Embed Size (px)
Citation preview

Synthesis and rearrangement of cyclononadienyllithium
Item Type text; Thesis-Reproduction (electronic)
Authors McCulloch, Charles Schindel, 1946-
Publisher The University of Arizona.
Rights Copyright © is held by the author. Digital access to this materialis made possible by the University Libraries, University of Arizona.Further transmission, reproduction or presentation (such aspublic display or performance) of protected items is prohibitedexcept with permission of the author.
Download date 23/05/2018 01:25:37
Link to Item http://hdl.handle.net/10150/348204

SYNTHESIS AND REARRANGEMENT OFCYCLONONADIENYLLITHIUM/ - ...
byCharles Schindel McCulloch
A Thesis Submitted to the Faculty of theDEPARTMENT OF CHEMISTRY
In Partial Fulfillment of the Requirements For the Degree of
, MASTER OF SCIENCEIn the Graduate CollegeTHE UNIVERSITY OF ARIZONA
19 7 7

STATEMENT BY AUTHOR
This thesis has been submitted in partial fulfillment of requirements for an advanced degree at The University of Arizona and is deposited in the University Library to be made available to borrowers under rules of the Library.
Brief quotations from this thesis are allowable without special permission, provided that accurate acknowledgment of source is made. Requests for permission for extended quotation from or reproduction of this manuscript in whole or in part may be granted by the head of the major department or the Dean of the Graduate College when in his judgment the proposed use of the material is in the interests of scholarship. In all other instances, however, permission must be obtained from the author.
APPROVAL BY THESIS DIRECTOR This thesis has been approved on the date shown below:
R. B. Bates . DateProfessor of Chemistry

ACKNOWLEDGMENTS
The author wishes to acknowledge Dr. R. B. Bates for his suggestions and guidance throughout the preparation of this work and Mrs. Ann Yabusaki for the final typing of the manuscript.
iii

TABLE OF CONTENTS
' PageLIST OF ILLUSTRATIONS ....... . . . . . vLIST OF TABLES .......... viABSTRACT ... ........ . . . . . . . . . . . . . . . . . . . . . viiINTRODUCTION ............... . . . 1EXPERIMENTAL . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
1,4-Cyclononadiene (IV)......................... 41,3-Cyclononadiene and 1,5-Cyclononadiene. ................ 5Cyclononadienyllithium (V) ........ 5Protonation of Cyclononadienyllithium (V).................. 5cisf Bicyclo [4.3.0] non- 8 - ene (VII) .......... 8Bicyclo [4.3.0] non-1 (9) - ene (IX),
Bicyclo [4.3.0]non-1(2)-ene (X),and Bicyclo[4.3.0]non-1(6)-ene (XI) ....... 8
Equilibration of Isomers VII, IX, X, XI . . . . . . 9Photolysis of V ............. . 9
DISCUSSION......................... 13Nmr Studies of Cyclononadienyllithium (V) .......... 13Rearrangement of Cyclononadienyllithium (V). . ..............17
LIST OF REFERENCES........... 25
iv

LIST OF ILLUSTRATIONS
Figure Page1. Nmr spectrum of 1,4-cyclononadiene . ......... 62. Nmr spectrum of 1,3-cyclononadiene ..... 63. Nmr spectrum of 1,5-cyclononadiene . .............. . . . 74. Nmr spectrum of cyclononadienyllithium .......... 75. Nmr spectrum of bicyclo [4.3.0]non-8-ene...................106. Nmr spectrum of bicyclo [4.3.0] non-1 (9) - ene................. 107. Nmr spectrum of bicyclo[4.3.0]non-1(2)-ene . . . . . . . . . . 118 . Nmr spectrum of bicyclo [4.3.0] non-1 (6) - ene . . ............ 119. Complete isomerization sequence resulting from thermal
cyclization of anion V . . ........ . . 2010. Spin decoupled spectra of anion V ............. 23
v

LIST OF TABLES
Table Page1. Nmr data for some cyclic pentadienyl anions
in hexane................ 142. Diene yields from protonation of cyclic pentadienyl
anions . .................... . . . . . . . . . . . . 18
vi

