Embed Size (px)
Citation preview

REVIEW ARTICLE
The application of proteomic approaches to the studyof mammalian spermatogenesis and sperm functionGraham MacLeod and Susannah Varmuza
Department of Cell & Systems Biology, University of Toronto, ON, Canada
Keywords
interactomes; phosphoproteomics; protein
phosphatase; proteomics; reproductive
biology; spermatogenesis
Correspondence
S. Varmuza, 25 Harbord Street, Toronto,
ON, Canada M5S 3G5
Fax: 416-978-8532
Tel: 416-978-2759
E-mail: [email protected]
(Received 22 February 2013, revised 4 July
2013, accepted 26 July 2013)
doi:10.1111/febs.12461
Spermatogenesis is the process by which terminally differentiated sperm are
produced from male germline stem cells. This complex developmental pro-
cess requires the coordination of both somatic and germ cells through
phases of proliferation, meiosis, and morphological differentiation, to pro-
duce the cell responsible for the delivery of the paternal genome. With
infertility affecting ~ 15% of all couples, furthering our understanding of
spermatogenesis and sperm function is vital for improving the diagnosis
and treatment of male factor infertility. The emerging use of proteomic
technologies has played an instrumental role in our understanding of sper-
matogenesis by providing information regarding the genes involved. This
article reviews existing proteomic literature regarding spermatogenesis and
sperm function, including the proteomic characterization of spermatogenic
cell types, subcellular proteomics, post-translational modifications, interact-
omes, and clinical studies. Future directions in the application of proteo-
mics to the study of spermatogenesis and sperm function are also
discussed.
Introduction
Mammalian spermatogenesis is a precisely regulated
biological process resulting in the production of sper-
matozoa, one of the most unique and highly differenti-
ated cell types. Spermatogenesis consists of three
distinct phases within the seminiferous epithelium, all
of which are associated with the somatic Sertoli cells.
The first phase, the proliferative phase, refers to the
mitotic division of spermatogonia, which serves to pro-
vide an increased number of germ cells for differentia-
tion and to repopulate the stem cell niche. Next is the
meiotic phase, in which tetraploid spermatocytes
undergo meiotic division to produce haploid spermat-
ids. The final phase is the differentiation phase, known
as spermiogenesis, wherein the spermatids undergo a
series of dramatic morphological changes, leading to
functional sperm. Although the stages of spermatogen-
esis are well characterized at the cellular level, the pre-
cise biological mechanisms regulating this process are
not entirely understood. Enhancing our understanding
of spermatogenesis will prove useful in improving the
diagnosis and treatment of male factor infertility, a
condition that negatively affects the quality of life for
over 100 million couples worldwide.
To better understand the process of spermatogene-
sis, we must uncover which genes are involved, what
roles they play, and how they are regulated. To date,
targeted mutagenesis studies have produced ~ 400 dif-
ferent knockout mouse models with reproductive
defects [1], not limited to the ~ 4% of all mouse genes
revealed by transcriptome analysis to be specifically
Abbreviations
2DE, two-dimensional electrophoresis; AP-MS, affinity purification MS; HSP, heat shock protein; IMAC, immobilized metal ion affinity
chromatography; MRM, multiple reaction monitoring; PPP1, phosphoprotein phosphatase 1; PTM, post-translational modification;
SYCP, synaptonemal complex protein; TAP, tandem affinity purification.
FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS 5635

expressed in the postmeiotic male spermatogenic cells
[2]; both testis-specific and ubiquitously expressed
genes can be found in the list of targeted mutations
that only affect spermatogenesis, the latter probably
reflecting functional redundancy in most tissues of par-
alogous genes. This collection of data underscores the
complex nature of spermatogenesis in mammals and
our need for an increased understanding of the pro-
cess. In contrast, numerous attempts over the past
10 years, since the publication of the human genome
sequence, to identify mutations linked to male infertil-
ity affecting ~ 5% of men have been unsuccessful in
uncovering good candidates for clinical genetic screen-
ing; the only tests routinely used in andrology clinics
are for Y microdeletions, chromosome abnormalities,
and cystic fibrosis transmembrane conductance regula-
tor mutations, which affect ~ 10% of patients [3,4].
Emerging proteomic technologies can provide a num-
ber of useful tools for studying mammalian spermato-
genesis. The use of proteomics is particularly
important for spermatogenesis, because the semiquan-
titative correlation between RNA and protein expres-
sion is lower in the testis than in other tissues [5],
indicating that oligonucleotide microarray and geno-
mic studies are less informative in this context. One
factor that complicates the comparison between RNA
and protein expression in the testis is the transcrip-
tional silencing found late in spermatogenesis, which
necessitates the storage of earlier-produced transcripts
for later use. This was illustrated in a recent isobaric
tags for relative and absolute qauntitation-based quan-
titative proteomics study that identified a large number
of proteins for which this is the case, especially during
the spermatocyte to round spermatid transition [6].
Compounding these difficulties further is the abun-
dance of tissue-specific alternative splicing observed in
the testis [7], one prominent example being the Ppp1 cc
gene, which is essential for the completion of sper-
matogenesis and encodes both the ubiquitous
PPP1CC1 and testis-specific PPP1CC2 isoforms [8].
Despite its importance, we have only recently begun to
scratch the surface of the potential of proteomic
research application to spermatogenesis. As an increas-
ing number of researchers have made use of such
technologies, it is not possible to discuss all of the
excellent research in the space available. Likewise, it is
not our aim to cover the technical details of proteomic
methodologies and data analysis. Many of the studies
described in this review have utilized model organisms,
most prominently the mouse and rat. The precise
level of conservation in the testis/sperm proteomes
between these species and humans remains unknown,
because complete proteome coverage has not been
accomplished. However, comparative studies have
revealed, that for many genes, there are spermatogene-
sis-associated homologs that have similar expression
patterns even over large evolutionary distances [9],
and, in general, conservation throughout mammals is
considered to be high. Comparative studies between
mouse and human reproductive proteins found good
correlations between mice and humans in general, but
also that proteins arising from the seminal vesicles
were showing a higher rate of divergence [10]. Another
comparison of published sperm proteome datasets
from a number of species revealed that a number of
functionally linked protein groups were conserved
throughout mammals [11]. This review will highlight
key studies that demonstrate the potential of
proteomic research in a number of different
contexts – including the proteomic characterization of
different spermatogenic cell types and subcellular com-
ponents, post-translational modifications (PTMs), clin-
ical studies, and protein–protein interaction networks.
Proteomic studies of spermatogenesis serve as an
important line of inquiry that complements genomic,
transcriptomic and epigenetic studies, which are
beyond the scope of this review but, together, hold the
key to understanding gene regulation during this pro-
cess. Furthermore, we hope that, by drawing attention
to the wide range of currently available datasets, this
review will serve as a useful resource for researchers
interested in the process of spermatogenesis.
Proteomic characterization ofspermatogenic cells
Spermatogenesis includes a number of different cell
types, many of which are in close contact in the semi-
niferous epithelium, the site of spermatogenesis within
the testis (Fig. 1). Each spermatogenic cell type repre-
sents a step towards the production of sperm, and
thus, by characterizing the proteome of the different
cell types, we can gain insights into the genes and pro-
teins involved in each step. Current estimates suggest
that the human sperm proteome contains approxi-
mately 2500–3000 proteins [11]; however, less differen-
tiated spermatogenic cells may contain a much higher
number [6], as much of the cytoplasmic material,
including organelles, is removed during the final stages
of spermiogenesis in order to streamline the cell for
motility and fertilization.
A number of studies have utilized whole testis pro-
tein extracts to examine protein expression throughout
the entire testis in a variety of species. In the mouse,
both fetal [12] and sexually mature whole testis
extracts [13] have been examined by two-dimensional
5636 FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS
Proteomic research in mammalian spermatogenesis G. MacLeod and S. Varmuza

electrophoresis (2DE) followed by MS. At least three
studies have examined the human testis proteome, with
varying methodologies [14–16]. Two of these studies
are from the laboratory of Sha and colleagues, where,
in 2008, with SDS/PAGE followed by LC-MS/MS,
1430 testis proteins were identified [14], and in 2010,
2DE followed by MALDI-TOF MS analysis identified
462 unique proteins [15]. In a more recent study, Li
et al. used 2DE and MALDI-TOF MS analysis to
identify 725 unique proteins in human testis protein
extracts [16]. Although these studies represent some of
the most extensive human testis proteome studies to
date, < 200 annotated proteins have been found in all
three datasets. This suggests not only that experimen-
tal variation can result in the generation of very differ-
ent datasets, but that we are probably very far from
reliably characterizing the entire testis proteome, as
these studies clearly do not approach the required
level of coverage. This highlights key limitations to
2DE-based approaches. Owing to more stringent pro-
tein size and dynamic range constraints, and lower
resolving power, they are less suitable for the identifi-
cation of large and diverse sets of proteins than
LC-MS/MS-based techniques, and are subject to a
greater degree of experimental variation. Conse-
quently, most laboratories are now using LC-MS/MS-
based methods for such studies, often in conjunction
with a suite of prefractionation and/or differential
labeling strategies [6,17].
Although studies of the whole testis proteome can
yield a considerable amount of data, they tell us little
about the roles of specific proteins in spermatogenesis,
as no information regarding the spatiotemporal pat-
tern of expression is obtained. To gain such informa-
tion, different approaches are needed, such as
following changes in expression during the first wave
of spermatogenesis at the onset of puberty, which is
synchronous, affording the opportunity to investigate
relatively homogeneous cell populations. In contrast,
in the adult testis, spermatogenesis has a wave-like
pattern along the seminiferous tubules, such that all
spermatogenic cell types are represented simulta-
neously in the testis. Thus, by examining gene expres-
sion patterns at different time points, we can gain
spatiotemporal information and insights into the
potential roles of genes in different aspects of sper-
matogenesis. Several groups have used such an
approach in a variety of different species. One recent
study, by Huang et al., used 2DE to analyze changes
in testis protein expression patterns in boar spermato-
genesis at three time points – 1 week (Sertoli cells and
spermatogonia only), 3 months (onset of spermatogen-
esis), and 1 year (maturity) [18]. The authors were then
able to identify 90 differentially expressed proteins via
MS. Several studies have used a similar approach in
mice, including one that utilized 2DE followed by
MALDI-TOF/TOF MS to identify 257 proteins that
were differentially expressed between six different time
points in the first wave of mouse spermatogenesis (0,
7, 14,21, 28 and 60 days) [19]. The authors then
applied clustering analysis of their data, and found six
distinct expression patterns that were each enriched for
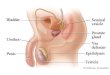
Fig. 1. The testis is a complex and dynamic tissue. A cross-sectional view is shown of a single mouse testis visualized by light microscopy
with periodic acid–Schiff and hematoxylin staining. Left: a cross-sectional view showing several seminiferous tubules, each with different
complements of developing spermatogenic cells. Spermatogenesis progresses in a wave-like pattern along the length of the seminiferous
tubule, meaning that different segments of the tubules (cross-section) show different stages of spermatogenic cells. Right: a closer look at
a single seminiferous tubule, showing spermatogenic cells at various stages in development. The complex architecture and mixture of cell
types at various stages of development makes the testis a challenging tissue to analyze.
FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS 5637
G. MacLeod and S. Varmuza Proteomic research in mammalian spermatogenesis

