Embed Size (px)
Citation preview

135
Aprile-Giugno 2016 • Vol. 46 • N. 182 • Pp. 135-148 Prospettive in Pediatria
Oncologia pediatrica
Tumori cerebrali: quali sfide, quali progressi
Elisabetta Schiavello Veronica Biassoni Maura Massimino
Pediatria Oncologica Fondazione IRCCS Istituto Nazionale dei
Tumori, Milano
Brain tumours are the most frequent solid tumours in childhood, being the first cause of cancer-related death in the paediatric population. A variable number of cases, increasing in the last years due to the improvement of diagnostic tools, occurs in children affected by predisposing brain tumour syndromes.Low-grade gliomas and embryonal tumours are the most frequent paediatric pathologies, accounting for 50% and 20% of childhood brain tumours, respectively, followed by epend-ymomas and malignant gliomas.Even if the diagnostic and therapeutic innovations of the last two decades have improved overall survival of patients with brain tumours, with 70% still alive at 5 years after diagno-sis, there are still some diseases (malignant gliomas, atypical teratoid-rhabdoid tumour, DIPG and metastatic tumours) that are associated with a dismal prognosis. Many efforts have been devoted in recent years, and are still ongoing, to discover new therapeutic ap-proaches in the context of international cooperative trials. The main purpose of this paper is to elucidate the most recent and promising findings in paediatric neuro-oncology, to-gether with the future and desirable therapeutic tools and the challenge still represented by the most dreadful diseases.
Summary
I tumori cerebrali rappresentano le neoplasie solide più frequenti nell’infanzia e la prima causa di morte per tumore in età pediatrica. Una percentuale variabile di neoplasie del si-stema nervoso centrale, in costante aumento negli ultimi anni per via dell’affinamento delle tecniche diagnostiche, si verifica nel contesto di sindromi predisponenti. I gliomi a basso grado e i tumori embrionari sono gli istotipi più frequenti in età pediatrica, rappresentando il 50% e 20% rispettivamente di tutte le neoplasie cerebrali, seguiti da ependimomi e gliomi maligni.Se è vero che le innovazioni diagnostico-terapeutiche degli ultimi due decenni hanno mi-gliorato la sopravvivenza globale dei bambini affetti da tumore cerebrale, portandola a oltre il 70% per tutte le istologie, è altrettanto vero che per alcuni istotipi (gliomi maligni, tumore teratoide-rabdoide atipico, DIPG) e in caso di malattia metastatica, i tassi di sopravvivenza rimangono insoddisfacenti. Pertanto molti sforzi sono stati fatti negli ultimi anni e si stanno ancora compiendo per valutare l’efficacia di nuovi approcci terapeutici, soprattutto nel con-testo di trials clinici internazionali. Scopo della presente trattazione è quello di illustrare, partendo da quanto è noto circa le principali neoplasie pediatriche, le novità più recenti e promettenti, le prospettive terapeutiche future o auspicabili e le sfide ancora aperte, soprat-tutto per quelle neoplasie per le quali la prognosi rimane ancora invariabilmente infausta.
Riassunto
Metodologia della ricerca bibliografica effettuataLa metodologia della ricerca è consistita in:• discussionetragliautoriperidentificareiprogres-
si negli ultimi 5 anni relativi alle principali patologie
oncologiche pediatriche del sistema nervoso cen-trale;
• iprincipaliargomentisuddivisiperlepatologiepiùrilevanti sono stati oggetto di una ricerca bibliogra-fica su PubMed;
• ladescrizionerelativaallamessaapuntodinuovi

136
E. Schiavello et al.
trials deriva soprattutto dalla attiva partecipazione a gruppi di lavoro cooperativi italiani e internazio-nali (SIOP, AIEOP) in questo ambito.
IntroduzioneLe neoplasie del sistema nervoso centrale (SNC) rappresentano il secondo tumore pediatrico dopo le leucemie e il tumore solido più frequente nell’infanzia, pari a circa il 25% di tutti i tumori in età pediatrica. Sono la prima causa di morte per tumore in que-sta fascia di età. L’incidenza è di 2,4 nuovi casi per 100.000/bambini/anno, con circa 2.200 nuovi casi/anno in pazienti di età inferiore ai 20 anni negli USA, e circa 350-400 nuovi casi in Italia. L’età alla diagnosi è variabile con un picco tra i 4 e gli 8 anni e una lieve prevalenza nel sesso maschile (CBTRUS, 2012; Gat-ta et al., 2014). L’eziologia della maggior parte delle neoplasie cerebrali è tuttora sconosciuta, anche se il miglioramento delle tecniche di citogenetica e se-quenziamento del DNA ha permesso di identificare alcune sindromi predisponenti. È fondamentale che il pediatra oncologo, e più in generale il pediatra, cono-scano queste sindromi e siano in grado di sospettar-ne la presenza quando il tumore cerebrale ne sia la prima manifestazione. Per questo scopo sono molto importanti un’attenta anamnesi familiare e la cono-scenza di altri eventuali segni/manifestazioni legate alla sindrome. Mentre nell’adulto gli istotipi prevalenti sono gli astro-citomi ad alto grado di malignità, nel bambino pre-valgono i gliomi a basso grado e i tumori embrionari (PNET/medulloblastoma) che rappresentano rispetti-vamente il 50% e il 20% dei tumori cerebrali nei bam-bini di età inferiore ai 15 anni (Fig. 1). Nonostante l’impiego di varie strategie atte a intensi-ficare la chemioterapia (CT) e radioterapia (RT) con-
venzionali, la sopravvivenza dei pazienti con malattia ad alto rischio rimane invece insoddisfacente. Per questo motivo e in considerazione della rarità di que-sti tumori, si sono formati gruppi di lavoro cooperativi volti a uniformare e migliorare la diagnostica anato-mo-patologica e strumentale e il trattamento, a valu-tare i meccanismi biologico-molecolari alla base delle patologie, a studiare terapie mirate secondo specifici gruppi di rischio prognostici valutando, quando possi-bile, l’introduzione di nuove terapie target e garantire un più attento follow-up delle sequele. Nell’ambito di questa revisione verranno presentati lo stato dell’arte relativo alla terapia delle principali neoplasie cerebrali pediatriche e le novità in ambito diagnostico e terapeutico. Verranno inoltre considera-te le principali sindromi predisponenti allo sviluppo di neoplasie del SNC.Segnaliamo che nel Maggio 2016 è stata pubblica-ta la nuova classificazione WHO delle neoplasie del SNC che ha ripreso e in parte modificato quella del 2007. Allo scopo della presente trattazione è stata im-piegata principalmente la classificazione del 2007 in quanto più adatta allo scopo “didattico” (Louis et al., 2016).
MedulloblastomaIl medulloblastoma è la più comune neoplasia ma-ligna pediatrica del SNC, ne rappresenta infatti cir-ca il 15-20%. È classificato come tumore primitivo neuro-ectodermico (PNET) cerebellare (Louis et al., 2007). L’età alla diagnosi è compresa tra i 2-8 anni, ma esistono forme anche nel neonato, nel lattante, nell’adolescente e nel giovane adulto, con un rappor-to maschi/femmine di 2:1. Origina nella fossa poste-riore, principalmente dal verme cerebellare, nel tetto del IV ventricolo. Come tutti i PNET, ha la tendenza
Figura 1. Incidenza delle neoplasie cerebrali pediatriche suddivise per istologia.

