Embed Size (px)
Citation preview

(CANCER RESEARCH 56, 2452-2457. May 15, 1996]
Tumor Necrosis Factor a Interferes with the Cell Cycle of Normal andPapillomavirus-immortalized Human Keratinocytes1
Katia B. L. Vieira, David J. Goldstein, and Luisa L. Villa2
Ludwig Institute for Cancer Research, Sà o Paulo, 01509-010, Brazil ¡K.B. L. V., L. L. V.J; Department of Biochemistry, Instituto de QuÃmica, Universidad« de Sao Paulo,CP 20.780 Sao Paulo, Brazil [K. B. L V.]; Department of Obstetrics and Gvnecology, Lombardi Cancer Center, Georgetown University Medical School, Washington DC20007 ¡D.J. G.ÃŒ
ABSTRACT
We have shown that normal and human papillomavirus (HPV) type 16immortalized human foreskin keratinocytes are growth inhibited by tumor necrosis factor a (TNF-a), whereas HPV-18- and SV40-immortalized
keratinocytes are resistant to this cytokine (1). In this report, we investigated the expression of mitotic regulatory proteins, such as cyclin A, cyclinB, and p34cdc2. After exposure to TNF-a, normal and HPV-16-immortaI-
ized cells exhibited a dramatic decrease in the expression of these proteins.In contrast, no alteration in the levels of these proteins was observed aftertreatment of the resistant cell lines, as well as two HPV-positive cervical
carcinoma cell lines. Expression of cyclin E does not seem to be modulatedby TNF-a in any of the cells tested. On the other hand, cyclin D, expression is slightly increased in normal keratinocytes and in the HPV-16-immortalized cells, whereas no alteration was observed in the HPV-18-
transfected cells. The phosphorylation state of pRb correlated with cellgrowth; sensitive cells, which accumulate in G0-G, after exposure toTNF-a, exhibited an accumulation of hypophosphorylated pRb, whereasno effect on pRb phosphorylation was observed for HPV-18-immortalizedcells. These results clearly correlate with TNF-a-induced growth arrest in
INTRODUCTION
Proliferation, differentiation, and immortalization of human keratinocytes can be modulated in vitro by different positive and negativeregulators. Among them, TGF-ß3and TNF-a are potent inhibitors of
keratinocyte proliferation that may have an important role in the invivo control of keratinocyte growth. TNF-a is a Mr 17,000 protein
with a wide range of biological activities, including the induction ofhemorrhagic necrosis of certain mouse and human tumors (2, 3). It hasbeen reported that TNF is cytocidal and cytostatic for some tumor celllines in tissue culture, including leukemic cell lines (4) and tumor-derived epithelial cells (5), as well as normal fibroblasts (6), endo-thelial cells (7), and normal epithelial cells (8). Like TGF-/3 and IFN,the action of TNF-a requires specific binding to high-affinity recep
tors that are expressed on cells of most tissues, as well as theirmalignant derivatives (9). Two types of TNF-a receptors have been
characterized, R55 and R75; both are responsible for different effectsof TNF-a (10, 11). In normal keratinocytes, the binding of TNF to its
receptor causes a reversible inhibition of proliferation that is accompanied by stimulation of differentiation (8).
HPVs are associated with a variety of neoplasms, the most commonbeing cervical carcinomas (12, 13). The high-risk HPV types 16 and
Received 10/2/95; accepted 3/12/96.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
1 K. B. L. V. was supported by fellowships from the Fundaçaode Amparo à Pesquisa
do Estado de Sao Paulo (FAPESP 92/1453-0) and from Conselho Nacional de Desen-volvimento Técnicoe CientÃfico(CNPq 200955/94-2).
2 To whom requests for reprints should be addressed, at Ludwig Institute for CancerResearch, R. Prof. Antonio Prudente 109, 4th Floor, 01509-010 Sao Paulo, SP, Brazil.Phone: (55-11) 270-4922; Fax: (55-11) 270-7001; E-mail: [email protected].
3 The abbreviations used are: TGF-/3, transforming growth tactor-ß;TNF-a, tumornecrosis factor a; HPV. human papillomavirus: cdk, cyclin-dependent kinase; NHFK,normal human foreskin keratinocytes: FACS, fluorescence-activated cell sorting: K-SFM.keratinocyte-serum-free medium.
18 can immortalize normal cells in culture (14, 15), and it has beenestablished that the E6 and E7 proteins of these viruses are sufficientfor the efficient immortalization of human keratinocytes (16, 17).These oncoproteins interact with cellular proteins such as pRb (18),p53 (19), and pl07 (20), leading to their inactivation. The HPV-16 E7
protein has also been found to associate with cyclin A/cdk2 (21) andwith histone H l kinase during the G2-M phase of the cell cycle (20).
It was demonstrated that the immortalization of normal human keratinocytes with the E6 and E7 genes leads to an increase in theexpression of certain mitotic control proteins (22).
Human foreskin keratinocytes immortalized with HPV-16, HPV-18, or SV40 have been shown to be resistant to the growth-inhibitoryeffect of TGF-ß(23). In contrast, normal keratinocytes were growth
inhibited in the presence of this cytokine, a process that was accompanied by down-regulation of the c-myc gene (23). Genetic studies
have demonstrated that the HPV E7 oncoprotein is responsible for thisphenomenon, presumably via its interaction with the tumor suppressorprotein pRb (24). We reported that HPV-16-immortalized cells wereas sensitive as normal keratinocytes to the growth-inhibitory effectexerted by TNF-a in subconfluent cell cultures (1). In contrast,HPV-18- and SV40-immortalized keratinocytes were resistant. Interestingly, no alteration in the expression of c-myc was observed in cellsexposed to TNF-a, suggesting that a different pathway was involvedin TNF-mediated keratinocyte growth arrest.
