Embed Size (px)
Citation preview

THE JOURNAL OF B~OLOQICAL CHEMISTRY Vol. 242, No. 21,Ifisue of November 10, pp. 4987-4993.1967
Printed in U.S.A.
Studies on Succinate Dehydrogenase
XIII. REVERSIBLE ACTIVATION OF THE MAMMALIAN ENZYME*
(Received for publication, May 3, 1967)
TOKUJI KIMURA,~ J. HAUBER, AND THOMAS P. SINGER
Frown the Division of Molecular Biology, Veterans Administration Hospital, San Francisco, California 9Qf21, and the Department of Biochemistry, University of California School of Medicine, San Francisco, California 94122
SUMMARY
Mammalian succinate dehydrogenase exists in unactivated and activated states. Transition of the unactivated enzyme to the activated form appears to be a conformation change initiated by combination of the protein with substrates or competitive inhibitors and results in a many-fold increase in catalytic activity. This activation has now been shown to be reversible on removal of the bound activating agent by gel exclusion in the case of the soluble enzyme and by repeated washing and centrifugation or gel exclusion in the case of particles. While the activation has a very high energy of activation and does not proceed at observable rates at 15”, the deactivation is rapid even at temperatures near 0”. The ease of deactivation depends on the tightness of the binding of the activator. With tightly bound activators, such as malonate, replacement of the bound activator with succinate, followed by removal of the latter on Sephadex columns, has been found to be particularly effective. A modest degree of activation of succinate dehydrogenase occurs in particulate and soluble preparations also without added activating agents at 38’. This type of temperature activation may be due to the presence of traces of bound activator in the preparation. The activation-deactivation cycle has major effects on suc- cinoxidase activity and on all types of succinate dehydrogen- ase assays, but the fumarate-FMNHz reaction (fumarate reductase activity) is unchanged by these treatments. The spectral shifts accompanying the activation are not reversed by removal of the activator and consequent deactivation. It appears, however, that the colored enzyme-competitive inhibitor complexes form only with the fully activated en-
* This work was supported by Grants HE 01995 and HE 10027 from the National J%art Institute, Grants G 204571 and GB 3762 from the National Science Foundation, and Contract Nonr 5024 (00) between the Office of Naval Research and the Uni- versity of California. For the previous paper in this series see Reference 1.
i Present address, Department of Biochemistry, St. Paul’s University, Tokyo, Japan.
zyme, although the same competitive inhibitors combine with the unactivated form in a colorless complex.
The activation1 of succinate dehydrogenase, discovered by Kearney, Singer, and Zastrow (2) and Kearney (3), is a con- formational change in the protein initiated by the combination of the enzyme with succinate, fumarate, malonate, or any sub- stance capable of combining at the active center, which results in the transformation of the enzyme from a form of low (or no) catalytic activity to one of high activity. While the formation of enzyme-substrate and enzyme-competitive inhibitor com- plexes is very rapid, the activation is relatively slow and its rate is profoundly affected by temperature (activation energy = 35,000 cal mole-’ (3).
The phenomenon occurs in all types of soluble preparations of the heart enzyme, regardless of the method of isolation, in respiratory chain preparations, and in intact mitochondria (3-5). The increased catalytic activity consequent on incu- bation of the enzyme with an activator has been measured by the phenazine methosulfate assay, by oxidation via the complete respiratory chain, and by the use of various artificial electron acceptors. In general, the more nearly a given assay method registers the catalytic activity of the dehydrogenase, the more profound is the difference between inactivated and activated samples (3). Although the process has been studied intensively only in preparations from heart, its occurrence has been noted in mitochondria from a variety of animal tissues and, recently, also from aerobic yeast cells (6).
1 The terminology used in this paper is as follows. “Unacti- vated” refers to a preparation which has not been activated or which has been “deactivated” by removal of the activating agent under controlled conditions; “activated” refers to samples which have been incubated with an activating agent at temperatures of 20-38” until the activity no longer increased.
4987
by guest on January 9, 2020http://w
ww
.jbc.org/D
ownloaded from

4988 Xtudies on Xuccinate Dehydrogenase. XIII Vol. 242, No. 21
The studies of Kearney (3) were confirmed and extended by Thorn (4), who found that, in addition to the activation elicited by substrates and competitive inhibitors, the dehydrogenase is also activated to a lesser extent by merely raising the tem- perature and that this type of activation was readily reversed by cooling the preparation. Thorn suggested that the activation by substrates and that observed in the absence of substances capable of binding at the active center represent the same process.
In earlier studies (3), dialysis was the only practical method available for testing the reversibility of the activation of soluble preparations. Since the prolonged contact with succinate required in dialysis experiments leads to irreversible inactivation of the enzyme, dialysis of malonate-activated samples was used to test the reversibility. Under these conditions, the increased activity due to activation by malonate was not di- minished on dialysis and, hence, the activation appeared to be irreversible. Since malonate forms a very tight complex with the dehydrogenase, the possibility could not be eliminated (7) in experiments of this type that enzyme-bound malonate re- mained attached to the protein and was responsible for retaining the dehydrogenase in the activated state. Following the report of Thorn (4) on the reversibility of the temperature activation and with the advent of rapid and efficient methods for the separation of small solutes from protein (e.g. gel filtration), Kimura, Hauber, and Singer (5) re-examined the reversibility of activation by substrates and competitive inhibitors.
