Embed Size (px)
Citation preview

British lournal of Haematology, 1987. 66, 349-354
Type IIB von Willebrand’s disease with probable autosomal recessive inheritance and presenting as thrombocytopenia in infancy
MIKAEL DONNER. LARS HOLMBERG AND INGA MARIE NILSSON* Department of Paediatrics, University Hospital, Lund, and *Coagulation Laboratory, MaZmB General Hospital, MalmB, Sweden
Received 8 September 1986; accepted for publication 8 January 1987
Summary. von Willebrand’s disease (vWD) is a congenital bleeding disorder that exists in two main forms. In the classic form, type I, the concentration of the von Willebrand factor (vWF) in plasma is decreased. In type II vWD, the vWF is structurally altered. Type I1 can be further divided into at least six subtypes (A, B, C, D. E and F). In type IIB the vWF, in contrast to other variants of vWD, shows an increased
affiity for platelets. IIB vWD is generally believed to be inherited in an autosomal dominant manner. We describe two families with three affected children in whom an autosomal recessive inheritance is more likely. Thrombocy- topenia, constant or variable, was present from early infancy in all three cases. Type IIB vWD should thus be included in the differential diagnosis of congenital thrombocytopenia.
von Willebrand’s disease (vWD) is a congenital bleeding disorder, of which symptoms are often evident early in childhood. The basic defect in vWD is a quantitative or qualitative deficiency of the von Willebrand factor (vWF), a large glycoprotein normally found in plasma, platelets and vessel walls (Bloom et al, 1973; Nachman et al. 1977). In plasma, the vWF occurs as a series of multimers of different molecular weight (large, medium and small multimers) (Zimmerman & Ruggeri, 1983). vWD can be divided into two main types, I and II (Zimmerman & Ruggeri, 1983; Holmberg & Nilsson, 1985). In type I the concentration of vWF is decreased, in type I1 the protein is qualitatively altered. Type II can be further divided into six subgroups, namely II A, B, C, D, E and F (Zimmerman et al, 1986, Mannucci et al, 1986). a classification based on changes in the multimeric composi- tion of vWF in plasma and platelets, on the properties of the vWF in functional tests, and on the mode of inheritance.
In type IIB vWD the large vWF multiiers are absent from plasma but present in platelets (Ruggeri & Zimmerman, 1980). In contrast to all other forms of vWD. type IIB vWF shows an enhanced interaction with platelets. This can be demonstrated by examining platelet aggregation in response to addition of the antibiotic ristocetin at varying concentra- tions to platelet-rich plasma. In type W. platelets aggregate at a much lower ristocetin concentration than that needed in normal platelet rich-plasma (Ruggeri et al. 1980).
Correspondence: Professor L. Holmberg. Department of Paediatrics, University Hospital, S-22 1 8 5 Lund, Sweden.
Increased platelet-vWF interaction has also recently been demonstrated in the absence of ristocetin (Holmberg et al, 1983, 1985: DeMarco et al. 1985). Some patients with type W vWD have thrombocytopenia obviously owing to abnor- mal vWF-platelet interaction and platelet aggregation in vivo. Thrombocytopenia can also be induced by DDAVP treat- ment, which increases the plasma vWF content (Holmberg et al, 1983).
Most forms of vWD. including type W, are inherited as autosomal dominant traits. We here report three children, from two families, each of whom exhibited all the laboratory characteristics of type IIB and severe thrombocytopenia in infancy but in whom autosomal recessive inheritance was more likely.
MATERIALS AND METHODS
Blood collection, platelet count, bfeeding time, F VH1:C, vWF: Ag, ristocetin cofactor activity (Rcos), crossed immunoelec- trophoresis (CIE). For further details see Holmberg et al (1986).
Washed platelets. The platelets in citrated PRP were gently centifuged (800 8. 30 min) onto a cushion of 35% bovine serum albumin (BSA) in 0.01 M phosphate buffer, 0.1 M NaCI. pH 6.6. The platelet layer was aspirated, the platelets re-suspended in the same buffer and washed three times in the same way. Finally, the platelets were re-suspended in Tyrode’s buffer at a concentration of 4 x lon platelets/ml.
Platelet lysates. These were prepared as described pre-
349

350 M. DonnCr, L. Holmberg and I . M. Nilsson viously (Holmberg et al, 1986). The amount of von Wille- brand factor antigen (vWF: Ag) in the lysate was determined by immunoradiometric assay (IRMA), and further analysed by multimeric sizing (see below).