cellular processes specific for particular stages of sper-
matogenesis (stem cell properties, mitosis, meiosis,
spermiogenesis, and fertilization). This analysis
allowed the authors to link a number of proteins with
unknown functions to specific stages of spermatogene-
sis, such as heat shock protein (HSP)27 (meiosis) and
peroxiredoxin-4 (spermiogenesis), which would not
have been possible if they had been looking at sperm
alone. The results of these studies show how the nat-
ure of the first wave of mammalian spermatogenesis
can be used to gain information regarding a protein’s
possible function, and allow us to infer potential cell
type-specific/enriched expression patterns. However, to
truly characterize specific spermatogenic cell types, it is
necessary to analyze them in isolation.
To date, there have been a number of studies that
have sought to characterize the proteomes of isolated
germ cell populations. This is done by separating cell
suspensions on the basis of size or DNA content with
a number of methods – fluorescence-activated cell
sorting (DNA content) [20], gravity sedimentation in a
StaPut apparatus [21], or centrifugal elutriation [22].
All of the major types of spermatogenic cells –spermatogonia, spermatocytes, spermatids, and
sperm – have been isolated and subjected to analysis
in order to characterize their proteomes. Some experi-
ments have examined even narrower classes of sper-
matogenic cells, such as those described by Delbes
et al., who performed a proteomic analysis of elon-
gated spermatids [23]. Many of these datasets are pub-
licly available, and provide a valuable resource for
other research groups. The first spermatogenic cells to
appear in the mammalian testis, the spermatogonia,
have, in fact, the least characterized proteome, proba-
bly because of technical difficulties in the isolation of
purified cell populations. Two early studies from the
Pineau laboratory identified 53 and 102 nonredundant
proteins from Staput-isolated rat spermatogonia, with
19 proteins in common between the two datasets
[24,25], and another study performed a proteomic
analysis on isolated spermatogonial stem cells from
adult mice that had been cultured in conditions
designed to maintain stem cell-like behavior; although
the number of proteins identified was limited, the
authors observed minimal differences in the proteomes
of the two cell types, which they hypothesized reflects
their similar developmental competence [26]. In sper-
matocytes, the largest proteomic dataset currently
available was produced by Guo et al. [27], who identi-
fied 3625 unique proteins (3427 unique Entrez genes)
in fluorescence-activated cell sorting-isolated primary
spermatocytes, including almost 400 testis-specific pro-
teins and 172 proteins associated with meiosis. These
included 28 different proteins that had previously been
identified as being essential for completion of male
meiosis, including the prominent synaptonemal com-
plex proteins synaptonemal complex protein (SYCP)1,
SYCP2, and SYCP3. Further analysis revealed a large
number of proteins known to be involved in DNA
repair and transcription, corresponding to the peak in
transcriptional activity that is known to occur in
pachytene spermatocytes. The same group used a simi-
lar approach to identify 2116 spermatid proteins map-
ping to 1924 unique genes, with ~ 300 testis-specific
proteins represented [28]. The spermatid proteome was
found to contain a large number of vesicle-related pro-
teins, reflective of the development of the acrosome in
these cells, and the authors identified a novel protein,
vesicle-associated mmebrane protein 4, that they
linked to this process. As mentioned above, one group
isolated a more specific spermatid population – elon-
gated spermatids – and were able to confidently iden-
tify 632 proteins with two or more unique peptides
[23]. Recently, one highly informative study featured a
quantitative proteomic comparison of isolated mouse
spermatogonia, pachytene spermatocytes, round sper-
matids, and elongating spermatids [6], which identified
2008 different proteins, over half of which belonged to
one of four expression pattern clusters reflecting
important aspects of spermatogenesis, such as mitotic
proliferation, meiosis, and spermiogenesis. Found in
the cluster of proteins with higher expression in hap-
loid spermatogenic cells were protein phosphatases
and kinases, including the testis-specific phosphatase
phosphoprotein phosphatase 1 (PPP1)CC2, which
plays key roles in spermatogenesis. The authors’ com-
parison of proteomic changes and transcriptomic data
further classified genes into five different regulatory
mechanisms, including prominent post-transcriptional
regulation of gene expression in the testis, illustrating
the benefit of considering these types of data side-by-
side. This study provides a wealth of information
regarding mechanisms for regulation of gene expres-
sion during spermatogenesis in general, as well as the
ability to see how many individual genes are regulated.
In addition, this study represents the largest spermato-
gonia proteomic dataset published to date [6]. Consid-
ering all of these studies together, we can see that
different spermatogenic cell types contain different
proteomic complements, reflecting the different biolog-
ical processes involved at different times in germ cell
maturation.
In contrast to immature spermatogenic cells, mature
sperm do not require any specialized isolation proce-
dures, and thus can easily be collected even from
human subjects. As a consequence, there have been
5638 FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS
Proteomic research in mammalian spermatogenesis G. MacLeod and S. Varmuza

significantly more studies of the proteome of mature
sperm than of the proteomes of other spermatogenic
cell types. The largest number of human sperm pro-
teins identified in a single dataset was 1760, published
by Johnson et al. in 2005 [29]; however, the authors
did not make their dataset publicly available. The larg-
est publicly available human sperm proteome datasets
consist of 1056 proteins comprising Triton-X-soluble
and insoluble fractions [30], and 1429 proteins in dis-
sociated head and tail fractions [31]. Similarly, the
same group has published high-quality mature sperm
proteomic datasets for mouse [32] and rat [33] that
represent excellent resources for those researching
spermatogenesis. Subsequent bioinformatic analysis of
these three sperm proteomes revealed considerable
overlap, despite the fact that the sperm proteome has
yet to be fully covered [11]. In other species, one very
large dataset has identified thousands of proteins in
mature bull sperm [34]. However, when high-fertility
and low-fertility groups were compared, only 20%
overlap was observed, a strikingly low number for
such a large dataset. A second examination of this
dataset by Baker and Aitken has shown that a large
number of the identified proteins are represented by
only a single unique peptide, which can lead to a high
incidence of false positives, bringing the total number
of identified proteins into question [11]. Nonetheless,
the dataset can still be a useful resource, provided that
certain caveats are considered during analysis. The
issue of single-peptide identifications is key when
examining proteomic data, and one should always
look deeper into the data before interpreting any pro-
tein identification as absolute.
The utility of proteomic analysis of whole testes or
isolated germ cell populations goes beyond simply cat-
aloging the proteome of the cell types. Proteomic
approaches have also been used to ask specific biologi-
cal questions by an increasing number of groups. For
example, the elongating spermatid proteome investiga-
tion by Delbes et al. described above compared the
proteomic profiles of wild-type and Paip2a/Paip2b
double-knockout mice, and identified 29 differentially
expressed proteins, several of which were subsequently
verified by western blot analysis [23]. As the PAIP2
proteins are important translational repressors in sper-
matogenesis, differential expression of proteins in this
knockout background revealed genes that are regu-
lated post-transcriptionally by this pathway. To exam-
ine the effects of Sertoli cell conditional Dicer1
deletion on protein expression throughout the early
postnatal testis, Papaioannou et al. performed a quan-
titative analysis of 130 testis proteins, and found that
a large proportion (~ 38%) were upregulated [35],
although this study would have been more informative
had the authors compared conditional Sertoli-specific
Dicer null testes, which completely lack germ cells,
owing to Sertoli cell dysfunction, with wild-type testes
from mice treated with busulfan, a germ cell toxin that
eliminates all germ cells except the stem cells; both are
Sertoli cell-only testes, but the former lack Dicer-medi-
ated transcriptional and translational regulation.
Another group used a proteomic analysis of enriched
mouse spermatocytes to test the effect of androgen
deprivation and replacement, and identified 88 differ-
entially regulated proteins, including several with
known roles in meiosis, such as HSPA2 [36]. Compar-
ative proteomics has been used to identify proteins
that are differentially regulated throughout spermato-
genesis by comparing the proteomes of isolated sper-
matogonia, pachytene spermatocytes and early
spermatids via 2D difference in gel electrophoresis.
Subsequently, 123 proteins that were differentially
expressed between the three stages of rat spermatogen-
esis were identified [37], including the protein phospha-
tase PPP1CC2, which is known to be essential for
completion of spermatogenesis in mice [8]. These four
studies are representative examples of how proteomic
analysis of spermatogenic cells has the potential to go
far beyond the generation of ‘protein lists’, to answer
biological questions regarding male fertility.
Subcellular proteomics inspermatogenesis
Despite significant advances in the resolution of prote-
omic technologies, to date no one protein extraction/
identification method can identify all proteins in the
cell, owing to technological limitations, and differences
in dynamic range and solubility. Furthermore, the
presence of a protein in a cell type does not necessarily
tell us much about its function. By breaking the cell
down into its constituent parts (be they fractions,
structural elements, or organelles), better coverage of
the proteome and the subcellular localization of pro-
teins within the spermatogenic cell types can be
obtained, which helps to provide functional insights.
In fact, the sperm are uniquely suited to this form of
analysis. Being arguably the most differentiated cell
type in the body, they contain a number of different
component parts that can be readily isolated and sub-
jected to proteomic analysis.
Several reproductive biology research groups have
taken a subcellular proteomic approach to the study
of spermatogenesis. The bulk of this research has
focused on sperm, because of their relative ease of iso-
lation and unique morphology (see Table 1 for a list
FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS 5639
G. MacLeod and S. Varmuza Proteomic research in mammalian spermatogenesis

