Embed Size (px)
Citation preview

Type 1 Gaucher Disease Presenting With ExtensiveMandibular Lytic Lesions: Identification andExpression of a Novel Acid b-Glucosidase Mutation
Melissa P. Wasserstein,1,2* John A. Martignetti,1,2 Robert Zeitlin,3 Harry Lumerman,4Marshall Solomon,5 Marie E. Grace,1 and Robert J. Desnick1,2
1Department of Human Genetics, Mount Sinai School of Medicine, New York, New York2Department of Pediatrics, Mount Sinai School of Medicine, New York, New York3Department of Surgery, Kingsbrook Jewish Medical Center, Brooklyn, New York4Department of Pathology, Mount Sinai School of Medicine, New York, New York5Department of Pathology, State University of New York Health Science Center at Brooklyn, Brooklyn, New York
The finding of extensive lytic lesions in themandible of a 19-year-old Ashkenazi Jewishwoman led to the diagnosis of Type 1 Gau-cher disease. She had extensive skeletal in-volvement, marked hepatosplenomegaly,and deficient acid b-glucosidase activity.Mutation analysis identified heteroallelismfor acid b-glucosidase mutations N370S andP401L, the latter being a novel missense mu-tation in exon 9. Expression of the P401L al-lele resulted in an enzyme with a reducedcatalytic activity (specific activity based oncross-reacting immunological material∼0.21), which was similar to that of the mildN370S mutant enzyme. The expression stud-ies predicted a mild phenotype for theproposita’s N370S/P401L genotype whichwas inconsistent with her severe diffuseskeletal disease and organ involvement.Since lytic mandibular lesions may be com-plicated by osteomyelitis, pathologic frac-ture, and tooth loss, regular dental assess-ments in Type 1 Gaucher patients should beperformed. Am. J. Med. Genet. 84:334–339,1999. © 1999 Wiley-Liss, Inc.
KEY WORDS: Gaucher; mandible; geno-type; P401L missense muta-tion
INTRODUCTION
Gaucher disease (GD) is an inborn error of glyco-sphingolipid metabolism caused by the deficient activ-ity of the lysosomal enzyme, acid b-glucosidase(E.C.3.2.1.45) [Brady et al., 1965, Desnick et al., 1982].The enzyme’s substrate, glucosylceramide, accumu-lates in cells of the monocyte/macrophage system. Thedisease occurs in three distinct phenotypic subtypeswhich are delineated by the absence (Type 1) or pres-ence and severity of neurological involvement (Types 2and 3). Most patients (∼95%) have Type 1 GD, the non-neuronopathic form characterized by hepatospleno-megaly, secondary hypersplenism, and skeletal in-volvement [Cox and Schofield, 1997]. In contrast, pa-tients with the neurologic forms, acute infantile Type 2or subacute late-infantile or juvenile Type 3, are rare.The age of onset, the degree of organomegaly, and theextent of skeletal involvement varies widely amongTypes 1 and 3 patients [Beutler and Grabowski, 1995].
Prediction of the disease subtype and severity basedon analysis of the patient’s genotype has been limited,in part due to the occurrence of numerous family-specific (or private) acid b-glucosidase mutations[Grabowski and Horowitz, 1997; Beutler and Gelbart,1998]. However, certain of these mutations have beenexpressed and characterized providing correlations be-tween their residual specific activities based on cross-reacting immunological material (CRIM SA) and sta-bilities with the disease phenotypes [Grace et al., 1991,1994, 1997; Pasmanik-Chor, et al., 1997]. For example,homoallelism for the L444P mutation which encodes avery low level of CRIM SA usually results in neurono-pathic disease [Mansuno et al., 1990; Eto et al., 1993;Tsuji et al., 1987] while homoallelism for the severe84GG frameshift allele is a fetal lethal [Levy-Lahadand Zimran, 1997]. The presence of one N370S allelewhich encodes a protein with significant CRIM SA is“neuroprotective,” precluding the development of neu-rological manifestations and resulting in the Type 1phenotype [Zimran et al., 1989; Sibille et al., 1993].
