Embed Size (px)
DESCRIPTION
Geochemistry thermodynamics textbook.
Citation preview

Thermodynamics For Geologists 2012
1. Introduction and definitions
The mineralogies of rocks sampled at the Earth's surface reflect the pressures and temperatures at
which the rocks were formed and their chemical compositions. The methods of chemical
thermodynaics can be used to predict the most stable, or equilibrium, mineral assemblage of a rock
under and given pressure-temperature conditions.
It has long been apparent that chemical equilibirum is approached at some time during the
crytallization history of many metamorphic (e.g., Eskola, 1920) and slowly cooled igneous rocks, and
even during rapid cooling of volcanic rocks (e.g., Carmichael, 1967). The application of
thermodynamic principles to these rocks can therefore provide valuable insight into their histories.
In such cases it may be valid to assume equilibrium and to apply thermodynamic considerations in
order to determine physical conditions (pressure, temperature, etc.) of crystallization. It is apparent that
the volume of rock which is in equilibrium may be small because of the preservation of original
compositional difference on the cm scale even during high-grade metamorphism. Given the
fundamental assumption of chemical equilibrium we shall begin by defining the important
thermodynamic variables.
1.1 System
A system is simply a group of atoms, minerals, or rocks which are under consideration. The exact
position of the boundary of a system may be fixed at will. For example, it may be convenient to choose
as the system a whole outcrop; or a hand specimen, or a single mineral or even a fixed volume of ocean
water at a given depth. The boundary of a system is generally fixed in such a way that the minerals,
fluids (or gases—more on this distinction later) and melts within it may be regarded as having the
potential to be at equilibrium.
Changes that take place within a system may or may not involve interaction with surrounding material.
An isolated system is one which cannot exchange energy or matter with its surroundings. A closed
1