Ristocetin-induced platelet aggregation (RIPA) was measured in platelet-rich plasma as described by Ruggeri et d (1 980). within 30 min after blood collection. The ristocetin concen- tration necessary to induce aggregation with an initial velocity of 20 mm/min was extrapolated from the aggreg- ometer tracings at a number of Werent concentrations. In normal PRP (n= 7), this concentration ranged from 1-00 to 1.70 mg/ml. Thus, aggregation (20 mm/min) occurring at a ristocetin concentration below 1.00 mg/ml was taken as evidence of increased ristocetin sensitivity.
Multimeric sizing. The mdtimeric distribution of vWF:Ag in platelet-poor plasma and platelet lysates was analysed by low and high resolution sodium dodecyl sulphate (SDS)- agarose electrophoresis (1.9% and 2.5% agarose concentra- tion, respectively) (Ruggeri & Zimmerman, 1981; Mannucci et al, 1985). The bands corresponding to the multimers were identified in the gels by reaction with a 1251-labelled mouse monoclonal antibody (Holmberg et d. 1986).
Binding of vWF:Ag to platelets. Washed platelets from a normal person were incubated with normal or patient plasma for 15 min at room temperature without stirring. Platelets were separated, and residual vWF:Ag was mea- sured in the supernatant by IRMA and expressed as a percentage of the starting value. Patient platelets for mixing experiments with normal plasma could not be obtained from the patient in Family I1 because of a low platelet count and spontaneous platelet aggregation in vitro during separation.
PATIENTS
Family I (Fig 1). The proband in this family is a girl born in February 1977. Delivery was terminated by vacuum extrac- tion. The newborn child had a haematoma as large as an orange in the lumbar region. During the first year of life she bruised readily. After a severe nose bleeding at the age of 9
months, she was given a thorough examination. The patient has been followed for 8 years. She has had frequent minor nasal bleedings, and now and then more severe nasal bleedings. Laboratory Rndings have been consistent except for her platelet count (see below). Both parents are healthy and have never had any bleeding symptoms. They are not blood relatives, the father W i g British, the mother Swedish. In 1979 they had a second child, a girl who also had bleeding symptoms. Apart from these two girls, no other family members were found to have any bleeding manifestations.
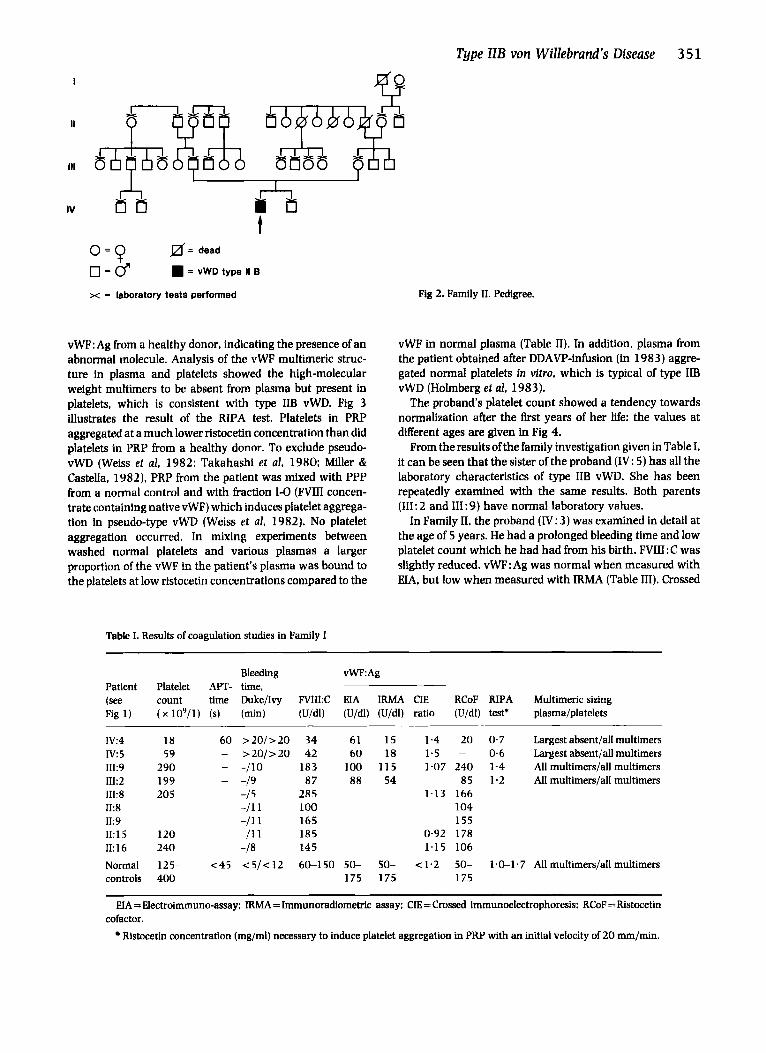
Family II (Fig 2). The proband is a boy born in August 1979. Because of maternal toxaemia of pregnancy he was delivered by caesarean section. During the neonatal period he showed a marked bleeding tendency at blood sampling. A low platelet count was discovered. The thrombocytopenia remained constant and the patient had to be admitted to hospital several times because of severe nose bleedings requiring blood transfusion. The condition was 6rst inter- preted as ITP, and corticosteroids were prescribed, but neither platelet count nor bleeding symptoms improved. At the age of 5 years he was re-investigated. Both parents are healthy without any bleeding problems. They are not blood relatives. Apart from the proband, no other family members were found to be affected.
