Embed Size (px)
Citation preview

Annu. Rev. Biophys. Biomol. Struct. 1992.21:441~3Copyright ~ 1992 by Annual Re~iews Inc. All rights reserved
STRUCTURE AND MECHANISMOF ALKALINE PHOSPHATASE
Joseph E. Coleman
Department of Molecular Biophysics and Biochemistry, Yale University,New Haven, Connecticut 06510
KEY WORDS~ zinc enzymes, enzyme mechanisms (alkaline phosphatase), crystalstructure (alkaline phosphatase), 31p NMR (alkaline phospha-tase), 1~3Cd NMR (alkaline phosphatase)
CONTENTS
INTRODUCTION ............................................................................................................. 442
GENERAL STRUCTURE OF E. COLI ALKALINE PHOSPHATASE .............................................. 443
SUMMARY OF SUBSTRATE SPECIFICITY AND KINETICS OF ALKALINE PHOSPHATASE ........... 443THE METALLOENZYME NATURE OF ALKALINE PHOSPHATASE ........................................... 453
COORDINATION CHEMISTRY AT THE ACTIVE CENTER OF ALKALINE PHOSPHATASE ............ 456Zn l ( A ) Coordination ............................................................................................... 460Zn2(B) Coordination ............................................................................................... 460M#3(C) Coordination .............................................................................................. 460Enzyme-Bound Phosphate in the E" P Intermediate ................................................... 461Enzyme-Bound Phosphate in the E-P Intermediate ................................................... 461
CHANGES IN ACTIVITY OF ALKALINE PHOSPHATASE ON SUBSTITUTING CADMIUM, COBALT,OR MANGANESE FOR THE NATIVE ZINC ION .......................................................... 463
CONCLUSIONS ON STRUCTURE AND MECHANISM OF ALKALINE PHOSPHATASE DERIVEDFROM MULTINUCLEAR NMR ................................................................................ 464
CORRELATION OF STRUCTURE AND MECHANISM ............................................................. 468Michaelis Complex with a Phosphate Monoester ...................................................... 468The Phoshoseryl Intermediate ................................................................................... 469Hydrolysis of the Phosphoseryl Intermediate ............................................................ 469Dissociation of the Product, Inorganic Phosphate, the Rate-Limiting Step ................ 470The Phosphotransferase Reaction ............................................................................. 471
SUMMARY OF THE MECHANISM OF ALKALINE PHOSPHATASE AS BASED ON SOLUTION DATAAND THE CRYSTAL STRUCTURE ........................................................................... 473
SITE-D~RECTED MUTANTS OF e. COLZ ALKALINE PHOSPHATASE ......................................... 476Serl02 ~ Cysl02, .4la102, Leul02 ........................................................................... 477Arg166 ~ Lys166, Glu166, Ser166, ,4la166 .............................................................. 477Lys328 ~ His328, Ala328 ........................................................................................ 478
SUMMARY ..................................................................................................................... 480
4411056-8700/92/0610-0441 $02.00
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

442 COLEMAN
INTRODUCTIONAlkaline phosphatase is often cited as the most frequently referencedenzyme (55). This fact relates more to the widespread use of alkalinephosphatase activity in human serum as an enzymatic signal for a varietyof disease states involving particularly the liver and bone, than to a greaternumber of investigations directed at the molecular properties of theenzyme. The emergence in the literature of the enzyme alkaline phos-phatase began around 1907 when Suzuki et al first suggested that phos-phatases constituted a separate class of eukaryotic enzymes (70). By 1912,the enzyme we now know as alkaline phosphatase was defined by the workof Grosser & Husler (40) and von Euler (75), who showed that while was present in a variety of tissues, the enzyme, which could hydrolyzeglycerophosphate and fructose 1--6 diphosphate, was present in highestamount in intestinal mucosa, von Euler & Funke (76) used the wordphosphatase for the first time in 1912. The enzyme from intestinal mucosa,particularly calf intestine, became the prototype for investigators exploringthe properties of the enzyme itself.
Not until 1961 did Engstrom discover that the intestinal enzyme formeda phosphoseryl residue when incubated with phosphate esters at low pH(27, 28). The enzyme’s catalysis of 180 exchange into inorganic phosphatestrongly supported the notion that the phosphoserine is a significant inter-mediate on the catalytic pathway (3, 65, 69). The demonstration thattransfer of phosphate by the enzyme from an ester to a second alcoholleads to retention rather than inversion of configuration around the phos-phorous (46) supported the conclusion derived from much previous evi-dence that the mechanism involves two sequential in-line nucleophilicattacks at phosphorous, the first by the hydroxyl of Ser 102 on the incomingphosphomonoester and the second on the phosphoseryl intermediate bysolvent water or an alcohol acceptor (32-34, 38).
In the late 1950s and early 1960s, investigators discovered that Esch-erichia coli possessed an alkaline phosphatase that was derepressible byphosphate starvation (21, 31, 42, 72). Its gene, phoA, was part of the phoregulon consisting of the group of genes in E. coli whose products areinvolved in phosphate transport and metabolism (4, 22, 38, 53) (for bib-liography, see 22, 38). The E. coli enzyme had similar catalytic properties,similar pH-rate profile, and formed the same phosphoseryl intermediate asthe intestinal enzyme (66, 67). The amino acid sequences of the mammalianenzymes, derived from their cDNA sequences, can be fit into the primarystructure of the bacterial enzyme, with the proper adjustments for someinsertions and deletions (47). With such adjustments, most of the criticalactive-site residues described below are conserved between the eukaryoticand bacterial enzymes.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE443
In 1962, the E. coli alkaline phosphatase joined the class of zinc metallo-enzymes with the demonstration by Plocke et al (62) that the enzymecontained stoichiometric amounts of zinc, a finding also confirmed in laterstudies of the calf intestinal enzyme (30). These early studies showed thatZn(II) is required for activity, and many subsequent studies have confirmedthis observation, including the demonstration that the metal is required forinitial phosphate binding and thus for the formation of the phosphoserylintermediate (3). The great stability over a wide range of pH of the metal-free apophosphoryl enzyme, which can be formed by removal of the metalion from the Cd(II) enzyme, demonstrates that the metal ion is requiredfor dephosphorylation of the phosphoseryl intermediate as well (18).
The most detailed information on the structure and function of theenzyme is available for the E. coli enzyme. This review is a synthesis ofthe solution data and the recently completed crystal structures of the E.coli alkaline phosphatase and its two phosphoenzyme intermediates at 2.0-/~ and 2.5-A resolution, respectively (48). The phosphoenzymes are E’ the noncovalent complex formed between inorganic phosphate and theenzyme, and E-P, the covalent or phosphoseryl intermediate, formed bythe phosphorylation of Serl02.
GENERAL STRUCTURE OF E. COLI ALKALINEPHOSPHATASEAlkaline phosphatase exists in the periplasmic space of E. coli as a dimerof identical subunits each containing 429 amino acids (11). The fourCys residues are present as two intrachain disulfides. The monomers aresynthesized as a preenzyme containing a Leu-rich signal peptide of 22residues (7, 45, 56, 57). Processing occurs via a signal peptidase aftersecretion through the membrane (17). Recent data suggest that formationof active enzyme upon dimerization may be a complex process involvingsome modulator molecules (J. F. Chlebowski, personal communication).A comparison of the amino acid sequences of the phosphorylated peptidesisolated from the enzyme with the complete amino acid sequence showedthat the phosphorylated residue was Serl02. Figure la shows the overallshape and polypeptide conformation found in the crystal structure of thedimer. Figure lb shows a ribbon diagram of the secondary structure ofthe monomer.
SUMMARY OF SUBSTRATE SPECIFICITY ANDKINETICS OF ALKALINE PHOSPHATASEAlkaline phosphatase is thought to be strictly a phosphomonoesterase,although one recent investigation found a low phosphodiesterase activity
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.
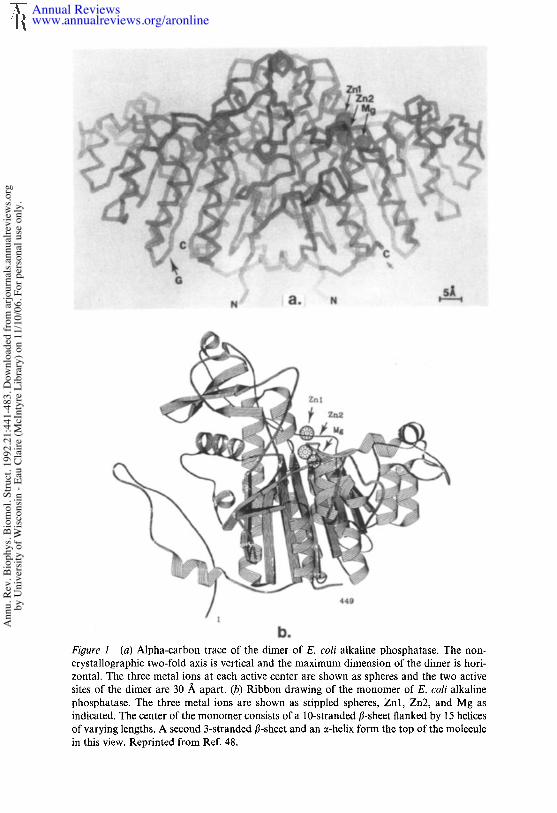
Figure 1 [a) Alpha-carbon trace of the dimer of E. coli alkaline phosphatase. The non- crystallographic two-fold axis is vertical and the maximum dimension of the dimer is hori- zontal. The three metal ions at each active center are shown as spheres and the two active sites of the dimer are 30 A apart. (b) Ribbon drawing of the monomer of E. coli alkaline phosphatase. The three metal ions are shown as stippled spheres, Znl, Zn2, and Mg as indicated. The center of the monomer consists of a 10-stranded 8-sheet flanked by 15 helices of varying lengths. A second 3-stranded 8-sheet and an a-helix form the top of the molecule in this view. Reprinted from Ref. 48.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 445
(J. F. Chlebowski, personal communication). The enzyme hydrolyzes notonly oxyphosphate monoesters (23, 29, 41, 63), but also a variety of (58, 59) and S-phosphorothioates (19), phosphoramidates (63), thiophos-phate, and phosphate (3, 19, 65, 69); hydrolysis of the latter group reflected by the catalysis of the exchange of 180 from H2180 into inorganicphosphate (3, 65, 69) or the release of H2S from thiophosphate (19). enzyme has an alkaline pH maximum, and the rate follows an approxi-mately sigmoid pH-rate profile with an apparent pK, of ~ 7.5 (19, 34, 50,63). Table 1 summarizes substrate structure, reaction products, and k~tvalues. In the case of oxyphosphate monoesters, the koat is apparentlyindependent of the R group, which can vary from a large protein moleculeto a methyl group (63). This finding reflects the fact that either the de-phosphorylation of E-P or the dissociation of the product, Pi, is the rate-controlling step depending on pH. A variety of NMR methods havedemonstrated that the latter step is rate limiting at alkaline pH (38, 44).In hydrolysis of Pi (180 exchange) or release of H2S, however, the phos-phorylation of Serl02 by the substrate appears to be so slow as to be ratecontrolling (9, 19, 63).
Alkaline phosphatase reactions can be fit to the general kinetic for-mulation given in Scheme 1,
Hydrolysis
RO-_ H20 E.P ,~E + Pi
E ÷ ROP ~ E-ROP ~ E-P. ¯ H20k_1 k.2~ ~ WO°k’3’ ~k3, k4"
H20 ~J" E-R’OP ~ E + R’OPk-4’Phosphotransferase
Scheme 1
which includes the covalent phosphoseryl intermediate, E-P, formed whenSerl02 is phosphorylated, and the noncovalent complex, E’P, which isformed with the product, Pi. At pH 5.5 and below, the E-P ~-E. P equi-librium favors E-P such that Pi can phosphorylate Serl02 to form highequilibrium concentrations of E-P (for summary, see 63). Scheme 1 alsoincludes the finding that solvent, H20, is not the only acceptor for thephosphate from E-P. Almost any alcohol will serve as an acceptor at pHvalues above 9 and acceptor concentrations near 1 M as shown by recent3~p NMR assays (38). Traditionally, amino alcohols with the amino group
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

