Embed Size (px)
Citation preview

Chemistry in Australia26 |
Drug developmentis a fragmented taleof hits, leads,reactions, protectionand a moleculecalled Janet.
Drugs prescribed bymedical professionals arebig business, withworldwide spending on
prescription drugs tipped to exceedUS$1 trillion by 2015, according to theIMS Institute for HealthcareInformatics. Developing a new drug isan expensive long-term propositioninvolving laboratory research,approved animal trials, clinical studiesand strict regulatory requirements thataim to protect the consumer.
‘It takes about 20 years and over$1.5 billion to get a drug from theconcept stage to the market,’ says DrJerome Wielens, a senior researcher instructural biology at St Vincent’sInstitute of Medical Research inMelbourne.
Many potential drug candidates failbecause of problems with side effectssuch as toxicity, or issues associatedwith oral delivery. Tablets arepreferred by consumers, but thatmeans a drug must negotiate thebody’s digestive system andmetabolic processes without losing itspotency. On its voyage through thehuman body, a typical drug moleculemust also be absorbed into thebloodstream, travel to where it isneeded, cross the cell membrane,deliver its intended impact, and thenbe quickly eliminated from the body.
Drug development programsrequire expertise in areas such as
chemistry, biology, crystallography,modelling and pharmacology. A drugdevelopment team will design,synthesise, assess and modifyhundreds of compounds – or more –during the development of a singledrug.
Selecting the right targetA team with a viral or bacterialdisease in its sights begins byidentifying a key protein or otherbiomolecule that is crucial to theorganism’s ability to survive, invadenew cells or hosts, or replicate.
The challenge is then to find orsynthesise a molecule that can blockthe action of the target protein so thatthe organism’s survival is severelycompromised and the human body’snatural defence mechanisms canfinish it off. This involves discoveringone or more initial ‘hits’, compoundsthat bind well to a site on the targetthat will directly or indirectly modulatethe target’s activity. The hits aredeveloped into ‘lead’ compounds with‘good potency in biological assaysthat reflect the targeted mechanism’(Murray C.W., Rees D.C. 2009, NatureChemistry 1, 187–92). The leadsundergo more extensive assessmentand further modification by medicinalchemists to optimise or introducesuitable drug-like properties.
Drug designers use severalapproaches. They might modify an
The drug
designersBY NANCY MILLS

existing drug or lead, work on a naturalproduct known to be biologicallyactive, use high-throughput screeningmethods to investigate as many asseveral million related compounds, orapply the principles of structure-baseddrug design or fragment-based drugdiscovery.
A fragmented approachFragment-based drug discovery is arelatively recent development. It isclosely related to structure-baseddrug design, a method of designingdrugs that uses the three-dimensionalshape of a protein target to developcompounds that inhibit or otherwisemodulate the activity of the protein toobtain a desired response.
A ‘fragment’ is a relatively simple,low-molecular-weight compound.Fragment-based drug discoveryfocuses on determining which types offunctional groups have an affinity for
sub-pockets within a binding site onthe target protein. The technique relieson having high-resolution structuralinformation for the protein and theprotein–fragment complexes, which iswhere synchrotron X-raycrystallography comes in.
‘Fragment screening enables you toidentify molecules you’d otherwisenever find,’ says Professor MichaelParker, Deputy Director of St Vincent’sInstitute of Medical Research. ‘Butyou’ll need a synchrotron becauserobotic sample-handling capabilities,intense X-ray beams and high-resolution data are essential.’
At St Vincent’s Institute, JeromeWielens identifies hit compounds byscreening a library of compounds,using NMR spectrometry to measuretheir binding affinity to the targetprotein.
‘I use synchrotron radiationwhenever we can crystallise the target
Chemistry in Australia 27|
Dr Rachel Williamson (AS) checks the microcrystallography beamline, one of two dedicated X-ray crystallography beamlines at
the facility.
Fragment-baseddrug discoveryfocuses ondeterminingwhich types offunctional groupshave an affinityfor sub-pocketswithin a bindingsite on the targetprotein.
Sta
te G
ove
rnm
en
t V
icto
ria
Chemistry in Australia28 |
protein bound to the hit compounds,’Jerome says. ‘With synchrotron X-raycrystallography data, we can developthree-dimensional models to show inatomic detail how the hit compoundsinteract with the target protein.’
