
pelvienne (et non pas la solidité de la structure). Ceci explique que quand on retire l’utérus, cela
risque de déstabiliser la statique pelvienne des femmes, et on a des pathologies (des prolapsus,
des procidences) qui sont liées à cette modification de la statique.
Chez l’homme, tout est extrêmement serré il y a très peu de place et le noyau central du périnée
est extrêmement fort, beaucoup plus solide, et les zones orificielles sont beaucoup plus petites en
taille.
Précision : sur le schéma, il est important de bien retenir le ligament anococcygien, le cul-de-sac de
Douglas, la position des sphincters et de l’urètre.

61
Longtemps on a fait de l’anatomie avec des coupes cadavériques car, notamment pour les coupes
sagittales, les reconstructions tridimensionnelles scanner datent facilement des années 2000, et les
coupes sagittales n’étaient que très peu accessibles (les coupes transversales datent des années 80)
(problème de puissance informatique). Pendant très longtemps, on a utilisé les coupes cadavériques,
et à chaque fois qu’il y avait des progrès du scanner ou de l’IRM, on faisait des corrélations entre
imagerie et les cadavres. Travaux qui sont encore pour certains d’actualités même si les cadavres ne
sont plus très utilisés. Pour ce faire, on utilisait les gens qui donnent leur corps à la science dans les
centres du don des corps qui sont au nombre de 26 en France, et qui sont tous sous la dépendance des
universités et le plus souvent des anatomistes, sauf celui de l’assistance publique rue du fer-à-moulin
qui est le seul centre hospitalier (particularisme parisien).
Sur la coupe anatomique, on retrouve la courbure sacrale, avec le promontoire, la première
vertèbre sacrée qui est quasiment horizontale, puis les vésicules séminales dans le cul-de-sac de
Douglas, ainsi que le fascia de Denonvilliers.
C. Vascularisation
i. Les artères
Les artères du rectum expliquent pourquoi on a longtemps considéré rectum et canal anal comme
appartenant au même organe : leur vascularisation est similaire.
L’aorte donne trois branches, impaires et médianes : le tronc cœliaque en T12, l’artère
mésentérique supérieure en L1, et l’artère mésentérique inférieure en L3, parfois à la jonction
entre L3 et L4, qui est la plus petite et qui est très souvent bouchée par l’athérosclérose. L’artère
mésentérique inférieure donne l’artère colique gauche puis le tronc des artères sigmoïdiennes
et enfin l’artère rectale supérieure (sa branche terminale) qui va se diviser au niveau de la
jonction recto-sigmoïdienne en deux branches, une branche droite et une branche gauche, et cette
artère rectale supérieure va être l’artère principale du rectum.
Il existe des branches dites secondaires, notamment l’artère rectale moyenne qui a un certain
degré d’inconstance, qui va vers le rectum sous-péritonéal, passe latéralement et va vasculariser
aussi mais de façon secondaire le rectum.

62
Enfin, on trouve l’artère rectale inférieure qui est une branche de l’artère pudendale (elle-
même branche de l’iliaque interne), qui contourne l’épine sciatique, passe dans la fesse puis
repasse dans le périnée par le canal pudendal. Cette artère est visible à la face interne de l’ischion,
où elle donne l’artère rectale inférieure qui va vasculariser l’anus.
Ces trois artères (rectale supérieure, moyenne et inférieure) sont anastomosées entre elles, ce
qui permet de faire de la chirurgie : si on lie l’artère mésentérique inférieure, le rectum reste
vascularisé par la rectale moyenne et/ou par la rectale inférieure.
La nomenclature est très marquée par l’histoire, et par l’histoire des sociétés. L’artère pudendale
vascularisant des zones considérées comme honteuses, elles étaient nommées artères honteuses car
elles vascularisaient des zones dont il ne fallait pas parler. Maintenant on les appelle pudendales car
elles vont vers le périnée, donc le pudendum.
ii. Les lymphatiques
Les lymphatiques suivent à rebours les artères. La principale voie de drainage lymphatique est
la mésentérique inférieure, où les ganglions vont jusqu’en latéro-aortique. Lors d’une chirurgie
du cancer du rectum, la ligature de la mésentérique inférieure permet de faire ce qui s’appelle un
curage en bloc où on emmène l’artère mésentérique inférieure ainsi que tout le drainage
lymphatique. Mais il existe un drainage minoritaire, le long de la rectale moyenne vers l’iliaque
interne ou vers la sacrale moyenne (encore plus rare).
Lorsqu’on est au niveau de l’anus, les cancers du très bas rectum ou les cancers de l’anus (en
général épidermoïdes transmis par les infections au papillomavirus) se drainent au niveau de la rectale inférieure, mais les lymphonoeuds, les ganglions ne se drainent pas dans le canal pudendal.
Ils se drainent en superficie vers le trigone fémoral. Palper le trigone fémoral fait donc partie de
l’examen clinique, dans le cas d’une infection de l’anus ou des voies génitales, on a des
adénopathies au niveau du trigone fémoral, tout comme dans le cas d’un cancer de l’anus.
La voie minoritaire (sacrale médiane et iliaque interne) est intéressante dans le cas d’une RCP
(Réunion de Concertation Pluridisciplinaire en cancérologie) puisque le drainage eut s’y effectuer et
surprendre beaucoup de médecins. Dans le curage habituel du cancer du mésorectum, on enlève la
loge du mésorectum, on coupe la mésentérique inférieure, mais on ne va pas chercher les ganglions
iliaques internes. Pour les tumeurs gynécologiques ou urologiques, ces adénopathies iliaques internes
sont le drainage principal, il y aura donc des curages iliaques.
iii. Les veines
Concernant le système veineux, il existe un double drainage : un drainage porte, et un
drainage cave (ainsi que des légendes urbaines). Le drainage veineux est parallèle à l’irrigation
artérielle. Les veines rectales inférieures vont récupérer les veines sigmoïdiennes puis récupérer
la veine colique gauche pour former la veine mésentérique inférieure et aller se jeter soit
directement dans la veine mésentérique supérieure avec la veine splénique, soit dans la veine
splénique, ce qui correspond au drainage porte, drainage principal. Quand on fait une exérèse du
rectum, on lie en même temps l’artère mésentérique inférieure (à un centimètre de l’aorte) et la
veine mésentérique inférieure (au bord inférieur du pancréas). Il existe aussi un drainage cave,

63
accessoire, par les veines rectales moyennes. Enfin, un drainage profond, par la veine rectale
inférieure puis la veine pudendale, qui se jette dans la veine iliaque interne.
On a donc des anastomoses porto-caves au niveau de la paroi du rectum puisque ces veines,
comme les artères, sont anastomosées.
On raconte traditionnellement que chez le malade cirrhotique, il y a une très forte pression veine
portale, où il y a un développement de pathologies hémorroïdaires. Il est vrai qu’on trouve des
varices œsophagiennes et rectales dues à des anastomoses porto-caves, mais l’immense majorité
des pathologies hémorroïdaires (appelées ainsi car les artères et veines rectales étaient autrefois
appelées artères hémorroïdales) est due aux plexus artérioveineux. Quand un malade saigne des
hémorroïdes, il saigne rouge vif, c’est un saignement artériel. Ce n’est donc pas le développement
des veines qui fait qu’on saigne des hémorroïdes, il s’agit d’une pathologie artérioveineuse qui
peut mener à des complications hémorragiques artérielles lors d’une opération. En résumé : oui
il y a des anastomoses porto-caves, peut-être que ces anastomoses favorisent l’apparition de
varices rectales chez le cirrhotique, mais la maladie hémorroïdaire est due à une pathologie des
plexus artérioveineux liée à ces anastomoses.
On retrouve un drainage veineux profond de l’anus qui se fait par la veine rectale inférieure, mais
le drainage superficiel de la peau de l’anus se fait vers le trigone fémoral, comme le drainage
lymphatique.
En conclusion, on retrouve une artère et une veine par étage (anus avec l’artère et la veine rectale
inférieure, rectum sous-péritonisé avec l’artère et la veine rectale moyenne, rectum péritonisé
avec l’artère et la veine rectale supérieure).
/ !\ Attention, ces artères et ces veines sont paires (une de chaque côté).
III. Le canal anal

64
Comme la première coupe, celle-ci correspond soit à une personne sans muscle, soit à une personne
pliée au maximum, permettant l’alignement entre rectum et anus.
Le canal anal était autrefois appelé rectum périnéal.
On rappelle que le tube digestif contient une couche circulaire interne et une couche
longitudinales externe qui donne par épaississement au niveau du colon les tænia coli. La couche
circulaire interne va s’épaissir au niveau de l’anus pour former le sphincter interne. Ce sphincter
s’ouvre dès que les matières arrivent dans le rectum (pas très utile …). Par ailleurs, on a la couche
longitudinale externe qui va se terminer au niveau de l’anus dans le ligament de Parks qui va venir
passer à travers les faisceaux des sphincters externes (sphincters de muscles striés volontaires).
Ce sont eux qui seront responsables de la continence. On retrouve aussi à la fois un sphincter
strié (sous commande volontaire) et un sphincter lisse (sous commande neuro-végétative) à
l’extrémité de l’urètre.
La ligne ano-rectale est juste au-dessus de la ligne pectinée, juste au-dessus du sphincter
interne et juste au-dessus du plancher pelvien. Lors d’un toucher rectal, on sent un angle, le
plancher rectal, cela nous indique que nous sommes à la jonction ou ligne ano-rectale.
La ligne pectinée est le vestige de la membrane cloacale qui fait la différence entre entoblaste et
ectoblaste chez l’embryon et qui explique la double origine de l’anus : la partie supérieure
correspond à de l’entoblaste et la partie inférieure à de l’ectoblaste. La muqueuse est proche de la
muqueuse rectale au niveau de la ligne pectinée alors que dans le canal anal pur on est face à une
structure malpighienne.
Sous la ligne pectinée, on trouve la ligne ano-cutanée qui est en regard de l’espace entre le
sphincter interne et le faisceau superficiel du sphincter externe.
Lorsque que le malade est en position de la taille, ou position gynécologique, on touche le sillon.
Ce sillon correspond à la marge anale, est poilu et au-dessus on se situe dans le canal anal. Le
sphincter interne est au contact du canal anal (par opposition au sphincter externe).

65
Le sphincter externe est traversé par des fibres issues de la couche longitudinale externe qui
rattachent le sphincter à la partie profonde de la peau. C’est ce qui fait qu’on a des plis au niveau
de l’anus (les plis radiés de l’anus), le sphincter est fixé par ces fibres à la peau, c’est un faisceau
sous-cutané.
On peut aussi voir sur la coupe le faisceau profond du sphincter externe (le muscle le plus latéral).
IV. L’appareil sphinctérien externe
La description de cet appareil a été popularisée par un Egyptien qui s’appelle Shafik. Il s’est intéressé
aux sphincters de l’anus, à la valve iléo-caecale.
L’appareil sphinctérien externe est composé de plusieurs éléments :
- Le faisceau sous-cutané du sphincter externe
- Le faisceau superficiel du sphincter externe (plus profond que le sous-cutané) qui se
rattache au ligament anococcygien
- Le faisceau pubo-rectal du muscle élévateur de l’anus
Ces trois composantes sont striées sous contrôle volontaire.
La continence anale est facilitée par :
- L’angle anorectal : la pression abdominale appuie sur la partie inférieure du rectum ce
qui crée un effet sifflet où les deux parois sont collées l’une à l’autre à cause de la
pression. L’angle anorectal est permis par le tonus du muscle puborectal ;
- Le tonus des sphincters (interne et externe) ;
- La consistance des selles (notre corps n’est pas fait pour retenir des selles liquides).
V. Physiologie de la défécation

66
On voit sur le schéma en haut à gauche la moelle lombale (qui donne les nerfs sympathiques) et
la moelle sacrée (qui donne les nerfs parasympathiques). Certains nerfs partent de la corne
intermédio-latérale de la corne lombale qui font relai dans le tronc sympathique lombal puis
donnent des fibres vers le plexus pelvien latéral et donner des fibres effectrices vers le sphincter
interne. De la même façon, on retrouve un système parasympathique, qui aura un rôle antagoniste
au système sympathique afin de créer un équilibre.
Parallèlement, on trouve des récepteurs conscients :
- R1, récepteurs extéro-ceptifs de la marge anale
- R2, récepteurs de la zone cutanée lisse (au-dessus de la ligne ano-cutanée, sous la ligne
pectinée)
- R3, barorécepteurs rectaux (de la partie inférieure du rectum)
Ces récepteurs conscients passent par le nerf anal qui est une branche du nerf pudendal qui
passe par le canal pudendal avec l’artère et la veine rectales inférieures. Ce nerf est conscient. Les
voies sensitives vont nous informer que des choses se passent dans notre bas rectum. Ces
récepteurs nous indiquent aussi la consistance (solide, liquide, gazeux). R2 nous indique que
« c’est en train de descendre » et R3 nous dit « c’est sorti ».
Parallèlement, parmi les voies effectrices, le système sympathique va induire une contraction du
sphincter interne lisse, le parasympathique une action de relâchement. Par contre, les nerfs
pudendal et anal qui passent tous deux par le canal pudendal sont des nerfs somatiques
volontaires donc nous permettent de contracter le pubo-rectal, le faisceau superficiel et le faisceau
sous-cutané du sphincter externe. Le sphincter interne est donc sous commande autonome
tandis que le sphincter externe est sous commande volontaire.
Lors d’un remplissage du rectum, le réflexe recto-anal inhibiteur végétatif va aboutir au
relâchement du sphincter interne et donc à une descente du contenu rectal qui va aller affleurer
les récepteurs de la partie basse du rectum afin de porter à notre conscience le contenu du rectum.
Ce réflexe est absent dans la maladie de Hirschsprung.

67
Ensuite, on a soit relâchement, défécation, position demi-assise pour permettre le relâchement du
pubo-rectal, soit au contraire acte volontaire de contraction.
Pour se souvenir que la parasympathique relâche le sphincter interne, il faut se souvenir qu’un
malaise dit vagal (par stimulation vagale) aboutit à un ralentissement du cœur, une perte d’urine et
parfois une perte de matières (rare).
VI. Les pathologies
A. Les hémorroïdes
Les hémorroïdes sont des plexus artérioveineux de deux types :
- Liés aux anastomoses entre les branches artérielle et veineuse pudendales
- Liés aux anastomoses rectales moyennes et supérieures
Au niveau de la partie basse du rectum et de la ligne pectinée, on retrouve le développement de
ces plexus artérioveineux hémorroïdaires. On peut aussi observer un développement de ces
anastomoses au niveau externe, entre les branches interne, moyenne et supérieure.
On dit classiquement qu’il y a trois paquets hémorroïdaires avec des branches terminales de
l’artère rectale supérieure qui vont aller à 3h, 8h et 11h (cf schéma) s’anastomoser avec les autres
branches. C’est vrai en clinique mais en faisant des Dopplers, on découvre qu’il y a plus de
branches que ça.
Les hémorroïdes internes sont traitées en priorité, les externes sont moins importants.

68
B. Les fistules
Les fistules anales sont liées à l’infection de glandes anales (d’Hermann et Desfosses) qui
s’ouvrent dans le fond des cryptes de la ligne pectinée. Ces glandes sont situées profondément
dans la paroi de l’anus, le cœur de ces glandes se situe dans la zone entre sphincter interne et
sphincter externe.
Personne ne sait pourquoi ces glandes se bouchent, toutes les explications proposées n’ont jamais
été vérifiées (aucune corrélation avec aucun mode de vie ou d’alimentation).
Si ces glandes provoquent une infection, elle va diffuser possiblement vers l’extérieur et
provoquer ce qu’on appelle un abcès de la marge de l’anus (si on trouve un abcès on doit
rechercher la glande qui en serait la cause)
La fistule anale est une maladie royale, Louis XIV avait une fistule anale. Il avait été opéré (sans
anesthésie) après bien sûr un entraînement sur quelques prisonniers. Son abcès suintait par
communication avec l’anus. Il a été opéré à vif par des chirurgiens qui étaient à l’époque distincts des
médecins puisqu’ils appartenaient au corps des barbiers avant Louis XIV.
Un des traitements est de couper le trajet (entre l’orifice primaire et l’orifice secondaire), mais
ceci implique de couper du sphincter. S’il s’agit de couper du sphincter interne, le risque d’avoir
de l’incontinence est assez faible étant donnée sa fonction. Par contre si le trajet englobe tout le
faisceau sous-cutané du sphincter on aura des troubles de la continence.
Par chance 70% des fistules passent entre les deux faisceaux du sphincter.
VII. Le muscle élévateur de l’anus

69
Le muscle élévateur de l’anus a plusieurs faisceaux, notamment un faisceau sphinctérien qu’on
appelle le faisceau pubo-rectal qui va permettre de maintenir le cap anal et de participer à la
continence. Il y a aussi un faisceau sur les côtés qui sert de frontière entre le pelvis et le périnée.
VIII. Conclusion
Le rectum est péritonisé ou sous-péritonéal, le canal anal est la partie du tube digestif qui est au
niveau du périnée. Il existe une unité de vascularisation qui fait que l’artère du rectum est l’artère
mésentérique inférieure et l’artère de l’anus est un peu la mésentérique inférieure et surtout
l’artère rectale inférieure, l’artère rectale moyenne étant inconstante.

70
FICHE RECAPITULATIVE : Anatomie du rectum et du canal anal
Le rectum et l’anus constitue la partie terminale du tube digestif. /!\ Anus (dans le périnée) ≠ rectum (dans le petit bassin = cavité pelvienne) ; séparés par le muscle élévateur de l’anus Le rectum comporte 2 étages : - Péritonisé - Sous péritonéal Au niveau périnéal c’est le canal anal (délimité par la ligne ano-rectale) Délimitation de la loge rectale (rectum sous péritonéal) : Sur les faces latérales du rectum, on a les lames sacro-recto-génito-pubienne contenant le plexus hypogastrique ou pelvien latéral = innervation végétative. En arrière du rectum, on a le fascia pré-sacral qui sépare en avant la loge rectale de l’espace pré-sacré (contient des veines et artères) Chez la femme on a le cul de sac péritonéal dit de Douglas (recto-vaginal) et 3 loges (rénale, génitale et rectale) Chez l’homme on a le fascia prostatopéritonéal Denonvilliers (entre rectum et prostate) et 2 loges (uro-génitale et rectale) Le rectum et le canal anal sont vascularisés par trois pédicules (comportant artères, veines et système lymphatique) Vascularisé par 3 artères (anastomosées entre elles) : - Artère Rectale supérieure née de l’artère mésentérique inférieure - Artère Rectale moyenne née de l’artère iliaque interne (paire) - Artère Rectale inférieure née de l’artère pudendale (paire) Drainé par 3 veines : - Veine Rectale supérieure vers veine mésentérique inférieure (système porte) - Veine Rectale moyenne vers veine iliaque interne (système cave) (paire) - Veine Rectale inférieure vers veine pudendale (système cave) (paire) Les lymphatiques suivent à rebours les artères, principalement la mésentérique inférieure. Canal anal (= rectum périnéal) = zone où se trouvent les sphincters Sphincters : Un sphincter interne lisse (= épaississement de la couche circulaire interne du rectum) végétatif et un sphincter externe strié volontaire Il y a 3 faisceaux au sphincter strié externe : - Le faisceau superficiel - Le faisceau profond (= faisceau pubo-rectal du muscle élévateur de l’anus) permettant le maintien de l’angle entre rectum et anus - Le faisceau sous-cutané (donne les plis radiés de l’anus) Physiologie de la défécation : 1) Récepteurs +++ sensibles à la distension du sphincter interne et de la partie inférieure du rectum. Ces informations provenant du plexus hypogastrique qui passe par les lames sacro-recto-génito-pubiennes sont transmises au cerveau. 2) Innervation de 2 types : -somatique, motrice : muscle élévateur de l’anus - végétative défécation et continence des selles (une part somatique (volontaire) et végétative (involontaire)). Les nerfs anaux et pudendaux (somatique) amènent une contraction du pubo-rectal qui va maintenir l’angle anorectal, le tonus des faisceaux sous-cutané et superficiel du sphincter pour maintenir une continence volontaire mais épuisable.