446 COLEMAN
Table 1 Values of kcat for phosphate monoesters hydro-lyzed by alkaline phosphatase
kc,t, pH 8.0Substrate (conditions) (s-~)
ROPO~- (0.1 M Tris) 8.5aROPO]- (1.0 M Tris) 13-45ROPO~- (I.0 M Tris), Co(II) ~2RSPO~- (1.0 M Tris) 30RNHPO~- (I.0 M Tris) 28ROPSO~- (0.! M Tris) 0.005ROPSO2_ (1.0 M Tris) 0.09ROPSO~ (1.0 M Tris), Co(II) 0.17
HSPO]- 0.26HOPO~- O. 154).2
~ When a single kc,, is given, it stands for a representative valuefor substrates that have been the subject of relatively few studies.If a range is given, it reflects a substrate for which a great manyvalues of Vm,x are available in the literature.
on the carbon adjacent to that carrying the accepting OH, e.g. Tris andethanolamine, have been used to demonstrate this phosphotransferaseactivity (63). These acceptors not only have enhanced aeceptor activity,but show maximum transferase activity around pH 8 (63). The rate rapidlyfalls off at higher pH values, pH 8-11 (38). Analysis of the high-resolutioncrystal structure does not as yet suggest the reason for this special reactivityof amino alcohols. The mechanism of the phosphotransferase activitybased on a variety of NMR evidence is postulated to involve the co-ordination of the alcoxide ion to one of the zinc ions at the active siteinstead of a water molecule (38).
Since the original isolation of a phosphorylated serine from alkalinephosphatase by Engstr6m’s laboratory (27, 28), kinetic analyses of theenzyme rcaction have included steps for the formation and dephos-phorylation of the serylphosphate. Rapid-flow kinetic methods applied toexamine the initial phases of the hydrolysis of nitrophenyl phosphates bythe enzyme revealed that the enzyme produced a relatively rapid burst ofphenolate product followed by a steady-state rate at acid pH, but no burstwas observed at alkaline pH, where the enzyme was maximally active. Theacid burst was readily explained by the finding that dephosphorylation ofE-P was very slow, 0.1 s-1, and rate limiting. The possible rate-limitingstep at alkaline pH was less clear. The rate of phosphorylation of the serinehydroxyl at pH 5.5, calculated from the burst rate, ranged from 17 to 30s-~ as assembled from numerous studies. In 1973, Bloch & Schlessinger
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PttOSPHATASE 447
(8) demonstrated that the rapid-flow kinetics were badly distorted phosphate that was bound to the native enzyme and carried along throughmost standard isolation procedures. When phosphate-free enzyme wasemployed for rapid-flow measurements, they observed instantaneousbursts of RO- (within the 3-ms dead time of the instrument) at both 5.5 and 8.0. The readdition of phosphate abolished the burst at alkalinepH and slowed down the burst rate at acid pH. Thus, contaminatingphosphate can abolish the burst at alkaline pH by the prior formation ofE-P, but in general cannot completely abolish the burst at pH 5.5, becauseE-P is not 100% formed. Even more importantly, most stock enzymeswere diluted from neutral pH where no E-P was present at the time ofmixing. Thus, HOPO24- simply competed with ROPO]- binding at acidpH. Because phosphorylation from the monoester is so much more rapidthan from phosphate, the rate of release of RO- slowed during the tran-sient phase, despite the fact that the ester still carried out most of thephosphorylation of the enzyme. An important fact emerging from thesestudies is that phosphorylation from ROPO~-, even at acid pH, is amuch more rapid process than previously believed. Since the bursts at bothpH 5.5 and pH 8.0 are instantaneous, k2, the phosphorylation rate inScheme 1, must be at least 300 s-~ throughout the pH range from pH 5.5to 8.0 and may be even faster.
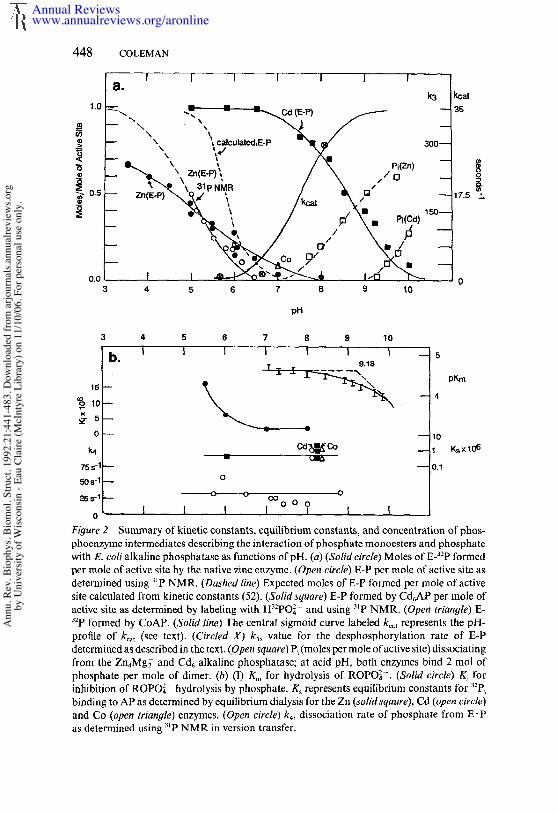
The values of many of the kinetic and equilibrium constants describingthe alkaline phosphatase reaction (Scheme 1) are plotted in Figure 2 as function of pH. The pH-rate profile, expressed as kcat, is presented as asigmoid function corresponding to a single pK, of 7.5 (Figure 2a). Aftercollection of most of the published data on pH-rate profiles for the E. colienzyme, this curve was chosen to represent kcat (see 19, 50, 58, 65). Althoughnot all pH-rate profiles fit the theoretical curve for a single ionization, themost extensive analyses of pH-rate profile data using Dixon plots (log Vma~vs pH) show that the pH-rate profile can be adequately fit by a singlepKa (50). This finding suggests, but does not prove, that a single protondissociation is involved. Under conditions usually employed for alkalinephosphatase assays, the pKa for Vmax is ~7.5; however, depending onwhether neutral buffers, cationic buffers or added organic solvents arepresent, the apparent PKa has been observed to vary from 6.58 to 7.55(50). Kr~ values for phosphate monoesters remain constant as a functionof pH until above pH 8, where an increase in the magnitude of Km can befit with an apparent pKa near 9 (Figure 2b) (50). Thus, Vmax/Kr, plots arebell-shaped if high pH values are included. The increase in K.~ at high pHmay be connected with a phenomenon of phosphate dissociation at highpH observed using 3~p NMR and discussed further below.
Direct measurements of k3, the rate for dephosphorylation of the phos-
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.
448 COLEMAN
1.0
0.03
1 I I I I I Ik3 kcat
17.5
0
p~
3 4 5 6 7 8 9 10
9.18
..... ~", pKm
%~ 10x~- 5
0-- --10C,d "~ CO -- 1 Ksx106
75 s-1 ~ 0.1050s-1 --
~’0-*"~-~ 025 s’1 ~ 0 0 0o I I I I ~ I
Figure 2 Summary of kinetic constants, equilibrium constants, and concentration of phos-phoenzyme intermediates describing the interaction of phosphate monoesters and phosphatewith E. col~ alkaline phosphatase as functions of pH. (a) (Solid circle) Moles of E-~-~P formedl~r mole of active ~ite by the n~tive ~inc enzyme. (Open circle) ~-P per mole of ~ctive ~ited¢tc~ined usin~ "~P N~R. (~sh~# li~) Expected moles of B-P foxed per mole of active~ite calculated from kinetic constants (~2). (~M ~r~) B-P formed by Cd~AP per mole ~ctive site ~s dete~ined by labelin$ with Hs:PO~- and usin$ s’P N~R. (O~ ~r~g/e) "~P foxed by CoAP. (~l~ lm~) The central sismoid curve labeled ~, represents theprofile of ~,~ (~ee text). (Circled ~ ~, wlue for the desphosphorylation rate of B-Pdetermined as desc~bed in the text, (O~c~ ~q~r~) P~ (moles per mole oractiw site) dissociatin~from the Zn,~g~ and Cd~ alkaline phosphat~se; at acid pH, both enzymes bind 2 tool ofphosphate per mole of 4imcr. (h) (1) K~ for hy@olysis of ROPO~-. (~oli# c~rdc) inhibition of ROPO~- hydrolysis by phosphate. K~ represents ~quilibrium constants forbindin$ to AP as determined by equilibrium dialysis for the Zn (,wlMs~,r~), Cd (o~ circle)and Co (~ m~#lc) enzymes. (Op~ circle) ~, dissociation rate of phosphate from as detc~ined usin8 s~P NMR in version tr,nsfer.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 449
phoseryl residue (E-P), are difficult to obtain. The relatively few values the literature are those calculated in order to fit reaction profiles observedin rapid flow kinetics. Because k_3/k3 = E-P/E-P, many reasonably accur-ate values of this ratio are available. Estimates of k3 vs pH taken fromkinetic analyses of the alkaline phosphatase reaction are plotted in Figure2a. Because most rapid-flow kinetics studied prior to 1973 were distortedby the presence of contaminating inorganic phosphate bound to theenzyme (8), some modification of the estimates of the magnitude of k3 arerequired, especially at alkaline pH. One can determine a lower limit for k3from the dead time of the stopped-flow instrument because an instan-taneous burst of ~ 1 mol of RO- per mole of active site is observed atalkaline pH for the phosphate-free enzyme and because this burst resultsfrom the slow, 35 s-t, rate-limiting dissociation of E’P. Therefore, asnoted above, k3 must be ~> 300 s ~ at pH 8.0; the scale for k3 in Figure 2ahas been adjusted to reflect this.
The equilibrium concentration of E-P (1.0 represents maximum possibleformation of phosphoserine) for the Zn, Co, and Cd enzymes has beenplotted from data obtained by 32p labeling of the enzyme by incubationwith H32po24- ~ H232po~- , followed by manual quenching (3, 65). The curvemarked "calculated" is the equilibrium concentration of E-P formed as afunction of pH predicted by Wilson and his colleagues from the kineticconstants for the E-P~E’P~E+P~ reaction as assembled from theextensive kinetic data (52). These investigators suggested that the reasonthe predicted curve is steeper than the observed values obtained fromseveral laboratories using 32p labeling is the difficulty of quenching theenzyme rapidly enough to trap all the phosphoserine.
The E-P ~ E. P equilibrium vs pH also has been determined using 3~pNMR to avoid perturbing the equilibrium by the method of detection.The 3~p NMR of the enzyme-bound phosphate can quantitate these twointermediates from pH 10 to 5. Between pH 7 and 10, no detectable E-Pis present at equilibrium. Between pH 7 and 5, the ratio of E-P/E" P risesalong a sigmoid curve and reaches a value of 1 at pH 5. Below pH 5, thezinc enzyme is unstable. Because the ratio is determined accurately by 31pNMR at close intervals over half the pH function, one can extrapolate asigmoid function for the E-P/E. P ratio with a midpoint at pH 5 as shownin Figure 2a. Values of k~ for dissociation of Pi remain relatively constantfrom pH 5.7 to 8.8 as determined using 31p NMR inversion transfer(Figure 2b) (Table 2) (38). Values of the equilibrium constant for phosphatebinding, Ks, as determined by equilibrium dialysis using H32po2~- areshown for pH 6 and pH 8.8, the latter for the Zn, Co, and Cd enzymes(3). For the Zn enzyme, Ki values for phosphate kinetically determined Wilson’s laboratory are shown for pH values of 5.5, 6.0, 7.0, and 8.0 (52).
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

450 COLEMAN
Table 2 Dissociation rates of the product, inorganic phosphate, fromthe active site of alkaline phosphatase as a function of the metal ioncomposition of the enzymea
koffEnzyme (s- ~)
Zn(II),~AP (pH 8.8)Zn(II)4AP (pH 5.7)Zn(II)4AP+ 1.0 M CI (pH 8.0)[Zn(II)~Mg(II)d2AP (pH [Zn(II)AMg(II)B] 2AP + 0.1 M CI- (pH 9.0)[Zn(lI)AMg(ll)B]2AP + 1 M C1- (pH 9.0)[Zn(II)^Cd(II)n]2AP (pH CO(II)6AP (pH 9.0)Cd(II)6AP+I M C1 (pH 9.0)
353360-260b
1.8~15~2<1
~10
a All samples were in 0.01 M Tris-acetate (38).b Too fast to measure directly by 3 tp NMR inversion transfer; limits given are
based on the inversion transfer measurement at 162 MHz.
The value of k3, the dephosphorylation rate of E-P, falls rapidly frompH 8.0 (>~ 300 s- 1) to pH 6.3 (~ 1.3 s-1), over the same pH range in whichkca t falls by ,-~90% (Figure 2a). Thus, the pH-rate profile (from alkalineto acid pH) is coincident with a change in the rate-limiting step fromphosphate dissociation to dephosphorylation of the phosphoserine. At pH8, k3 exceeds the rate of dissociation of the product phosphate, k4, by ~ 10-fold. At pH 6.3, however, the relationship is reversed; k4 is 26-fold fasterthan k3 and is no longer rate limiting. On the other hand, k3 is not yet asslow as k_3, the rate of phosphorylation of Serl02 from E’P, and thussignificant equilibrium concentrations of E-P are not observed at pH 6.3.At pH 5, k3 has decreased further such that k3 = k 3 and [E-P] = [E-P]at equilibrium (Figure 2a). Judging by 180 exchange experiments, bothconstants must be on the order of 0.I s- ~. The possible identity of theAH~---A +H÷ equilibria governing k3 and the pH-rate profile are dis-cussed in the section on structure and mechanism.
Phosphate is both a substrate and competitive inhibitor of the enzyme.Phosphate binding appears to be essentially pH independent between pH5.5 and 8.0 if the values for Ki and K~ are taken as a guide (Figure 2b) (3,52, 63). In support of this conclusion is the fact that the dissociation rateof inorganic phosphate from the enzyme, k4, is also pH independent frompH 5.5 to 8.8 (38). A pH independence of phosphate binding to alkalinephosphatase is surprising because two proton dissociations of phosphoricacid may fall in the pH range 5 to 10, H~PO2 ~---HPO] +H÷ (pKa = 6.8)and HPO42- ~ PO43- +H+ (normal pK, ~ 12). The latter could affect the
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 451
AP-phosphate equilibrium if E" P forces formation of the trianion as somereasoning suggests (see below). An enzyme active center, which excludesall but specific water molecules, may limit proton access from the bulksolvent as well. Even in the apophosphoryl enzyme, which is formed byremoving Cd from the E-P form of the enzyme, the 31p chemical shiftshows that the dianion form of the phosphoseryl group is not titrated tobecome the monoanion until below pH 4.0 after the enzyme unfolds (34).
At variance with the kinetic constants that suggest that phosphate bind-ing is pH independent is the observation, made using 3tp NMR, that whileZn4AP remains saturated with phosphate to pH 7.0, above pH 7.0 boundphosphate significantly decreases such that [E’P] = [Pi] at pH 10 (Figure2a) when enzyme is 2 mM and phosphate 4 mM (33). The latter findingappears paradoxical, since the measurement of k4 by NMR inversiontransfer shows that the dissociation rate remains constant at ,-~35 s-tunder the same conditions and over the same pH range (Figure 2b). Whileone could postulate that the "on" constant for phosphate binding changesat high pH, this seems unlikely. If a water molecule on ZnA becomes Zn--OH at alkaline pH, phosphate binding will compete with hydroxidebinding to ZnA. In support of this postulate is the observation that phos-phate does not begin to dissociate from the cadmium enzyme until wellabove pH 9.0 (Figure 2a) (33). However, if both bound phosphates on enzyme were competing equally with OH, then k4 should increase as itdoes in the case of C1- competition (Table 2). One possible explanationof this paradox is negative cooperativity of phosphate binding, i.e. phos-phate at one active site is bound less tightly than at the other at pH valuesabove 7.0. Then most of the remaining E" P will be at the tight bindingsite and the inversion transfer will be weighted in favor of this site.
Negative cooperativity has not been discussed thus far in this review.The phenomenon formed a significant part of earlier discussions of thealkaline phosphatase reaction because many early experiments measuringH32po4~- binding or burst magnitude showed that only a single boundphosphate per dimer or a single active site was phosphorylated. Whensample-preparation techniques were changed such that samples with a fullcomplement of metal ions were prepared, two E-P or two E" P complexesper dimer were easily formed under most conditions, especially at pHvalues below 7. Note that the Cd6AP retains 1 mol of E-P per active siteor 2 mol/dimer until the pH rises above 7.0 (Figure 2a). From pH 7 to the sum of E-P+E-P also remains 2/dimer for the Cd6 enzyme (32).Likewise, when enzymes uncontaminated with phosphate were employedin rapid-flow experiments, most burst stoichiometries approached 2/dimer(8).
Before we discard negative cooperativity in alkaline phosphatase as an
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