Contract grant sponsor: National Center for Research Re-sources; Contract grant number: 5 M01 RR00071; Contract grantsponsor: March of Dimes Birth Defects Foundation; Contractgrant number: F-FY96-1062; Contract grant sponsor: Mount Si-nai Child Health Research Center; Contract grant number: 5 P30HD28822.
*Correspondence to: Melissa P. Wasserstein, M.D., Depart-ment of Human Genetics, Box 1497, Mount Sinai Medical Center,One Gustave L. Levy Place, New York, NY 10029. E-mail:[email protected]
Received 22 June 1998; Accepted 26 January 1999
American Journal of Medical Genetics 84:334–339 (1999)
© 1999 Wiley-Liss, Inc.
Patients homoallelic for the N370S mutation have mildGD [Zimran et al., 1989].
The most frequent clinical presentations of Type 1GD are thrombocytopenia or hepatosplenomegaly. Lesscommonly, patients present with skeletal complica-tions such as avascular necrosis or pathologic frac-tures. In this communication we describe a 19-year-oldwoman in whom the diagnosis of Type 1 GD was madefollowing the discovery of lytic lesions of the mandible.Analysis of her acid b-glucosidase alleles identified anovel missense mutation, P401L, in exon 9 and thecommon N370S lesion.
CLINICAL DESCRIPTION
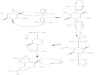
A 12-year-old girl of Ashkenazi Jewish descent wasseen by her oral surgeon (R.Z.) for extraction of severalprimary teeth prior to orthodontic therapy. A pan-oramic film showed no apparent pathologic changes,and the teeth were extracted without complication.Seven years later, she returned with complaints of painand swelling of the right jaw. Oral examination dem-onstrated a tissue-impacted right mandibular thirdmolar with erythema and edema in the region of theimpacted tooth. Radiologic examination of the jawshowed large radiolucent lesions in both sides of themandible (Fig. 1A). The lesions involved most of the leftand right body regions, and extended from the corticalplate of the inferior borders superiorly invaginating be-tween the teeth. As shown in Fig. 1B, a Dentascanconfirmed the extent of the intraosseous lesions anddocumented that they were multilocular. A multiplesite biopsy was performed which showed sheets of am-phophilic histiocytes with the characteristic “wrinkledtissue paper” appearance of the cytoplasm observed inGaucher cells (Fig. 2).
The patient was treated with a soft diet to decreasethe risk of a pathologic fracture, and was referred forfurther evaluation. She denied bleeding abnormalities,easy bruising, bony pains, or fractures. She had a ton-sillectomy at age 5 without complication. Her parentswere of Ashkenazi Jewish descent, and no relativeswere known to have GD.
On physical examination, there was no swelling, ten-derness, or erythema over the jaw. She had no cervicallymphadenopathy. The spleen was palpable 7 cm belowthe left costal margin, and the liver 2 cm below theright costal margin. Both organs were firm, smooth,and nontender. There was no point tenderness over herlong bones, and there was full range of motion at alljoints. No ecchymoses or petechiae were noted.