ABSTRACT
The synthesis of the lithium salt of 1,4-cyclononadiene is reported. Nuclear magnetic resonance studies of the distribution of negative charge in cyclononadieny 11 ithium showed unexpectedly a considerable variation from the distribution in cyclooctadienyl1ithium.
The disrotatory cyclization of cis,cis-cyclononadienyllithium and subsequent protonation resulted in cis-bicyclo[4.5.0]non-8-ene aspredicted by Woodward and Hoffmann, cis - Bicyclo [4.5.0] non- 8 - ene was
oobserved to rearrange at 120 in the presence of strong base to an equilibrium mixture of bicyclo[4.3.0]nonene isomers, which contained 97% bicyclo[4.3.0]non-1(6)-ene, 3% bicyclo[4.3.0]non-l(2) - ene, and trace amounts (>.3%) bicyclo[4.3.0]non-1(9)-ene.

INTRODUCTION
The preparation and characteristics of cycloheptadienyllithium (I) and cyclooctadienyllithium (II) have previously been studied (McCombs 1969). Both can be readily prepared by proton abstraction from the respective 1,4 diene with butyllithium and tetrahydrofuran. McCombs
(I) (II) (III)
found that while the cyclooctadienyl anion (II) cyclized on standing at 35° to cis-bicyclo[3.3.0]oct-2-ene (III), the cycloheptadienyl anion (I) was stable up to 100° with respect to cyclization, showing only a slight tendency to decompose. Furthermore, the fact that II is stable at -78° in sunlight for long periods indicates the thermal nature of the cyclization. It was the purpose of this work to prepare and characterize cyclononadienyllithium (V) and study its thermal and photochemical reactions .
(IV) (V)
1

2It seemed much more likely that V could be cyclized photochemi-
cally to products with trans ring fusions (i.e., VIb and Vllb) than II, since while trans-fused 5,5-ring systems exhibit considerable strain, trans-fused 6,5-ring systems are less strained.
of 1,3-, 1,4-, and 1,5-cyclononadienes has been observed (Devaprabha- kara, Cardenas, and Gardner 1963). This method was used to prepare1,4-cyclononadiene (IV) which was subsequently metalated to cyclonon- adienyllithium (V). Devaprabhakara et al., reported strong evidence that both olefinic bonds in IV were cis. If IV formed by this isomerization were in fact composed predominantly of a single stereoisomer, then the stereochemistry of the cyclized product(s) could be predicted, assuming V were to cyclize.
(Woodward and Hoffmann 1965), if the 6ire cyclononadienyl anion (V) were formed from cis,cis 1̂,4-cyclononadiene (IV) then thermal cyclization should occur by a disrotatory pathway to cis-bicyclononadienyl anion
Base-catalyzed isomerization of 1,2-cyclononadiene to a mixture
(V) (Via) (Vila) (Villa)
(VIb) (VI lb) (VUIb)
Assuming Woodward-Hoffmann rules for electrocyclic reactions

(Via). Photochemical cyclization should proceed by a conrotatory pathway to trans-bicyclononadienyl anion (VTb). Protonation of the anions would give the corresponding cis (Vila) and trans (VI lb) bicyclo[4.3.0]non-8-enes which could be hydrogenated and compared with the commercially available isomeric hydrindanes (Villa) and (VIIlb).If the anions Via and VIb were sufficiently stable to be observed, as was the case with the bicyclooctadienyl anion, then these could also be characterized.

EXPERIMENTAL
All gas chromatography was done on a Varian Aerograph Model 90-P with a Model 20 recorder. Nmr spectra were obtained on a Varian T-60 nmr spectrometer at 60MHz with TMS as an internal standard. Mass spectral data were obtained on a Hitachi-Perkin Elmer RMU-6E Double Focusing Mass Spectrometer. Photochemical studies were done with a Hanovia Medium Pressure Mercury lamp.
1,4-Cyclononadiene (IV)The procedure of Devaprabhakara et al. (1963) was used. Into a
100 ml round-bottomed flask containing a magnetic stirring bar andfitted with a reflux condenser and drying tube were placed 2.7 g (0.024mole) of potassium t-butoxide, 24 ml of dimethyl sulfoxide, and 7 ml(0.05 mole) of commercial (Willowbrook Laboratories) 1,2-cyclononadiene.
oThe. solution was placed in an oil bath and heated at 70 with stirring for 3-1/2 hr. The crude mixture was washed with 100 ml of 1 N sodium bisulfate and two 100 ml portions of distilled water and dried over calcium chloride. The dried mixture was then distilled and the fraction boiling at 164-171° collected. Nmr analysis of the distillate indicated approximately 30% 1,4-cyclononadiene (IV) as evidenced by the methylene absorption at x7.2. Pure IV was separated from the distillate by gas chromatography on a 10 ft column of 20% Carbowax 20 M on non-acid- washed 30/60 Chromosorb P. Of the three components of the mixture,1,3-, 1,4-, and 1,5-cyclononadienes, 1,4-cyclononadiene (IV) was the