It was reported in a preliminary paper that Sephadex passage of soluble preparations or repeated centrifugation of particulate ones results in extensive deactivation of samples previously activated by succinate (5). Malonate-activated preparations were only partially deactivated by these treatments. De- activated preparations could be again fully activated by incu- bation with succinate or malonate. The present paper is a full account of these studies.
Another problem which has been explored in this study concerns the relation of the activation process to the spectral changes which accompany it. Kearney (3) found that, during the activation of soluble succinate dehydrogenase by malonate, changes in the absorption spectrum occurred, which suggested that the conformational changes caused by activation might affect the absorption characteristics of nonheme iron-protein bonds. Dervartanian and Veeger (8) recently proposed that the spectral changes elicited by malonate and other competitive inhibitors are due to the formation of enzyme-inhibitor complexes and are “explainable in terms of changes in polarity near the flavin prosthetic group.” Although no evidence was presented to support this view, activation was stated to be of secondary importance in this connection. It will be shown in the present paper that only the activated form of the enzyme exhibits spectral changes on combination with inhibitors and, hence, that activation is a prerequisite for the formation of spectro- photometrically detectable enzyme-inhibitor complexes and, further, that the combination of malonate at the active center does not in itself cause changes in the absorption spectrum of the enzyme.
EXPERIMENTAL PROCtiDURE
ETP2 and Keilin-Hartree preparations were made from bovine heart by procedures previously described (9). Soluble succinate
2 The abbreviations used are: DCI, 2,6-dichloroindophenol;
dehydrogenase was isolated either by the method of Bernath and Singer (10) or by the procedure of Wang, TSOU, and Wang (11) except for the omission of cyanide and with the precautions detailed in a previous paper (12). Covalently bound flavin was determined by the method of Singer, Hauber, and Kearney (13), fumarate reductase activity by the manometric FMNHz assay (lo), and succinate dehydrogenase activity by the PMS-DC1 method (14). In the dehydrogenase assay a recorder speed of 12 inches per min was routinely used at all concentrations of PMS and the log converter was adjusted to 1.0 absorbance unit for full deflection of the recorder. The accuracy of all activity determinations was within 2%.
In order to prevent activation during the preparation and assay of samples, spectrophotometer cells and Warburg vessels are filled at 0”, 0.067 M imidazole buffer (pH 7.60 at 0”) was substituted for phosphate in all assays, and activity was deter- mined at 15”.
Deactivation of batches of enzyme were performed by passage through a column of Sephadex G-25, which had a diameter- height ratio of 10, at a flow rate of approximately 20 ml per hour. The volume applied was 2 ml, which represented 20% of the total column volume. Activation of aliquots immediately prior to dehydrogenase assay (denoted as “assay activation”) involved the incubation of the complete reaction mixture, including succinate, but without dyes for 7 min3 at 38”. The cuvettes were then cooled3 to 15” and the reaction was started by the addition of DC1 and PMS. Samples assayed without activation were maintained for a similar length of time at 15”. In order to control activation by temperature alone, the assay mixture, less succinate and dyes, was kept for 7 min at 38” and was then cooled to 15”. The reaction was started by the suc- cessive addition of succinate, DCI, and of PMS.
Spectra were measured with a Cary model 11 spectropho- tometer and kinetic experiments on spectral changes were followed with a Gilford type recording spectrophotometer modified to permit 1% accuracy on a 0.05 absorbance scale, with a lag of <l set and instantaneous variation of chart speed in the range of 0.75 to 12 inches per min.
RESULTS
Reversibility of Activation by &cc&ate and Malonate-In particles as well as soluble, purified preparations, succinate dehydrogenase is usually present in a partially activated state. In order to assure that the enzyme is fully activated, two tech- niques are available: activation in batches and activation of the aliquot in the actual assay procedure.
Once activated, the enzyme remains in that state unless the activating agent is completely removed by passage through Sephadex G-25. In addition, particles can be more extensively deactivated by repeated washing and centrifugation. The batchwise activation and deactivation of particles and of samples of the soluble enzyme by succinate has already been documented in a preliminary note (5). Reversible activation by malonate
ETP, electron transport particle; PMS, N-methylphenazonium methysulfate (phenazine methosulfate).
3 Cuvettes were filled at 0”. It took about 3 min to reach 38”, and the cuvettes were then maintained an additional 4 min at 38”. These conditions assure full activation of dilute enzyme preparations in the presence of 20 mM succinate. The cuvettes were then transferred to a bath regulated at 15” and were main- tained there for 10 min in order to assure temperature equili- bration.