system can exchange energy but not mass with its surroundings, e.g., isochemical metamophism. An
open system can exchange both energy and mass with its surroundings, e.g. if the system is sediments
at the base of a water column matter can move between the water and the sediments.
A system is made up of one or more phases. A phase is a restricted part of the system with distinct
physical and chemical properties. When considering rocks on often identifies several mineral phases,
e.g. all the olivine crystals constitute the olivine phase, all of the plagioclase crystals constitute the
plagioclase phase and so on. In addition there may be a molten phase and a fluid phase containing
H2O, CO2, S, F, Cl and CO.
1.2 Components
Each phase in a system may be considered to be composed of one or more components. For any
particular phase the components may be defined in a number of different ways. For example, in an
(Mg,Fe)2SiO4 olivine solid solution some of the possible sets of components are: (a) Mg2SiO4 and
Fe2SiO4, (b) MgO, FeO, SiO2, and (c) Mg2+, Fe2+, Si4+ and O2-. The choice of components is arbitrary
and depends upon the type of thermodynamic problem under consideration. Generally, if is most
convenient to take the end-members of solid solution series as the components of complex mineral
phases:
Olivine: Mg2SiO4, Fe2SiO4
Plagioclase: CaAl2Si2O8, NaAlSi3O8, KalSi3O8
Clinopyroxene: CaMgSi2O6, Mg2Si2O6, Fe2Si2O6, CaFeSi2O6, NaAlSi2O6
The reason behind the choice of these end-member formula units is that thermodynamic data are not
available for all possible choices of components. For example, the thermodynamic properties of pure
Mg2SiO4 olivine and NaAlSi3O8 plagioclase are well known, but those of Mg2+ in olivine and Na+ in
plagioclase are not; this is because the ions cannot be studied in isolation.
1.3 Chemical potential, µ
In order to determine the directions of chemical change in rocks it is necessary to introduce the concept
of the chemical potentials, µs, of the components present in phases. A chemical potential is analogous
2
to an electrical or gravitational potential in that by measuring the µs of components in different parts of
the system, the tendency (or opposite) of material to flow or react can be deduced. The direction of
flow of components is always from regions of high chemical potential to those of low chemical
potential so that the system as a whole approaches its lowest possible energy state.
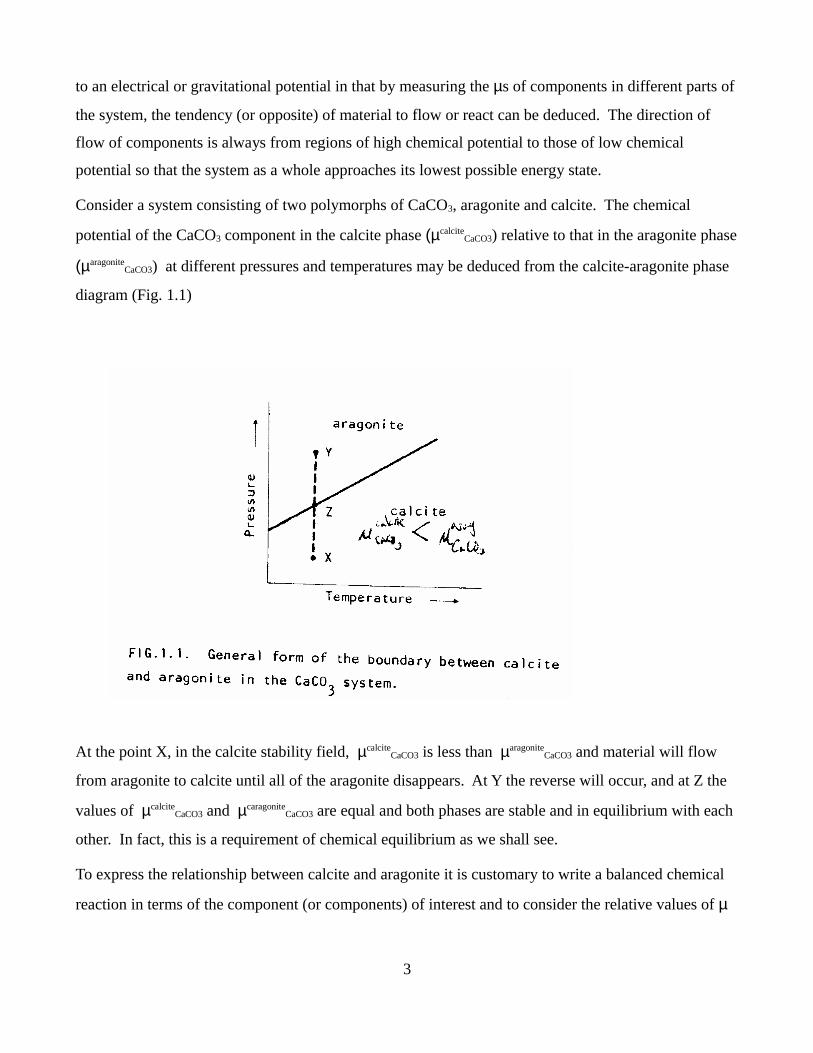
Consider a system consisting of two polymorphs of CaCO3, aragonite and calcite. The chemical
potential of the CaCO3 component in the calcite phase (µcalciteCaCO3) relative to that in the aragonite phase
(µaragoniteCaCO3) at different pressures and temperatures may be deduced from the calcite-aragonite phase
diagram (Fig. 1.1)
At the point X, in the calcite stability field, µcalciteCaCO3 is less than µaragonite
CaCO3 and material will flow
from aragonite to calcite until all of the aragonite disappears. At Y the reverse will occur, and at Z the
values of µcalciteCaCO3 and µcaragonite
CaCO3 are equal and both phases are stable and in equilibrium with each
other. In fact, this is a requirement of chemical equilibrium as we shall see.
To express the relationship between calcite and aragonite it is customary to write a balanced chemical
reaction in terms of the component (or components) of interest and to consider the relative values of µ
3