RESULTS
The laboratory findings in the two families are given in Tables I and m.
In Family I the proband was 6rst examined in detail at the age of 9 months. She had prolonged bleeding time and activated partial thromboplastin time. Platelet count was 18 x 109/1. Megakaryocytes were abundant in the bone marrow aspirate. Young forms with conspicuous basophilia predominated, indicating an active megakaryopoiesis. PVIII: C was decreased and vWF: Ag was low when measured by IRMA, but normal when measured by electroimmuno- assay (HA). Crossed immunoelectrophoresis of vWF : Ag showed increased anodal mobility, as compared with
I
I11
I V
O=? o=d
0 = ~ W O type IIB x = laboratory tests performed
6 Fig 1. Family I. Pedigree.
Type ZZB von Willebrand's Disease 351
1
I I
I I I I
t 0 = 9 a= dead
0-0" = vWD type II B
x = laboratory teats performed
vWF : Ag from a healthy donor, indicating the presence of an abnormal molecule. Analysis of the vWF multimeric struc- ture in plasma and platelets showed the high-molecular weight multimers to be absent from plasma but present in platelets, which is consistent with type IIB vWD. Fig 3 illustrates the result of the RIPA test. Platelets in PRP aggregated at a much lower ristocetin concentration than did platelets in PRP from a healthy donor. To exclude pseudo- vWD (Weiss et al, 1982: Takahashi et al, 1980; Miller & Castella, 1982), PRP from the patient was mixed with PPP from a normal control and with fraction 1-0 (FVIII concen- trate containing native vWF) which induces platelet aggrega- tion in pseudo-type vWD (Weiss et al, 1982). No platelet aggregation occurred. In mixing experiments between washed normal platelets and various plasmas a larger proportion of the vWF in the patient's plasma was bound to the platelets at low ristocetin concentrations compared to the
Table 1. Results of coagulation studies in Family I
Fig 2. Family II. Pedigree.
vWF in normal plasma (Table II). In addition, plasma from the patient obtained after DDAVP-infusion (in 1983) aggre- gated normal platelets in vitro. which is typical of type IIB vWD (Holmberg et al, 1983).