by guest on January 9, 2020http://w
ww
.jbc.org/D
ownloaded from

Issue of November 10, 1967 1’. Kimura, J. Hauber, and T. P. Singer 4989
TABLE I
Reversibility of activation by malonate
The ETP was a preparation which had been washed four times succinate for 20 min at 38” to exchange malonate for succinate, by centrifugation for 20 min at 144,000 X g in 0.25 M sucrose-3 mM and after cooling the sample was again centrifuged and passed imidazole (pH 7.6 at 0”)) and then passed through Sephadex G-25 through Sephadex G-25. In Experiments 5 to 7 the soluble prep- equilibrated with sucrose-imidazole (approximately 15 min). aration of Wang et al. (11) at the 0.55-saturated (NH&SOI precip- This was the material used in Experiment 1. Batch activation itate stage was passed through Sephadex G-25 at 0” and an ali- was in the presence of 5 mM malonate, pH 7.6, at 38” for 20 min quot (9.5 mg of protein per ml) was batch-activated with 2 mM under Nt at a protein concentration of 6.3 mg per ml. After cool- malonate at 25” for 15 min under Nt and then cooled to 0”. In ing to O”, the particles were collected by centrifugation and re- Experiment 7 this material was passed at 0” through a column of suspended in sucrose-imidaxole (Experiment 2). In Experiment Sephadex G-25 (equilibrated with 0.05 M imidazole, pH 7.6) to re- 3 this material was passed through a column of Sephadex G-25 at move malonate. Assays were performed at 15” by the standard O”, equilibrated with sucrose-imidazole, in order to remove bound PMS-DC1 method (14), except in the case of ETP in which imi- malonate and thus deactivate the enzyme. In Experiment 4 the dazole buffer, pH 7.6 (at 15”), was substituted for phosphate. sample from Experiment 3 was further incubated with 20 mM The values given are V,,, with respect to PMS.
Preparation
ETP ETP ETP ETP
Soluble dehydrogenase Soluble dehydrogenase Soluble dehydrogenase
Treatment
Specific activity at 15’
Not activated Activated with in assay”
Temperature succinate in assay6 activated in assayC
None 0.097 0.798 0.225 Malonate-activated 0.799d 0.799d 0.799d Same after gel exclusion 0.403 0.805 0.403 Same as Experiment 3 after incubation 0.039 0.644 0.099
with succinate and second gel exclu- sion
None 0.19 2.18 Malonate-activated 2.16d 2.16d Same after gel exclusion 0.90 2.19
5 Reaction mixture, less succinate and dyes, was equilibrated in spectrophotometer cuvette for 10 min at 15” and the reaction was started by adding succinate and dyes.
b Reaction mixture, including succinate but not dyes, was kept at 38” for 7 min and was then cooled at 15” for 10 min. Reaction was started with dyes.
c Reaction mixture without succinate or dyes was kept for 2 min at 38” and was then cooled as in the mixture not activated in assay.
d Corrected for the inhibition by the malonate carried over.
is shown in Table I. The column labeled “Not activated in assay” gives the apparent succinate dehydrogenase activity after the particular treatment. The column labeled “Activated with succinate in assay” refers to an identical aliquot which had been briefly incubated with succinate immediately before the start of the catalytic reaction. Since this treatment produces complete activation and full catalytic activity is measured, these samples serve as controls for any inactivation which may have occurred during the batch treatment. It may be noted that passage of the soluble enzyme and, occasionally, of particles (Table I) through Sephadex G-25 causes irreversible inactivation.
Reversal of the activation by succinate to the point where only 1 to 3qi’, of the activity remains is readily accomplished (5). Deactivation beyond this stage even by repeated passage through Sephadex and centrifugation has not been observed. The malonate-activated enzyme is much more difficult to de- activate extensively (Table I, Experiment 3), as might be expected from the fact that malonate is more tightly bound to the active center. This probably explains failure to reverse the activation by malonate in earlier experiments in which dialysis was used to reduce the malonate concentrations (3). Extensive deactivation of the malonate-activated enzyme may be ac- complished, however, with the use of succinate to displace the malonate and then removing the succinate on Sephadex (Table I, Experiment 4).
Tem-perature Activation-Thorn (4) has observed an activation
of succinate dehydrogenase in Keilin-Hartree preparations on incubation at 20-37” in the absence of added phosphate or succinate, although the degree of activation reached was con- siderably less than that obtained with phosphate. On cooling the preparation from 37” to 20”, a slow deactivation occurred.
The finding of Thorn that a modest degree of activation is attained on incubation at 38” in the absence of added substrate or competitive inhibitor has been confirmed (Tables I and II). The apparent thermal activation is quite rapid at 38’ and is complete within 2 min under the experimental conditions (Table III). The activation reached in the absence of succinate or malonate, however, was far less than the maximal activation attained in the presence of added substrates or competitive inhibitors (Tables I and II). It should be mentioned that no reversal of the thermal activation occurred in the brief period required for cooling the samples from 38” to 15”, the temperature of the assay, as judged by controls maintained and assayed at 38”.
I f the deactivation shown in Table I is the consequence of the dissociation and removal of the activating agent (succinate or malonate), as appears to be the simplest interpretation, it should be possible to examine the effect of temperature on the reversal of activation by succinate or malonate by varying the temperature at which chromatography on Sephadex is performed. Table III presents data on this point. It may be seen that major deactivation occurs both at 2” and at 25”, although enzyme
by guest on January 9, 2020http://w
ww
.jbc.org/D
ownloaded from

4990 Xtudies on Succinate Dehydrogenase. XIII Vol. 242, No. 21
ETP ETP
ETP
Keilin-Hartree Keilin-Hartree
Keilin-Hartree
_-
-
pecific activity
T.4BLE 11
Activation of succinate dehydrogenase at 38" without added activators
An ETP preparation (3 ml, 104 mg of protein) was deactivated by washing four times by centrifugation for 15 min at 144,000 X g in 20 ml of 0.25 M sucrose-2 mM imidazole, pH 7.4. The residue was resuspended in sucrose-imidazole to a protein concent,ration of 9.58 mg per ml. The Keilin-Hartree preparation was deacti- vated similarly except that the buffer used for washing was 0.05 M imidazole, pH 7.6 (at O”), and the residue was resuspended to a protein concentration of 21.3 mg per ml.
