Embed Size (px)
Citation preview


EE - EP1663216 B1
Takroliimust sisaldavad modifitseeritud vabanemisega kompositsioonid
Käesolev leiutis hõlmab ravimkoostist ja/või doseerimise vorme, eelistatult
suukaudseid ühekordset annust sisaldavaid doseerimise vorme, mis sisaldavad
modifitseeritud vabanemisprofiiliga takroliimust, kui ühend allutatakse
tavapärasele lahustamismeetodile, mille puhul usutakse, et see peegeldab aktiivse 5
koostisosa vabanemise tegelikku kiirust ja ajastust in vivo; uudne kompositsioon
vähendab või isegi väldib efektiivselt CYP3A4 metabolismi toimeid.
LEIUTISE TAUST
Takroliimus, samuti tuntud kui FK-506 või FR-900506, omab allpool väljatoodud
tritsüklilist keemilist struktuuri: 10
,
mis vastab valemile C44H69NO12. Takroliimus esineb valgete kristallide või
kristalliinpulbri vormis. See on vees praktiliselt lahustumatu, vabalt lahustuv
etanoolis ning väga lahustuv metanoolis ja kloroformis.
Takroliimuse valmistamine on kirjeldatud dokumendis EP-A-0 184 162 ning 15
takroliimuse analoogid on avalikustatud nt. dokumentides EP-A-0 444 659 ja US
6,387,918.
Takroliimus on makroliidne ühend, mis omab kasulikku immunosupressiivset
toimet, antimikroobset toimet ning teisi farmakoloogilisi toimeid ning on väärtuslik

EE - EP1663216 B1 2
organite või kudede transplantatsioonijärgsete äratõukereaktsioonide,
transplantaat-peremehe-vastu haiguse, autoimmuunsete haiguste ja
infektsioossete haiguste ravis või ennetamises. Takroliimus pikendab
peremeesorganismi ning transplanteeritud siiriku elulemust loomade peal läbi
viidud maksa, neeru, südame, luuüdi ja peensoole ning pankrease, kopsu ja 5
trahhea, naha, sarvkesta ja jäseme transplantatsioonimudelites.
Loomade peal on demonstreeritud takroliimuse omadust supresseerida mõningast
humoraalset immuunsust ning suuremas ulatuses rakulise immuunsuse poolt
vahendatud reaktsioone, nagu näiteks allografti äratõukereaktsiooni, hilist tüüpi
ülitundlikkusreaktsioone, kollageen-indutseeritud artriiti, eksperimentaalset 10
allergilist entsefalomüeliiti ning transplantaat-peremehe-vastu haigust.
Takroliimus inhibeerib T-lümfotsüütide aktivatsiooni, kuigi selle toime täpne
mehhanism ei ole teada. Eksperimentaalsed andmed näitavad, et takroliimus
seostub intratsellulaarse valguga FKBP-12. Seejärel moodustub takroliimus-
FKBP-12, kaltsiumi, kalmoduliini ja kaltsineuriini kompleks ning kaltsineuriini 15
fosfataasne aktiivsus on inhibeeritud. Antud efekt võib vältida defosforülatsiooni ja
aktiveeritud T-rakkude tuumafaktori translokatsiooni; arvatakse, et tuuma
komponent initsieerib lümfokiinide moodustumiseks vajaliku geenitranskriptsiooni.
Lõpptulemuseks on T-lümfotsüüdi aktivatsiooni inhibeerimine, st.
immunosupressioon. 20
Takroliimus metaboliseeritakse suures ulatuses CYP3A4 isoensüümi poolt
sooleseinas ja maksas. Seetõttu võivad antud isoensüümi mõjutavad ravimid
mõjutada takroliimuse absorbtsiooni ja sellele järgnevat süsteemselt imendunud
takroliimuse eliminatsiooni. CYP3A4 inhibiitorid võivad tõsta takroliimuse taset,
samas kui CYP3A4 indutseerijad võivad suurendada takroliimuse metabolismi ja 25
vähendada takroliimuse taset. Sellele vastavalt võib takroliimust manustada koos
ühe või enama CYP3A4 inhibiitoriga, et parandada selle üleüldist biosaadavust.
Tavaliselt manustatakse takroliimust suukaudselt ning seetõttu imendub see
gastrointestinaaltraktist. On täheldatud, et samaaegne toidu seedimine mõjutab
takroliimuse absorptsiooni negatiivselt. Seetõttu olid takroliimuse absorptsiooni 30
kiirus ja ulatus kõige suuremad paastumise tingimustes.

EE - EP1663216 B1 3
Üldiselt on teada, et terapeutiliselt aktiivse aine absorptsioon ja biosaadavus
võivad suukaudsel manustamisel olla mõjutatud erinevate faktorite poolt. Antud
faktorite hulka kuuluvad toidu olemasolu gastrointestinaaltraktis ning üleüldiselt on
ravimi maos olemise aeg tunduvalt pikem toidu juuresolekul kui paastumise
tingimustes. Kui ravimühendi biosaadavus on gastrointestinaaltraktis oleva toidu 5
juuresoleku tõttu teatud punktist alates mõjutatud, väljendab antud ravimühend
toiduga seotud efekti. Toiduga seotud efektid on olulised, kuna ravimi
absorptsioon ning seeläbi plasmatase varieerub suurtes piirides olenevalt toidu
tarbimisest. Imendumine vereringesse võib ebasoodsalt olla mõjutatud punktini,
millal patsientidel tekib oht ebapiisavaks absorptsiooni tekkeks, ravimaks 10
seisundit, mille jaoks ravimit manustati. Teisest küljest võivad paastumise
tingimustes aeg-ajalt täheldatud väga kõrged tippkontsentratsioonid samuti esile
kutsuda märkimisväärseid nefro- või neurotoksilisi, samuti gastrointestinaaltrakti ja
teiste organsüsteemidega seotud kõrvaltoimeid.
Takroliimuse absorptsioon gastrointestinaaltraktist pärast suukaudset manustamist 15
on kiire, keskmine aeg suukaudsest manustamisest tervetele subjektidele või
neeru- või maksa transplantaadiga patsientidele tippkontsentratsiooni (tmax)
saavutamiseni on ligikaudu 1-2 tundi, kuid absorptsioon on mittetäielik ja
varieeruv. Biosaadavus on üldiselt madal – enamasti umbes 20% suukaudsel
manustamisel. 20
Kõige sagedamini täheldatavad kõrvaltoimed on oksendamine ja iiveldus, kuid
samuti on täheldatud teisi kõrvaltoimeid, nagu näiteks treemor, peavalu,
hüpertensioon, neeru düsfunktsioon, hüperkaleemia, hüpomagneseemia,
hüperglükeemia, insomnia, kõhulahtisus, kõhukinnisus, kõhuvalu, nefrotoksilisus
ja neurotoksilisus. 25
Suukaudse manustamise jaoks pakendatakse ja turustatakse takroliimust hetkel
pehmete želatiinkapslitena, mis sisaldavad 0,5; 1 või 5 mg veevaba takroliimust
ning seda turustatakse kaubamärgi all Prograf® ja Protropic®. Soovituslik algne
suukaudne doos patsientidele on alates umbes 0,1 kuni 0,2 mg/kg/päevas. Doosi
eesmärgiks on saavutada teatud minimaalne plasmakontsentratsioon väärtusega 30
alates ligikaudu 5 kuni 20 ng/ml. Prograf® on näidustatud allogeense maksa- või

EE - EP1663216 B1 4
neerutransplantaadi saanud patsientidele organi äratõukereaktsiooni
profülaktikaks.
Siiski esineb vajadus uudse farmatseutilise kompositsiooni ja/või annusvormide
järele, mis sisaldavad parema biosaadavusega takroliimust. Suurenenud
biosaadavus võib võimaldada patsiendi poolt võetavate manustamise ühikute arvu 5
vähendamist, nt. dooside arvu vähendamist ühekordse doosini päevas, ning võib
samuti vähendada või kaotada vajadust manustada doosiga üheaegselt toitu ning
seeläbi võimaldades patsientidele suuremat vabadust ravimi manustamise
ajastamisel. Lisaks peetakse võimalikuks, et plasmakontsentratsioonide
kõikumised ajaprofiilis võivad oluliselt väheneda. Lisaks võib paranenud 10
biosaadavuse tulemuseks olla paremini vabanemisprofiil korratav (st. vähem
varieeruv võrreldes Prograf®’i vabanemisprofiiliga).
Dokument WO 01/37808 A samuti avalikustab formulatsiooni, mis on saavutatav
takroliimuse, PEG-24-kolesterooli eetri (Solulan C-24), monoglütseriidide ja
deoksühoolhappe orgaanilises lahustis lahuse piserdamisega non-pareil 15
helmestele ning lisaks on see seotud suukaudse biosaadavuse parandamisega.
Dokument EP-A-1064942 avalikustab stabiilse vabanemisega formulatsioonid, mis
on saavutatavad takroliimuse lahustamisel sulatatud glütserooli monostearaadis
või tetraglütseriini trirasvhappe estris ning segades seda HPMC või laktoosiga.
Dokument WO 03/004001 A avalikustab kontrollitud aglomeratsiooni meetodi, 20
parandamaks vees halvasti lahustuvate ühendite biosaadavust tahketes lahustes
või dispersioonides.
Artikkel: Honbo jt.: „The oral dosage form of FK-506“ väljaandes Transplantation
Proceedings, 1987, köide 19, nr. 5 lisa 6, lk. 17-22 avalikustab kapslid, mis
sisaldavad takroliimuse tahket dispersiooni HPMC-s, mis on valmistatud lahuse 25
meetodil.
Avastuse tegijad on leidnud, et takroliimuse biosaadavus suureneb tunduvalt, kui
takroliimust manustatakse imetajale modifitseeritud või kontrollitud vabanemisega
kompositsioonina, mis võimaldab kontrollida aktiivse toimeaine vabanemise kiirust

EE - EP1663216 B1 5
ja ajastust, st. in vivo vabanemisprofiili, vähendades efektiivselt või isegi vältides
CYP3A4 metabolismi mõjusid.
Usutakse, et tavapärased in vitro lahustamise meetodid korreleeruvad või
vähemalt peegeldavad tegelikku in vivo modifitseeritud vabanemisprofiili inimese
puhul. Sellega kooskõlas olevana annab käesolev leiutis esimese aspektina tahke 5
farmatseutilisi kompositsiooni, mis sisaldab takroliimust, mille puhul vähem kui
20% (massifraktsioonist) takroliimust vabastatakse 0,5 tunni jooksul, kui viiakse
läbi in vitro lahustuvustest, kasutades USP labameetodit ja kasutades
lahustumiskeskkonnana 0,9 N HCl ning vähem kui 50% (massifraktsioonist)
takroliimust vabastatakse 8 tunni jooksul, kui viiakse läbi in vitro lahustuvustest, 10
kasutades USP Paddle meetodit ning vesilahustuvuskeskkonda, mis on
kohaldatud pH 4,5 ja 0,005% hüdroksüpropüültselluloosile.
Antud modifitseeritud vabanemisprofiil saadakse farmatseutilise kompositsiooni
kaudu, mis sisaldab takroliimuse tahket dispersiooni või tahket lahust hüdrofiilses
või vees segunevas keskkonnas ning ühte või enamat modifitseerivat 15
vabanemisainet, mis on valitud vees segunevate polümeeride, vees lahustumatute
polümeeride, õlide ja õlimaterjalide hulgast, ning keskkond sisaldab
polüetüleenglükooli ja polüksameeri proportsioonides vahemikus 1:3 ja 10:1.
Lisaks hõlmab käesolev leiutis tahkeid annusvorme, eriti suukaudse annusvorme,
mis sisaldavad käesoleva leiutise alla kuuluvaid kompositsioone; tahked 20
annusvormid omavad modifitseeritud vabanemisprofiili. Takroliimuse
vabanemisaja pikendamine duodeenumi distaalses osas võib vähendada ravimiga
seotud gastrointestinaalseid kõrvaltoimeid ning võrdlemisi kõrget metabolismi
määra gastrointestinaaltrakti proksimaalses osas (CYP3A4 vahendatud
metabolism). Selle võib saavutada ilma süsteemset biosaadavust kaotamata, tänu 25
käesoleva leiutise alla kuuluvatele unikaalsetele kompositsioonidele, eelistatult
kompositsioonidele, mis sisaldavad aktiivset koostisosa, mis on täielikult või
osaliselt lahustatud keskkonnas, et moodustada tahke dispersioon ja/või tahke
lahus muutuva temperatuuri juures.

EE - EP1663216 B1 6
Lisaks hõlmab käesolev leiutis käesoleva farmatseutilise kompositsiooni
kasutamise meditsiinilistes preparaatides või medikamentides, eriti kasulike
tahkete doseerimise vormide valmistamises.
LEIUTISE DETAILNE KIRJELDUS
Mõisted 5
Siinkohal kasutatud mõisted „aktiivne koostisosa“ või „aktiivne farmatseutiline
koostisosa“ hõlmavad iga koostisosa, mille eesmärgiks on väljendada
farmakoloogilist aktiivsust või mõnda teist otsest efekti haiguse diagnoosile, ravile,
leevendamisele, käsitlusele või ennetamisele, või mõjutada inimese või teise
looma keha struktuuri või ükskõik millist funktsiooni. Antud mõiste hõlmab neid 10
komponente, mis võivad läbida keemilise muutuse ravimtoote tootmise protsessi
juures ning mis esinevad ravimtootes modifitseeritud vormina eesmärgiga
väljendada määratud aktiivsust või efekti.
Mõiste „hüdrofiilne“ kirjeldab käesolevas kontekstis, et millelegi „meeldib vesi“, st.
hüdrofiilne molekul või molekuli osa on tavaliselt elektriliselt polaarne ning 15
võimeline moodustama veemolekulidega vesiniksidemeid, võimaldades sellel
lahustuda lihtsamalt vees kui õlis või teistes mittepolaarsetes lahustites.
Käesolevas kontekstis kirjeldab mõiste „amfifiilne“ molekuli (kui surfaktanti), millel
on polaarne vees lahustuv rühm, mis on kinnitunud vees lahustuva
süsivesinikahela külge. Seetõttu on üks molekuli ots hüdrofiilne (polaarne) ning 20
teine ots on hüdrofoobne (mittepolaarne).
Käesolevas kontekstis märgib mõiste „hüdrofoobne“ ühendit, mis on tavaliselt
elektriliselt neutraalne ja mittepolaarne ning seetõttu eelistab teisi neutraalseid ja
mittepolaarseid lahusteid või molekulaarseid keskkondi.
Siinkohal kasutatud mõiste „keskkond“ hõlmab farmatseutilises tootes esinevat iga 25
lahustit või kandjavedelikku, millel ei ole ühtegi farmakoloogilist rolli. Näiteks vesi
on keskkonnaks ksilokaiinile ning propüleenglükool on keskkonnaks paljudele
antibiootikumidele.

EE - EP1663216 B1 7
Käesolevas kontekstis märgib mõiste „tahke dispersioon“ ravimit või aktiivset
koostisosa või substantsi, mis on pihustatud teatud tasemele inertses keskkonnas,
kandjas, lahustis või tahkes olekus maatriksis, st. tavaliselt peen teatud
dispersioon.
Käesolevas kontekstis märgib mõiste „tahke lahus“ ravimit või aktiivset koostisosa 5
või substantsi, mis on lahustunud molekulaartasemel inertses keskkonnas,
kandjas, lahustis või tahkes olekus maatriksis.
Siinkohal kasutatud mõiste „analoog“ tähendab keemilist ühendit, mis on
strukruurilt sarnane teisega.
Mõiste „ravim“ tähendab ühendit, mis on mõeldud kasutamiseks haiguse 10
diagnoosimisel, ravimisel, leevendamisel, käsitlemisel või ennetamisel inimese või
teiste loomade puhul.
Antud kontekstis tähendab mõiste „annusvorm“ vormi, milles ravim toimetatakse
patsiendini. See võib olla parenteraalne, toopiline, tablett, suukaudne (vedel või
lahustunud pulber), suposiit, inhalatsioon, transdermaalne jne. 15
Siinkohal kasutatud mõiste „biosaadavus“ märgib määra, mille juures ravim või
teine ühend muutub pärast manustamist saadavaks sihtmärkkudedele. Siinkohal
kasutatud mõiste „bioekvivalentsus“ märgib teaduslikku alust, mille põhjal
geneerilisi ravimeid ja kaubamärgiravimeid võrreldakse üksteisega. Näiteks on
ravimid bioekvivalentsed, kui nad sisenevad ringlusesse sama kiirusega, kui neid 20
on manustatud samasugustes doosides samade tingimuste juures.
Bioekvivalentsuse uuringutes tihti kasutatavad parameetrid on tmax, Cm, AUC0-infinity,
AUC0-t. Teised olulised parameetrid võivad olla W50, W75 ja/või MRT. Vastavalt
võib vähemalt ühte nendest parameetritest kasutada bioekvivalentsuse olemasolu
määramisel. Lisaks peetakse käesolevas kontekstis kahte kompositsiooni 25
bioekvivalentseteks, kui ühe kasutatava parameetri väärtus on vahemikus 80-
125% Prograft®’i omast või testis kasutatud sarnase kommertsiaalselt
saadaoleva takroliimust sisaldava toote omast.
Käesolevas kontekstis märgib „tmax“ aega, mis kulub pärast manustamist
maksimaalse plasmakontsentratsiooni (Cmax) saavutamiseni; AUC0-infinity märgib ala 30

EE - EP1663216 B1 8
plasmakontsentratsiooni-aja kõvera all olevat ala aja väärtusest 0 kuni
lõpmatuseni; AUC0-t märgib plasmakontsentratsiooni-aja kõvera all olevat ala aja
väärtusest 0 kuni väärtuseni t; W50 märgib aega, millal plasmakontsentratsioon on
Cmax’i väärtusest 50% või enam; W75 märgib aega, millal plasmakontsentratsioon
on Cmax’i väärtusest 75% või enam; ning MRT märgib takroliimuse (ja/või selle 5
analoogi) keskmist eliminatsiooniaega.
Antud kontekstis tähendab mõiste „ravim“ ühendit, mida kasutatakse haiguse,
vigastuse või valu ravimiseks. Ravim jaotub vastavalt mõistete „profülaktiline“, st.
tervise säilitamise kunsti, ning „terapeutiline“, st. tervise taastamise kunsti, alla.
Käesolevas kontektsis on mõisted „kontrollitud vabanemine“ ja „modifitseeritud 10
vabanemine“ samasuguse tähendusega, mis hõlmavad takroliimuse ükskõik millist
tüüpi vabanemist käesoleva leiutise alla kuuluvast kompositsioonist, mis on sobilik
pärast subjektile manustamist spetsiifilise terapeutilise või profülaktilise vastuse
saamiseks. Vastavas valdkonnas kogenud spetsialist teab, kuidas kontrollitud
vabanemine/modifitseeritud vabanemine erineb tavalistest tablettidest või 15
kapslitest vabanemisest. Mõisted „vabanemine kontrollitud viisil“ või „vabanemine
modifitseeritud viisil“ omavad samasuguse tähendust, nagu on ülalpool välja
toodud. Mõisted hõlmavad aeglase vabanemisega (mille tulemuseks on madalam
Cmax ja hilisem tmax, kuid t½ on muutumatu), pikendatud vabanemisega (mille
tulemuseks on madalam Cmax ja hilisem tmax, kuid nähtav t½ on pikem); hilinenud 20
vabanemisega (mille tulemuseks on muutumatu Cmax, kuid viivitusaeg ning
vastavalt on ka tmax hilisem ning t½ on muutumatu), samuti ka pulseeriva
vabanemisega, järsu vabanemisega, ühtlase vabanemisega, pikendatud
vabanemisega, ajaliselt optimeeritud vabanemisega, kiire vabanemisega (et
saavutada toime paranenud algus) meetodeid. Mõistete alla kuulub samuti näiteks 25
kehas esinevate spetsiifiliste tingimuste, nt. erinevate ensüümide või pH muutuste
kasutamine, et kontrollida ravimaine vabanemist.
Antud kontekstis tähendab mõiste „erosioon“ või „erodeeriv“ materjali või struktuuri
pinna järk-järgulist lagunemist, näiteks tableti või tableti katte lagunemist.
Käesolev leiutis annab farmatseutilised kompositsioonid ja tahked annusvormid 30
takroliimuse ravile vastavate seisundite parandatud raviks, eriti kompositsioonid ja

EE - EP1663216 B1 9
annusvormid, mis võimaldavad aktiivse koostisosa moditiseeritud vabanemist, et
parandada selle biosaadavust.
Leiutise alla kuuluvate kompositsioonide aktiivseks koostisosaks on takroliimus
(tuntud ka kui FK-506 või FR-900506). Siiski esineb käesoleva leiutise ulatuses
takroliimus ükskõik millises füüsikalises vormis (kristallidena, amorfse pulbrina, 5
ükskõik millise võimaliku polümorfina, ükskõik millise võimaliku lahusena,
sealhulgas hüdraadina, anhüdraadina, nende kompleksidena jne.). Siia hulka
kuuluvad ka ükskõik milline takroliimuse derivaat või aktiivne metaboliit,
farmatseutiliselt sobivad soolad, lahused, kompleksid ja vastavad eelravimid.
Seetõttu annab käesolev leiutis eelistatud teostuse korral tahke farmatseutilise 10
kompositsiooni, mis sisaldab takroliimust, mille puhul vähem kui 20%
(massifraktsioonist) aktiivset koostisosa vabastatakse 0,5 tunni jooksul, kui kui
viiakse läbi in vitro lahustuvustest, kasutades USP labameetodit ja kasutades
lahustumiskeskkonnana 0,1 N HCl ning; eelistatult mille puhul vähem kui 20%
(massifraktsioonist), veelgi eelistatumalt vähem kui 10% (massifraktsioonist) 15
aktiivset koostisosa vabastatakse 3 tunni jooksul.
Arvatakse, et antud vabanemisprofiil parandab imetajate puhul tunduvalt
takroliimuse biosaadavust, kuna kas kõik või suurem osa aktiivsest koostisosast
vabastatakse gastrointestinaaltraktis sellisel viisil, et CYP3A4 metabolismi
välditakse oluliselt või vähemalt selle põju on tunduvalt vähenenud. Lisaks 20
peetakse võimalikuks, et antud efekt korreleerub või vähemalt peegeldab
käesoleva leiutise alla kuuluva tahke farmatseutilise kompositsiooni ja/või
annusvormi in vitro lahustumise profiili; profiil on lihtsalt leitav, kui kompositsioon
ja/või doseerimise vormi peal läbi viia tavapärane in vitro lahustuvuse meetod, mis
vastab näiteks USP-le. Arvatakse, et ükskõik milline USP in vitro lahustuvuse 25
meetod on kasulik käesoleva eesmärgi jaoks.
Näiteks vabastab käesoleva leiutise alla kuuluv tahke farmatseutiline
kompositsioon aktiivsest koostisosast vähemalt 50% (massifraktsioonist) 4 tunni
jooksul, eelistatult 2,5 tunni jooksul, kui viiakse läbi in vitro lahustuvustest,
kasutades USP labameetodit ja kasutades lahustumiskeskkonnana 0,1 N HCl 30

