Embed Size (px)
Citation preview

UNIVERSIDADE FEDERAL DO CEARÁ
CENTRO DE TECNOLOGIA
DEPARTAMENTO DE ENGENHARIA QUÍMICA
ANTONINO FONTENELLE BARROS JUNIOR
ESTIMAÇÃO DE PARÂMETROS, MODELAGEM E SIMULAÇÃO DA
SÍNTESE DE FISCHER-TROPSCH EM REATOR TUBULAR DE
LEITO FIXO COM CATALISADOR DE COBALTO
FORTALEZA
2013

ANTONINO FONTENELLE BARROS JUNIOR
ESTIMAÇÃO DE PARÂMETROS, MODELAGEM E SIMULAÇÃO DA
SÍNTESE DE FISCHER-TROPSCH EM REATOR TUBULAR DE LEITO
FIXO COM CATALISADOR DE COBALTO
Tese submetida à Coordenação do Curso de Pós-
Graduação em Engenharia Química, da
Universidade Federal do Ceará, como requisito
parcial para obtenção do grau de Doutor em
Engenharia Química.
Orientador: Prof. Dr. Fabiano André Narciso
Fernandes
FORTALEZA
2013

Dados Internacionais de Catalogação na Publicação
Universidade Federal do Ceará
Biblioteca de Pós-Graduação em Engenharia - BPGE
B274e Barros Junior, Antonino Fontenelle. Estimação de parâmetros, modelagem e simulação da síntese de Fischer-Tropsch em reator
tubular de leito fixo com catalisador de cobalto / Antonino Fontenelle Barros Junior. – 2013
116 f. : il. color., enc. ; 30 cm.
Tese (doutorado) – Universidade Federal do Ceará, Centro de Tecnologia, Departamento de
Engenharia Química, Programa de Pós-Graduação em Engenharia Química, Fortaleza, 2013.
Área de Concentração: Processos químicos e Bioquímicos.
Orientação: Prof. Dr. Fabiano André Narciso Fernandes.
1. Engenharia Quimica. 2. Catalisadores. I. Título.
CDD 660

ANTONINO FONTENELLE BARROS JUNIOR
ESTIMAÇÃO DE PARÂMETROS, MODELAGEM E SIMULAÇÃO DA
SÍNTESE DE FISCHER-TROPSCH EM REATOR TUBULAR DE LEITO
FIXO COM CATALISADOR DE COBALTO
Tese submetida à Coordenação do Curso de Pós-
Graduação em Engenharia Química, da
Universidade Federal do Ceará, como requisito
parcial para a obtenção do grau de Doutor em
Engenharia Química.
Aprovada em 01/03/2013
BANCA EXAMINADORA

À Luiza Helena, minha mãe.

AGRADECIMENTOS
À minha mãe, Luiza Helena, pelo estabelecimento de princípios éticos e morais, e
pela dedicação, apoio e ensinamentos fornecidos durante toda a minha vida.
À minha mulher, Fabyane, e às minhas filhas Melissa e Marcela, pelo carinho e
compreensão dispensados nos momentos de elaboração deste trabalho.
Ao meu orientador, Prof. Dr. Fabiano André Narciso Fernandes, por toda atenção
dispensada e pelas críticas e sugestões que contribuíram para o aprimoramento desta
dissertação.
A todos os amigos e professores que direta ou indiretamente auxiliaram na
conclusão deste trabalho.

RESUMO
A reação de síntese de Fischer-Tropsch, que pode ser compreendida como uma
polimerização entre os gases monóxido de carbono e hidrogênio, mistura conhecida por gás
de síntese, com a formação de hidrocarbonetos parafínicos e olefínicos, ocorre na presença de
catalisadores heterogêneos, onde aqueles de cobalto aparecem como os mais promissores
quando se deseja produzir frações de hidrocarbonetos comercialmente mais favoráveis, como
gasolina, diesel e graxas. A reação já é encarada como alternativa ao petróleo, pois o gás de
síntese é gerado a partir de outras fontes, notadamente o gás natural. O conhecimento da
reação ainda é fundamentalmente experimental, e não existem mecanismos específicos que
expliquem com exatidão a formação dos produtos e sua distribuição ao longo de uma faixa de
hidrocarbonetos. Esse trabalho realiza inicialmente uma estimação de parâmetros,
enquadrados em uma modelagem cinética, que procuram explicar o desenvolvimento da
reação e a formação das parafinas e olefinas em reatores tubulares de leito fixo com
catalisadores de cobalto. De posse dos parâmetros, procurou-se um modelo matemático mais
adequado à operação do reator tubular, com a utilização de equações para a transferência de
massa e de calor. Essas simulações foram submetidas posteriormente a uma análise estatística
para a determinação de variáveis mais significativas para a reação.
Palavras-chave: Reação de Fischer-Tropsch. Reator de leito fixo. Modelagem. Simulação.
Catalisador de cobalto.

ABSTRACT
In this work, the reaction of the Fischer-Tropsch synthesis, which may be understood as a
polymerization between the gases carbon monoxide and hydrogen, mixture known as
synthesis gas, with the formation of paraffinic and olefinic hydrocarbons, occurs under
heterogeneous catalysis, where those of cobalt appear as the most promising when you want
to produce hydrocarbon fractions commercially more favorable, such as gasoline, diesel and
waxes. The reaction is already perceived as an alternative to petroleum, since the synthesis
gas is generated from other sources, notably natural gas. The knowledge of the reaction is still
essentially experimental, and there are no specific mechanisms that explain precisely the
formation of the products and their distribution over a range of hydrocarbons. This work
performs initial parameter estimation, framed in a kinetic modeling, which seek to explain the
development of the reaction and the formation of paraffins and olefins in tubular fixed bed
reactors with cobalt catalyst. In possession of the parameters, we tried to one better suited to
reality modeling of reactor operation, with the use of equations for mass transfer and heat.
These simulations were later subjected to a statistical analysis to determine the most
significant variables for the reaction.
Key-words: Fischer-Tropsch synthesis. Fixed-bed reactor. Modeling. Simulation. Cobalt
catalyst.

LISTA DE SÍMBOLOS
A Área da seção do reator (m
2)
AL Área lateral do reator (m2)
Ci Concentração da espécie i (mol.m-3
)
C G
i Concentração da espécie i na fase gasosa (mol.m
-3)
C L
i Concentração da espécie i na fase líquida (mol.m
-3)
Cp Capacidade calorífica em pressão constante (J.mol-1
.K-1
)
Di Coeficiente de difusividade da espécie i (m2.h
-1)
dh Diâmetro hidráulico (em m)
E Energia total (J.mol-1
)
H Entalpia molar (J.mol-1
)
J Fluxo molar (mol.h-1
)
kads Constante para readsorção de olefinas via mecanismo alquila (h-1
)
'
ik Iniciação do mecanismo alquila (g.m-3
.MPa0.5
)
k '
i2 Iniciação do mecanismo alquenila (g.mol-1
)
kM Coeficiente de transferência de massa gás-líquido (h-1
)
k '
met Terminação em metila (h-1.MPa
0.5)
kolef Terminação em olefina para mecanismo parafina (h-1
)
k '
olef2 Terminação em olefina para mecanismo olefínico (h-1
.MPa0.5
)
kO2 Terminação em etileno (g2.h.mol
-1.m
-3)
k'
p1 Propagação do mecanismo alquila para metil (g.mol-1
)
k'
p Propagação do mecanismo alquila para n > 1 (g.mol-1
)
k'
p2 Propagação do mecanismo alquenila para vinila (m3.mol
-1. h
-1)
k"'
p2 Propagação do mecanismo alquenila para vinila nas equações de propenila
(g.mol-1)
k"
p2 Propagação do mecanismo alquenila para n > 3 (g.mol-1)
k'
par Terminação em parafina (h-1
.MPa0.5
)
L Comprimento do reator (m)

PCO Pressão parcial de CO na mistura gasosa (MPa)
PH2 Pressão parcial de H2 na mistura gasosa (MPa)
P(n) Alcano com n carbonos
P=(n) Olefina com n carbonos
.
Q Fluxo de calor no reator (J.h-1
)
R Constante universal dos gases (8,314 J.mol-1
.K-1
)
RSFT Taxa de reação de síntese de Fischer-Tropsch (mol.g-1
.h-1
)
R(n) Grupo alquila de n carbonos
R”(n) Grupo alquenila de n carbonos
'
Ar Taxa de desaparecimento de espécie A por massa do catalisador (mol.g-1
.h-1
)
Re Número adimensional de Reynolds
Sc Número adimensional de Schmidt
Ta Temperatura do fluido de troca de calor (K)
U Coeficiente global de transferência de calor (J.m-2
.h-1
.K-1
)
We Número adimensional de Weber
.
WS Fluxo de trabalho de eixo no reator (J.h-1
)
XG Razão adimensional de Lockhart-Martinelli
ap Densidade aparente do catalisador (g.m-3
)
ν Velocidade de escoamento no leito reacional (m.h-1
)

SUMÁRIO
1. INTRODUÇÃO.............................................................................................................
2. REVISÃO BIBLIOGRÁFICA.....................................................................................
2.1. Síntese de Fischer-Trosch ..............................................................................
2.1.1. Histórico............................................................................................
2.1.2. Aspectos físico-químicos ..................................................................
2.1.3. Conversão do gás natural via síntese de Fischer-Tropsch..............
2.1.4. A questão ambiental.........................................................................
2.2. Gás natural.....................................................................................................
2.2.1. Oferta, uso e transporte do gás natural...........................................
2.3. Biomassa.........................................................................................................
2.4. Tipos de reatores.............................................................................................
2.4.1. Reator de leito fixo...........................................................................
2.4.2. Simulação em reatores de leito fixo.................................................
2.4.2.1. Método de diferenças finitas..............................................
2.5. Catalisadores..................................................................................................
2.5.1. Catalisadores de ferro......................................................................
2.5.2. Catalisadores de cobalto..................................................................
2.5.3. Catalisadores de rutênio..................................................................
2.6. Mecanismo da síntese de Fischer-Tropsch.....................................................
2.6.1. Formação de espécies de metileno: geração do monômero............
2.6.2. Mecanismo alquil para formação e propagação da cadeia............
2.6.3. Mecanismo alquenil para formação e propagação da cadeia........
2.6.4. Readsorção de olefinas.....................................................................
3. ESTIMAÇÃO DE PARÂMETROS E MODELAGEM PARA A REAÇÃO DE
SÍNTESE DE FISCHER-TROPSCH...............................................................................
3.1. Modelo matemático........................................................................................
3.2. Cálculos referentes ao modelo cinético adotado............................................
3.3. Resultados.......................................................................................................
3.3.1. Análise estatística..............................................................................
3.3.2. Validação do modelo........................................................................
4. MODELAGEM DINÂMICA PARA O REATOR DE LEITO FIXO........................
13
16
16
16
17
20
21
21
21
23
25
25
28
28
29
30
30
31
32
34
36
37
39
40
40
47
52
54
57
59

4.1. Discretização do reator...................................................................................
4.1.1. Discretização do reator com transferência de massa......................
4.1.2. Discretização do reator com transferência de massa e de calor.....
4.2. Simulações computacionais............................................................................
4.3. Discussão dos resultados.................................................................................
4.3.1. Simulações com o modelo simplificado............................................
4.3.1.1. Análise para as frações gases leves, gasolina e querosene
4.3.1.2. Análise para as frações diesel e graxa................................
4.3.1.3. Análise para a conversão em hidrocarbonetos.................
4.3.2. Simulações com o modelo com transferência de massa
interfásico......................................................................................................
4.3.2.1. Análise para as frações gases leves, gasolina e querosene
4.3.2.2. Análise para as frações diesel e graxa................................
4.3.2.3. Análise para a conversão em hidrocarbonetos.................
4.3.2.4. Comparação com a modelagem sem transferência de
massa..................................................................................................
4.3.3. Simulações com o modelo com transferência de calor....................
4.3.3.1. Análise para as frações gases leves, gasolina e querosene
4.3.3.2. Análise para as frações diesel e graxa................................
4.3.3.3. Análise para a conversão em hidrocarbonetos.................
4.3.3.4. Análise para a variável temperatura.................................
4.3.3.5. Comparação com a modelagem sem transferência de
calor....................................................................................................
5. CONCLUSÃO..................................................................................................................
6. REFERÊNCIAS...............................................................................................................
59
63
66
67
73
74
75
79
82
84
85
89
91
93
95
97
100
103
105
108
110
112

13
1. INTRODUÇÃO
A reação de síntese de Fischer-Tropsch (SFT) é conhecida desde o início do
século XX, mais precisamente na década de 1920. O ponto de partida ocorreu quando, em
1920, os cientistas alemães Franz Fischer e Hans Tropsch desenvolveram o processo de
conversão do gás de síntese, produzido a partir do carvão, em combustíveis líquidos. Após o
marco inicial, a reação de SFT só foi utilizada em escala comercial na Alemanha, durante a 2ª
Guerra Mundial, nos Estados Unidos na década de 1950 e na África do Sul a partir da década
de 1950 (Stranges, 1997).
A reação principal da síntese de Fisher-Tropsch (SFT) pode ser descrita pela
Equação (1.1):
CO + (1 + m/2n)H2 (1/n)CnHm + H2O (1.1)
onde n e m são números inteiros que relacionam a estequiometria de um alcano (o
número de átomos de hidrogênio é igual ao dobro mais duas unidades do número de átomos
de carbono) ou de um alceno (o número de átomos de hidrogênio é igual ao dobro do número
de átomos de carbono). Com a necessidade e a pressão da sociedade em relação a
combustíveis com menores capacidades poluentes, principalmente no tocante ao teor de
enxofre contido nos combustíveis fósseis, vem à tona a síntese de Fischer-Tropsch, que utiliza
gás de síntese produzido por uma rota que pode ser levada a minimizar o teor de enxofre a um
menor custo. O procedimento consiste em transformar o gás natural (associado ou não-
associado ao petróleo e praticamente isento de enxofre) em gás de síntese. Assim, o produto
final, o combustível líquido, alcançaria menor teor de enxofre e de compostos nitrogenados
(NOx).
O estudo da reação de SFT está incluso em uma abordagem mais ampla,
englobado pelas tecnologias gas to liquid, representada simplesmente por GTL, e biomass to
liquid (BTL). Pode significar a abertura de novas fronteiras para a utilização do gás natural e
da biomassa, agregando valor comercial ao converter gases reagentes em combustíveis
líquidos ou em parafinas, produtos mais valorizados na economia globalizada e em alta
demanda por fontes de energia. Outra possibilidade para a reação de SFT está relacionada ao
fato de se evitar o transporte de combustíveis em fase gasosa, que necessita de operações de
elevado custo quando o centro consumidor se situa a grande distância do centro produtor.

14
Assim, a transformação desses gases combustíveis em combustíveis líquidos, ou seja, o uso
da tecnologia GTL, pode baratear e facilitar o transporte do produto gerado. A utilização
dessa tecnologia, associada ao uso cada vez mais discutido de biocombustíveis, pode
simbolizar o esforço mundial em estabelecer fontes de energia renováveis e livres de toda a
geopolítica que cerca o comércio de petróleo (Barros Junior e Fernandes, 2011).
Verifica-se que empresas e universidades têm buscado a otimização constante
para os processos já existentes. Trabalhos recentes como os de Zennaro et al. (2000) e
Visconti et al. (2007), foram elaborados visando criar modelos matemáticos apropriados para
explicar a formação de produtos na reação de SFT. No entanto, devido ao elevado número de
variáveis existentes para a estimação de parâmetros e simulação da reação, muitos autores
optam por não abordá-los no total, ou por fornecer os resultados expressos apenas em termos
de conversão de CO em hidrocarbonetos, que pode omitir o fato que grande parte do
monóxido consumido se transforma em metano, de menor interesse comercial que produtos
mais pesados.
Nesse contexto, a pesquisa baseada em simulação se torna de grande importância,
pois estimula a descoberta de procedimentos teóricos que tenham por objetivo tornar
economicamente mais viável a reação de SFT, de acordo com os tipos de reatores em
operação e leitos catalíticos utilizados. A otimização dos processos via simulação ainda traz
as seguintes vantagens: pode-se prever antecipadamente determinados caminhos reacionais
mais adequados para se alcançar certos rendimentos com catalisadores e reatores específicos;
podem-se analisar ideias originais sem necessidade de alto investimento em equipamentos e
reagentes; e podem-se determinar caminhos teoricamente aceitáveis para a reação de SFT cuja
operação se baseia apenas em dados empíricos.
Esse trabalho foi elaborado com o intuito de se realizar uma modelagem dinâmica
para o reator de leito fixo, onde a quantidade de produtos formados pode ser estimada em
relação ao tempo de operação do reator e do estágio de discretização utilizado no reator. É
uma abordagem inovadora em comparação a outras existentes na literatura, que não utilizam o
tratamento dinâmico para a reação de SFT em reator de leito fixo. Foi desenvolvido um
modelo cinético para a reação de SFT e outro para o reator tubular de leito fixo. Os principais
objetivos estabelecidos para essa tese foram:
Desenvolvimento de modelo matemático para a descrição da reação de síntese de
Fischer-Tropsch (SFT) com cinética de reação adequada aos catalisadores de cobalto,

15
de acordo com padrões estatísticos em intervalos de confiança previamente
estabelecidos.
Estudo do processo de simulação de uma reação de síntese de SFT em reator de leito
fixo, para análise da importância estatística das variáveis independentes adotadas.
Análise das melhores condições de operação do reator em função dos resultados
obtidos para frações específicas e comercialmente mais atraentes, como diesel,
gasolina e graxas.

16
2. REVISÃO BIBLIOGRÁFICA
2.1. Síntese de Fischer-Tropsch
2.1.1. Histórico
A reação de polimerização entre monóxido de carbono (CO) e hidrogênio (H2),
produzindo água e hidrocarbonetos dos mais variados tamanhos de cadeia carbônica, é
chamada de síntese de Fischer-Tropsch (SFT) (Davis, 2003). Na verdade, a referida reação já
é conhecida há cerca de 80 anos, ao se considerar que os primeiros experimentos de Franz
Fischer datam do ano de 1925, em seu laboratório no Kaiser Wilhelm Institute for Coal
Research (atualmente Instituto Max Planck) (Schulz, 1999).
A reação em si é uma idéia bem-sucedida de uma catálise heterogênea, em que se
utilizam catalisadores sólidos de ferro, cobalto ou raramente rutênio, normalmente suportados
em sílica. Os reagentes iniciais são gases (CO e H2) e durante a reação são formados
hidrocarbonetos parafínicos e olefínicos, tanto gasosos quanto líquidos, e até mesmo sólidos,
dependendo do crescimento da cadeia do hidrocarboneto, o que mostra a necessidade de um
estudo apurado e cuidadoso para que se tenha certeza em se trabalhar com as melhores
condições de operação para obtenção de determinado produto.
A ideia de produção de combustíveis líquidos a partir do gás de síntese (uma
mistura de monóxido de carbono e hidrogênio) é útil não só pela atual conjuntura macro-
econômica, mas também como forma de melhorar o custo do transporte dos materiais, já que
o transporte de gases tem se mostrado mais caro e complicado que o de líquidos. No período
de 1925 a 1945, o estudo da síntese de Fischer-Tropsch transcendeu um pouco o aspecto
econômico. A necessidade de novas tecnologias para o processamento do carvão era o foco
para novas iniciativas, novas pesquisas e plantas industriais, principalmente na Alemanha.
No período de 1955 a 1970, o cenário mundial de energia estava baseado na
produção plena, barata e farta de petróleo, o que fez o interesse pela SFT restringir-se a um
nível acadêmico e de pesquisa, com exceção da África do Sul, devido ao aspecto político-
econômico de restrição à importação de petróleo e à disposição de carvão extremamente
barato. Nos anos 70 e 80, o interesse em SFT retomou o fôlego, principalmente devido ao
boicote dos países produtores de petróleo em relação à demanda mundial. Novas pesquisas
foram implementadas e novas plantas construídas visando uma menor dependência do

17
petróleo, majoritariamente financiadas pelos Estados Unidos da América, Japão e Europa, em
especial a Alemanha. Inovações surgiram nesse período, como plantas de processamento de
óleo pesado e carvão, com unidades integradas de gaseificação e purificação do gás de síntese
(Schulz, 1999).
Quando da descoberta de grandes campos de petróleo na Arábia Saudita, no
Alasca e no Mar do Norte, na década de 90, o preço do petróleo caiu e a produção de
combustíveis líquidos a partir do gás de síntese mostrou-se muito cara diante das matérias-
primas existentes na época. Mais uma vez a SFT poderia ser descartada como fonte de
energia, mas seu estudo atualmente está acima do aspecto econômico. Com a necessidade e a
pressão da sociedade em relação a combustíveis com menores capacidades poluentes,
principalmente no tocante ao teor de enxofre contido nos combustíveis fósseis, novamente
vem à tona a síntese de Fischer-Tropsch, que utiliza gás de síntese (mistura de monóxido de
carbono e hidrogênio) produzido por uma rota que pode ser levada a minimizar o teor de
enxofre a um menor custo. A ideia consiste em transformar o gás natural (associado ou não-
associado ao petróleo e praticamente isento de enxofre, conforme veremos mais adiante) em
gás de síntese. Assim, o produto final, o combustível líquido, alcançaria menor teor de
enxofre e de compostos nitrogenados (NOx) (Fernandes, 2007).
2.1.2. Aspectos físico-químicos
A reação principal da síntese de Fisher Tropsch (SFT) pode ser condensada por
(Fernandes, 2006):
CO + (1 + m/2n)H2 (1/n)CnHm + H2O (2.1)
onde n e m são números inteiros que relacionam a estequiometria de um alcano (o número de
átomos de hidrogênio é igual ao dobro mais duas unidades do número de átomos de carbono)
ou de um alceno (o número de átomos de hidrogênio é igual ao dobro do número de átomos
de carbono). Anteriormente, Anderson (1984) havia proposto um esquema reacional
semelhante, embora um pouco mais complexo:
nCO + (2n +1) H2 CnH2n+2 + nH2O (2.2)

18
onde n é definido como /(1 - ), enquanto é uma relação entre as taxas de terminação e de
propagação da cadeia.
Um aspecto importante da SFT a ser analisado é o uso de catalisadores
heterogêneos. Por exemplo: o uso de catalisadores de ferro suportados em sílica promove uma
reação secundária, conhecida por water gas shift (WGS). Nessa reação, o monóxido de
carbono, que deveria reagir com o hidrogênio, se combina com a água formada na reação
principal, gerando dióxido de carbono (CO2) e hidrogênio (H2). A estequiometria da reação de
WGS é dada por:
CO + H2O CO2 + H2 (2.3)
A ocorrência de uma reação secundária inibe a reação principal e diminui o
rendimento do processo. Portanto, um dos fatores que requerem análise mais cuidadosa,
quando se utiliza catalisadores de ferro, é a taxa com a qual a reação de WGS ocorre. O uso
de catalisadores de cobalto, também suportados, atenua o efeito da reação de WGS e
maximiza a produção de hidrocarbonetos.
A reação de SFT é bastante exotérmica e o processo deve ser cuidadosamente
estudado para que sejam alcançados projetos cada vez mais econômicos e financeiramente
rentáveis. O aumento da temperatura, além de inibir a ação catalítica pela produção de
carbono (principalmente com catalisador de ferro), pode causar o aumento da produção
seletiva de gás metano (Schulz, 1999). Isso se deve ao aumento das velocidades das reações
de propagação e terminação da cadeia carbônica com o aumento da temperatura. A partir de
resultados experimentais, conclui-se que as reações de terminação da cadeia sejam mais
favorecidas em maiores temperaturas, formando produtos de massa molecular mais baixa.
Esse aspecto referente à diminuição do comprimento da cadeia carbônica dos produtos
formados com o aumento de temperatura está diretamente relacionado com a rentabilidade do
processo, uma vez que se deseja obter frações mais pesadas que possam, por sua vez, serem
submetidas a processos de craqueamento para aumentar as conversões em diesel e gasolina,
frações comercialmente mais aceitas. Assim, deve-se tratar o aumento da taxa de reação de
SFT com o aumento da temperatura com cautela, para que não se perca rendimento em
produtos economicamente mais favoráveis, como os de maior cadeia (Vosloo, 2001).
Quando resultados experimentais ou de simulação para as reações de SFT são
encontrados na literatura, observa-se, como tendência geral, que a conversão total da reação é

19
expressa como simplesmente a transformação de monóxido de carbono em hidrocarbonetos,
sem detalhar o tamanho da cadeia do produto gerado. Essa análise superficial pode esconder
alguns pontos importantes, como a formação de grande quantidade de metano em detrimento
de frações mais importantes do ponto de vista econômico, como gasolina, diesel ou graxa. Em
relação à análise de resultados em simulação, a conversão total em hidrocarbonetos não deve
ser utilizada isoladamente como referência para a eficiência da reação ou do processo de
simulação.
A característica exotérmica da reação de SFT e a necessidade de troca de calor
abrem uma possibilidade de associação entre uma planta de SFT com outra que utilize o calor
liberado pela primeira. Essa integração entre plantas é útil tanto do ponto de vista econômico
quanto em termos de rendimento em frações mais rentáveis, pois, ao se utilizar o calor
liberado pela reação de SFT em outras finalidades, ocorre uma minimização do aumento da
temperatura do reator, e, por consequência, são esperadas maiores conversões em frações
mais pesadas. Algumas mudanças no setor de reforma podem ser efetuadas de forma a
aumentar a eficiência térmica do processo, tais como:
O uso de um reformador por troca de calor associado a outro reformador autotérmico.
É muito similar a uma reforma combinada, onde a maior diferença reside no fato de
que a energia do reformador a vapor não é suprida por um aquecedor a fogo, e sim
pelo gás de saída do reformador autotérmico. Esse procedimento gera uma economia
de 30% no consumo de oxigênio e melhora a eficiência térmica da planta em 4%
(Vosloo, 2001). Deve-se atentar para o problema técnico ocasionado pelo resíduo
metálico proveniente do reformador;
O uso da alimentação/produto do trocador de calor para a recuperação energética da
saída do reformador. Nesse caso, mesmo acarretando o mesmo impasse técnico
anterior, o consumo de oxigênio pode ser diminuído em 3,5% e o aumento da
produção de combustíveis líquidos pode aumentar em até 2,5% (Vosloo, 2001) com a
utilização no reformador de gás natural pré-aquecido.

20
2.1.3. Conversão do gás natural via Síntese de Fischer-Tropsch
A reação de SFT necessita de gás de síntese em diferentes proporções molares,
que podem ser obtidas a partir do carvão ou do gás natural. Essa última opção parece ser a de
maior interesse ambiental atualmente, embora em experiências anteriores, conforme
mencionado, tenha-se utilizado bastante o carvão. O processo para a conversão do gás natural
em produto líquido (GTL) via síntese de Fischer-Tropsch pode ser dividido em três etapas:
Geração do gás de síntese;
Conversão do gás de síntese (reação de SFT);
Hidroprocessamento (hidrocraqueamento).
Segundo Vosloo (2001), o uso combinado dessas três etapas, apesar de bem
estabelecidas e comercialmente viáveis, foi pouco utilizado no passado. Atualmente, sabe-se
que as plantas mais modernas de SFT já superam essa limitação e conseguem fazer o uso das
três etapas de forma a obter custos efetivos mais baixos. No entanto, para se fazer a tecnologia
GTL cada vez mais competitiva, o desafio vai além da otimização dos aspectos mais
conhecidos desta tecnologia, incluindo aspectos como desenvolvimento de catalisadores e
mecanismo de reação. Nesse contexto, a importância da simulação é percebida.
A Figura 2.1 mostra de forma sucinta todas as etapas do processo Fischer-Tropsch
desde a obtenção do gás natural até a fase final de hidrotratamento dos produtos.
Figura 2.1– Etapas do Processo de Fischer-Tropsch (Farias, 2007).
Gás NaturalReforma
do MetanoCOH2
SinteseFischer-Tropsch
Gases Leves
Separação GasolinaDiesel
HidrocraqueamentoHidroisomerização
GasolinaDiesel
Graxas
FraçãoLeve

21
2.1.4. A questão ambiental
A tecnologia de conversão de biomassa em combustíveis líquidos é outra opção
de fontes de energia potencialmente menos poluidoras. Os restos de madeira não
aproveitados, ou para citar um exemplo brasileiro, de bagaço da cana, que atualmente é
queimado para a produção de energia para caldeiras, podem ser convertidos em monóxido ou
dióxido de carbono e reaproveitado via SFT para a geração de combustíveis líquidos, ou
outras formas de hidrocarbonetos.
Novas concepções ambientais e novos regulamentos para combustíveis líquidos
que entraram em vigor em 2008 na Europa e nos Estados Unidos, trazem a necessidade do uso
de combustíveis líquidos “verdes”, que diminuem a emissão de óxidos de enxofre (SOx) e de
nitrogênio (NOx) (Fernandes, 2007). Diante desse fato, o custo para adequar os combustíveis
utilizados hoje em dia fará com que o combustível líquido (diesel principalmente) tenha custo
comparável ao obtido através da SFT.
2.2. Gás natural
2.2.1. Oferta, uso e transporte do gás natural
A oferta mundial de gás natural é crescente e atualmente parte deste é queimada
em flare. Esperam-se novas tecnologias que visem um melhor aproveitamento e uma mais
eficiente conversão do gás natural, ainda que diretamente nos campos de extração. O principal
problema em relação à utilização do gás natural é a grande distância existente entre os campos
de produção e os mercados consumidores de combustíveis, que gera um custo adicional em
relação ao transporte do gás.
Os grandes usuários de gás natural no mundo são as usinas termelétricas,
termelétricas de cogeração, as grandes indústrias e setores de comércio, serviços e o setor
domiciliar. As usinas termelétricas poderão reforçar a geração em centros de consumo já
saturados ou para atender áreas ainda sem rede de energia elétrica de origem hídrica. As
termelétricas de cogeração, por fornecerem energia elétrica a vapor com maior eficiência e
confiabilidade, permitirão reduzir os custos das refinarias e contribuir para o incremento do
consumo do gás natural (Farias, 2007).

22
O gás natural é tradicionalmente visto como uma fonte de combustível fóssil
abundante e menos poluidora para a geração de energia térmica e elétrica. Atualmente, 90%
do seu consumo global se destinam ao uso combustível ou energético e apenas 10% à
produção de amônia ou metanol cujos mercados têm tamanho limitado e consomem apenas
uma fração de todo gás natural disponível.
Recentemente foram descobertas várias reservas de gás natural em regiões
remotas, de difícil acesso e submarinas. A boa notícia é que, mesmo sendo extremamente
distantes, o tamanho destas reservas compensa o aproveitamento do gás natural nelas
contidas. Infelizmente, não é economicamente viável a construção de gasodutos ligando estas
reservas aos principais locais consumidores deste gás. É de se esperar que uma melhor
solução para o impasse a respeito do aproveitamento destas reservas seja a conversão do gás
em produtos líquidos de maior valor agregado e que tenham melhor facilidade de transporte.
Diante desses fatos, os processos de conversão do gás em combustíveis líquidos,
através da tecnologia GTL (gas to liquid), mostram-se como tendência natural a ser adotada,
porque além de econômica e financeiramente atraentes, são atividades que amenizam a
agressão ao meio ambiente.
Os produtos derivados da tecnologia GTL apresentam vantagens econômicas,
dentre as quais se podem citar:
O custo de transporte é menor do que o do gás natural, sendo que o gás natural
liquefeito (GNL) possui uma densidade energética bastante significativa (cerca de 600
vezes maior que o gás natural em condições padrões de temperatura e pressão (25ºC e
1atm), sendo transportado uma maior quantidade de massa num menor espaço;
Os produtos obtidos a partir desta tecnologia apresentam vantagens ambientais
importantes em relação aos produtos tradicionais, dentre os quais pode-se destacar: os
derivados gerados apresentam um teor de enxofre bem menor e não há presença de
óxidos de nitrogênio (genericamente representados por NOx), que poderiam ocasionar
problemas de ordem ambiental bastante severos.
Por estas razões, a participação mundial do gás natural na matriz energética
mundial (hoje em 22%) está crescendo a taxas superiores do que os dos derivados do petróleo
(Davis, 2002)

23
Apesar das diversas vantagens, o gás natural apresenta uma característica
desvantajosa bem destacada – o elevado custo no transporte – devido à dificuldade em se
armazenar e transportar sob pressão gases em recipientes fechados. Atualmente existem duas
alternativas práticas para o transporte de gás natural, o que o torna bem menos versátil, do
ponto de vista industrial, que o petróleo. Estas duas formas de transporte são através de
gasodutos ou mediante tanques metaneiros (também conhecidos como navios criogênicos).
Ambas as formas de transporte requerem tecnologia específica e geralmente de alto custo. Por
exemplo, em tanques criogênicos, apresenta-se como uma tecnologia muito cara por requerer
estações de liquefação e regaseificação nos pontos de envio e recepção, respectivamente
(Figura 2.2).
Figura 2.2 – Fluxograma da tecnologia GNL mediante tanques metaneiros (Farias, 2007).
2.3. Biomassa
Biocombustíveis têm sido uma fonte de energia para os seres humanos desde os
tempos mais antigos. O interesse na geração de energia a partir de biocombustíveis tem
aumentado atualmente devido à produção de gases “verdes”, abrindo novas possibilidades de
emprego e de independência energética. A utilização de biocombustíveis pode inclusive
minimizar a agressão ao meio ambiente causada pela queima de seus similares fósseis.
Estação de Liqüefação
Terminal e Regasificação
Cia.Distribuidora de Gás (LDC)
Térmica
Cia.Distribuidora de Energia Elétrica
Transporte do Gas
Produção de Gás

24
Biocombustíveis podem ser preparados a partir da biomassa por processos
bioquímicos ou termoquímicos. Dentre esses processos, destaca-se a reação de SFT. A
transformação da biomassa em combustíveis líquidos está englobada no que se chama
tecnologia BTL (biomass to liquid).
Atualmente, além dos combustíveis líquidos propriamente ditos, a conversão BTL
vem sendo utilizada para a produção de frações mais pesadas de hidrocarbonetos, como a
parafina. Empresas norte americanas, como a Rentech Incorporation (em Denver, Colorado),
estão se especializando na conversão de biomassa, como restos de madeira, em parafinas para
uso industrial.
As fontes de biomassa podem ser usadas para a geração de energia de diversas
formas. Uma distinção pode ser feita entre resíduos primários, secundários e terciários, que já
estão disponíveis como subprodutos de outras atividades ou como biomassa cultivada para
uso especificamente de geração de energia (Hoogwijk et al., 2003):
Resíduos primários são gerados durante a produção de culturas para alimentação e
produtos florestais, como por exemplo, desbastes de florestas comerciais e palha. Essa
biomassa está normalmente disponível no campo e necessita ser coletada para uso
posterior;
Resíduos secundários são produzidos durante o processamento da biomassa para a
produção de produtos alimentícios e produtos derivados de biomassa, e estão
tipicamente disponíveis em indústrias de comida e bebida, de papel, de beneficiamento
de cana de açúcar, etc;
Resíduos terciários se tornam disponíveis quando um derivado de biomassa já foi
utilizado, o que torna essa categoria uma diversidade de rejeitos, desde a fração
orgânica do resíduo sólido municipal, dos rejeitos de madeira e de madeira de
demolição, de lamas, etc.
Em geral, os resíduos de biomassa estão interligados com vários mercados.
Muitos rejeitos têm utilização como forragem, fertilizantes e acumuladores de solo, matérias-
prima para, por exemplo, papel reciclado e aglomerados tipo MDF. A avaliação dos preços
desses resíduos depende da demanda dos mercados local e internacional, bem como do

25
mercado internacional de matérias-prima e do tipo de tecnologia de tratamento desenvolvida
para o material remanescente (Faaij, 2004).
2.4. Tipos de reatores
A reação de síntese de Fischer-Tropsch (SFT) emprega reatores multifásicos ou
multipropósitos, já que duas ou mais fases são necessárias para promover a reação. No caso
específico da reação de SFT, a maioria dos reatores envolve fase líquida e gasosa em contato
com uma fase sólida, normalmente o catalisador.
Um dos tipos de reatores multifásicos muito utilizados atualmente são os reatores
de leito de lama (slurry beds) e os de leito fluidizado (fluidized beds), que têm sido
largamente usados na síntese de Fischer-Tropsch. Essa espécie de reator trifásico, que
promove o contato de dois reagentes gasosos com uma fase líquida inerte onde se encontra
disperso um catalisador sólido, é utilizado não só para a reação de SFT, como também em
outras aplicações recentes, como: remoção de gases poluentes (SO2 e H
2S) por oxidação numa
lama contendo carbono como catalisador; hidrogenação de acetileno; oxidação de etileno a
óxido de etileno (Ramachandran e Chaudhari, 1983). O reator tubular de leito fixo (fixed bed
tubular reactor), detalhado adiante por se tratar do reator escolhido para a execução do
processo de simulação matemática nesta tese, apresenta a vantagem de possuir maior controle
de variáveis, como temperatura, e menores custos de manutenção.
2.4.1. Reator de leito fixo
Um dos primeiros reatores projetados para a reação de SFT foi o reator tubular de
leito fixo. Após anos de desenvolvimento, foi refinado o conceito para o reator se tornar útil
para esse tipo de reação. Ele tem sido utilizado desde as primeiras operações da indústria
Sasol I. Os reatores da empresa Sasol contêm 2000 tubos preenchidos com catalisador de
ferro imersos em água para a remoção do calor (Spath e Dayton, 2003). O gás de síntese é
introduzido pelo topo do reator, flui através dos tubos, e os produtos e reagentes em excesso
são removidos pelo fundo do reator. As graxas constituem cerca de 50% em massa dos
produtos, e a taxa de conversão pode alcançar até 70% (em relação ao número de mol de
monóxido de carbono inicial). O reator opera numa faixa de pressão entre 20 e 30 bar e a
faixa de temperatura oscilava entre 220°C e 260°C. Um controle de temperatura adicional

26
pode ser obtido pelo uso de altas velocidades de escoamento gasoso, bem como a utilização
de reciclo. O gás reciclado é cerca de 2,5 vezes maior que a quantidade de gás alimentado
originalmente no reator. A vida do catalisador é estimada em 70 a 100 dias e sua remoção é
complicada.
A Figura 2.3 representa uma esquematização generalizada de um reator tubular de
leito fixo.
Figura 2.3 - Reator de leito fixo (Farias, 2007)
Uma descrição, de um modo mais geral, do reator tubular de leito fixo utilizado na
síntese de Fischer-Tropsch é importante nesse momento. O sistema que compõe o reator
consiste de um reator multitubular que é alimentado pela parte superior do mesmo e atravessa
o leito catalítico no interior dos tubos. O gás de síntese passa através do aparelho em regime
empistonado, e é necessária a utilização de água para resfriamento na casca do reator para que
se remova o calor liberado pela reação de SFT, para que a operação completa ocorra em
condições isotérmicas dentro do reator (Spath e Dayton, 2003). A natureza exotérmica da
reação de SFT combinada com a alta atividade de catalisadores que utilizam em sua matriz o
cobalto pode causar uma mudança de temperatura no reator de crucial importância, podendo
levar à desativação do catalisador. No caso do reator de leito fixo tubular, isto se torna ainda
mais problemático devido ao perfil de temperatura dentro do tubo. Este problema é então
contornado encontrando-se a estabilidade entre o diâmetro do tubo e o uso de resfriamento,
assim como a reciclagem de inertes. Esse tipo de reator, sendo uma das tecnologias de maior
competitividade atualmente, ocupa uma posição especial nas práticas industriais da reação de

27
SFT, como mostrado sistematicamente em operações comerciais em larga escala da empresa
Sasol (Dry, 2002) e da Shell (Sie, 1991).
As principais vantagens de um reator de leito fixo para a reação de SFT são:
Melhor controle de temperatura, pelo fato de apresentar melhor eficiência na
transferência de calor;
Menor custo operacional, por não apresentar partes móveis no reator;
Possibilidade de formação de cadeias maiores, que não ocorre em reatores de leito
fluidizado;
Não há necessidade de separação entre catalisador e produtos ao final da reação, como
nos reatores de leito de lama.
Processos industriais típicos para a SFT utilizando reatores de leito fixo produzem
uma vasta mistura de hidrocarbonetos, do metano até as graxas (Wang et al., 2003). Para
aumentar a área superficial e facilitar a remoção do calor, as partículas catalíticas são de
tamanho extremamente reduzido (geralmente poucos milímetros), o que ocasiona a existência
de limitações por difusão em macroporos intraparticular, já que a reação de SFT requer a
utilização dos microporos do catalisador, devido à necessidade de crescimento da cadeia.
Assim, nos poros da partícula catalítica pode ocorrer condensação por capilaridade e
frequentemente se forma uma camada estagnada de hidrocarbonetos pesados (graxas).
Entretanto, o modelo de um reator de leito fixo para reações de SFT pode ter sua modelagem
aproximada a partir de descrições quantitativas entre as interações da complexa cinética
química e do singular fenômeno de transporte envolvido.