Treatment
Unactivateda Activated with succinate at 38”
for 7 minb Maintained at 38’
0.061 0.643
Without succinate for 2 mint Without succinate for 5 mint Without succinate for 10 mint Without succinate for 20 mint
Unactivateda Activated with succinate
At 38” for 4 minb At 38” for 12 minb
Maintained at 38”
0.127 0.126 0.124 0.120 0.082
0.380 0.380
Without succinate for 2 mint Without succinate for 4 mint Without succinate for 6 mine Without succinate for 20 mint
0.109 0.109 0.104 0.101
a Complete reaction mixture, less dyes, at 0” was placed in a bath at 15” for 10 min.
b Complete reaction mixture, less dyes, at 0” was placed in a bath at 38” and was maintained at that temperature for the periods stated, at which time it was transferred to a 15” bath and was incubated at that temperature for 10 min. It took 23 to 3 min to raise the temperature from 0 to 38”.
c Same as above, except that succinate was not present during the exposure to 38”.
TABLE III
Effect of temperature on deactivation process
Experimental conditions were as in Table I, except that the Keilin-Hartree preparation was activated at 25’ for 30 min in the presence of 80 mM succinate. Passage through a column on Sephadex G-25 wk performed either at 25’ or at 2”. Assay condi- tions were as in Table I.
Specific activity at 15’
Treatment
None.................................. After activation by succinate. Succinate activated, then Sephadex pas-
sage at25”.......................... Succinate activated, then Sephadex pas-
sage at2”...........................
Not activated Activated in assay in assay
0.176 0.325 0.343 0.386
0.033
0.151
0.390
0.392
is more extensively deactivated at the higher temperature under the experimental conditions. Thus, while activation proceeds at measurable rates only above 15”, deactivation by removal of the activating agent is observed also at 2”. Table III also shows that, when the temperature is held constant at 25” during
the cycle of activation and deactivation, extensive reversal of the activation still occurs. This observation merits emphasis, since Thorn (4) has suggested that the activation process may be primarily an equilibrium, governed by temperature, between active and inactive forms and that substances capable of com- bining at the active center may act merely by stabilizing the activated form of the protein. This proposal is summarized in the following scheme where EU and Ea represent, respec- tively, unactivated and activated forms and C a substrate or competitive inhibitor which combines with and stabilizes EA, the activated enzyme
EU =EA & ‘-c EAC
According to Thorn (4) the conversion of EA to EU would be promoted by lowering the temperature. The experiments of Table III are not readily reconciled with this scheme, however, since deactivation was more extensive when the temperature was held constant at 25” during activation and deactivation than when Sephadex passage was performed at 2”. These experiments seem to suggest that on complete removal of the activating agent the enzyme reverts to the deactivated (or unactivated) state regardless of temperature.
Although it is conceivable that activation by temperature alone and by substances combining at the active center are analogous but causally unrelated processes, possibly temperature activation is actually due to substrates or competitive inhibitors present in the enzyme preparation. Some support for this suggestion came from an experiment in which activation at 38” without added succinate was some 40% less if the particles were first incubated with succinate to displace any tightly bound endogenous activator and then excluded on Sephadex G-25 than in untreated samples.
E$ect of Activation on Fumarate Reductase Activity-Kearney (3) and Thorn (4) have emphasized that the activation of mam- malian succinate dehydrogenase is evident in all assays of the forward reaction and is by no means restricted to the PMS assay. In general, the more nearly a given assay measures the full activity of the dehydrogenase, the more pronounced is the difference in measured activity between activated and un- activated preparations.
It has been briefly noted (10) but not previously documented that the fumarate-FMNHS (fumarate reductase) activity of mammalian succinate dehydrogenase is unaffected by activation. Recently Thorn (4) observed a modest degree of activation of the fumarate-FMNHz reaction in Keilin-Hartree preparations. In view of this discrepancy the problem was reinvestigated. Table IV compares the activation of the succinate-PMS and fumarate-FMNHz reactions with ETP, Keilin-Hartree prepa- rations, and the soluble enzyme as test systems. It is seen t.hat while succinate dehydrogenase activity is increased many-fold in each instance by incubation with succinate or malonate, fumarate reductase activity, at V,,, with regard to FMNH2, remains essentially unaffected.