EE - EP1663216 B1 10
esimese kahe tunni jooksul ning seejärel kasutades lahustumiskeskkonda, mille
pH on 6,8.
Kasutades vähem tavapärast lahustumiskeskkonda, vabastab käesoleva leiutise
alla kuuluv kompositsioon aktiivset koostisosast vähem kui 50%
(massifraktsioonist), eriti vähem kui 40w% (massifraktsioonist) 8 tunni jooksul, 5
eelistatult 15 tunni jooksul, kui viiakse läbi in vitro lahustuvustest, kasutades USP
labameetodit ning vesilahustuvuskeskkonda, mis on kohaldatud pH 4,5 ja 0,005%
hüdroksüpropüültselluloosile.
Farmatseutilise kompositsiooni soovitud modifitseeritud vabanemisprofiili võib
saavutada, kasutades farmatseutilist kompositsiooni, mis koosneb aktiivse 10
koostisosa, st. takroliimuse või selle analoogi tahkest dispersioonist või tahkest
lahusest hüdrofiilses või veega segunevas keskkonnas ja ühest või enamast
modifitseerivast vabanemisainest.
Käesoleva leiutise ühe teostuse puhul antakse modifitseeritud vabanemisega
takroliimust sisaldav farmatseutiline kompositsioon, mille puhul on aktiivne 15
koostisosa lahustatud või disperseeritud hüdrofiilses või veega segunevas
keskkonnas, mis sisaldab polüetüleenglükooli ja poloksameeri segu. Kasuliku
segu konkreetne näide on 70% (massifraktsioonist) polüetüleenglükool 6000
(PEG6000) ja 30% (massifraktsioonist) poloksameer 188 segu.
Käesoleva leiutise alla kuuluv kompositsioon võib vastavale imetajale suukaudsel 20
manustamisel väljendada vähemalt 1,3 AU-CIAUCPrograf® väärtust, AUC väärtused
on kindlaks määratud sarnastes tingimustes.
Nagu siin välja toodud näidete põhjal selgub, paraneb käesolevale leiutisele
vastava kompositsiooni manustamisele järgnevalt saadud biosaadavus
märkimisväärselt. Seetõttu on AUC/AUC Prograf® väärtus vähemalt ligikaudu 1,5, 25
nagu näiteks ligikaudu 1,75 või enam, ligikaudu 1,8 või enam, ligikaudu 1,9 või
enam, ligikaudu 2,0 või enam, ligikaudu 2,5 või enam, ligikaudu 2,75 või enam,
ligikaudu 3,0 või enam, ligikaudu 3,25 või enam, ligikaudu 3,5 või enam, ligikaudu
3,75 või enam, ligikaudu 4,0 või enam, ligikaudu 4,25 või enam, ligikaudu 4,5 või

EE - EP1663216 B1 11
enam, ligikaudu 4,75 või enam või ligikaudu 5,0 või enam; AUC väärtused on
kindlaks määratud sarnastes tingimustes.
Pärast käesolevale leiutisele vastava farmatseutilise kompositsiooni suukaudset
manustamist peetakse võimalikuks, et plasmakontsentratsioon-ajaprofiil näitab
pikendatud ajaperioodi, mille korral plasmakontsentratsiooni säilitatakse 5
terapeutilise akna (st. plasmakontsentratsiooni, mis viib terapeutilise efekti
saavutamiseni) piirides ilma tõsiste soovimatute kõrvaltoimete tekkimiseta.
Seetõttu jälgitakse samuti tippkontsentratsiooni vähenemist.
Käesoleva leiutise alla kuuluv kompositsioon võib suukaudsel manustamisel
vastavale imetajale vabastada takroliimust kontrollitud viisil ning väljendab Cmax, 10
mis moodustab kõige rohkem umbes 80% Prograf® tablettide Cmax väärtusest,
nagu näiteks kõige rohkem umbes 75%, kõige rohkem umbes 70%, kõige rohkem
umbes 65%, kõige rohkem umbes 60%, kõige rohkem umbes 55%, kõige rohkem
umbes 50%, kõige rohkem umbes 45% või kõige rohkem umbes 40%.
Käesolevas kontekstis on mõisted „kontrollitud vabanemine“ ja „modifitseeritud 15
vabanemine“ samatähenduslikud mõisted ning hõlmavad takroliimuse ükskõik
millist tüüpi vabanemist käesoleva leiutise alla kuuluvast kompositsioonist, mis on
sobilik pärast subjektile manustamist spetsiifilise terapeutilise või profülaktilise
vastuse saamiseks. Vastavas valdkonnas kogenud spetsialist teab, kuidas
kontrollitud vabanemine/modifitseeritud vabanemine erineb tavalistest tablettidest 20
või kapslitest vabanemisest. Mõisted „vabanemine kontrollitud viisil“ või
„vabanemine modifitseeritud viisil“ omavad samasugust tähendust, nagu on
ülalpool välja toodud.
Mõisted „kontrollitud vabanemine/modifitseeritud vabanemine“ hõlmavad aeglase
vabanemisega (mille tulemuseks on madalam Cmax ja hilisem tmax, kuid t½ on 25
muutumatu), pikendatud vabanemisega (mille tulemuseks on madalam Cmax ja
hilisem tmax, kuid nähtav t½ on pikem); hilinenud vabanemisega (mille tulemuseks
on muutumatu Cmax, kuid viivitusaeg ning vastavalt on ka tmax hilisem ning t½ on
muutumatu), samuti ka pulseeriva vabanemisega, järsu vabanemisega, stabiilse
vabanemisega, pikendatud vabanemisega, ajaliselt optimeeritud vabanemisega, 30
kiire vabanemisega (et saavutada toime paranenud algus) meetodeid. Mõistete

EE - EP1663216 B1 12
alla kuulub samuti näiteks kehas esinevate spetsiifiliste tingimuste, nt. erinevate
ensüümide või pH muutuste kasutamine, et kontrollida ravimaine vabanemist.
Et olla rohkem spetsiifiline: pärast käesolevale leiutisele vastavat farmatseutilise
kompositsiooni suukaudset manustamist imetajale, sealhulgas inimesele, mis
sisaldab 5 mg doosi takroliimust, vabastatakse takroliimus kontrollitud viisil ning 5
väljendab Cmax, mis on kõige rohkem ligikaudu 30 ng/ml, nagu näiteks kõige
rohkem ligikaudu 25 ng/ml või kõige rohkem ligikaudu 20 ng/ml.
Siiski ei pruugi tippkontsentratsiooni vähenemine viia terapeutilise efekti
vähenemiseni, senikaua kuni säilitatakse takroliimuse plasmakontsentratsioon
terapeutilise akna piirides. Vastavalt hõlmab käesolev leiutis farmatseutilist 10
kompositsiooni, kus W50 on vähemalt ligikaudu 2 tundi, nagu näiteks vähemalt
ligikaudu 3 tundi, vähemalt ligikaudu 4 tundi, vähemalt ligikaudu 5 tundi, vähemalt
ligikaudu 6 tundi, vähemalt ligikaudu 7 tundi, vähemalt ligikaudu 8 tundi, vähemalt
ligikaudu 9 tundi, ligikaudu 10 tundi või enam, ligikaudu 11 tundi või enam,
ligikaudu 12 tundi või enam, ligikaudu 13 tundi või ligikaudu 14 tundi või enam. 15
Lisaks omab käesoleva leiutise alla kuuluv kompositsioon Cdiff = [Cmax – Ct (t=12
tundi)], mis on samades tingimustes väiksem kui Prograf®’i oma. Kui Prograf®’i
Cdiff on seatud väärtusele 100, siis käesolevale leiutisele vastava kompositsiooni
Cdiff on tavaliselt 90 või vähem, nagu näiteks umbes 85 või vähem, umbes 75 või
vähem, umbes 70 või vähem, umbes 65 või vähem, umbes 60 või vähem, umbes 20
55 või vähem, umbes 50 või vähem, umbes 45 või vähem või umbes 40 või
vähem.
Konkreetsemalt: pärast käesoleva leiutise alla kuuluva farmatseutilise
kompositsiooni suukaudset manustamist imetajale, sealhulgas inimesele, mis
sisaldab 5 mg takroliimust, vabastatakse takroliimus kontrollitud viisil ning 25
väljendab Cdiff väärtusega ligikaudu 20 ng/ml või vähem, nagu näiteks ligikaudu 15
ng/ml või vähem, ligikaudu 13 ng/ml või vähem või ligikaudu 10 ng/ml või vähem.
Käesoleva leiutise alla kuuluv farmatseutiline kompositsioon vabastab takroliimust
kontrollitud viisil, et pikendada takroliimuse terapeutilist toimet. Ühe aspekti
kohaselt võib vabanemine olla sõltuv pH-st, st. vabanemine toimub peamiselt 30

EE - EP1663216 B1 13
pärast maost läbiminekut. Antud pH-sõltuv vabanemine tagatakse peamiselt
siinkohal kirjeldatud enteerilise kattematerjali teel. Vabanemine võib samuti olla
pH-st mittesõltuv, näiteks andes kompositsiooni, millel on kontrollitud
vabanemisega kate, nt. tselluloosil põhinev kate, nagu näiteks etüültselluloos, või
andes kompositsiooni maatrikskompositsiooni vormis, nagu näiteks hüdrofiilne 5
tselluloosi polümeermaatriksi tüüp, mis põhineb nt. HPMC-l. Loomulikult võib
samuti kasutada kombinatsiooni.
Üldiselt on biosaadavuse muutused ja/või teiste biosaadavusega seotud
parameetrite muutused tavaliselt määratud in vivo uuringutes, kus sobivatel
loommudelitel testistakse kõnealuseid kompositsioone koos nt. Prograf®’i või 10
teiste sarnaste kommertsiaalselt saadaolevate takroliimust sisaldavate toodetega.
Teatud formulatsioonide biosaadavuse tõestuse saamiseks kasutatavad
koeramudelid on farmatseutikatööstuses tavaline meetod.
Takroliimust hõlmavad uuringud on mitterandomiseeritud ristkatsed, mille puhul
iga koer on iseenda kontroll. Tavaliselt kasutatakse nelja koera ja nelja ravimit. 15
Kuna ei kasutata intravenoosseid süste, on saadud biosaadavused suhtelised.
Lisaks on üllatuslikult leitud, et samaaegne toidu manustamise vajadus, et
kindlustada takroliimuse piisav imendumine, on tunduvalt vähenenud või puudub
isegi täielikult.
Seetõttu annavad käesoleva leiutise alla kuuluvad farmatseutilised 20
kompositsioonid takroliimuse tunduvalt kõrgema biosaadavuse, mis võib
vähendada igapäevaste manustamise ühikute arvu ning vähendada või kaotada
vajaduse manustada ravimitega üheaegselt toitu, mis annab farmatseutilise
kompositsiooni saajale suurema vabaduse, ning sellest lähtuvalt võib patsientide
vastuvõtlikkus ja/või soostumus ravile märkimisväärselt paraneda. Lisaks 25
vähendavad kompositsioonid tunduvalt kõrvaltoimete esinemist, eriti
tippkontsentratsiooniga seotud kõrvaltoimete esinemist (nagu näiteks nefro- ja
neurotoksilisus, kõhulahtisus, kõhukinnisus, kühuvalu, iiveldus jne.) ning annavad
takroliimuse pikendatud vabanemise, mis viib parema ravitulemuseni.

EE - EP1663216 B1 14
Nagu ülalpool mainitud, on üks takroliimuse kompositsioonide moodustamist
puudutav peamisi väljakutseid vältida ebasoodsat toiduga seotud efekti. Üldiselt
imendub takroliimus palju paremini, kui seda manustatakse suukaudselt ilma
toiduta. Seetõttu täheldatakse pärast manustamist koos toiduga või ilma toiduta
suurt biosaadavuse varieerumist. Antud sõltuvus raskendab konkreetsete 5
juhtnööride, mis puudutavad manustatava doosi suurust, andmist ning lisaks see
nõuab patsiendile info andmist manustamise režiimi kohta. Käesoleva leiutise
eesmärgiks on anda kompositsioone, mille puhul on ebasoodne toiduga seotud
efekt vähenenud. Seetõttu annab käesolev leiutis kompositsiooni, mis ei väljenda
pärast kompositsiooni manustamsit vastavale imetajale märkimisväärset 10
ebasoodsat toiduga seotud efekti, mille tõestuseks on (AUCtoit/AUCpaast) väärtus,
mis on vähemalt ligukaudu 0,85, madalama usalduspiiri 90% juures vähemalt
0,75.
Konkreetsemalt: käesoleva leiutise alla kuuluva farmatseutilise kompositsiooni
(AUCtoit/AUCpaast) väärtus on ligikaudu 0,9 või enam, nagu näiteks ligikaudu 0,95 15
või enam, ligikaudu 0,97 või enam või ligikaudu 1 või enam, nagu näiteks kuni
umbes 1,1 või kuni umbes 1,2.
Käesoleva leiutise alla kuuluva kompositsiooni veel üheks eeliseks on see, et
võrreldes traditsioonilise suukaudse raviga on võimalik saavutada efektiivne
terapeutiline vastus vähendatud doosi suuruse juures. Vastavalt sellele vabastab 20
vastavale imetajale suukaudsel manustamisel käesolevale leiutisele vastav
farmatseutiline kompositsioon takroliimust või selle analoogi kontrollitud viisil ning
kompositsioon on põhiolemuselt bioekvivalentne Prograf®’i või sarnase
kommertsiaalselt saadaoleva takroliimust sisaldava tootega, kui seda
manustatakse doosis, mis moodustab Program® või sarnase kommertsiaalselt 25
saadaoleva takroliimust sisaldava toote vormis manustatud takroliimuse doosist
kõige rohkem ligikaudu 85% (massifraktsioonist), nagu näiteks kõige rohkem
ligikaudu 80% (massifraktsioonist), kõige rohkem ligikaudu 75%
(massifraktsioonist), kõige rohkem ligikaudu 70% (massifraktsioonist), kõige
rohkem ligikaudu 70% (massifraktsioonist), kõige rohkem ligikaudu 65% 30
(massifraktsioonist), kõige rohkem ligikaudu 60% (massifraktsioonist), kõige

EE - EP1663216 B1 15
rohkem ligikaudu 55% (massifraktsioonist) või kõige rohkem ligikaudu 50%
(massifraktsioonist).
Bioekvivalentsuse uuringutes tihti kasutatavad parameetrid on tmax, Cmax, AUC0-
infinity, AUC0-t. Teised olulised parameetrid võivad olla W50, W75 ja/või MRT.
Vastavalt sellele võib kasutada vähemalt ühte nendest parameetritest, et kindlaks 5
määrata bioekvivalentsuse olemasolu. Lisaks on käesolevas kontekstis kaks
kompositsiooni bioekvivalentsed, kui kasutatava parameetri väärtus on vahemikus
80-125% Prograft®’i omast või testis kasutatud sarnase kommertsiaalselt
saadaoleva takroliimust sisaldava toote omast.
Käesolevas kontekstis märgib „tmax“ aega, mis kulub pärast manustamist 10
maksimaalse plasmakontsentratsiooni (Cmax) saavutamiseni; AUC0-infinity märgib ala
plasmakontsentratsiooni-aja kõvera all olevat ala aja väärtusest 0 kuni
lõpmatuseni; AUC0-t märgib plasmakontsentratsiooni-aja kõvera all olevat ala aja
väärtusest 0 kuni väärtuseni t; W50 märgib aega, millal plasmakontsentratsioon on
Cmax’i väärtusest 50% või enam; W75 märgib aega, millal plasmakontsentratsioon 15
on Cmax’i väärtusest 75% või enam; ning MRT märgib takroliimuse (ja/või selle
analoogi) keskmist eliminatsiooniaega.
Kaks teist peamist takroliimusega ravi või profülaktikaga seotud puudust on
võrdlemisi kõrge kõrvaltoimete esinemise sagedus ning võrdlemisi kõrge
interindividuaalne variatsioon. Kujutatakse ette, et käesolevale leiutisele vastav 20
kompositsioon viib kõrvaltoimete esinemissageduse vähenemiseni. Vähenemine
võib kujutada endast kõrvaltoimete vähenenud sagedust või tõsiduse vähenemist.
Kõnealuste kõrvaltoimete hulka kuuluvad näiteks nefro- ja neurotoksilisus,
kõhulahtisus, kõhukinnisus, kõhuvalu, iiveldus jne. Käesoleva leiutise üks aspekt
puudutab teatud vormis esinevat farmatseutilist kompositsiooni, mis sisaldab 25
takroliimust või selle analoogi koos ühe või enama farmatseutiliselt sobiva
abiainega, mille puhul antud kompositsiooni suukaudsel manustamisel vastavale
imetajale vabaneb takroliimus või selle analoog kontrollitud viisil ning vähendab
kõrvaltoimeid võrreldes Prograf®’i puhul esinevatega, kui neid manustada
samades tingimustes ja doosis, mis toob kaasa võrdse terapeutilise efekti. 30

EE - EP1663216 B1 16
Biosaadavuse, kõveraaluse ala (the Area Under the Curve) suurendamine
vähendab intra- ja intervariaabelsust, mis on seotud ravimaine absorptsiooniga.
See on eriti tõene, kui madal ja halvenenud biosaadavus on halva vees
lahustuvuse tagajärg. Peetakse võimalikuks, et käesolevale leiutisele vastavad
kompositsioonid annavad variatsiooni koefitsiendi (Coefficient of Variation) 5
kõveraaluse ala andmete suhtes, mis on märkimisväärselt väiksemad kui
Prograf®’i ja teiste sarnaste toodete puhul.
Nagu eelnevalt mainitud, on käesoleva leiutise üks põhilisteks omadusteks see, et
käesoleva leiutise alla kuuluva kompositsiooni suukaudsel manustamisel on
võimalik saada biosaadavuse paranemine. Tavaliselt on ravimühendi suukaudsele 10
manustamisele järgnev madal biosaadavus ravimühendi kompositsiooni
kontrollitud või modifitseeritud vabanemise kujundatud barjäär, kuna pikema
ajaperioodi jooksul on peaaegu võimatu saavutada efektiivseid ravimi tasemeid.
Siiski on tänu praegusele tehnoloogiale võimalik saada märkimisväärselt
paranenud biosaadavust ning seetõttu on võimalik disainida kontollitud, 15
modifitseeritud või hilinenud vabanemisega kompositsioone.
Takroliimus metaboliseeritakse laialdaselt CYP3A4 isoensüümi poolt sooleseinas
ja maksas. Vastavalt sellele võib sobivaks kontrollitud vabanemisega
kompositsiooniks olla kompositsioon, mis on disainitud vabastama takroliimust
hilinenud viisil, et vältida või vähendada CYP3A4 metabolismi 20
gastrointestinaaltraktis.
Hilinenud vabanemine tagatakse peamiselt mingit tüüpi enteerilise katte abil.
Samal ajal kui poolläbilaskev kate tagab mingisuguse hilinenud vabanemise, ei
taga see piisavalt „hilinenud“ vabanemist. Lisaks see nõuab teatud ajavahemikku,
et vabastada sisu. Käesoleva leiutise jaoks otsitud kate on pH-st sõltuv kate. 25
Antud tüüpi kate on väga resistentne ravimi vabastamisele, kuni saavutatakse
teatud pH väärtus. pH väärtuste väga üksikute kümnendike piires muudab kile
omadusi ning muutub läbilaskvaks. pH-le tundlike polümeeride, mis on mao pH
juures suhteliselt lahustumatud ja mitteläbilaskvad, kuid mis on rohkem lahustuvad
ja läbilaskvad peensoole ja jämesoole pH juures, näidete hulka kuuluvad, kuid 30
nimekiri ei piirdu antud näidetega, järgmised ühendid: polüakrüülamiidid, ftalaadi
derivaadid, nagu näiteks süsivesinike happelised ftalaadid, amüloosatsetaatftalaat,