third collected. The structure was confirmed by nmr spectrometry (Fig. 1). The boiling point of IV was 171-172°.
1,3-Cyclononadiene and 1,5-Cyclononadiene1,3-Cyclononadiene and 1,5-Cyclononadiene were prepared for
reference purposes by the method described above. Reaction times were 2 hr and 30 hr, respectively. Products were identified by nmr (Figs. 2 and 3). Of the three components of the isomerized mixture collected by gas chromatography, 1,3-cyclononadiene and 1,5-cyclononadiene were the first and second, respectively.
Cyclononadienyllithium (V)Commercial (Alfa Products) n-butyllithium in hexane (0.175 ml,_ c7.8 x 10 mole) was added through a serum cap to 1,4-cyclononadiene
- 4(IV, 0.075 ml, 5.5 x 10 mole) and tetramethylethylenediamine (0.085 ml) in an nmr tube which had previously been heated at 140° to remove adsorbed moisture and flushed with argon. This mixture was heated in an oil bath at 50° for 4 hr. Formation of the anion, which was dark red, was monitored by nmr (Fig. 4). Addition of larger amounts of tetramethylethylenediamine caused faster formation of the anion, but also caused separation of the mixture into two layers with the anion on the bottom.
Protonation of Cyclononadienyllithium (V)The anion was prepared as described above. The nmr tube was
then placed in an ice bath and distilled water added slowly. The top layer of the two phase system was then analyzed by gas chromatography

6
Fig. 1. Nmr spectrum of 1,4-cyclononadiene.
«*-, 1 • • 1 " 1 , 1 , 500 *00
: O:!
j
1!
! u-------- ----- 1................. r ' ( ......... 1 " 1" ' T ........1 ' 1 .........., ■ 1 1 '300 200 100 0 Mi
. >'•*
: i r
jA i1 1 1 ■' I r V r rr1— . r l % \ f . ' I v l : d-:L : I v . . . l . : 1: : . . n 1 -
Fig. 2. Nmr spectrum of 1,3-cyclononadiene.

7
IMP
Fig. 3. Nmr spectrum of 1,5-cyclononadiene.
Fig. 4. Nmr spectrum of cyclononadienyllithium.

utilizing the previously described Carbowax column. The two components collected were identified by the nmr spectra and were, respectively,1,3-cyclononadiene (68%) and 1,4-cyclononadiene (IV 32%).
cis-Bicyclo[4.3.0]non-8-ene (VII)The anion (V) was prepared as previously described. The nmr
tube was then immersed to one-half its length in liquid nitrogen andsealed at the top with a gas/oxygen torch. Care was needed at this point to prevent air from entering the tube as a result of reduced internal pressure caused by the low temperature. The sealed tube was then heated in an oil bath at 120° for 1-1/2 hr. At the end of thistime the tube was cooled in an ice bath, the top cut off and distilledwater added slowly. The upper phase of the two phase system was inspected by gas chromatography. The alkene Vila was the second component collected and was identified by mass and nmr spectrometry (Fig. 5). Hydrogenation of (VII, 0.005 ml) in hexane at atmospheric pressure and 0° using a 5% Pd on barium sulfate catalyst yielded 97% cis-hydrindane as analyzed by gas chromatography, cis-Hydrindane was identified by . comparison of retention time with a known sample (Chemical Samples Co.).
Bicyclo [4.3.0]non-1(9)- ene (IX) ,Bicyclo[4.3.0]non-1(2)- ene (X)
and Bicyclo[4.3.0]non-1(6)-ene (XI)The anion V was prepared as previously described and the tube
sealed as in the preparation of VII. Heating the anion at 120° formed VII as previously described and VII isomerized under the reaction conditions to IX, X/ and XI, see Fig. 9, page 20. The isomerization was