in the manner discussed above:
CaCO3aragonite⇔CaCO3
calcite
Importantly, note how this chemical reaction is written. The reaction is written using components and
the phases in which the components are found are the superscripts. Chemical reactions are never
written as reactions between phases. Also note that the reaction is mass-balanced—there are equal
numbers of atoms on both sides of the reaction. In general, the components on the left hand side (lhs)
of reactions are considered to be the reactants (or the initial state) and those on the right hand side (rhs)
are the products (or final state). Following this convention the difference in µCaCO3 between products
and reactions can be calculated as
CaCO3=CaCO3products−CaCO3
reactants
or
CaCO3=CaCO3calcite
−CaCO3aragonite
Therefore in Figure 1 it can be seen that
at X, ∆µCaCO3 < 0
at Y, ∆µCaCO3 > 0
at Z, ∆µCaCO3 = 0
The last of these simple equations yields the fundamental condition for chemical equilibrium.
The chemical potentials of components are expressed in joules per mol (j mol-1). Because chemical
potential is a molar property it is independent of the actual number of moles in the system, this type of
thermodynamic variable, or property, is labeled intensive. Other examples of intensive properties are
temperature, pressure, and density.
The chemical potential is function of the state of the system. This means that the chemical potential of
any component i, µi, has a unique value if pressure, temperature and the bulk composition of the system
are fixed (which defines the state of the system). For example, consider Figure 1.1. If the system is
pure CaCO3, then at point X µcalciteCaCO3 and µaragonite
CaCO3 each have unique values. These values are
independent of the path the system followed to reach the temperature and pressure corresponding to
point X. The only way to change µcalciteCaCO3 and µaragonite
CaCO3 is to change the state of the system, i.e., by
4

changing pressure or temperature or adding other components.
1.4 Gibbs Free Energy and chemical potential
In the general case of phases and systems consisting of more than one component it often becomes
necessary to consider the thermodynamic properties of the phase (or system) as a whole rather than the
properties of it constituent components. The sum of the chemical potentials of each component times
the number of moles of each component for all components in a phase or system is defined as the
Gibbs Free Energy:
Gtotal=∑ii ni ,
where ni is the number of moles of component i in the phase of interest and the summation extends
over all of the components in the phase. For an olivine solid solution consisting of n1 moles of
Mg2SiO4 and n2 moles of Fe2SiO4 the free energy of the olivine phase is given by
Gtotalolivine
=n1Mg2 SiO2
olivinen2Fe2 SiO2
olivine
It is apparent that G is a function of the total number of moles of components present in the phase or
system of interest. Properties, or variables, that depend upon the amount of material present are
extensive (e.g., mass, volume, heat content) .
Since the free energy of a multicomponent phase is a function of its composition, the chemical
potential of any component i in a phase may be obtained by partial differentiation of G with respect to
ni for constant amounts of all other components, nj's
∂Gtotal
∂n i
P , T , nj
=i
where the subscripted variables on the right: Pressure, P, Temperature, T, and the nj's are all held
constant during the differentiation (more on this topic later).
For a pure, one-component phase the following relationship is evident
G=n=G /n
So for a pure phase the chemical potential of its constituent component is equal to the molar Gibbs Free
Energy of the phase. Note that from the above equations the units for G are Joules.
5

For most thermodynamic calculations it is convenient to work with molar properties of phases or of
components of phases (although there are some notable exceptions). For this reason Ga will refer to the
Gibbs Free Energy per mole of a phase a and µia will refer to the chemical potential of component i in
phase a. If phase a is composed purely of i then
Ga=ia .
Absolute values of Gibbs Free Energies of phases, or of components, cannot be determined. But,
thankfully this is not necessary. The Gibbs Free Energies of phases can be measured relative to an
arbitrary set of standard substances, usually either elements or oxides, and from this a scale of relative
Gibbs Free Energies or chemical potentials can be constructed. Since the equilibrium condition for
reactions is that the chemical potential difference is zero, it is only necessary to know the difference
between the µs of reactant and product components in order to determine whether the reaction proceeds
from right to left, or vice versa.
1.5 Equilibria involving more than one component
Equilibria in system containing more than one component can be considered in a similar manner that
that already discussed for the aragonite-calcite reaction. At high pressures, and moderate temperatures
anorthite decomposes to grossular, aluminosilicate and SiO2:
3 CaAl2Si2O8plagioclase ⇔ Ca3Al2Si3O12
garnet + 2 Al2SiO5kyanite + SiO2
quartz
Following the principles elucidated above ∆G can be defined:
G=Ca3 Al2 Si3 O12
garnet 2Al2 Si O5
kyanite SiO2
quartz−3CaAl2 Si2 O8
plagioclase
and at equilibrium where all components in the phases are stable
G=0 .
If ∆G is less than 0 the products are stable and if it is greater than 0 the reactants are stable. This is the
key to predicting thermodynamic equilibrium. Note in the above calculation of ∆G the chemical
potential of the individual components must be multiplied by the number of moles of the components
in the reaction.
However, in rocks the garnets are never pure grossular (Ca3Al2Si3O12) and the plagioclases are never
6