28
2.4.2. Simulação em reatores de leito fixo
Ao contrário da reação de SFT em reator de leito de lama, a literatura em
modelagem para a reação de SFT em reator de leito fixo é muito limitada (Jess e Kern, 2009).
Os estudos para a simulação com a utilização de reatores em leito fixo possuem seus
específicos pontos de partida, como considerar desprezíveis a transferência radial de calor e a
transferência de massa no poro do catalisador. O problema maior reside, no entanto, em não
procurar uma abordagem dinâmica para o reator, avaliando a formação de produtos e a
evolução da temperatura ao longo do reator e do tempo de operação.
Moutsoglou e Sunkara (2011) também realizaram a simulação de um reator de
leito fixo, mas com a utilização de ferro como catalisador. Fizeram a opção por uma
abordagem unidimensional, desconsiderando os efeitos de difusão radial de temperatura e de
reagentes, e a difusão no poro do catalisador.
Wu et al. (2010) realizaram outra simulação, desta vez bidimensional (com a
utilização de difusão no poro do catalisador para justificar sua inovação) para a reação de SFT
em reator de leito fixo. Foi utilizado o catalisador de cobalto, o mesmo usado nessa tese. A
modelagem matemática adotou o método de colocações ortogonais para a resolução das
equações.
2.4.2.1. Método de diferenças finitas
Com a necessidade de uma abordagem dinâmica para a reação de SFT em reator
tubular, capaz de avaliar a formação de produtos e evolução de temperatura ao longo do reator
e do tempo de operação do mesmo, foi preciso que métodos matemáticos apropriados fossem
utilizados para a resolução das equações propostas.
O método de diferenças finitas é uma das várias técnicas para a diferenciação de
uma função discreta, ou seja, um conjunto discreto de valores da variável dependente em
pontos conhecidos da variável independente. O cálculo do declive nada mais é que o cálculo
discreto de dy/dx num dado ponto x0.
Dessa forma o método se baseia na discretização de uma variável, por exemplo, o
comprimento de um reator, em pontos, onde é realizada uma diferenciação aproximada da
função. Quanto maior o número de pontos de discretização, melhor a aproximação da
diferenciação, mas, em contrapartida, maior o esforço computacional envolvido.

29
Normalmente adota-se uma divisão que seja útil em termos de resultados em simulação, como
seccionar o reator tubular em 10 pontos, além dos pontos inicial e final. Nessa metodologia, o
erro do procedimento é calculado através de diferenciações em graus cada vez maiores,
dependendo da precisão requerida.
Fujiwara (1989) já utilizou o método de diferenças finitas para a modelagem de
reatores de leito fixo, embora não o tenha feito para a reação de SFT.
2.5. Catalisadores
Os catalisadores para a reação de SFT geralmente são preparados por processos
como precipitação, impregnação ou troca iônica, que ocasionam a deposição de um precursor
do metal sobre a superfície do meio de suporte. A partir de então, o catalisador é submetido à
secagem, seguido de calcinação e depois, submetido à ativação com fluxo de gás, que pode
ser o hidrogênio, monóxido de carbono ou gás de síntese (mistura de monóxido de carbono e
hidrogênio), o que gera a fase metálica ativa. Assim, as correspondências entre as condições
da reação de SFT e a composição do catalisador determinam a seletividade e a atividade para
um dado conjunto de processos e parâmetros reacionais. A escolha do catalisador mais
eficiente é determinante para o sucesso prático da reação de SFT (Adesina, 1996).
Hoje em dia, vários metais podem ser utilizados com eficácia comprovada como
catalisadores para a reação de SFT. Destacam-se os de ferro, cobalto e rutênio. Dentre esses, o
catalisador de rutênio tem aplicações bem mais limitadas que os demais devido ao seu
elevado preço.
Presumivelmente, os catalisadores baseados em ferro e cobalto são aqueles mais
largamente utilizados em processos industriais. A principal diferença entre estes catalisadores
é na formação de produtos oxigenados durante o processo. No catalisador de cobalto, esses
derivados oxigenados são obtidos como subprodutos a partir da reação com o cobalto, e no
caso do catalisador de ferro, o subproduto formado é o dióxido de carbono e a água (Davis,
2003).
A análise da influência da temperatura na distribuição dos produtos da reação de
síntese de Fischer-Tropsch traz o mesmo resultado para todos os tipos de catalisadores
estudados. Logo, com o aumento da temperatura de operação do reator, espera-se uma
modificação na seletividade da formação de produtos, mostrando uma maior tendência à

30
geração de hidrocarbonetos de menor massa molecular, e, ao mesmo tempo, o conteúdo de
álcoois nos produtos diminui (Davis, 2003).
2.5.1. Catalisadores de ferro
Os catalisadores de ferro, visando alcançar maiores atividade e estabilidade, são
combinados a um promotor alcalino (metal do grupo I) (Schulz, 1999). Focando na produção
de hidrocarbonetos, esses catalisadores admitem duas rotas de seletividade. A de baixa
temperatura (240ºC), que permite utilizar o catalisador de ferro e obter boa atividade
catalítica, mesmo com o uso de baixas temperaturas reacionais, onde muitos dos
hidrocarbonetos produzidos se encontram na fase líquida nestas condições. A principal fração
do produto obtido é a parafina, onde esta é refinada para fins comerciais (Schulz e Cronjé,
1977). Porém, é experimentalmente comprovado que quando se deseja obter diesel de alta
qualidade, esta rota pode se tornar bastante seletiva para o hidrocraqueamento na produção do
combustível líquido. A outra rota, a de alta temperatura (270ºC), é voltada para a produção de
hidrocarbonetos olefínicos com baixo peso molecular, conforme esperado quando se utiliza
reatores de leito fluidizado (Sasol Synthol Process). Graças à temperatura de reação
proporcionalmente elevada, são obtidos produtos com altas frações em hidrocarbonetos de
baixa massa molecular média.
2.5.2. Catalisadores de cobalto
Os catalisadores de cobalto foram aplicados na primeira planta de Fischer-
Tropsch pela empresa alemã Ruhrchemie, companhia de exploração de carvão, em 1935
(Pichler et al.,1957). O desenvolvimento de catalisadores de cobalto de alto desempenho foi
descrito por Fischer e Meyer (1931) e Fischer e Koch (1932) como sendo bastante inovador.
Atualmente, os catalisadores de cobalto são utilizados para a produção de diesel
por Fischer-Tropsch a partir do gás natural proporcionando a máxima produção de graxas que
posteriormente passam por um processo de hidrocraqueamento. Essa combinação da reação
de SFT com hidrocraqueamento estabelece uma produção de diesel onde a conversão se
aproxima de 80% (Eisenberg et al., 1998).
A partir de corridas de até 400 horas, investigaram a estabilidade dos catalisadores
de cobalto para a SFT em reator tubular (Wu et al., 2010), demonstrando que reações

31
indesejáveis, como a de water-gas-shift (WGS), comum em catalisadores de ferro, é
inexistente. Também perceberam que o catalisador de cobalto suportado em sílica é somente
ativo em sua superfície, pois sua camada mais interna se encontra em um estado oxidado
inativo graças à produção de água.
Essa descoberta facilita a modelagem quando mostra não ser mais necessária a
difusão das partículas reagentes no sítio catalítico, uma vez que a reação (já em fase líquida)
necessita apenas da transferência de massa na superfície do catalisador.
Outro aspecto relevante na utilização dos catalisadores de cobalto para a reação de
SFT é a readsorção de olefinas (analisada com mais detalhes no item 2.6.4). Sabe-se que a
reação de SFT produz parafinas e olefinas com a insaturação no primeiro carbono, chamada
de α-olefinas. Com catalisadores de cobalto, essas olefinas sofrem readsorção na superfície
catalítica e se transformam em parafinas. Assim, esse efeito aumenta o rendimento em
parafinas, reduz em olefinas e aumenta a conversão em frações mais rentáveis
economicamente, como gasolina, diesel e graxas.
Nessa tese, essas informações foram decisivas para que a modelagem tenha sido
escolhida com o catalisador de cobalto. A menor difusão no poro catalítico, a inexistência de
reação de WGS e a readsorção de olefinas tornam o catalisador de cobalto mais atraente sob o
ponto de vista financeiro.
2.5.3. Catalisadores de rutênio
Estes catalisadores são bastante ativos (Vannice, 1975), pois trabalham a baixa
temperatura e produzem hidrocarbonetos de alto peso molecular (síntese de polimetileno). O
rutênio atua como catalisador na forma de metal puro (sem promotores). Isto provê um
sistema catalítico simples para a síntese de Fischer-Tropsch, no qual as conclusões do
mecanismo devem ser mais fáceis. Devido ao seu elevado preço e reservas mundiais
limitadas, este catalisador torna-se inviável para aplicações industriais. Um estudo sistemático
do mecanismo deste catalisador poderia contribuir substancialmente para uma maior
exploração dos fundamentos da síntese de Fischer-Tropsch (Schulz, 1999).

32
2.6. Mecanismo da síntese de Fischer-Tropsch
Tendo em vista a importância do potencial econômico da química de Fischer-
Tropsch, uma compreensão detalhada do mecanismo do processo é desejável. Tal
entendimento poderia possibilitar aplicações mais eficientes nas operações industriais
(Overett et al., 2000).
O mecanismo de reação da reação de síntese de Fischer-Tropsch (SFT) consiste
de uma polimerização de CO que leva a uma distribuição dos produtos com diferentes pesos
moleculares (Anderson, 1956), chamada distribuição Anderson-Schulz-Flory (ASF):
12)1( n
n nF (2.4)
Na Equação (2.4), o termo Fn se refere à fração de átomos de carbono livre dentro
de uma cadeia contendo n átomos de carbono e determina a distribuição do número de
carbono total dos produtos da reação de SFT. Esta distribuição determina uma relação entre o
rendimento do produto e o número de carbonos, mostrando uma seletividade de gases até
graxas.
Considerando o seguinte mecanismo de crescimento de cadeia apresentado na
Figura 2.4, tem-se:
11 *** n
r
n
r
n CCC PP
rt
nC
Figura 2.4 – Mecanismo de terminação e propagação de cadeia. (Schulz, 1999).
Nessa esquematização, o termo *Cn é um intermediário da reação, Cn é um
produto resultante de *Cn por uma reação de terminação (não envolvendo crescimento de
cadeia), no qual *Cn-1 é um precursor da reação.
Utilizando a aproximação de estado estacionário, pode-se escrever:
[*Cn ] = cte, (2.5)

33
Observe que o termo *Cn é formado na propagação da cadeia a partir do precursor
*Cn-1 e é consumido em dois caminhos competitivos, correspondendo às velocidades de
propagação da cadeia com n átomos de carbono e terminação da cadeia com n átomos de
carbono. Assim, o acúmulo do intermediário será nulo:
1
[* ][ ] ( )[* ] 0
dt
np n p t n
d Cr C r r C (2.6)
Rearranjando a Equação (2.6), obtem-se a seguinte relação:
1
[* ]
[ ]
pn
n p t
rC
C r r
(2.7)
Nestas relações [*Cn] é a concentração do intermediário cuja cadeia admite n
átomos de carbono, e rp e rt são pseudo-constantes de taxa de primeira ordem para as reações
de propagação e terminação da cadeia, respectivamente.
Deste modo, pode-se definir a constante como:
tP
P
rr
r
(2.8)
Esta razão nos mostra a probabilidade de haver propagação da cadeia em relação a
todas as possibilidades existentes (a cadeia, em regime estacionário, ou se propaga ou se
finda). Se o valor de é elevado, a propagação da cadeia prevalece, e produtos de maior
massa molecular devem ser esperados, como visto na Figura 2.5. O valor numérico de varia
entre 0 e 1. A relação da distribuição das frações mássicas dos hidrocarbonetos formados na
reação de SFT (eixo y) em função de (fator de probabilidade de crescimento da cadeia) é
mostrada na Figura 2.5.
Como os valores de dependem das condições operacionais da reação e do tipo
de catalisador utilizado, o conhecimento de leva a uma possibilidade de otimização do
processo visando produtos comercialmente mais desejados (van der Laan, 1999). Por
exemplo, com o objetivo de priorizar a produção de hidrocarbonetos líquidos (gasolina,
querosene e diesel) na reação de SFT, é necessária uma mudança na configuração das
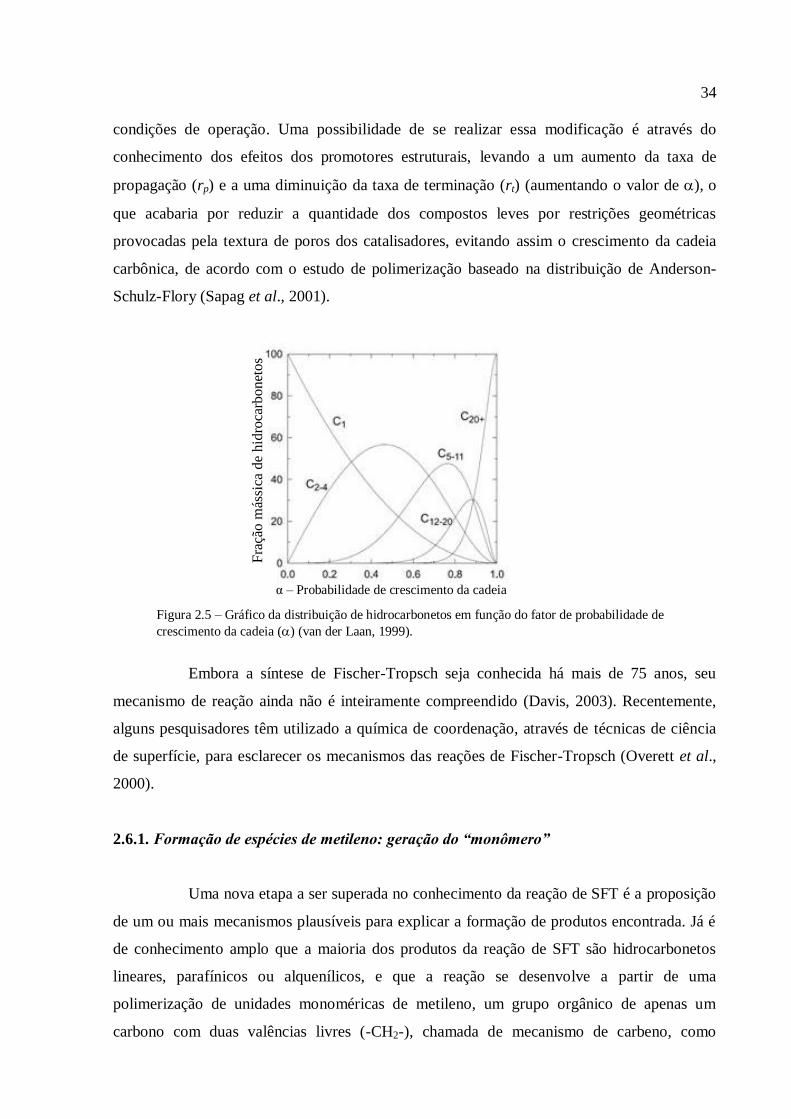
34
condições de operação. Uma possibilidade de se realizar essa modificação é através do
conhecimento dos efeitos dos promotores estruturais, levando a um aumento da taxa de
propagação (rp) e a uma diminuição da taxa de terminação (rt) (aumentando o valor de ), o
que acabaria por reduzir a quantidade dos compostos leves por restrições geométricas
provocadas pela textura de poros dos catalisadores, evitando assim o crescimento da cadeia
carbônica, de acordo com o estudo de polimerização baseado na distribuição de Anderson-
Schulz-Flory (Sapag et al., 2001).
Embora a síntese de Fischer-Tropsch seja conhecida há mais de 75 anos, seu
mecanismo de reação ainda não é inteiramente compreendido (Davis, 2003). Recentemente,
alguns pesquisadores têm utilizado a química de coordenação, através de técnicas de ciência
de superfície, para esclarecer os mecanismos das reações de Fischer-Tropsch (Overett et al.,
2000).
2.6.1. Formação de espécies de metileno: geração do “monômero”
Uma nova etapa a ser superada no conhecimento da reação de SFT é a proposição
de um ou mais mecanismos plausíveis para explicar a formação de produtos encontrada. Já é
de conhecimento amplo que a maioria dos produtos da reação de SFT são hidrocarbonetos
lineares, parafínicos ou alquenílicos, e que a reação se desenvolve a partir de uma
polimerização de unidades monoméricas de metileno, um grupo orgânico de apenas um
carbono com duas valências livres (-CH2-), chamada de mecanismo de carbeno, como
Fra
ção m
ássi
ca d
e hid
roca
rbo
net
os
α – Probabilidade de crescimento da cadeia
Figura 2.5 – Gráfico da distribuição de hidrocarbonetos em função do fator de probabilidade de
crescimento da cadeia () (van der Laan, 1999).

35
originalmente proposto por Fischer e Tropsch em 1926. Durante vários anos, diversos outros
mecanismos foram propostos, onde se destacam aqueles que envolvem espécies de
hidroxicarbeno (=CHOH) (Kummer e Emmet, 1953) e inserção de CO (Henrici-Olive e
Olive, 1976). A Figura 2.6 mostra uma representação esquemática desses três mecanismos
utilizados na reação de SFT:
CARBENO HIDROXICARBENO INSERÇÃO DE CO
Figura 2.6 – Representação esquemática de três mecanismos básicos de reação de Fischer-Tropsch: mecanismo
de carbeno, mecanismo de hidroxicarbeno e mecanismo de inserção de CO (Anderson, 1984).
Com a observação empírica de que a maioria dos catalisadores utilizados para a
reação de SFT também dissocia o monóxido de carbono (CO), pode-se concluir que na
temperatura utilizada para a reação, quando a molécula é adsorvida na área superficial ativa
dos catalisadores, os mecanismos de inserção do CO e hidroxicarbeno para crescimento de
cadeia (inserções de CO sucessivas e hidrogenações das espécies acil resultantes) tornaram-se
menos aplicados (Brodén et al., 1976). No entanto, para efeito de terminação de cadeia, a
etapa de inserção de CO ainda é usada com frequência para considerar a formação das
pequenas frações de produtos oxigenados. Esse mecanismo, baseado na adsorção e posterior
dissociação de moléculas de CO na superfície do catalisador, é apoiado por evidências
experimentais e estudos teóricos (Hoffmann e Sung, 1985). Ainda não está totalmente claro
como o H2 se dissocia na superfície de catalisador. Contudo, a maioria dos pesquisadores
aceita a dissociação do H2 para formar espécies híbridas na superfície. Reações de hidrogênio
4 H(ads)

36
com átomos de carbono na superfície (carboneto) levam consequentemente à formação de
grupos metino (CH) e metileno (=CH2). As unidades de metileno assim formadas são
unidades de monômero para a reação de polimerização global.
2.6.2. Mecanismo alquil para propagação e terminação de cadeia
Em 1980, os pesquisadores Brady e Pettit observaram que quando o diazometano
é passado sobre catalisadores como Ni, Pd, Fe, Co, Ru ou Cu, o produto primário obtido era o
etileno. Porém, quando H2 era misturado com diazometano na presença de um desses
catalisadores, uma vasta faixa de hidrocarbonetos similar à distribuição de produtos da reação
de SFT era obtida. Eles concluíram que existiria uma reação entre o metileno e o hidrogênio
superficial formando um metil na superfície do catalisador, e o crescimento da cadeia se daria
por sucessivas inserções de metileno na ligação metal-alquil. Diante dessas previsões, foi
proposto o mecanismo alquil para a reação de Fischer-Tropsch (Figura 2.7).
A etapa de terminação do crescimento da cadeia leva a uma eliminação do -
hidreto para formar -olefinas ou através de uma redução com hidreto superficial para formar
alcanos (parafinas). Observe a esquematização na Figura 2.8.
Figura 2.7 – Mecanismo alquil para iniciação e crescimento de cadeia na reação de Fischer-Tropsch (Overett et
al., 2000).

37
Figura 2.8 – Término do mecanismo alquil. a) Formação de alcanos através de uma redução com hidrogênio
superficial. b) Formação de -olefina através de uma reação de eliminação (Overett et al., 2000).
2.6.3. Mecanismo alquenil para propagação e terminação de cadeia
O mecanismo de alquenil foi proposto por Maitlis (1989), como uma alternativa
ao mecanismo alquil. Este mecanismo propõe que a reação de SFT ocorra como uma
polimerização de espécies metileno na superfície do catalisador, que por sua vez seriam
formadas pela quimisorção dissociativa do gás de síntese com a subsequente hidrogenação
das espécies carboneto na superfície do catalisador. A reação é iniciada pela formação da
espécie vinil na superfície (-CH=CH2) através da reação de uma espécie metino de superfície
(CH) e um metileno de superfície (=CH2). O crescimento da cadeia ocorre pela reação da
espécie vinílica com um metileno superficial para formar uma espécie alil (CH2CH=CH2). A
espécie alil sofre isomerização para então formar uma espécie alquenil (CH=CHCH3) que
posteriormente pode reagir. Essa isomerização que resulta na migração da dupla ligação para
a posição é ainda observada à medida que a cadeia cresce com a inserção de grupos
metileno. A etapa de terminação ocorre quando a espécie alquenil é reduzida pela reação com
um hidrogênio superficial formando uma -olefina. Note que esse mecanismo não explica a
formação de espécies parafínicas (hidrocarbonetos saturados). Observe o mecanismo
alquenila desenhado graficamente na Figura 2.9.

38
O problema sobre a produção de etileno (eteno) deve ser tratado separadamente. O
mecanismo alquil não forma etileno porque a propagação das espécies que poderiam formar
etileno, como o grupo etil (-CH2CH3), possui um grupo metil estável no final da cadeia que
não tem a tendência em doar hidrogênio para gerar a dupla ligação. Essa circunstância não
afeta grupos alquil de cadeias mais longas, como por exemplo, o grupo propil, -CH2CH2CH3,
que possui um -CH2- intermediário menos estável que pode facilmente doar seu hidrogênio
para formar a dupla ligação, resultando em uma olefina.
Figura 2.9 – Mecanismo alquenil para a reação de Fischer-Tropsch. (Overett et al, 2000).
O mecanismo alquenila também não forma etileno porque, se isso ocorresse,
haveria a reação entre a cadeia iniciada, -CH=CH2, e um hidrogênio de superfície. No entanto,
tal fato não ocorre, pois a cadeia carbônica do grupo vinil adsorvido ao metal possui uma
interação quimisortiva de elevada intensidade, praticamente anulando a tendência à redução
com hidrogênio de superfície, e assim nenhum etileno pode ser formado (Fernandes, 2005).
Da mesma forma que o mecanismo alquil, o alquenil só produz olefinas com número de
carbonos superior a 2. Assim, a produção de etileno fica restrita a um mecanismo diferente,
em que ocorre a reação entre dois grupos metileno. Devido a essa restrição e a
impossibilidade de produzi-lo por mecanismos alquil ou alquenil, se justifica a baixa fração
molar de etileno na distribuição de produtos.

39
2.6.4. Readsorção de Olefinas
O modelo de readsorção de olefinas baseia-se na reversibilidade para a desorção
(readsorção) em sítios ativos. O fato experimental que desencadeou essa nova forma de
observar o mecanismo da reação de SFT ocorreu quando se percebeu que olefinas, quando
adicionadas ao gás de síntese, poderiam iniciar o crescimento de cadeia (Claeys, et al., 1997;
Schulz e Claeys, 1999). Matematicamente, a reversibilidade de desorção de olefinas foi
incluída em modelos cinéticos de síntese de Fischer-Tropsch por Schulz e Claeys (1999). De
forma interessante e até certo ponto surpreendente, o efeito causa uma diminuição no
conteúdo de olefinas nos produtos de reação com aumento no número de carbonos na cadeia
carbônica do produto parafínico (Figura 2.10). Dessa forma, a reversibilidade de desorção de
olefinas modifica as equações de estado estacionário para o intermediário *Cn de reação. Já
que o mesmo passa a ser formado a partir do precursor *Cn-1, ele pode ser finalizado como
parafina Cn ou como olefina Cn, mas também prosseguir com o crescimento da cadeia,
gerando a espécie *Cn+1, ou ainda ser regenerado na reação reversível com a olefina. Assim, é
de se prever que haja uma maior probabilidade de aumento das cadeias carbônicas e aumento
da massa molecular média dos produtos. Este é um efeito vantajoso quando a reação é
direcionada para um máximo rendimento na produção de combustível diesel em processos
que incluem o hidrocraqueamento. Esse mecanismo de readsorção de olefinas é uma
característica dos catalisadores de cobalto, e não é observado em catalisadores de ferro.
*Cn*Cn-1
*Cn+1
kt,parkt,olef
ka
kp
CO/H2
(Cn) Parafinas (Cn) -Olefinas
Figura 2.10 – Esquema de reação do modelo de distribuição do produto para readsorção de olefinas (Van deer Laan, 1999).

40
3. ESTIMAÇÃO DE PARÂMETROS E MODELAGEM PARA A REAÇÃO DE
SÍNTESE DE FISCHER-TROPSCH
Um modelo matemático tem sido desenvolvido e implementado com o objetivo de
descrever o comportamento do reator para a reação de síntese de
Fischer-Tropsch. Os parâmetros relativos à cinética dessa reação foram estimados para que
um estudo abrangente do processo seja realizado avaliando as condições operacionais do
reator.
A presente seção será dividida em três etapas:
a modelagem matemática e cinética;
o desenvolvimento matemático (cálculos relacionados ao modelo);
discussão de resultados.
3.1. Modelo matemático
O modelo matemático abordado nesse estudo assume que:
o reator em que ocorre a reação de síntese de Fischer-Tropsch (SFT) opera em
condições isotérmicas, alcançada com uma eficiente troca de calor;
o gás atravessa o reator em regime empistonado, devido a um perfil constante de
velocidade;
a reação ocorre em estado estacionário;
Ainda são consideradas etapas cinéticas próprias dos catalisadores de cobalto da
reação de SFT, como o processo de adsorção de olefinas. Além disso, as fases sólida e líquida
estão em equilíbrio na saída do reator (característica de modelagem para reatores em estado
estacionário), e foram tomadas como desprezíveis as seguintes situações que poderiam
influenciar o modelo (Fernandes, 2005):
Resistência para a transferência de massa e calor entre a fase gasosa e o catalisador,
pois não há a formação de um filme muito viscoso ao redor do catalisador, tornando as
resistências às transferências de massa e calor negligenciáveis;

41
Gradiente radial de concentração e de temperatura no interior dos tubos do reator,
pois, para a alta velocidade de escoamento gasoso em que se opera o reator o seu
diâmetro é relativamente pequeno;
Gradiente radial de concentração e de temperatura no interior dos poros do catalisador,
já que o catalisador apresenta macroporos de pequeno diâmetro;
Perda de carga no reator referente ao leito fixo, porque o comprimento do reator
associado à velocidade de escoamento gasoso fazem com que o leito catalítico tenha
perda de carga muito pequena.
As características anteriores, inclusive a perda de carga, foram negligenciadas
tanto teórica quanto experimentalmente, pelo uso de um reator diferencial, onde o reator de
leito fixo é simplificado para as condições propostas considerando um comprimento
diferencial do reator. Contudo, trabalhos anteriores de simulação (Barros Junior, 2008)
demonstram que a influência da perda de carga nas frações de produtos obtidas é pequena.
Após análise da literatura, o modelo cinético adotado para as reações de síntese de
Fischer-Tropsch (SFT) com readsorção de olefinas e uso de catalisadores de cobalto, foi
aquele sugerido por Zennaro et al. (2000) e expresso na Equação (3.1), pois é o que apresenta
resultados que mais se aproximam de seus valores experimentais.
2
0,743
CO H
SFT 2
CO
1,1.10 .P . PR
1 0,040.P
(3.1)
onde o termo RSFT (expressa em mol.g-1
.h-1
) se refere à velocidade em que ocorrem as reações
de SFT em regime estacionário. A Equação (3.1) foi proposta para catalisadores em reatores
de leito fixo diferenciais com catalisadores de cobalto suportados em dióxido de titânio a 20
atm de pressão, para uma faixa de temperatura na faixa de 180°C a 240°C, proporção H2/CO
igual a 2,1 e velocidade espacial de 5000 h-1
. Os dados estão resumidos na Tabela 3.1:

42
Tabela 3.1. Condições de operação e parâmetros do reator. (Zennaro et al., 2000)
Condição de operação Valor/ intervalo
Temperatura (°C) 180 – 240
Pressão total (MPa) 2,0 MPa
Proporção H2/CO 2,1
Velocidade espacial 5000 h-1
O modelo cinético utilizado para realizar a simulação da reação de SFT em reator
tubular foi baseado na polimerização do grupo metileno (-CH2-), o qual se forma a partir do
monóxido de carbono e do hidrogênio adsorvidos na superfície do catalisador (mecanismo
carbeno), com a distribuição de produtos mostrando que os mecanismos alquil e alquenil
atuam juntos.
Na notação para a proposta de mecanismo que será discutida a seguir, o termo
R(n) designa um grupo alquil com n carbonos, P(n) determina o alcano (parafina) com n
átomos de carbono, R”(n) se refere ao grupo alquenil com n carbonos, enquanto P=(n) está
relacionado com a -olefina com n átomos de carbono. Logo, de acordo com a notação
adotada, R(1) é o grupo metil e P=(2) seria o etileno (eteno). O mecanismo alquila pode ser
representado pelas seguintes etapas:
2CH + H ikR(1) etapa de iniciação
R(1) + 2CH 1pkR(2) etapa de propagação do grupo metila
R(n) + 2CH pkR(n+1) etapa de propagação de um grupo qualquer
R(n) + H parkP(n) formação de parafina pelo hidrogênio de superfície
R(n) olefk P
=(n) formação de -olefina pela eliminação de -hidreto
P=(n) adsk
R(n) readsorção de olefinas (para o catalisador de cobalto)
O balanço molar para as espécies de propagação (R(n), onde n é o número de
carbonos na cadeia do grupo alquila) é afetada pela propagação da cadeia e pela terminação
por eliminação de -hidreto (para a formação de olefinas) e por redução (para a formação de
parafinas). Uma espécie de propagação, R(n), é formada a partir de uma espécie R(n – 1), e
consumida pela sua própria propagação formando espécies R(n + 1) e para formar uma
parafina (P(n)) ou uma olefina (P=(n)), com n átomos de carbono em sua cadeia. Assim, a taxa
de acúmulo das espécies de propagação R(n) é dada na Equação (3.2):

43
p 2 ads p 2 par olef
dk . CH . R(n 1) + k . P (n) k . CH . R(n) k . H . R(n) k . R(n)
dt
R(n)
(3.2)
Os balanços molares para os grupos metila e etila são diferenciados e aparecem
como expresso nas Equações (3.3) e (3.4):
i 2 p1 2 met
dk . CH . H k . CH . R(1) k . H . R(1)
dt
R(1) (3.3)
p1 2 ads p 2 par
dk . CH . R(1) + k . P (2) k . CH . R(2) k . H . R(2)
dt
R(2)
(3.4)
Em um processo de polimerização com a síntese de Fischer-Tropsch, o tempo de
vida das espécies em propagação com n carbonos (R(n)) é muito curto, e estas espécies são
formadas e consumidas constantemente. Essa característica conduz a uma concentração
praticamente constante de R(n), e a sua concentração pode ser considerada constante como
proposta de modelagem (teoria de estado quasi-estacionário). Se o estado quasi-estacionário é
aplicado a todas as espécies de propagação, sua taxa de acúmulo (Equação (3.2)) é nula e a
concentração das espécies de propagação, para o mecanismo alquil, é dada pela Equação
(3.5):
p 2 ads
p 2 par olef
k . CH . + k .R(n) =
k . CH k . H k
R(n 1) P (n)
(3.5)
Aplicando a condição de estado quasi-estacionário ao grupo metila, é obtida a
Equação (3.6):
i 2
p1 2 met
k . CH . HR(1) =
k . CH k . H
(3.6)
Para o grupo etila, tem-se a Equação (3.7):

44
p1 2 ads
p 2 par
k . CH . + k .R(2) =
k . CH k . H
R(1) P (2)
(3.7)
O balanço de massa para parafinas e olefinas produzidas é afetado somente pela
terminação de suas cadeias em propagação, chegando às Equações (3.8) e (3.9):
par
dk . H . R(n)
dt
P(n) (3.8)
olef ads
dk . R(n) k . P (n)
dt
P (n)
(3.9)
O mecanismo alquenila, pode ser representado pelas equações seguintes, onde
ocorre a reação inicial de metileno com metino, com as posteriores inserções de metileno e
isomerizações:
2CH + CH 2ik R”(2) etapa de iniciação
R”(2) + 2CH '
2pkR”(3) etapa de propagação de vinila
R”(n) + 2CH 2pkR”(n+1) etapa de propagação de grupo qualquer
R”(n) + H 2olefkP
=(n) formação de olefinas por redução
P=(n) adsk
R(n) readsorção de olefinas (para o catalisador de cobalto)
O balanço de massa, segundo o mecanismo alquenila, para as espécies de
propagação R”(n), é dependente de propagação das espécies e da terminação por redução.
Uma espécie de propagação, R”(n), é formada a partir de uma espécie R”(n – 1), e consumida
pela sua própria propagação formando espécies R”(n + 1) e para formar uma olefina (P=(n)),
com n átomos de carbono em sua cadeia. Assim, a taxa de acúmulo das espécies de
propagação R”(n) é dada por (Fernandes, 2005):
p2 2 p2 2 olef2
dk . CH . R"(n 1) k . CH . R"(n) k . H . R"(n)
dt
R"(n) (3.10)