The proliferation of most differentiated higher eukaryotic cells iscontrolled by factors that regulate the expression of specific genes.There are a number of genes that encode regulatory proteins, theexpression of which is coupled to the control of cell cycle progression,including the product of the retinoblastoma tumor suppressor gene,cyclins, and their associated cdks. These proteins also may be regulated by a complex process of phosphorylation and dephosphorylation(25, 26). We report that the expression of different cell cycle regulatory proteins, such as cyclin A, cyclin B, and p34cdc2, is substantially
decreased in normal keratinocytes and in an HPV-16-immortalizedcell line after TNF-a treatment. Cyclin D, levels were slightly in
creased in these cells, whereas no changes in the expression of cyclinE were observed. In the presence of TNF-a, cells containing HPV-16were predominantly in G0-G, and showed a reduction in S phase.These growth-inhibited cells contained mainly hypophosphorylatedpRb. In striking contrast, HPV-18- or SV40-immortalized cell lines,as well as two HPV-positive cervical carcinoma-derived cell lines thatare resistant to the growth-inhibitory effect exerted by TNF-a, showed
no alterations in the levels of the cell cycle proteins tested or in pRbphosphorylation. These results indicate that TNF-a inhibits cell proliferation of normal and HPV-16-immortalized cell lines by blockingcell growth at G0-G,.
MATERIALS AND METHODS
Cell Culture. Primary cultures of NHFK were isolated from newbornforeskin by trypsin treatment as described previously (27). These cells weremaintained in K-SFM (Life Technologies, Inc., Gaithersburg, MD) containing
epidermal growth factor and bovine pituitary extract and were passaged two tosix times. Human keratinocyte cell lines immortalized by SV40 (HFSV40),
2452
on May 10, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CELL CYCLE CONTROL IN TNF-A-TREATED HUMAN KERATINOCYTES
HPV-16 (HF698), and HPV-18 (HFISNco) were grown in 3+1 mediumconsisting of a mixture of 3 parts K-SFM and 1 part DMEM supplementedwith 10% FCS. These HPV-immortalized cell lines are independent clonal
isolates and were obtained by transfection of NHFK derived from differentforeskins, as described previously (27, 28). SiHa and HeLa cell lines (American Type Culture Collection, Rockville, MD) were cultivated in DMEM
containing 10% FCS.Protein Extraction and Immunoblotting. Cells were grown in 100-mm
tissue culture dishes to 40-60% confluence. After treatment with 1 nM ofhuman recombinant TNF-a (Boehringer Mannheim, Indianapolis, IN) for
different periods of time, cells were lysed directly on culture dishes with coldlysis buffer [1% Triton X-100, 50 mM HEPES (pH 7.5), 5 mM EDTA, 50 HIM
NaCl, 10 mM NaPP¡,50 mM sodium fluoride, 1 mM sodium vanadate, and 1 mMphenylmethylsulfonyl fluoride] and 1 mg/ml aprotinin and leupeptin for 30min on ice. Lysates were clarified by centrifugation at 4°C,and 75-150 ju.g
aliquots were separated in polyacrylamide-SDS gels and electroblotted onto
polyvinylidene difluoride membranes (Immobilen PVDF; Millipore, Bedford,MA) in the presence of 20% methanol. Membranes were reacted with thefollowing antibodies: cyclin A (Ab-2; 3 /xg/ml), cyclin B (Ab-2; 3 /xg/ml),p34cdc2 (Ab.,; 3 /lg/ml)i cycijn E (Ab-i; 2.5 /xg/ml) (all from Oncogene
Science, Uniondale, NY), cyclin D, -HD 11 (5 Mg/ml; Santa Cruz Biotechnology, Santa Cruz, CA), and pRb340 (1:100; Life Technologies, Inc., Gaithers-burg, MD), and developed with the Western-Light Chemiluminescent detection system (Tropix, Bedford, MA), according to the manufacturer's
instructions. After exposure to X-ray films (Reflection NEF-491; DuPont,Wilmington, DE), the intensity of the bands were evaluated by laser densi-
tometry (Ultrascan XL; Pharmacia, Uppsala, Sweden).[/neffcy/-3H]Thymidine Incorporation. Around 2 X IO4cells were plated
into Lab-Tek Tissue Culture Chamber/Slide (Nunc, Inc., Naperville, IL) and
grown until 40% confluence. Cultures were treated with different concentrations of TNF-a ( 1 and 5 nM), and after 60 h, the medium was supplementedwith 6 nCi/ml [mer/iy/-3H]thymidine (25 Ci/mmol; Amersham, Aylesbury,
United Kingdom). After 16 h, the slides were washed twice with PBS, fixed in100% ethanol, and exposed to photographic emulsion (autoradiography emulsion NTB3; Eastman Kodak, Rochester, NY) for 12 days. The slides weredeveloped and counterstained with H&E to analyze the number of labelednuclei by optic microscopy.
Flow Cytometry. Cells were plated into 100-mm dishes and treated with 1nM TNF-a for 60 h after they reached 40-60% confluence. Cells weretrypsinized, and IO6cells were resuspended in 100 /nl of citrate buffer [250 mM
sucrose, 40 mM Tris-sodium citrate, and 5% DMSO (pH 7.6)] and stored at—¿�70°Cuntil staining, as described previously (29). In brief, IO6 cells in citrate
buffer were treated with 3% trypsin. After trypsin inhibition and RNase A ( 100^ig/ml), cells were stained with 400 /xg/ml propidium iodide. Samples were runbetween 15 min and 3 h after staining in a FACStarplus flow cytometer (Becton
Dickinson). Results were analyzed using the ModFit DNA Distribution Modeling software for cell cycle phase analysis.