pure anorthite (CaAl2Si2O8). In both cases these multicomponent, solid solutions are of widely variable
compositions; the composition of the phase affects the chemical potential of the component.
Nevertheless, whenever there is equilibrium between the plagioclase solid-solution, the garnet solid-
solution, kyanite (almost always nearly pure Al2SiO5) and quartz (also almost always nearly pure SiO2)
then the ∆G for the reaction is zero. This condition applies regardless of the complexity of the phases
in the rocks and the type and number of other components and other phases in the rocks.
Although the chemical potentials of components in solid solutions are not the same as in pure phases
(more on this later), the reactions and equilibrium conditions may be considered in terms of simple
end-member components and the equilibrium condition. The consideration of equilibrium relations
between the various simple components in a multicomponent natural rock is an extremely powerful
tool when applied to geological problems because often multiple reactions between multiple
components can be written and used to constrain the conditions at which the rock formed.
1.6 Determination of µ and G
In order to determine the equilibrium conditions for a reaction between components the numerical
values of µ and G must be known for the components and phases of interest. Although the absolute
values of these quantities cannot be determined, the relative values can be obtained by specifying an
arbitrary reference. The fundamental equation that specifies the Gibbs Free Energy of a system or a
phase is:
G=H−TS ,
where H is the enthalpy, or heat content of the system or phase, T is the absolute temperature (on the
Kelvin scale) and S is its entropy.
It is important to recognize the difference between the heat content and temperature, which are often
used interchangeably in everyday language. The heat content, that is enthalpy, is a measure of the
internal energy, E (sometimes U is also used) plus the pressure, P, multiplied by the volume, V, of the
material:
H=EPV .
Its units are in J or J mol-1. On the other hand, temperature is a measure of the hotness or coldness of
7

an object. One of the more rigorous relationships defining temperature from statistical mechanics
relates it to the average kinetic energy of a gas divided by Boltzmann's constant, k:
T=m v2
3 k,
where m is the mass of a gas molecule and v is its velocity. Classical thermodynamics provides other
definitions of temperature that need not be dealt with at this time.
The enthalpy of the system is usually expressed relative to the enthalpies of the constituent elements,
which by convention are taken to be zero at 1 bar and the temperature of interest (i.e., any temperature).
Thus a scale of relative enthalpies of compounds of elements can be established (and looked up in
tables). Consider the enthalpy of pure forsterite, Mg2SiO4. Because the heat contents of Mg (crystal),
Si (crystal) and O2 (gas) at 1 bar are defined to be zero, the enthalpy of Mg2SiO4 is obtained from the
reaction
2 Mg + Si + 2 O2 ⇔ Mg2SiO4
and calculated by subtracting the values of the reactants from those of the products:
∆Hreaction = Hforsterite – (2 HMg + Hsi+ 2HO2)
= Hforsterite – 0 = -2175.68 kJ at 1 bar, 298 K
But how is Hforsterite determined? Conceptually the measurement of Hforsterite is simple: Dissolve one
mole of Mg2SiO4 in acid, or a solvent composed of an oxide melt, at a specific temperature and
measure the heat generated; that heat is Hforsterite. However, the experimental procedure is vastly more
complex than this sentence implies. Thankfully, in many, if not most, cases the necessary values for
thermodynamic calculations have already been measured. Note that the negative sign for Hforsterite in the
reaction indicates that heat is given off during the formation of forsterite from its consituent oxides; this
type of reaction in which heat is liberated is an exothermic reaction, a reaction in which heat is
absorbed is an endothermic reaction.
The entropy of a system (or phase), S, may be regarded as a measure of the degree of randomness or
disorder in the system. The greater the degree of disorder the higher the value of the entropy. As an
example let us consider a system of a large volume containing a small amount of a gaseous species.
The probability of finding a molecule of the gas within any given volume of the system is very small
because a few molecules are dispersed throughout a large volume. The degree of randomness of the
8