45
Quando se aplica o estado quasi-estacionário, a termo diferencial fornecido pela
Equação (3.10) se anula e a concentração das espécies de propagação é dada pela Equação
(3.11):
p2 2
p2 2 olef2
k . CH .R"(n) =
k . CH k . H
R"(n 1)
(3.11)
A formação de espécies de propagação (que corresponde à etapa de iniciação para
o mecanismo alquenil) ocorre pela reação de um grupo metileno com uma espécie metino de
superfície, que é seguido pela sua propagação. Sua taxa de acúmulo é fornecida na Equação
(3.12):
'
i2 2 p2 2
dk . CH .[ CH] k . CH . R"(2)
dt
R"(2) (3.12)
Aplicando a condição de estado quasi-estacionário à etapa de iniciação, a Equação
(3.13) é encontrada:
i2 2 i2
' '
p2 2 p2
k . CH . CH k . CHR"(2) =
k . CH k
(3.13)
Para o grupo propenila, a equação de acúmulo também é diferenciada e é dada
pela Equação (3.14):
'
p2 2 p2 2 olef2
dk . CH . R"(2) k . CH . R"(3) k . H . R"(3)
dt
R"(3) (3.14)
Aplicando a condição de estado quasi-estacionário à etapa de iniciação, a Equação
(3.15) é encontrada:
'
p2 2
p2 2 olef2
k . CH .R"(3) =
k . CH k . H
R"(2)
(3.15)

46
O balanço de massa para as olefinas produzidas pelo mecanismo alquenil é
afetado somente pela terminação de suas cadeias em propagação, expresso na Equação (3.16):
olef2 ads
dk . H . R"(n) k
dt
P (n). P (n)
(3.16)
Metano, etano e eteno são formados por reações diferentes daquelas estudadas
para parafinas e olefinas superiores. Suas reações são as seguintes:
R(1) + H metkP(1) formação de metano por redução com hidreto de superfície
R(2) + H etkP(2) formação de etano por redução com hidreto de superfície
2CH + 2CH 1OkP
=(2) etapa de terminação de metileno
A modelagem consiste em um conjunto de equações diferenciais bem como de
equações algébricas baseadas em balanços de massa (Fernandes, 2005). Metano, etano e eteno
são formados por reações que não envolvem espécies em propagação e têm, portanto,
equações diferentes em seus balanços. Metano é formado pela reação de terminação entre a
espécie metila 3CH , anteriormente chamado R(1), que reage com o hidrogênio superficial
em velocidade superior à de terminação da espécie R(n) quando n > 2. De forma similar
ocorre para o etano (que no caso seria o produto P(2) originário do grupo R(2)). O eteno é
formado pela reação entre duas espécies metileno em preferência à terminação de espécies
R(2) ou R”(2), que não ocorrem. Assim, para o reator tubular, tem-se as Equações (3.17) a
(3.19):
met
dk . H . R(1)
dt
P(1) (3.17)
et
dk . H . R(2)
dt
P(2) (3.18)
O1 2 2
dk . CH . CH
dt
P (2)
(3.19)

47
3.2. Cálculos referentes ao modelo cinético adotado
Algumas concentrações utilizadas no modelo são de espécies intermediárias,
como H , CH e 2CH , o que dificulta suas estimativas e não estão diretamente
disponíveis como condições de operações reacionais. Entretanto, uma boa estratégia consiste
em substituir algebricamente essas concentrações por outras de maior facilidade de medição,
como as pressões parciais dos componentes presentes no meio reacional. Quando possível, a
taxa de reação global de SFT foi utilizada no lugar das concentrações das espécies
intermediárias, já que essa taxa é conhecida para muitos catalisadores e depende das pressões
parciais dos componentes. Para o catalisador de cobalto, foi usada a Equação (3.1).
A taxa global de reação (RSFT) reflete principalmente a velocidade de propagação
da cadeia de hidrocarbonetos, correspondendo à velocidade de consumo da espécie 2CH .
Esse comportamento se manifesta na Equação (3.20):
SFT p 2 ap
n 1
R k . CH . R(n) .(1/ρ )
(3.20)
onde ρap se refere à densidade aparente do catalisador. Assumindo a condição de estado
estacionário, o termo R(n) pode ser considerado constante e RSFT deve ser proporcional à
concentração de metileno no sistema, o que representa a velocidade de formação de 2CH .
Daí chega-se à conclusão que a concentração das espécies 2CH é proporcional à velocidade
de reação global.
A concentração das espécies H , de acordo com o mecanismo de adsorção de H2
ao catalisador, é proporcional à raiz quadrada da sua taxa de adsorção, ou seja, proporcional à
raiz quadrada da sua pressão parcial no meio reacional. Assim, é possível alcançar as
Equações (3.21) até (3.23) a seguir:
SFT p 2 ap SFT1 2
n 1
R k . CH . R(n) .(1/ρ ) k . CH
(3.21)
SFT2
SFT1
RCH
k (3.22)
2
0.5
HH k'. P (3.23)

48
onde os termos k’ e SFT1k se referem a constantes de proporcionalidade que expressam,
respectivamente, a dependência da pressão parcial de H2 e da velocidade de reação para a
concentração de cada intermediário.
A partir dos balanços de massa para a reação de SFT, com as mudanças referentes
à substituição das espécies intermediárias com base na aproximação do estado estacionário e
em cinética de polimerização, obtemos as equações a seguir. Substituindo as Equações (3.22)
e (3.23) na Equação (3.6), e chamando k '
i = i SFT1k'.k / k , k
'
p1 = p1 SFT1k /k e k '
met = metk'.k
obtém-se a Equação (3.24):
2
2
' 0.5
i SFT H
' ' 0.5
p1 SFT met H
k .R .PR(1) =
k .R k .P (3.24)
A equação final para o grupo etila, R(2), via mecanismo alquila, é obtido a partir
da Equação (4.7). Observe que o grupo R(2) não forma etileno, e, portanto não há terminação
em olefina. Substituindo os valores referentes às Equações (3.22) e (3.23), e chamando k'
p =
p SFT1k / k e k'
par = park'.k , é obtida a Equação (3.25):
2
p SFT ads
0.5
p SFT par H
=k' .R . + k .
R(2) = k' .R k' .P
R(1) P (2)
(3.25)
Considerando que a taxa de formação de metino de superfície, CH , seja
semelhante à de formação de grupo metileno no mecanismo alquenila, 2CH , substituindo a
Equação (3.22) na Equação (3.13), e denominando k '
i2 = i2 SFT1k / k , pode-se deduzir a
Equação (3.26) a seguir:
SFT
'
p2
'
i2k .RR"(2)
k (3.26)
Substituindo as Equações (3.22) e (3.23) na Equação (3.15) e denominando k"'
p2 =
'
p2 SFT1k / k , k"
p2 = p2 SFT1k / k e k '
olef2 = olef2k'.k , pode-se deduzir a Equação (3.27) a seguir:

49
2
"'
p2 SFT
" 0.5
p2 SFT olef2 H
k .RR"(3) . R"(2)
k .R k' .P
(3.27)
Tomando a Equação (3.5), substituindo os valores referentes às Equações (3.22) e
(3.23), e chamando k'
p = p SFT1k / k e k'
par = park'.k , é obtida a Equação (3.28):
2
p SFT ads
0.5
p SFT par H olef
=k' .R . k .
R(n)k' .R k' .P k
R(n 1) P (n)
(3.28)
Agora, observando a Equação (3.12), substituindo os valores referentes às
Equações (3.22) e (3.23), e chamando k"
p2 = p2 SFT1k / k e k '
olef2 = olef2k'.k , é obtida a Equação
(3.29):
2
"
p2 SFT
" 0.5
p2 SFT olef2 H
k .RR"(n) . R"(n 1)
k .R k' .P
(3.29)
As equações diferenciais para os produtos formados, divididas de acordo com o
número de carbonos de cada hidrocarboneto, são fornecidas a seguir. Ao se substituir as
relações obtidas na Equação (3.23) na Equação (3.17), e chamando k '
met = metk'.k , pode-se
chegar à Equação (3.30):
2
0.5
met H
dk' .P . R(1)
dt
P(1) (3.30)
Quando a Equação (3.22) é substituída na Equação (3.19), e se denomina k O2 =
2
O1 SFT1k / k , se obtém a Equação (3.31):
2
O2 SFT
dk .R
dt
P (2)
(3.31)
Substituindo a Equação (3.23) na Equação (3.8), e realizando a operação k'
par =
park'.k , pode-se obter a Equação (3.32), para n > 1:

50
2
0.5
par H
dk' .P . R(n)
dt
P(n) (3.32)
Finalmente, ao substituir as Equações (3.22) e (3.23) no somatório das Equações
(3.10) e (3.16), que corresponde à formação de olefinas via mecanismos alquila e alquenila,
respectivamente, e escrevendo k '
olef2 = olef2k'.k , se deduz a Equação (3.33):
2
0.5 =
olef olef2 H ads
d P (n)k . R(n) k' .P . R"(n) k . P (n)
dt
(3.33)
O programa, executado em linguagem Fortran, muito eficiente em simulações que
necessitem de cálculos matemáticos simultâneos, é iniciado com a leitura dos dados
experimentais e a introdução dos valores iniciais dos parâmetros. A seguir, o método de
Levenberg-Maquardt executa testes em função de valores de tolerância (estabelecidos em
função da margem de erro tolerada) para as equações descritas para o modelo cinético da
reação de SFT voltado para o reator tubular. Caso o erro se encontre dentro de intervalos
aceitáveis, a sub-rotina não é mais executada e o programa imprime os resultados. Caso
contrário, o programa sugere novos valores de parâmetros e reinicia o procedimento. Esse
procedimento cíclico é repetido até que os parâmetros se adequem à tolerância utilizada. A
cada teste para um conjunto de parâmetros, a programação verifica se os valores obtidos estão
descrevendo corretamente o comportamento do reator tubular de leito fixo onde ocorre a
reação de SFT. Para essa finalidade, foram utilizados os dados experimentais obtidos por
Visconti et al. (2007). Ao final, os parâmetros são impressos. Um resumo do procedimento
adotado para a execução do programa de simulação é mostrado no algoritmo a seguir (Figura
3.1):
Figura 3.1 – Algoritmo para o programa de estimação de parâmetros
Leitura e entrada
de dados
Estimativa de
parâmetros do
modelo usando o
método de
Levenberg-
Maquardt
Impressão dos
resultados
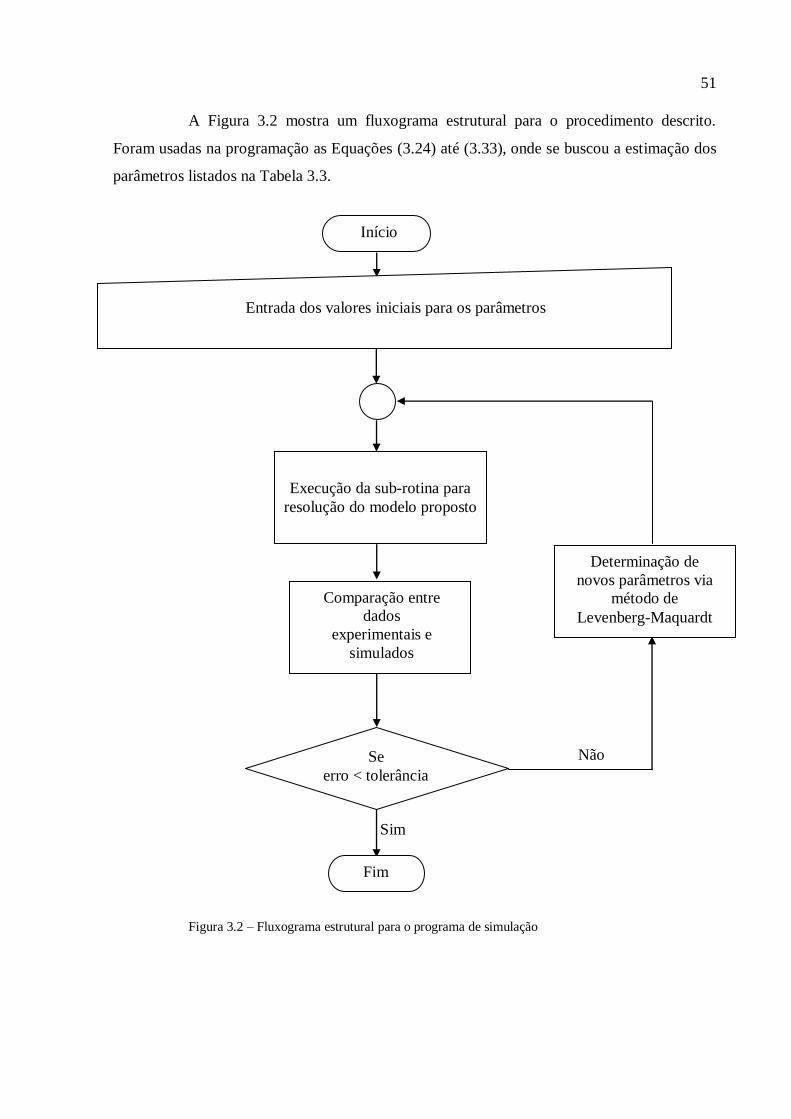
51
A Figura 3.2 mostra um fluxograma estrutural para o procedimento descrito.
Foram usadas na programação as Equações (3.24) até (3.33), onde se buscou a estimação dos
parâmetros listados na Tabela 3.3.
Figura 3.2 – Fluxograma estrutural para o programa de simulação
Determinação de
novos parâmetros via
método de
Levenberg-Maquardt
Sim
Fim
Execução da sub-rotina para
resolução do modelo proposto
Se
erro < tolerância
Não
Início
Entrada dos valores iniciais para os parâmetros
Comparação entre
dados
experimentais e
simulados

52
A estimação de parâmetros baseou-se na função objetivo que calcula minimiza a
diferença entre o valor simulado e o valor observado (min(Ysim – Yobs)). Foi utilizado o
método numérico dos mínimos quadrados para a resolução do modelo. A inicialização das
equações foi realizada adotando-se as seguintes condições:
De z = 0 até z = L, em t = 0: as concentrações de CO e H2 são iguais às
concentrações iniciais.
Em z = 0 para t qualquer: as concentrações de CO e H2 são iguais às
concentrações iniciais.
3.3. Resultados
O modelo matemático foi avaliado estatisticamente e comparado aos dados
experimentais (Visconti et al., 2007). Os dados foram gerados em um reator diferencial, que
consiste de um reator tubular em que os efeitos de dispersão axial e radial, bem como efeitos
de perda de carga, podem ser desprezados.
A distribuição de produtos em termos de fração mássica em uma reação de síntese
de Fischer-Tropsch (SFT) gera uma curva de formato característico, conforme apresentado na
Figura 3.3. A maior parte do monóxido reagente é convertida em gás metano (CH4), e ocorre
uma menor conversão em hidrocarbonetos com 2 carbonos, graças à terminação em olefinas
ser tratada de forma diferenciada. Para o número de carbonos n > 2, a fração mássica diminui
gradativamente com o aumento da cadeia carbônica, gerando os produtos de maior apelo
comercial. O uso do catalisador de cobalto, com a existência da readsorção de olefinas,
diminui a conversão em olefinas, favorecendo a produção de parafinas. A Figura 3.3 compara
os resultados obtidos no modelo matemático com os valores obtidos por Visconti et al.
(2007).
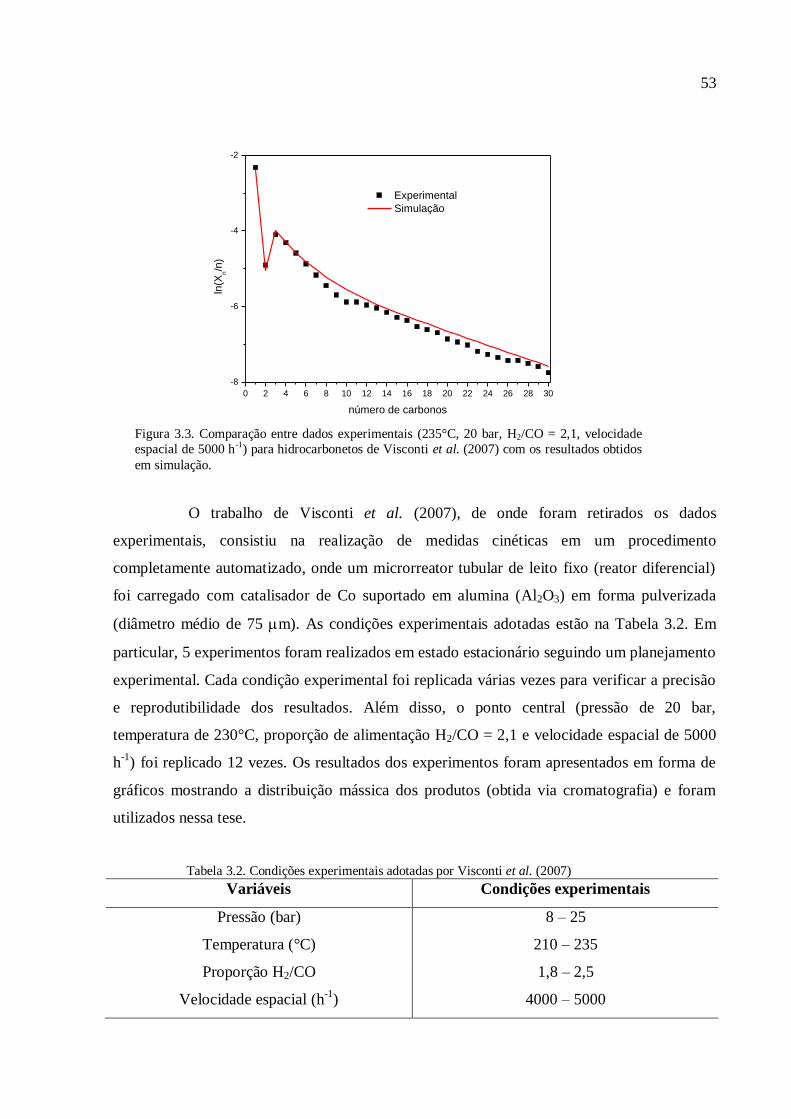
53
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
-8
-6
-4
-2
Experimental
Simulaçãoln
(Xn/n
)
número de carbonos
O trabalho de Visconti et al. (2007), de onde foram retirados os dados
experimentais, consistiu na realização de medidas cinéticas em um procedimento
completamente automatizado, onde um microrreator tubular de leito fixo (reator diferencial)
foi carregado com catalisador de Co suportado em alumina (Al2O3) em forma pulverizada
(diâmetro médio de 75 m). As condições experimentais adotadas estão na Tabela 3.2. Em
particular, 5 experimentos foram realizados em estado estacionário seguindo um planejamento
experimental. Cada condição experimental foi replicada várias vezes para verificar a precisão
e reprodutibilidade dos resultados. Além disso, o ponto central (pressão de 20 bar,
temperatura de 230°C, proporção de alimentação H2/CO = 2,1 e velocidade espacial de 5000
h-1
) foi replicado 12 vezes. Os resultados dos experimentos foram apresentados em forma de
gráficos mostrando a distribuição mássica dos produtos (obtida via cromatografia) e foram
utilizados nessa tese.
Tabela 3.2. Condições experimentais adotadas por Visconti et al. (2007)
Variáveis Condições experimentais
Pressão (bar) 8 – 25
Temperatura (°C) 210 – 235
Proporção H2/CO 1,8 – 2,5
Velocidade espacial (h-1
) 4000 – 5000
Figura 3.3. Comparação entre dados experimentais (235°C, 20 bar, H2/CO = 2,1, velocidade
espacial de 5000 h-1) para hidrocarbonetos de Visconti et al. (2007) com os resultados obtidos
em simulação.

54
Os parâmetros cinéticos foram estimados por um programa computacional
elaborado em linguagem Fortran que fez uso do método de Levenberg-Marquardt para
minimizar as somas dos quadrados dos desvios, de acordo com os conceitos em análise
estatística apropriados. Os parâmetros estão apresentados na Tabela 3.3:
Tabela 3.3. Parâmetros cinéticos para polimerização de CO em catalisador de cobalto
Parâmetros Estimativas
k'
i 0,0656 (g.m-3
.MPa0.5
)
k'
i2 35,815 (g.mol
-1)
k'
p1 1,318.10-4 (g.mol
-1)
k'
p 1,506.10-4 (g.mol
-1)
k"
p2 509,64 (g.mol-1
)
k'
p2 470,33 (m3.mol
-1.h
-1)
k"'
p2 521,52 (g.mol-1
)
k'
met 1.8.10-7 (h
-1.MPa
0.5)
kO2 1,500 (g2.h.mol
-1.m
-3)
kolef 2,74.10-9 (h
-1)
k'
olef2 0,1913 (h-1
.MPa0.5
)
k'
par 8.67.10-9
(h-1
.MPa0.5
)
kads 7,500.10-4
(h-1
)
3.3.1. Análise estatística
Os testes estatísticos do modelo adotado com os parâmetros apresentados para os
dados experimentais presentes na literatura (Visconti et al. (2007)) estão na Tabela 3.4.
Observa-se inicialmente o teste estatístico chi-quadrado (2) para os experimentos 1 a 4:
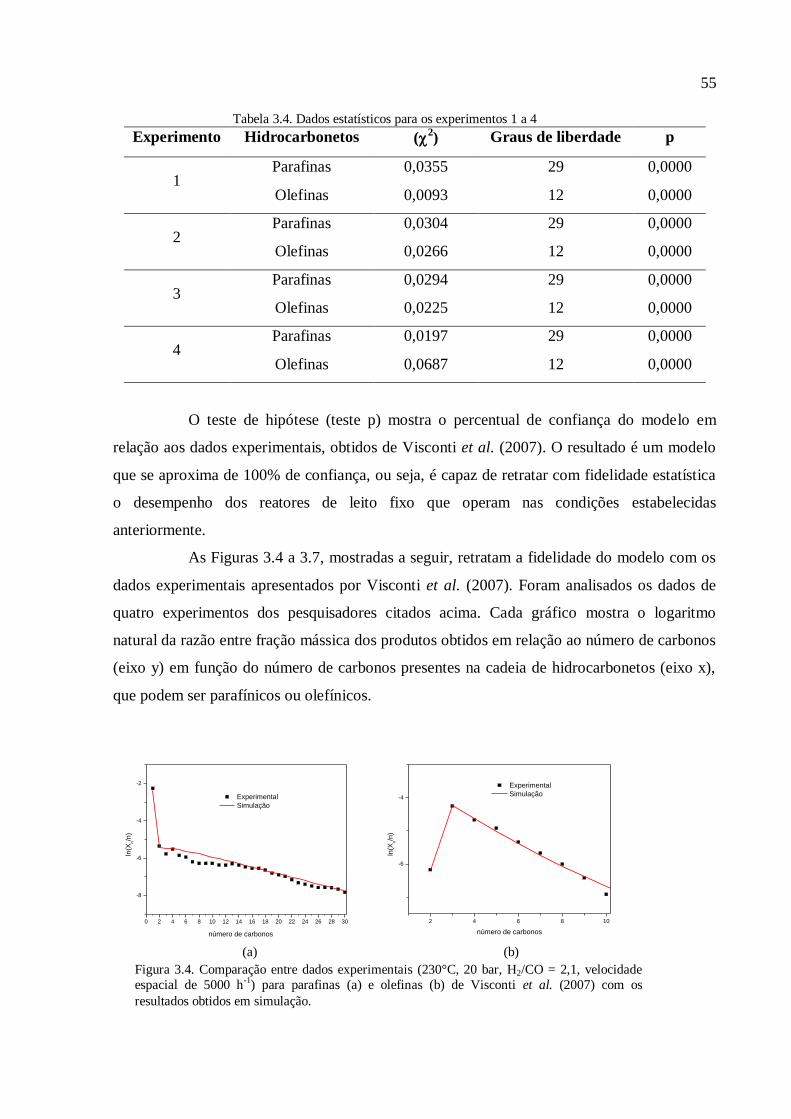
55
Tabela 3.4. Dados estatísticos para os experimentos 1 a 4
Experimento Hidrocarbonetos (2) Graus de liberdade p
1 Parafinas 0,0355 29 0,0000
Olefinas 0,0093 12 0,0000
2 Parafinas 0,0304 29 0,0000
Olefinas 0,0266 12 0,0000
3 Parafinas 0,0294 29 0,0000
Olefinas 0,0225 12 0,0000
4 Parafinas 0,0197 29 0,0000
Olefinas 0,0687 12 0,0000
O teste de hipótese (teste p) mostra o percentual de confiança do modelo em
relação aos dados experimentais, obtidos de Visconti et al. (2007). O resultado é um modelo
que se aproxima de 100% de confiança, ou seja, é capaz de retratar com fidelidade estatística
o desempenho dos reatores de leito fixo que operam nas condições estabelecidas
anteriormente.
As Figuras 3.4 a 3.7, mostradas a seguir, retratam a fidelidade do modelo com os
dados experimentais apresentados por Visconti et al. (2007). Foram analisados os dados de
quatro experimentos dos pesquisadores citados acima. Cada gráfico mostra o logaritmo
natural da razão entre fração mássica dos produtos obtidos em relação ao número de carbonos
(eixo y) em função do número de carbonos presentes na cadeia de hidrocarbonetos (eixo x),
que podem ser parafínicos ou olefínicos.
Figura 3.4. Comparação entre dados experimentais (230°C, 20 bar, H2/CO = 2,1, velocidade
espacial de 5000 h-1) para parafinas (a) e olefinas (b) de Visconti et al. (2007) com os
resultados obtidos em simulação.
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
-8
-6
-4
-2
Experimental
Simulação
ln(X
n/n
)
número de carbonos 2 4 6 8 10
-6
-4
Experimental
Simulação
ln(X
n/n
)
número de carbonos
(a) (b)
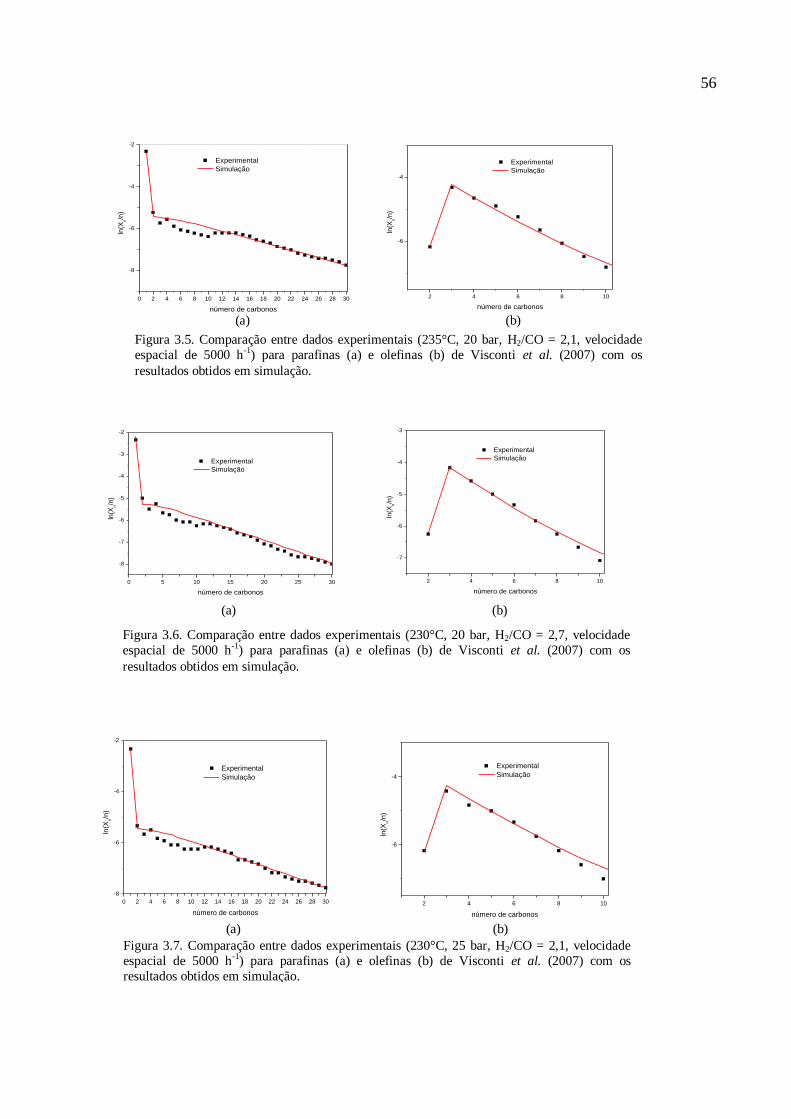
56
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
-8
-6
-4
-2
Experimental
Simulação
ln(X
n/n
)
número de carbonos
2 4 6 8 10
-6
-4
Experimental
Simulação
ln(X
n/n
)
número de carbonos
(a) (b)
Figura 3.5. Comparação entre dados experimentais (235°C, 20 bar, H2/CO = 2,1, velocidade
espacial de 5000 h-1) para parafinas (a) e olefinas (b) de Visconti et al. (2007) com os
resultados obtidos em simulação.
0 5 10 15 20 25 30
-8
-7
-6
-5
-4
-3
-2
Experimental
Simulação
ln(X
n/n
)
número de carbonos
2 4 6 8 10
-7
-6
-5
-4
-3
Experimental
Simulação
ln(X
n/n
)
número de carbonos (a) (b)
Figura 3.6. Comparação entre dados experimentais (230°C, 20 bar, H2/CO = 2,7, velocidade
espacial de 5000 h-1) para parafinas (a) e olefinas (b) de Visconti et al. (2007) com os
resultados obtidos em simulação.
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
-8
-6
-4
-2
Experimental
Simulação
ln(X
n/n
)
número de carbonos 2 4 6 8 10
-6
-4
Experimental
Simulação
ln(X
n/n
)
número de carbonos
Figura 3.7. Comparação entre dados experimentais (230°C, 25 bar, H2/CO = 2,1, velocidade
espacial de 5000 h-1) para parafinas (a) e olefinas (b) de Visconti et al. (2007) com os
resultados obtidos em simulação.
(a) (b)
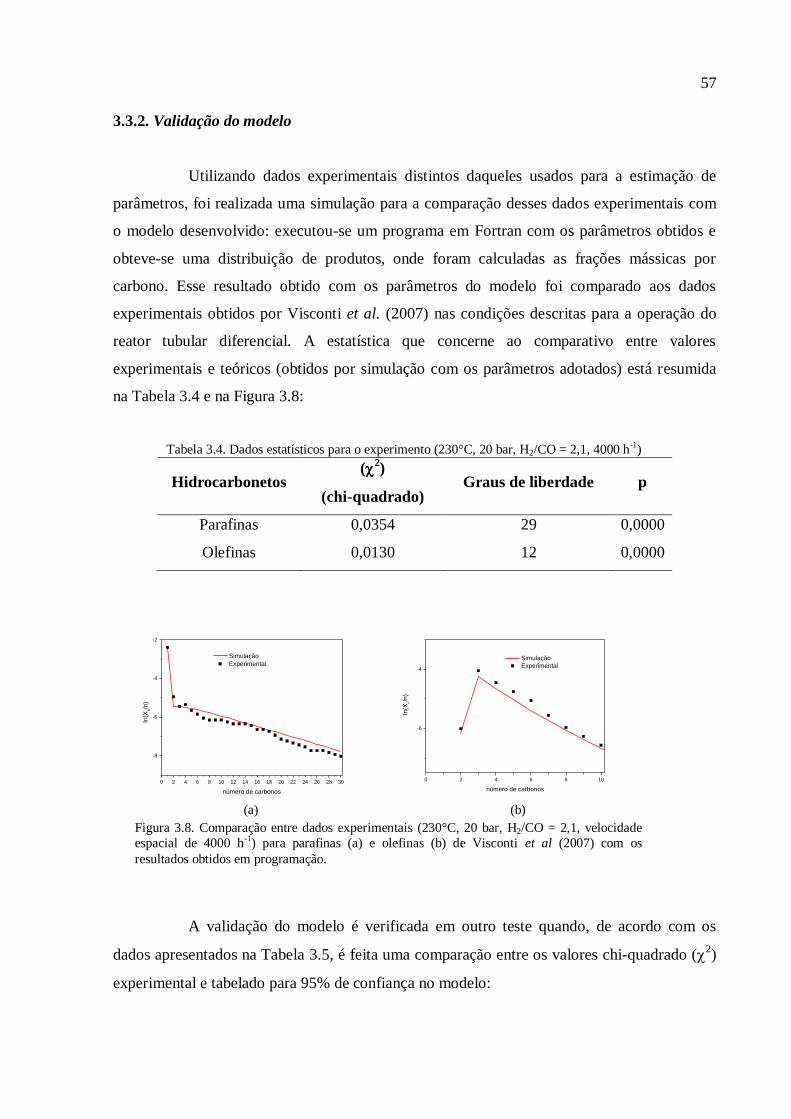
57
3.3.2. Validação do modelo
Utilizando dados experimentais distintos daqueles usados para a estimação de
parâmetros, foi realizada uma simulação para a comparação desses dados experimentais com
o modelo desenvolvido: executou-se um programa em Fortran com os parâmetros obtidos e
obteve-se uma distribuição de produtos, onde foram calculadas as frações mássicas por
carbono. Esse resultado obtido com os parâmetros do modelo foi comparado aos dados
experimentais obtidos por Visconti et al. (2007) nas condições descritas para a operação do
reator tubular diferencial. A estatística que concerne ao comparativo entre valores
experimentais e teóricos (obtidos por simulação com os parâmetros adotados) está resumida
na Tabela 3.4 e na Figura 3.8:
Tabela 3.4. Dados estatísticos para o experimento (230°C, 20 bar, H2/CO = 2,1, 4000 h-1)
Hidrocarbonetos (
2)
(chi-quadrado) Graus de liberdade p
Parafinas 0,0354 29 0,0000
Olefinas 0,0130 12 0,0000
A validação do modelo é verificada em outro teste quando, de acordo com os
dados apresentados na Tabela 3.5, é feita uma comparação entre os valores chi-quadrado (2)
experimental e tabelado para 95% de confiança no modelo:
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
-8
-6
-4
-2
Simulação
Experimental
ln(X
n/n
)
número de carbonos
0 2 4 6 8 10
-6
-4
Simulação
Experimental
ln(X
n/n
)
número de carbonos
(a) (b)
Figura 3.8. Comparação entre dados experimentais (230°C, 20 bar, H2/CO = 2,1, velocidade
espacial de 4000 h-1) para parafinas (a) e olefinas (b) de Visconti et al (2007) com os
resultados obtidos em programação.

58
Tabela 3.5. Comparação entre os testes chi-quadrado (2) experimental e tabelado:
Hidrocarbonetos (2) (experimental) (
2) (tabelado)
Parafinas 0,0354 17,706
Olefinas 0,0130 3,940
O valor de 2 experimental bastante inferior ao tabelado garante que o modelo
simulado esteja dentro do intervalo de confiança estabelecido.