Each model is visualised on acomputer where it can be rotated andmodified to investigate differentaspects of binding. Jerome uses hisknowledge of chemistry and drugdevelopment to suggest changes tothe compound that should improve itspotency. He also identifies whichregions of a hit compound can beused to engineer other properties intothe drug, for example to improve itsability to pass through the digestivesystem unscathed.
Dr Martin Stoermer from theInstitute for Molecular Bioscience(IMB) at the University of Queenslandis developing drugs for infectiousdiseases, particularly flaviviruses suchas dengue, West Nile, kunjin, Japaneseencephalitis and yellow fever virusesthat are spread by mosquitoes.Previously dismissed as ‘tropical’,these diseases are becoming moresignificant in Australia as globalwarming expands the range of theirmosquito vectors.
As a medicinal chemist, Martinuses structure-based drug design as acritical tool in his research. He worksclosely with experiencedcollaborators such as IMB’s Professor
Jenny Martin, who regularly visits theAustralian Synchrotron or uses theremote access capabilities of its high-throughput macromolecular andmicrocrystallography beamlines.
Dr Tom Peat from CSIRO MaterialsScience and Engineering in Melbourneis also a regular visitor to the AustralianSynchrotron. He uses drug designmethods to tackle a range of infectiousdiseases and cancers. Targetmolecules include HIV integrase (seeRhodes D.I. et al. 2011, Antiviral Chem.Chemother. 21, 155–68), which plays akey role in incorporating viral DNA intothe host cell’s DNA, and hepatitis Cpolymerase, which is essential for viralreplication.
Tom begins with structuralinformation on the target (generally aprotein) and any small molecules,substrates or products known to bindto the target. His group usually alsohas binding affinities obtained bysurface plasmon resonance and eitherin vitro or cell-based assays.
‘We might just start with themolecules that bind best and forwhich we have structural information,’Tom says. ‘We use synchrotron X-raycrystallography at an early stage forfragment screening and then at everystep going forward to improve ourknowledge of structure-activityrelationships (SARs) – and often untilwe have a candidate drug inpreclinical development.’
Refining the hit list‘Deciding which hit compounds towork with first can be difficult,’ JeromeWielens says. ‘We have to considerpotency, chemical tractability andavailability of starting materials alongwith many other factors.’
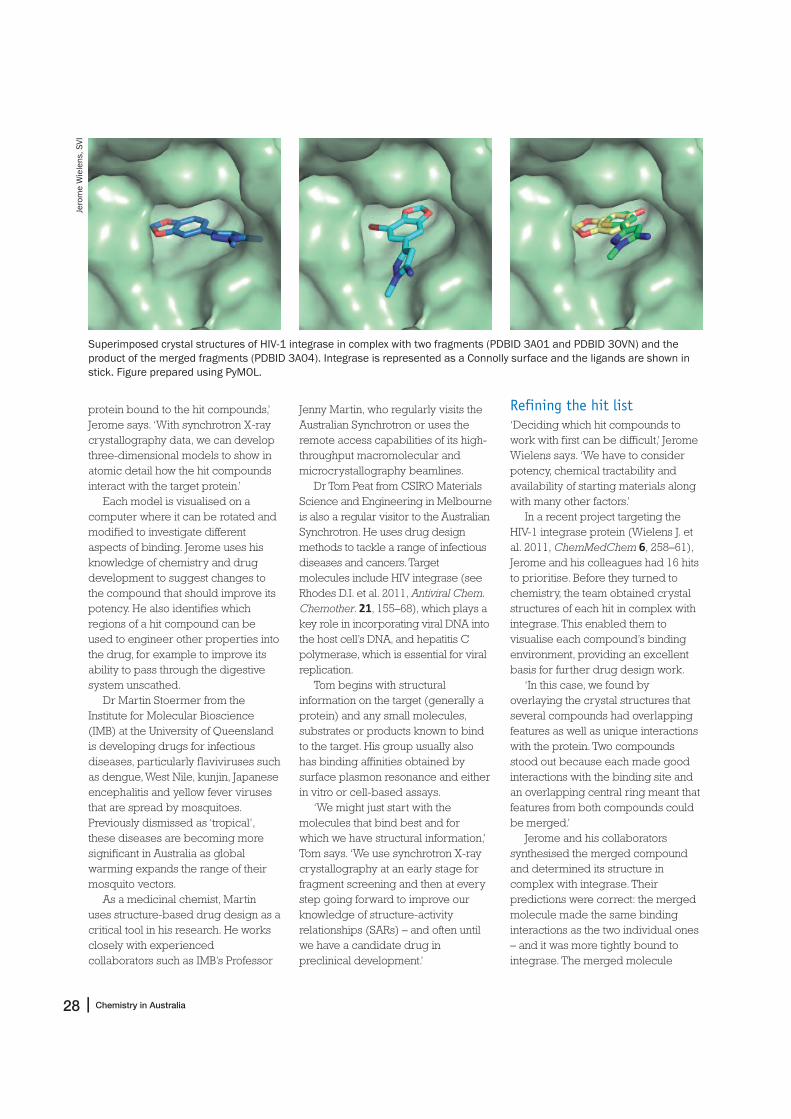
In a recent project targeting theHIV-1 integrase protein (Wielens J. etal. 2011, ChemMedChem 6, 258–61),Jerome and his colleagues had 16 hitsto prioritise. Before they turned tochemistry, the team obtained crystalstructures of each hit in complex withintegrase. This enabled them tovisualise each compound’s bindingenvironment, providing an excellentbasis for further drug design work.