137
Tumori cerebrali: quali sfide, quali progressi
a metastatizzare per via liquorale, alla diagnosi infat-ti la presentazione è metastatica nel 35% dei casi. Sono descritte localizzazioni extra SNC perlopiù nei pazienti adulti.Le seguenti varianti istologiche sono riconosciute dal-la classificazione WHO del 2007:• classico68-80%;• desmoplastico/nodulare7%;• conestesanodularità3%;• agrandicellule/anaplastico10-22%;Queste ultime due varianti hanno mostrato un certo grado di sovrapposizione e in numerosi studi vengono classificate in un unico gruppo denominato a grandi cellule/anaplastico (LCA) (Gilbertson e Ellison, 2008).Il trattamento con la radioterapia craniospinale e la chemioterapia dopo la resezione chirurgica ha radi-calmente trasformato una malattia fatale in una malat-tia in cui la probabilità di guarigione è intorno al 70%, anche se per circa un terzo dei pazienti rimane ancora una patologia incurabile. Nelle ultime due decadi si è cercato di adattare il trattamento stratificando i pazien-ti in base a fattori di rischio prognostici basati sull’età, sull’entità della resezione chirurgica e sulla presenza di malattia metastatica, portando a un miglioramento delle percentuali di guarigione e della qualità di vita rispetto ai trial degli anni ’80-90 (Packer et al., 2003). I pazienti con resezione totale/subtotale (residuo tu-morale < 1,5 cm2, valutato in RMN sul piano assiale) e malattia non metastatica, di età superiore a 3 anni alla diagnosi, sono stati prognosticamente classificati a rischio intermedio/standard. Per questa categoria di pazienti è stato possibile diminuire la dose totale di RT craniospinale da 36 a 23,4 Gy, aggiungendo la CT adiuvante con sopravvivenze globali a 5 anni attorno all’80% (Clifford et al., 2015). Questi pazienti ricevono RT craniospinale con sovradose sulla fossa cranica posteriore/letto tumorale fino a 54 Gy, in associazio-ne a chemioterapia contenente derivati del platino e nitrosuree. I pazienti classificati invece “ad alto rischio”, perché con malattia a istologia anaplastica o metastatica, hanno una sopravvivenza a 5 anni di circa il 60-70% dopo trattamenti identificabili con una RT a dosi più
intense e CT ad alte dosi/mieloablativa. Non ci sono al momento evidenze sufficienti per stabilire se alte dosi di radioterapia, possibilmente secondo tecnica iperfrazionata/accelerata (HART), insieme alla CT sequenziale ad alte dosi/mieloablativa possano es-sere il trattamento più efficace nella malattia ad alto rischio. Sono in atto valutazioni nei gruppi cooperati-vi internazionali per valutarne l’efficacia e la tossicità all’interno di uno studio di fase 3 che verrà aperto in Europa sotto l’egida della SIOP (International Society of Pediatric Oncology). Per i pazienti minori di 3 anni la prognosi in passato era considerata sfavorevole per via del ritardo dia-gnostico, dei rischi chirurgici maggiori, dell’aumen-tata tossicità relativa alla radioterapia, dei trattamenti meno intensi e di una maggiore aggressività biologi-ca. Dagli anni ’80 è stata identificata come cut-off l’età minore di 3-5 anni per mettere a punto piani cura in cui la RT venisse omessa o dilazionata nel tempo, allo scopo da ridurre le inaccettabili sequele legate all’età (deficit neurologici motori/sensitivi, endocrino-logici, cognitivi e neuropsicologico-comportamentali) (Rutkowski et al., 2010; von Bueren et al., 2011). Da una metanalisi europea che ha analizzato i dati di so-pravvivenza di 270 bambini di età inferiore ai 5 anni alla diagnosi, trattati con CT sequenziale ad alte dosi, inclusa la terapia mieloablativa senza RT, è emerso che la sopravvivenza può variare dal 14% per il sotto-tipo LCA, al 42% per il classico, al 76% per il desmo-platico-nodulare.Dati recenti hanno dimostrato che sia i sottotipi istologici sia alcuni fattori biologici possono influenzare la progno-si e quindi devono essere utilizzati nella stratificazione del rischio. Infatti uno studio recente (Taylor et al., 2012) ha messo in evidenza che il medulloblastoma è un’enti-tà molto eterogenea caratterizzata da quattro principali sottogruppi molecolari: WNT, Sonic Hedgehog (SHH), gruppo 3 e gruppo 4 (Tab. I). Si è pertanto iniziato a valutare la possibilità di diversificare i trattamenti anche in base alle caratteristiche biologiche identificate dai sot-togruppi molecolari. In particolare:• sottogruppoWNT:aprognosipiùfavorevole,com-
prende i pazienti idealmente candidabili alla ridu-
Tabella I. Principali caratteristiche cliniche dei sottogruppi molecolari di medulloblastoma (da Taylor et al., 2012, mod.).
WNT SHH GRUPPO 3 GRUPPO 4
Sesso : : : : Età
Istologia Classico,raramente
LCA
Desmoplastico,classico,
LCA
Classico,LCA
Classico,LCA
Prognosi Eccellente Intermedia Severa Intermedia
Tipologia di recidiva – Locale Metastatico Metastatico
138
E. Schiavello et al.
zione dei trattamenti allo scopo di minimizzarne gli effetti collaterali. Sono in corso di valutazione infatti, all’interno di studi, ulteriori riduzioni della dose di RT craniospinale e del campo del boost (che dalla fossa cranica posteriore passa a inte-ressare il letto tumorale), associati a una riduzione del numero di cicli di CT adiuvante;
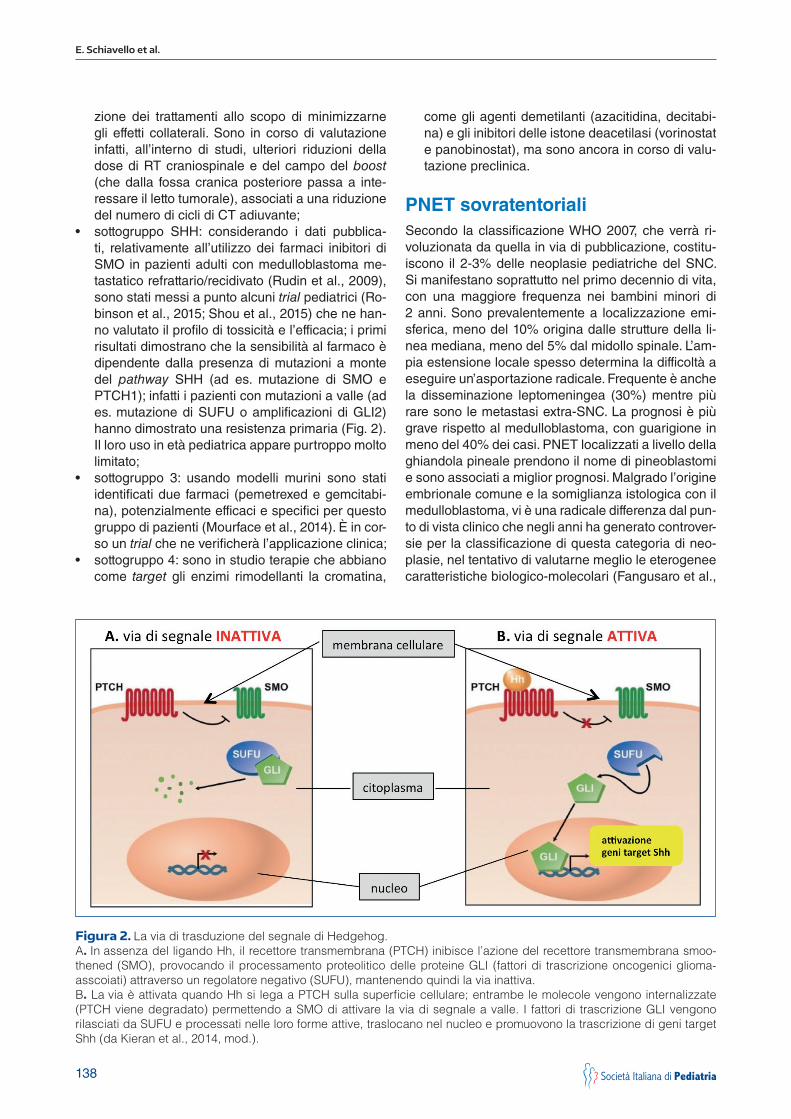
• sottogruppo SHH: considerando i dati pubblica-ti, relativamente all’utilizzo dei farmaci inibitori di SMO in pazienti adulti con medulloblastoma me-tastatico refrattario/recidivato (Rudin et al., 2009), sono stati messi a punto alcuni trial pediatrici (Ro-binson et al., 2015; Shou et al., 2015) che ne han-no valutato il profilo di tossicità e l’efficacia; i primi risultati dimostrano che la sensibilità al farmaco è dipendente dalla presenza di mutazioni a monte del pathway SHH (ad es. mutazione di SMO e PTCH1); infatti i pazienti con mutazioni a valle (ad es. mutazione di SUFU o amplificazioni di GLI2) hanno dimostrato una resistenza primaria (Fig. 2). Il loro uso in età pediatrica appare purtroppo molto limitato;
• sottogruppo 3: usandomodelli murini sono statiidentificati due farmaci (pemetrexed e gemcitabi-na), potenzialmente efficaci e specifici per questo gruppo di pazienti (Mourface et al., 2014). È in cor-so un trial che ne verificherà l’applicazione clinica;
• sottogruppo 4:sonoinstudioterapiecheabbianocome target gli enzimi rimodellanti la cromatina,
come gli agenti demetilanti (azacitidina, decitabi-na) e gli inibitori delle istone deacetilasi (vorinostat e panobinostat), ma sono ancora in corso di valu-tazione preclinica.
