Embed Size (px)
Citation preview

UNIVERSITÄTSKLINIKUM HAMBURG-EPPENDORF
Zentrum für Experimentelle Medizin
Institut für Klinische Pharmakologie
Prof. Dr. Rainer H. Böger
Einfluss von Einzelnukleotid-Polymorphismen (SNPs) in der
Dimethylarginin-Dimethylaminohydrolase 1 und 2 bei Präeklampsie
Dissertation
zur Erlangung des Grades eines Doktors der Medizin
an der Medizinischen Fakultät der Universität Hamburg.
vorgelegt von:
Pia Anne Schmidt-Ropertz
aus Marburg
Hamburg 2014

Angenommen von der
Medizinischen Fakultät der Universität Hamburg am: 29.07.2015
Veröffentlicht mit Genehmigung der
Medizinischen Fakultät der Universität Hamburg.
Prüfungsausschuss, der/die Vorsitzende:
Prof. Dr. med. Rainer H. Böger
Prüfungsausschuss, zweite/r Gutachter/in:
Univ.-Prof. Dr. med. Kurt Hecher
Prüfungsausschuss, dritte/r Gutachter/in:
Prof. Dr. rer. nat. Hans-Jürgen Kreienkamp

Für meine Eltern und meine Großmutter Anneliese Ropertz

4
Inhalt 1. Hypothesen und Fragstellungen ......................................................................... 6
2. Einleitung ............................................................................................................ 7
2.1. Definition/ Diagnosekriterien Präeklampsie .................................................... 8
2.2. Risikofaktoren .............................................................................................. 12
2.3. Differentialdiagnosen .................................................................................... 13
2.4. Die Pathophysiologie der Präeklampsie ....................................................... 14
2.4.1. Theorie der anormalen Umformungen der Spiralarterien der
Plazenta ............................................................................................................. 14
2.4.2. NO / ADMA / DDAH ............................................................................... 16
2.5. Therapie ....................................................................................................... 23
3. Methoden .......................................................................................................... 27
3.1. Eingeschlossene Patientinnen und Probandinnen ....................................... 27
3.1.1. Gruppe A - Gesunde Schwangere ......................................................... 27
3.1.2. Gruppe B - Patientinnen mit Präeklampsie ............................................ 27
3.2. Klinische Daten ............................................................................................ 28
3.3. Bestimmung von ADMA-, SDMA- und L-Arginin im Plasma ......................... 29
3.4. Genotypisierung ........................................................................................... 31
3.5. DDAH-Genexpressionsanalyse .................................................................... 34
3.6. Statistische Auswertung ............................................................................... 37
4. Ergebnisse ........................................................................................................ 38
4.1. Basischarakteristika ..................................................................................... 38
4.2. Laborwerte ................................................................................................... 43
4.3. Dimetyhlarginine .......................................................................................... 44
4.4. Korrelationen ................................................................................................ 47
4.4.1. Fetale ADMA/SDMA-Konzentrationen und Fetomaternaler Gradient .... 49
4.4.2. Arteriovenöse Differenz ......................................................................... 50
4.5. SNPs ............................................................................................................ 51
4.6. Genexpression ............................................................................................. 54
5. Diskussion ........................................................................................................ 55
5.1. Dimethylarginine .......................................................................................... 56

5
5.1.1. Fetale Dimethylarginine, fetomaternaler Gradient und arteriovenöse
Differenz ............................................................................................................. 61
5.2. SNPs ............................................................................................................ 62
5.3. Genexpressionsanalysen ............................................................................. 65
5.4. Klinische Relevanz der Ergebnisse und Pharmakologische Ziele ................ 67
6. Zusammenfassung ........................................................................................... 68
Anhang ...................................................................................................................... 70
7. Literaturverzeichnis ........................................................................................... 70
8. Abkürzungsverzeichnis ..................................................................................... 88
9. Abbildungsverzeichnis ...................................................................................... 91
10. Tabellenverzeichnis ........................................................................................... 92
11. Materialien ........................................................................................................ 93
11.1. Chemikalien und Reagenzien ................................................................... 93
12. Studienorganisation .......................................................................................... 96
12.1. Einverständnis Studienteilnahme .............................................................. 96
12.2. Einverständnis Gewebe- und Nabelschnurblutentnahme ......................... 97
12.3. Einverständniserklärung Genetische Untersuchung ................................. 98
12.4. Patientinnen-Information -1- ...................................................................... 99
12.5. Patientinnen-Information -2- .................................................................... 100
12.6. Patientinnenerfassungsbogen ................................................................. 101
13. Danksagung .................................................................................................... 103
14. Lebenslauf .......................................................... Error! Bookmark not defined.
15. Eidesstattliche Versicherung ........................................................................... 105

6
1. Hypothesen und Fragstellungen
Die Präeklampsie ist eine häufige Erkrankung in der Schwangerschaft und
erhöht die perinatale Mortalität und Morbidität. Die Ursachen dieser
Erkrankungen sind bislang nicht geklärt, so dass ein kardiovaskulärer
Risikofaktor, das Asymmetrische Dimethylarginin unter anderem zum
Gegenstand der Forschung wurde. Ausgehend von den Befunden
vorausgegangener Studien, vermuten wir, dass erhöhte Konzentrationen von
Asymmetrischem Dimethylarginin bei Patientinnen mit Präeklampsie vorliegen
könnten. In diesem Zusammenhang sind Polymorphismen der abbauenden
Enzyme DDAH 1 und 2 in der Plazenta besonders interessant im Rahmen der
Fragestellung genetischer Aspekten dieser Erkrankung. Sodass folgenden
Fragen in der vorliegenden Arbeit beantwortet werden sollten:
Besteht ein Zusammenhang zwischen Polymorphismen in den DDAH-
Genen und mütterlichen ADMA-Konzentrationen.
Bestehen Unterschiede in der Häufigkeit der SNPs zwischen gesunden
Schwangeren und Präeklampsie-Patientinnen.

7
2. Einleitung
Die Präeklampsie gehört zu den hypertensiven Schwangerschaftserkrankungen
und tritt bei zwei bis acht Prozent aller Schwangerschaften auf (Duley 2009).
Neben thromboembolischen Ereignissen sind sie eine der häufigsten
maternalen Todesursachen weltweit und machen 10-15% der maternalen
Todesfälle aus. Je nach Ausprägungsgrad sind sie mit frühzeitiger Geburt oder
intrauterinen Wachstumsstörungen (IUGR) verbunden und somit perinataler
Morbidität und Mortalität eng verknüpft (Khan 2005; Duley 2009; Steegers
2010).
Ein großes Forschungsgebiet beschäftigt sich mit den pathophysiologischen
Hintergründen, um die Entwicklung einer Präeklampsie vorhersagen und
frühzeitig behandeln zu können. Bisher sind kaum ausreichend valide
Screenings zur Risikobestimmung für die Entwicklung einer Präeklampsie
bekannt. Biomarker oder spezifische Laborwerte, die man bisher zur
Vorhersage diskutiert hat, können nicht eindeutig die Entwicklung einer
Präeklampsie vorhersagen oder waren nicht spezifisch genug um in die
generelle Schwangerschaftsvorsorge etabliert zu werden. Überdies hinaus ist
der genetische Einfluss der Präeklampsie unumstritten (Williams 2012), jedoch
findet sich zurzeit kein definitives genetisches Korrelat in den Patientinnen, was
die Betrachtung sogenannter Einzelnukleotidpolymorphismen in dieser Hinsicht
interessant macht.
Ein neuer Risikomarker kardiovaskulärer Erkrankungen rückte in den
Vordergrund einiger Studien, ein endogener Inhibitor der NO-Synthese im
Endothel: das Asymmetrisches Dimehtylarginin (ADMA) (Vallance 1992). Bis
heute haben viele klinische und experimentelle Studien gezeigt, dass erhöhte
Serum-ADMA-Konzentrationen als Risikofaktor für einige kardiovaskuläre
Erkrankungen, wie essentielle Hypertonie, Arteriosklerose, koronare
Herzerkrankungen und Hypercholesterinämie von Bedeutung sind (Böger 1996,
Böger 1998; Surdacki A 1999; Böger 2004b; Valkonen 2005; Böger 2006). Man
geht ebenfalls von einer ADMA-Erhöhung bei Diabetes mellitus Typ 2 aus
(Abbasi F. 2001).

8
1993 berichtete Fickling et al. zum ersten Mal von einer erhöhten Serum-
ADMA-Konzentration in der Schwangerschaft bei Präeklampsiepatientinnen,
während in gesunden Schwangerschaften die Serum-ADMA-Konzentrationen
im Vergleich zu Nichtschwangeren sanken (Fickling 1993). Diese Ergebnisse
konnten in mehreren Studien reproduziert werden, die ebenfalls erhöhte ADMA-
Serumkonzentrationen bei Präeklampsie-Patientinnen fanden (Böger 2010;
Slaghekke 2006; Speer 2008; Braekke 2009; Mao 2010).
2.1. Definition/ Diagnosekriterien Präeklampsie
Die Präeklampsie gehört zu den Schwangerschaftshypertonien. Unterschieden
wird die Schwangerschaftshypertonie von der Präeklampsie, der
Propfpräeklampsie, der chronischen Hypertonie, dem HELLP-Syndrom und der
Eklampsie (Tabelle 1).
Erkrankung Ausprägung
Gestationshypertonie RR > 140/90 mmHg, nach 20. SSW bis 6 Wochen post partum
PräeklampsieGestationshypertonie + Proteinurie (>300 mg/24h-Urin) +
Ödeme mgl.
Chronsische HypertonieBereits mind. 6 Wochen vor der SS bestehende Hypertonie
oder über 6 Wochen nach der SS andauernd
PropfpräeklampsieChronischer Hypertonus/ chronische Niereninsuffizienz (o.ä.) und
Proteinurie (>300 mg/24h Urin)
HELLP-SyndromPräeklampsie+ Thrombozytopenie u./o. Leberenzymerhöhung u./o.
Hämolysezeichen
Eklampsie Tonisch- klonische Anfälle + Präeklampsie (mgl.)
Hyperthyreose
Kollagenosen
Hypertensive Entgleisung
Formen der Gestationshypertonie
Tabelle 1 Formen der Gestationshypertonie
Quelle: DGGG, AWMF-Leitlinie hypertensive Schwangerschaftserkrankung, Aug 2010
Die Präeklampsie ist definiert als eine persistente Hypertonie von >140/90
mmHg, mit einer simultanen Proteinurie von >300 mg Eiweiß im 24h-Urin, mit