system is therefore large and the molar entropy of the gas is also large. As the volume of the system is
decreased the pressure of the gas increases, pressure being inversely proportional to volume, and
intermolecular distances decrease. By decreasing the total volume of the system the probability of
finding a molecule in any given volume of gas is increased and entropy or randomness is therefore
decreased. If pressure is increased sufficiently, the gas may liquefy or even solidify. Using this
argument, it is apparent that molecules in the liquid state with low volume and high density should
have lower molar entropy than those in the gaseous state, and the the molar entropy in the solid state
will, in general, be lower still. In addition to the increased molecular density in liquid and solid states
relative to the gaseous state, the translational and rotational degrees of freedom of molecules (also
related to randomness) are much lower in the liquid and solid states than in the gaseous state because of
the existence of stronger intermolecular forces.
As a rule of thumb, the following relative entropy scale may be constructed:
(Sgas)low P > (Sgas)high P > Sliquid > Ssolid
Since G is an extensive property both H and S are extensive properties. However, because it is
intended to use molar values of G throughout the rest of this book, Ha and Sa will henceforward be
taken to refer to the enthalpy per mole and entropy per mole of phase a. The units of Ha are J mol-1
and of Sa are J mol-1 K-1. The analogous expression to that for G, but in this case for µ is
ia=H
_
ia−T S
_
ia ,
where H_
ia is the partial molar enthalpy of i in phase a and S
_
ia is the partial molar entropy of i in
phase a. The partial molar enthalpy of component i is the enthalpy per mole of this component in the
phase of composition a. If phase a only consists of i, as with G and µ, the molar enthalpy of a is equal
to the partial molar enthalpy of i in a. But, if phase a is a mixture of components, this equality does not
apply (see Chapter 3). The same definition and relations also exist for the partial molar entropy.
Consider the reaction of forsterite and quartz to form enstatite (all phases are pure):
Mg2SiO4forsterite + SiO2
quartz ⇔ Mg2Si2O6enstatite
The change in enthalpy of the system produced by reacting 1 mole of forsterite with 1 mole of quartz is
given by:
9

∆H = Henstatite – Hquartz – Hforsterite
For example at 900 K, 1 bar:
∆H = -3 091 930 – (905 422 + 2 172 360) = - 14 142 J
Note the negative sign indicating an exothermic reaction that gives off heat (which remember is energy
). ∆H is the heat evolved (or absorbed) as a reaction takes place at constant pressure. In a similar
manner the entropy change for the reaction is:
∆S = Senstatite – Squartz – Sorsterite,
and at 900 K, 1 bar:
∆S = 360.33 – (109.16 + 258.91) = -7.74 J K-1
Note that the results of the calculations do not have mols in the units. This is because when each of the
values of H and S in the above equations are multiplied by the stoichiometric coefficients (number of
mols) of the components involved in the reactions. Thus for a general reaction of the type:
x A + y B ⇔ z C + a D
∆H = zHC + aHD – (xHA + yHB)
∆S = zSC + aSD – (xSA + ySB),
∆G = zGC + aGD – (xGA + yGB)
Note the general rule of “Products – Reactants” in these and all thermodynamic calculations.
In order to determine whether or not forsterite would react with quartz to form enstatite or vice versa at
a given set of P, T conditions it is necessary to know ∆G at those chosen conditions:
G=Mg2 Si2 O6
enstatite−Mg2 Si O4
forsterite−Si O2
quartz
and because in this example the phases are all pure:
G=Genstatite−GforsteriteGquartz =H−T S
10