‘In this case, we found byoverlaying the crystal structures thatseveral compounds had overlappingfeatures as well as unique interactionswith the protein. Two compoundsstood out because each made goodinteractions with the binding site andan overlapping central ring meant thatfeatures from both compounds couldbe merged.’
Jerome and his collaboratorssynthesised the merged compoundand determined its structure incomplex with integrase. Theirpredictions were correct: the mergedmolecule made the same bindinginteractions as the two individual ones– and it was more tightly bound tointegrase. The merged molecule
Superimposed crystal structures of HIV-1 integrase in complex with two fragments (PDBID 3A01 and PDBID 3OVN) and the
product of the merged fragments (PDBID 3A04). Integrase is represented as a Connolly surface and the ligands are shown in
stick. Figure prepared using PyMOL.
Jero
me
Wie
len
s, S
VI

became the basis for further designwork to improve its potency anddevelop it into a drug.
Enter the chemists‘Once a hit or ‘active’ is known to havethe desired pharmacological activity,it’s the chemist’s job to optimise thecompound’s potency, physicalproperties and pharmacokineticprofile,’ says Dr Michael Harding, asenior medicinal chemist at Biota.
For example, a chemist mightdecide to modify the activecompound’s shape and size tomaximise binding affinity for the targetprotein. They could also increase ordecrease side-chain lengths and ringsizes or add new groups to optimiseor pick up new hydrogen-bonding orhydrophobic interactions. Addingpolar groups such as amines andalcohols can also improve compoundsolubility.
Bioisosteric replacement is acommonly used technique thatreplaces a functional group with
another of similar biological activitybut enhanced physical orpharmacokinetic properties, such assubstituting a tetrazole for acarboxylic acid. The tetrazole has acomparable pKa (dissociationconstant) but greater lipophilicity,resulting in improved absorption.Hydrogen is often replaced withfluorine, which is a similar size but willblock positions prone to oxidation andconsequently increase metabolicstability.
‘The ideal synthetic procedure isgeneral, robust, uses readily availablereagents, proceeds under mildconditions and produces a high yield,’Michael says.
Commonly used reactions includealkylations, condensations (amidecouplings, heterocycle formation),palladium-catalysed couplings(Suzuki, Buchwald–Hartwig,Sonogashira) and reductiveaminations. Solid-phase reagents,automated flash chromatography andmass-directed HPLC systems have
been developed to speed up thework-up of these reactions andthe purification of the finalproducts.
Michael says a useful strategyis to synthesise a library ofcompounds that will explorestructure–activity relationshipsand guide further design. Amedicinal chemist might aim tomake around 20 milligrams offinal compound for a biologicalassessment and somepreliminary in vitro and in vivopharmacokinetic profiling.
More chemistry, please‘Our throughput is mostdefinitely not in the combichemballpark,’ Martin Stoermer says.‘We don’t believe that the way tofind a needle in a haystack is tomake the haystack bigger!
‘We would rather make smalltargeted libraries of up to 20compounds in a structure–activity relationship series. Over
the lifetime of a typical NHMRC grantof 3 years, on a particularly heavymed chem project we might make100–200 carefully targetedcompounds.’