9
ansonsten unauffälligem Urinstatus. Sie tritt überwiegend in bzw. nach der 20.
SSW auf, das heißt während der zweiten Schwangerschaftshälfte, und sollte
post partum innerhalb von circa sechs Wochen regredient sein (AWMF-Leitline
2013).
Die Präeklampsie wird durch verschiedene Ausprägungsgrade der klinischen
Zeichen in eine leichte und eine schwere Form eingeteilt. Blutdruckwerte von
140-159 mmHg systolisch und 90-109 mmHg diastolisch werden zu der leichten
Form, Werte systolisch >160 mmHg und diastolisch von >110 mmHg zur
schweren Form gezählt, ebenso Patientinnen mit >5 g Protein im 24h Urin
(Phyllis 2010). Diese Einteilung gilt für vor der Schwangerschaft normotensive
Patientinnen. Eine schwere Präeklampsie ist ebenso bei einer Hypertonie von
>140/90 mmHg und Proteinurie und einer der folgenden Kriterien zu
diagnostizieren (mit schwieriger Abgrenzung zum HELLP-Syndrom):
Beeinträchtigung der Nierenfunktion (Kreatinin >0,9 mg/dl, Oligurie
<500 ml/24h)
Eingeschränkter Leberfunktion
Lungenödem
Thrombozytopenie (< 100.000/μl)
Hämolyse (erhöhtes LDH/ erniedrigtes Haptoglobin)
Fetale Wachstumsrestriktion (Seck 2009; AWMF-Leitline 2013)
Je nach pathophysiologischem Hintergrund kann man die Präeklampsie auch in
eine früh-auftretende und eine spät-auftretende Präeklampsie einteilen (early-
and lateonset). Eine früh-auftretende Präeklampsie ist häufig assoziiert mit
einer schweren Präeklampsie („severe“), sowie einer gestörten Plazentation.
Eine spät-auftretende Präeklampsie, oft als milde Präeklampsie ausgeprägt, ist
mit kardiovaskulären und metabolischen Risikofaktoren vergesellschaftet
(Steegers 2010).
Die einzige nützliche Methode zur Risikoprofilerstellung bei Patientinnen mit
Prädisposition, die klinisch bedeutsam ist, ist die Doppler-Sonografie ab dem
zweiten Trimenon. Laut der DGGG-Mutterschaftsrichtlinien sind sie nur

10
empfehlenswert bei Risikopatientinnen (Deutsche Gesellschaft für Gynäkologie
und Geburtshilfe eV 2012). Mit Hilfe des Spektral-Dopplers und Colour-Doppler-
Imaging-Methode können die Gefäße dargestellt, der Blutfluss gemessen und
die Widerstandsindices der Ateriae uterina, der Arteria umbilicalis und der
Arteria cerebri mediae als Quotienten erfasst werden. Die Werte sind vom
Gestationsalter abhängig und müssen mit entsprechenden Normverteilungen
abgeglichen werden (Seck 2009). Dieser diagnostischen Maßnahme werden
bisher nur Schwangeren mit auffälligen Ultraschallbefunden (wie Risiko für
intrauterine Wachstumsretadierung, Hypertonus oder Diabetes) oder im
Rahmen einer erneuten Feindiagnostik in der 22. bis 27.
Schwangerschaftswoche, durchgeführt. Neuere Untersuchungen zeigen, dass
das dopplersonographische Screening auch in Nichtrisikokollektiven nützen
könnte. Eine bilaterale Veränderung in Form von hohem Widerstand in den
Gefäßen und veränderte Flussgeschwindigkeiten können richtungweisend sein
(Papageorghiou 2001). Vor allem die maternalen Arteriae Uterina sind dabei
von Bedeutung. Dort wird der uteroplazentare Druck gemessen, in der Regel
sinkt dieser bis zur 24. SSW. Bis zu diesem Zeitpunkt bestehen in den Aa.
Uterina hohe systolische und niedrige diastolische Flussgeschwindigkeiten, die
zu einer postsystolischen Inzisur (notch) führen. Diese Inzisur weist auf
Pulsreflexionen hin, die Ausdruck der noch unvollständigen
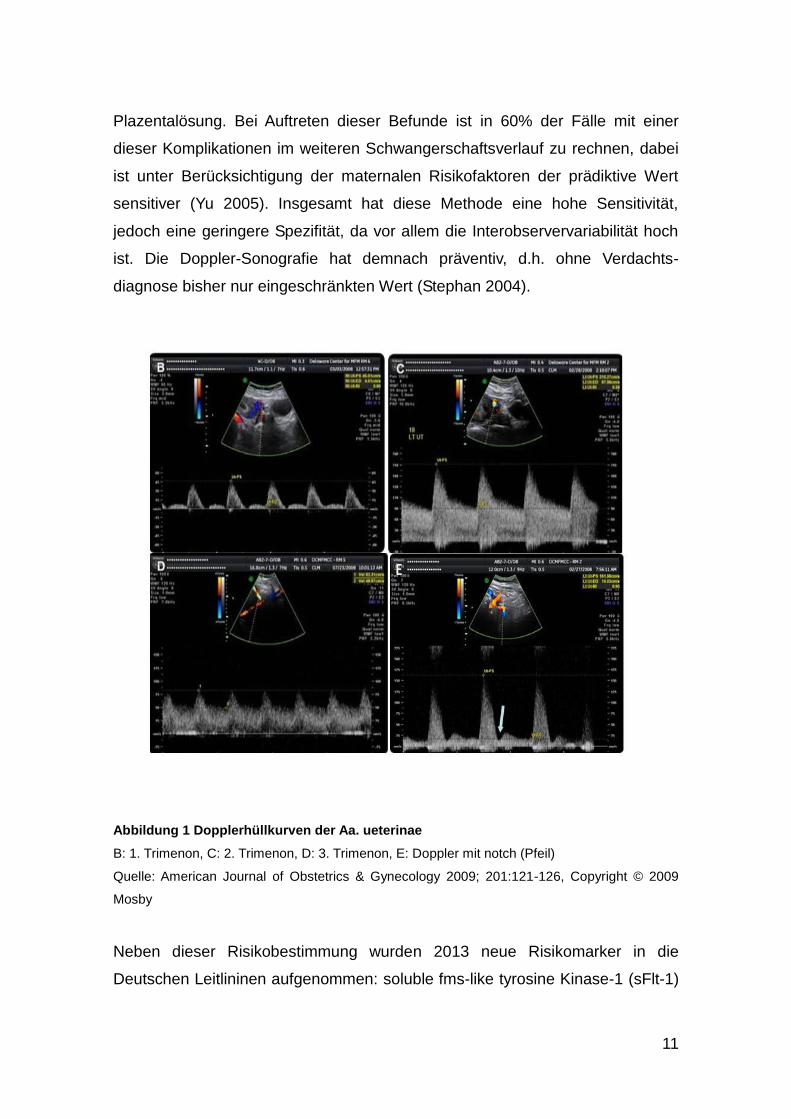
Trophoblasteninvasion sein könnten (Lin S. 1995) (Abbildung 1, B). Im weiteren
Verlauf der Schwangerschaft invadieren die Trophoblasten die maternalen
Gefäße, die diastolische Strömungsgeschwindigkeit nimmt zu und der notch
verschwindet (Abbildung 1, C) (Thaler I. 1990). Bleibt der Widerstand in den Aa.
Uterina hoch und lässt sich weiterhin eine postsystolische Inzisur nachweisen,
kann dies Zeichen einer gestörten Trophoblasteninvasion mit ungenügender
Erweiterung der Spiralarterien sein (Abbildung 1, E). Als Folge dessen kann
sich eine Minderperfusion des Uterus mit einer gestörten plazentaren
Entwicklung ausbilden (Lees 1998). Ein nachgewiesener postsystolischer notch
oder eine stark reduzierte diastolische Flussgeschwindigkeit hat einen
prädiktiven Wert hinsichtlich der Entstehung einer Schwangerschaftshypertonie,
einer intrauterinen Wachstumsretadierung (=IUGR) oder vorzeitigen
11
Plazentalösung. Bei Auftreten dieser Befunde ist in 60% der Fälle mit einer
dieser Komplikationen im weiteren Schwangerschaftsverlauf zu rechnen, dabei
ist unter Berücksichtigung der maternalen Risikofaktoren der prädiktive Wert
sensitiver (Yu 2005). Insgesamt hat diese Methode eine hohe Sensitivität,
jedoch eine geringere Spezifität, da vor allem die Interobservervariabilität hoch
ist. Die Doppler-Sonografie hat demnach präventiv, d.h. ohne Verdachts-
diagnose bisher nur eingeschränkten Wert (Stephan 2004).
Abbildung 1 Dopplerhüllkurven der Aa. ueterinae
B: 1. Trimenon, C: 2. Trimenon, D: 3. Trimenon, E: Doppler mit notch (Pfeil)
Quelle: American Journal of Obstetrics & Gynecology 2009; 201:121-126, Copyright © 2009
Mosby
Neben dieser Risikobestimmung wurden 2013 neue Risikomarker in die
Deutschen Leitlininen aufgenommen: soluble fms-like tyrosine Kinase-1 (sFlt-1)