452 COLEMAN
artifact, however, I must point out that in the alkaline pH range severalsensitive NMR techniques can detect unequal binding constants for thetwo enzyme-bound phosphates. Detailed 3~p NMR titrations of the Zn4APand the Cd6AP with Pi show that at pH 6 both enzymes titrate linearly theformation of 2 tool of enzyme-bound phosphate per mole of dimer. TheCd6AP does the same at pH 8, but the Zn4AP titrates the formation ofonly 1 mol of E’P under the NMR conditions employed (33). The samephenomenon is apparent when phosphate binding to the enzyme is evi-denced by 35Cl line broadening resulting from the binding of two 35Cl-ions to each Zna. At pH 6, the phosphate ions displace one chloride fromeach ZnA site, i.e. half the C1-. In contrast, at pH 8 one phosphate displacesonly one quarter of this C1- in a stoichiometric fashion; displacement ofthe other quarter requires up to six phosphates per dimer (37). Thus,inequivalent phosphate binding affinities may be involved in productrelease.
The rate-limiting phosphate product dissociation accounts for burstkinetics observed at alkaline pH by stopped-flow methods when a phos-phate-free enzyme is employed (5, 20). Because the enzyme does not appearto recognize the R group of a phosphomonoester, researchers have oftenassumed that phosphate ester binding affinity was similar to that of phos-phate. Indeed, the best measurement of Km values have been ~ 10 -6 M forROPO~-, although they increase to almost 10-4 M for an O-phos-phorothioate, ROPSO~- (19, 58). When an acceptor alcohol is present, therate of the phosphotransferase activity is always additive to that of theinitial phosphohydrolase as has been confirmed using accurate 31p-NMRmeasurement techniques (38). Because the dissociation of P~ is rate limitingfor the hydrolysis reaction under these conditions, the dissociation of thenew product ester in the transferase reaction is clearly considerably morerapid than phosphate dissociation. Km varies as a function of the metal-ion species, Zn or Co, at the active center (19), but the K~ for phosphateis rather similar for the Zn, Co, and Cd enzymes [(0.75_+ 0.25) x -6 M](3). If the dissociation rate constant for ROPO3z- were ~ 35 s-1 as it is forphosphate, then Kn, would not represent k_ like, since the phosphorylationrate at pH 8 is at least 102 s-1 and probably even faster.
In comparing the kinetic pattern observed for phosphate esters as sub-strates and the information derived from using phosphate as substrate toform E-P, one should keep in mind that phosphorylation of the enzymeby HOPO~2- has become rate-limiting, as concluded from the fact that the180 exchange rate, 0.1 to 0.2 s-1, is slower than the dephosphorylation ofE-P over most of the pH range and is always slower than the dissociationof P~. The 180 exchange reaction is pH independent, evidence that theapparent pKa reflected in the normal pH-rate profile is that of a group
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 453
primarily affecting a step following formation of the phosphoseryl inter-mediate (see below). This pH independence, presumably controlled by slow phosphorylation, is confirmed by the hydrolysis of thiophosphate,which releases H2S (19). The rate of release of H2S from thiophosphate the same at pH 5.5 and 8.0 and is only slightly more rapid than the rateof JsO exchange into phosphate catalyzed by the enyzme (Table 1).
The above background is useful in considerations of the nature of thetransition state in the alkaline phosphatase mechanism and whether thereaction is primarily associative via the initial attack of the Ser-O- onthe phosphorous or whether the enzyme mechanism involves significantdissociative features via activation of the leaving group as in non-enzyme-catalyzed phosphomonoester hydrolysis (6, 49, 78). Recent investigationshave probed this question and confirmed that kcat/Km values for a varietyof substrates as functions of the pKa of the leaving group show fl valuesof ~ 0.1 rather than the value of ,-~ 1 expected for a dissociative reaction(51, 78). Although these newer findings and earlier, less rigorous kineticshave been interpreted to mean that the enzyme mechanism is purely nucleo-philic in character, forming a five-coordinate intermediate, the high res-olution crystal structure of E" P reveals interesting structural arrangementsof metal and phosphate that bear on this view of the mechanism, asdiscussed in the next section of this perspective.
Using the phosphotransferase reaction to capture as a second chiralphosphate ester the phosphate from E-P initially formed by a chiral phos-phomonoester containing 160, 170, and 180, Jones et al (46) demonstratedthat the alkaline phosphatase reaction proceeds with retention of con-figuration around phosphorus. This result suggested that the reaction pathconsists of two in-line nucleophilic attacks; the first by the Ser-OH (or Set-O-) and the second by solvent H20 (or -OH).
THE METALLOENZYME NATURE OF ALKALINEPHOSPHATASE
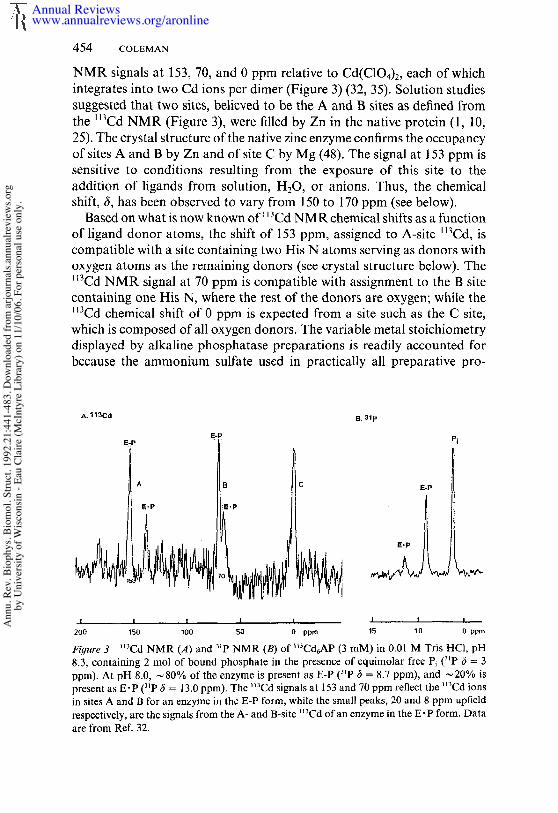
The original analytical data reporting that alkaline phosphatase was a Znmetalloenzyme showed two Zn ions per dimer (62). Subsequent deter-minations of the zinc content by the original laboratory and other inves-tigators showed that with different preparation techniques the Zn: proteindimer ratio was generally 4 and under some circumstances could reach 6(1, 10, 25, 60). Numerous studies contribute to these conclusions; it is nowcertain that a fully active native alkaline phosphatase contains four Znand two Mg ions, the Zn occupying sites designated A and B, while Mgoccupies sites designated as C. The A, B, and C sites were first unequi-vocally demonstrated using I13Cd NMR. The 113Cd6 enzyme shows three
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.
454 COLEMAN
NMR signals at 153, 70, and 0 ppm relative to Cd(C104)2, each of whichintegrates into two Cd ions per dimer (Figure 3) (32, 35). Solution studiessuggested that two sites, believed to be the A and B sites as defined fromthe ll3Cd NMR (Figure 3), were filled by Zn in the native protein (1, 25). The crystal structure of the native zinc enzyme confirms the occupancyof sites A and B by Zn and of site C by Mg (48). The signal at 153 ppm sensitive to conditions resulting from the exposure of this site to theaddition of ligands from solution, H20, or anions. Thus, the chemicalshift, (5, has been observed to vary from 150 to 170 ppm (see below).
Based on what is now known of l~Cd NMR chemical shifts as a functionof ligand donor atoms, the shift of 153 ppm, assigned to A-site ll3Cd, iscompatible with a site containing two His N atoms serving as donors withoxygen atoms as the remaining donors (see crystal structure below). The~3Cd NMR signal at 70 ppm is compatible with assignment to the B sitecontaining one His N, where the rest of the donors are oxygen; while the~3Cd chemical shift of 0 ppm is expected from a site such as the C site,which is composed of all oxygen donors. The variable metal stoichiometrydisplayed by alkaline phosphatase preparations is readily accounted forbecause the ammonium sulfate used in practically all preparative pro-
B. :31p
~ ~ I I I I I I200 150 100 50 0 ppm 15 10 0 ppm
Figure 3 ~t~Cd NMR (A) and ~tp NMR (B) of ~Cd6AP (3 raM) in 0.01 M Tris HCI, 8.3, containing 2 tool of bound phosphate in the presence of equimolar free Pi (31p c5 = 3ppm). At pH 8.0, ~80% of the enzyme is present as E-P (~P 6 = 8.7 ppm), and ~20% present as E’P (3~P 6 = 13.0 ppm). The ~3Cd signals at 153 and 70 ppm reflect the ~t3Cd ionsin sites A and B for an enzyme in the E-P form, while the small peaks, 20 and 8 ppm upfieldrespectively, are the signals from the A- and B-site "~Cd of an enzyme in the E-P form. Dataare from Ref. 32.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 455
cedures can easily remove Zn from the B site as well as from the A site athigh pH. In fact, the best way to prepare the apoenzyme is to dialyze theenzyme at pH 9 against buffered 2 M ammonium sulfate (33).
The relationship between occupancy of the three metal-ion binding sites,A, B, and C, on each monomer and phosphatase activity was not an easyone to define, but required enzymes with known metal:protein stoi-chiometry as well as knowledge of which sites were occupied (33, 34, 38).Mixed-site occupancy occurs easily when metal ions are present at less thanfull stoichiometry. While a (ZnAZnBMgc)2 enzyme has maximal activity, enzyme containing only two Zn ions, one at each A site, is not inactive.Such a two-Zn enzyme binds phosphate and catalyzes phosphorylation ofSerl02 (2). In fact, if Mg occupies both the B and C sites, the activity the (ZnAMgBMgc)2AP is near normal in the phosphotransferase reaction(Table 3) (21). In contrast, the normal hydrolysis reaction is depressed in the (ZnAMgBMgc)2AP (Table 3) (21, 38). Because standard
Table 3 Effect of metal-ion species on thc E-P ~ E- P equilibrium at thc active center ofalkaline phosphatase (AP) and the kca t values
3~p chemicalshift, ppm
Metal site occupancy at pH at whichactive center kc~, s- t [E-P] = [E- pin E-P E" P
[Zn(II)AZn(II)n]2AP 35 5.0 8.6 4.3[Zn(II)AZn(II)nMg(II)c] 35 5.0 8.6 3.4[Zn(II)AMg(II)~]2AP (0.01 M 1 4.0 8.5 1.8[Zn(II)AMg(II)B]2AP (1 M 30 -- -- --[Zn(II)ACd(II)a]2AP 2 6 8.0 12.6[Cd(II)ACd(II)a]2AP 0.001b 10 8.4 13.4[Cd (II)ACd(II)~Cd(II)c]2AP <0.1e 8.7 8.7 13.0[Cd(II)AZn(II)n]2AP (unstable) -- 7 9.3ApoPhosphorylAP -- -- 5.8[Co(II)ACO(II)a]~AP 2 ~ 5d ND~[Mn(II)AMn(II)a]2AP 0.2~ ~ 6.5~ ND
~ These values were determined from 3 ~P-NMR of the enzyme vs pH unless otherwise indicated."This is the rate of phosphorylatien from E’P taken from the NMR experiment shown in Figure 7.
Since the major intermediate accumulating at pH 8 is E-P, dephosphorylation of E-P, k3, cannot be faster.Thus the turnover, k~, cannot be faster than 0.001 s- ~.
~The lower limit for turnover determined by the standard colorimetric assay; i.e. it represents thebackground from zinc contamination of the best apoenzyme preparations assayed in metal-free buffersand substrates. The value 0.2 s- ~ is readily detected above the apoenzyme background under metal-freeassay conditions (64).
dTaken from 32p labeling data (3).° Not detected due to line broadening by the paramagnetic metal ions. Data are from Ref. 16 and 66.
Assays were at pH 8, 0.01 M Tris unless otherwise stated.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