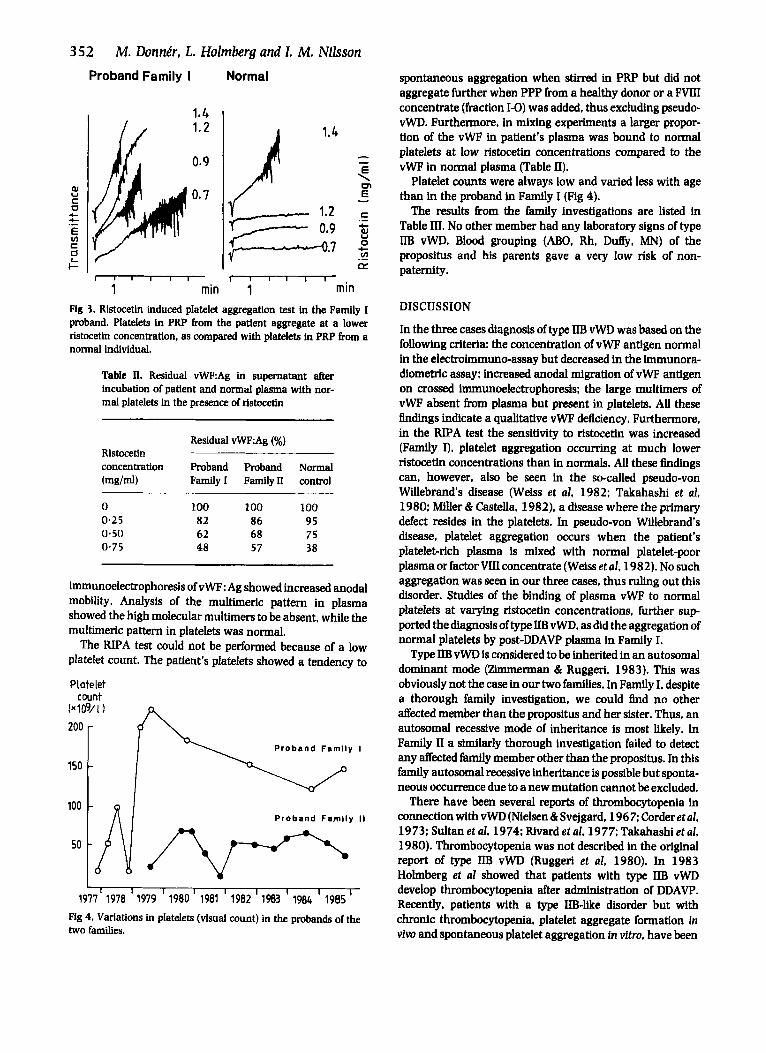
The proband's platelet count showed a tendency towards normalization after the first years of her life: the values at different ages are given in Fig 4.
From the results of the family investigation given in Table I, it can be seen that the sister of the proband (IV: 5) has all the laboratory characteristics of type IIB vWD. She has been repeatedly examined with the same results. Both parents (111: 2 and 111: 9) have normal laboratory values.
In Family 11, the proband (IV: 3) was examined in detail at the age of 5 years. He had a prolonged bleeding time and low platelet count which he had had from his birth. FV1II:C was slightly reduced. vWF:Ag was normal when measured with EIA, but low when measured with IRMA (Table III). Crossed
Bleeding vWF:Ag Patient Platelet APT- time, (see count time Duke/Ivy FV1II:C HA IRMA CIE RCoF RIPA Multimeric sizing Fig 1) ( x loy / ] ) (s) (min) (U/dl) (U/dl) (U/dl) ratio (U/dl) test* plasma/platelets
Iv:4 I V 5 III:9 III:2 III:8 II:8 II:9 II:15 11: 1 6
18 60 > 2 0 / > 2 0 34
290 - -/lo 183 199 - -19 8 7 205 -/ 5 285
-11 1 1 0 0 -/11 165
120 -111 185 240 -/a 145
59 - > 2 0 / > 2 0 42 61 15 1 .4 2 0 0 .7 Largest absent/all multimers 6 0 18 1.5 - 0 . 6 Largest absent/all multimers
100 11 5 1.07 240 1 . 4 All multimers/all multimers 88 5 4 85 1.2 All multimers/all multimers
1.13 166 104 155
0.92 178 1.15 106
Normal 125- <45 < 5 / < 1 2 6(tl50 50- 50 - ~ 1 . 2 50- 1.0-1.7 Allmultimers/allmultimers controls 400 175 175 175
EIA = Electroimmuno-assay: IRMA = ImmunoradiomeMc assay: CIE = Crossed immunoelectrophoresis: RCoF = Ristocetin
Ristocetin concentration (mg/ml) necessary to induce platelet aggregation in PRP with an initial velocity of 2 0 mm/min. cofactor.
352 M. Donnkr, L. Holmberg and 1. M. Nilsson
200
150
100
50
Proband Family I Normal
- Proband Faml ly I
-
- Proband Fami ly I I
-
I 1*4 I
I I I I I I - 1 min 1 min
Fig 3. Ristocetin induced platelet aggregation test in the Family I proband. Platelets in PRP from the patient aggregate at a lower ristocetii concentration, as compared with platelets in PRP from a normal individual.
Table II. Residual vWP:Ag in supernatant after incubation of patient and nonnal plasma with nor- mal platelets in the presence of ristocetin
Residual vWF:Ag (96) Ristocetin concentration Proband Proband Nonnal (mg/mU Family1 Familyn control
0 100 100 100 0.25 82 86 95 0.50 62 68 75 0-75 48 57 38
immunoelectrophoresis of vWF : Ag showed increased anodal mobility. Analysis of the multimeric pattern in plasma showed the high molecular multimers to be absent, while the multimeric pattern in platelets was normal.