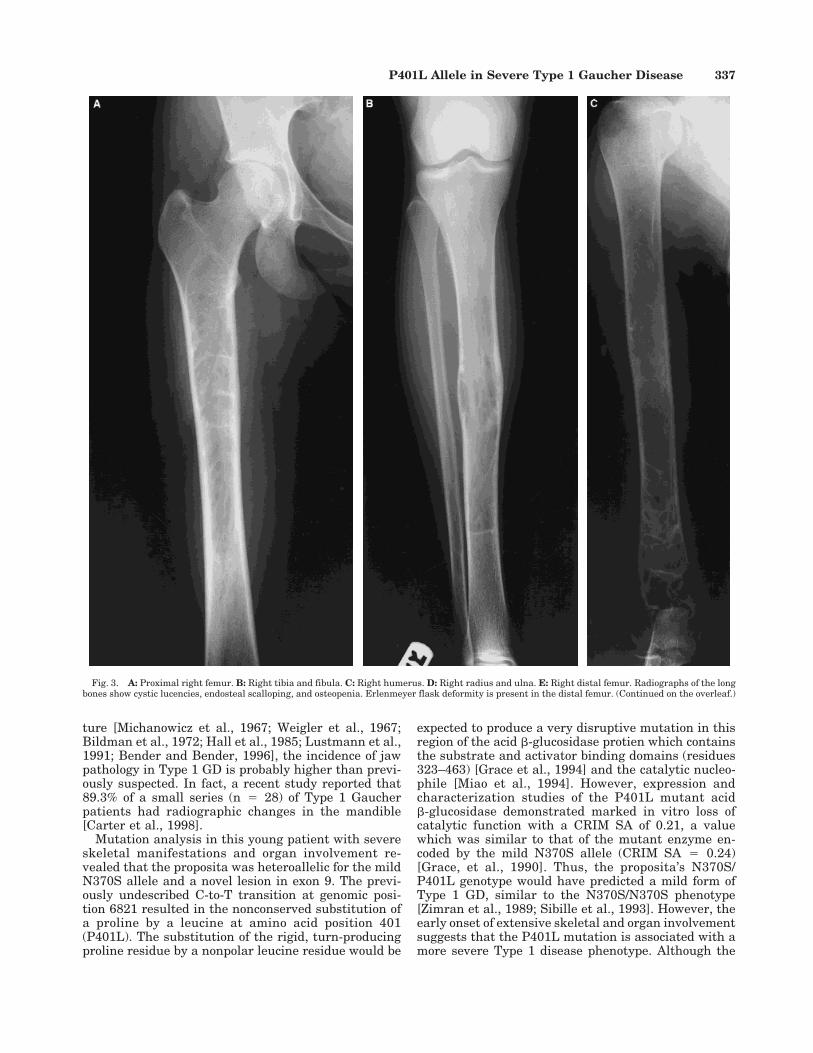
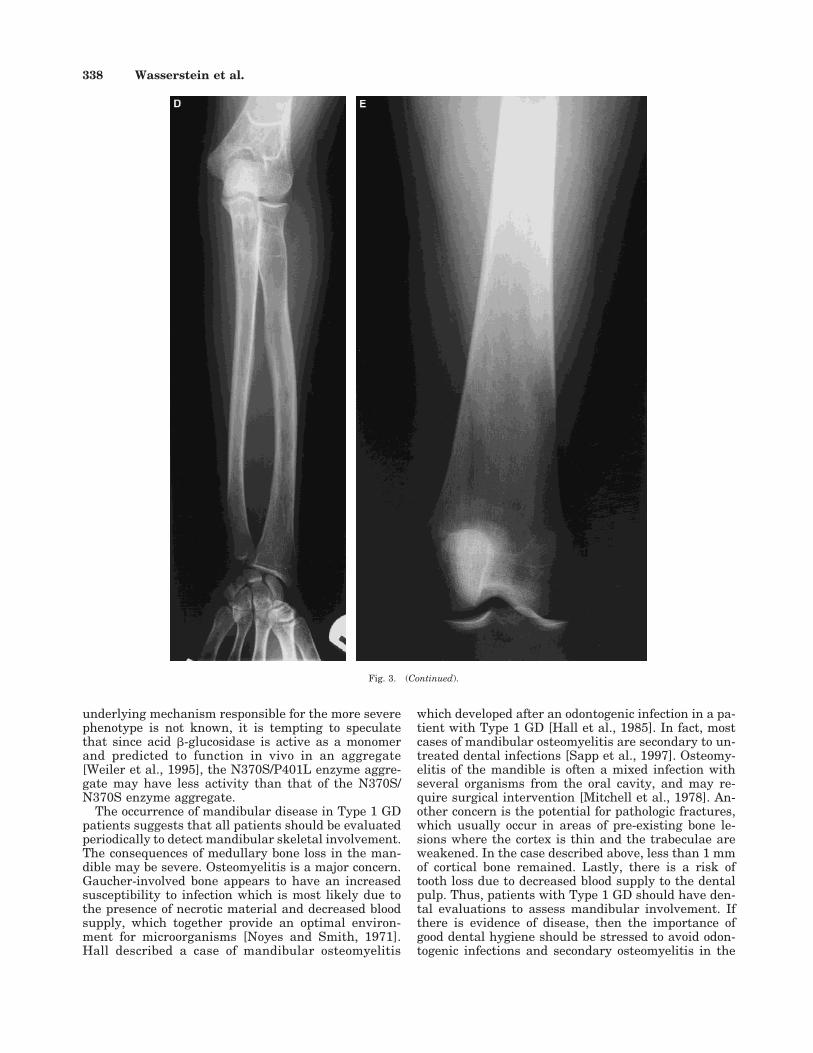
Her hemoglobin was low at 10.5 g/dl, her plateletcount was normal at 165,000/mm3, and her white bloodcell count was 5,700/mm3. The angiotensin-convertingenzyme, a biochemical marker of Gaucher activity, wasmarkedly elevated at 222.4 units/ml (normal range8–52 U/ml). A skeletal survey revealed osteopenia, Er-lenmeyer flask deformities of the distal femora, andsymmetric medullary expansion of the diaphysis ofboth the tibia and humerus. Cystic lucencies were seenin all long bones (Fig. 3). Bone density of the lumbarvertebrae, left radius and ulna, and total left hip wasmeasured at 65%, 68%, and 75% of age-matched nor-
mals, respectively. Quantitative magnetic resonanceimaging scans showed a hepatic volume of 2,368 cc (1.7times normal) and a splenic volume of 1,108 cc (10.2times normal).
Her acid b-glucosidase activity in isolated leukocyteswas markedly deficient (1.19 nmol/hr/mg protein; nor-mal individuals 3.0–8.5 nmol/hr/mg protein), confirm-ing the diagnosis of GD. Mutation analysis of her acidb-glucosidase gene documented that she was heteroal-lelic for the mild N370S mutation and a novel mutationin exon 9, P401L. Enzyme replacement therapy withimiglucerase (CerezymeR, Genzyme Therapeutics,Cambridge, MA) was initiated at 30 units/kg biweekly.
METHODSDNA Isolation, Polymerase Chain Reaction
Amplification, and Mutation Detection
Genomic DNA was isolated from peripheral bloodleukocytes using standard techniques [Sambrook et al.,1989]. The sample was initially screened for four com-mon Ashkenazi Jewish mutations (N370S, L444P,