71
Maladie de Hirschsprung => absence de réflexe recto-anal inhibiteur => Ce réflexe ouvre spontanément le sphincter interne lorsque les matières arrivent = incontinence. Malaise vagal => risque d’incontinence (stimulation vagale) Les Pathologies : Les hémorroïdes = naissent de plexus artérioveineux Les fistules = infection des glandes anales (situées entre le sphincter interne et externe), diffuse vers l’extérieur et forme un abcès

72
UE6 – SD – Histologie - n°2
16/01/2018
Patrick BARBET
RT : Audrey Cochennec
RL : Laurent Onfroy
Histologie du tube digestif (2ème partie)
Plan :
I. Généralités histologiques du tube digestif
A. La musculeuse digestive
B. L’innervation de la paroi digestive
II. Variations topographiques de la paroi
A. L’œsophage
B. L’estomac
C. L’intestin grêle

73
I. Généralités histologiques du tube digestif
A. La musculeuse digestive
Dans la paroi digestive on retrouve 2 zones musculaires : la musculaire muqueuse et la
musculeuse avec ses 2 couches, circulaire interne et longitudinale externe. Entre ces 2 couches
on trouve le plan myentérique qui possède les plexus nerveux myentériques assurant le
contrôle intrinsèque de la contraction musculaire. Ces éléments musculaires sont
essentiellement lisses sauf dans la partie haute du système digestif (de l’oropharynx jusqu’au
1/3 supérieur de l’œsophage) et une partie du rectum où il y a une musculature striée.
Au niveau de l’intestin, la musculaire muqueuse (qui peut elle-même être organisée à différents
niveaux d’une circulaire interne et d’une longitudinale externe), notamment dans l’intestin grêle,
peut émettre des expansions verticales jusqu’au sommet de la muqueuse des villosités
intestinales, ce sont les muscles de Brüucke.
Dans le tube digestif on trouve des sphincters dont le sphincter inférieur de l’œsophage et le
pylore (=sphincter musculaire lisse) qui encadrent l’estomac, ce dernier correspond à un
épaississement progressif de la circulaire interne dans toute la région antro-pylorique.
Puis à la fin du tube digestif on trouve le sphincter ano-rectal qui fait suite à la paroi digestive
où l’on a une musculeuse avec une circulaire interne et une longitudinale externe. Contrairement
au pylore on a un épaississement local de la circulaire interne avec des faisceaux de fibres
musculaires lisses empilés qui ont tendance à s’aplatir, il forme le sphincter interne lisse à
contraction involontaire. En complément, à la périphérie du sphincter interne, on trouve un
ensemble de structures musculaires striées du sphincter strié à contraction volontaire
possédant une particularité histologique, la longitudinale externe envoie des expansions
musculaires lisses à l’intérieur du sphincter strié. Il y a donc une intrication de muscle lisse et
de muscle strié pour former une structure composite.
B. Innervation de la paroi digestive
La motricité digestive est entretenue grâce à un système nerveux intrinsèque de la paroi
digestive. On va observer dans ce système nerveux différents types de cellule :
Des neurones (=cellules ganglionnaires) dont les corps cellulaires sont situés à des
endroits très précis qu’on appelle des plexus (zones où il y a les corps cellulaires des
neurones). Ces neurones vont former des prolongements qui vont donner un véritable
maillage. Au microscope on observe que les neurones sont arrondis avec un noyau bien
développé et un cytoplasme basophile (neurotransmetteur)
Des cellules gliales tout autour qui servent de protection (soutien) comme les cellules
de Schwann présentes autour des prolongements
On trouve en plus de cellules particulières : les cellules de Cajal qui ont un rôle de
pacemaker et permettent une contraction coordonnée. Elles se trouvent dans le
plexus myentérique

74
Le tube digestif est innervé par deux plexus différents situés dans deux couches différentes. La
première région est le plan myentérique, on les appelle les plexus myentériques (= plexus
d’Auerbach). La deuxième région est la sous muqueuse, on les appelle les plexus de Meissner.
Il y a également des constituants nerveux dans la muqueuse.
Il y a une particularité dans le sphincter ano-rectal, l’innervation se raréfie, c’est donc une zone
hypo ganglionnaire Chez un nourrisson, lors du diagnostic de la maladie de Hirschsprung (=
absence d’innervation de la partie terminale du colon), si l’on veut vérifier l’innervation de la
paroi il faut aller 3 cm plus haut que le sphincter ano-rectal lors de la biopsie.
III. Variations topographiques de la paroi digestive
A. L’Œsophage
La muqueuse œsophagienne : possède un épithélium pavimenteux malpighien non
kératinisé. De plus on observe dans cette couche des glandes muqueuses exocrines ; en
particulier au 1/3 supérieur de l’œsophage et à la partie basse de l’œsophage. Ces glandes qui se
trouvent à la jonction de l’œsophage et de l’estomac (=cardia), sont appelées les glandes
cardiales.
La sous-muqueuse : au 1/3 moyen de la sous-muqueuse de l’œsophage on trouve des glandes
exocrines que l’on appelle glandes œsophagiennes essentiellement muqueuses qui sont
drainées par des canaux excréteurs. Autour de ces canaux on peut observer des cellules
lymphocytaires (=tissu lymphoïde).
La musculeuse : Elle est divisée en 3 parties distinctes :
On a au 1/3 supérieur, dans le prolongement du plan pharyngien, un muscle strié à
contraction volontaire
Au 1/3 moyen il y a une transition progressive d’un muscle strié vers un muscle lisse
(il y a donc des fibres musculaires lisses et des striées)
Au 1/3 inférieur on trouve un muscle lisse à contraction involontaire
B. L’Estomac
La muqueuse : est composée d’un épithélium de surface prismatique simple à pole muqueux
fermé qui sert de protection chimique. A la partie superficielle de la muqueuse, on aperçoit des
cryptes gastriques (=invagination de la muqueuse) dont la paroi est constituée de l’épithélium
de surface sans glandes. C’est dans la partie profonde, au prolongement des cryptes qu’il y a les
glandes.
Dans l’estomac il y a 2 territoires principaux qui se différencient par la nature des glandes que
l’on trouve dans le chorion de la muqueuse : le fundus et l’antro-pylore.
Au niveau du fundus on observe des glandes tubuleuses simples, rectilignes, allongées avec un
aspect bigarré, ce sont les glandes fundiques. Elles sont composées de différentes régions : on a

75
d’abord l’isthme, puis le col ou collet, et enfin le fond. De plus ces glandes sont constituées
plusieurs populations cellulaires :
Les cellules principales (les plus nombreuses) qui sont prismatiques avec un gros
noyau actif au centre et un cytoplasme mal coloré, elles sécrètent la pepsinogène. Ce
sont des cellules exocrines qui sécrètent par un mode mérocrine classique avec
quelques microvillosités à la surface apicale
Les cellules bordantes (les plus volumineuses) sont fortement actives avec un
cytoplasme éosinophile dû à la richesse en mitochondrie. Ce sont des cellules
glandulaires de type exocrine qui participent à la formation d’HCl et de facteur
intrinsèque. A la surface apicale de ces cellules on trouve de nombreuses microvillosités
et un espace d’invagination de la membrane plasmique de part et d’autre du noyau, on
définit cet espace comme une zone de canalicules sécrétoires
Les cellules mucoïdes au niveau du collet
Les cellules neuroendocrines sont mélangées avec les autres populations cellulaires,
notamment au niveau des glandes, elles permettent une synthèse hormonale (non
spécifique à l’estomac) via les vaisseaux du chorion interglandulaire
Des cellules précurseurs ou « souches » au niveau du collet qui permettent le
renouvellement au sein de l’épithélium qui aboutit vers le haut à des cellules à pôle
muqueux fermé (=épithélium de surface) et vers le bas à des cellules principales, ou
bordantes, mucoïdes ou neuroendocrines.
Dans le territoire antro-pylorique on a des glandes plus courtes et ovalaires, on les appelle les
glandes muqueuses pures. Au plan histologique la transition entre le fundus et l ‘antro-pylore
est progressive, c’est la zone de transition.
La musculeuse : elle est renforcée dans certains territoires par une troisième couche oblique
interne. Elle sera donc composée de 3 couches : l’oblique interne, la circulaire moyenne et la
longitudinale externe.
C. L’Intestin grêle
La muqueuse intestinale : est composée d’un épithélium de surface de type prismatique
simple avec des cellules striées, caliciformes, neuroendocrines, « souches » et des cellules de
Paneth.
On trouve dans le chorion de la muqueuse (que ce soit dans le grêle ou le colon) les glandes de
Lieberkuhn qui sont une invagination de l’épithélium de surface où l’on voit essentiellement
des entérocytes et des cellules caliciformes, mais observe également d’autres populations
cellulaires comme les cellules de Paneth au fond de la glande (principalement dans l’intestin
grêle) et des cellules souches situées près des cellules de Paneth sur les bords latéraux qui
permettent le renouvellement de l’épithélium vers le haut pour donner des entérocytes, des
cellules caliciformes ; et vers le bas pour donner des cellules de Paneth.
Dans l’intestin grêle, on retrouve 2 territoires qui se distinguent par la présence ou l’absence de
villosité. Le relief des villosités augmente la surface (X10) ce qui favorise les échanges sous
forme d’absorption intestinales. Une villosité intestinale est une expansion dans la lumière de la

76
muqueuse entièrement recouverte d’un épithélium d’absorption. Mais une villosité n’est pas
qu’une simple expansion structurelle, c’est une vraie unité fonctionnelle qui se caractérise par
3 éléments morphologiques :
Des expansions musculaires lisses qui proviennent de la musculaire muqueuse et qui
va jusqu’au sommet de la villosité, c’est le muscle de Brüucke. Les villosités ne sont
donc pas inertes, on décerne ainsi deux types de mouvements involontaires : soit
pendulaire soit de raccourcissement
Chaque villosité comporte une anse vasculaire qui lui est propre : une artériole va
donner deux branches qui vont se diviser pour donner un réseau de capillaires à la
villosité qui sera ensuite drainée par une veinule. Il y a également une branche
anastomotique sans voie d’échange en amont du capillaire.
Il y a un réseau circulatoire complémentaire, c’est la circulation lymphatique. Ce
réseau circulatoire possède 1 ou 2 (2 le plus souvent) vaisseaux chylifères qui partent du
sommet de la villosité et qui assurent donc un drainage lymphatique depuis la villosité
jusqu’au tissu conjonctif sous-jacent.
Ainsi à la partie profonde de la muqueuse, on décrit 2 types d’endroit : le socle de la villosité et
les espaces intervillositaires avec les glandes de Lieberkuhn. On trouve une continuité entre le
revêtement des glandes et du revêtement des villosités. Ceci nous permet de définir une unité
fonctionnelle : l’entéron composé d’une villosité intestinale et des glandes de Lieberkuhn à son
contact.

77
Et 2 schémas en plus pour comprendre ! (RD : Clémence Gleize)

78
Fiche récapitulative : Histologie du tube digestif (2ème
partie)
I. Généralités histologiques de la paroi digestive
A. La musculeuse digestive, essentiellement lisse (sauf partie haute et rectum à fibres striées) avec 2 zones : - la musculaire muqueuse, avec les muscles de Brücke au niveau de l'intestin (grêle ++) - la musculeuse avec les couches circulaires interne, le plan myentérique (dont les plexus assurent le contrôle intrinsèque de la contraction) et la longitudinale externe Les sphincters : - inférieur de l'œsophage - le pylore = sphincter musculaire lisse = épaississement progressif de la circulaire interne dans la région antro-pylorique - ano-rectal : interne lisse à contraction involontaire = épaississement local de la circulaire interne + sphincter strié à contraction volontaire en périphérie avec des expansions de la longitudinale externe => intrication entre lisse et strié
B. Innervation => système nerveux intrinsèque avec comme types cellulaires - les neurones avec des corps cellulaires regroupés en plexus et prolongements en maillage - les cellules gliales pour le soutien (ex:ex : cellules de Schwann) - les cellules de Cajal = pacemaker, dans le plan myentérique => contraction coordonnée 2 plexus : plexus myentérique (= d'Auerbach) et plexus de Meissner dans la sous-muqueuse Le sphincter ano-rectal = hypoganglionaire (attention lors d'une biopsie du colon chez un nourrisson)
II. Variations topographiques de la paroi digestive
A. Œsophage
Muqueuse = Epithélium pavimenteux malpighien non kératinisé avec glandes muqueuses
exocrines au 1/3 sup et à la partie basse (cardiales)
Sous-muqueuse : glandes œsophagiennes muqueuses au 1/3 moyen avec canaux excréteurs et
tissus lymphoïde autour
Musculeuse :
-1/3 sup = strié volontaire
-1/3 moyen = transition progressive
-1/3 inf = lisse involontaire
B. Estomac
Muqueuse = épithélium de surface prismatique à pôle muqueux fermé => protection chimique
A la partie superficielle : cryptes (invaginations) sans glandes
2 zones selon nature des glandes chorioniques dans la profondeur, avec transition entre les 2:2 :
- Glandes fundiques = tubuleuses simples allongées bigarrées, avec comme régions l'isthme puis
le col et le fond, et comme populations cellulaires :
>Principales : sécrètent pepsinogène parpepsinogène par un mode mérocrine

79
>Bordantes : sécrètent HCl et facteur intrinsèque avec canalicules sécrétoires
>Endocrines : sécrètent hormones peptidiques
>Au niveau du col : mucoïdes et précurseurs (renouvellement)
-Glandes antro-pyloriques = courtes ovalaires "muqueuses pures"
Musculeuse parfois en 3 couches : oblique interne / circulaire moyenne / longitudinale externe
C. Intestin grêle
Muqueuse intestinale = épithélium prismatique simple avec cellules :
- Striées - "souches"
- Caliciformes - de Paneth
- Neuroendocrines Glandes de Lieberkuhn dans le chorion = invaginations de l'epithéliumépithélium de surface
avec ses populations cellulaires (caliciforme ++) dont les souches sur les bords (renouvellement)
Les villosités = expansion dans la lumière de la muqueuse => augmentation surface
d'absorption (x 10)
Ce sont de vrais unités fonctionnellesvraies unités fonctionnelles avec :
Le muscle de Brücke = expensionexpansion musculaire lisse de la musculaire muqueuse
=> mouvements involontaires (pendulaires ou de raccourcissement)
Une anse vasculaire propre : une artériole donne un réseau capillaire qui se draine dans
une veinule + une branche anastomotique
Une circulation lymphatique avec 1-2 vaisseau(x) chylifère(s) => drainage du sommet
jusqu'au TC sous-jacent
Partie profonde avec 2 territoires : le socle de la villosité et les espaces intervillositaires
(glandes)
L'entéron = villosité + glandes de LieberkhunLieberkuhn à son contact

80
UE6 – Système digestif – Histologie –
cours n°3
16 janvier 2018
Patrick Barbet
RT : Marine COCHEREL
RL : Garance DOMBRET
Histologie du tube digestif (3e partie) et embryologie
du tube digestif
I. Histologie : l’intestin grêle
A. Renouvellement cellulaire
B. La fonction d’absorption
C. Le système lymphoïde
D. Variations topographiques
II. Histologie : le gros intestin
A. Côlon – Rectum
B. Appendice
III. Embryologie : l’intestin primitif
A. L’intestin antérieur
B. L’intestin moyen
C. L’intestin postérieur

81
I. Histologie : l’intestin grêle
A. Renouvellement cellulaire Dans le chorion de la muqueuse intestinale se trouvent les glandes de Lieberkühn issues de
l’invagination de l’épithélium de surface dans l’espace intervillositaire, on a une continuité de
revêtement. Cependant, cette définition est incomplète puisqu’on y trouve les mêmes cellules
qu’en surface :
- Des entérocytes à plateau strié
- Des cellules caliciformes : plus on s’éloigne du duodénum et on se rapproche de l’extrémité
du rectum, plus leur nombre augmente.
- Des cellules neuroendocrines : ouvertes ou fermées suivant qu’elles s’ouvrent ou non vers
la lumière des glandes, elles sécrètent différentes hormones. On les regroupe dans le
système des paraneurones (neurones particuliers décrits par Fujita)
Mais aussi d’autres types cellulaires :
- Des cellules précurseurs ou « souches ».
- Des cellules de Paneth : situées au fond des glandes, elles sont réactionnelles et pleines de
granulations éosinophiles.
L’entéron est une unité fonctionnelle qui correspond à une villosité avec les glandes de
Lieberkühn qui l’entourent.
C’est à la partie moyenne de ces glandes que l’on trouve la zone germinative avec ses cellules
précurseurs qui vont permettre le renouvellement de toutes les cellules épithéliales en « glissant »
et en se différentiant :
- Soit vers le haut surtout pour les entérocytes, les cellules caliciformes au niveau des
villosités (= zone fonctionnelle)
- Soit en profondeur pour notamment les cellules de Paneth
Ce cycle dure environ 1 semaine.
B. La fonction d’absorption
Dans le territoire intestinal où il y a de l’absorption (essentiellement la partie proximale du grêle),
un certain nombre de mécanismes anatomiques ou histologiques contribuent à augmenter la
surface d’échange :
- Les valvules conniventes (structure anatomique) : replis semi-circulaires nombreux
dans la partie proximale de l’IG à partir de D3 car absentes de D1 et D2. Ils impliquent à la
fois la sous-muqueuse, la musculaire muqueuse et la muqueuse intestinale.
Augmentent la surface d’échange (x3)
- Les villosités (sur le sommet de la valvule) : unité fonctionnelle, expansion d’autant plus
haute qu’on est dans un territoire d’absorption importante (jéjunum proximal ++)
Augmentent la surface d’échange (x10)
- Les microvillosités au niveau du plateau strié à la surface de l’entérocyte : possèdent une
armature avec un axe de filament d’actine au centre relié à la partie superficielle du
cytosquelette = mur terminal (mais pas de mouvement !). De plus, des molécules assurent
le lien entre les filaments d’actine et les structures de voisinages : villine et fimbrine.

