Embed Size (px)
Citation preview

Copyright 0 1997 by the Genetics Society of America
The Evolution of Ribosomal DNA Divergent Paralogues and Phylogenetic Implications
Edward S. Buckler IV, Anthony Ippolito and Timothy P. Holtsford
Division of Biologzcal Sciences, University of Missouri, Columbia, Missouri 65211 Manuscript received June 13, 1996
Accepted for publication November 9, 1996
ABSTRACT Although nuclear ribosomal DNA (rDNA) repeats evolve together through concerted evolution, some
genomes contain a considerable diversity of paralogous rDNA. This diversity includes not only multiple functional loci but also putative pseudogenes and recombinants. We examined the occurrence of diver- gent paralogues and recombinant.2 in Gossypium, Nicotiana, Tripsacum, Winteraceae, and Zea ribosomal internal transcribed spacer (ITS) sequences. Some of the divergent paralogues are probably rDNA pseudogenes, since they have low predicted secondary structure stability, high substitution rates, and many deamination-driven substitutions at methylation sites. Under standard PCR conditions, the low stability paralogues amplified well, while many high-stability paralogues amplified poorly. Under highly denaturing PCR conditions (i.e., with dimethylsulfoxide), both low- and high-stability paralogues ampli- fied well. We also found recombination between divergent paralogues. For phylogenetics, divergent ribosomal paralogues can aid in reconstructing ancestral states and thus serve as good outgroups. Divergent paralogues can also provide companion rDNA phylogenies. However, phylogeneticists must discriminate among families of divergent paralogues and recombinants or suffer from muddled and ~.
inaccurate organismal phylogenies.
R IBOSOMAL DNA (rDNA) is used to estimate spe- cies phylogenies for many organisms (HILLIS and
DIXON 1991). Plant rDNA consists of thousands of cop- ies o r paralogues, and these paralogues are generally located in one to several arrays. Intragenomic rDNA diversity is generally low, and this low diversity results from concerted evolution within and between ribo- somal loci (ARNHEIM 1983). However, if concerted evo- lution is slower than speciation, then a single genome will contain divergent paralogues (descendants of a du- plicated ancestral gene) (BALDWIN et al. 1995). Uniden- tified paralogous relationships and infrequent recombi- nation between paralogues can result in erroneous species phylogenies (SANDERSON and DOYLE 1992). In plants, divergent rDNA paralogues were found in stud- ies of internal transcribed spacers (ITS) and intergenic spacers (IGS) (DVORAK 1990; SUH et al. 1993; DUBCOV- SKY and DVORAK 1995; BUCKLER and HOLTSFORD 1996b). We investigated the structure and evolution of divergent paralogue DNA sequences, differential PCR amplification of the different classes of paralogues, and the phylogenetic implications of intragenomic rDNA diversity.
We characterized the divergent paralogues by exam- ining ITS secondary structure stability, substitution rates, and substitution at methylation sites in order to test the hypothesis that some of the divergent para-
Currrsponding author: Edward S. Buckler IV, Division of Biological Sciences, 105 Tucker Hall, University of Missouri, Columbia, MO 6521 1. E-mail: [email protected]
Genetics 145 821-832 (March, 1997)
logues were pseudogenes. Pseudogenes have been re- ported for many gene systems, and lately they have been isolated from the rDNA of plants and animals (MUNRO et al. 1986; LINARES et al. 1994; KELLOCG and APPELS
1995; BUCKLER and HOLTSFORD 1996a,b). rDNA para- logues might become pseudogenes when a ribosomal locus becomes inactive or when a solitary rDNA copy is dispersed to other genomic regions (an orphon, CHILDS et al. 1981). Since active ITS regions have func- tional constraints, the characteristics of functional ITS regions can help discriminate between functional and pseudogene ITS sequences. Functional ITS regions are thought to act as biological springs with many hairpins (VENKATESWARLU and NAZAR 1991) and compact, stable secondary structures (VAN NUES et al. 1995). ITS pseu- dogenes should accumulate random substitutions at high rates, which will destabilize hairpins and reduce secondary structure stability. Pseudogenes are often characterized by cytosine mutations at methylation sites, as these sites are highly mutable (LI et nl. 1984). Plant ribosomal pseudogenes might also exhibit espe- cially high levels of cytosine deamination because selec- tive constraints (possibly for chromatin condensation) have maintained a high density of methylation sites in plant rDNA (GARDINER-GARDEN et al. 1992; BUGKLER and HOL~TSFORD 1996a), and solitary orphoned rDNA is likely to be released from chromatin condensation selection. These deaminations might be the proximate cause for ITS pseudogene instability.
Although recombination between divergent para- logues is infrequent (BALDWIN et al. 1995), ITS recombi-
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

822 E. S. Buckler, A. Ippolito and T. P. Holtsford
nants or chimeric paralogues have been reported (VAN HOUTEN et al. 1993; BALDWIN et al. 1995; WENDEL et al. 1995b). Like divergent paralogues, recombinants are likely to cause phylogenetic errors (MCDADE 1992). We also consider recombination between divergent ITS pa- ralogues and identified them using several phylogenetic methods.
We examined ITS sequences for five plant groups with good sequence alignability, putative recombinant paralogues, and/or putative divergent paralogues: Gos- sypium (WENDEL et al. 1995b), Nicotiana (A. IPPOLITO and T. HOLTSFORD, unpublished results), Tripsacum (BUCKL~ER and HOLTSFORD 1996b), Winteraceae (SUH et nl. 1993), and Zea (BUCKLEK and HOLTSFORD 1996b).
MATERLALS AND METHODS
Sampling and DNA manipulation: Zea and Tripsacum in- ternal transcribed spacer region sampling, cloning, and se- quencing is detailed in BUCKLER and HOLTSFORD (1996b). Briefly, 13 Zea ITS clones were PCR amplified by using 7- deaza-2"deoxyguanosine triphosphate (c'dGTP), which aids denaturation only after incorporation (INNIS 1990). Fifty- three Zea ITS clones and all 10 Tripsacum clones were PCR amplified with 10% dimethylsulfoxide (DMSO) to facilitate denaturation throughout PCR (VARADARAJ and SJSINNER 1994). tising the same conditions and primers, we cloned 12 sequences from PCR amplification without any denaturants and cloned 16 sequences from PCR with DMSO. The clones came from the following Nicotiana species: acuminata, alata, bonariensis, forgetiann, langsdorfl, noctajlora, otophora, plumbapn- folia, ruslica, stocktonii. Some of the Nicotiana sequences did not have a complete sequence for the 5.8s region; therefore these sites were excluded from analyses involving the 5.8s. The outgroup (Anthocercis viscosa) sequence was kindly sup- plied by Dr. R. PRICE (University of Georgia).
To test whether the two predominant classes of paralogues were preferentially amplified during PCR (Figure l ) , PCR w a s carried out with known quantities of cloned template. Cloned rDNA template (0.1 ng) was amplified under standard Nicoti- ana PCR conditions, and PCR was run both with and without DMSO.
We also examined Gossypium and Winteraceae ITS se- quences (SUH et al. 1993; WENDEI. et al. 1995a,b). These se- quences were PCR amplified without denaturants. The Gos- sypium PCR products were sequenced directly, while the Winteraceae PCR products were cloned prior to sequencing.
Phylogeny reconstruction: Sequences were aligned using Clustal (as implemented by DNASTAR Inc., Madison, WI) and refined by eye. We estimated the phylogeny for each genus using fastDNAml (version 1.0.8, O1,SEN et al. 1994), a maximum likelihood algorithm for DNA sequence data (FELSENSI'EIN 1981). We searched for the best tree using the global branch swapping option. Putative recombinants were excluded from the final phylogenetic reconstructions.