456 CO~.nM~
assays employ 1 M Tris buffer, this difference is not routinely detected.Surprisingly, a large activation of the hydrolysis ÷ transferase reaction byMg is only observed for the two-Zn species; Mg occupancy of the C sitein a 4-Zn enzyme, (ZnAZn~c)2AP, enhances total activity relatively littleif at all. In all the studies of E. coli alkaline phosphatases of defined metal-ion stoichiometry, detecting significant effects of the C-site metal ion onstructure or function has proved difficult. This statement may not applyto the mammalian enzymes, which undergo much more dramatic acti-vation by Mg even when maximum Zn ions are present (22).
The above brief introduction to the properties of alkaline phosphataseis intended only to set the background for the main purpose of thisperspective, which is an attempt to synthesize the findings of the recent2.0-A crystal structure of the enzyme and its two phosphoenzyme inter-mediates with the extensive solution data bearing on the catalytic mech-anism of alkaline phosphatase. The resultant synthesis gives the clearestpicture yet of the active center and the intermediates on the reaction pathof alkaline phosphatase. For detailed descriptions of the earlier kinetic,spectroscopic, and other physicochemical studies of the enzyme, earlierreviews should be consulted (23, 34, 63).
COORDINATION CHEMISTRY AT THE ACTIVECENTER OF ALKALINE PHOSPHATASE
The coordination chemistry at the active center of alkaline phosphatase isbest presented by a detailed description of the structure around each ofthe three metal ions at the active center of the native enzyme that has themetal composition ZnAZnBMgc. Multinuclear NMR investigations of theenzyme done in the past have used the designations A, B, and C for thethree sites; the crystal structure uses the designations 1, 2, and 3. Figure4a presents the general structure of the active center by a stereo view ofthe immediate region of the active center including the three metals (48).Figure 4b shows a computer-graphics representation of the E" P complexof the native Zn4Mg2 enzyme that includes the metal-ligand bonds, theslowly exchanging water molecules, the hydrogen bonds, and the aminoacid side chains located within the immediate region of the active center(48). Both representations are taken from the structure of the E- P complex,i.e. with phosphate bound noncovalently at the active center by soakingcrystals of the (ZnlZn2Mg3)2AP in phosphate at neutral pH. This struc-ture has been determined at 2-/~ resolution and all information suggests itshould represent the authentic E’P intermediate. The three metal ionsform a cluster in which the metal-to-metal distances trace a triangle of3.94 × 4.88 × 7.09 ~. Table 4 lists the metal-metal distances, the ligands
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 457
Figure 4 (a) Stereo drawing of the active site of the E. coli alkaline phosphatase,(ZnlZn2Mg3)2AP plus 2 mM HPO~-, pH 7.5. Atoms are shaded by atom type. Someresidues and water molecules are omitted for clarity. (b) The active-site region of the E-Pcomplex (2-,~ resolution) including all the atoms within 10/~ of the phosphorus atom. Watermolecules are labeled W. Hydrogen bonds are shown as broken lines.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.
458 COLEMAN
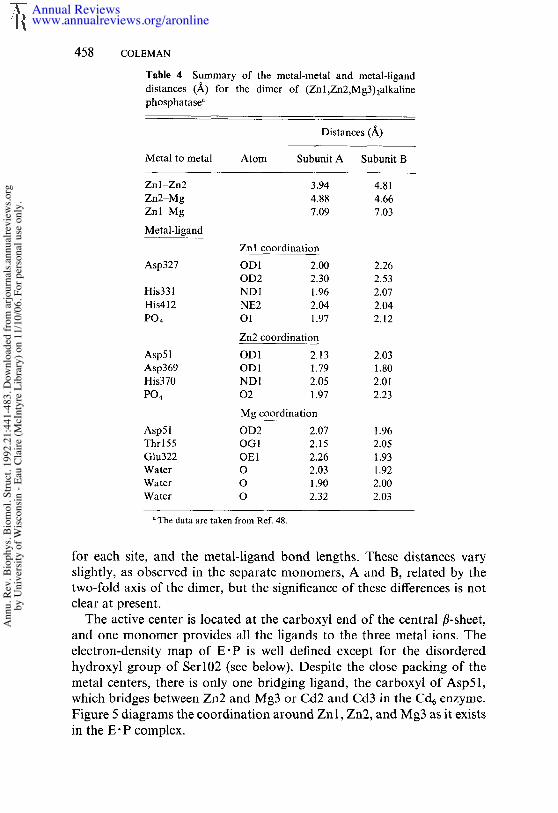
Table 4 Summary of the metal-metal and metal-liganddistances (/~) for the dimer (Zn 1 ,ZnZ,Mg3) 2alkalinephosphatase"
Distances (~_)
Metal to metal Atom Subunit A Subunit B
Zn 1-Zn2 3.94 4.81Zn2-Mg 4.88 4.66Znl Mg 7.09 7.03
Metal-ligandZnl coordination
Asp327 ODI 2.00 2.26OD2 2.30 2.53
His331 NDI 1.96 2.07His412 NE2 2.04 2.04PO4 O1 1.97 2.12
Zn2 coordinationAsp51 OD 1 2,13 2.03Asp369 OD1 1,79 1.80His370 ND1 2.05 2.01PO4 02 1.97 2.23
Mg coordinationAsp51 OD2 2,07 1.96Thr 155 OG 1 2.15 2.05Glu322 OE1 2.26 1.93Water O 2.03 1.92Water O 1.90 2.00Water O 2.32 2.03
The data are taken from Ref. 48.
for each site, and the rnetal-ligand bond lengths. These distances varyslightly, as observed in the separate monomers, A and B, related by thetwo-fold axis of the dimer, but the significance of these differences is notclear at present.
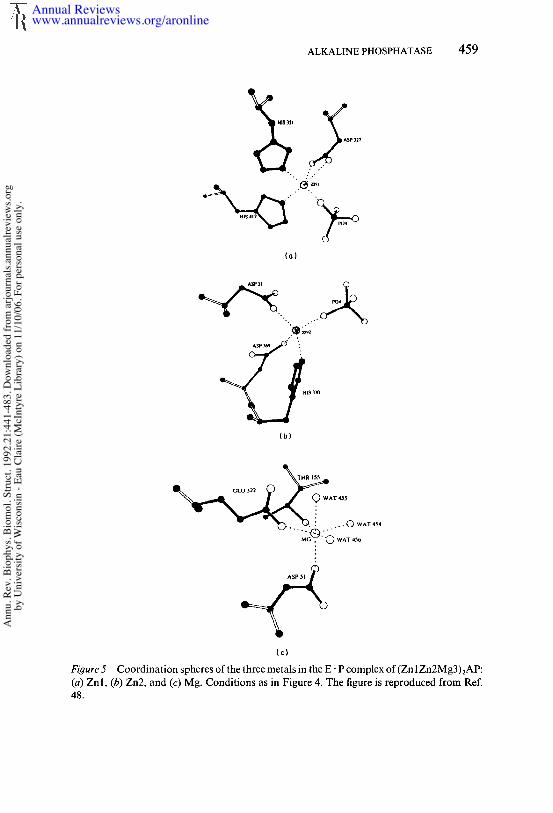
The active center is located at the carboxyl end of the central fl-sheet,and one monomer provides all the ligands to the three metal ions. Theelectron-density map of E" P is well defined except for the disorderedhydroxyl group of Serl02 (see below). Despite the close packing of themetal centers, there is only one bridging ligand, the carboxyl of Asp51,which bridges between Zn2 and Mg3 or Cd2 and Cd3 in the Cd6 enzyme.Figure 5 diagrams the coordination around Znl, Zn2, and Mg3 as it existsin the E° P complex.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.
ALKALINE PHOSPHATASE 459
(o)
(b)
TI4R 155GLU 322~ AT 455
- "~O-.._ ~S.. --- 0 ,,,’^~
Figure 5 Coordination spheres of the three metals in the E- P complex of(Zn 1Zn2Mg3)~AP:(a) Znl, (b) Zn2, and (c) Mg. Conditions as in Figure 4. The figure is reproduced from 48.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

460 COLEMAN
ZnI (A) CoordinationThe A site or Zn 1, which upon substitution with first-transition metal ionsshows spectroscopic properties of a typical Zn(II) metalloenzyme (2, 34),has four ligands from the protein, which include both carboxyl oxygensof Asp327, the N3 of His331, and the N3 of His412. In the absence ofHPO]-, water relaxation data (64) and 35C1- NMR data (37) suggest the coordination sphere of the A site is completed by two H20 molecules.In the E.P complex, one of the phosphate oxygens forms a typical co-ordinate bond with Znl, with a Znl-O bond length of 1.97/~ and a Zn-O-P bond angle of 120° (Figure 5a). Water-relaxation data using the enzyme show that one of the water molecules coordinated to the A-site metal is displaced by phosphate (64), 35C1 NMR reveals a similardisplacement of a monodentate ligand at A-site, i.e. CI-, by phosphatebinding to the Zn enzyme (37). The Znl coordination in E.P can best described as pseudotetrahedral, where both carboxyl oxygens of Asp327occupy one of the apices. His372, which was originally thought to co-ordinate Znl, is not a direct ligand; it is 3.8 A away from Znl, and the N3of the imidazole ring forms a hydrogen bond with one of the coordinatedcarboxyl oxygens of Asp327 (Figure 4b).
Zn2 ( B) Coordination
In the E- P complex, Zn2 is coordinated tetrahedrally by the N3 of His370,one of the carboxyl oxygens of the bridging Asp51, and one of the carboxyloxygens of Asp369 (Figure 5b). The tetrahedral coordination is completedby one of the phosphate oxygens. While the Zn2-O bond length is 1.97 A,identical to that for the Znl-O bond, the Zn2-O-P bond angle is nearlylinear (175°). The OH of Serl02 is disordered in the E-P complex butappears to form a coordinate bond with Zn2 in the phosphate-free enzyme(see below).
M93 ( C) Coordina t ionThe Mg site can be described best as a slightly distorted octahedronconsisting of the second carboxyl oxygen of the bridging Asp51, one ofthe carboxyl oxygens of Glu322, and the hydroxyl of Thr155, while therest of the coordination sites are filled by three slowly exchanging watermolecules (numbered 454 to 456) (Figure 5c). Asp153 is not a direct ligandas orginally believed (68, 80), but is an indirect ligand in that the carboxylgroup forms hydrogen bonds with two coordinated water molecules (454and 455) that are the direct ligands to Mg (Figure 4b). The Mg site
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 461
does not appear close enough to participate directly in the hydrolysismechanism, but could of course contribute to the shape of the electrostaticpotential around the active center (see below).
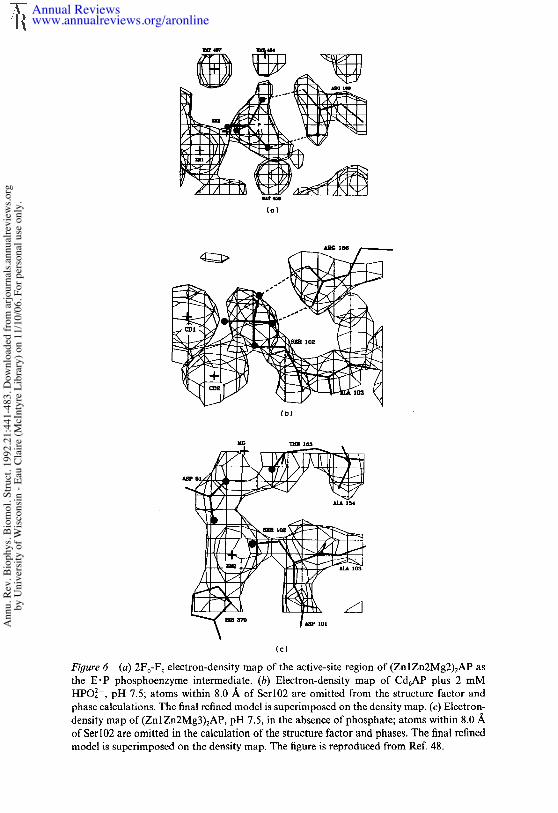
Enzyme-Bound Phosphate in the E’P Intermediate
Because the relationship between the amino acid side chains and thephosphate in the E. P intermediate forms the basis for the initial discussionof the relationship of the crystal structure to solution studies of the mech-anism, additional details of the structure are outlined here. In addition tooxygen-metal bonds to Znl and Zn2 made by two of the oxygen atoms ofphosphate, the other two phosphate oxygens in E" P hydrogen bond to theguanidinium group of Arg166 (Figure 4a). Figure 6a shows the electrondensity map on which this structure is based. The guanidinium group isinvolved in an additional hydrogen-bond network that includes hydrogenbonds to Aspl01 and a water molecule (Wat459), the latter held by Asp153and possibly also by Tyr 169. Of the two phosphate oxygens involved withthe guanidino group, one is further hydrogen bonded to the amide ofSerl02, and the second seems to be involved with two of the slowlyexchanging water molecules. One of these molecules (Wat454) is coor-dinated to Mg, while the second (Wat457) forms a bridge between thephosphate oxygen and Lys328 (Figure 4b). Znl, O1, P, 02, and Zn2 arenearly coplanar.
Enzyme-Bound Phosphate in the E-P Intermediate
As shown in Figure 2, the substitution of Cd for Zn shifts the pH stabilityof the covalent phosphoryl enzyme well into the alkaline pH range. Ser 102of the Cd6 enzyme remains 90% phosphorylated at pH 7.5; hence, crystalsof the Cd6 enzyme when soaked in phosphate can be used to determine astructure of the E-P intermediate. The electron density map at 2.5-Aresolution of the E-P intermediate formed by Cd6AP is shown in Figure6b and should be compared to the electron density map of the E- P complexof the Zn4Mg2 enzyme at 2-A resolution shown in Figure 6a. An esterbond between the phosphate and the hydroxyl of Serl02 is clearly present.Cd2 also appears to form a coordinate bond with the ester oxygen. Thephosphate group has moved slightly deeper into the active-center cavity,and whether an oxygen of the phosphate remains close enough to Cdl toretain a coordinate bond is difficult to determine from the electron densityalone (see discussion of mechanism below). The two hydrogen bondsbetween phosphate oxygens and the guanidino group of Arg166 are main-tained in the E-P complex.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.
(o)
(¢)Figure6 (a) 2Fo-Fc electron-density map of the active-site region of (ZnlZn2Mg2)2AP the E’P phosphoenzyme intermediate. (b) Electron-density map of Cd6AP plus 2 HPO]-, pH 7.5; atoms within 8.0 ~. of Serl02 are omitted from the structure factor andphase calculations. The final refined model is superimposed on the density map. (e) Electron-density map of (Zn 1Zn2Mg3)zAP, pH 7.5, in the absence of phosphate; atoms within 8.0/~of Serl02 are omitted in the calculation of the structure factor and phases. The final refinedmodel is superimposed on the density map. The figure is reproduced from Ref. 48.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 463
CHANGES IN ACTIVITY OF ALKALINEPHOSPHATASE ON SUBSITUTING CADMIUM,COBALT, OR MANGANESE FOR THE NATIVEZINC ION
The substitution of the native Zn ions in sites A(1) and B(2) with otherfirst transition or liB metal ions significantly changes kc~t as well as therate constants for individual steps. While the structural reasons for allthese changes are not immediately obvious, the results have been helpfulin understanding specific aspects of the mechanism.
The most dramatic change is the fall in kcat upon Cd substitution from~ 35 s-1 to a rate impossible to detect using standard assay techniques.Both 32p labeling at low pH and NMR detection of E-P and E" P, however,show that the Cd enzyme forms both of these phosphoenzyme inter-mediates. Inversion transfer from 31p~ to the E" 3~p complex on the Cd6enzyme show that k4, the dissociation of the product phosphate, hasslowed to less than I s-1 as a result of the Cd substitution (Table 2). Thedissociation of phosphate, however, is no longer the rate-limiting step forthe Cd enzyme. By following the formation of E-P via observation of its31p NMR resonance, the phosphorylation rate of the cadmium enzymefrom HOPO3~-, k_3, is ~ 10-3 s -1 at pH 9.0 (see Figure 7 below). Overmuch of the pH range, however, k3, the dephosphorylation of E-P, mustbe even slower because the major equilibrium species of intermediate is E-P until the pH is above 9 (Figure 2a). One of the possible explanations forthe latter remarkable fall in k3 is that the solvent nucleophile required forthe dephosphorylation step is coordinated to the A-site metal, and its pK~has been shifted far to alkaline pH by the Cd substitution. Although theNMR studies show the Cd enzyme following all the steps of the nativeenzyme, it is essentially an enzyme in slow motion.
Substitution of Co, on the other hand, causes far less drastic changes.While kca t falls significantly, 2-10 s-1 depending on preparation (Table 3)(19), the pH-rate profile for Vmax remains nearly the same as that for thezinc enzyme, with an apparent pKa of ~7.5 (19). Compatible with thisfinding is the observation that significant equilibrium concentrations of E-32p form only below pH 7. The acid instability of the Co enzyme preventsdetermination of the complete pH dependence of the E-P/E-P ratio (3).The Co substitution, however, is accompanied by a dramatic switch in therelative magnitudes of phosphate dissociation, k4, vs the rephosphorylationrate of Serl02 from E" P, k 3- 31p NMR can be used to accurately measurethe exchange of 180 out of Hp~80]- as catalyzed by alkaline phosphatase,since an isotope shift in the 31p signal is induced proportional to the numberof ’80 oxygens in the phosphatc (9). The exchangc catalyzed by the native
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