Fig. 1. A: Panoramic radiograph shows large multilocular radiolucentlesions of the left and right mandible extending from the cortical plate ofthe inferior borders and superiorly invaginating between the teeth.B: Computed tomographic scan of the mandible documents symmetric ill-defined lucent expansile lesions involving the mandible with sparing onlyof the central portion of the mandible in the incisor region.
P401L Allele in Severe Type 1 Gaucher Disease 335

IVS2+1, 84insG) by polymerase chain reaction (PCR)amplification and restriction digestion [Tsuji et al.,1987; Beutler and Gelbart, 1990; He et al., 1992]. Todetect the unknown acid b-glucosidase mutation, thecomplete coding region and adjacent intron-exonboundaries were amplified from genomic DNA (500 ng)using biotinylated sense (exons 1+2, 3, 4, 6, 7) and an-tisense (exons 5, 8+9, and 10+11) primers as previouslydescribed [Sibille et al., 1993]. The primers used for thespecific amplification of exon 9 from genomic DNAwere: sense 58-AGGCTAATGTGGGAGGATC-38 andantisense 58-GTAGGAGATGATAGGCCTGGTA-38.The genomic designation (6821C→T) is based on theupdated acid b-glucosidase GenBank sequence [Horo-witz et al., 1989; Beutler et al., 1992], accession num-ber J03059 (8/95). The first nucleotide of exon 1 is atgenomic position 1230.
Construction And Purification ofExpression Plasmids
The point mutation for P401L was introduced intothe acid b-glucosidase cDNA by an M13mp-19-basedoligonucleotide-directed site-specific mutagenesis pro-cedure (Sculptor™ In Vitro Mutagenesis System (Am-ersham, Arlington, IL) as previously described [Graceet al., 1994, 1997]. The mutant cDNAs were thencloned into the EcoR1 site of the baculovirus expressionvector pAc1392. Recombinant baculovirus containingacid b-glucosidase cDNA with the P401L substitutionwere produced in cloned Spodoptera frugiperda (Sf9)cells as described [Grace et al., 1994, 1997].