12
und Placental Growth Factor (PlGF) (AWMF-Leitlinie 2013). PlGF als
Wachstumsfaktor für die Angiogenese der Plazenta und sFlt-1 zur Rückbildung
dieser. Ein Ungleichgewicht, bzw. der Quotient dieser beiden
Angiogenesefaktoren soll eine Präeklampsie ab der 25. SSW vorhersagen und
ihren Schwerdegrad abschätzen können (Levine 2004).
2.2. Risikofaktoren
Es sind einige prognostisch wichtige Risikofaktoren für die Entwicklung einer
Präeklampsie bekannt. Zur Risikogruppe zählen Patientinnen, die zum ersten
Mal schwanger sind oder eine Präeklampsie in einer früheren Schwangerschaft
hatten. Das Risiko eines erneuten Auftretens in der nächsten Schwangerschaft
ist umso größer, je früher die Erkrankung aufgetreten ist, d.h. sie liegt über
60%, wenn die Präeklampsie in der vorhergegangen Schwangerschaft bereits
vor der 28. SSW auftrat. Ein erhöhtes Risikoprofil haben außerdem Frauen mit
chronischen Nierenerkrankungen, chronischer Hypertonie, Diabetes mellitus
Typ 1 oder familiärer Belastung, was ein Hinweis auf eine genetische
Disposition sein kann (Steinhard 1999).
Das Risiko an einer Präeklampsie zu erkranken, wird nach Sibai et al., durch
einen hohen body mass index (BMI) für Erstgebärdende wesentlich erhöht
(Sibai 2005). Sie stellten außerdem dar, dass eine Gewichtszunahme im letzten
Trimenon in der Schwangerschaft über einem Kilo pro Woche mit einem
erhöhten Risiko für die Erkrankung assoziiert ist, dies wurde als Risikofaktor
einer schweren Präklampsie in die deutschen Leitlinien übernommen (Sibai
1997; AWMF-Leitline 2013). Bodnar et al. zeigte, dass ein BMI von >26 kg/m2
das Risiko für die Entwicklung einer Präeklampsie verdoppelt und ein BMI von >
30 kg/m2 es sogar verdreifachen kann (Bodnar 2005).
Weitere Risikofaktoren sind ein vor der Schwangerschaft bereits bestehende
Insulinresistenz, Mehrlingsschwangerschaften, ein präexistenter Hypertonus,
vaskuläre Erkrankungen und Thrombophilien, wie das Antiphospholipid-
Syndrom. Als weiterer Risikofaktor werden Schwangere im Alter von über 40
Jahren oder ausgedehnte Intervalle zwischen zwei Schwangerschaften

13
diskutiert, sowie das Polyzystische-Ovar-Syndrom und rezidivierende Aborte
(Sibai 1997).
2.3. Differentialdiagnosen
Eine schwere Präeklampsie ist vom HELLP-Syndrom abzugrenzen. Das
HELLP- Syndrom („hemolysis, elevated liver enzymes, low platelets“), eine
Sonderform der Präeklampsie, betrifft 0,3-0,8% aller Schwangerschaften. Im
Median tritt das HELLP zwischen der 32. und 34. Schwangerschaftswoche auf,
in 20% der Fälle kann dies noch postpartal der Fall sein (Kirkpatrick 2010;
AWMF-Leitline 2013). Dabei besteht zusätzlich eine Hämolyse, mit Erhöhung
der Lactatdehyrogenase (LDH) und Erniedrigung des Haptoglobins. Die
Leberwerte der Aspartat-Aminotransferase (ASAT) und der Alanin-
Aminotransferase (ALAT) als Marker einer Leberschädigung können ebenfalls
ansteigen. Im Rahmen des HELLP-Syndroms können Symptome, wie
Kopfschmerzen, verschwommenes Sehen, Skotome und Oberbauchschmerzen
entstehen (Seck 2009).
Der Thrombozytenumsatz kann gesteigert sein, es kommt dadurch zur
Thrombozytopenie. Mikrothromben sind auch ohne Proteinurie ein Hinweis auf
eine Präeklampsie. Gefürchtete Komplikationen sind eine Disseminierte
intravasale Gerinnung (DIC), intermittierende oder persistierende
Niereninsuffizienz, sowie eine postpartal persistierende Leberinsuffizienz.
Häufig ist das HELLP-Syndrom mit einer Plazentainsuffizienz, bzw. mit einer
vorzeitigen Plazentalösung assoziiert (Kirkpatrick 2010).
Die Grenzen zwischen Präeklampsie und HELLP-Syndrom sind nicht eindeutig
definiert, abzugrenzen sind beide jedoch von der Eklampsie. Die Eklampsie ist
gekennzeichnet durch tonisch-klonische Krämpfe, nach Ausschluss organischer
Ursachen einer Epilepsie oder intrazerebralen Raumforderungen. Symptome
der Präeklampsie können hierbei gering ausgeprägt sein oder gänzlich fehlen,
ebenso wie Prodromalsymptome, wie Kopf-, Oberbauchschmerzen und
Augenflimmern (AWMF-Leitline 2013).

14
2.4. Die Pathophysiologie der Präeklampsie
Die Pathophysiologie der Präeklampsie bleibt weiterhin nicht endgültig geklärt.
Viele Erklärungsansätze sprechen für eine multifaktorielle Genese, aus
vaskulären, immunologischen, genetischen und Umweltfaktoren, deren
gemeinsamer Nenner die endotheliale Dysfunktion ist. Die Genese scheint
sowohl durch maternale, fetale und auch plazentare Faktoren beeinflusst zu
sein (Kim 2013).
In der Schwangerschaft nimmt das Herzzeitvolumen um 30-40% zu und
gleichzeitig der periphere Gefäßwiderstand ab (Lees 1967). Der arterielle Druck
sinkt zunächst adaptiv, bis Entbindungsbeginn steigt er wieder. Die renale
Hyperfiltration bei gesunden Schwangeren entsteht durch erhöhten renalen
Plasmafluss, die Werte des Serumkreatinins fallen, die Kreatininclearance
steigt.
Bei Patientinnen mit Präeklampsie verlaufen einige dieser Veränderungen in
der Schwangerschaft gegensätzlich.
Oxidativer Stress spielt in vielen Theorien zur Pathophysiologie eine
entscheidende Rolle (Wang Y. 1998; Many 2000; Brahmarshi 2012; Mistry
2013). Dieser entsteht durch ungenügenden Abbau, bzw. vermehrter Bildung
von ständig im Körper entstehenden Sauerstoffspezies. Diese haben vielfältige
Angriffsmöglichkeiten, ähnlich wie in der Pathophysiologie der Artherosklerose.
Sauerstoffradikale und Lipidperoxide, die als zell- und membranschädigende
Substanzen das vaskuläre Endothel angreifen, führen zu einer vaskulären
endothelialen Dysfunktion. Stabile Lipidperoxidationsprodukte und oxidierte
Fragmente aus dem Synzytiothrophoblasten, die in die systemische Zirkulation
gelangen und Zytokine, die aus der hypoxischen Plazenta freigesetzt werden,
könnten so zur mütterlichen Symptomatik führen (Roberts 2009).
Im Folgenden betrachte ich die zwei relevantesten Theorien für meine
Hypothesen.
2.4.1. Theorie der anormalen Umformungen der Spiralarterien der Plazenta
Der Blutfluss durch einen nicht-schwangeren Uterus beträgt wenige Milliliter pro
Minute. Ab dem zweiten Trimenon vervielfacht sich dieser, bis er zum Ende der