464 COLEMAN
Zn(II) enzyme is compatible with a kinetic scheme in which E" P dissociatesat least 10 times more rapidly than E-P is reformed, in agreement withthe rate constants given above, i.e. k4 = ,,~35 s-1 (Table 2), while therephosphorylation rate, k_a, is ~0.2 s-1 for Zn. On the other hand, Co(II)catalyzes the exchange of approximately three phosphate oxygens perturnover, showing that several rephosphorylations of the Serl02 occurbefore E" P can dissociate. The multiple 180 exchanges from E" P catalyzedby the Co enzyme allow the conclusion that E P is a dynamic complex inwhich the phosphate oxygens interchange their positions on a reasonablyfast time scale, probably between 10 and 100 times per second to accom-modate both the ~80 exchange data and the k~, observed for the Coenzyme. Although paramagnetic broadening of the bound phosphate byCo(lI) prevents the measurement of k4 for the Co enzyme by the accurateNMR methods, similar equilibrium dissociation constants measured forphosphate binding to the Co and Zn enzymes (3) suggest that the phos-phate dissociation rates may be similar. Thus Co appears to catalyzephosphorylation of Serl02 from E" P more rapidly than Zn. While preciseexplanations of the above phenomena are not possible, even with the 2-Amap of the E. P complex, they do emphasize the close interactions betweenthe phosphate and the metal ions.
Substitution of Mn for Zn produces an enzyme with far less activity,but one that is nevertheless detectable by the standard assays with p-nitrophenyl phosphate and estimated to be 0.2 s-~ (64). Although verylow, the latter is significantly greater than the turnover number for the Cdenzyme. While the kinetics of the Mn enzyme have not been investigatedin detail, part of the fall in kca t must reflect a fall in k3, the dephos-phorylation rate of E-P, because significant equilibrium concentrations ofE-P are formed by the Mn enzyme at pH 8 and above (3).
CONCLUSIONS ON STRUCTURE AND M ECHANISMOF ALKALINE PHOSPHATASE DERIVED FROMMULTINUCLEAR NMR
Investigators used ~3Cd NMR of the ~t3Cd6AP to originally identify thepresence of three separate metal-ion binding sites, A, B, and C, on eachmonomer of alkaline phosphatase (32). The chemical shifts of two of thesesites, A and B, depend on which phosphoenzyme intermediate, E;P or E-P, is present (Figure 3), suggesting originally that both CdA and CdB werenear the phosphate binding site as the structure now shows. 31p NMRsignals can be detected from both E" P and E-P intermediates and havebeen valuable probes of the mechanism. The 3~p NMR signal of E" P has
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 465
provided the demonstration by saturation and inversion transfer thatthe dissociation of inorganic phosphate, kd ~-, 35 s-~, is the slowest, andtherefore the rate-limiting, step in the mechanism (38). The chemical shiftsof the 31p NMR resonances of the two alkaline phosphatase intermediatesas well as the pH at which [E" P] = [E-P] for each metalloderivative of theenzyme are summarized in Table 3 and suggest several general conclusions.
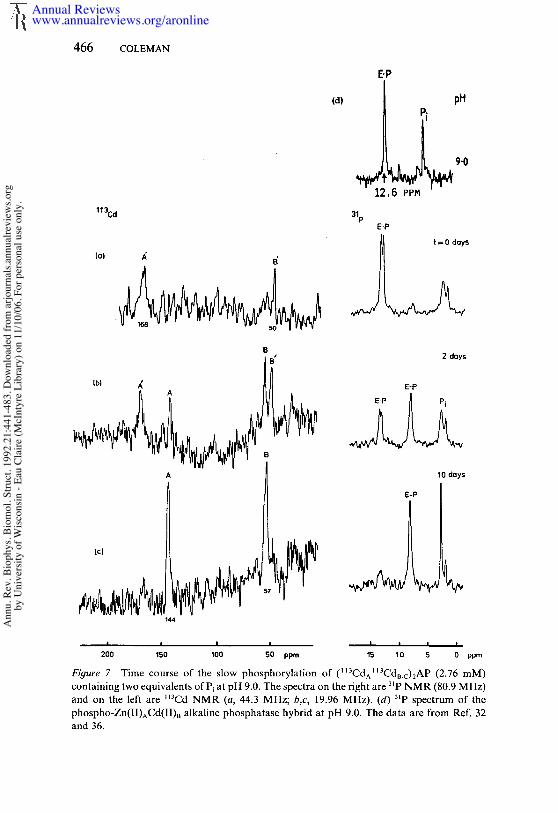
For the native (ZnAZn~Mgc)2 enzyme, E-P has a resonance at 4 ppm,while E-P resonates 8 ppm downfield of phosphoric acid. The chemicalshift of the E-P signal is relatively insensitive to the substitution of thevarious metal-ion species at sites A or B. In marked contrast, the chemicalshift of the E" P signal is highly sensitive to the nature of the metal ion inboth sites, shifting from the most upfield position of 1.8 ppm in the(ZnAMgBMgc)2AP to 13 ppm in the (CdACdBCdc)2AP. This great sitivity of the 6 of E-P to the nature of the metal ion suggested that thephosphate of E" P was coordinated to one or both metal ions. In the caseof the (CdACdBCdc)zAP, the phosphate of E. P appeared to be coordinatedto only one of the active-center 113Cd ions based on the observation thatthe 31p signal for E" P is a doublet showing a single 30-Hz 31p_~3Cd coupling(Figure 7a) (33). Heteronuclear decoupling shows that this coupling comesfrom the A-site 1~3Cd, a conclusion supported by the disappearance of thiscoupling in the (ZnACdB)2AP hybrid enzyme (Figure 7d) (36). Althoughthe coupling disappears, upon Zn substitution for the A-site ll3Cd, theunusual downfield chemical shift of 12.6 ppm is maintained in a (ZnACda)2hybrid (Table 3, Figure 7d), which leads to the unexpected conclusion thatCd in the B site rather than the A site induces the unusual downfield shiftof the E" 3~p signal. This observation became less surprising when thecrystal structure ofE" P showed that a second phosphate oxygen is as closeto Zna(2) as the more normally coordinated oxygen is to ZnA(1) (Figure4b).
In contrast to the changes in chemical shift of the E.3~P signal, theresonance of the phosphoserine, E-3~P, remains between 8.6 and 8.4 ppmno matter what metal ions are in the A, B, and C sites (Table 3). Likewise,in heteronuclear-decoupling experiments, no change in the linewidth ofthe E-3~P singlet is observed on irradiation of either the ll3CdA or II3CdBsignals. These characteristics of the phosphorous nucleus in the E-P com-plex led to the suggestion that at least on the appropriate NMR timeaverage, the phosphate of E-P is not coordinated to either A-site or B-sitemetal ions. However, if the Cd~(2) is coordinated to the ester oxygen the electron density map suggests, a I~3Cd-O-3~P coupling may not beresolved. The structure in the immediate vicinity of the phosphoseryl-102is significantly influenced by the metal ions, since removal of the Cd ionsfrom the E-P form of the Cd6 enzyme results in an apophosphoseryl
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.
466 COLEMAN
E’P
113Cd
(d) pH
~ #~9.012,6 PPH
31pE.P
B 2 days
10 days
200 150 100 50 ppm 15 10 5 0 ppm
Figure 7 Time course of the slow phosphorylation of (II3CdAII3CdI~.c)2AP (2.76 mM)containing two equivalents ofPi at pH 9.0. The spectra on the right are 3~p NMR (80.9 MHz)and on the left are M3Cd NMR (a, 44.3 MHz; b,c, 19.96 MHz). (d) 3~p spectrum of phospho-Zn(II)ACd(II)B alkaline phosphatase hybrid at pH 9.0. The data are from Ref. and 36.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 467
enzyme that has a 31p chemical shift that moves upfield to 5.8 ppm from8.7 ppm (Table 3).
The above NMR characterizations are graphically illustrated by both~t3Cd and 31p NMR signals during the phosphorylation of (~13CdA~3CdB_c)2AP by inorganic phosphate. The E" P species (3~p doublet at ppm) forms rapidly (Figure 7a). This species slowly phosphorylates enzyme over a period of days (Figures 7b,c). At two days, half of theenzyme has become E-P (3~p singlet at 8.7 ppm). Simultaneously, the l l3Cdsignals have each split into two, reflecting the different values for CdA andCdB in the E" P and E-P forms of the enzyme, respectively. Perhaps reflect-ing the release of the coordinate bond between the phosphate and ~3CdAin E’P, the shift of the 113Cd signal from site A on formation of E-P is 20ppm, while that from ll3Cda is only 8 ppm (Figure 7b). The metal-ionspecies at site A largely determines the pH at which [E’P] = [E-P] or thepH at which k3 = k_3 (21, 24, 33). This pH shifts from 5 for ZnnAP to 8.7for Cd6AP, while remaining at pH 6 for the (ZnACda)2AP hybrid (Table3). Because k 3 appears to be pH independent, based on the ~80 exchangeout of HP18024- catalyzed by the enzyme, this extreme change in the pHdependence of k3 must relate to an effect of CdA on the dephosphorylationof E-P.
Dephosphorylation of E-P is absolutely metal-ion dependent, and Cdis a "softer" metal than Zn and therefore is expected to affect loweringthe pKa of a coordinated water less. If the nucleophile required for thedephosphorylation of E-P were a metal-hydroxide formed from a solventH20 coordinated to ZnA, then k3 would vary with the Zn’OH2~Zn’-OH+H+ dissociation. The titration data in Figure 2 indicatethat the pK~ of a water coordinated to ZnA would have to be ~7.5 inorder to match both the pH dependence of k3 and the pH dependence ofkcat. Because below the pKa of the Zn’OH2, the concentration of theZn--OH nucleophile would continue to fall by 10-fold for each 1-unitfurther drop in pH, k3 would fall as well. Thus, when the pH reaches avalue where k3 matches k_3, E-P will build up along a sigmoid curve whosemidpoint is the pH at which k3 = k_3 (Scheme 1). Because CdA moves thepH at which k3 = k_3 toward the alkaline by ,-~ 3.7 pH units, the postulateof a metalA-hydroxide as the nucleophile in the dephosphorylation ofSerl02 requires that the pK~ of the Cd- OH2 ~--- Cd" -OH + H÷ equilibriumbe at least 3.7 pH units higher than that for the corresponding zincequilibrium. How much above pH 8.7 the pKa of the Cd’OH2 is dependson the value of k_3, which we do not know for the Cd enzyme.
The nucleophile in both the hydration and esterase reactions catalyzedby carbonic anhydrase is a Zn-hydroxide at the active center of the enzyme,and the Zn’OH2~---Zn" OH+H+ equilibrium is described by a pKa of
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

468 COLEMAN
6.8 (71). The substitution of Cd for the native Zn ion shifts this pKa 9.3, and the apparent pK~ of the sigmoid pH profile of k~,t for esteraseactivity shifts from 6.8 to 9.3 (71). Thus, a shift in the pKa of a coordinatedsolvent molecule has a precedent in another zinc metalloenzyme and isone of the few changes expected of a Cd substitution that could accountfor such a dramatic shift in the pH dependence of a rate constant for anenzyme reaction.
CORRELATION OF STRUCTURE AND MECHANISM
Michael& Complex with a Phosphate Monoester
The electron density map of the active center in the absence of phosphateindicates a bond between the oxygen of Serl02 and Zn2 (Figure 6e),supporting the notion that one of the functions of Zn2(A) is to activatethe Ser hydroxyl to Ser-O-. However, the 2-/~ map of the E" P complexfrom which nucleophilic attack of the Set O- would occur shows that thiscoordination position is occupied by one of the phosphate oxygens, leavingthe serlne side chain free in the cavity (disordered). Nevertheless, someactivation may be conferred by the positive charge density, and in the E" Pcomplex, protons may not have access to the Set-O-. The structure of theE" P complex with the (ZnAZn~Mgc)2AP indicates that the four oxygensof the phosphate are each in contact with a positively charged center, onewith ZnA, tWO in a hydrogen bond network with the guanidino group ofArg166, and the fourth with ZnB through the unusual P-O-Zn bond angleof almost 180° described earlier. Perhaps this unusual bond, rather thanthe lack of a bond, in the E" P complex of the cadmium enzyme accountsfor the inability to observe detectable ~3Cd-3~P coupling to the 113Cd atthe B site.
The Znl-O-P-O-Zn2 bridge and the two hydrogen bonds between thetwo other phosphate oxygens places the oxygen of Serl02 in the expectedposition for nucleophilic attack on the phosphorous nucleus, a positionthat would become one of the apical positions if a five-coordinate inter-mediate were formed. The oxygen coordinated to Znl would occupy theopposite apical position. These relationships are best seen in Figure 4a. Ifthe E" P structure is extrapolated to that of E- ROP (with which E" P mustshare at least some features in common), then the oxygen coordinated toZn;, must be the ester oxygen. None of the other positions would allowspace for the attachment of an R group, which can range in size fromCH3 to a macromolecule. This observation suggests that in the normalhydrolysis reaction, ZnA is an electrophile activating the leaving group,much as protonation of the ester oxygen does in the model systems. Thus,the alkaline phosphatase mechanism may have as significant a dissociative
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 469
component provided by activation of the leaving group by ZnA as it doesan associative component provided by the attack of Serl02, the latteractivated by the second Zn ion. An electrophilic activation by the Znwould explain why the fl values for k~,t/Km observed for substrates thatcannot protonate the leaving group (phosphopyridines) do not differ sig-nificantly compared with those that can (oxyesters) (51); both fl values equally small (51). A dissociative character of the alkaline phosphatasereaction has been suggested by the low magnitude of secondary isotopeeffects when the nonbridging oxygens of the substrate are labeled with 180(77). This is compatible with the observation that an alkoxide, RO-,appears to be the species coordinated to ZnA(1) in the phosphotransferasemechanism (38). Thus a ZnA-coordinated alkoxide as the immediate leav-ing group in normal hydrolysis is entirely possible.
The Phosphoseryl IntermediateAs noted above, the very large alkaline shift in the pH at which [E-P] = [E-P], induced by Cd at the A site (Table 3), provides a means forvisualizing the E-P complex in the crystal structure. A normal-appearingester bond has formed with Serl02 and the phosphate is slightly deeper inthe active center cavity than in the case of the E" P complex (Figure 6b).The electron-density map of the E-P complex shows that Cd2 forms acoordinate bond with the ester oxygen of the seryl phosphate (Figure 6b).The latter would suggest that the metal in site B(2) has the same functionin the second half of the mechanism as the metal in site A(1) has in thefirst half, i.e. activation of the leaving group. The possibility of a phosphateoxygen remaining in contact with Cd 1 in the E-P complex is not clear fromthe electron-density map (Figure 6b). Mechanistic considerations suggestthat if the nucleophile in the final hydrolysis reaction is a solvent co-ordinated to metal A(I), then the coordination of an electron-donatinggroup of the substrate to the same metal ion as the nucleophile would notfacilitate this step.
Hydrolysis of the Phosphoseryl IntermediateThe crystal structure of the E-P complex indicates that a solvent moleculein position to be a nucleophile adding to the phosphate nucleus from theapical position opposite to that occupied by the seryl ester would clearlybe placed in the vicinity of Zn(1). An obvious choice is to place the solventas one coordinated to Zn 1 in the approximate position originally occupiedby the coordinated phosphate ester oxygen in the E. P complex (Figure 4).For the reasons described in the previous section, the ionization of acoordinated solvent molecule on metal A is the simplest explanation for
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

