Embed Size (px)
Citation preview

Inorganic Chemistry Communications 11 (2008) 935–938
Contents lists available at ScienceDirect
Inorganic Chemistry Communications
journal homepage: www.elsevier .com/ locate/ inoche
Targeted binding of a platinum(II)–methionine complex to the disulfide linkageof a nonapeptide oxytocin
Tingting Chen a, Xiaoyong Wang b,*, Jiafei Mao a, Haiying Wei a, Zijian Guo a,*
a State Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing 210093, PR Chinab State Key Laboratory of Pharmaceutical Biotechnology, School of Life Sciences, Nanjing University, Nanjing 210093, PR China
a r t i c l e i n f o a b s t r a c t
Article history:Received 15 February 2008Accepted 3 May 2008Available online 8 May 2008
Keywords:Disulfide bondMethionineOxytocinPlatinum(II) complex
1387-7003/$ - see front matter � 2008 Elsevier B.V. Adoi:10.1016/j.inoche.2008.05.007
* Corresponding authors. Tel.: +86 25 83594549; faE-mail addresses: [email protected] (X. Wang), z
The electrospray mass spectrometry and NMR spectroscopy techniques reveal that the platinum(II)–methionine complex [Pt(Met)Cl2] binds to the disulfide bond between Cys1 and Cys6 residues of oxytocin(OT). The major adducts identified are [Pt(Met)(OT)]Cl2 species where OT forms five- or six-memberedchelates with Pt(II) center. The study suggests that even the oxidized disulfide in oligopeptide still showsa high affinity for platinum complexes, which may be associated with the ubiquity of sulfur-related sideeffects in platinum anticancer chemotherapy.
� 2008 Elsevier B.V. All rights reserved.
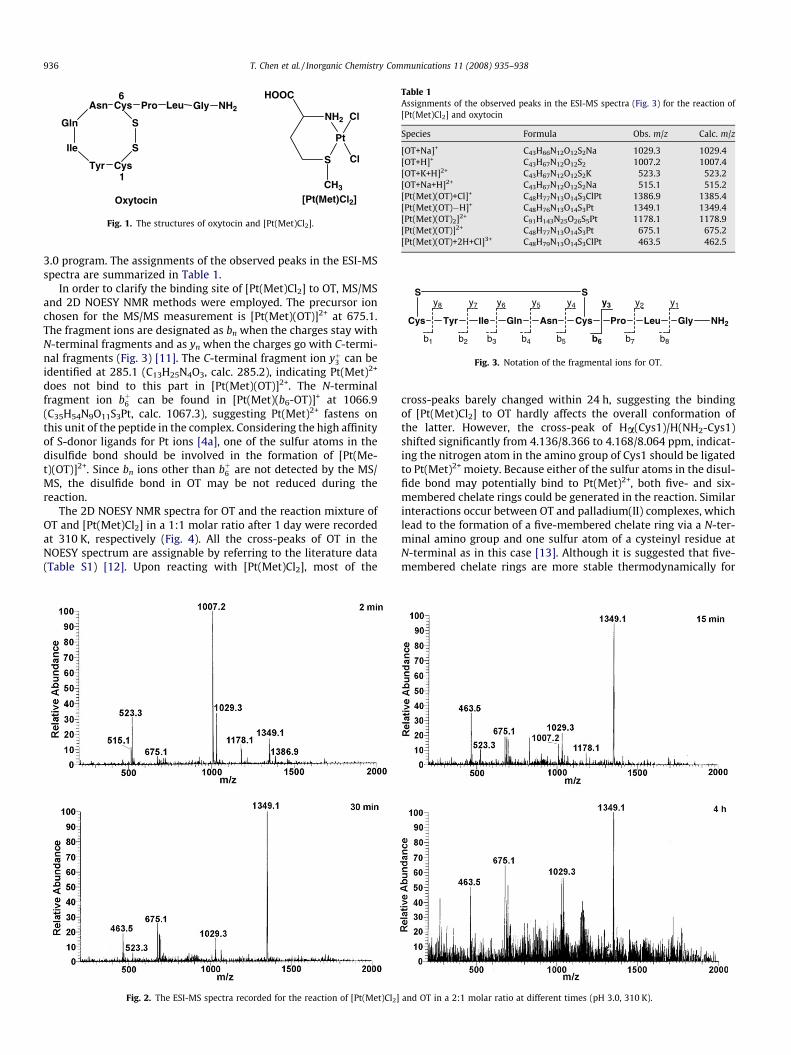
Interactions between platinum complexes and sulfur-contain-ing biomolecules play important roles in the metabolism of plati-num anticancer drugs [1]. Strong and irreversible binding ofplatinum center to intracellular S-donor ligands such as glutathi-one [2] and methionine [3] would prevent platinum drugs reachingtheir biological target DNA, and cause detoxification, drug resis-tance, and nephrotoxicity in clinical application [4]. The reactivityof platinum drugs towards small sulfur-containing molecules hasbeen well studied; nevertheless, reactions with large biomoleculessuch as proteins are poorly understood [1]. Oxytocin (OT) is a pep-tide hormone that stimulates smooth muscle contractions in theuterus during labor and the mammary glands during lactation[5], and may play a crucial role in sustaining affinity in mammals[6]. OT consists of nine amino acid residues, i.e. Cys-Tyr-Ile-Gln-Asn-Cys-Pro-Leu-Gly, with an amidated C-terminus and a disulfidebridge between Cys1 and Cys6 residues (Fig. 1) [7]. We previouslydemonstrated that a model compound of cisplatin, [Pt(Met)Cl2],can promote the cleavage of disulfide bond in oxidized glutathione(GSSG) [8]. In this communication, we extend our study to the non-apeptide OT where the environment of disulfide bond is differentfrom that of GSSG but similar to that in proteins. Such a studyought to provide deeper insights into the reactivity of platinum(II)complexes towards disulfide bonds in different surroundings.