59
4. MODELAGEM DINÂMICA PARA O REATOR DE LEITO FIXO
Foi desenvolvido um modelo matemático para um reator tubular de leito fixo para
a reação de síntese de Fischer-Tropsch (SFT). Devido à necessidade de integração numérica
do tempo de processamento e da posição no reator, foi desenvolvido um modelo dinâmico
com a utilização da técnica de discretização de diferenças finitos, para reduzir o modelo
matemático de equações diferenciais parciais para equações diferenciais ordinárias.
A modelagem sugerida é inovadora porque avalia o comportamento dinâmico do
reator em função das variáveis tempo e comprimento do reator. Assim, pode-se obter as
frações mássicas de saída para as variáveis dependentes em função do tempo decorrido para a
reação e do comprimento do reator.
O objetivo final é obter um modelo matemático que represente o reator, avaliando
a necessidade de se considerar os fenômenos de transferência de massa gás-líquido e a
transferência de calor no reator, e suas influências na distribuição final de produtos.
4.1. Discretização do reator
Seja o elemento de volume dV (Ac.dz) em um reator tubular mostrado na Figura
4.1:
Figura 4.1 – Esquematização para discretização do reator.
O balanço molar para o reagente H2 no segmento de reator entre z e (z + z) na
Figura 4.1, em função do fluxo molar J e da taxa de acúmulo da espécie H2 no elemento de
volume, é:
2
2 2 2
H'
H H c cz
CJ J . . A .Δz . A .Δz
tH ap
z zr
(4.1)
z z + z

60
Dividindo a Equação (4.1) por cA .Δz e adotando o limite para z 0, a equação
anterior adquire a forma:
2 2
2
H H'C C
. .z t
H apv r
(4.2)
Usando o mesmo procedimento para o reagente CO, a equação diferencial é:
'CO COC C. .
z tCO apv r
(4.3)
Em relação à formação de produtos, a equação diferencial para a água gerada na
reação é:
2 2
2
H O H O'
H O
C C. .
z tapv r
(4.4)
Em relação aos hidrocarbonetos produzidos, genericamente representados por
P(n), para as parafinas, e P=(n), para as olefinas, onde n simboliza o número de carbonos na
cadeia linear do hidrocarboneto formado:
P(n) P(n)'
P(n)
C C. .
z tapv r
, para 1 ≤ n ≤ 30 (4.5)
P (n) P (n)'
P (n)
C C. .
z tapv r
, para 2 ≤ n ≤ 30 (4.6)
Decidiu-se discretizar a coordenada espacial (z) do reator em 10 pontos (o uso de
um número maior de pontos mostrou a obtenção de resultados semelhantes) para estudar o
comportamento do reator, de acordo com a Figura 4.2:

61
Figura 4.2 – Discretização do reator tubular em 10 pontos.
Ao se aplicar o método de diferenças finitas para discretização do reator em
relação ao comprimento z, em 10 subdivisões, para as espécies j, sejam reagentes ou produtos.
O ponto inicial, correspondente ao valor i = 0, é o ponto de entrada do reator, e o valor de i =
10 corresponde à saída do reator. São obtidas as equações seguintes:
ponto inicial: j j,1 j,0
0
C C C
z Δzi
(4.7)
pontos centrais: j j,2 j,0
1
C C C
z 2.Δzi
(4.8)
j j,3 j,1
2
C C C
z 2.Δzi
(4.9)
j j,4 j,2
3
C C C
z 2.Δzi
(4.10)
j j,5 j,3
4
C C C
z 2.Δzi
(4.11)
j j,6 j,4
5
C C C
z 2.Δzi
(4.12)
j j,7 j,5
6
C C C
z 2.Δzi
(4.13)
j j,8 j,6
7
C C C
z 2.Δzi
(4.14)
j j,9 j,7
8
C C C
z 2.Δzi
(4.15)
j j,10 j,8
9
C C C
z 2.Δzi
(4.16)
i = 0 i = 1 i = 2 i = 3 i = 4 i = 5 i = 6 i = 7 i = 8 i = 9 i = 10

62
ponto final: j j,10 j,9
10
C C C
z Δzi
(4.17)
Quando é realizada a discretização do reator em relação ao comprimento, também
se realiza uma diferenciação em relação ao tempo simultaneamente. Assim, pode-se afirmar
que, para o componente j no ponto inicial do reator (i = 0):
j
0
C0
zi
(4.18)
As condições inicial e de contorno para as espécies são, respectivamente:
De z = 0 até z = L, em t = 0: os valores de Cj são iguais às concentrações
iniciais.
Em z = 0 para t qualquer: os valores de Cj são iguais às concentrações iniciais.
Quando se analisa o primeiro ponto central (i = 1), a taxa de acúmulo da espécie j
é:
j j '
1 1
C C. .
t zj ap
i i
v r
(4.19)
Na Equação (4.19) o termo j
1
C
zi
é fornecido pelas equações de discretização em
relação ao comprimento.
Com argumento semelhante, é possível prever equações similares para os outros
pontos centrais e para o ponto final do reator.
O termo '
jr se refere à taxa de reação, em relação à espécie j, em mol por hora
pela massa de catalisador. Em relação ao consumo de CO, a velocidade pode ser expressa, em
cada ponto i de discretização do reator (Zennaro et al., 2000), por:
2
0,743
CO H
SFT 2
CO
1,1.10 . C ( ).R.T . C ( ).R.TR ( )
1 0,040. C ( ).R.T
i ii
i
(4.20)

63
Na Equação (4.20) o termo R se refere à constante universal dos gases e T é a
temperatura de operação do reator.
Conforme relatado, a reação de síntese de Fischer-Tropsch é dada, de forma geral,
pela Equação (2.1):
CO + (1 + m/2n)H2 (1/n)CnHm + H2O (2.1)
Assim, a taxa de reação ('
SFTr ) é dada em função do consumo de CO ('
COr ). A
partir da proporção estequiométrica, a taxa de consumo de H2 (2
'
Hr ) é, aproximadamente, o
dobro da de consumo de CO, e a taxa de formação de água é idêntica à de consumo de CO.
4.1.1. Discretização do reator com transferência de massa
Com a implementação dos fenômenos de transferência de massa no leito
reacional, considerou-se que a reação de polimerização de Fischer-Tropsch ocorre apenas em
fase líquida, apesar de os reagentes (H2 e CO) serem alimentados em fase gasosa. É
necessário que a transferência de massa entre as fases gasosa e líquida seja considerada para
os gases hidrogênio e monóxido de carbono. Portanto, a equação anterior deve ser
desmembrada em duas novas equações: uma para a fase líquida e outra para a fase gasosa,
expressas pelas Equações (4.21) e (4.22).
Em relação ao gás hidrogênio, tem-se:
Para a fase gasosa: 2 2
2 2
G G
H HG L
H H
C C. . C C
z tMv k
(4.21)
Para a fase líquida: 2 2
2 2 2
L L
H HG L '
H H
C C. . C C .
z tLM apH
v k r
(4.22)
As Equações (4.21) e (4.22), bem como as Equações (4.23) até (4.29), utilizam a
compreensão matemática do conceito de balanço molar: o acúmulo do componente em
relação ao tempo é obtido pela adição do termo de fluxo molar entre as fases (representado
pelo 2º termo da equação) com o que estabelece a transferência de massa propriamente dita.

64
Nas equações em fase líquida, o termo de consumo devido à reação química também consta
dos balanços molares (representado pelo 3º termo da equação).
Usando o mesmo procedimento para o reagente CO, as equações diferenciais são
dadas pelas Equações (4.23) e (4.24):
Para a fase gasosa: G G
G LCO COCO CO
C C. . C C
z tMv k
(4.23)
Para a fase líquida: L
L LG L 'CO COCO CO CO
C C. . C C .
z tM apv k r
(4.24)
Em relação à formação dos produtos orgânicos (parafinas e olefinas) e água
(produtos da reação de polimerização de Fischer-Tropsch), considera-se que sua geração
ocorre em fase líquida para todas as frações de produtos, sem a necessidade de transferência
de massa entre as fases líquida e gasosa. Assim, a equação diferencial para a água gerada na
reação se refere apenas ao balanço molar obtido inicialmente na Equação (4.4).
Em relação aos hidrocarbonetos produzidos, genericamente representados por
P(n), para as parafinas, e P=(n), para as olefinas, onde n simboliza o número de carbonos na
cadeia linear do hidrocarboneto formado também não será considerada a transferência de
massa entre fase líquida e gasosa, uma vez que os produtos orgânicos já são obtidos em fase
líquida, e serão repetidas as Equações (4.5) e (4.6).
A aplicação do método de diferenças finitas para a discretização do reator segue o
mesmo procedimento utilizado nas Equações (4.7) até (4.18).
Quando se analisa o primeiro ponto central (i = 1), a taxa de acúmulo da espécie j
em relação ao tempo é fornecida pelas Equações (4.25) até (4.29):
para o hidrogênio em fase gasosa: 2 2
2 2
G G
H H G L
M H H
1 1
C C. . C C
t zi i
v k
(4.25)
para o hidrogênio em fase líquida: 2 2
2 2 2
L L
H H G L '
M H H
1 1
C C. . C C .
t zL apH
i i
v k r
(4.26)
para CO em fase gasosa: G G
G LCO COM CO CO
1 1
C C. . C C
t zi i
v k
(4.27)

65
para CO em fase líquida: L
L LG L 'CO CO
M CO CO CO
1 1
C C. . C C .
t zap
i i
v k r
(4.28)
para os produtos (água e hidrocarbonetos): j j '
1 1
C C. .
t zj ap
i i
v r
(4.29)
Nas Equações (4.25) até (4.29) o termo j
1
C
zi
é fornecido pelas equações de
discretização em relação ao comprimento. Com argumento semelhante, é possível prever
equações similares para os outros pontos centrais e para o ponto final do reator.
O coeficiente de transferência de massa gás-líquido kM, tanto para CO quanto para
H2, usados nas Equações (4.21) até (4.28), foram estimados por interação a partir da Equação
(4.30), proposta por Guettel e Turek (2008):
0,38
0,2 0,25 0,3iL L G L2
h
D0,091. . Re .We . 6.X .Sc
dMk
(4.30)
Na expressão da Equação (4.30), são utilizados os números adimensionais de
Reynolds (ReL), de Weber (WeL) e de Schmidt (ScL), além da razão adimensional de
Lockhart-Martinelli (XG). O termo dh (nesse trabalho utilizou-se 4,6 cm) se refere ao diâmetro
hidráulico do reator, calculado em função do diâmetro interno do reator e de sua porosidade.
O termo Di se refere ao coeficiente de difusividade de H2 ou de CO, disponíveis na Tabela
4.1:
Tabela 4.1 – Coeficientes de difusividade para CO e H2 (Guettel e Turek, 2008)
Difusividade de H2 Difusividade de CO
45,5.10-9
m2.s
-1 17,2.10
-9 m
2.s
-1
As equações para a taxa de reação ( '
jr se refere à velocidade de reação, em relação
à espécie j, em mol por hora pela massa de catalisador) em relação ao consumo de CO e, por
consequência, em relação aos demais componentes da reação de SFT continua seguindo a
Equação (4.20) (Zennaro et al., 2000), modificada para o uso de reagentes apenas em fase
líquida, em cada ponto i de discretização do reator, conforme aparece na Equação (4.31):

66
2
0,743 L L
CO H
SFT 2L
CO
1,1.10 . C ( ).R.T . C ( ).R.TR ( )
1 0,040. C ( ).R.T
i ii
i
(4.31)
Na Equação (4.31), assim como na Equação (4.20), o termo R é a constante
universal dos gases e T é a temperatura de operação do reator.
4.1.2. Discretização do reator com transferência de massa e de calor
A troca de calor é incluída no estudo da extensão da reação de SFT a partir da
Equação (4.32), que mostra o balanço de energia para uma reação em um reator:
. .SIST
j0 j0 j j
dEQ W J .H J .H
dt
n n
S
j j
(4.32)
Na Equação (4.32) acima os termos . .
Q e WS indicam os fluxos de calor e de
trabalho de eixo com o tempo, j0 j0J .H mostra o produto do fluxo molar com a entalpia
(inicialmente) e SISTdE
dt traduz a variação da energia dos componentes em relação com o
tempo.
O desenvolvimento das equações posteriores baseou-se nas Equações (4.21) a
(4.31), incluídas na seção 4.1.1, onde já abordavam a utilização do conceito de transferência
de massa. Dessa forma, percebe-se que a transferência de calor se constitui em uma melhoria
para a modelagem do reator em busca de respostas estatísticas mais próximas do
comportamento real.
Sabe-se que, para o reator tubular em estudo, o termo .
WS é nulo, pois não há uso
de agitadores ou turbinas, ou algo que possa transformar energia em trabalho de eixo.
Substituindo .
Q por L aU.A . T T , em que U é o coeficiente global de transferência de calor,
AL é a área lateral do reator, aqui considerada constante, e Ta é a temperatura do fluido
utilizado como trocador de calor, e conhecendo os termos de fluxo e entalpia em função da
velocidade de reação e da capacidade calorífica média, a Equação (4.32) é reescrita na forma
da Equação (4.33):

67
'
L a R FTS ap i i i
dTU.A . T T H . .V.ρ J .Cp . T Cp .
dt
n n
i i
r (4.33)
Na Equação (4.33), '
FTSr é a taxa de reação de SFT, expressa em mol por tempo
pela massa de catalisador. Como '
FTSr está expresso em função da massa de catalisador, é
necessário o termo de elemento de volume (A.dz, onde A é a área seccional do reator,
considerada constante, e dz o comprimento do elemento de volume relacionado à Figura 4.1)
seja multiplicado pela densidade aparente do leito catalítico, com a finalidade de tornar a
Equação (4.33) dimensionalmente coerente e fisicamente compreensível. O elemento de
volume é dado pela discretização do reator como L/10 (um décimo do comprimento do
reator).
Para avaliar a evolução da temperatura ao longo do eixo do reator e ao longo do
tempo de reação, que caracteriza a natureza dinâmica da proposta de modelagem abordada
nessa tese, é necessário o uso da discretização do reator já mencionada. Assim, usando a
Equação (4.33) para H2, CO, H2O e produtos orgânicos, e adotando-se que Ji é o produto da
concentração pela área e pela velocidade de escoamento (Ji = Ci.A.), tem-se a Equação
(4.34):
'
L a R FTS ap i i i
dT dTU.A . T T H . .A. L/10 .ρ C . .Cp . Cp .
dz dt
n n
i i
r (4.34)
4.2. Simulações computacionais
Foram selecionadas três variáveis independentes para que, em função delas, se
avaliasse a fração mássica para as variáveis dependentes no tempo final de operação e no
estágio final do reator. A escolha das variáveis independentes ocorreu por serem de
importância destacada em projetos de reatores tubulares de leito fixo para a reação de SFT em
escala industrial. São elas:
Proporção molar H2/CO na entrada do reator
Pressão total dos gases na entrada do reator
Velocidade inicial de escoamento do gás.

68
Cada fator selecionado pode variar de acordo com o planejamento experimental
estatístico sugerido, dentro de valores estabelecidos. As variações estão listadas na Tabela 4.2:
Tabela 4.2 – Faixa estudada de cada fator analisado para a simulação
Variável independente Valor mínimo Valor máximo
Proporção molar H2/CO 1,2 2,0
Pressão total dos gases 15 atm 35 atm
Velocidade de escoamento 0,45 m.s-1
0,65 m.s-1
As variáveis dependentes, cujas frações mássicas (e percentuais no caso da
conversão) são obtidas como respostas para a programação em Fortran, estão listadas na
Tabela 4.3:
Tabela 4.3 – Respostas para o programa de simulação
Respostas na simulação
Conversão em hidrocarbonetos
Gases leves
Gasolina
Querosene
Diesel
Graxas
A conversão total em hidrocarbonetos, calculada em função da quantidade de
matéria de carbono que entra no reator, na forma de CO, subtraindo-se da quantidade de
matéria que sai do reator, também na forma de CO, é calculada através da Equação 4.36:
inicial final
inicial
CO CO
CO
n nconversão em hidrocarbonetos =
n
(4.36)
Nesse cálculo, as conversões de CO em outras espécies que não sejam
hidrocarbonetos, como a transformação até carbeto e compostos oxigenados, foram
desprezadas, pois as quantidades em questão são negligenciáveis.
Um planejamento experimental tipo composto central teve 8 corridas diferentes,
com valores máximos e mínimos das variáveis independentes pré-estabelecidos na Tabela 4.2,
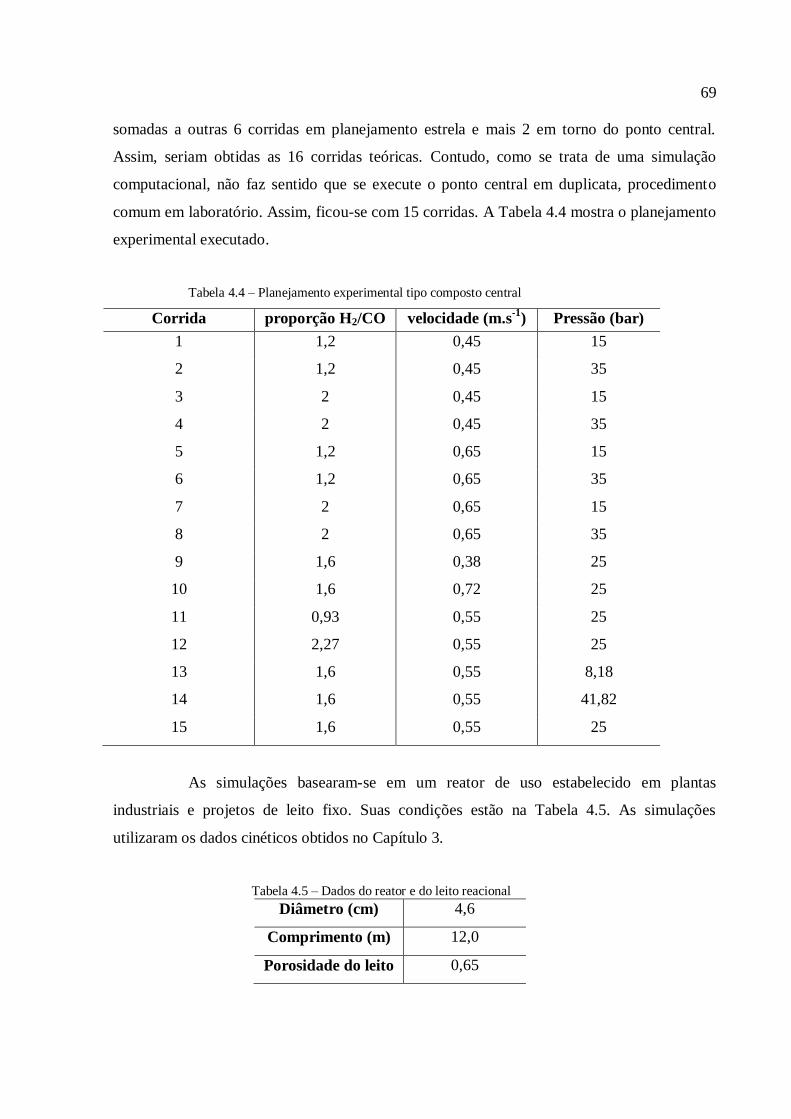
69
somadas a outras 6 corridas em planejamento estrela e mais 2 em torno do ponto central.
Assim, seriam obtidas as 16 corridas teóricas. Contudo, como se trata de uma simulação
computacional, não faz sentido que se execute o ponto central em duplicata, procedimento
comum em laboratório. Assim, ficou-se com 15 corridas. A Tabela 4.4 mostra o planejamento
experimental executado.
Tabela 4.4 – Planejamento experimental tipo composto central
Corrida proporção H2/CO velocidade (m.s-1
) Pressão (bar)
1 1,2 0,45 15
2 1,2 0,45 35
3 2 0,45 15
4 2 0,45 35
5 1,2 0,65 15
6 1,2 0,65 35
7 2 0,65 15
8 2 0,65 35
9 1,6 0,38 25
10 1,6 0,72 25
11 0,93 0,55 25
12 2,27 0,55 25
13 1,6 0,55 8,18
14 1,6 0,55 41,82
15 1,6 0,55 25
As simulações basearam-se em um reator de uso estabelecido em plantas
industriais e projetos de leito fixo. Suas condições estão na Tabela 4.5. As simulações
utilizaram os dados cinéticos obtidos no Capítulo 3.
Tabela 4.5 – Dados do reator e do leito reacional
Diâmetro (cm) 4,6
Comprimento (m) 12,0
Porosidade do leito 0,65

70
As variáveis dependentes obtidas como resposta na programação para a simulação
dinâmica do reator tubular são mostradas na Tabela 4.6.
Tabela 4.6: Convenções sobre cada fração-resposta no programa de simulação
Respostas na simulação Explicação
Conversão em hidrocarbonetos Conversão do CO inserido em
hidrocarbonetos apenas
Gases leves Conversão do CO inserido em
hidrocarbonetos entre 1 e 5 carbonos
Gasolina Conversão do CO inserido em
hidrocarbonetos de 6 a 8 carbonos
Querosene Conversão do CO inserido em
hidrocarbonetos de 9 a 11 carbonos
Diesel Conversão do CO inserido em
hidrocarbonetos de 12 a 20 carbonos
Graxas Conversão do CO inserido em
hidrocarbonetos de 21 a 30 carbonos
As Equações (4.2) a (4.20), em associação ao modelo cinético desenvolvido no
Capítulo 3, foram resolvidas com o uso de programação em Fortran através de integração
numérica com a utilização do método de Runge-Kutta de 5ª ordem. Foram obtidas as frações
mássicas de cada variável dependente em função do tempo de reação e do estágio do reator.
Mais uma vez, executou-se um programa em linguagem Fortran para realizar as
simulações, sejam com o uso ou não de transferências de massa e de calor. Na programação
em Fortran, a variável tempo já é intrínseca ao programa. Só é necessário que se defina a
variável comprimento do reator. Assim, se consegue calcular as frações mássicas dos
produtos em relação ao tempo de operação e ao estágio do reator. Um resumo do
procedimento adotado para a execução do programa de simulação é mostrado no algoritmo a
seguir (Figura 4.3):