82
Augmentent la surface d’échange (x20) entre l’entérocyte et le film de mucus
(=glycocalyx formé par les cellules caliciformes) qui constitue un véritable
microclimat : zone de passage plus lent des substances, qui laisse le temps aux
échanges et permet finalement la fonction d’absorption.
On y trouve également des enzymes qui peuvent poursuivre la digestion.
NB : Les entérocytes sont moins fragiles que les autres cellules : il peut y avoir des destructions de
la membrane plasmique qu’il peut réparer lui-même => résistance aux agressions.
La surface théorique de la lumière digestive au niveau de l’IG est de 0,5 m² mais multiplier
par 600 (x3x10x20) : on dispose tous d’environ 300 m² de surface d’absorption.
C. Le système lymphoïde
Il existe un système lymphoïde associé à la muqueuse qui se manifeste partout.
- Lymphocytes physiologiquement présents à l’intérieur de l’épithélium intestinal =
lymphocytes intra-épithéliaux.
Nombre normal : 1 lymphocyte pour 5 cellules épithéliales
Nombre qui peut augmenter dans certaines maladies : dans la maladie cœliaque c’est
l’un des 1ers signes histologiques.
- A côté, il y a des cellules lymphoïdes notamment dans le chorion interglandulaire qui
constituent un infiltrat physiologique normal => diffus.
D’autre part on trouve à certains endroits des structures cette fois-ci réactionnelles = follicules
lymphoïdes
Organisés
Zone où le relief de surface de la muqueuse est particulier (pas de villosité au niveau
du grêle) et où l’épithélium possède des cellules particulières = cellules M
(présentatrices d’antigène)
A la partie terminale de l’iléon on peut trouver une volumineuse et très importante
formation lymphoïde = plaque de Peyer
Ainsi les zones particulières lymphoïdes du GALT (Gut-Associated Lymphoid Tissue) sont :
Les plaques de Peyer
L’appendice

83
D. Variations topographiques
- Duodénum histologique = D1 et D2 : zone où en plus des glandes de Lieberkühn on
trouve soit dans le chorion de la muqueuse, soit surtout dans la sous-muqueuse, des
glandes muqueuses pures = Glandes de Brunner.
NB : ≠ duodénum anatomique (D1- D2 – D3 – D4)
Peu de cellules caliciformes
Villosités relativement courtes (villosités les plus hautes sont dans la zone d’absorption
maximal => au début de l’IG) et foliées (aplaties). Pour mesurer leur hauteur, on évalue la hauteur de la villosité à proprement parlé par rapport à
la hauteur des glandes de Lieberkühn.
Dans le duodénum : rapport de 2 à 3/1
- La vraie structure d’absorption : à partir de D3, l’essentiel du jéjunum et la partie
initiale de l’iléon.
Villosités hautes et digitiformes en majorité (rapport : 3 à 5/1)
- Iléon terminal : absorption diminuée
Villosités courtes (rapport : 3/1)
Très nombreuses cellules caliciformes
On peut trouver soit un tissu lymphoïde peu abondant ou de volumineuses plaques de
Peyer.
II. Histologie : le gros intestin
A. Côlon – Rectum La structure de type colique est caractérisée en principe par une lumière assez large et une paroi
fine.
Elle possède plusieurs caractéristiques propres :
Pas de villosité
Pas de muscle de Brücke ni de chylifère central
On ne trouve que les invaginations des glandes de Lieberkühn (en coupe elles forment
le « champ de marguerite »)
Beaucoup de cellules caliciformes
Réaction locale : tissu lymphoïde représenté par des nappes lymphoïdes diffuses auxquelles
s’associent parfois, surtout au niveau du rectum, des macrophages (peuvent se charger de lipides).
B. Appendice
Dans le gros intestin se trouve un appendice vermiculaire => formation de type colique associée
au caecum avec une paroi digestive en 5 couches, bordée par une muqueuse intestinale

84
atrophique (peu développée) avec une lumière étroite (contrairement au reste de la structure
colique).
De plus, on trouve des formations lymphoïdes à cheval entre la muqueuse et la sous muqueuse
qui sont si riches qu’on a du mal à distinguer la musculaire muqueuse.
Enfin, on trouve l’adventice en périphérie que l’on peut suivre partout jusqu’à une zone où la
partie digestive est reliée au reste par une lame de TC avec des vaisseaux sanguins = méso-
appendice
III. Embryologie : l’intestin primitif
La mise en place des structures de base pendant la période de l’organogénèse se poursuit pendant
la période fœtale par la croissance et la différenciation, et pour le tube digestif par la mise en place
de structures (activité enzymatique, villosités, etc.) qui continuera après la naissance.
L’appareil digestif est constitué à partir de cellules qui proviennent de différents feuillets : Entoblaste => cellules épithéliales
Mésoblaste => cellules conjonctives, musculaires lisse, éléments vasculaires
Crêtes neurales => cellules nerveuses du système nerveux périphérique
L’intestin primitif se divise en trois territoires : antérieur – moyen – postérieur.
A. L’intestin antérieur (crânial)
Il correspond à tout le territoire entre membrane pharyngienne et la région des bourgeons
hépatique et pancréatique et se subdivise en deux parties :
- Une partie crâniale = intestin pharyngien : il contribue à la formation des structures qui
correspondent
En haut : à la région faciale et cervicale
En bas : à la partie supérieure du médiastin dominée par les éléments vasculaires
(aorte et artère pulmonaire).
L’entoblaste évolue pour donner naissance à des structures digestives (l’œsophage,
estomac, première partie du duodénum) et du diverticule respiratoire ainsi qu’aux cellules
épithéliales qui tapissent les voies excrétrices du foie et du pancréas.
L’évolution des structures ne va pas conserver une organisation sagittale : l’estomac ne
reste pas fusiforme ou tubulaire sinon c’est une malformation congénitale (pour le tube
digestif, l’élément clé sont les vitesses de croissance de telle ou telle partie de l’estomac,
et non pas les contraintes extérieures).
- Une partie caudale
B. L’intestin moyen
Il correspond à l’anse intestinale primitive, qui part du milieu de D2 et se termine à la jonction
partie mobile – partie fixe du colon transverse. Elle est centrée par l’artère mésentérique
supérieure.

85
Développement par des mouvements de croissance différentielle ++ (surtout à la
partie crâniale)
La partie crâniale va donner le jéjunum et essentiel de l’iléon
Correspondance précise entre le territoire de l’artère mésentérique supérieure et le
territoire de l’intestin moyen.
C. L’intestin postérieur (caudal)
Il correspond au territoire entre l’intestin moyen et la membrane cloacale.
La partie anale est en partie formée par une structure d’origine épiblastique =
proctodéum (revêtement malpighien chez l’adulte qui n’est donc pas issu de
l’entoblaste)
Mise en place du système nerveux intrinsèque : migration de cellules provenant des
crêtes neurales céphaliques selon un gradient cranio-caudal.
Exemple d’anomalie du développement, la maladie de Hirschsprung = forme simple
d’obstruction intestinale à cause d’une zone mal innervée. On observe une dilatation en amont de
la zone obstruée => mégacôlon congénital.
Parmi les causes de cette maladie il y a des défauts de migration des cellules nerveuses
ganglionnaires
Dans le cas d’une forme complète : le défaut de migration des neuroblastes est plus ou
moins étendu ce qui peut conduire à une transplantation intestinale.
Abréviations :
IG : Intestin grêle
TC : Tissu conjonctif

86
Fiche récapitulative : Histologie du tube digestif (3e partie)
et embryologie du tube digestif
Histologie
I. L’intestin grêle
A. Renouvellement cellulaire
Muqueuse : entérocytes, cellules caliciformes, cellules neuroendocrines, cellules précurseurs et
cellules de Paneth.
La zone germinative se trouve à la partie moyenne des glandes de Lierberkühn, elles migrent soit
vers le haut (zone fonctionnelle – villosités) soit vers le bas (cellules de Paneth). Les cellules de la
muqueuse ont une durée de vie d’environ 7 jours.
B. La fonction d’absorption
Grande surface d’échange de 300 m² assurée par :
- Les valvules conniventes : à partir de D3, replis de la sous-muqueuse. (x3)
-Les villosités : unité fonctionnelle. (x10)
-Les microvillosités : du plateau strié des entérocytes (x20).
La couche de mucus (glycocalyx) permet aussi une amélioration de la fonction d’absorption.
C. Le système lymphoïde
-Lymphocytes intra-épithéliaux : Diffus ; 1 pour 5 cellules épithéliales (augmente dans la maladie
cœliaque) ; accompagnés de cellules lymphoïdes formant ainsi un infiltrat physiologique normal.
-Follicules lymphoïdes : structures réactionnelles, organisées, avec un relief de surface sans
villosités et avec des cellules M présentatrices d’Ag. Les plaques de Peyer sont des formations
lymphoïdes volumineuses situées à la partie distale de l’iléon.
D. Variations topographiques
- Duodénum histologique (D1, D2) : glandes de Brunner ; villosités foliées ; rapport
villosités/glandes de Lieberkûhn (V/G) = 2-3/1
-De D3 à la partie initiale de l’iléon : villosités hautes et digitiformes ; rapport V/G = 3-5/1
-Iléon terminal : rapport V/G = 3/1 ; cellules caliciformes et plaques de Peyer.
II. Le gros intestin
A. Côlon –Rectum
Ils ont une lumière large, une paroi fine, pas de villosités, pas de muscle de Brücke. On y trouve
des glandes de Lieberkühn et beaucoup de cellules caliciformes.
B. Appendice

87
Appendice vermiculaire associé au caecum. La paroi comporte 5 couches avec quelques
particularités :
-muqueuse atrophique avec une lumière étroite
-musculaire muqueuse : difficile à distinguer à cause des formations lymphoïdes.
-L’adventice se poursuit en méso-appendice (lame de TC)
Embryologie
L’intestin primitif
Il est composé de cellules de l’entoblaste, du mésoblaste et des crêtes neurales.
A. L’intestin antérieur
De la membrane pharyngienne aux bourgeons hépatique et pancréatique. On a :
-une partie crâniale (intestin pharyngien) : Correspond aux régions faciale et cervicale et à la
partie supérieure du médiastin. L’entoblaste va donner les structures digestives, le diverticule
respiratoire et les épithéliums des voies excrétrices du foie et du pancréas
-une partie caudale : l’estomac ne reste pas tubulaire il se développe grâce à des vitesses de
croissances différentes selon les parties.
B. L’intestin moyen
Anse intestinale primitive : de D2 au colon transverse. Elle se développe par une croissance à
vitesses différentielles. L’intestin moyen correspond au territoire de l’artère mésentérique
supérieure.
C. L’intestin postérieur
De l’intestin moyen à la membrane cloacale.
-Le proctodéum à la partie anale vient de l’épiblaste. (revêtement malpighien)
Le système nerveux intrinsèque vient des crêtes neurales céphaliques. Les neuroblastes migrent
ensuite suivant un gradient cranio-caudal.
La maladie de Hirschsprung correspond à un défaut de migration des neuroblastes => mégacôlon
congénital.

88
UE6 – SD Physiologie- Cours n°3
17/01/18
Pascal HOUILLIER [email protected]
RT : Laure COËZ
RL : Elise POITRINAL
Physiologie digestive : digestion et absorption
Plan :
III. Les hydrates de carbones A. Généralités B. Digestion et absorption C. Syndrome de malabsorption
IV. Les protéines
A. Généralités B. Digestion et absorption C. Déficit d’absorption
V. Les vitamines hydrosolubles A. Vue d’ensemble B. La vitamine B12 C. La vitamine C
VI. Les lipides A. Généralités B. Digestion et absorption
VII. Les vitamines liposolubles
VIII. Les points clés
Mot du RT : Un cours dense, mais pas compliqué. J’ai essayé de l’alléger le plus possible, et de choisir les slides illustrant au mieux l’idée générale. N’hésitez pas à jeter un œil au powerpoint qui est bien fait. Bon courage !

89
Introduction Le rôle de l’alimentation est de fournir à notre organisme des nutriments qui vont être utilisés pour assurer notre survie : ce qu’on ingère n’est, pour la plupart, pas directement absorbable et doit passer par un phénomène préalable de digestion. Il existe néanmoins des éléments directement absorbables comme le glucose. Les protéines doivent être digérées et transformées en acides aminés par les enzymes luminales (souvent issue du pancréas, elles ne sont pas produites par le tube digestif), ainsi que par des enzymes de digestion exprimées à la surface des entérocytes (synthétisées par le tube digestif). L’absorption concerne aussi les petits oligopeptides, dont la digestion se poursuit alors à l’intérieur de la cellule entérocytaire avec des hydrolases les réduisant en acides aminés individuels. Le saccharose doit lui aussi être digéré à l’aide d’une enzyme résidente à la paroi des cellules épithéliales, qui le réduit en glucose et en fructose. Les triglycérides sont réduits en glycérol, AG et cholestérol, puis après absorption dans le cytosol de l’entérocyte sont resynthétisés sous forme de triglycérides, avant d’être exportés dans la circulation sanguine, via la circulation porte.
Il y a une distribution horizontale ou transversale des fonctions de digestion qui va être variable selon ce qu’on a besoin de digérer dans le tube digestif. Il y a aussi une distribution axiale ou longitudinale des fonctions d’absorption : tout n’est pas absorbé au même endroit, il existe des sites majeurs spécifiques aux différents nutriments.

90
L’épaisseur de la flèche indique l’intensité d’absorption. Conséquences majeures sur les personnes dont les segments du tubes digestifs seraient altérés (ex : l’excision du duodénum cause des problèmes d’absorption du fer).
I. Les hydrates de carbone
A. Généralités - Consommation : 200 à 800g/j - Présents sous forme : 1/d’amidon : apporté par l’alimentation, existe sous 2 formes : l’amylopectine (80% - PM sup à 10^6) et l’amylose (20% - PM sup. à 10^5 - Ne pas confondre avec la maladie du même nom). L’amidon est un ensemble de molécules de glucose attachées les unes aux autres via des liaisons linéaires alpha 1-4 ou ramifiées alpha 1-6. L’amylopectine présente les 2 types de liaisons, et l’amylose exclusivement les alpha 1-4. 2/de cellulose : présente des liaisons beta. Nous ne sommes pas équipés pour digérer ce type de liaisons, la cellulose va donc être éliminé sous forme de cellulose. Certaines espèces animales (comme les ruminants) possèdent cependant la beta amylase et sont donc capables de la digérer. 3/de disaccharides : saccharose, lactose 4/de monosaccharides : glucose, fructose

91
B. Digestion et absorption
i. Dans le tube digestif L’alpha amylase humaine (fabriquée par la salive et par le pancréas)n agit sur l’amidon par sa capacité à hydrolyser toutes les liaisons linéaires alpha 1-4, sauf les terminales présentes en bout de chaine. Après digestion par l’enzyme, on obtient du : - Maltose (dimères de glucose) - Maltotriose (trimères de glucose) - Dextrines (oligomères de glucose). Aucune de ces molécules n’est directement absorbable. Elles comportent toutes des liaisons alpha 1-6 et des liaisons alpha 1-4 terminales.
ii. Le compartiment juxtamembranaire Pour poursuivre la digestion et transformer ces molécules en glucoses, on utilise des enzymes résidentes de la membrane apicale des entérocytes, à savoir : - les lactases : digèrent le lactose, hétérodimère composé d’un glucose et d’un galactose
libérés après hydrolyse..
- la sucrase alpha dextrinase ou isomaltase : complexe enzymatique possédant une double
propriété. La sucrase est capable de digèrer le saccharose en libérant du fructose et du
glucose. L’alpha dextrinase ou l’isomaltase est capable de digérer les dextrines, en
hydrolysant les liaisons alpha-1-6 (rares sont les enzymes capables d’hydrolyser les liaisons
alpha 1-6).
- la maltase : elle digère le maltose et le maltotriose. Capable d’hydrolyser les liaisons alpha-1-
4 conduisant à des oligomères de glucose sans liaisons alpha-1-4.
A la suite de l’intervention de ces enzymes, on obtient alors des molécules de :
- fructose
- glucose
- galactose
… sous forme de monomères et directement absorbables par l’épithélium intestinal. Les
membranes plasmiques des entérocytes ne sont pas naturellement perméables au
glucose, galactose et fructose ; il faut des voies de passages qui sont des transporteurs.
Ces transporteurs sont de 2 types (majoritaires dans le duodénum):
Co-transporteur sodium/glucose & sodium/galactose (SGLT1) : le glucose et le
galactose entrent dans la cellule grâce au gradient électrochimique de sodium généré
grâce à la Na/K/ATPase basolatérale.
Transporteur de fructose GLUT5 : La destinée des dextrines produites par

92
l’amylase est l’hydrolyse un par un des glucoses par ces 3enzymes, on obtient
finalement du maltotriose.
L’absorption est le transport vectoriel à travers l’épithélium des produits de la digestion et
nécessite donc leur sortie de la cellule pour gagner la circulation.
Les trois produits de digestion (glucose/fructose/galactose) quittent la cellule par le
transporteur GLUT2, à la membrane basolatérale.
Quand tout fonctionne bien, les hydrates de carbone sont majoritairement absorbés dans le grêle proximal, 80% étant déjà passés à la fin du duodénum. L’iléon est non concerné par l’absorption des hydrates.
NB : Certains hydrates ne sont pas capable d’être absorbés : c’est le cas du cellulose et sorbitol qui osmotiquement actifs, déclenchent la diarrhée.
C. Syndrome de malabsorption des hydrates de carbone
Déficit en lactase : - Acquis, qui se développe au cours de la vie - Diffère de l’intolérance au lactose - Génétiquement déterminé : Orient et Afrique +++ / rare chez les européens
- Lors de la prise de lactose (ex : produits laitiers), le lactose n’est pas digéré et reste dans
l’iléon terminal où il rencontre des bactéries qui le métabolisent et produisent de l’eau
et des électrolytes. A la suite d’une prise de lactose, on observe alors un ballonnement et
une distension intestinale, accompagné de bruits de filtrations abdominaux (borborygmes)
puis de diarrhée due à l’accélération du transit par les métabolites (accompagné d’une
sécrétion d’eau et d’electrolytes).