EE - EP1663216 B1 17
tselluloosatsetaatftalaat, teised tselluloosesterftalaadid, tselluloosesterftalaadid,
hüdroksüpropüültselluloosftalaat, hüdroksüpropüületüültselluloosftalaat, hüdroksü-
propüülmetüültselluloosftalaat, metüültselluloosftalaat, polüvinüülatsetaatftalaat,
polüvinüülatsetaatvesinikftalaat, naatriumtselluloosatsetaatftalaat, tärklise
happeline ftalaat, stüreenmaleiinhappe bibutüülftalaadi kopolümeer, stüreen-5
maleiinhappe polüvinüülatsetaatftalaadi kopolümeer, stüreen- ja maleiinhappe
kopolümeerid, polüakrüülhappe derivaadid, nagu näiteks akrüülhape ja akrüülestri
kopolümeerid, polümetakrüülhape ja selle estrit, polüakrüülmetakrüülhappe
kopolümeerid, šellak ning vinüülatsetaadi ja krotoonhappe kopolümeerid.
Konkreetset huvi pakkuvate pH-tundlike polümeeride hulka kuuluvad šellak, 10
ftalaadi derivaadid, eriti tselluloosatsetaatftalaat, polüvinüülatsetaatftalaat ning
hüdroksüpropüülmetüültselluloosftalaat, polüakrüülhappe derivaadid, eriti
polümetüülmetakrülaat segatuna akrüülhappe ja akrüülestri kopolümeeridega,
ning vinüülatsetaadi ja krotoonhappe kopolümeerid.
Hilinenud vabanemisega katet omavatest kompositsioonidest aktiivse aine 15
vabanemine võib samuti olla ensümaatiline reaktsioon, kui kattematerjalidena
kasutatakse näiteks Zein või mono-/diglütseriidi segusid.
Vastavale imetajale, sh inimesele suukaudsel manustamisel vabastab käesolevale
leiutisele vastav kontrollitud vabanemisega farmatseutiline kompositsioon
takroliimust sellisel viisil, et saavutatakse plasmakontsentratsioon väärtusega 20
vähemalt ligikaudu 5 ng/ml, nagu näiteks vähemalt ligikaudu 7,5 ng/ml või
vähemalt ligikaudu 10 ng/ml ajavahemikuks vähemalt ligikaudu 24 tundi.
Käesoleva leiutise teatud aspekti kohaselt on tipp-plasmakontsentratsiooni ja 24
tundi pärast manustamist mõõdetud plasmakontsentratsiooni vahe kõige rohkem
umbes 20 ng/ml, nagu näiteks kõige rohkem umbes 10 ng/ml, kõige rohkem 25
umbes 7,5 ng/ml või kõige rohkem umbes 5 ng/ml.
Käesoleva leiutise alla kuuluv kompositsioon on disainitud omama takroliimuse
moditiseeritud vabanemist ning võib teatud vormis, sisaldades takroliimust koos
ühe või enama farmatseutiliselt sobiva abiainega, suukaudsel manustamisel
vastavale imetajale omada takroliimuse ja/või selle analoogi hilinenud vabanemist, 30
nii et kõige rohkem 10% (massifraktsioonist), nagu näiteks kõige rohkem ligikaudu

EE - EP1663216 B1 18
7,5% (massifraktsioonist) või kõige rohkem ligikaudu 5% (massifraktsioonist)
takroliimuse täiskogusest või selle analoogist vabastatakse esimese kahe tunni
jooksul, nagu näiteks esimese nelja tunni jooksul pärast manustamist.
Järgmised tingimused võivad olla täidetud, pidades silmas in vitro lahustuvustesti,
mis viiakse läbi happelistes tingimustes: 5
i) kõige enam umbes 30% (massifraktsioonist), nagu näiteks kõige enam umbes
25% (massifraktsioonist), kõige enam umbes 20% (massifraktsioonist), kõige
enam umbes 15% (massifraktsioonist) või kõige enam umbes 10%
(massifraktsioonist) takroliimusest vabastatakse 2 tunni jooksul in vitro
lahustuvustestis, milles kasutatakse lahustumiskeskkonda, mille pH on kõige 10
enam umbes 5, nagu näiteks kõige enam umbes 4,5, kõige enam umbes 4, kõige
enam umbes 3,5, kõige enam umbes 3, kõige enam umbes 2 või kõige enam
umbes 1,5;
ii) kõige enam umbes 10% (massifraktsioonist), nagu näiteks kõige enam umbes
7,5% (massifraktsioonist), kõige enam umbes 5% (massifraktsioonist) või kõige 15
enam umbes 2,5% (massifraktsioonist) takroliimust vabastatakse 2 tunni jooksul in
vitro lahustuvustestis, milles kasutatakse lahustumiskeskkonda, mille pH on kõige
enam umbes 5, nagu näiteks kõige enam umbes 4,5, kõige enam umbes 4, kõige
enam umbes 3,5, kõige enam umbes 3, kõige enam umbes 2 või kõige enam
umbes 1,5; 20
iii) kõige enam umbes 60% (massifraktsioonist), nagu näiteks kõige enam umbes
50% (massifraktsioonist), kõige enam umbes 40% (massifraktsioonist) või kõige
enam umbes 30% (massifraktsioonist) takroliimust vabastatakse 15 tunni jooksul,
nagu näiteks umbes 12 tunni jooksul, testimise korral in vitro lahustuvustestis,
milles kasutatakse lahustumiskeskkonda, mille pH on kõige enam umbes 4,5, 25
nagu näiteks kõige enam umbes 4,0, kõige enam umbes 3,5, kõige enam umbes
3, kõige enam umbes 2 või kõige enam umbes 1,5;
iv) kõige enam umbes 40% (massifraktsioonist), nagu näiteks kõige enam umbes
30% (massifraktsioonist), kõige enam umbes 25% (massifraktsioonist) või kõige
enam umbes 20% (massifraktsioonist) takroliimust vabastatakse 6 tunni jooksul in 30
vitro lahustuvustestis, milles kasutatakse lahustumiskeskkonda, mille pH on kõige

EE - EP1663216 B1 19
enam umbes 4,5, nagu näiteks kõige enam umbes 4,0, kõige enam umbes 3,5,
kõige enam umbes 3, kõige enam umbes 2 või kõige enam umbes 1,5 ja/või
v) kõige enam umbes 30% (massifraktsioonist), nagu näiteks kõige enam umbes
25% (massifraktsioonist), kõige enam umbes 20% (massifraktsioonist) või kõige
enam umbes 15% (massifraktsioonist) takroliimust vabastatakse 4 tunni jooksul in 5
vitro lahustuvustestis, milles kasutatakse lahustumiskeskkonda, mille pH on kõige
enam umbes 4,5, nagu näiteks kõige enam umbes 4,0, kõige enam umbes 3,5,
kõige enam umbes 3, kõige enam umbes 2 või kõige enam umbes 1,5
Peale takroliimuse võib käesoleva leiutise alla kuuluv kompositsioon samuti
sisaldada teisi terapeutiliselt, profülaktiliselt ja/või diagnostiliselt aktiivseid aineid. 10
Märkimisväärselt huvi pakuvad vähemalt ühte järgmist aktiivset ainet sisaldavad
takroliimuse kombinatsioonid: ained, mis on näidustatud kasutamiseks seoses
organite transplantatsiooniga, nagu näiteks steroidid, kaltsineuriini inhibiitorid ja/või
antiproliferatiivsed ained. Konkreetsete näidete hulka kuuluvad prednisoon,
prednisoloon, metüülprednisoon, tsüklosporiin, mükofenolaat, asatiopriin, 15
siroliimus, everoliimus, mükifenolaatnaatrium ning FTY720 (Novartis).
Farmatseutilisi kombinatsioone võib valmistada ükskõik millisel tavapärasel
meetodil, nagu näiteks granuleerimise, segamise, pihustuskuivatamise jne. teel.
Eriti kasulik meetod on dokumendis WO 03/004001 kirjeldatud meetod. Siinkohal
on kirjeldatud konkreetse materjali valmistamise protsess, mis hõlmab kontrollitud 20
aglomeratsiooni meetodit, st. meetodit, mis võimaldab osakese suuruse
kontrollitud kasvu. Meetod hõlmab näiteks takroliimusest ja eelnevalt sulatatud
kandjast koosneva esimese kompositsiooni pihustamist teisele tahkele
kandjakeskkonnale. Tavaliselt on sulava kandja sulamispunktiks vähemalt 5˚C,
kuid madalam kui takroliimuse sulamispunkt. Kandja sulamispunkt võib olla 25
vahemikus 10˚C kuni 150˚C, nagu näiteks vahemikus 30˚C kuni 100˚C või
vahemikus 40˚C kuni 50˚C, mis on kõige eelistatum variant.
Tavalise antud alal praktiseerija kompetentsi kuulub sobiva kandja valimine, mis
oleks farmatseutliselt sobilik, võimeline lahustuma või vähemalt osaliselt
lahustuma takroliimuses ning omama soovitud vahemikus sulamistemperatuuri, 30
kasutades üldteadmisi ning rutiinset eksperimenteerimist. Kandjate sobivad

EE - EP1663216 B1 20
kandidaadid on kirjeldatud dokumendis WO 03/004001, mis on siin välja toodud
viitena.
Käesolevas kontekstis on sobivateks kandjateks näiteks need, mis on mainitud kui
õlid või õlisarnased materjalid (nagu siin on hiljem arutletud), samuti need, mis on
avalikustatud dokumendis WO 03/004001. 5
Dokumendis WO 03/004001 kirjeldatud kontrollitud aglomeratsiooni meetodi
kasutamise eeliseks on see, et selle puhul on võimalik kasutada võrdlemisi suurt
kogust konkreetse materjali sulamit, ilma et selle tulemuseks oleks osakese
suuruse ebasoovitav kasv. Vastavalt sellele omab käesoleva leiutise alla kuuluva
farmatseutilise kompositsiooni konkreetne materjal geomeetrilise kaalu keskmist 10
diameetrit dqw väärtusega ≥10 µm, nagu näiteks ≥20 µm, alates ligikaudu 20 kuni
ligikaudu 2000, alates ligikaudu 30 kuni ligikaudu 2000, alates ligikaudu 50 kuni
ligikaudu 2000, alates ligikaudu 60 kuni ligikaudu 2000, alates ligikaudu 75 kuni
ligikaudu 2000, nagu näiteks alates ligikaudu 100 kuni ligikaudu 1500 µm, alates
ligikaudu 100 kuni ligikaudu 1000 µm või alates ligikaudu 100 kuni ligikaudu 700 15
µm või kõige rohkem 400 µm või kõige rohkem 300 µm, nagu näiteks alates
ligikaudu 50 kuni ligikaudu 400 µm, nagu näiteks alates ligikaudu 50 kuni ligikaudu
350 µm, alates ligikaudu 50 kuni ligikaudu 300 µm, alates ligikaudu 50 kuni
ligikaudu 250 µm või alates ligikaudu 100 kuni ligikaudu 300 µm, kui valmistamisel
kasutatakse kontrollitud aglomeratsiooni meetodit. 20
Ülalmainitud meetodi kohaselt saadud konkreetsel materjalil on sobivad
omadused, pidades silmas voolavust ja/või kokkusurutavust ning on seetõttu
sobilik edasiseks töötlemiseks farmatseutilisteks annusvormideks.
Takroliimuse tahke dispersioon ja/või tahke lahus
Tahke dispersioon või käesoleva leiutise eelistatud teostuse puhul kasutatav tahke 25
dispersioon sisaldavad takroliimust, mis on pihustatud või lahustatud hüdrofiilses
või veega segunevas keskkonnas, mille sulamispunkt (külmumispunkt või
voolavuspunkt) on vähemalt 20˚C kontsentratsiooni juures vahemikus umbes
0,01% (massifraktsioonist) ja umbes 15% (massifraktsioonist), ning mille

EE - EP1663216 B1 21
dispersioon moodustab tahke dispersiooni või tahke lahuse muutuva temperatuuri
juures (toatemperatuuril).
Aktiivse koostisosa kontsentratsioon hüdrofiilses või veega segunevas
keskkonnas on kõige rohkem 15% (massifraktsioonist), eelistatult kõige rohkem
10% (massifraktsioonist), eelistatult kõige rohkem 8% (massifraktsioonist), veelgi 5
eelistatumalt kõige rohkem 6% (massifraktsioonist), veelgi eelistatumalt kõige
rohkem 5% (massifraktsioonist), kõige rohkem 4% (massifraktsioonist), eriti kõige
rohkem 3% (massifraktsioonist), konkreetsemalt kõige rohkem 2%
(massifraktsioonist); ja/või on vähemalt ligikaudu 0,05% (massifraktsioonist),
eelistatult vähemalt ligikaudu 0,1% (massifraktsioonist), veelgi eelistatumalt 10
vähemalt ligikaudu 0,5% (massifraktsioonist), eriti vähemalt ligikaudu 0,7%
(massifraktsioonist), konkreetselt vähemalt ligikaudu 1% (massifraktsioonist).
Füüsikaliselt võivad aktiivne koostisosa ja keskkond moodustada kas tahke
dispersiooni, st. aktiivne koostisosa on pihustatud keskkonda teatud vormis, või
võivad moodustada tahke lahuse, st. aktiivne koostisosa on lahustatud 15
keskkkonnas molekulaartasemel. Aktiivne koostisosa ja keskkond võivad samuti
moodustada tahke dispersiooni, milles on osa aktiivsest koostisosast lahustunud
molekulaartasemel. Dispersiooni ja/või lahuse füüsikalist seisundit võib kindlaks
määrata, kasutades valikuliselt erinevaid meetodeid, nagu näiteks HSM (Hot
Stage Microscopy) mikroskoopiat, DSC (Differential Scanning Calorimetry) 20
kalorimeetriat, skaneerivat elektronmikroskoopiat (SEM) kombinatsioonis EDX
(Energy Dispersive X-ray) analüüsiga ning pulbri röntgendifraktsioonianalüüsiga.
Eelistatud teostuse puhul on aktiivne koostisosa täielikult keskkonans lahustunud,
et moodustada muutuva temperatuuri juures tahke lahuse.
Käesoleva leiutise kohaselt kasutatavate kasulike hüdrofiilsete või veega 25
segunevate keskkondade näited valitakse järgmise rühma hulgast:
polüetüleenglükoolid, poloksameerid ning nende segud.
Käesoleva leiutise eelistatud teostuse puhul on keskkond polüetüleenglükool
(PEG), eriti PEG, mille keskmine molekulaarmass on vähemalt 1500, eelistatult
vähemalt 3000, veelgi eelistatumalt vähemalt 4000, eriti vähemalt 6000, segatuna 30
poloksameeriga proportsioonis (kaal/kaal põhjal) vahemikus 1:3 ja 10:1, eelistatult

EE - EP1663216 B1 22
vahemikus 1:1 ja 5:1, veelgi eelistatumalt vahemikus 3:2 ja 4:1, eriti vahemikus 2:1
ja 3:1, konkreetselt umbes 7:3. Kasuliku segu konkreetseks näiteks on PEG6000
ja poloksameer 188 segu suhtega 7:3.
Polüetüleenglükoolide (PEG) sulamispunkt (külmumispunkt või voolavuspunkt)
suureneb, kui keskmine molekulaarmass suureneb. Näiteks PEG 400 on 5
vahemikus 4-8˚C, PEG 600 on vahemikus 20-25˚C, PEG1500 on vahemikus 44-
48˚C, PEG2000 on ligikaudu 52˚C, PEG4000 on ligikaudu 59˚C, PEG6000 on
ligikaudu 65˚C ning PEG8000 on ligikaudu 61˚C.
Kasulikud poloksameerid (samuti märgitud polüoksüpropüleen-polüoksüetüleen
plokk-kopolümeerid) on näiteks poloksameer 188, poloksameer 237, poloksameer 10
338 või poloksameer 407 või teised etüleenoksiidi ja propüleenoksiidi plokk-
kopolümeerid, nagu näiteks Pluronic® ja/või Tetronic® seeriad. Sobivate
Pluronic® seeriasse kuuluvate plokk-kopolümeeride hulka kuuluvad polümeerid,
mille molekulaarmass on umbes 3000 ja enam, nagu näiteks alates ligikaudu 4000
kuni ligikaudu 20000 ja/või viskoossusega (Brookfield) alates ligikaudu 200 kuni 15
ligikaudu 4000 cps, nagu näiteks alates ligikaudu 250 kuni ligikaudu 3000 cps.
Sobivate näidete hulka kuuluvad Pluronic® F38, P65, P68LF, P75, F77, P84, P85,
F87, F88, F98, P103, P104, P105, F108, P123, F123, F127, 10R8, 17R8, 25R5,
25R8 jne. Sobivate Tetronic® seeriasse kuuluvate plokk-kopolümeeride hulka
kuuluvad polümeerid, mille molekulaarmass on umbes 8000 või enam, nagu 20
näiteks alates ligikaudu 9000 kuni ligikaudu 35000 ja/või viskoossusega
(Brookfield) alates ligikaudu 500 kuni ligikaudu 45000 cps, nagu näiteks alates
ligikaudu 600 kuni ligikaudu 40000. Ülalpool antud viskoossused määratakse 60˚C
juures ainete puhul, mis on toatemperatuuril pastad, ning 77˚C juures ainete
puhul, mis on toatemperatuuril tahked. 25
Käesoleva leiutise eelistatud teostuse puhul on poloksameeriks poloksameer 188,
mille keskmine molekulaarmass on ligikaudu 8400 ning sulamispunkt on ligikaudu
50-54˚C.

EE - EP1663216 B1 23
Farmatseutiliselt sobivad abiained
Käesolevale leiutisele vastavates kompositsioonides või tahketes annusvormides
kasutatavate sobilike abiainete näidete hulka kuuluvad täidisained, diluendid,
disintegrandid, sidujad, lubrikandid ja teised sarnased ained ning nende segud.
Kuna käesolevale leiutisele vastavaid kompositsioone või tahkeid annusvorme 5
võib kasutada erinevateks eesmärkideks, arvestatakse abiainete valikul tavaliselt
antud erinevaid kasutusvõimalusi. Teised kasutamiseks sobilikud farmatseutiliselt
sobivad abiained on näiteks hapestavad ained, leelistavad ained, säilitusained,
antioksüdandid, puhverdavad ained, šelatained, värvained, komplekseerivad
ühendid, emulgeerivad ja/või lahustavad ained, maitseained ja lõhnaained, 10
humektandid, magusained, niisutavad ained ja teised sarnased ühendid.
Sobivate täidisainete, diluentide ja/või sidujate hulka kuuluvad laktoos (nt.
pihustuskuivatatud laktoos, α-laktoos, β-laktoos, Tabletose®, erinevad
Pharmatose® astmed, Microtose® või Fast-Floc®), mikrokristalliline tselluloos
(erinevad Avicel® astmed, Elcema®, Ming Tai® või Solka-Floc®), 15
hüdroksüpropüültselluloos, L-hüdroksüpropüültselluloos (madala asendusega),
hüdroksüpropüülmetüültselluloos (HPMC) (nt. Methocel E, F ja K, Metolose SH
firmalt Shin-Etsu, Ltd., nagu näiteks Methocel E ja Metolose 60 SH 4000 cps
ühikut, Methocel F ja Metolose 65 SH, 4000 4000 cps ühikut, Methocel K 15000 ja
100000 cps ühikut, ning Metolose 90 SH 4000, 15000, 39000 ja 100000 ühikut), 20
metüültselluloosi polümeerid (nagu näiteks Methocel A, Methocel A4C, Methocel
A15C, Methocel A4M), hüdroksüetüültselluloos, naatriumkarboksü-
metüültselluloos, karboksümetüleen, karboksümetüülhüdroksüetüültselluloos ja
teised tselluloosi derivaadid, sukroos, agaroos, sorbitool, mannitool, dekstriinid,
maltodekstriinid, tärklised või modifitseeritud tärklised (sealhulgas kartulitärklis, 25
maisitärklis ja riisitärklis), kaltsiumfosfaat (nt. baasiline kaltsiumfosfaat,
kaltsiumvesinikfosfaat, dikaltsiumfosfaathüdraat), kaltsiumsulfaat,
kaltsiumkarbonaat, naatriumalginaat, kollageen jne.
Diluentide konkreetsed näited on nt. kaltsiumkarbonaat, dibaasiline
kaltsiumfosfaat, tribaasiline kaltsiumfosfaat, kaltsiumsulfaat, mikrokristalliline 30
tselluloos, pulbristatud tselluloos, dekstraanid, dekstriin, dekstroos, fruktoos,

EE - EP1663216 B1 24
kaoliin, lakroos, mannitool, sorbitool, tärklis, eelželatiniseeritud tärklis, sukroos,
suhkur jne.
Disintegrantide konkreetsete näidete hulka kuuluvad nt. alginiinhape või
alginaadid, mikrokristalliline tselluloos, hüdroksüpropüültselluloos ja teised
tselluloosi derivaadid, kroskarmelloosnaatrium, krospovidoon, polakriliinkaalium, 5
naatriumtärklisglükolaat, tärklis, eelželatiniseeritud tärklis, karboksümetüültärklis
(nt. Primogel® ja Explotab®) jne.
Sidujate konkreetsed näited on nt. akaatsia, alginiinhape, agar-agar,
kaltsiumkarrageen, naatriumkarboksümetüültselluloos, mikrokristalliline tselluloos,
dekstriin, etüültselluloos, želatiin, vedel glükoos, guaarkummi, 10
hüdroksüpropüülmetüültselluloos, metüültselluloos, pektiin, PEG, povidoon,
eelželatiniseeritud tärklis jne.
Kompositsioonile võib samuti lisada glidante ja lubrikante. Näidete hulka kuuluvad
steaarhape, magneesiumstearaat, kaltsiumstearaat või teine metalliline stearaat,
talk, vahad ja glütseriidid, kerge mineraalõli, PEG, glütserüülbehenaat, kolloidne 15
ränidioksiid, hüdrogeenitud taimeõlid, maisitärklis, naatriumstearüülfumaraat,
polüetüleenglükoolid, alküülsulfaadid, naatriumbensoaat, naatriumatsetaat jne.
Teised abiained, mida võib lisada käesoleva leiutise alla kuuluvasse
kompositsiooni või tahkesse annusvormi, on nt. maitseained, värvained, maitset
maskeerivad ained, pH-d kohandavad ained, puhverdavad ained, säilitusained, 20
stabiliseerivad ained, antioksüdandid, niisutavad ained, niiskust reguleerivad
ained, pindaktiivsed ained, suspendeerivad ained, absorptsiooni parandavad
ained, ained modifitseeritud vabanemise jaoks jne.
Teised käesolevale leiutisele vastavas kompositsioonis või tahkes annusvormis
olevad lisaained võivad olla antioksüdandid, nagu näiteks askorbiinhape, 25
askorbüülpalmitaat, butüleeritud hüdroksüanisool, butüleeritud hüdroksütolueen,
hüpofosforhape, monotioglütserool, kaaliummetabisulfit, propüülgallaat,
naatriumformaldehüüdsulfoksülaat, naatriummetabisulfit, naatriumtiosulfaat,
vääveldioksiid, tokoferool, tokoferoolatsetaat, tokoferoolhemisuktsinaat, TPGS või
teised tokoferooli derivaadid jne. Kandja kompositsioon võib samuti sisaldada nt. 30