PNET sovratentorialiSecondo la classificazione WHO 2007, che verrà ri-voluzionata da quella in via di pubblicazione, costitu-iscono il 2-3% delle neoplasie pediatriche del SNC. Si manifestano soprattutto nel primo decennio di vita, con una maggiore frequenza nei bambini minori di 2 anni. Sono prevalentemente a localizzazione emi-sferica, meno del 10% origina dalle strutture della li-nea mediana, meno del 5% dal midollo spinale. L’am-pia estensione locale spesso determina la difficoltà a eseguire un’asportazione radicale. Frequente è anche la disseminazione leptomeningea (30%) mentre più rare sono le metastasi extra-SNC. La prognosi è più grave rispetto al medulloblastoma, con guarigione in meno del 40% dei casi. PNET localizzati a livello della ghiandola pineale prendono il nome di pineoblastomi e sono associati a miglior prognosi. Malgrado l’origine embrionale comune e la somiglianza istologica con il medulloblastoma, vi è una radicale differenza dal pun-to di vista clinico che negli anni ha generato controver-sie per la classificazione di questa categoria di neo-plasie, nel tentativo di valutarne meglio le eterogenee caratteristiche biologico-molecolari (Fangusaro et al.,
Figura 2. La via di trasduzione del segnale di Hedgehog. A. In assenza del ligando Hh, il recettore transmembrana (PTCH) inibisce l’azione del recettore transmembrana smoo-thened (SMO), provocando il processamento proteolitico delle proteine GLI (fattori di trascrizione oncogenici glioma-asscoiati) attraverso un regolatore negativo (SUFU), mantenendo quindi la via inattiva.B. La via è attivata quando Hh si lega a PTCH sulla superficie cellulare; entrambe le molecole vengono internalizzate (PTCH viene degradato) permettendo a SMO di attivare la via di segnale a valle. I fattori di trascrizione GLI vengono rilasciati da SUFU e processati nelle loro forme attive, traslocano nel nucleo e promuovono la trascrizione di geni target Shh (da Kieran et al., 2014, mod.).

139
Tumori cerebrali: quali sfide, quali progressi
2010). Le valutazioni di next generation sequencing hanno completamente rivoluzionato la classificazione dei PNET (Sturm et al., 2016). Un esempio è l’ETAN-TR (embryonal tumor with abundant neuropil and true rosettes), riconosciuto come variante istologica a prognosi peggiore. Nei PNET la RT sul letto tumorale appare inevitabile anche nei bambini di età inferiore ai 3 anni, associata a CT (i farmaci più utilizzati sono nitrosouree, procarbazina, ciclofosfamide, vincristina, cisplatino e carboplatino). La HART e la CT mielo-ablativa sembrerebbero avere un impatto favorevole sulla prognosi (Massimino et al., 2013). È stata recen-temente identificata una nuova entità molecolare e clinico-patologica chiamata ETRM (embryonal tumor with multilayered-rosettes), che include ependimobla-stoma, medulloepitelioma ed ETANTR, è associata soprattutto a pazienti molto giovani ed è caratterizzata da prognosi severa (Korshunov et al., 2010).
EpendimomaL’ependimoma rappresenta circa il 10% dei tumori pe-diatrici del SNC. Deriva dall’ependima ventricolare o dai residui ependimali intraparenchimali. Frequente-mente localizzato nella fossa cranica posteriore con origine dal pavimento/tetto del IV ventricolo o dall’an-golo ponto-cerebellare, più raramente può presentar-si a livello sovratentoriale o nel midollo spinale; rari sono anche gli ependimomi extrassiali. La WHO distingue:• ependimomamixopapillare(grado IWHO,localiz-
zazione midollare);• ependimomaclassico(gradoIIWHO);• ependimomaanaplastico(gradoIIIWHO);• subependimoma (grado I WHO, presente quasi
esclusivamente nell’adulto).La radicalità chirurgica ha importante significato pro-gnostico con sopravvivenza a 5 anni, che passa dal 70-80% in caso di resezioni quasi complete, al 30% in presenza di residui macroscopici di malattia (Mas-simino et al., 2009). Data la frequente infiltrazione del pavimento del IV ventricolo una chirurgia radicale si associa a morbilità maggiore, pertanto la radicalità chirurgica può anche essere ottenuta in più sedute operatorie. L’istologia anaplastica sembra correlata a una prognosi peggiore. Evidenze cliniche indica-no che l’ependimoma comprenderebbe in realtà un gruppo eterogeneo di neoplasie; emerge pertanto la necessità di stratificare il trattamento in base a grup-pi prognostici di rischio che tengano conto anche di fattori biologico-molecolari, soprattutto valutandoli in differenti gruppi di età e a seconda delle sedi di in-sorgenza dal momento che la distinzione prognostica secondo il grado istologico non sempre rispecchia l’andamento clinico della malattia (Mansur, 2013).Lo standard terapeutico negli ependimomi intracra-nici è la RT focale alla dose di 59,4 Gy sul letto tu-morale; nel protocollo SIOP attualmente in corso è
in studio anche l’applicazione di una sovradose sul residuo di malattia, già sperimentata nel protocollo italiano AIEOP (Associazione Italiana Ematologia ed Oncologia Pediatrica) (manoscritto in pubblicazione) e una riduzione a 54 Gy per i pazienti < 18 mesi. Il ruolo della CT, valutato nell’ambito di trial clinici in ag-giunta alla RT, sarebbe quello di ridurre le dimensioni della neoplasia o di ottenere una migliore definizione dal parenchima sano circostante tale da consentire un reintervento possibilmente radicale. La CT è stata inoltre usata nel contesto di specifici trial nei pazienti < 3 anni allo scopo di evitare/posticipare la RT, ma i risultati sono stati inferiori all’atteso (Massimino et al., 2011). Attualmente è in corso uno studio randomizza-to COG (Children’s Oncology Group), volto a valutare il ruolo della CT adiuvante in pazienti con ependimo-ma trattati con chirurgia e RT. In Europa è in corso dal 2015 uno studio SIOP con stratificazione dei pazienti in tre gruppi di rischio: • gruppoI:pazienti> 1annoconresezionecomple-
ta ed ependimoma grado II-III candidati a ricevere RT conformazionale e successiva randomizzazio-ne alla CT di mantenimento (vincristina, ciclofosfa-mide, etoposide, cisplatino) o solo follow-up;
• gruppoII:pazienti> 1 annoconresiduodimalat-tia i quali ricevono CT adiuvante (randomizzati per l’utilizzo di Metotrexate ad alte dosi), successivo reintervento sul residuo se fattibile e quindi RT con eventuale boost;
• gruppo III:bambini< 1 annochericevono laCTstandard (vincristina, ciclofosfamide, etoposide, cisplatino) e vengono randomizzati all’aggiunta di valproato (inibitore dell’istone deacetilasi).
Studi recenti hanno identificato sottotipi molecolari di-stinti di ependimoma (Witt et al., 2011; Pajtler et al., 2015) come specificato nella Figura 3 (Gaijar et al., 2015; Parker et al., 2014). Il trattamento alla recidiva, che può essere anche tardiva (oltre i 5-10 anni), vede come principale strumento la chirurgia completa se-guita eventualmente da re-irradiazione (Bouffet et al., 2012).
Gliomi
Gliomi maligniIncludono l’astrocitoma anaplastico (grado III WHO) e il glioblastoma multiforme (grado IV WHO). Costi-tuiscono il 10% delle neoplasie del SNC in età pedia-trica, con una sopravvivenza nel bambino del 25% a 5 anni, di poco superiore a quella degli adulti e una aspettativa di vita media non superiore ai 20 mesi dal-la diagnosi (MacDonald et al., 2011). Spesso insorgono in sede sovratentoriale, più rara-mente a livello cerebellare, con un pattern di crescita infiltrante, elevata velocità di accrescimento e frequenti recidive dopo il trattamento. Per via di questa modalità di accrescimento, una chirurgia radicale spesso non è