71
Figura 4.3 – Algoritmo para o programa de simulação
A Figura 4.4 mostra um fluxograma estrutural para o procedimento descrito. O
termo L descreve o passo correspondente a cada metro do reator tubular:
Leitura de parâmetros
e entrada de dados
Execução da sub-rotina para
os cálculos correspondentes
às equações do modelo
(podendo ou não utilizar as equações de transferência de
massa e/ou de calor)
Impressão dos
resultados para o valor
correspondente de L
(comprimento do reator)
Atualização do valor de L até que se
alcance o final do reator
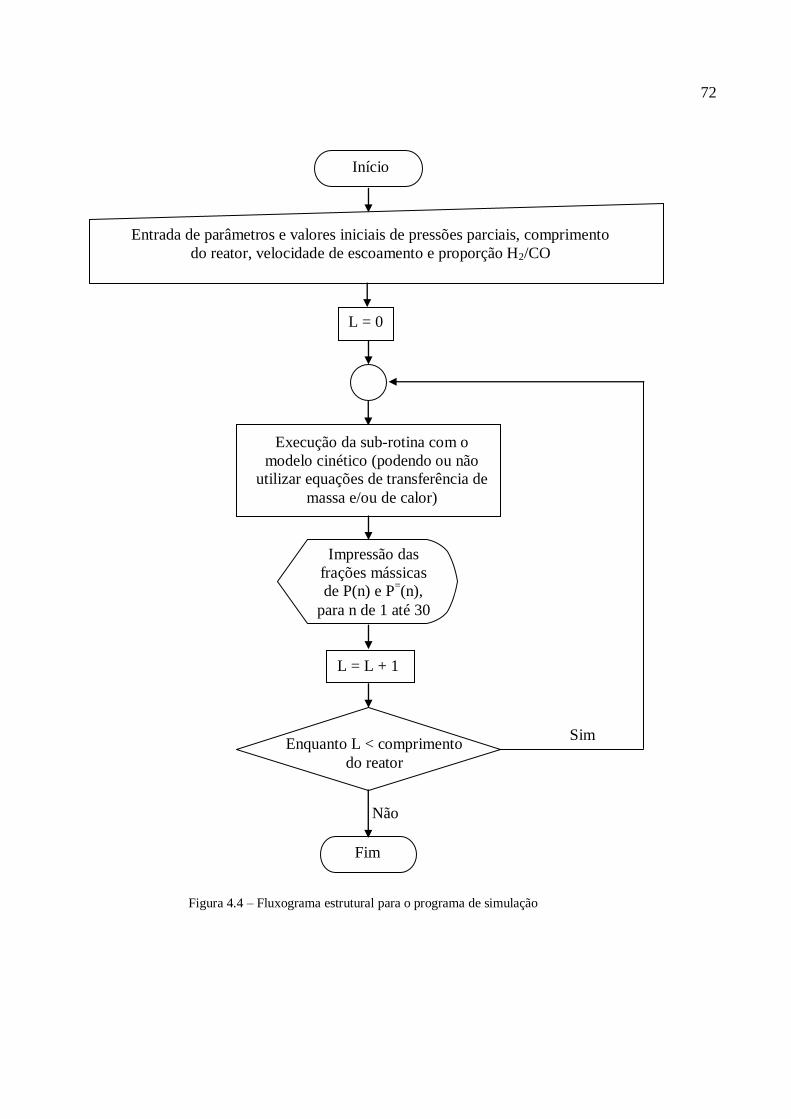
72
Figura 4.4 – Fluxograma estrutural para o programa de simulação
Início
L = 0
L = L + 1
Enquanto L < comprimento
do reator
Sim
Não
Fim
Impressão das
frações mássicas
de P(n) e P=(n),
para n de 1 até 30
Execução da sub-rotina com o
modelo cinético (podendo ou não
utilizar equações de transferência de
massa e/ou de calor)
Entrada de parâmetros e valores iniciais de pressões parciais, comprimento
do reator, velocidade de escoamento e proporção H2/CO
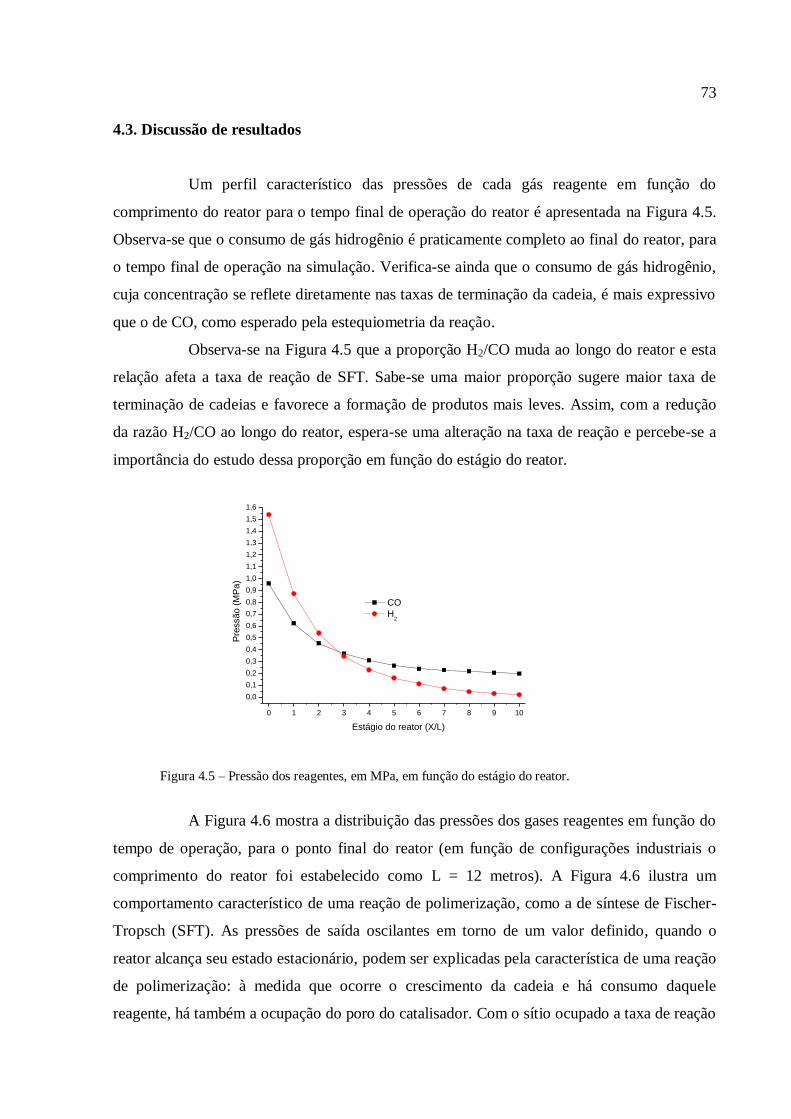
73
4.3. Discussão de resultados
Um perfil característico das pressões de cada gás reagente em função do
comprimento do reator para o tempo final de operação do reator é apresentada na Figura 4.5.
Observa-se que o consumo de gás hidrogênio é praticamente completo ao final do reator, para
o tempo final de operação na simulação. Verifica-se ainda que o consumo de gás hidrogênio,
cuja concentração se reflete diretamente nas taxas de terminação da cadeia, é mais expressivo
que o de CO, como esperado pela estequiometria da reação.
Observa-se na Figura 4.5 que a proporção H2/CO muda ao longo do reator e esta
relação afeta a taxa de reação de SFT. Sabe-se uma maior proporção sugere maior taxa de
terminação de cadeias e favorece a formação de produtos mais leves. Assim, com a redução
da razão H2/CO ao longo do reator, espera-se uma alteração na taxa de reação e percebe-se a
importância do estudo dessa proporção em função do estágio do reator.
Figura 4.5 – Pressão dos reagentes, em MPa, em função do estágio do reator.
A Figura 4.6 mostra a distribuição das pressões dos gases reagentes em função do
tempo de operação, para o ponto final do reator (em função de configurações industriais o
comprimento do reator foi estabelecido como L = 12 metros). A Figura 4.6 ilustra um
comportamento característico de uma reação de polimerização, como a de síntese de Fischer-
Tropsch (SFT). As pressões de saída oscilantes em torno de um valor definido, quando o
reator alcança seu estado estacionário, podem ser explicadas pela característica de uma reação
de polimerização: à medida que ocorre o crescimento da cadeia e há consumo daquele
reagente, há também a ocupação do poro do catalisador. Com o sítio ocupado a taxa de reação
0 1 2 3 4 5 6 7 8 9 10
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,1
1,2
1,3
1,4
1,5
1,6
CO
H2
Pre
ssã
o (
MP
a)
Estágio do reator (X/L)
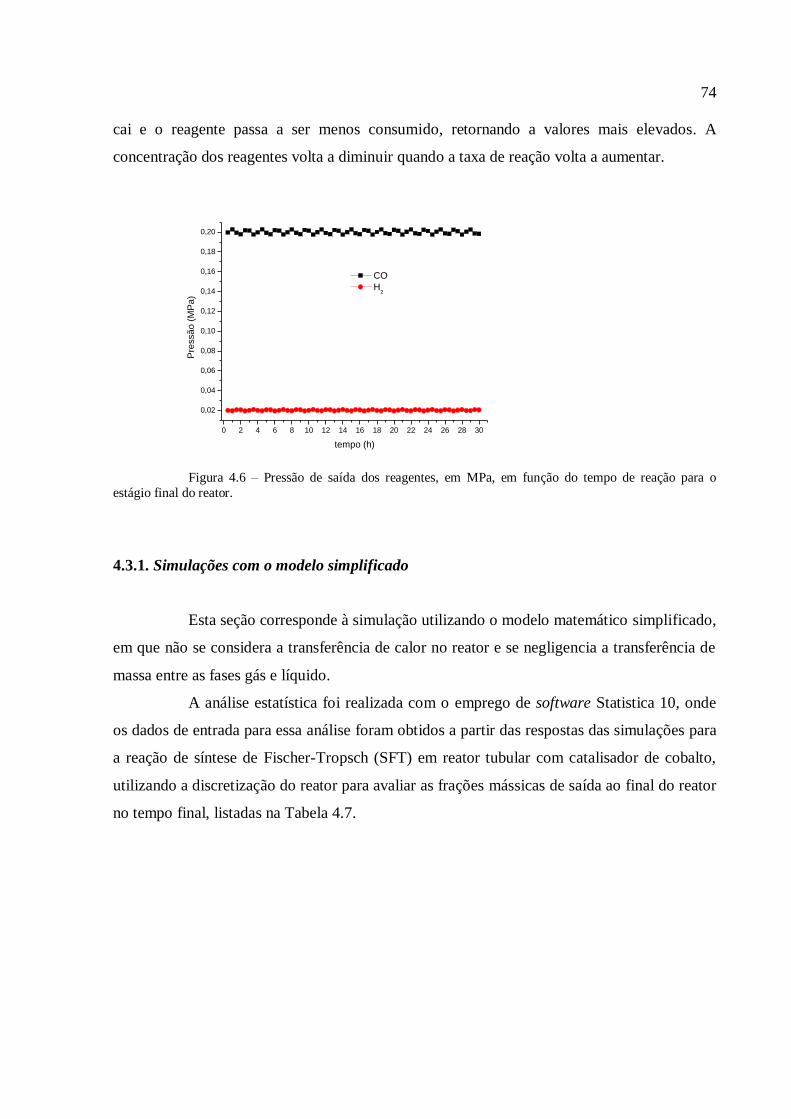
74
cai e o reagente passa a ser menos consumido, retornando a valores mais elevados. A
concentração dos reagentes volta a diminuir quando a taxa de reação volta a aumentar.
0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30
0,02
0,04
0,06
0,08
0,10
0,12
0,14
0,16
0,18
0,20
CO
H2
Pre
ssã
o (
MP
a)
tempo (h)
Figura 4.6 – Pressão de saída dos reagentes, em MPa, em função do tempo de reação para o
estágio final do reator.
4.3.1. Simulações com o modelo simplificado
Esta seção corresponde à simulação utilizando o modelo matemático simplificado,
em que não se considera a transferência de calor no reator e se negligencia a transferência de
massa entre as fases gás e líquido.
A análise estatística foi realizada com o emprego de software Statistica 10, onde
os dados de entrada para essa análise foram obtidos a partir das respostas das simulações para
a reação de síntese de Fischer-Tropsch (SFT) em reator tubular com catalisador de cobalto,
utilizando a discretização do reator para avaliar as frações mássicas de saída ao final do reator
no tempo final, listadas na Tabela 4.7.
75
Tabela 4.7: Frações mássicas para a reação de SFT no programa de simulação
Corrida Leves Gasolina Querosene Diesel Graxa Conversão
1 50,28 10,65 9,13 20,70 18,35 0,598
2 26,46 9,20 10,16 27,10 37,23 0,616
3 50,90 8,35 6,51 15,39 25,34 0,886
4 34,87 10,23 10,50 26,21 28,67 0,940
5 47,57 10,51 9,20 21,34 20,56 0,578
6 23,80 8,68 9,88 26,81 40,69 0,615
7 60,62 9,36 7,20 15,45 14,56 0,835
8 32,13 9,74 10,07 25,14 32,97 0,915
9 41,62 10,83 10,37 24,43 23,10 0,807
10 36,28 9,70 9,98 25,93 28,07 0,774
11 30,59 9,95 10,51 27,05 32,38 0,482
12 46,56 10,30 9,02 21,47 21,65 0,942
13 56,76 5,00 3,62 9,13 29,09 0,707
14 24,45 8,87 10,13 27,22 39,53 0,802
15 39,49 9,93 10,46 24,85 25,72 0,793
4.3.1.1. Análise para as frações gases leves, gasolina e querosene
São apresentados os gráficos de superfície para cada fração de hidrocarbonetos
produzida ao final do reator no tempo final de operação. Os fatores que apresentam relevância
significativa do ponto de vista estatístico para as frações gases leves, gasolina e querosene,
são:
Pressão
Proporção H2/CO
O valor p é um teste de hipótese e fornece a probabilidade de a hipótese nula ser
rejeitada. São considerados significantes os fatores (variáveis) cujos valores de p são
inferiores ao valor aceito como limite para erro tipo I (rejeitar a hipótese nula), que foi
adotado como 5% (ou 0,05) nessa tese. A análise de efeitos para a fração gases leves está na
Tabela 4.8.
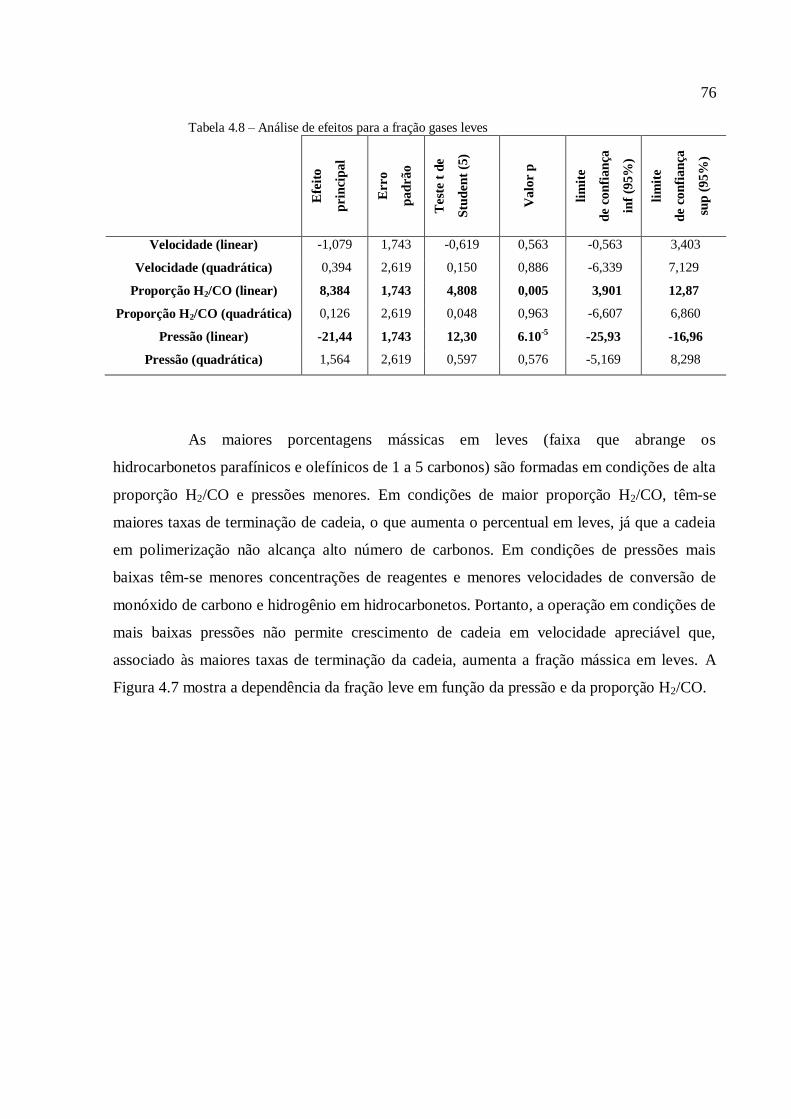
76
Tabela 4.8 – Análise de efeitos para a fração gases leves
Efe
ito
prin
cip
al
Erro
pad
rão
Test
e t
de
Stu
den
t (5
)
Valo
r p
lim
ite
de c
on
fian
ça
inf
(95%
)
lim
ite
de c
on
fian
ça
sup
(95%
)
Velocidade (linear) -1,079 1,743 -0,619 0,563 -0,563 3,403
Velocidade (quadrática) 0,394 2,619 0,150 0,886 -6,339 7,129
Proporção H2/CO (linear) 8,384 1,743 4,808 0,005 3,901 12,87
Proporção H2/CO (quadrática) 0,126 2,619 0,048 0,963 -6,607 6,860
Pressão (linear) -21,44 1,743 12,30 6.10-5
-25,93 -16,96
Pressão (quadrática) 1,564 2,619 0,597 0,576 -5,169 8,298
As maiores porcentagens mássicas em leves (faixa que abrange os
hidrocarbonetos parafínicos e olefínicos de 1 a 5 carbonos) são formadas em condições de alta
proporção H2/CO e pressões menores. Em condições de maior proporção H2/CO, têm-se
maiores taxas de terminação de cadeia, o que aumenta o percentual em leves, já que a cadeia
em polimerização não alcança alto número de carbonos. Em condições de pressões mais
baixas têm-se menores concentrações de reagentes e menores velocidades de conversão de
monóxido de carbono e hidrogênio em hidrocarbonetos. Portanto, a operação em condições de
mais baixas pressões não permite crescimento de cadeia em velocidade apreciável que,
associado às maiores taxas de terminação da cadeia, aumenta a fração mássica em leves. A
Figura 4.7 mostra a dependência da fração leve em função da pressão e da proporção H2/CO.
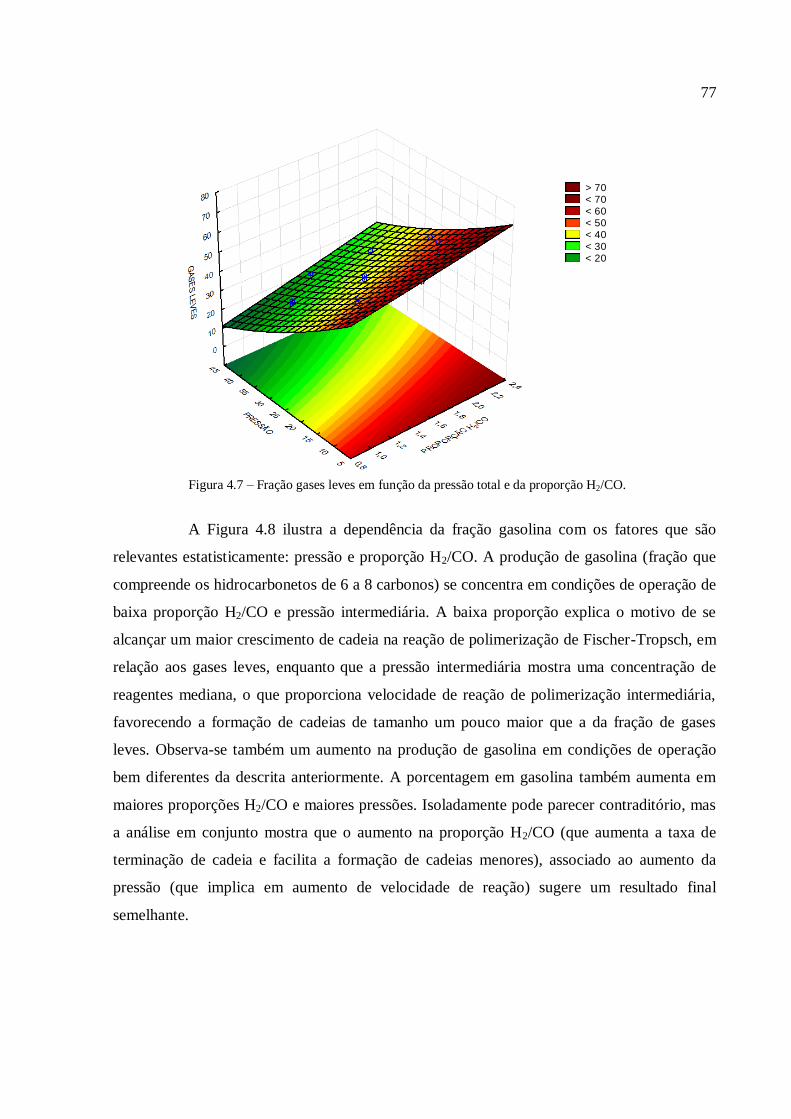
77
Figura 4.7 – Fração gases leves em função da pressão total e da proporção H2/CO.
A Figura 4.8 ilustra a dependência da fração gasolina com os fatores que são
relevantes estatisticamente: pressão e proporção H2/CO. A produção de gasolina (fração que
compreende os hidrocarbonetos de 6 a 8 carbonos) se concentra em condições de operação de
baixa proporção H2/CO e pressão intermediária. A baixa proporção explica o motivo de se
alcançar um maior crescimento de cadeia na reação de polimerização de Fischer-Tropsch, em
relação aos gases leves, enquanto que a pressão intermediária mostra uma concentração de
reagentes mediana, o que proporciona velocidade de reação de polimerização intermediária,
favorecendo a formação de cadeias de tamanho um pouco maior que a da fração de gases
leves. Observa-se também um aumento na produção de gasolina em condições de operação
bem diferentes da descrita anteriormente. A porcentagem em gasolina também aumenta em
maiores proporções H2/CO e maiores pressões. Isoladamente pode parecer contraditório, mas
a análise em conjunto mostra que o aumento na proporção H2/CO (que aumenta a taxa de
terminação de cadeia e facilita a formação de cadeias menores), associado ao aumento da
pressão (que implica em aumento de velocidade de reação) sugere um resultado final
semelhante.
Fitted Surface; Variable: Var1
3 factors, 1 Blocks, 15 Runs; MS Residual=10,38257
DV: Var1
> 70 < 70 < 60 < 50 < 40 < 30 < 20
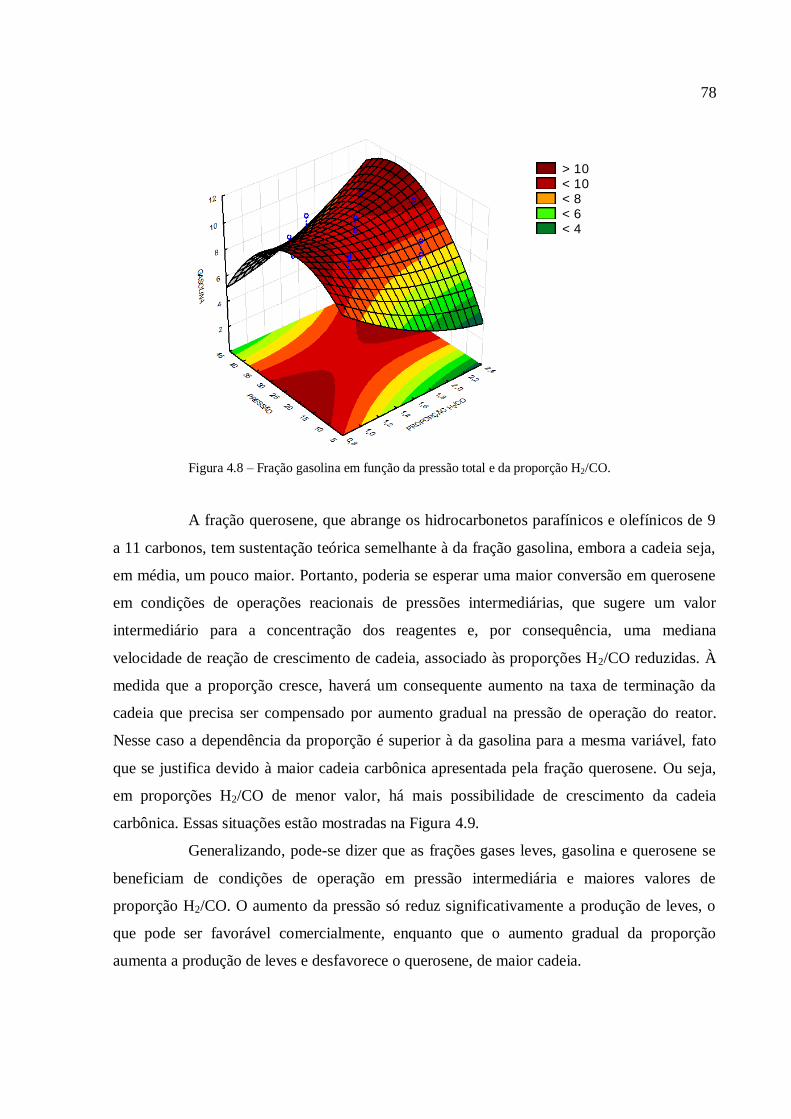
78
Figura 4.8 – Fração gasolina em função da pressão total e da proporção H2/CO.
A fração querosene, que abrange os hidrocarbonetos parafínicos e olefínicos de 9
a 11 carbonos, tem sustentação teórica semelhante à da fração gasolina, embora a cadeia seja,
em média, um pouco maior. Portanto, poderia se esperar uma maior conversão em querosene
em condições de operações reacionais de pressões intermediárias, que sugere um valor
intermediário para a concentração dos reagentes e, por consequência, uma mediana
velocidade de reação de crescimento de cadeia, associado às proporções H2/CO reduzidas. À
medida que a proporção cresce, haverá um consequente aumento na taxa de terminação da
cadeia que precisa ser compensado por aumento gradual na pressão de operação do reator.
Nesse caso a dependência da proporção é superior à da gasolina para a mesma variável, fato
que se justifica devido à maior cadeia carbônica apresentada pela fração querosene. Ou seja,
em proporções H2/CO de menor valor, há mais possibilidade de crescimento da cadeia
carbônica. Essas situações estão mostradas na Figura 4.9.
Generalizando, pode-se dizer que as frações gases leves, gasolina e querosene se
beneficiam de condições de operação em pressão intermediária e maiores valores de
proporção H2/CO. O aumento da pressão só reduz significativamente a produção de leves, o
que pode ser favorável comercialmente, enquanto que o aumento gradual da proporção
aumenta a produção de leves e desfavorece o querosene, de maior cadeia.
Fitted Surface; Variable: Var2
3 factors, 1 Blocks, 15 Runs; MS Residual=1,376026
DV: Var2
> 10 < 10 < 8 < 6 < 4
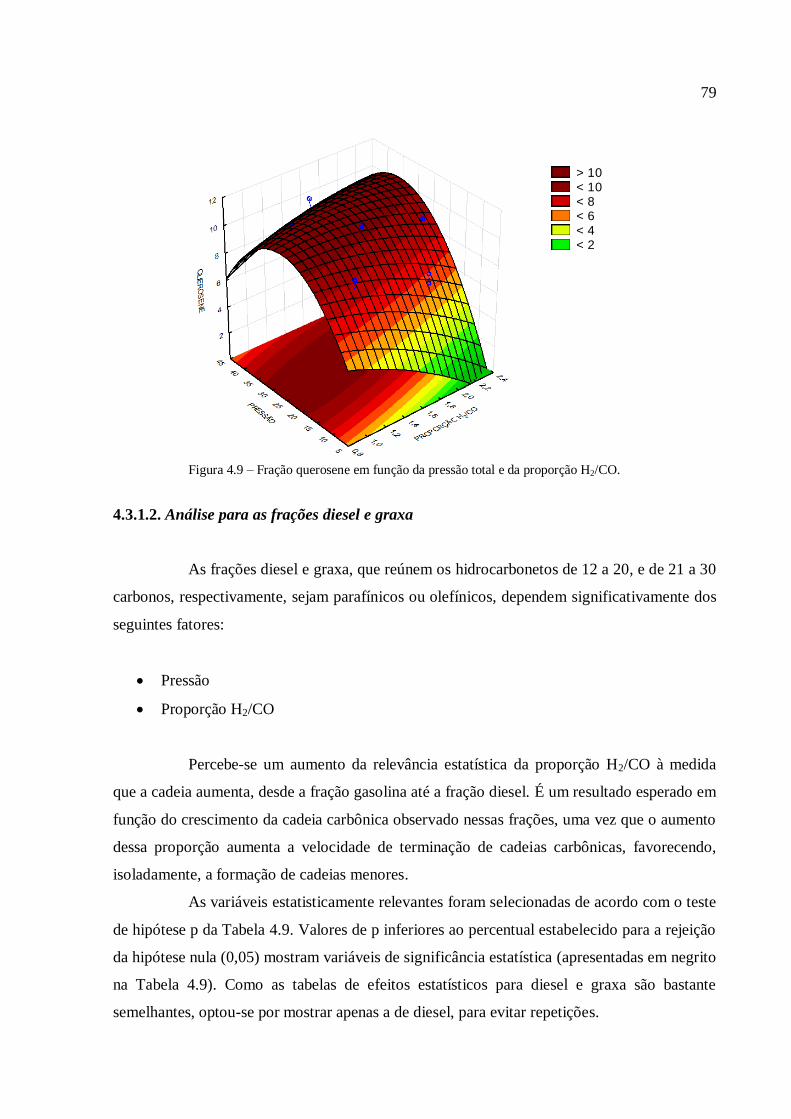
79
Figura 4.9 – Fração querosene em função da pressão total e da proporção H2/CO.
4.3.1.2. Análise para as frações diesel e graxa
As frações diesel e graxa, que reúnem os hidrocarbonetos de 12 a 20, e de 21 a 30
carbonos, respectivamente, sejam parafínicos ou olefínicos, dependem significativamente dos
seguintes fatores:
Pressão
Proporção H2/CO
Percebe-se um aumento da relevância estatística da proporção H2/CO à medida
que a cadeia aumenta, desde a fração gasolina até a fração diesel. É um resultado esperado em
função do crescimento da cadeia carbônica observado nessas frações, uma vez que o aumento
dessa proporção aumenta a velocidade de terminação de cadeias carbônicas, favorecendo,
isoladamente, a formação de cadeias menores.
As variáveis estatisticamente relevantes foram selecionadas de acordo com o teste
de hipótese p da Tabela 4.9. Valores de p inferiores ao percentual estabelecido para a rejeição
da hipótese nula (0,05) mostram variáveis de significância estatística (apresentadas em negrito
na Tabela 4.9). Como as tabelas de efeitos estatísticos para diesel e graxa são bastante
semelhantes, optou-se por mostrar apenas a de diesel, para evitar repetições.
Fitted Surface; Variable: Var3
3 factors, 1 Blocks, 15 Runs; MS Residual=,5604172
DV: Var3
> 10 < 10 < 8 < 6 < 4 < 2
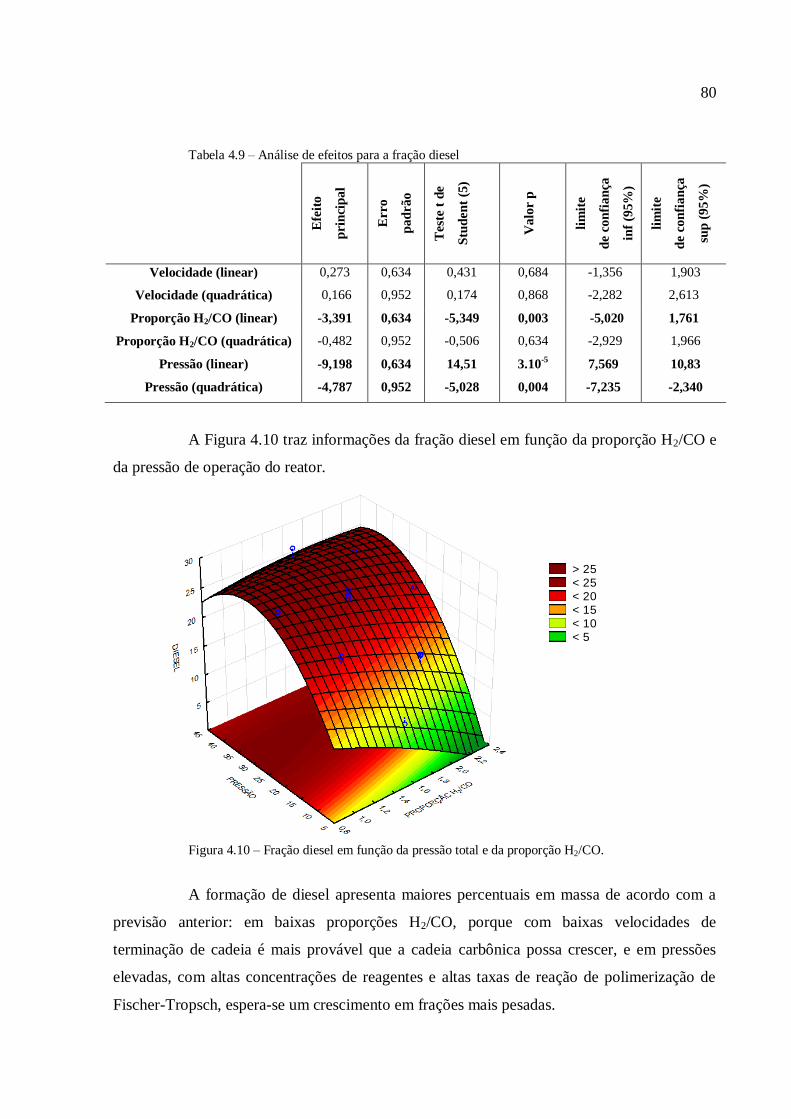
80
Tabela 4.9 – Análise de efeitos para a fração diesel
Efe
ito
prin
cip
al
Erro
pad
rão
Test
e t
de
Stu
den
t (5
)
Valo
r p
lim
ite
de c
on
fian
ça
inf
(95%
)
lim
ite
de c
on
fian
ça
sup
(95%
)
Velocidade (linear) 0,273 0,634 0,431 0,684 -1,356 1,903
Velocidade (quadrática) 0,166 0,952 0,174 0,868 -2,282 2,613
Proporção H2/CO (linear) -3,391 0,634 -5,349 0,003 -5,020 1,761
Proporção H2/CO (quadrática) -0,482 0,952 -0,506 0,634 -2,929 1,966
Pressão (linear) -9,198 0,634 14,51 3.10-5
7,569 10,83
Pressão (quadrática) -4,787 0,952 -5,028 0,004
-7,235 -2,340
A Figura 4.10 traz informações da fração diesel em função da proporção H2/CO e
da pressão de operação do reator.
Figura 4.10 – Fração diesel em função da pressão total e da proporção H2/CO.
A formação de diesel apresenta maiores percentuais em massa de acordo com a
previsão anterior: em baixas proporções H2/CO, porque com baixas velocidades de
terminação de cadeia é mais provável que a cadeia carbônica possa crescer, e em pressões
elevadas, com altas concentrações de reagentes e altas taxas de reação de polimerização de
Fischer-Tropsch, espera-se um crescimento em frações mais pesadas.
Fitted Surface; Variable: Var4
3 factors, 1 Blocks, 15 Runs; MS Residual=1,372001
DV: Var4
> 25 < 25 < 20 < 15 < 10 < 5
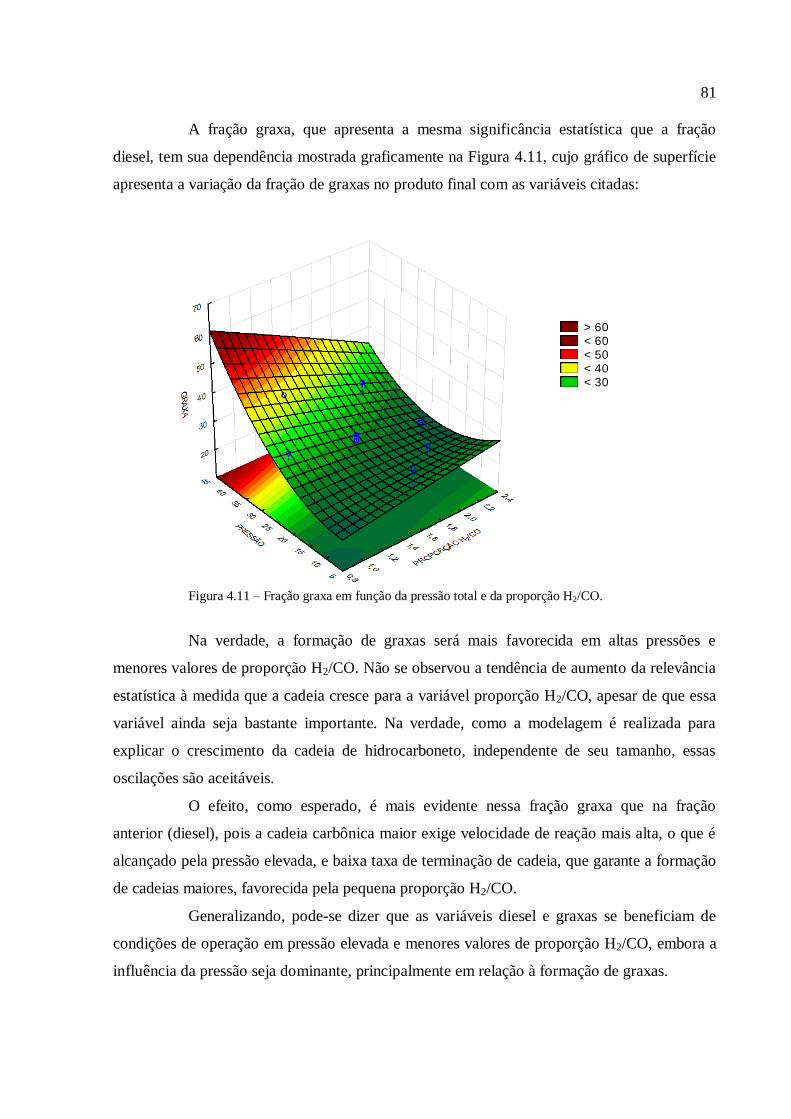
81
A fração graxa, que apresenta a mesma significância estatística que a fração
diesel, tem sua dependência mostrada graficamente na Figura 4.11, cujo gráfico de superfície
apresenta a variação da fração de graxas no produto final com as variáveis citadas:
Figura 4.11 – Fração graxa em função da pressão total e da proporção H2/CO.
Na verdade, a formação de graxas será mais favorecida em altas pressões e
menores valores de proporção H2/CO. Não se observou a tendência de aumento da relevância
estatística à medida que a cadeia cresce para a variável proporção H2/CO, apesar de que essa
variável ainda seja bastante importante. Na verdade, como a modelagem é realizada para
explicar o crescimento da cadeia de hidrocarboneto, independente de seu tamanho, essas
oscilações são aceitáveis.
O efeito, como esperado, é mais evidente nessa fração graxa que na fração
anterior (diesel), pois a cadeia carbônica maior exige velocidade de reação mais alta, o que é
alcançado pela pressão elevada, e baixa taxa de terminação de cadeia, que garante a formação
de cadeias maiores, favorecida pela pequena proporção H2/CO.
Generalizando, pode-se dizer que as variáveis diesel e graxas se beneficiam de
condições de operação em pressão elevada e menores valores de proporção H2/CO, embora a
influência da pressão seja dominante, principalmente em relação à formação de graxas.
Fitted Surface; Variable: Var5
3 factors, 1 Blocks, 15 Runs; MS Residual=23,1007
DV: Var5
> 60 < 60 < 50 < 40 < 30
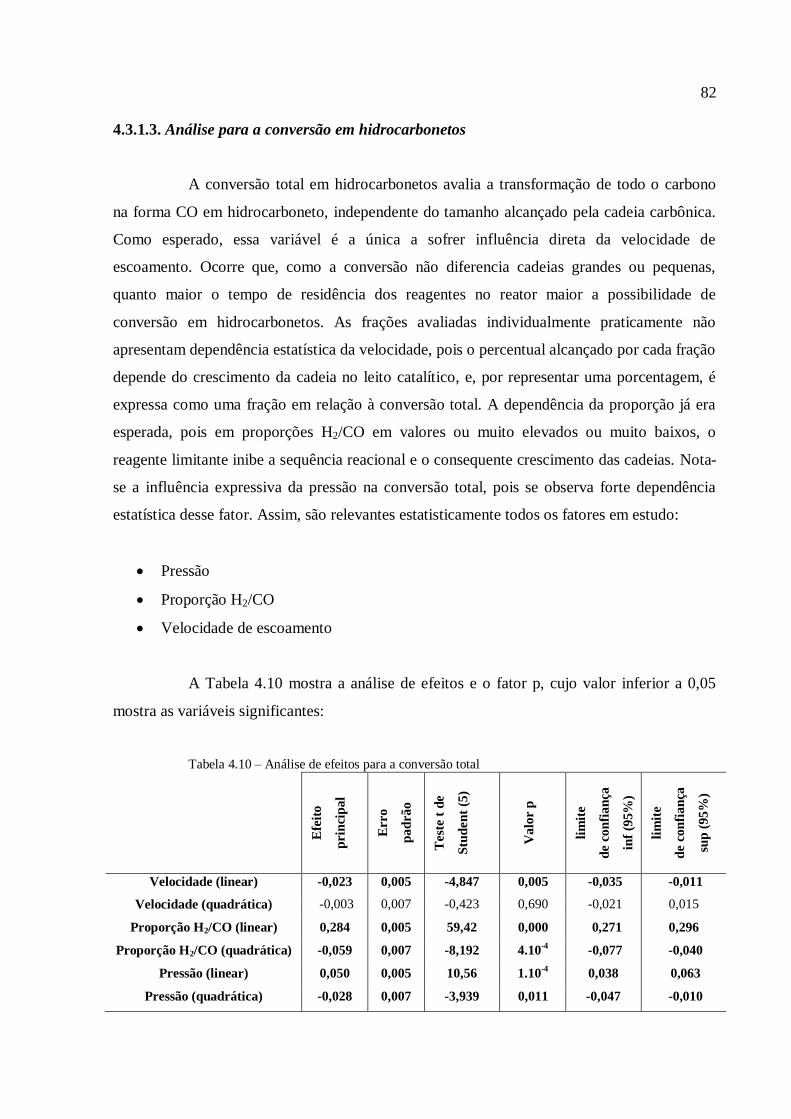
82
4.3.1.3. Análise para a conversão em hidrocarbonetos
A conversão total em hidrocarbonetos avalia a transformação de todo o carbono
na forma CO em hidrocarboneto, independente do tamanho alcançado pela cadeia carbônica.
Como esperado, essa variável é a única a sofrer influência direta da velocidade de
escoamento. Ocorre que, como a conversão não diferencia cadeias grandes ou pequenas,
quanto maior o tempo de residência dos reagentes no reator maior a possibilidade de
conversão em hidrocarbonetos. As frações avaliadas individualmente praticamente não
apresentam dependência estatística da velocidade, pois o percentual alcançado por cada fração
depende do crescimento da cadeia no leito catalítico, e, por representar uma porcentagem, é
expressa como uma fração em relação à conversão total. A dependência da proporção já era
esperada, pois em proporções H2/CO em valores ou muito elevados ou muito baixos, o
reagente limitante inibe a sequência reacional e o consequente crescimento das cadeias. Nota-
se a influência expressiva da pressão na conversão total, pois se observa forte dependência
estatística desse fator. Assim, são relevantes estatisticamente todos os fatores em estudo:
Pressão
Proporção H2/CO
Velocidade de escoamento
A Tabela 4.10 mostra a análise de efeitos e o fator p, cujo valor inferior a 0,05
mostra as variáveis significantes:
Tabela 4.10 – Análise de efeitos para a conversão total
Efe
ito
prin
cip
al
Erro
pad
rão
Test
e t
de
Stu
den
t (5
)
Valo
r p
lim
ite
de c
on
fian
ça
inf
(95%
)
lim
ite
de c
on
fian
ça
sup
(95%
)
Velocidade (linear) -0,023 0,005 -4,847 0,005 -0,035 -0,011
Velocidade (quadrática) -0,003 0,007 -0,423 0,690 -0,021 0,015
Proporção H2/CO (linear) 0,284 0,005 59,42 0,000 0,271 0,296
Proporção H2/CO (quadrática) -0,059 0,007 -8,192 4.10-4
-0,077 -0,040
Pressão (linear) 0,050 0,005 10,56 1.10-4
0,038 0,063
Pressão (quadrática) -0,028 0,007 -3,939 0,011
-0,047 -0,010
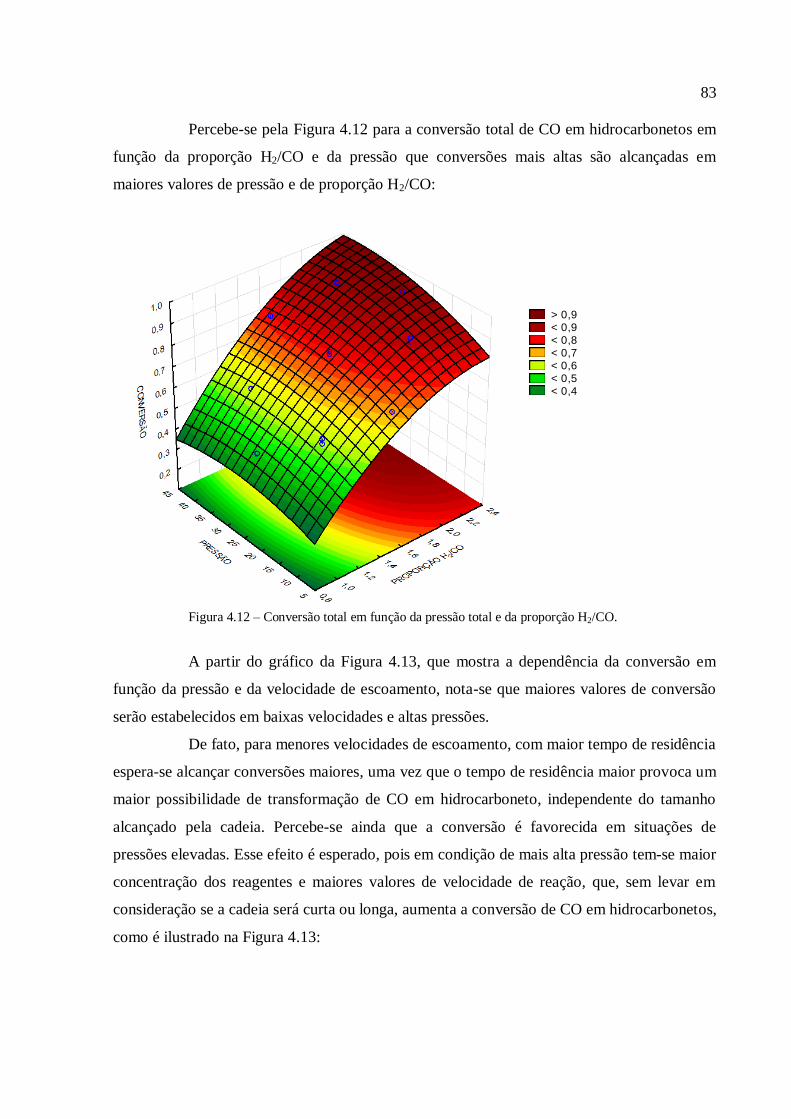
83
Percebe-se pela Figura 4.12 para a conversão total de CO em hidrocarbonetos em
função da proporção H2/CO e da pressão que conversões mais altas são alcançadas em
maiores valores de pressão e de proporção H2/CO:
Figura 4.12 – Conversão total em função da pressão total e da proporção H2/CO.
A partir do gráfico da Figura 4.13, que mostra a dependência da conversão em
função da pressão e da velocidade de escoamento, nota-se que maiores valores de conversão
serão estabelecidos em baixas velocidades e altas pressões.
De fato, para menores velocidades de escoamento, com maior tempo de residência
espera-se alcançar conversões maiores, uma vez que o tempo de residência maior provoca um
maior possibilidade de transformação de CO em hidrocarboneto, independente do tamanho
alcançado pela cadeia. Percebe-se ainda que a conversão é favorecida em situações de
pressões elevadas. Esse efeito é esperado, pois em condição de mais alta pressão tem-se maior
concentração dos reagentes e maiores valores de velocidade de reação, que, sem levar em
consideração se a cadeia será curta ou longa, aumenta a conversão de CO em hidrocarbonetos,
como é ilustrado na Figura 4.13:
Fitted Surface; Variable: NewVar
3 factors, 1 Blocks, 15 Runs; MS Residual=,0000778
DV: NewVar
> 0,9 < 0,9 < 0,8 < 0,7 < 0,6 < 0,5 < 0,4
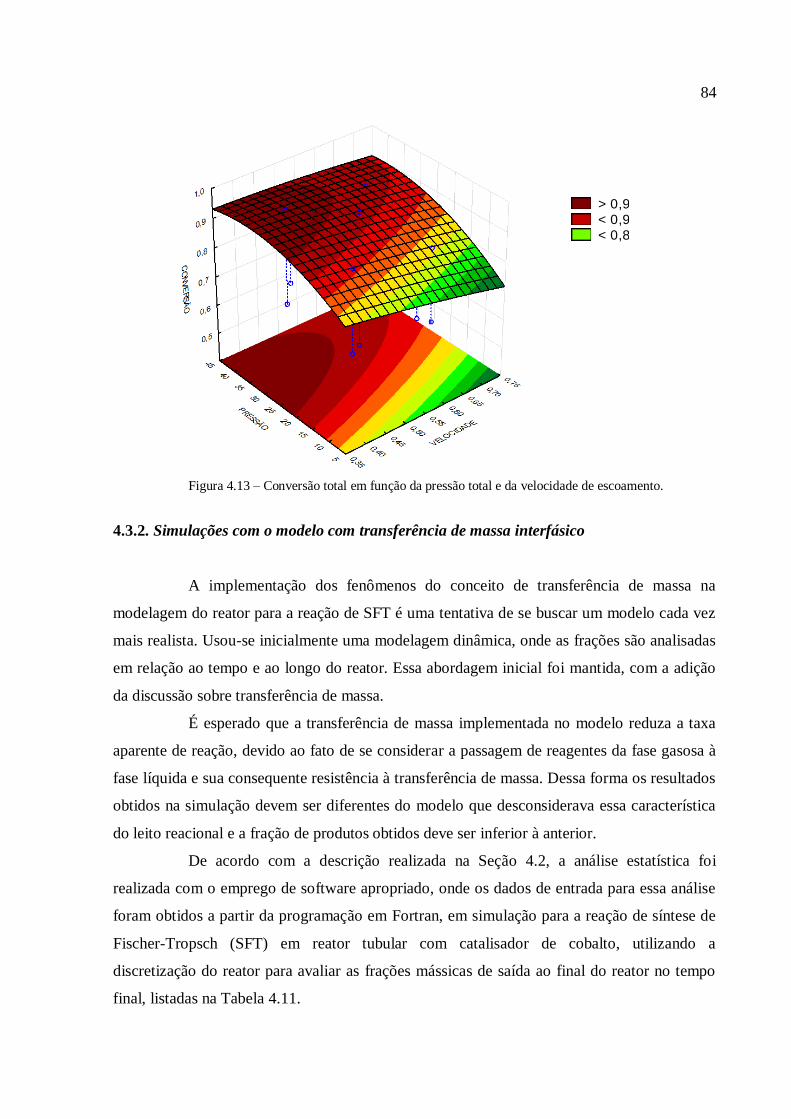
84
Figura 4.13 – Conversão total em função da pressão total e da velocidade de escoamento.
4.3.2. Simulações com o modelo com transferência de massa interfásico
A implementação dos fenômenos do conceito de transferência de massa na
modelagem do reator para a reação de SFT é uma tentativa de se buscar um modelo cada vez
mais realista. Usou-se inicialmente uma modelagem dinâmica, onde as frações são analisadas
em relação ao tempo e ao longo do reator. Essa abordagem inicial foi mantida, com a adição
da discussão sobre transferência de massa.
É esperado que a transferência de massa implementada no modelo reduza a taxa
aparente de reação, devido ao fato de se considerar a passagem de reagentes da fase gasosa à
fase líquida e sua consequente resistência à transferência de massa. Dessa forma os resultados
obtidos na simulação devem ser diferentes do modelo que desconsiderava essa característica
do leito reacional e a fração de produtos obtidos deve ser inferior à anterior.
De acordo com a descrição realizada na Seção 4.2, a análise estatística foi
realizada com o emprego de software apropriado, onde os dados de entrada para essa análise
foram obtidos a partir da programação em Fortran, em simulação para a reação de síntese de
Fischer-Tropsch (SFT) em reator tubular com catalisador de cobalto, utilizando a
discretização do reator para avaliar as frações mássicas de saída ao final do reator no tempo
final, listadas na Tabela 4.11.
Fitted Surface; Variable: NewVar
3 factors, 1 Blocks, 15 Runs; MS Residual=,0000778
DV: NewVar
> 0,9 < 0,9 < 0,8
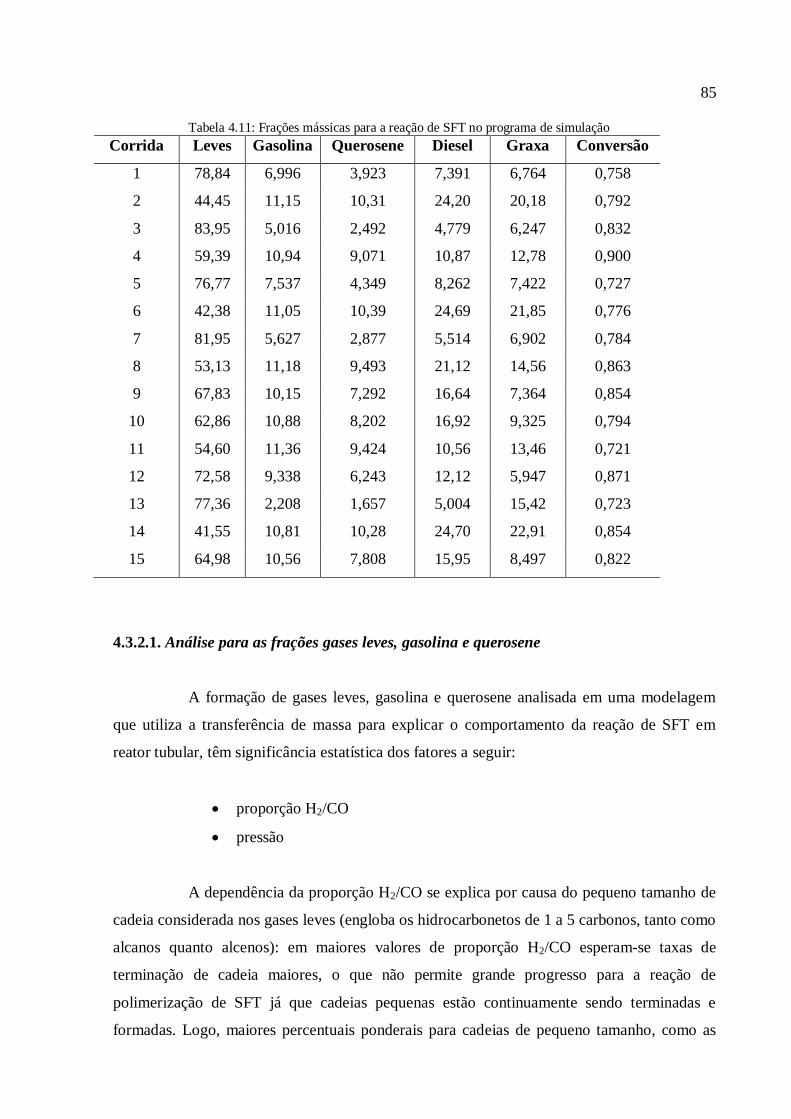
85
Tabela 4.11: Frações mássicas para a reação de SFT no programa de simulação
Corrida Leves Gasolina Querosene Diesel Graxa Conversão
1 78,84 6,996 3,923 7,391 6,764 0,758
2 44,45 11,15 10,31 24,20 20,18 0,792
3 83,95 5,016 2,492 4,779 6,247 0,832
4 59,39 10,94 9,071 10,87 12,78 0,900
5 76,77 7,537 4,349 8,262 7,422 0,727
6 42,38 11,05 10,39 24,69 21,85 0,776
7 81,95 5,627 2,877 5,514 6,902 0,784
8 53,13 11,18 9,493 21,12 14,56 0,863
9 67,83 10,15 7,292 16,64 7,364 0,854
10 62,86 10,88 8,202 16,92 9,325 0,794
11 54,60 11,36 9,424 10,56 13,46 0,721
12 72,58 9,338 6,243 12,12 5,947 0,871
13 77,36 2,208 1,657 5,004 15,42 0,723
14 41,55 10,81 10,28 24,70 22,91 0,854
15 64,98 10,56 7,808 15,95 8,497 0,822
4.3.2.1. Análise para as frações gases leves, gasolina e querosene
A formação de gases leves, gasolina e querosene analisada em uma modelagem
que utiliza a transferência de massa para explicar o comportamento da reação de SFT em
reator tubular, têm significância estatística dos fatores a seguir:
proporção H2/CO
pressão
A dependência da proporção H2/CO se explica por causa do pequeno tamanho de
cadeia considerada nos gases leves (engloba os hidrocarbonetos de 1 a 5 carbonos, tanto como
alcanos quanto alcenos): em maiores valores de proporção H2/CO esperam-se taxas de
terminação de cadeia maiores, o que não permite grande progresso para a reação de
polimerização de SFT já que cadeias pequenas estão continuamente sendo terminadas e
formadas. Logo, maiores percentuais ponderais para cadeias de pequeno tamanho, como as
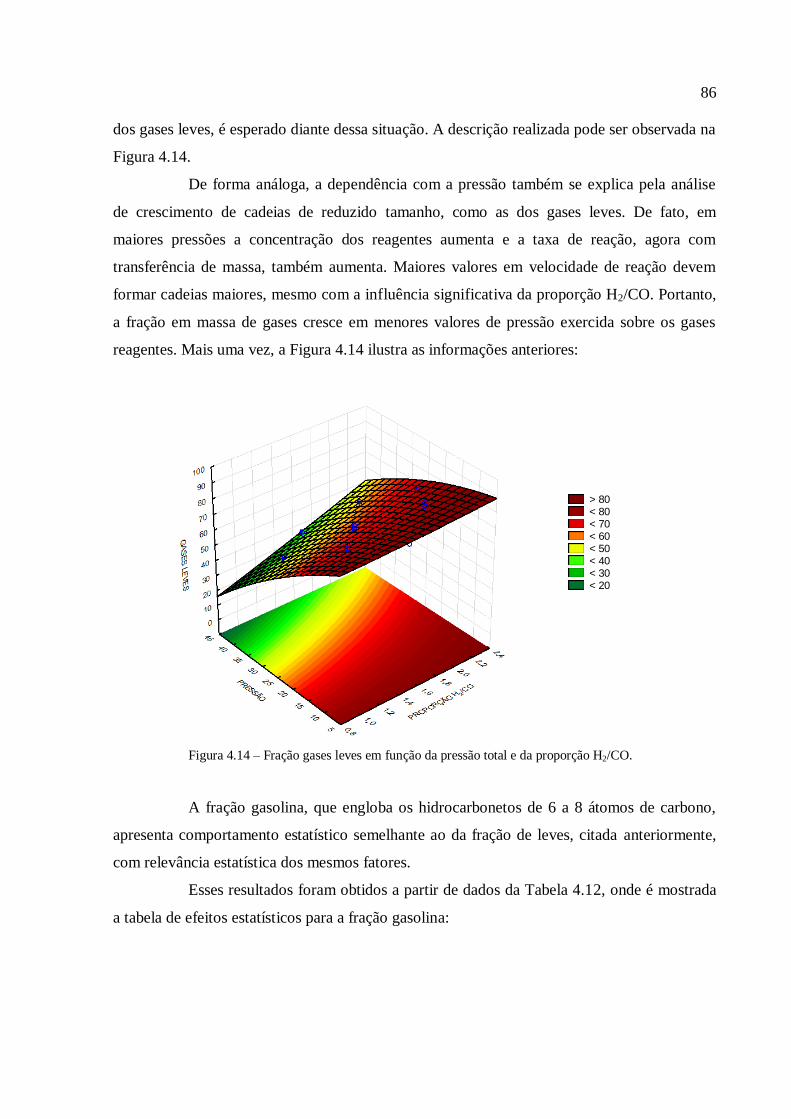
86
dos gases leves, é esperado diante dessa situação. A descrição realizada pode ser observada na
Figura 4.14.
De forma análoga, a dependência com a pressão também se explica pela análise
de crescimento de cadeias de reduzido tamanho, como as dos gases leves. De fato, em
maiores pressões a concentração dos reagentes aumenta e a taxa de reação, agora com
transferência de massa, também aumenta. Maiores valores em velocidade de reação devem
formar cadeias maiores, mesmo com a influência significativa da proporção H2/CO. Portanto,
a fração em massa de gases cresce em menores valores de pressão exercida sobre os gases
reagentes. Mais uma vez, a Figura 4.14 ilustra as informações anteriores:
Figura 4.14 – Fração gases leves em função da pressão total e da proporção H2/CO.
A fração gasolina, que engloba os hidrocarbonetos de 6 a 8 átomos de carbono,
apresenta comportamento estatístico semelhante ao da fração de leves, citada anteriormente,
com relevância estatística dos mesmos fatores.
Esses resultados foram obtidos a partir de dados da Tabela 4.12, onde é mostrada
a tabela de efeitos estatísticos para a fração gasolina:
Fitted Surface; Variable: Var1
3 factors, 1 Blocks, 15 Runs; MS Residual=18,87783
DV: Var1
> 80 < 80 < 70 < 60 < 50 < 40 < 30 < 20
87
Tabela 4.12 – Análise de efeitos para a fração gasolina
Efe
ito
prin
cip
al
Erro
pad
rão
Test
e t
de
Stu
den
t (5
)
Valo
r p
lim
ite
de c
on
fian
ça
inf
(95%
)
lim
ite
de c
on
fian
ça
sup
(95%
)
Velocidade (linear) 0,369 0,177 2,088 0,091 -0,085 0,823
Velocidade (quadrática) -2,218 0,265 -0,820 0,449 -0,900 0,465
Proporção H2/CO (linear) -1,079 0,177 -6,107 0,002 -1,533 -0,625
Proporção H2/CO (quadrática) -0,335 0,265 -1,262 0,262 -1,017 0,347
Pressão (linear) 4,922 0,177 27,85 1.10-6
4,468 5,376
Pressão (quadrática) -3,050 0,265 11,49 8,8.10-5
-3,733 -2,368
São considerados significantes os fatores (variáveis) cujos valores de p são
inferiores ao valor aceito como limite para erro tipo I (rejeitar a hipótese nula), que foi
adotado como 5% (ou 0,05) nessa tese. Os fatores relevantes estatisticamente para a fração
graxa estão em negrito na Tabela 4.12.
A explicação para a dependência em relação à proporção H2/CO segue mesmo
raciocínio já analisado: cadeias menores (como as de gases leves e de gasolina) têm sua
formação favorecida em condições de maiores proporções H2/CO, para aumentar a taxa de
terminação das cadeias. Além disso, a dependência da pressão para a fração gasolina é ainda
mais significativa que para os gases leves, o que se justifica pelo fato de a cadeia da fração
gasolina, em média, ser maior que a da fração gases leves. Observa-se que o aumento de
pressão provoca o crescimento das cadeias para a fração gasolina, mas não para leves. De
fato, para cadeias maiores que as dos leves, como as da gasolina e as do querosene, fração que
será analisada adiante, o aumento da pressão, que aumenta a velocidade de reação, favorece a
formação de cadeias maiores. Contudo, observa-se que a Figura 4.15 possui um máximo para
a fração gasolina, o que mostra que a cadeia é um pouco maior que as dos leves, mas não é
tão extensa para que se aumente a pressão sem prejuízo na conversão. A Figura 4.15 ilustra
essas informações.
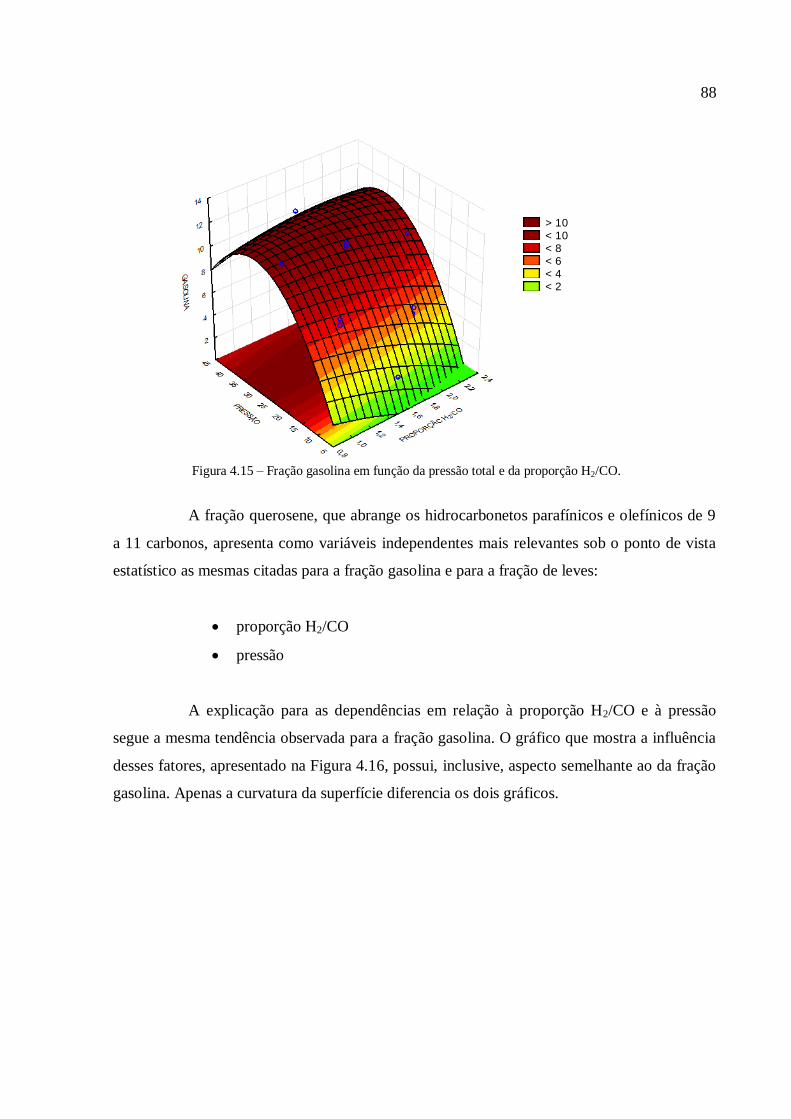
88
Figura 4.15 – Fração gasolina em função da pressão total e da proporção H2/CO.
A fração querosene, que abrange os hidrocarbonetos parafínicos e olefínicos de 9
a 11 carbonos, apresenta como variáveis independentes mais relevantes sob o ponto de vista
estatístico as mesmas citadas para a fração gasolina e para a fração de leves:
proporção H2/CO
pressão
A explicação para as dependências em relação à proporção H2/CO e à pressão
segue a mesma tendência observada para a fração gasolina. O gráfico que mostra a influência
desses fatores, apresentado na Figura 4.16, possui, inclusive, aspecto semelhante ao da fração
gasolina. Apenas a curvatura da superfície diferencia os dois gráficos.
Fitted Surface; Variable: Var2
3 factors, 1 Blocks, 15 Runs; MS Residual=,1066644
DV: Var2
> 10 < 10 < 8 < 6 < 4 < 2
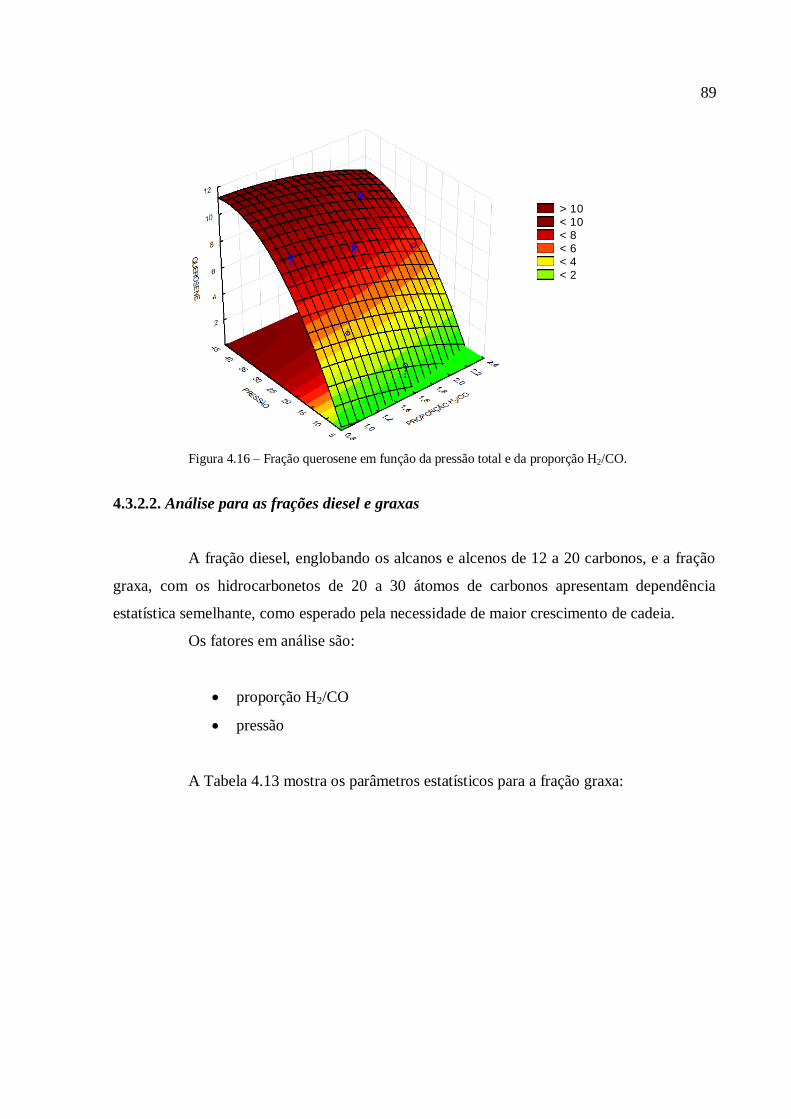
89
Figura 4.16 – Fração querosene em função da pressão total e da proporção H2/CO.
4.3.2.2. Análise para as frações diesel e graxas
A fração diesel, englobando os alcanos e alcenos de 12 a 20 carbonos, e a fração
graxa, com os hidrocarbonetos de 20 a 30 átomos de carbonos apresentam dependência
estatística semelhante, como esperado pela necessidade de maior crescimento de cadeia.
Os fatores em análise são:
proporção H2/CO
pressão
A Tabela 4.13 mostra os parâmetros estatísticos para a fração graxa:
Fitted Surface; Variable: Var3
3 factors, 1 Blocks, 15 Runs; MS Residual=,5271037
DV: Var3
> 10 < 10 < 8 < 6 < 4 < 2
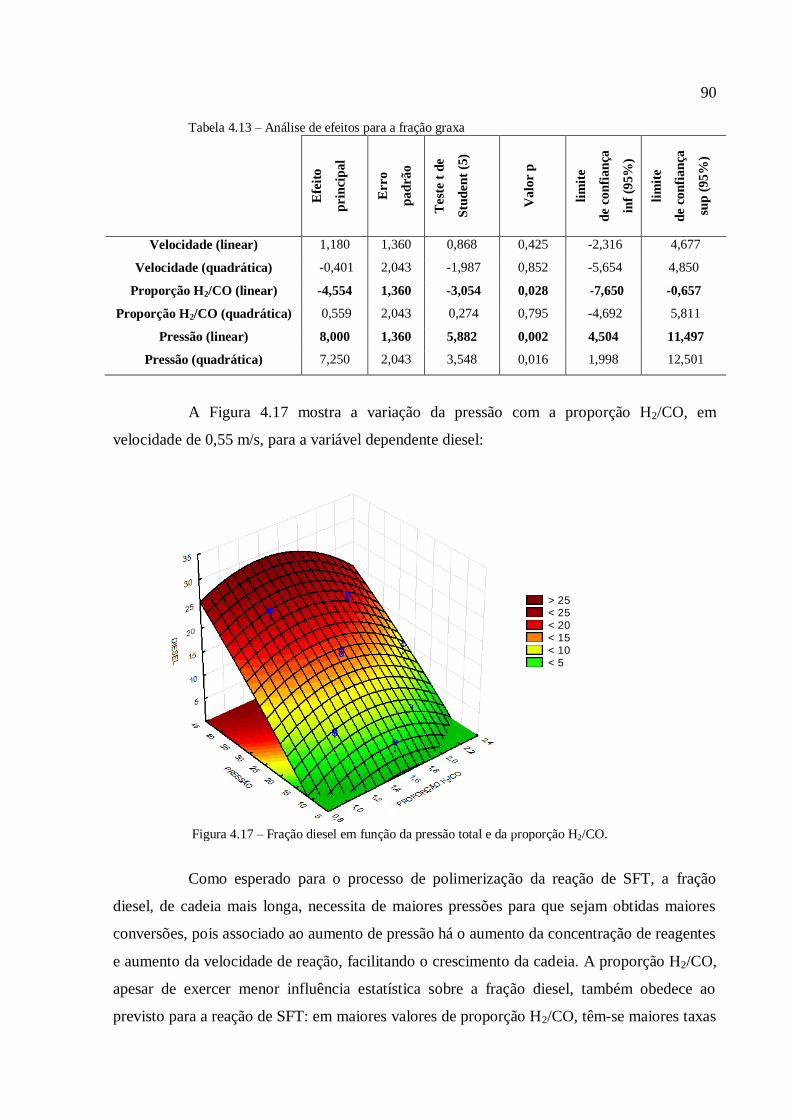
90
Tabela 4.13 – Análise de efeitos para a fração graxa
Efe
ito
prin
cip
al
Erro
pad
rão
Test
e t
de
Stu
den
t (5
)
Valo
r p
lim
ite
de c
on
fian
ça
inf
(95%
)
lim
ite
de c
on
fian
ça
sup
(95%
)
Velocidade (linear) 1,180 1,360 0,868 0,425 -2,316 4,677
Velocidade (quadrática) -0,401 2,043 -1,987 0,852 -5,654 4,850
Proporção H2/CO (linear) -4,554 1,360 -3,054 0,028 -7,650 -0,657
Proporção H2/CO (quadrática) 0,559 2,043 0,274 0,795 -4,692 5,811
Pressão (linear) 8,000 1,360 5,882 0,002 4,504 11,497
Pressão (quadrática) 7,250 2,043 3,548 0,016 1,998 12,501
A Figura 4.17 mostra a variação da pressão com a proporção H2/CO, em
velocidade de 0,55 m/s, para a variável dependente diesel:
Figura 4.17 – Fração diesel em função da pressão total e da proporção H2/CO.
Como esperado para o processo de polimerização da reação de SFT, a fração
diesel, de cadeia mais longa, necessita de maiores pressões para que sejam obtidas maiores
conversões, pois associado ao aumento de pressão há o aumento da concentração de reagentes
e aumento da velocidade de reação, facilitando o crescimento da cadeia. A proporção H2/CO,
apesar de exercer menor influência estatística sobre a fração diesel, também obedece ao
previsto para a reação de SFT: em maiores valores de proporção H2/CO, têm-se maiores taxas
Fitted Surface; Variable: Var4
3 factors, 1 Blocks, 15 Runs; MS Residual=6,141179
DV: Var4
> 25 < 25 < 20 < 15 < 10 < 5
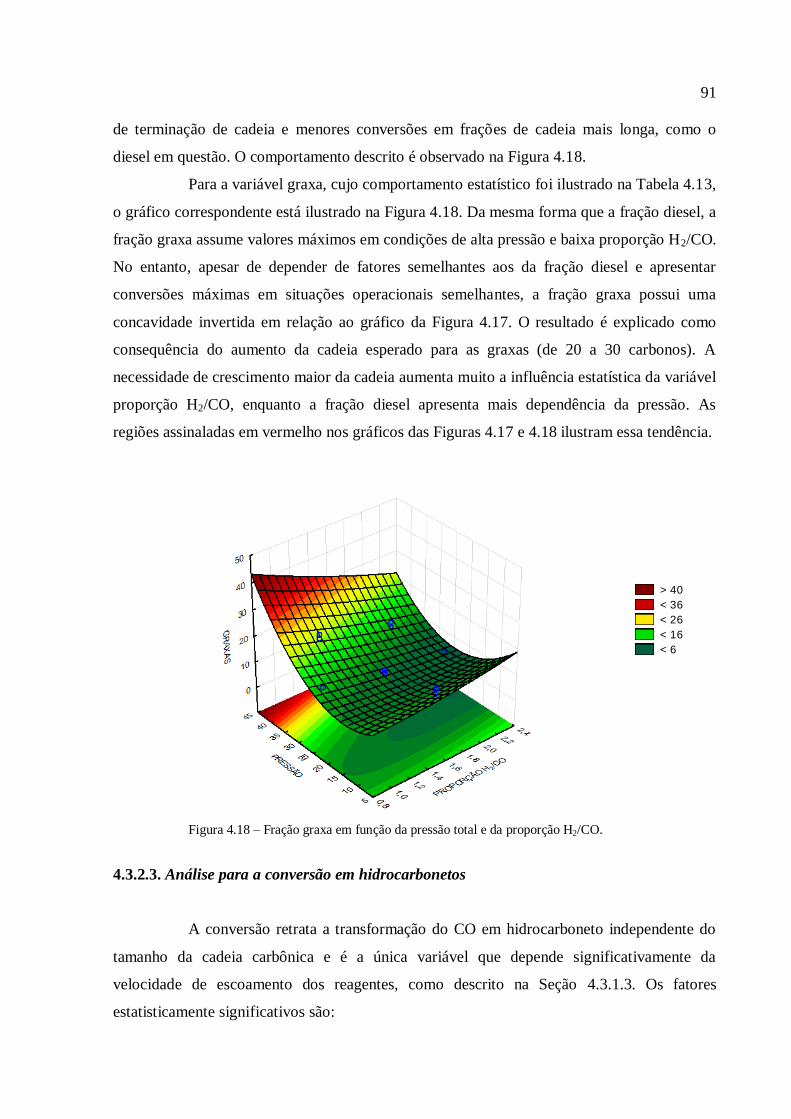
91
de terminação de cadeia e menores conversões em frações de cadeia mais longa, como o
diesel em questão. O comportamento descrito é observado na Figura 4.18.
Para a variável graxa, cujo comportamento estatístico foi ilustrado na Tabela 4.13,
o gráfico correspondente está ilustrado na Figura 4.18. Da mesma forma que a fração diesel, a
fração graxa assume valores máximos em condições de alta pressão e baixa proporção H2/CO.
No entanto, apesar de depender de fatores semelhantes aos da fração diesel e apresentar
conversões máximas em situações operacionais semelhantes, a fração graxa possui uma
concavidade invertida em relação ao gráfico da Figura 4.17. O resultado é explicado como
consequência do aumento da cadeia esperado para as graxas (de 20 a 30 carbonos). A
necessidade de crescimento maior da cadeia aumenta muito a influência estatística da variável
proporção H2/CO, enquanto a fração diesel apresenta mais dependência da pressão. As
regiões assinaladas em vermelho nos gráficos das Figuras 4.17 e 4.18 ilustram essa tendência.
Figura 4.18 – Fração graxa em função da pressão total e da proporção H2/CO.
4.3.2.3. Análise para a conversão em hidrocarbonetos
A conversão retrata a transformação do CO em hidrocarboneto independente do
tamanho da cadeia carbônica e é a única variável que depende significativamente da
velocidade de escoamento dos reagentes, como descrito na Seção 4.3.1.3. Os fatores
estatisticamente significativos são:
Fitted Surface; Variable: Var5
3 factors, 1 Blocks, 15 Runs; MS Residual=6,316403
DV: Var5
> 40
< 36
< 26
< 16
< 6 0,81,01,21,41,61,82,02,22,4proporção H2/CO51015202530354045pressão01020304050graxa
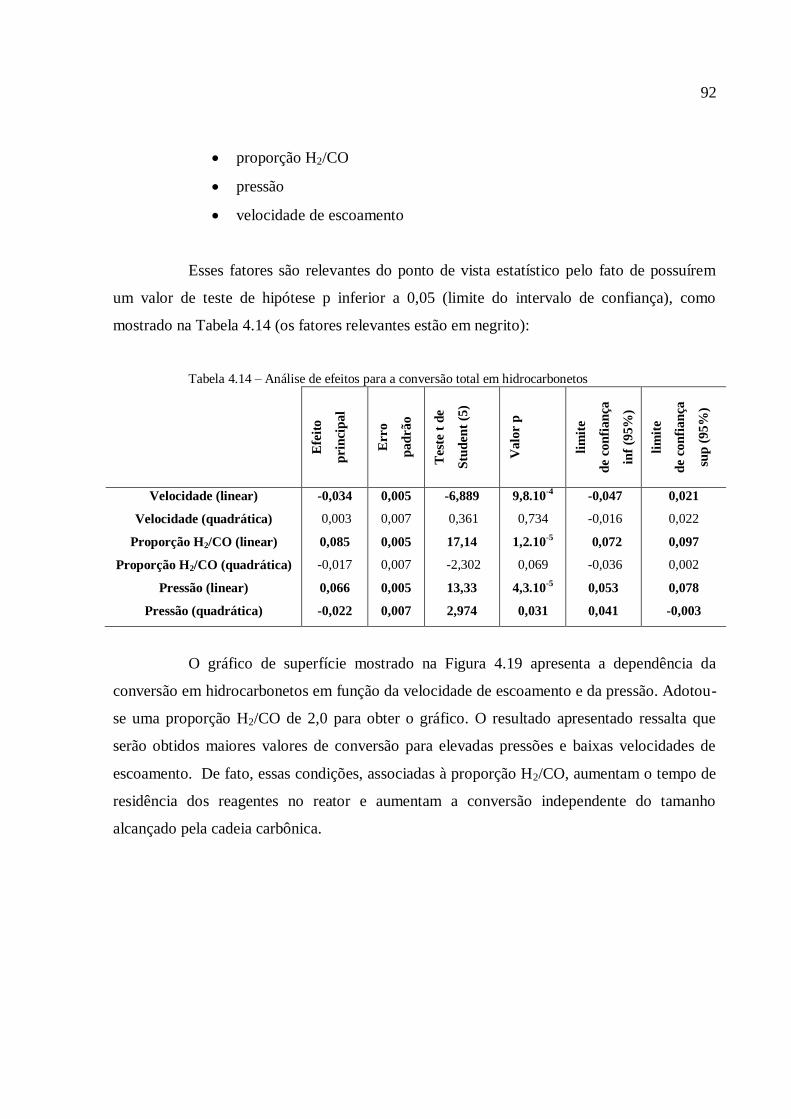
92
proporção H2/CO
pressão
velocidade de escoamento
Esses fatores são relevantes do ponto de vista estatístico pelo fato de possuírem
um valor de teste de hipótese p inferior a 0,05 (limite do intervalo de confiança), como
mostrado na Tabela 4.14 (os fatores relevantes estão em negrito):
Tabela 4.14 – Análise de efeitos para a conversão total em hidrocarbonetos
Efe
ito
prin
cip
al
Erro
pa
drão
Test
e t
de
Stu
den
t (5
)
Va
lor
p
lim
ite
de c
on
fian
ça
inf
(95%
)
lim
ite
de c
on
fian
ça
sup
(9
5%
)
Velocidade (linear) -0,034 0,005 -6,889 9,8.10-4
-0,047 0,021
Velocidade (quadrática) 0,003 0,007 0,361 0,734 -0,016 0,022
Proporção H2/CO (linear) 0,085 0,005 17,14 1,2.10-5
0,072 0,097
Proporção H2/CO (quadrática) -0,017 0,007 -2,302 0,069 -0,036 0,002
Pressão (linear) 0,066 0,005 13,33 4,3.10-5
0,053 0,078
Pressão (quadrática) -0,022 0,007 2,974 0,031 0,041 -0,003
O gráfico de superfície mostrado na Figura 4.19 apresenta a dependência da
conversão em hidrocarbonetos em função da velocidade de escoamento e da pressão. Adotou-
se uma proporção H2/CO de 2,0 para obter o gráfico. O resultado apresentado ressalta que
serão obtidos maiores valores de conversão para elevadas pressões e baixas velocidades de
escoamento. De fato, essas condições, associadas à proporção H2/CO, aumentam o tempo de
residência dos reagentes no reator e aumentam a conversão independente do tamanho
alcançado pela cadeia carbônica.
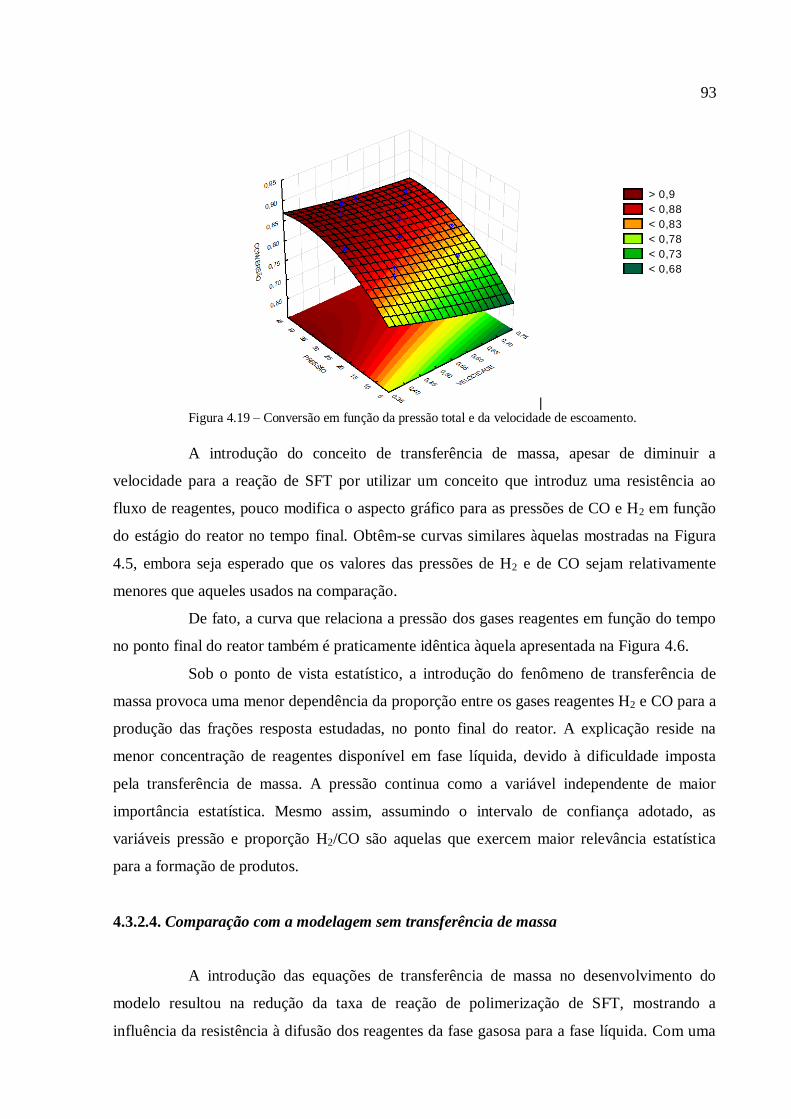
93
Figura 4.19 – Conversão em função da pressão total e da velocidade de escoamento.
A introdução do conceito de transferência de massa, apesar de diminuir a
velocidade para a reação de SFT por utilizar um conceito que introduz uma resistência ao
fluxo de reagentes, pouco modifica o aspecto gráfico para as pressões de CO e H2 em função
do estágio do reator no tempo final. Obtêm-se curvas similares àquelas mostradas na Figura
4.5, embora seja esperado que os valores das pressões de H2 e de CO sejam relativamente
menores que aqueles usados na comparação.
De fato, a curva que relaciona a pressão dos gases reagentes em função do tempo
no ponto final do reator também é praticamente idêntica àquela apresentada na Figura 4.6.
Sob o ponto de vista estatístico, a introdução do fenômeno de transferência de
massa provoca uma menor dependência da proporção entre os gases reagentes H2 e CO para a
produção das frações resposta estudadas, no ponto final do reator. A explicação reside na
menor concentração de reagentes disponível em fase líquida, devido à dificuldade imposta
pela transferência de massa. A pressão continua como a variável independente de maior
importância estatística. Mesmo assim, assumindo o intervalo de confiança adotado, as
variáveis pressão e proporção H2/CO são aquelas que exercem maior relevância estatística
para a formação de produtos.
4.3.2.4. Comparação com a modelagem sem transferência de massa
A introdução das equações de transferência de massa no desenvolvimento do
modelo resultou na redução da taxa de reação de polimerização de SFT, mostrando a
influência da resistência à difusão dos reagentes da fase gasosa para a fase líquida. Com uma
Fitted Surface; Variable: Var6
3 factors, 1 Blocks, 15 Runs; MS Residual=,0000834
DV: Var6
> 0,9
< 0,88
< 0,83
< 0,78
< 0,73
< 0,68 0,350,400,450,500,550,600,650,700,75velocidade51015202530354045pressão0,650,700,750,800,850,900,95conversão