15
Schwangerschaft circa 700 ml/ min beträgt. Ein minimaler Teil dessen versorgt
das Myometrium des Uterus, der größte Teil wird im intervillösen Raum für den
maternofetalen Stoffaustausch benötigt. Diese drastische Steigerung der
Durchblutung ist nur möglich, wenn der Widerstand in den uteroplazentaren
Gefäßen während der Schwangerschaft abnimmt (Kiechle 2007).
Während der Plazentation wandern Trophoblasten in die Decidua und einen Teil
des Myometriums ein. Am Anfang des ersten Trimenons werden vor allem die
kleinen maternalen Arteriolen zu Gefäßen mit größerer Kapazität und kleinerem
Widerstand verändert. Ab dem zweiten Trimenon werden die Spiralarterien
umgebaut (siehe Abbildung 3). Diese sind die Endarterien des uteroplazentaren
Kreislaufs, welche Blut direkt in den intervillösen Raum führen, der den
Stoffaustausch zwischen Mutter und Feten ermöglicht (Redman 2005). Ihre
Endothelstruktur wird komplett von Trophoblastenzellen ersetzt, die muskulären
Elemente und die Elastica interna werden aufgelöst (Pijnenborg 1980). Die
englumigen Uterusgefäße der Schwangeren entwickeln sich zu weiten
Gefäßen, mit viel Kapazität. Der dadurch bedingte Abfall des
Gefäßwiderstandes in den uterinen Gefäßen ist Voraussetzung für die
Steigerung des Zustroms des mütterlichen Bluts in die Plazenta. Demnach wird
der mütterliche Blutdruck zum regulierenden Einfluss auf den uteroplazentaren
Kreislauf. Diese Widerstandsabnahme vermindert den Druck mit dem das
maternale Blut in dem intervillösen Raum die fetalen Zotten erreicht, so dass es
zu einer gleichmäßigen Strömung kommt. Der niedrige intervillöse Druck ist
Voraussetzung für eine ungestörte fetale Perfusion der Zotten, die ebenfalls
einen relativ geringen Druck aufweisen. Dieser Vorgang ist eine notwendige
anatomisch-physiologische Adaptation an einen vermehrten Durchblutungs-
bedarf in der Schwangerschaft. Dies führt zu einer kontinuierlichen Zunahme
der diastolischen Strömungsgeschwindigkeit (Schneider 2006).
Bei Präeklampsie-Patientinnen kann dieser Vorgang gänzlich oder zum Teil
ausbleiben. Die trophoblastischen Zellen wandern in die Spiralarterien in der
decidualen Region ein, aber unvollständig in das Myometrium. Die
Spiralarterien bleiben klein, mit wenig Kapazität, was zu Hypoxie und Ischämie
in der Plazenta führt. Dies kann zu intrauteriner Mangelentwicklung des Feten
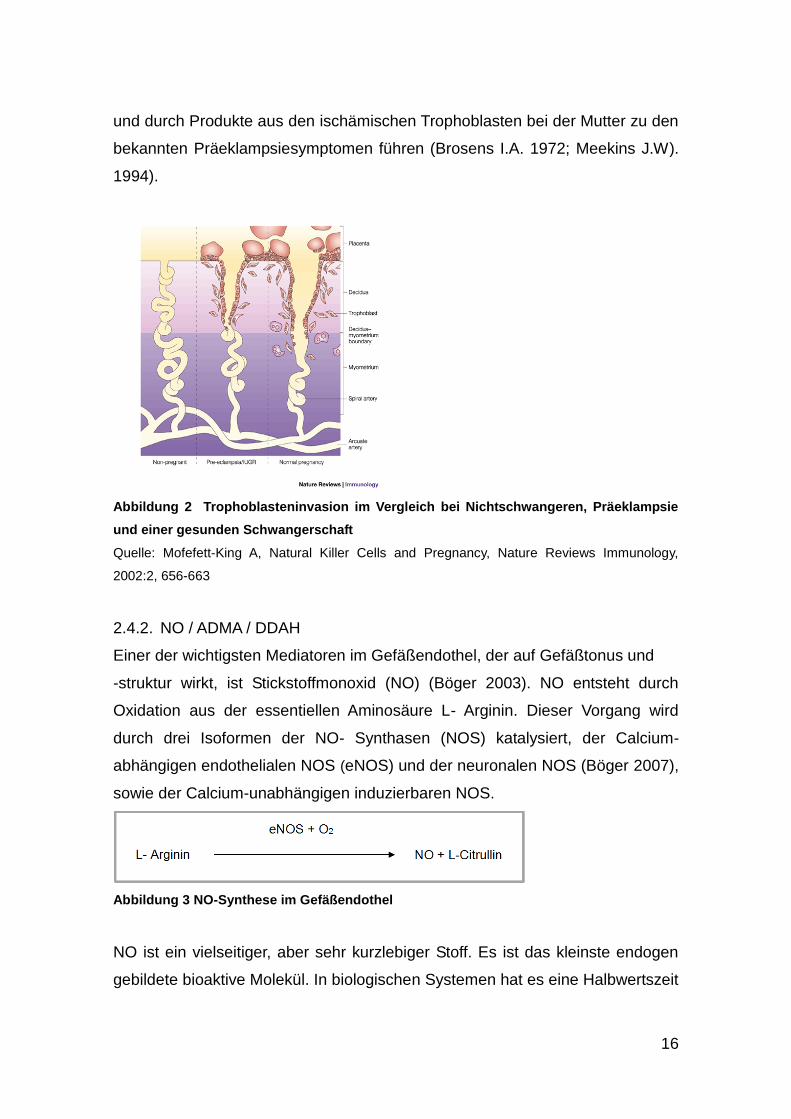
16
und durch Produkte aus den ischämischen Trophoblasten bei der Mutter zu den
bekannten Präeklampsiesymptomen führen (Brosens I.A. 1972; Meekins J.W).
1994).
Abbildung 2 Trophoblasteninvasion im Vergleich bei Nichtschwangeren, Präeklampsie
und einer gesunden Schwangerschaft
Quelle: Mofefett-King A, Natural Killer Cells and Pregnancy, Nature Reviews Immunology,
2002:2, 656-663
2.4.2. NO / ADMA / DDAH
Einer der wichtigsten Mediatoren im Gefäßendothel, der auf Gefäßtonus und
-struktur wirkt, ist Stickstoffmonoxid (NO) (Böger 2003). NO entsteht durch
Oxidation aus der essentiellen Aminosäure L- Arginin. Dieser Vorgang wird
durch drei Isoformen der NO- Synthasen (NOS) katalysiert, der Calcium-
abhängigen endothelialen NOS (eNOS) und der neuronalen NOS (Böger 2007),
sowie der Calcium-unabhängigen induzierbaren NOS.
Abbildung 3 NO-Synthese im Gefäßendothel
NO ist ein vielseitiger, aber sehr kurzlebiger Stoff. Es ist das kleinste endogen
gebildete bioaktive Molekül. In biologischen Systemen hat es eine Halbwertszeit

17
von nur wenigen Sekunden. Es nimmt eine zentrale Rolle als Mediator für
Regulationsmechanismen des kardiovaskulären Systems ein. Neben der
Regulation des Gefäßtonus ist NO ein Thrombozytenaggregationhemmer,
verhindert die Leukozytenadhäsion am Gefäßendothel (Böger 2006) und die
Proliferation von glatten Muskelzellen (Böger 2004a). NO wird vom Endothel
freigesetzt und trägt zur Regulation der Durchblutung bei, indem es die Wirkung
von vasokonstriktorisch-wirkende Substanzen im Blut antagonisiert, wie
beipspielsweise Angiotensin-II, Katecholaminen und Endothelin (Böger 1996).
NO bindet die Häm-Gruppe der löslichen Isoform der Guanylcyclase in der
glatten Gefäßmuskulatur, was über die Bildung des second messengers cGMP
die biologischen Effekte vermittelt (Aktories; Förstermann; Hofmann; Starke
2009).
NO ist ein wesentlicher Faktor für die Vasodilatation der Gefäße in der
Schwangerschaft, mit der Bildung im Syncytiotrophoblasten und dem
fetoplazentaren Endothel hat es dort großen Einfluss auf den niedrigen
fetoplazentaren Widerstand. Zusätzlich wird der Einfluss auf die Einwanderung
der Trophoblastenzellen in Hinblick auf Implantation, Differenzierung, Invasion,
Motilität und Apoptose diskutiert (Lyall F. 1998).
Der kompetitive NO-Synthase-Inhibitor ADMA (siehe Abbildung 5), erstmals von
Nakajima et al. beschrieben, entsteht bei der Methylierung von Arginin-
Seitenketten innerhalb von Proteinen und Polypeptiden durch Protein-Arginin-
Methyltransferasen (PRMTs) (Nakajima 1971). Als Methylgruppendonator
fungiert hierbei S- Adenosylmethionin (SAM), welches aus dem
Homocysteinstoffwechsel stammt. Dieser Prozess findet ubiquitär im Körper
während der posttranslationalen Modifikation statt. Die Rolle des Methylierens
von Proteinen ist nicht vollständig geklärt, jedoch ist sicher, dass dieser Prozess
eine regulative Funktion bei der Bindung von RNA, während der Transkription,
bei der DNA-Reparatur, in der Interaktion zwischen Proteinen und bei der
Signaltransduktion besitzt (Vallance 2004). Es gibt acht PRMTs, die in zwei
Typen eingeteilt werden. Durch Katalysation mit Hilfe von PRMT Typ 1 entsteht
durch Dimethylierung einer Stickstoffgruppe und nachfolgendem proteolytischen
Abbau freies ADMA. PRMT Typ 1 wurde im Herzen, in glatten Muskelzellen und

18
endothelialen Zellen gefunden. PRMT Typ 2 dagegen methyliert zwei Guanin-
Stickstoffgruppen, daraus resultiert SDMA (Abbildung 5). Beide Typen der
PRMTs, von denen noch weitere Isoformen existieren, können auch nur eine
Methylgruppe übertragen, so entsteht NG- Monometylarginin (L-NMMA) (Siroen
2006). Es ist nicht klar, ob die Bildung von ADMA von der PRMT-Aktivität oder
vom Proteinturnover abhängt. Es zeigte sich jedoch, dass die ADMA-
Konzentration mit der PRMT-Expression korreliert (Ogawa 1987).
Weiter konnte gezeigt werden, dass ADMA und L-NMMA, jedoch nicht SDMA,
konzentrationsabhängig die endotheliale und neuronale NOS hemmen
(Vallance 2004). Erstmals wurde die NOS-Inhibition durch asymmetrisches
Dimethylarginin (ADMA) als den Vertreter aus dieser Substanzgruppe, der in
den höchsten Konzentrationen nachweisbar war von Vallance und Mitarbeitern
beschrieben (Vallance 1992). Böger konnte demonstrieren, dass diese
Substanz nach ihrer Isolation aus humanem Urin in vitro zu einer signifikanten
und konzentrationsabhängigen Hemmung der NO-Bildung führte. Im Gegensatz
zu ADMA bewirkt sein Isomer, symmetrisches Dimethylarginin (SDMA) keine
Hemmung der NO-Synthese (Abbildung 4) (Böger 2000).
Abbildung 4 Methylariginine
Quelle: http://www.intechopen.com/source/html/40284/media/image1.png
Täglich wird etwa 300 µmol ADMA im Körper freigesetzt (Achan 2003). Die
Elimination aus dem Körper geschieht zu 80% über den metabolischen Abbau
zu L- Citrullin und Dimethylamin über Hydrolyse durch die Dimethylarginin-
Dimethylaminohydrolase (DDAH) (Achan 2003). Es existieren nach heutigem
Kenntnisstand 2 Isoformen. Leiper et al. konnten mittels eines Nothern blots die