The RIPA test could not be performed because of a low platelet count. The patient's platelets showed a tendency to
Platelet count
L
1977' 1978 I 1979 I 1980 I 1981 I 1982 I 1983 ' 1984 I 1985 ' Fig 4. Variations in platelets (visual count) in the probands of the two families.
spontaneous aggregation when stirred in PRP but did not aggregate further when PPP from a healthy donor or a FWII concentrate (fraction 1-0) was added, thus excluding pseudo- vWD. Furthermore, in mixing experiments a larger propor- tion of the vWF in patient's plasma was bound to normal platelets at low rlstocetin concentrations compared to the vWF in normal plasma (Table II).
Platelet counts were always low and varied less with age than in the proband in Family I (Fig 4).
The results from the family investigations are listed in Table III. No other member had any laboratory signs of type IIB vWD. Blood grouping ( N O , Rh, Du&, h4N) of the propositus and his parents gave a very low risk of non- paternity.
DISCUSSION
In the three cases diagnosis of type IIB vWD was based on the following criteria: the concentration of vWP antigen normal in the electroimmuno-assay but decreased in the immunora- diometric assay: increased anodal migration of vWF antigen on crossed immunoelectrophoresis; the large multiiers of vWF absent from plasma but present in platelets. All these findings indicate a qualitative vWF deficiency. Furthermore, in the RJPA test the sensitivity to ristocetin was increased (Family I), platelet aggregation occurring at much lower ristocetin concentrations than in normals. All these findings can, however, also be seen in the so-called pseudo-von Willebrand's disease (Weiss et al, 1982; Takahashi et al. 1980: Miller & Castella, 1982). a disease where the primary defect resides in the platelets. In pseudo-von Willebrand's disease, platelet aggregation occurs when the patient's platelet-rich plasma is mixed with normal platelet-poor plasma or factor Wconcentrate (Weiss et al. 1982). No such aggregation was seen in our three cases, thus ruling out this disorder. Studies of the binding of plasma vWF to normal platelets at varying rlstocetfn Concentrations, further sup ported the diagnosis of type IIB vWD, as did the aggregation of normal platelets by post-DDAVP plasma in Family I.
Type ID3 vWD is considered to be inherited in an autosomal dominant mode (Zlmmerman & Ruggeri. 1983). This was obviously not the case in our two families. In Family I, despite a thorough family investigation. we could find no other d a t e d member than the propositus and her sister. Thus, an autosomal recessive mode of inheritance is most likely. In Family 11 a similarly thorough investigation failed to detect any affected family member other than the propositus. In this family autosomal recessive inheritance is possible but sponta- neous occurrence due to a new mutation cannot be excluded.