Immunoblotting
Immunoelectroblotting using a polyclonal anti-human acid b-glucosidase antibody was performed aspreviously described [Grace et al., 1994, 1997]. Specificactivities based on the amount of CRIM SA were de-termined as described [Grace et al., 1990, 1991]. Therelative amount of CRIM for the expressed mutantP401L was determined by computer analysis of thescanned immunoblots using the NIH Image™ pro-gram, version 1.60, and referenced to that of the nor-mal enzyme.
Enzyme Assays
Aliquots of the crude sonicates were assayed for acidb-glucosidase activity using the fluorescent-labelednatural substrate, NBD-glucopyranosylceramide(NBD-GC; Signa Chemical Co., St Louis, MO). Back-ground levels were determined by comparison to re-sults obtained from Sf9 cells infected with recombinantbaculovirus containing the cDNA for acid b-glucosidasein the antisense direction (Rev). One unit of acid b-glu-cosidase is the amount of enzyme that hydrolyzes onemmol of substrate per min at 37°C.
RESULTS
Initial screening of genomic DNA isolated from pe-ripheral blood leukocytes from the proposita for thefour common GD mutations (N370S, L444P, 84insG,IVS2+1) identified one N370S allele. Sequence analysisof the entire coding region and all exon/intron bound-aries in the acid b-glucosidase gene was performed inorder to identify the second mutant allele. A C-to-Ttransition at genomic position 6821 (cDNA position1319) was discovered resulting in the nonconservativesubstitution of proline by leucine at amino acid position401 (P401L). Sequence analysis showed that the mu-tation was not derived from a recombinant event withthe downstream pseudogene. The patient’s mother wasfound to carry the N370S allele; her father was de-ceased and no samples were available for testing.
To determine the function of the P401L mutation,the cDNA for acid b-glucosidase containing this pointmutation was expressed in Sf9 cells using the baculo-virus expression system and the resultant mutant pro-tein was characterized. The P401L mutant displayedsignificantly reduced activity when compared to a nor-mal acid b-glucosidase control. The normal acid b-glu-cosidase allele expressed in Sf9 cells produced approxi-mately 8.4 × 10−2 units of activity per milligram of totalprotein toward the fluorescently labeled natural sub-strate, NBD-glucosylceramide, while the P401L alleleproduced only 1.9 × 10−2 units of activity per milligramof total protein. An antisense acid b-glucosidase controldisplayed essentially no activity.
The level of catalytic efficiency (kcat) for the mutantenzyme was determined by calculating its CRIM SA(amount of activity per milligram of acid b-glucosidaseprotein) relative to that of the expressed normal en-zyme, where relative amounts of acid b-glucosidaseprotein produced were determined from immunoblotsusing a polyclonal anti-acid b-glucosidase antibody.The P401L mutant protein had a CRIM SA value of0.21 (Table I) indicating a catalytic efficiency approxi-mately 5 times less than that of the normal enzyme.
DISCUSSION
Skeletal involvement is the most frequent cause ofpain and disability in Type 1 GD [Stowens et al., 1985,Hermann et al., 1986]. There is a broad spectrum ofskeletal disease, ranging from mild osteopenia to avas-cular necrosis and pathologic fractures [Beighton et al.,1982; Mankin et al., 1990]. Although mandibular in-volvement has only been reported in the dental litera-
Fig. 2. Microscopic demonstration in mandibular biopsy specimen ofsheets of amphophilic histiocytes with “wrinkled” tissue paper cytoplasmicappearance.
336 Wasserstein et al.
ture [Michanowicz et al., 1967; Weigler et al., 1967;Bildman et al., 1972; Hall et al., 1985; Lustmann et al.,1991; Bender and Bender, 1996], the incidence of jawpathology in Type 1 GD is probably higher than previ-ously suspected. In fact, a recent study reported that89.3% of a small series (n 4 28) of Type 1 Gaucherpatients had radiographic changes in the mandible[Carter et al., 1998].