stopped after 3, 7-1/2, and 22 hr, respectively, for IX, X, and XI. by the same procedure used for VII. The products were collected by gas chromatography (on the Carbowax 20 M column previously described) and their relative retention times increased in the order IX, VII, X, and XI. Samples were identified by nmr and mass spectrometry (Figs. 6, 7, 8).
Equilibration of Isomers VII, IX, X, XIThis experiment was done starting with two different isomers in
order to make a more accurate assumption regarding the approximation of an equilibrium mixture of isomers. Into each of two m r tubes commercial (Alfa Products) n-butyllithium in hexane (0.070 ml) and tetra- methylethylenediamine (0.040 ml) was added. Into one tube an additional (0.025 ml) of VII was added and into the other (0.025 ml) of XI was. added. The tubes were sealed as mentioned previously and heated at 120° in an oil bath for 258 hr. At the end of this time, both were cooled, the tops of the nmr tubes cut off and distilled water added. The top phase of each was examined by gas chromatography and identical mixtures of 97% XI, 3% X, and a trace of IX were observed. Isomers were identified by comparison with previously prepared samples.
Photolysis of VThe anion was again prepared as described above, with the excep
tion that a quartz tube instead of an nmr tube was used. The tube was then subjected to ultraviolet radiation from a water cooled medium pressure mercury lamp for 240 hr. During this time the temperature in the lamp compartment was kept belqw 40° to prevent thermal cyclization.

10
Fig. 5. Nmr spectrum of bicyclo[4.3.0]non-8-ene.
Fig. 6. Nmr spectrum of bicyclo[4.3.0]non-l(9)-ene.

11
Fig. 7. hftnr spectrum of bicyclo[4.3.0]non-l(2)-ene.
V1MV,wVVVrVFm'T^rrT^wrrr, i *** V rVr̂ s***^-;vsVy^.V(vv^^^ir̂
Fig. 8. Nmr spectrum of bicyclo[4.3.0]non-l(6)-ene.

12After irradiation the tube was cooled in an ice bath and distilled water added carefully. The top layer of the two phase system was examined by gas chromatography. It was found that the major alkenes present were 1,3- and 1,4-cyclononadiene (IV) from protonation of the cyclic anion. Very small amounts of X and XI (~1% each) were also observed. Samples were identified by comparison of retention times with known compounds.

DISCUSSION
The starting diene IV was prepared by base-catalyzed isomerization of 1,2-cyclononadiene, but in slightly better yield than previously reported. Nmr analysis of the crude mixture indicated 30% of IV as opposed to 27% previously reported. Carbon magnetic resonance indicated, five absorptions in the ratio 2:2:2:2:1 for diene IV, which is in agreement with both the cis,cis and trans,trans structures, each of which has a plane of symmetry. The proposed configuration of IV is cis,cis on the basis of angle strain and earlier spectral observations (Devaprabhakara, Cardenas, and Gardner 1963). Following this, it is proposed that the structure of the anion V is cis,cis, with which m r data appear to correlate (see below).
Nmr Studies of Cyclononadienyllithium (V): The nmr spectrum of cyclononadienyllithium (V) consisted of four
distinguishable groups of signals as indicated in Table 1; the signals for the other two methylene groups were obscured by. solvent. Anion V was determined to be roughly U-shaped by the coupling constants Jab and Jbc as compared to those of 6,6-dimethylcyclohexadienyllithium (Bates, Gosselink, and Kaczinski 1967). There is, however, a striking difference in the chemical shifts of the end and center protons in the two systems. Whereas in 6,6-dimethylcyclohexadienyllithium the end and center protons absorb at t6.6 and 6.1, respectively, those in V are found at t6.16 and 7.15. This suggests an increase in the amount of double
13