There have been several reports of thrombocytopenia in connection with vWD (Nielsen & Svejgard, 1967: Corder et al. 1973; Sultan et d, 1974; Rivard et nl, 1977; Takahashi et al, 1980). Thrombocytopenia was not described in the original report of type IIB vWD (Ruggeri et al, 1980). In 1983 Holmberg et al showed that patients with type W vWD develop thrombocytopenia afler administration of DDAVP. Recently, patients with a type IIB-like disorder but with chronic thrombocytopenia, platelet aggregate formation in viw and spontaneous platelet aggregation in vitro, have been
Type ZZB von Willebrand’s Disease 353 Table m. Results of coagulation studies in Family I1
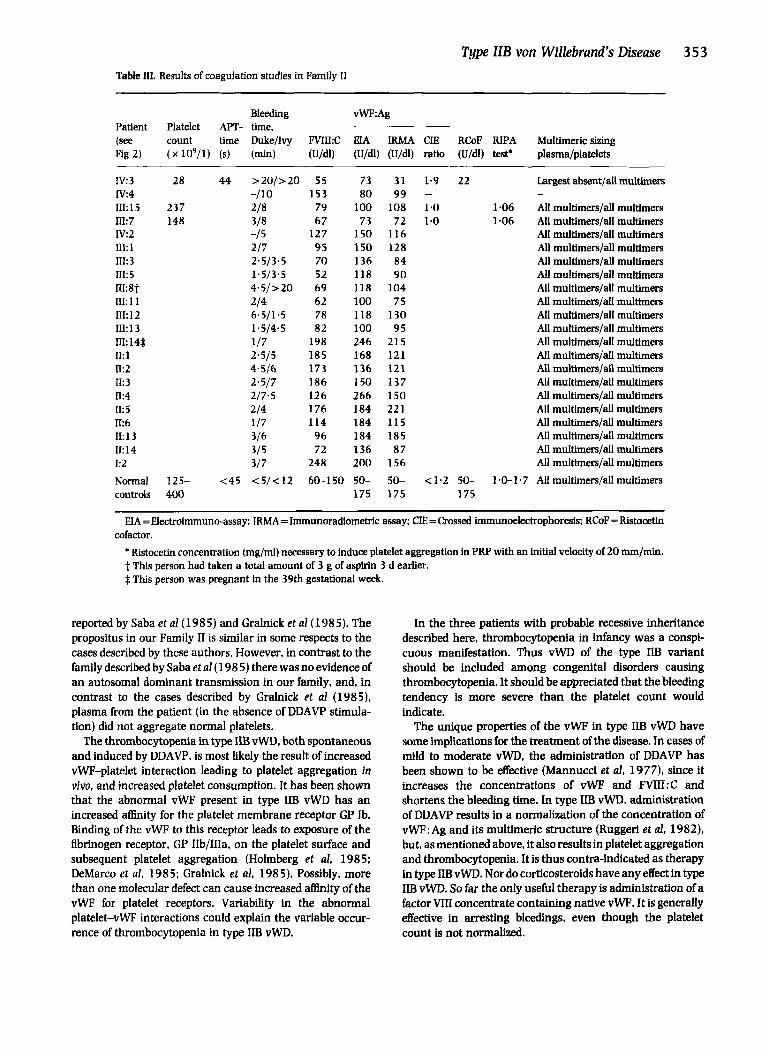
Bleeding vWF:Ag Patient Platelet APT- time. (see count time Duke/Ivy FVIkC EIA IRMA CIE RCoF RIPA Multimericsizing Fig 2) ( x 109/1) (s) (min) (U/dl) (U/dl) (U/dl) ratio (U/dl) test’ plasma/platelets
I V 3 28 44 >20/>20 55 73 31 1.9 22 Largest absent/all multimers
III:15 237 218 79 100 108 1-0 1.06 All multimers/all multimers In: 7 148 318 67 73 72 1.0 1 -06 AU multimers/all multimers IV:2 -/ 5 127 1 5 0 116 All multimers/all multimers m:i 2/7 95 150 128 All multimers/all multimers n1:3 2.5/3,5 70 136 84 All multimers/all multirners m:s 1.5/3.5 52 118 9 0 All multimers/all multimers n1:8t 4 .5 />20 69 118 104 All multimers/d multimers 111: 1 1 214 62 100 7 5 All multimers/all multimers 111: 12 6.5/1.5 78 118 130 All multimers/all multimers m:i 3 1.5/4.5 82 100 95 All multimers/all multimers n1:14$ 1/7 198 246 215 All multimers/all multimers n: 1 2.5/5 185 168 121 All multimers/all multimers n:2 4*5/6 173 136 121 All multimers/all multimers II: 3 2-5/7 186 150 137 AU muItimers/all rnultimers n:4 2/7.5 126 266 150 All multimers/all multimers 11:s 2/4 176 184 221 All multimers/all multimers k 6 1/7 114 184 115 All multimers/d multimers II:13 3/6 96 184 185 All multimers/all multimers 11: 14 315 72 136 87 All multimers/all multimers I:2 3/7 248 200 156 All multimers/all multimers Normal 125- <45 <5/<12 60-150 5 0 - 5 0 - ~ 1 . 2 50- 1.0-1.7 Allmultimers/allmultimers controls 400 175 175 175
IV:4 -/lo 153 80 99 - -
H A =Electroimmuno-assay: IRMA =Immunoradiometric assay; CIE = Crossed immunoelectrophoresis: RCoF = Ristocetin
* Ristocetii concentration (mg/ml) necessary to induce platelet aggregation in PRP with an initid velocity of 20 mm/min. t This person had taken a total amount of 3 g of aspirin 3 d earlier. 4 This person was pregnant in the 39th gestational week.
cofactor.
reported by Saba et a1 (1985) and Gralnick et a1 (1985). The propositus in our Family I1 is similar in some respects to the cases described by these authors. However, in contrast to the family described by Saba et al(198 5) there was no evidence of an autosomal dominant transmission in our family, and, in contrast to the cases described by Gralnick et d (1985). plasma from the patient (in the absence of DDAVP stimula- tion) did not aggregate normal platelets.