Mutation analysis in this young patient with severeskeletal manifestations and organ involvement re-vealed that the proposita was heteroallelic for the mildN370S allele and a novel lesion in exon 9. The previ-ously undescribed C-to-T transition at genomic posi-tion 6821 resulted in the nonconserved substitution ofa proline by a leucine at amino acid position 401(P401L). The substitution of the rigid, turn-producingproline residue by a nonpolar leucine residue would be
expected to produce a very disruptive mutation in thisregion of the acid b-glucosidase protien which containsthe substrate and activator binding domains (residues323–463) [Grace et al., 1994] and the catalytic nucleo-phile [Miao et al., 1994]. However, expression andcharacterization studies of the P401L mutant acidb-glucosidase demonstrated marked in vitro loss ofcatalytic function with a CRIM SA of 0.21, a valuewhich was similar to that of the mutant enzyme en-coded by the mild N370S allele (CRIM SA 4 0.24)[Grace, et al., 1990]. Thus, the proposita’s N370S/P401L genotype would have predicted a mild form ofType 1 GD, similar to the N370S/N370S phenotype[Zimran et al., 1989; Sibille et al., 1993]. However, theearly onset of extensive skeletal and organ involvementsuggests that the P401L mutation is associated with amore severe Type 1 disease phenotype. Although the
Fig. 3. A: Proximal right femur. B: Right tibia and fibula. C: Right humerus. D: Right radius and ulna. E: Right distal femur. Radiographs of the longbones show cystic lucencies, endosteal scalloping, and osteopenia. Erlenmeyer flask deformity is present in the distal femur. (Continued on the overleaf.)
P401L Allele in Severe Type 1 Gaucher Disease 337
underlying mechanism responsible for the more severephenotype is not known, it is tempting to speculatethat since acid b-glucosidase is active as a monomerand predicted to function in vivo in an aggregate[Weiler et al., 1995], the N370S/P401L enzyme aggre-gate may have less activity than that of the N370S/N370S enzyme aggregate.
The occurrence of mandibular disease in Type 1 GDpatients suggests that all patients should be evaluatedperiodically to detect mandibular skeletal involvement.The consequences of medullary bone loss in the man-dible may be severe. Osteomyelitis is a major concern.Gaucher-involved bone appears to have an increasedsusceptibility to infection which is most likely due tothe presence of necrotic material and decreased bloodsupply, which together provide an optimal environ-ment for microorganisms [Noyes and Smith, 1971].Hall described a case of mandibular osteomyelitis
which developed after an odontogenic infection in a pa-tient with Type 1 GD [Hall et al., 1985]. In fact, mostcases of mandibular osteomyelitis are secondary to un-treated dental infections [Sapp et al., 1997]. Osteomy-elitis of the mandible is often a mixed infection withseveral organisms from the oral cavity, and may re-quire surgical intervention [Mitchell et al., 1978]. An-other concern is the potential for pathologic fractures,which usually occur in areas of pre-existing bone le-sions where the cortex is thin and the trabeculae areweakened. In the case described above, less than 1 mmof cortical bone remained. Lastly, there is a risk oftooth loss due to decreased blood supply to the dentalpulp. Thus, patients with Type 1 GD should have den-tal evaluations to assess mandibular involvement. Ifthere is evidence of disease, then the importance ofgood dental hygiene should be stressed to avoid odon-togenic infections and secondary osteomyelitis in the
Fig. 3. (Continued).
338 Wasserstein et al.

involved bone. In addition, a soft diet should be recom-mended to avoid pathologic fractures of the jaw.
ACKNOWLEDGMENTS
The authors thank Ms. Katie Gonzalez for excellenttechnical assistance. This work was supported in partby a grant to the Mount Sinai General Clinical Re-search Center (5 M01 RR00071) from the NationalCenter for Research Resources, and a grant (F-FY96-1062) from the March of Dimes Birth Defects Founda-tion (M.G.). Dr. Wasserstein is the recipient of a YoungInvestigator Award from the Mount Sinai Child HealthResearch Center (5 P30 HD28822).
REFERENCESBeighton P, Goldblatt J, Sacks S. 1982. Bone involvement in Gaucher
disease. In Desnick RJ, Gatt S, Grabowski, editors. Gaucher disease: acentury of delineation and research. New York: Alan R. Liss. p107–129.