Complex [Pt(Met)Cl2] was synthesized according to the litera-ture method [9] and OT was purchased from Fluka. The testingsample was prepared by mixing [Pt(Met)Cl2] (26.6 lL, 7.5 mM)
ll rights reserved.
x: +86 25 [email protected] (Z. Guo).
and OT (18.2 lL, 5.5 mM) in a 2:1 molar ratio and diluting the solu-tion to 100 lL with doubly distilled water, which was incubated at37 �C for an appropriate time and ESI-MS spectra were subse-quently recorded.
The reaction between [Pt(Met)Cl2] and OT proceeded quitequickly. As Fig. 2 shows, notable Pt–OT products were detected2 min after the starting of reaction, including [Pt(Met)(OT)+Cl]+ at1386.9, [Pt(Met)(OT)�H]+ at 1349.1, [Pt(Met)(OT)2]2+ at 1178.1,and [Pt(Met)(OT)]2+ at 675.1. The peaks at 1029.3, 1007.2, 523.3,and 515.1 are attributed to the unreacted OT species [OT+Na]+,[OT+H]+, [OT+K+H]2+, and [OT+Na+H]2+, respectively. Theseunreacted species significantly decreased or disappeared by15 min and most OT transformed into above assigned Pt–OTspecies plus [Pt(Met)(OT)+Cl+2H]3+ at 463.5. As a result, [Pt(Met)-(OT)]2+ increased considerably and [Pt(Met)(OT)�H]+ became thedominant species at the time. The observation indicates that theoriginally formed [Pt(Met)(OT)+Cl]+ and [Pt(Met)(OT)2]2+ are notstable in solution and can further convert into other species. Theintermediate [Pt(Met)(OT)2]2+ and the unreacted [OT+H]+ disap-peared and the reaction approached completion at 30 min. Someunreacted species [OT+Na]+ was still present in the reaction after4 h. However, the absolute abundance of the Pt–OT peaksdecreased evidently at the time, which may be due to the poor sol-ubility of the formed platinum complexes. Prolongation of thereaction time resulted in the polymerized platinum species thatare undetectable using the current spectrometer. Such specieshave been encountered previously [8,10] and they are extremelydifficult to be isolated from each other.
The isotopic distribution of all platinum(II) species in the ESI-MS spectra are consistent with the simulated patterns by IsoPro
b8
y1
b7
y2
b6
y3
b5
y4
b4
y5
b3
y6
b2
y7
b1
y8
Cys Tyr Ile Gln Asn Cys Pro Leu Gly
S S
NH2
Fig. 3. Notation of the fragmental ions for OT.
[Pt(Met)Cl2]Oxytocin
Gln
Ile
Tyr Cys
S
S
CysAsn Pro Leu Gly NH2
1
6
S
Pt
NH2
HOOC
CH3
Cl
Cl
Fig. 1. The structures of oxytocin and [Pt(Met)Cl2].
Table 1Assignments of the observed peaks in the ESI-MS spectra (Fig. 3) for the reaction of[Pt(Met)Cl2] and oxytocin
Species Formula Obs. m/z Calc. m/z
[OT+Na]+ C43H66N12O12S2Na 1029.3 1029.4[OT+H]+ C43H67N12O12S2 1007.2 1007.4[OT+K+H]2+ C43H67N12O12S2K 523.3 523.2[OT+Na+H]2+ C43H67N12O12S2Na 515.1 515.2[Pt(Met)(OT)+Cl]+ C48H77N13O14S3ClPt 1386.9 1385.4[Pt(Met)(OT)�H]+ C48H76N13O14S3Pt 1349.1 1349.4[Pt(Met)(OT)2]2+ C91H143N25O26S5Pt 1178.1 1178.9[Pt(Met)(OT)]2+ C48H77N13O14S3Pt 675.1 675.2[Pt(Met)(OT)+2H+Cl]3+ C48H79N13O14S3ClPt 463.5 462.5
936 T. Chen et al. / Inorganic Chemistry Communications 11 (2008) 935–938
3.0 program. The assignments of the observed peaks in the ESI-MSspectra are summarized in Table 1.