The thrombocytopenia in type IIB vWD, both spontaneous and induced by DDAVP, is most likely the result of increased vWF-platelet interaction leading to platelet aggregation in viva and increased platelet consumption. It has been shown that the abnormal vWF present in type IIB vWD has an increased a h i t y for the platelet membrane receptor GP Ib. Binding of the vWF to this receptor leads to exposure of the fibrinogen receptor, GP IIb/IIIa, on the platelet surface and subsequent platelet aggregation (Holmberg et al, 1985; DeMarco et al, 1985; Gralnick et al, 1985). Possibly, more than one molecular defect can cause increased aflinity of the vWF for platelet receptors. Variability in the abnormal platelet-vWF interactions could explain the variable occuc- rence of thrombocytopenia in type IIB vWD.
In the three patients with probable recessive inheritance described here, thrombocytopenia in infancy was a conspi- cuous manifestation. Thus vWD of the type IIB variant should be included among congenital disorders causing thrombocytopenia. It should be appreciated that the bleeding tendency is more severe than the platelet count would indicate.
The unique properties of the vWF in type IIB vWD have some implications for the treatment of the disease. In cases of mild to moderate vWD. the administration of DDAVP has been shown to be effective (Mannucci et d, 1977), since it increases the concentrations of vWF and FVIII:C and shortens the bleeding time. In type IIB vWD, administration of DDAVP results in a normalization of the concentration of vWF:Ag and its multimeric structure (Ruggeri et d, 1982), but, as mentioned above, it also results in platelet aggregation and thrombocytopenia. It is thus contra-indicated as therapy in type W vWD. Nor do corticosteroids have any effect in type IIB vWD. So far the only useful therapy is administration of a factor Vm concentrate containing native vWF. It is generally effective in arresting bleedings, even though the platelet count is not normalized.

354 M. Donnir, L. Holmberg and I . M. Nilsson ACKNOWLEDGMENT
This study was supported by grants from the Swedish Medical Research Council (Nos. 04997 and 00087). and the Faculty of Medicine, University of Lund.
REFERENCES
Bloom, A.L., Giddings, J.C. & Wilks. C.J. (1973) Factor WII on the vascular intima: possible importance in haemostasis and thrombo- sis. Nature; New Biology, 241, 217-219.
Corder, M.P.. Culp. N.W. & Barrett, 0.. Jr (1973) Familial Occurrence of von Willebrand’s disease, thrombocytopenia, severe gastroin- testinal bleeding. American Journal of Medical Sciences, 265, 219- 223.
DeMarco. L., Girolami, A.,Zimmerman,T.S. &Ruggeri. Z.M. (1985) Interaction of puriEed type IIB von Wfflebrand factor with the platelet membrane glycoprotein IIb induces Rbrinogen binding to the glycoprotein IIb/IIIa complex and initiates aggregation. Pro- ceedings of the National Academy of Sciences of the United States of America, 82, 7424-7428.
Gralnick. H.R., Williams. S.B.. McKeown, L.P.. Rick, M.E.. Maison- neuve. P., Jenneau. C. & Sultan, Y. (1985) von Willebrand’s disease with spontaneous platelet aggregation induced by an abnormal plasma von Willebrand factor. Journal of Clinical Investi- gation, 76, 1522-1529.
Holmberg, L., Berntorp. E., Donnhr, M. & Nilsson, LM. (1986) von Willebrand’s disease characterized by increased rlstocetin sensiti- vity and the presence of all von Willebrand factor multimers in plasma. Blood, 68, 668-672.
Holmberg, L., Kristoffemon, A.C.,Lamme, S., Nilsson, LM.. Awidl, A. & Solum. N.O. (1985) Platelet-von Willebrand factor interactions in type W von Wfflebrand’s disease. Scandinavian Journal 01 Haematology, 35. 305-3 14.
Holmberg, L. & Nilsson. LM. (1 98 5) von Willebrands disease. Clinics in Haematologu, 14, 461488 .
Holmberg. L., Nilsson, I.M., Borge, L., Gunnarsson. M. & Sjorin, E. (1983) Platelet aggregation induced by ldesadno-8-u-arginine vasopressin (DDAVP) in type IIB von Willebrand’s disease. New EnglandJoumal of Medicine. 309, 816-821.
Mannucci. P.M., Abildgaard, C.F.. Gralnick. H.R., Hffl, F.G.H., Hoyer. L.W.. Lombardi. R., Nilsson, LM., Tuddenham, E. & Meyer, D. (1985) Multicenter cornparision of von Wfflebrand factor rnul- timer sizing techniques. Thrombosis and Hemostasis, 54,873.