14
Table 1. Nmr data for some cyclic pentadienyl anions in hexane.
Chemical CouplingShift (t) Constant (Hz) Anion
Ha 6.1 Hb 4.1 He 6.6
Jab 6.4 Jbe 7.5 Jac 1.7
He (Bates, Goss el ink 11 © I] and Kaczinski
Hb IS*?)Ha
Ha 6.9 Hb 4.4 He 6.2 Hd 7.6
Jab 7 Jbe 10 Jed 5 Jac 0
Hd
U ^ © y V Hc I (McCombs 1969)IT Hb Ha
Ha 7.7 Hb 4.4 He 7.1 Hd 7.3
Jab 6.8 Jbe 9.0 Jed — Jac 0
^ ^ H d
(\ /)— He II (McCombs X X y / 1969)
Ha
Ha 7.15 Hb 4.27 He 6.16 Hd 7.90
Jab 8 Jbe 12 Jed 7.5 Jac 0
y-- X Hd
(vO/y-'Hc v] ^ H bHa

15
bond character at the ends of the pentadienyl system for anion V compared with 6,6-dimethylcyclohexadienyllithium. Together with the observation that there is no apparent long-range coupling between the central and end protons of the pentadienyl system, these data indicate a significant departure from a perfect U-shaped planar anion. With the central and end protons in the same plane, a maximum long-range coupling of 1.7Hz, as is observed in 6,6-dimethylcyclohexadienyllithium, is predicted for these types of protons (Barfield 1964).
Additional information on the changes in structural configuration of cyclic pentadienyl anions caused by the addition of methylene groups was obtained from the nmr data in Table 1. In considering this data it was necessary to recall the effect of increasing ring size on the coupling constants of cis-olefinic protons in cyclic alkenes as shown below (Silverstein and Bassler 1967).
Jab 3-4Hz Jab 6-9Hz Jab 10-13Hz
As the size of the ring increases the olefinic protons are forced closer together with a resulting larger observed coupling constant (assuming twisting about the double bond to be negligible). Addition of methylene groups to the cyclic pentadienyl anions in Table 1 did not result in a uniform increase of coupling constants, however. Indeed, the nmr parameters of anion V more closely resemble those of

cycloheptadienyllithium (I) than those of cyclooctadienyl1ithium (II). Addition of a methylene group to the planar 6,6-dimethylcyclohexadienyl- lithium anion did result in the expected increase in coupling constants Jab and Jbc but also resulted in the disappearance of the long-range coupling constant Jac. This was apparently due to both an opening of the U-shape in the plane of the anion and a slight out of plane twisting of the anion which caused the long-range coupling to diminish. It was also noted that this addition resulted in a decrease of negative charge at the end carbons of the cyclic pentadienyl anion as indicated by their downfield shift. Addition of a methylene group to anion I to give anion II showed a quite different effect than that observed above. The coupling constants Jab and Jbc for anion II were less than those for anion I, indicating that the pentadienyl system of anion II was con-
- siderably more twisted than that of anion I. Addition of one more methylene group to give anion V resulted in a large increase in coupling
. constants Jab and Jbc, with Jac too small to be observed. The lack of long-range coupling (Jac ~0) indicated that anion V was still somewhat twisted. From these data it was concluded that the cyclic pentadienyl system of anion V was considerably opened from a U-shape in the plane of the anion and less twisted than anion II. This implied that the decrease of negative charge at the end carbons of cyclic pentadienyl anions I and V was caused more by opening the U-shaped anion in the plane of the anion than by twisting out-of-plane.
It is also proposed that the lower cyclization temperature of ianion II as compared to anion V is due to the twisting of anion II which presumably brings the bond forming end carbons of the cyclic pentadienyl

17anion into closer proximity than in the more nearly planar anion V. By the use of molecular models with flexible bonds it was observed that models of anions I and V formed more nearly planar anions with relatively small angle distortions in the pentadienyl system than anion II which was more twisted.
Table 2 compares the results of protonation of anion V, 6,6- dimethylcyclohexadienyllithium, cycloheptadienyllithium (I), and cyclooctadienyllithium (McCombs 1969). It was not possible to correlate protonation data with relative changes in negative Charge as indicated by nmr data. It is possible that under conditions used for - protonation the kinetically controlled product was not obtained exclusively.
The spin decoupled spectra of anion V are included after page 22 as Fig* 10.
Rearrangement of Cyclononadienyllithium (V)Although anion V begins to cyclize at temperatures above 50°
and the resulting alkene VII isomerizes under the reaction conditions at this temperature, these reactions are fairly slow at temperatures under 100°. For this reason studies were carried out at 120°, at which temperature significant reaction occurred within a matter of hours.
After heating anion V at 120° for a few hours and then quenching and examining the mixture by gas chromatography, several compounds were identified. These compounds, discussed below, were the result of thermal cyclization of anion V and the subsequent isomerization of the cyclized product under the reaction conditions, These compounds and