140
E. Schiavello et al.
fattibile e vengono invece effettuate asportazioni par-ziali o, in alcuni casi, solo una biopsia. Il trattamento standard, indipendentemente dalla sede di insorgen-za, è la RT focale (50-55 Gy). La RT craniospinale è indicata solo in presenza di metastasi midollari/liquo-rali. L’utilizzo della CT dà ancora risultati deludenti, sebbene qualche beneficio sia stato ottenuto con l’as-sociazione di vincristina/nitrosuree o procarbazina/antimetaboliti, documentando un miglioramento della sopravvivenza rispetto alla sola associazione chirur-gia + RT. L’associazione di temozolomide (TMZ) + RT, ampiamente impiegata nei gliomi maligni dell’adulto, viene adottata con minor successo anche in ambito pediatrico, malgrado la differente metilazione dell’enzi-ma metilguanina-metiltransferasi, la cui inattività è alla base della risposta alla terapia nell’adulto. Un recente studio statunitense (Cohen et al., 2011) ha documen-tato come l’associazione RT + TMZ non migliori l’out-come rispetto a studi precedenti, nei quali venivano impiegati diversi regimi di CT. Sono attualmente in fase di elaborazione i dati relativi a uno studio multicentrico internazionale (HERBY BO2504) coordinato in Italia dal nostro Istituto, nel quale pazienti pediatrici affetti da glioma maligno venivano randomizzati a ricevere, oltre al trattamento standard, (RT/TMZ) bevacizumab (anti-corpo monoclonale anti-VEGF) durante la RT e nella fase post-radiante. L’utilizzo di farmaci a dosi mieloa-blative, seguito da rescue con cellule staminali perife-riche, è oggi riservato a bambini < 3 anni, se inseriti in
protocolli specifici. Recentemente sono stati identificati almeno 6 sottogruppi epigeneticamente distinti di glio-blastoma dell’età pediatrica (per approfondimento cfr Tab. II), con caratteristiche clinico-biologiche proprie, sulla base di specifiche alterazioni genomiche/epige-netiche (Tab. II), per le quali non esistono ancora al momento terapie specifiche. Dati preliminari da case report di singole istituzioni hanno però confermato l’ef-ficacia dell’inibitore della proteina mutata BRAF-V600 in pazienti pediatrici con recidive di gliomi maligni di basso/alto grado, che presentino questa mutazione (Bautista et al., 2014). È in fase di apertura anche pres-so il nostro Istituto uno studio multicentrico di fase II, con un inibitore di BRAF-V600 in pazienti con tumori solidi in stadio avanzato positivi alla mutazione. Infine, sempre nell’ambito delle terapie target, è in apertura uno studio multicentrico di fase I, per valutare afatinib (inibitore tirosinchinasico del recettore del fattore di crescita epidermoidale (EGFR) e del recettore epider-moidale 2 dell’uomo (HER2) che, a differenza degli ini-bitori di prima generazione, inibisce la tirosinchinasi in maniera irreversibile) nei bambini con tumori recidivati/refrattari.
Gliomi del tronco dell’encefaloRappresentano il 10-15% dei tumori intracranici e il 20% dei tumori della fossa posteriore. Sono in gran parte astrocitomi a diverso grado di malignità, che in-
Figura 3. Sottotipi di ependimoma (da Gaijar et al., 2015, mod.).
141
Tumori cerebrali: quali sfide, quali progressi
filtrano estesamente il tron-co cerebrale. Si distinguono principalmen-te tre tipi di tumore:• diffuso, infiltrante il tron-
co in maniera omogenea (DIPG – diffuse intrinsic pontine glioma). Rap-presenta il 10% delle ne-oplasie pediatriche del SNC e costituisce l’80% dei gliomi del tronco en-cefalico;
• localizzato (focale) conun nodulo iperdenso o cistico;
• esofitico, aggettante nellume del IV ventricolo o situato all’angolo ponto-cerebellare o cervicomi-dollare.
La terapia dipende dalla lo-calizzazione anatomica. I tumori esofitici, non diffusi né infiltranti, sono spesso gliomi a basso grado di ma-lignità e possono essere cu-rati con la sola chirurgia. CT e RT vengono impiegate nei gliomi del tronco non-DIPG in casi ristretti e selezionati (Vanan e Eisenstat, 2015). Per i DIPG invece, dato l’ele-vato rischio chirurgico anche correlato alla sola biopsia, senza alcuna modificazione della prognosi per exeresi più ampie, è indicato tratta-re solo con RT (54 Gy). Per questi pazienti non esiste un trattamento standard, fatta eccezione per la RT, la cui dose è mutuata da altre ma-lattie, quali i gliomi maligni non-pontini, l’ependimoma o la sovradose del medullo-blastoma.La CT associata alla RT ri-mane tuttora in fase speri-mentale, poiché né la CT adiuvante a dosi standard, né quella mieloablativa sono state in grado di modificar-ne la prognosi (Massimino et al., 2008). Approcci spe-rimentali, come la HART o l’immunoterapia con inter-feron, sono stati deludenti. Ta
bel
la II
. Sot
togr
uppi
di g
liom
i mal
igni
ped
iatri
ci b
asat
i sui
risu
ltati
di m
etila
zion
e de
l Ger
man
Can
cer R
esea
rch
Cen
ter (
DFK
Z) (d
a G
ajja
r et a
l., 2
015,
mod
.).
So
tto
gru
pp
i di g
liom
i m
alig
ni
K27
G34
IDH
RT
K-I
(rece
ptor
tyro
sine
ki
nase
I)
Mes
ench
imal
eP
XA
(x
anto
astr
oci
tom
a p
leo
mo
rfo
)-L
IKE
Età
di i
nsor
genz
aB
ambi
niA
dole
scen
ti/gi
ovan
i adu
ltiA
dole
scen
ti/gi
ovan
i adu
ltiB
ambi
ni,
adol
esce
nti/g
iova
ni
adul
ti
Ado
lesc
enti/
giov
ani a
dulti
Bam
bini
Sed
iS
trut
ture
line
a m
edia
na:
cerv
elle
tto, p
onte
, tal
amo
e m
idol
lo
Em
isfe
ri ce
rebr
ali
Em
isfe
ri ce
rebr
ali
(>fron
tale/
parie
tale
)
Em
isfe
ri ce
rebr
ali
Em
isfe
ri ce
rebr
ali
Em
isfe
ri ce
rebr
ali
Prin
cipa
li al
tera
zion
iH
3.3
o H
3.1
Mut
azio
ne K
27M
utaz
ione
TP
53M
utaz
ione
AT
RX
Am
plifi
cazi
one
PD
GF
RA
Mut
azio
ne A
CV
R1
(pon
te)
Mut
azio
ne F
GF
R1
(tal
amo)
H3.
3M
utaz
ione
G
34M
utaz
ione
T
P53
Mut
azio
ne
ATR
X
Mut
azio
ne IH
D1
o ID
H2
Mut
azio
ne T
P53
Mut
azio
ne
ATR
X
Am
plifi
cazi
one
PD
GF
RA
Mut
azio
ne T
P53
Del
ezio
ne C
DK
N2A
/C
DK
N2B
Am
plifi
cazi
one
EG
FR
Mut
azio
ne N
F1
Mut
azio
ne T
P53
Del
ezio
ne C
DK
N2A
/CD
KN
2BA
mpl
ifica
zion
e E
GF
RA
mpl
ifica
zion
e P
DG
FR
A
Mut
azio
ne B
RA
F
V60
0ED
elez
ione
CD
KN
2A
Sop
ravv
iven
za m
edia
na6
mes
iA
nno
> 2ann
i1
anno
1 an
no> 4 ann
i
Freq
uent
emen
te
glio
mi s
econ
dari
La p
rese
nza
dell’
ampl
ifica
zion
e di
EG
FR
, P
DG
FR
e M
YC
N è
ass
ocia
ta
in tu
tti i
grup
pi a
un
outc
ome
pegg
iore
.

142
E. Schiavello et al.
I risultati delle terapie ad oggi disponibili rimangono comunque sconfortanti con i migliori valori di soprav-vivenza globale mediana pari a 12 mesi. Senza RT, la sopravvivenza mediana dei bambini affetti da DIPG è di circa 4 mesi. Allo scopo di migliorare la sopravvi-venza di questo gruppo di pazienti si stanno facendo molti sforzi, a livello nazionale e internazionale. Per quanto riguarda la nostra esperienza istituzionale dal 2006 al 2009, abbiamo partecipato a uno studio in-ternazionale per i DIPG alla diagnosi, adottando la combinazione Nimotuzumab (anticorpo monoclona-le umanizzato anti-EGFR/ERBB1 che risulta overe-spresso/amplificato nei DIPG e con capacità di oltre-passare la barriera emato-encefalica) + RT. Abbiamo trattato 37 bambini con risultati paragonabili a quelli già riportati in letteratura. Dal 2009 a livello istituzio-nale adottiamo l’associazione nimotuzumab/vinorel-bina in combinazione con la RT e la re-irradiazione dei pazienti in progressione, nel contesto di uno stu-dio pilota di fase II non randomizzato, con risultati mi-gliori rispetto all’atteso con una sopravvivenza libera da progressione (PFS) mediana di 8,5 mesi e una so-pravvivenza (OS) mediana di 15 mesi (Massimino et al., 2014). L’unione degli sforzi a livello internazionale ha dato vita a un gruppo cooperativo europeo (SIO-Pe-DIPG-network), volto a valutare retrospettivamen-te ciò che storicamente è stato fatto in ciascun paese per i bambini affetti da DIPG e a promuovere studi prospettici. Studi di whole exome/genome sequen-cing hanno documentato che più dell’80% dei DIPG presentano la mutazione negli istoni H3.1 e H3.3 (analogamente ad alcuni gliomi maligni talamici) che comportano ipometilazione di K27, H3K27M. Questa mutazione sarebbe l’evento oncogenico iniziale nei DIPG. Oltre a studi preclinici e clinici volti a indagare l’efficacia di terapie mirate rivolte ai principali target patogenetici evidenziati, sono attualmente in fase di valutazione nell’ambito di trial clinici l’immunoterapia (allo scopo di indurre una risposta immune sistemica contro il tumore, attraverso l’inoculo di un vaccino di-retto contro antigeni tumore-specifici), la convection-enhanced delivery (somministrazione intratumorale mediante cateteri posizionati nel tronco encefalico di agenti chemioterapici o radioattivi) e frazionamenti di radioterapia non convenzionali (Tisnado et al., 2016; Nikbakht et al., 2016; Vitanza e Cho, 2016).