Mannucci, P.M., Lombardi, R., Federici, A.B.. Dent, J.A.. Zimmer- man, T.S. & Ruggeri. Z.M. (1986) A new variant of Type II von Wfflebrand disease with aberrant multimeric structure of plasma but not platelet von Wdebrand factor (Type IF). Blood. 68,269- 274.
Mannucci. P.M.. Ruggeri, ZM., Pareti, F.I. & Capitano, A. (1977) 1- Desamino-8-warginine vasopressin: A new pharmacological approach to the management of haemophilia and von Wiile- brand’s disease. Lancet, I, 869-872.
Miller, J.L. & Castella. A. (1982) Platelet-type von Willebrand’s
disease: characterization of a new bleeding disorder. Blood, 60, 7 9 0- 7 9 4.
Nachman. R.. Levtne, R. & J d e , E.A. (1977) Synthesis of factor VIII antigen by cultured guinea pig megakaryocytes. Journal ofClinicu1 Investigation, 60, 914-921.
Nielsen. E.G. & Svejgard, A. (1967) von Willebrand’s disease associated with intermittent thrombocytopenia. Lancet. ii, 966- 968.
Ruggeri, Z.M.. Mannucci. P.M., Lornbardi. R.. Federici, A.B. & Zimmerman, T.S. (1982) Multimeric composition of factor W I / von Willebrand factor following administration of DDAVP Impli- cations for pathophysiology and therapy of von Willebrand’s disease subtypes. Blood, 59,1272-1278.
Ruggeri. Z.M., Pareti, F.L, Mannucci. P.M., Ciavarella. N. & Zimmer- man, T.S. (1980) Heightened interaction between platelets and factor Vm/von Willebrand factor in a new subtype of von Willebrands disease. New England lournal ofMedicine. 302,1047- 1051.
Ruggeri. Z.M. & Zimmerman, T.S. (1980) Variant von Willebrand’s disease: Characterization of two subtypes by analysis of multimeric composition of factor VIII/von Willebrand factor in plasma and platelets. Journal of Clinical Investigation, 65, 1318-1325.
Ruggeri. Z.M. & Zimmennan, T.S. (1981) The complex multimeric composition of factor VIII/von Willebrand factor. Blood, 57,1140- 1143.
Rivard. G.E., Daviault, M.B., Brault, N., D’Aragon, L. & Raymond, R. (1977) von Wiltebrand’s disease associated with thrombocytope- nia and a fast migrating factor VIII related antigen. Thrombosis Research, 11, 507-516.
Saba. H.I., Saba. S.R., Dent. J., Ruggeri, Z.M. & Zimmerman. T.S. (1 985) Type W Tampa: A variant of von Willebrand disease wtih chronic thrombocytopenia. circulating platelet aggregates and spontaneous platelet aggregation. Blood, 66, 282-286.
Sultan. Y., Bemal-Hoyos. E.J., Levy-Toledano. S.. Jenneau. C. & Caen, J.P. (1974) Dominant inherited familial factor VllI deRciency (von Wfflebrand dlsease) associated with thrombo-cytopathic thrornbo- cytopenia (biologic and genetic implications). Pathologie et Biologie,
Takahashi. H.. Sakuragawa. N. & Shibata. A. (1980) von Willebrand disease with an increased rlstocetin-induced platelet aggregation and a qualitative abnormality of the factor Vm protein. American Journal of Hematology, 8, 299-308.
Weiss, H.J., Meyer, D.. Rabinowitz. R.. et al (1982) Pseudo-von Willebrand’s disease: an intrinsic platelet defect with aggregation by unmodieed human factor VIII/von Willebrand factor and enhanced adsorption of its high-molecular-weight multimers. New England Journal of Medicine, 306, 326-3 3 3.
Zimmerman. T.S., Dent, J.A.. Ruggeri. Z.M., et al (1986) Subunit composition of plasma von Wdebrand factor. Cleavage is present, in normal individuals. increased in IIA and IIB von Willebrand. disease, but minimal in variants with aberrant structure (types IIC. LID and IIE). Journal of Clinical Investigation, 77, 947-951.
Zimmerman, T.S. & Ruggeri, Z.M. (1983) von Willebrand’s disease. Clinics in Haematology. 12, 175-200.
22.27-36.





