of studies that have examined subcellular sperm frac-
tions). The sperm surface has been one subcellular
locale of great interest for proteomic analysis, because
of its dynamic nature during epididymal maturation
and capacitation, and its importance to fertilization
[38–43]. Recently, two groups have published studies
that have used different biochemical approaches to iso-
late sperm membrane fractions containing lipid
rafts – dynamic, sterol-enriched and sphingolipid-
enriched microdomains that are believed to play key
roles in regulating sperm function at the membrane.
Asano et al. used a detergent-free approach to isolate
three distinct types of mouse sperm membrane raft, as
well as a ‘nonraft’ membrane fraction [38]. Following
gel-based separation, in-gel trypsin digestion, and
LC-MS/MS analysis, they were able to identify 190
proteins between the three sperm raft subtypes, as well
as a number of additional ‘nonraft’ proteins on the
sperm surface. Nixon et al., conversely, used a mild
detergent-based protocol to isolate proteins from the
detergent-resistant membrane fraction from capaci-
tated human [42] and mouse [41] sperm, which were
then subjected to 2DE and LC-MS/MS analysis. From
this analysis, 124 and 100 proteins, respectively, were
identified. Interestingly, there were more proteins in
common between the two mouse datasets with differ-
ent methodologies (35) than between the human and
mouse datasets (14) that were obtained with the same
mild detergent-based method for detergent-resistant
membrane isolation. This discrepancy is interesting,
but perhaps not surprising, as interspecies proteomic
studies have shown that sperm surface proteins are
subject to more rapid evolutionary change than other
sperm proteins [44]. These studies all identified known
zona pellucida-binding proteins, as well as novel cell
adhesion and signaling molecules that may play impor-
tant roles in sperm function and fertilization. Also, the
comparison of these datasets illustrates that different
strategies for preparation of subcellular fractions pro-
duce different pools of proteins – each with their own
contaminants and losses. Although they may have con-
siderable overlap, these differences must be taken into
account when proteomic data from different sources
are analyzed. Furthermore, datasets such as these do
not account for quantitative differences between
subcellular fractions, or differences in PTMs between
fractions.
In addition to the sperm surface, a number of other
subcellular fractions of sperm have been examined
with proteomic methodologies, such as the acrosomal
matrix, a critical subcellular compartment during fer-
tilization in which > 1000 proteins were identified [45].
This number is surprisingly large, which could indicateTable
1.Recentexamplesofsubcellularproteomic
studiesofsperm
.AKAP4,A-kinaseanchorprotein
4;IZUMO1,Izumosperm
–eggfusion1;MFGE8,milk
fatglobule
epiderm
algrowth
factor8;ODF1,outerdensefiber1;PRM2,protamine2;SPACA1,sperm
acrosome-associated1;TSSK6,testis-specificserinekinase6;ZPBP1,zonapellucida-bindingprotein
1.
Sperm
subcellular
fraction
Species
Protein
separation
method
MS/M
Smethod
Numberof
proteins
identified
Example
protein
andbiological
function
Reference
Membrane
Mouse
1D
SDS/PAGE
LC-M
S/M
S190
MFGE8,fertilization
Asanoetal.[38]
Membrane
Human
2D
SDS/PAGE
LC-M
S/M
S124
ZPBP1,bindingofsperm
tozona
pellucida
Nixonetal.[42]
Membrane
Mouse
2D
SDS/PAGE
MALDI-TOF+
nanoLC-M
S/M
S
100
IZUMO1,fusionofsperm
toegg
plasmamembrane
Nixonetal.[41]
Tail
Human
1D
SDS/PAGE
LC-M
S/M
S1049
ODF1,sperm
atogenesis
Amaraletal.[46]
Tail
Human
1D
SDS/PAGE
LC-M
S/M
S901
AKAP4,sperm
motility
Bakeretal.[31]
Head
Human
1D
SDS/PAGE
LC-M
S/M
S704
TSSK6,sperm
chromatin
condensation
Bakeretal.[31]
Nucleus
Human
1D
SDS/PAGE,
2D
SDS/PAGE
LC-M
S/M
S403
PRM2,DNA
packaging
deMateoetal.[49]
Acrosomal
matrix
Mouse
1D
SDS/PAGE
LC-M
S/M
S1026
SPACA1,acrosomeassembly
Guyonnetetal.[45]
5640 FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS
Proteomic research in mammalian spermatogenesis G. MacLeod and S. Varmuza

a much more complicated subcellular compartment
than initially thought, or conversely, a high degree of
false-positive identification/contamination during frac-
tionation. The sperm tail proteome has been investi-
gated intensively in two recent studies. Amaral et al.
produced the largest dataset, with a total of 1049 pro-
teins identified in the human sperm tail [46], including
a surprisingly high number of peroxisomal proteins,
given that the peroxisome is an organelle that is
thought to be largely absent in mature sperm. Concur-
rently, Baker et al. published a study wherein the head
and tail portions of human sperm were separated, and
each fraction was subjected to proteomic analysis [31].
The resultant dataset identified 900 proteins in the
sperm tail and 700 proteins in the sperm head, with
just 159 proteins being found in both subcellular frac-
tions. This relatively low degree of overlap points to
the specialized nature of the sperm subcellular struc-
tures, each with a specific role in the transmission of
the paternal genome. Almost half (46%) of the sperm
tail proteins identified in the Baker et al. dataset were
also identified by Amaral et al. Although this is clearly
a significant amount of overlap, it is lower than one
might expect, given the size of these datasets in rela-
tion to the hypothesized size of the entire sperm prote-
ome, signifying that a substantial portion of the
proteome has been missed in all of the experiments
conducted to date.
The sperm nucleus is another subcellular structure
of great interest in reproductive biology research [47].
During spermiogenesis, the spermatid nucleus under-
goes dramatic chromatin condensation. This process is
regulated by proteins in and around the nucleus, and
is commonly found to be disrupted by gene deletions,
resulting in male infertility [48]. It is not surprising
that several groups have turned to proteomics to ana-
lyze the nuclear content of sperm. One recent study
isolated human sperm nuclei to 99.9% purity, and, fol-
lowing gel fractionation of the protein content, identi-
fied 403 proteins by LC-MS/MS analysis [49].
Strikingly, more than 50% of the identified proteins
had never been reported in human sperm, despite the
availability of a number of such datasets (see above).
This illustrates one of the benefits of subcellular frac-
tionation prior to proteomic analysis – simplification
of the sample leads to an increased depth of coverage
and, thus, a greater amount of useful data. To look
even closer at the proteins involved in DNA packaging
during spermatogenesis, Govin et al. [50] devised a
strategy to extract proteins with either the potential to
bind DNA (basic proteins) or capable of binding basic
proteins (acidic proteins) from isolated mouse
stage 12–16 spermatid nuclei. This analysis identified
70 proteins, which were putative DNA-packaging pro-
teins and their chaperones in spermatogenesis. Fur-
thermore, this study highlighted a clear link between
proteomics and epigenomics, as the proteins identified
provide a clear link to epigenetic regulation of the
postmeiotic nucleus. A comparison of five different
subcellular human sperm proteomic datasets reveals
that a large number of proteins appear to be unique to
each individual dataset (Fig. 2). This demonstrates the
increased proteome coverage that can be achieved
when individual fractions are analyzed, as well as the
specialized nature of sperm components. However, as
a caveat, some of this interstudy variation could result
from differences in analytical strategies, especially with
regard to LC-MS/MS analysis and peptide database
searching. The amount of this variation that arises
from true proteomic differences in subcellular com-
partments and experimental error remains an open
question that future studies should strive to address.
On the basis of our analysis, only five proteins were
common to all human sperm subcellular fractions
(Table 2; see Tables S1 and S2 for full data on overlap
between datasets, and a list of proteins common to
three or more human sperm datasets). The fact that
these proteins are found in all five subcellular prote-
ome datasets does not necessarily imply that they have
a role in spermatogenesis or sperm function, and they
could simply represent prominent housekeeping genes.
In fact, if a protein is restricted to only one highly
Fig. 2. Overlap in protein identification between subcellular
fractions of human sperm. A Venn diagram is shown, depicting the
number of proteins that are unique and shared between the
datasets from human sperm listed in Table 1. The diagram shows
that a considerable number of proteins appear to be unique to
each fraction, whereas a few proteins are identified in all five
datasets. The Venn diagram was generated with the VIB/UGent
Bioinformatics & Evolutionary Genomics web-based tool available
at http://bioinformatics.psb.ugent.be/webtools/Venn/.
FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS 5641
G. MacLeod and S. Varmuza Proteomic research in mammalian spermatogenesis

