Embed Size (px)
Citation preview

doi:10.1016/j.jmb.2006.02.034 J. Mol. Biol. (2006) 358, 810–828
Structural and Biochemical Study of Effector MoleculeRecognition by the E. coli Glyoxylate and AllantoinUtilization Regulatory Protein AllR
John R. Walker1,2, Svetlana Altamentova1, Alexandra Ezersky1,3,Graciela Lorca4, Tatiana Skarina1, Marina Kudritska1, Linda J. Ball5
Alexey Bochkarev2,4 and Alexei Savchenko1,4*
1Ontario Center for StructuralProteomics, Best Institute112 College St., TorontoOntario, M5G1L6 Canada
2Structural GenomicsConsortium, 112 College St.Toronto, Ontario, M5G1L6Canada
3Department of MedicalBiophysics, University ofToronto, Toronto, OntarioM5G2M9 Canada
4Banting and Best Departmentof Medical Research, 112 CollegeSt., Toronto, Ontario, M5G1L6Canada
5Structural GenomicsConsortium, Botnar ResearchCentre, University of OxfordOX3 7LD, UK
0022-2836/$ - see front matter q 2006 E
Abbreviations used: wHTH, wingSV, sedimentation velocity; qRT-PCtime polymerase chain reaction; PDEMSA, electrophoretic mobility shif
E-mail address of the [email protected]
The interaction of Escherichia coli AllR regulator with operator DNA isdisrupted by the effector molecule glyoxylate. This is a general, yetuncharacterized regulatory mechanism for the large IclR family oftranscriptional regulators to which AllR belongs. The crystal structures ofthe C-terminal effector-binding domain of AllR regulator and its complexwith glyoxylate were determined at 1.7 and 1.8 A, respectively. Residuesinvolved in glyoxylate binding were explored in vitro and in vivo. Alteringthe residues Cys217, Ser234 and Ser236 resulted in glyoxylate-independentrepression by AllR. Sequence analysis revealed low conservation of aminoacid residues participating in effector binding among IclR regulators,which reflects potential chemical diversity of effector molecules, recog-nized by members of this family. Comparing the AllR structure to that ofThermotoga maritima TM0065, the other representative of the IclR familythat has been structurally characterized, indicates that both proteinsassume similar quaternary structures as a dimer of dimers. Mutations inthe tetramerization region, which in AllR involve the Cys135–Cys142region, resulted in dissociation of AllR tetramer to dimers in vitro and werefunctionally inactive in vivo. Glyoxylate does not appear to functionthrough the inhibition of tetramerization. Using sedimentation velocity,glyoxylate was shown to conformationally change the AllR tetramer aswell as monomer and dimer resulting in altered outline of AllR molecules.
q 2006 Elsevier Ltd. All rights reserved.
Keywords: transcriptional regulation; IclR family; allantoin and glyoxylateutilization; effector-binding domain; X-ray crystallography
*Corresponding authorIntroduction
During anaerobic growth Escherichia coli andother enterobacteriaceae are able to use allantoin asa nitrogen source.1,2 Allantoin is first converted toureidoglucolate, which is further processed toeither glyoxylate or oxaluric acid by ureidoglyco-nate hydrolase or ureidoglyconate dehydrogenase,respectively.1 Glyoxylate enters central metabolismvia the glycerate pathway,3,4 while oxaluric acid is
lsevier Ltd. All rights reserve
ed helix-turn-helix;R, quantitative realB, Protein Data Bank;t assay.ing author:
converted to oxamate and carbamoyl phosphate byoxamate transcarbamoylase.1
In E. coli 12 of the genes coding for the enzymesinvolved in allantoin and glycerate catabolism areclustered together and arranged in five transcrip-tional units: allS, allA, allR, gcl-hyi-glxR-0484-allB-o433-glxK and allD-f411-f261.2,5 The expression ofthe genes allS, allA and gcl is induced in thepresence of allantoin and glyoxylate during aerobicand anaerobic growth,6 while allD expression isinduced by these compounds only in anaerobicconditions.2
The induction of the allS, allA and gcl operons isdependent on the product of the allR gene,2,7 which isconstitutively expressed in aerobic and anaerobicgrowth conditions and serves as a negative regulatorfor the allantoin regulon.2,8 AllR, also called GclR,8
d.

Structure and Biochemistry of AllR 811
recognizes an almost perfect palindrome sequenceoverlapping the promoter regions of the allS, allA andgcl genes. The proximity of the AllR binding site andthe start site of transcription suggests that thisregulator prevents the initiation of transcription bythe RNA polymerase by steric hindrance or bycontact inhibition.9
In the presence of glyoxylate, binding of the AllRregulator to the operator DNA is inhibited andtranscription of the allantoin regulon is de-repressed.7 Inactivation of the AllR gene productcauses constitutive expression of the allS, allA andgcl promoters.2 Glyoxylate appears to be a selectiveeffector; structurally related compounds such asglyconate or D-lactate do not have any effect on theability of the protein to bind DNA in vitro.7
AllR is a member of the large IclR family oftranscriptional regulators and shares 42% sequenceidentity with the founding member of this family,IclR (E. coli isocitrate lyase regulator10–14). TheE. coli K12 genome15 contains eight members ofIclR family, while almost 450 bacterial and archaealmembers of this family have been identified in othersequenced genomes (8 and Pfam1614†).
The IclR family members have a conserveddomain architecture illustrated by high sequencesimilarity with signature helix-turn-helix DNAbinding motive in the N terminus. Small moleculebinding was proposed16 as a main function for theC-terminal part of the IclR regulators. Effectorsranging from 2-keto-3-deoxy-glyconate (Erwiniachrysanthemi KdgR17–19) to p-hydroxybenzoate(Acinetobacter PobR regulator16,20,21), protocate-chuate (Acinetobacter PcaU regulator22–24) andhomogentisate (Pseudomonas putida HmgR25) areidentified as stimuli for members of this family. TheIclR proteins are also highly selective; p-hydro-xybenzoate and protocatechuate are structurallysimilar ligands, yet protocatechuate has no effect onthe family member PobR, which responds top-hydroxybenzoate.16
We provided first insights on domain compo-sition and the ligand-binding mechanisms of IclRfamily members with the crystal structure of theIclR protein family representative, Thermotogamaritima TM0065 regulator.26 The structure con-firmed the presence of an N-terminal winged helix-turn-helix (wHTH) DNA-binding domain and alsorevealed the presence of a large C-terminal domainwith significant structural similarity to the well-characterized, small molecule-binding GAF27–29
and PAS30–32 domains. However, because the ligandof the TM0065 protein has not yet been identified,details of ligand binding by this protein couldnot be deduced from the structure. Here, we presentthe structure of the ligand-binding domain ofthe E. coli AllR protein together with its naturalligand, glyoxylate, and investigate the effectsof ligand binding on the interaction of the proteinwith DNA.
† http://www.sanger.ac.uk/Software/Pfam/
Results
Characterization of E. coli AllR C-terminaldomain
The structure of Tm0065 regulator revealed thepresence of two distinct domains: an N-terminalwHTH DNA-binding domain (Figure 1, amino acidresidues 1 to 63) and a large C-terminal domain(Figure 1, amino acid residues 76 to 246), which arelinked together by an a-helix (Figure 1, amino acidresidues 63 to 74). The structural architecture of theC-terminal domain of TM0065 regulator26 is similarto that of GAF/PAS domains, which bind smallmolecules in a large variety of signaling proteins.27
To determine if this is a common feature of IclRregulators and whether there is functional homo-logy between the GAF/PAS domains and the IclRregulator C-terminal domain we purified theC-terminal domain of E. coli AllR for structuraland functional studies.
Three different versions of the AllR C-terminaldomain, differing by a few amino acid residues atthe N terminus, were prepared (see Materials andMethods), based on multiple sequence alignments(Figure 1), secondary structure predictions andinsights from the TM0065 structure. In each casethe first residue of the domain was selected to bepositioned between the connecting a-helix and thefirst structural element of C-terminal domain(Figure 1). All three constructs led to the expressionof soluble polypeptides of the expected sizes (datanot shown). An AllR fragment correspondingto amino acid residues 97 to 271 (Figure 1), wasselected for further study and called C-AllR.
Both full-length AllR and C-AllR were purifiedto homogeneity and submitted to crystallizationtrials. The crystals obtained for the full-length AllRregulator were not suitable for structure determi-nation. The structure of the C-AllR was determinedin the presence (PDB code 1T9L) and absence(PDB code 1TF1) of its effector molecule, glyoxylate,to resolutions of 1.7 A and 1.8 A, respectively(Figure 2 and Table 1). The structure of C-AllR incomplex with glyoxylate was determined by singleanomalous diffraction (SAD) from selenomethio-nine-containing crystals.
Like the TM0065 structure, the C-AllR domain isan a/b domain, with a centrally located six-strandedantiparallel b-sheet, surrounded on one side by twolong a-helices (a5 and a9), and the other by threeshorter a-helices (a6–a8) (Figure 2(a)). The b-sheet isstrongly curved with the shortest helix (a6) fitting onthe inside of the half-barrel. The secondary structureelements are numbered according to those of theT. maritima 0065 structure (PDB code 1MKM).
The C-AllR structure superimposes well withC-terminal domain of the full-length T. maritima0065, the only full-length structurally characterizedAllR homolog, with a mean rmsd of 1.8 A over 173Ca positions (Figure 2(b)). Each of the secondarystructural elements is preserved with the exception

Figure 1. Multiple sequence alignment of AllR against other E. coli members of the IclR family YagI (GI1786468), KgdR(GI1736468), IclR (GI1790449), YiaJ (GI1789999), YjhI (GI1790752), YfaX (GI1799601), MhpR (GI1786541), AcinetobacterPobR (GI3172124) and PcaU (GI3264839) and T. maritima Tm0065 regulators. The Tm0065 and C-AllR secondarystructure elements are represented as arrows for b-strands and cylinders for a-helices, shown in blue and red,respectively. The TM0065 N-terminal DNA-binding domain and inter-domain helix (a5) elements are colored in lightblue, while C-terminal domain elements are colored dark blue. The first residue of each of the three versions(corresponding to Ala91, Asn95 or Glu97 to Pro271) of C-AllR domain is indicated with an arrow. AllR residues that havebeen mutated for biochemical studies have been boxed. The amino acid residues in YfaX, YiaJ, MhpR and PobRregulators, corresponding to AllR mutations are underlined. Residues that are within 4.2 A from glyoxylate in AllRstructure are highlighted in green. Residues that are involved in inter-subunit interactions are highlighted in gray: darkgray represents residues that make contacts within 4.2 A in all subunits, light gray indicates residues that make contactin a subset of the four subunits. Amino acid sequence alignments were carried out using the program Clustal W.71
812 Structure and Biochemistry of AllR
of a 310 helix observed in the TM0065 structure, andthe intersubunit antiparallel b-sheet b4a: b4a,residues 139–141 observed in the C-AllR structure(Figures 1 and 2). Thus the properties of the C-AllRdomain probably reflect its properties in the contextof the full-length transcriptional factor.
C-AllR interdomain interaction: functionalimplications
The palindromic nature of the AllR binding sitesuggests that the functional unit of AllR is anoligomer. To determine the oligomeric state of AllR,we subjected full-length and C-AllR preparations togel filtration analysis. Based on calculated masses,the purified full-length AllR regulator was pre-dominantly a tetramer, while C-AllR was a dimer(data not shown). While the TM0065 protein was
found in predominantly dimeric form in solutionit formed tetramers in crystal structure.26 In theTm0065 structure, dimers were formed throughinteractions in N-terminal DNA-binding domainsand inter-domain a-helixes (a-helix 5 in Figure 1). Incontrast, TM0065 tetramers formed as a dimer ofdimers connected through C-terminal domains.Thus in Tm0065 N-terminal and C-terminaldomains are involved in separate intersubunitinteractions. In the C-AllR structure, AllRC-terminal domains dimerise through the b4astrand and the loops adjacent to this strand thatcovers 1282 A2 (Figure 3(a)). This corresponds to theinterface observed between the C-terminal domainsof TM0065 (1209 A2). Therefore although the C-AllRstructure lacks an AllR N-terminal domain, we caninfer that the full-length AllR oligomerizationarrangement is similar to that of Tm0065.