and by analogy with the case of calcite and aragonite:
∆G < 0 reaction proceed to right
∆G > 0 reaction proceed to left
∆G = 0 reaction at equilibrium
For this specific example at 900 K, 1 bar:
∆G = - 7 176 J
and the reaction proceeds to the right and enstatite is produced from forsterite and quartz.
Hess's law
A useful property of thermodynamic calculations is that they can be mathematically manipulated. Such
manipulations allow us to calculate thermodynamic values that cannot be directly measured. A good
example is the formation of diamond from graphite because of the high temperatures and pressures
necessary for:
Cgraphite ⇔ Cdiamond ∆H1
However, we can calculate ∆H1 from other reactions involving the reactants and products. Consider the
two simple-to-perform reactions:
Cgraphite + O2, gas ⇔ CO2, gas ∆H2
Cdiamond + O2, gas ⇔ CO2, gas ∆H3 .
We can reverse the second reaction
CO2, gas ⇔ Cdiamond + O2, gas -∆H3
and then add the graphite combustion reaction with the reversed diamond combustion reaction to make
Cgraphite + CO2, gas + O2, gas ⇔ Cdiamond + O2, gas + CO2, gas
∆H = ∆H2 – ∆H3
But each side of the reaction contains CO2, gas + O2, gas and so, similarly to mathematical equations, they
cancel each other out to produce the final reaction:
11

Cgraphite ⇔ Cdiamond ∆H1 = ∆H2 – ∆H3.
This is know as Hess's law, and the concept is frequently used to calculate values that cannot be
directly measured. All extensive variables (G, S, H, and V, or volume) are additive and they can all be
determined in analogous indirect ways.
1.7 Temperature dependence of H and S, µ and G
If a substance is heated at constant pressure, then its heat content, or enthalpy, increases proportionally
to temperature. The relationship between the temperature increase and enthalpy increase is different
for different substances and is expressed by a characteristic constant of proportionality, the heat