These observations are reminiscent of the effect of cyanide on succinate dehydrogenase (15). Incubation of the particulate enzyme with cyanide results in loss of succinate-PMS, succinate- methylene blue, and of fumarate-leucomethylviologen activities, but the fumarate-FMNHz reaction remains unaffected. As an explanation of this differential inactivation it was suggested (15) that cyanide might affect the nonheme iron moieties of the enzyme which therefore would be required in those catalytic
by guest on January 9, 2020http://w
ww
.jbc.org/D
ownloaded from
Issue of November 10, 1967 T. Kimura, J. Hauber, and T. P. Singer 4991
activities which are cyanide-sensitive, while the reaction between FMNHs and the bound flavin of the dehydrogenase may be a direct one, not requiring functional iron. Kearney (3) originally suggested that the conformation change in the transition of the unactivated enzyme to the active form may affect the region of nonheme iron in the protein. If the conformational change were limited to this region, the failure of the cyclic activation- deactivation process to influence reactivity with FMNHt might be explained, since it is probable that FMNHz reacts, indeed, directly with the flavin moiety of the dehydrogenase.
Relation of Activation to Spectral Changes-Kearney (3) reported that the addition of malonate to succinate dehydro- genase causes marked changes in the absorption spectrum, including the appearance of a new peak at 510 rnp. This ob- servation was confirmed and extended to other competitive inhibitors by Dervartanian and Veeger (8). Since the ap- pearance of the spectral shift accompanied activation of the enzyme, Kearney suggested that the spectral changes were a consequence of the activation process. Deactivation of the soluble enzyme under the conditions given in Table I does not reverse the spectral changes in the 500 to 510 rnp region which accompany activation of the enzyme by malonate. This raised the question as to what relation, if any, the activation process has to the spectral changes which accompany it.
The addition of malonate to the deactivated soluble enzyme at O-15” (where no activation occurs) caused no spectral change in the 500 to 510 rnp region. Since such samples have
TABLE IV
Effect of activation on the fumarate-FMNHS reaction
The ETP preparation was washed four times by centrifugation with 0.05 M imidazole buffer, pH 7.4, and was resuspended in the same buffer at a protein concentration of 19.8 mg per ml. For batch activation, a 5-ml aliquot was incubated with 25 ~1 of 1 M
malonate under Nz for 30 min at 38”. After cooling tb O”, aliquots of both the malonate-activated and unactivated samples were assayed without further activation. The Keilin-Hartree prepara- tion was similarly treated, except that the protein concentration during malonate activation was 24.9 mg per ml. The preparation of Wang et al. (11) was the gel eluate, precipitated with 0.55- saturated (NHJSOt and desalted on Sephadex G-25 (equilibrated with 0.02 M imidazole buffer, pH 7.5 at 0”). A 1.56-ml aliquot (8.55 mg of protein per ml) was incubated with 0.04 ml of 0.2 M
succinate, pH 7.6, for 15 min at 25” for batch activation. In the fumarate reductase assay imidazole buffer was substituted for phosphate. Activities are corrected for inhibition by residual malonate, where present.
ETP, washed four times.. Same, activated by 5 mM mal-
onate....................... Keilin-Hartree, washed four
times. . . Same, activated by 2 mM mal-
onate....................... Soluble enzyme, Wang et al.
(11) preparation, . Same, activated by 5 mM suc-
cinat(e . . . . . .
Temperature of assay
25”
25”
25”
25”
15”
15”
Specific activity
Succc;te- Fkmu&te-
reaction reactiol: ~___
0.100 0.032
0.942 0.036
0.124 0.023
0.404 0.018
0.123 0.022
1.31 0.022
; 0.008 - 5 t
i: 0.006 -
-1 9 0 0.004 - B u E NO MALONATE
1 I I I I 100 200 300
VbJ FUMARATE
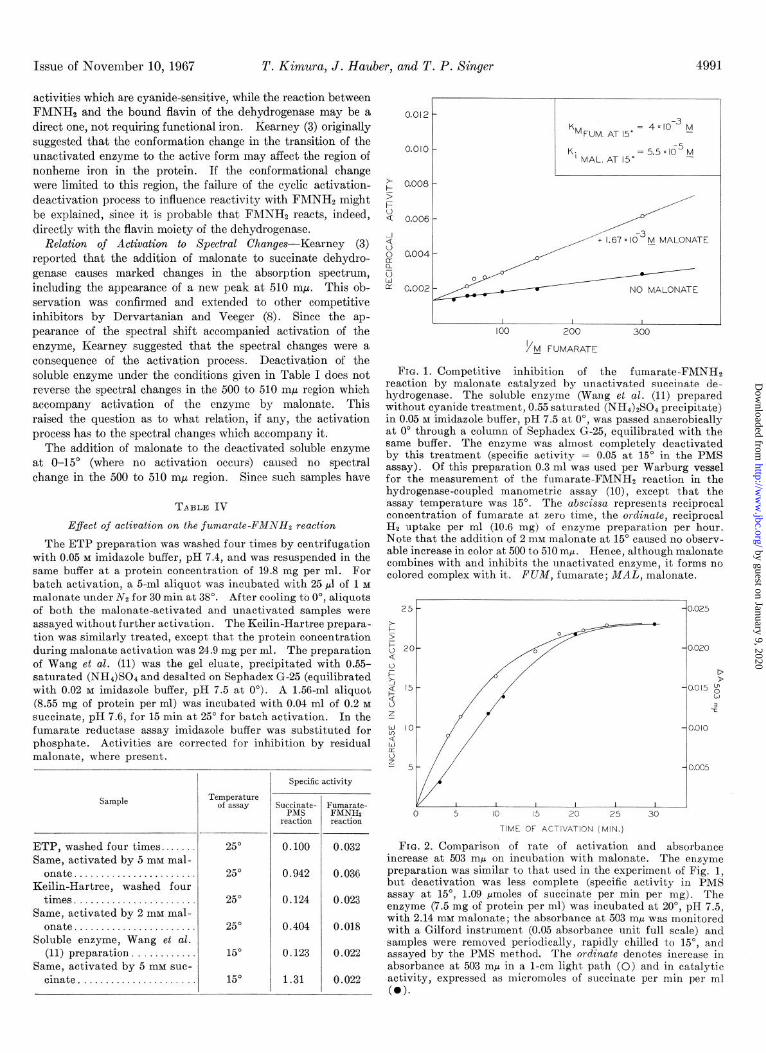