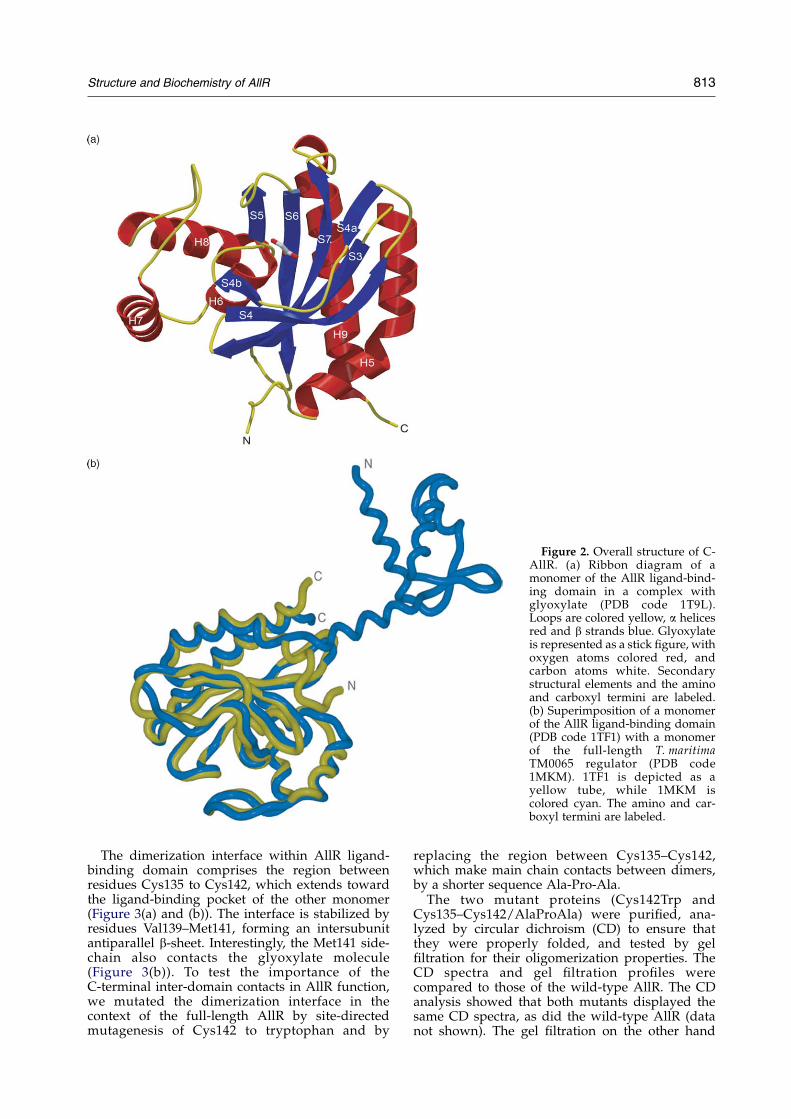
Figure 2. Overall structure of C-AllR. (a) Ribbon diagram of amonomer of the AllR ligand-bind-ing domain in a complex withglyoxylate (PDB code 1T9L).Loops are colored yellow, a helicesred and b strands blue. Glyoxylateis represented as a stick figure, withoxygen atoms colored red, andcarbon atoms white. Secondarystructural elements and the aminoand carboxyl termini are labeled.(b) Superimposition of a monomerof the AllR ligand-binding domain(PDB code 1TF1) with a monomerof the full-length T. maritimaTM0065 regulator (PDB code1MKM). 1TF1 is depicted as ayellow tube, while 1MKM iscolored cyan. The amino and car-boxyl termini are labeled.
Structure and Biochemistry of AllR 813
The dimerization interface within AllR ligand-binding domain comprises the region betweenresidues Cys135 to Cys142, which extends towardthe ligand-binding pocket of the other monomer(Figure 3(a) and (b)). The interface is stabilized byresidues Val139–Met141, forming an intersubunitantiparallel b-sheet. Interestingly, the Met141 side-chain also contacts the glyoxylate molecule(Figure 3(b)). To test the importance of theC-terminal inter-domain contacts in AllR function,we mutated the dimerization interface in thecontext of the full-length AllR by site-directedmutagenesis of Cys142 to tryptophan and by
replacing the region between Cys135–Cys142,which make main chain contacts between dimers,by a shorter sequence Ala-Pro-Ala.
The two mutant proteins (Cys142Trp andCys135–Cys142/AlaProAla) were purified, ana-lyzed by circular dichroism (CD) to ensure thatthey were properly folded, and tested by gelfiltration for their oligomerization properties. TheCD spectra and gel filtration profiles werecompared to those of the wild-type AllR. The CDanalysis showed that both mutants displayed thesame CD spectra, as did the wild-type AllR (datanot shown). The gel filtration on the other hand

Table 1. AllR site-directed mutagenesis primers
Mutation Primer sequence
Met141Ala AGTGTAAATCGATGGTCAGGGCGTGTGCGCCACTGGGCAGTCCys142Trp AATCGATGGTCAGGATGTGGGCGCCACTGGGCAGTCGTCCys142_Cys153 GGTATTAATTGGTCAGTTAGAGGCTCCAGCTGCGCCACTGGGCAGTCGTCTGCLeu149Met GCGCCACTGGGCAGTCGTATGCCACTGCATGCTTCCGAsp207Asn GAACTGGGCTATACCGTAAATAAAGAAGAGCATGTTGHis211Asp TACCGTAGATAAAGAAGAGGATGTTGTAGGTCTGAATTGLeu215Ala GAAGAGCATGTTGTAGGTGCGAATTGCATAGCTTCAGCCys217Ala GAGCATGTTGTAGGTCTGAATGCCATAGCTTCAGCAATTTACGATCys217Ser GAGCATGTTGTAGGTCTGAATTGCATAGCTTCAGCAATTTACGATSer234Ala TAGTGTTGTTGCCGCTATCGCCATCTCCGGGCCTTCATCSer234Asn AGTGTTGTTGCCGCTATCAACATCTCCGGGCCTTCATCSer236Ala GTTGCCGCTATCTCCATCGCCGGGCCTTCATCAAGACTGSer236Met GTTGCCGCTATCTCCATCATGGGGCCTTCATCAAGACTG
Mutagenesis primers listed are designated for non-coding strand. Primers for the coding strand are the reverse complements of thecorresponding non-coding strand primers. Both strand primers were used for each PCR reaction. The sequence for the mutant aminoacid is underlined. Modified nucleotides are in italic.
814 Structure and Biochemistry of AllR
showed that, contrary to the wild-type AllR, theCys142Trp and Cys135–Cys142/AlaProAlamutants were now dimers (data not shown).
The Cys142Trp and Cys135–Cys142 dimerizationmutants were then tested for their ability to repressthe expression of the gcl and allA genes in vivo. Forthese studies an allRK E. coli strain was constructedand complemented by either the wild-type ormutant AllR regulators expressed from a low copynumber vector, pBAD33, using an arabinose-inducible promoter. The transcript levels of twoknown target genes, allA and gcl, were used as theread-out. Transcript levels were measured byquantitative real time polymerase chain reaction(qRT-PCR), in cells grown on the minimal mediumcontaining xylose as a carbon source with orwithout added glyoxylate. The expression profilesfor both allA and gcl genes had the same pattern andthus the profile of the gcl gene only is presented.The allRK E. coli strain carrying the pBAD33 vectoralone was used as a control.
As summarized in Figure 4(a), expression of gcl inthe BW25113 E. coli increases tenfold in the presenceof glyoxylate compared to growth on xylose alone.Deletion of allR (allRK graphs) in this strain causeddramatic derepression (5000-fold) of gcl, confirminga major role of AllR in transcription regulation ofthis gene. This deletion experiment highlighted thatglyoxylate induction only partially releases AllR-mediated repression of gcl.
As further evidence of a glyoxylate effect on theAllR regulator, growth on glyoxylate containingmedium had no effect on the gcl gene expression ina allRK E. coli strain.
gcl repression in the allRK strain was restored bycomplementation with plasmid-born wild-typeAllR (Figure 4(a), WT graphs). Similar to the resultsobtained for the BW25113 strain, a tenfold increasein gcl expression was observed during the growth ofallRK strain harboring wild-type AllR on glyoxy-late. On the other hand, complementation of allRK
strain with Cys135–Cys142 deletion and Cys142TrpAllR mutants was severely diminished compared tothe wild-type AllR. The Cys135–Cys142 deletion
mutant lacked the ability to repress gcl, with anotable effect on gcl expression compared to theallRK strain. While the Cys142Trp AllR mutantcaused 50-fold repression of gcl expressioncompared to allRK strain, that nevertheless wassignificantly less than the effect of wild-type AllRregulator (5000-fold repression during the grown inthe absence of glyoxylate). No considerable changein gcl expression was observed in allRK strainscarrying either of mutant AllRs during growth onglyoxylate.
The ability of Cys135–Cys142 deletion andCys142Trp mutants to bind operator DNA wastested in vitro by electrophoretic mobility shiftassays (EMSA). Purified Cys135–Cys142 deletionand Cys142Trp AllR mutants were compared withwild-type AllR in a binding assay with a 26 bpoperator found in the promoter region of the gclgene7 (Figure 4(b)). In agreement with in vivostudies EMSA results (Figure 4(b)) confirmed asignificant (sevenfold at the 25 nM protein concen-tration) decrease in the binding ability of Cys142Trpmutant protein. No significant mobility shift wasobserved in the presence of up to 100 nM Cys135–Cys142 deletion mutant, suggesting a dramatic lossof DNA binding ability by this mutant protein.
Despite the fact that Cys142Trp mutant proteinwas predominantly dimeric in solution in contrastto wild-type AllR, which formed tetramer, bothprotein complexes with operator DNA had similarmigration rate in EMSA (Figure 4(b)). This simi-larity indicated that Cys142Trp mutant proteinmight regain the ability to form tetramers whenbound to the operator DNA.
The results summarized in Figure 4 clearlydemonstrate that the ability of AllR to represstranscription through binding to operator DNA wasseverely weakened by the Cys142Trp mutation andCys135–Cys142 deletion. These results confirm thebiological relevance of inter-domain contacts inC-AllR crystals and highlight the importance ofAllR tetramerization for binding of operator DNA.
In the C-AllR crystal structure, another dimeri-zation interface was also observed (Figure 3(a)).
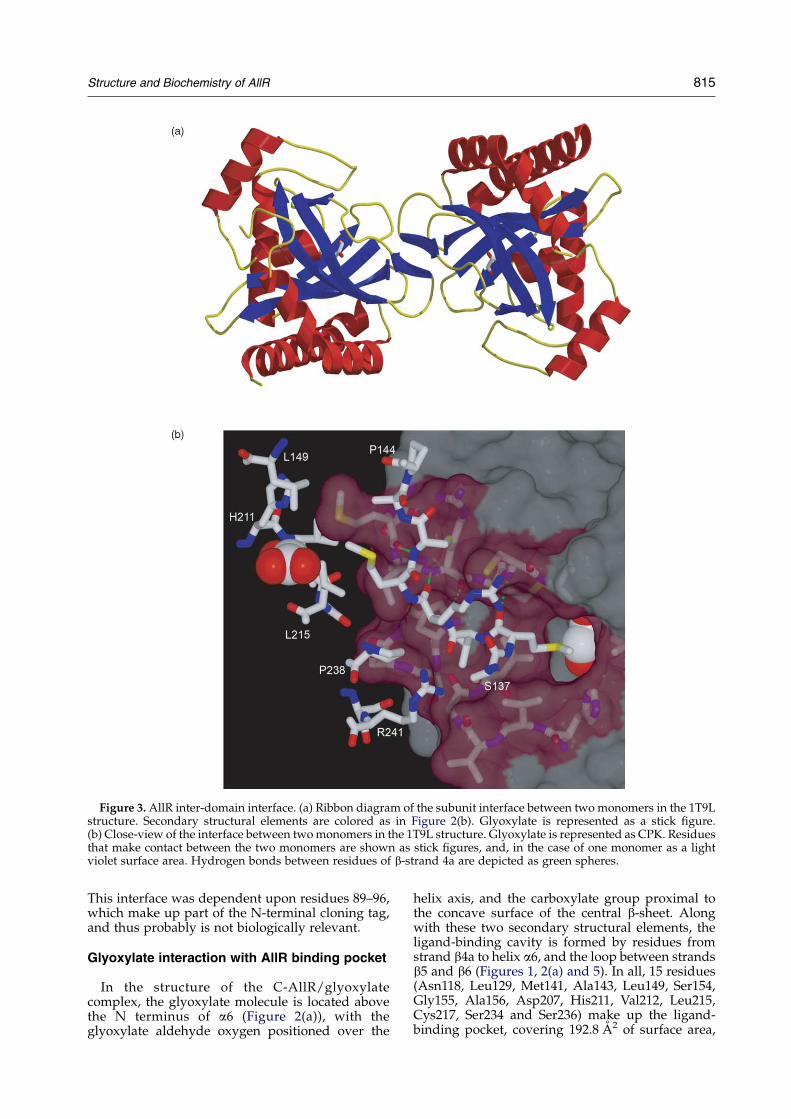
Figure 3. AllR inter-domain interface. (a) Ribbon diagram of the subunit interface between two monomers in the 1T9Lstructure. Secondary structural elements are colored as in Figure 2(b). Glyoxylate is represented as a stick figure.(b) Close-view of the interface between two monomers in the 1T9L structure. Glyoxylate is represented as CPK. Residuesthat make contact between the two monomers are shown as stick figures, and, in the case of one monomer as a lightviolet surface area. Hydrogen bonds between residues of b-strand 4a are depicted as green spheres.