Gliomi a basso gradoPotenzialmente insorgono in qualsiasi zona del SNC, ma nei bambini le sedi più frequenti sono la fossa po-steriore (astrocitoma cerebellare) e le vie ottiche. La resezione completa, generalmente possibile per le localizzazioni emisferiche o cerebellari, è sempre auspicabile, poiché curativa nella quasi totalità dei casi. Le neoplasie emisferiche profonde/della linea mediana, le lesioni ottico-ipotalamiche e del tronco cerebrale possono invece essere asportate solo par-zialmente, in alcuni casi è fattibile solo una biopsia. La
CT è utilizzata soltanto nei gliomi che crescono dopo resezione chirurgica o in quelli considerati non opera-bili, previa diagnosi istologica. I farmaci generalmente impiegati sono: carboplatino/vincristina, cisplatino/etoposide, vinblastina.La RT non è il trattamento di prima scelta sia per l’otti-ma prognosi di questi pazienti, pur in assenza di trat-tamento radiante, sia per la possibilità di insorgenza di secondi tumori, soprattutto quando i gliomi a basso grado compaiono nel contesto di sindromi genetiche, quali le facomatosi. La RT deve pertanto essere ri-servata a casi particolari e ben selezionati. I gliomi a basso grado hanno un’ottima prognosi, con una OS a 5anni> 90%eunaEFSa10anni> 70%(NageswaraRao e Packer, 2014).
Gliomi delle vie otticheCostituiscono lo 0,5-5% dei tumori cerebrali pe-diatrici, con un’incidenza di 1:100.000 e picco tra i 4-6 anni. Nel 15-20% dei pazienti si associano a neu-rofibromatosi. In genere si tratta di tumori astrocitari a basso grado, che possono comportarsi aggressi-vamente producendo strabismo, cecità e, talvolta, esoftalmo progressivo. In casi selezionati vi è indica-zione alla CT, raramente alla RT. Un atteggiamento di attesa è indicato perlomeno inizialmente, poiché si tratta di tumori che possono rimanere stazionari per un lungo periodo soprattutto quando associati a NF1. Il trattamento deve essere effettuato alla progressio-ne o quando la lesione diventa sintomatica; la CT ha permesso di rimandare o omettere definitivamente la RT, largamente impiegata in passato. Qualora si ren-da necessario il trattamento radiante, per la progres-sione della malattia, le tecniche attuali permettono di conformare la dose (ridotta a 45 Gy) con relativo risparmio di tessuti sani (Fried et al., 2013). La so-pravvivenzaa5anniè> 80%,masonoconsiderevolile sequele nei pazienti più giovani.
Astrocitoma cerebellareRappresenta il 20% dei tumori del SNC pediatrici e 1/3 dei tumori della fossa posteriore; esordisce tra i 3-10 anni, spesso si tratta di un astrocitoma pilocitico. È localizzato in un emisfero cerebellare e può invade-re il verme cerebellare, infiltrare i peduncoli e occlu-dere l’acquedotto di Silvio, determinando idrocefalo. La chirurgia completa, se tecnicamente possibile, è curativa. Studi recenti hanno documentato che i gliomi a bas-so grado hanno alterazioni genomiche che correla-no con un loro tropismo per diverse aree anatomiche del SNC e quindi con la possibilità di ottenere una radicalità chirurgica. I principali istotipi con le relative caratteristiche cliniche e genetico-molecolari sono ri-assunte nella Tabella III.
Terapie future per i gliomi a basso grado• terapietarget dirette alla duplicazione di BRAF o della
via di MAPK/ERK: in corso studi di fase II (con se-lumetinib) sui gliomi a basso grado alla recidiva/pro-
143
Tumori cerebrali: quali sfide, quali progressi
Tabella III. Principali varianti istopatologiche dei gliomi a basso grado e relative caratteristiche cliniche, topografiche e molecolari.
Grado WHO
Caratteristiche Sede Mutazioni
Astrocitomi a basso grado
Astrocitoma Pilocitico I Prognosi eccellente, non recidiva dopo chirurgia completa, possibile progressione del residuo, rara disseminazione e trasformazione maligna
Più frequenti: vie ottiche (30-50% dei pazienti con NF1) e fossa posteriore(>sporadici)
Mutazione germinale NF1 (nei pazienti con NF)Duplicazione BRAF e formazione trascritto di fusione KIAA1549-BRAF(> 90%degliastrocitomipilocitici cerebellari non NF1)Alterazioni attivanti il pathway RAS-RAF-MEK-ERK
Astrocitoma Pilomixoide II Variante dell’astrocitoma pilocitico con decorso più aggressivo
Come astrocitoma pilocitico
Duplicazione BRAF e trascritto di fusione KIAA1549-BRAF
Xantoastrocitoma Pleomorfo II Raro Corticale (> temporale)
Mutazione BRAF V600EDelezione CDKN2A
Astrocitoma Diffuso II Pattern infiltranteFigure mitotiche rare/assenti (se numerose à astrocitoma anaplastico)Geneticamente distinto da quello dell’adulto
Fossa posteriore, regioni sovratentoriali
Rare mutazioni IDH1-IDH2 (a differenza dell’adulto)Riarrangiamento MYB-MYBL1 (localizzazioni emisferiche)Mutazione BRAFV600EAlterazioni FGFR1Mutazione H3K27M (nei DIPG e nei gliomi maligni della linea mediana)I gliomi K27M + hanno decorso più aggressivo a prescindere dall’istologia osservata
Astrocitoma Subependimale a cellule giganti (SEGA)
I Non infiltranti.Sempre associati alla sclerosi tuberosa (20% dei pazienti con sclerosi tuberosa)
In prossimità dei ventricoli laterali
Mutazioni germinali TAC1 o TAC2 con conseguente attivazione della via di mTOR
Oligodendrogliomi a basso grado
II Profilo genetico diverso da quello degli adulti
Ubiquitari Le mutazioni tipiche dell’adulto (IDH1-IDH2, codelezione 1p19q, mutazione TERT) sono assenti nei bambini ma presenti negli adolescenti/giovani adulti.Mutazioni caratteristiche poco note, data la rarità pediatrica (duplicazione gene FGFR1 in piccole coorti)
Tumori glioneuronali
Tumore neuronale disembrioplastico (DNET)
I Misto: componenti gliali + neuronali
Emisferi cerebrali (>lobotemporale)e giunzione cervico-midollare
Mutazione BRAFV600EAlterazione FGFR1
Ganglioglioma I Misto: componenti gliali + neuronali
Emisferi cerebrali (>lobotemporale)e giunzione cervico-midollare
Mutazione BRAFV600E

144
E. Schiavello et al.
gressione. Farmaci target contro BRAF V600E sono attualmente oggetto di uno studio clinico multicentrico;
• farmaci target contro AKT/mTOR (everolimus). Everolimus è già approvato dalla US-FDA per il trattamento dei SEGA (astrocitomi subependimali a cellule giganti) nei pazienti con sclerosi tubero-sa. La sua efficacia è stata valutata nell’ambito di uno studio di fase II in pazienti con gliomi a basso grado alla progressione/recidiva, nel quale è stata documentata una risposta > 25%. Studi ulterioridovranno documentarne l’efficacia in prima linea.
Considerando la buona prognosi con le terapie attual-mente a disposizione, gli sforzi futuri dovranno essere rivolti a ridurre le tossicità tardive; le terapie target po-tranno trasformare quella porzione di gliomi a basso grado, che tendono a progredire/recidivare in malattie croniche (Gaijar et al., 2015; Garcia et al., 2016).