Bender IB, Bender AL. 1996. Dental observations in Gaucher’s disease.Review of the literature and two case reports with 13- and 60-yearfollow ups. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 82:650–659.
Beutler E, Gelbart T. 1990. Gaucher disease associated with a uniqueKpn1 restriction site: identification of the amino acid substitution. AnnHum Genet 54:149–153.
Beutler E, West C, Gelbart T. 1992. Polymorphisms in the human gluco-cerebrosidase gene. Genomics 12:795–800.
Beutler E, Grabowski GA. 1995. Gaucher disease. In: Scriver CR, BeaudetAL, Sly WS, Valle D, editors. The metabolic and molecular bases ofinherited disease. 7th ed. New York: McGraw-Hill. p 2641–2670.
Beutler E, Gelbart T. 1998. Hematologically important mutations: Gau-cher disease. Blood Cells Mol Dis 24:2–8.
Bildman B, Martinez M, Robinson LH. 1972. Gaucher’s disease discoveredby mandibular biopsy: report of a case. J Oral Surg 30:510–512.
Brady RO, Kanfer JN, Shapiro D. 1965. Metabolism of glucocerebrosides.II. Evidence of an enzymatic deficiency in Gaucher’s disease. BiochemBiophys Res Commun 18:221–225.
Carter LC, Fischman SL, Mann J, Elstein D, Stabholz A, Zimran A. 1998.The nature and extent of jaw involvement in Gaucher disease: obser-vations in a series of 28 patients. Oral Surg Oral Med Oral Pathol OralRadiol Endod 85:233–239.
Cox TM, Schofield JP. 1997. Gaucher’s disease: clinical features and natu-ral history. Clin Haematol 10:657–689.
Desnick RJ, Grabowski GA, Gatt S, editors. 1982. Gaucher disease: a cen-tury of delineation and research. New York: Alan R. Liss, Inc. p 1–30.
Eto Y, Kawame H, Hasegawa Y, Ohashi T, Ida H, Tokoro T. 1993. Molecu-lar characteristics in Japanese patients with lipidosis: novel mutationsin metachromatic leukodystrophy and Gaucher disease. Mol Cell Bio-chem 119:179–184.
Grabowski GA, Horowitz H. 1997. Gaucher’s disease: molecular, geneticand enzymological aspects. Clin Haematol 10:635–656.
Grace ME, Graves P, Smith F, Grabowski GA. 1990. Analysis of catalytic
activity and inhibitor binding of human acid b-glucosidase by site-directed mutagenesis. J Biol Chem 265:6827–6835.
Grace ME, Berg A, He GS, Goldberg L, Horowitz M, Grabowski GA. 1991.Gaucher disease: heterologous expression of two alleles associated withneuronopathic phenotypes. Am J Hum Genet 49:646–655.
Grace ME, Newman KM, Scheinker V, Berg-Fussman A, Grabowski GA.1994. Analysis of human acid b-glucosidase by site-directed mutagen-esis and heterologous expression. J Biol Chem 269:2283–2291.
Grace ME, Desnick RJ, Pastores GM. 1997. Identification and expressionof acid beta-glucosidase mutations causing severe type 1 and neuro-logic type 2 Gaucher disease in non-Jewish patients. J Clin Invest99:2530–2537.
Hall MB, Brown RW, Baughman RA. 1985. Gaucher’s disease affecting themandible. J Oral Maxillofac Surg 43:210–213.
He GS, Grace ME, Grabowski GA. 1992. Gaucher disease: four rare allelesencoding F2131, P289L, T323I, and R463C in type 1 variants. HumMutat 1:423–427.
Hermann G, Goldblatt J, Levy RN, Goldsmith SJ, Desnick RJ, GrabowskiGA. 1986 Gaucher’s disease type 1: assessment of bone involvement byCT and scintigraphy. Am J Roentgenol 147:943–948.
Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. 1989.The human glucocerebrosidase gene and pseudogene: structure andevolution. Genomics 4:87–96.
Levy-Lahad E, Zimran A. 1997. Gaucher’s disease: genetic counselling andpopulation screening. Clin Haematol 10:779–792.