Secondary structure: Minimum-energy secondary struc- tures were estimated for the ITS 1 and 2 regions of all se- quences with the computer program mFold (ZUKER 1989). We estimated stability at both 37" (default temperature) and 72" (PCR extension temperature). The sum ITS1 AG and ITS2 A G (at 37") was used for stability comparisons. A Kmeans test of the estimated AG (ITS1 plus ITS2, WIIXINSON 1989) divided the alleles into two classes, class I (high secondary structure stability and low free energy) and class I1 (low sec- onday strllctllre stability and high free energy). To provide
a basis for comparison of secondary structure AG (LEICHT et al. 1995), we estimated the AG of random sequences with the base composition and length of the actual ITS sequences. For each ITS sequence the bases were randomly shuffled, and then the minimum AG was found for this random sequence using mFold. We then averaged these random AGs for each genus and compared this value to the observed A&.
Methylation-related substitutions: Deamination-like substi- tutions (C -+ T and G "* A) were examined at cytosine sites along both coding and noncoding strands. Possible sites of methylation were determined for each ingroup by compari- son with outgroup alleles (BUCKLER and HOI.TSFORD 1996a). A potential methylation site (CpG or CpNpG, GARDINER-GAR- DEN et al. 1992) that existed in 75% of the outgroup alleles was considered ancestral and substitutions characteristic of methyl-cytosine deaminations to thymine were tabulated. The following outgroups were used for each genus: Gossypium (outgroup: G. robinsoniz and G. sturtianum), Nicotiana sect. Alatae (ingroup: N. longjora, N. langsdo$i, N. alata, N. bona- riensis, N. forgetiana, and N. plumbaginfolia; outgroup: Anthoc- eris viscosa, N. attenuata, N. acuminata, N. stockonii and N. ms- t ica), Tripsacum class I alleles (outgroup: Zea alleles), Tripsacum class I1 alleles (outgroup: Coix and Bothriochloa), Winteraceae (outgroup: Tasmania, Drimys, Pseudowinterd), Zea (outgroup: Tripsacum class I alleles). The expected num- ber of deaminations at methylation sites was estimated from the frequency of deamination-like substitutions at nonmethyl- ation sites.
Substitution rates and patterns: We estimated relative sub- stitution rates between class I and I1 alleles of the same species with Kimura distances (KIMURA 1980) and determined sig- nificance with the chi-square rate test (1D method of TAJIMA 1993). The parsimony methods of MacClade 3.01 (MADDISON and MADDISON 1992) traced unambiguous character-state changes onto the phylogeny. Substitution frequencies were standardized for base content (LI et al. 1984). Gtests of inde- pendence were used to evaluate substitution frequency differ- ences between class I and I1 alleles (SOKAL and ROHLF 1981). The standardized substitution frequencies for the entire ITS region provided the expected substitution frequencies for the two conserved ITS regions (LIU and SCHARDL 1994) and the 5.8s region. A Gtest of heterogeneity compared the observed number of substitutions between the conserved and less con- served regions. A single outgroup taxon was used for these substitution analyses: Gossypium ( G . sturliunum), Nicotiana (Anthoceris viscosa), Tripsacum class I alleles [ Zea peranis clone #03 from BUCKLER and HOLTSFORU ( 1996b) 1, Tripsa- cum class I1 alleles (Coix #93 and Bothriochloa #74 were both used because no single taxon was a very close outgroup), Winteraceae (Pseudowintera axillaris 1 ) , and Zea ( Tripsacurn dactyloides #84). Because Oryza was an outgroup to both Trip- sacum's class I and I1 alleles, it was used in those relative rate tests; however, Oryza was not used to estimate character states changes since closer outgroups were available for that analysis.
Identification of putative recombinants: We used several phylogenetic tools to recognize recombination events be- tween highly diverged paralogues. First, we compared ITS1 us. ITS2 phylogenies to look for recombination events in the 5.8s. Second, recombinants are likely to cause many homopla- sies in a phylogeny (FUNK 1985), and putative recombinants should exhibit higher homoplasy relative to the observed number of character differences. Therefore to find a putative recombinant, we estimated homoplasy and observed charac- ter differences between every pair of taxa for the most parsi- monious tree(s) [using PAUP 3.1 of SWOFFORD (1993)l. The taxa, which had high homoplasy to observed character differ- ences ratios, were identified with Grubbs test for outliers (SO- KAL and ROHLF 1981). Then to find the most likely parental
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

rDNA Divergent Paralogues 823
sequences for a putative recombinant, we found the pair of sequences whose character states matched the maximum number of sites in the proposed recombinant. This pair of sequences will be referred to as “parental”; however, the sequences are probably the descendants of the actual parents.
To test the likelihood of the recombination hypothesis, a maximum likelihood phylogeny was estimated with two partial sequences replacing the proposed recombinant. To produce the partial sequences, the informative sites of the recombi- nant were classified as synapomorphies (recombinant shared a character state with one parent but not the outgroup) or plesiomorphies (recombinant shared a character state with one parent and the outgroup). If a synapomorphy or plesio- morphy was shared with parent 1, then that region was as- signed to partial sequence 1 and coded as missing for partial sequence 2. The region between contrasting informative sites was split at the midpoint. Likelihood ratio tests compared the likelihood of the recombinant hypothesis, two partial se- quences, us. the nonrecombinant hypothesis, one complete sequence (KISHINO and HASECAWA 1989). This likelihood does not account for the likelihood of correctly identifylng the parental sequences.
RESULTS
Paralogue characteristics: Based on ITS stability, there were two distinct classes of ribosomal alleles (Fig- ure 1). A b e a n s test of AG (ITS1 plus ITS2) divided the alleles into class I (high stability) and class I1 (low stability) ( P < 0.0001 for all groups, WILKINSON 1989). We found five low-stability sequences in Gossypium, eight in Nicotiana, two in Tripsacum, three in the Win- teraceae, and five in Zea. G. triphyllum had intermediate AG, but it was included in the class I1 alleles because of its phylogenetic position. In all groups, the class I alleles were substantially more stable than the confor- mations of randomized sequences (Figure 1). However with the exception of most Gossypium alleles and one Zea allele, the class I1 alleles were generally no more stable than the conformations of randomized se- quences.
Substitutions to A or T were more frequent in class I1 than in class I alleles (Table 1). Roughly 60% (50% if corrected for base content) of all class I1 substitutions were characteristic of deaminations (C -+ T and G +
A), except for the Winteraceae. For the class I1 alleles many of the deamination-type substitutions occurred at inferred ancestral methylation sites (Figure 1, Table 1). We tested the hypothesis that the increase in C + T and G + A transitions were part of a regional change in base composition, by determining whether there were more N -+ (AI T) compared to N + (C I G) substitutions for the class I1 relative to class I while excluding C + T and G + A transitions. In all groups, substitutions to A or T were more common than substitutions to C or G, but only in Zea and Tripsacum was the trend significant (Gtest of independence, 0.01 < P < 0.05).
For many groups, the class I1 alleles had higher rela- tive substitution rates in the ITS and 5.8s subunit than class I alleles (Table 1). However, both class I and I1
alleles exhibited fewer substitutions in highly conserved regions, especially in the 5.8s region (Table 1).
While many class I1 paralogues were identified, Nico- tiana also had a pair of stable divergent class I para- l o p e s (Figure 1). Two class I divergent paralogues were amplified in both N. alata and langsdo$i, and they will be referred to as locus 1 and 2.
Phylogenetic analysis indicated that the class I1 alleles were often basal to the class I alleles of the species from which they were cloned (Zea, Nicotiana, and Wintera- ceae in Figure 1 ) . The class I1 alleles diverged multiple times: once in Gossypium, three times in Nicotiana, once in Tripsacum, once in the Winteraceae, and four times in Zea (Figure 1). In the Winteraceae and Nicoti- ana, the phylogenies of the class I1 divergent paralogues partially mirror the class I phylogenies.