Neoplasie rare
Tumori dei plessi corioideiI tumori dei plessi corioidei sono patologie rare (cir-ca 3% delle neoplasie cerebrali pediatriche), posso-no verificarsi a ogni età, ma rappresentano il 10-20% delle neoplasie nei bambini < 1 anno. Possono esse-re benigni (papillomi), borderline (papillomi atipici) o maligni (carcinomi). Spesso esordiscono con idroce-falo; si localizzano più frequentemente nei ventrico-li laterali, ma possono svilupparsi anche nel quarto ventricolo e infiltrare il parenchima circostante. Il papilloma dei plessi corioidei, più frequente del carcinoma, rappresenta meno dell’1% dei tumori intracranici del bambino e in genere ha andamento benigno; la sua intensa vascolarizzazione determina l’elevato rischio emorragico della chirurgia. Dato che il principale fattore prognostico è rappresentato dalla radicalità chirurgica, essa deve essere raggiunta an-che in più sedute considerando l’estensione iniziale di malattia e il rischio emorragico. La resezione comple-ta è curativa per i papillomi e per la maggioranza dei papillomi atipici; per questi ultimi occorre comunque un attento follow-up, dato il maggior rischio di recidive (Sun et al., 2014). Per quanto riguarda il carcinoma dei plessi, poiché la maggior parte dei pazienti ha meno di 3 anni, il trattamento consiste, dopo la chirurgia, nella sommi-nistrazione di CT adiuvante per tutti i pazienti e RT a seconda dell’età e dell’estensione post-chirurgica della malattia. La prognosi dei carcinomi dei plessi co-rioidei è poco favorevole con sopravvivenza a cinque anni attorno al 40% per i pazienti con residuo.Il carcinoma dei plessi corioidei è associato in circa il 50% dei casi alla sindrome di Li-Fraumeni (Tabori et al., 2010), per cui si raccomanda di raccogliere un’at-tenta anamnesi familiare e patologica remota in modo da considerare, ove possibile e nei soggetti con muta-zione, di non effettuare la RT, così da ridurre il rischio
di tumori radioindotti. Occorre inoltre attuare program-mi di screening per la diagnosi precoce di altri tumori per il paziente e per i familiari affetti.
Tumore teratoide-rabdoide atipico (AT/RT)È una neoplasia maligna molto aggressiva di de-rivazione embrionaria, caratteristica del bambino < 3 anni. Insorge prevalentemente in fossa cranica posteriore e negli emisferi cerebrali, la presentazione è spesso metastatica alla diagnosi con localizzazioni leptomeningee o liquorali. È associato alla delezione del cromosoma 22 e alla conseguente inattivazione somatica biallelica di SMARCB1-INI, gene oncosop-pressore regolatore della proliferazione/differenzia-zione cellulare. La perdita dell’espressione nucleare della proteina valutata in immunoistochimica consen-te la diagnosi differenziale con le altre neoplasie di origine embrionaria. La chirurgia deve mirare all’exe-resi completa, quando possibile. L’impiego di schemi di trattamento intensi comprendenti CT intratecale e sistemica ad alte dosi/mieloablativa e RT ha permes-so di ottenere un miglioramento della prognosi con una sopravvivenza attuale > 50% (Benesch et al.,2014).Circa un terzo dei pazienti con AT/RT presenta una mutazione germinale di SMARCB1 che definisce la sindrome da predisposizione ai tumori rabdoidi; sebbene la maggior parte di queste mutazioni in-sorgano de novo, sono riportati alcuni casi familia-ri di trasmissione costituzionale della mutazione di SMARCB1. Sono fondamentali un’accurata anam-nesi e la ricerca di tale mutazione nei pazienti affetti, in modo da stabilire la necessità di testare eventual-mente i familiari e di mettere in atto un adeguato programma di screening (Sredni e Tomita, 2015). La sopravvivenza globale è inferiore a 12 mesi con un outcome peggiore per i bambini più piccoli, in pre-senza di metastasi o di sindrome da predisposizione al tumore rabdoide. Sono attualmente in fase di valu-tazione farmaci target, tra cui gli inibitori delle aurora-chinasi all’interno di uno studio di fase II per pazienti con AT/RT ricaduto/refrattario.
Neoplasie germinaliSi suddividono in:• germinomipuri;• tumoriacellulegerminalinongerminomatosi(tu-
more del sacco vitellino, carcinoma embrionale, coriocarcinoma, teratoma maturo/immaturo, tumo-ri misti).
Rappresentano il 3,5% delle neoplasie cerebrali pe-diatriche, con un’età media di insorgenza attorno ai 12 anni. Insorgono prevalentemente lungo la linea mediana, nella sede della ghiandola pineale, in re-gione sovrasellare e nel terzo ventricolo. L’interessa-mento del peduncolo ipotalamo-ipofisario determina spesso la comparsa di deficit endocrinologici fino al panipopituitarismo. A differenza delle neoplasie ger-

145
Tumori cerebrali: quali sfide, quali progressi
minali extra-SNC, in quelle cerebrali si distinguono neoplasie secernenti da quelle non secernenti in base alla presenza/assenza di livelli patologici di αFP e/o βHCG nel siero e/o nel liquor. Le neoplasie germinali secernenti sono tra le poche neoplasie per le quali la diagnosi viene formulata sulla base dei dosaggi dei marcatori sierici/liquorali. Per le neoplasie germinali non secernenti la diagnosi istologica è invece man-datoria e consiste nella sola biopsia data la chemio-radiosensibilità. La chirurgia può essere anche indi-rizzata al trattamento dell’idrocefalo, in relazione alla sede di insorgenza del tumore e costituisce inoltre un momento fondamentale nel trattamento dei teratomi maturi non radio-chemioresponsivi e, raramente, per i residui neoplastici al termine del trattamento medico. La CT, quando indicata, utilizza farmaci mutuati dal trattamento dei germinomi extracerebrali (derivati del platino, etoposide e bleomicina). Poiché la RT focale è stata gravata da un eccesso di ricadute ventricolari, attualmente è consigliata l’associazione di CT + RT sul sistema ventricolare con sopravvivenze superio-ri al 90%. Per i tumori germinali secernenti alla RT craniospinale va associata la CT. In caso di malattia metastatica, nei tumori germinali non secernenti, la RT craniospinale da sola è curativa. Mentre i germi-nomihannotassidisopravvivenza> 90%,ipazien-ti affetti da neoplasie germinali non germinomatose hanno una sopravvivenza a 5 anni tra il 60 e il 70% (Cheng et al., 2016). Considerata la buona prognosi di queste patologie, gli sforzi attuali sono rivolti alla ridu-zione delle sequele tardive, ad esempio impiegando moderne tecniche di radioterapia conformazionale a intensità modulata per definire meglio il volume target risparmiando i tessuti circostanti. Oggetto di studio è la terapia con protoni (Kortmann, 2014).
Sindromi predisponenti allo sviluppo di neoplasie cerebrali (Tab. IV)
Neurofibromatosi Tipo 1 (NF1)L’astrocitoma pilocitico è il tumore più frequente, ve-rificandosi nel 15% dei pazienti affetti da NF1, con sede preferenziale a livello delle vie ottiche, o in percentuale minore nel tronco encefalico. Anche l’a-strocitoma diffuso può presentarsi in questi pazienti. Il trattamento oncologico dell’astrocitoma pilocitico nei pazienti con NF1 va attentamente ponderato, in considerazione della possibile insorgenza di lesioni gliali successive nel tempo, distinguendo le lesioni di natura gliale dalle alterazioni tipiche dei pazienti con neurofibromatosi (UBOs) che non necessitano di un trattamento oncologico e riservando la chirurgia ed eventualmente la CT nei casi in sicura progressione evolutiva sintomatica. La RT riveste un ruolo ancora più marginale, in quanto è documentato un aumen-tato rischio dei pazienti con NF1 di sviluppare gliomi radio-indotti e tumori maligni delle guaine nervose pe-
riferiche (MPNST). Tra i tumori benigni si riscontrano neurofibromi spinali (40%) il cui trattamento si avvale eventualmente della sola chirurgia.
Neurofibromatosi Tipo 2 (NF2)Virtualmente tutti i pazienti affetti da NF2 sviluppano schwannomi (grado I WHO) bilaterali entro i 30 anni di età, con conseguente ipoacusia o sordità. Gli schwannomi vestibolari sono solitamente responsivi, specialmente per quanto riguarda la funzione uditiva, al trattamento con anticorpo monoclonale anti VEG-FR (bevacizumab). Circa il 60% dei pazienti sviluppa tumori spinali, nella maggior parte dei casi schwan-nomi. Circa il 50% dei pazienti con NF2 sviluppa meningiomi che, a differenza degli schwannomi, non sembrerebbero beneficiare della terapia con Beva-cizumab. Nei pazienti con NF2 sono descritte anche lesioni astrocitarie ed ependimomi.
Sindrome da predisposizione al tumore rabdoideÈ caratterizzata dalla comparsa di tumori rabdoidi al di fuori del SNC o a livello dello stesso (AT/RT) che si svi-luppano nei primi anni di vita e hanno, rispetto ai casi sporadici, maggiore aggressività. Circa un terzo dei pa-zienti affetti da AT/RT ha una mutazione germinale nel gene SMARCB1. In presenza di una mutazione germi-nale nel paziente è giustificata la consulenza genetica.