Lustmann J, Ben-Yehuda D, Somer M, Ulmansky M. 1991. Gaucher’s dis-ease affecting the mandible and maxilla: report of a case. Int J OralMaxillofac Surg 20:7–8.
Mankin HJ, Doppelt SH, Rosenberg AE, Barranger JA. 1990. Metabolicbone disease in patients with Gaucher disease. In : Avioli LV, KraneSM, editors. Metabolic bone disease. 2nd ed. Philadelphia: W.B. Saun-ders. p 730–752.
Mansuno M, Tomatsu S, Sukegawa K, Orii T. 1990. Non-existence of atight association between a 444leucine to proline mutation and pheno-types of Gaucher disease: high frequency of an NciI polymorphism inthe non-neuronopathic form. N Engl J Med 316:470–575.
Miao S, McCarter JD, Grace ME, Grabowski GA, Aebersold R, Withers SG.1994. Identification of Glu340 as the active-site nucleophile in humanglucocerebrosidase by use of electrospray tandem mass spectrometry. JBiol Chem 269:10975–10978.
Michanowicz AE, Michanowicz JP, Stein GM. 1967. Gaucher’s disease:report of a case. Oral Surg Oral Med Oral Pathol 23:36–41.
Mitchell DR, Standish SM, Fast TB. 1978. Diseases of the jaw bones. In:Oral diagnosis/oral medicine. Philadelphia: Lea & Febiger. p 460–461.
Noyes FR, Smith WS. 1971. Bone crises and chronic osteomyelitis in Gau-cher’s disease. Clin Orthop 79:132–140.
Pasmanik-Chor M, Madar-Shapiro L, Stein OE, Arets H, Gatt S, HorowitzM. 1997. Expression of mutated glucocerebrosidase alleles in humancells. J Med Mol Genet 6:887–895.
Sambrook J, Fritsch E, Maniatis T. 1989. Molecular cloning Cold SpringHarbor, NY: Cold Spring Harbor Laboratory Press. p 9.16–9.23
Sapp JP, Eversole LR, Wysocki GP. 1997. Infections of teeth and bone. In:Contemporary oral and maxillofacial pathology. St. Louis: Mosby. p80–82.
Sibille A, Eng CM, Kim SJ, Pastores G, Grabowski GA. 1993. Phenotype/genotype correlations in Gaucher disease type 1: clinical and therapeu-tic implications. Am J Hum Genet 52:1094–1101.
Stowens DW, Teitelbaum SL, Kahn AJ, Barranger JA. 1985. Skeletal com-plications in Gaucher disease. Medicine 64:310–322.
Tsuji S, Choudary PV, Martin BM, Stubblefield BK, Mayor JA, BarrangerJA, Ginns EI. 1987. A mutation in the human glucocerebrosidase genein neuronopathic Gaucher’s disease. N Engl J Med 316:570–575.
Weigler JM, Seldin R, Minkowitz S. 1967. Gaucher’s disease involving themandible: report of a case. J Oral Surg 25:158–163.
Weiler S, Kishimoto Y, O’Brien JS, Barranger JA, Tomich JM. 1995. Iden-tification of the binding and activating sites of the sphingolipid activa-tor protein, saposin C, with glucocerebrosidase. Protein Sci 4:756–764.
Zimran A, Sorge J, Gross E, Kubitz M, West C, Beutler E. 1989. Predictionof severity of Gaucher’s disease by identification of mutations at DNAlevel. Lancet 2:349–352.
TABLE I. Characterization of Acid b-Glucosidases Expressedin Sf9 Cells
cDNASpecific activitya
(mmol/min/mg) CRIM SAb 1/CRIM SA
Normal 8.4 × 10−2 1.0 1.0Antisense 0.0 No CRIM —P401L 1.9 × 10−2 0.21 4.8N370S 1.8 × 10−2 0.23c 4.2L444P 3.9 × 10−3 0.05c 20
aUsing NBD-labeled glucosylceramide as substrate.bCRIM SA, total units of specific activity per cross-reacting immunologicalmaterial.cAverage of data from this and earlier studies [Grace et al., 1990, 1994].
P401L Allele in Severe Type 1 Gaucher Disease 339