capacity, Cp (strictly speaking this is the heat capacity at constant pressure; Cv, the heat capacity at
constant volume also exists but we shall not be using it, much . . . ). If the substance is heated through
an infinitesimally small temperature interval of dT then the infinitesimally small increase in enthalpy,
dH is rigorously defined by:
dH = Cp dT.
Because enthalpies of reactants and products change with temperature, the enthapy change of a reaction
(e.g., A ⇔ Β) is also temperature dependent:
dHA = (Cp)A dT; dHB = (Cp)B dT;
d∆H = (Cp)B dT - (Cp)A dT = ∆Cp dT
where ∆H = ΣΗproducts - ΣHreactants and ∆Cp = Σ(Cp)products – Σ(Cp)reactants. In tables of thermodynamic data it
is common to give enthalpies of materials at a reference temperatures, usually, although not always, at
298.15 K. In order to obtain the enthalpy at higher temperatures the above expression for dH must be
integrated from the reference temperature to the temperature of interest:
∫T
298dH= ∫
T
298C p dT or HT=H298 ∫
T
298C p dT .
If Cp is constant and therefore independent of temperature (which it rarely is) it can be removed from
the intregral and
HT=H298Cp T−298 .
However, in most cases Cp is a complex function that goes to 0 at 0 K:
12
To express such a function are typically expressed as a polynomial. A simple example (e.g., Kelley
1960) would be:
Cp=abTc
T 2,
where the a, b and c are experimentally determined constants for the phase (usually pure) of interest
and T is the temperature in K (as usual for thermodynamics). Thus to determine the enthalpy of most
substances the polynomial must be integrated:
HT=H298 ∫T
298abT
c
T2dT ,
which when integrated yields
HT=H298[a12
bT 2−
cT ]
298
T
Example/Exercise 1. Determine the enthalpy of (low) albite at 900 K and 1 bar given the value at 298
K and 1 bar as well as the other data in the following table.
13
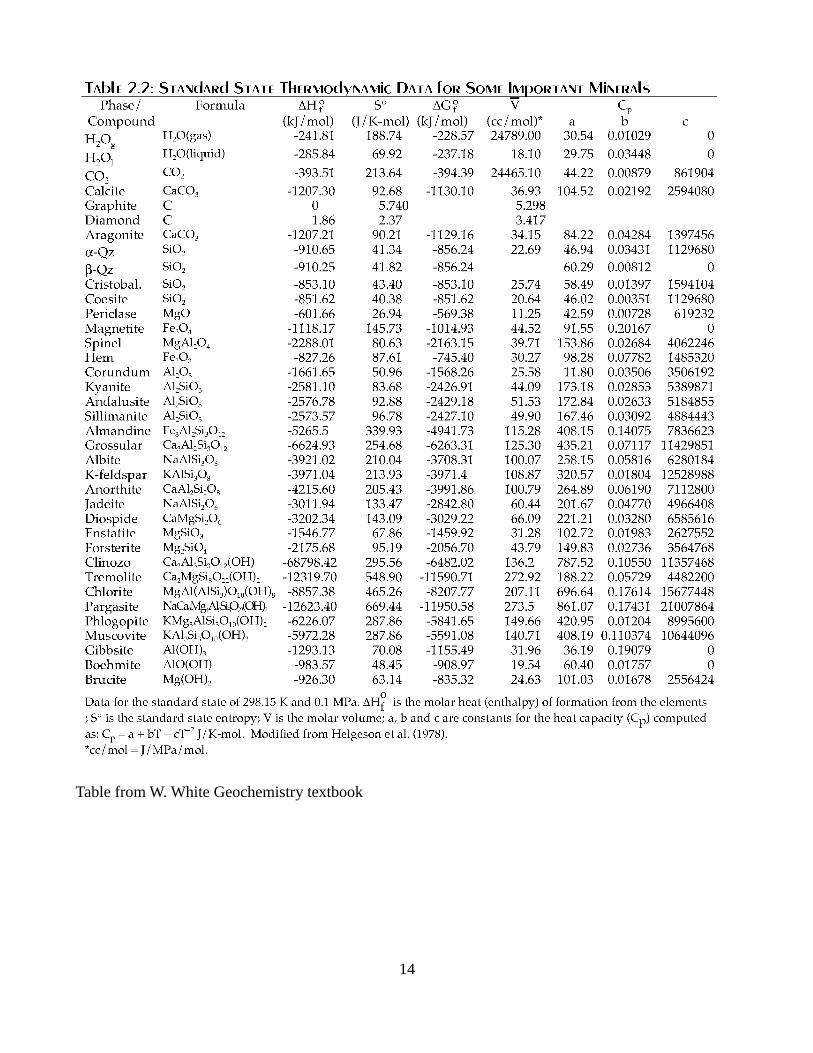
Table from W. White Geochemistry textbook
14