Table 2. Diene yields from protonation of cyclic pentadienyl anions.
1,3-diene 40% 72% 50% 62%
1,4-diene 60% 28% 50% 38%

the isomerization scheme are shown in Fig. 9. That this is a thermaland not a photochemical cyclization is clear since exposure of anion Vto ultraviolet light for 240 hr gives only a minute yield of bicyclo-
o[4.3.Ojnonene isomers whereas heating at 120 for 22 hr gives approximately 80% conversion to these isomers. .
Although in the analogous thermal cyclization of cycloocta- dienyllithium at 35°, the closed anion could be observed by nmr spectrometry (McCombs 1969), there was no spectral evidence for the closed anion VI in this study. This is probably due to the formation of anion VI at a higher temperature which would cause it to abstract a proton - more rapidly from some constituent of the reaction medium than would the bicyclo[3.3.0]octenyl anion at 35°.
The nmr spectrum of VII (Fig. 5) shows a sharp singlet at t4.3 which integrates for two protons as its distinguishing characteristic. The configuration of VII as isolated from this thermal cyclization was determined to be cis on the basis of palladium-catalyzed hydrogenation at 0° and comparison with a known sample of cis - hydr indane (Villa).The small amount (~2-3%) of trans - hydr indane observed was attributed to isomerization on the catalyst surface; indeed, when the hydrogenation was carried out at 23°, the percent of trans-hydrindane more than doubled. The observation that VII obtained from the thermal cyclization of V has a cis-ring fusion is in agreement with the thermally .. favored disrotatory cyclization of V prepared from cis,cis-1,4- cyclononadiene (Woodward and Hoffmann 1965).

IV
< s >
V VI
X
V
XI
VIIIX
VII cis-bicyclo[4.3.0]non-8-ene IX bicyclo[4.3.0]non-1(9)-ene X bicyclo[4.3.0]non-1(2)-ene XI bicyclo[4.3.0]non-1(6)-ene
Fig. 9. Complete isomerization sequence resulting from thermal cyclization of anion V.

The nmr spectrum of IX (Fig. 6) shows a sharp singlet at t4.21 which integrates to one proton as its distinguishing feature. The nmr spectrum was used to differentiate IX from X. The appearance of this downfield olefinic proton can be observed as a broadened singlet at t4.73 for X (Fig. 7). This was in agreement with the observed chemical shift of a similar olefinic proton in bicyclo[4.4.0]dec-1-ene which was observed at t4.71 (R. H. Camighan, unpublished results, n.d.).
VII IX Xx4.15 x4.21 t4.73 t4.71(sharp) (sharp) (broad) (broad)
Furthermore, there is a significant downfield shift of the methylene absorptions in the t8 region when the double bond is in the six-membered ring as would be predicted. Supporting evidence for the assignments of IX and X comes from the earlier appearance of the former in the isomerization reaction and the greater stability of the latter (see below). A separate synthesis of X was observed when the rearrangement of bicyclo[6.1.0]nona-2,4,6-triene in a potassium metal in dimethoxyethane solution at -78° was observed (W. Beavers, unpublished results, 1973). The rearranged product had the proposed structure X and nmr spectrometry showed the two substances to be identical.
The nmr spectrum of XI shows no absorptions below x7.3 indicating its lack of olefinic protons. Its structure assignment was based