In order to clarify the binding site of [Pt(Met)Cl2] to OT, MS/MSand 2D NOESY NMR methods were employed. The precursor ionchosen for the MS/MS measurement is [Pt(Met)(OT)]2+ at 675.1.The fragment ions are designated as bn when the charges stay withN-terminal fragments and as yn when the charges go with C-termi-nal fragments (Fig. 3) [11]. The C-terminal fragment ion yþ3 can beidentified at 285.1 (C13H25N4O3, calc. 285.2), indicating Pt(Met)2+
does not bind to this part in [Pt(Met)(OT)]2+. The N-terminalfragment ion bþ6 can be found in [Pt(Met)(b6-OT)]+ at 1066.9(C35H54N9O11S3Pt, calc. 1067.3), suggesting Pt(Met)2+ fastens onthis unit of the peptide in the complex. Considering the high affinityof S-donor ligands for Pt ions [4a], one of the sulfur atoms in thedisulfide bond should be involved in the formation of [Pt(Me-t)(OT)]2+. Since bn ions other than bþ6 are not detected by the MS/MS, the disulfide bond in OT may be not reduced during thereaction.
The 2D NOESY NMR spectra for OT and the reaction mixture ofOT and [Pt(Met)Cl2] in a 1:1 molar ratio after 1 day were recordedat 310 K, respectively (Fig. 4). All the cross-peaks of OT in theNOESY spectrum are assignable by referring to the literature data(Table S1) [12]. Upon reacting with [Pt(Met)Cl2], most of the
Fig. 2. The ESI-MS spectra recorded for the reaction of [Pt(Met)Cl2]
cross-peaks barely changed within 24 h, suggesting the bindingof [Pt(Met)Cl2] to OT hardly affects the overall conformation ofthe latter. However, the cross-peak of Ha(Cys1)/H(NH2-Cys1)shifted significantly from 4.136/8.366 to 4.168/8.064 ppm, indicat-ing the nitrogen atom in the amino group of Cys1 should be ligatedto Pt(Met)2+ moiety. Because either of the sulfur atoms in the disul-fide bond may potentially bind to Pt(Met)2+, both five- and six-membered chelate rings could be generated in the reaction. Similarinteractions occur between OT and palladium(II) complexes, whichlead to the formation of a five-membered chelate ring via a N-ter-minal amino group and one sulfur atom of a cysteinyl residue atN-terminal as in this case [13]. Although it is suggested that five-membered chelate rings are more stable thermodynamically for
and OT in a 2:1 molar ratio at different times (pH 3.0, 310 K).

Fig. 4. The 2D NOESY NMR spectra for OT and the reaction mixture of OT and[Pt(Met)Cl2] in a 1:1 molar ratio after 1 day (500 MHz, DMSO/DCl, 310 K). The cross-peak of Ha(Cys1)/H(NH2-Cys1) undergoes a significant shift from 4.136/8.366 to4.168/8.064 ppm.
T. Chen et al. / Inorganic Chemistry Communications 11 (2008) 935–938 937
Pd(II)–OT complexes [14], six-membered rings cannot be excludedfor Pt(II)–OT complexes.
On the basis of above results, a possible pathway for the reac-tion of [Pt(Met)Cl2] with OT can be proposed in Scheme 1. When[Pt(Met)Cl2] reacts with OT, one of the chlorine atoms is replacedby one of the sulfur atoms of the disulfide bond in OT; and subse-
T
C
(m/z 1386.9 or 463.5)
(major species: m/z 1
+
1
6
C
HC CH2
SSH2N Cys
Asn
Gln
IleTyr
O
1
6
C
HC
SH2N Cys
Asn
Gln
IleTyrO
S
H2C
GlnIle
Tyr
Cys
S S
Cys
Asn
1 6 Pro Leu Gly NH2
Pt
GlnIle
Tyr
Cys
S S
Cys
Asn
1 6 Pro Leu Gly NH2
OR
OR
S
Pt
NH2
HOOC
CH3
Cl
Cl
H2N S CH3
HOOC
Cl
ClPt
H2N S CH3
HOOC PtH2N S CH3
HOOC
PtH2N S CH3
HOOC
Scheme 1. A proposed pathway for the reaction of [Pt(Met)Cl2] and OT in a
quently another chlorine is substituted by the nitrogen atom ofthe terminal amino group of Cys1, forming [Pt(Met)(OT)+Cl]+,[Pt(Met)(OT)�H]+, [Pt(Met)(OT)2]2+ and [Pt(Met)(OT)]2+, with[Pt(Met)(OT)�H]+ and [Pt(Met)(OT)]2+ being the major species.Among them, [Pt(Met)(OT)+Cl]+ and [Pt(Met)(OT)2]2+ are relativelyunstable.