Structure and Biochemistry of AllR 815
This interface was dependent upon residues 89–96,which make up part of the N-terminal cloning tag,and thus probably is not biologically relevant.
Glyoxylate interaction with AllR binding pocket
In the structure of the C-AllR/glyoxylatecomplex, the glyoxylate molecule is located abovethe N terminus of a6 (Figure 2(a)), with theglyoxylate aldehyde oxygen positioned over the
helix axis, and the carboxylate group proximal tothe concave surface of the central b-sheet. Alongwith these two secondary structural elements, theligand-binding cavity is formed by residues fromstrand b4a to helix a6, and the loop between strandsb5 and b6 (Figures 1, 2(a) and 5). In all, 15 residues(Asn118, Leu129, Met141, Ala143, Leu149, Ser154,Gly155, Ala156, Asp207, His211, Val212, Leu215,Cys217, Ser234 and Ser236) make up the ligand-binding pocket, covering 192.8 A2 of surface area,
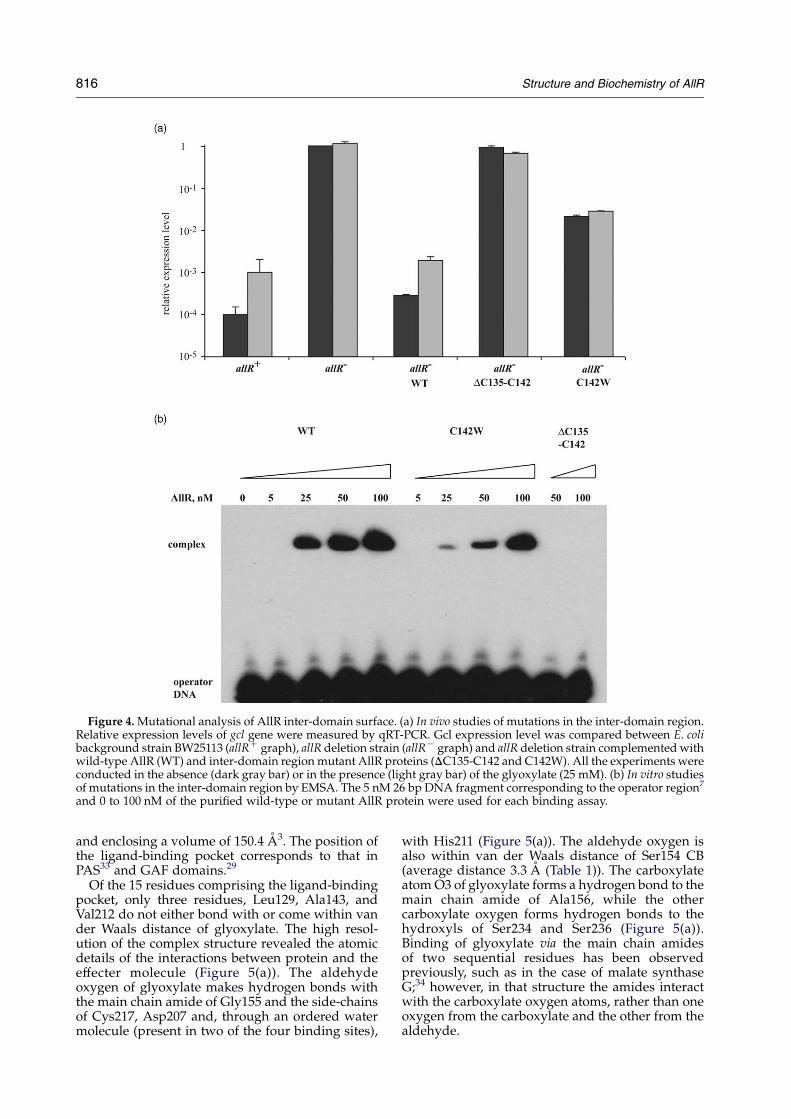
Figure 4. Mutational analysis of AllR inter-domain surface. (a) In vivo studies of mutations in the inter-domain region.Relative expression levels of gcl gene were measured by qRT-PCR. Gcl expression level was compared between E. colibackground strain BW25113 (allRC graph), allR deletion strain (allRK graph) and allR deletion strain complemented withwild-type AllR (WT) and inter-domain region mutant AllR proteins (DC135-C142 and C142W). All the experiments wereconducted in the absence (dark gray bar) or in the presence (light gray bar) of the glyoxylate (25 mM). (b) In vitro studiesof mutations in the inter-domain region by EMSA. The 5 nM 26 bp DNA fragment corresponding to the operator region7
and 0 to 100 nM of the purified wild-type or mutant AllR protein were used for each binding assay.
816 Structure and Biochemistry of AllR
and enclosing a volume of 150.4 A3. The position ofthe ligand-binding pocket corresponds to that inPAS33 and GAF domains.29
Of the 15 residues comprising the ligand-bindingpocket, only three residues, Leu129, Ala143, andVal212 do not either bond with or come within vander Waals distance of glyoxylate. The high resol-ution of the complex structure revealed the atomicdetails of the interactions between protein and theeffecter molecule (Figure 5(a)). The aldehydeoxygen of glyoxylate makes hydrogen bonds withthe main chain amide of Gly155 and the side-chainsof Cys217, Asp207 and, through an ordered watermolecule (present in two of the four binding sites),
with His211 (Figure 5(a)). The aldehyde oxygen isalso within van der Waals distance of Ser154 CB(average distance 3.3 A (Table 1)). The carboxylateatom O3 of glyoxylate forms a hydrogen bond to themain chain amide of Ala156, while the othercarboxylate oxygen forms hydrogen bonds to thehydroxyls of Ser234 and Ser236 (Figure 5(a)).Binding of glyoxylate via the main chain amidesof two sequential residues has been observedpreviously, such as in the case of malate synthaseG;34 however, in that structure the amides interactwith the carboxylate oxygen atoms, rather than oneoxygen from the carboxylate and the other from thealdehyde.
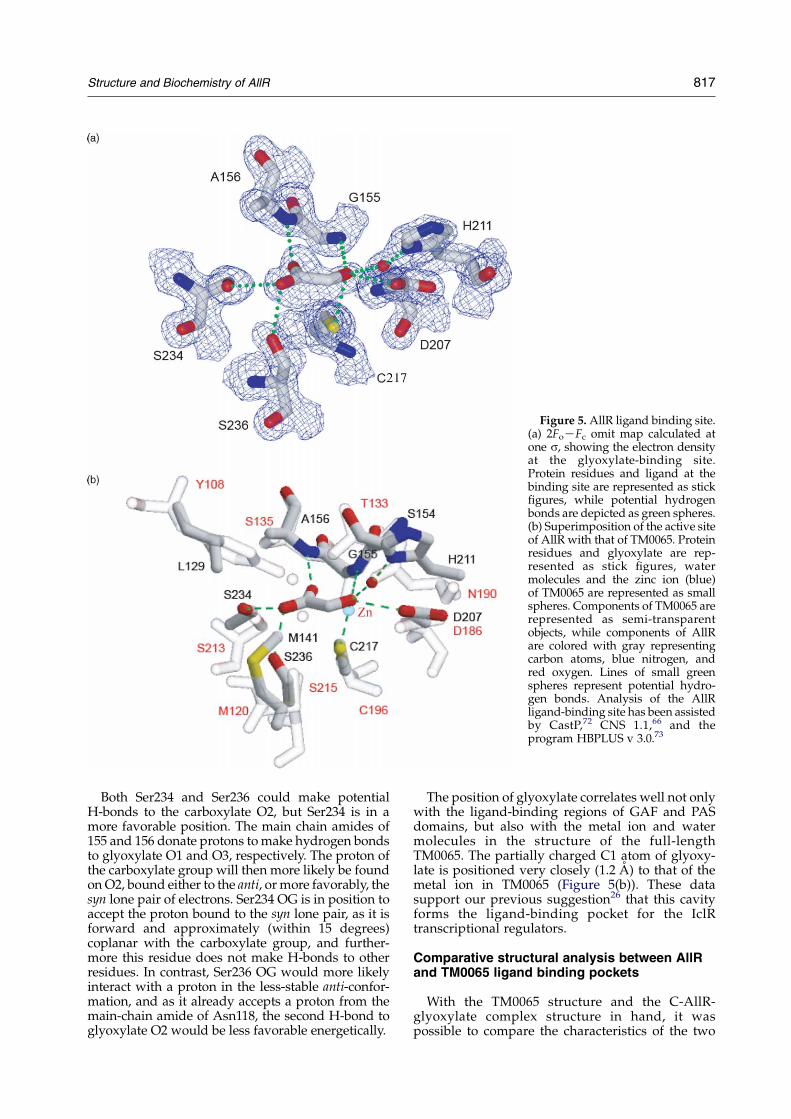
Figure 5. AllR ligand binding site.(a) 2FoKFc omit map calculated atone s, showing the electron densityat the glyoxylate-binding site.Protein residues and ligand at thebinding site are represented as stickfigures, while potential hydrogenbonds are depicted as green spheres.(b) Superimposition of the active siteof AllR with that of TM0065. Proteinresidues and glyoxylate are rep-resented as stick figures, watermolecules and the zinc ion (blue)of TM0065 are represented as smallspheres. Components of TM0065 arerepresented as semi-transparentobjects, while components of AllRare colored with gray representingcarbon atoms, blue nitrogen, andred oxygen. Lines of small greenspheres represent potential hydro-gen bonds. Analysis of the AllRligand-binding site has been assistedby CastP,72 CNS 1.1,66 and theprogram HBPLUS v 3.0.73
Structure and Biochemistry of AllR 817
Both Ser234 and Ser236 could make potentialH-bonds to the carboxylate O2, but Ser234 is in amore favorable position. The main chain amides of155 and 156 donate protons to make hydrogen bondsto glyoxylate O1 and O3, respectively. The proton ofthe carboxylate group will then more likely be foundon O2, bound either to the anti, or more favorably, thesyn lone pair of electrons. Ser234 OG is in position toaccept the proton bound to the syn lone pair, as it isforward and approximately (within 15 degrees)coplanar with the carboxylate group, and further-more this residue does not make H-bonds to otherresidues. In contrast, Ser236 OG would more likelyinteract with a proton in the less-stable anti-confor-mation, and as it already accepts a proton from themain-chain amide of Asn118, the second H-bond toglyoxylate O2 would be less favorable energetically.
The position of glyoxylate correlates well not onlywith the ligand-binding regions of GAF and PASdomains, but also with the metal ion and watermolecules in the structure of the full-lengthTM0065. The partially charged C1 atom of glyoxy-late is positioned very closely (1.2 A) to that of themetal ion in TM0065 (Figure 5(b)). These datasupport our previous suggestion26 that this cavityforms the ligand-binding pocket for the IclRtranscriptional regulators.
Comparative structural analysis between AllRand TM0065 ligand binding pockets
With the TM0065 structure and the C-AllR-glyoxylate complex structure in hand, it waspossible to compare the characteristics of the two
818 Structure and Biochemistry of AllR
ligand-binding pockets (Figures 1 and 5(b)) as wellas to use multiple sequence alignment to comparethe characteristics of the potential ligand-bindingresidues in the other IclR family members.
The closest interactions of the AllR protein withthe glyoxylate are between Cys217 of AllR and C1 ofthe glyoxylate; the distance (averaged among thefour molecules in the structure) between the sulfuratom and glyoxylate is 2.6 A. The ligand interactionwith Cys217 is mimicked in TM0065 by theinteraction of the co-crystallized metal ion withthe corresponding cysteine at position 196. Theobservation that in TM0065 the cysteine can binda metal ion, and previous mass spectroscopicanalysis8 that shows that this residue is capable ofmaking a covalent bond to b-mercaptoethanol,indicates that this residue is particularly reactive.A cysteine residue is found at the correspondingposition in a large number of IclR regulators,suggesting that it is a key feature of the ligand-binding pockets.