Increasing the heat content of a phase also leads to an increase in the entropy. If the amount of heat,
dH is supplied at constant pressure then:
dS=dHT
and because dH = CP dT we find
dS=Cp dT
T
which yields
ST=S298 ∫T
298
C p
TdT
or, using the previous temperature dependent expression for Cp
ST=S298 ∫T
298
aTb
c
T 3dT
which when integrated produces
ST=S298[a lnTbT−c
2T 2 ]298
T
.
Example/Exercise 2 Calculate ∆H800, ∆S800 and ∆G800 for the following reaction at 1 bar:
NaAlSi3O8albite ⇔ NaAlSi2O6
jadeite + SiO2quartz
using the information in the table above. In which direction will the reaction proceed? Compare the
calculated ∆G800 from what you would calculate if ∆Cp = 0. (Note that this does not mean that the
invividual Cp's of the reactants and products are zero, but that their sum is equal to zero. Also, the
results you find for this example are applicable to virtually all solid-solid reactions, i.e., those only
involving minerals and not involving gases, fluids, or melts.)
1.8 Pressure dependence of µ and G
At constant temperature T, the effect of pressure on the free energy of phase a (Ga) is given by
15

∂Ga
∂ P
T
=V a .
Similarly the change in chemical potential of component i in phase a is equal to the partial molar
volume of component i in phase a:
∂i
a
∂P
T
= V ia .
For a reaction involving a volume change, ∆V, where
V=V products−V reactants
the analogous expression is
∂G∂P
T
=V
Integration of these expressions from a reference pressure (almost always 1 bar) at a given temperature
high pressure allows the calculation of Ga, µi and ∆G at the temperature and pressure of interest:
GP ,T=G1 bar ,T ∫P
1 barV dP .
1.9 Temperature and pressure dependence of µ and G
The Gibbs Free Energy of a reaction at any temperature and pressure may be calculated by combining
the equations for the temperature dependence with that for the pressure dependence:
GP ,T=H 1 bar, T ∫T
298C p dT−T S1 bar, T ∫
T
298
Cp
TdT ∫
P
1 barV dP
Note that this expression implicitly assumes that ∆Cp is pressure independent.
For solid-solid reactions the simplifying assumption that is commonly made (and is often an excellent
approximation) is that the ∆V is independent of temperature and pressure:
∫P
1 barV dP=P−1V
However, situations exist in which the effects of isobaric thermal expansion, α, and isothermal
compressibility, β, on the reactants and products must be taken into account; the isobaric thermal
16

expansion is defined as:
=1VdVdT
P
and the isothermal compressibility as:
=−1V
dVdP
T
In these cases
dV =V dTdP
and
V=V 1 bar, 298T−298P−1
Unfortunately, α and β are not constants and the integration of the above equation can be complicated.
1.10 Calculation of a reaction boundary
Consider the reaction
NaAlSi3O8albite ⇔ NaAlSi2O6
jadeite + SiO2quartz .
At equilibrium we know that :
G=NaAlSi 2 O6
jd SiO2
qz −NaAlSi 3O 8
ab =0
which, if all phases are pure (more about impure phases later), is equivalent to
G=G jdGqz−Gab=0
Using the table of data given above we calculate:
∆H1 bar, 298 = -1.569 kJ and ∆S1 bar, 298 = -35.23 J K-1
Note that enthalpies are almost always in kJ and entropies in J. It is recommended that enthalpies be
converted to J in order to avoid errors in subsequent calculations. Furthermore, numerical values
should not be rounded off until the final calculation is performed.
We can now determine which side of the reaction is stable at 1 bar, 298 K, but we want to calculate the
equilibrium of this reaction at various temperatures and pressures. Thus, we need to perform the
17

integrations discussed above to convert from these values at 1 bar, 298 K to high pressures and
temperatures.
If we were interested in the equilibrium at 298 K and high pressure the appropriate equation is:
GP , 298=H 1 bar, 298−298S1 bar, 298 ∫P
1 barV dP=0
We can compare this expression to that for a point that is infinitesimally higher it temperature, T + dT,
and in pressure, P + dP :
GP+dP,T+dT=H 1 bar, 298C pdT−298dT S1 bar, 298C p
298dTdT ∫
P+dP
1 barV dP=0
Equating these last two equations (because they are infinitesimally different in P and T) yields
− S dTV dP=0
or, the famous Clausius-Clapeyron equation:
dPdT
=SV
which gives us the slope of the reaction in P-T space at any specified temperature and pressure.
A simplifying assumption that ∆Cp =0 implies that ∆H and ∆S are constant for all temperatures and
allows the simple calculation of the approximate reaction boundary. Thus, at equilibrium and 1 bar:
G=H 1 bar−T SH 1 bar=T S
T=−1569 J
−35.23 J K−1=44.5 K
At higher pressure the boundary can be calculated by:
G=0=H 1 bar−T SP−1V
or, the P,T point at 1 bar (T = 44.5 K) can be extrapolated to higher pressure using the Clausius-
Clapeyron equation and assuming no change in ∆S or ∆V with pressure and temperature, which for this
particular example reaction results in:
P=1dPdT
T−44.5 .
18

This assumption is often good for solid solid reactions, but bad for reactions involving melts and
terrible for reactions involving gases or fluids. For these reactions, and if we want the most precise
calculations for solid-solid reactions the complete equation, with all of its integrals must be solved, i.e.,
GP , T=H 1 bar, T ∫T
298C p dT−T S1 bar, T ∫
T
298
C p
TdT ∫
P
1 barV dP
at various temperature and pressure conditions. Thankfully, this is now easily done by computer.
19





