Differential amplification of paralogues: In Zea and Nicotiana, we amplified genomic DNA with and without DMSO to estimate the effect of the amplification method. The average AG stability at 37” significantly differed between clones amplified with and without the denaturant DMSO (Zea PCR, c’dGTP us. DMSO: F = 15.51, d.f. = 1, 64, P = 0.0002; Nicotiana PCR, normal vs. DMSO: F = 31.56, d.f. = 1,24, P < 0.0001). In those two groups, all of the class I1 alleles were amplified when DMSO was not included in the PCR (Table 1). Zea ITS conformations were so stable that they could not be amplified without denaturants. In Zea, alleles with both moderately and highly stable secondary struc- tural conformations were amplified with c’dGTP incor- poration, while only alleles with highly stable conforma- tions were amplified with DMSO. In all five groups, the more stable ITS conformations (AG < -33 kcal/mol at 72”) amplified only with denaturants (c’dGTP or DMSO).
PCR amplification experiments comparing class I us. class I1 templates confirmed the bias for various tem- plates (Figure 2). Only low stability templates (class 11) amplified well with normal PCR conditions, while both class I and I1 amplified well in the presence of DMSO. Competition experiments between equal amounts of class I and I1 template in a single DMSO PCR reaction had inconsistent results (data not shown).
rDNA class I1 alleles appear to be ubiquitous yet un- common in these plants. In Zea, digestion with a diag- nostic restriction enzyme indicated no presence of class I1 alleles in DMSO-amplified PCR products (BU<:KI.ER and HOLTSFORD 199613).
Recombination: Phylogenetic analysis of the ITS1 us. the ITS2 trees suggested four sequences were discor- dant: T. dactyloides, Bubbia comptonii, N. otophora, and N. data 3. Five sequences were outliers for high homoplasy relative to observed character differences (Grubbs P < 0.05): T. dactyloides, B. comptonii, N. otophorn, G. dauid- sonii, and 2. mays ssp. huehuetenangensis #65. The recom- binant likelihood test gave strong support for the Tripsa- cum dactyloides and Bubbia recombinants (Table 2 and
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

824 E. S. Buckler, A. Ippolito and T. P. Holtsford
Gossypium 3 G. & G. davidsonii, kloeschianum, shcrtianum ' \ raimondii, thurberi & trilobum D -
F 2- A 4 G. G. gossypioides longicalyp\' D t \ \ ;~ \ G. barbadense, danvinii, A -
0- - 0 0 G. herbaceum A t 8
-0
lobatum & harknessii & turnen D G. ardium, laxum, G. amourianum,
G. mustelinum AD t schwe~emanii D -1-
& tomentosum AD U 1-0 G. triphyllum B t w g - 0 . 0 0 A i
A i G. arboreum A t 0
- - =1% divergence -2 I I I I I
A A
N. bonariensis t Nicotiana
N. bonariensis, foreetiana & alata 1
-1 0 1 2 3 z-score (AG)
I I I I I I I -2 -1 0 1 2 3
=5% divergence . , " z-score (AG)
Tripsacum
n 2- b A -
% W l - 2 - 3 0- ? - 0 . N eo 0
O . 0 -1 I I I I I -1 0 1
" -5% divergence z-score (AG) FIGURE 1 .-Maximum likelihood trees for five taxonomic groups' ITS sequences. Recombinants were excluded. Class I alleles
are unmarked, class I1 alleles are indicated by t, and outgroups are in bold. The graphs illustrate the z-score of the free energy (AG) of the ITS1 plus the AG of the ITS2 [z-score (AG)] us. the z-score of the number of C + T substitutions at standard methylation sites (CpG and CpNpG) minus nonmethylation site C "+ T [z-score (mC > T)]. The dashed line is the mean AG of the randomized sequences. Class I alleles are plotted with circles, class I1 alleles with triangles, e.g., sequences plotted taxa in the lower left corner have low free energy (high secondary structure stability) and few C -+ T at methylation sites. Gossypium's genome is designated by the letter following its species epithet (WENDEL et al. 199513). Nicotiana has divergent paralogues within the class I alleles, and these are designated by the number following its species epithet.
Figure 3), which both appear to be simple recombina- tions between class I and I1 alleles. G. dauidsonii and N. otophora had weaker evidence for recombination (Table 2 and Figure 3). The N. otophora sequence was class I- like with a stable ITS region and expected levels of methylation-type substitutions. The N. otophora se- quence might be the result of multiple recombination or gene conversion events between Nicotiana locus 1
and 2. In Zea, a high level of homoplasy was found throughout the phylogeny, which probably resulted from the many intraspecific comparisons and the segre- gating polymorphism among the closely related taxa. There was no apparent pair of parental sequences for 2. mays ssp. huehuetenangensis #65, which exhibited high homoplasy. .
The Nicotiana alata 3 sequence has very little phyloge-
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

rDNA Divergent Paralogues 825
Tasmannia, Drimys, Winteraceae and Pseudowintera \ \
\ \ Bubbia
I ir: A
Exospermum t V E v
Exospermum Z balansae
& bicolor t acsmithii Belliolum & z Z bicolor - =1% divergence
2 -
1 - A & -
0- 0 0 -
a0 0 -1 -1 0 1 2
I I I I I
z-score (AG) Zea
5 - A A i Ai s t b -
n 4-
V I A 3-
W E 2- A
Ai
z mays - =1% divergence I I I I I I I I I l l I
- 2 - 1 0 1 2 3 4 Z-SCOE (AG)
FIGURE 1. - Continued
netic support for recombination (three synapomor- phies), but the distribution of substitutions favor a re- combinant hypothesis (Figure 3 and Table 2). N. data 3 and N. data 1 of locus 1 only differed by two deami- nated methylation sites in the first 180 bases but by 24 bases in the next 281 ITS sites (a heterogeneity test compared substitution frequencies for first 180 sites vs. next 281 sites: all substitutions: G = 14.06, d.f. = 1, P = 0.0002; nonmethylation-related: G = 11.06, d.f. = 1, P = 0.0009; methylation-related: G = 4.99, d.f. = 1, P = 0.026). Relative rate tests between the putative recom- binant N. aluta 3 and other locus 1 Nicotiana suggested no significant rate differences for the first 180 bases, but the rest of the ITS evolved 2.9 times faster ( P < 0.05 in 50% of tests). Relative to all class I Nicotiana the putative recombinant N. alata 3 had lower ITS2 secondary-structure stability and more C + Ts at methyl- ation sites. Together this suggests N. alata 3 is a recombi- nant between a class I locus 1 Nicotiana and a sequence similar to the class I1 alleles from N. bonariensis.
WENDEL et al. (1995b) suggested G. gossy$ioides was a D genome species with rDNA of recombinant A and D origin. But G. gossypioides rDNA had a low homoplasy to character difference ratio and shared one homoplasy with the AD genome species. The D genome origin of the rDNA is supported by zero synapomorphies and 12 plesiomorphies, while the class I1 clade (A-genome like)
origin is supported by 10 synapomorphies and five plesi- omorphies. Since only the class I1 clade has synapomor- phic support, this analysis suggested that G. gossypioides’s rDNA is entirely of class I1 clade origin.
The simple recombinants could have been produced by ‘Ijumping” of the PCR reaction (P-o et al. 1990). In PCR jumping, Taq polymerase prematurely termi- nates extension, and frequently adds an adenosine to the product. In the next round, this premature product can act as a primer and recombine with other se- quences. The PCR recombinants should result in auta- pomorphic As or Ts at the point of recombination. In the three simple recombinants these substitutions were not found. Considering the frequency of A being added by Tag when strand jumping (88%, P-0 et al. 1990) and the frequency of A or T in the sequences (roughly 22% per strand), the probability of missing the A inser- tion from a Taq recombination is roughly 32% for each sequence. The probability that all three sequences are the result of cryptic Tag recombination is roughly 3%.