We reported recently that [Pt(Met)Cl2] can cleave the disulfidebond in GSSG, giving rise to mono- and dinuclear platinum com-plexes with the cleaved fragments [8]. In this study, we demon-strated that [Pt(Met)Cl2] can also react readily with the disulfidebond in OT to form Pt–OT complexes. This reaction proceeds fasterthan that with GSSG possibly because the disulfide bond in OT issterically more accessible than that in GSSG. However, no evidencefor the cleavage of the disulfide bond can be found in the reaction,which may be due to the formation of platinum chelates is morepreferable in the presence of neighboring amino group of Cys1.Therefore, the manner in which the platinum center reacts withthe disulfide containing biomolecules is likely to be dependenton the nature of the environment of the disulfide bond. Our studiesof this series explicitly show that cisplatin-like complexes can re-act with sulfur-containing biomolecules of different types, includ-ing protein-like molecules. The strong affinity of S-donors forplatinum may be relevant to the prevalence of sulfur-related sideeffects in platinum anticancer chemotherapy.
Acknowledgements
We are grateful for the financial support from the National Nat-ural Science Foundation of China (Nos. 20231010, 20631020,90713001, and 20721002).
GlnIle
yr
ys
S S
Cys
Asn
(m/z 1178.1)
349.1 or 675.1)
1 6
Pro Leu Gly NH2
Pro Leu Gly NH2
Pro Leu Gly NH2
GlnIle
Tyr
Cys
S S
Cys
Asn
1 6 Pro Leu Gly NH2
GlnIle
Tyr
Cys
S S
Cys
Asn
1 6 Pro Leu Gly NH2
S
Pt
H2N
HOOC
CH3
2:1 molar ratio. Charges and possible isomers are omitted for clarity.

938 T. Chen et al. / Inorganic Chemistry Communications 11 (2008) 935–938
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.inoche.2008.05.007.
References
[1] X.Y. Wang, Z.J. Guo, Anti-Cancer Agents Med. Chem. 7 (2007) 19–34.[2] M.A. Fuertes, C. Alonso, J.M. Pérez, Chem. Rev. 103 (2003) 645–662.[3] O. Vrana, V. Brabec, Biochemistry 41 (2002) 10994–10999.[4] [a] J. Reedijk, Chem. Rev. 99 (1999) 2499–2510;
[b] G. Giaccone, Drugs 59 (Suppl. 4) (2000) 9–17.[5] G. Gimpl, F. Fahrenholz, Physiol. Rev. 81 (2001) 629–683.
[6] J.T. Winslow, N. Hastings, C.S. Carter, C.R. Harbaugh, T.R. Insel, Nature 365(1993) 545–548.
[7] H. Shojo, Y. Kaneko, Mol. Genet. Metab. 71 (2000) 552–558.[8] H.Y. Wei, X.Y. Wang, Q. Liu, Y.H. Mei, Y. Lu, Z.J. Guo, Inorg. Chem. 44 (2005)
6077–6081.[9] H.C. Freeman, M.L. Glomb, J. Chem. Soc. Dalton Trans. (1970) 1523–1524.
[10] Q. Liu, H.Y. Wei, J. Lin, L.G. Zhu, Z.J. Guo, J. Inorg. Biochem. 98 (2004) 702–712.[11] X.J. Sun, C. Jin, Y.H. Mei, G.S. Yang, Z.J. Guo, L.G. Zhu, Inorg. Chem. 43 (2004)
290–296.[12] T. Kato, S. Endo, T. Fujiwara, K. Nagayama, J. Biomol. NMR 3 (1993) 653–673.[13] X.M. Luo, W. Huang, Y.H. Mei, S.Z. Zhou, L.G. Zhu, Inorg. Chem. 38 (1999)
1474–1480.[14] H. Wei, X.M. Luo, Y.B. Wu, Y. Yao, Z.J. Guo, L.G. Zhu, J. Chem. Soc. Dalton Trans.
(2000) 4196–4200.





























![Dietary supplementation with free methionine or methionine … · 2019. 6. 27. · with MHA or DL-methionine in heat stress-exposed broilers [23, 24]. In this study, we hypothesize](https://img.pdfslide.net/doc/110x75/60e337666b3f9a31a45a96d1/dietary-supplementation-with-free-methionine-or-methionine-2019-6-27-with-mha.jpg)