RESULTS
A differential inhibition of growth proliferation by TNF-a betweennormal and HPV-immortalized cell lines has been reported previously(1). These findings were confirmed by [3H]thymidine incorporation
where the number of labeled nuclei was indeed markedly decreasedafter treatment of both normal and HPV-16-immortalized humanforeskin keratinocytes (HF698 cells) with 1 nM TNF-a (Table 1). Incontrast, keratinocytes immortalized by either HPV-18 (HFISNco) orby SV40 (HFSV40), which are both resistant to TNF-a growthinhibition, did not show any changes in [3H]thymidine incorporation,
even when the concentration of TNF-a was raised to 5 nM (data not
shown).The nuclei-labeling frequencies closely reflect alterations in cell
cycle progression, as measured by flow cytometry (Table 1). Theseexperiments revealed that upon treatment with TNF-a, a marked
decrease in the frequency of HF698 cells in S phase was observed; onthe other hand, HPV-18-immortalized cells were not affected by this
Table 1 Effect of TNF-a in the proliferation of normal and HPV-immi)rlalized
human keratinnc\tes
Cell lines TNF-atreatment"NHFKHF698HFISNcoNo1
nMNo1
nMNo1
DM|3H]Thymidine
incorporation65
4158
284544Cells
in S phase'(%)ND''
ND19.5
5.327.7
24.4
number of labeled nucleinumber of unlabeled nuclei
X 100;
" 60 h.
* [3//]Thymidine incorporation =
mean values of two experiments.' By flow cytometry.'' ND, not determined.
cytokine. Therefore, the TNF-induced cell cycle arrest was restrictedto normal as well as HPV-16-immortalized keratinocytes.
Expression of Mitotic Regulatory Proteins after TNF-a Treat
ment. To analyze the expression of cell cycle regulatory proteins,normal and HPV-transfected human foreskin keratinocytes, as well ascervical carcinoma-derived cell lines, were exposed to TNF-a for
increasing periods of time. Total protein extracts were prepared andanalyzed by SDS-PAGE, followed by immunoblot using antibodies
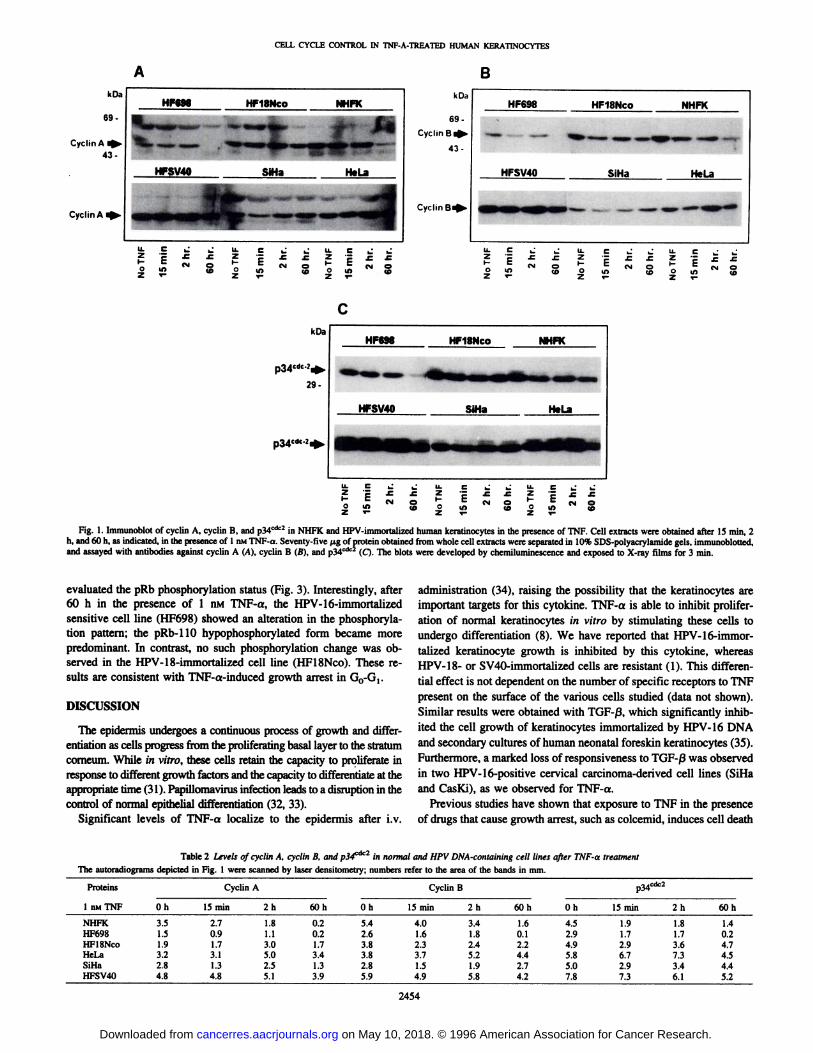
directed against cell cycle proteins. Following 60 h exposure to 1 nMTNF-a, the HF698 cell line and normal keratinocytes exhibited adramatic reduction (between 7- and 25-fold) in the levels of cyclin A,cyclin B, and p34cdc2 (Fig. 1 and Table 2). In contrast, the HPV-18-
and SV40-immortalized keratinocytes and the HPV-containing cervi
cal carcinoma cell lines, SiHa and HeLa, did not exhibit any significant alteration in the expression of these proteins when treatedsimilarly. Because these cell lines display different doubling times inculture, we repeated the assays after 36 h in the absence of growthfactors or serum deprivation before TNF-a treatment. The same
decrease in protein levels was observed in the HF698 cell line and innormal keratinocytes (data not shown). These results indicate thatTNF-a is able to interfere with the expression of mitotic regulatory
proteins in cells that are sensitive to the inhibitory effects exerted bythis cytokine. However, resistant cell lines, either immortalized ortumorigenic, showed an unaltered expression of these cyclins in thepresence of this cytokine.