FIG. 1. Competitive inhibition of the fumarate-FMNHS reaction by malonate catalyzed by unactivated succinate de- hydrogenase. The soluble enzyme (Wang et al. (11) prepared without cyanide treatment, 0.55 saturated (NH&S04 precipitate) in 0.05 M imidazole buffer, pH 7.5 at O”, was passed anaerobically at 0” through a column of Sephadex G-25, equilibrated with the same buffer. The enzyme was almost completely deactivated by this treatment (specific activity = 0.05 at 15” in the PMS assay). Of this preparation 0.3 ml was used per Warburg vessel for the measurement of the fumarate-FMNHS reaction in the hydrogenase-coupled manometric assay (lo), except that the assay temperature was 15”. The abscissa represents reciprocal concentration of fumarate at zero time, the ordinate, reciprocal Hz uptake per ml (10.6 mg) of enzyme preparation per hour. Note that the addition of 2 mM malonate at 15” caused no observ- able increase in color at 500 to 510 mp. Hence, although malonate combines with and inhibits the nnactivated enzyme, it forms no colored complex with it. FUM, fumarate; MAL, malonate.
25t -(0.025
/ 0.020
I ?
0015 g w
t3
0.010
0.005
0 5 IO 15 20 25 30
TIME OF ACTIVATION (MIN.)
FIG. 2. Comparison of rate of activation and absorbance increase at 503 mp on incubation with malonate. The enzyme preparation was similar to that used in the experiment of Fig. 1, but deactivation was less complete (specific activity in PMS assay at 15”, 1.09 Mmoles of succinate per min per mg). The enzyme (7.5 mg of protein per ml) was incubated at 20”, pH 7.5, with 2.14 mM malonate; the absorbance at 503 rnr was monitored with a Gilford instrument (0.05 absorbance unit full scale) and samples were removed periodically, rapidly chilled to 15”, and assayed by the PMS met’hod. The ordinate denotes increase in absorbance at 503 rnp in a l-cm light path (0) and in catalytic activity, expressed as micromoles of succinate per min per ml (0).
by guest on January 9, 2020http://w
ww
.jbc.org/D
ownloaded from

Studies on Xuccinate Dehydrogenase. XIII Vol. 242, No. 21
TABLE V Activation of succinate dehydrogenase in bovine heart mitochondria
Mitochondria were isolated as previously described (10). Washing was performed by homogenization (glass-Teflon homog- enizer) in 0.25 M sucrose-10 mM phosphate, pH 7.4, and sedimenta- tion for 15 min at 144,000 X g. Batch activation was carried out at 38” for 15 min under Nz in 80 mM succinate. Succinate dehy- drogenase assays were according to Arrigoni and Singer (14) and included 9 mM n-cysteinesulfinate to provide continuous removal of oxalacetate by transamination, except that 0.05 M imidazole, pH 7.6 (at 0”), was used in lieu of phosphate. In order to over- come the permeability barrier to PMS, 0.75 mM Ca++ was present during the assay. Assays with and without activation by suc- cinate were performed as in Tables I and II.
Specific activity at 15’
Mitochondria treatment Not activated Activated by
succinate in assay
Unwashed............................ 0.078 0.42 Washed once with sucrose-phosphate.. 0.045 0.38 Washed five times with sucrose-phos-
phate.............................. 0.017 0.45 Unwashed, batch activated with 80 mM
succinate........................... 0.42 0.43 Washed five times, activated with suc-
cinate.............................. 0.40 0.46
full fumarate reductase activity, as noted in the previous section, it was possible to show that malonate nevertheless combines with the enzyme in the usual enzyme-inhibitor complex under these conditions (Fig. 1). When the unactivated enzyme- malonate complex was incubated at 20” and both increase in succinate dehydrogenase activity and in absorbance at 503 rnE.1 were monitored, a fair degree of parallelism was observed (Fig. 2). Further, when the dependence of both processes on malonate concentration was studied with other factors held constant,4 half-maximal activation occurred at 0.14 mM malonate, while half-maximal color development required 0.13 mM malonate. These observations suggest that activation is a prerequisite for the appearance of the spectral shifts; i.e. that only the acti- vated form of the enzyme forms colored complexes of this type with competitive inhibitors.