specialized structure, it may have a specific and essen-
tial role in the structure or function of that structure.
Collectively, these studies account for > 2200 proteins,
approaching the estimated size of the sperm proteome.
It is clear from the examples presented above that
there is a wealth of information to be obtained when
subcellular fractionation is combined with proteomic
analysis. Not only can localization information be
gained, but increased depth of coverage can also be
achieved by simplifying the sample and decreasing the
dynamic range. Despite the progress to date, there are
still other structures and cell types to be analyzed, and
even among those for which datasets exist, much of
the subcellular proteome remains undiscovered. For
example, many male infertility mutations show similar
phenotypes, despite their range of gene ontology func-
tions, often starting at the round spermatid stage,
which is characterized by the evolving development of
the acrosome. These mouse mutations often mimic the
testicular failure phenotypes observed in infertile men.
Proteomic analysis of the developing acrosome would
be a useful addition to the repertoire.
PTMs in spermatogenic cells
Another area of proteomics that has garnered
increased interest over the past several years is the
study of PTMs. Once translated in the cell, proteins
can be covalently modified in a number of different
ways, which can govern the activity of proteins and
signaling networks. Identifying PTMs of testis proteins
provides a more detailed picture of how those proteins
exist in the tissue, and can offer clues regarding their
functions. Also, by characterizing changes in PTM sta-
tus in response to different stimuli or at different time
points in spermatogenesis, we can gain even more
insights into the regulation of a protein’s activity. The
analysis of PTMs is especially relevant in mature
sperm, as they are both transcriptionally and transla-
tionally silent [51], meaning that the bulk of cellular
functions must be governed by post-translational mod-
ulation of protein function.
Regulated by the opposing activity of kinases and
phosphatases, phosphorylation is by far the most stud-
ied PTM in spermatogenesis. Following recent techno-
logical advances in the isolation of phosphorylated
peptides, such as those by Larsen et al. [52], the field
of phosphoproteomics has experienced significant
growth over the past several years. The study of sper-
matogenesis has been no exception to this, as an
increasing number of groups have performed phospho-
proteome analysis in sperm and spermatogenic cells.
However, the largest existing testis phosphoproteomic
datasets were not produced from laboratories focusing
directly on spermatogenesis, but from large-scale stud-
ies aiming to map phosphorylation sites across multi-
ple tissues. One large-scale analysis of nine tissues
from 3-week-old mice identified > 2500 phosphopro-
teins in the testis corresponding to > 10 000 phosphor-
ylation sites, following immobilized metal ion affinity
chromatography (IMAC) phosphopeptide enrichment
[53]. Another phosphoproteomic study utilized tita-
nium dioxide (TiO2) phosphopeptide enrichment on 14
different tissues and organs in the rat, and also identi-
fied > 10 000 phosphorylation sites in the testis,
including over 200 testis-specific phosphorylation sites,
and provided quantitative data by the use of extracted
ion chromatograms [54]. These studies, although not
directly focused on spermatogenesis, have provided a
wealth of data to the research community, and insights
into the post-translational regulation of spermatogene-
sis. As an example, both studies found that the testis
was second only to the brain in the number of tissue-
specific phosphorylation sites (17% of all identified
sites in one experiment [53]), which suggests the possi-
bility that protein phosphorylation may be particularly
important in spermatogenesis and the regulation of
sperm function.
Aside from these large-scale analyses of the mamma-
lian testis, most groups have chosen to focus on sperm
Table 2. Proteins common to five human sperm subcellular proteome datasets. Testis/sperm specificity data were compiled from
www.uniprot.org, and information regarding the existence of infertile mouse models was compiled from http://www.informatics.jax.org.
Official gene
symbol
Uniprot
accession Name
Testis/sperm-
specific?
Infertile
mouse
model?
RAB2A B2R5W8 RAB2A, a member of the RAS
oncogene family
No No
HSPA2 Q9UE78 Heat shock 70-kDa protein 2 No Yes
VCP Q969G7 Valosin-containing protein No No
SPANXB1 Q5JYZ7 SPANX family, member B1 Yes No
LTF Q8IU92 Lactotransferrin No No
5642 FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS
Proteomic research in mammalian spermatogenesis G. MacLeod and S. Varmuza

for phosphoproteomic analyses, owing to their ease of
retrieval and transcriptional/translational silent status.
Baker et al. have published two separate examinations
of changes in protein phosphorylation during rat epi-
didymal maturation, using TiO2 phosphopeptide
enrichment, and have identified 53and 22 differentially
phosphorylated proteins [55,56]. Perhaps the most
extensively studied aspect of sperm maturation via
phosphoproteomic analysis is capacitation, which has
been examined in the mouse [57], rat [58], and human
[59]. Interestingly, no common phosphorylation event
was found between species, which points to key meth-
odological differences, as well as the probability that a
large portion of the phosphoproteome remains unex-
amined. This may also reflect evolutionary divergence;
analysis of phosphosites, both predicted and known,
across a range of species may yield insights into those
sites that are biologically relevant (see, for example,
www.phosphonet.ca). Another investigation even per-
formed phosphoproteomic analysis of clinical samples,
comparing sperm from fertile individuals with those
suffering from asthenozoospermia (reduced sperm
motility), and identified 66 differentially phosphory-
lated peptides [60]. In contrast to those groups examin-
ing sperm, our group has focused on the response of
developing spermatogenic cells in the testis to the loss
of the protein phosphatase PPP1CC, and identified 10
proteins that are hyperphosphorylated in response to
this deletion [61]. Presently, we are undertaking a larger
phosphoproteomic analysis of the developing mouse
testis, which has identified > 700 phosphorylated pro-
teins, with the ultimate aim of identifying candidate
substrates of PPP1CC2, the testis-specific isoform
(G. MacLeod, P. Taylor, L. Mastropaolo, S. Varmuza,
in preparation). However, a key challenge in such
experiments is differentiating between changes resulting
from perturbation of direct phosphorylation/dephos-
phorylation events, and indirect effects such as com-
pensatory changes, and or downstream signaling
events.
In addition to the growing number of phosphopro-
teomic studies relating to spermatogenesis and sperm
function, a series of other types of PTM have also
been catalogued (Table 3). Similarly to the aforemen-
tioned phosphoproteomic study, Lundby et al.
recently quantified > 15 000 lysine acetylation sites on
> 4500 proteins across a number of rat tissues,
including almost 2000 proteins in the testis [62].
Lysine acetylation is of particular interest in sper-
matogenesis, owing to its well-characterized involve-
ment in the modification of histones, which have a
central role in spermatogenesis. Protein glycosylation
in the mouse testis has been examined in at least two
large-scale studies, which identified 239 and 634
unique glycoproteins [63,64]. One of these studies
identified four glycoproteins that were dominantly
expressed in the testis over any other tissue – dipepti-
dase 3, zona pellucida 3 receptor, TEX101, and Dick-
kopf-like 1 [63]. Other PTMs that have been studied
in human sperm include S-nitrosylation, by Lefi�evre
et al. (240 modified proteins) [65] and, most recently,
SUMOylation by Vigodner et al. (55 modified pro-
teins) [66]. Another PTM of interest is ubiquitination;
however, to our knowledge, no large-scale analysis of
the ubiquitin-modified proteome has been conducted
in the mouse testis or sperm, and a recently published
survey of several mouse tissues analyzed only liver,
kidney, heart, muscle, and brain [67]. However, the
spermatogenesis defects in mice lacking functional
ubiquitin-conjugating enzyme E2B, a ubiquitin ligase,
is a clear sign that this PTM is critical to germline
development [68].
Table 3. Recent examples of large-scale studies characterizing PTMs in mammalian testes or sperm. Studies listed are only those that
published a complete list of mapped PTMs (i.e. not only those showing significant change). AAL, Aleuria aurantia lectin; ConA,
concanavalin A; RCA120, Ricinis communis agglutinin-120; SPEG, solid-phase extraction of N-linked glycopeptides.
PTM Cell/tissue Species Enrichment method
No. of modified
testis proteins Reference
Phosphorylation Testis Mouse IMAC 2714 Huttlin et al. [53]
Phosphorylation Testis Rat TiO2 3430 Lundby et al. [54]
Phosphorylation Testis Mouse IMAC + TiO2 755 MacLeod, Taylor, Mastropaolo and
Varmuza (unpublished results)
Phosphorylation Sperm Human TiO2 120 Baker et al. [58]
Lysine acetylation Testis Rat Anti-acetyl-lysine immunoprecipitation 1941 Lundby et al. [62]
N-Glycosylation Testis Mouse SPEG 239 Tian et al. [63]
N-Glycosylation Testis Mouse Lectin columns (ConA, RCA120, AAL) 634 Kaji et al. [64]
S-Nitrosylation Sperm Human Biotin-switch assay 240 Lefi�evre et al. [65]
SUMOylation Sperm Human Anti-SUMO immunoprecipitation 55 Vigodner et al. [66]
FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS 5643
G. MacLeod and S. Varmuza Proteomic research in mammalian spermatogenesis