DISCUSSION
Divergent paralogues were common in four of these five groups. These divergent paralogues pose a serious problem for reconstructing a species phylogeny from a small sampling of rDNA. Different PCR conditions can
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021
826 E. S. Buckler, A. Ippolito and T. P. Holtsford
TABLE 1
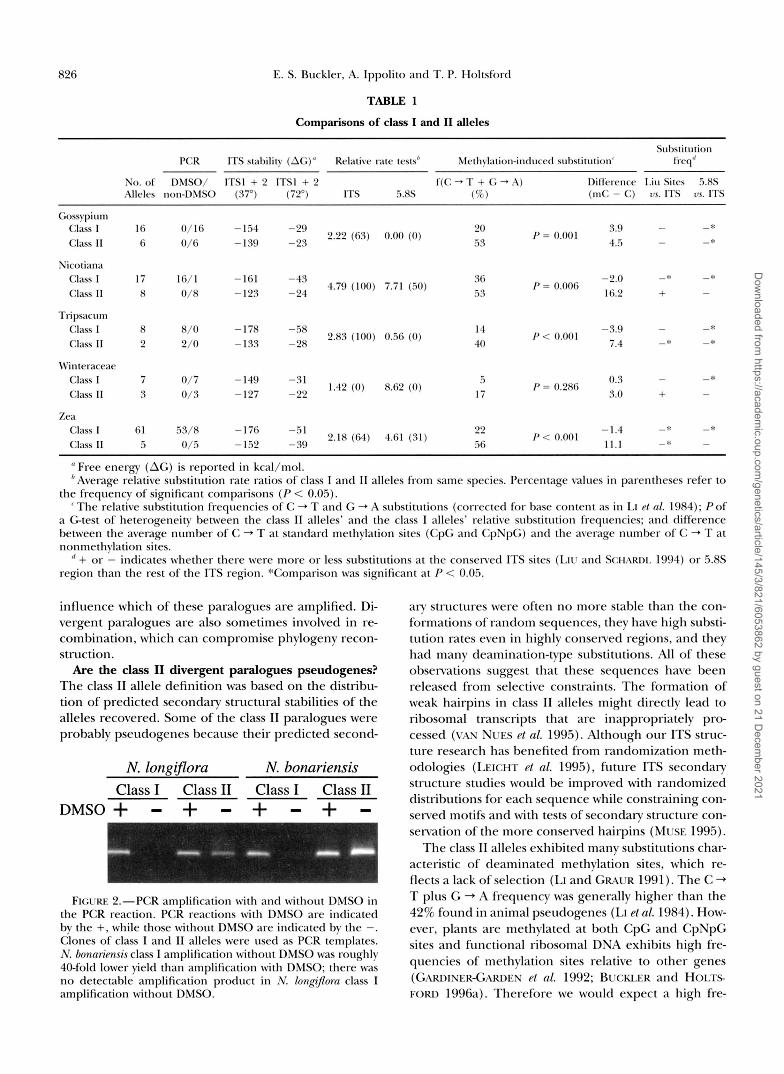
Comparisons of class I and I1 alleles
Suhstitution PCR ITS stability (AG)" Relative late tests" Mcth~lation-induccd substitution' frcq"
No. of DMSO/ ITS1 + 2 ITS1 + 2 f(C -, T + G -,A) I~ilrerence I.iu Sites 5.8s Alleles non-DMSO ( 3 7 ) (72') ITS 5.8s (5%) (mC - C) 7~ ITS us. ITS
Clak I 16 0/16 -1.54 -29 Class II 6 0/6 -139 -23
2.22 (63) 0.00 (0)
Nicotiana Class 1 17 16/1 -161 -43 36 -2.0 -* Class 11 8 0/8 -123 -24 .5 3 16.2 + - 4.79 (100) 7.71 (.io)
-* P = 0.006
Tripsacum Class I 8 8/0 -178 -58 Class I1 2 2/0 -133 -28
2.83 (100) 056 (0)
M'interaceae Class I 7 0/7 -149 -31 Class 11 3 0/3 -127 -22
1.42 (0) 8.62 (0)
Zea Class I 61 .53/8 -176 -51 -1.4 -* -* 22 Class 11 5 0/.5 -1.52 -39 56 1 1 . 1 -* -
2.18 (64) 4.61 (31) P < 0.001
" Free energy (AG) is reported in kcal/mol. 'Average relative substitution rate ratios of class I and 11 alleles from same species. Percentage values in parentheses refer to
the frequency of significant comparisons ( P < 0.05). 'The relative substitution frequencies of C + T and G + A substitutions (corrected for base content as in LI el nl. 1984); Pof
a Gtest of heterogeneity between the class I1 alleles' and the class I alleles' relative substitution frequencies; and difference between the average number of C + T at standard methylation sites (CpG and CpNpG) and the average number of' C + T at nonmethylation sites.
" + or - indicates whether there were more or less substitutions at the conserved ITS sites (LIV and SCHARDI. 1994) or 5.8s region than the rest of the ITS region. "Comparison was significant at P < 0.05.
influence which of these paralogues are amplified. Di- vergent paralogues are also sometimes involved in re- combination, which can compromise phylogeny recon- struction.
Are the class I1 divergent paralogues pseudogenes? The class I1 allele definition was based on the distribu- tion of predicted secondary structural stabilities of the alleles recovered. Some of the class I1 paralogues were probably pseudogenes because their predicted second-
N. longiflora N. bonariensis Class I Class I1 Class I Class I1
DMSO+ - + - + - + -
I FIGLIRE 2.-PCR amplification with and without DMSO in
the PCR reaction. PCR reactions wi th DMSO are indicated by the +,while those without DMSO are indicated by the -. Clones of class I and I1 alleles were used as PCR templates. N. honnrimsi.~ class I amplification without DMSO was roughly 40-fold lower yield than amplification with DMSO; there was no detectable amplification product in N. lonpiflorn class I amplification without DMSO.
ary structures were often no more stable than the con- formations of random sequences, they have high substi- tution rates even in highly conserved regions, and they had many deamination-type substitutions. All of these observations suggest that these sequences have been released from selective constraints. The formation of weak hairpins in class I1 alleles might directly lead to ribosomal transcripts that are inappropriately pro- cessed (VAN NUES et al. 1995). Although our ITS struc- ture research has benefited from randomization meth- odologies (LEICHT et al. 1995), ftlture ITS secondary structure studies would be improved with randomized distributions for each sequence while constraining con- served motifs and with tests of secondary structure con- servation of the more conserved hairpins (MUSE 1995).
The class I1 alleles exhibited many substitutions char- acteristic of deaminated methylation sites, which re- flects a lack of selection (LI and GRAUR 1991). The C +
T plus G + A frequency was generally higher than the 42% found in animal pseudogenes (LI et al. 1984). How- ever, plant5 are methylated at both CpG and CpNpG sites and functional ribosomal DNA exhibits high fre- quencies of methylation sites relative to other genes (GARDINER-GARDEN d nl. 1992; BUCKLER and HOLTS FORD 1996a). Therefore we would expect a high fre-
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

rDNA Divergent Paralogues 827
TABLE 2
Phylogenetic tests of recombination hypotheses
Change in Null hypothesis" Alternate hypotheses" tree length A LnL' SDc Pd
Bubbial not recombinant Bubbial divided in parts 1 and 2 -5 -47.81 14.10 0.0007 G. davidsonii not recombinant G. davidsonii divided in parts 1 and 2 - 3e - 16.20 8.83 0.0666' N. data not recombinant N. alatu divided in parts 1 and 2 -1 -6.87 5.97 0.2497 N. otophora not recombinant N. otophora divided in parts 1 and 2 -6 -21.45 12.49 0.0859 T. dactyloides not recombinant T. dactyloides divided in parts 1 and 2 -15 -116.01 19.77 0.0000
* pl and p2 are the parts of the recombinant depicted in Figure 2. ' A LnL is the difference in natural log likelihood scores between the null and alternate hypotheses. 'The standard deviation of the A LnL. 'The probability of rejecting the null hypothesis. This probability is conditional on the correct identification of the parental
G. klotzschainum, G. dauidsonii's sister taxon, was excluded from these analyses as some of the homoplasies are shared between sequences.