G, Regulatory Protein Expression after TNF-a Treatment.Because an increase was observed in the number of cells in G, afterTNF-a treatment of HF698 and normal keratinocytes, the expression
of proteins related to this cell cycle phase was evaluated. Proteinextracts were prepared after 60 h in the presence of 1 nM TNF-a and
analyzed by immunoblot. Expression of cyclin E was not affected byTNF-a treatment in both sensitive and resistant cell lines (Fig. 2 and
Table 3). Analyses of the expression of cyclin D, revealed that thisprotein is slightly increased (about 2-fold) in HF698 cells and innormal cells in the presence of TNF-a (Fig. 2; Table 3). However, nochange in the level of this cyclin was observed in the HPV-18-
immortalized cell line (HFISNco). We were unable to evaluate thelevels of expression of cyclin D, in SiHa, HeLa, and HFSV40 cellsbecause these cells express extremely low levels of this protein, afinding which is in agreement with studies published previously (30).
pRb Phosphorylation in the Presence of TNF-a. pRb acts as a
cell cycle oscillator in the sense that it is dephosphorylated as cellsexit mitosis, is mainly present in the hypophosphorylated form duringG0, and then is rephosphorylated during late G, (26). Both growth-promoting and -inhibitory signals impinge on pRb by affecting its
phosphorylation (26). Using a monoclonal antibody that is able todetect the multiple forms of this phosphoprotein (pi 10-114), we
2453
on May 10, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
CELL CYCLE CONTROL IN TNF-A-TREATED HUMAN KERATINOCYTES
BkOa
69-
Cyclin A i43-
CyclinA i
HF698 HF18NCO NHFK
HFSV40 SiHa HeLa
k Da
69 -
CyclmBi
43-
CyclinB«
HF698 HF18NCO NHFK
HFSV40 SiHa HeLa
É EO LO
Z T-
H Eo i«
Z T-
kOa
29-
HF698 HF18NCO NHFK
HFSV40 SiHa HeLa
U. C2 e*- EO IO
_z"-O
e ij'£•=
N5
Fig. 1. Immunoblot of cyclin A, cyclin B, and p34cac2 in NHFK and HPV-immortalized human keratinocytes in the presence of TNF. Cell extracts were obtained after 15 min, 2
h, and 60 h, as indicated, in the presence of 1 nMTNF-a. Seventy-five (ig of protein obtained from whole cell extracts were separated in 10% SDS-polyacrylamide gels, immunoblotted,and assayed with antibodies against cyclin A (A), cyclin B (B), and p34cdc2 (Q. The blots were developed by chemiluminescence and exposed to X-ray films for 3 min.
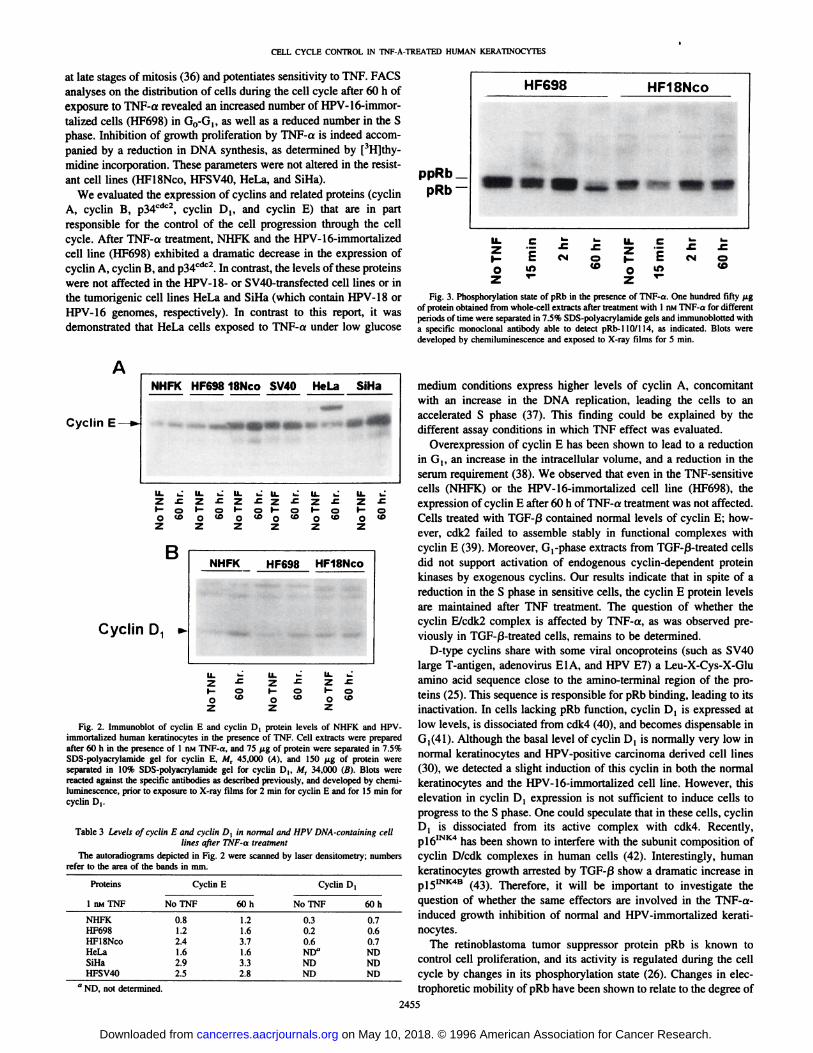
evaluated the pRb phosphorylation status (Fig. 3). Interestingly, after60 h in the presence of 1 nM TNF-a, the HPV-16-immortalized
sensitive cell line (HF698) showed an alteration in the phosphorylation pattern; the pRb-110 hypophosphorylated form became more
predominant. In contrast, no such phosphorylation change was observed in the HPV-18-immortalized cell line (HFlSNco). These results are consistent with TNF-a-induced growth arrest in GO-G!.
DISCUSSION
The epidermis undergoes a continuous process of growth and differentiation as cells progress from the proliferating basal layer to the stratumcomeum. While in vitro, these cells retain the capacity to proliferate inresponse to different growth factors and the capacity to differentiate at theappropriate time (31). Papillomavirus infection leads to a disruption in thecontrol of normal epithelial differentiation (32, 33).
Significant levels of TNF-a localize to the epidermis after i.v.
administration (34), raising the possibility that the keratinocytes areimportant targets for this cytokine. TNF-a is able to inhibit prolifer
ation of normal keratinocytes in vitro by stimulating these cells toundergo differentiation (8). We have reported that HPV-16-immor-
talized keratinocyte growth is inhibited by this cytokine, whereasHPV-18- or SV40-immortalized cells are resistant (1). This differen
tial effect is not dependent on the number of specific receptors to TNFpresent on the surface of the various cells studied (data not shown).Similar results were obtained with TGF-ß,which significantly inhibited the cell growth of keratinocytes immortalized by HPV-16 DNA
and secondary cultures of human neonatal foreskin keratinocytes (35).Furthermore, a marked loss of responsiveness to TGF-ßwas observedin two HPV-16-positive cervical carcinoma-derived cell lines (SiHaand CasKi), as we observed for TNF-a.