It is clear from these studies that the nature of a
protein is not fully determined when it is trans-
lated – PTMs confer an extremely high diversity of
protein forms within a given cell type, a complexity
that must be better understood for deciphering of a
protein’s function. Fortunately, technological improve-
ments in MS instrumentation and protein enrichment
are moving us closer to this goal.
Protein–protein interaction networks(interactomes) in spermatogenesis
Proteins rarely, if ever, function in isolation – it is the
interaction between numerous protein species that
results in functions being performed. Thus, to fully
understand a process as complex as spermatogenesis,
we must look not only at what proteins are present in a
given space and how they are modified, but at which
proteins they interact with – the interactome. By char-
acterizing interactomes, i.e. the full complement of pro-
tein–protein interactions for a given protein, we have a
much better chance of understanding a protein’s func-
tion than if we look at each in isolation. In the past,
the most common approach to studying interactomes
was the yeast two-hybrid assay. Although this
approach has been quite successful in identifying pro-
tein–protein interactions, even with regard to genes
that are important to spermatogenesis (e.g. [69–71]), ithas several prominent limitations, owing to the artifi-
cial nature of the system (e.g. the absence of species/tis-
sue-specific PTMs) and the fact that it looks only at
binary interactions. Currently, the yeast two-hybrid
system has been largely replaced by affinity purifica-
tion/MS (AP-MS)-based approaches, featuring either
single or tandem affinity purification (TAP) tags. Other
commonly employed techniques for uncovering
protein–protein interactions, such as coimmunoprecipi-
tation, size-exclusion chromatography, and immunohis-
tochemistry, have been successfully used in the testis;
however, they are less amenable to large-scale and
high-throughput analysis than AP-MS. These
approaches are typically applied to mammalian tissue
culture systems, and can simultaneously identify a large
number of protein–protein interactions, including mul-
tiprotein complexes. AP-MS, when applied with the
appropriate controls, also results in the identification
of fewer false-positive interactions. Additionally, the
use of a mammalian system is, for obvious reasons,
preferable to the use of a yeast-based system (see [72]
for an overview of such systems). Although tissue cul-
ture-based AP-MS systems have been used with some
success to examine the interactomes of proteins
involved in spermatogenesis [73], they still suffer limita-
tions, owing to their artificial nature. This limitation is
particularly important in the study of spermatogenesis.
As outlined in the preceding sections, the testis is
particularly abundant in tissue-specific protein expres-
sion as well as PTMs, and the complex architecture of
the testis cannot be modeled in culture. Thus, if the in-
teractome of a protein involved in spermatogenesis is
defined by the use tissue culture, it is likely that a large
amount of information will be missed. For this reason,
it is important to conduct interactome studies directly
in the testis when possible. This prospect is signifi-
cantly more demanding than tissue culture-based
approaches, because of the need to have either highly
specific antibodies or the ability to generate a trans-
genic line with an affinity-tagged version of the gene
of interest. Despite these technological demands, a
number of groups have successfully performed interac-
tome studies in the testis. In 2009, Chen et al. used
immunoprecipitation followed by gel-free LC-MS/MS
to characterize the interactomes of MIWI and MILI in
the mouse testis [74]. The authors then went on to per-
form a reciprocal experiment using one of their identi-
fied interaction partners, TDRKH, and characterized a
multiprotein interaction network in the testis. Simi-
larly, another experiment used a modified immunopre-
cipitation and MS approach to identify an additional
member of the CATSPER complex in the mouse testis
[75]. The authors of this study had undertaken this
approach because of their inability to successfully
express the CATSPER complex in any other system,
which underscores the importance of examining pro-
tein interactions in their natural environment. Other
groups have generated transgenic mouse lines in order
to perform TAP directly in the testis. To identify novel
interactors involved in Bardet–Biedl syndrome, a com-
mon feature of which is male infertility, Seo et al. gen-
erated a mouse line expressing a green fluorescent
protein-coupled and S-tag-coupled Bardet–Biedl syn-
drome 4 construct, and performed TAP in transgenic
testes, successfully identifying a novel protein complex
member [76]. Another TAP experiment in the testis
using a mouse expressing TAP-tagged 14-3-3f identi-
fied a large number of novel protein–protein interac-
tions, although the lack of appropriate control
experiments leaves the total number of true interactors
in question [77].
The generation of transgenic mouse lines expressing
affinity-tagged genes is not a trivial process, and
requires a significant amount of time for design and
production. For this reason, systems have been devel-
oped to aid in the production of affinity-tagged trans-
genic lines in a more efficient manner. For example,
the Floxin vector system can be used to derive knock-
5644 FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS
Proteomic research in mammalian spermatogenesis G. MacLeod and S. Varmuza

in embryonic stem cell lines from gene-trap lines in
just a few relatively basic steps [78]. This system has
the added benefit of allowing for the introduction of
the transgene into the endogenous locus to minimize
any overexpression/mis-expression-related artefacts.
Our laboratory has used the Floxin vector backbone
to generate a streptavidin-binding peptide–3 9 FLAG
tandem affinity tagged knock-in vector that can be
used to produce N-terminally tagged knock-ins of any
gene [73]. This system was used to create an embryonic
stem cell line expressing streptavidin-binding peptide–3 9 FLAG–PPP1CC2, which was used to identify a
novel interacting protein, dolichyl-diphosphooligosac-
charide–protein glycosyltransferase, with a proposed
role in spermatogenesis. However, this system has not
yet been used to successfully generate a transgenic
mouse for interactome studies. Although culture-based
AP-MS strategies remain a powerful tool for interac-
tome discovery, the full power of such approaches can
only be harnessed when they are used in a more bio-
logically relevant context. With technological advances
in the generation of transgenic animals and AP-MS
methodologies, such approaches will probably become
more common in the coming years.
The utilization of proteomics inclinical studies of male infertility
The ultimate aim in furthering our understanding of
spermatogenesis is to improve the ability to diagnose
and treat male infertility. In recent years, a number of
investigators have applied proteomic analysis to clini-
cal samples in an effort to go beyond basic research.
By analyzing the genital tract proteomes of infertile
men, it may be possible to detect aberrations that
explain impairments and evaluate the feasibility and
course of future treatment. Although it is difficult to
obtain a sufficient amount of starting material from
spermatogenic cells in the testes of infertile men, ejacu-
lated sperm and seminal fluid are much easier to
obtain, and have thus been the subject of the bulk of
the studies to date.
The proteome of sperm from infertile donors has
been examined by a number of groups, to date primar-
ily by 2DE followed by identification of differentially
expressed protein spots via MS. Experiments have
identified proteins that are differentially expressed in
patients showing generalized infertility [49,79], low
sperm count and motility [80], asthenozoospermia [81–83], Sertoli cell-only syndrome [84], in vitro fertilization
failure [85], diabetes, and obesity [86]. Collectively,
these studies have been able to identify a large number
of candidate biomarkers for sperm defects that could
be clinically relevant in the coming years. However,
these types of study have had limited impact at the
clinical level, in part because of the heterogeneity of
the disease paradigms, and because of the difficulty in
parsing datasets from tissues that are catastrophically
different from controls.
The seminal plasma provides a protective and facili-
tative environment for sperm transit that is critical in
a number of ways for proper sperm function. There-
fore, a number of clinical studies have applied proteo-
mic analysis in the hopes of finding useful biomarkers
for defects of the male reproductive system. According
to Batruch et al. [87], the seminal plasma is an excel-
lent fluid for clinical research, given its ease of collec-
tion and the fact that it contains secreted and shed
proteins originating from several different tissues,
which can offer clues to the origin of clinical defects.
Many mouse mutations resulting in testicular failure
are characterized by the exfoliation of immature germ
cells into the seminiferous lumen, which would then
contribute breakdown products to the seminal fluid as
they transit the epididymis. The same feature can be
seen in infertile men whose ejaculate contains imma-
ture germ cells, which are sometimes used for intracy-
toplasmic sperm injection in the treatment of male
factor infertility. Although examinations of seminal
plasma proteins have been performed for decades, in
recent years the characterization of the fluid has
reached new heights, owing to the increased use of
LC-MS/MS technologies, leading to the identification
of ~ 3000 seminal plasma proteins. In 2006, Pilch and
Mann, using Fourier transform MS, identified > 900
proteins in seminal plasma from a single individual,
representing, at the time, the largest catalog of pro-
teins in that fluid [88]. Since then, a number of groups
have used proteomic approaches to identify novel bio-
markers for male infertility [87,89–93]. One investiga-
tion, by Wang et al., utilized one-dimensional SDS/
PAGE followed by LC-MS/MS to identify 741 seminal
plasma proteins from normal fertile donors and those
suffering from asthenozoospermia, including 101 that
were differentially expressed between the two groups
[93]. The most comprehensive proteomic analyses of
the seminal plasma published to date are represented
by a series of papers from Jarvi and colleagues, which
include label-free quantitative analysis [87,89,91,94].
Collectively, among five published accounts, > 2500
seminal plasma proteins have been identified in sam-
ples from fertile donors, postvasectomy patients, and
those suffering from nonobstructive azoospermia and
prostatitis. From these studies, a large number of
potential biomarkers have been identified, including
those useful in discrimination between obstructive and
FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS 5645
G. MacLeod and S. Varmuza Proteomic research in mammalian spermatogenesis