EE - EP1663216 B1 25
stabiliseerivaid aineid. Kandjakompositsioonis oleva antioksüdandi ja/või
stabiliseeriva aine kontsentratsioon on tavaliselt alates ligikaudu 0,1%
(massifraktsioonist) kuni ligikaudu 5% (massifraktsioonist).
Käesolevale leiutisele vastav kompositsioon või tahke annusvorm võib samuti
sisaldada ühte või enamat surfaktanti või aineid, millel on pindaktiivsed omadused. 5
Peetakse võimalikuks, et antud ained on seotud kergelt lahustuva aktiivse
koostisosa niisutamisega ning seetõttu aitavad kaasa aktiivse koostisosa
paranenud lahustuvusomadustele.
Käesolevale leiutisele vastavates kompositsioonides või tahketes annusvormides
kasutamiseks sobilikud abiained on surfaktandid, nagu näiteks amfifiilsed 10
surfaktandid, mis on avalikustatud dokumendi WO 00/50007 Lipocine, Inc. nime
all. Sobilike surfaktantide näideteks on järgmised ühendid:
i) polüetoksüleeritud rasvhapped, nagu näiteks polüetüleenglükooli mono- või
diestrid rasvhapetega või nende segud, nagu näiteks polüetüleenglükooli mono-
või diestrid lauriinhappega, oleiinhappega, steaarhappega, müristiinhappega, 15
ritsinoolhappega, ning polüetüleenglükool võib valida järgmiste ühendite hulgast:
PEG 4, PEG 5, PEG 6, PEG 7, PEG 8, PEG 9, PEG 10, PEG 12, PEG 15, PEG
20, PEG 25, PEG 30, PEG 32, PEG 40, PEG 45, PEG 50, PEG 55, PEG 100,
PEG 200, PEG 400, PEG 600, PEG 800, PEG 1000, PEG 2000, PEG 3000, PEG
4000, PEG 5000, PEG 6000, PEG 7000, PEG 8000, PEG 9000, PEG 1000, PEG 20
10000, PEG 15000, PEG 20000, PEG 35000,
ii) polüetüleenglükoolglütserooli estrid rasvhapetega, st. ülalpool mainitud estritega
sarnased estrid, kuid idividuaalsete rasvhapete glütserüülestrite vormis;
iii) glütserooli, propüleenglükooli, etüleenglükooli, PEG’i või sorbitooli estrid näiteks
taimeõlidega, nagu näiteks hüdrogeenitud kastoorõli, mandliõli, palmituumaõli, 25
kastoorõli, aprikoosituumaõli, oliiviõli, maapähkliõli, hüdrogeenitud palmituumaõli
ja teiste sarnastega,
iv) polüglütseeritud rasvhapped, nagu näiteks polüglütseroolstearaat,
polüglütseroololeaat, polüglütseroolritsinoleaat, polüglütseroollinoleaat,
v) propüleenglükooli estrid rasvhapetega, nagu näiteks 30
propüleenglükoolmonolauraat, propüleenglükoolritsinoleaat ning teised sarnased,

EE - EP1663216 B1 26
vi) mono-ja diglütseriidid, nagu näiteks glütserüülmonooleaat, glütserooldioleaat,
glütserüülmono- ja/või –dioleaat, glütserüülkaprülaat, glütserüülkapraat jne.;
vii) sterool ja sterooli derivaadid;
viii) polüetüleenglükoolsorbitaani estrid rasvhapetega (PEG-sorbitaani estrid
rasvhapetega), nagu näiteks ülalpool viidatud erineva molekulaarmassiga PEG’i 5
estrid ning erinevad Tween® seeriad;
ix) polüetüleenglükoolalküülestrid, nagu näiteks PEG oleüüleeter ja PEG
leurüüleeter;
x) suhkru estrid, nagu näiteks sukroosmonopalmitaat ja sukroosmonolauraat;
xi) polüetüleenglükoolalküülfenoolid, nagu näiteks Triton® X või N seeriad; 10
xii) polüoksüetüleen-polüoksüpropüleen plokk-kopolümeerid, nagu näiteks
Pluronic® seeriad, Synperonic® seeriad, Emkalyx®, Lutrol®, Supronic® jne.
Antud polümeeride geneerimine mõiste on „poloksameerid“ ning käesolevas
kontekstis on olulised näited järgmised: Poloxamer 105, 108, 122, 123, 124. 181,
182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 15
331, 333, 334, 335, 338, 401, 402, 403 ja 407;
xiii) sorbitaani estrid rasvhapetega, nagu näiteks Span® seeriad või Ariacel®
seeriad, nagu näiteks sorbinaanmonolauraat, sorbitaanmonopalmitaat,
sorbitaanmonooleaat, sorbitaanmonostearaat jne.;
xiv) madalama alkoholi estrid rasvhapetega, nagu näiteks oleaat, 20
isopropüülmüristaat, isopropüülpalmitaat jne.;
xv) ioonsed surfaktandid, sealhulgas katioonsed, anioonsed ja zwitterioonsed
surfaktandid, nagu näiteks rasvhapete soolad, sapisoolad, fosfolipiidid,
fosforhappe estrid, karboksülaadid, sulfaadid ja sulfonaadid jne.
Kui käesoleva leiutise alla kuuluvas kompositsioonis või tahkes annusvormis 25
esineb surfaktant või surfaktantide segu, on surfaktandi/surfaktantide
kontsentratsioon tavaliselt vahemikus alates ligikaudu 0,1 – 80%
(massifraktsioonist), nagu näiteks alates ligikaudu 0,1 kuni ligikaudu 20%
(massifraktsioonist), alates ligikaudu 0,1 kuni ligikaudu 15% (massifraktsioonist),
alates ligikaudu 0,5 kuni ligikaudu 10% (massifraktsioonist) või alternatiivselt 30
alates ligikaudu 0,10 kuni ligikaudu 80% (massifraktsioonist), nagu näiteks alates
ligikaudu 10 kuni ligikaudu 70% (massifraktsioonist), alates ligukaudu 20 kuni

EE - EP1663216 B1 27
ligikaudu 60% (massifraktsioonist) või alates ligikaudu 30 kuni ligikaudu 50%
(massifraktsioonist).
Ühest või enamast farmatseutiliselt sobivast abiainest võib valida ühe ühendi, mis
valitakse järgmiste ühendite hulgast: ränihape või selle derivaat või sool,
sealhulgas silikaadid, ränidioksiid ja selle polümeerid; magneesiumaluminosilikaat 5
ja/või magneesiumalumunometasilikaat, bentoniit, kaoliin, magneesiumtrisilikaat,
montmorilloniit ja/või saponiit.
Antud materjalid on eriti kasulikud sorptsioonimaterjalidena õlide või õlisarnaste
materjalide jaoks ravimites, kosmeetikas ja/või toidukaupades. Konkreetse
teostuse puhul kasutatakse materjali ravimites õlide või õlisarnaste materjalide 10
sorptsiooniks. Materjalile, millel on võime funktsioneerida õlide või õlisarnaste
materjalide sorptsioonimaterjalina, viidatakse ka kui „õli sorptsioonimaterjalile“.
Lisaks kasutatakse käesolevas kontekstis mõistet „sorptsioon“, et märkida
„absorptsiooni“ ning samuti ka „adsorptsiooni“. Peaks olema selge, et millal iganes
kasutatakse ükskõik kumba mõistet, on selle eesmärgiks katta mõistega nii 15
absorptsiooni kui adsorptsiooni fenomen.
Nimelt võib farmatseutiliselt sobilik abiaine sisaldada ränihapet või selle derivaati
või soola, nagu näiteks silikoondioksiidi või selle polümeeri kui farmatseutiliselt
sobiliku abiainena. Sõltuvalt kasutatava silikoondioksiidi kvaliteedist võib seda
kasutada lubrikandina või võib see olla õli sorptsioonimaterjal. Viimast funktsiooni 20
täitvad omadused näivad olevat kõige tähtsamad.
Käesolevale leiutisele vastav kompositsioon või tahke annusvorm võib sisaldada
farmatseutiliselt sobilikku abiainet, mis on silikoondioksiidi toode, millel esinevad
Aeroperl® 300-le (saadaval: Degussa, Frankfurt, Saksamaa) vastavad omadused.
Käesolevale leiutisele vastavates kompositsioonides või annusvormides õli 25
sorptsiooni materjalide kasutamine on väga kasulik farmatseutiliste, kosmeetiliste,
toite- ja/või toidukompositsioonide valmistamisel, mille puhul kompositsioon
hõlmab õli või õlisarnast materjali. Üks eelistest on see, et on võimalik kaasata
võrdlemisi suurt kogust õli ja õlisarnast materjali ning endiselt omada tahket
materjali. Seetõttu on võimalik valmistada tahkeid kompositsioone võrdlemisi 30

EE - EP1663216 B1 28
kõrge doosi õli või õlisarnaste materjalide kasutamisega, kasutades käesolevale
leiutisele vastavat õli sorptsiooni materjali. Farmatseutilisel alal on võrdlemisi
suure koguse õli või õlisarnase materjali kasutamine tahkes kompositsioonis
eeliseks, eriti situatsioonides, kus aktiivne aine ei oma sobilikke omadust
arvestades vees lahustuvust (nt. halba vees lahustuvust), stabiilsust 5
vesikeskkonnas (st. vesikeskkonnas toimub lagunemine), suukaudne biosaadavus
(nt. madal biosaadavus) jne. või nendes situatsioonides, kus soovitakse
modifitseerida aktiivse aine vabanemist kompositsioonist, et saavutada aktiivse
aine kontrollitud, hilinenud, stabiilne ja/või pulseeriv kohaleviimine. Seetõttu
kasutatakse seda teatud teostuste puhul farmatseutiliste kompositsioonide 10
valmistamisel.
Tahkete kompositsioonide töötlemisel kasutatav õli sorptsiooni materjal tavaliselt
absorbeerib umbes 5% (massifraktsioonist) või enam, nagu näiteks umbes 10%
(massifraktsioonist) või enam, umbes 15% (massifraktsioonist) või enam, umbes
20% (massifraktsioonist) või enam, umbes 25% (massifraktsioonist) või enam, 15
umbes 30% (massifraktsioonist) või enam, umbes 35% (massifraktsioonist) või
enam, umbes 40% (massifraktsioonist) või enam, umbes 45% (massifraktsioonist)
või enam, umbes 50% (massifraktsioonist) või enam, umbes 55%
(massifraktsioonist) või enam, umbes 60% (massifraktsioonist) või enam, umbes
65% (massifraktsioonist) või enam, umbes 70% (massifraktsioonist) või enam, 20
umbes 75% (massifraktsioonist) või enam, umbes 80% (massifraktsioonist) või
enam, umbes 85% (massifraktsioonist) või enam, umbes 90% (massifraktsioonist)
või enam või umbes 95% (massifraktsioonist) või enam õli või õlist materjali ning
on endiselt tahke materjal.
Teine käesoleva leiutise aspekt puudutab õli või õlist materjali sisaldavate 25
kompositsioonide või tahkete annusvormidega.
Käesolevas kontekstis kasutatakse mõisteid „õli ja õlised materjalid“ väga laial
skaalal, mille hulka kuuluvad õlid, vahad, pooltahked materjalid ning materjalid,
mida tavaliselt kasutatakse lahustitena (nagu näiteks orgaanilised lahustid) või
kaaslahustid, mida kasutatakse farmaatsiatööstuses, ning mõiste hõlmab samuti 30
terapeutiliselt ja/või profülaktiliselt aktiivseid aineid, mis on muutuva temperatuuri
juures vedelas olekus; lisaks hõlmab antud mõiste emulsioone, nagu näiteks

EE - EP1663216 B1 29
mikroemulsioonid ja nanoemulsioonid ning suspensioonid. Õlid ja õlisarnased
materjalid, mida on võimalik absorbeerida, on tavaliselt muutuva või kõrgenenud
temperatuuri juures vedelas olekus (praktilistel põhjustel on maksimaalne
temperatuur umbes 250˚C). Nad võivad olla hüdrofiilsed, lipofiilsed, hüdrofoobsed
ja/või amfifiilsed materjalid. 5
Käesolevas kontekstis kasutamiseks sobilikud õlid ja õlisarnased materjalid on
ained või materjalid, mille sulamispunkt on vähemalt umbes 0˚C ning kõige
rohkem umbes 250˚C.
Käesoleva leiutise teatud teostuste puhul on õli või õlisarnase materjali
sulamispunkt umbes 5˚C või enam, nagu näiteks umbes 10˚C või enam, umbes 10
15˚C või enam, umbes 20˚C või enam või umbes 25˚C või enam.
Käesoleva leiutise teiste teostuste puhul on õli või õlisarnase materjali
sulamispunkt vähemalt umbes 25˚C, nagu näiteks vähemalt 30˚C, vähemalt 35˚C
või vähemalt 40˚C. Praktilistel põhjustel võib sulamispunkt olla tavaliselt liiga
kõrge, seetõttu on õli või õlisarnase materjali sulamispunkt kõige enam umbes 15
300˚C, nagu näiteks kõige enam umbes 250˚C, kõige enam umbes 200˚C, kõige
enam umbes 150˚C või kõige enam umbes 100˚C. Kui sulamispunkt on kõrgem,
võib võrdlemisi kõrge temperatuur aidata kaasa näiteks oksüdatsioonile või mõnda
teist tüüpi aktiivse aine lagunemisprotsessile nendel juhtudel, kus nt. terapeutiliselt
ja/või profülaktiliselt aktiivne aine on kaasatud. 20
Käesolevas kontekstis on sulamispunkt määratud DSC (Differential Scanning
Calorimetry) poolt. Sulamispunkt on määratud kui temperatuur, mille juures DCS
kõvera lineaarne tõus lõikub temperatuuriteljega.
Huvipakkuvad õlid või õlisarnased materjalid on tavalised ühendid, mida
kasutatakse ravimite tootmises nii-öelda sulami sidujatena või tahkete lahustitena 25
(tahkes annusvormis) või kaaslahustitena või toopiliseks kasutamises mõeldud
ravimite koostisosadena.
See võib olla hüdrofiilne, hüdrofoobne ja/või omada pindaktiivseid omadusi.
Üldiselt on hüdrofiilsed ja/või hüdrofoobsed õlid või õlisarnased materjalid
sobilikud farmatseutiliste kompositsioonide tootmises kasutamiseks, mis 30

EE - EP1663216 B1 30
sisaldavad terapeutiliselt ja/või profülaktiliselt aktiivset ainet, millel on võrdlemisi
madal vesilahustuvus ja/või kui aktiivse aine vabanemine farmatseutilisest
kompositsioonist on määratud olema kohene või mittemodifitseeritud.
Hüdrofoobseid õlisid või õlisarnaseid materjale kasutatakse teisest küljest
modifitseeritud vabanemisega farmatseutilise kompositsiooni tootmisel. Ülalpool 5
väljatoodud kaalutlused on lihtsustatud, et illustreerida üldiseid põhimõtteid, kuid
esineb palju juhtumeid, mille puhul on teised õlide või õlisarnaste materjalide
kombinatsioonid olulised ning seetõttu ei tohiks antud ülalpool väljatoodud näited
mingil viisil piirata käesolevat leiutist.
Tavaliselt valitakse sobilik hüdrofiilne õli või õlisarnane materjal järgmiseid 10
ühendeid sisaldava rühma hulgast: polüeeterglükoolid, nagu näiteks
polüetüleenglükoolid, polüpropüleenglükoolid, polüoksüetüleenid,
polüoksüpropüleenid; poloksameerid ja nende segud; või neid võib valida
järgmiseid ühendeid sisaldava rühma hulgast: ksülitool, sorbitool,
kaaliumnaatriumtartraat, sukroostribehenaat, glükoos, ramnoos, laktitool, 15
beheenhape, vesinikkinoonmonometüüleeter, naatriumatsetaat, etüülfumaraat,
müristhape, sidrunhape, Gelucire 50/13, teised Gelucire tüübid, nagu näiteks
Gelucire 44/14 jne, Gelucire 50/10, Gelucire 62/05, Sucro-ester 7, Sucro-ester 11,
Sucro-ester 15, maltoos, mannitool ja nende segud.
Sobilik hüdrofoobne õli või õlisarnane materjal valitakse järgmiseid ühendeid 20
sisaldava rühma hulgast: sirge ahelaga küllastunud süsivesinikud, sorbitaani
estrid, parafiinid; rasvad ja õlid, nagu näiteks kakaovõi, veiserasv, searasv,
polüeeterglükooli estrid; kõrgemad rasvhapped, nagu näiteks steaarhape,
müristhape, palmithape, kõrgemad alkoholid, nagu näiteks tsetanool,
stearüülalkohol, madala sulamispunktiga vahad, nagu näiteks 25
glütserüülmonostearaat, glütserüülmonooleaat, hüdrogeenitud rasv,
müristüülalkohol, stearüülalkohol, asendatud või asendamata monohglütseriidid,
asendatud ja/või asendamata diglütseriidid, asendatud ja/või asendamata
triglütseriidid, kollane mesilasvaha, valge mesilasvaha, karbaubavaha,
kastoorvaha, jaapani vaha, atsetülaatmonoglütseriidid; NVP polümeerid, PVP 30
polümeerid, akrüülpolümeerid või nende segud.

EE - EP1663216 B1 31
Huvipakkuva teostuse korral on õliks või õlisarnaseks materjaliks
polüetüleenglükool, mille keskmine molekulaarmass on vahemikus alates
ligikaudu 400 kuni ligikaudu 35000, nagu näteks alates ligikaudu 800 kuni
ligikaudu 35000, alates ligikaudu 1000 kuni ligikaudu 35000, nagu näiteks
polüetüleenglükool 1000, polüetüleenglükool 2000, polüetüleenglükool 3000, 5
polüetüleenglükool 4000, polüetüleenglükool 5000, polüetüleenglükool 6000,
polüetüleenglükool 7000, polüetüleenglükool 8000, polüetüleenglükool 9000,
polüetüleenglükool10000, polüetüleenglükool 15000, polüetüleenglükool 20000 või
polüetüleenglükool 35000. Teatud situatsioonide korral võib kasutada
polüetüleenglükooli, mille molekulaarmass on alates ligikaudu 35000 kuni 10
ligikaudu 100000.
Teise huvipakkuva teostuse korral on õliks või õlisarnaseks materjaliks
polüetüleenoksiid, mille keskmine molekulaarmass on vahemikus alates ligikaudu
2000 kuni ligikaudu 7000000, nagu näteks alates ligikaudu 2000 kuni ligikaudu
100000, alates ligikaudu 5000 kuni ligikaudu 75000, alates ligikaudu 10000 kuni 15
ligikaudu 60000, alates ligikaudu 15000 kuni ligikaudu 50000, alates ligikaudu
20000 kuni ligikaudu 40000, alates ligikaudu 100000 kuni ligikaudu 7000000, nagu
näiteks alates ligikaudu 100000 kuni ligikaudu 1000000, alates ligikaudu 100000
kuni ligikaudu 600000, alates ligikaudu 100000 kuni ligikaudu 400000 või alates
ligikaudu 100000 kuni ligikaudu 300000. 20
Teise teostuse puhul on õliks või õlisarnaseks materjaliks poloksameer, nagu
näiteks Poloxamer 188, Poloxamer 237, Poloxamer 338 või Poloxamer 407 või
teised etüleenoksiidi ja propüleenoksiidi plokk-kopolümeerid, nagu niteks
Pluronic® ja/või Tetronic® seeriad. Pluronic® seeriate hulka kuuluvate sobilike
plokk-kopolümeeride hulka kuuluvad polümeerid, mille molekulaarmass on 25
ligikaudu 3000 või enam, nagu näiteks alates ligikaudu 4000 kuni ligikaudu 20000
ja/või viskoossusega (Brookfield) alates ligikaudu 200 kuni ligikaudu 4000 cps,
nagu näiteks alates ligikaudu 250 kuni ligikaudu 3000 cps. Sobilike näidete hulka
kuuluvad Pluronic® F38, P65, P68LF, P75, F77, P84, P85, F87, F88, F98, P103,
P104, P105, F108, P123, F123, F127, 10R8, 17R8, 25R5, 25R8 jne. Tetronic® 30
seeriate hulka kuuluvate sobilike plokk-kopolümeeride hulka kuuluvad polümeerid,
mille molekulaarmass on ligikaudu 8000 või enam, nagu näiteks alates ligikaudu