Previous studies have shown that exposure to TNF in the presenceof drugs that cause growth arrest, such as colcemid, induces cell death
Table 2 Levels of cyclin A, cyclin B, and p3<fic2 in normal and HPV DNA-containing cell lines after TNF-a treatment
The autoradiograms depicted in Fig. 1 were scanned by laser densitometry; numbers refer to the area of the bands in mm.
Proteins1
niuTNFNHFKHF698HFlSNcoHeLaSiHaHFSV40Cyclin
AOh3.51.51.93.22.84.815
min2.70.91.73.11.34.82h1.81.13.05.02.55.160h0.20.21.73.41.33.9Oh5.42.63.83.82.85.9Cyclin
Bp34cdc215
min4.01.62.33.71.54.92h3.41.82.45.21.95.860h1.60.12.24.42.74.2Oh4.52.94.95.85.07.815min1.91.72.96.72.97.32h1.81.73.67.33.46.160h1.40.24.74.54.45.2
2454
on May 10, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
CELL CYCLE CONTROL IN TNF-A-TREATED HUMAN KERATINOCYTES
at late stages of mitosis (36) and potentiates sensitivity to TNF. FACSanalyses on the distribution of cells during the cell cycle after 60 h ofexposure to TNF-a revealed an increased number of HPV-16-immor-
talized cells (HF698) in G0-G,, as well as a reduced number in the Sphase. Inhibition of growth proliferation by TNF-a is indeed accompanied by a reduction in DNA synthesis, as determined by [3H]thy-
midine incorporation. These parameters were not altered in the resistant cell lines (HFlSNco, HFSV40, HeLa, and SiHa).
We evaluated the expression of cyclins and related proteins (cyclinA, cyclin B, p34cdc2, cyclin D,, and cyclin E) that are in part
responsible for the control of the cell progression through the cellcycle. After TNF-a treatment, NHFK and the HPV-16-immortalized
cell line (HF698) exhibited a dramatic decrease in the expression ofcyclin A, cyclin B, and p34cdc2. In contrast, the levels of these proteins
were not affected in the HPV-18- or SV40-transfected cell lines or inthe tumorigenic cell lines HeLa and SiHa (which contain HPV-18 orHPV-16 genomes, respectively). In contrast to this report, it wasdemonstrated that HeLa cells exposed to TNF-a under low glucose
ppRbpRb
HF698 HFlSNco
cI O
ID
ZKO
C
iIO
CM OCD
Fig. 3. Phosphorylation state of pRb in the presence of TNF-a. One hundred fifty ¿tgof protein obtained from whole-cell extracts after treatment with I nMTNF-a for differentperiods of time were separated in 7.5% SDS-polyacrylamide gels and immunobiotted witha specific monoclonal antibody able to detect pRb-llO/H4, as indicated. Blots weredeveloped by chemiluminescence and exposed to X-ray films for 5 min.
Cyclin E
NHFK HF69818NCO SV40 HeLa SiHa
oz
o•¿�ooZ
B
Cyclin D, »-
NHFK HF698 HFlSNco
oz
o«o
-C
O
Fig. 2. Immunoblot of cyclin E and cyclin D! protein levels of NHFK and HPV-immortalized human keratinocytes in the presence of TNF. Cell extracts were preparedafter 60 h in the presence of I nM TNF-a, and 75 (¿gof protein were separated in 7.5%SDS-polyacrylamide gel for cyclin E. Mr 45.000 (A}, and 150 /xg of protein wereseparated in 10% SDS-polyacrylamide gel for cyclin D,, M, 34,000 (B). Blots werereacted against the specific antibodies as described previously, and developed by chemiluminescence, prior to exposure to X-ray films for 2 min for cyclin E and for 15 min for
cyclin D,.
Table 3 Levels of cyclin E and cyclin D¡in normal and HPV DNA-containing celllines after TNF-a treatment
The autoradiograms depicted in Fig. 2 were scanned by laser densitometry: numbersrefer to the area of the bands in mm.
Proteins1
nMTNFNHFKHF698HFlSNcoHeLaSiHaHFSV40Cyclin
ENo
TNF0.81.22.41.62.92.560h1.21.63.71.63.32.8Cyclin
D,No
TNF0.30.20.6ND"NDND60h0.70.60.7NDNDND
" ND, not determined.
medium conditions express higher levels of cyclin A, concomitantwith an increase in the DNA replication, leading the cells to anaccelerated S phase (37). This finding could be explained by thedifferent assay conditions in which TNF effect was evaluated.
Overexpression of cyclin E has been shown to lead to a reductionin G,, an increase in the intracellular volume, and a reduction in theserum requirement (38). We observed that even in the TNF-sensitivecells (NHFK) or the HPV-16-immortalized cell line (HF698), theexpression of cyclin E after 60 h of TNF-a treatment was not affected.Cells treated with TGF-ßcontained normal levels of cyclin E; how
ever, cdk2 failed to assemble stably in functional complexes withcyclin E (39). Moreover, G,-phase extracts from TGF-ß-treated cellsdid not support activation of endogenous cyclin-dependent protein
kinases by exogenous cyclins. Our results indicate that in spite of areduction in the S phase in sensitive cells, the cyclin E protein levelsare maintained after TNF treatment. The question of whether thecyclin E/cdk2 complex is affected by TNF-a, as was observed previously in TGF-ß-treated cells, remains to be determined.