94
menor concentração de reagentes, tem-se menor adsorção de reagentes nos sítios catalíticos e
uma menor taxa de formação de produtos.
De acordo com as Tabelas 4.8 e 4.12, a introdução das equações de transferência
de massa na modelagem aumenta a proporção de leves no produto final do reator em seu
estágio final. O aumento da fração de leves aumenta também a conversão total em
hidrocarbonetos, um fato aparentemente contraditório, já que a transferência de massa diminui
a taxa de reação. No entanto, o cálculo da conversão não diferencia cadeias maiores de
menores, e o aumento na conversão em leves, principalmente metano, pode causar a ilusão de
que a conversão está maior, quando, na verdade, a conversão em frações comercialmente mais
procuradas, como gasolina, diesel e graxas, foi reduzida, conforme esperado.
A conversão total em hidrocarbonetos tem sua análise de efeitos mostrados nas
Tabelas 4.10 e 4.14. Evidencia-se que a velocidade de escoamento passa a exercer maior
influência estatística com a transferência de massa sendo levada em consideração, além da
pressão e da proporção H2/CO. Esse resultado está em sintonia com a discussão anterior. Para
que a conversão em leves aumente é preciso uma maior taxa de terminação de cadeias para
impedir que a cadeia cresça e alcance maiores tamanhos. Menores valores de velocidade
resultam em maiores tempos de residência e favorecem maiores cadeias carbônicas. Assim, é
compreensível que a variável velocidade de escoamento passe a exercer maior influência
estatística para a transferência de massa.
Em relação às frações de maior interesse comercial, como gasolina, diesel e
graxas, analisadas separadamente, a utilização das equações de transferência de massa na
modelagem aumenta a dependência estatística da pressão para o resultado final obtido. Da
mesma forma, percebe-se que a dependência da proporção H2/CO diminui, embora essa
redução ainda se encontre dentro do intervalo de confiança estabelecido. Esta conclusão pode
ser percebida a partir da análise das Tabelas 4.8, 4.9, 4.12 e 4.13. A dependência estatística da
velocidade de escoamento mantém-se em patamares pouco significantes sob o ponto de vista
estatístico.
A dependência da pressão continua a mais importante estatisticamente, uma vez
que pressões maiores resultam em maiores taxas de reação, e o aumento da pressão compensa
em parte a utilização das equações de transferência de massa. Por sua vez, o aumento da
proporção H2/CO aumenta a taxa de terminação das cadeias carbônicas, que justifica o fato de
que, sob o ponto de vista estatístico, exerça uma menor influência com a utilização da
transferência de massa na modelagem. A própria transferência de massa já aumenta a fração