G. klotzchainum and G. davidsonii, if included these results would be less significant.
quency of C + T and G + A substitutions if the class I1 alleles were released from selection for methylation- induced chromatin condensation (BUCKLER and HOLTSFORD 1996a). But other substitutions to A o r T were also more frequent in class I1 alleles so that changes in regional base composition may also play a role in the divergence of class I1 alleles. The orphoning of rDNA might be the process through which the changes in base composition could occur.
The class 11 5.8s regions generally evolved faster than class I 5.8s regions. However, the conserved regions in the ITS and the 5.8s of class I1 alleles had fewer substitutions than surrounding ITS regions. Two theo- ries may explain why the class I1 5.8s is not evolving at the ITS rate. First, when two functional rDNA arrays diverge the ITS regions will diverge faster than the 5.8s (BALDWIN et al. 1995). Subsequently, if one array be- comes transcriptionally inactive its 5.8s will begin to diverge rapidly. For example, the T. dactyloides class I1 alleles have a highly conserved 5.8s; these class I1 alleles
might derive from a recently inactivated array, as this species is polymorphic for nucleoli number (RANDOLPH 1955). Second, the base composition substitution model for the ITS us. 5.8s comparisons might be too simple. Although the density of methylation sites was approximately the same for the ITS and 5.8s regions (BUCKLER and HOLTSFORD 1996a), the differences in base composition between the ITS and 5.8s could sub- stantially modify substitution patterns (MORTON 1995).
Overall, the strength of support for the class I1 alleles being pseudogenes varied with the groups as follows (most pseudogene-like to least, see Table 3): Nicotiana, Zea, Winteraceae, Tripsacum, and Gossypium. Pseu- dogene evidence is strongest for those alleles that have long, independent divergences such as those of Nicoti- ana. In contrast, the class 11 Gossypium alleles exhibited little evidence for being pseudogenes (Tables 1 and 3). Further, restriction analysis of genomic DNA and direct PCR analyses suggest the class I1 alleles are representa- tive of the rDNA present in the genome (WENDEL et al.
Bubbia
G. davidsonii
N. otophora . n I I * I 1. n I I
N. alata * * * t
T. dactyloides Conserved
5.8s Conserved ITS1 Site ITS2 Site
FIGURE 3.-Recombination evidence for five ITS sequences. The filled circles were synapomorphies with parental sequence 1, while the open circles were synapomorphies with parental sequence 2. Thin lines indicate the hypothesized origin for the ITS regions based on both synapomorphic and pleisiomorphic characters; regions within outlined boxes have ambiguous origin. The solid boxes indicate the conserved ITS regions. Hypothesized parental sequences: Bubbia (1, Winteraceae class 11; 2, class I Bubbia) ; G. davidsonii (1, G. raimondii, thuberi and trilobum; 2, G. arboreum) ; N. otophmu (1, locus 1 Nicotiana; 2, locus 2 Nicotiana); N. alutu (1, locus 1 N. aluta; 2, Nicotiana basal to locus 1 N. alata) ; and T. dactyloides (1, Tripsacum class I1 alleles; 2, Tripsacum class I alleles). The recombinant N. d a t a had a spatially skewed distribution of substitutions. The stars indicate differences between the recombinant and one putative parent, the N. alutu locus 1 allele.
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

828 E. S. Buckler, A. Ippolito and T. P. Holtsford
TABLE 3
Relative support for the class I1 alleles being pseudogenes
Analyses" Nicotiana Zea Winteraceae Tripsacum Gossypium
Secondary structure Phylogenetic C + T substitutions Methylation site C -+ T Relative rates Conserved regions
The analyses used for this summary table come from Table 1 and Figure 1. + indicates that the analysis conforms to a pseudogene hypothesis for the specified genera, - indicates the analysis is contrary
to the pseudogene hypothesis; no symbol indicates the analysis is equivocal. The pseudogene hypothesis support criteria: secondary structure, class I1 alleles' AG little different from AG of random sequences; phylogenetic, divergent basal paralogues; C + T substitutions 2 40% of all substitutions; methylation sites, strong class I1 bias for C + T at methylation sites; relative rates, class I1 higher for both ITS and 5.8s; conserved regions, neither class I1 conserved regions had significantly low substitution frequencies.
1995a). In cases such as this where the pseudogene evidence is weak, the substitution characteristics might result from changes in regional base composition. The Gossypium case demonstrates that individual class I1 characteristics are insufficient for pseudogene identifi- cation, rather pseudogenes should exhibit a suite of characteristics. Future high sensitivity rDNA expression studies would be needed to demonstrate that putative pseudogenes are definitively not transcribed or are not functional.
How do class I1 alleles compare with ribosomal diver- gent paralogues in animals? A ribosomal 28s orphoned pseudogene has been recovered from humans (MUNRO et al. 1986). The orphon only has a partial 28s region, and restriction mapping suggests no neighboring rDNA sequences. We examined the substitution differences between the normal human rDNA and the orphon. Most of the 37 substitutions were C + T and G + A (59%), and most of those deamination type substitu- tions occurred at putative mammalian methylation sites (91 %). The substitution patterns of this human orphon and pseudogene is very similar to that found in the class I1 alleles.
Divergent ribosomal paralogues have been reported among the small number of rDNA copies in inverte- brates (GUNDEMON et al. 1987; CARRANZA et al. 1996). In both cases, the clustering of substitutions in less con- served regions of the 18s suggested both sets of para- logues were functional. In Plasmodium, two paralogues have been shown to be expressed at distinct develop- mental stages, while in Dugesia only one of the para- logues has been found to be expressed. We found that the unexpressed paralogue's predicted free energy was 7.3% higher (less stable) than the expressed paralogue, and unexpressed paralogue evolved 3.6-fold faster than the expressed paralogue (Crenobia outgroup, Tajima rate test P < 0.001). There was no suggestion of deami- nation-type substitutions, but most invertebrates have no rDNA methylation (BIRD and TAGGART 1980). We
speculate that the unexpressed Dugesia paralogue might be a pseudogene from a recently inactivated array (like the Tripsacum class I1 alleles).
Are the class I1 divergent paralogues orphons? Since divergent paralogues were common in these groups, concerted rDNA evolution may have a limited genomic scope and rate. Chromosomal location may explain some of the limitations in concerted evolution. Al- though the chromosomal location of these ribosomal paralogues is currently unknown, class I1 alleles proba- bly reside in one of three chromosomal positions: First, since interchromosomal recombination of nonhomolo- gous loci occurs but is probably infrequent (ARNHEIM
1983; DUBCOVSKY and DVORAK 1995), rDNA paralogues could diverge from major functional loci at minor loci (loci with few rDNA copies). Second, solitary para- logues can be dispersed throughout the genome as or- phons (CHILDS et al. 1981). Third, the terminal regions of tandem arrays will maintain anomalous paralogues since homologous regions for crossovers will b- e rare (SMITH 1976; LINARES et al. 1994). If the rDNA tran- scripts of the minor loci, orphons, or terminal regions were selectively neutral, these paralogues would be- come pseudogenes primarily through deamination- driven mutation pressure.