In Martin’s protease inhibitorresearch for the West Nile virus, theteam started with the natural peptidesequences that the target enzymeprocesses. These natural peptidesequences had a lysine–arginine motifin a particular position, and a thirdbasic residue (Lys–Lys–Arg) wasadded to optimise the substrates. Theteam then terminated the tripeptidesequence with an organic moiety atthe nitrogen terminus and an aldehydeat the carbon terminus to create a lownanomolar inhibitor.
Martin says his ‘bread and butter’reactions are similar to those outlinedin a recent Journal of MedicalChemistry review by Roughley andJordan (2011, 54, 3451–79), includingheteroatom substitutions, acylation, C–C bond formation, heterocycleformation, protection, deprotection,
Chemistry in Australia 29|
drug design
Shown here bound to its target protein, this molecule could lead to a potential drug for
West Nile virus. Capable of causing severe neurological illness, the mosquito-borne West
Nile virus could spread to Australia with global warming.
Ma
rtin
Sto
erm
er,
IM
B U
Q

Chemistry in Australia30 |
reduction, oxidation and addition orinterconversion of functional groups.
Tom Peat and his colleagues use‘all the regular synthetic/medicinalchemistry tools’ to modify compoundsto get more specific binding andbetter inhibition. In the course ofdeveloping a potential new drug, theytypically go through hundreds ofmolecules – such as the moleculeshown above bound to its target HIVintegrase protein – usually workingwith quantities ranging from tens tohundreds of milligrams.
Dr John Deadman from JDJBioservices, the chemist who workedon the molecule shown above,describes it as a 3-indanone linked toa 2-carboxybenzodioxolane.
‘There isn’t a common name assuch, although sometimes we call it“Janet”,’ John says.
A fragment screen identified thebenzodioxolane and carboxylic acidcomponents and indicated thatanother aromatic ring could beincorporated. John chose theindanone. Starting with the
benzodioxolane as the 5-aldehydecomponent, he used metallation,carboxylation, esterification,Knoevenagel condensation anddeprotection reactions to convert itinto the final molecule.
Technique, technique, techniqueSynchrotron X-ray crystallography isan essential part of drug developmentwhen the target and potential drugmolecules are available as crystalslarger than a few microns across.
When the molecules cannot beprepared as crystals of sufficient size,or cannot be crystallised at all, othersynchrotron techniques such aspowder diffraction, X-ray absorptionspectroscopy and small angle X-rayscattering may be useful.
Powder diffraction can providehigh-resolution diffraction data frompolycrystalline materials, solve thestructure of pharmaceuticals andevaluate the quantity of constituents ina tablet, for example. X-ray absorptionspectroscopy yields useful data on
chemical speciation, and small angleX-ray scattering provides informationon the shapes of molecules in solution.Dr Ben Boyd from the Monash Instituteof Pharmaceutical Sciences recentlyused small angle X-ray scattering toexamine how conditions in the humandigestive system affect the geometryof self-assembled lipid structures. Benand his colleagues are using theirsynchrotron data to help identifystructure–function relationships thatcould lead to more-effective drugproducts.
Think local, aim global‘Pharmaceutical companies aroundthe world see structural biology,particularly crystallography, as amandatory part of drug development,’says Michael Parker. ‘The AustralianSynchrotron has been absolutelyessential for many of the drugdevelopment activities of Australianmedical research institutes incollaboration with commercialcompanies such as Biota and with theCRC for Cancer Therapeutics.
‘All major biotech andpharmaceutical companies activelydeveloping drugs in Australia now useor aspire to use the AustralianSynchrotron.’
Nancy Mills <[email protected]> isthe Australian Synchrotron’s science writer. Shethanks Jerome Wielens, Michael Parker, MartinStoermer, Michael Harding, Tom Peat, JohnDeadman, Jenny Martin and the synchrotroncrystallography team for their generous assistancewith this article. The researchers acknowledge thesupport of various government grants, Avexa Limitedand the Bio21 Collaborative Crystallisation Centre.
In the course of developing a potentialnew drug, they typically go throughhundreds of molecules...
(Z)-5-((2-Oxoindolin-3-ylidene)methyl)benzo[d][1,3]dioxole-4-carboxylic acid, also known as ‘Janet’, is one of the lead com-
pounds undergoing further investigation in a project that targets HIV integrase.
Tom
Pe
at,
CS
IRO





