19
unterschiedliche Expression von DDAH 1 und 2 in menschlichen Geweben
nachweisen. Eine hohe DDAH 1 Expression fanden sie im Gehirn, Pankreas,
Nieren und Leber. Eine hohe DDAH 2 Expression konnten sie im Herzen, in den
Nieren und der Plazenta nachweisen (Leiper 1999).
ADMA wird durch eine nukleophile Reaktion am Guanidinorest durch DDAH
abgebaut. Cystein hält das Enzym im aktiven Zentrum in der tertiären Struktur,
wenn Cystein durch einen Serinrest ausgetauscht wird, verliert es seine
Aktivität.
Cystein ist anfällig für Oxidation und wird reguliert durch Stickstoffmonoxid, es
wird diskutiert, ob oxidativer Stress eine dauerhafte Inhibition der DDAH-
Aktivität auslösen könnte.
Eine hohe NO-Produktion vermindert die Aktivität von DDAH, speziell durch
Hochregulation der induzierbaren NOS wird DDAH nitrosiert, jedoch ist die S-
Nitrosylierung reversibel. Dies lässt vermuten dass hinter der NO-Produktion
ein homöostatischer Mechanismus steht, der bei hoher NO-Konzentration die
weitere Produktion verhindert (Abbildung 6) (Vallance 2004).
Die SDMA-Konzentration steigt bei abnehmender Nierenfunktion an, da es
ausschließlich über die Niere ausgeschieden wird. ADMA dagegen steigt
währenddessen weniger stark an, weil ADMA zum großen Teil über DDAH
metabolisiert wird, denn nur die etwa 20% des zirkulierenden ADMAs werden
über die Nieren ausgeschieden (Ogawa 1987). Zudem können sowohl ADMA,
als auch SDMA über die AGXT2 abgebaut werden (Ogawa 1990; Rodionov
2010).
ADMA ist stabil und kann zwischen Zellen diffundieren, es kann in einer Zelle
die NOS inhibieren, darüber hinaus erreicht ADMA eine Inhibition nicht nur in
dieser Zelle, sondern kann auch in weiteren Zellen eine gleichwertige
Hemmung induzieren. Dies wurde bei Makrophagen und endothelialen Zellen
gezeigt, ebenso könnte dies für glatte Muskelzellen und endotheliale Zellen
gelten (Maas 2007). Das Hinzufügen von ADMA in diesen Zellen führt zudem in
vitro zu einer endothelialen Dysfunktion (Calver 1993).
1998 berichteten Fickling und Holden von der Erhöhung der Serum-ADMA-
Konzentration bei Präeklampsie (Fickling 1993). Sie zeigten, dass der Blutdruck
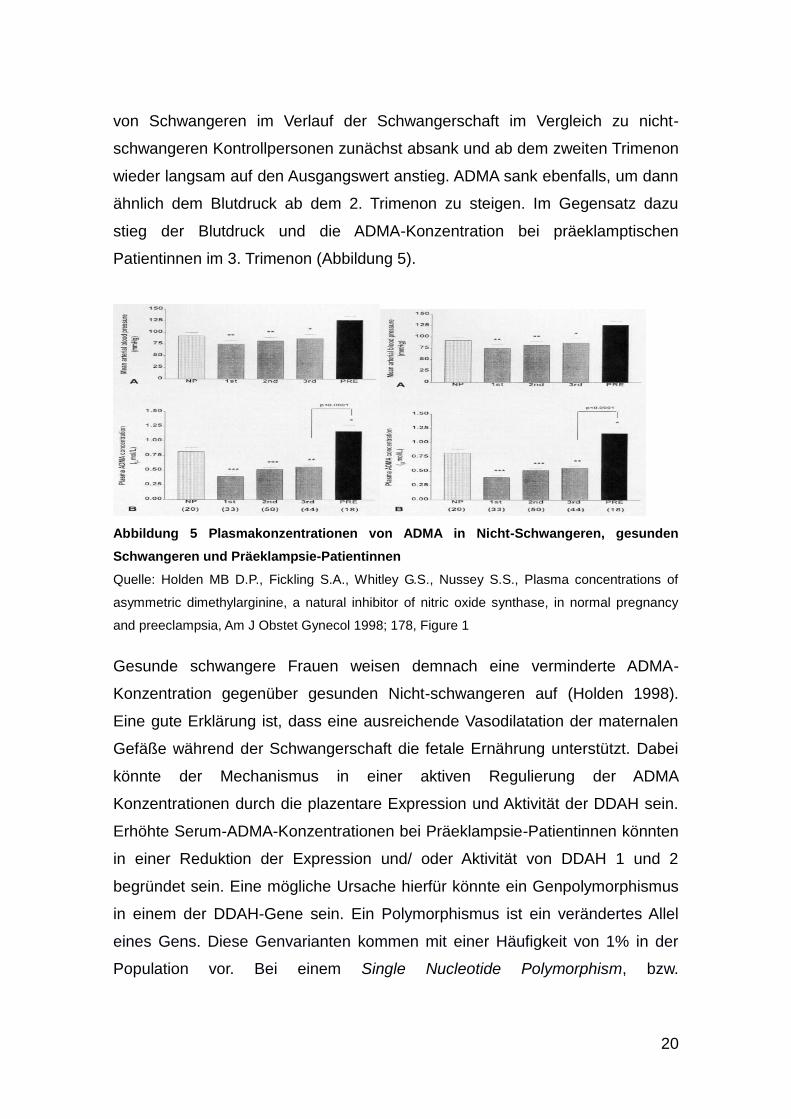
20
von Schwangeren im Verlauf der Schwangerschaft im Vergleich zu nicht-
schwangeren Kontrollpersonen zunächst absank und ab dem zweiten Trimenon
wieder langsam auf den Ausgangswert anstieg. ADMA sank ebenfalls, um dann
ähnlich dem Blutdruck ab dem 2. Trimenon zu steigen. Im Gegensatz dazu
stieg der Blutdruck und die ADMA-Konzentration bei präeklamptischen
Patientinnen im 3. Trimenon (Abbildung 5).
Abbildung 5 Plasmakonzentrationen von ADMA in Nicht-Schwangeren, gesunden
Schwangeren und Präeklampsie-Patientinnen
Quelle: Holden MB D.P., Fickling S.A., Whitley G.S., Nussey S.S., Plasma concentrations of
asymmetric dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal pregnancy
and preeclampsia, Am J Obstet Gynecol 1998; 178, Figure 1
Gesunde schwangere Frauen weisen demnach eine verminderte ADMA-
Konzentration gegenüber gesunden Nicht-schwangeren auf (Holden 1998).
Eine gute Erklärung ist, dass eine ausreichende Vasodilatation der maternalen
Gefäße während der Schwangerschaft die fetale Ernährung unterstützt. Dabei
könnte der Mechanismus in einer aktiven Regulierung der ADMA
Konzentrationen durch die plazentare Expression und Aktivität der DDAH sein.
Erhöhte Serum-ADMA-Konzentrationen bei Präeklampsie-Patientinnen könnten
in einer Reduktion der Expression und/ oder Aktivität von DDAH 1 und 2
begründet sein. Eine mögliche Ursache hierfür könnte ein Genpolymorphismus
in einem der DDAH-Gene sein. Ein Polymorphismus ist ein verändertes Allel
eines Gens. Diese Genvarianten kommen mit einer Häufigkeit von 1% in der
Population vor. Bei einem Single Nucleotide Polymorphism, bzw.

21
Einzelnukleotidpolymorphismus liegt ein Austausch eines einzelnen Nukleotids
im DNA-Molekül vor. Betrifft dies den kodierenden Bereich eines Gens, kann es
zu einer veränderten, bzw. fehlerhaften Genexpression führen. Nach Williams
ist die Präeklampsie eine sehr komplexe genetische Erkrankung, das Ergebnis
verschiedener genetischer Varianten auf unterschiedlichsten Loci, die individuell
wenig Effekt, aber wohlmöglich zusammen die individuelle Anfälligkeit für die
Erkrankung erhöhe (Williams 2012).
So sind Genpolymorphismen Gegenstand für weitere Forschungsvorhaben
geworden. Diese wurde auch für die DDAH 1 und DDAH 2 bereits beschrieben,
die möglicherweise zu einer verringerten Proteinexpression bzw. zur Expression
eines veränderten DDAH Proteins führen können (Akbar 2004; Valkonen 2005;
Maas 2009).
Lüneburg et al. betrachteten in einer Metaanalyse sechs SNPs im DDAH 1-
Locus und fand eine hohe Assoziation mit erhöhten Plasma-ADMA-
Konzentrationen (Lüneburg 2011).
Ein anderer Genpolymorphimus wurde bislang in Zusammenhang mit
Hypertonie untersucht. Maas et al. fanden eine Assoziation von zwei DDAH 2-
Promotor-(-1151 A/C und -449 G/C) Polymorphismen (rs805304 und rs805305)
mit einer erhöhten Prävalenz der Hypertonie (Maas 2009). In einer finnischen
Studie wurden SNPs im DDAH 1 bei Präeklampsiepatientinnen und gesunden
Kontrollen untersucht. Es zeigten sich keine signifikanten Unterschiede in der
Häufigkeit der Genotypen, jedoch konnten sie in Verbindung mit zwei
Hochriskio-Haplotypen eine Prädisposition für eine Präeklampsie zeigen und
zwei weitere, die sich statistisch signifikant präventiv gegenüber einer
Präeklampsie auswirken (Akbar 2004).
Siroen et al. untersuchten die plazentare DDAH 1-Aktivität bei gesunden
Schwangeren und Präeklampsiepatientinnen und fanden keine signifikante
Hochregulierung der DDAH-Aktivität oder -Expression in Schwangeren.
Vielmehr fanden sie keinen Unterschied der ADMA-Konzentration zwischen
gesunden Schwangeren und Präeklampsiepatientinnen. Sie fanden nur bei
HELLP-Patientinnen erhöhte ADMA-Werte und bei diesen im Vergleich zur