Multiple rDNA loci probably exist in most of the exam- ined groups. The Nicotiana spp. with class I1 alleles have nine or 10 chromosomes, two satellite constrictions, and two nucleoli per haploid genome (GOODSPEED 1954). These two loci are putatively represented by the two di- verged type I alleles for both N. alata and l a n g s w i (Fig- ure 1). Interestingly, N. plumbagin~ofolzu, the only Nicotiana taxon known to have only one nucleolus (GOODSPEED 1954), has class I1 alleles which are more recently di- verged than other Nicotiana class I1 alleles. Perhaps the N. plumbaginifolza class I1 alleles are from a recently inacti- vated locus. In Tripsacum, chromosomal number and morphological characters suggest the genus is allote- traploid (base chromosome n = 18, GALINAT et al. 1964).
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

rDNA Divergent Paralopes 829
Nucleoli are generally found on one of two different chromosomal loci in Tripsacum (MANGELSDORF 1974). However, some populations of T. dactyloides have nucleo- lar organizers at both of these loci, and the class I1 alleles came from this species (RANDOLPH 1955). In Gossypium, in situ hybridizations indicate two rDNA loci in the d i p loid species and four rDNA loci in the AD tetraploids (CRANE et al. 1993). Most Winteraceae, including those with the class I1 alleles, are likely ancient polyploids ( n = 43, inferred ancestral state n = 7 , SUH et al. 1993).
According to Zea chromosomal hybridizations, al- most all rDNA exists at one locus (PHILLIPS et al. 1979). This result suggests that class I1 alleles are orphons or occur in the terminal regions of the main rDNA array. Recent mapping experiments have found a putative ri- bosomal orphon on another chromosome (G. JOHAI, and J. GRAY, personal communication).
Tripsacum class I1 alleles provide a possible example of a functional locus only recently becoming nonfunc- tional, as the Tripsacum class I1 alleles were monophy- letic with the prevalent alleles from other groups (Figure 1). The small divergence of the conserved motifs of the ITS and the 5.8s support this argument (Table 1).
Some of the substitution patterns might be explained if the class I1 alleles were orphons. The tendency of class I1 alleles toward a greater A and T composition suggest these alleles were in different chromosomal en- vironments as might be the case for an orphon. The high rate of deamination-type substitutions suggests methylation induced chromatin condensation is less im- portant in class I1 alleles, as it would be for orphons. Further mapping research will be needed to determine where these paralogues reside.
PCR amplification of rDNA Small differences in the probability of amplification during each PCR cycle may result in major differences in the frequency of amplified product after many PCR cycles. This PCR selection has been observed in other gene families (WAGNER et al. 1994). Class I1 alleles have numerous substitutions that result in less stable hairpins, which lead to class I1 alleles being more successfully amplified under standard non- denaturing conditions than hairpin-ridden class I al- leles (Figure 2 ) . In contrast, we suspect that under dena- turing conditions the most numerous alleles were predominantly amplified, which were the class I alleles or the putative functional rDNA. For example under normal PCR extension temperatures (72"), the pre- dicted secondary structures of class I Nicotiana alleles were 80% more stable than those of the class I1 alleles. Without DMSO, 88% of the Nicotiana alleles amplified were class I1 alleles, while no class I1 alleles were ampli- fied under denaturing conditions. However, PCR selec- tion was probably weak in other groups such as Gossyp- ium, since hybrids between G. nrboreum and G. thurbm. exhibited the polymorphisms of both parents (WENDEL et al. 1995a). PCR selection might have only been im-
portant in the high G+C content sequences of Zea, Tripsacum, and Nicotiana.
BALDWIN and DONOCHUE [unpublished data cited in BALDWIN et al. (1995)] directly sequenced some of the Winteraceae samples but found no evidence for the Winteraceae locus 1, which we argue are pseudogenes. However, BALDWIN and DONOCHUE used different PCR conditions and primers than those used by SUH et al. (1993), again illustrating the importance of PCR condi- tions when sampling the diversity of rDNA.
PCR selection can also be a serious problem with low levels of contamination. Leaf tissue may contain both algae and fungi, which could be preferentially PCR am- plified. This appears to have happened in a Mimulus (Scrophulariaceae) study (RITLAND et al. 1993), where the amplified sequences were more similar to the Chlo- rophyta (green algae) than to higher plants (M. HERSH- KOVITZ, unpublished results). While attempting PCR amplification of Zea ITS sequences without DMSO or c'dGTP, we also once amplified a chlorophyte contami- nant. These algae could be a pernicious problem, be- cause they have low-stability ITS conformations and therefore may provide better PCR templates. We pre- dict that the real Mimulus ITS sequences have high- stability secondary structures.
The variety of rDNA within an organism is masked when rDNA is sampled with standard amplification and direct sequencing methods. Direct sequencing of rDNA PCR product is rapid and provides a useful survey of many paralogues. However, systematic differences in amplification probabilities between types of paralogues can result in biased sampling and an oversimplified view of the gene tree. PCR primers specific to various loci could be developed to limit investigations to single rDNA arrays.
Recombination among rDNA paralogues: Four groups evinced varying support for recombination between di- vergent paralogues. Recombination between class 1 and I1 alleles happened in Tripsacum and N'interaceae and perhaps in Nicotiana (N. alata 3, Figure 3 ) . Recombina- tion involved both simple cross-overs ( T . ductyloides, B u k bia, and N. data 3) and a possibly more complex gene conversion and/or multiple crossover events in N. oto- phora. Recombination was probably prevalent among the closely related Zea alleles, and the use of more sophisti- cated models of recombination could elucidate the levels of recombination. The simple crossover events could have resulted either from organismal or PCR-based re- combination. However, for many phylogenetic purposes the source of recombination is inconsequential, rather the elimination of recombinants before the phylogenetic reconstruction is most important.
In plant systematic studies, ribosomal recombinants must be recognized, since multiple paralogous ribo- somal loci and interspecific hybrids are common (FUNK 1985; DUBCOVSKY and DVORAK 1995). Recombinants of distantly related ITS paralogues must be excluded from
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

830 E. S. Buckler, A. Ippolito and T. P. Holtsford
phylogenies, because their inclusion can result in major topological changes (MCDADE 1992). When the recom- binants N. otophora and T. dactyloihs were included, the phylogenetic topologies of the taxa were significantly altered.
Recombinant identification should rely on apomor- phies for identifying reticulation, as plesiomorphic character states could result from the retention of the ancestral state not recombination (FUNK 1985). How- ever, two ITS studies have inferred recombinants with the use of both apomorphic and plesiomorphic charac- ters (VAN HOUTEN et al. 1993; WENDEI. et al. 199513). The evidence that G. gossypioides's ITS is a recombinant rests almost entirely on plesiomorphic characters, as does the ITS evidence for introgression of annual Mi- croseris species into the allotetraploid M. scapigma. Most evidence suggests G. gossypioides is from the D genome clade except for its rDNA ITS, which is a member of the class I1 clade (A genome clade of WENDEL et 01. 1995b). We believe that while G . gossypioides may be a hybrid taxon, its ITS sequence is not a recombinant, and we suggest that the class I1 clade sequence may have entirely replaced G. gossypioide.<s original D ge- nome rDNA. Perhaps this is an example of biased gene conversion (HILLIS et ul. 1991).
The homoplasy ratio test, likelihood ratio tests of the recombinant hypothesis, and separate ITS1 and ITS2 phylogenies were useful but rudimentary methods for identifying reticulation. While our methods did not sur- vey and test the entire range of possible recombination events, more complete approaches are still in need of development. The RETICIAD work of RIESEBERG and MOREFELL) (1995) is a more methodical approach to identifying recombinants through parsimony; however the present algorithm only works with binary characters and has an antiquated implementation. Forest phyloge- netics (HYPERPARS 0.2 of A. DICKERMAN, personal communication) is theoretically promising, but the cur- rent implementation still needs improvement for mod- erate size datasets such as these. Hopefully future re- search in this area will allow us to more rigorously identify recombinants and uncover the true phyloge- nies.