nonobstructive azoospermia, which provide a noninva-
sive diagnostic alternative to the current practices of
testis biopsy and histology. Another study incorpo-
rated gene expression data from publicly available
databases alongside seminal plasma proteomic data to
identify biomarkers for pathologies of the male genetic
tract [95]. Similar proteomic approaches are being con-
templated for the diagnosis of other male reproductive
tract disorders and diseases, such as prostate cancer.
The flip side to these clinical needs is the development
of a nonsteroidogenic male contraceptive; one promis-
ing compound interferes reversibly with the action of
the testis-specific chromatin regulator BRDT to
temporarily interrupt spermatogenesis [47].
One factor that requires further investigation in clin-
ical studies such as those mentioned above is the
extent of interindividual variability in the proteomic
content of sperm and/or seminal plasma. One study by
Milardi et al., which examined the seminal plasma
proteomes of five different fertile men, found only 83
proteins in common between all individuals in a study
that identified between 919 and 1487 proteins per sam-
ple [92]. This number seems surprisingly low, and per-
haps points to interexperiment variability as much as
interindividual variability. The fact remains, however,
that until we better understand what ‘normal’ varia-
tion in proteomic content constitutes, it will be difficult
to identify a full complement of phenotype-related
abnormalities. Despite this limitation, the routine use
of proteomic analysis in a clinical setting may be prac-
tical in the near future. Alternatively, experiments such
as those listed above can be used as discovery tools to
produce smaller biomarker panels that are simpler to
use and interpret, less expensive, and thus easier
to institute in a clinical setting [94].
Emerging proteomic applications andthe study of spermatogenesis
In the preceding sections, future applications of sper-
matogenesis research have been highlighted for differ-
ent proteomic subdisciplines. If information from all
of these types of studies are considered together, a
great deal can be learned about a protein’s function in
spermatogenesis. As currently existing technologies
improve and new ones emerge, their application to the
study of spermatogenesis will follow. Other methods,
such as antibody microarrays, have the potential to
generate useful information when applied to the study
of spermatogenesis, although this approach has yet to
gain purchase in the field (see, for example, [96]).
One emerging proteomic technology that will probably
be beneficial to the study of spermatogenesis consti-
tutes the interface between imaging and proteo-
mics – MALDI imaging MS. This technique allows
for the identification and detection of proteins directly
in tissue slices which offers a wealth of spatiotemporal
information. This technique has been applied to both
the rat testis [97] and the mouse epididymis [98], in
both cases allowing the mapping of a number of dif-
ferent proteins throughout those tissues. Despite a
high amount of promise, this technique has been held
back by a series of technological hurdles; once these
have been resolved, this technique could become an
extremely powerful new tool.
Technological advances in label-free quantitative
proteomics, such as selective reaction monitoring,
multiple reaction monitoring, and MS1 filtering, could
possibly constitute the most important development
in the proteomic study of spermatogenesis in the near
future. These technologies give researchers the ability
not only to determine which proteins are present, but
also to accurately quantitate them in a variety of bio-
logical contexts at a resolution far exceeding that
obtained with spectral counting-based approaches.
Furthermore, label-free quantitative approaches are
more amenable for use on tissue samples than label-
ing strategies such as stable isotope labeling with
amino acids in cell culture, and thus may find more
widespread use in reproductive biology research. The
development of cross-platform, open-source software
packages such as SKYLINE for label-free quantitative
analysis should allow more researchers to have access
to these quantitative proteomic methods, as well as
allow for increased comparison of results across mul-
tiple laboratories [99]. In fact, our laboratory has
recently used this software in a quantitative phospho-
proteomic study of the mouse testis (G. MacLeod,
P. Taylor, L. Mastropaolo, S. Varmuza, in prepara-
tion). Quantitative proteomics should allow for more
accurate assessment of sperm and seminal plasma sam-
ples in cases of male infertility as well; either in global
proteomic analysis of these samples to identify quanti-
tative changes, or, perhaps more likely, to validate
potential biomarkers identified in larger studies. For
example, Drabovich et al. [94] used selective reaction
monitoring with labeled internal standards to reanalyze
20 candidate biomarkers that discriminate between
fertile, postvasectomy and nonobstructive azoospermia
patients. These approaches could facilitate pilot studies
to identify more specific subsets of proteins that could
then be quantitated on larger samples with less
expensive technologies such as ELISA.
As the technologies discussed in this article continue
to improve, data are being generated at an increasing
rate. Thus, a pressing issue in the future of proteomics
5646 FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS
Proteomic research in mammalian spermatogenesis G. MacLeod and S. Varmuza

is how to integrate the data from a variety of sources,
including genetic studies. A need exists for improved
methods for turning ‘protein lists’ into testable hypoth-
eses. Also, questions of the accuracy of much of the
data in existing proteomic databases have been raised;
a particularly contentious issue is the field of PTM site
assignment. However, despite these questions, it is
clear that the use of proteomic technologies to study
biological processes such as spermatogenesis and
sperm function is becoming more and more prevalent
and powerful. The field of reproductive biology as a
whole will benefit from this, and, as our understanding
of these processes improves, our ability to diagnose
and treat male infertility will be greatly enhanced.
References
1 Matzuk MM & Lamb DJ (2008) The biology of
infertility: research advances and clinical challenges.
Nat Med 14, 1197–1213.
2 Schultz N, Hamra FK & Garbers DL (2003) A
multitude of genes expressed solely in meiotic or
postmeiotic spermatogenic cells offers a myriad of
contraceptive targets. Proc Natl Acad Sci USA 100,
12201–12206.
3 Massart A, Lissens W, Tournaye H & Stouffs K (2012)
Genetic causes of spermatogenic failure. Asian J Androl
14, 40–48.
4 Carrell DT & Aston KI (2011) The search for SNPs,
CNVs, and epigenetic variants associated with the
complex disease of male infertility. Syst Biol Reprod
Med 57, 17–26.
5 Cagney G, Park S, Chung C, Tong B, O’Dushlaine C,
Shields DC & Emili A (2005) Human tissue profiling
with multidimensional protein identification technology.
J Proteome Res 4, 1757–1767.
6 Gan H, Cai T, Lin X, Wu Y, Wang X, Yang F & Han
C (2013) Integrative proteomic and transcriptomic
analyses reveal multiple post-transcriptional regulatory
mechanisms of mouse spermatogenesis. Mol Cell
Proteomics. 12, 1144–1157.
7 Yeo G, Holste D, Kreiman G & Burge C (2004)
Variation in alternative splicing across human tissues.
Genome Biol 5, R74.
8 Varmuza S, Jurisicova A, Okano K, Hudson J,
Boekelheide K & Shipp EB (1999) Spermiogenesis is
impaired in mice bearing a targeted mutation in the
protein phosphatase 1cc gene. Dev Biol 205, 98–110.
9 Bonilla E & Xu EY (2008) Identification and
characterization of novel mammalian spermatogenic
genes conserved from fly to human. Mol Hum Reprod
14, 137–142.
10 Dean MD, Clark NL, Findlay GD, Karn RC, Yi X,
Swanson WJ, MacCoss MJ & Nachman MW (2009)
Proteomics and comparative genomic investigations
reveal heterogeneity in evolutionary rate of male
reproductive proteins in mice (Mus domesticus). Mol
Biol Evol 26, 1733–1743.
11 Baker MA & Aitken RJ (2009) Proteomic insights into
spermatozoa: critiques, comments and concerns. Expert
Rev Proteomics 6, 691–705.
12 Wilhelm D, Huang E, Svingen T, Stanfield S, Dinnis D
& Koopman P (2006) Comparative proteomic analysis
to study molecular events during gonad development in
mice. Genesis 44, 168–176.
13 Zhu Y, Cui Y, Guo X, Wang L, Bi Y, Hu Y, Zhao X,
Liu Q, Huo R, Lin M et al. (2006) Proteomic analysis
of the effect of hyperthermia on spermatogenesis in
adult male mice. J Proteome Res 5, 2217–2225.
14 Guo X, Zhang P, Huo R, Zhou Z & Sha J (2008)
Analysis of the human testis proteome by mass
spectrometry and bioinformatics. Proteomics Clin Appl
2, 1651–1657.
15 Guo X, Zhao C, Wang F, Zhu Y, Cui Y, Zhou Z, Huo
R & Sha J (2010) Investigation of human testis protein
heterogeneity using 2-dimensional electrophoresis. J
Androl 31, 419–429.
16 Li J, Liu F, Liu X, Liu J, Zhu P, Wan F, Jin S, Wang
W, Li N, Liu J et al. (2011) Mapping of the human
testicular proteome and its relationship with that of the
epididymis and spermatozoa. Mol Cell Proteomics 10,
M110.004630.
17 Geiger T, Wehner A, Schaab C, Cox J & Mann M
(2012) Comparative proteomic analysis of eleven
common cell lines reveals ubiquitous but varying
expression of most proteins. Mol Cell Proteomics 11,
M111.014050.
18 Huang S, Lin J, Teng S, Sun HS, Chen Y, Chen H,
Liao J, Chung M, Chen M, Chuang C et al. (2011)
Differential expression of porcine testis proteins during
postnatal development. Anim Reprod Sci 123, 221–233.
19 Huang X, Guo X, Shen J, Wang Y, Chen L, Xie J,
Wang N, Wang F, Zhao C, Huo R et al. (2008)
Construction of a proteome profile and functional
analysis of the proteins involved in the initiation of
mouse spermatogenesis. J Proteome Res 7, 3435–3446.
20 Clausen OPF, Purvis K & Hansson V (1977)
Application of micro-flow fluorometry to studies of
meiosis in the male rat. Biol Reprod 17, 555–560.
21 Lam DMK, Furrer R & Bruce WR (1970) The
separation, physical characterization and differentiation
kinetics of spermatogonial cells of the mouse. Proc Natl
Acad Sci USA 65, 192–199.
22 Grabske RJ, Lake S, Gledhill BL & Meistrich ML
(1975) Centrifugal elutriation: separation of
spermatogenic cells on the basis of sedimentation
velocity. J Cell Physiol 86, 177–189.
23 Delbes G, Yanagiya A, Sonenberg N & Robaire B
(2011) PABP interacting protein 2A (PAIP2A) regulates
FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS 5647
G. MacLeod and S. Varmuza Proteomic research in mammalian spermatogenesis