EE - EP1663216 B1 32
9000 kuni ligikaudu 35000 ja/või viskoossusega (Brookfield) alates ligikaudu 500
kuni ligikaudu 45000 cps, nagu näiteks alates ligikaudu 600 kuni ligikaudu 40000
cps. Ülalpool välja toodud viskoossused määratakse 60˚C juures ainete puhul, mis
on toatemperatuuril pastad, ning 77˚C juures ainete puhul, mis on toatemperatuuril
tahked. 5
Õliks või õlisarnaseks materjaliks võib samuti olla sorbitaanester, nagu näiteks
sorbitaandiisostearaat, sorbiraandioleaat, sorbitaanminilauraat,
sorbitaanmonoisostearaat, sorbitaanmonooleaat, sorbitaanmonopalmitaat,
sorbitaanmonostearaat, sorbitaanseskvi-isostearaat, sorbitaanseskvioleaat,
sorbitaanseskvistearaat, sorbitaanitriisostearaat, sorbitaantrioleaat, 10
sorbitaantristearaat või nende segud.
Õli või õlisarnane materjal võib loomulikult sisaldada erinevate õlide või õlisarnaste
materjalide segu, nagu näiteks hüdrofiilsete ja/või hüdrofoobsete materjalide segu.
Teisteks sobilikeks õlideks või õlisarnasteks materjalideks võivad olla lahustid või
pooltahked abiained, nagu näiteks propüleenglükool, polüglükoliseeritud 15
glütseriidid, sealhulgas Gelucire 44/14, komplekssed taimset päritolu
rasvmaterjalid, sealhulgas teobroomi õli, karnaubavaha, taimeõlid, nagu näiteks
mandliõli, kookospähkliõli, maisiõli, puuvillaseemneõli, seesamiõli, sojaõli, oliiviõli,
kastoorõli, palmituumaõli, maapähkliõli, rapsiõli, viinamarjaseemneõli jne.,
hüdrogeenitud taimeõlid, nagu näiteks hüdrogeenitud maapähkliõli, hüdrogeenitud 20
palmituumaõli, hüdrogeenitud puuvillaseemneõli, hüdrogeenitud sojaõli,
hüdrogeenitud kastoorõli, hüdrogeenitud kookospähkliõli; naturaalsed loomset
päritolu rasvmaterjalid, sealhulgas mesilasvaha, lanoliin, rasvalkoholid, sealhulgas
tsetüül-, stearüül-, lauriin-, mürist-, palmit-, steaarrasvalkoholid; estrid, sealhulgas
glütseroolstearaat, glükoolstearaat, etüüloleaat, isopropüülmüristaat; vedelad 25
interesterifitseeritud poolsünteetilised glütseriidid, sealhulgas Miglycol 810/812;
amiid või rasvhappe alkoholamiidid, sealgulgas steaaramiidetanool, dietanoolamiid
või rasvased kookospähkli happed, atseethappe estrid mono- ja diglütseriididega,
sidrunhappe estrid mono- ja diglütseriididega, piimhappe estrid mono- ja
diglütseriididega, mono- ja diglütseriidid, polüglütserooli estrid rasvhapetega, 30
polüglütserool polüritsinoleaat, propüleenglükooli estrid rasvhapetega,
sorbitaanmonostearaadid, sorbitaantristearaadid, naatriumstearoüüllaktülaadid,

EE - EP1663216 B1 33
kaltsiumstearoüüllaktülaadid, diatsetüültartaarhappe estrid mono- ja
diglütseriididega jne.
Tavaliselt on käesolevale leiutisele vastava farmatseutilise kompositsiooni või
tahke annusvormi puhul õli või õlisarnase materjali kontsentratsioon
kompositsioonid umbes 5% (massifraktsioonist) või enam, nagu näiteks umbes 5
10% (massifraktsioonist) või enam, umbes 15% (massifraktsioonist) või enam,
umbes 20% (massifraktsioonist) või enam, umbes 25% (massifraktsioonist) või
enam, umbes 30% (massifraktsioonist) või enam, umbes 35% (massifraktsioonist)
või enam, umbes 40% (massifraktsioonist) või enam, umbes 45%
(massifraktsioonist) või enam, umbes 50% (massifraktsioonist) või enam, umbes 10
55% (massifraktsioonist) või enam, umbes 60% (massifraktsioonist) või enam,
umbes 65% (massifraktsioonist) või enam, umbes 70% (massifraktsioonist) või
enam, umbes 75% (massifraktsioonist) või enam, umbes 80% (massifraktsioonist)
või enam, umbes 85% (massifraktsioonist) või enam, umbes 90%
(massifraktsioonist) või enam või umbes 95% (massifraktsioonist) või enam. 15
Käesoleva leiutise alla kuuluvas kompositsioonis või tahkes annusvormis oleva õli
või õlisarnase materjali kontsentratsioon võib olla vahemikus ligikaudu 20% kuni
ligikaudu 80% (massifraktsioonist), nagu näiteks alates ligikaudu 25% kuni
ligikaudu 75% (massifraktsioonist).
Üks eeliseid on see, et on võimalik kasutada võrdlemisi suurt kogust õli ja 20
õlisarnast materjali ning endiselt omada tahket materjali. Seetõttu on võimalik
valmistada tahkeid kompositsioone, kasutades käesoleva leiutisega vastavuses
olevalt õli sorptsiooni materjalina võrdlemisi suurt kogust õli või õlisarnaseid
materjale. Farmatseutilisel alal on võrdlemisi suure koguse õli või õlisarnase
materjali kasutamine tahkes kompositsioonis eeliseks, eriti situatsioonides, kus 25
aktiivne aine ei oma sobilikke omadust arvestades vees lahustuvust (nt. halba
vees lahustuvust), stabiilsust vesikeskkonnas (st. vesikeskkonnas toimub
lagunemine), suukaudne biosaadavus (nt. madal biosaadavus) jne. või nendes
situatsioonides, kus soovitakse modifitseerida aktiivse aine vabanemist
kompositsioonist, et saavutada aktiivse aine kontrollitud, hilinenud, stabiilne ja/või 30
pulseeriv kohaleviimine.

EE - EP1663216 B1 34
Veel üheks eeliseks on see, et konkreetne saadud materjal on vabalt voolav
pulber ning seetõttu on seda lihtne töödelda näiteks tahketeks annusvormideks,
nagu näiteks tabletid, kapslid või kotikesed. Tavaliselt on konkreetsel materjalil
omadused, mis on sobilikud tablettide valmistamiseks otsese kompressiooni teel,
ilma suurte koguste lisaainete lisamiseta. Konkreetsete materjalide voolavuse 5
testimiseks sobilik test on väljaandes Ph.Eur. kirjeldatud meetod ning mõõtes
materjali voolukiirust 10,0 mm diameetriga otsiku (piluga) torust väljumisel.
Vähemalt osa takroliimust esineb kompositsioonis tahke lahuse vormis, sealhulgas
molekulaarse dispersiooni ja tahke dispersiooni vormis. Tavaliselt 10% või enam,
nagu näiteks 20% või enam, 30% või enam, 40% või enam, 50% või enam, 60% 10
või enam, 70% või enam, 80% või enam, 90% või enam, nagu näiteks 96% või
enam või ligikaudu 100% (massifraktsioonist) takroliimusest esineb
kompositsioonis tahke dispersiooni vormis.
Tahke dispersioon võidakse saada erinevaid teid pidi, näiteks kasutades
orgaanilisi lahusteid või pihustades või lahustades aktiivse aine teises sobivas 15
keskkonnas (nt. õlid või õlisarnased materjalid, mis on toatemperatuuril või
kõrgenenud temperatuuril vedelas vormis).
Tahkeid dispersioone (lahuse meetod) võib näiteks valmistada lahustades aktiivse
aine (nt. ravimaine) ja kandja füüsilise segu tavalises orgaanilises lahustis, millele
järgneb lahusti aurustamine. Kandja on tihti hüdrofiilne polümeer. Sobivate 20
orgaaniliste lahustite hulka kuulub farmatseutiliselt sobiv lahusti, milles aktiivne
aine on lahustuv, nagu näiteks metanool, etanool, metüleenkloriid, kloroform,
etüülatsetaat, atsetoon või nende segud.
Sobivate vees lahustuvate kandjate hulka kuuluvad polümeerid, nagu näiteks
polüetüleenglükool, poloksameerid, polüoksüetüleenstearaadid, polü-ε-25
kaprolaktoon, polüvinüülpürrolidoon (PVP), polüvinüülpürrolidoon-
polüvinüülatsetaadi kopolümeer PVP-PVA (Kollidon VA64), polümetakrüüli
polümeerid (Eudragit RS, Eudragit RL, Eudragit NE, Eudragit E) ja
polüvinüülalkohol (PVA), hüdroksüpropüültselluloos (HPC),
hüdroksüpropüülmetüültselluloos (HPMC), metüültselluloos ning 30
polü(etüleenoksiid) (PEO).

EE - EP1663216 B1 35
Happelisi funktsionaalrühmi sisaldavad polümeerid võivad olla sobilikud tahkete
dispersioonide jaoks, mis vabastavad aktiivset ainet sobivas pH vahemikus, mis
võimaldab sooles sobiva absorptsiooni toimumist. Antud polümeerideks võib olla
üks või enam ühendit järgmisest rühmast: hüdroksüpropüülmetüültselluloosftalaat
(HMPCP), polüvinüülatsetaatftalaat (PVAP), hüdroksüpropüülmetüültselluloos-5
atsetaatsuktsinaat (HPMCAS), alginaat, karbomeer, karboksümetüültselluloos,
metakrüülhappe kopolümeer (Eudragit L, Eudragit S), šellak, tselluloos-
atsetaatftalaat (CAP), tärklisglükolaat, polakrüliin, metüültselluloosatsetaatftalaat,
hüdroksüpropüültselluloosatsetaatftalaat, tselluloosatsetaattereftalaat, tselluloos-
atsetaatisoftalaat ja tselluloos-atsetaatrimellitaat. 10
Tahkes dispersioonis esineva aktiivse aine ja polümeeri kogusega on seotud
aktiivse aine kaalusuhe polümeeriga, mis võib olla vahemikus alates ligikaudu 3:1
kuni ligikaudu 1:20. Siiski võib samuti kasutada kitsamat vahemikku alates
ligikaudu 3:1 kuni ligikaudu 1:5, nagu näiteks alates ligikaudu 1:1 kuni ligikaudu
1:3. 15
Tahke dispersioon moodustub eelistatult pihustuskuivatamismeetodi, kontrollitud
aglomeratsiooni, külmkuivatamise või kandjaosakeste katmise või teiste lahusti
eemaldamise protsesseide teel. Kuivatatud produkt sisaldab aktiivset ainet, mis
esineb tahke dispersiooni vormis, sealhulgas molekulaarse dispersiooni ja tahke
lahuse vormis. 20
Lisaks orgaaniliste lahustite kasutamisele võib alternatiivsel viisil ravimit ja
polümeeri purustada või ekstrudeerida kõrgendatud temperatuuri juures (sulami
ekstrudeerimine).
Farmatseutilisi kompositsioone, mis sisaldavad takroliimust vähemalt osaliselt
tahke dispersiooni või lahuse vormis, võib põhimõtteliselt valmistada kasutades 25
ükskõik millist sobivat protseduuri, et valmistada käesolevas valdkonnas tuntud
farmatseutilisi kompositsioone.
Lisaks orgaanilisel lahusel baseeruvale meetodile võib saada takroliimuse tahkeid
dispersioone või tahkeid lahuseid, pihustades ja/või lahustades takroliimust
kandjakompositsioonis, mida kasutatakse kontrollitud aglomeratsiooni meetodi 30

EE - EP1663216 B1 36
puhul. Lisada võib stabiliseerivaid aineid jne., et kindlustada tahke
dispersiooni/lahuse stabiilsus.
Teisest küljest hõlmab käesolev leiutis meetodit käesolevale leiutisele vastava
farmatseutilise kompositsiooni valmistamiseks. Üldiselt võib kasutada ükskõik
millist farmatseutilisel alal esinevat sobilikku meetodit. Siiski, et võimaldada 5
võrdlemisi suure koguse õli või õlisarnase materjali kasutamist, on kasulikuks
osutunud dokumendis WO 03/004001 kirjeldatud meetod. Meetod hõlmab vedelas
vormis oleva esimese kompositsiooni, antud kompositsioon sisaldab esimest
keskkonda või kandjat ning kompositsiooni sulamispunkt on üle 5˚C, pihustamist
teisele kompositsioonile, mis sisaldab teist kandjat või kandjamaterjali, antud teine 10
kompositsioon on nt. vedeldatud olekus ning selle temperatuur on allpool esimese
keskkonna või kandja sulamispunkti. Aktiivne aine võib esineda esimeses
keskkonnas või kandjakompositsioonis ja/või teises kandjas või
kandjakompositsioonis. Siiski on juhtudel, kus takroliimus esineb vähemalt
osaliselt tahke dispersiooni vormis, kasulik sisestada või lahustada takroliimus 15
esimeses keskkonnas või kandjakompositsioonis.
Tahked annusvormid
Käesolevale leiutisele vastav farmatseutiline kompositsioon on konkreetses vormis
ning seda võib sellisena kasutada. Siiski on paljudel juhtudel palju mugavam
omada kompositsiooni graanulite, pelletite, mikrosfääride, nanoosakeste ja teiste 20
sarnaste olekute vormis või tahke annusvormina, mille hulka kuuluvad näiteks
tabletid, kapslid, kotikesed ja teised sarnased. Käesolevale leiutisele vastav tahke
annusvorm võib olla ühekordset annust sisaldav annusvorm või see võib olla
polüdepoo vormis, mis sisaldab mitmeid ühekordseid annuseid, nagu näiteks
pelleteid, helmeid ja/või graanuleid. 25
Tavaliselt on käesoleva leiutise alla kuuluv farmatseutiline kompositsioon või tahke
annusvorm mõeldud suukaudsel, bukaalsel või sublingvaalsel teel
manustamiseks.
Käesolev leiutis hõlmab samuti ülalpool mainitud esitlemise vorme. Käesoleva
leiutise ulatuses on kompositsioonid/tahked annusvormis, mis on mõeldud 30

EE - EP1663216 B1 37
vabastama takroliimust ja/või selle analoogi kiire vabanemise, hilinenud
vabanemise või modifitseeritud vabanemise teel. Kõiki antud viise peetakse
kontrollitud vabanemise viisiks. Lisaks kuulub mõiste „kontrollitud viis“ alla ka pH-
sõltuv vabanemine.
Käesolevale leiutisele vastav tahke annusvorm sisaldab farmatseutilist 5
kompositsiooni ülalpool kirjeldatud konkreetses vormis. Antud käesoleva leiutise
peamiste aspektide all avalikustatud detailid ja üksikasjad kehtivad mutatis
mutandis (vajalike muudatustega) leiutise teiste aspektide kohta. Vastavalt sellele
on siinkohal konkreetses vormis esinevate farmatseutiliste kompositsioonide jaoks
kirjeldatud ja/või nõutud biosaadavuse tõusu, biosaadavuse parameetrite muutusi, 10
ebasoodsa toiduga seotud efekti vähenemise ning samuti takroliimuse ja/või selle
analoogiga jne. seotud omadused analoogid käesolevale leiutisele vastava tahke
annusvormiga.
Tavaliselt on teatud vormis esineva farmatseutilise kompositsiooni
kontsentratsioon vahemikus alates ligikaudu 5 kuni 100% (massifraktsioonist), 15
nagu näiteks alates ligikaudu 10% kuni ligikaudu 90% (massifraktsioonist), alates
ligikaudu 15% kuni ligikaudu 85% (massifraktsioonist), alates ligikaudu 20% kuni
ligikaudu 80% (massifraktsioonist), alates ligikaudu 25% kuni ligikaudu 80%
(massifraktsioonist), alates ligikaudu 30% kuni ligikaudu 80% (massifraktsioonist),
alates ligikaudu 35% kuni ligikaudu 80% (massifraktsioonist), alates ligikaudu 40% 20
kuni ligikaudu 75% (massifraktsioonist), alates ligikaudu 45% kuni ligikaudu 75%
(massifraktsioonist) või alates ligikaudu 50% kuni ligikaudu 70%
(massifraktsioonist) doseerimise vormist. Näiteks on konkreetses vormis esineva
farmatseutilise kompositsiooni kontsentratsioon 50% (massifraktsioonist) või enam
doseerimise vormist. 25
Käesolevale leituisele vastav tahke annusvorm saadakse käesolevale leiutisele
vastava konkreetse materjali töötlemisel antud erialal kogenud isikule
teadaolevate meetodite abil. Tavaliselt see hõlmab ühe või enama siinkohal
mainitud farmatseutiliselt sobiva abiaine edasist lisamist.
Käesolevale leiutisele vastavat kompositsiooni või tahket annusvormi võib 30
disainida,et vabastada takroliimust ja/või selle analoogi ükskõik millisel sobival

EE - EP1663216 B1 38
viisil, tingimusel et esineb biosaadavuse tõus. Seetõttu võib aktiivse aine
vabastamine toimuda võrdlemisi kiirelt, et saada paranenud toime algus; seda võib
vabastada, järgides null- või esimese järgu kineetikat või võib seda vabastada
modifitseeritud teel, et saada vabanemine ettemääratud mustri järgi. Kõiki antud
viise peetakse kontrollitud viisideks. Käesoleva leiutise ulatuse alla kuuluvad 5
samuti tavalised formulatsioonid.
Prograf®’i soovituslik manustamise vahemik on 0,1 kuni 0,2 mg/kg/päevas, antuna
iga 12 tunni järel, jaotatuna kaheks doosiks. Kõige olulisem on jälgida tasemeid
veres.
Tavaline tase 1-3 kuu jaoks on 7-20 ng/ml ning 4-12 kuu jaoks peaksid tasemed 10
olema 5-15 ng/ml. Need on ainult juhendavad väärtused ning võivad varieeruda
sõltuvalt transplantaadi tüübist ning rahvusest.
Neeru transplantaadi patsientide puhul saadi järgnevad andmed:
Heledanahalised, n=114 Mustanahalised, n=56
Aeg pärast
transplan-
teerimist
Doos
(mg/kg)
Miinimum-
kontsentratsioonid
(ng/ml)
Doos (mg/kg) Miinimum-
kontsentratsioonid
(ng/ml)
7. päev 0,18 12,0 0,23 10,9
1. kuu 0,17 12,8 0,26 12,9
6. kuu 0,14 11,8 0,24 11,5
12. kuu 0,13 10,1 0,19 11,0
Käesoleva leiutise alla kuuluvate toodete oodatud dooside soovitused on alates
0,02 mg/kg/päevas kuni 0,15 mg/kg/päevas, manustatuna üks kord päevas. 15
Käesolevale leiutisele vastavat kompositsiooni või tahket annusvormi võib samuti
katta kilekattega, enteerilise kattega, modifitseeritud vabanemise kattega, kaitsva
kattega, antiadhesiivse kattega jne.
Käesolevale leiutisele vastavat tahket annusvormi võib samuti katta, et saada
sobivad omadused, nt. pidades silmas aktiivse aine kontrollitud vabanemist. Katta 20

EE - EP1663216 B1 39
võib ühekordset annust sisaldavaid annusvorme (nt. tablette, kapsleid) või
polüdepoo annusvorme või selle individuaalseid ühikuid.
Sobivateks kattematerjalideks on näiteks metüültselluloos, hüdroksüpropüül-
metüültselluloos, hüdroksüpropüültselluloos, akrüülpolümeerid, etüültselluloos,
tselluloosatsetaatftalaat, polüvinüülatsetaatftalaat, hüdroksüpropüülmetüül-5
tselluloosftalaat, polüvinüülalkohol, naatriumkarboksümetüültselluloos,
tselluloosatsetaat, tselluloosatsetaatftalaat, želatiin, metakrüülhappe kopolümeer,
polüetüleenglükool, šellak, sukroos, titaandioksiid, karnaubavaha, mikrokristalliline
vaha, glütserüülmonostearaat, maisivalk.
Kattematerjalile võib lisada plastitseerijaid ja teisi koostisosi. Samuti võib lisada 10
kattematerjalile sama või erinevat aktiivset ainet.
Käesoleva leiutise eelistatud teostuste puhul on tahked annusvormid disainitud
vabastama takroliimust ja/või selle analoogi kontrollitud viisil. Käesolevas
kontekstis hõlmab mõiste „kontrollitud viis“ kõiki vabanemise tüüpe, mis erinevad
tavaliste tablettide puhul saadud vabanemisest. Seetõttu hõlmab antud mõiste 15
järgmiseid mõisteid: „kontrollitud vabanemine“, „modifitseeritud vabanemine“,
„stabiilne vabanemine“, „pulseeriv vabanemine“, „pikendatud vabanemine“, „järsk
vabanemine“, „aeglane vabanemine“, „pikendatud vabanemine“ ning samuti
mõisted „hilinenud vabanemine“ ning pH-sõlvuva vabanemise. Siiski seostub
käesoleva leiutise teatud aspekt hilinenud vabanemisega kompositsiooni või 20
annusvormiga, mis antud kontekstis märgib kompositsiooni või annusvormi, mis
kõige rohkem vabastab 10% (massifraktsioonist) aktiivset ainet esimese 2 tunni
jooksul pärast manustamist ja/või pärast lahustuvustesti algust, mis hõlmab
lahustuvuskeskkonda, mille pH on kõige enam umbes 3.
Modifitseeritud vabanemisega süsteemid 25
Esimene modifitseeritud vabanemisega süsteem hõlmab maatrikssüsteeme, mille
puhul on takroliimus sisestatud või pihustatud teise materjali maatriksile, mille
eesmärgiks on viivitada takroliimuse vabanemist vesikeskkonda (st.
gastrointestinaaltrakti valendiku vedelikku). Kui takroliimus pihustatakse sellist
tüüpi maatriksile, toimub ravimi vabanemine peamiselt maatriksi pinnalt. Seetõttu 30