The C1 of the glyoxylate also comes into van derWaals contact with Leu149 and Leu215. Multiplesequence alignments demonstrate that hydro-phobic amino acid residues predominate at thecorresponding positions in the members of IclRfamily: TM0065 has valine and isoleucine in therespective positions (Figure 1).
Overall, the TM0065 ligand-binding pocket isslightly larger than that of C-AllR, with 245.5 A2 ofsurface area, and enclosing a volume of 234.7 A3.Five amino acid residues (Met141, Asp207, Cys217,Ser234 and Ser236) that interact with glyoxylate inthe C-AllR are conserved in the ligand-bindingpocket of TM0065 (Figures 1 and 5(b)). Anadditional four AllR residues in the ligand-bindingpocket, Leu149, Ser154, His211, Leu215, are sub-stituted by chemically similar residues in TM0065(Val128, Thr133, Asp190 and Ile194, respectively).
Figure 6 (legen
The amino acid residues Gly155 and Ala156 havebeen substituted in TM0065 with alanine andserine, respectively, and the hydroxyl oxygenatoms of Thr133 and Ser135 extend further intothe ligand-binding pocket compared to their C-AllRcounterparts. Leu129 is replaced in TM0065 withTyr128, which also points its hydroxyl into theligand-binding pocket. Ile99 also protrudes intothe pocket, but Val128 is recessed compared to itsC-AllR counterpart Leu149. Sequence analysisbased on positions of AllR and TM0065 ligand-binding cavity residues reveals several interestingpreferences for these positions in the IclR family.Small or hydrophobic residues dominate thepositions corresponding to AllR Met141, Leu149and Leu215, while hydroxyl or amine amino acidsoccupy positions corresponding to Ser234 andSer236. The amino acids corresponding to Asn118,Asp207 and His211 demonstrate the widest rangeof variation among IclR regulators.
Mutagenesis analysis of AllR glyoxylate-bindingpocket
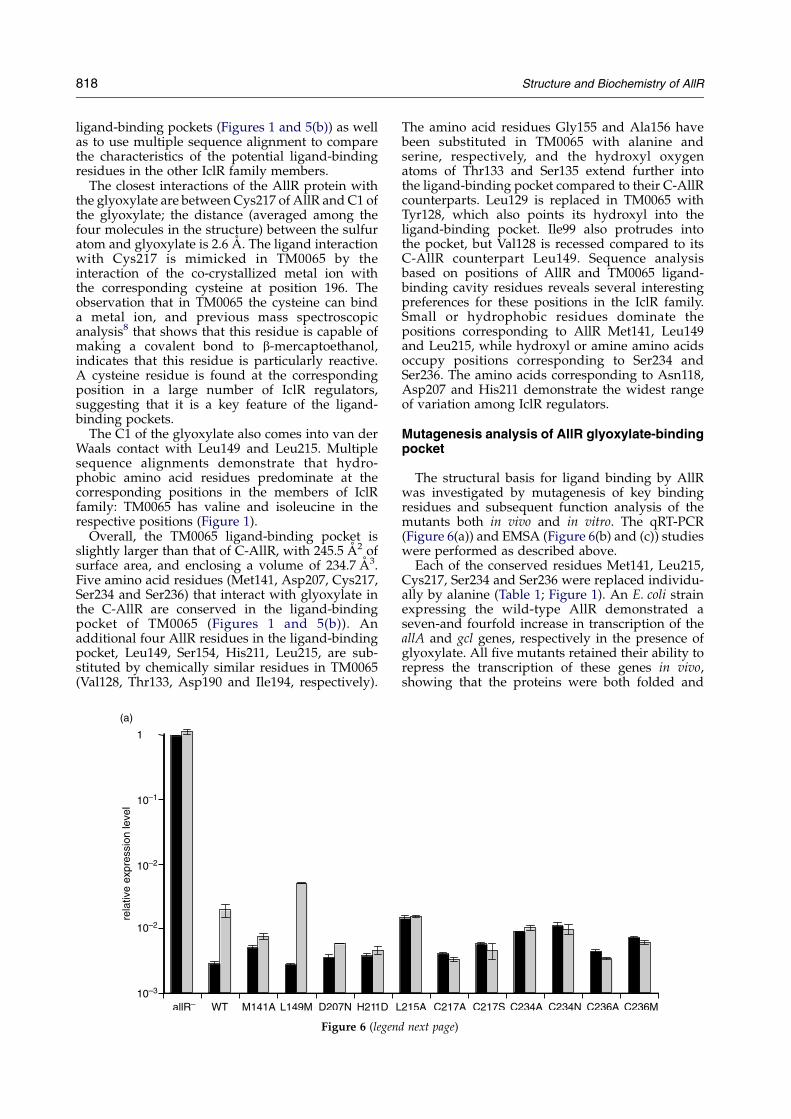
The structural basis for ligand binding by AllRwas investigated by mutagenesis of key bindingresidues and subsequent function analysis of themutants both in vivo and in vitro. The qRT-PCR(Figure 6(a)) and EMSA (Figure 6(b) and (c)) studieswere performed as described above.
Each of the conserved residues Met141, Leu215,Cys217, Ser234 and Ser236 were replaced individu-ally by alanine (Table 1; Figure 1). An E. coli strainexpressing the wild-type AllR demonstrated aseven-and fourfold increase in transcription of theallA and gcl genes, respectively in the presence ofglyoxylate. All five mutants retained their ability torepress the transcription of these genes in vivo,showing that the proteins were both folded and
d next page)
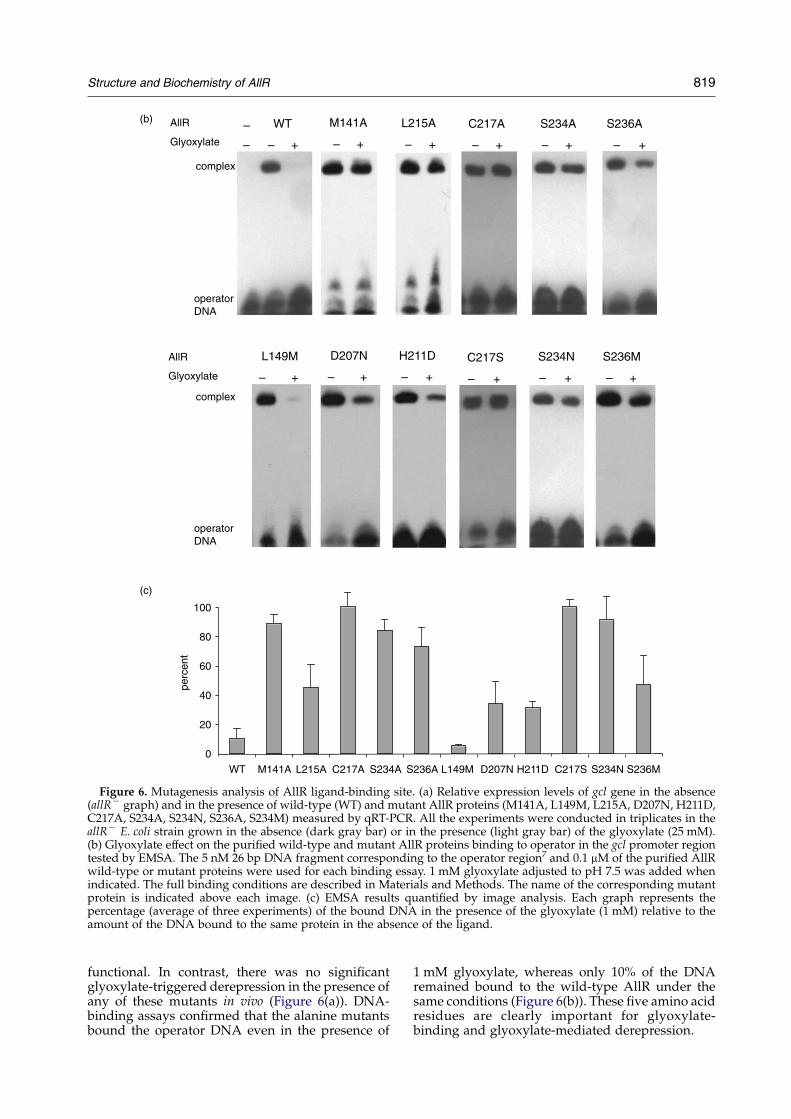
Figure 6. Mutagenesis analysis of AllR ligand-binding site. (a) Relative expression levels of gcl gene in the absence(allRK graph) and in the presence of wild-type (WT) and mutant AllR proteins (M141A, L149M, L215A, D207N, H211D,C217A, S234A, S234N, S236A, S234M) measured by qRT-PCR. All the experiments were conducted in triplicates in theallRK E. coli strain grown in the absence (dark gray bar) or in the presence (light gray bar) of the glyoxylate (25 mM).(b) Glyoxylate effect on the purified wild-type and mutant AllR proteins binding to operator in the gcl promoter regiontested by EMSA. The 5 nM 26 bp DNA fragment corresponding to the operator region7 and 0.1 mM of the purified AllRwild-type or mutant proteins were used for each binding essay. 1 mM glyoxylate adjusted to pH 7.5 was added whenindicated. The full binding conditions are described in Materials and Methods. The name of the corresponding mutantprotein is indicated above each image. (c) EMSA results quantified by image analysis. Each graph represents thepercentage (average of three experiments) of the bound DNA in the presence of the glyoxylate (1 mM) relative to theamount of the DNA bound to the same protein in the absence of the ligand.
Structure and Biochemistry of AllR 819
functional. In contrast, there was no significantglyoxylate-triggered derepression in the presence ofany of these mutants in vivo (Figure 6(a)). DNA-binding assays confirmed that the alanine mutantsbound the operator DNA even in the presence of
1 mM glyoxylate, whereas only 10% of the DNAremained bound to the wild-type AllR under thesame conditions (Figure 6(b)). These five amino acidresidues are clearly important for glyoxylate-binding and glyoxylate-mediated derepression.

820 Structure and Biochemistry of AllR
A second series of mutagenesis studies wasdesigned to test whether replacement of theresidues involved in glyoxylate binding (Leu149,Asp207, His211, Cys217, Ser234 and Ser236) withthe corresponding residues from E. coli YiaJ, YfaXand MhpR and Acinetobacter PobR regulators(Figure 1) would affect binding. The amino acidswere replaced by methionine, asparagine, asparticacid, serine, asparagine and methionine (Figure 1),respectively.
Similarly to the Cys217Ala mutation, the replace-ment of Cys217 by serine destroyed the ability ofAllR to respond to glyoxylate in vivo (Figure 6(a)).No dissociation of the Cys217Ser complex withoperator DNA in the presence of glyoxylate couldbe detected in DNA-binding assays (Figure 6(b) and(c)). These results underline the critical role ofCys217 in ligand binding.
Mutations of the other residues to their mates inother IclR family members were not as deleterious.In the case of the Leu149 mutation to methionine theglyoxylate derepression was almost three timesmore efficient compared to the wild-type AllR. Thisresult was confirmed by the in vitro DNA binding(Figure 6(b)). Modeling of methionine into theC-AllR–glyoxylate structure (data not shown)demonstrated an improved van der Waals inter-action between methionine and glyoxylate,compared with that of leucine and glyoxylate. Thedistance between the glyoxylate C1 and methionineCE atoms was now 2.99 A compared to 4.04 Abetween C1 and leucine CD1.
The Asp207Asn mutation reduced the effects ofglyoxylate on the derepression of the gcl genetwofold, while other mutants (His211Asp, Ser234-Asn, Ser236Met) showed no response to glyoxylatein vivo (Figure 6(a)). Interestingly, these last threemutants (Asp207Asn, His211Asp, Ser236Met) wereable to respond to glyoxylate in assays of DNA-binding, releasing 67, 69 and 53% of the operatorDNA in the presence of the ligand (Figure 6(b) and(c)). The residual activity of the Asp207Asn mutantmay arise from the fact that both aspartate andasparagine would be predicted to contribute to theformation of important hydrogen bonds.
Effect of glyoxylate binding on AllRconformation
The binding of glyoxylate reduces the affinity ofAllR for its operator DNA sequence. In otherprokaryotic inducer-regulator systems containingwHTH and inducer-binding domains,35–38 thebinding of the inducer in the signaling domainhas been shown to trigger a conformational changethat alters the DNA-binding domains often througha change in the oligomeric state of the regulatorprotein.39–41 There was no gross conformationalchange in the C-terminal domain of AllR caused byglyoxylate binding; the structures are completelysuper-imposable. Since crystallographic studiesmay not have allowed us to observe ligand-inducedchanges in the AllR oligomerization, we turned to
analytical ultracentrifugation to investigate theeffect of glyoxylate on AllR protein conformationin solution.