95
em produtos mais leves, o que reduz a importância da proporção H2/CO. Essa redução, como
já comentado, não modifica a relevância da variável, pois a mesma ainda se encontra dentro
do intervalo de confiança adotado (95%).
4.3.3. Simulações com o modelo com transferência de calor
A análise estatística seguiu o mesmo procedimento adotado na Seção 4.2, com a
utilização das mesmas variáveis dependentes e independentes, descritas nas Tabelas 4.2 e 4.4,
bem como com a utilização do planejamento experimental mostrado na Tabela 4.3. A
novidade reside em incluir a variável temperatura como uma variável dependente das
condições experimentais.
Os dados termodinâmicos usados para a transferência de calor estão na Tabela
4.15. A capacidade calorífica a pressão constante (Cp) foi considerada praticamente inalterada
diante das mudanças da composição no leito reacional ao longo do comprimento do reator e
do tempo. Essa simplificação é procedente diante da pouca diferença de composição,
principalmente em relação aos produtos orgânicos, de um estágio para outro.
Tabela 4.15: Dados termodinâmicos utilizados na simulação do reator para a reação de SFT para a
temperatura de 240°C
H (kJ.mol-1
) – 165
Coeficiente global de transferência de calor
(U, em J.m-2
.s-1
.K-1
) 1,31.10
6
Área seccional do reator (cm2) 66,44
área de troca de calor por unidade de volume (cm-1
) 0,425
capacidade calorífica média a pressão constante
(Cp, em J.mol-1
.K-1
) 0,424
Executou-se a simulação sugerida para um reator tubular operando nas condições
descritas, com a utilização das equações para a transferência de massa e de calor. A variável
temperatura, tanto a do leito reacional como a do líquido trocador de calor, ambas tratadas
como variáveis dependentes, foi analisada de acordo com a Tabela 4.16:
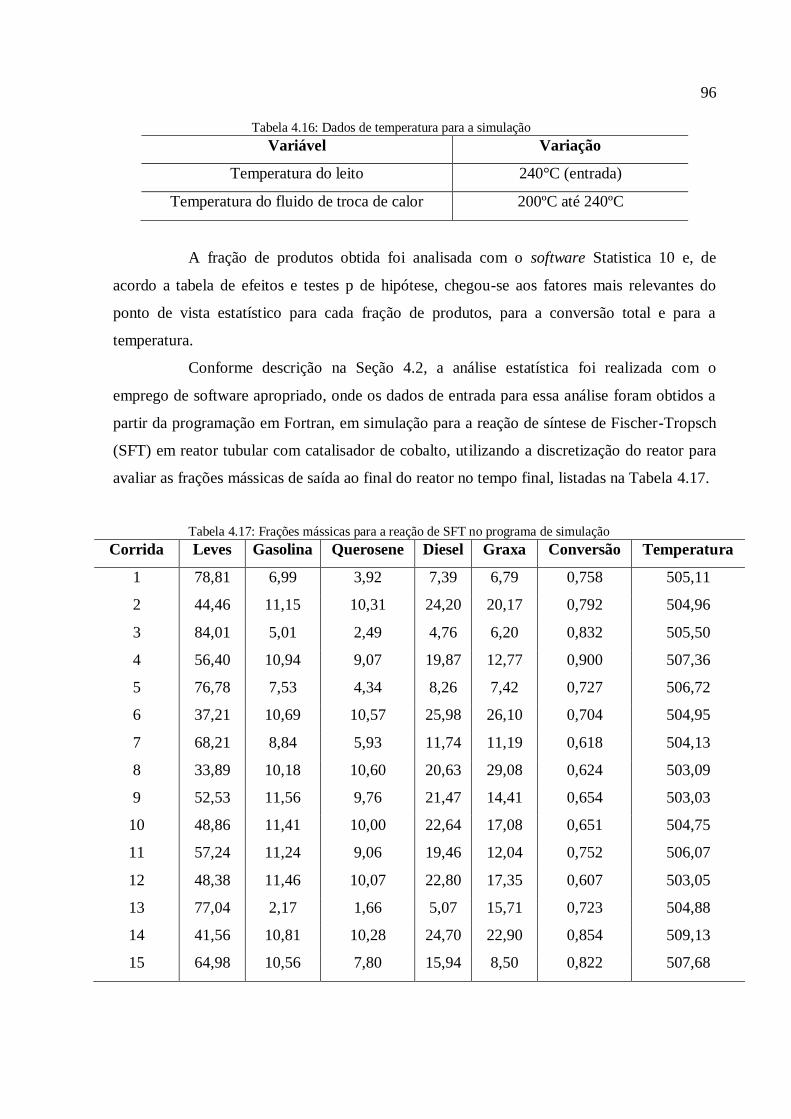
96
Tabela 4.16: Dados de temperatura para a simulação
Variável Variação
Temperatura do leito 240°C (entrada)
Temperatura do fluido de troca de calor 200ºC até 240ºC
A fração de produtos obtida foi analisada com o software Statistica 10 e, de
acordo a tabela de efeitos e testes p de hipótese, chegou-se aos fatores mais relevantes do
ponto de vista estatístico para cada fração de produtos, para a conversão total e para a
temperatura.
Conforme descrição na Seção 4.2, a análise estatística foi realizada com o
emprego de software apropriado, onde os dados de entrada para essa análise foram obtidos a
partir da programação em Fortran, em simulação para a reação de síntese de Fischer-Tropsch
(SFT) em reator tubular com catalisador de cobalto, utilizando a discretização do reator para
avaliar as frações mássicas de saída ao final do reator no tempo final, listadas na Tabela 4.17.
Tabela 4.17: Frações mássicas para a reação de SFT no programa de simulação
Corrida Leves Gasolina Querosene Diesel Graxa Conversão Temperatura
1 78,81 6,99 3,92 7,39 6,79 0,758 505,11
2 44,46 11,15 10,31 24,20 20,17 0,792 504,96
3 84,01 5,01 2,49 4,76 6,20 0,832 505,50
4 56,40 10,94 9,07 19,87 12,77 0,900 507,36
5 76,78 7,53 4,34 8,26 7,42 0,727 506,72
6 37,21 10,69 10,57 25,98 26,10 0,704 504,95
7 68,21 8,84 5,93 11,74 11,19 0,618 504,13
8 33,89 10,18 10,60 20,63 29,08 0,624 503,09
9 52,53 11,56 9,76 21,47 14,41 0,654 503,03
10 48,86 11,41 10,00 22,64 17,08 0,651 504,75
11 57,24 11,24 9,06 19,46 12,04 0,752 506,07
12 48,38 11,46 10,07 22,80 17,35 0,607 503,05
13 77,04 2,17 1,66 5,07 15,71 0,723 504,88
14 41,56 10,81 10,28 24,70 22,90 0,854 509,13
15 64,98 10,56 7,80 15,94 8,50 0,822 507,68
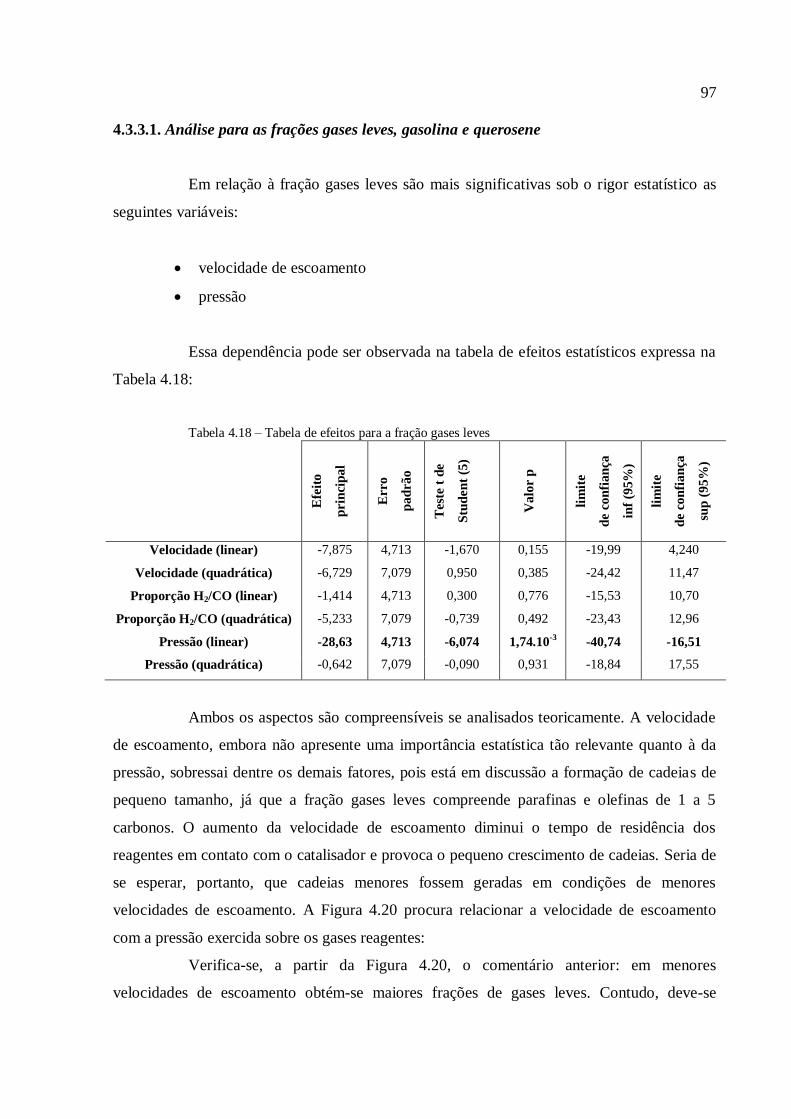
97
4.3.3.1. Análise para as frações gases leves, gasolina e querosene
Em relação à fração gases leves são mais significativas sob o rigor estatístico as
seguintes variáveis:
velocidade de escoamento
pressão
Essa dependência pode ser observada na tabela de efeitos estatísticos expressa na
Tabela 4.18:
Tabela 4.18 – Tabela de efeitos para a fração gases leves
Efe
ito
prin
cip
al
Erro
pa
drã
o
Test
e t
de
Stu
den
t (5
)
Va
lor
p
lim
ite
de c
on
fia
nça
inf
(95%
)
lim
ite
de c
on
fia
nça
sup
(9
5%
)
Velocidade (linear) -7,875 4,713 -1,670 0,155 -19,99 4,240
Velocidade (quadrática) -6,729 7,079 0,950 0,385 -24,42 11,47
Proporção H2/CO (linear) -1,414 4,713 0,300 0,776 -15,53 10,70
Proporção H2/CO (quadrática) -5,233 7,079 -0,739 0,492 -23,43 12,96
Pressão (linear) -28,63 4,713 -6,074 1,74.10-3
-40,74 -16,51
Pressão (quadrática) -0,642 7,079 -0,090 0,931 -18,84 17,55
Ambos os aspectos são compreensíveis se analisados teoricamente. A velocidade
de escoamento, embora não apresente uma importância estatística tão relevante quanto à da
pressão, sobressai dentre os demais fatores, pois está em discussão a formação de cadeias de
pequeno tamanho, já que a fração gases leves compreende parafinas e olefinas de 1 a 5
carbonos. O aumento da velocidade de escoamento diminui o tempo de residência dos
reagentes em contato com o catalisador e provoca o pequeno crescimento de cadeias. Seria de
se esperar, portanto, que cadeias menores fossem geradas em condições de menores
velocidades de escoamento. A Figura 4.20 procura relacionar a velocidade de escoamento
com a pressão exercida sobre os gases reagentes:
Verifica-se, a partir da Figura 4.20, o comentário anterior: em menores
velocidades de escoamento obtém-se maiores frações de gases leves. Contudo, deve-se
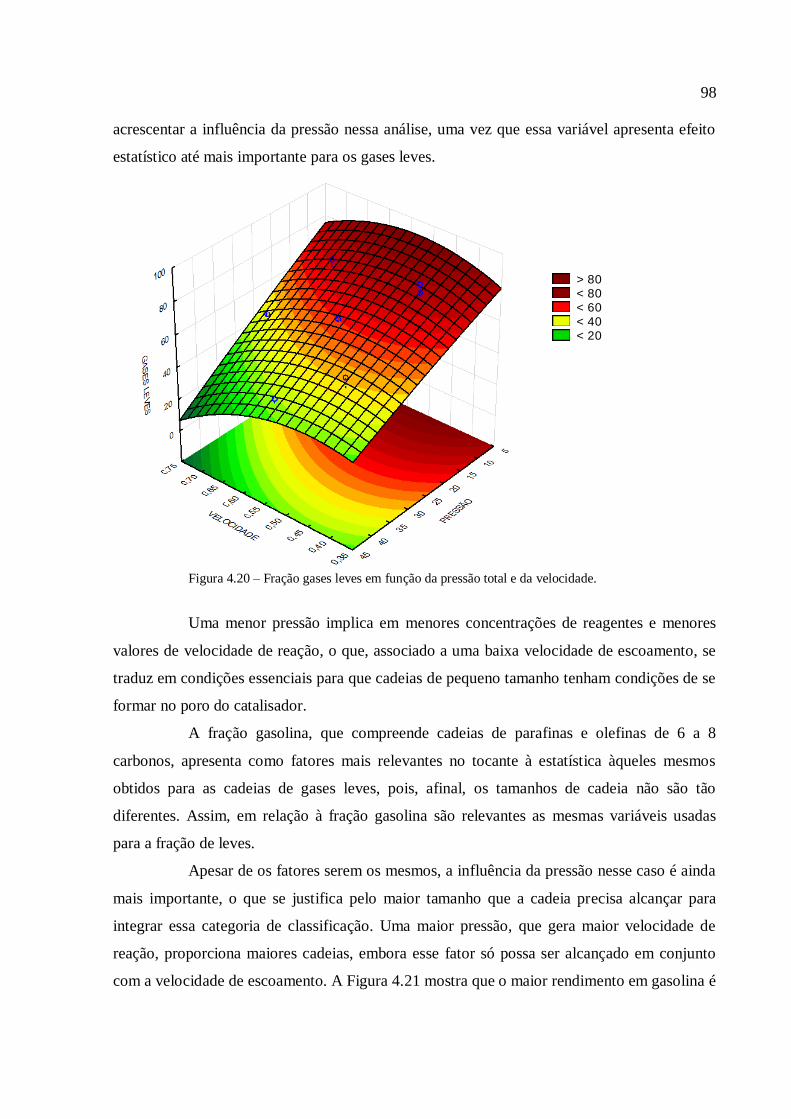
98
acrescentar a influência da pressão nessa análise, uma vez que essa variável apresenta efeito
estatístico até mais importante para os gases leves.
Figura 4.20 – Fração gases leves em função da pressão total e da velocidade.
Uma menor pressão implica em menores concentrações de reagentes e menores
valores de velocidade de reação, o que, associado a uma baixa velocidade de escoamento, se
traduz em condições essenciais para que cadeias de pequeno tamanho tenham condições de se
formar no poro do catalisador.
A fração gasolina, que compreende cadeias de parafinas e olefinas de 6 a 8
carbonos, apresenta como fatores mais relevantes no tocante à estatística àqueles mesmos
obtidos para as cadeias de gases leves, pois, afinal, os tamanhos de cadeia não são tão
diferentes. Assim, em relação à fração gasolina são relevantes as mesmas variáveis usadas
para a fração de leves.
Apesar de os fatores serem os mesmos, a influência da pressão nesse caso é ainda
mais importante, o que se justifica pelo maior tamanho que a cadeia precisa alcançar para
integrar essa categoria de classificação. Uma maior pressão, que gera maior velocidade de
reação, proporciona maiores cadeias, embora esse fator só possa ser alcançado em conjunto
com a velocidade de escoamento. A Figura 4.21 mostra que o maior rendimento em gasolina é
Fitted Surface; Variable: Var1
3 factors, 1 Blocks, 15 Runs; MS Residual=75,84829
DV: Var1
> 80 < 80 < 60 < 40 < 20
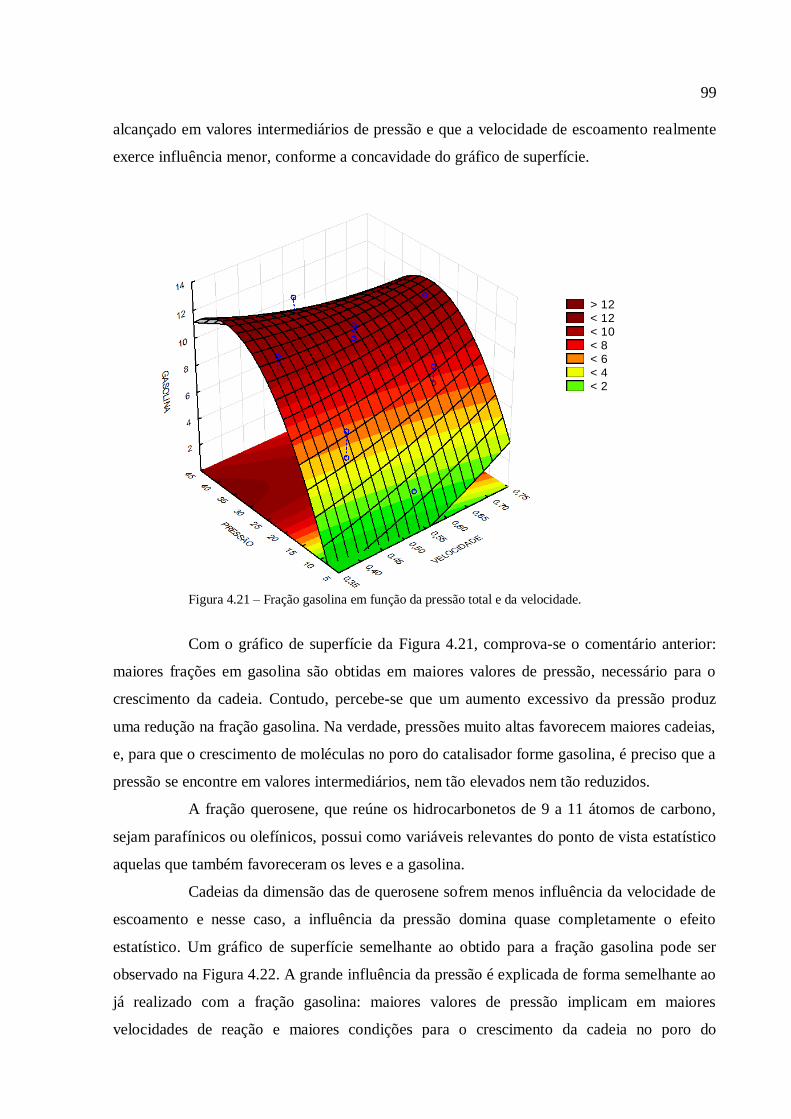
99
alcançado em valores intermediários de pressão e que a velocidade de escoamento realmente
exerce influência menor, conforme a concavidade do gráfico de superfície.
Figura 4.21 – Fração gasolina em função da pressão total e da velocidade.
Com o gráfico de superfície da Figura 4.21, comprova-se o comentário anterior:
maiores frações em gasolina são obtidas em maiores valores de pressão, necessário para o
crescimento da cadeia. Contudo, percebe-se que um aumento excessivo da pressão produz
uma redução na fração gasolina. Na verdade, pressões muito altas favorecem maiores cadeias,
e, para que o crescimento de moléculas no poro do catalisador forme gasolina, é preciso que a
pressão se encontre em valores intermediários, nem tão elevados nem tão reduzidos.
A fração querosene, que reúne os hidrocarbonetos de 9 a 11 átomos de carbono,
sejam parafínicos ou olefínicos, possui como variáveis relevantes do ponto de vista estatístico
aquelas que também favoreceram os leves e a gasolina.
Cadeias da dimensão das de querosene sofrem menos influência da velocidade de
escoamento e nesse caso, a influência da pressão domina quase completamente o efeito
estatístico. Um gráfico de superfície semelhante ao obtido para a fração gasolina pode ser
observado na Figura 4.22. A grande influência da pressão é explicada de forma semelhante ao
já realizado com a fração gasolina: maiores valores de pressão implicam em maiores
velocidades de reação e maiores condições para o crescimento da cadeia no poro do
Fitted Surface; Variable: Var2
3 factors, 1 Blocks, 15 Runs; MS Residual=1,193479
DV: Var2
> 12 < 12 < 10 < 8 < 6 < 4 < 2
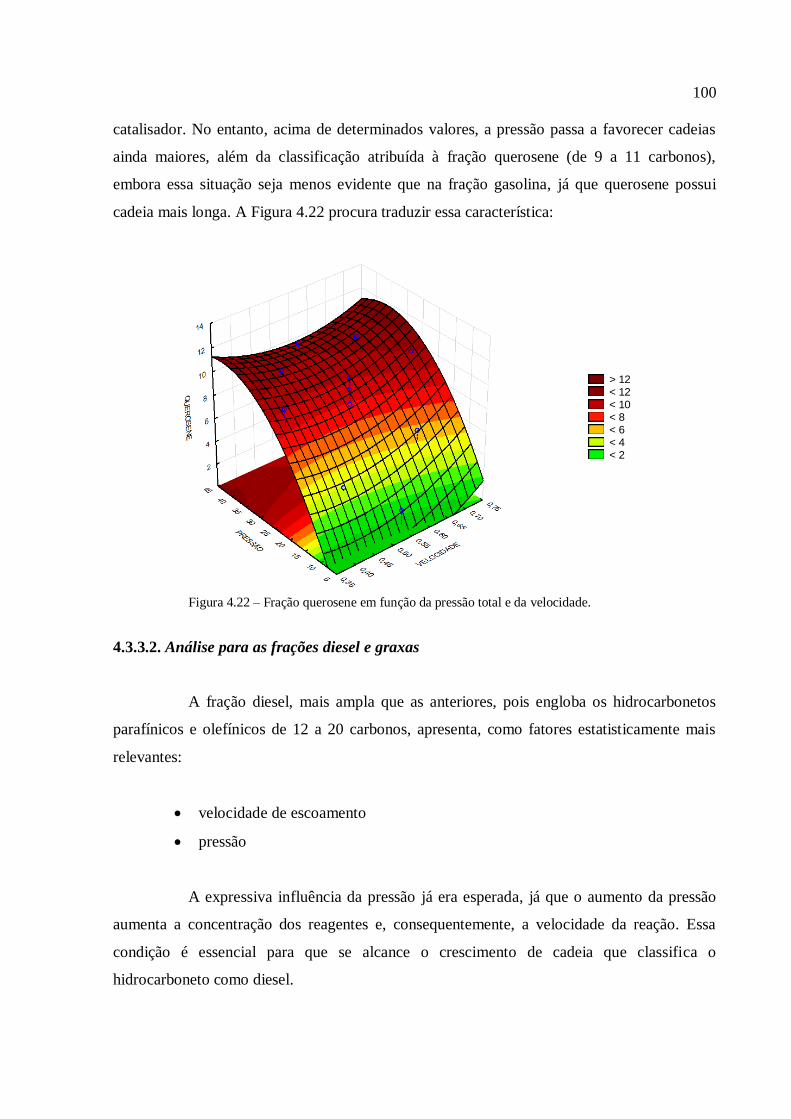
100
catalisador. No entanto, acima de determinados valores, a pressão passa a favorecer cadeias
ainda maiores, além da classificação atribuída à fração querosene (de 9 a 11 carbonos),
embora essa situação seja menos evidente que na fração gasolina, já que querosene possui
cadeia mais longa. A Figura 4.22 procura traduzir essa característica:
Figura 4.22 – Fração querosene em função da pressão total e da velocidade.
4.3.3.2. Análise para as frações diesel e graxas
A fração diesel, mais ampla que as anteriores, pois engloba os hidrocarbonetos
parafínicos e olefínicos de 12 a 20 carbonos, apresenta, como fatores estatisticamente mais
relevantes:
velocidade de escoamento
pressão
A expressiva influência da pressão já era esperada, já que o aumento da pressão
aumenta a concentração dos reagentes e, consequentemente, a velocidade da reação. Essa
condição é essencial para que se alcance o crescimento de cadeia que classifica o
hidrocarboneto como diesel.
Fitted Surface; Variable: Var3
3 factors, 1 Blocks, 15 Runs; MS Residual=1,777964
DV: Var3
> 12 < 12 < 10 < 8 < 6 < 4 < 2
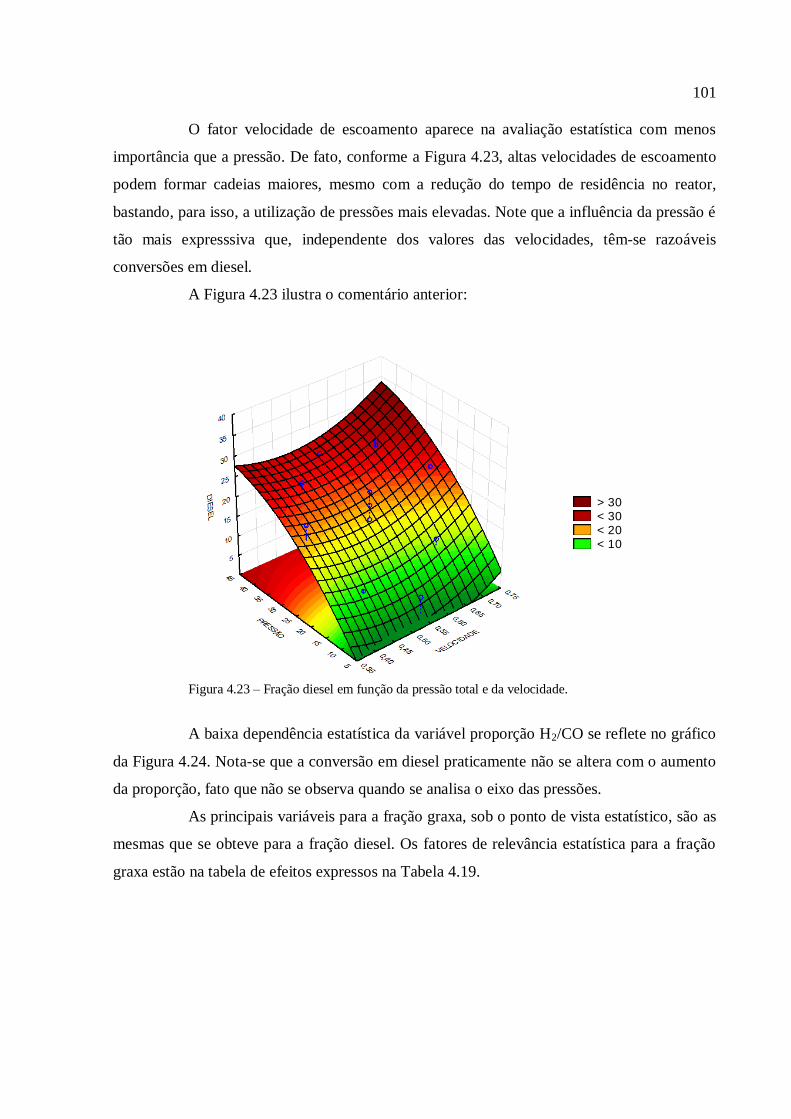
101
O fator velocidade de escoamento aparece na avaliação estatística com menos
importância que a pressão. De fato, conforme a Figura 4.23, altas velocidades de escoamento
podem formar cadeias maiores, mesmo com a redução do tempo de residência no reator,
bastando, para isso, a utilização de pressões mais elevadas. Note que a influência da pressão é
tão mais expresssiva que, independente dos valores das velocidades, têm-se razoáveis
conversões em diesel.
A Figura 4.23 ilustra o comentário anterior:
Figura 4.23 – Fração diesel em função da pressão total e da velocidade.
A baixa dependência estatística da variável proporção H2/CO se reflete no gráfico
da Figura 4.24. Nota-se que a conversão em diesel praticamente não se altera com o aumento
da proporção, fato que não se observa quando se analisa o eixo das pressões.
As principais variáveis para a fração graxa, sob o ponto de vista estatístico, são as
mesmas que se obteve para a fração diesel. Os fatores de relevância estatística para a fração
graxa estão na tabela de efeitos expressos na Tabela 4.19.
Fitted Surface; Variable: Var4
3 factors, 1 Blocks, 15 Runs; MS Residual=14,01634
DV: Var4
> 30 < 30 < 20 < 10
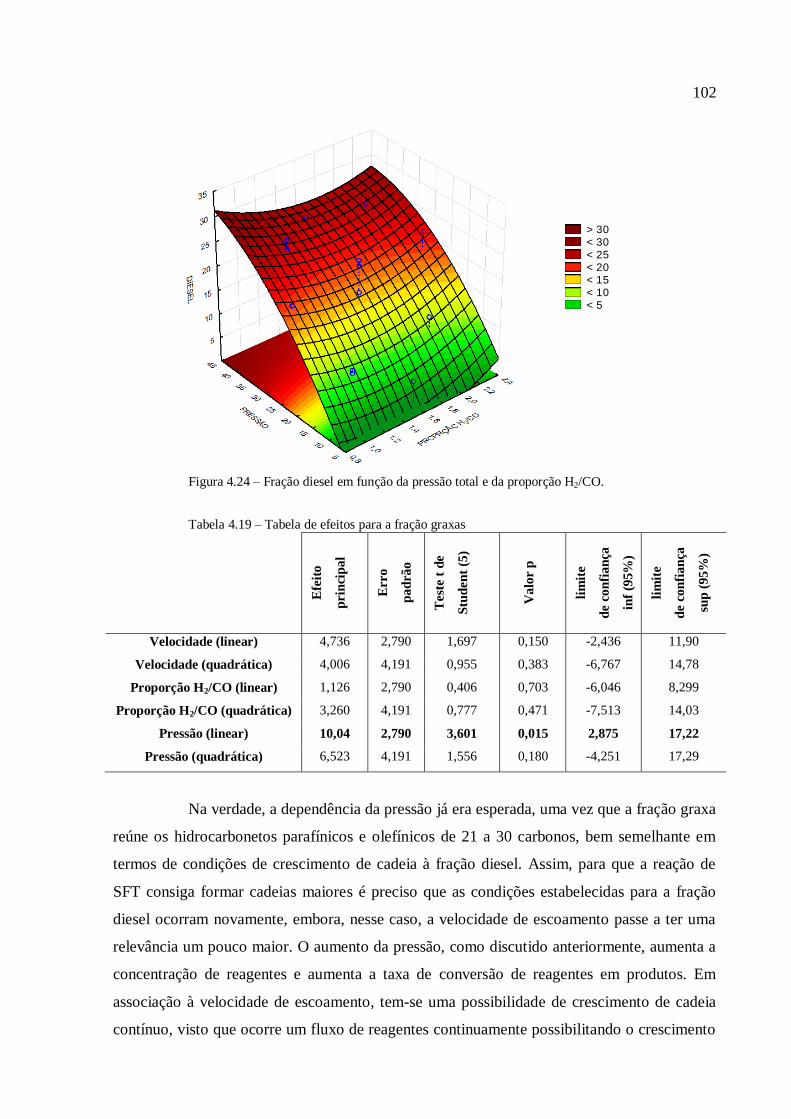
102
Figura 4.24 – Fração diesel em função da pressão total e da proporção H2/CO.
Tabela 4.19 – Tabela de efeitos para a fração graxas
Efe
ito
prin
cip
al
Erro
pa
drã
o
Test
e t
de
Stu
den
t (5
)
Va
lor
p
lim
ite
de c
on
fia
nça
inf
(95%
)
lim
ite
de c
on
fia
nça
sup
(9
5%
)
Velocidade (linear) 4,736 2,790 1,697 0,150 -2,436 11,90
Velocidade (quadrática) 4,006 4,191 0,955 0,383 -6,767 14,78
Proporção H2/CO (linear) 1,126 2,790 0,406 0,703 -6,046 8,299
Proporção H2/CO (quadrática) 3,260 4,191 0,777 0,471 -7,513 14,03
Pressão (linear) 10,04 2,790 3,601 0,015 2,875 17,22
Pressão (quadrática) 6,523 4,191 1,556 0,180 -4,251 17,29
Na verdade, a dependência da pressão já era esperada, uma vez que a fração graxa
reúne os hidrocarbonetos parafínicos e olefínicos de 21 a 30 carbonos, bem semelhante em
termos de condições de crescimento de cadeia à fração diesel. Assim, para que a reação de
SFT consiga formar cadeias maiores é preciso que as condições estabelecidas para a fração
diesel ocorram novamente, embora, nesse caso, a velocidade de escoamento passe a ter uma
relevância um pouco maior. O aumento da pressão, como discutido anteriormente, aumenta a
concentração de reagentes e aumenta a taxa de conversão de reagentes em produtos. Em
associação à velocidade de escoamento, tem-se uma possibilidade de crescimento de cadeia
contínuo, visto que ocorre um fluxo de reagentes continuamente possibilitando o crescimento
Fitted Surface; Variable: Var4
3 factors, 1 Blocks, 15 Runs; MS Residual=14,01634
DV: Var4
> 30 < 30 < 25 < 20 < 15 < 10 < 5
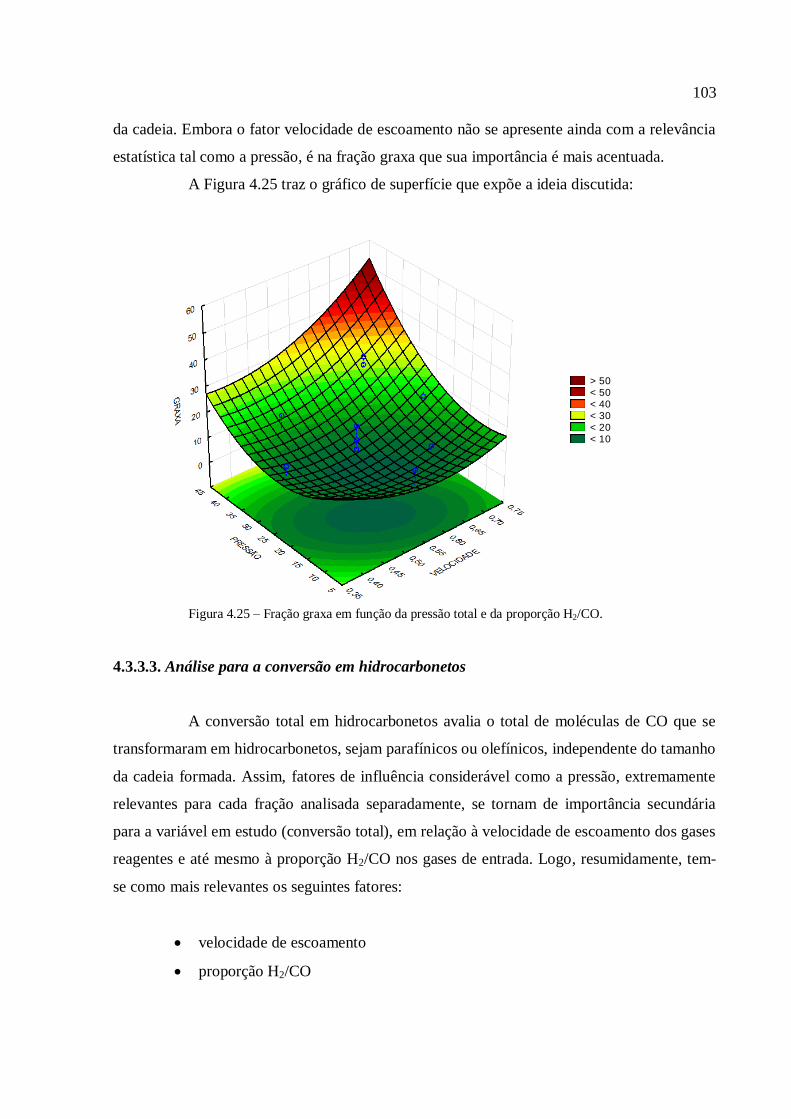
103
da cadeia. Embora o fator velocidade de escoamento não se apresente ainda com a relevância
estatística tal como a pressão, é na fração graxa que sua importância é mais acentuada.
A Figura 4.25 traz o gráfico de superfície que expõe a ideia discutida:
Figura 4.25 – Fração graxa em função da pressão total e da proporção H2/CO.
4.3.3.3. Análise para a conversão em hidrocarbonetos
A conversão total em hidrocarbonetos avalia o total de moléculas de CO que se
transformaram em hidrocarbonetos, sejam parafínicos ou olefínicos, independente do tamanho
da cadeia formada. Assim, fatores de influência considerável como a pressão, extremamente
relevantes para cada fração analisada separadamente, se tornam de importância secundária
para a variável em estudo (conversão total), em relação à velocidade de escoamento dos gases
reagentes e até mesmo à proporção H2/CO nos gases de entrada. Logo, resumidamente, tem-
se como mais relevantes os seguintes fatores:
velocidade de escoamento
proporção H2/CO
Fitted Surface; Variable: Var5
3 factors, 1 Blocks, 15 Runs; MS Residual=26,58351
DV: Var5
> 50 < 50 < 40 < 30 < 20 < 10
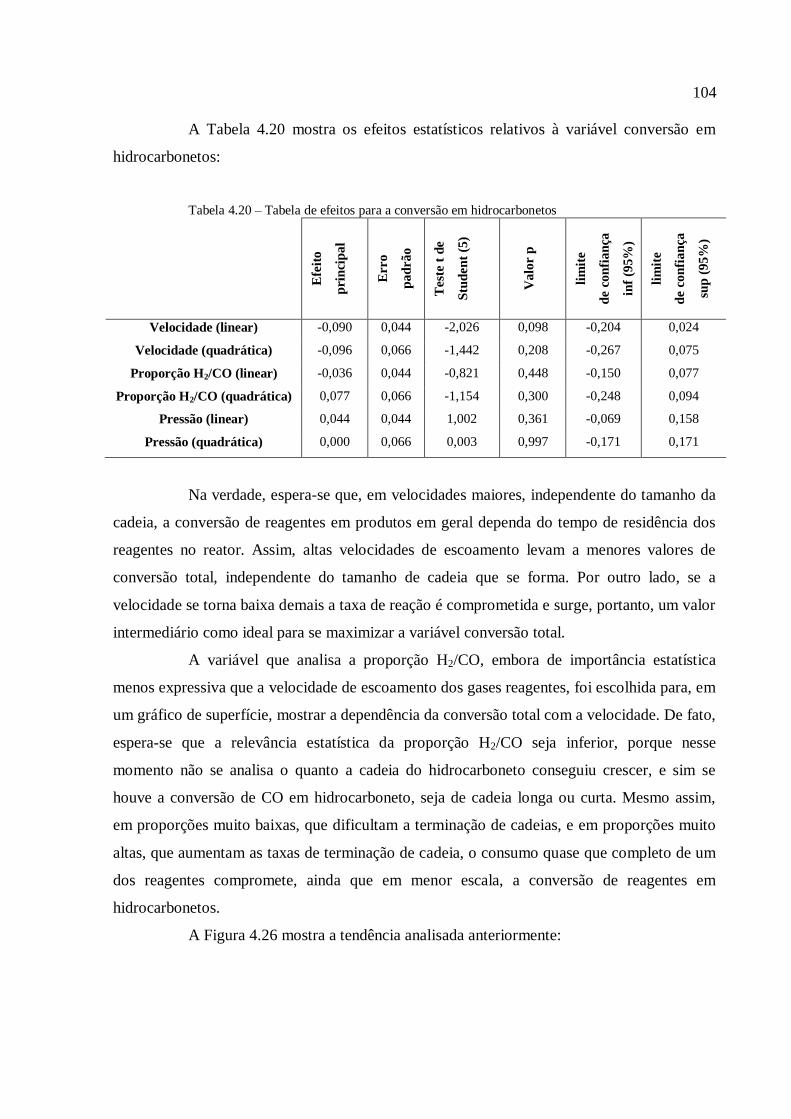
104
A Tabela 4.20 mostra os efeitos estatísticos relativos à variável conversão em
hidrocarbonetos:
Tabela 4.20 – Tabela de efeitos para a conversão em hidrocarbonetos
Efe
ito
prin
cip
al
Erro
pad
rão
Test
e t
de
Stu
den
t (5
)
Valo
r p
lim
ite
de c
on
fian
ça
inf
(95%
)
lim
ite
de c
on
fian
ça
sup
(95%
)
Velocidade (linear) -0,090 0,044 -2,026 0,098 -0,204 0,024
Velocidade (quadrática) -0,096 0,066 -1,442 0,208 -0,267 0,075
Proporção H2/CO (linear) -0,036 0,044 -0,821 0,448 -0,150 0,077
Proporção H2/CO (quadrática) 0,077 0,066 -1,154 0,300 -0,248 0,094
Pressão (linear) 0,044 0,044 1,002 0,361 -0,069 0,158
Pressão (quadrática) 0,000 0,066 0,003 0,997 -0,171 0,171
Na verdade, espera-se que, em velocidades maiores, independente do tamanho da
cadeia, a conversão de reagentes em produtos em geral dependa do tempo de residência dos
reagentes no reator. Assim, altas velocidades de escoamento levam a menores valores de
conversão total, independente do tamanho de cadeia que se forma. Por outro lado, se a
velocidade se torna baixa demais a taxa de reação é comprometida e surge, portanto, um valor
intermediário como ideal para se maximizar a variável conversão total.
A variável que analisa a proporção H2/CO, embora de importância estatística
menos expressiva que a velocidade de escoamento dos gases reagentes, foi escolhida para, em
um gráfico de superfície, mostrar a dependência da conversão total com a velocidade. De fato,
espera-se que a relevância estatística da proporção H2/CO seja inferior, porque nesse
momento não se analisa o quanto a cadeia do hidrocarboneto conseguiu crescer, e sim se
houve a conversão de CO em hidrocarboneto, seja de cadeia longa ou curta. Mesmo assim,
em proporções muito baixas, que dificultam a terminação de cadeias, e em proporções muito
altas, que aumentam as taxas de terminação de cadeia, o consumo quase que completo de um
dos reagentes compromete, ainda que em menor escala, a conversão de reagentes em
hidrocarbonetos.
A Figura 4.26 mostra a tendência analisada anteriormente:
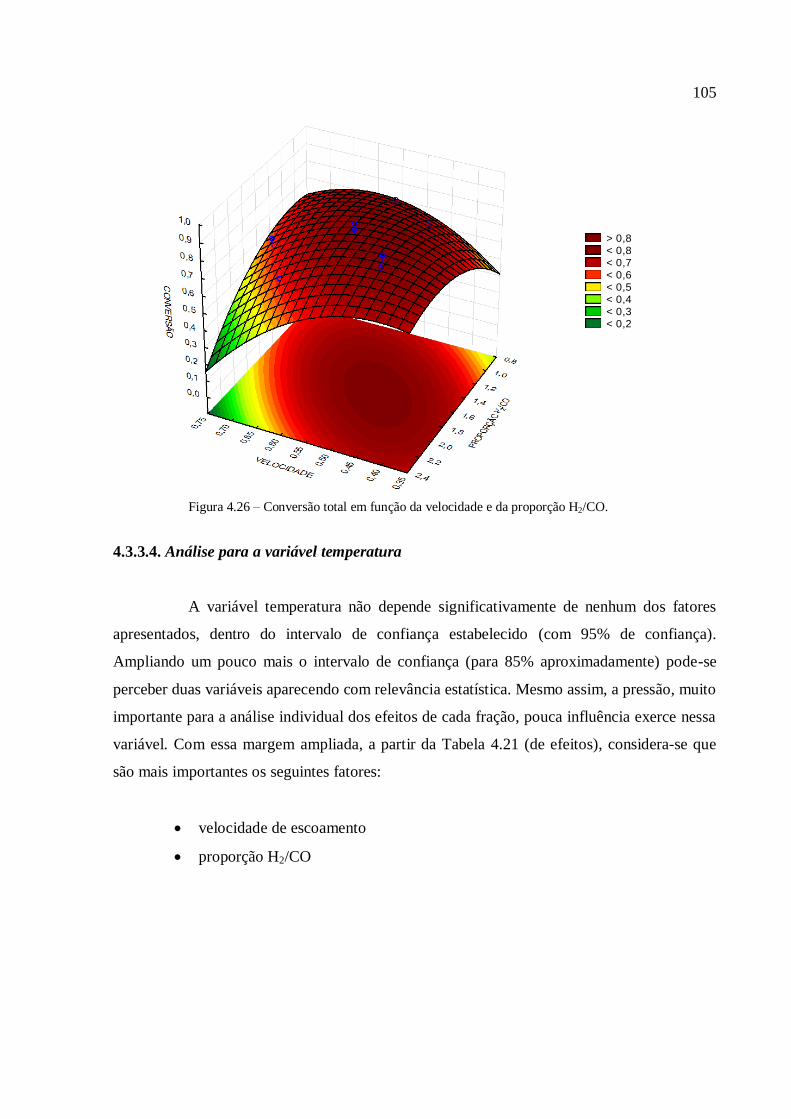
105
Figura 4.26 – Conversão total em função da velocidade e da proporção H2/CO.
4.3.3.4. Análise para a variável temperatura
A variável temperatura não depende significativamente de nenhum dos fatores
apresentados, dentro do intervalo de confiança estabelecido (com 95% de confiança).
Ampliando um pouco mais o intervalo de confiança (para 85% aproximadamente) pode-se
perceber duas variáveis aparecendo com relevância estatística. Mesmo assim, a pressão, muito
importante para a análise individual dos efeitos de cada fração, pouca influência exerce nessa
variável. Com essa margem ampliada, a partir da Tabela 4.21 (de efeitos), considera-se que
são mais importantes os seguintes fatores:
velocidade de escoamento
proporção H2/CO
Fitted Surface; Variable: Var6
3 factors, 1 Blocks, 15 Runs; MS Residual=,0067436
DV: Var6
> 0,8 < 0,8 < 0,7 < 0,6 < 0,5 < 0,4 < 0,3 < 0,2
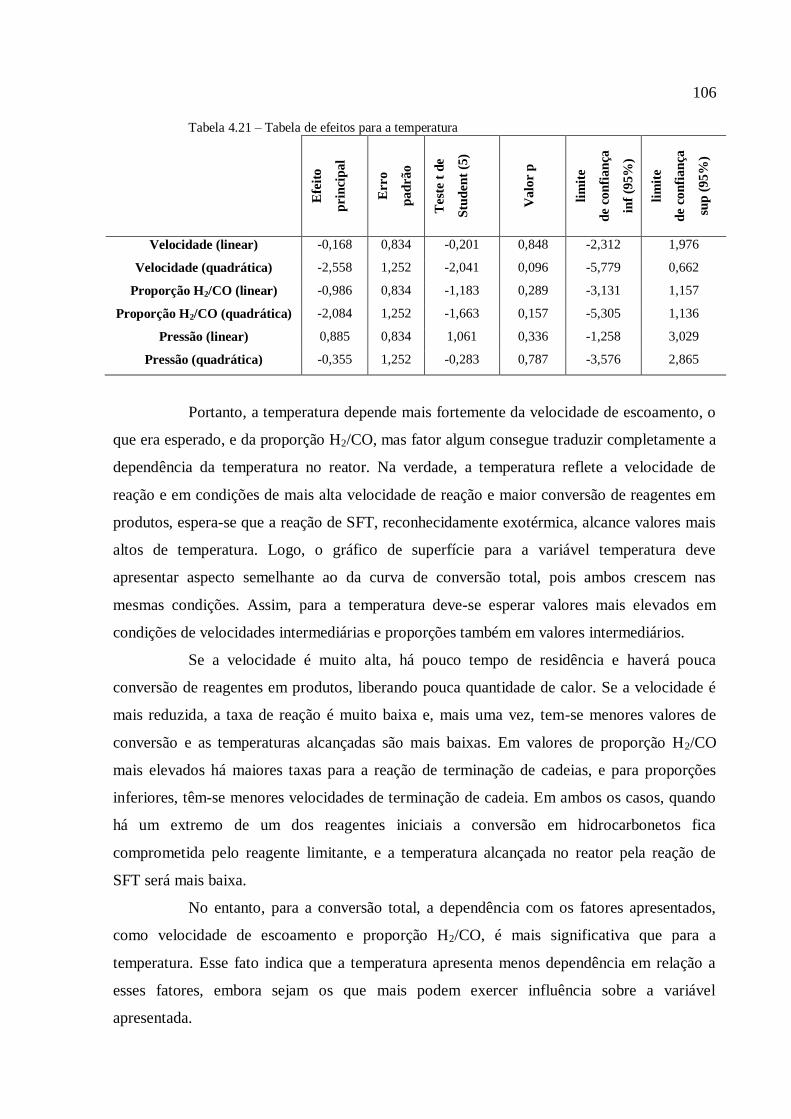
106
Tabela 4.21 – Tabela de efeitos para a temperatura
Efe
ito
prin
cip
al
Erro
pad
rão
Test
e t
de
Stu
den
t (5
)
Valo
r p
lim
ite
de c
on
fian
ça
inf
(95%
)
lim
ite
de c
on
fian
ça
sup
(95%
)
Velocidade (linear) -0,168 0,834 -0,201 0,848 -2,312 1,976
Velocidade (quadrática) -2,558 1,252 -2,041 0,096 -5,779 0,662
Proporção H2/CO (linear) -0,986 0,834 -1,183 0,289 -3,131 1,157
Proporção H2/CO (quadrática) -2,084 1,252 -1,663 0,157 -5,305 1,136
Pressão (linear) 0,885 0,834 1,061 0,336 -1,258 3,029
Pressão (quadrática) -0,355 1,252 -0,283 0,787 -3,576 2,865
Portanto, a temperatura depende mais fortemente da velocidade de escoamento, o
que era esperado, e da proporção H2/CO, mas fator algum consegue traduzir completamente a
dependência da temperatura no reator. Na verdade, a temperatura reflete a velocidade de
reação e em condições de mais alta velocidade de reação e maior conversão de reagentes em
produtos, espera-se que a reação de SFT, reconhecidamente exotérmica, alcance valores mais
altos de temperatura. Logo, o gráfico de superfície para a variável temperatura deve
apresentar aspecto semelhante ao da curva de conversão total, pois ambos crescem nas
mesmas condições. Assim, para a temperatura deve-se esperar valores mais elevados em
condições de velocidades intermediárias e proporções também em valores intermediários.
Se a velocidade é muito alta, há pouco tempo de residência e haverá pouca
conversão de reagentes em produtos, liberando pouca quantidade de calor. Se a velocidade é
mais reduzida, a taxa de reação é muito baixa e, mais uma vez, tem-se menores valores de
conversão e as temperaturas alcançadas são mais baixas. Em valores de proporção H2/CO
mais elevados há maiores taxas para a reação de terminação de cadeias, e para proporções
inferiores, têm-se menores velocidades de terminação de cadeia. Em ambos os casos, quando
há um extremo de um dos reagentes iniciais a conversão em hidrocarbonetos fica
comprometida pelo reagente limitante, e a temperatura alcançada no reator pela reação de
SFT será mais baixa.
No entanto, para a conversão total, a dependência com os fatores apresentados,
como velocidade de escoamento e proporção H2/CO, é mais significativa que para a
temperatura. Esse fato indica que a temperatura apresenta menos dependência em relação a
esses fatores, embora sejam os que mais podem exercer influência sobre a variável
apresentada.
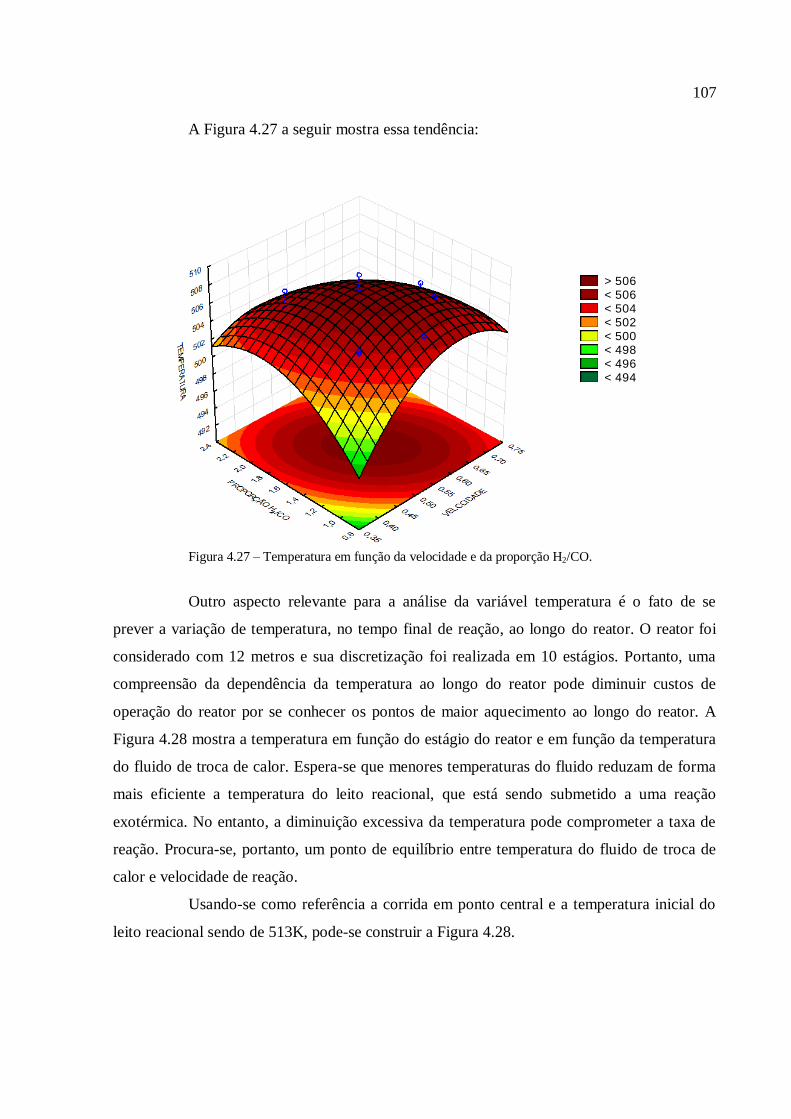
107
A Figura 4.27 a seguir mostra essa tendência:
Figura 4.27 – Temperatura em função da velocidade e da proporção H2/CO.
Outro aspecto relevante para a análise da variável temperatura é o fato de se
prever a variação de temperatura, no tempo final de reação, ao longo do reator. O reator foi
considerado com 12 metros e sua discretização foi realizada em 10 estágios. Portanto, uma
compreensão da dependência da temperatura ao longo do reator pode diminuir custos de
operação do reator por se conhecer os pontos de maior aquecimento ao longo do reator. A
Figura 4.28 mostra a temperatura em função do estágio do reator e em função da temperatura
do fluido de troca de calor. Espera-se que menores temperaturas do fluido reduzam de forma
mais eficiente a temperatura do leito reacional, que está sendo submetido a uma reação
exotérmica. No entanto, a diminuição excessiva da temperatura pode comprometer a taxa de
reação. Procura-se, portanto, um ponto de equilíbrio entre temperatura do fluido de troca de
calor e velocidade de reação.
Usando-se como referência a corrida em ponto central e a temperatura inicial do
leito reacional sendo de 513K, pode-se construir a Figura 4.28.
Fitted Surface; Variable: Var7
3 factors, 1 Blocks, 15 Runs; MS Residual=2,375538
DV: Var7
> 506 < 506 < 504 < 502 < 500 < 498 < 496 < 494
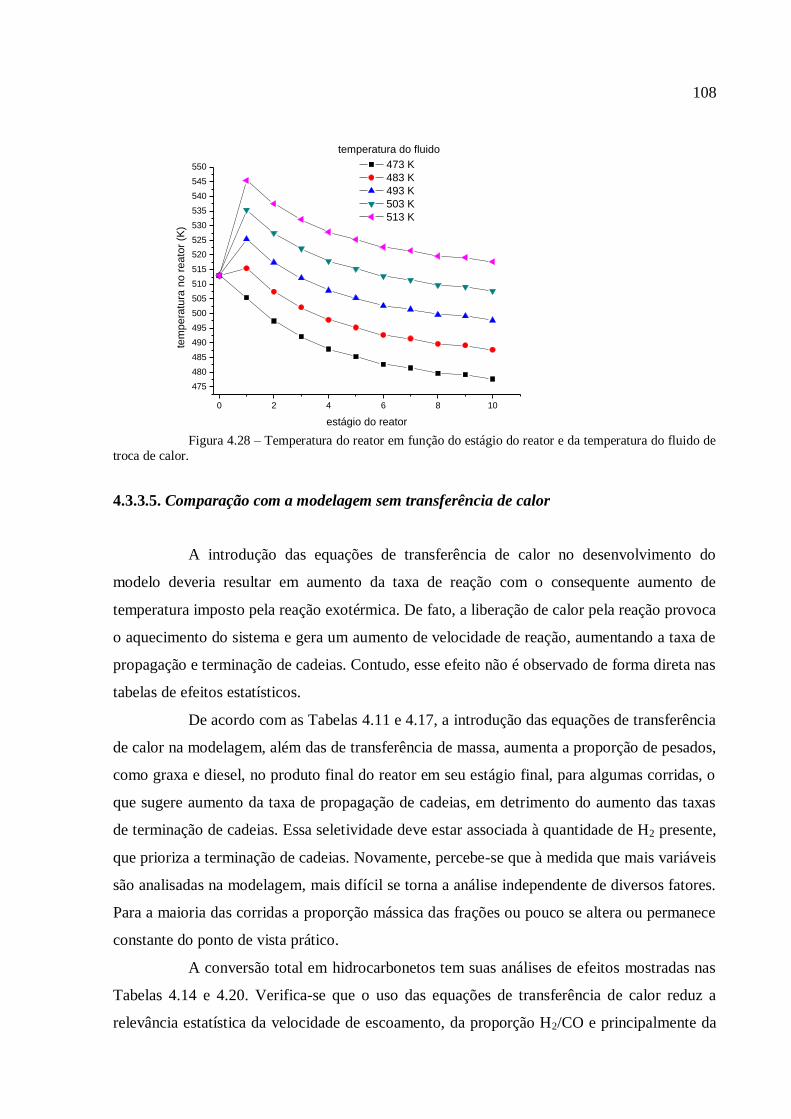
108
Figura 4.28 – Temperatura do reator em função do estágio do reator e da temperatura do fluido de
troca de calor.
4.3.3.5. Comparação com a modelagem sem transferência de calor
A introdução das equações de transferência de calor no desenvolvimento do
modelo deveria resultar em aumento da taxa de reação com o consequente aumento de
temperatura imposto pela reação exotérmica. De fato, a liberação de calor pela reação provoca
o aquecimento do sistema e gera um aumento de velocidade de reação, aumentando a taxa de
propagação e terminação de cadeias. Contudo, esse efeito não é observado de forma direta nas
tabelas de efeitos estatísticos.
De acordo com as Tabelas 4.11 e 4.17, a introdução das equações de transferência
de calor na modelagem, além das de transferência de massa, aumenta a proporção de pesados,
como graxa e diesel, no produto final do reator em seu estágio final, para algumas corridas, o
que sugere aumento da taxa de propagação de cadeias, em detrimento do aumento das taxas
de terminação de cadeias. Essa seletividade deve estar associada à quantidade de H2 presente,
que prioriza a terminação de cadeias. Novamente, percebe-se que à medida que mais variáveis
são analisadas na modelagem, mais difícil se torna a análise independente de diversos fatores.
Para a maioria das corridas a proporção mássica das frações ou pouco se altera ou permanece
constante do ponto de vista prático.
A conversão total em hidrocarbonetos tem suas análises de efeitos mostradas nas
Tabelas 4.14 e 4.20. Verifica-se que o uso das equações de transferência de calor reduz a
relevância estatística da velocidade de escoamento, da proporção H2/CO e principalmente da
0 2 4 6 8 10
475
480
485
490
495
500
505
510
515
520
525
530
535
540
545
550 473 K
483 K
493 K
503 K
513 K
tem
pe
ratu
ra n
o r
ea
tor
(K)
estágio do reator
temperatura do fluido