EE - EP1663216 B1 40
vabastatakse ravim seadme pinnalt, mis sisaldab maatriksit pärast seda, kui see
difundeerub läbi maatriksi või kui seadme pealispind erodeerub, paljastades
ravimi. Osade teostuste puhul võivad mõlemad mehhanismid toimida üheaegselt.
Maatrikssüsteemid võivad olla suured, st. tablett suurusega umbes 1cm või
väikesed (<0,3cm). Süsteem võib olla ühene (nt. boolus), võib olla jaotatud, kuna 5
koosneb mitmetest alaühikutest (näiteks mitmed kapslid, mis moodustavad ühe
doosi), mida manustatakse sisuliselt üheaegselt, või võivad sisaldada osakeste
paljusust, millele viidatakse ka kui mitmeosakeselisusele. Mitmeosakeselisel
süsteemil võib olla mitmeid formulatsiooni kasutusi. Näiteks võib mitmeosakeselist
süsteemi kasutada pulbrina, et täita kapsli kesta või kasutada seda orginaalsel 10
kujul toiduga segamise jaoks, et kergendada manustamist.
Kasulik mitmeosakeseline maatriks sisaldab mitmesuguseid takroliimust
sisaldavaid osakesi; iga osake sisaldab takroliimust ja/või selle analoogi näiteks
tahke lahuse/dispersiooni vormis koos ühe või enama abiainega, mis on valitud
sellise maatriksi moodustamise jaoks, mis on võimeline kontrollima takroliimuse 15
lahustuvuse määra vesikeskkonnas. Maatriksi materjalid on üldiselt hüdrofoobsed
materjalid, nagu näiteks vahad, mõned tselluloosi derivaadid või teised
hüdrofoobsed polümeerid. Vajadusel võib maatriksi materjale formuleerida
hüdrofoobsete materjalidega, mida saab kasutada sidujate või parandajatena.
Antud annusvormise tootmiseks kasulikud maatriksi materjalid on näiteks 20
järgmised ühendid: etüültselluloos, vahad, nagu näiteks parafiin, modifitseeritud
taimeõlid, karnaubavaha, hüdrogeenitud kastoorõli, mesilasvaha ja teised
sarnased, samuti sünteetilised polümeerid, nagu näiteks polü(vinüülkloriid),
polü(vinüülatsetaat), vinüülatsetaadi ja etüleeni kopolümeerid, polüstüreen ning
teised sarnased. Vees lahustuvate või hüdrofiilsete sidujate või vabanemist 25
modifitseerivate ainete hulka, mida võib valikuliselt formuleerida maatriksi hulka,
kuuluvad hüdrofiilsed polümeerid, nagu näiteks hüdroksüpropüütselluloos (HPC),
hüdroksüpropüümetüültselluloos (HPMC), metüültselluloos, polü (N-vinüül-2-
pürrolidinoon) (PVP), polü(etüleenoksiid) (PEO), polü(vinüülalkohol) (PVA),
ksantaankummi, karragenaan ja teised sarnased looduslikud ja sünteetilised 30
materjalid. Lisaks sisaldavad materjalid, mis funktsioneerivad kui vabanemist
modifitseerivad ained, vees lahustuvaid materjale, nagu näiteks suhkrud või
soolad. Eelistatud vees lahustuvate materjalide hulka kuuluvad laktoos, sukroos,

EE - EP1663216 B1 41
glükoos ja mannitool, samuti hüdrofiilsed polümeerid, nagu näiteks HPC, HPMC
jaPVP.
Mitmeosakeselist toodet võib samuti töödelda kontrollitud aglomeratsiooni teel.
Antud juhul on takroliimus lahustunud või osaliselt lahustunud sobivas sulavas
kandjas ning pritsitud kandjaosakestele, mis sisaldavad maatriksainet. Sobivad 5
sulavad kandjad on eelnevalt mainitud.
Alternatiivselt on takroliimus lahustunud orgaanilises lahustis koos maatriksainega
ning pihustuskuivatatud või kantud kandjaosakestele, võrdle allpool. Antud
protsessi juures tavaliselt kasutatavate lahustite hulka kuuluvad atsetoon, etanool,
isopropanool, etüülatsetaat ning kahe või enama ühendi segud. 10
Moodustumise korral võib takroliimuse mitmeosakeselist maatriksi segada
kokkusurutavate abiainetega, nagu näiteks laktoosi, mikrokristallilise tselluloosi,
dikaltsiumfosfaadi ja teiste sarnaste ühenditega ning suruda segu kokku, et
valmistada tablett. Samuti võib kasulikul viisil kasutada disintegrante, nagu näiteks
naatriumtärklisglükolaati või ristsidemetega polü(vinüülpürrolidooni). Antud 15
meetodil valmistatud tabletid lagunevad, kui nad asetada vesikeskkonda (nagu
näiteks gastrointestinaaltrakti), seeläbi paljastades mitmeosakeselise maatriksi,
mis vabastab sealt takroliimuse.
Maatrikssüsteem võib samuti olla hüdrofiilse maatrikstahvli vormis, mis sisaldab
mitmeosakeselise tootena takroliimust ja/või selle analoogi (nt. tahke dispersiooni 20
vormis) ning hüdrofiilse polümeeri kogust, mis on piisav, et tagada takroliimuse
lahustuvuse üle kasulik kontrolli aste. Maatriksi moodustamise jaoks kasulike
hüdrofiilsete polümeeride hulka kuuluvad hüdroksüpropüümetüültselluloos
(HPMC), hüdroksüpropüültselluloos (HPC), polü(etüleenoksiid),
polü(vinüülalkohol), ksantaankummi, karbomeer, karragenaan ja zooglan. 25
Eelistatud materjaliks on HPMC. Samuti võib kasutada teisi sarnaseid
hüdrofiilseid polümeere. Kasutamise korral neelab vesi hüdrofiilse materjali, mis
pikapeale lahustub vees.Takroliimus vabaneb maatriksist nii difusiooni teel kui
maatriksi erosiooni teel. Antud hüdrofiilsete maatrikstahvlite takroliimuse
lahustuvuse määr võib olla kontrollitud kasutatava hüdrofiilse polümeeri koguse, 30
molekulaarmassi ja geeli tugevuse poolt. Üldiselt vähendab suurema koguse

EE - EP1663216 B1 42
hüdrofiilse polümeeri kasutamine lahustuvuse määra, samuti vähendab
lahustuvuse määra kõrgema molekulaarmassiga polümeeri kasutamine. Väiksema
molekulaarmassiga polümeeri kasutamine tavaliselt suurendab lahustuvuse
määra. Maatrikstahvel koosneb tavaliselt umbes 20 kuni 90 kaaluprotsendi
ulatuses takroliimusest ning umbes 80 kuni 10 kaaluprotsendi ulatuses 5
polümeerist.
Eelistatud maatrikstahvel koosneb kaalu arvestades umbes 30% kuni umbes 80%
tahkest dispersioonist, mis sisaldab takroliimust ja/või selle analoogi, umbes 15%
kuni umbes 35% maatriksi moodustajast (nagu näiteks HPMC), 0% kuni 35%
laktoosist, 0% kuni umbes 20% mikrokristallilisest tselluloosist ning umbes 0,25% 10
kuni umbes 2% lubrikandist (nagu näiteks magneesiumstearaat).
Maatrikssüsteemid klassina väljendavad tihti ravimi mittepidevat vabanemist
maatriksist. See tulemus võib olla ravimi vabanemise difusioonil põhineva
mehhanismi tagajärjeks, ning võib kasutada annusvormi geomeetria
modifikatsioone, mille eeliseks on ravimi vabanemise määra muutmine rohkem 15
ühtlaseks.
Teine käesoleva leiutise alla kuuluva takroliimuse vabanemise kontrolliga
annusvormide klass hõlmab membraan-modereeritud või reservuaarsüsteeme.
Antud klassi puhul on takroliimuse reservuaar näiteks tahkes
lahuses/dispersioonis mitmeosakeselise tootena ümbritsetud vabanemise määra 20
piirava membraaniga.Takroliimus läbib membraani antud valdkonnas hästi tuntud
massitranspordi mehhanismide abil, sisaldades, kui mitte piiratud membraanis
toimuva lahustumisega ning sellele järgneva difusiooniga läbi membraani või
difusiooniga läbi membraanis paiknevate vedelikuga täidetud pooride. Antud
individuaalsed reservuaarsüsteemiga annusvormid võivad olla suured, nagu tableti 25
puhul, mis sisaldab ühtainsat suurt reservuaari, või mitmeosakeselised, nagu
kapsli või polüdepoo tablettide puhul, mis sisaldavad suurt hulka
reservuaariosakesi, millest igaüks on membraaniga kaetud. Kate võib olla
mittepoorne, mis on siiski takroliimuse jaoks läbitav (näiteks takroliimus võib
difundeerida otse läbi membraani), või võib see olla poorne. Nagu teiste käesoleva 30
leiutise teostuste puhul, ei ole antud konkreetne transportmehhanism ülioluline.

EE - EP1663216 B1 43
Vastavas valdkonnas tuntud stabiilse vabanemisega katteid võib kasutada, et
valmistada membraani, eriti polümeeri katteid, nagu näiteks tselluloosi ester või
eeter, akrüülpolümeer või polümeeride segu. Eelistatud materjalide hulka kuuluvad
etüültselluloos, tselluloosatsetaat ja tselluloosatsetaatbutüraat. Polümeeri võib
kasutada lahusena või orgaanilises lahustis või vesidispersioonina või lateksina. 5
Katmise protsessi võib läbi viia standardvarustusega, nagu näiteks keevkihiga
(fluid bed)katjaga, Wurster katjaga või pöörleva keevkihiga katjaga.
Soovi korral võib katte läbitavust kohandada, segades kaks või enam materjali.
Eriti kasulik protsess katte poorsuse muutmiseks hõlmab eelnevalt kindlaks
määratud koguse peenelt jaotatud vees lahustuva materjali, nagu näiteks suhkrute 10
või soolade või vees lahustuvate polümeeride lisamist kasutatava membraani
moodustava polümeeri lahusele või dispersioonile (nt. vesilateks). Kui annusvorm
neeldub gastrointestinaaltrakti vesikeskkondda, imbuvad antud vees lahustuvad
membraani lisandid membraanist välja, jättes alles poorid, mis kergendavad ravimi
vabanemist. Membraani katet võib samuti modifitseerida vastavas valdkonnas 15
tuntud plastitseerijate lisamisega.
Eriti kasulik membraani katte asetamise protsessi variatsioon hõlmab
kattepolümeeri lahustamist lahustite segust, mis on valitud nii, et kui kate kuivab,
toimub asetatud kattelahuses faasinihe, mille tulemuseks on poorse struktuuriga
membraan. 20
Üldiselt ei ole vaja tuge membraani mehhaaniliseks tugevdamiseks.
Membraani morfoloogia ei ole kriitilise tähtsusega, senikaua kuni siinkohal
loetletud läbitavuse omadused on täidetud. Membraan või olla amorfne või
kristalliline. Sellel võib esineda ükskõik millise konkreetse protsessi teel
moodustunud ükskõik milline morfoloogia kategooria ning võib olla näiteks 25
kahekihiliselt polümeriseeritud membraan (mis sisaldab õhukest vabanemise
määra piiravat kihti või poorset tuge), poorne hüdrofiilne membraan, poorne
hüdrofoobne membraan, hüdrogeel-membraan, ioonmembraan ning teised
sarnased materjalid, mida iseloomustab takroliimuse kontrollitud läbitavus.

EE - EP1663216 B1 44
Eesmärgiks on vähendada ülemise gatrointestinaaltrakti ekspositsiooni
takroliimuse kõrgetele kontsentratsioonidele. Vastavalt sellele hõlmab sobilik
annusvorm selliseid vorme, mis hõlmavad spetsiifilist viivitust enne takroliimust
kontrollitud vabanemise algust. Näidisteostus võib olla illustreeritud tableti (või
konkreetse materjali poolt), mis sisaldab takroliimust sisaldavat südamikku, mis on 5
esmase kihina kaetud takroliimuse stabiilse vabanemise jaoks kasuliku tüübi
polümeerse materjaliga ning teise kihina kaetud ravimite hilinenud vabanemise
jaoks kasuliku tüübiga, kui annusvorm seeditakse. Esimene kiht kantakse peale
ning ümbritseb tabletti või individuaalseid osakesi. Teine kiht kantakse peale ning
see ümbritseb esimest kihti. 10
Tabletti võib valmistada antud valdkonnas üldtuntud meetodite abil ning sisaldab
terapeutiliselt kasulikku kogust takroliimust pluss selliseid abiaineid, mis on
vajalikud antud meetodite abil tableti moodustamiseks.
Esimene kiht peab olema antud valdkonnas üldtuntud stabiilse vabanemisega
kate, eriti polümeerkatted, et valmistada sellise membraani, nagu eelnevalt arutati 15
reservuaarsüsteemide juures. Või võib see olla kontrollitud vabanemisega
maatriksi südamik, mis on kaetud teist korda hilinenud vabanemisega materjaliga.
Tableti teise kihi valmistamiseks kasulikud materjalid on polümeerid, mis on antud
valdkonnas tuntud kui enteerilised katted ravimite hilinenud vabanemiseks. Kõige
tavapärasemalt on nad pH-tundlikud materjalid, nagu näiteks 20
tselluloosatsetaatftalaat, tselluloosatsetaattrimellitaat, hüdroksüpropüül-
metüültselluloosftalaat, polü(vinüülatsetaatftalaat) ning akrüülpolümeerid, nagu
näiteks Eudragit L-100 (Röhm Pharma) ning seotud materjalid, nagu on
detailsemalt kirjeldatud allpool olevas peatükis „Hilinenud vabanemine“. Hilinenud
vabanemisega katte paksust kohandatakse, et saavutada soovitud 25
hilinemisomadused. Üldiselt on paksemad katted resistentsemad erosioonile ning
selle tagajärjel väljendavad pikemat ja efektiivsemat hilinemist. Eelistatud katted
varieerivad alates umbes 30 µm paksusest kuni umbes 3 mm paksuseni.
Kui kasutatakse hüdrofoobset maatriksmaterjali, nagu näiteks
glütserüülmonostearaat, ei ole hilinemist võimaldav kate vajalik. Tablett ei vabasta 30

EE - EP1663216 B1 45
takroliimust, kuni on jõutud ensümaatilise lagundamise alani, täpsemalt pärast
duodeenumi.
Seedimise ajal läbib kahekordse kattega tablett mao, kus teine kate aitab vältida
takroliimuse vabanemist seal valitsevates happelistes tingimustes. Kui tablett
väljub maost ning siseneb peensoolde, kus pH on kõrgem, teine kate erodeerub 5
või lahustub vastavalt valitud materjali füsikokeemilistele omadustele. Teise katte
erosioonil või lahustumisel aitab esimene kate vältida takroliimuse kohest või kiiret
vabanemist ning moduleerib vabanemist, et vältida tippkontsentratsioonide
saavutamist ning seeläbi minimeerides kõrvaltoimeid.
Teine näide on mitmeosakeseline süsteem, mille puhul on iga osake kahekordselt 10
kaetud viisil, nagu on ülalpool tablettide katmise juures kirjeldatud; esimene kiht on
polümeeriga, mis on disainitud vabastama takroliimust stabiilselt, ning seejärel
kaetud polümeeriga, mis on disainitud viivitama vabanemise algust
gastrointestinaaltrakti keskkonda, kui annusvormi seeditakse.
Takroliimuse vabanemise määr stabiilse vabanemise katet omavast 15
mitmeosakelisest süsteemist (st. mitmeosakeseline süsteem enne, kui nad saavad
hilinenud vabanemise katte) ning katte modifitseerimise meetodid on samuti
kontrollitud eelnevalt reservuaarsüsteemi takroliimuse mitmeosakeselise süsteemi
juures arutletud faktorite poolt.
Teine kahekordse kattega mitmeosakeselise süsteemi katmise jaoks mõeldud 20
membraan on hilinenud vabanemise kate, mis asetatakse esimese stabiilse
vabanemise katte peale, nagu on ülalpool tablettide juures avalikustatud, ning
võivad olla valmistatud samadest materjalidest. Peaks tähele panema, et
niinimetatud „enteeriliste“ materjalide kasutamine antud teostuse läbiviimiseks
erineb märkimisväärselt nende tavapäraste enteeriliste annusvormide 25
valmistamise jaoks mõeldud kasutusest. Tavapäraste enteeriliste vormide puhul
on eesmärgiks viivitada ravimi vabanemist, kuni annusvorm on läbinud mao ning
seejärel viiakse doos duodeenumisse. Takroliimuse otseselt ja täielikult
duodeenumisse doseerimine võib olla siiski ebasoovitav, kuna antud leiutise
eesmärgiks on kõrvaltoimete minimeerimine või vältimine. Seetõttu võib antud 30
teostuse puhul tavapäraste enteeriliste polümeeride kasutamise korral olla vajalik

EE - EP1663216 B1 46
neid tarvitada oluliselt tihedamalt kui tavapärase praktika puhul, et viivitada ravimi
vabanemist, kuni annusvorm jõuab alumisse gastrointestinaaltrakti. Siiski on
samuti võimalik teostada takroliimuse stabiilset või kontrollitud kohaleviimist,
pärast seda, kui hilinenud vabanemise kate on lahustunud või erodeerunud;
seetõttu võivad antud teostuse eelised väljenduda hilinenud vabanemisega 5
elemendi ja stabiilse vabanemisega elemendi õige kombinatsiooni korral, ning
hilinenud vabanemisega osa üksi võib, aga ei pruugi ilmtingimata vastavuses olla
USP enteeriliste kriteeriumitega. Hilinenud vabanemisega katte paksust
kohandatakse, et saavutada soovitud hilinemine. Üldiselt on paksemad katted
resistentsemad erosioonile ning selle tulemusena omavad pikemat 10
hilinemisperioodi.
Esimene hilinenud vabanemisega annusvorm on „pH-sõltuva kattega annusvorm“,
nagu näiteks tablett või kapsel. Tableti puhul see hõlmab tableti südamikku, mis
sisaldab takroliimust näiteks tahke lahuse/dispersioonina mitmeosakeselise
tootena, kontrollitud vabanemise maatriksi, näiteks HPMC, disintegranti, lubrikanti 15
ning ühte või enamat farmatseutlilist kandjat; antud südamik on kaetud
materjaliga, eelistatult polümeeriga, mis sisuliselt lahustumatu ja mao pH juures
mitteläbitav, ning mis on rohkem lahustuv ja läbitav peensoole pH juures.
Eelistatult on kattepolümeer sisuliselt lahustumatu ja läbimatu pH<5,0 juures ning
vees lahustuv pH>5,0 juures. Tableti südamik võib olla kaetud polümeeri 20
kogusega, mis on piisav kindlustamaks, et annusvormist ei toimu praktiliselt mitte
mingit takroliimuse vabanemist, kuni annusvorm on maost väljunud ning viibinud
peensooles umbes 15 minutit või enam, eelistatult umbes 30 või enam, seeläbi
kindlustades, et duodeenumisse vabaneb minimaalne takroliimuse kogus. Samuti
võib kasutada pH-tundlike polümeeride segusid vees lahustuva polümeeriga. 25
Tabletid on kaetud polümeeri kogusega, mis sisaldab alates umbes 10 kuni 80
kaaluprotsenti takroliimust sisaldava tableti südamiku kaalust. Eelsitatud tabletid
on kaetud polümeeri kogusega, mis sisaldab alates umbes 15 kuni 50
kaaluprotsenti takroliimust sisaldava tableti südamiku kaalust.
pH-tundlike polümeeride hulka, mis on väga lahustumatud ja läbitamatud mao pH 30
juures, kuid mis on rohkem lahustuvad ja läbitavad peensoole ja jämesoole pH
juures, kuuluvad polüakrüülamiidid, ftalaadi derivaadid, nagu näiteks süsivesinike

EE - EP1663216 B1 47
happelised ftalaadid, amüloosatsetaatftalaat, tselluloosatsetaatftalaat, teised
tselluloosi estri fralaadid, tselluloosi eetri ftalaadid,
hüdroksüpropüültselluloosftalaat, hodroksüpropüületüültselluloosftalaat, hüdroksü-
propüülmetüültselluloosftalaat, metüültselluloosftalaat, polüvinüülatsetaatftalaat,
polüvinüülatsetaatvesinikftalaat, naatriumtselluloosatsetaatftalaat, tärklise 5
happeline ftalaat, stüreen-maleiinhappe dibutüülftalaadi kopolümeer, stüreen-
maleiinhappe polüvinüülatsetaadftalaadi kopolümeer, stüreeni ja maleiinhappe
kopolümeerid, polüakrüülhappe derivaadid, nagu näiteks akrüülhape ja akrüülestri
kopolümeerid, polümetakrüülhape ja selle estrid, polüakrüülmetakrüülhappe
kopolümeerid, šellak ja vinüülatsetaat ja krotoonhappe kopolümeerid. 10
Eelistatud pH-tundlike polümeeride hulka kuuluvad šellak, ftalaadi derivaadid, eriti
tselluloosatsetaatftalaat, polüvinüülatsetaatftalaat ning hüdroksüpropüül-
metüültselluloosftalaat; polüakrüülhappe derivaadid, eriti polümetüülmetakrülaat
segatuna akrüülhappe ja akrüülestri kopolümeeridega; ning vinüülatsetaat ja
krotoonhappe kopolümeerid. 15
Hilinemisaega enne takroliimuse vabastamist pärast seda, kui „pH-sõltuva kattega
tablett“ annusvormina on väljunud maost, võib kontrollida vastavate koguste
Eudragit-L® ja Eudragit-S® valikuga kattes ning katte paksuse valikuga. Eudragit-
L® kiled lahustuvad pH>6,0 väärtuste juures ning Eudragit-S® kiled lahustuvad
väärtuste juures >7,0, ning segud lahustuvad vahepealsete pH väärtuste juures. 20
Kuna duodeenumi pH on ligikaudu 6,0 ning jämesoole pH on ligikaudu 7,0,
pakuvad Eudragit-L® ja Eudragit-S® segudest valmistatud katted duodeenumi
kaitset takroliimuse eest. Kui soovitakse viivitada takroliimuse vabanemisega kuni
takroliimust sisaldav „pH-sõltuva kattega tablett“ on jõudnud jämesoolde, võib
kasutada kattematerjalina Eudragit-S®, nagu on kirjeldatud väljaandes Dew jt. (Br. 25
J. Clin. Pharmac. 13 (1982) 405-408). Et viivitada takroliimuse vabanemisega
umbes 15 minutit või enam, eelistatult 30 minutit või enam pärast seda, kui
annusvorm on väljunud maost, sisaldavad eelistatud katted alates umbes 9:1 kuni
umbes 1:9 Eudragit-L® / Eudragit-S®, eelistatult alates umbes 9:1 kuni umbes 1:3
Eudragit-L® / Eudragit-S®. Kate võib sisaldada alates ligikaudu 3 kuni ligikaudu 70 30
kaaluprotsenti katmata tableti südamiku kaalust. Eelistatult sisaldab kate ligikaudu
5 kuni ligikaudu 50 kaaluprotsenti katmata tableti südamiku kaalust.