93
- Solution : éviction du lactose de l’alimentation Intolérance congénitale au lactose : - Déficit génétique sévère ou total en lactase - Présent dès la naissance (diffère du cas précédent) - Rare et grave, potentiellement mortelle
- Responsable de diarrhée profuse conduisant à la dénutrition et la déshydratation du
nouveau- né - On doit nourrir les nourrissons avec du lait sans lactose
Déficit en sucrase isomaltase - Autosomique récessive - +++ chez les esquimaux / rare chez les Nord Américains - rend impossible la digestion du sucre
- Syndrome clinique d’intolérance aux hydrates de carbone : distension abdominale,
borborygmes, diarrhée...
Syndrome de malabsorption de glucose galactose
- Pathologie rare et grave, due à un déficit en transporteur glucose/galactose SGLT1
- Ce qui n’est pas digéré sera utilisé par les bactéries provoquant les symptômes
susmentionnés
II. Digestion des protéines
A. Généralités - Le besoin nutritionnel est de 0,7g/kg/j, les recommandations sont autour de 1g/kg/j, mais on a tendance à consommer plus de protéines que ce qui est recommandé dans notre société actuelle. - Elles proviennent majoritairement de l’alimentation - Certaines protéines proviennent aussi du tube digestif qui les sécrète (10 à 30g/j) - D’autres proviennent de la desquamation des cellules épithéliales intestinales (1 à 30g/j) - Au total = 60-120g/j de protéines
- L’absorption des protéines par le grêle est particulièrement efficace, Il n’y a pas
d’élimination de protéines significatives dans les selles.
B. Digestion et absorption des protéines
i. La lumière intestinale
La digestion des protéines commence dans l’estomac grâce aux enzymes protéolytiques. La
pepsine et les pepsinogènes, activées par le pH acide (inf à 3) sont responsables d’une partie
de la digestion des protéines dès l’estomac. Cette digestion est minoritaire (15 à 20%).

94
La majorité du travail va être assurée par les enzymes protéolytiques pancréatiques qui vont
rentrer en contact des aliments au niveau de l’abouchement des canaux pancréatiques dans le
duodénum. Les AA peuvent ainsi emprunter la circulation porte.
Les enzymes pancréatiques sont les suivantes (retenir seulement le nom des enzymes):
o Trypsine : endopeptidase (Lys, Arg), produit des oligopeptides
o Chymotrypsine : endopeptidase (AA aromatiques, AA neutres), produit des oligopeptides
o Elastase : endopeptidase (AA aliphatiques), produit des oligopeptides
o Carboxypeptidase A : exopeptidase (AA aromatiques)
o Carboxypeptidase B : exopeptidase (Arg, Lys)
Le rôle des enzymes pancréatiques est donc de réduire la taille des protéines pour obtenir des
oligopeptides et des acides aminés.
La suite de la digestion est effectuée par des enzymes digestives exprimées à la membrane apicale des entérocytes, dont la densité est maximale dans le duodénum et le jéjunum proximal.
Ces enzymes membranaires sont de natures variées :
Dipeptidyl aminopeptidase: coupe les extrémités amino-terminales d’une
chaine peptidique, création de dipeptides.
Aminopeptidase : hydrolyse les dimères ou trimères pour libérer des acides
aminés.
Aminooligopeptidase : agissent sur des séquences plus longues d’acides
aminés (3 à 8) et les hydrolysent par leurs extrémités amino-terminales en premier.
Les protéines et grands peptides (plus de 4 acides aminé) ne peuvent pas être absorbés.
Seules les formes L sont absorbées. Grace à ces enzymes (pancréatiques + digestives), on
obtient des acides aminés et des petits oligopeptides, et idéalement des di et tri-peptides,
qui sont suffisamment petits pour être absorbés directement.

95
ii. Le compartiment intracellulaire
Au pôle basal de l’entérocyte ne peuvent sortir que les acides aminés. Dipeptides et tripeptides n’ont pas de transporteur à la membrane baso-latérale : la digestion doit donc se poursuivre dans le cytosol afin de les transformer en acides aminés. Au pôle apical : Le transporteur de dipeptides et tripeptides est pept1 : c’est un cotransport d’un ion hydrogène et d’un di/tripeptide. Ce transporteur fonctionne grâce à la faible concentration de sodium intracellulaire ainsi qu’à la polarisation membranaire due à la Na/K ATPase qui crée un gradient favorable à la réabsorption des ions hydrogènes.
Mesure de la vitesse apparition d’un acide aminé dans le sang : la glycine. On administre la même quantité de glycine sous forme de monomères et de dimères. On voit alors apparaître beaucoup plus rapidement la glycine dans le sang de l’individu (donc une fois qu’elle a

96
été absorbée), quand on l’a donnée sous forme de dipeptide que sous forme de monomère de glycine. L’efficacité de pept1 est plus grande que celle des transporteurs d’acide aminé individuel.
Pour expliquer cette préférence pour les polymères, on a découvert il y a quelques années que
l’activité de transport est hormonalement contrôlée (vrai pour les hydrates de carbones, les acides
aminés et les lipides), par la leptine par exemple.Quand la leptine (hormone de la satiété) se lie
à son récepteur (OB) dans l’épithélium intestinal, elle active le transporteur de polymères PEPT1
et favorise l’absorption d’AA sous forme de polymère.
Au-delà des transporteurs de polymères comme PEPT1 il y a aussi les transporteurs d’AA
individuels.
Transporteurs d’acides aminés (pas à mémoriser, mais illustre la variété)
• Transporteurs apicaux
1/ Na dépendants
B : AA neutres
XAG : AA acides
IMINO : Pro, OHPro
2/ Na indépendants
. bO,+ : AA neutres et basiques, cystine
. y+ : AA basiques
Transporteurs basolatéraux
1/ Na dépendants
• A : AA neutres, Pro, OHPro
• ASC : Ala, Ser, Cys
2/ Na indépendants
• asc : AA neutres, Pro,OHPro
• y+ : AA basiques

97
• L : AA neutres hydrophobes.
L’absorption des protéines, se produit essentiellement dans le grêle proximal
(duodénum/jéjunum).
C. Déficit d’absorption des AA Maladie de Hartnup -Rare, génétiquement déterminée -Déficit transport intestinal et rénal des AA neutre : B -Peu de conséquences nutritionnelles
-Mais conséquences dans le rein
Cystinurie : -Déficit de transport cystine, arginine, ornitine, lysine -Peu de conséquences nutritionnelles : la cystéine peut être absorbée par d’autres cotransporteurs -Conséquences rénales : deux molécules de cystéine forment de la cystine peu soluble dans l’urine, ce qui entraine des cristaux de cystine puis des calculs. Ces calculs sont très récidivants, engendrent des complications cliniques comme les colites néphrétiques ou de l’IR chronique. Prolinurie : Déficit transport proline et hydroxy proline
III. Les vitamines hydrosolubles
A. Vue d’ensemble

98
(Ce tableau n’est pas à connaître pas cœur) Les deux grandes catégories sont les vitamines hydrosolubles et liposolubles. Les sites d’absorption et transporteurs d’une vitamine à l’autre sont variables.
B. Principale vitamine hydrosoluble : la vitamine B12 - Site principal d’absorption : iléon terminal - Capacité d’absorption et besoin très proche - Fournie par l’alimentation surtout, et transport lié à des protéines
- Dans l’environnement acide de l’estomac, les protéines qui l’accompagnaient sont digérées
par les pespines (protéases). La vitamine B12 se fixe alors sur un autre chaperon, l’haptocorrine (ou protéine R) fabriqué par l’estomac.
- Dans le duodénum : les enzymes protéolytiques délient l’haptocorrine de B12, et laissent la
place au facteur intrinsèque qui se lie à son tour à la vitamine. - Le couple poursuit son chemin le long de l’intestin grêle, jusqu'à l’iléon, où il trouve son port
d’attache : la cubiline.
- Dès lors, la vitamine B12 rompt avec le facteur intrinsèque pour se lier à la transcobalamine
de type 2. C’est sous cette forme que la vitamine B12 va quitter l’entérocyte pour rejoindre
la circulation et aller dans le foie et être distribuée aux organes qui ont besoin d’elle
(moelle osseuse et système nerveux central).
Lorsqu’une étape est défaillante c’est tout le processus qui est impacté, attention aux carences et
conséquences neuro/hématologiques !
Le facteur intrinsèque et la cubiline sont indispensables à l’absorption de la vitamine B12 dans
l’iléon. Si l’un ou l’autre manquent, la capacité d’absorption de la vitamine B12 va être
dramatiquement diminuée. Et comme la capacité d’absorption est proche du besoin quotidien,
on est très rapidement carencé en vitamine B12.
C. La vitamine C
- Capacité d’absorption très largement supérieur aux besoins de l’organisme
- Iléon principal site d’absorption
-Carence entraine le scorbut, mais rare (différent de B12)
-Cas de la prise de vitamine C avant les partiels : absorption de la vitamine C, puis métabolisation en acide oxalique. On doit alors éliminer cet excès d’acide dans l’urine qui contient du calcium. Or l’oxalate ADORE le Ca2+ pour former un complexe très peu soluble… donc tu pensais t’être dopé pour tout déchirer, mais tu es juste en train de préparer une belle colite néphrétique pour le soir du dernier jour des partiels… ;)

99
IV. Les lipides
A. Généralités - Quantité ingérée très variable d’un individu et d’un jour à l’autre, entre 25 et 150g/j - Majoritairement sous forme de triglycérides / minoritairement sous forme d’esters de
cholestérol et de phospholipides - Pas directement absorbables
- Doivent être réduits en AG, glycérol, cholestérol et lysophospholipides, qui présentent une très bonne capacité d’absorption, nous faisons profit d’à peu près tous les lipides
B. Digestion et absorption
i. Dans l’estomac
Les lipides sont difficiles à digérer à cause de leur caractère non hydrophile, alors que le tube
digestif est un environnement aqueux. Dans l’estomac, les lipides surnagent au dessus des
autres aliments et forment une couche de graisse qui n’est que peu ou pas mélangée avec les
autres. Même si les mouvements de brassage de l’estomac essayent de former une émulsion, ce
n’est pas une émulsion stable, et la quantité de lipides attaquée lors de cette première
phase de la digestion est dérisoire.
ii. Dans le tube digestif
Rappel :
Triglycérides : une molécule de glycérol + 3 AG
Ester de cholestérol : 1 molécule de cholestérol + 1 AG
Phospholipides ou lécithines : donnent des molécules d’acide gras attachées à une chaine que l’on
ne peut absorber. On les transforme en lysolécithine, absorbable.
Aucune de ces molécules n’est absorbable, et doit être transformée en AG, cholestérol, glycérol et lysophospholipides. Dans le duodénum, les acides biliaires arrivent et créent le manteau d’une structure capable de rester dans un environnement hydrophile, tout en préservant un noyau lipophile. Cela conditionne les lipides dans ces gouttelettes d’émulsion (diamètre = 1 micron), et augmente considérablement le rapport surface/volume. Les enzymes lipolytiques pancréatiques vont ainsi pouvoir attaquer les lipides conditionnés dans ces gouttelettes. Cas des triglycérides La lipase pancréatique hydrolyse les chaines d’AG glycérides mono glycérides + libération de 2 AG absorption Pour agir, la lipase a besoin d’un pH > 6 et d’une coenzyme indispensable la colipase d’origine duodénale.

100
Cas des esters de cholestérol Même principe, l’enzyme clé est la cholestérol-ester-hydrolase qui donne une molécule de cholestérol + une molécule d’AG libre Cas des phospholipides Idem, l’enzyme est la phospholipase A2 qui fabrique un lysophospholipide et une molécule d’AG. Les produits obtenus sont alors directement absorbables, mais on n’oublie pas qu’ils sont des produits hydrophobes dans un environnement aqueux ces produits de digestion sont de nouveaux conditionnés en micelles (diamètre 5nm) qui contiennent 20 à 30 molécules de digestion (AG, glycérol, lisophospholipides…) par les acides biliaires. C’est sous la forme de micelles que les produits de digestion des lipides viennent s’approcher de la membrane plasmique des entérocytes, par des mouvements de brassage. Par ailleurs, la membrane plasmique exprime un échangeur Na/H+ et est ainsi capable de sécréter un environnement acide sur une petite épaisseur, suffisante pour déclencher la rupture des micelles contenu libéré au voisinage de la membrane absorption à la membrane par FABP (Fatty Acide Binding Protein) et SCP (Sterol Carrier Protein) entrée dans l’entérocyte.
iii. Dans les entérocytes Dans le RE des entérocytes, il y a un travail de resynthèse des triglycérides/cholestérol/phospholipides. De nouveaux, les lipides sont empaquetés, ici dans des chylomicrons très volumineux, grâce à des apolipoprotéine (par ex, la béta lipoprotéine) puis vont sortir par exocytose et rentrer dans la circulation pour être amenés jusqu’au foie. Récapitulatif des conditions nécessaires à l’absorption Ph duodénal >6 qui permet l’action des enzymes pancréatiques Enzymes lipolytiques Sels biliaires Mouvement de brassage de la paroi du grêle Un épithélium intègre Bétalipoprotéine Chacune de ces conditions est limitante !
Restent alors dans la lumière, les sels biliaires : ils sont réabsorbés (à 95%) dans l’iléon
terminal, en étant conjugués ou non, par des transporteurs présents dans la membrane, et sont
ensuite liés à des molécules de transport qui les ramènent vers la circulation porte où ils sont
réutilisés par les hépatocytes.
V. Les vitamines liposolubles - Vitamines : A, D, E, K - Absorbées en même temps que les lipides - Pas de processus de digestion, directement absorbables

101
VI. Les points clés
Quelques QCMs type :
QCM1 :
• La maltase peut digérer – A- l’amidon – B- le saccharose – C- le lactose – D- le glucose – E- des oligomères de glucose
Réponse : E QCM2 :
• La digestion des protéines par les enzymes pancréatiques – A- est exclusivement luminale – B- produit majoritairement des acides aminés – C- prédomine dans la première partie de l’intestin grêle – D- nécessite la présence de sels (acides) biliaires – E- est maximale à pH acide
Réponse: A et C

102
QCM3 :
• Les lipides alimentaires – A- sont en majorité des esters de cholestérol – B- sont surtout digérés dans le colon – C- sont directement absorbables par l’épithélium intestinal – D- doivent être conditionnés en gouttelettes d’émulsion pour être digérés – E- sont digérés à pH acide (< 6)
Réponses : D QCM 4 :
• Quelles sont les vitamines liposolubles? – A- vitamine A – B- vitamine B1 – C- vitamine B12 – D- vitamine D – E- vitamine E
Réponse : A, D et E QCM 5 :
• La digestion des triglycérides produit – A- des acides gras – B- du cholestérol – C- des phospholipides – D- des monoglycérides – E- du maltose
Réponses : A, D QCM 6 : La digestion de l’amidon produit : - A – du glucose - B – du fructose - C - du galactiose - D - du maltose - E - du maltotriose
Réponses : D, E

103
Fiche récapitulative : physiologie digestive – digestion et absorption
La majeure partie de ce qu’on ingère passe par une phase de digestion afin d’être absorbé puis utilisé par notre organisme. Les enzymes qui permettent la digestion sont soit luminales (elles sont présentes dans le tube digestif mais pas produites par les entérocytes), soit forment partie intégrante du tube digestif car elles sont produites par les entérocytes. Il y a une distribution axiale ou longitudinale : les différents nutriments ne sont pas absorbés aux mêmes endroits dans le tube digestif. Les hydrates de carbone : conso : 200 à 800g/j Existent sous forme : d’amidon (amylopectine et amylose), de cellulose, de disaccharides (saccharose, lactose), de monosaccharides (glucose, fructose). Dans le tube digestif, l’alpha amylase hydrolyse l’amidon au niveau des liaisons alpha 1-4 non terminales et donne : du maltose, du maltotriose, des dextrines (pas directement absorbables). Dans le compartiment juxtamembranaire, les lactases, la sucrase alpha dextrinase ou isomaltase et la maltase hydrolysent respectivement le lactose, le saccharose et le maltose. Elles forment des molécules de fructose, de glucose, et de galactose qui sont directement absorbables par l’épithélium intestinal via des transporteurs (cotransporteur sodium/glucose et sodium/galactose (SGLT1) et le transporteur de fructose GLUT5). Ils quittent la cellule par un transporteur GLUT2 à la membrane basolatérale. Ils sont majoritairement absorbés au niveau du grêle proximal Syndromes de malabsorption des hydrates de carbone : Déficit en lactase, Intolérance congénitale au lactose, Déficit en sucrase isomaltase, Syndrome de malabsorption de glucose galactose. Les protéines : conso : 60 à 120g/j – on a tendance à en consommer plus que les recommandations (1g/kg/j). L’alimentation n’est pas la seule source d’apports protéiques ; certaines protéines proviennent d’une sécrétion entérocytaire (10 à 30g/j) ou de la desquamation des cellules (1à 30g/j). La digestion commence dans estomac avec enzymes protéolytiques (pepsine et pepsinogènes) activées par un pH inf à 3. Digestion minoritaire (15 à 20%). La majeure partie de la digestion est assurée dans l’intestin grêle par des enzymes protéolytiques pancréatiques (trypsine, chymotrypsine, elastase, carboxypeptidase A et B) présentes au niveau de l’abouchement des canaux pancréatiques dans le duodenum. La digestion se poursuit avec des enzymes digestives (dipeptidyl aminopeptidase, aminopeptidase, aminooligopeptidase) au niveau de la membrane apicale des entérocytes qui forment des di, des tripeptides transformés dans le cytosol des entérocytes en AA. Les di et tripeptides entrent dans la cellule via le transporteur pepT1. Un même acide aminé entre plus rapidement dans le sang sous forme de dimère ou de trimère PepT1 est plus efficace que les transporteurs d’AA individuels. PepT1 peut être activé par la liaison de la leptine (une hormone) à son récepteur OB sur la membrane apicale des entérocytes. Il y a des transporteurs d’AA dans la membrane basolatérale et apicale, certains sont dépendants d’autres indépendants du sodium, ils ont différentes particularités. L’absorption des protéines se produit exclusivement dans le grêle proximal. Déficit d’absorption des AA : Maladie de Hartnup, cystinurie, prolinurie

104
Les vitamines hydrosolubles : Les sites d’absorption des transporteurs sont variables d’une vitamine à l’autre. On s’intéresse surtout à B12 (cobolamine). Site principal d’absorption : iléon terminal. Capacité d’absorption supérieure aux besoins. Mécanisme de digestion de B12 : Dans l’estomac, B12 a pour chaperon l’haptocorrine (ou protéine R) fabriqué par l’estomac. Dans le duodénum, le facteur intrinsèque prend la place de l’haptocorrine et chaperonne B12 jusqu’à l’iléon ou elle trouve son port d’attache : la cubiline. Puis B12 rompt avec le facteur intrinsèque et se lie à la transcobolamine de type 2. Sous cette forme B12 rejoint la circulation et le foie pour être distribuée aux organes qui en ont besoin (SNC et moelle osseuse). La vitamine C : Capacité d’absorption très supérieure aux besoins de l’organisme. Principalement absorbée dans l’iléon. Carence entraine scorbut. Les lipides : quantité ingérée très variable (de 25 à 150 g/j) – maj sous forme de triglycérides, moins sous forme d’esters de cholesterol et de phospholipides. Doivent être réduits en AG, glycérol et lysophospholipides pour être absorbés. Digestion et absorption : Dans l’estomac, formation d’une émulsion stable de lipides. Dans le duodénum, les acides biliaires conditionnent les lipides en gouttelettes d’émulsion (1micron de diamètre). Les enzymes lipolytiques (actives si PH>6) peuvent alors attaquer les lipides conditionnés dans ces gouttelettes. Les produits obtenus sont de nouveau conditionnés en micelles (5nm de diamètre – 20 à 30 molécules de digestion). Les micelles s’approchent de la membrane apicale et un échangeur Na/H+ sécrète un environnement qui permet l’ouverture des micelles. Elles sont absorbées à la membrane par FABP et SCP. Dans le RE des entérocytes, il y a une resynthèse de triglycérides, de cholesterol et de phospholipides qui vont former des chylomicrons volumineux grâce à des apolipoprotéines. Les vitamines liposolubles : vitamines A, D, E, K – sont absorbés en même temps que les lipides ; Elles sont directement absorbables.