470 COLEMAN
the pH-rate profile and for the behavior of the pH-dependent E-P ~--- E" Pequilibrium upon the Cd for Zn substitution.
Dissociation o~f the Product, Inorganic Phosphate, the Rate-Limiting StepAs discussed above, at alkaline pH (8 to 9) where alkaline phosphatase maximally active, dissociation of the product, P~, is rate limiting for thenative (ZnnZnBMgc)2 enzyme, as most easily demonstrated by NMRmethods. For the native enzyme, the reported values for k4 (Scheme l)have ranged from 20 to 50 s-1 (19, 44). This value is approximately therange reported in the literature for kcat, 13-?0 s -1, under a variety ofconditions 09, 29, 41, 63). At acid pH, the rate-limiting step changes todephosphorylation of the phosphoseryl intermediate, primarily because ofa dramatic fall in the rate constant, k3, for this reaction from ~> 300 s-I(pH 8) to ~0.5 -1 (pH 5.5) 0 9). T his f all i n k3 i s c ompatible with tnotion that the nucleophile for this dephosphorylation is a Zn-hydroxideat the A site that becomes protonated in the acid form of the enzyme, thusmarkedly reducing the concentration of active nucleophile present (21).
As outlined in the earlier section on kinetics, both the rate of phosphatedissociation, k4, and the rate of phosphorylation of Serl02 by E’P, k_3,depend on the species of metal ion present at both A and B sites. Bothmetal sites seem to be involved in the regulation of the phosphate dis-sociation because the rate for the ZnACdB hybrid enzyme shows a lessdramatic fall in k4 to ~2 s ~, compared with the CdACdB enzyme in whichk4 is < l s- ~ (Table 2) (38). The structure of the E" P complex as revealedby the crystal structure does not directly suggest a reason for this metal-ion dependence of k4. Because metal-phosphate bonds to both A- and B-site metal ions are required for phosphate binding, dissociation probablydepends critically on the detailed charge distribution at the sites as well ason subtle aspects of the coordination geometry. The latter may be some-what different for the larger Cd(II) ion, i.e. 0.9?/~ compared with 0.?4 for Zn(II). It is not clear, however, why the larger Cd(II) ion results slower dissociation of phosphate.
Early kinetic studies called attention to the fact that an activation ofalkaline phosphatase caused by increasing ionic strength should be dis-tinguished from the "apparent" activation caused by the additive trans-ferase activity (61). Because 1 M concentrations of Tris usually carrysignificant CI- concentrations as well, these separate effects can be confus-ing. The ionic strength effect (activation), e.g. from the addition of NaCI,can be accounted for by the fact that the anion, CI-, enhances the dis-sociation rate for phosphate as shown in Table 2. Even the relatively slowphosphate dissociation from the (ZnAMgBMgc):AP can be enhanced ~ 15-
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 471
fold by 1 M C1- (Table 2). The molecular mechanism of this effect appearsadequately explained by the 35C1 NMR (Zn4Mg2 enzyme) and ~3Cd NMR(Cd6 enzyme) observations that C1- can occupy two coordination sites the A-site metal ion (37). Thus, C1- is in competition with one of thephosphate-oxygen-metal bonds and can thus labilize the bound phosphateof E.P.
Because of the apparent lack of effect of pH on phosphate binding tothe enzyme through the range of the second pKa of phosphate, inves-tigators have always argued that the phosphate is forced to bind to theenzyme active center as the dianion (63). The phosphate-dissociation rateis independent of pH from pH 8.8 to pH 5.5 as has been shown directlywith 3~p NMR inversion transfer for Zn4AP (38) (Figure 2). All forms E-P show 3~p chemical shifts more downfield than the dianion of freephosphoserine, for which the 6 = 4 ppm (Table 3). The 3~p chemical shiftsof all forms of E" P, with the exception of that for (ZnAMgBMgc)2AP,are downfield of that for the dianion forms of free Pi, 6 = 3 ppm. The(ZnAMg~Mgc)2AP enzyme has an unusually far upfield 3~P resonance forE’P (Table 3) and a very slow off rate for phosphate, k4 = 1.0-1.8 -~(Table 2). This slowing of the rate-limiting step in the hydrolysis reactionis an adequate explanation ibr the fact that in the presence of an acceptorlike Tris, where the majority of the reaction is phosphotransferase ratherthan hydrolysis, kcat does not decrease for the (ZnAMgBMgc)2AP, while no acceptor is present, kc,t is 10% of that for the native enzyme. Thisassertion follows because the dissociation of an ester, e.g. R’OP as thetransferase product, is much faster than HOP as discussed in more detailbelow.
The Phosphotransferase ReactionThe transfer of the phosphate from E-P to an acceptor alcohol by alkalinephosphatase was discovered early in kinetic studies of the enzyme (26, 63).The fact that a two- to fourfold enhancement in the rate of RO- releasecould be observed by adding an acceptor like Tris(tris-hydroxymethylaminomethane) (Table 1) led to the routine inclusion of 1 M Tris in standard assay of alkaline phosphatase. The enhanced rate of releaseofp-nitrophenolate made the assay significantly more sensitive. Productanalysis showed that the enhanced activity resulted almost entirely fromthe formation of a new phosphate monoester, Tris-phosphate. The rateof formation of the original hydrolysis product, phosphate, remainedrelatively unaffected (63, 79).
Isolation of nonchromophoric phosphate esters is difficult, and relativelylittle quantitative data on the phosphotransferase reaction have accumu-lated over the years. Recently, 3~P NMR proved to be a convenient method
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.
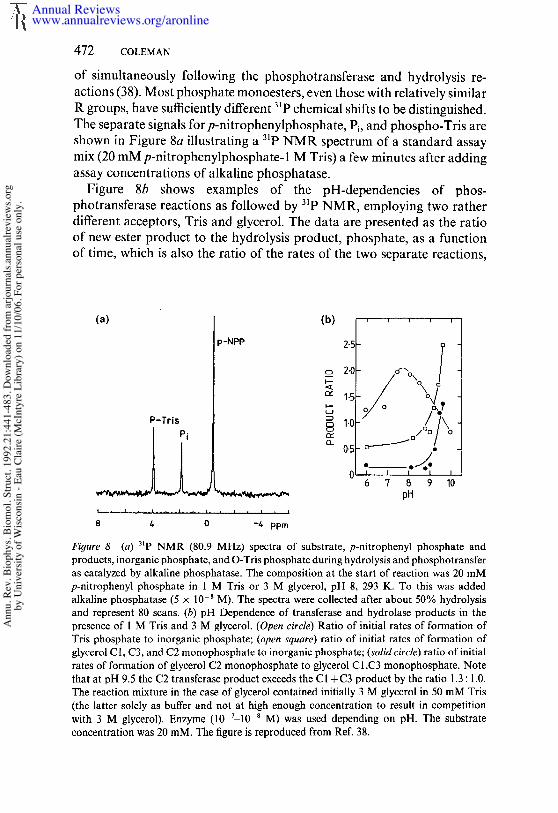
472 COLEMAN
of simultaneously following the phosphotransferase and hydrolysis re-actions (38). Most phosphate monoesters, even those with relatively similarR groups, have sufficiently different ~lp chemical shifts to be distinguished.The separate signals for p-nitrophenylphosphate, P~, and phospho-Tris areshown in Figure 8a illustrating a 31p NMR spectrum of a standard assaymix (20 mM p-nitrophenylphosphate- 1 M Tris) a few minutes after addingassay concentrations of alkaline phosphatase.
Figure 8b shows examples of the pH-dependencies of phos-photransferase reactions as followed by 3~P NMR, employing two ratherdifferent acceptors, Tris and glycerol. The data are presented as the ratioof new ester product to the hydrolysis product, phosphate, as a functionof time, which is also the ratio of the rates of the two separate reactions,
(a) (b) .....
)-NPP 2.5
o_ 2.0.O’~ox /
c~ ~.5
P-Tris ~
6 7 8 9 I0~ pH
8 4 0 -4 ppm
Figure 8 (a) ~P NMR (80.9 MHz) spectra of substrate, p-nitrophenyl phosphate products, inorganic phosphate, and O-Tris phosphate during hydrolysis and phosphotransferas catalyzed by alkaline phosphatase. The composition at the start of reaction was 20 mMp-nitrophenyl phosphate in 1 M Tris or 3 M glycerol, pH 8, 293 K. To this was addedalkaline phosphatase (5 x I0-~ M). The spectra were coll~ted after about 50% hydrolysisand represent 80 scans. (b) pH Dependence of transferase and hydrolase products in thepresen~ of I M Tris and 3 M gly~rol. (Open circle) Ratio of initial rates of formation ofTris phosphate to inorganic phosphate; (open square) ratio of initial rates of formation ofglycerol C I, C3, and C2 monophosphate to inorganic phosphate; (solid circle) ratio of initialrates of formation of glycerol C2 monopbosphate to glycerol CI,C3 monophosphate. Notethat at pH 9.5 the C2 transferase product exceeds the CI +C3 product by the ratio 1.3: 1.0.The reaction mixture in the ease of glycerol contained initially 3 M glycerol in 50 mM Tris(the latter solely as buffer and not at high enough concentration to result in competitionwith 3 M glycerol). Enzyme (I0 v-10 ~ M) was used depending on pH. The substrateconcentration was 20 raM. The figure is reproduced from ReE 38.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 473
phosphotransferase/phosphohydrolyase. The two curves for glycerol rep-resent the formation of the equivalent C 1 + C3 monophosphates and theC2 monophosphate. All three hydroxyl groups of glycerol are approxi-mately equally phosphorylated by the enzyme. Tris is a much more effectiveacceptor in the neutral pH range than glycerol. The phosphotransferasereaction also increases in rate as the pH approaches the pKa of the aminogroup, ,-~ 8 for Tris (63). This observation led to the suggestion that theneutral species of the amino alcohols were the most effective acceptors(63). The transferase activity of Tris, however, peaks at pH 8, fallingrapidly to higher pH values (Figure 8b). At pH 9 and above, nonaminoalcohols like glycerol, ethanol, and propanol are substantially moreefficient acceptors than Tris (Figure 8b).
The rapid increase in the phosphate transfer to aliphatic alcohols abovepH 9 is compatible with an arrangement in which the accepting alcohol isin the alcoxide form when bound at the active center and accepts thephosphoryl group from the seryl phosphate intermediate (38). If the oxygenof the acceptor coordinates ZnA, as seems likely (38), then the apparentpK~ of the -C-OH will be lowered. An alcoxide has also been postulatedto be the form of ethanol bound to alcohol dehydrogenase (82), anotherzinc enzyme in which an open coordination position on a zinc ion isthe driving force for the formation of the alcoxide below its pKa. Atconcentrations 0.5 M or greater, Tris and glycerol shift the it 3Cd resonancefrom the A site upfield by 14 and 6 ppm, respectively, at pH 9 (38). Thesel~3Cd chemical shift changes may represent the displacement of a waterligand by the alcoxide of the acceptor (38). Both the enhanced acceptorefficiency of the amino alcohols below the pKa of the amino group andthe rapid decline upon deprotonation of the amino group (Figure 8b) couldresult from the lowering of the pKa of the -CHz-OH group caused by theadjacent -C-NH~ group such that the alcoxide of the amino alcohols isformed more readily (has a lower pK.) below the pKa of the amino groupthan when the neutral form of the amino group is present.
SUMMARY OF THE MECHANISM OF ALKALINEPHOSPHATASE AS BASED ON SOLUTION DATA ANDTHE CRYSTAL STRUCTURE
Figure 9 shows the Michaelis complex of the dianion of a phosphatemonoester bound to the active center of Ser 102 along with the steps leadingto the phosphorylation of Serl02 and its subsequent dephosphorylation.The probable structural relationships are taken from the crystal structureof the E’P complex of the native zinc enzyme (Figure 4b) and the E-Pcomplex of the cadmium enzyme (Figure 6b). ZnA coordinates the ester
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.
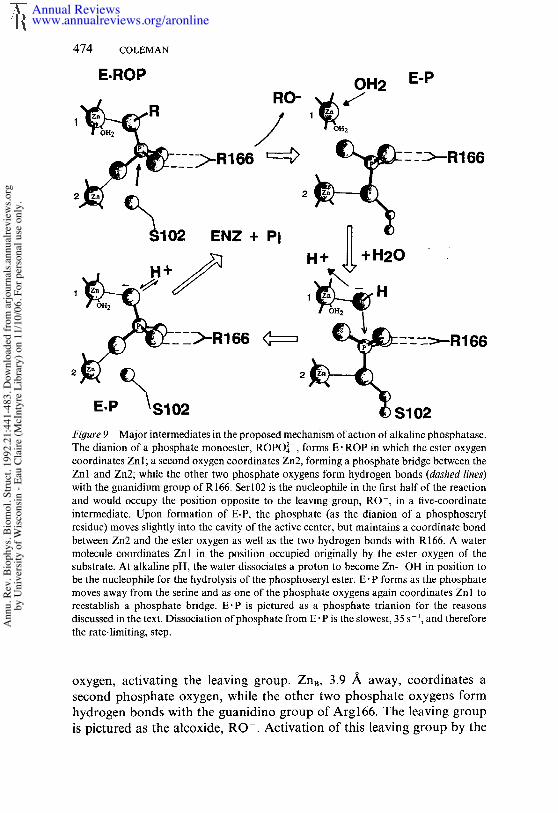
474 COLEMAN
E.ROP OH2 E-P
Figure 9 Major intermediates in the proposed mechanism of action of alkaline phosphatase.The dianion of a phosphate monoester, ROPO4~-, forms E. ROP in which the ester oxygencoordinates Zn 1; a second oxygen coordinates Zn2, forming a phosphate bridge between theZnl and Zn2; while the other two phosphate oxygens form hydrogen bonds (dashed lines)with the guanidium group of R166. Serl02 is the nucleophile in the first half of the reactionand would occupy the position opposite to the leaving group, RO-, in a five-coordinateintermediate. Upon formation of E-P, the phosphate (as the dianion of a phosphoserylresidue) moves slightly into the cavity of the active center, but maintains a coordinate bondbetween Zn2 and the ester oxygen as well as the two hydrogen bonds with R166. A watermolecule coordinates Znl in the position occupied originally by the ester oxygen of thesubstrate, At alkaline pH, the water dissociates a proton to become Zn--OH in position tobe the nucleophile for the hydrolysis of the phosphoseryl ester. E" P forms as the phosphatemoves away from the serine and as one of the phosphate oxygens again coordinates Znl toreestablish a phosphate bridge. E’P is pictured as a phosphate trianion for the reasonsdiscussed in the text. Dissociation of phosphate from E" P is the slowest, 35 s-t, and thereforethe rate-limiting, step.
oxygen, activating the leaving group. ZnB, 3.9 ~ away, coordinates asecond phosphate oxygen, while the other two phosphate oxygens formhydrogen bonds with the guanidino group of Arg166. The leaving groupis pictured as the alcoxide, RO-. Activation of this leaving group by the
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 475
A-site metal ion via a bond with the ester oxygen appears to be one of thefunctions of this ion in the first stage of the alkaline phosphatase reaction.The oxygen of Serl02 is in the other apical position to form the new esterbond with the phosphorous. Dissociation of RO- and formation of thephosphoseryl intermediate is associated with movement of the phosphatefarther into the cavity (Figure 6b). Hence, When the alcoxide leaves, itscoordination site on ZnA can be occupied by a water molecule that mustdissociate a proton and become a Zn--OH. The strongest evidence forthis metal-induced proton dissociation is the large alkaline shift (3.7 pHunits) in the pH dependence of the E" P ~- E-P equilibrium when the muchsofter metal ion, Cd(II), is substituted for Zn(II). The Zn--OH, placed the apical postion opposite to the seryl ester, occupies the place of theoriginal leaving group and thus is ideally positioned to be the nucleophilein the second stage of the hydrolysis reaction (Figure 9).
The second stage of the reaction is nearly a mirror image of the firststage. ZnB now activates the leaving group by forming a coordinate bondwith the ester oxygen of the phosphoseryl residue (Figure 9). The hydroxylcoordinated to ZnA attacks the phosphorous from the other apicalposition, and the transition state will decay into E P. The issue of whetherthe new phosphate oxygen remains negatively charged is discussed below,but the dissociation of phosphate anion in the E. P intermediate is remark-ably slow, 35 s-~.
As discussed in connection with the phosphotransferase reaction, whythe dissociation of R2OPO3~- is more rapid than that of HOPO32- if thelatter is bound as the dianion is unclear. One possibility is that coordinationof phosphate to both Zn ions as well as hydrogen bonding of the othertwo oxygens to the guanidino group of Arg166 forces the formation of thetrianion, OPO~-. On the other hand, the bound ROPO~3- can only be adianion. ZnA does seem to be capable of lowering the pKa of bound waterby several pH units as well as inducing the alcoxide in the phos-photransferase reaction. Thus, lowering the third pKa of phosphate maynot be so unexpected as it might at first appear. Such a postulate wouldprovide an explanation as to why dissociation of phosphate from the activecenter is so much slower than that for the dianion of a phosphate ester. Thehigher negative-charge density would also explain why phosphorylation ofSerl02 by both phosphate and thiophosphate is so slow compared tophosphorylation by any phosphate ester. The pKa of the leaving group forthiophosphate, H2S, is 6.9 compared to a pKa of 15.7 for HOH, the leavinggroup in phosphorylation from phosphate. Yet in the presence of rate-controlling phosphorylation, the difference in the phosphorylation ofSerl02 by these two agents is at most only twofold (Table 1), despite thelarge ApKa between the leaving groups. If both substrates were bound as
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