22on its lack of olefinic protons, the observed parent peak at 122 in the mass spectrum as with the other isomers, the nature of its origin, and its hydrogenation with 5% palladium on carbon to yield a mixture of cis- and trans-hydrindanes.
Equilibration of these bicyclo[4.3.0]noriene isomers at 120° in the isomerizing medium gave a mixture of isomers containing 97% XI, 3%X, and trace amounts (̂ .3%) of IX. This was in agreement with the general observation that tetrasubstituted alkenes are more stable than those less highly substituted and also with the observation that double bonds are more stable exocyclic to a five-membered ring and endocyclic to a six-membered ring where the choice is available.
The conrotatory photochemical cyclization of V to give the trans isomer of VII may fail because anion V does not absorb strongly enough to overcome considerable steric and angle strain in the formation of VIlb.
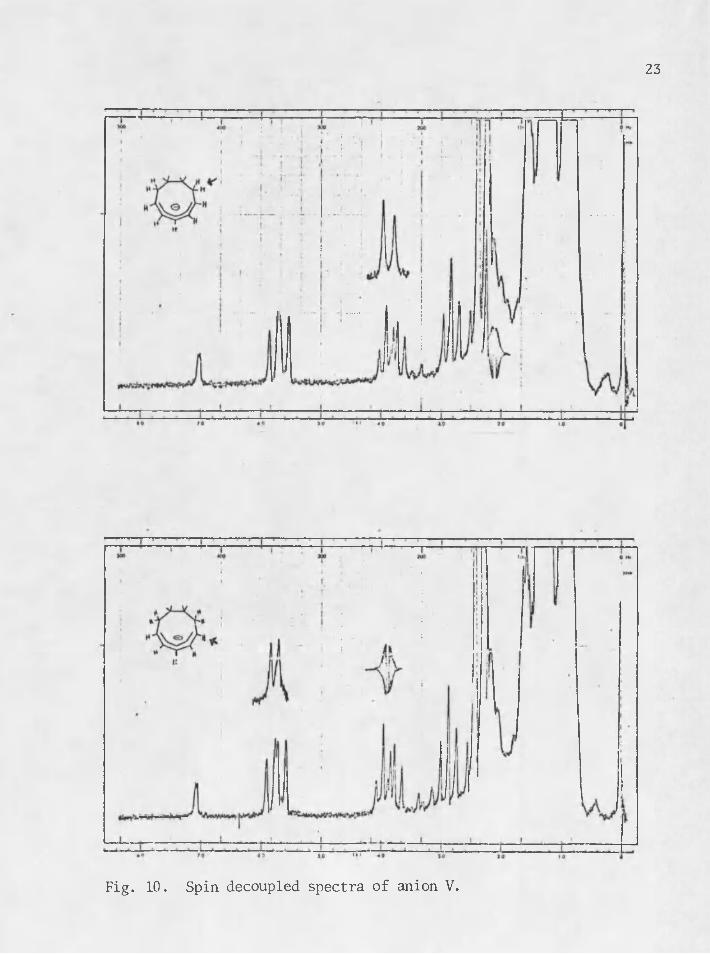
23
Fig. 10. Spin decoupled spectra of anion V.

Fig. 10, continued.

LIST OF REFERENCES
Barfield, Michael. J. Chem. Phys., 4d, 3825 (1964).Bates, Robert B., D. W. Gosselink, and J. A. Kaczinski. Tetrahedron
Lett.i 205 (1967).Bates, Robert B., and D. A. McCombs. Tetrahedron Lett., 977 (1969).Beavers, W. Unpublished experimental results. Department of Chemistry,
University of Arizona (1973).Camighan, R. H. Unpublished experimental results. Department of
Chemistry, University of Arizona (n.d.).Devaprabhakara, D., C. G. Cardenas, P. D. Gardner. J. Am. Chem. Soc.,
85, 1553 (1963).McCombs, Douglas A. Ph.D. Dissertation, University of Arizona (1969).Silverstein, R. M., and G. C. Bassler. Spectrometric Identification of
Organic Compounds, Wiley and Sons, New York, 145 (1967).Woodward, R. B., and R. Hoffmann. J. Am. Chem. Soc., 87, 395 (1965).
25







![Prebiotic Synthesis of Adenine. Hydrogen Atom Tunneling in the Virtual [1,7]-Sigmatropic Rearrangement of Monocyclic HCN-Pentamer Rainer Glaser, a, * Jian](https://img.pdfslide.net/doc/110x75/5513c5f55503463a298b4bd6/prebiotic-synthesis-of-adenine-hydrogen-atom-tunneling-in-the-virtual-17-sigmatropic-rearrangement-of-monocyclic-hcn-pentamer-rainer-glaser-a-jian.jpg)






