105

106
UE6 – SD –Sémiologie - n°4
19/01/18
Benoît BORDACAHAR
RT : Guillemette COLLE
RL : Amaury POQUE
Sémiologie et exploration de l’intestin grêle, du côlon
et du rectum
Plan :
I. L’intestin grêle
A. Sémiologie de l’intestin grêle
B. Explorations morphologiques de l’intestin grêle
i. Fibroscopie et coloscopie
ii. Transit du grêle
iii. Vidéocapsule
iv. Entéroscopie
v. TDM et IRM
II. Le côlon
A. Sémiologie du côlon
B. Explorations morphologiques du côlon
i. Anuscopie et rectoscopie
ii. Coloscopie
iii. Facteurs de risque
iv. Méthodes de dépistage
Abréviations :

107
TD : tube digestif
AEG : Altération de l’état général
I. L’intestin grêle L’intestin grêle est le plus long segment du tube digestif (entre 3 et 5m). Il est composé du
duodénum, du jéjunum et de l’iléon.
Il a un rôle majeur étant responsable de l’absorption des nutriments, de l’eau et des électrolytes
mais il a également un rôle de motricité pour l’avancée du bol alimentaire.
Il y a différentes façons d’explorer l’intestin grêle :
- Morphologiques (imagerie, scanner, IRM, endoscopie)
- Histologiques (notamment prise de biopsies)
- Fonctionnelles (dosages biologiques pour vérifier que l’intestin grêle peut réaliser son
rôle dans l’absorption des nutriments)
A. Sémiologie de l’intestin grêle Signes cliniques :
Ils ne sont pas spécifiques et très variables. Il faut toutefois rechercher :
- Le syndrome occlusif : se manifeste par des nausées, vomissements, douleurs abdominales, un arrêt du transit. On suspecte alors une sténose. Si elle est complète il y a
une intolérance alimentaire totale.
- L’anémie par carence en fer : il faut alors réaliser un bilan martial (dosage du fer sérique
et de la ferritine). Peut être due à une ulcération de l’intestin grêle. (=dermabrasion de la
muqueuse qui devient atrophique et ne remplit plus son rôle, elle peut d’autre part
entraîner un saignement provoquant une anémie.)
- Le syndrome de malabsorption : il peut être accompagné ou non d’une diarrhée
graisseuse (stéatorrhée) ainsi que d’une carence vitaminique et de signes cliniques
comme la fragilité des phanères (perte des cheveux, ongles cassants par exemple)
- Signes non spécifiques : (que l’on peut observer dans toutes les atteintes de l’intestin
grêle) douleurs abdominales.
- Hémorragie digestive basse : il faut également rechercher une ulcération.
Nb : Pour différencier si une hémorragie digestive est haute ou basse : en cas d’hématémèse (fait
de vomir du sang) ou de méléna (sang noir, sort par voie basse avec une odeur nauséabonde) on
parle d’une hémorragie digestive haute. En cas de rectorragie (sang rouge) on parle d’une
hémorragie digestive basse. L’angle duodénojéjunal est un repère anatomique pour différencier
TD haut du TD bas. (Pas toujours bon : une lésion du caecum peut parfois entraîner un méléna …)
La sémiologie est donc très riche mais non spécifique du type de lésion, on a donc besoin
d’explorer le tube digestif par la suite.

108
B. Explorations morphologiques de l’intestin grêle Les explorations morphologiques peuvent être soit externes par des examens de radiologie
(entéroscanner, IRM, …) mais cela ne permet pas de voir la lumière du tube digestif, soit par une
endoscopie digestive.
Lors d’une endoscopie digestive, on introduit un appareil contrôlé par des béquillages permettant
de l’orienter dans les quatre directions cardinales. Des prélèvements peuvent également être
effectués.
Les différents examens sont :
- Fibroscopie avec biopsie de D2
- Coloscopie avec iléoscopie (biopsie de l’iléon terminal)
- Transit du grêle
- Entéroscopie
- Vidéocapsule
- TDM ou Entéro Scanner ou IRM
i. Fibroscopie gastroduodénale/gastroscopie
La gastroscopie permet d’observer l’œsophage, l’estomac et d’aller jusqu’au 2ème duodénum.
Objectif : visualiser les lésions mais surtout faire des prélèvements (biopsies en D2)
Indications :
- Diarrhée chronique : si la coloscopie et l’examen parasitologique des selles (ou coproculture)
sont normaux, on fait une biopsie duodénale pour rechercher une atrophie villositaire pouvant
évoquer une maladie cœliaque.
- Devant tout signe clinique ou biologique de malabsorption ou de carence martiale, dans le
cas où on ne trouve pas d’explication lors de la gastroscopie ou de la coloscopie.
Image : On voit une muqueuse duodénale qui apparaît lisse par endroit alors qu’on doit voir de
petites franges lorsque le tissu est sain. La muqueuse est ici érythémateuse avec un aspect crânelé. Cela évoque un diagnostic de maladie cœliaque à confirmer par une biopsie du 2ème duodénum.

109
Maladie cœliaque :
3 signes histologiques :
- Hypertrophie des cryptes
- Atrophie des villosités
- Infiltration lymphocytaire au niveau de l’épithélium duodénal. (LT CD4)
2 types de facteurs : - prédisposition génétique avec mutation HLA DQ2 ou DQ8 (99% des cas)
- Facteur environnemental (provoque le déclenchement de la maladie) : alimentation, un
régime riche en gluten peut déclencher la maladie cœliaque. On en trouve dans les céréales
(SABO : Seigle, Avoine, Blé, Orge)
Les symptômes apparaissent dès l’enfance le plus souvent, ils sont +/- spécifiques : douleur
abdominale, diarrhée, carence en fer et en vitamines. On réalise une gastroscopie avec des biopsies duodénales puis on prescrit à ces patients un régime sans gluten. L’atrophie et les
symptômes disparaissent alors.
ii. Transit du grêle
C’est un examen radiologique qui consiste à faire ingérer au patient un produit de contraste pour,
par la suite, visualiser des zones de rétrécissement de l’intestin grêle en réalisant un cliché 30min
après l’ingestion.
C’est un examen ancien peu utilisé aujourd’hui, il a été remplacé par l’entéroscanner ou l’IRM
qui dépistent des signes avec une plus grande sensibilité.
iii. Vidéocapsule
La vidéocapsule fait la taille d’un gros comprimé, elle
comprend une caméra miniature à l’avant permettant une
vision à 180° du tube digestif du patient. Elle prend au total
57 000 images, de bonne qualité, retransmises sur un petit
boîtier porté par le patient.
Sa prise nécessite une légère préparation : le patient doit
suivre un régime sans résidu et avaler une préparation lui

110
permettant de vider ses intestins pour éviter que l’observation ne soit gênée par des résidus
alimentaires.
Cette technique est peu invasive mais est uniquement diagnostique, elle ne permet pas de réaliser
un traitement.
On peut observer :
- du sang en cas d’hémorragie de l’intestin grêle
- Des lésions inflammatoires (notamment des ulcères qui peuvent aller jusqu’à des
perforations)
Cela permet d’estimer la position des lésions dans l’intestin grêle pour faire une entéroscopie
haute ou basse selon les cas et ainsi traiter la lésion.
iv. Entéroscope à « double ballon »
L’endoscope est entouré d’un overtube dans lequel il coulisse. Il y a deux ballons pour
permettre à l’endoscope d’avancer : un à l’extrémité de l’endoscope et un sur l’overtube.
On gonfle l’un après l’autres les deux ballons pour progresser dans l’intestin grêle
(« comme pour enfiler une chaussette »).
Avantage :
- on peut effectuer une biopsie ou un traitement en cas de saignement via un canal
opérateur permettant de passer des instruments.
- Elle permet une exploration plus étendue que la fibroscopie (jusqu’à 2 ou 3m)
Inconvénient : technique lente et fastidieuse -> réservée au traitement et non au
diagnostic (contrairement à la vidéocapsule).
v. TDM et IRM
Ce sont des techniques de radiologie et non d’endoscopie.
Il y a deux types de maladies inflammatoires digestives : maladie de Crohn (peut se situer à
n’importe quel endroit du tube digestif) et rectocolite hémorragique (ne touche que le rectum et
le côlon).
Entéro IRM :
- en première intention si maladie de Crohn suspectée.
- Privilégiée pour voir des maladies inflammatoires, permet de différencier les sténoses
inflammatoires des sténoses fibreuses.
- Surtout non irradiant (++) : permet de faire des surveillances régulières chez des
patients atteints de la maladie de Crohn.
La maladie de Crohn se manifeste souvent par des lésions de sténose inflammatoire de l’iléon
terminal. En IRM on voit :
- La sténose avec un épaississement de la paroi (hypersignal)
- Rétrodilatation de l’iléon sain en amont

111
- Hypervascularisation (car inflammation)
Entéroscanner :
- En première intention en cas d’occlusion ou de tumeur.
- Il nécessite l’ingestion d’un produit de contraste pour visualiser si les anses se dilatent
correctement. (présence de sténose ou non)
II. Le côlon
A. Sémiologie du côlon Les signes cliniques sont très peu spécifiques, on ne peut pas savoir à quel niveau se situe la
lésion. Les signes peuvent être :
- Anémie par carence en fer
- Hémorragie digestive basse
- Troubles du transit intestinal (constipation, diarrhée, alternance des deux)
- Signes non spécifiques : douleurs abdominales, ballonnements, gaz, …
- Syndrome occlusif du côlon, perforation colique
Ces signes vont conduire à une exploration du côlon.
B. Exploration morphologique du côlon
i. Anuscopie et rectoscopie
L’examen proctologique commence par un
examen clinique :
- Observation de la marge anale : recherche
d’hémorroïdes ou de marisques (cicatrices
d’hémorroïdes antérieurs)
- Toucher rectal : il donne des informations
sur l’existence de lésions (tumeur par
exemple) ou de saignement.
Méthode : Utilisation d’un appareil rigide après le
toucher rectal.
Avantages :
- Permet la visualisation de l’intégralité du canal anal et du bas du rectum pour visualiser les
hémorroïdes internes.
- Permet le diagnostic d’hémorroïdes.
ii. Coloscopie
La coloscopie permet de visualiser des polypes (lésion de la muqueuse pouvant évoluer vers un
cancer du côlon) et de les retirer. Elle a donc également une visée thérapeutique.

112
2 types de polypes :
- Pédiculé : très facile à enlever, on coupe au niveau du pied à l’aide d’une anse.
- Sessile (=plane) : beaucoup plus difficile à retirer car plane, nécessite l’injection d’un
produit entre la muqueuse et la sous muqueuse pour soulever la lésion et la retirer sans risque de perforation.
Indications de la coloscopie (à savoir +++)
- Signes fonctionnels digestifs (mal de ventre, AEG, saignements, troubles du transit, …)
- Dépistage des lésions pré-néoplasiques.
- Surveillance et dépistage des patients à risque élevé de cancer du côlon
Risque moyen : population générale de 50 à 74 ans, sans antécédents personnels ou familiaux de cancer du côlon, polype ou maladies prédisposantes.
Test de dépistage proposé (cf après) mais pas de coloscopie.
Risque élevé : risque relatif multiplié par 2 ou 4
Antécédent personnel d’adénome (= polype avec dysplasie), du cancer du côlon ou du rectum ;
Parent au 1er degré avec un cancer du côlon avant 65 ans OU 2 parents au 1er degré avec un cancer du côlon quel que soit l’âge de survenue.
Adénome avancé (+ d’1cm ou avec dysplasie de haut grade).
Maladie inflammatoire chronique intestinale (maladie de Crohn ou
rectocolite hémorragique) qui évolue depuis plus de 10 ans et qui atteint plus que le côlon gauche (c’est-à-dire pas que le rectum et
partie terminale du côlon)
Mucoviscidose
Acromégalie
Coloscopie reproposée à 3 ou 5 ans
Risque très élevé : prédisposition génétique
PAF (Polype Adénomateuse Familiale) avec une mutation du gène
APC -> induit un cancer du côlon de façon certaine
Syndrome HNPCC (syndrome de Lynch), c’est un syndrome de
prédisposition mais il n’est pas polyposique.
Coloscopie toujours proposée voire une colectomie.
iii. Méthodes de dépistage Le cancer du côlon est le 3ème cancer en France par son incidence et le 2ème en ce qui concerne la
mortalité. Il est souvent détecté tard c’est pourquoi il est nécessaire de le dépister. Dans la
population générale cela se fait en deux temps :
1- Test Hemoccult (simple, sans risque, peu coûteux) : est proposé tous les 2 ans pour les
personnes de 50 à 74 ans. Il consiste à rechercher du sang occulte (non visible) dans les
selles.
Il a été remplacé en 2015 par le test FIT (Test Immunologique Fécal), beaucoup plus
sensible qui contient un anticorps dirigé contre l’hémoglobine humaine.
2- Coloscopie en cas de test positif.
Ces tests de dépistage doivent avoir plusieurs caractéristiques pour être efficaces :
- Il doit être acceptable par la population (simple, pas trop contraignant)
- Eviter les faux négatifs (+++)

113
- Avoir un faible coût financier. (Moins élevé que le coût du traitement)
Mot du RT : Le prof a rajouté à la fin une partie sur la constipation et la diarrhée qui ne rentre
pas trop dans le plan, je vous la mets comme ça :
Il y a deux types de constipation :
- Terminale : le transit colique est normal mais le contenu du côlon reste bloqué à cause du
sphincter anal trop résistant
Manométrie ano-rectale
- Transit : celui-ci est ralenti tout le long du côlon. On réalise des mesures du temps de
transit (localisation de marqueurs ingérés via ASP : si trajet n’est pas fini au bout de 48h :
constipation)
Il existe également deux types de mécanismes pour la diarrhée :
- Motrice : test au rouge carmin pour mesurer le temps d’évacuation. Ingestion d’un
colorant rouge par le patient, on regarde en combien de temps il est évacué dans les selles.
- Malabsorption : on observe une stéatorrhée.

114
Fiche récapitulative : Sémiologie et exploration de l’intestin
grêle, du colon et du rectum
I) L’intestin grêle
Les signes cliniques liés aux maladies de l’intestin grêle sont non-spécifiques et très variables :
- Syndrome occlusif : suspicion de sténose intolérance alimentaire
- Anémie : suspicion d’ulcère de l’intestin grêle bilan martial
- Hémorragie digestive basse : suspicion d’ulcération
- Syndrome de malabsorption
- Douleurs abdominales
La maladie cœliaque a pour signes histologiques une hypertrophie des cryptes, une atrophie
des villosités et une infiltration lymphocytaire au niveau de l’épithélium duodénal, elle est
favorisée par la mutation HLA DQ2 ou DQ8 et une alimentation riche en gluten (SABO).
Les explorations morphologiques de l’intestin grêle peuvent être soit internes :
- Fibroscopie gastroduodénale : observation de l’œsophage, l’estomac et jusqu’à D2,
possibilité de faire des prélèvements et indiqué en cas de diarrhée chronique (biopsie pour
confirmer une maladie cœliaque) ou en cas de malabsorption ou de carence martiale.
- Vidéocapsule : technique non-invasive mais seulement observatoire (sang ou lésions). C’est
un comprimé avec une caméra miniature qui transmet les clichés pris à un boitier. Nécessité
d’une légère préparation (régime sans résidu + avaler une préparation).
- Entéroscope à double ballon : exploration de 2 à 3 mètres d’appareil digestif, possibilité de
biopsie ou d’action locale à l’aide d’un canal opérant.
Soit externes :
- Transit du grêle : examen de radiologie du tube digestif après avoir avalé le produit de
contraste afin de repérer des zones de rétrécissements. Technique désormais peu utilisée.
- Entéro IRM : non-irradiant, pour suspicion ou suivi de maladie de Crohn et maladies
inflammatoires.
- Entéroscanner : irradiant, nécessité de produit de contraste, prescris en cas d’occlusion ou
de tumeur.
II) Le colon
Encore une fois, les signes cliniques des maladies du colon sont très peu spécifiques :
- Anémie par carence en fer
- Hémorragie digestive basse (= rectorragie)

115
- Trouble du transit intestinal (constipation, diarrhée, ...)
- Syndrome occlusif du côlon, perforation du côlon
- Signes non spécifiques : douleurs abdominales, ballonnements gaz, …
Les méthodes d’exploration morphologiques du colon sont ici encore, soit internes :
- L’anuscopie et la rectoscopie : Examen clinique avec observation des marges anales puis
toucher rectal. On insère ensuite un anuscope ou un rectoscope afin de visualiser l’intégralité
du canal anal et le début du rectum. Cela permet le diagnostic d’hémorroïdes.
- La coloscopie : insertion d’un endoscope par voie anale. Cela permet de visualiser et retirer
des polypes (pédiculé ou sessile). Cet examen est indiqué en cas de signes fonctionnels
digestifs (maux de ventre, AEG, saignements, ...), pour le dépistage de lésions pré-
néoplasiques ou pour la surveillance et le dépistage du cancer du côlon.
Soit externes par dépistage, tout d’abord par test FIT (Test Immunologique Fécal) qui contient
un Ac dirigé contre l’hémoglobine humaine, puis, si le test est positif, une coloscopie. Pour être
efficace, un test de dépistage doit être acceptable par la population, avoir un faible coup
financier et éviter les faux négatifs.
Point Transit :
- Une constipation peut être terminale, le sphincter anal est trop résistant ou bien de
transit lorsque ce dernier est trop ralenti dans le côlon.
- Une diarrhée, quant à elle, peut être motrice, on effectue alors un test au rouge carmin pour
mesurer le temps d’évacuation, ou bien due à une malabsorption, auquel cas on observe
une stéatorrhée.
Risque moyen (Test de dépistage proposé)
Risque élevé Coloscopie reproposée à 3 ou 5 ans)
Risque très élevé (Coloscopie toujours proposée voir colectomie)
- Population
de 70 à 74
ans sans
antécédent
- Antécédent personnel d’adénome
ou cancer.
- 1 parent du 1er degré avec cancer
du colon avant 65 ans ou 2 parents
avec cancers du colon
- Maladie inflammatoire chronique
intestinale depuis plus de 10 ans
- Mucoviscidose
- Acromégalie
- Polype
Adénomateuse
Familiale (APF)
avec mutation du
gène APC
- Syndrome HNPCC
(dit de Lynch)