476 COLEMAN
trianions, poor leaving groups (probably requiring protonation first) andhigh charge density around the phosphorous could account for the veryslow phosphorylation step as well as slow dissociation of the noncovalentlybound species.
In the case of most enzyme-catalyzed phosphorylation or phospho-transferase reactions, investigators have tended to assume that such bio-logical reactions were nucleophilic and associative and formed classic five-coordinate intermediates, the leaving group departing from the oppositeapical position. This assumption was made despite the fact that in modelsystems most phosphate mono- and diester hydrolyses apparently proceedby a dissociative mechanism with the transient production of the highlyelectrophilic metaphosphate, [PO~-] (6, 49, 78). Only phosphate triesterhydrolysis follows a nucleophilic path in model systems (6, 49, 78).Enzymes could presumably make the nucleophilic path more favorable bycreating protein-binding sites that successfully reduced the negative chargearound phosphate, a significant inhibitory factor in mono- and diesterhydrolysis in model systems. Determination of the predicted chiralitychanges around phosphate during enzyme-catalyzed phosphate incor-porations, i.e. inversion if no covalent enzyme intermediate was formedand retention if a covalent enzyme intermediate was formed as in alkalinephosphatase, tended to solidify the nucleophilic point of view. However,the finding that the Zn ions activate the leaving group in both the initialphosphorylation of the active-center serine and its dephosphorylationsuggests that there may be significant dissociative character to bothreactions. While the enzyme might hold a metaphosphate rigid enough toprevent scrambling of chirality, the evidence that the phosphate oxygensexchange positions relatively rapidly in the Co(II) enzyme does not favorthe existence of free PO ~- at the active site. Whether a classic five-coordinateintermediate is a true transition state in the alkaline phosphatase reactionremains an open question.
SITE-DIRECTED MUTANTS OF E. COLI ALKALINEPHOSPHATASE
Cloning of the phoA gene has allowed researchers to employ site-directedmutagenesis to change several of the amino acid side chains at the activecenter of alkaline phosphatase. The results of mutations at Serl02 andArg166 have been most informative relative to mechanism, whilemutations of several surrounding residues have revealed subtle influenceson the rate. The results of these mutations are summarized below.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 477
Set102 ~ Cysl02, Alal02, Leul02The Serl02~Cysl02 mutant shows between 30 and 90% of normalactivity against p-nitrophenyl phosphate and 2,4-dinitrophenyl phosphatedepending on conditions. The range of turnover values results both fromthe fact that the phosphotransferase/phosphohydrolase ratio is increasedand that kca t depends on the nature of the leaving group. The Cysl02enzyme has a kcat of 4.6 S-1 with 4-nitrophenyl phosphate and a kcat of 15s-l with 2,4-dinitrophenyl phosphate under conditions where kcat for thewild-type enzyme is 18 s-1 (39). In addition, the pH profile of the Cysl02enzyme shows no clearly apparent pKa of activity. Lastly, a covalent S-phosphorothioate cannot be isolated from the enzyme by quenching atlow pH (39). Most of the above findings on the Cysl02 mutant canbe explained if the rate-limiting step has changed to phosphorylationthroughout the pH range. Throughout the pH range, phosphorylation ofthe native enzyme by ROPO~4- occurs at the rate of 102 to 103 s-l; thus, thephosphorylation rate can fall by 10- or even 100-fold without significantlylowering kcat. These findings also suggest that dephosphorylation of the S-phosphorothioate formed with Cysl02 is occuring more rapidly than forthe normal covalent intermediate. A detailed kinetic study is required tosort out the rate constants of the Cysl02 mutant. Phosphotransfer froma ~60-, ~70-, and ~80-labeled phosphate ester to an acceptor catalyzedby the Cysl02 mutant revealed retention of configuration around thephosphorous, proving that the reaction pathway has not been substantiallychanged (12).
The Serl02 ~ Alal02 or Leul02 mutants are not completely devoid ofactivity. The kcat values are reported to be ~ 1/1000 and ~ 1/500 of thosefor the wild-type enzyme (14). Km values for ROPO4~- and Ki values for Piare reported to be normal as is the pH-rate profile. Because residue 102 isnot directly involved in the structure of E’P, the lack of alteration insubstrate and phosphate binding is not surprising. Since ZnA carries twocoordinated H20 molecules, as shown using NMR relaxation methods(37, 64), a water remaining on ZnA may be in a position to directly attackthe ester of E" ROP at a low rate. An alcoxide, RzO-, could do the sameand account for the detection of some phospho-Tris formed by thesemutants (14).
Arg166 -~ Lys166, Glu166, Ser166, Ala166All four mutations of Arg166 result in enzymes that remain active butdemonstrate an increase in K~ and a decrease in phosphate affinity (13,15). Although a decrease in phosphate binding affinity might increase the
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

478 COLEMAN
rate of phosphate dissociation, k4, and therefore kent, the fact that theopposite occurs suggests that the loss of a positively charged center, helpingto reduce the negative charge density at phosphorous, is the more impor-tant function. This fact also suggests that despite a significant dissociativeaspect to the mechanism, the alkaline phosphatase reaction must retainsignificant aspects ofa nucleophilic hydrolysis mechanism. This conclusionis supported by the finding for these mutants that when the nucleophile iswater (hydrolysis), a relatively poor nucleophile, kca t is reduced more bythe mutation than when RO- (phosphotransfer), a better nucleophile, the nucleophile in the second half of the reaction. Likewise, the leavinggroup in the first half of the mechanism, phosphorylation of Ser102, hasa more pronounced effect on the observed kcat of the Arg166 mutants, e.g.kc,t decreases 10-fold on shifting from p-nitrophenolate as the leavinggroup to phenolate for both the Lys and Glu mutants. The effect of theLys mutant must result from the different topological placement of thepositive charge by the shorter lysyl side chain.
The fact that changes related to the bond-making and -breaking stepsare now reflected in kc,t suggest that these mutations may have changedthe relative values of the rate constants such that phosphate departure isno longer the slowest and therefore exclusively rate-limiting step. Furtherinvestigation is needed. While reduced in affinity, the phosphate bindingis nevertheless adequately maintained in the Arg166 mutants by the inter-actions with the metal ions and the further hydrogen bonds with bridgingwater molecules to Mgc and Lys328 (Figure 4b). E. E. Kim & H. Wyckoff (in preparation) have determined the crystal structure of theArg166-~ Ala166 mutant, which shows no changes in the active centerexcept for the truncation of the density at the B-carbon of residue 166.Thus, the changes in the reaction parameters described above can beattributed exclusively to the functions of the Arg166 side chain.
Lys328 -~ His328, Ala328Despite the conservation of many of the active-site residues found atthe active center of the E. eoli enzyme in several mammalian alkalinephosphatase isozymes, it has been puzzling why the mammalian enzymesuniformly show kcat values at least an order of magnitude larger than thatof the bacterial enzyme (for review, see 22). One intriguing structuralfinding at the active center of the bacterial enzyme is that Lys328, replacedby a His in all mammalian alkaline phosphatases, is bridged to the boundphosphate in E. P through a water molecule (Figure 4b). The latter watermolecule is also hydrogen bonded to one carboxyl oxygen of Asp327, thebidentate ligand to ZnA. The other oxygen of Asp327 makes a hydrogenbond with the N3 of His372 as discussed in the section on structure.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 479
Because of the difference in the nature of residue 328 between the mam-malian and bacterial enzymes and the close association of the Lys328 sidechain with the A-site metal ligands as well as with phosphate via hydrogenbonds, two single site mutations of this residue have been examined,Lys328 ~ His328 and Lys328 -~ Ala328 (54, 81).
At pH 10.3, the Lys328 ~ Ala328 mutant shows a 14-fold increase inkca t in the presence of Tris as an acceptor and a 6-fold increase in kca t inthe absence of the acceptor (15). At pH 8.0 in 1 M Tris, kcat for the mutantenzyme was 81 s -~ and for the wild-type enzyme was 16 s-~. The Lys328 -~His328 is similar, but the enhancement of kcat is not as large. Both mutantenzymes show reduced affinity for phosphate. Km for ROPO~- is increasedfrom 21 #M for the wild-type enzyme to 159/~M for the Ala328 mutantand 58 #M for the His328 mutant. Some significant changes are observedin the alkaline pH-rate profile of the mutant enzymes both in the presenceand absence of a phosphate acceptor as well as in the magnitude and rateof the burst observed at both acid and alkaline pH, suggesting that therelative values of k3 (dephosphorylation of E-P) and k4 (phosphate dis-sociation) may have been changed by the mutation. Xu & Kantrowitz (81)suggest these changes might result from a change in the pH dependenceof k3, reflecting a change in the pKa of the solvent H20 coordinated toZnA. We lack sufficient detail on the pH dependence of the individual rateconstants to be certain of the precise origin of the observed changes in pH-dependent functions. In another zinc enzyme, carbonic anhydrase, a seriesof detailed studies of rate-constant changes vs pH for several site mutationsand a crystal structure of an isozyme carrying three of the side-chainchanges show that changes in residues near the active center that alter thehydrogen-bonding patterns of the ordered water molecules near the Zn-coordinated solvent are far more effective in altering the pKa of the co-ordinated solvent than are mutations of charged residues in the active sitecavity that do not alter the hydrogen bond structure (73).
Several other site mutations of residues near the active center increasekca t. These include Aspl01 ~ Alal01, which increases kca t 4.2-fold (80), well as several mutations in the polypeptide chain region from residue 99to 109. Mandecki et al (54) screened for "up" mutants in this region andfound that V99A, T100V, A103D, A103C, and T107V all increase kcat two-to fourfold. Residues 101 to 112 form a helical stem that holds Serl02 inplace at the active center. Thus, alterations in kcat are not surprising. Whatis perhaps unexpected is that most changes enhance kcat. The assays are in50 mM Tris; thus, the increases in kcat primarily relate to the hydrolysisreaction. In the case of the E. coli enzyme, in which the dissociation of thephosphate is a very slow and rate-limiting step at alkaline pH, severalslight perturbations of the topology at the active center can enhance
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