Phylogenetic implications: At the outset of phylog- eny reconstruction, rDNA paralogues and recombi- nants must be identified. Divergent paralogues, includ- ing pseudogenes and recombinants, are probably ubiquitous, as polyploidization is common in angio- sperms (30-80% of species, FUNK 1985). This can cre- ate both obstacles and opportunities for phylogenetic reconstruction of plant lineages.
Paralogues provide more information about the evolu- tionaly history of rDNA sequences because the addi- tional branches help reconstruct the gene trees. Once divergent paralogues are identified, they can also im- prove species phylogenies. For systematic studies, para- logues can serve as better outgroups than sister species
(e.& BUCKLER and HOLTSFORD 1996b), especially for groups without closely related extant taxa. Additionally, paralogous loci can provide additional gene trees for the resolution of species trees (e.g., Nicotiana in Figure 1, Winteraceae in SUH et al. 1993). However, because pseu- dogenes have long branches and high homoplasy at methylation sites, the phylogeny must be reconstructed with an appropriate method. The parsimony method has significant problems with the unequal rates of substitu- tion, while the current maximum likelihood algorithms deal well with rate differences and are robust to substitu- tion model violations (KUHNER and FELSENSTEIN 1994; HUELSENBECK 1995). Substitutions at pseudogene meth- ylation sites should also be down-weighted as they are more mutable than other sites. In the future, different base change matrices could be used for various types of paralogues. However, the first step should be to discrimi- nate between paralogous rDNA, as paralogues may cause species to appear para- or polyphyletic if a gene tree is interpreted as a species tree.
Although divergent paralogues can be advantageous, divergent paralogues can cause two types of phyloge- netic complications: First, there can be many minor loci, and these minor loci may convert to major loci occasionally, while the old major loci may disappear (Duucovsw and DVORAK 1995). In this case we would expect many class 11-type substitutions along a single branch, if the minor locus were unexpressed for a pe- riod of time (this is not the case of Gossypium). If different minor loci gave rise to the current major loci in two species, the divergence indicated by a gene tree would be older than the organismal divergence. Sec- ond, simulations indicate that phylogenies with inter- mediate levels of recombination or gene conversion between paralogues rarely generate the correct species tree (SANDERSON and DOYLE 1992). Especially among diverged paralogues (as in the case of allopolyploids), the rate of interlocus recombination or gene conver- sion could be low to intermediate. If not recognized, recombination of divergent paralogues could result in serious problems for recovering the correct phyloge- netic tree. However, recognition of divergent para- logues and pseudogenes will provide phylogeneticists more outgroup opportunities, better phylogenetic re- constructions, and the option to exploit multiple rDNA phylogenies.
We thank C. KEITH BUCKLER for discussing these topics almost daily for the last year. We also thank J. WENDEI., E. ZIMMPR,J. SCHWPZRI'L,
M . KEl.I .ER, K. MCCUE, A. WlPD, D. BPKGSTROM, T. W.l.l.O<X;, and two anonymous 1-eviews for reading previous drafts of the manuscript and critical discussion. This work was supported by a National Science Foundation Predoctoral Fellowship and a University of Missouri Maize Training Program Fellowship (a unit of. the DOE/NSF/USDA Collaborative Research in Plant Biology Program) to E.S.B. and Uni- versity o f Missouri Research Board award 93-060.
LITERATURE CITED ARNHFTM, N., 1983 Concerted evolution of multigene families, pp.
38-61 in Etdution of C ~ P C and Protein, edited by M . NEI and R. K. KOEI~N. Sinauer Associates, Sunderland, MA.
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

rDNA Divergent Paralogues 831
BALDWIN, B. G., M. J. SANDERSON, J. M. PORTER, M. F. WOJCIECHOW- SKI, C. S. CAMPBELL et al., 1995 The ITS region of nuclear ribo- somal DNA a valuable source of evidence on angiosperm phylog- eny. Ann. Missouri Bot. Garden 82: 247-277.
BIRD, A. P., and M. H. TAWART, 1980 Variable patterns of total DNA and rDNA methylation in animals. Nucleic Acids Res. 8: 1485-1497.
BUCKLER, E. S., IV, and T. P. HOLTSFORD, 1996a Zea ribosomal re- peat evolution and substitution patterns. Mol. Biol. Evol. 13:
BUCKLER, E. S., IV, and T. P. HOI~TSFORD, 1996b Zea systematics: ribosomal ITS evidence. Mol. Biol. Evol. 13: 612-622.
CARRANZZ, S., G. GIRIBET, C. RIBERA, J. BAGUNA and M. RIUTORT, 1996 Evidence that two types of 18s rDNA coexist in the genome of hges ia (Schmidtea) mditerranea (Platyhelminthes, Turbellaria, Tricladida). Mol. Biol. Evol. 13: 824-832.
CHIIDS, G., R. MAXSON, R. H. COHN and L. KEDES, 1981 Orphons: dispersed genetic elements derived from tandem repetitive genes of eucaryotes. Cell 23: 651-663.
CRANE, C. F., H. J. PRICE, D. M. STEILY, D. G. CLESCHIN, JR. and T. D. MCKNIGHT, 1993 Identification of a homeologous chromo- some pair by in situ DNA hybridization to ribosomal RNA loci in meiotic chromosomes of cotton (Cossypium hirsutum). Ge- nome 36: 1015-1022.
DUBCOVSKY, J., and J. DVORAK, 1995 Ribosomal RNA multigene loci: nomads of the Triticeae genomes. Genetics 140: 1367-1377.
DVORAK, J., 1990 Evolution of multigene families: the ribosomal RNA loci of wheat and related species, pp. 83-97 in Plant Popula- tion Genetics, Breeding, and Cmetic Resources, edited by A. H. D. BROWN and M. T. CLEGG. Sinauer Assoc., Sunderland, MA.
FELSENSTEIN, J., 1981 Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17: 368-376.
FL:NK, V. A,, 1985 Phylogenetic patterns and hybridization. Ann. Missouri Bot. Garden 72: 681-715.
G A I m A - r , W. C., R. S. K. CHACANTI and F. D. HACXR, 1964 Tripsa- cum as a possible amphidiploid of wild maize and Manisuris. Botanical Museum Leaflets, Harvard University 20: 289-316.
~ARI)lNER-GARDEN, M., J. A. SVEI) and M. FROMMER, 1992 Methyla- tion sites in angiosperm genes. J. Mol. Evol. 34: 219-230.
GOOIXPEED, T. H., 1954 Chromosome number and morphology, pp. 139-166 in The &us Nicotiana. Chronica Botanica Com- pany, Waltham, MA.
GUNDERSON, J. H., M. L. SOGIN, G. WOLLETT, M. HOI.I.INCDAI.E, V. F. 1)E 1-4 CRUZ et al., 1987 Structurally distinct stage-specific ribo- somes occur in Plasmodium. Science 238: 933-937.
HIILIS, D. M., and M. T. DIXON, 1991 Ribosomal DNA molecular evolution and phylogenetic inference. Q. Rev. Biol. 66: 411-453.
HIILIS, D. M., C . MORITZ, C. A. PORTER and R. J. BAW,R, 1991 Evi- dence for biased gene conversion in concerted evolution of ribo- somal DNA. Science 251: 308-310.
HUEISENBECK, J. P., 1995 Performance of phylogenetic methods in simulation. Syst. Biol. 44: 17-48.