116
UE6 – SD –Physiologie- n° 4
19/01/2018
Pascal HOUILLIER
RT : Marie COLLIN
RL : Eléonore MIRICI
Physiologie du Système Digestif Transport intestinal d’eau et d’électrolytes, en
situation normale ou anormale.
I. Introduction
II. Sécrétion et absorption d’eau et d’électrolytes en situation
normale
A. L’eau
B. Le sodium
C. Le potassium
D. Le chlore
E. Le bicarbonate
III. Phénotypes cellulaires d’absorption et sécrétion
A. Présentation de l’épithélium intestinal
B. Cellules villositaires de l’intestin grêle
C. Cellules cryptiques de l’intestin grêle
D. Cellules cryptiques et de surface du côlon
IV. Physiopathologie des diarrhées
A. Déterminants des diarrhées

117
B. Mécanismes des diarrhées
V. Deux cations bivalents
A. Le calcium
B. Le fer
VI. Microbiote digestif
Objectifs : -Connaitre et comprendre les mécanismes et les déterminants des transports (absorption et ou sécrétion) de l’eau et des électrolytes dans le tube digestif. -Connaitre les principaux mécanismes qui peuvent conduire à la parte intestinale d’eau et d’électrolytes (diarrhée).
I. Introduction
En terme d’homéostasie de l’eau et des électrolytes, un être humain est un sac étanche dans lequel on trouve des cellules qui baignent dans un environnement hydro-électrolytique (c.à.d. que le milieu intérieur = le milieu extracellulaire). Il faut donc maintenir la composition de ce milieu dans des limites acceptables. Ce sac étanche est percé de part en part par un tuyau (=le tube digestif). Plusieurs fois par jour, on fait rentrer des éléments dans ce tuyau et une partie, voire la totalité, va disparaitre de la lumière du tube digestif et apparaitre dans l’organisme. Cette activité de nutrition et d’apport de substances qui vont rentrer dans le tube digestif est dangereuse pour la stabilité de la composition du milieu intérieur. Il nous faut donc un organe qui puisse maintenir constant la composition de ce liquide intracellulaire : c’est le rein qui contrôle la composition du milieu intérieur en contrôlant ce qui sort de l’organisme (cet organe ne sera pas traiter dans ce cours).
II. Sécrétion et absorption d’eau et d’électrolytes en situation
normale
A. L’eau :
Un sujet sain ingère quotidiennement 2000mL d’eau et les selles en recèlent 100mL. Mais l’intestin n’absorbe pas que 1900mL d’eau car il faut y ajouter les sécrétions diverses (salivaires = 1500 mL, gastrique = 2000mL, pancréatiques = 1500mL, intestinales = 1500mL et biliaires = 500mL). Au total, l’intestin absorbe en moyenne 8500mL d’eau par jour.

118
B. Le Sodium :
Pour le sodium : (absorption d’environ 150mmol/j) on n’en trouve pas dans les selles donc tout est absorbé par l’intestin, mais il absorbe aussi le sodium venant des sécrétions (de type salivaires, gastriques, biliaires, pancréatiques et intestinales). La plus grande partie du sodium va être absorbée dans l’intestin grêle et notamment dans l’iléon +++, et une petite partie dans le côlon. ATTENTION : Cela ne signifie pas que la capacité d’absorption du côlon est inférieure à celle de l’intestin grêle. Mais pour les doses ingérées quotidiennement, le côlon n’a pas besoin d’absorber beaucoup puisque le grêle le fait en majeure partie. Il peut y avoir un contrôle hormonal de l’absorption du sodium. En effet, l’Aldostérone va agir sur le côlon en favorisant l’absorption intestinale du Sodium.
C. Le Potassium :
Pour le Potassium : Pour une ingestion quotidienne de 100mmol de K+ par jour, les selles contiennent 10mmol de potassium. De même, on ajoute à l’ingestion le K+ celui venu des sécrétions digestives. Il est majoritairement absorbé par l’intestin grêle (minoritaire dans le côlon). Dans le côlon, il peut y avoir une absorption de K+ et une sécrétion de K+. La sécrétion est favorisée par l’aldostérone. Ainsi, elle va permettre en cas de surcharge en K+ (Hyperkaliémie) d’en éliminer une partie. L’aldostérone est une hormone produite dans la corticosurrénale à partir du cholestérol. Sa synthèse et sa sécrétion ont lieu en réponse à deux déterminants majeurs : -l’Angiotensine 2 (qui dépend de la sécrétion de rénine) -la concentration de K+ dans le sang Quand on se surcharge en K+, on induit, quelques soient le volume cellulaire ou la concentration de rénine, la sécrétion d’aldostérone qui va pouvoir agir sur ces organes cibles : -le côlon -le rein L’aldostérone va ainsi favoriser la sécrétion de K+ par le côlon et par le rein. Donc : L’Aldostérone agit sur le côlon, en favorisant : -la sécrétion intestinale de Potassium -l’absorption intestinale du Sodium

119
La prise d’un Laxatif : entraîne une hyperaldostéronisme qui va agir sur le côlon, en favorisant la perte intestinale de K+.
D. Le Chlore :
Pour le chlore, c’est pareil, il est absent des selles. On absorbe à la fois le chlore de l’alimentation (ingestion de 200mmol/j) et des sécrétions digestives (encore une fois majoritairement par le grêle et moins par le côlon). En pathologie : Des sécrétions massives intestinales de chlore peuvent être stimulées par certaines toxines, notamment par l’exposition à la toxine du choléra par exemple. Elle peut aussi être induite par des hormones (prostaglandines, VIP…), par des acides biliaires, …
E. Le Bicarbonate :
Pour le bicarbonate : il est très peu présent dans l’alimentation occidentale (ingestion de 0mmol/j de bicarbonates). Notre alimentation est plus acide qu’alcaline. C’est en train de changer vers une alimentation végétarienne donneuse d’alcalins, contrairement à l’alimentation carnivore donneuse d’acides. Cependant les sécrétions digestives contiennent des bicarbonates. Au final, le bicarbonate, provenant presque uniquement des sécrétions digestives, est absorbé puisqu’on n’en retrouve pas dans les selles. Il est absorbé à peu près autant par le grêle et par le côlon. On absorbe moins que ce qu’on sécrète, car le bicarbonate sera utilisé pour neutraliser l’acidité sécrétée par l’estomac.

120
III. Phénotypes cellulaires d’absorption et sécrétion
L’épithélium du grêle et du côlon est organisé de façon fonctionnelle : c’est là où se passe le flux de l’eau et des électrolytes.
A. Présentation de l’épithélium intestinal :
C’est un épithélium villositaire directement en contact avec la lumière du tube digestif. Il y a donc des endroits où l’on est à la profondeur (cryptes) et d’autres où l’on est au sommet (villosités). Les cellules épithéliales des cryptes sont principalement sécrétrices de chlore par voie trans-cellulaire. Parallèlement, il va y avoir une sécrétion de sodium (pour des raisons d’électro-neutralité) passive par voie para-cellulaire (entre les cellules qui sont perméables au sodium). En plus du sodium, il y a une sécrétion d’eau. Concernant les cellules des villosités, ce sont des cellules qui absorbent, du chlore, de l’eau, … Or les cellules des villosités sont une évolution des cellules nées au fond des cryptes, zone germinative de l’intestin grêle, dont les cellules sont migratrices. En fonction des cellules qui ont l’activité la plus intense, l’épithélium va être plutôt absorbant ou sécrétant. En situation normale, c’est les cellules absorbantes qui prédominent. L’épithélium digestif est capable de communiquer avec le centre nerveux via des neurones. Ces neurones sont sensibles à des médiateurs, substances chimiques et contraintes mécaniques. Ils vont ainsi fournir des informations, sur ce qu’il se passe dans l’intestin, au système nerveux entérique. Donc, si on agresse l’épithélium de l’intestin, des signaux, envoyés par les neurones, seront intégrés par le système nerveux entérique. Ce dernier envoie ensuite une réponse de sauvegarde pour chercher à faire fuir l’agresseur et transforme l’épithélium absorbeur en épithélium sécréteur, provoquant une diarrhée.
B. Cellules villositaires de l’intestin grêle :

121
-Les cellules épithéliales de surface du duodénum et du jéjunum expriment à leur membrane apicale : de nombreux co-transporteurs de Na+ comme : SGLT1 (co-transport de sodium / glucose), co-transport Na+/AA, ainsi que des échangeurs de Na+/protons. Le chlore est absorbée via l’échangeur Cl- / HCO3-. Les cellules villositaires absorbent donc le sodium et le chlore à son pôle apical. Cette absorption s’accompagne d’une excrétion de HCO3- et de H+ ce qui donne de l’eau. Et à membrane basolatérale : le transporteur Na+/K+-ATPase. -Les cellules de l’iléon ne sont pas bien différentes, on voit apparaitre les co-transporteur Na+/acides biliaires (à leur pôle apical).
C. Cellules cryptiques de l’intestin grêle :
La cellule du fond des cryptes est sécrétrice de chlore par un canal CFTR (sur leur membrane apicale) activé par le VIP (hormone produite par l’épithélium du tube digestif activant la voie AMPc/PKA).

122
Les cellules cryptiques présentent de même à leur membrane baso-latérale le co-transport Na+/K+/2Cl-. Ce dernier alimente la cellule en Cl- ce qui permet son activité sécrétoire par CFTR.
Cette sécrétion est accompagnée d’une diffusion passive para-cellulaire de sodium, ce qui permet une sécrétion de NaCl à un débit inférieur à celui de l’absorption par les cellules villositaires. Les cellules cryptiques présentent donc un phénotype de sécrétion.
D. Cellules cryptiques et de surface du colon :
La différence avec les cellules coliques, c’est qu’il n’y a pas de villosités.
Sinon, on retrouve bien un épithélium de surface à phénotype absorbant et des cryptes à
phénotype sécréteur. De même que pour l'épithélium intestinal, les cellules naissent au niveau
des cellules souches cryptiques puis migrent vers la surface, tout en passant d'un phénotype
sécréteur à un absorbant.
Mais pour ce qui concerne les cellules épithéliales de surface, il existe une différence entre côlon droit et côlon gauche. En effet l’absorption du NaCl diffère entre le côlon proximal (droit) et le côlon distal (gauche).
-Dans le colon proximal : on a le couplage entre les échangeurs Na+/H+ et HCO3-/Cl- dans les
cellules de surface qui expriment peu CFTR, d'où un phénotype d’absorption de NaCl. Au fur et à
mesure que l'on descend dans les cryptes, CFTR est plus exprimé et le phénotype est donc de
sécrétion. La réabsorption dans le colon proximal est électroneutre.

123
-Dans le colon distal : on a toujours un phénotype absorbeur au niveau des cellules de surface et
sécréteur pour les cryptes, cependant l'absorption du Na+ est réalisée par un canal : ENaC
(Epithelial Na Channel). C’est une structure multimérique composée de 3 sous-unités. La ré-
absorption du Na+ est électrogénique : elle dépolarise la membrane apicale, favorisant la
sécrétion de K+ par des canaux potassiques. L’épithélium du colon distal a donc un phénotype
sécréteur de potassium.
Dans le côlon comme dans le grêle, le phénotype global est celui d’un épithélium absorbant (sauf s’il reçoit un signal d’agression où dans ce cas la sécrétion va prédominer afin d’évacuer l’agresseur et provoque donc une diarrhée). Beaucoup de facteurs sont capables de modifier les transports ioniques dans le côlon. La majorité d’entre eux favorisent la sécrétion : • Eicosanoïdes (PGE2, PGF2a, PGI2, PGD2, HETEs) : Favorisent la sécrétion • Amines (sérotonine, Ach, histamine) : Favorisent la sécrétion • Amines (adrénaline, noradrénaline) : Favorisent l’absorption • Purines (ATP, Adénosine) : Favorisent la sécrétion • Peptides (VIP, GRP, endothéline,…) : Favorisent la sécrétion • Peptides (substance P, NPY, somatostatine, opioïdes, … ) : Favorisent l’absorption • Gaz (NO) : Favorisent la sécrétion •Toxines bactériennes (toxine cholérique, toxine d’E. coli, toxine A de C. difficile,…) : Favorisent la sécrétion L’augmentation de la sécrétion d’eau et d’électrolytes par les toxines bactériennes est un mécanisme de défense visant à éliminer les bactéries en augmentant la vitesse du flux dans le TD.
La sécrétion se fait au dépend du volume extracellulaire, ce qui peut mettre en péril l’individu.
IV. Physiopathologie des diarrhées :
A. Déterminants des diarrhées :
Quand on parle d’anomalies de transport de l’intestin, dans conditions de vie normale, on n’utilise pas du tout les pleines capacités du côlon à absorber de l’eau et des électrolytes. On délivre entre 1,5 et 2L de liquide qui n’a pas été absorbé par le grêle. Mais si la capacité du grêle diminue, et qu’au lieu d’absorber la majorité de ce qu’il reçoit il en absorbe la moitié ou moins, il délivre par exemple 5L au côlon. Et bien le côlon est capable de faire face à cela, car cela ne dépasse par ses capacités d’absorption. On ne voit donc pas de signes cliniques (=pas de diarrhée). Cependant si la maladie du grêle progresse, on va dépasser les capacités coliques et on verra une diarrhée. D’autre part, quand on a une maladie du côlon, on voit des signes cliniques beaucoup plus vite puisqu’il n’y a rien derrière pour corriger le tir. On se rend plus rapidement compte des maladies coliques. NB : certaines capacités du grêle ne sont pas partagées avec le côlon mais l’incapacité d’absorber des aa par exemple ne se traduira pas brutalement, contrairement à une diarrhée liée à une maladie du côlon. La temporalité est différente. Exemple d’exposition à la toxine cholérique (facteur pathogène de la maladie du choléra) :
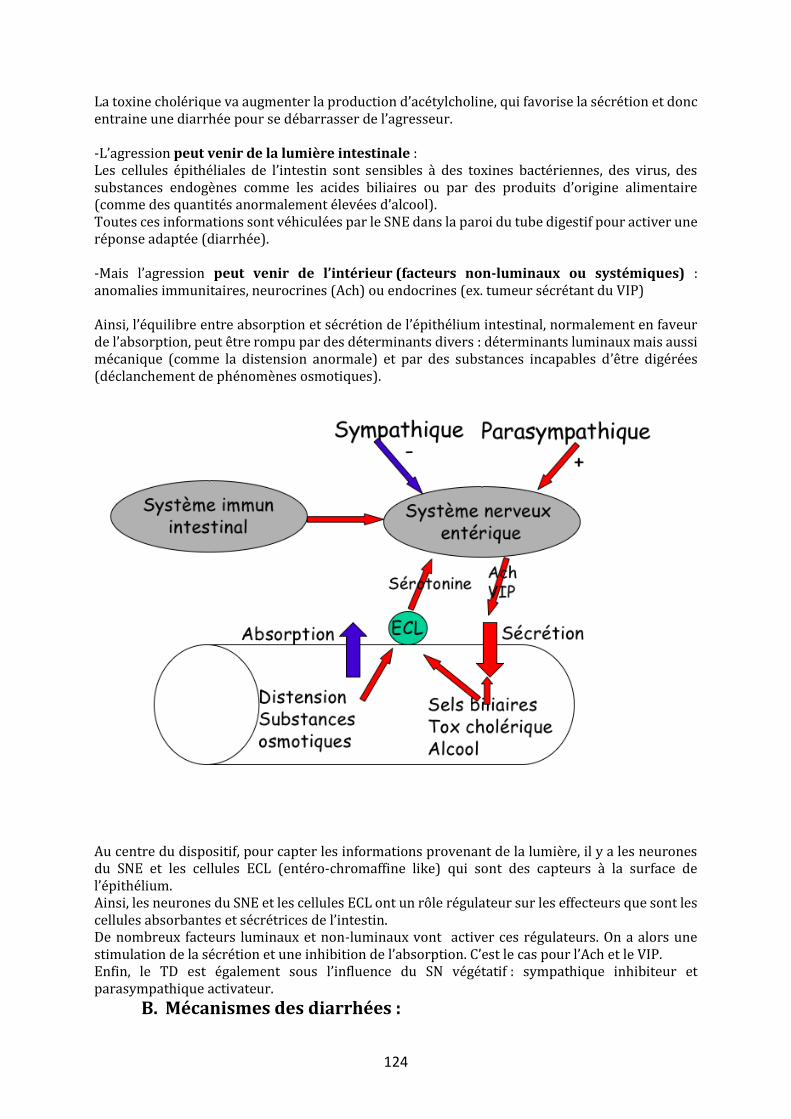
124
La toxine cholérique va augmenter la production d’acétylcholine, qui favorise la sécrétion et donc entraine une diarrhée pour se débarrasser de l’agresseur. -L’agression peut venir de la lumière intestinale : Les cellules épithéliales de l’intestin sont sensibles à des toxines bactériennes, des virus, des substances endogènes comme les acides biliaires ou par des produits d’origine alimentaire (comme des quantités anormalement élevées d’alcool). Toutes ces informations sont véhiculées par le SNE dans la paroi du tube digestif pour activer une réponse adaptée (diarrhée). -Mais l’agression peut venir de l’intérieur (facteurs non-luminaux ou systémiques) : anomalies immunitaires, neurocrines (Ach) ou endocrines (ex. tumeur sécrétant du VIP) Ainsi, l’équilibre entre absorption et sécrétion de l’épithélium intestinal, normalement en faveur de l’absorption, peut être rompu par des déterminants divers : déterminants luminaux mais aussi mécanique (comme la distension anormale) et par des substances incapables d’être digérées (déclanchement de phénomènes osmotiques).
Au centre du dispositif, pour capter les informations provenant de la lumière, il y a les neurones du SNE et les cellules ECL (entéro-chromaffine like) qui sont des capteurs à la surface de l’épithélium. Ainsi, les neurones du SNE et les cellules ECL ont un rôle régulateur sur les effecteurs que sont les cellules absorbantes et sécrétrices de l’intestin. De nombreux facteurs luminaux et non-luminaux vont activer ces régulateurs. On a alors une stimulation de la sécrétion et une inhibition de l’absorption. C’est le cas pour l’Ach et le VIP. Enfin, le TD est également sous l’influence du SN végétatif : sympathique inhibiteur et parasympathique activateur.
B. Mécanismes des diarrhées :

125
Les diarrhées sont classées selon leur mécanisme.
1-Les diarrhées osmotiques : résultent par la prise de substances non absorbables telles que
certains sels de magnésium inorganiques (MgSO4, MgO, Mg(OH)2) ou de sodium (phosphate et
sulfate de Na), certains sucres non digestibles (sorbitol, mannitol, lactulose) auxquels peuvent
s'ajouter le fructose ou lactose lors d'intolérances.
Comment savoir que c’est une diarrhée osmotique ?
Il faut recueillir les sels et mesurer les concentrations d’électrolytes (on ne s’amuse pas trop à le
faire en pratique).
Selles normales :
-On trouve environ 40mM de sodium et 90mM de potassium.
-Les selles ont la même osmolalité que le plasma normal (290mOsm/kg), on cherche la part
osmotique qui n'est pas due aux ions Na+ et K+ (cations majoritaires dans les selles) et aux
anions (qui les accompagnent forcément pour des raisons d'électro neutralité.).
On cherche donc :
Δosm = 290 – (Na + K) x 2.
Δosm = 290 – 130 x 2 = 30mOsm/kg.
Avec :
Δosm : différence d’osmolalité des selles et du plasma=trou osmotique
On parle de diarrhée osmotique si Δosm > 50mOsm/kg.
2-Les diarrhées sécrétoires : Les diarrhées « sécrétoires » sont les diarrhées observées en présence de bactéries, de concentration anormale d’acides biliaires dans le côlon, de laxatifs, de concentrations anormales d’hormones, ou d’adénome villeux (tumeurs peu fréquentes mais qui transportent des ions et des électrolytes en grande quantité). 3-Les diarrhées peuvent aussi provenir d’une altération de la structure de l’intestin : dues par exemple à des maladies inflammatoires chroniques de l’intestin, à une atrophie villositaire (Sprue) ou aussi à une Tuberculose iléale.