109
pressão. Percebe-se que a introdução de novas equações torna o modelo mais dependente da
velocidade e da proporção, embora em menor escala que antes. Essa situação reflete o
comportamento mais realista da modelagem do reator, onde a conversão é muito mais
dependente do tempo de residência dos reagentes e de suas concentrações em sítios
catalíticos, a partir de certos valores para a pressão exercida.
Em relação às frações de maior interesse comercial, como gasolina, diesel e
graxas, analisadas separadamente, a utilização das equações de transferência de calor na
modelagem aumenta a dependência estatística da pressão para o resultado final obtido, uma
vez que as outras variáveis analisadas (velocidade e proporção H2/CO) perdem relevância na
análise de efeitos. Esse resultado mostra que, ao se analisar separadamente cada fração de
produto obtida, a dependência da pressão apresenta a tendência a ser aquela que mais exercerá
influência na produção da fração em estudo. Esta conclusão pode ser percebida a partir da
análise das Tabelas 4.12, 4.13, 4.18 e 4.19.
A proporção H2/CO foi a variável que mais perdeu importância estatística com a
consideração dos efeitos de transferência de calor. A velocidade de escoamento dos gases
reagentes já não apresentava alta dependência sem a transferência de calor. Uma vez que o
aumento da proporção H2/CO aumenta a taxa de terminação das cadeias carbônicas, que
justifica o fato de que, sob o ponto de vista estatístico, exerça uma menor influência com a
utilização da transferência de calor nas equações da modelagem. A própria transferência de
calor, que ocasiona ganho na taxa de reação e proporciona maiores taxas de crescimento de
cadeias carbônicas, já aumenta a fração em produtos mais pesados, o que reduz a importância
da proporção H2/CO, fazendo com a mesma não se mantenha no intervalo de confiança
adotado (95%).

110
5. CONCLUSÃO
Neste trabalho, o objetivo principal foi a realização de uma modelagem
matemática, com posterior estimação de parâmetros e simulação para a reação de SFT em
reator tubular de leito fixo utilizando catalisador de cobalto. O procedimento inovador na
simulação consistiu em tratar o reator de forma dinâmica, uma vez que a simulação procurou
calcular a concentração dos componentes da reação, sejam produtos ou reagentes, ao longo do
reator e em função do tempo de operação do reator.
A modelagem cinética para a reação de SFT foi realizada para ser a mais geral
possível, podendo ser utilizada tanto para catalisadores à base de cobalto, quanto para
catalisadores à base de ferro. O modelo mostrou-se muito eficaz na representação de
catalisadores à base de cobalto.
A estimativa de parâmetros foi obtida para a cinética de reação prevista na
modelagem matemática, ou seja, pode ser utilizada tanto para reações com catalisadores à
base de cobalto, quanto para outros à base de ferro. Novamente, a representação para
catalisadores à base de cobalto mostrou-se muito boa.
Na busca por uma modelagem mais realista foram utilizadas equações que
avaliassem a transferência de massa no reator. Houve uma mudança significativa em relação
ao resultado obtido sem o uso dessas equações. As simulações realizadas mostraram que é
essencial se considerar a transferência de massa entre as fases gás e líquido, e que a
consideração de uma fase pseudo-homogênea não pode ser aplicada para reatores utilizados
para SFT.
Percebeu-se uma redução nas taxas de reação e uma maior formação de leves.
Nessa situação o estudo por simulação se torna mais importante visando a maximização dos
produtos de maior relevância econômica, como gasolina, diesel e graxas.
A utilização de equações de transferência de calor pouco modificou o resultado
obtido sem sua utilização. Concluiu-se que a transferência de calor, em associação a um
eficiente fluido para troca de calor, pouco influencia no estudo para a modelagem da reação e
do reator.
Utilizando os resultados obtidos para as simulações mediante planejamento
experimental estabelecido, pode-se sintetizar condições de operação do reator tubular de leito
fixo com catalisador de cobalto, para maior produção das frações economicamente mais
rentáveis:

110
Para maior rendimento percentual em massa de gasolina, o reator deve operar em
condições de proporções H2/CO moderadas (entre 1,5:1 e 2:1), relativamente altas
velocidades de escoamento (superiores a 0,55 m.s-1
), e para compensar os menores
tempos de residência, alta pressão gasosa total à entrada do reator (próximo a 40 atm),
que aumenta a concentração dos reagentes e facilita a colisão entre as espécies
reagentes responsáveis pelo crescimento da cadeia.
Para maior rendimento percentual em massa de diesel deve-se operar o reator de leito
fixo em condições de pressão elevada (próximo a 40 atm), com velocidade de
escoamento dos gases reagentes não muito reduzidas (abaixo de 0,55 m.s-1
), para
aumentar o tempo de residência e favorecer o crescimento da cadeia, com a proporção
H2/CO suficiente para manter a reação sustentável.
Para maior rendimento percentual em graxas o reator deve operar em condições
semelhantes às indicadas para a busca por maiores rendimentos em diesel, ou seja,
condições de pressão elevada (próximo a 40 atm), com velocidade de escoamento dos
gases reagentes abaixo de 0,55 m.s-1
, para aumentar o tempo de residência e favorecer
o crescimento da cadeia, e com a proporção H2/CO suficiente para manter a reação
sustentável.
111

112
6. REFERÊNCIAS
ADESINA, A.A. Hydrocarbon Synthesis via Fischer-Tropsch Reaction: Travails and
Triumphs. Applied Catalysis A: General, v.138, p.345–367, 1996.
ANDERSON, R.B. Catalysts for the Fischer-Tropsch synthesis. New York: Van
Nostrand Reinhold, 1956, v. 4.
ANDERSON, R.B. The Fischer–Tropsch Synthesis, 1. ed., New York: Academic Press,
1984.
BARROS JUNIOR, A. F.; FERNANDES, F. A. N. Comprehensive Polymerization Model
for Fischer-Tropsch Synthesis. Chemical Engineering Technology, v. 34, p. 963–971,
2011.
BARROS JUNIOR, A. F. Modelagem e Simulação da Síntese de Fischer-Tropsch em
Reator Tubular de Leito Fixo com Catalisador de Ferro. Dissertação (Mestrado em
Engenharia Química) – Centro de Tecnologia, Universidade Federal do Ceará, Fortaleza,
2008.
BRADY R.C.; PETIT, R. Reactions Of Diazomethane On Transition-Metal Surfaces And
Their Relationship To The Mechanism Of The Fischer-Tropsch Reaction. Journal Of
The American Chemical Society, v. 102, p. 6181–6182, 1980.
BRODÉN, G.; RHODIN, T.N.; BRUCKER, C. Synchrotron Radiation Study Of
Chemisorptive Bonding Of Co On Transition-Metals - Polarization Effect On Ir (100).
Surface Science, v. 59, p. 593–611, 1976.
CLAEYS, M.; SCHULZ H.; HARMS, S., Effect of Water Partial Pressure on Steady State
Fischer-Tropsch Activity and Selectivity of a Promoted Cobalt Catalyst. Studies in
Surface Science and Catalysis, v. 107, p. 193–200, 1997.

113
DAVIS, B.H. Overview of reactors for liquid phase Fischer-Tropsch synthesis. Catalysis
Today, v. 71, p. 249–300, 2002.
DAVIS, B.H. Fischer-Tropsch Synthesis: Relationship between Iron Catalyst
Composition and Process Variables. Catalysis Today, v. 84, p. 83–98, 2003.
DRY, M.E. The Fischer-Tropsch process: 1950–2000. Catalysis Today, p. 227–241,
2002.
EISENBERG, B.; FIATO, R. A; MAULDIN, C. H. Exxon's advanced gas-to-liquids
technology. Studies in Surface Science And Catalysis, v. 119, p. 943–948, 1998.
FAAJI, A.P.C. Bio-energy in Europe: Changing Technology Choices. Energy Policy, p.
1–2, 2004.
FARIAS, F.E.M. Produção de Combustíveis Líquidos através da Síntese de Fischer-
Tropsch. Dissertação (Mestrado em Engenharia Química) – Centro de Tecnologia,
Universidade Federal do Ceará, Fortaleza, 2007.
FERNANDES, F.A.N. Polymerization Kinetics of Fischer-Tropsch Reaction on Iron
Based Catalysts and Product Grade Optimization. Chem. Eng. Tecnol., v. 28, p. 1–9,
2005.
FERNANDES, F.A.N. Modeling and Product Grade Optimization of Fischer-Tropsch
Synthesis in a Slurry Reactor. Ind. Eng. Chem. Res., v. 45, p.1047–1057, 2006a.
FERNANDES, F.A.N.; TELES, U.M. Modeling and Optimization of Fischer–Tropsch
Products Hydrocracking. Fuel Processing Technology, v. 88, p. 207–214, 2007.
FISCHER, F.; MEYER, K. Brennstoff-Chem., v. 12, p. 225, 1931.
FISCHER, F.; KOCH, H. Brennstoff-Chem., v. 13, p. 61, 1932.

114
FUJIWARA, M. M. Simulação de Modelos Bidimensionais de Reatores Catalíticos de
Leito Fixo por Elementos Finitos. Dissertação (Mestrado em Engenharia Química) –
Faculdade de Engenharia de Campinas, Universidade Estadual de Campinas, Campinas,
1989.
GUETTEL, R.; TUREK, T. Comparison of Different Reactor Types for Low-
Temperature Fischer-Tropsch Synthesis: a Simulation Study. Chemical Engineering
Science, v. 64, p. 955–964, 2009.
HENRICI-OLIVE G.; OLIVE S. Fischer–Tropsch Synthesis: Molecular–Weight
Distribution of Primary Products and Reaction Mechanism. Angewandte Chemie –
International Edition (in English), v. 15, p. 136–141, 1976.
HOFFMANN, R.; SUNG, S. How Carbon-Monoxide Bonds To Metal-Surfaces. Journal
of the American Chemical Society, v. 107, p. 578–584, 1985.
HOOGWIJK, M.; FAAJI, A.; VAN DEN BROEK, R.; BERNDES, G.; GIELEN, D.;
TURKENBURG, W. Exploration of the Ranges of the Global Potential of Biomass for
Energy. Biomass and Bioenergy, v. 25 (2), p. 119–133, 2003.
JESS, A.; KERN, C. Modeling of Multi-Tubular for Fischer-Tropsch Synthesis.
Chemical Engineering Technology, v. 32, p. 1164-1175, 2009.
KUMMER J.T.; EMMETT P.H. Fischer-Tropsch Synthesis Mechanism Studies - The
Addition Of Radioactive Alcohols To The Synthesis Gas. Journal of The American
Chemical Society, v. 75 (21): p. 5177–5183, 1953.
MAITLIS, P.M. A New View of the Fischer-Tropsch Polymerization Reaction. Pure
and Applied Chemistry, v. 61 (10), p. 1747–1754, 1989.
MOUTSOGLOU, A.; SUNKARA, P. P. Fischer-Tropsch Synthesis in a Fixed Bed
Reactor. Energy and Fuels, v. 25, p. 2242–2257, 2011.

115
OVERETT, J.M.; HILL, R.O.; MOSS, J.R. Organometallic Chemistry and Surface
Science: Mechanistic Models for the Fischer-Tropsch Synthesis. Coordination
Chemistry Reviews, p. 206–207 e 581–605, 2000.
PICHLER, H.; ROELEN, O.; SCHNUR, F. Kohlenoxidhydrierung. In: Ullmanns
Enzyklopadie der technischen Chemie, 3. ed., Berlin: Urban a. Schwarzenbrg Munchen,
v. 9, p. 685, 1957.
RAMACHANDRAN, P.A., CHAUDHARI, R.V. Three-Phase Catalytic Reactors, New
York: Gordon and Breach Science Publisher, 1983.
SAPAG K.; ROJAS S.; GRANADOS M.L.; FIERRO, J. L.G.; MENDIOROZ, S. CO
Hydrogenation with Co Catalyst Supported on Porous Media. Journal of Molecular
catalysis A: Chemical, v. 167 (1–2), p 81–89, 2001.
SCHULZ, H.; CRONJÉ, J.H. Fischer-Tropsch Synthesis, in: Ullmanns Enzyklopadie
der technischen Chemie. 4. ed., Weinheim: Verlag Chemie, v. 14, p. 329, 1977.
SCHULZ, H. Short History and Present Trends of Fischer-Tropsch synthesis. Applied
Catalysis A: General, v. 186, p. 3–12, 1999.
SCHULZ, H.; CLAEYS, M. Kinetic Modeling of Fischer-Tropsch Product Distributions.
Applied catalysis A: General, v. 186, p. 91–107, 1999.
SIE, S.T.; SENDEN, M.M.G.; VAN WECKEN, M.M.H. Conversion of Natural Gas to
Transportation Fuels via the Shell Middle Distillate Synthesis Process (SMDS). Catalysis
Today, v. 8, p. 371–394, 1991.
SPATH, P.L.; DAYTON, D.C. Preliminary Screening – Technical and Economic
Assessment of Synthesis Gas to Fuels and Chemicals with Emphasis on the Potential
for Biomass-Derived Syngas. National Renewable Energy Laboratory, p. 107–160, 2003.

116
STRANGES, A. N. The US bureau of mine’s synthetic fuel program, 1920–1950: German
connections and American Advances. Annals of Science, v. 54, p. 29–68, 1997.
VAN DER LAAN, G.P. Kinetics, Selectivity and Scale Up of the Fischer-Tropsch
Synthesis. University of Groningen (Thesis), 1999.
VAN DER LAAN, G.P.; BEENACKERS, A.A.C.M. Kinetics and Selectivity of the
Fischer-Tropsch Synthesis: a Literature Review. Catalysis Reviews-Science And
Engineering, v. 41, p. 255–318, 1999.
VANNICE, M.A. Catalytic Synthesis of Hydrocarbons from H2–CO Mixtures over
Group-8 Metals .1. Specific Activities and Product Distributions of Supported Metals.
Journal Catalysis, v. 37, p. 449–461, 1975.
VISCONTI, C. G.; TRONCONI, E.; LIETTI, L.; ZENNARO, R.; FORZATTI, P.
Development of a Complete Kinect Model for the Fischer-Tropsch Synthesis over
Co/Al2O3 Catalysts. Chemical Engineering Science, v. 62, p. 5338–5343, 2007.
VOSLOO A.C. Fischer–Tropsch: a Futuristic View. Fuel Processing Technology, v. 71,
p. 149–155, 2001.
WANG, Y.N.; XU, Y.Y.; LI, Y. W.; ZHAO, Y. L.; ZHANG, B.J. Heterogeneous
Modeling for Fixed-Bed Fischer–Tropsch Synthesis: Reactor Model and its Applications.
Chemical Engineering Science, v. 58, p. 867–875, 2003.
WU, J.; ZHANG, H.; YING, W.; FANG, D. Simulation and Analysis of a Fixed-Bed
Fischer-Tropsch Synthesis Reactor with Co-Based Catalyst. Chemical Engineering
Science, v. 33, p. 1083–1092, 2010.
ZENNARO, R.; TAGLIABUE, M.; BARTHOLOMEW, C. H. Kinetics of Fischer-
Tropsch Synthesis on titania-supported cobalt. Catalysis Today, v. 58, p. 309-319, 2000.





