DISCUSSION
The first part of this discussion will consider generalizations concerning the occurrence, possible physiological role, and prac- tical importance of the activation process. Activation of succinate dehydrogenase has been observed in a variety of rat, beef, and pig tissues and in yeast mitochondria (3, 4, 16, 17). Thus, it appears to be a general characteristic of the aerobic type or mitochondrial succinate dehydrogenase (6). The anaerobic type of enzyme, or fumarate reductase, from obligate anaerobes (18) and yeast cytoplasm (16) does not seem to require activation.
The most interesting aspect of the phenomenon is undoubtedly its possible physiological significance. In this connection the
4 The preparation was that of Wang et al. (11) modified according to Kimura et al. (12). It was used after the (NHJzS04 precipita- tion. The enzyme (30 PM) was incubated under Nz for 30 min at 21”. Dehydrogenase activity was measured at 15”, increase in ab- sorbance at 505 rnp at 0”.
first question which arises is in what state the enzyme exists in mitochondria. Experiments in which bovine heart mitochondria were isolated and immediately assayed prior to any washing procedure which might remove bound activator always showed the enzyme to be only partly in the activated state (Table V). Repeated washing by centrifugation in isotonic sucrose caused further deactivation. Similar data have been obtained in polarographic assays of succinoxidase activity in yeast mito- chondria, as documented elsewhere (16).
It seems entirely possible that only that fraction of the dehy- drogenase which has substrate or competitive inhibitor attached to its active center is at any moment in the activated state. The hypothetical bound activator is not likely to be succinate or fumarate, since these substances are readily removed by sedi- mentation of particles, while the substance which appears to be bound to the enzyme in heart muscle particles is removed only with difficulty.
Whatever the physiological role of the activation process may be, it has manifest practical importance in the measurement of succinate oxidation. Thorn (4) has emphasized and docu- mented that ignoring the activation process in the assay of succinoxidase is conducive to misleading results. Actually, both the manometric and spectrophotometric methods used by Thorn involved relatively long reaction periods, permitting activation to take place. In more rapid procedures, such as stopped-flow methods at relatively low tempertaure or EPR procedures5 in conjunction with rapid freezing techniques, the difference between true rate after activation and that observed without activation may be many times greater than reported by Thorn.
Despite emphasis of the importance of activation in assays of the enzyme by Thorn (4) and others (5, 10, 17), in many current papers dealing with this enzyme no provisions are made for activation. One reason for this may be erroneous impres- sions that certain types of preparation of the enzyme are acti- vatable, others are not, or that some preparations are activated by succinate as well as phosphate, others only by succinate (11, 19, 20). It is desirable to examine these reports in order to clarify the situation. Kearney (3) has shown that all prepa- rations examined are activated by phosphate, succinate, and malonate by the same mechanism: the maximal activation reached was the same, although activation by phosphate was markedly less efficient than by malonate or succinate. Subse- quently, Wang et al. (11, 21) confirmed activation of the enzyme by succinate in their preparation (isolated from pig heart) but suggested that the effect of phosphate was one of removing trace metals, since with their preparation phosphate and EDTA in- creased the activity to similar extent. This lead to the assump- tion (19) that the preparations of Wang et al. (11) and of Singer et al. (22) behave dissimilarly. More recently, it has been stated (8) that the former is already fully activated since it is prepared in the presence of succinate.
Thorn (4) has found that particles from pig heart are activated by phosphate in the same manner as those from bovine heart. In this laboratory no activation of the soluble or particulate
6 The theoretical problems posed by the activation process in the measurement of the rate of appearance of EPR signals at (I = 1.94 have been discussed in a recent review (17). In this con- nection. Dr. H. Beinert has kindlv examined samnles of unacti- vated and fully activated ETP and found that aciivation per se does not give rise to an observable EPR signal at g = 1.94.
by guest on January 9, 2020http://w
ww
.jbc.org/D
ownloaded from

Issue of November 10, 1967 T. Kimura, J. Hauber, and T. P. Singer 4993
enzyme from bovine or pig heart by EDTA has been observed, possibly because rigorous care was taken to remove trace metals from all reagents. On the other hand, activation by both phosphate and succinate have been confirmed with the original preparation of Wang et al. (11) and with modifications thereof, at all stages beyond the first (NH4)804 precipitation, provided that (NH&SO( was removed prior to assay either by gel fil- tration or brief dialysis.6 Hence, there appear to be no grounds for assuming basic differences among soluble preparations in regard to activability.
As to the mechanism of the activation, the present studies suggest a compromise between the earlier scheme of Kearney (3) and that of Thorn (4). Using Thorn’s terminology, pre- sentfed earlier in this paper, the events may be visualized as proceeding by the initial rapid combination of unactivated enzyme and activator, followed by a slower reversible conversion of the complex to the activated form. In the direction of activation it would have a high energy barrier (3, 4) and would rapidly revert. to the unactivated form on dissociation and removal of the activating agent, which may stabilize the acti- vated state. The conversion is apparently a conformation change in the protein, but one which does not seem to influence the flavin site directly, since the fumarate-FMNH2 reaction remains unaffected.