22
Aktivität in der Leber eine 20fach verminderte Aktivität von DDAH im
Plazentagewebe (Siroen 2006).
Die DDAH-Aktivität und Expression scheint nicht nur in diesem Kontext
interessant zu sein, sondern könnte viel früher in der Schwangerschaft eine
Rolle spielen. Ayling et al. zeigten in ihrer Studie, dass die fetalen
Trophoblastenzellen während der Plazentation DDAH 1 exprimieren. Sie
überexprimierten DDAH-1 und konnten eine 8-fach erhöhte Enzymaktivität
messen, sowie eine 59%ige Verminderung der ADMA-Konzentration und als
Folge dessen eine fast 2-fach angestiegene NO-Produktion (Ayling 2006). NO
und HGF (Hepatocyte growth factor) wirkten sich in einem funktionellen Assay
signifikant auf die Trophoblasteninvasion und -motilität aus. Daher könnte dies
einer der Mechanismen sein, die physiologische Adaptation der Spiralarterien
zu gewährleisten und bei mit verminderter DDAH-Expression oder –Aktivität zu
Präeklampsie zu führen (Cartwright 2002).
Um den Metabolismus von ADMA in der Plazenta genauer zu bestimmen,
wollten wir ebenfalls die fetalen ADMA-Konzentrationen betrachten um den
Ursprung der erhöhten Konzentrationen in der Plazenta zu finden. Braekke et
al. und Maeda et al. zeigten, dass die fetalen Konzentrationen von ADMA in
Feten mit gesunden und an Präeklampsie-erkrankten Müttern sich nicht
unterschieden (Braekke 2009; Maeda 2003). Sie fanden in beiden Kollektiven
einen maternofetalen Gradienten der ADMA-Konzentration, diese ist bei den
Feten gegenüber ihrer Mütter circa dreifach erhöht. Braekke et al. entdeckten
zudem eine venös-arterielle Differenz. ADMA war in den venösen-umbilikalen
Gefäßen höher als in den arteriellen, so dass sie darauf schlossen, dass eine
Erhöhung der maternalen ADMA-Konzentration seine Ursache in der Plazenta
und nicht im Feten haben muss. Dies könnte ein indirekter Hinweis auf eine
Metabolisierungsstörung der Plazenta sein, beispielsweise eine Funktions-
einschränkung der DDAH , da diese eine bedeutende Rolle im Abbau der
höheren fetalen ADMA-Konzentrationen spielen könnte, um die mütterlichen
Serumkonzentrationen niedrig zu halten.
Anderssohn et. al. konnten in der vorherigen Studie ebenfalls ca. 3-fach höhere
ADMA-Konzentrationen in den fetalen Serumproben gegenüber denen der
23
Mütter finden, ohne Unterschied zwischen Kontroll- und Präeklampsiegruppe.
Der feto-maternale Gradient war allerdings signifikant niedriger in der
Präeklampsiegruppe, als in den Kontrollen (Anderssohn 2012). Eine erhöhte
ADMA- Konzentration im umbilikalen Blut konnte in anderen Studien ebenfalls
nachvollzogen werden (Kul 2009; Vida 2007; Maeda 2003). Dieses Gebiet
bedarf allerdings weiterer Forschung.
In der Literatur finden sich einige Studien zum Vergleich der ADMA-
Konzentration bei Präeklampsie-Patientinnen und gesunden Schwangeren, die
allerdings grundsätzlich durch unterschiedliche Messmethoden schwer
vergleichbar scheinen (
Tabelle 2).
Autoren Jahr Messtechnik n Median ADMA SSW n Median ADMA SSW p < 0,05
Ehsanipoor et al. 2013 ELISA 23 2, 14 ± 0,33 µmol/l 39.6 ± 1.1 20 1,88 ± 0,19 µmol/l 37.6 ± 2.2 *
Sandrim et al. 2010 ELISA 47 2.112 µmol/l (SD 0.012) 35.2 ± 3.5 47 2.199 µmol/l (SD 0.016) 35.9 ± 3.7 *
Ellis et al. 2001 HPLC 19 0.56 µmol/l (0.36 - 1.10) 24-32 12 0.68 µmol/l (0.51 - 0.82) 24-32 *
Ellis et al. 2001 HPLC 16 0,53 µmol/l (0.40 - 0.73) 36-40 32 0.68 µmol/l (0.43 - 1.70) 36-40 *
Fickling et al. 1993 HPLC 10 0,46 µmol/l 3. Trimenon 8 1,23 µmol/l 3. Trimenon *
Holden et al. 1998 HPLC 44 0.56 µmol/l (SD 0.23) 32.6 (3,8) 18 0.56 µmol/l (SD 0.23) 32.7 (3,2) *
Kim et al. 2004 HPLC 13 0.28 µmol/l (IQR 0.17 - 0.33) 39.1 ± 2.2 16 0.21 µmol/l (IQR 0.16 - 0.24) 35.9 ± 3.9
Lopez et al. 1996 HPLC 22 1,28 µM 40+2 ± 1,4 22 0,97 µM 38+1 ± 1,8
Maas et al. 2004 HPLC 93 0.42 µmol/l (IQR 0.29 - 0.55) 39.0 (38.1-40.3) 67 0.43 µmol/l(IQR 0.31 - 0.56) 36.0 (33.8-38.8)
Mao et al. 2010 HPLC 30 0.82 µmol/l (SD 0.11) 3. Trimenon 62 1.27 µmol/l(SD 0.31) 3. Trimenon *
Pettersson et al. 1998 HPLC 12 0.36 µmol/l (SEM 0.01) 32-40 12 0.55 µmol/l(SEM 0.02) 35.6 ± 0,8 *
Powers et al. 2008 HPLC 31 0.49 µmol/l (SD 0.08) 39.5 ± 1.4 15 0.55 µmol/l(SD 0.07) 35.9 ± 3.8 *
Siroen et. al. 2006 HPLC 15 0,37 ± 0,06 µmol/l 41.3 ± 1.1 16 0,40 ± 0,06 µmol/l 37.1 ± 2.9
Speer et al. 2008 HPLC 31 0,34 ± 0,08 µmol/l 18,3 ± 1,6 15 0.45 ± 0.09 µmol/l 16,9 ± 3,2 *
Speer et al. 2008 HPLC 31 0,49 ± 0,08 µmol/l 39,5 ± 1,4 15 0.55 ± 0,07 µmol/l 35,5 ± 4,2 *
Anderssohn et al. 2012 LC-Ms/Ms 28 0,42 ± 0,07 µmol/l 36.6 ± 3.5 18 0,51 ± 0,15 µmol/l 33.9 ± 3.3 *
Braekke et al. 2009 LC-Ms/Ms 40 0.39 µmol/l (IQR 0.33 - 0.43) 32.9 (24.9–38.7) 43 0.44 µmol/l (IQR 0.38 - 0.50) 38.7 (34.4–41.7) *
Messmethoden von Asymmetrischem Dimethylarginin in Präeklampsie-Patientinnen
Kontrollen Präeklampsie
Tabelle 2 Messmethoden von ADMA in Präeklampsie-Patientinnen
modifiziert nach AA Khalil, D Tsikas, R Akolekar, J Jordan, KH Nicolaides. Asymmetric
dimethylarginine, arginine and homoarginine at 11-13 weeks’ gestation and preeclampsia: a
case-control study. Journal of Human Hypertension (2011) 1-6
2.5. Therapie
Nach der aktuellsten Deutschen Leitlinie der AWMF sind Antihypertensiva bei
schweren Formen der Präeklampsie empfohlen, bei Patientinnen, die
Blutdruckspitzen über >170 mmhg systolisch und >110 mmHg diastolisch
aufweisen bei Propfpräeklampsie bereits ab >160/100 mmHg (AWMF-Leitline
2013).

24
Die erste Wahl zur Langzeitbehandlung ist dabei α-Methyldopa (Presinol ®),
das eine positive Auswirkung auf den Hypertonus hat ohne zu verminderter
uteroplazentarer Perfusion oder zu fetalen Wachstumsretadierungen zu führen.
Für die Akutbehandlung oder bei Blutdruckspitzen wird als 1. Wahl nach der
deutschen Leitlinie Nifedipin (Adalat) verabreicht (AWMF-Leitline 2013).
Bei behandlungsbedürftiger Präeklamspie mit oder ohne HELLP-Syndrom wird
aktuell außerdem eine Therapie mit Glukokortikosteroiden diskutiert, da einige
Studien einheitlich gute Auswirkungen auf die prä- und postpartalen klinischen
und biochemischen Verlauf hatten, auch wenn eine abschließende Beurteilung
noch aussteht (Fischer 1999; Magann 1994). Eine prophylaktische Gabe von
Magnesiumsulfat ist laut Leitlinie bei schwerer Präeklampsie indiziert, die in
51% der Fälle das Eklampsie-Risiko senken konnte (Altmann 2012; AWMF-
Leitline 2013).
Die einzige Therapie mit Heilung der Präeklampsie ist bisher die frühzeitige Ent-
bindung, die von folgenden mütterlichen Indikationen bedingt wird:
Schwere therapierefraktäre Hypertonie
Niereninsuffizienz
Akutes Lungenödem
Disseminierte intravasale Gerinnung (DIC)
Eklampsie
Jenseits der vollendeten 37. Schwangerschaftswoche kann eine Entbindung,
bei ausführlicher Überwachung auch durch eine Einleitung, ohne relevante
Erhöhung der fetalen Morbidität durchgeführt werden. Zwischen der 34. und 37.
Schwangerschaftswoche ist die frühzeitige Entbindung ebenfalls empfohlen, da
eine Prolongation für die fetale Morbidität kein besseres Ergebnis zeigte. Ab der
24. Schwangerschaftswoche ist die Patientin in einem Perinatalzentrum
aufzunehmen und wenn möglich frühestens nach vollständiger Induktion der
Lungenreife zu entbinden. Präeklampsie-Patientinnen vor der 24.
Schwangerschaftswoche sind individuell zu behandeln, dabei ist eine hohe
fetale Mortalität und Morbidität zu beachten, die die mütterliche Morbidität in
den Vordergrund stellt (Gaugler-Senden 2006).

25
Altun et al. untersuchte im Tierversuch an Ratten, in denen durch Stress
präeklamptischen Symptome induziert wurden, die Effekte einer L-Arginin-
Supplementation. Diese Studie konnte zwar eine Blutdrucksenkung unter der
Substitution von L-Arginin zeigen, jedoch hatten die Ratten der gesunden
Kontrollgruppe nach wie vor einen um 33% niedrigeren Blutdruck (Altun 2008).
Es gibt einige Medikamente und diätetische Substanzen, die seit mehreren
Jahren auf ihre präventive Wirkung hin überprüft werden, jedoch ergeben sich
sehr heterogene Ergebnisse in folgenden Studien.
Acetylsalicylsäure (ASS) könnte in das pathologische Ungleichgewicht der
Prostacyclin-/Thromboxanfreisetzung eingreifen, indem es die Cyclooxygenase
inaktiviert und damit konsekutiv die Synthese von TXA2 hemmt. Deswegen
wurde die präventive Wirkung von ASS in zahlreichen Studien mit
präeklamptischen Patientinnen getestet (Duley L. 2007; Papageorghiou 2003;
Becker 2013). 2001 wurde eine Analyse der Cochrane-Daten veröffentlicht
(Duley 2001), die zeigte, dass die Präeklampsieinzidenz, Frühgeburtlichkeit und
perinatale Morbidität mit einer ASS-Therapie gesenkt werden konnte. Dieser
Effekt war umso erfolgreicher, je früher der Therapiebeginn und je höher die
Dosis war (Leitich H 1997). Die Ergebnisse einiger Studien lassen vermuten,
dass eine ASS-Dosis von 100mg/Tag und ein Therapiebeginn zwischen der 12.
und 16. Schwangerschaftswoche die beste präventive Wirkung haben soll
(CLASP Study 1994). Mit der vollendeten 34. SSW kann die Therapie abgesetzt
werden. Der frühzeitige Verschluss des Ductus Botalli beim Feten, der im
Zusammenhang mit dieser Therapie erwähnt wurde, konnte bei
niedrigdosierten Gaben von ASS nicht bestätigt werden (Di Sessa 1994).
In neueren Studien wird außerdem die alleinige oder die Applikation in
Kombination mit ASS mit niedermolekularem Heparin empfohlen. In einer
kanadischen Studie wurden Frauen, die in der vorherigen Schwangerschaft an
einer schweren Präeklampsie, einem ungeklärten Intrauterinen Fruchttod,
einem IUGR oder einer vorzeitigen Plazentalösung gelitten hatten, mit
niedermolekularem Heparin behandelt. Bei den mit Heparin-behandelten
Schwangeren wurde deutlich seltener eine Präeklampsie im Verlauf der neuen