D-type cyclins share with some viral oncoproteins (such as SV40large T-antigen, adenovirus EIA, and HPV E7) a Leu-X-Cys-X-Gluamino acid sequence close to the amino-terminal region of the pro
teins (25). This sequence is responsible for pRb binding, leading to itsinactivation. In cells lacking pRb function, cyclin D, is expressed atlow levels, is dissociated from cdk4 (40), and becomes dispensable inG,(41). Although the basal level of cyclin D, is normally very low innormal keratinocytes and HPV-positive carcinoma derived cell lines
(30), we detected a slight induction of this cyclin in both the normalkeratinocytes and the HPV-16-immortalized cell line. However, this
elevation in cyclin D, expression is not sufficient to induce cells toprogress to the S phase. One could speculate that in these cells, cyclinD[ is dissociated from its active complex with cdk4. Recently,pl6INK4 has been shown to interfere with the subunit composition of
cyclin D/cdk complexes in human cells (42). Interestingly, humankeratinocytes growth arrested by TGF-ßshow a dramatic increase inp]5iNK4B ^43j Therefore, it will be important to investigate the
question of whether the same effectors are involved in the TNF-a-induced growth inhibition of normal and HPV-immortalized kerati
nocytes.The retinoblastoma tumor suppressor protein pRb is known to
control cell proliferation, and its activity is regulated during the cellcycle by changes in its phosphorylation state (26). Changes in elec-
trophoretic mobility of pRb have been shown to relate to the degree of
2455
on May 10, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CELL CYCLE CONTROL IN TNF-A-TREATED HUMAN KERATINOCYTES
phosphorylation (44). In the presence of TNF-a, the HPV-16-immor-
talized cell line showed a difference in the phosphorylated patternwhen compared with the untreated cells; these cells clearly containmainly the pRb-110 hypophosphorylated form after 60 h of treatment.
No alteration in the level of pRb phosphorylation was observed for theHPV-18-resistant cell line. In quiescent cells (G0), pRb is present inthe hypophosphorylated form (pRb-110), but proliferating cells in G,contain a significant proportion of phosphorylated pRb (112-114;
Refs. 26 and 45). It was also shown previously that different cell typesarrested in G, by negative growth factors, such as TGF-ßor a-IFN,contain predominantly pRb-110 (45, 46). Our results on the phospho
rylation status of pRb correlated strictly with the cell cycle analysis byFACS in which TNF-a-treated cells are predominantly arrested in G,(data not shown). Because these cells express HPV-16 E7 and, there
fore, lack a functional pRB, it is reasonable to speculate that theirphosphorylation would not correlate with levels of cdks or cyclins orthe formation of stable cdk/cyclin complexes (41). Furthermore, theobserved inconsistencies with levels of pRB phosphorylation andexpression of cell cycle proteins may be the consequence of alteredexpression of cdk inhibitors, particularly p21WAFl/CIP1and pl6INK4.
In a recent study, Butz et al. (47) have shown that, when exposedto genotoxic agents, several HPV-positive cell lines with a functional
p53 protein express higher WAF1/CIP1 mRNA levels. However, theability to undergo G, arrest under these conditions differed betweenprimary cells and HPV-positive cervical cancer cell lines. In the latter,it is possible that endogenous E6 is interfering with the p53-mediatedgrowth arrest. It is possible that the differential sensitivity to TNF-aobserved in the HPV-immortalized cells tested in our study mayreflect different levels of p2lWAF1/CIP1.Work is in progress to inves
tigate this hypothesis.Although the differential susceptibility to TNF-a of normal and
HPV DNA-containing cell lines has been correlated to specific
changes in cell cycle progression, the molecular basis for this difference is not yet known. Some interesting data were obtained withmalignant HPV-18-positive cervical carcinoma cells (HeLa) and theirnontumorigenic segregants (HeLa-fibroblast hybrids; Ref. 48). InHeLa cells exposed to TNF-a, a marked decrease in monocyte che-moattractant protein-1 is observed. However, this protein, encoded bythe JE gene, is induced strongly by TNF-a in the nontumorigenicsegregants, which is accompanied by down-regulation of HPV-18transcription (48). These authors suggest that the functional inactiva-tion of chemokines such as monocyte chemoattractant protein-1 mayhave an important role in growth regulation of HPV-positive cells invivo. It is also possible that the 10-fold greater transcriptional activityof the HPV-18 promoter (49) could effect higher levels of the HPV
oncoproteins and thereby alter the cellular phenotype, including resistance to inhibitory cytokines. Interestingly, HPV-18 DNA-contain
ing cervical carcinomas seem to exhibit a more aggressive clinicalbehavior (50, 51).
Cancer is a multistep process often involving the acquisition ofresistance to many cellular inhibitory factors. The phenotypic difference between tumorigenicity of HPV DNA-containing cell lines may
be linked to the acquired resistance to the inhibitory effects of certaincytokines. The characterization of the mechanism of TNF-a inhibitionof primary and HPV-16-immortalized keratinocytes should help to
identify the cellular factors responsible for the aggressiveness ofcertain cervical tumors.
ACKNOWLEDGMENTS
We are grateful to Dr. Richard Schlegel for helpful discussions and forproviding some reagents. We also thank Karen Creswell for technical assistance during the FACS analyses.
REFERENCES
16.
17.
18.
19.
20.
22.
23.
24.
27.
29.
30.
Villa. L. L.. Vieira. K. B. L., Pei. X-F.. and Schlegel. R. Differential effect of tumornecrosis factor on proliferation of primary human keratinocytes and cell lines containing human papillomavirus types 16 and 18. Mol. Carcinog., 6.- 5-9, 1992.Old, L. J. Tumor necrosis factor (TNF). Science (Washington DC), 230: 630-632,
1985.Beutler, B., and Cerami, A. Cachelin: more than a tumor necrosis factor. N. Engl. J.Med., 316: 379-385, 1987.
Kronke, M.. Schluter, C., and Pfizenmaier, K. Tumor necrosis factor inhibits mycexpression in HL-60 cells at the level of mRNA transcription. Proc. Nati. Acad. Sci.USA, 84: 469-473, 1987.Yarden, A., and Kim., in. A. Tumor necrosis factor reduces c-myc expression andcooperates with INF--y in HeLa cells. Science (Washington DC), 234: 1419-1421,
1986.Tsujimoto, M.. Yip. Y. K., and Vilcek, J. Tumor necrosis factor: specific binding andinternalization in sensitive and resistant cells. Proc. Nati. Acad. Sci. USA, 82:7627-7630. 1985.