Eo + C = EuC = EAC
Although the structure of the colored complexes formed between activated succinate dehydrogenase and competitive inhibitors remains somewhat obscure, the experiments presented clearly show that only the activated form of the enzyme is capable of forming complexes of this type, although the unacti- vated form binds, and is inhibited by, competitive inhibitors. This interpretation is in essential agreement with the views of Dervartanian and Veeger (8), although they state that their views differ fundamentally from those of the present authors. If there is a difference in interpretation, it would concern the structure of the colored enzyme-inhibitor complex. If it is, indeed, a charge-transfer complex, as has been suggested (8), it is clear that this complex differs in structure from that which
B At the gel eluate stage, the enzyme is still nearly fully acti- vated. As expected, it is the gel filtration step which removes the bound succinate. We have discussed elsewhere (17) why under the particular assay conditions used by Wang et al. (11, 21) the slow activation by phosphate would have escaped detection.
is initially formed with the unactivated enzyme, and its for- mation may hinge on conformation changes during activation.
Acknowledgment-The authors are grateful to Dr. Edna B. Kearney for her advice throughout this study.
REFERENCES 1. KEARNEY, E. B., J. Biol. Chem., 236, 865 (1960). 2. KEARNEY, E. B.., SINGER, T. P., AND ZASTROW, N., Arch.
Biochem. Biovhus.. 66, 579 (1955). 3. KEARNEY, E. B., 3. sioi. Chem., 2i9, 363 (1957). 4. THORN, M. B., Biochem. J., 86, 116 (1962). 5. KIMURA, T., HAUBER, J., AND SINGER, T. P., Biochem. Biophys.
Res. Commun., 11, 83 (1963). 6. SINGER, T. P., in T. E. KING, H. S. MASON, AND M. MORRISON
(Editors), International symposium on oxidases and related oxidation-reduction systems, Vol. 1, John Wiley and Sons, New York, 1965, p. 448.
7. KEARNEY, E. B., in K. ICHIHARA (Editor), Proceedings of the international symposium on enzyme chemistry, Tokyo and Kyoto, 1967, M&&en, Tokyo, 1958, p. 340. -’ _
8. DERVARTANIAN. D. V.. AND VEEGER. C.. Biochim. Biovhus. Acta, 92, 233 11964). ’
, , 1 ”
9. RINGLER, R. L., MINAKAMI, S., AND SINGER, T. P., J. Biol. Chem., 238, 801 (1963).
10. BERNATH, P., AND SINGER, T. P., in S. P. COLOWICK AND-N. 0. KAPLAN (Editors), Methods in enzymology, Vol. 6, Academic Press, New York, 1962, p. 597.
11. WANG, T. Y., Tsou, C. L., AND WANG, Y. L., Sci. Sinica (Peking), 6, 73 (1956).
12. KIMURA, T., HAUBER, J., AND SINGER, T. P., Nature, 198, 362 (1963).
13. SINGER, T. P., HAUBER, J., AND KEARNEY, E. B., Biochem. Biophys. Res. Commun., 9, 146 (1962).
14. ARRIGONI, O., AND SINGER, T. P., Nature, 193, 1256 (1962). 15. GIUDITTA, A., AND SINGER, T. P., J. Biol. Chem., 234, 666
(1958). 16. SINGER, T. P., ROCCA, E., AND KEARNEY, E. B., in E. C.
SLATER (Editor), International symposium on Jlavins and Jlavoproteins, American Elsevier Publishing Company, Inc., New York, 1966, p. 391.
17. SINGER, T. e., in’&f. FLORKIN AND E. H. STOTZ (Editors), Comprehensive biochemistry, Vol. 14, American Elsevier Publishing Company, Inc., New York, 1966, p. 127.
18. WARRINGA, M. G. P. J., SMITH, 0. H., GIUDITTA, A., AND SINGER. T. P.. J. Biol. Chem.. 230. 97 11958).
19. KING, T. ‘E., B&him. Biophys.‘A&, 47,‘4%i1961). 20. MCDONALD, J. K., ANDERSON, J. A., CHELDELIN, V. H., AND
KING, T. E., Biochim. Biophys. Acta, 73, 533 (1963). 21. WANG, T. Y., Tsou, C. L., AND WANG, Y. L., Sci. Sinica
(Peking), 7, 65 (1958). 22. SINGER, T. P., KEARNEY, E. B., AND BERNATH, P., J. Biol.
Chem., 223, 599 (1956).
by guest on January 9, 2020http://w
ww
.jbc.org/D
ownloaded from

Tokuji Kimura, J. Hauber and Thomas P. SingerTHE MAMMALIAN ENZYME
Studies on Succinate Dehydrogenase: XIII. REVERSIBLE ACTIVATION OF
1967, 242:4987-4993.J. Biol. Chem.
http://www.jbc.org/content/242/21/4987Access the most updated version of this article at
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/242/21/4987.full.html#ref-list-1
This article cites 0 references, 0 of which can be accessed free at
by guest on January 9, 2020http://w
ww
.jbc.org/D
ownloaded from

















![Succinate Dehydrogenase-a Comparative Review · Membrane-bound succinate dehydrogenase [SDH; E.C.1.3.99.1 succinate:(acceptor) oxido-reductase] is present in all aerobic cells. Ever](https://img.pdfslide.net/doc/110x75/5e54371d86904d694572eef0/succinate-dehydrogenase-a-comparative-review-membrane-bound-succinate-dehydrogenase.jpg)