The sedimentation velocity (SV) studies wereperformed on the full-length AllR in the absenceand presence of a 100-fold molar excess ofglyoxylate (Figure 7). The experiments were carriedout at two different protein concentrations (25 and50 mM) to assess whether AllR self-association wasalso concentration-dependent. Fitting the data as acontinuous distribution c(s)42,43 provided excellentsensitivity and resolution, enabling a clear distinc-tion between different sedimenting species.
At both tested protein concentrations, with andwithout inducer, three main AllR oligomeric specieswere identified (Figure 7(a)). Their sedimentationcoefficients (s) corresponded to expected values formonomeric, dimeric and tetrameric forms of AllRprotein (29,607 Da per monomeric polypeptidechain as measured by mass spectrometry; data notshown). Not surprisingly, all peaks are less wellresolved at the higher protein concentration, with orwithout glyoxylate present, due to higher molecularcrowding and spontaneous formation of higheraggregates (Figure 7(b)).
The addition of the ligand causes a dramaticincrease (Figure 7(a) and (b)) in the percentage oftetrameric AllR present in the sedimenting mixtureand a corresponding decrease in the populations ofmonomer and dimer species, showing that bindingto glyoxylate stabilizes and favors the tetramericstate. In addition, the presence of glyoxylate leads tosharper, more discrete peaks for all three oligomers,as well as small shifts towards higher s values foreach. In the absence of ligand, calculated Svedbergconstants (S) were: 1.3 S, 2.1 S and 3.3 S at 50 mMAllR concentration, and 1.3 S, 2.1 S and 3.3 S at25 mM AllR concentration, for monomer, dimer andtetramer species, respectively. In the presence ofligand, these values increased slightly to: 1.4 S, 2.3 Sand 3.6 S at 50 mM AllR concentration, and 1.4 S,2.2 S, 3.5 S at 25 mM AllR concentration (1 SZ10K13 s). These effects indicate that the confor-mations of each oligomer become more compactupon glyoxylate binding, and that this is mostnotable for the tetramer.
Discussion
The AllR repressor belongs to the large but poorlyinvestigated family of IclR bacterial transcriptionalfactors, which regulate transcription in response toa variety of metabolites. The structural scaffold ofthe family representatives was revealed in ourearlier studies of the T. maritima IclR regulatorTM0065.26 Here, we investigate the molecularbasis for ligand binding and its effects on regulatoractivity by defining the apo and ligand-boundstructure of E. coli member of this family, AllRrepressor.
Although the two proteins share relatively littlesequence identity (26%), the structure of AllR
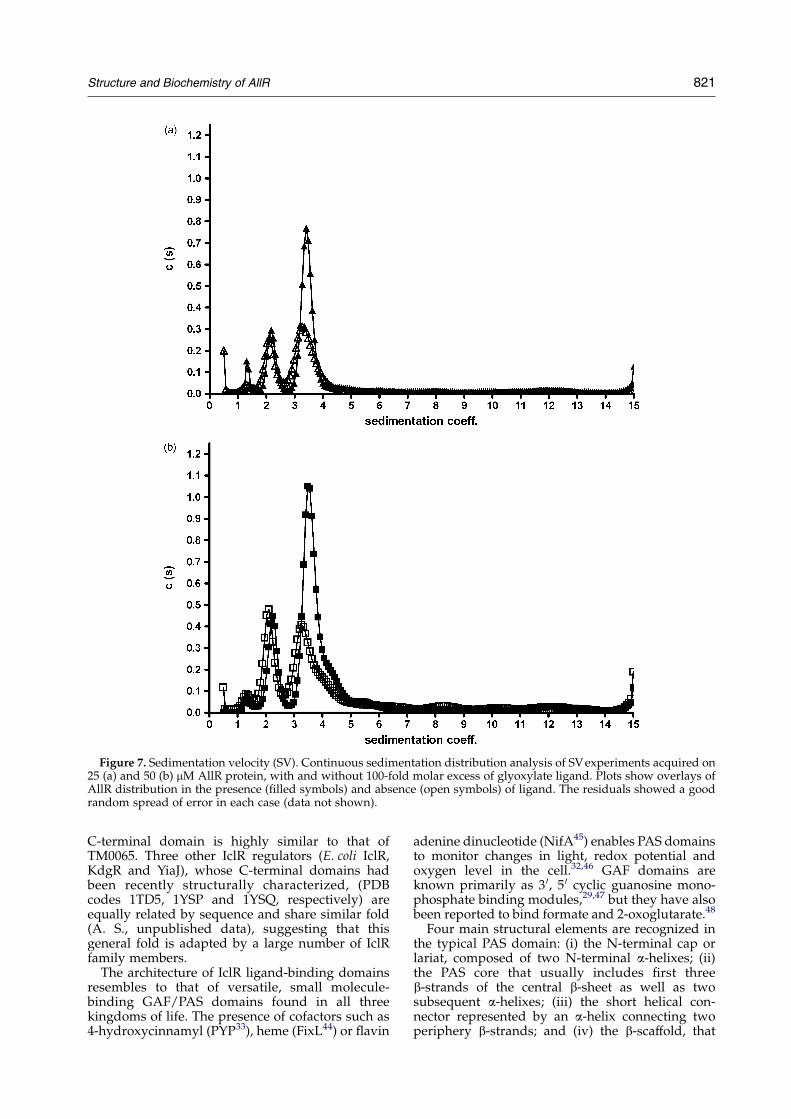
Figure 7. Sedimentation velocity (SV). Continuous sedimentation distribution analysis of SV experiments acquired on25 (a) and 50 (b) mM AllR protein, with and without 100-fold molar excess of glyoxylate ligand. Plots show overlays ofAllR distribution in the presence (filled symbols) and absence (open symbols) of ligand. The residuals showed a goodrandom spread of error in each case (data not shown).
Structure and Biochemistry of AllR 821
C-terminal domain is highly similar to that ofTM0065. Three other IclR regulators (E. coli IclR,KdgR and YiaJ), whose C-terminal domains hadbeen recently structurally characterized, (PDBcodes 1TD5, 1YSP and 1YSQ, respectively) areequally related by sequence and share similar fold(A. S., unpublished data), suggesting that thisgeneral fold is adapted by a large number of IclRfamily members.
The architecture of IclR ligand-binding domainsresembles to that of versatile, small molecule-binding GAF/PAS domains found in all threekingdoms of life. The presence of cofactors such as4-hydroxycinnamyl (PYP33), heme (FixL44) or flavin
adenine dinucleotide (NifA45) enables PAS domainsto monitor changes in light, redox potential andoxygen level in the cell.32,46 GAF domains areknown primarily as 3 0, 5 0 cyclic guanosine mono-phosphate binding modules,29,47 but they have alsobeen reported to bind formate and 2-oxoglutarate.48
Four main structural elements are recognized inthe typical PAS domain: (i) the N-terminal cap orlariat, composed of two N-terminal a-helixes; (ii)the PAS core that usually includes first threeb-strands of the central b-sheet as well as twosubsequent a-helixes; (iii) the short helical con-nector represented by an a-helix connecting twoperiphery b-strands; and (iv) the b-scaffold, that

Figure 8. Homology between AllR and PAS/GAFdomains. Superimposition of the AllR ligand-bindingdomain (PDB code 1T9L) with the structure of thephotoactive yellow protein (PDB code 3PYP). AllR isdepicted as a cyan worm, while the four segmentsof PYP32 are depicted with the following colors: theN-terminal cap, yellow; the PAS core, orange; the helicalconnector, violet; and the b-scaffold, green. Respectiveligands (glyoxylate, 4-hydroxycinnamic acid) are shownin stick format. The amino and carboxyl termini arelabeled, black for AllR, red for PYP. Figures 1, 2, 4 and 6were prepared with a combination of SPOCK,74
Molscript,75 and Raster3D.76
822 Structure and Biochemistry of AllR
includes three b-strands that makes the second halfof the central b-sheet (Figure 8). In all known casesthe cofactor is invariably located in the cavity (so-called “active site”) of the PAS core, thus making itthe most functionally relevant element of the PASdomain. The PAS core is also known to harborresidues involved in protein–protein interactions.The b-scaffold provides the structural support forthe PAS core and completes the central b-sheet andcontains a number of highly conserved residues,constituting a so-called “PAC sequence motive”,while the N-terminal cap is the least conservedelement of the PAS domain and can be replacedwith other structural elements in different PASdomains.49
The architecture of both the PAS core and theb-scaffold of the PAS domain are clearly recognizedin the C-terminal domains of AllR and TM0065,with the location of glyoxylate contact correspond-ing to the PAS core “active site” (Figure 8). The PASN-terminal cap appeared to be replaced withhelixes 5 and 9 in C-AllR and TM0065. The C-AllRa-helices (number 7 and 8) do not have anequivalent in the PAS domain fold. Interestingly,the representatives of GAF/PAS domains shareno significant sequence similarity with any IclRregulator. The C-AllR–glyoxylate complex struc-tural analysis also demonstrates that the similaritiesbetween C-AllR and PAS/GAF domains do not
extend into the chemical nature and content ofamino acids involved in the ligand binding. Theequivalent amino acids involved in cofactor bind-ing in the PAS/GAF domains are not found in theIclR regulators. While the strongest interaction ofglyoxylate with C-AllR is through the cysteineresidue (Cys217), the position of this residue doesnot correspond to that of cysteine that forms acovalent bond with ligand in the PAS domain ofPYP. Such comparative analysis demonstrates thatIclR regulators form a new separate group in thediverse family of proteins containing PAS/GAFsignaling domain and shows new functionalcapabilities of this domain as small-molecule-binding fold.
Based on C-AllR and TM0065 structures anal-ysis the minimal functional unit of this family ofproteins appears to be a tetramer. The tetrameriza-tion of these proteins arises from the association ofdimers. In TM0065 structure the stable dimer isformed by the hydrophobic interactions betweena-helix 1 of the N-terminal DNA-binding domainsand a-helix 4 that links N and C-terminal domainsof each monomer.26 Tm0065 dimers then tetra-merize through interactions between the ligand-binding domains, which involves the loopbetween the b4 and b4b strands. From C-AllRstructure analysis, it appears that AllR uses asimilar oligomeric arrangement, except that thetetramerization interface contains an additionalshort b4a strand. The variation in the tetrameriza-tion interface may be important to minimize non-specific association among the various IclR para-logs (eight in E. coli) that are expressed in the samecell.
The functional relevance of tetramerization foundboth in TM0065 and AllR for their activity wasdefined by mutational analysis. The AllR dimersthat were unable to tetramerize due to mutation oralteration in the b4a strand were functionallyinactive in vivo. Thus the association of at leastfour wHTH domains was required for efficientbinding of AllR to its operator region in thepromoter regions of gcl and allA genes featuringthe inverted repeat T-8T-7G-6G-5A-4A-3A-2A-1
A0T1(A/T)2T3T4C5C6A7A8.7 Similarly most of theexperimentally identified operator regions of theIclR family regulators contain palindromic orpseudo-palindromic sequences,22,50–52 underliningthe symmetric nature of binding of these regulatorsto DNA. The founding member of the family, E. coliIclR, also is thought to bind its operator as atetramer.8,14 Interestingly the promoter regionsunder control of IclR family members such asPobR, PcaU and PcaR feature threefold repetitionsequences;24,53 this may imply a more complexoligomeric nature of functional units for theseregulators. Further studies of AllR interactionswith its operator will have a general impact onour understanding of IclR regulators interactionswith DNA.
In in vitro experiments, a major part of the AllR–operator DNA complex was dissociated in the

Structure and Biochemistry of AllR 823
presence of 1 mM glyoxylate (Figure 6(b)). Suchsignificant AllR–operator DNA dissociation in vivowould result in dramatic derepression of genescontrolled by this regulator. However, only atenfold increase in the gcl gene transcription wasdetected during E. coli strain growth in the presenceof 25 mM glyoxylate (Figure 4(a)). At the same time,the deletion of allR gene prompted a 5000-foldincrease in the transcription of this gene. Thissignificant difference between in vivo and in vitroeffects of glyoxylate on AllR function suggests thateven in the presence of millimolar extracellularconcentration of this compound, the intracellularconcentration may be too low for substantialinactivation of intracellular pool of AllR regulators.Although no experimental data are available on thelevels of glyoxylate in E. coli cells, a recent study54
on its close relative Salmonella typhimurium deter-mined that during growth in either 22 mM glucoseor 135 mM acetate the intracellular concentration ofglyoxylate was only 11 and 120 mM, respectively. Asan important intermediate glyoxylate can berapidly incorporated into the central metabolismby the combined action of malate synthase and theD-glycerate pathway,3,4 or alternatively it can bereduced to glycolate by constitutive glyoxylatereductase activity.4 Low fluctuation of intracellularglyoxylate concentration would result in tightregulation of allantoin and glyoxylate degradationgenes by AllR repressor.