Robaye, B., Mosselmans, R.. Fiers, W., Dumont, J. E.. and Galand, P. Tumor necrosisfactor induces apoptosis (programmed cell death) in normal endothelial cells in vitro.Am. J. Pathol.. 138: 447-453, 1991.Pillai, S., Bikle, D. D.. Eessalu, T. E.. Aggarwal, B. B., and Elias, P. M. Binding andbiological effects of tumor necrosis factor-a on cultured human neonatal foreskinkeratinocytes. J. Clin. Invest., 83: 816-821. 1989.
Loetscher. H.. Steinmetz. M., and Lesslauer, W. Tumor necrosis factor: receptors andinhibitors. Cancer Cells. 3: 221-226, 1991.Brockhaus, M.. Schoenfeld. H-J., Schlaeger, E-J., Hunziker, W., Lesslauer, W., and
Loetscher. H. Identification of two types of tumor necrosis factor receptors on human celllines by monoclonal antibodies. Proc. Nati. Acad. Sci. USA, 87: 3127-3131, 1990.Tartaglia. L. A.. Weber. R. F., Figari, I. S.. Reynolds, C., Palladino. M. A., Jr., andGoeddel. D. V. The two different receptors for tumor necrosis factor mediate distinctcellular responses. Proc. Nati. Acad. Sci. USA, 88: 9292-9296, 1991.
zur Hausen. H. Papillomaviruses in anogenital cancer as a model to understand therole of viruses in human cancers. Cancer Res., 49: 4677-4681, 1989.
Howley, P. M.. and Schlegel, R. The human papilloma viruses: an overview. Am. J.Med.. 85 (Suppl. 2A): 155-158, 1988.Pirisi, L., Yasumoto, A., Feller, M., Doniger, J., and DiPaolo, J. A. Transformation ofhuman fibroblasts and keratinocytes with human papillomavirus type 16 DNA. J.Virol., 61: 1061-1066, 1987.Durst. M., Dzarlieva-Petrusevska. R. T.. Boukamp. P., Fusenig, N. E., and Gissmann,
L. Molecular and cytogenetic analysis of immortalized human primary keratinocytesobtained after transfection with human papillomavirus type 16 DNA. Oncogene, 1:251-256, 1987.Munger, K., Phelps. W. C.. Bubb, V., Howley, P. M., and Schlegel, R. The E6 andE7 genes of the human papillomavirus type 16 together are necessary and sufficientfor transformation of primary human keratinocytes. I. Virol.. 63: 4417-4421. 1989.
Hudson, I. B., Bedell. M. A.. McCance, D. J., and Laimins. L. A. Immortalization andaltered differentiation of human keratinocytes in vitro by the E6 and E7 open readingframes of human papillomavirus type 18. J. Virol.. 64: 519-526, 1990.Dyson, N.. Howley. P. M., Munger. K., and Harlow. E. The human papillomavirus-16
E7 oncoprotein is able to bind to the retinoblastoma gene product. Science (Washington DC), 243: 934-937. 1989.Wemess. B. A.. Levine, A. J., and Howley. P. M. Association of human papillomavirustypes 16 and 18 E6 proteins with p53. Science (Washington DC), 248: 76-79. 1990.
Davies, R., Hicks, R.. Crook. T., Morris, J.. and Vousden, K. Human papillomavirustype 16 E7 associates with a histone HI kinase and with pl07 through sequencesnecessary for transformation. J. Virol., 67: 2521-2528, 1993.
Tommasino, M., Adamczewski, J. P., Carlolti, F., Barth. C. F.. Manetti, R.. Contorni,M., Cavalieri, F., Hunt, T., and Crawford, L. HPV-16 E7 associates with the proteinkinase p33cdk2 and cyclin A. Oncogene. 8: 195-202, 1993.
Steinmann, K. E., Pei, X-F.. Stoppler, H.. Schlegel, R., and Schlegel, R. Elevatedexpression and activity of mitotic regulatory proteins in human papillomavirus-immortalized keratinocytes. Oncogene, 9: 387-394, 1994.
Pietenpol. I. A., Holt. J. T.. Stein. R. W., and Moses, H. L. Transforming growthfactor-^, suppression of c-myc gene transcription: role in inhibition of keratinocyteproliferation. Proc. Nati. Acad. Sci. USA, 87: 3758-3762, 1990.
Pietenpol, J. A.. Stein. R. W., Moran, E., Yaciuk, P., Schlegel. R.. Lyons. R. M.,Pittelkow, R. M., Munger. K.. Howley, P. M.. and Moses, H. L. TGF-/3, inhibition ofc-myc transcription growth in keratinocytes is abrogated by viral transforming proteins with pRb binding domains. Cell. 61: 777-785, 1990.Sherr, C. J. Mammalian G, cyclins. Cell, 73: 1059-1065, 1994.
Sherr, C. J. The ins and outs of RB: coupling gene expression to the cell cycle clock.Trends Cell Biol., 4: 15-18. 1994.Schlegel. R., Phelps, W. C.. Zhang. Y. L.. and Barbosa, M. S. Quantitative keratinocyteassay detects two biological activities on human papillomavirus DNA and identifies viraltypes associated with cervical carcinomas. EMBO J., 7: 3181-3187, 1988.Barbosa, M. S., and Schlegel. R. The £6and £7genes of HPV-18 are sufficient forinducing two-stage in vitro transformation of human keratinocytes. Oncogene, 4:1529-1532. 1989.Vindelov, L. L., Christensen. I. J., and Nissen, N. I. A detergent-trypsin method forpreparation of nuclei for flow cytometric DNA analysis. Cytometry, 3: 323-327,
1983.Tarn. S. W.. Theodoras, A. M., Shay. J. W., Draetta, G. F., and Pagano, M.Differential expression and regulation of cyclin D, protein in normal and tumorhuman cells: association with Cdk4 is required for cyclin D, function in G, progression. Oncogene, 9: 2663-2674, 1994.