specific key proteins during spermiogenesis in the
mouse. Biol Reprod 86,95: 1–8.
24 Guillaume E, Dupaix A, Moertz E, Courtens J, J�egou
B & Pineau C (2000) Proteome analysis of
spermatogonia: identification of a first set of 53
proteins. Proteome 1, 1–20.
25 Com E, Evrard B, Roepstorff P, Aubry F & Pineau C
(2003) New insights into the rat spermatogonial
proteome. Mol Cell Proteomics 2, 248–261.
26 Dihazi H, Dihazi GH, Nolte J, Meyer S, Jahn O,
M€uller GA & Engel W (2009) Multipotent adult
germline stem cells and embryonic stem cells:
comparative proteomic approach. J Proteome Res 8,
5497–5510.
27 Guo X, Zhang P, Qi Y, Chen W, Chen X, Zhou Z &
Sha J (2011) Proteomic analysis of male 4C germ cell
proteins involved in mouse meiosis. Proteomics 11,
298–308.
28 Guo X, Shen J, Xia Z, Zhang R, Zhang P, Zhao C,
Xing J, Chen L, Chen W, Lin M et al. (2010)
Proteomic analysis of proteins involved in
spermiogenesis in mouse. J Proteome Res 9, 1246–1256.
29 Johnston DS, Wooters J, Kopf GS, Qiu Y & Roberts
KP (2005) Analysis of the human sperm proteome.
Ann N Y Acad Sci 1061, 190–202.
30 Baker MA, Reeves G, Hetherington L, M€uller J, Baur I
& Aitken RJ (2007) Identification of gene products
present in Triton X-100 soluble and insoluble fractions
of human spermatozoa lysates using LC-MS/MS
analysis. Proteomics Clin Appl 1, 524–532.
31 Baker MA, Naumovski N, Hetherington L, Weinberg
A, Velkov T & Aitken RJ (2013) Head and flagella
sub-compartmental proteomic analysis of human
spermatozoa. Proteomics 13, 61–74.
32 Baker MA, Hetherington L, Reeves GM & Aitken RJ
(2008) The mouse sperm proteome characterized via
IPG strip prefractionation and LC-MS/MS
identification. Proteomics 8, 1720–1730.
33 Baker MA, Hetherington L, Reeves G, M€uller J &
Aitken RJ (2008) The rat sperm proteome characterized
via IPG strip prefractionation and LC-MS/MS
identification. Proteomics 8, 2312–2321.
34 Peddinti D, Nanduri B, Kaya A, Feugang J, Burgess S
& Memili E (2008) Comprehensive proteomic analysis
of bovine spermatozoa of varying fertility rates and
identification of biomarkers associated with fertility.
BMC Syst Biol 2, 19.
35 Papaioannou MD, Lagarrigue M, Vejnar CE, Rolland
AD, K€uhne F, Aubry F, Schaad O, Fort A, Descombes
P, Neerman-Arbez M et al. (2011) Loss of dicer in
Sertoli cells has a major impact on the testicular
proteome of mice. Mol Cell Proteomics 10, M900587–
MCP200.
36 Stanton PG, Sluka P, Foo CFH, Stephens AN, Smith
AI, McLachlan RI & O’Donnell L (2012) Proteomic
changes in rat spermatogenesis in response to in vivo
androgen manipulation; impact on meiotic cells. PLoS
One 7, e41718.
37 Rolland AD, Evrard B, Guitton N, Lavigne R, Calvel
P, Couvet M, J�egou B & Pineau C (2007) Two-
dimensional fluorescence difference gel electrophoresis
analysis of spermatogenesis in the rat. J Proteome Res
6, 683–697.
38 Asano A, Nelson JL, Zhang S & Travis AJ (2010)
Characterization of the proteomes associating with
three distinct membrane raft sub-types in murine sperm.
Proteomics 10, 3494–3505.
39 Belleannee C, Belghazi M, Labas V, Teixeira-Gomes A,
Gatti JL, Dacheux J & Dacheux F (2011) Purification
and identification of sperm surface proteins and
changes during epididymal maturation. Proteomics 11,
1952–1964.
40 Byrne K, Leahy T, McCulloch R, Colgrave ML &
Holland MK (2012) Comprehensive mapping of the
bull sperm surface proteome. Proteomics 12, 3559–3579.
41 Nixon B, Bielanowicz A, Mclaughlin EA, Tanphaichitr
N, Ensslin MA & Aitken RJ (2009) Composition and
significance of detergent resistant membranes in mouse
spermatozoa. J Cell Physiol 218, 122–134.
42 Nixon B, Mitchell LA, Anderson AL, Mclaughlin EA,
O’Bryan MK & Aitken RJ (2011) Proteomic and
functional analysis of human sperm detergent resistant
membranes. J Cell Physiol 226, 2651–2665.
43 Gu B, Zhang J, Wu Y, Zhang X, Tan Z, Lin Y,
Huang X, Chen L, Yao K & Zhang M (2011)
Proteomic analyses reveal common promiscuous
patterns of cell surface proteins on human embryonic
stem cells and sperms. PLoS One 6, e19386.
44 Dorus S, Wasbrough ER, Basby J, Wilkin EC & Karr
TL (2010) Sperm proteomics reveals intensified
selection on mouse sperm membrane and acrosome
genes. Mol Biol Evol 27, 1235–1246.
45 Guyonnet B, Zabet-Moghaddam M, SanFrancisco S &
Cornwall GA (2012) Isolation and proteomic
characterization of the mouse sperm acrosomal matrix.
Mol Cell Proteomics 11, 758–774.
46 Amaral A, Castillo J, Estanyol JM, Ballesca JL,
Ramalho-Santos J & Oliva R (2012) Human sperm tail
proteome suggests new endogenous metabolic
pathways. Mol Cell Proteomics 12, 330–342.
47 Matzuk MM, McKeown MR, Filippakopoulos P, Li Q,
Agno JE, Lemieux ME, Picaud S, Yu RN, Qi J, Knapp
S et al. (2012) Small-molecule inhibition of BRDT for
male contraception. Cell 150, 673–684.
48 Yan W (2009) Male infertility caused by spermiogenic
defects: lessons from gene knockouts. Mol Cell
Endocrinol 306, 24–32.
49 de Mateo S, Castillo J, Estanyol JM, Ballesc�a JL &
Oliva R (2011) Proteomic characterization of the
human sperm nucleus. Proteomics 11, 2714–2726.
5648 FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS
Proteomic research in mammalian spermatogenesis G. MacLeod and S. Varmuza

50 Govin J, Gaucher J, Ferro M, Debernardi A, Garin J,
Khochbin S & Rousseaux S (2012) Proteomic strategy
for the identification of critical actors in reorganization
of the post-meiotic male genome. Mol Hum Reprod 18,
1–13.
51 Boerke A, Dieleman SJ & Gadella BM (2007) A
possible role for sperm RNA in early embryo
development. Theriogenology 68 (Suppl 1), S147–S155.
52 Larsen MR, Thingholm TE, Jensen ON, Roepstorff P
& Jørgensen TJD (2005) Highly selective enrichment of
phosphorylated peptides from peptide mixtures using
titanium dioxide microcolumns. Mol Cell Proteomics 4,
873–886.
53 Huttlin EL, Jedrychowski MP, Elias JE, Goswami T,
Rad R, Beausoleil SA, Vill�en J, Haas W, Sowa ME &
Gygi SP (2010) A tissue-specific atlas of mouse protein
phosphorylation and expression. Cell 143, 1174–1189.
54 Lundby A, Secher A, Lage K, Nordsborg NB,
Dmytriyev A, Lundby C & Olsen JV (2012) Quantitative
maps of protein phosphorylation sites across 14 different
rat organs and tissues. Nat Commun 3, 876.
55 Baker MA, Smith ND, Hetherington L, Pelzing M,
Condina MR & Aitken RJ (2011) Use of titanium
dioxide to find phosphopeptide and total protein
changes during epididymal sperm maturation.
J Proteome Res 10, 1004–1017.
56 Baker MA, Hetherington L, Weinburg A, Naumovski
N, Velkov T, Pelzing M, Dolman S, Condina MR &
Aitken RJ (2012) Analysis of phosphopeptide changes
as spermatozoa acquire functional competence in the
epididymis demonstrates changes in the post-
translational modification of Izumo1. J Proteome Res
11, 5252–5264.
57 Platt MD, Salicioni AM, Hunt DF & Visconti PE
(2009) Use of differential isotopic labeling and mass
spectrometry to analyze capacitation-associated changes
in the phosphorylation status of mouse sperm proteins.
J Proteome Res 8, 1431–1440.
58 Baker MA, Smith ND, Hetherington L, Taubman K,
Graham ME, Robinson PJ & Aitken RJ (2010) Label-
free quantitation of phosphopeptide changes during rat
sperm capacitation. J Proteome Res 9, 718–729.
59 Ficarro S, Chertihin O, Westbrook VA, White F, Jayes
F, Kalab P, Marto JA, Shabanowitz J, Herr JC, Hunt
DF et al. (2003) Phosphoproteome analysis of
capacitated human sperm. J Biol Chem 278,
11579–11589.
60 Parte PP, Rao P, Redij S, Lobo V, D’Souza SJ,
Gajbhiye R & Kulkarni V (2012) Sperm
phosphoproteome profiling by ultra performance liquid
chromatography followed by data independent analysis
(LC–MSE) reveals altered proteomic signatures in
asthenozoospermia. J Proteomics 75, 5861–5871.
61 Henderson H, MacLeod G, Hrabchak C & Varmuza S
(2011) New candidate targets of protein phosphatase-
1c-gamma-2 in mouse testis revealed by a differential
phosphoproteome analysis. Int J Androl 34, 339–351.
62 Lundby A, Lage K, Weinert B, Bekker-Jensen D,
Secher A, Skovgaard T, Kelstrup C, Dmytriyev A,
Choudhary C, Lundby C et al. (2012) Proteomic
analysis of lysine acetylation sites in rat tissues reveals
organ specificity and subcellular patterns. Cell Rep 2,
419–431.
63 Tian Y, Kelly-Spratt KS, Kemp CJ & Zhang H (2010)
Mapping tissue-specific expression of extracellular
proteins using systematic glycoproteomic analysis of
different mouse tissues. J Proteome Res 9, 5837–5847.
64 Kaji H, Shikanai T, Sasaki-Sawa A, Wen H, Fujita M,
Suzuki Y, Sugahara D, Sawaki H, Yamauchi Y,
Shinkawa T et al. (2012) Large-scale identification of
N-glycosylated proteins of mouse tissues and
construction of a glycoprotein database, GlycoProtDB.
J Proteome Res 11, 4553–4566.
65 Lefi�evre LL, Chen Y, Conner SJ, Scott JL, Publicover
SJ, Ford WCL & Barratt CLR (2007) Human
spermatozoa contain multiple targets for protein
S-nitrosylation: an alternative mechanism of the
modulation of sperm function by nitric oxide?
Proteomics 7, 3066–3084.
66 Vigodner M, Shrivastava V, Gutstein LE, Schneider J,
Nieves E, Goldstein M, Feliciano M & CallawayM (2013)
Localization and identification of sumoylated proteins in
human sperm: excessive sumoylation is a marker of
defective spermatozoa.Hum Reprod 28, 210–223.
67 Wagner SA, Beli P, Weiner BT, Sch€olz C, Kelstrup
CD, Young C, Nielsen ML, Olsen JV & Choudhary C
(2012) Proteomic analyses reveal divergent
ubiquitylation site patterns in murine tissues. Mol Cell
Proteomics 11, 1578–1585.
68 Baarends WM, Wassenaar E, Hoogerbrugge JW,
van Cappellen G, Roest HP, Vreeburg J, Ooms M,
Hoeijmakers JH & Grootegoed JA (2003) Loss of
HR6B ubiquitin-conjugating activity results in damaged
synaptonemal complex structure and increased
crossing-over frequency during the male meiotic
prophase. Mol Cell Biol 23, 1151–1162.
69 Fardilha M, Esteves SLC, Korrodi-Greg�orio L, Vint�em
AP, Domingues SC, Rebelo S, Morrice N, Cohen
PTW, da Cruz e Silva OA & da Cruz e Silva EF (2011)
Identification of the human testis protein phosphatase 1
interactome. Biochem Pharmacol 82, 1403–1415.
70 Hrabchak C & Varmuza S (2004) Identification of the
spermatogenic zip protein Spz1 as a putative protein
phosphatase-1 (PP1) regulatory protein that specifically
binds the PP1cgamma2 splice variant in the mouse
testis. J Biol Chem 279, 37079–37086.
71 Hrabchak C, Henderson H & Varmuza S (2007) A
testis specific isoform of endophilin B1, endophilin B1t,
interacts specifically with protein phosphatase-
1cgamma2 in mouse testis and is abnormally expressed
FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS 5649
G. MacLeod and S. Varmuza Proteomic research in mammalian spermatogenesis