126
V. Deux cations bivalents :
A. Le Calcium :
L’alimentation est la seule source de calcium de l’organisme, il n’est pas capable d’en fabriquer. On en a besoin de façon significative au minium pour minéraliser le squelette mais pas seulement. Une des caractéristiques de ce cation bivalent est que son comportement intestinal est particulier : Quand on prend 25mmol de calcium par jour (dose recommandée chez l’adulte), on se rend compte que la majorité n’est pas absorbée (on en retrouve 20mmol dans les selles). Une des raisons de cette absorption non totale du calcium peut être que ce cation est réactif avec beaucoup d’anions. Or sous forme liée à ces anions, il ne peut pas franchir les membranes.
La relation entre la quantité de calcium consommée et celle absorbée est curvilinéaire : Si l’apport est très faible, on voit plutôt une absorption négative donc une sécrétion de calcium. Donc une éviction prolongée du calcium alimentaire abouti forcément à une déminéralisation. Pour avoir une absorption nulle au minimum voire positive, il faut un apport de calcium supérieur à 10 mg/kg/jour, ce qui correspond à 700mg de calcium/jour pour un individu normal de 70 kg. On trouve deux mécanismes de transport du calcium à travers l’épithélium intestinal : -L’absorption trans-cellulaire du calcium : les cellules villositaires du duodénum et du jéjunum (et d’une moindre mesure de l’iléon) se fait par des canaux TRPV5/6. Une fois qu’il est entré dans l’entérocyte, on ne peut pas le laisser libre dans le cytosol car c’est un élément de nombreuses voies de signalisation. Il faut donc lier le calcium à des molécules de transport intracellulaire de calcium : les calcium banding proteins. Ce sont des navettes qui viennent chercher le calcium dans la cellule et le relarguent au voisinage des mécanismes de sorties à travers la membrane basolatérale. Ces mécanismes de sortie sont : -une Ca2+-ATPase, pompe primaire : PMCA 1b -un échangeur 3 Na+/Ca2+ (NCX1).

127
Enfin, ces mécanismes de transport du calcium sont contrôlés par un métabolite de la vitamine D : la 1,25 (OH)2-vitamine D ou calcitriol. C’est une hormone stéroïde lipophile qui agit par l’intermédiaire d’un récepteur nucléaire qui, en se liant à l’ADN, se comporte comme un FT pour activer l’absorption intestinale de calcium. Ainsi, il favorise la synthèse du canal apical, des transporteurs basolatéraux et des calciums binding proteins. -Une partie du calcium traverse l’épithélium qui, lui, est perméable par voie paracellulaire passive (dans le sens du gradient). Déterminants de l’absorption intestinale de Ca2+ :
Stimulants : Inhibiteurs :
- 1,25 (OH)2 vit D
- Carence calcique
- Croissance (GH)
- Grossesse
- Œstrogènes
- Vieillissement - Glucocorticoïdes - Hormones thyroïdiennes - Phytates - Oxalate
NB : -Lors de la grossesse, il faut minéraliser le fœtus. Il y a donc augmentation de la synthèse de vitamine D produite par le placenta. Lors de l’allaitement, le calcium présent dans le lait maternel est issu de la déminéralisation de la mère, il n’y a pas d’augmentation de la synthèse de calcitriol. La mère se reminéralise dans les mois qui suivent l’arrêt de l’allaitement. -Phytates et Oxalates sont retrouvés dans l’alimentation et sont des anions capables de se lier au Ca2+ dans la lumière intestinale, ce qui inhibe son absorption (seul le calcium libre peut être absorbé).

128
Les apports nutritionnels conseillés sont différents selon l’âge car les besoins ne sont pas les mêmes. Les classes d’âge où les besoins sont les plus élevés sont : les adolescents (jusqu’à ses 20 ans où il y a une diminution de sa capacité de minéralisation) et les femmes de plus de 55 ans (ménopause). En France, dans les années 90, la majorité des enfants (3-9 ans) avaient des apports calciques égaux ou supérieurs aux ANC (apports nutritionnels conseillés), mais les apports diminuent avec les générations, alors que les ANC augmentent. Dès l’adolescence, les apports pour la majorité des 10-18 ans étaient inférieurs aux ANC, de même chez les adultes. C’était encore pire pour les plus de 55 ans dont les ANC étaient particulièrement élevés. On dispose aujourd’hui des enquêtes INCA (fin 90’s) et INCA 2 (milieu des années 2000). En comparant les deux enquêtes, on a remarqué que les apports de calcium ont augmentés. La consommation de calcium est nettement supérieure chez les hommes. Cette consommation est toujours insuffisante pour les femmes.
B. Le fer :
Le fer est majoritairement contenu dans les érythrocytes. Les macrophages, les hépatocytes (compartiment de stockage) ainsi que la moelle érythroïde en contiennent également. Il faut un apport de fer de l’ordre de 1 à 2 mg/j. Notre seule source de fer c’est l’alimentation. Il y a un cycle du fer : le fer circulant est utilisé par les cellules de la moelle érythroïde pour former les érythrocytes, plus tard dégradés par les macrophages. On n’est pas capable de recycler efficacement tout le fer donc il faut en absorber pour maintenir notre capital. L’absorption se fait au niveau des cellules villositaires duodénales et au niveau du jéjunum proximal.

129
Une enzyme de la membrane apicale transforme le fer ferrique en fer ferreux (Fe3+ Fe2+).
Ce fer ferreux passe la membrane grâce à un co-transporteur Fer/H+ à la membrane apicale
(DCT1). Il se lie ensuite à une protéine intracellulaire, la mobilferrin qui l'amène à la membrane
basale où la molécule IREG1 permet sa sortie de la cellule et son passage dans la circulation.
Cette absorption est contrôlée en fonction des besoins par une hormone : l'hepcidine. Celle- ci réprime l’absorption : plus sa concentration est basse, plus l’absorption du fer est favorisée. La forme liée à l’hème est absorbée directement puis dégradée, ce qui libère le fer. Si l’on retire le gène de l’hepcidine, on observe une hémochromatose. (conséquence d’une surcharge en fer). On peut aussi extraire le fer du sang du tube digestif et lui faire rejoindre la circulation générale. En cas d’anémie, l’absorption intestinale de fer augmente, et en cas de surcharge, elle diminue.
C. Le Microbiote digestif
On héberge des milliards de bactéries dans notre tube digestif, regroupées à différents étages. Ces groupes de bactéries sont différents selon les étages du tube digestif. La découverte de ce microbiote a permis de s’intéresser à son rôle : Il a des fonctions métaboliques, il intervient dans la différenciation et la prolifération cellulaire, il produit de l’énergie et des vitamines, il a une fonction de barrière, il pourrait avoir une fonction immune et antibactérienne… Bref on ne peut pas vivre sans microbiote.

130
Points clés : - L’épithélium intestinal absorbe et secrète de l’eau et des ions.
- Les (certains) transports ioniques sont contrôlés par des facteurs nerveux et endocrines.
- En situation normale, l’absorption domine la sécrétion et le résultat est une absorption nette.
- Une diarrhée est le résultat de l’augmentation de la sécrétion qui devient supérieure à l’absorption.
QCM : 1.L’absorption intestinale de NaCl est favorisée par :
A) La sérotonine
B) Les opiacés
C) L’acétylcholine
D) L’ATP
E) Les toxines bactériennes
2. Le segment du TD absorbant la plus grande quantité de sodium est : A) l’estomac B) le duodénum C) le jéjunum D) l’iléon E) le colon ascendant 3. Puisque le glucose est absorbé via un co-transport Na/glucose (SGLT1) un régime sans sodium

131
entraine une malabsorption du glucose (V/F)Faux 4. La salive ne contient pas de sodium (V/F) Faux 5. L’alcool en excès peut provoquer une diarrhée (V/F) Vrai 6. Les opiacés stimulent l’absorption intestinale d’électrolytes (et d’eau) (V/F) Vrai (l’opium est un composé anti-diarrhéique) Abréviations : TD=tube digestif AA= Acides aminés SNE=système nerveux entérique Cellules ECL= entérochromatine like FT=facteur de transcription

132
Fiche récapitulative : Transport intestinal d’eau et d’électrolytes, en situation normale ou anormale
Sécrétion et absorption d’eau et d’électrolytes en situation normale :
eau Na+ K+ Cl- HCO3-
Ingestion oui oui oui oui Très peu Sécrétions (salivaires, intestinales, …)
oui oui oui oui Source principale de bicarbonate
Selles En faible quantité par rapport à ce qui est absorbé
absence 10mmol pour 100mmol ingérés
absence absence
Absorption Iléon>colon Iléon>>colon Intestin grêle>>colon
Intestin grêle>colon
Intestin grêle=colon
Particularité Absorption intestinale augmentée par l’aldostérone
Sécrétion augmentée par l’aldostérone
Sécrétion stimulée par certaines toxines (choléra)
On absorbe moins que ce qu’on sécrète
Phénotype cellulaire : Intestin grêle : épithélium avec villosités. Il y a donc des cellules en profondeur (cryptes)
et des cellules de surface (villositaires). Ces cellules ont une structure et une fonction
différentes :
Cellules de surface : elles ont principalement une fonction d’absorption de l’eau et des électrolytes.
Cellules cryptiques : elles font principalement de la sécrétion. Le Cl- est sécrété en
transcellulaire (canal CFTR) et le Na+ en paracellulaire. Il y a aussi sécrétion d’eau. Ces
cellules ont pour vocation de migrer et de changer leur phénotype pour devenir les
cellules de surface.
En situation normale, la fonction d’absorption de l’épithélium l’emporte sur la sécrétante. Mais lors d’agression, le phénotype des cellules peut changer pour s’adapter : l’épithélium devient alors majoritairement sécrétant (diarrhée). Colon : Il n’y a PAS de villosité. Sinon l’organisation reste la même (cellules absorbantes
en surface et sécrétantes en profondeur)
Beaucoup de facteurs sont susceptibles de modifier les transports ioniques dans le colon (amines, purines…). Diarrhées :

133
Dans les conditions normales, nous n’utilisons pas les pleines capacités du colon. Si on diminue les capacités de l’intestin grêle, on ne voit pas tout de suite de signes clinique car le colon arrive à y faire face (jusqu’à un certain seuil). Alors que si les capacités du colon diminuent, on verra plus rapidement l’apparition de signes cliniques (diarrhées). Lors de l’exposition au choléra, il y a augmentation de la production d’acétylcholine, ce qui stimule la sécrétion. Cette agression peut venir de la lumière intestinale ou de l’intérieur. Il existe plusieurs types de diarrhées :
Les diarrhées osmotiques qui résultent de la prise de substances non absorbables. Elles
s’observent si le trou osmotique Δosm>50mOsm/kg.
Les diarrhées sécrétoires
Provenant d’une altération de structure de l’intestin
Deux cations ambivalents : Calcium : Uniquement fourni par l’alimentation car le calcium n’est pas synthétisé par
l’organisme. Il est nécessaire pour minéraliser les os. Mais on remarque que son
absorption n’est pas totale car il ne peut pas franchir les membranes sous forme liée. De
plus, si l’apport en calcium est faible ou nul, on observera une sécrétion de calcium. Il
faut donc que sa consommation soit supérieure à 10 mg/kg/jour. Au niveau du transport,
il peut traverser les cellules par voie transcellulaire ou paracellulaire passive. Il existe des
facteurs qui stimulent (croissance, oestrogènes…) et d’autres qui inhibent ce transport
(vieillissement, glucocorticoïdes…).
Les apports nutritionnels conseillés sont différents selon les âges. Fer : Il est majoritairement contenu dans les érythrocytes. Ici encore, il vient uniquement
de l’alimentation. Il existe un cycle du fer mais nous ne sommes pas capables de le recycler
totalement, d’où la nécessité d’en ingérer. Son absorption se fait au niveau des cellules
villositaires duodénales et du jéjunum proximal. L’absorption du fer est contrôlée par
l’hepcidine qui la réprime.
Le microbiote digestif : Nous hébergeons dans notre système digestif des milliards de bactéries dont les populations sont différentes en fonction des étages. C’est notre microbiote. Il est impliqué dans des fonctions métaboliques, de barrière, de protection et de fonction immune. Le microbiote est indispensable à la vie.

134
UE6 – Système digestif –
Pharmacologie - n°2
17/01/2018
Laurent Chouchana
RT : Astrid d’Ussel
RL : Luc Muratet
Pharmacologie : hépatites médicamenteuses
I.Généralités, épidémiologie
II.Caractéristiques cliniques
III.Principaux mécanismes
A. Toxicité directe ou via un métabolite réactif B. Toxicité mitochondriale C. Hépatite immuno-allergique D. Hépatite auto-immune E. Réactivations virales
IV. Facteurs prédisposants
V. Evaluation d’une atteinte hépatique
VI. Prédiction du risque
Abréviations : HTM : hépatotoxicité médicamenteuse HM : hépatite médicamenteuse
IH : insuffisance hépatique HA : hépatite aiguë
Mot du RT : Le professeur a précisé que le nom des médicaments n’était pas à savoir sauf les
plus importants : paracétamol, halothane.

135
I. Généralités, épidémiologie
L’hépatotoxicité des médicaments représente un enjeu important pour :
- les patients : HTM potentiellement grave voire très grave. C’est en effet la 1ère cause d’hépatite fulminante en France et dans le monde. Si elle est accompagnée d’ictère et d’atteinte cytolytique (destruction des hépatocytes), on comptabilise 10% de décès. On note également un risque de chronicité de l’atteinte.
L’HTM est difficile à diagnostiquer (souvent par exclusion des diagnostics différentiels).
Cette atteinte hépatique pose également le problème du traitement ultérieur (si suspicion d’un traitement indispensable pour le patient, que fait-on ? Arrêt ? Diminuer la dose ?)
Une étude américaine sur 2000 patients adultes avec une insuffisance hépatique aiguë a montré que pour 46% des patients, cette IH était causée par le paracétamol (toxicité directe pour le foie à partir d’une certaine dose) et 11% par d’autres médicaments (=>environ 60% des IH sont médicamenteuses).
Une autre étude française (1997-2001) recherche les causes de transplantation hépatique : pour les 2/3 d’entre eux, la transplantation est due à une hépatite fulminante (hépatite fulminante causée dans 18% des cas par la toxicité d’un médicament) et pour le 1/3 restant, cette transplantation est une seconde greffe (retransplantation).
=> L’HTM est une cause importante d’hépatite fulminante et de greffe hépatique.
- la collectivité : elle représente un coût important. La prévalence des hépatites aiguës sévères est de 1/10 000 individus et l’incidence est de 14 à 24 / 100 000 patients-années. L’HA est responsable de 3-17 hospitalisations sur 10 000, de 10% des causes d’hépatite, de 10% des signalements à un CRPV. On considère qu’il y a environ 1 200 médicaments suspects d’hépatotoxicité (paracétamol en tête de liste).
Lors d’une étude française (1997-2000) sur 81 000 patients observés, 95 cas ont été suspectés d’HM dont 34 cas jugés probables et 2 décès. Cette étude a également montré une sous-notification importante (en considérant cette sous-notification, l’HM serait responsable de 500 décès tous les ans.)
- l’industrie pharmaceutique : elle peut aboutir au retrait d’un médicament du marché. C’est un enjeu crucial pour les industriels de savoir si leur médicament est hépatotoxique. L’industriel réalise ainsi des screenings sur des modèles animaux et est souvent contraint d’arrêter le développement. Mais n’étant pas toujours prédictif, la détection est difficile avant l’AMM. L’HTM est donc responsable de 20% des causes de retrait du marché (certains traitements sont retirés quelques mois seulement après la mise sur le marché comme le ximélagatran).

136
II. Caractéristiques cliniques
Les HM peuvent mimer l’ensemble du spectre clinique et anatomopathologique des maladies aiguës ou chroniques du foie. Tous les types de pathologies du foie sont possibles.
Toutes les structures du foie peuvent être atteintes notamment les hépatocytes +++ (cytolyse, cholestaste, cirrhose, stéatose,…), les cholangiocytes +++ (cholangite aiguë ou chronique), les cellules endothéliales (maladie veino-occlusive) ou encore les cellules Ito (fibrose périsinusoidale).
L’atteinte la plus fréquente est la cytolyse hépatique (destruction des hépatocytes) qui peut être aiguë (paracétamol, isoniazide, halothane…) ou chronique.
Une autre atteinte fréquente est la cholestase, elle aussi aiguë (augmentin) ou chronique.
Entre la cytolyse et la cholestase, on a l’atteinte hépatique mixte, causée par différents médicaments.
L’hépatotoxicité survient après différentes phases :
Initiation (avec des métabolites réactifs et des molécules se fixant covalemment sur les protéines des hépatocytes qui deviennent inactives entrainant la lyse des hépatocytes)
Progression (implique des cellules de l’immunité)
Lésion secondaire
Réparation Décès
Les mécanismes généraux des hépatites aiguës médicamenteuses peuvent être des hépatites toxiques ; des hépatites idiosyncrasiques ; des hépatites immunologiques (autoimmune ou immunoallergique). Les caractéristiques cliniques dépendent du mécanisme d’action.
- Hépatites toxiques : le médicament ou son métabolite va directement porter atteinte aux
hépatocytes.
Incidence élevée. Caractère potentiellement reproductible (chez un patient différent ou
chez le même patient à un temps différent). Délai de survenue court (pour la toxicité
directe) ou retardé (via le métabolite). La gravité ne change pas après la réintroduction
du médicament (même intensité de réponse). Pas de signes d’hypersensibilité (fièvre,
douleurs articulaires,…). Dose dépendante.