2456
on May 10, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

CELL CYCLE CONTROL IN TNF-A-TREATED HUMAN KERATINOCYTES
31. Watt, F. M. Selective migration of terminally differentiating cells from basal layer ofcultured human epidermis. J. Cell Biol., 98: 16-21, 1984.
32. Stoler, M. H., and Broker, T. R. In situ hybridization detection of human papilloma-
virus DNA and messenger RNA in genital condylomas and a cervical carcinoma.Hum. Pathol., 17: 1250-1258, 1986.
33. Merrick, D. T., Blanton, R. A., Gown, A. M., and McDougall, J. K. Alteredexpression of proliferation and differentiation markers in human papillomavirus 16and 18 immortalized epithelial cells grown in organotypic culture. Am. J. Pathol..140: 167-177, 1992.
34. Beutler, B. A., Milsark, I. W., and Cerami, A. Cachetin/tumor necrosis factor: production,distribution, and metabolic fate in vivo. 3. Immunol., 135: 3972-3977, 1985.
35. Braun, L., Durst. M., Mikumo, R., and Gruppuso, P. Differential response of nontu-morigenic human papillomavirus type 16-positive epithelial cells to transforminggrowth factor-0,. Cancer Res., 50: 7324-7332, 1990.
36. Darzynkiewicz, Z., Williamson, B., Carswell, E. A., and Old, L. Cell cycle specificeffects of tumor necrosis factor. Cancer Res., 44: 83-90, 1984.
37. Volland. S., Amtmann, E., and Sauer, G. TNF accelerates the S phase of the cell cyclein tumor cells. Int. J. Cancer, 56: 698-705, 1994.
38. Ohtsubo, M.. and Roberts. J. M. Cyclin-dependent regulation of G, in mammalianfibroblasts. Science (Washington DC). 259: 1908-1912, 1993.
39. Koff, A.. Ohtsuki, M.. Polyak. K., Roberts, J. M., and Massagué,J. Negativeregulation of G[ in mammalian cells: inhibition of cyclin E-dependent kinase byTGF-0. Science (Washington DC), 260: 536-539, 1993.
40. Bates, S., Parry. D., Bonetta, L., Vousden, K., Dickson, C., and Peters, G. Absenceof cyclin D/cdk complexes in cells lacking functional retinoblastoma protein. Oncogene, 9: 1633-1640, 1994.
41. Lukas, J., Muller, H., Bartkova, J., Spitkowsky, D.. Kjerulff, A. A., Jansen-Durr. P..Strauss, M., and Bartek, J. DNA tumor virus oncoproteins and retinoblastoma genemutations share the ability to relieve the cell's requirement for cyclin D, function in
G,. J. Cell Biol., 125: 625-638, 1994.42. Peters, G. Stifled by inhibitions. Nature (Lond.). 371: 204-205, 1994.
43. Hannon. G. J.. and Beach, D. pl51NK4B is a potential effector of TGF-induced cellcycle arrest. Nature (Lond.), 371: 257-261, 1994.
44. Furukawa, Y., Piwnica-Worms, H., Ernst, T. J., Kanakura, Y., and Griffin, J. D. cdc2gene expression at the G, to S transition in human T lymphocytes. Science (Washington DC), 250: 805-808, 1990.
45. Burke, L. C., Bybee, A., and Thomas, N. S. B. The retinoblastoma protein is partiallyphosphorylated during early G, in cycling cells but not in G, cells arrested witha-INF. Oncogene, 7: 783-788, 1992.
46. Munger, K., Pietenpol, J. A., Pittelkow. M. R.. Holt, J. T., and Moses, H. L.Transforming growth factor-ß, regulation of c-myc expression, pRB phosphoryl-
ation, and cell cycle progression in keratinocytes. Cell Growth & Differ., 3:291-298, 1992.
47. Butz, K., Shahabedin, L., Geisen, C., Spitkowsky, D., Ullmann, A., and Hoppe-Seyler, F. Functional p53 protein in human papillomavirus-positive cancer cells.Oncogene, IO: 927-936, 1995.
48. Rosi, F., Lengert, M., Albrecht, J., Kleine, K., Zawatsky, R., Schraven, B., and zurHausen, H. Differential regulation of the JE gene encoding the monocyte chemoat-tractant protein (MCP-1 ) in cervical carcinoma cells and derived hybrids. J. Virol.,68: 2142-2150, 1994.
49. Romanczuk, H., Villa, L. L.. Schlegel, R., and Howley, P. M. The viral transcriptionalregulatory region upstream of the E6 and E7 genes is a major determinant of thedifferential immortalization activities of human papillomavirus type 16 and 18. J.Virol., 65: 2739-2744, 1991.
50. Barnes, W., Delgado, G., Kurman, R. J., Petrilli, E. S., Smith, D. M., Ahmed, S.,Lorincz, A. T., Temple, G. F., Jenson, A. B., and Lancaster, W. D. Possibleprognostic significance of human papillomavirus type in cervical cancer. Gynecol.Oncol., 29: 267-273, 1988.
51. Kurman, R. J., Schiffman, M. H., Lancaster, W. D., Reid, R., Jenson, A. B., Temple,G. F., and Lorincz, A. T. Analysis of individual human papillomavirus types incervical neoplasia: a possible role for type 18 in rapid progression. Am. J. Obstet.Gynecol., /59: 293-296, 1988.
2457
on May 10, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from

1996;56:2452-2457. Cancer Res Katia B. L. Vieira, David J. Goldstein and Luisa L. Villa Normal and Papillomavirus-immortalized Human Keratinocytes
Interferes with the Cell Cycle ofαTumor Necrosis Factor
Updated version
http://cancerres.aacrjournals.org/content/56/10/2452
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/56/10/2452To request permission to re-use all or part of this article, use this link
on May 10, 2018. © 1996 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from





