26
Schwangerschaft verzeichnet (1,8% vs. 14,5% in der unbehandelten
Kontrollgruppe) (Rey 2009).
In Bezug auf die Hypothesen der vorliegenden Arbeit wäre die Verabreichung
von NO-Donatoren eine nahe liegende Therapieoption. Als Donatoren kommen
Glyceroltrinitrat (GTN), Isosorbidnitrat (ISDN), Isosorbidmononitrat (ISMN) und
S-Nitrosoglutathion (GSNO) in Frage. In Tierversuchen konnten GTN- und
ISDN-Therapien deutliche Senkungen des arteriellen Drucks und verbesserte
Uterusdurchblutung erzielen. Studien an präeklamptischen Patientinnen zeigten
bei einer intavenösen GTN- oder transdermalen GTN-/ISDN- Therapie eine
signifikante Blutdrucksenkung und meist auch eine Verbesserung der
uteroplazentaren Perfusion (Beinder 1999). Allerdings konnte gezeigt werden,
dass bei normotensiven Schwangeren mit auffälligem notch in der
Dopplersonographie durch die präventive Anwendung von Glyceryltrinitrat-
Pflaster keine Reduktion der Präeklampsie-Inzidenz erzielt werden konnte
(Lees 1998). Zudem ist das fetale und embryonale Nebenwirkungsspektrum
von NO-Donatoren noch nicht genügend geklärt.

27
3. Methoden
3.1. Eingeschlossene Patientinnen und Probandinnen
Für die vorliegende klinische Studie wurden Patientinnen rekrutiert, die von
ihren FrauenärztInnen das Universitätsklinikum Hamburg-Eppendorf oder in die
Asklepiosklinik Hamburg-Barmbek eingewiesen-, bzw. überwiesen wurden. Die
Patientinnen hatten nach den Diagnosekriterien der DGGG eine Präeklampsie
oder ein HELLP-Syndrom. Auf Grund dessen bekamen sie dort zu späterem
Zeitpunkt eine elektive Sectio caesarea oder die Geburt wurde unter
Überwachung im Kreißsaal per Misoprostol (Off-label-Use) eingeleitet. Die
Patientinnen wurden nach Aufklärung und Einwilligung in die Studie
eingeschlossen.
3.1.1. Gruppe A - Gesunde Schwangere
Einschlusskriterien:
komplikationslose Einlingsgravidität zur elektiven Sectio caesarea oder
zur Geburtseinleitung mit Misoprostol in das Universitätsklinikum
Eppendorf, bzw. die Asklepiosklinik Hamburg-Barmbek
Matching anhand des BMI, maternalem und Gestationsalter mit den
präeklamptischen Patientinnen
Ausschlusskriterien:
Mehrlingsgravidität
Patientinnen mit schweren akuten oder chronischen Erkrankungen, die
eine Medikamenteneinnahme erfordern
bekannter Alkohol-, Drogen- oder Medikamentenabusus
3.1.2. Gruppe B - Patientinnen mit Präeklampsie
Einschlusskriterien:
Nach Diagnosekriterien der DGGG/National Institute of Health
Neuauftreten eines Blutdrucks >140 mmHg systolisch und/ oder >90
mmHg diastolisch und Proteinurie von über >300 mg/ 24-Stunden Urin

28
bei einer Schwangeren, nach der 20. Schwangerschaftswoche/ 2-fach
positiver Urinstix als Indiz für eine Proteinurie aus
Patientinnen mit einer Propf-Präeklampsie, Eklampsie oder HELLP-
Syndrom
Alter zwischen 18 und 45 Jahren
mindestens in der 20. SSW
Ausschlusskriterien:
Mehrlingsgravidität
Patientinnen mit schweren akuten oder chronischen Erkrankungen, die
eine Medikamenteneinnahme erfordern
bekannter Alkohol-, Drogen- oder Medikamentenabusus
3.2. Klinische Daten
Die folgenden klinischen Daten der Patientinnen wurden aus der Patientenakte
dokumentiert und oder bei der Patientin erfragt:
Anzahl der bisherigen Schwangerschaften inklusive der aktuellen
Schwangerschaft (Gravida)
Anzahl der bisherigen Geburten (Para)
Schwangerschaftswoche
Alter
Systolischer Blutdruck in mmHg bei Aufnahme
Diastolischer Blutdruck in mmHg bei Aufnahme
Größe in m
Gewicht vor der Schwangerschaft in kg
BMI vor der Schwangerschaft in kg/m2
Aktuelles Gewicht in kg
Vorhandensein von pathologischen uterinen Dopplerbefunden
Vorhandensein von Ödemen
Schwangerschaftswoche bei Entbindung/Sectio
Geschlecht des Kindes

29
Geburtsgewicht des Kindes
Gestationsaltersadaptierte Perzentile des Geburtsgewichts
Körperlänge des Kindes bei Geburt
Außerdem wurden folgende Laborwerte, soweit vorhanden, aus bereits
entnommenen Blutproben vor der Entbindung dokumentiert:
ASAT und ALAT
Haptoglobin
Harnsäure
Harnstoff
Kreatinin
LDH
PTT
Quick
Urinalbumin/ 24 h
3.3. Bestimmung von ADMA-, SDMA- und L-Arginin im Plasma
Bei Aufnahme, wurden den Teilnehmerinnen circa 2 ml Blut in einer EDTA-
Monovette entnommen. Zusätzlich wurden nach der Entbindung in einer EDTA-
Monovette Nabelschnurblut, aus den Vena umbilicalis und sofern möglich auch
aus einer der Arteriae umbilicalis entnommen. Die Blutproben wurden bei 2000
rpm und vier Grad Celsius für 20 Minuten zentrifugiert. Zwei Aliquots von 500-
600 µl der zentrifugierten Blutprobe wurden in zwei Eppendorfgefäße pipettiert,
ebenso wie zwei Aliquots des Nabelschnurbluts von 200 µl. Der Blutkuchen der
beiden Proben wurde in der EDTA-Monotvette belassen. Anschließend wurde
alles bei -80 Grad Celsius bis zur weiteren Analyse gelagert.
Um die ADMA-Serum-Konzentration in der venösen Plasmaprobe der
Schwangeren und des Nabelschnurbluts zu bestimmen, wurde die im eigenen
Institut etablierte Liquid-Tandem- Massenspektrometrie (Liquid Chromatography
mass spectrometrie (LC-Ms/Ms)) verwendet (Schwedhelm 2007).
Nach Butylierung lagen ADMA, SDMA, und Arginin als entsprechende Ester vor.
Für die Bestimmung von ADMA, SDMA und Arginin wurde in einem „Multiple
Reaction Monitoring“ (MRM) der Übergang der Ionen m/z 259.3 zu m/z 214,

30
m/z 259.3 zu m/z 228, m/z 245.3 zu m/z 172 und m/z 231.3 zu m/z 70 verfolgt.
Die Quantifizierung erfolgte durch Zusatz eines internen deuterierten Standards.
Als interne Standards wurden d6-ADMA, und d7-Arginin zugesetzt.
Die Stocklösung des internen Standards wurde 1:1000 mit Methanol verdünnt.
Eine MultiScreen 96well-Filterplatte wurde auf einer 96well-„Unterplatte“
positioniert. Vom verdünnten Standard wurden jeweils 100 µl in die Vertiefungen
der MultiScreen 96well-Platte pipettiert, 25 µl Kalibrierlösung mit Plasma
zugegeben und die Platte bei Raumtemperatur 15 min geschüttelt.
Die MultiScreen 96well-Platte zusammen mit der Polypropylenplatte wurde bei
2000 rpm in einer Plattenzentrifuge für 15 min zentrifugiert. Nach Zentrifugation
wurde der Erfolg optisch kontrolliert. Die Polypropylenplatte wurde auf den
Heizblock gestellt, der zuvor auf 75°C geheizt wurde. Nach ca. 30 min (wenn
die Flüssigkeit komplett verdampft ist) wurde je 100 µL butanolische Salzsäure
zugegeben. Die Platte wurde mit einer Abdeckmatte verschlossen. Die
verschlossene Platte wurde bei 65°C 30 min lang auf dem Heizblock geheizt.
Danach wurde die Platte für 1 min bei 2500 rpm (4°C) zentrifugiert und die
Abdeckmatte wieder entfernt. Die butanolische Salzsäure wurde bei 75°C für
ca. 60 min auf der Heizplatte verdampft. Nach Abkühlen auf Raumtemperatur
wurde die Platte mit einer Abdeckmatte verschlossen und bei –20°C bis zur
Messung gelagert.
Vor der Messung wurde mit einer Mehrkanalpipette in jedes Well 100 µl
Methanol/Wasser 50/50 (v/v), pH 5 (gepuffert mit HCOOH/NH3), gegeben. Für
das Lösen der Analyten wurden die 96well-Platten mit einer Klebefolie
verschlossen und für 30 min (RT) geschüttelt. Anschließend wurden die Proben
auf eine neue MultiScreen 96well-Platte pipettiert (Mehrkanalpipette) und 5 min
bei 2000 rpm (4°C) zentrifugiert. Die Messung auf dem Varian L1200 MS/MS
Massenspektrometer erfolgte isokratisch mit den HPLC Varian ProStar
Pumpen, 66 % Acetonitril, 33 % (0,1 %ige) Ameisensäure.
Die Berechnung erfolgte durch die StarWorkstation. Der Variationskoeffizient
des Assays beträgt ca. 3%. Die Nachweisgrenze liegt bei 0.05 µM.