Sindrome del carcinoma basocellulare nevoide (Sindrome di Gorlin)Le caratteristiche oncologiche correlate a questa sin-drome sono la presenza di multipli carcinomi baso-cellulari e cheratocisti mascellari (più frequenti nei pazienti con mutazione di PTCH1) e un aumentato rischio di sviluppare medulloblastoma in età pedia-trica, in particolare nei primi due anni di vita, rischio più elevato nei pazienti con mutazione SUFU (30%) rispetto ai pazienti PTCH1 mutati (2%). Gli istotipi più rappresentati sono il medulloblastoma desmoplasti-co/nodulare e ad estesa nodularità; emerge quindi l’indicazione a proporre una valutazione genetica in tutti i pazienti affetti da uno di questi istotipi ad esordio nei primi tre anni di vita. Inibitori della via di trasdu-zione di Hedgehog vengono impiegati in pazienti con sindrome di Gorlin per trattare carcinomi basocellulari non operabili.
Sclerosi tuberosaCaratterizzata dalla comparsa di amartomi benigni nel SNC, cute, retina, cuore, polmone e rene; le lesio-ni SNC sono le principali cause di morbidità e mor-talità in questi pazienti. I tuberi corticali sono spesso già presenti alla nascita mentre i noduli subependi-mali si sviluppano nei primi anni di vita; inoltre 6-14% dei pazienti sviluppa SEGA. Il quadro neurologico di questi bambini è spesso caratterizzato da epilessia,
146
E. Schiavello et al.
autismo e ritardo mentale. I tuberi corticali e i SEGA possono essere trattati chirurgicamente quando le dimensioni e/o i sintomi a essi correlati non siano di-versamente gestibili. Poiché i geni TSC1/2 agiscono come soppressori indiretti di mTOR (mammalian tar-get of rapamycin), farmaci inibitori di m-TOR (everoli-mus) sono stati studiati e recentemente approvati per il trattamento dei SEGA.
Sindrome di Von Hippel-LindauCaratterizzata dalla formazione di emangioblastomi, tumori vascolari di basso grado (grado I WHO), a li-vello cerebellare, del tronco cerebrale, del midollo e retina. Il trattamento di elezione è la chirurgia mentre
la RT è riservata a casi selezionati, non passibili di resezione chirurgica e sintomatici.
Sindrome di Li-FraumeniLa metà dei pazienti sviluppa un tumore nei primi 30 anni di vita e il 90% prima dei 60 anni. Complessiva-mente il 14% dei pazienti con sindrome di Li-Frau-meni sviluppa una neoplasia cerebrale, con maggior rischio nei primi 10 anni di vita. Gli istotipi maggior-mente rappresentati sono le neoplasie astrocitarie, il medulloblastoma (prevalentemente la variante SHH) e il carcinoma dei plessi coroidei (il 36-50% dei pa-zienti con carcinoma dei plessi è portatore di una mutazione germinale di p53). Poiché il carcinoma dei
Tabella IV. Sindromi predisponenti lo sviluppo di neoplasie del SNC: caratteristiche genetiche e tumori extra-SNC as-sociati (da Johansson et al., 2015, mod.). AD autosomica dominante, AR autosomica recessiva, GIST tumore gastro-intestinale stromale.
Sindrome Trasmissione Incidenza Gene Cromosoma Neoplasie extra-SNC
Neurofibromatosi 1 AD(50%
sporadici)
1/2500 NF1 17q11.2 Neurofibromi, MPNST, amartoma dell’iride, leucemia, GIST, feocromocitoma
Neurofibromatosi 2 AD(50%
sporadici)
1/33000 NF2 22q12 amartomi renali, schwannomi, neurofibromi
Sindrome da predisposizione al tumore rabdoide
AD Prevalenza < 1/1000000
SMARCB1SMARCA4
22q11.2119p13.2
Tumore rabdoide renale
Sindrome di Gorlin AD 1/56-164000 PTCH1
SUFU
9q22.3
10q24.3
Carcinoma basocellulare, cisti mandibolari
Carcinoma basocellulare
Sclerosi tuberosa AD(75%
sporadici)
1/6-12000 TSC1TSC2
9p3416p13.3
Amartomi cute, retina, cuore, polmone e rene
Sindrome Von Hippel LIndau
AD 1/43000 VHL 3p25.3 Emangioblastoma retinico, carcinoma renale, feocromocitoma
Sindrome di Li-Fraumeni AD(7-20%
sporadici)
1/5000 TP53 17p13.1 Sarcomi tessuti molli, tumore mammario, carcinoma adrenocorticale. Rischio di tumore: 70% uomini, 100% donne
Sindrome di Turcot tipo 1 AD 50/100000 MLH1MSH2PMS2MSH6
3p21.32p21
7p22.22p16
Carcinoma colorettale non poliposico, tumori ematologici. Rischio di tumore: 100%
Sindrome di Turcot tipo 2 AD 2-3/100000 APC 5q21 Poliposi adenomatosa del colon familiare
Sindrome melanoma-astrocitoma
AD Circa 200000 negli USA
CDKN2 9p21.3 Melanomi

147
Tumori cerebrali: quali sfide, quali progressi
Bibliografia
Bautista F, Paci A, Minard-Colin V, et al. Vemurafenib in pediatric patients with BRAF V600E mutated high-grade gliomas. Pediatr Blood Cancer 2014;61:1101-3.
Benesch M, Bartelheim K, Fleischhack G, et al. High-dose chemotherapy (HDCT) with auto-SCT in children with atypical teratoid/rhabdoid tumors (AT/RT): a report from the European Rhabdoid Registry (EU-RHAB). Bone Marrow Transplant 2014;49:370-5.
Bouffet E, Hawkins CE, Ballourah W, et al. Survival benefit for pediatric patients with recurrent ependymoma treated with
reirradiation. Int J Radiat Oncol Biol Phys 2012;83:1541-8.
CBTRUS Central Brain Tumor Registry of the United States (2012) Statistical Report 2004-2008.
Cheng S, Kilday JP, Laperriere N, et al. Outcomes of children with central nervous system germinoma treated with multi-agent chemotherapy followed by reduced radiation. J Neurooncol 2016;127:173-80.
Clifford SC, Lannering B, Schwalbe EC, et al. Biomarker-driven stratification of disease-risk in non-metastatic medul-loblastoma: results from the multi-center
HIT-SIOP-PNET4 clinical trial. Oncotarget 2015;6:38827-39.
** Articolo molto rilevante circa la stra-tificazione del rischio prognostico nella terapia del medulloblastoma, consideran-do anche fattori molecolari in uno studio multicentrico europeo.
Cohen KJ, Pollack IF, Zhou T, et al. Temozolo-mide in the treatment of high-grade gliomas in children: a report from the Children’s Oncology Group. Neuro Oncol 2011;13:317-23.
Fangusaro J, Massimino M, Rutkowski S, et al. Non-cerebellar primitive neuroectoder-mal tumors (PNET): summary of the Milan
Box di orientamento
• Cosa sapevamo primaLa prognosi delle neoplasie pediatriche del SNC è influenzata dal grading istologico, dall’entità dell’exe-resi chirurgica e dalla presenza di metastasi. La chirurgia e quando indicate la radioterapia e la chemiote-rapia hanno costituito i capisaldi del trattamento delle neoplasie pediatriche del SNC e sono responsabili del miglioramento della sopravvivenza globale dei bambini affetti da tumore cerebrale, portandola ad oltre il 70% per tutte le istologie.
• Cosa sappiamo adessoLe indagini genomiche rese possibili dalla creazione di gruppi cooperativi internazionali hanno docu-mentato l’esistenza di entità clinico-prognostiche diverse all’interno dello stesso istotipo. Queste impor-tanti osservazioni hanno aperto la strada all’impiego di approcci terapeutici mirati a ciascun sottotipo molecolare di malattia anziché al singolo istotipo, nell’ambito delle principali neoplasie SNC pediatriche (medulloblastoma, gliomi maligni e a basso grado, ependimomi e PNET).Emerge inoltre in modo significativo il ruolo delle sindromi predisponenti nei bambini con tumori cerebrali.
• Per la pratica clinicaLe target therapy sono attualmente oggetto di numerosi studi clinici cooperativi pediatrici i cui risultati, at-tesi negli anni a venire, ne chiariranno la reale efficacia. Attualmente non costituiscono ancora lo standard terapeutico e sono riservate, nella maggior parte dei casi, a pazienti alla recidiva o in progressione dopo le terapie standard. Nell’affrontare il bambino con tumore cerebrale e la sua famiglia è molto importante la ricerca di segni che orientino per la presenza di una sindrome nel paziente e nella famiglia allo scopo di una corretta diagnosi, di un’adeguata impostazione dell’iter oncologico e in termini di prevenzione.
plessi insorge spesso nei primi anni di vita, è molto importante proporre a tutti i pazienti con questa dia-gnosi una consulenza genetica a prescindere dalla presenza di familiarità, anche allo scopo di impostare correttamente il trattamento oncologico.