Here and in our previous studies26 we haveidentified residue positions that are important forligand binding. Extension of these positions to therest of the IclR family members shows a widevariation of the amino acid residues involved in theligand binding. While only five out of 12 AllRamino acid residues involved in glyoxylate bindingare conserved in TM0065, none of these 12 aminoacid residues involved in the interaction (Figures 1and 5(b)) including Cys217 is absolutely conservedacross the family. This feature indicates thatdramatic changes in chemical environment of theligand-binding pockets dictated by the variationamong the ligands can be expected from structu-rally similar members of the IclR family. At thesame time the IclR family members with conservedgroup of amino acid residues corresponding to theones involved in the glyoxylate binding in AllR canbe expected to bind chemically similar ligands.
According to the functional model, glyoxylatebinding reduces the affinity of the regulator for itsoperator, thereby releasing the expression of theregulated genes. This effect must propagate fromC-terminal ligand-binding domain to N-terminalDNA-binding domain. Our analytical ultracentri-fugation results suggest that signal is not propa-gated by destabilization of the tetramer required forDNA binding, suggesting that the signal may bedirectly propagated via the linker region. TheTM0065 structure exhibits two alternate confor-mations of the linker region between the N andC-terminal domains (residues 62–79), resulting indifferent relative orientations of the ligand-binding
domains with respect to the wHTH dimer.26 Thisconformational flexibility may be important forsignal propagation between the N and C-terminaldomains.
Another explanation of high conformationalsimilarity between C-AllR and C-AllR-glyoxylatestructures may be that the AllR apo- and glyoxylate-bound forms adapt partially overlapping ensem-bles of conformations. The crystal structures of each“freeze-out” the same, presumably highly sampledconformation that is shared by both forms. Theanalytical ultracentrifugation profile may reflectthis ensemble. Glyoxylate binding also stabilizesAllR tetramer versus the two other oligomer states ofthis protein. The latter effect is probably due to theclose proximity of the ligand-binding pocket andthe AllR tetramerization interface and the increasednumber of inter-subunit contacts seen in theeffector-bound form.
In summary the AllR studies presented herereveal for the first time the details of the effectorbinding by IclR regulator. Nevertheless details ofthe promoter recognition by IclR regulators and thederepression mechanism upon the effector-bindingremain to be determined.
Materials and Methods
DNA manipulations and cloning
Standard methods were used for site-directed muta-genesis, chromosomal DNA isolation, restriction enzymedigestion, agarose gel electrophoresis, ligation, andtransformation.55 Plasmids were isolated using spinminiprep kits (Qiagen, USA), and PCR products werepurified using Qiaquick purification kits (Qiagen, USA).
The allR gene was amplified from wild-type E. coliBW25113 chromosomal DNA by PCR using the followingprimers (restriction sites KpnI and HindIII are italizedand the Shine Dalgarno sequence added is underlined)allRefw: GAGCTCGGTACCAGGAGGAAACTATGACGGAAGTTAGACGGCGC and allRerv: GCATGCAAGCTTTTATGGATGTGCTTTCAGTCC. The PCR products werepurified, treated with KpnI and HindIII, and then clonedinto pBAD33.56 For over-expression, full-length allR andthree C-terminal domain constructs (corresponding toAla91, Asn95 or Glu97–Pro271) were subcloned into theNdeI and BamHI sites of a modified form of pET15b(EMD Biosciences, USA), in which a tobacco etch vitus(TEV) protease1 cleavage site replaced the thrombincleavage site and a double stop codon was introduceddownstream from the BamHI site. This constructprovides for an N-terminal hexahistidine tag separatedfrom the protein by a TEV protease recognition site(ENLYFQYG). Site-directed mutants were generated byQuikChange Site-Directed Mutagenesis Kit (Stratagene,USA) using the primers listed in Table 1.
Protein purification
The fusion proteins were over-expressed in E. coli BL21-Star(DE3) (Stratagene, USA) harboring an extra plasmidencoding three rare tRNAs (AGG and AGA for Arg, ATAfor Ile). The cells were grown in LB at 37 8C to an A600nm
w0.6 and expression induced with 0.4 mM IPTG.

824 Structure and Biochemistry of AllR
After addition of IPTG, the cells were incubated withshaking at 15 8C overnight. The cells were harvested,resuspended in binding buffer (500 mM NaCl, 5% (v/v)glycerol, 20 mM Tris (pH 9), 5 mM imidazole), flash-frozenin liquid N2 and stored at K70 8C. The thawed cells werelysed by sonication after the addition of 0.5% NP-40 and1 mM each of PMSF and benzamidine. The lysate wasclarified by centrifugation (30 min at 17,000 rpm; BeckmanCoulter Avanti J-25 centrifuge, JA-17 rotor) and passedthrough a DE52 column pre-equilibrated in binding buffer.The flow-through fraction was then applied to a metalchelate affinity-column charged with Ni2C. After thecolumn was washed, the protein was eluted from thecolumn in elution buffer (binding buffer with 500 mMimidazole). The hexa-histidine tag was then cleaved fromthe protein by treatment with recombinant His-taggedTEV protease. The cleaved protein was then resolved fromthe cleaved His-tag and the His-tagged protease byflowing the mixture through a second Ni2C-column.
The purified proteins were dialyzed against 10 mM Tris(pH 9.0), 500 mM NaCl, and concentrated using a BioMaxconcentrator (Millipore, USA). Before crystallization, anyparticulate matter was removed from the sample bypassage through a 0.2 mm Ultrafree-MC centrifugalfiltration device (Millipore, USA).
Selenomethionine-labeled proteins were expressedusing the same vector and host strain but in sup-plemented M9 media.57 The sample was preparedunder the same conditions as the native protein exceptfor the addition of 5 mM b-mercaptoethanol to thepurification buffers.
Crystallization and data collection
The primary crystallization condition was determinedby a sparse crystallization matrix (Hampton Research
Table 2. Data collection and refinement statistics
Data set 1TF1Space group C2221
Cell constants (A) 92.80 97.54Data set range (A) 19.73 1.80Completeness (%)a 97.9 (98.8)Redundancy 3.0 (3.1)Rmerge (%)b 8.7 (27.7)Refinement statisticsResolution range (A) 19.73 1.8Unique reflectionsWorking set 56,530 (92.6%)Test set 3033 (5.1%)
Completeness (%) 97.7 (98.5)Rfactor (%)c 21.4 (24.2)Rfree (%)d 25.7 (28.4)Total atoms 6056Protein 5461Glyoxylate 0Solvent 595
Root mean square deviationBonds (A) 0.014Angles (degrees) 1.70Dihedrals (degrees) 23.1
RamachandranMost favored (%) 92.4Allowed (%) 7.5Generous (%) 0.2Disallowed 0.0
a Data in parentheses represent data in the highest resolution shelb RmergeZSjIKhIijSI, where I is the observed intensity of an indivc RfactorZSjFoKFcjSFo, where Fo and Fc are observed and calculad RfreeZthe cross-validated Rfactor computed with the test set of re
kits: Crystal Screen 1 and PEG/Ion Screen), at roomtemperature using the sitting-drop vapor-diffusiontechnique in 96-well plates. This condition was modifiedby varying the pH and the concentration of the solutes.The best condition was obtained using hanging drops(2 ml protein: 2 ml precipitant ratio) in crystallizationcondition: 0.1 M Hepes (pH 6.8), 1.9 M ammoniumsulfate, 4% 2-methyl-2,4-pentanediol (MPD) in two tofive days at room temperature. For diffraction studies, thecrystals were flash-frozen with the crystallization bufferplus 40% MPD as the cryoprotectant. For the complexstructure 1 mM glyoxylate was added to the proteinsolution prior to crystallization.
Diffraction data of crystals of C-AllR complexed withglyoxylate were collected at 100 K at the 19BM beam lineof the Structural Biology Center at the Advanced PhotonSource, Argonne National Laboratory. The absorptionpeak and the rising inflection point were determined bycalculating and plotting f 0 and f 0 values against energy58
from the fluorescence spectrum. The three-wavelengthMAD data were collected from a Se-Met substitutedprotein crystal using an inverse-beam strategy. Allcrystallographic data were measured with the custom-built 3!3 tiled CCD (charge-coupled device) detector59
with a 2108–210 mm2 active area and fast duty cycle(w1.7 s). The experiment, data collection, and visualiza-tion were controlled with d*TREK60 and all data wereintegrated and scaled with the program packageHKL2000.61 Data collection and processing statistics areprovided in Table 2.
Data corresponding to the peak wavelength wasprocessed with HKL2000,61 and then input into theprogram SOLVE.62 SOLVE located 23 of the possible 32selenium sites (including two in the N-terminal histidinetag), and gave a mean figure of merit of 0.39 (0.28 inthe highest resolution bin). Density modification and
1T9LC2221
144.49 92.97 97.68 144.3650.0 1.6891.2 (56.3)2.9 (2.3)7.6 (47.1)
33.7 1.70
64,895 (89.4%)3456 (5.1%)94.5 (85.0)19.2 (22.3)23.2 (26.0)5981545720504
0.0171.8023.4
92.47.50.20.0
l.idual reflection, and hIi is the mean intensity of that reflection.
ted structure factors, respectively.flections.

Structure and Biochemistry of AllR 825
automatic model building using RESOLVE63,64 resultedin excellent maps and partial models for each of the foursubunits. The models were manually completed with theaid of the 4-fold non-crystallographic symmetry and thegraphics program O.65 Further sessions with O65 andrefinement of atomic positions and individual B-factorsusing the program CNS-1.166 resulted in a R-factor of19.2% (Rfree 23.2) for data from 33.7 to 1.7 A. The finalmodel comprises 5981 protein atoms (four C-AllRligand-binding domains), 20 ligand atoms (four glyox-ylate molecules) and 504 water molecules. The modelhas excellent stereochemistry as judged by PRO-CHECK,67 with no Ramachandran violations. All resi-dues of the ligand-binding domain, with the exception ofthe three C-terminal amino acid residues (two in the caseof subunit A), along with several residues of theN-terminal tag were located in the experiment.
To determine the ligand-free structure of the AllRligand-binding domain, a data set was collected oncrystals formed in the absence of glyoxylate using anR-AxisIVCC detector and a MicroMax-007 generator(Rigaku MSC, USA). Improvement of the input model(1T9L, without glyoxylate or water molecules), wascarried out by rigid-body refinement, energy minimiz-ation and B-factor refinement using CNS-1.1,66 along withmanual rebuilding in O65 (aided with omit maps). Thefinal model comprises 5461 protein atoms and 595 watermolecules, and has an R-factor of 21.4% (Rfree 25.7%) fordata from 19.7 to 1.8 A. All residues of the ligand-bindingdomain, with the exception of the three C-terminal aminoacid residues (two in the case of subunit A), along withseveral residues of the N-terminal tag were located in theexperiment, and the model has excellent stereochemistryas judged by PROCHECK,67 with no Ramachandranviolations.
Construction of allR deletion mutant
All the in vivo studies were conducted in the geneticbackground of strain BW25113.68 Deletion mutants weregenerated by the methods described by Datsenko &Wanner.68 To prepare competent cells for transformation,BW25113 containing pKD46 was cultured at 30 8C in SOBbroth55 containing 100 mg of ampicillin per ml. When theabsorbance at 600 nm (A600) reached 0.5, the culture wascentrifuged at 4000 rpm for 5 min, and the cells werewashed three times with cold water before beingresuspended in a minimal volume of water (1% of theoriginal culture volume). The kanamycin resistance gene(km) was amplified by PCR from pKD4 by using theprimers allRFw: GCACAGGCGTTAGAGCGGGGAATTGCGATTCTGCAATATTTGGGTGTAGGCTGGAGCTGCTTC and allRrv: GACCAGCTCACCCTGACTGACAAAACGATCTTCTGTCAGTCTTGATCATATGAATATCCTCCTTA.