480 COLEMAN
this step. What evolutionary advantage prevented such mutations fromevolving naturally is unclear. Perhaps phosphotransferase (not inhibitedby phosphate departure) is a much more important reaction to E. colithan we currently appreciate because the natural acceptor has not beenidentified.
SUMMARY
Alkaline phosphatase was the first zinc enzyme to be discovered in whichthree closely spaced metal ions (two Zn ions and one Mg ion) are presentat the active center. Zn ions at all three sites also produce a maximallyactive enzyme. These metal ions have center-to-center distances of 3.9/~(Znl-Zn2), 4.9 ,~ (Zn2-Mg3), and 7.1 /~ (Znl-Mg3). Despite the packing of these metal centers, only one bridging ligand, the carboxyl ofAsp51, bridges Zn2 and Mg3. A crystal structure at 2.0-~ resolution ofthe noncovalent phosphate complex, E" P, formed with the active centershows that two phosphate oxygens form a phosphate bridge between Znland Zn2, while the two other phosphate oxygens form hydrogen bondswith the guanidium group of Arg166. This places Serl02, the residueknown to be phosphorylated during phosphate hydrolysis, in the requiredapical position to initiate a nucleophilic attack on the phosphorous. Extra-polation of the E-P structure to the enzyme-substrate complex,E.ROPO~4-, leads to the conclusion that Znl must coordinate the esteroxygen, thus activating the leaving group in the phosphorylation of Ser 102.Likewise, Zn2 appears to coordinate the ester oxygen of the seryl phos-phate and activate the leaving group during the hydrolysis of the pho-sphoseryl intermediate. Both of these findings suggest that there may bea significant dissociative character to each of the two displacements atphosphorous catalyzed by alkaline phosphatase. A water molecule (orhydroxide) coordinated to Znl following formation of the phosphoserylintermediate appears to be the nucleophile in the second step of the mech-anism. Dissociation of the product phosphate from the E" P intermediateis the slowest, 35 s- ~, and therefore the rate-limiting, step of the mechanismat alkaline pH.
Since the determination of the initial crystal structure of alkaline phos-phatase, two other crystal structures of enzymes involved in phosphateester hydrolysis have been completed that show a triad of closely spacedzinc ions present at their active centers. These enzymes are phospholipaseC from Bacillus cereus (structure at 1.5-/~ resolution) (43) and P1 nucleasefrom Penicillium citrinum (structure at 2.8-,~ resolution) (74). enzymes hydrolyze phosphodiesters. Substrates for phospholipase C arephosphatidylinositol and phosphatidylcholine, while P1 nuclease is an
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

ALKALINE PHOSPHATASE 481
endonuclease hydrolyzing single stranded ribo- and deoxyribonucleotides.P1 nuclease also has activity as a phosphomonoesterase against 3’-terminalphosphates of nucleotides. The Zn ions in both enzymes form almostidentical trinuclear sites. Znl and Zn3 are ,-~ 3.2 ~ apart and are linkedby a bridging carboxylate of an Asp residue as well as a bridging H20 or-OH ion. Zn2 is an isolated site ~ 5.8/~ from Znl and ~ 4.7/~ from Zn3.
Are the three enzymes with trinuclear Zn sites at their active centersrelated? The general arrangement of the zinc ions is similar, but alkalinephosphatase and the other two enzymes are significantly different. Allthree metal sites in P1 nuclease and phospholipase C are typical Zn bindingsites with mixed oxygen and nitrogen donors, while the third site in alkalinephosphatase is made up of all oxygen donors, which may explain itspreference for Mg. The aspartate carboxylate bridging two of the Zn ionsappears to define a binuclear zinc site in the two diesterases. In contrast,the bridging aspartate carboxylate defines the Zn-Mg pair in native alkalinephosphatase. As discussed in this review, the two nonbridged Zn ions, 3.9~ apart in alkaline phosphatase, appear to form the major functionalpair forming the phosphate bridge. In contrast, the few substrate analogbinding studies done on the diesterases suggest that the isolated Zn2 siteinteracts with the phosphate diester (43). On the other hand, differencemaps for phosphate bound to both P1 nuclease and phospholipase C showthat an interaction with inorganic phosphate can take place involvingcoordination to all three zinc ions and raises the possibility that all three Znions could be catalytically involved at some stage of a complete mechanism.
Alkaline phosphatase is not closely related structurally to the other twoenzymes. Alkaline phosphatase has a relatively small amount of a-helicalstructure on both faces of a large core of parallel and antiparallel fl-sheets.The secondary structure of the two diesterases is composed almost entirelyof c~-helices, and their structures are closely related (43, 74). In addition,alkaline phosphatase forms a phosphoseryl intermediate, while both P1nuclease and phospholipase C catalyze single-step hydrolyses, invertingthe chirality around phosphate. While it is too early to state whether thetrinuclear zinc sites found in the three phosphohydrolases reflect relatedfunctions, the data available at present suggest that the presence of multipleZn ions located at a single enzyme active center add many alternatives tothe reaction pathways available to zinc metalloenzymes.
ACKNOWLEDGMENT
I thank Harold W. Wyckoff and Eunice Kim for many helpful discussionsand suggestions. The crystallography of alkaline phosphatase was sup-ported by Grant GM22778. Original work in the author’s laboratory wassupported by NIH Grant DK09070.
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

482 COLEMAN
Literature Cited
1. Anderson, R. A., Bosron, W. F.,Kennedy, F. S., Vallee, B. L. 1975. Proc.Natl. ,4cad. Sci. USA ?2:2989-93
2. Applebury, M. L., Coleman, J. E. 1969.J, Biol. Chem. 244:709-18
3. Applebury, M. L., Johnson, B. P., Cole-man, J. E. 1970. J. Biol. Chem. 245:4968-76
4. Bachman, B. J., Low, K. B., Taylor, A.L. 1976. Bacteriol. Rev, 40:116~7
5. Bale, J. R. 1978. Fed. Proc. 37:12876. Benkovic, S. J., Schray, K. J. 1973.
Enzymes 8:201-387. Benson, S. A., Hall, M. N., Silhavy, T.
J. 1985. Biochemistry 54:101-347a. Bertini, I., Luchinat, C., Maret, W.,
Zeppezaur, M., eds. 1986. Zinc Enzymes.Basel: Birkh/iuser
8. Bloch, W. A., Schlessinger, M. J. 1973.J. Biol. Chem. 248:5794-58059. Bock, J. L., Cohn, M. 1978. J. Biol.
Chem. 253:4082-8510. Bosron, W. F., Anderson, R. A., Falk,
M. C., Kennedy, F. S., Vallee, B. L.1977. Biochemistry 16:610-14
11. Bradshaw, R. A., Cancedda, F., Erics-son, L. H., Neuman, P. A., Piccoli, S. P.,et al. 1981. Proc. Natl. Acad. Sci. USA78:3473-77
12. Butler-Ransohoff, J. E., Kendall, D. A.,Freeman, S., Knowles, J. R., Kaiser, E.T. 1988. Biochemistry 27:4777-80
13. Butler-Ransohoff, J. E., Kendall, D. A.,Kaiser, E. T. 1988. Proc. Natl. Acad. Sc’i.USA 85:4276-78
14. Butler-Ransohoff, J. E., Rokita, S. E.,Kendall, D. A., Banzon, J. A., Carano,K. S., Kaiser, E. T., Matlin, A. R. 1992.J. Or.q. Chem. 57:142-45
15. Chaidaroglou, A., Brezinski, J. D., Mid-dlcton, S. A., Kantrowitz, E. R. 1988.Biochemistry 27:8338-43
16. Chaidaroglou, A., Kantrowitz, E. R.1989. Protein Eng. 3:127 32
17. Chang, C. N., Inouye, H., Model, P.,Beckwith, J, 1980. J. Bacteriol. 142:726
18. Chlebowski, J. F., Armitage, I. M.,Tusa, P. P., Coleman, J. E. 1976. J. Biol.Chem. 251:1207-16
19. Chlebowski, J. F., Coleman, J. E. 1974.J. Biol. Chem. 249:7192-7202
20. Chlebowski, J. F., Coleman, J. E. 1976.J. BioL Chem. 251:12024
21. Coleman, J. E. 1987. In PhosphateMetabolism and Cellular Regulation inMicroorganisms, ed. A. Torriani-Gorini,F. G. Rothman, S. Silver, A. Wright,E. Yagil, pp. 127-38. Washington, DC:Am. Soc. Microbiol.
22. Coleman, J. E., Besman, M. J. A. 1987.
In Hydrolytic Enzymes, ed. A.Neuberger, K. Brocklehurst, pp, 377-406. New York: Elsevier
23. Coleman, J. E., Chlebowski, J. F. 1979.Adv. Inor9. Biochem. 1:1-66
24. Coleman, J. E., Gettins, P. 1986. SeeRef. 7a, pp. 77-99
25. Coleman, J. E., Nakamura, K., Chle-bowski, J..F. 1983. J. Biol. Chem. 258:386-95
26. Dayan, J., Wilson, I. B. 1964. Biochim.Biophys. Acta 81:620-28
27. Engstr6m, L. 1961. Biochim. Biophys.Acta 52:49-59
28. Engstr6m, L., Agren, G. 1962. Biochim.Biophys. Acta 56:606-7
29. Fernley, H. N., Walker, P. G. 1969.Biochem. J. 11 I: 187-94
30. Fossett, M., Chappelet-Tordo, D., Laz-dunski, M. 1974. Biochemistry 13: 1783-88
31. Garen, A., Levinthal, C. 1960. Biochim.Biophys. Acta 38:470-83
32. Gettins, P., Coleman, J. E. 1983. J. Biol.Chem. 258:396-407
33. Gettins, P., Coleman, J. E. 1983. J. Biol.Chem. 258:408-16
34. Gettins, P., Coleman, J. E. 1983. Adv.Enzymol. 55:381-452
35. Gettins, P., Coleman, J. E. 1982. Fed.Proc. 41:2966-73
36. Gettins, P., Coleman, J. E. 1984. J. Biol.Chem. 259:4991-97
37. Gettins, P., Coleman, J. E. 1984. J. Biol.Chem. 259:11036-40
38. Gettins, P., Metzler, M., Coleman, J. E.1985. J. Biol. Chem. 260:2875-83
39. Ghosh, S. S., Back, S. C., Rokita, S. E.,Kaiser, E. T. 1986. Science 231:145-48
40. Grosser, P., Husler, J. 1912. Biochem. Z.39:1
41. Halford, S. E., Bennett, N. G.,Trentham, D. R., Outfreund, H. 1969.Biochem. J. 114:243 51
42. Horiuchi, T., Horiuchi, S., Mizuno, D.1959. Nature (London) 183:1529-30
43. Hough, E., Hansen, L. K., Birknes, B.,Jynge, K., Hansen, S., et al. 1989. Nature(London) 338:357-60
44. Hull, W. E., Halford, S. E., Gutfreund,H., Sykes, B. D. 1976. Biochemistry 15:1547~51
45. lnouye, H., Barnes, W., Beckwith, J.1982. J. Bacteriol. 149:434~39
46. Jones, S. R., Kindman, L. A., Knowles,J. R. 1978. Nature (London) 257: 564-65
47. Kim, E. E., Wyckoff, H. W. 1989. Clin.Chim. Acta 186:175-88
48. Kim, E. E., Wyckoff, H. W. 1991. J.Mol. Biol. 218:449-64
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

49. Knowles, J. R. 1980. Annu. Rev.Biochem. 49:877-919
50. Krishnaswamy, M., Kenkare, U. W.1970. J. Biol. Chem. 245:3956~53
51. Labow, B. I. 1989. Mechanisms ofphos-phoryl transfer by alkaline phospha-tase from E. coli. MS thesis. BrandeisUniv.
52. Levine, D., Reid, T. W., Wilson, I. B.1969. Biochemistry 8:2374~0
53. Ludke, D., Bernstein, J., Hamilton, C.,Torriani, A. 1984. J. Baeteriol. 159: 19-25
54. Mandecki, W., Shallcross, M. A.,Sowadski, J., Tomazic-Allen, S. 1991.Protein En9. 4:801-4
55. McComb, R. B., Bowers, G. N., Posen,S. 1979. Alkaline Phosphatase. NewYork: Plenum
56. Michaelis, A., Beckwith, J. 1982. Annu.Rev. Microbiol. 36:435-65
57. Muller, M., Blobel, G. 1984. Proc. Natl.Aead. Sei. USA 81:7737-41
58. Mushak, P., Coleman, J. E. 1972. Bio-chemistry 11:201-5
59. Neumann, H. 1968. J. Biol. Chem. 243:4671-76
60. Petitclerc, C., Lazdunski, C., Chappelet,D., Moulin, A., Lazdunski, M. 1970.Eur. J. Biochem. 14:301-8
61. Plocke, D. J., Vallee, B. L. 1962. Bio-chemistry 1:1039-43
62. Plocke, D. J., Levinthal, C., Vallee, B.L. 1962. Biochemistry 1:373-78
63. Ried, T. W., Wilson, I. B. 1971. Enzymes4:373-415
64. Schulz, C., Bertini, I., Viezzoli, M. S.,Brown, R. D., Koenig, S. H., Coleman,J. E. 1989. Inorganic Chem. 28:1490-96
65. Schwartz, J. H. 1963. Proe. Natl. Aead.Sci, USA 49:871-78
ALKALINE PHOSPHATASE 48366. Schwartz, J. H., Lipmann, F. 1961. Proc.
Natl. Acad. Sci. USA 47:1996-200567. Schwartz, J. H., Crestfield, A. M., Lip-
mann, F. 1963. Proc. Natl. Acad. Sci.USA 49:722-29
68. Sowadski, J. M., Handschumacher, M.D., Murthy, H. M. K., Foster, B. A.,Wyckoff, H. W. 1985. J. Mol. Biol. 186:417 33
69. Stein, S. S., Koshland, D. E. 1952. Arch.Biochem. Biophys. 39:22%30
70. Suzuki, U., Yoshimura, K., Takaishi,M. 1907. Bull. Coll. Argric. Tok. Imp.Univ. 7:503
71. Tashian, R. E., Hewett-Emmett, D. Ann.New York Acad. Sci. 429:1440
72. Torriani, A. 1960. Biochim. Biophys.Acta 38:46049
73. Tu, C., Paranawithana, S. R., Jewell, D.A., Tanhauser, S. M., LoGrasso, P. V.,et al. 1990. Biochemistry 29:6400-5
74. Volbeda, A., Lahm, A., Sakiyama, F.,Suck, D. 1991. EMBO J. 10:1607-18
75. Von Eu!er, H. 1912. Z. Physiol. Chem.79:375
76. Von Euler, H., Funke, Y. 1912. Z.Physiol. Chem. 77:488
77. Weiss, P. M., Cleland, W. W. 1989. J.Am. Chem. Soc. 111:1928-29
78. Westheimer, F. H. 1968. Acc. Chem.Res. 1:70-78
79. Wilson, 1. B., Dayan, J., Cyr, K. 1964.J. Biol. Chem. 239:418~85
80. Wyckoff, H. W., Handschumacher, M.D., Murthy, H. M. K., Sowadski, J. M.1983. Adv. Enzymol. Relat. Areas Mol.Biol. 55:453-80
81. Xu, X., Kantrowitz, E. R. 1991. Bio-chemistry 30:7789-96
82. Zeppezauer, M. 1986. See Ref. 7a, pp.417-34
Annual Reviewswww.annualreviews.org/aronline
Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.

Ann
u. R
ev. B
ioph
ys. B
iom
ol. S
truct
. 199
2.21
:441
-483
. Dow
nloa
ded
from
arjo
urna
ls.an
nual
revi
ews.o
rgby
Uni
vers
ity o
f Wisc
onsin
- Ea
u Cl
aire
(McI
ntyr
e Li
brar
y) o
n 11
/10/
06. F
or p
erso
nal u
se o
nly.





