137
- Hépatites idiosyncrasiques ou immunologiques : implique des mécanismes immuno
allergiques. Incidence faible. Peu ou pas reproductible (car différence de système
immunitaire). Délai de survenue très variable (souvent beaucoup plus retardé). Après
réintroduction du médicament, gravité de la réponse majorée avec un délai plus court
(système déjà sensibilisé). Signes d’hypersensibilité évocateurs mais inconstants.
Non dose dépendant.
III. Principaux mécanismes
a. L’atteinte hépatique directe
Exemple typique du paracétamol (forte consommation). Le médicament hépatotoxique typique. 1ère cause d’hépatite fulminante médicamenteuse. La dose toxique est chez le sujet sain de 10g (c’est pour cela que toutes les boites de paracétamol en France contiennent au maximum 8g de médicament. Cette norme n’est pas appliquée dans les pays anglo-saxons). Cette dose toxique est beaucoup plus faible pour les personnes alcooliques, dénutris (les jeûnes de 24-48h) ou consommant un traitement inducteur enzymatique (3-10g). => On recommande de ne pas dépasser la dose journalière de 3g.
Cytolyse hépatique pure avec potentiellement des atteintes rénales
Délai d’apparition retardée : 24-48h
Mortalité peut être importante si on a une encéphalopathie et un facteur V (facteur de la coagulation) diminué de 50%.
Traitement : N-acétylcystéine permettant une régénération du glutathion et une réduction de la mortalité.
Rappel sur le métabolisme du paracétamol : en dose thérapeutique, une grande partie du médicament passe par une voie de glucuronoconjugaison (85%) formant des métabolites non toxiques et une infime partie (4%) du traitement passe par un cytochrome : CYP2E1 produisant un métabolite réactif toxique pour le foie : le NAPQI (le NAPQI peut se fixer sur des protéines hépatiques et entrainer une lyse hépatique). En situation thérapeutique (<4g), la faible dose de NAPQI est entièrement conjuguée au glutathion par une glutathion transférase ce qui aboutit à un métabolite non toxique. Le glutathion est la molécule protectrice du foie.
En situation toxique, on assiste à la saturation de la voie de glucuronoconjugaison et à un épuisement des concentrations du foie en glutathion. La concentration en NAPQI augmente, responsable d’une mort cellulaire hépatique.
Chez les alcooliques, la voie métabolique passant par le cytochrome est potentialisée.
Concernant les autres médicaments, la molécule est directement toxique ou la toxicité passe par les métabolites (souvent via le CYP450). Réaction in situ entre les

138
métabolites et les protéines des hépatocytes avec atteinte préférentielle de zones centrolobulaires.
Le mécanisme de la toxicité hépatique peut être du à une peroxydation lipidique, une déplétion en glutathion, une oxydation des protéines (altération cytosquelette, atteinte mitochondriale) ou encore une fixation covalente aux protéines.
De manière préventive, on peut protéger un foie par inactivation du cytochrome CYP45 ou par formation de métabolites stables (par ajout de glutathion S-transférases ou d’époxydes hydrolases) => protection contre les espèces réactives et la peroxydation des lipides.
b. Toxicité mitochondriale
Les médicaments peuvent atteindre les mitochondries notamment au niveau du foie. On aura alors des stéatoses (surcharge de graisse) microvésiculaires +++ et très rarement des hépatites cytolytiques.
Le pronostic des stéatoses microvésiculaires est très différent des macro-vacuolaires.
C’est une stéatose prolongée. Elle présente un tableau clinique insidieux (pas d’augmentation des transaminases) avec hépatomégalie. Elle peut se chroniciser et aboutir à une insuffisance hépatocellulaire avec fibrose et cirrhose. Sa principale cause est la diminution de la β-oxydation mitochondriale des acides gras (d’où leur accumulation).
Un exemple de médicament avec une toxicité mitochondriale : l’acide valproïque.
c. L’hépatite immuno-allergique
C’est un mécanisme allergique classique : le médicament, son métabolite ou le métabolite fixé à une protéine hépatique est reconnu par un macrophage (comme un antigène). Le macrophage l’absorbe et présente l’antigène au lymphocyte TCD4 (qui va activer les lymphocytes TCD8-> expansion clonale).
Un exemple de médicament est l’Halothane. Ce traitement entraine une augmentation des transaminases chez 20% des patients et provoque une hépatite fulminante chez 1/10 000 patients. L’halothane est métabolisé par un cytochrome et son métabolite se fixe à une protéine hépatique. C’est l’ensemble de ce complexe qui est reconnu par les Ac et les lymphocytes.
d. L’hépatite auto-immune
Les anticorps ne reconnaissent pas le médicament mais les protéines hépatiques. La réaction continue même après retrait du médicament.
Par exemple, on peut citer l’acide tiénillique qui par métabolisme du cytochrome CYP2C9 va former un métabolite réactif qui forme une liaison covalente avec ce cytochrome. Le système immunitaire reconnait alors le cytochrome CYP2C9.

139
e. Réactivations virales
Certains médicaments immunomodulateurs ou immunosuppresseurs vont entrainer une réactivation virale (du VHB).
Par exemple une hépatite B chronique va pouvoir se réactiver en hépatite B fulminante.
Ainsi, avant de commencer un traitement précis, un dépistage systématique et obligatoire est réalisé (pour éviter une réactivation virale pendant le traitement).
III. Facteurs prédisposants
Pourquoi certains patients développent une hépatite aiguë et d’autres non ?
Cela est dû à des facteurs dits de susceptibilité : le potentiel hépatotoxique de la molécule + des facteurs génétiques (au niveau des cytochromes, du système HLA ou du système immunitaire) + des facteurs prédisposants (âge, sexe, maladies sous-jacentes, alcool, dénutrition,…)
Parmi les facteurs prédisposants physiologiques on retrouve l’âge (<40 ans : 10-20% des
hépatites alors que >75 ans : 80-90% des hépatites médicamenteuses), le sexe
(uniquement au-delà de 50 ans où il y a 2 fois plus d’hépatites médicamenteuses chez les
femmes que chez les hommes et on observe des différences hommes/femmes selon les
types de médicaments), la grossesse (certains médicaments deviennent hépatotoxique
avec la grossesse).
Il existe également des facteurs prédisposants pathologiques :
- Nutritionnels : jeûne (contribue à une carence en glutathion), la dénutrition, l’alcool.
- Le nombre de médicaments (addition des risques si prise de plusieurs médicaments
hépatotoxiques)
Une hépatopathie sous-jacente n’augmente pas le risque d’HM mais augmente potentiellement sa sévérité.
Il existe également des facteurs génétiques avec notamment les polymorphismes
touchant le cytochrome P450. Selon l’activité des cytochromes, on aura une
augmentation ou une diminution de la production de métabolites.
En effet, on aura une augmentation de la production de métabolites hépatotoxiques avec les métaboliseurs ultra-rapides (via le cytochrome CYP2D6) ou par induction enzymatique (alcool ou médicament. Exemple de l’isoniazide avec la rifampicine qui potentialise la voie métabolique passant par le cytochrome et donc augmente la concentration en métabolites réactifs). =>

140
La variation d’activité des cytochromes peut moduler le potentiel hépatotoxique chez un individu.
On aura de l’autre côté une réduction du métabolisme s’accompagnant d’une accumulation de la molécule mère hépatotoxique chez les métaboliseurs lents.
D’autres facteurs génétiques interviennent au niveau des mécanismes de défense notamment un déficit en glutathion, en époxyde hydroxylase ou encore en carbocystéine sulfoxidation.
L’hépatite médicamenteuse représente la résultante de nombreux facteurs de
susceptibilité.
IV. Evaluation d’une atteinte hépatique
On utilise les mêmes tests que pour une maladie hépatique.
Les tests les plus pertinents sont les transaminases : ALAT (plus spécifique) et les ASAT. On peut également mesurer les phosphatases alcalines (PAL) et la bilirubine totale.
(Les transaminases sont des enzymes présentes particulièrement dans les cellules du muscle et du foie. Une augmentation de leur concentration est le signe d’une destruction des cellules hépatiques. Les PAL sont des enzymes présentes en grande quantité dans le foie.)
D’autres tests peuvent être utilisés mais sont peu sensibles et très spécifiques : les γ-GT et la 5-nucléotidase.
Pour l’interprétation des tests, on parle :
- d’anomalie des tests hépatiques quand on a une augmentation modérée des transaminases (entre 2 et 3 fois la normale = 2-3N).
- d’atteinte hépatique quand on a une augmentation des transaminases > 2-3N, Au-delà de 5-6N, on commence à s’inquiéter et arrêter le médicament.
Pour caractériser l’atteinte hépatique, on utilise le taux d’ALAT, de PAL et le ratio R=N ALAT/N PAL (avec N ALAT et N PAL : le nombre de fois où la valeur est au-delà de la normale.)

141
Ainsi, on a une atteinte cytolytique pure si on a que ALAT>3N ; on a une atteinte cholestatique pure si on a que PAL≥2N. Si on note une augmentation des 2 paramètres, on calcule le rapport et si R est compris entre 2 et 5 alors on a une atteinte mixte ; si R≤2 alors on a une atteinte cholestatique et si R≥5 alors on a une atteinte cytolytique.
D’autre part, l’atteinte est dite chronique quand elle dure plus de 3 mois et aiguë si elle
dure moins de 3 mois.
Entre 0,1 et 10% des patients auront seulement une augmentation des transaminases <
3N ce qui n’est pas inquiétant. Au-delà de cette valeur, on parle d’atteinte hépatique aiguë
et cela concerne 0,01 et 1% des patients. Finalement, on aura des patients en défaillance
hépatique avec un décès ou une transplantation (entre 0,0001 et 0,01%).
L’atteinte aiguë cytolytique est cliniquement proche des hépatites virales
(transaminases peuvent monter très haut) mais peut parfois être asymptomatique. Il
n’y a pas de relation entre l’importance de l’élévation de ALAT et la gravité.
Si les transaminases sont rapidement > 7 000 Ul/L, on penche plus vers une atteinte
hépatite directe (plutôt qu’un mécanisme immuno-allergique) ou une ischémie
hépatique (bas débit hépatique, foie mal irrigué dû le plus souvent à un problème
cardiaque). A
la biopsie hépatique, on peut noter une nécrose hépatocytaire prédominante (surtout
dans l’espace centrolobulaire) ainsi qu’un infiltrat inflammatoire et des éosinophiles
(plus présent dans les réactions immuno-allergiques). Avec
l’arrêt du traitement, on a une guérison plutôt rapide (plus l’arrêt est précoce plus la
guérison est rapide). Il peut y avoir une hépatite fulminante si l’arrêt est tardif. La
guérison peut être lente et décalée dans le temps si l’arrêt est tardif.
Les facteurs de gravité d’une atteinte hépatique cytolytique sont l’ictère, le TP (taux de
prothrombine) ou facteur V < 50% (si TP diminué, il faut le confirmer avec le facteur V
qui est le seul facteur hépatique non vitamine K dépendant et qui traduit donc la fonction
hépatique) et l’encéphalopathie hépatique.
Si on a une survenue rapide de l’encéphalopathie hépatique et du TP ou facteur V<50%, on parle d’hépatite fulminante.

142
Il peut y avoir d’autres signes associés : une hypoglycémie, une hypertension portale, une insuffisance rénale, un désordre métabolique, un syndrome de détresse respiratoire.
Quelques exemples de médicaments responsables d’une atteinte aiguë cytolytique : par hypersensibilité (Halothane, Sulfamides) non immunologique (Paracétamol,…).
La cholestase pure se traduit par un ictère (de la peau et des muqueuses), des urines
foncées, du prurit (tout cela est dû à l’augmentation de le bilirubine). On a également une
augmentation du taux de PAL. La biopsie peut montrer des dépôts de bilirubine dans
les hépatocytes. On observe aussi une dilatation des voies biliaires.
Cependant, les cholestases vraiment pures sont rarement médicamenteuses.
L’atteinte hépatique mixte ou pseudo-cholangite présente une élévation
prédominante des PAL. On observe à la biopsie une nécrose et un infiltrat
inflammatoire. La régression est beaucoup plus lente que dans la cytolyse hépatique.
C’est souvent une atteinte peu grave.
V. Prédiction du risque
Devant une hépatite médicamenteuse, il faut identifier les médicaments en cause en se
faisant aider de la pharmacovigilance : CRPV (faire un interrogatoire policier pour savoir
quand les traitements ont été commencés, arrêtés ; penser à toutes les molécules sans oublier
les compléments alimentaires ou la phytothérapie). Il faut bien sûr arrêter tous les
médicaments suspects (notamment dès que les transaminases dépassent 5-7N) car il persiste
un risque d’atteinte fulminante.
Les arguments cliniques en faveur d’une hépatite médicamenteuse sont l’âge (au-delà de 50
ans), la survenue de l’hépatite au cours du 1er mois de traitement, une consommation
médicamenteuse importante et la présence d’autres signes associés (notamment signes
immuno-allergiques : fièvre, arthralgies, éruption cutanée, hyperéosinophilie, thrombopénie,
hémolyse).
On peut également rechercher la présence d’auto-anticorps spécifiques hépatiques, faire des
tests in vitro (aujourd’hui à l’état de recherche), dosage de médicaments, aspects
histologiques : nécrose centrolobulaire + infiltrat inflammatoire + stéatose microvésiculaire
sont spécifiques des hépatites médicamenteuses.
Le diagnostic de l’hépatite médicamenteuse se fait principalement par exclusion, élimination
des diagnostics différentiels : virale (sérologies VHA/B/C ou autres), les pathologies
hépatobiliaires (notamment les calculs biliaires => transaminase élevée et atteinte
hépatique ; faire une échographie), les hépatites auto-immunes (par la recherche de
différents anticorps), l’alcool (hépatite alcoolique aiguë), la stéato-hépatite non alcoolique
(dû à l’obésité, l’hyperlipidémie, le diabète), la cholestase gravidique (chez la femme

143
enceinte), la maladie veino-occlusive (mais assez rare), les maladies métaboliques ou
génétiques et enfin éliminer l’ischémie hépatique.
L’incidence de l’ischémie hépatique est mal connue (doute avec l’hépatite médicamenteuse).
Elle survient rapidement après un épisode de bas débit (suite à un état de choc et
d’hypoxémie artérielle) qui est le plus souvent asymptomatique. Le foie va être mal irrigué et
cela peut suffire pour entrainer une cytolyse hépatique non spécifique.
D’un point de vue biologique, on a des transaminases qui montent très vite très
précocement (dans les heures suivant l’épisode, elles montent jusqu’à 20N) ; ce phénomène
est plus visible avec les ASAT (par rapport aux ALAT). Les PAL et la bilirubine sont peu
augmentées. Il peut y avoir en plus des troubles de la coagulation et une insuffisance rénale
fonctionnelle. Le plus souvent, l’évolution aboutit à la guérison rapide.
Cet évènement peut survenir chez des patients avec un problème cardiaque ou bien au décours
d’une chirurgie (où il y aurait eu un épisode de bas débit).
Les facteurs de risque de cette atteinte concernent principalement la cardiopathie sous-
jacente.
La biopsie hépatique n’est pas indispensable au diagnostic d’une hépatite médicamenteuse
mais sert surtout à prouver une cause non médicamenteuse (histologie de l’HTM non
spécifique). Elle permet de rechercher des lésions évocatrices (nécrose, stéatose, lésions
pseudoalcooliques, hypertrophie des cellules de Ito, hépatocytes géants), d’identifier un
infiltrat inflammatoire (non spécifique).
La confrontation entre la clinique et la biopsie permet de faire le diagnostic d’une hépatite médicamenteuse.

144
Fiche récapitulative : Hépatites médicamenteuses
HTM : 1ère cause d’hépatite fulminante dans le monde, jusqu’à 10% de décès (de plus, risque
de chronicité de l’atteinte)
Diagnostic différentiel +++ : pas de signes spécifiques.
Paracétamol : 1ère cause d’HM
Hépatite aigüe : 0,1% des hospitalisations (10/10 000 en moyenne)
HM trop peu notifiée
HTM est souvent responsable d’un arrêt de développement d’un médicament et est la
cause d’un retrait d’AMM sur 5
HM touche les hépatocytes et les cholangiocytes +++ —> action par cytolyse hépatique
(atteinte des hépatocytes) ou cholestase, pouvant être aigües/chroniques
3 phases de l’HTM : progression -> lésion 2aire -> réparation/décès
3 types d’HM : toxique, idiosyncratique, ou immunologique
HM toxique : Paracétamol : dose limite toxique de 10g (moins pour les alcooliques/dénutris),
dose au delà de laquelle le glutathion ne peut plus fixer le NAPQI
Atteinte mitochondriale : stéatose microvésiculaire +++ (pronostic très différent des
macro-vacuolaires)
HM immuno-allergiques : exemple de l’halothane dont le métabolite se fixe à un
cytochrome, le complexe sera ensuite reconnu par le SI
HM auto-immun : exemple de l’acide tiénillique dont le métabolite va se lier au cytochrome
CYP2C9, qui sera ensuite reconnu par le SI
/ ! \ réactivations virales (médicaments immunomodulateurs/suppresseurs) : par exemple,
risque du passage de l’hépatite B de l’état chronique à fulminante —> obligation de
dépistage avant tout début de traitement !
Facteurs de prédisposition : âge (>40ans) ; sexe (2* plus de femmes atteintes au delà de
50ans) ; alcool ; dénutrition ; jeûne ; et sur-induction du CYP450 (plus de métabolites
toxiques produits)
Évaluation d’une atteinte : calcul du rapport ALAT/PAL
L’atteinte peut être chronique (>3 mois) ou aigüe (<3 mois)
Atteinte aigüe cytolytique : clinique peut être proche des hépatites virales (transaminases
montent très haut) ou peut être asymptomatique. La biopsie montre une nécrose
centrolobulaire +++, et plus le traitement est arrêté tôt, plus la guérison sera rapide.
Les HM sont souvent confondues avec les ischémies hépatiques (pour qui les
transaminases montent aussi très précocement, et pour qui la guérison est rapide)
La biopsie aide avant tout pour le diagnostic différentiel.

145
Les missions ont repris ! Suivez bien les actualités sur les différents groupes
des missions. Donner ses cheveux, afin qu’ils soient revendus et qu’ils
financent des perruques pour les personnes en chimiothérapie ? C’est possible et ça s’appelle SOLEMèche ! Aller rejoindre le groupe Facebook pour en savoir plus !
Une très belle mission pour la Saint Valentin vous attend après le ski…
Bonne semaine à tous on se retrouve après le ski !
ACTUALITES





