The PCR products were purified with a Qiagen kit,treated with DpnI, and repurified by electrophoresis. Thekm gene was transformed into BW25113-competent cellsby electroporation (Gene Pulser; pulse controller at 200 U,capacitance at 250 mF, and voltage at 25 kV). Afterelectroporation, the cells were grown with shaking in1 ml of SOC medium at 37 8C for 1 h, and the cultureswere plated onto Luria-Bertani (LB) agar containing 25 mgof kanamycin per ml. The Kmr transformants werepurified on new kanamycin-LB plates. The mutantsin which the target genes were replaced by the km genewere verified by PCR using the primers allRcfw:TTGCGATTCTGCAATATTTGG and allRcrv: CGATCTTCTGTCAGTCTTGAT. To delete the km gene from the
chromosome, pKD46 was removed from the cells bygrowing the bacteria at 37 8C, and then pCP20, expressingthe FLP recombinase, was introduced by transformation.The transformants containing pCP20 were grown over-night with shaking at 42 8C, and the cultures were platedon LB agar without antibiotics. Colonies were tested forsensitivity to kanamycin and ampicillin.
qRT-PCR studies
Bacterial cells were cultured in Mops minimal media69
with 0.2% (w/v) xylose as carbon source. When the A600
reached 0.5, cells were collected by centrifugation at 4 8C.Total RNA was subsequently isolated with the RNeasyMini Kit (Qiagen, USA) in accordance with themanufacturer’s protocol. Residual DNA present in theRNA preparations was removed by RNase-free DNase(Fermentas, Lithuania). cDNAs were synthesized withthe superscript first-strand synthesis kit (Invitrogen,USA) in accordance with the manufacturer’s instructionsand stored at K20 8C prior to use. Real-time quantitativePCR (qRT-PCR) was carried out on the AppliedBiosystems 7300 apparatus (Applied Biosystems, USA)using Platinumw SYBRw Green qPCR SuperMix UDG(Invitrogen, USA) in accordance with the manufacturer’srecommended protocol. Primers used for the RT-PCRwere as follows: for allA, GTGGAGCGTTACCACGATTTand GGTCTGGTTTGTCGTCACCT; for gcl, GCAAAATGGCGGTTACAGTT and TCGGCCTGGATTAACATTTC; for rrsC, CAGCCACACTGGAACTGAGA andGTTAGCCGGTGCTTCTTCTG. The relative expressionvalues were normalized using the number of cyclesobtained for the house-keeping gene rrsC and thenexpressed in relation to the allRK strain under repressingconditions.
Electrophoretic mobility shift assays (EMSA)
Electrophoretic mobility shift assays for AllR andmutant proteins were performed in triplicates usingproteins purified and concentrated using the proceduresdescribed above.
The oligonucleotides corresponding to AllR recog-nition region in gcl-promoter region (5 0-AAAGTTGGAAAAATTTTCCAATAAAT-3 0) were labeled andannealed by Biotin 3 0End DNA labeling kit (Pierce,USA) according to the manufacturers instructions.Binding assays were performed at 37 8C for 20 min inthe binding buffer (10 mM Tris–HCl (pH 7.5), 50 mM KCl,1 mM DTT) supplied by LightShift ChemiluminescentEMSA Kit (Pierce, USA) in the presence of 50 ng/ml ofpoly(dI-dC) non-specific competitor DNA. The bindingreaction also contained 0.1 mM of purified protein, 5 mMMgCl2, 5 nM Biotin-labeled oligonucleotide.
EMSA were performed in a BioRad Protein II apparatususing 6% (w/v) polyacrylamide/0.5! TBE gels. Aliquotsof 20 ml of the above binding reaction were loaded per lane.Electrophoresis was performed at 100 V using ice-cold0.5! TBE as a running buffer (5! TBE is 450 mM Tris(pH 8.3), 450 mM boric acid, 10 mM EDTA). Biotin-labeledoligonucleotides and oligonucleotide-protein complexeswere transferred from the polyacrylamide gel to theBiodyne B Positive Nylon Membrane (Pierce,USA) in0.5!TBE immediately after electrophoresis by electro-blotting at 380 mA for 40 min. Transferred DNA was cross-linked for 15 min using UV cross-linker equipped with312 nm bulbs. The oligonucleotide-protein complexeswere detected by horseradish peroxidase/Super Signal

† www.rcsb.org
826 Structure and Biochemistry of AllR
Detection System. Membranes were exposed to KodakX-ray film for 1 min and quantified by image analysis.
Circular dichroism (CD) spectroscopy
CD spectra were collected at 25 8C on an AVIV 62D CDSpectrophotometer from 200 to 260 nm using a 0.1 mmpath length cell, with a scan rate of 100 nm/min, timeconstant 1.0 s, bandwidth of 1 nm, and sensitivity of 100mdeg. Each spectrum was averaged from ten scans. Afterbuffer subtraction, the spectra were calibrated in units ofmolar ellipticity.
Size-exclusion chromatography
FPLC size-exclusion chromatography was performedon a Superdex-200 10/30 column (GE Biosciences) pre-equilibrated with 10 mM Tris (pH 9), 0.5 M NaCl, 0.5 mMTris (2-carboxyethyl) phosphine hydrochloride (TCEP).The column was calibrated with cytochrome c (12.4 kDa),carbonic anhydrase (29 kDa), bovine serum albumin(66 kDa), alcohol dehydrogenase (150 kDa), b-amylase(200 kDa). A 20 ml of protein sample at a 5 mg mlK1
concentration or premixed with standard proteins wascentrifuged at 14,000 rpm for 10 min before being injectedinto the column through a 20 ml injection loop. Filtrationwas carried out at 4 8C at a flow rate of 0.5 ml minK1. Theeluted proteins were detected by measuring the absor-bance at 280 nm.
Analytical ultracentrifugation
Sedimentation velocity (SV) experiments were per-formed with a Beckman Xl-I analytical ultracentrifuge(Beckman-Coulter UK Ltd, High Wycombe, UK) using aneight-hole An-50 Ti rotor. Cells were equipped with12 mm double sector, charcoal-filled Epon centerpiecesand quartz windows. Measurements were carried outusing the absorption optics of the instrument at suitablewavelengths for detection of the concentration gradient.
Full-length AllR protein was extensively dialyzedagainst 20 mM Tris–HCl buffer (pH 9.0) containing0.5 M NaCl, 0.5 mM TCEP and 5% glycerol. SV exper-iments were recorded at two concentrations of AllR (25and 50 mM) in the presence and absence of 100-fold molarexcess of glyoxylate (Sigma). An aliquot of the samplebuffer was used as a reference for all measurements. Thepartial specific volume ( �v) of AllR was calculated from theprotein sequence using the program Sednterp v.1.08 byDavid Hayes and John Philo.70 The density and viscosityof the buffer were calculated from the buffer compositionalso using this software.
SVexperiments were performed at 10 8C and 50,000 rpmwith 400 ml of protein solution in each cell. Scans wereacquired every 180 s at a single wavelength (280 nm) usinga radial step size of 0.003 cm. Sedimentation coefficientswere computed using the program Sedfit v.8.9g,42,43 whichcombines finite element solutions of the Lamm equationfor a large number of discrete species, with maximumentropy regularization, to represent a continuous size-distribution. This method of processing the SV datayielded the highest resolution and sensitivity.
Protein Data Bank accession numbers
The atomic coordinates and structure factors havebeen deposited in the Protein Data Bank (accessioncodes 1TF1 and 1T9L), Research Collaboratory for
Structural Bioinformatics, Rutgers University, NewBrunswick, NJ†.
Acknowledgements
We thank all members of the SBC at ANL for theirhelp in conducting experiments and Aled Edwardsfor critical reading of the manuscript. This workwas supported by National Institutes of Healthgrant GM62414-01, by the Ontario Research andDevelopment Challenge Fund and by a grant fromthe Canadian Institutes of Health Research Grant.
References
1. Vogels, G. D. & Van der Drift, C. (1976). Degradationof purines and pyrimidines by microorganisms.Bacteriol. Rev. 40, 403–468.
2. Cusa, E., Obradors, N., Baldoma, L., Badia, J. &Aguilar, J. (1999). Genetic analysis of a chromosomalregion containing genes required for assimilation ofallantoin nitrogen and linked glyoxylate metabolismin Escherichia coli. J. Bacteriol. 181, 7479–7484.
3. Kornberg, H. L. (1966). The role and control of theglyoxylate cycle in Escherichia coli. Biochem. J. 99, 1–11.
4. Ornston, L. N. & Ornston, M. K. (1969). Regulation ofglyoxylate metabolism in Escherichia coli K-12.J. Bacteriol. 98, 1098–1108.
5. Chang, Y. Y., Wang, A. Y. & Cronan, J. E., Jr (1993).Molecular cloning, DNA sequencing, and biochemi-cal analyses of Escherichia coli glyoxylate carboligase.An enzyme of the acetohydroxy acid synthase-pyruvate oxidase family. J. Biol. Chem. 268, 3911–3919.
6. Pellicer, M. T., Fernandez, C., Badia, J., Aguilar, J.,Lin, E. C. & Baldom, L. (1999). Cross-induction of glcand ace operons of Escherichia coli attributable topathway intersection. Characterization of the glcpromoter. J. Biol. Chem. 274, 1745–1752.
7. Rintoul, M. R., Cusa, E., Baldoma, L., Badia, J., Reitzer, L.& Aguilar, J. (2002). Regulation of the Escherichia coliallantoin regulon: coordinated function of the repressorAllR and the activator AllS. J. Mol. Biol. 324, 599–610.
8. Donald, L. J., Hosfield, D. J., Cuvelier, S. L., Ens, W.,Standing, K. G. & Duckworth, H. W. (2001). Massspectrometric study of the Escherichia coli repressorproteins, Ic1R and Gc1R, and their complexes withDNA. Protein Sci. 10, 1370–1380.
9. Muller-Hill, B. (1998). Some repressors of bacterialtranscription. Curr. Opin. Microbiol. 1, 145–151.
10. Maloy, S. R. & Nunn, W. D. (1981). Role of gene fadRin Escherichia coli acetate metabolism. J. Bacteriol. 148,83–90.
11. Sunnarborg, A., Klumpp, D., Chung, T. & LaPorte, D. C.(1990). Regulation of the glyoxylate bypass operon:cloning and characterization of iclR. J. Bacteriol. 172,2642–2649.
12. Negre, D., Cortay, J. C., Old, I. G., Galinier, A.,Richaud, C., Saint Girons, I. & Cozzone, A. J. (1991).Overproduction and characterization of the iclR geneproduct of Escherichia coli K-12 and comparison withthat of Salmonella typhimurium LT2. Gene, 97, 29–37.

Structure and Biochemistry of AllR 827
13. Donald, L. J., Chernushevich, I. V., Zhou, J.,Verentchikov, A., Poppe-Schriemer, N., Hosfield,D. J. et al. (1996). Preparation and properties of pure,full-length IclR protein of Escherichia coli. Use of time-of-flight mass spectrometry to investigate the pro-blems encountered. Protein Sci. 5, 1613–1624.
14. Yamamoto, K. & Ishihama, A. (2003). Two differentmodes of transcription repression of the Escherichiacoli acetate operon by IclR. Mol. Microbiol. 47, 183–194.
15. Blattner, F. R., Plunkett, G., III, Bloch, C. A., Perna,N. T., Burland, V., Riley, M. et al. (1997). The completegenome sequence of Escherichia coli K-12. Science, 277,1453–1474.
16. Kok, R. G., D’Argenio, D. A. & Ornston, L. N. (1998).Mutation analysis of PobR and PcaU, closely relatedtranscriptional activators in acinetobacter. J. Bacteriol.180, 5058–5069.
17. Nasser, W., Reverchon, S. & Robert-Baudouy, J. (1992).Purification and functional characterization of theKdgR protein, a major repressor of pectinolysis genesof Erwinia chrysanthemi. Mol. Microbiol. 6, 257–265.
18. Thomson, N. R., Nasser, W., McGowan, S., Sebaihia, M.& Salmond, G. P. (1999). Erwinia carotovora has twoKdgR-like proteins belonging to the IciR family oftranscriptional regulators: identification and character-ization of the RexZ activator and the KdgR repressor ofpathogenesis. Microbiology, 145, 1531–1545.
19. Rodionov, D. A., Gelfand, M. S. & Hugouvieux-Cotte-Pattat, N. (2004). Comparative genomics of the KdgRregulon in Erwinia chrysanthemi 3937 and othergamma-proteobacteria. Microbiology, 150, 3571–3590.
20. DiMarco, A. A., Averhoff, B. & Ornston, L. N. (1993).Identification of the transcriptional activator pobR andcharacterization of its role in the expression of pobA,the structural gene for p-hydroxybenzoate hydroxylasein Acinetobacter calcoaceticus. J. Bacteriol. 175, 4499–4506.
21. Quinn, J. A., McKay, D. B. & Entsch, B. (2001).Analysis of the pobA and pobR genes controllingexpression of p-hydroxybenzoate hydroxylase inAzotobacter chroococcum. Gene, 264, 77–85.
22. Gerischer, U., Segura, A. & Ornston, L. N. (1998). PcaU,a transcriptional activator of genes for protocatechuateutilization in Acinetobacter. J. Bacteriol. 180, 1512–1524.