Sindrome di Turcot-Tipo 1Causata dalla mutazione biallelica di geni implicati nel controllo e nella riparazione degli errori di repli-cazione del DNA e correlati alla HNPCC (hereditary nonpolyposis colorectal cancer). Oltre allo sviluppo di HNPCC, si manifestano neoplasie ematologiche e tumori cerebrali, in particolare gliomi maligni, nelle prime decadi di vita.
Sindrome di Turcot-Tipo 2È caratterizzata dalla comparsa di neoplasie cerebrali
nei primi 20 anni di vita, tipicamente il medulloblasto-ma, in pazienti con poliposi adenomatosa familiare.
Sindrome Melanoma-AstrocitomaÈ caratterizzata dall’associazione familiare di melanoma maligno e tumori SNC. I tumori più frequenti sono me-dulloblastoma, glioblastoma multiforme, ependimoma, glioma, meningioma, neuroblastoma/PNET, schwanno-ma e neurinoma del nervo acustico. Tutti questi tumo-ri originano dalla cresta neurale oppure hanno origine mesenchimale. È necessaria un’attenta sorveglianza dei pazienti e dei loro parenti di primo e secondo grado. Nella Tabella IV sono indicate le principali caratteristi-che genetiche delle sindromi sopra descritte e i tumo-ri extra-SNC a esse associati.

148
E. Schiavello et al.
consensus and state of the art workshop on marrow ablative chemotherapy with hemato-poietic cell rescue for malignant brain tumors of childhood and adolescents. Pediatr Blood Cancer 2010;54:638-40.
Fried I, Tabori U, Tihan T, et al. Optic pathway gliomas: a review. CNS Oncol 2013;2:143-59.
Gaijar A, Bowers Daniel C, Karajannis Mat-thias A, et al. Pediatric brain tumors: inno-vative genomic information is transforming the diagnostic and clinical landscape. JCO 2015;33:2986-9.
** Articolo completo sulle più recenti in-novazioni circa la diagnostica e la terapia del-le principali neoplasie cerebrali pediatriche.
Garcia MA, Solomon DA, Haas-Kogan DA. Exploinig molecular biology for diagnosis and targeted management of pediatric low-grade gliomas. Future Oncology 2016 Apr.
Gatta G, Botta L, Rossi S, et al. Childhood cancer survival in Europe 1999-2007: results of EUROCARE-5 – a population-based study. Lancet Oncol 2014;15:35-47.
Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Annu Rev Pathol 2008;3:341-65.
Johansson G, Andersson U, Melin B. Re-cent developments in brain tumor predispos-ing sindrome. Acta Oncol 2016;55:401-11.
** Interessante revisione sulle principali sindrome predisponenti i tumori cerebrali e implicazioni cliniche.
Kieran MW. Targeted treatment for sonic hedgehog-dependent medulloblastoma. Neuro Oncol 2014;16:1037-47.
Korshunov A, Remke M, Gessi M, et al. Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumors with ependymoblastic ro-settes. Acta Neuropathol 2010;120:253-60.
Kortmann RD. Current concepts and fu-ture strategies in the management of intra-cranial germinoma. Expert Rev Anticancer Ther 2014;14:105-19.
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97-109.
Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Clas-sification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131:803-2.
MacDonald TJ, Aguilera D, Kramm CM. Treatment of high-grade glioma in children and adolescents. Neuro Oncol 2011;13:1049-58.
Mansur DB. Multidisciplinary manage-ment of pediatric intracranial ependymoma. CNS Oncol 2013;2:247-57.
Massimino M, Biassoni V, Miceli R, et al. Results of nimotuzumab and vinorel-bine, radiation and re-irradiation for diffuse pontine glioma in childhood. J Neurooncol 2014;118:305-12.
Massimino M, Buttarelli FR, Antonelli M, et al. Intracranial ependymoma: factors affec-ting outcome. Future Oncol 2009;5:207-16.
Massimino M, Gandola L, Barra S, et al. Infant ependymoma in a 10-year AIEOP (Associazione Italiana Ematologia Oncologia Pediatrica) experience with omitted or defer-red radiotherapy. Int J Radiat Oncol Biol Phys 2011;80:807-14.
Massimino M, Gandola L, Biassoni V, et al. Evolving of therapeutic strategies for CNS-PNET. Pediatr Blood Cancer 2013;60:2031-5.
Massimino M, Spreafico F, Biassoni V, et al. Diffuse pontine gliomas in children: changing strategies, changing results? A mono-institutional 20-year experience. J Neuroncol 2008;87:355-61.
Morfouace M, Shelat A, Jacus M, et al. Pemetrexed and gemitabine as combination therapy for the treatment of group 3 medul-loblastoma. Cancer Cell 2014;25:516-29.
Nageswara Rao AA, Packer RJ. Advances in the management of low-grade gliomas. Curr Oncol Rep 2014;16:39.
Nikbakht H, Panditharatna E, Mikael L, et al. Spatial and temporal homogeneity of driver mutations in diffuse intrinsic pontine glioma. Nat Commun 2016;7:11185.
Packer RJ, Rood BR, MacDonald TJ. Me-dulloblastoma: present concepts of stratifi-cation into risk groups. Pediatr Neurosurg 2003;39:60-7.
Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all cns compartments, histopathological grades, and age groups. Cancer Cell 2015;27:728-43.
Parker M, Mohankumar KM, Punchihewa C, et al. C11orf-RELA fusions drive oncoge-ninic NF-kappaB signalling in ependymoma. Nature 2014;506:451-5.
** Importante articolo circa una nuova entità clinico-molecolare di ependimoma so-pratentoriale.
Robinson GW, Orr BA, Wu G, et al. Vismo-degib exerts targeted efficacy against recur-rent sonic hedgehog-subgroup medulloblas-toma: results from Phase II pediatric brain Tumor consortium studies PBTC-025B and PBTC-032. J Clin Oncol 2015;33:2646-54.
Rudin CM, Hann CL, Laterra J, et al. Treat-ment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009;361:1173-8.
*Articolo capostipite per la terapia con far-maci target nel medulloblastoma.
Rutkowski S, von Hoff K, Emser A, et al. Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol 2010;28:4961-8.
Shou Y, Robinson DM, Amakye DD, et al. A five-gene hedgehog signature developed as a patient preselection tool for hedgehog inhibi-tor therapy in medulloblastoma. Clin Cancer Res 2015;21:585-93.
Sredni ST, Tomita T. Rhabdoid tumor pre-disposition syndrome. Pediatr Dev Pathol 2015;18:49-58.
Sturm D, Orr BA, Toprak UH, et al. New brain tumor entities emerge from mo-lecular classification of CNS-PNETs. Cell 2016;164:1060-72.
** Interessante descrizione della nuova classificazione dei PNET, considerando i fattori bio-molecolari, frutto di una proficua collaborazione tra biologi, patologi e clinici.
Sun MZ, Ivan ME, Oh MC, et al. Effects of adjuvant chemotherapy and radiation on overall survival in children with choroid plex-us carcinoma. J Neurooncol 2014;120:353-60.
Tabori U, Shlien A, Baskin B, et al. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tu-mors. J Clin Oncol 2010;28:1995-2001.
* Articolo rilevante per le implicazioni clini-che legate alla diagnosi di patologie genetiche predisponenti le neoplasie.
Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblasto-ma: the current consensus. Acta Neuropathol 2012;123:465-72.
** Articolo fondamentale per la nuova classificazione in sottogruppi molecolari del medulloblastoma.
Tisnado J, Young R, Peck RY, et al. Con-ventional and advanced imaging of diffuse intrinsic pontine glioma. J Child Neurol 2016 Apr 12.
Vanan MI, Eisenstat DD. dipg in children – what can we learn from the past? Front On-col 2015;5:237.
Vitanza NA, CHO YJ. Advances in the biology and treatment of pediatric central nervous system tumors. Curr Opin Pediatr 2016;28:34-9.
von Bueren AO, von Hoff K, Pietsch T, et al. Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol 2011;13:669-79.
Witt H, Mack SC, Ryzhova M, et al. Delinea-tion of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 2011;20:143-57.
Corrispondenza
Elisabetta SchiavelloSC Pediatria, Fondazione IRCCS Istituto Nazionale dei Tumori, via Venezian 1, 20133 Milano - E-mail: [email protected]




