EE - EP1663216 B1 48
Kasutused
Käesoleva leiutise alla kuuluvat farmatseutilist kompositsiooni võib kasutada tahke
suukaudse annusvormi, nagu näiteks tablettide, kapslite või kotikeste
valmistamiseks; või graanulite, pelletite, mikrosfääride või nanoosakeste
valmistamiseks. 5
Eelistatult kasutatakse farmatseutilist kompositsiooni kohese vabanemisega tahke
annusvormi või hilinenud vabanemisega tahke annusvormi valmistamiseks.
Käesoleva leiutise alla kuuluva kompositsiooni veel üheks eeliseks on võimalus
saavutada efektiivne terapeutiline vastus vähenenud doosi juures võrreldes
tavalise suukaudse raviga. Seetõttu peetakse võimalikuks, et käesoleva leiutise 10
tahke annusvorm on suukaudsel manustamisel vastavale imetajale doosis, mis on
kõige enam umbes 85% (massifraktsioonist), nagu näiteks kõige rohkem ligikaudu
80% (massifraktsioonist), kõige rohkem ligikaudu 75% (massifraktsioonist), kõige
rohkem ligikaudu 70% (massifraktsioonist), kõige rohkem ligikaudu 70%
(massifraktsioonist), kõige rohkem ligikaudu 65% (massifraktsioonist), kõige 15
rohkem ligikaudu 60% (massifraktsioonist), kõige rohkem ligikaudu 55%
(massifraktsioonist) või kõige rohkem ligikaudu 50% (massifraktsioonist) Prograf®
või sarnase kommertsiaalselt saadaoleva takroliimust sisaldava toote vormis
manustatud takroliimuse doosist põhiolemuselt bioekvivalentne Prograf®’i või
sarnase kommertsiaalselt saadaoleva takroliimust sisaldava tootega. 20
Ükskõik millised käesoleva leiutise alla kuuluvad takroliimust sisaldavad
annusvormid ja kompositsioonid võivad parandada seisundite ravi, mis vastavad
takroliimuse ravile.
Takroliimus on näidustatud (või on see soovitatav) selliste haiguste raviks, nagu
näiteks erinevate organite või kudede (nt. süda, neer, maks, luuüdi, nahk, 25
sarvkest, kops, pankreas, peensool, jäse, lihas, närv, intervertebraalketas,
trahhea, müoblast, kõhr jne.) transplantaatide äratõukereaktsioonid; transplantaat-
peremehe-vastu reaktsioonid, mis järgnevad luuüdi transplantatsioonile;
autoimmuunsed haigused, nagu näiteks reumatoidartriit, süsteemne
erütematoosne luupus, Hashimoto türeoidiit, multiipelne skleroos, myasthenia 30

EE - EP1663216 B1 49
gravis, 1. tüüpi diabeet jne.; infektsioonid, mis on põhjustatud patogeensete
mikroorganismide poolt (nt. Aspergillus fumigatus, Fusarium oxysporum,
Trichphyton asteroides jne.); põletikulised või hüperproliferatiivsed nahahaigused
või immunoloogiliselt vahendatud haiguste nahamanifestatsioonid (nt. psoriaas,
atoopiline dermatiit, kontaktdermatiit, eksematoidne dermatiit, seborröiline 5
dermatiit, lame lihhen, pemfiigus, bulloosne pemfigoid, bulloosne epidermolüüs,
urtikaaria, angioödeem, vaskuliidid, erüteem, dermaalne eosinofiilia,
erütematoosne luupus, akne ja koldeline juuste väljalangemine); silma
autoimmuunsed haigused (nt. keratokonjuntiviit, vernaalne konjunktiviit, Bechet’
tõvega seotud uveiit, keratiit, herpeskeratiit, keratokoonus, sarvkesta epiteliaalne 10
düstroofia, keratoleukoom, okulaarne pemfiigus, Mooren’i haavand, skleriit,
Graves’i oftalmopaatia, Vogt-Koyanagi-Harada sündroom, keratoconjunctivitis
sicca (kuiv silm), phlyctenule konjunktiviit, iridotsükliit, sarkoidoos, endokriinne
oftalmopaatia jne); pöörduvad obstruktiivsed hingamisteede haigused (astma, nt.
bronhiaalastma, allergiline astma, mitteallergiline astma, allergiline astma ja 15
tolmuastma, eriti krooniline või kauaaegne astma, nt. hiline astma ja
hingamisteede hüperreaktiivsus, bronhiit jne.; mukoossed või vaskulaarsed
põletikud (nt. maohaavand, isheemiline või trombootiline vaskulaarne vigastus,
isheemilised soolehaigused, enteriit, nekrotiseeriv enterokoliit, soolevigastused,
mis on seotud kuumapõletustega, leukotrieen B4-vahendatud haigused); 20
soolepõletikud/allergiad (nt. tsöliaakiahaigused, proktiit, eosinofiilne gastroenteriit,
mastotsütoos, Crohn’i tõbi ja haavandiline koliit); toiduga seotud allergilised
haigused koos gastrointestinaaltraktiväliste sümptomaatiliste manifestatsioonidega
(nt. migreen, riniit ja ekseem); neeruhaigused (nt. inerstitsiaalne nefriit,
Goodpasture’i sündroom, hemolüütilis-ureemiline sündroom ja diabeetiline 25
nefropaatia); närvisüsteemi haigused (nt. multiipelne müosiit, Guillain-Barre
sündroom, Meniere’i haigus, multiipelne neuriit, solitaarne neuriit, ajuinfarkt,
Alzheimer’i tõved, Parkinsoni tõved, amüotroopne lateraalne skleroos (ALS) ja
radikulopaatia); aju isheemilised haigused (nt. peavigastus, ajuhemorraagia, nt.
subarahnoidaalne hemorraagia, intratserebraalne hemorraagia, ajutromboos, 30
ajuembolism, südameseiskus, insult, mööduv isheemiline atakk (TIA),
hüpertensiivne entsefalopaatia, ajuinfarkt); endokriinsed haigused (nt.
hüpertüreoidism ja Basedow’ tõbi); hematoloogilised haigused (nt. erütrotsüütide

EE - EP1663216 B1 50
aplaasia, aplastiline aneemia, hüpoplastiline ameenia, idiopaatiline
trombotsütopeeniline purpur, autoimmuunne hemolüütiline aneemia,
agranulotsütoos, pernitsioosne aneemia, megaloblastiline aneemia ja
anerütroplaasia), luuhaigused (nt. osteoporoos); hingamisteede haigused (nt.
sarkoidoos, pulmonaarfibroos ja idiopaatiline interstitsiaalne pneumoonia); 5
nahahaigused (nt. dermatomüosiit, leukoderma, tavaline soomustõbi,
fotosensitiivsus ja kutaanne T-rakk lümfoom); vereringehaigused (nt.
arterioskleroos, ateroskleroos, aortiidisündroom, nodoosne polüarteriit ja
müokardiit), kollageenhaigused (nt. skleroderma, Wegeneri granuloom ja Sjögren’i
sündroom); adipoossus, eosinofiilne fastsiit, periodontaalsed haigused (nt. 10
igemekahjustus, periodontiit, alveolaarluu või substantia ossea dentis), nefrootiline
sündroom (nt. glomerulonefriit); androgeenne alopeetsia, seniilne alopeetsia;
lihasdüstroofia, püoderma ja Sezary sündroom; kromosoomide häiretega seotud
haigused (nt. Downi sündroom); Addisoni tõbi; aktiivsed hapniku poolt vahendatud
haigused (nt. organkahjustus, nt. organite (nt. südame, maksa, neeru, seedetrakti 15
jne.) isheemilised tsirkulatsioonihäired, mis on seotud säilitamise,
transplantatsiooni või isheemiliste haigustega (nt. tromboos, südameinfarkt jne.),
soolestiku haigused (nt. endotoksiinidest põhjustatud šokk, pseudomembranoosne
koliit ja ravimite või radiatsiooni poolt indutseeritud koliit); neeruhaigused (nt.
isheemiline äge neerupuudulikkus, krooniline neerupuudulikkus); kopsuhaigused 20
(nt. toksikoos, mis on põhjustatud kopsu hapniku või ravimite (nt. Paracort,
bleomütsiin) poolt, kopsuvähk ja kopsuemfüseem); silmahaigused (nt. katarakt,
raualadestushaigused (siderosis bulbi), pigmentoosne retiniit, vanaduskae,
klaaskeha armistumine, sarvkesta alkaalne põletus); dermatiit (nt. multiformne
erüteem, lineaarne immunoglobuliin A bulloosne dermatiit, tsementdermatiit); ning 25
teised haigused (nt. gingiviit, periodontiit, sepsis, pankreatiit ning haigused, mis on
põhjustatud keskkonnasaaste poolt (nt. õhusaaste), vananemine, kartsinogeen,
kartsinoomi metastaasid ja hüpobaropaatia); histamiini vabanemise või leukotrieen
C4 vabanemisega seotud haigused; angioplastikale ja postkirurgilise adhesioonile
järgnev koronaararteri restenoos; autoimmuunsed haigused ja põletikulised 30
seisundid (nt. primaarne mukoosne ödeem, autoimmuunne atroofiline gastriit,
enneaegne menopaus, mehe striilsus, juveniilne diabeet, pemphigus vulgaris,
pemfigoid, sümpaatiline oftalmia, läätse poolt indutseeritud uveiit, idiopaatiline

EE - EP1663216 B1 51
leukopeenia, aktiivne krooniline hepatiit, idiopaatiline tsirroos, diskoidne
erütematoosne luupus, autoimmuunne orhiit, artriit (nt. arthritis deformans), või
polükondriit); inimese immuunpuudulikkuse viirus (HIV) infektsioon, AIDS;
allergiline konjunktiviit; trauma, põletuse või kirurgia tõttu tekkinud hüpertroofiline
arm ja keloid. 5
Lisaks on tritsüklilistel makroliididel, nagu näiteks takroliimus, regeneratiivne toime
maksale ja/või hepatotsüütide hüpertroofiat ja hüperplaasiat stimuleerivad toimed.
Seetõttu on käesoleva leiutise alla kuuluv farmatseutiline kompositsioon kasulik
maksahaiguste (näiteks immunoloogilised haigused, nt. kroonilised
autoimmuunsed maksahaigused, nagu näiteks autoimmuunsed maksahaigused, 10
primaarne biliaarne tsirroos või skleroseeriv kolangiit), osalise maksa resektsiooni,
ägeda maksa nekroosi (nt. toksiinide, viirushepatiidi, šoki või anoksia poolt
põhjustatud nekroosi), B-hepatiidi, mitte-A-mitte-B-hepatiidi, hepatotsirroosi ja
maksapuudulikkuse (nt. fulminantse hepatiidi, hilise algusega hepatiidi ja
ägenenud kroonilise maksapuudulikkuse (äge maksapuudulikkus krooniliste 15
maksahaiguste foonil)) ravi ja/või profülaktika efekti tõstmise jaoks
Lisaks on käesoleva leiutise alla kuuluv kompositsioon kasulik erinevate haiguste
ennetamise ja/või ravi efekti tõstmiseks tritsükliliste makroliidide kasuliku
farmakoloogilise aktiivsuse tõttu, nagu näiteks kemoterapeutilise efekti
suurendamine, tsütomegaloviirusinfektsiooni, põletikuvastase aktiivsuse, 20
peptidüül-prolüül isomeraasi või rotamaasi vastase aktiivsuse inhibeerimine,
malaariavastane aktiivsus, kasvajavastane aktiivsus jne.
Materjalid ja meetodid
Materjalid
Takroliimus (varustatud Eurotrade'i poolt); partii nr. RD 03-111 25
Laktoosmonohüdraat 200 (DMV poolt)
Granuleeritud ränimoksiid, Aeroperl® 300, (Degussa)
Polüetüleenglükool, Pluracol® E6000 (BASF poolt)
Poloksameer 188, Pluronic® F-68 (BASF poolt)

EE - EP1663216 B1 52
Glütserüülmonostearaat, Rylo® MD50, (Danisco Cultor'i poolt), Ph.Eur.; partii nr.
4010056276
Avicel PH200 (mikrokristalliline tselluloos) (FMC poolt)
Laktoos DCL 11 (DMV poolt)
Magneesiumstearaat 5
Kroskarmelloosnaatrium, Ac-Di-Sol® (FMC poolt)
Eudragit® L30D.55 (Degussa poolt)
Trietüültsitraat (Merck poolt)
Vahuvastane emulsioon (Unikem'i poolt)
Mikrotalk 10
HPMC viitab ShinEtsu poolt toodetud Metalose 90SH-le (tüüp 2910, 2208) or
Metolose 60SH-le (tüüp 2910), mis on saadaolev erinevate polümerisatsiooni
astmetena (viskoosus 3-100,000cP).
Tabletid, kapslid või graanulid võivad olla enteeriliselt kaetud erinevat tüüpi
polümeeride poolt, nagu näiteks hüdroksüpropüülmetüül-15
tselluloosatsetaatsuktsinaat (Aqoat), tselluloosatsetaatftalaat CAP,
hüdroksüpropüülmetüültselluloosftalaat HP-MCP või metakrüülhappe
kopolümeerid, nagu näiteks Eugragit L30D, Eudragit 100/S, Eudragit 100/L.
Võrdlus enne takroliimuse formulatsiooni:
Prograf® Hard Gelatin Capsules (kõvad želatiinikapslid), toodetud Fujisawa 20
Ireland Ltd. poolt
Koostisosad mg
Takroliimus, anhüdroosne 1,0
Želatiin 6,9
Hüpromelloos 1,0 25
Laktoosmonohüdraat 24,7
Magneesiumstearaat 0,3
Šellak q.s.
Sojaletsitiin q.s.
Raudoksiid punane (E 172) q.s. 30
Titaandioksiid (E171) q.s.

EE - EP1663216 B1 53
Dimetikoon (E900) q.s.
Meetodid
Kaaluvariatsiooni määramine
Siinkohal välja toodud Näidetes valmistatud tablettide peal viidi läbi test
kaaluvariatsiooni määramiseks, mis teostati kooskõlas Ph.Eur.-iga. 5
Keskmise tableti kõvaduse määramine
Siinkohal välja toodud Näidetes valmistatud tablettide peal viidi läbi test tableti
kõvaduse määramiseks, kasutades Schleuniger Model 6D aparatuuri ja teostati
kooskõlas aparaadi üldintruktsioonidega kooskõlas.
Lagunemisaja määramine 10
Aeg, mis kulus tableti lagunemiseks, st. osakesteks või aglomeraatideks
lagunemiseks, määratu kooskõlas Ph.Eur.-iga.
Geomeetrilise kaalu keskmise diameetri dgw määramine
Geomeetrilise kaalu keskmine diameeter määrati, kasutades laserdifraktsiooni,
mis pihustas saadud konkreetse materjali (või stardimaterjali) õhku. Mõõtmised 15
viidi läbi 1-baarilise dispersioonirõhu juures Sympatec Helos aparatuuriga, mis
salvestab ekvivalentse sfäärilise diameetri jaotuvuse. Antud jaotuvus sobitatakse
logaritmilisse normaalsesse maht-suurus jaotusesse.
Siinkohal kasutatud „geomeetrilise kaalu keskmine diameeter“ tähendab
logaritmilise normaalse maht-suurus jaotuse keskmist diameetrit. 20
In vitro lahustuvustestid
Käesoleva leiutise alla kuuluvate kompositsioonide ja annusvormide peal viidi läbi
järgmised testimeetodid.
Test 1: 25

EE - EP1663216 B1 54
In vitro lahustuvustest vastavalt USP meetodile A, hilinenud vabanemisega
osakesed (USP labameetod; pöörlemiskiirus: 50 rpm; 37˚C; pärast 2 tundi
happelises keskkonnas, muudetakse keskkond fosfaatpuhvriks, mille pH on 6,8).
Test 2:
In vitro lahustuvustest vesilahustuvuskeskkonnas, mille pH oli kohandatud 5
väärtuseni 4,5 (900 ml vett koos 0,005% HPC (hüdroksüpropüütselluloosiga)
kohandatud pH väärtuseni 4,5; 37˚C; USP labameetod; pöörlemiskiirus: 50 rpm).
Käesoleva leiutise alla kuuluvad farmatseutilised kompositsioonid ja annusvormid
on näitlikustatud näidetes 1-7. Käesoleva leiutise alla kuuluvate kompositsioonide
ja annusvormide in vitro lahustuvustesti tulemused on näites 8. 10
NÄIDE 1
Modifitseeritud vabanemisega polüdepoo kapslid, mis põhinevad
hüdroksüpropüütselluloosi vesinikkolloidmaatriksi paisumisel
Aine % mg
Takroliimus 0,50 1,00
HPMC 20,00 40,00
Laktoos 200 30,00 60,00
PEG 6000 34,65 69,30
Poloksameer 188 14,85 29,70
Kokku 100,00 200,00
Takroliimus lahustati polüetüleenglükool 6000-s ja poloksameer 188-s (70:30 15
(massifraktsiooni) suhe) 70˚C juures. Lahus pihustati 150 g laktoosi ja 100 g
HPMC segule keevkihis Strea-1. Granulaarne toode sõeluti läbi 0,7 mm sõela ning
sisestati kõvadesse želatiinkapslitesse (200 mg).
NÄIDE 2
Modifitseeritud vabanemisega maatriksi tableid, mis põhinevad 20
hüdroksüpropüütselluloosi vesinikkolloidmaatriksi paisumisel

EE - EP1663216 B1 55
Aine % mg
Takroliimus 0,50 1,00
HPMC 19,90 40,00
Laktoos 200 29,85 60,00
PEG 6000 34,48 69,30
Poloksameer 188 14,78 29,70
Magneesiumstearaat 0,50 1,01
Kokku 100,00 201,01
Takroliimus lahustati polüetüleenglükool 6000-s ja poloksameer 188-s (70:30
(massifraktsiooni) suhe) 70˚C juures. Lahus pihustati 250 g laktoosile keevkihis
Strea-1. Granulaarne toode sõeluti läbi 0,7 mm sõela ja segati HPMC ja
magneesiumstearaadiga 0,5 minutit Turbula segajas.
Segu suruti kokku 8 mm tablettideks, mis sisaldasid 1 mg aktiivset koostisosa (200 5
mg tablett), tableti kuju nõgus. Keskmine lagunemisaeg: 20 min. Kõvadus: 45 N
NÄIDE 3
Enteeriline kate
Näidete 1 ja 2 kapslid ja tableid kaeti seejärel järgmiste enteeriliste katetega, et
saada aktiivse koostisosa hilinenud vabanemine pärast manustamist. 10

EE - EP1663216 B1 56
Koostisosad %
Eudragit® L30D 40
Puhastatud vesi 52
Trietüülatsetüültsitraat 1,8
Vahuvastane emulsioon 0,2
Talk 6
Kokku 100
Kattesuspensioon valmistati, segades trietüülatsetüültsitraadi, vahuvastase
emulsiooni ja puhastatud vee Ultra Turrax aparatuuris pöördekiirusega 9500 rpm
30 minuti jooksul. 1 minuti pärast lisati talk. Segu viidi läbi sõela nr. 300 ja segati
magnetsegajaga. Eudragit viidi läbi sõela nr. 300 ja lisati segule, mida segati 5 5
minutit.
Katmisprotsessi läbiviimise tingimused olid järgmised: sisendsüsteemi
temperatuur 40˚C, väljundsüsteemi temperatuur 31˚C, õhu sissevool 140 cm3
tunnis ning katmisaeg ligikaudu 50 minutit (300 g kattematerjali). Kaeti ligikaudu
400 g tablette või 200 g kapsleid. 10
Kilega kaetud tablette ja kapsleid hoiti 48 tundi 30 ˚C juures enne lahustuvusteste.
NÄIDE 4
Enteerilise kattega tablett, mille südamik põhineb PEG 6000/Poloksameer 188-l
ning enteeriline kate, mis põhineb Eudragit L30D 55-l.
Tableti südamiku koostis: 15
Ühend % mg
Takroliimus 1,98 2,00
Laktoosmonohüdraat,
Laktoos 200
40,50 40,91
PEG 6000 33,26 33,60
Poloksameer 188, Lutrol
68
14,40 14,40

EE - EP1663216 B1 57
Magneesiumstearaat 0,50 0,51
Talk 4,50 4,55
Kroskarmelloosnaatrium,
Ac-di-sol
5,00 5,05
100,00 101,01
Takroliimuse tableti südamik toodeti, lahustades PEG 6000-s temperatuuril üle
80˚C. Lisati poloksameer 188 ning lahus kuumutati temperatuurile üle 80˚C. Lahus
pihustati toiteühikule Phast FS1.7 200 g laktoosmonohüdraadile keevkihile Phast
FB100. Tulemusena saadud graanulid sõeluti läbi Comill sõela 1397, 4500 rpm
ning segati kroskarmelloosnaatriumiga 3 minutit Turbula segajas. 5
Magneesiumstearaat ja talk sõeluti läbi sõela nr. 300 ja segati Turbula segajas 3
minutit. Graanulid segati magneesiumstearaadi ja talgiga (1:9) 0,5 minutit Turbula
segajas.
Saadud segu pressiti 6 mm tablettideks, mis sisaldasid 2 mg aktiivset koostisosa
(100 mg tablett), tableti kuju nõgus. 10
Keskmine lagunemisaeg: 7 minutit. Kõvadus: 65 N
Enteeriline kate:
Enteeriline kate põhineb akrüülpolümeer Eudragit L30D-55-l. Eudragit L30D
esineb vesilateksi suspensiooniga, mis tekitab vees lahustumatu kile, kui katmise
ajal vesi aurustatakse. Polümeer on lahustumatu pH väärtuste juures alla 5,0 ning 15
lahustub kergesti, kui pH väärtused on üle 6,0. Kilekatte koostis on:
Ühend %
massifraktsioonist
Eudragit L30D-55 40
Vesi 52
Trietüültsitraat 1,8
Vahuvastane
emulsioon
0,2
Talk (mikro) 6
Kokku 100

EE - EP1663216 B1 58
Kasutatud kilepolümeeri (Eudragit) kogus põhines järgmisel arvutusel: mg
kilepolümeeri tabletipinna cm2 kohta. Enteerilise katte paksus oli 80 µm. Kasutatud
kile paksuse tõestus põhines tableti kõrguse suurenemise mõõtmisel digitaalse
mikromeetriga. Kilega katmise protsess viidi läbi Phast FB100 keevkihil, millel oli
Wurster’i-sarnane sisend. Protsessi tingimused olid järgmised: sisendsüsteemi 5
õhutemperatuur 50˚C, õhu sissevool 100 cm3 tunnis; toote temperatuur 38˚C;
toitekiirus 15g/min.
Pärast katmist nõuab kile moodustumine kaetud tablettide hoiustamist, st. 30˚C 48
tunni jooksul ahjus. Alternatiivselt võib efektiivsemaks tulemuseks hoida kaetud
tablette 40˚C juures 24 tundi. 10
NÄIDE 5
Kontrollitud vabanemisega PEG 6000/Poloksameer 188 tablett, mis põhineb
HPMC maatriksil.
Tableti koostis:
Ühend % mg
Takroliimus 1,21 2,00
Lakroosmonohüdraat, Laktoos
200
24,75 40,91
PEG 6000 20,33 33,60
Poloksameer 188, Lutrol 68 8,71 14,40
Magneesiumstearaat 0,50 0,83
Talk 4,50 7,44
Hüdroksüpropüülmetüültselluloos,
Metolose 90SH 15000
40,00 66,12
100,00 165,29
Takroliimus lahustati PEG 6000-s temperatuuril üle 80˚C. Lisatakse poloksameer 15
188 ja lahus kuumutatakse temperatuurile üle 80˚C. Lahus pihustatakse
toiteühikule Phast FS1.8 200g laktoosmonohüdraadile keevkihile Phast FB100.
Tulemusena saadud graanulid sõeluti läbi Comill sõela 1397, 4500 rpm ning segati
hüdroksüpropüülmetüültselluloosiga 3 minutit Turbula segajas.