23. Trautwein, G. & Gerischer, U. (2001). Effects exertedby transcriptional regulator PcaU from Acinetobactersp. strain ADP1. J. Bacteriol. 183, 873–881.
24. Popp, R., Kohl, T., Patz, P., Trautwein, G., Gerischer, U.& Differential, D. N. A. (2002). binding of transcrip-tional regulator PcaU from Acinetobacter sp. strainADP1. J. Bacteriol. 184, 1988–1997.
25. Arias-Barrau, E., Olivera, E. R., Luengo, J. M.,Fernandez, C., Galan, B., Garcia, J. L. et al. (2004).The homogentisate pathway: a central catabolicpathway involved in the degradation of L-phenyl-alanine, L-tyrosine, and 3-hydroxyphenylacetate inPseudomonas putida. J. Bacteriol. 186, 5062–5077.
26. Zhang, R. G., Kim, Y., Skarina, T., Beasley, S.,Laskowski, R., Arrowsmith, C. et al. (2002). Crystalstructure of Thermotoga maritima 0065, a member ofthe IclR transcriptional factor family. J. Biol. Chem. 277,19183–19190.
27. Ho, Y. S., Burden, L. M. & Hurley, J. H. (2000).Structure of the GAF domain, a ubiquitous signalingmotif and a new class of cyclic GMP receptor. EMBO J.19, 5288–5299.
28. Martinez, S. E., Beavo, J. A. & Hol, W. G. (2002).GAF domains: two-billion-year-old molecularswitches that bind cyclic nucleotides. Mol. Interv. 2,317–323.
29. Martinez, S. E., Wu, A. Y., Glavas, N. A., Tang, X. B.,Turley, S., Hol, W. G. & Beavo, J. A. (2002). The twoGAF domains in phosphodiesterase 2A have distinctroles in dimerization and in cGMP binding. Proc. NatlAcad. Sci. USA, 99, 13260–13265.
30. Repik, A., Rebbapragada, A., Johnson, M. S.,Haznedar, J. O., Zhulin, I. B. & Taylor, B. L. (2000).PAS domain residues involved in signal transductionby the Aer redox sensor of Escherichia coli. Mol.Microbiol. 36, 806–816.
31. Narikawa, R., Okamoto, S., Ikeuchi, M. & Ohmori, M.(2004). Molecular evolution of PAS domain-contain-ing proteins of filamentous cyanobacteria throughdomain shuffling and domain duplication. DNA Res.11, 69–81.
32. Taylor, B. L. & Zhulin, I. B. (1999). PAS domains:internal sensors of oxygen, redox potential, and light.Microbiol. Mol. Biol. Rev. 63, 479–506.
33. Genick, U. K., Borgstahl, G. E., Ng, K., Ren, Z.,Pradervand, C., Burke, P. M. et al. (1997). Structure of aprotein photocycle intermediate by millisecond time-resolved crystallography. Science, 275, 1471–1475.
34. Smith, C. V., Huang, C. C., Miczak, A., Russell, D. G.,Sacchettini, J. C. & Honer zu Bentrup, K. (2003).Biochemical and structural studies of malate synthasefrom Mycobacterium tuberculosis. J. Biol. Chem. 278,1735–1743.
35. van Aalten, D. M., DiRusso, C. C. & Knudsen, J.(2001). The structural basis of acyl coenzymeA-dependent regulation of the transcription factorFadR. EMBO J. 20, 2041–2050.
36. Huffman, J. L. & Brennan, R. G. (2002). Prokaryotictranscription regulators: more than just the helix-turn-helix motif. Curr. Opin. Struct. Biol. 12, 98–106.
37. Ramos, J. L., Martinez-Bueno, M., Molina-Henares,A. J., Teran, W., Watanabe, K., Zhang, X. et al. (2005).The TetR family of transcriptional repressors. Micro-biol. Mol. Biol. Rev. 69, 326–356.
38. Rangachari, V., Marin, V., Bienkiewicz, E. A.,Semavina, M., Guerrero, L., Love, J. F. et al. (2005).Sequence of ligand binding and structure change inthe diphtheria toxin repressor upon activation bydivalent transition metals. Biochemistry, 44, 5672–5682.
39. Chen, S. & Calvo, J. M. (2002). Leucine-induceddissociation of Escherichia coli Lrp hexadecamers tooctamers. J. Mol. Biol. 318, 1031–1042.
40. Schleif, R. (2003). AraC protein: a love-hate relation-ship. Bioessays, 25, 274–282.
41. Vilar, J. M. & Saiz, L. (2005). DNA looping in generegulation: from the assembly of macromolecularcomplexes to the control of transcriptional noise. Curr.Opin. Genet. Dev. 15, 136–144.
42. Schuck, P., Perugini, M. A., Gonzales, N. R., Howlett,G. J. & Schubert, D. (2002). Size-distribution analysisof proteins by analytical ultracentrifugation: strat-egies and application to model systems. Biophys. J. 82,1096–1111.
43. Schuck, P. (2000). Size-distribution analysis of macro-molecules by sedimentation velocity ultracentrifuga-tion and lamm equation modeling. Biophys. J. 78,1606–1619.
44. Gong, W., Hao, B. & Chan, M. K. (2000). Newmechanistic insights from structural studies ofthe oxygen-sensing domain of Bradyrhizobium japoni-cum FixL. Biochemistry, 39, 3955–3962.
45. Soderback, E., Reyes-Ramirez, F., Eydmann, T.,Austin, S., Hill, S. & Dixon, R. (1998). The redox-and fixed nitrogen-responsive regulatory proteinNIFL from Azotobacter vinelandii comprises discrete

828 Structure and Biochemistry of AllR
flavin and nucleotide-binding domains. Mol. Micro-biol. 28, 179–192.
46. Gilles-Gonzalez, M. A. & Gonzalez, G. (2004). Signaltransduction by heme-containing PAS-domain pro-teins. J. Appl. Physiol. 96, 774–783.
47. Yamazaki, M., Li, N., Bondarenko, V. A., Yamazaki,R. K., Baehr, W. & Yamazaki, A. (2002). Binding ofcGMP to GAF domains in amphibian rod photo-receptor cGMP phosphodiesterase (PDE). Identifi-cation of GAF domains in PDE alphabeta subunitsand distinct domains in the PDE gamma subunitinvolved in stimulation of cGMP binding to GAFdomains. J. Biol. Chem. 277, 40675–40686.
48. Little, R. & Dixon, R. (2003). The amino-terminal GAFdomain of Azotobacter vinelandii NifA binds 2-oxoglu-tarate to resist inhibition by NifL under nitrogen-limiting conditions. J. Biol. Chem. 278, 28711–28718.
49. Pellequer, J. L., Wager-Smith, K. A., Kay, S. A. &Getzoff, E. D. (1998). Photoactive yellow protein: astructural prototype for the three-dimensional fold ofthe PAS domain superfamily. Proc. Natl Acad. Sci.USA, 95, 5884–5890.
50. Nasser, W., Reverchon, S., Condemine, G. & Robert-Baudouy, J. (1994). Specific interactions of Erwiniachrysanthemi KdgR repressor with different operatorsof genes involved in pectinolysis. J. Mol. Biol. 236,427–440.
51. DiMarco, A. A. & Ornston, L. N. (1994). Regulation ofp-hydroxybenzoate hydroxylase synthesis by PobRbound to an operator in Acinetobacter calcoaceticus.J. Bacteriol. 176, 4277–4284.
52. Pan, B., Unnikrishnan, I. & LaPorte, D. C. (1996). Thebinding site of the IclR repressor protein overlaps thepromoter of aceBAK. J. Bacteriol. 178, 3982–3984.
53. Guo, Z. & Houghton, J. E. (1999). PcaR-mediatedactivation and repression of pca genes from Pseudo-monas putida are propagated by its binding to both the-35 and the -10 promoter elements. Mol. Microbiol. 32,253–263.
54. Epelbaum, S., LaRossa, R. A., VanDyk, T. K., Elkayam,T., Chipman, D. M. & Barak, Z. (1998). Branched-chainamino acid biosynthesis in Salmonella typhimurium: aquantitative analysis. J. Bacteriol. 180, 4056–4067.
55. Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989).Molecular Cloning: A Laboratory Manual, 2nd edit.,3 vols, Cold Spring Harbor Laboratory Press, ColdSpring Harbor, NY.
56. Guzman, L. M., Belin, D., Carson, M. J. & Beckwith, J.(1995). Tight regulation, modulation, and high-levelexpression by vectors containing the arabinose PBADpromoter. J. Bacteriol. 177, 4121–4130.
57. Van Duyne, G. D., Standaert, R. F., Karplus, P. A.,Schreiber, S. L. & Clardy, J. (1993). Atomic structuresof the human immunophilin FKBP-12 complexes withFK506 and rapamycin. J. Mol. Biol. 229, 105–124.
58. Evans, G. & Pettifer, R. F. (2001). CHOOCH: a programfor deriving anomalous-scattering factors from X-rayfluorescence spectra. J. Appl. Crystallog. 34, 82–86.
59. Westbrook, E. M. & Naday, I. (1997). Charge-coupleddevice-based area detectors. Methods Enzymol. 276,244–268.
60. Pflugrath, J. W. (1999). The finer things in X-raydiffraction data collection. Acta Crystallog. sect. D, 55,1718–1725.
61. Otwinowski, Z. & Minor, W. (1997). Processing ofX-ray diffraction data collected in oscillation mode. InMethods in Enzymology (Carter, C. W. & Sweet, R. M.,eds), vol. 276, pp. 307–326, Academic Press, NewYork.
62. Terwilliger, T. C. & Berendzen, J. (1999). AutomatedMAD and MIR structure solution. Acta Crystallog. sect.D, 55, 849–861.
63. Terwilliger, T. C. (2003). Automated side-chain modelbuilding and sequence assignment by templatematching. Acta Crystallog. sect. D, 59, 45–49.
64. Terwilliger, T. C. (2002). Automated structure sol-ution, density modification and model building. ActaCrystallog. sect. D, 58, 1937–1940.
65. Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M.(1991). Improved methods for building protein modelsin electron density maps and the location of errors inthese models. Acta Crystallog. sect. A, 47, 110–119.
66. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano,W. L., Gros, P., Grosse-Kunstleve, R. W. et al. (1998).Crystallography & NMR system: a new software suitefor macromolecular structure determination. ActaCrystallog. sect. D, 54, 905–921.
67. Laskowski, R. A., MacArthur, M. W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein structures.J. Appl. Crystallog. 26, 283–291.
68. Datsenko, K. A. & Wanner, B. L. (2000). One-stepinactivation of chromosomal genes in Escherichia coliK-12 using PCR products. Proc. Natl Acad. Sci. USA,97, 6640–6645.
69. Neidhardt, F. C., Bloch, P. L. & Smith, D. F. (1974).Culture medium for enterobacteria. J. Bacteriol. 119,736–747.
70. Laue, T. M., Shah, B. D., Ridgeway, T. M. & Pelletier,S. L. (1992). Computer-aided interpretation of ana-lytical sedimentation data for proteins. In AnalyticalUltracentrifugation in Biochemistry and Polymer Science(Harding, S. E., Rowe, A. J. & Horton, J. C., eds), pp.90–125, Royal Society of Chemistry, Cambridge.
71. Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994).CLUSTAL W: improving the sensitivity of progressivemultiple sequence alignment through sequenceweighting, position-specific gap penalties and weightmatrix choice. Nucl. Acids Res. 22, 4673–4680.
72. Liang, J., Edelsbrunner, H. & Woodward, C. (1998).Anatomy of protein pockets and cavities: measure-ment of binding site geometry and implications forligand design. Protein Sci. 7, 1884–1897.
73. McDonald, I. K. & Thornton, J. M. (1994). Satisfyinghydrogen bonding potential in proteins. J. Mol. Biol.238, 777–793.
74. Christopher, J. A. (1998). SPOCK: The StructuralProperties Observation and Calculation Kit (ProgramManual). The Center for Macromolecular Design,Texas A&M University.
75. Kraulis, P. J. (1991). MOLSCRIPT: a program toproduce both detailed and schematic plots of proteinstructures. J. Appl. Crystallog. 24, 946–950.
76. Merritt, A. E. & Murphy, E. P. M. (1994). Raster3Dversion 2.0, a program for photorealistic moleculargraphics. Acta Crystallog. sect. D, 50, 869–873.
Edited by K. Morikawa
(Received 18 November 2005; received in revised form 9 February 2006; accepted 12 February 2006)Available online 3 March 2006





