INNIS, M. A,, 1990 PCR with 7-deaza-2’deoxyguanosine triphos- phate, pp. 54-59 in PCR Protorols: A G u i d e to Methods and Applica- tions, edited by M. A. INNIS, D. H. GELFAND, J. J. SNlNswand T. J. WHITE. Academic Press, Inc., San Diego.
KEI.I.o<:c;, E. A,, and R. APPEIS, 1995 Intraspecific and interspecific variation in 5s RNA genes are decoupled in diploid wheat rela- tives. Genetics 140: 325-343.
KIMURA, M., 1980 A simple model for estimating evolutionary rates of base substitutions through comparative studies of nucleotide
WSHINO, H., and M. HASE(:AWA, 1989 Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J. Mol. Evol. 29: 170-179.
KUHNER, M. IC, and J. FELSENSTEIN, 1994 A simulation comparison of phylogeny algorithms under equal and unequal evolutionary rates. Mol. Biol. Evol. 11: 459-468.
LEI(:HT, B. G., S. V. MUSE, M. HANCZYC and A. G. CIARK, 1995 Con- straints on intron evolution in the gene encoding the myosin alkali light chain in Drosophila. Genetics 139: 299-308.
LI, W.-H., and D. GRAUR, 1991 Fundamentals of Molecular Euolution. Sinauer Associates, Sunderland, M A .
1.1, W.-H., C.-I. WC and C.-C. LUO, 1984 Nonrandomness of point
623-632.
sequences. J. Mol. Evol. 16: 1 1 1 - 120.
mutation as reflected in nucleotide substitutions in pseudogenes and its evolutionary implications. J. Mol. Evol. 21: 58-71.
LINARES, A. R., T. BOWEN and G. A. DOVER, 1994 Aspects of nonran- dom turnover involved in the concerted evolution of intergenic spacers within the ribosomal DNA of Drosophila melanoguster. J.
LIU, J. S., and C. L. SCHARDI., 1994 Aconserved sequence in internal transcribed spacer-1 of plant nuclear ribosomal-RNA genes. Plant Mol. Biol. 26: 775-778.
MADDISON, W. P., and D. R. MADDISON, 1992 MacClade: Analysis of Phylogeny and ChaructmEvolution. Sinauer Associates, Sunderland, MA.
MANGELSDOW, P. C., 1974 Corn. Its origzn Evolution and Improvement. Harvard University Press, Cambridge, MA.
MCDADE, L. A., 1992 Hybrids and phylogenetic systematics 11. The impact of hybrids on cladistic analysis. Evolution 46: 1329-1346.
MORTON, B. R., 1995 Neighboring base composition and transver- sion/transition bias in a comparison of rice and maize chloro- plast noncoding regions. Proc. Natl. Acad. Sci. USA 92: 9717- 9721.
MUNRO, J., R. H. BUKDON and D. P. LEADER, 1986 Characte-ization of a human orphon 28s ribosomal DNA. Gene 48: 65-70.
MUSE, S. V., 1995 Evolutionary analyses of DNA sequences subject to constraints on secondary structure. Genetics 139: 1429-1439.
OLSEN, G. J., H. MATSUDA, R. HACSTROM and R. OVERBEEK, 1994 fastDNAml: a tool for construction of phylogenetic trees of DNA sequences using maximum likelihood. Comput. Appl. Biosci. 10:
P ~ o , S., D. M. IRWIN and A. C . WILSON, 1990 DNA damage pro- motes jumping between templates during enzymatic amplifica- tion. J. Biol. Chem. 265: 4718-4721.
PHII.I.IPS, R. L., A. s. WANC, I. RUBENSTEIN and w. D. PARK, 1979 Hybridization of ribosomal DNA to maize chromosomes. May- dica 24: 7-21.
RANDOLPH, L. F., 1955 Cytogenetic aspects ofthe origin and evolu- tionary history of corn, pp. 16-61 in Corn and Corn Improvement, edited by G. F. SPRAGUE. Academic Press, New York.
RIESEBERG, L. H., and J. D. MOREFIELD, 1995 Character expression, phylogenetic reconstruction, and the detection of reticulate evtr lution, pp. 333-353 in Experimental and Molerular Approaches to Plant Biosystaatirs, edited by P. C. HOCH and A. G. STEPHENSON. Monographs in Systematic Botany at the Missouri Botanical Gar- den, St. Louis, MO.
RITLAND, C. E., K. RITIAND and N. A. STRAUS, 1993 Variation in the ribosomal internal transcribed spacers (ITS1 and ITS2) among eight taxa of the Mimulus guttatus species complex. Mol. Biol. Evol. 10: 1273-1288.
SANDERSON, M. J., and J.J. DOYLE, 1992 Reconstruction of organis- mal and gene phylogenies from data on multigene families: con- certed evolution, homoplasy, and confidence. Syst. Biol. 41: 4- 17.
SMITH, G. P., 1976 Evolution of repeated DNA sequences by un- equal crossover. Science 191: 528-535.
SOKAI., R. R., and F. J. ROHLF, 1981 Biometry. W. H. Freeman and Co., San Francisco.
SUH, Y., L. B. TIIIEN, H. E. REEVE and E. A. ZIMMER, 1993 Molecular evolution and phylogenetic implications of internal transcribed spacer sequences of ribosomal DNA in Winteraceae. Am. J. Bot. 80: 1042-1055.
SWOFFORD, D. L., 1993 PAUP: Phylogenetic Analysis Using Parsimony, Version 3.1. Computer program distributed by the Illinois Natural History Survey, Champaign, IL.
TAIIMA, F., 1993 Simple methods for testing the molecular evolu- tionary clock hypothesis. Genetics 135: 599-607.
VAN HOUTEN, W. H. J., N. SCARLETT and K. BACHMANN, 1993 Nu- clear DNA markers of the Australian tetraploid MicrosPris srapigera and i t s North American diploid relatives. Theor. Appl. Genet. 87: 498-505.
VAN NUES, R. W., J. VENEMA, J. M. J. RIENTIES, A. DIRKSMUI.DER and H. A. RAUE, 1995 Processing of eukaryotic pre-rRNA: the role of the transcribed spacers. Biochem. Cell Biol. 73: 789-801.
VARADARAJ, K., and D. M. SKINNER, 1994 Denaturants or cosolvents improve the specificity of PCR amplification of a G+C-rich DNA using genetically-engineered DNA-polymerases. Gene 140: 1-5.
VENKATESWARLU, K., and R. NAZAR, 1991 A conserved core structure
Mol. Evol. 39: 151-159.
41-48.
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021

83 2 E. S. Buckler, A. Ippolito and T. P. Holtsford
in the 18-25s rRNA intergenic region from tobacco, Nicotiana rustica. Plant Mol. Biol. 17: 189-194.
WAGNER, A,, N. BIACKSTONE, P. CARTWRIGHT, M. DICK, B. MISOF et aZ., 1994 Surveys of gene families using polymerase chain reaction: PCR selection and PCR drift. Syst. Biol. 43: 250-261.
WENDEI., J. F., A. SCHNABEI. and T. SEEIANAN, 1995a Bidirectional interlocus concerted evolution following allopolyploid speciation in cotton (Gossypium). Proc. Natl. Acad. Sci. USA 9 2 280-284.
WENDEI., J. F., A. SCHNABEI. and T. SEELANAN, 1995b An unusual
ribosomal DNA sequence from Gossypium gossypioides reveals an- cient, cryptic, intergenomic introgression. Mol. Phylo. Evol. 4
WILKINSON, L., 1989 SYSTAT: The System for Statistics. SYSTAT, Inc.,
ZUKER, M., 1989 On finding all suboptimal foldings of an RNA
298-313.
Evanston, IL.
molecule. Science 244: 48-52.
Communicating editor: A. G. CIARK
Dow
nloaded from https://academ
ic.oup.com/genetics/article/145/3/821/6053862 by guest on 21 D
ecember 2021