EE - EP1663216 B1 59
Magneesiumstearaat ja talk sõeluti läbi sõela nr. 300 ja segati Turbula segajas 3
minutit. Graanulid segati magneesiumstearaadi ja talgiga (1:9) 0,5 minutit Turbula
segajas.
Saadud segu pressiti 8 mm tablettideks, mis sisaldasid 2 mg aktiivset koostisosa
(165 mg tablett), tableti kuju nõgus. 5
Keskmine lagunemisaeg: 2 tundi 34 minutit. Kõvadus: 50 N
NÄIDE 6
Kontrollitud vabanemisega tableti formulatsioon, mis põhineb erodeeruval HPMC
maatriksil; HPMC on lisatud kui osa intragranulaarsest faasist.
Sulamisgranulatsioon. 10
Tableti koostis:
Koostisosa mg
Takroliimus 2
Laktoos 80
PEG 6000 15
Poloksameer 188 6
Metoloos SH 90 80
Avicel PH200 60
Magneesiumstearaat 2
Kokku 245
Tableti formulatsioon põhines sulamisgranulatsioonil suure lõikejõuga segajas
Pellmix 1/8. Mikroniseeritud takroliimus segati 640 g laktoos 125 mesh ja 120 g
polüetüleenglükool 6000, 48 g poloksameer 188 ja 640 g
hüdroksüpropüülmetüültselluloos Metolose SH 90 15000 cP suure lõikejõuga 15
segajas. Segamisnõu kate kuumutati 80˚C juurde ning segu kuumutati laba
pöörlemiskiiruse juures 1000 rpm, kuni PEG-i ja poloksameeri sulamispunktini.
Pärast sulamist jätkati muljumist 4 minutit pöördekiirusel 800 rpm. Graanulid
sõeluti läbi sõela suurusega 0,7 mm ja jahutati kandikul. Graanulid segati 480 g
Avicel PH200 3 minutit ning 16 g magneesiumstearaadi lisamise jaoks ning pärast 20

EE - EP1663216 B1 60
lisamist veel 0,5 minutit. Segu suruti tablettidesse ühelöögilise tabletimasina Diaf
TM20 abil. Tableti diameeter: 8 mm. Tableti kuju: ümmargune, nõgus.
NÄIDE 7
Enteerilise kattega tableti formulatsioon (sulamisgranulatsioon ja enteerilise
kattega tabletid) 5
Tableti koostis:
Koostisosa mg
Takroliimus 2
Laktoos 80
PEG 6000 15
Poloksameer 188 6
Avicel PH200 60
Magneesiumstearaat 2
Kokku 165
Tableti formulatsioon põhines sulamisgranulatsioonil suure lõikejõuga segajas
Pellmix 1/8. Mikroniseeritud takroliimus segati 640 g laktoos 125 mesh ja 120 g
polüetüleenglükool 6000, 48 g poloksameer 188 suure lõikejõuga segajas.
Segamisnõu kate kuumutati 80˚C juurde ning segu kuumutati laba 10
pöörlemiskiiruse juures 1000 rpm, kuni PEG-i ja poloksameeri sulamispunktini.
Pärast sulamist jätkati muljumist 4 minutit pöördekiirusel 800 rpm. Graanulid
sõeluti läbi sõela suurusega 0,7 mm ja jahutati kandikul. Graanulid segati 480 g
Avicel PH200 3 minutit ning 16 g magneesiumstearaadi lisamise jaoks ning pärast
lisamist veel 0,5 minutit. Segu suruti tablettidesse ühelöögilise tabletimasina Diaf 15
TM20 abil. Tableti diameeter: 7 mm. Tableti kuju: ümmargune, nõgus.
NÄIDE 8
In vitro lahustumise andmed
Eelnevatele näidetele vastavate kompositsioonide ja annusvormide peal viidi läbi
in vitro lahustuvustestid, kasutades kahte erinevat lahustuvus-keskkonda/testi. 20
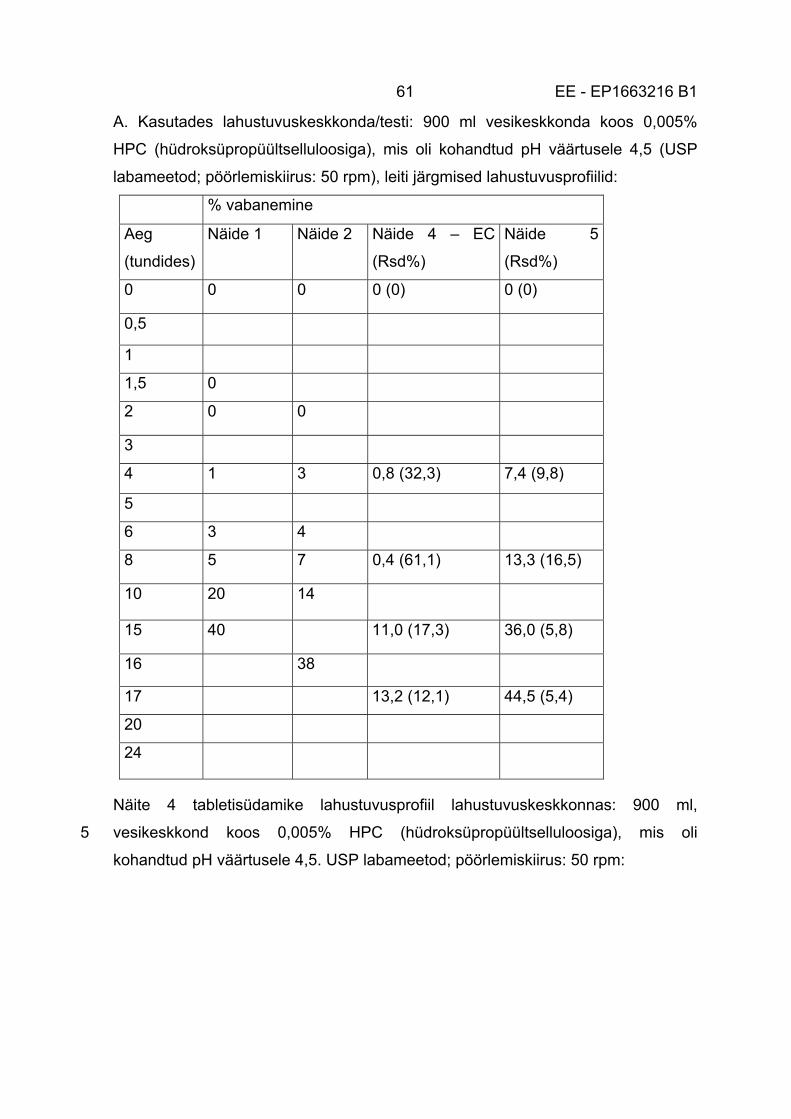
EE - EP1663216 B1 61
A. Kasutades lahustuvuskeskkonda/testi: 900 ml vesikeskkonda koos 0,005%
HPC (hüdroksüpropüültselluloosiga), mis oli kohandtud pH väärtusele 4,5 (USP
labameetod; pöörlemiskiirus: 50 rpm), leiti järgmised lahustuvusprofiilid:
% vabanemine
Aeg
(tundides)
Näide 1 Näide 2 Näide 4 – EC
(Rsd%)
Näide 5
(Rsd%)
0 0 0 0 (0) 0 (0)
0,5
1
1,5 0
2 0 0
3
4 1 3 0,8 (32,3) 7,4 (9,8)
5
6 3 4
8 5 7 0,4 (61,1) 13,3 (16,5)
10 20 14
15 40 11,0 (17,3) 36,0 (5,8)
16 38
17 13,2 (12,1) 44,5 (5,4)
20
24
Näite 4 tabletisüdamike lahustuvusprofiil lahustuvuskeskkonnas: 900 ml,
vesikeskkond koos 0,005% HPC (hüdroksüpropüültselluloosiga), mis oli 5
kohandtud pH väärtusele 4,5. USP labameetod; pöörlemiskiirus: 50 rpm:

EE - EP1663216 B1 62
Aeg
(minutites)
%
vabanemine
Rsd %
0 0 0
5 27,2 15,1
10 49,1 10,9
20 80,7 8,0
35 98,9 5,4
42 102,7 3,6
52 104,9 2,0
Näite 4 enteerilise kattega tablettide lahustuvusprofiil lahustuvuskeskkonnas
vastavalt USP meetodile A, hilinenud vabanemisega osakesed. USP labameetod.
Pöörlemiskiirus: 50 rpm:
Aeg
(minutites)
%
vabanemine
Rsd
%
0 0 NA
120 0 NA
155 84,8 12,8
165 102,9 NA
175 101,0 3,5
5

EE - EP1663216 B1 63
Patendinõudlus
1. Tahke ravimkoostis, mis sisaldab takroliimuse tahket dispersiooni või tahket
lahust hüdrofiilses või veega segunevas keskkonnas ning ühte või enamat
vabanemist modifitseerivat ainet, mis valitakse järgmise rühma hulgast: veega
segunevad polümeerid, vees lahustumatud polümeerid, õlid ja õlised materjalid, 5
mille puhul (i) vähem kui 20 massiprotsenti takroliimust vabastatakse 0,5 tunni
jooksul, kui viiakse läbi in vitro lahustuvustest, kasutades USP labameetodit ning
kasutades lahustuvuskeskkonnana 0,1 N HCl , (ii) vähem kui 50 massiprotsenti
takroliimust vabastatakse 8 tunni jooksul, kui viiakse läbi in vitro lahustuvustest,
kasutades USP labameetodit ja vesilahustuvuskeskkonda, mille pH on kohaldatud 10
väärtuseni 4,5, koos 0,005% hüdroksüpropüültselluloosiga ning (iii) keskkond, mis
sisaldab polüetüleenglükooli ja poloksameeri proportsioonis vahemikus 1:3 ja
10:1.
2. Koostis vastavalt punktile 1, mis erineb selle poolest, et vähem kui 40
massiprotsenti aktiivset farmatseutilist koostisosa vabastatakse 8 tunni jooksul, kui 15
viiakse läbi in vitro lahustuvustest, kasutades USP labameetodit ja
vesilahustuvuskeskkonda, mille pH on kohaldatud väärtuseni 4,5, koos 0,005%
hüdroksüpropüültselluloosiga.
3. Koostis vastavalt punktile 1 või 2 membraan-modereeritud süsteemi vormis, mis
sisaldab takroliimuse reservuaari tahkes dispersioonis mitmeosakeselise tootena, 20
mida ümbritseb vabanemise määra piirav membraan.
4. Koostis vastavalt punktile 1, mis erineb selle poolest, et polüetüleenglükooli
keskmine molekulaarmass on vähemalt 1500.
5. Koostis vastavalt punktile 1, mis erineb selle poolest, et poloksameeriks on
poloksameer 188. 25
6. Koostis vastavalt punktile 4, mis erineb selle poolest, et polüetüleenglükooli
keskmine molekulaarmass on 6000 (PEG 6000).
7. Koostis vastavalt punktile 1 osakeste vormis.

EE - EP1663216 B1 64
8. Koostis vastavalt punktile 1, mis erineb selle poolest, et vees lahustumatu
polümeer valitakse järgmise rühma hulgast: etüültselluloos, tselluloosatsetaat,
tselluloosnitraat ja nende segud.
9. Koostis vastavalt punktile 1, mis erineb selle poolest, et õli või õline materjal on
hüdrofiilne ning valitakse järgmise rühma hulgast: polüeeterglükoolid, nagu 5
polüpropüleenglükoolid; polüoksüetüleenid; polüoksüpropüleenid; poloksameerid;
polüglükoliseeritud glütseriidid ja nende segud.
10. Koostis vastavalt punktile 1, mis erineb selle poolest, et õli või õline materjal
on hüdrofoobne ning valitakse järgmise rühma hulgast: sirge ahelaga küllastunud
süsivesinikud, sorbitaanestrid, parafiinid, rasvad ja õlid, nagu kakaovõi, veiserasv, 10
searasv, polüeeterglükooli estrid; kõrgemad rasvhapped, nagu steaarhape,
müristhape, palmithape, kõrgemad alkoholid, nagu tsetanool, stearüülalkogol,
madala sulamispunktiga vahad, nagu glütserüülmonostearaat,
glütserüülmonooleaat, hüdrogeenitud rasv, müristüülalkohol, stearüülalkohol,
asendatud ja/või asendamata monoglütseriidid, asendatud ja/või asendamata 15
diglütseriidid, asendatud ja/või asendamata triglütseriidid, kollane mesilasvaha,
valge mesilasvaha, karnaubavaha, kastoorvaha, jaapani vaha,
atsetülaatmonoglütseriidid; NVP polümeerid, PVP polümeerid, akrüülpolümeerid ja
nende segud.
11. Koostis vastavalt punktile 1, mis erineb selle poolest, et õli või õlise 20
hüdrofoobse materjali sulamispunkt on vähemalt 20˚C.
12. Koostis vastavalt punktile 1, mis erineb selle poolest, et veega segunevaks
polümeeriks on tselluloosi derivaat, mis valitakse järgmise rühma hulgast:
hüdroksüpropüülmetüültselluloos (HPMC), hüdroksüpropüültselluloos (HPC),
metüültselluloos, naatriumkarboksümetüültselluloos, hüdroksüetüültselluloos, 25
poloksameerid, polüoksüetüleenstearaadid, polü-ε-kaprolactoon,
polüvinüülpürrolidoon (PVP), polüvinüülpürrolidoon-polüvinüülatsetaadi
kopolümeer PVP-PVA, polümetakrüüli polümeerid ja polüvinüülalkohol (PVA),
polü(etüleenoksiid) (PEO) ning nende segud.

EE - EP1663216 B1 65
13. Koostis vastavalt punktile 1, mis on enteerilise kattega, kasutades veega
segunevat polümeeri, millel on pH-sõltuv vees lahustuvus.
14. Koostis vastavalt punktile 1, mis erineb selle poolest, et veega segunev
polümeer valitakse järgmise rühma hulgast: polüakrüülamiidid; ftalaadi derivaadid,
nagu näiteks süsivesinike happelised ftalaadid, sealhulgas amüloosatsetaatftalaat, 5
tselluloosatsetaatftalaat, tselluloosatsetaattereftalaat, tselluloosatsetaatisoftalaat,
teised tselluloosesterftalaadid, tselluloosesterftalaadid, hüdroksüpropüül-
tselluloosftalaat, hüdroksüpropüültselluloosatsetaatftalaat, hüdroksüpropüül-
etüültselluloosftalaat, hüdroksüpropüülmetüültselluloosftalaat (HMPCP),
metüültselluloosftalaat, metüültselluloosatsetaatftalaat, polüvinüülatsetaatftalaat, 10
polüvinüülatsetaatvesinikftalaat, naatriumtselluloosatsetaatftalaat, tärklise
happeline ftalaat; ftalaadid või teised ühendid, sealhulgas polüvinüülatsetaatftalaat
(PVAP); teised tselluloosi derivaadid, sealhulgas hüdroksüpropüül-
metüültselluloosatsetaatsuktsinaat (HPMCAS), karboksümetüültselluloos,
tselluloosatsetaattrimellitaat; alginaadid; karbomeerid; polüakrüülhappe 15
derivaadid, nagu näiteks akrüülhape ja akrüülestri kopolümeerid,
polümetakrüülhape ja selle estrid, polüakrüülmetakrüülhappe kopolümeerid,
metakrüülhappe kopolümeerid, stüreen-maleiinhappe dibutüülftalaadi kopolümeer,
stüreen-maleiinhappe polüvinüülatsetaatftalaadi kopolümeer, stüreeni ja
maleiinhappe kopolümeerid; šellak, tärklisglükolaat; polakrüliin; vinüülatsetaat ja 20
krotoonhappe kopolümeerid ja nende segud.
15. Koostis vastavalt punktile 1, mis lisaks sisaldab ühte või enamat
farmatseutiliselt sobivat abiainet, mis valitakse järgmise rühma hulgast: täitjad,
diluendid, disintegrandid, sidujad ja lubrikadid.
16. Koostis vastavalt punktile 15 osakeste vormis, mis erineb selle poolest, et 25
osakeste geomeetrilise kaalu keskmine diameeter dgw on alates 10 µm kuni 2000
µm.
17. Koostis vastavalt punktile 15 osakeste vormis, mis erineb selle poolest, et
osakeste geomeetrilise kaalu keskmine diameeter dgw on alates 50 µm kuni 300
µm. 30

EE - EP1663216 B1 66
18. Suukaudne tahke annusvorm, mis sisaldab patendinõudlusele 15 vastavat
farmatseutilist kompositsiooni.
19. Patendinõudlusele 18 vastav annusvorm, mis on ühekordset annust sisaldav
annusvorm.
20. Annusvorm vastavalt punktile 18, mis lisaks sisaldab farmatseutiliselt sobivat 5
lisandit, mis on valitud järgmise rühma hulgast: maitseained, värvained, maitset
maskeerivad ained, pH-d kohandavad ained, puhverdavad ained, säilitusained,
stabiliseerivad ained, antioksüdandid, niisutavad ained, niiskust reguleerivad
ained, pindaktiivsed ained, suspendeerivad ained ja absorptsiooni parandavad
ained. 10
21. Annusvorm vastavalt punktile 20, mis erineb selle poolest, et vähemalt üks
farmatseutiliselt sobiv abiaine valitakse järgmise rühma hulgast: ränihape või selle
derivaat või sool, sealhulgad silikaadid, ränidioksiid ja selle polümeerid;
magneesiumaluminosilikaat ja/või magneesiumaluminometasilikaat, betoniit,
kaoliin, magneesiumtrisilikaat, montmorilloniit ja/või saponiit. 15
22. Annusvorm vastavalt punktile 21, mis erineb selle poolest, et vähemalt üks
farmatseutiliselt sobiv abiaine on ränidioksiid või selle polümeer.
23. Tahke koostise vastavalt punktile 1 kasutamine suukaudse annusvormi,
eelistatult tablettide, kapslite või kotikeste valmistamiseks.
24 Tahke koostise vastavalt punktile 1 kasutamine graanulite, pelletite, 20
mikrosfääride või nanoosakeste valmistamiseks.
25. Tahke koostise vastavalt punktile 1 kasutamine kontrollitud või modifitseeritud
vabanemisega tahke annusvormi valmistamiseks.
26. Tahke koostise vastavalt punktile 1 kasutamine hilinenud vabanemisega tahke
annusvormi valmistamiseks. 25
27. Annusvorm vastavalt ükskõik millisele punktile 18 kuni 22 kasutamiseks
patsiendi raviks, mille puhul on takroliimuse annus alates 0,02 mg/kg/päevas kuni
0,15 mg/kg/päevas, manustatuna üks kord päevas.

EE - EP1663216 B1 67
28. Annusvorm vastavalt ükskõik millisele punktile 18 kuni 22 kasutamiseks
takroliimusele reageerivate seisundite parandatud ravis.
29. Meetod koostise vastavalt punktile 1 valmistamiseks, mis hõlmab etappi, kus
takroliimus lahustatakse või pihustatakse keskkonda, et saada muutuva
temperatuuri juures tahke lahus või dispersioon. 5





