in PP1cgamma null mice. Biochemistry (NY) 46,
4635–4644.
72 Dunham WH, Mullin M & Gingras AC (2012) Affinity-
purification coupled to mass spectrometry: basic
principles and strategies. Proteomics 12, 1576–1590.
73 MacLeod G & Varmuza S (2012) Tandem affinity
purification in transgenic mouse embryonic stem cells
identifies DDOST as a novel PPP1CC2 interacting
protein. Biochemistry 51, 9678–9688.
74 Chen C, Jin J, James DA, Adams-Cioaba MA, Park
JG, Guo Y, Tenaglia E, Xu C, Gish G, Min J et al.
(2009) Mouse Piwi interactome identifies binding
mechanism of Tdrkh Tudor domain to arginine
methylated Miwi. Proc Natl Acad Sci USA 106,
20336–20341.
75 Chung J, Navarro B, Krapivinsky G, Krapivinsky L &
Clapham DE (2011) A novel gene required for male
fertility and functional CATSPER channel formation in
spermatozoa. Nat Commun 2, 153.
76 Seo S, Zhang Q, Bugge K, Breslow DK, Searby CC,
Nachury MV & Sheffield VC (2011) A novel protein
LZTFL1 regulates ciliary trafficking of the BBSome
and Smoothened. PLoS Genet 7, e1002358.
77 Puri P, Acker-Palmer A, Stahler R, Chen Y, Kline D &
Vijayaraghavan S (2011) Identification of testis 14-3-3
binding proteins by tandem affinity purification.
Spermatogenesis 1, 354–365.
78 Singla V, Hunkapiller J, Santos N, Seol AD, Norman
AR, Wakenight P, Skarnes WC & Reiter JF (2010)
Floxin, a resource for genetically engineering mouse
ESCs. Nat Methods 7, 50–52.
79 Xu W, Hu H, Wang Z, Chen X, Yang F, Zhu Z,
Fang P, Dai J, Wang L, Shi H et al. (2012)
Proteomic characteristics of spermatozoa in
normozoospermic patients with infertility.
J Proteomics 75, 5426–5436.
80 Thacker S, Yadav SP, Sharma RK, Kashou A, Willard
B, Zhang D & Agarwal A (2011) Evaluation of sperm
proteins in infertile men: a proteomic approach. Fertil
Steril 95, 2745–2748.
81 Zhao C, Huo R, Wang F, Lin M, Zhou Z & Sha J
(2007) Identification of several proteins involved in
regulation of sperm motility by proteomic analysis.
Fertil Steril 87, 436–438.
82 Siva AB, Kameshwari DB, Singh V, Pavani K,
Sundaram CS, Rangaraj N, Deenadayal M & Shivaji S
(2010) Proteomics-based study on asthenozoospermia:
differential expression of proteasome alpha complex.
Mol Hum Reprod 16, 452–462.
83 Mart�ınez-Heredia J, de Mateo S, Vidal-Taboada JM,
Ballesc�a JL & Oliva R (2008) Identification of
proteomic differences in asthenozoospermic sperm
samples. Hum Reprod 23, 783–791.
84 Li J, Guo W, Li F, He J, Yu Q, Wu X, Li J & Mao X
(2012) HnRNPL as a key factor in spermatogenesis:
lesson from functional proteomic studies of
azoospermia patients with sertoli cell only syndrome.
J Proteomics 75, 2879–2891.
85 Pixton KL, Deeks ED, Flesch FM, Moseley FLC,
Bj€orndahl L, Ashton PR, Barratt CLR & Brewis IA
(2004) Sperm proteome mapping of a patient who
experienced failed fertilization at IVF reveals altered
expression of at least 20 proteins compared with fertile
donors: case report. Hum Reprod 19, 1438–1447.
86 Kriegel TM, Heidenreich F, Kettner K, Pursche T,
Hoflack B, Grunewald S, Poenicke K, Glander H &
Paasch U (2009) Identification of diabetes- and obesity-
associated proteomic changes in human spermatozoa by
difference gel electrophoresis. Reprod Biomed Online 19,
660–670.
87 Batruch I, Lecker I, Kagedan D, Smith CR, Mullen BJ,
Grober E, Lo KC, Diamandis EP & Jarvi KA (2011)
Proteomic analysis of seminal plasma from normal
volunteers and post-vasectomy patients identifies over
2000 proteins and candidate biomarkers of the
urogenital system. J Proteome Res 10, 941–953.
88 Pilch B & Mann M (2006) Large-scale and high-
confidence proteomic analysis of human seminal
plasma. Genome Biol 7, R40.
89 Batruch I, Smith CR, Mullen BJ, Grober E, Lo KC,
Diamandis EP & Jarvi KA (2012) Analysis of seminal
plasma from patients with non-obstructive azoospermia
and identification of candidate biomarkers of male
infertility. J Proteome Res 11, 1503–1511.
90 Davalieva K, Kiprijanovska S, Noveski P, Plaseski T,
Kocevska B, Broussard C & Plaseska-Karanfilska D
(2012) Proteomic analysis of seminal plasma in men
with different spermatogenic impairment. Andrologia
44, 256–264.
91 Kagedan D, Lecker I, Batruch I, Smith C, Kaploun I,
Lo K, Grober E, Diamandis E & Jarvi K (2012)
Characterization of the seminal plasma proteome in
men with prostatitis by mass spectrometry. Clin
Proteomics 9, 2.
92 Milardi D, Grande G, Vincenzoni F, Messana I,
Pontecorvi A, De Marinis L, Castagnola M & Marana
R (2012) Proteomic approach in the identification of
fertility pattern in seminal plasma of fertile men. Fertil
Steril 97, 67–73.
93 Wang J, Wang J, Zhang H, Shi H, Ma D, Zhao H, Lin
B & Li R (2009) Proteomic analysis of seminal plasma
from asthenozoospermia patients reveals proteins that
affect oxidative stress responses and semen quality.
Asian J Androl 11, 484–491.
94 Drabovich AP, Jarvi K & Diamandis EP (2011)
Verification of male infertility biomarkers in seminal
plasma by multiplex selected reaction monitoring assay.
Mol Cell Proteomics 10, M110.004127.
95 Rolland AD, Lavigne R, Dauly C, Calvel P, Kervarrec
C, Freour T, Evrard B, Rioux-Leclercq N, Auger J &
5650 FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS
Proteomic research in mammalian spermatogenesis G. MacLeod and S. Varmuza

Pineau C (2013) Identification of genital tract markers
in the human seminal plasma using an integrative
genomics approach. Hum Reprod 28, 199–209.
96 Yamamoto T, Nakayama K, Hirano H, Tomonaga T,
Ishihama Y, Yamada T, Kondo T, Kodera Y, Sato Y,
Araki N et al. (2013) Integrated view of the human
chromosome X-centric proteome project. J Proteome
Res 12, 58–61.
97 Lagarrigue M, Becker M, Lavigne R, Deininger S,
Walch A, Aubry F, Suckau D & Pineau C (2011)
Revisiting rat spermatogenesis with MALDI imaging at
20-lm resolution. Mol Cell Proteomics 10,
M110.005991.
98 Chaurand P, Fouch�ecourt S, DaGue BB, Xu BJ,
Reyzer ML, Orgebin-Crist M & Caprioli RM (2003)
Profiling and imaging proteins in the mouse epididymis
by imaging mass spectrometry. Proteomics 3,
2221–2239.
99 Schilling B, Rardin MJ, MacLean BX, Zawadzka AM,
Frewen BE, Cusack MP, Sorensen DJ, Bereman MS,
Jing E, Wu CC et al. (2012) Platform-independent and
label-free quantitation of proteomic data using MS1
extracted ion chromatograms in skyline: application to
protein acetylation and phosphorylation. Mol Cell
Proteomics 11, 202–214.
Supporting information
Additional supporting information may be found in
the online version of this article at the publisher’s web
site:Table S1. Full data from the Venn diagram in Fig. 2.
Table S2. A list of proteins found in three or more
human sperm subcellular proteomic datasets.
FEBS Journal 280 (2013) 5635–5651 ª 2013 FEBS 5651
G. MacLeod and S. Varmuza Proteomic research in mammalian spermatogenesis