31
3.4. Genotypisierung
Für die Genotypisierung der mütterlichen und kindlichen Blutproben wurde aus
dem Blutkuchen mittels des Kits „NucleoSpin®- Blood Quick Pure“ (Macherey-
Nagel, Deutschland, Düren) die DNA isoliert.
Abbildung 6 NucleoSpin Blood Quick Pure
Quelle: http://www.mn-net.com/tabid/1344/default.aspx
Im ersten Schritt wurden 100 µl Blutkuchen mit 100 µl PBS verdünnt.
Jede Probe wurde anschließend in einem 1,5 ml Eppendorf Gefäß mit 25 µl
Proteinase K versetzt. Das Gemisch wurde mittels 200 µl BQ1-Puffer lysiert und
für 20 Sekunden gevortext. Nach einer 15-minütigen Inkubationszeit bei 70°
Grad waren die Blutproben lysiert. Es wurde jeweils 200 µl 96-100% Ethanol
hinzugefügt und 20 Sekunden gevortext.
Dieses Gemisch wurde auf die NucleoSpin®- Blood Quick Pure Säulen
gegeben und für eine Minute bei 11.000xg zentrifugiert.
Das Auffanggefäß mit dem Durchfluss wurde verworfen. Zur Auswaschung
wurden die Säulen in ein neues Gefäß eingesetzt und mit jeweils 350 µl BQ2
Puffer gewaschen und dann für drei Minuten bei 11.000xg zentrifugiert, dieser
Vorgang beinhaltetete auch die Trocknung der Silica Membran. Im letzten
Schritt wurde die DNA mit jeweils 50 µl Elution-Puffer, der bei 70° Grad
vorgewärmt wurde, in ein neues 1,5 ml Eppendorf Gefäß von der Membran
gelöst. Nach Inkubationszeit von einer Minute wurde es nochmals eine Minute
bei 11.000 x g zentrifugiert.

32
Danach wurde der DNA-Gehalt der Proben in ng/µl per Nano Drop ND-1000
Spectrophotometer ermittelt.
Die Proben wurden auf eine 96-Well-Platte aufgetragen, verdünnt jeweils zu
einer DNA-Konzentration von 100 ng/µl. Anschließend wurden die Proben für
die Amplifikation per RT-PCR auf eine 96-Well-Platte mit einer DNA-
Konzentration von 5 ng/µl verdünnt und aufgetragen.
Die Real-Time-quantitative PCR, ist eine Vervielfältigungsmethode basierend
auf der klassischen Methode, die zusätzlich die Quantifizierung der zu
amplifizierenden DNA erlaubt. In einer Polymerase-Kettenreaktion wird die zu
amplifizierende DNA (Template), eine thermostabile DNA- Polymerase mit
spezifischen Oligonucleotid-Primern, Puffer und Desoxynucleotidtriphosphate
benötigt. Es gibt drei PCR-Zyklen: Die Denaturierung des DNA-Doppelstrangs,
die Hybridisierung der Oligonucleotid-Primer an die einzelsträngige Template-
DNA (Annealing) und Verlängerung der Primer durch die Polymerase
(Elongation). Pro Zyklus wird die Template-DNA verdoppelt (Holland 1991). Um
zwischen den Allelen zu unterscheiden, sind unterschiedliche
Fluoreszenzfarbstoffe, wie FAM und VIC-Farben am 5’- Ende platziert. Die
allelische Diskriminierung hat zum Ziel die Proben als homozygot für Allel X,
homozygot für Allel Y oder als heterozygot zu klassifizieren. Die Auswertung der
Allelic-Discrimination-Plots wurde mit der Software SDS 2.2 (Fa. Applied
Biosystems) mittels einer aktivierten Auto-Caller-Funktion (CI: 95%)
durchgeführt. Das Ergebnis ist ein graphisches Koordinatensystem, welches
Allel X versus Allel Y dargestellt. Bei erfolgreicher Reaktion sind 3 Gruppen bzw.
Cluster zu sehen: jene der Homozygoten von Allel X oder Y und die
Heterozygoten XY. Die Zuordnung zur tatsächlichen Bezeichnung (Bsp. CC,
GG, CG) wurden vorher bestimmt.
Ein Beipspiel des Allelic-Discrimination-Plot anhand des SNPs DDAH1 rs
1146381 ist in Abbildung 7 zu sehen.
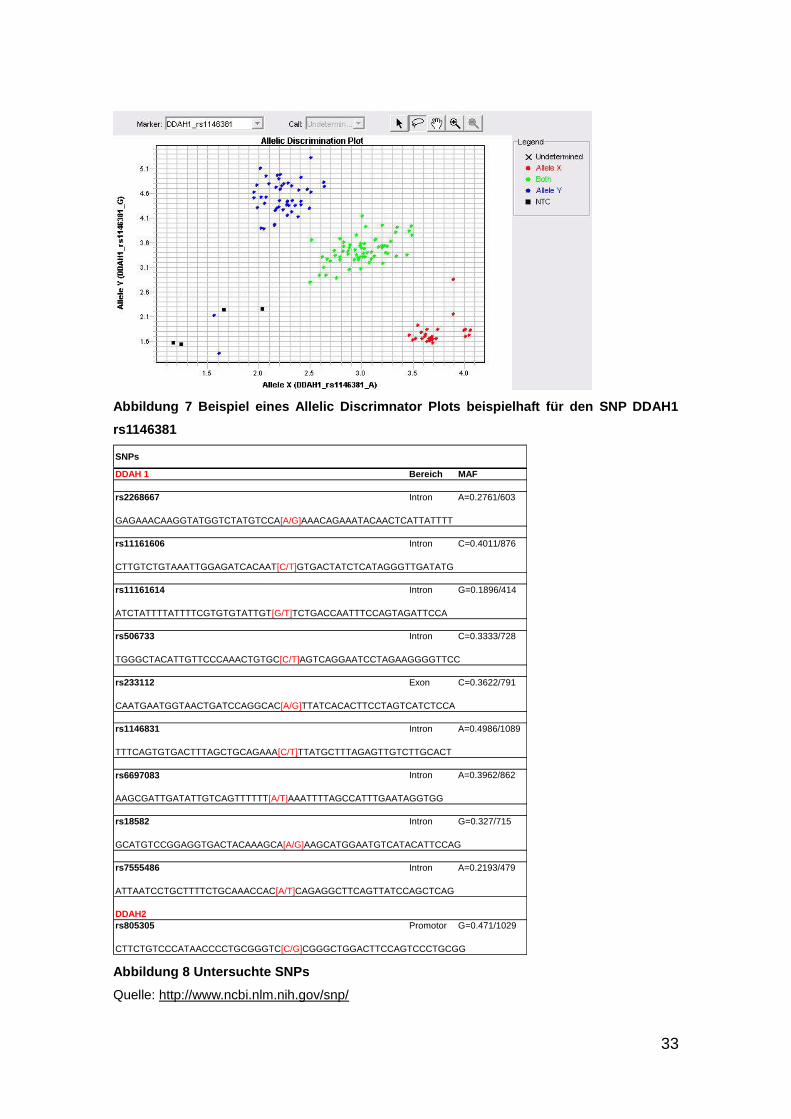
33
Abbildung 7 Beispiel eines Allelic Discrimnator Plots beispielhaft für den SNP DDAH1
rs1146381
DDAH 1 Bereich MAF
rs2268667 Intron A=0.2761/603
GAGAAACAAGGTATGGTCTATGTCCA[A/G]AAACAGAAATACAACTCATTATTTT
rs11161606 Intron C=0.4011/876
CTTGTCTGTAAATTGGAGATCACAAT[C/T]GTGACTATCTCATAGGGTTGATATG
rs11161614 Intron G=0.1896/414
ATCTATTTTATTTTCGTGTGTATTGT[G/T]TCTGACCAATTTCCAGTAGATTCCA
rs506733 Intron C=0.3333/728
TGGGCTACATTGTTCCCAAACTGTGC[C/T]AGTCAGGAATCCTAGAAGGGGTTCC
rs233112 Exon C=0.3622/791
CAATGAATGGTAACTGATCCAGGCAC[A/G]TTATCACACTTCCTAGTCATCTCCA
rs1146831 Intron A=0.4986/1089
TTTCAGTGTGACTTTAGCTGCAGAAA[C/T]TTATGCTTTAGAGTTGTCTTGCACT
rs6697083 Intron A=0.3962/862
AAGCGATTGATATTGTCAGTTTTTT[A/T]AAATTTTAGCCATTTGAATAGGTGG
rs18582 Intron G=0.327/715
GCATGTCCGGAGGTGACTACAAAGCA[A/G]AAGCATGGAATGTCATACATTCCAG
rs7555486 Intron A=0.2193/479
ATTAATCCTGCTTTTCTGCAAACCAC[A/T]CAGAGGCTTCAGTTATCCAGCTCAG
DDAH2
rs805305 Promotor G=0.471/1029
CTTCTGTCCCATAACCCCTGCGGGTC[C/G]CGGGCTGGACTTCCAGTCCCTGCGG
SNPs
Abbildung 8 Untersuchte SNPs
Quelle: http://www.ncbi.nlm.nih.gov/snp/
34
3.5. DDAH-Genexpressionsanalyse
Nach der Geburt der Plazenta wurden mittels Einmalskalpell drei ca. 1-2 cm
große Biopsate der Plazenta aus makroskopisch unauffälligen Bereichen
entnommen. Die Entnahme erfolgte jeweils von der maternalen Seite, von der
fetalen Seite und mittels eines Querschnitts durch das gesamte
Plazentagewebe. Die Biopsate wurden umgehend in flüssigem Stickstoff
eingefroren und anschließend bei -80 Grad Celsius gelagert. Im Mittel wurde
die Plazenta innerhalb von 30 min eingefroren. Aus logistischen Gründen war
dies nicht immer möglich, so dass wir in diesen Fällen ein
Konservierungsmedium (RNAlater, Qiagen- Deutschland, Hilden) benutzten,
mittels dessen man RNA im Gewebe stabilisieren kann. Die Biopsate wurden in
10 µl RNAlater pro 1 mg Gewebe in einem Eppendorfgefäß mindestens über
Nacht bei 2-8 Grad Celsius gelagert. Anschließend wurde das Gewebe aus
dem Medium entnommen und in einem weiteren Eppendorfgefäß bei -80 Grad
Celsius bis zur weiteren Verarbeitung gelagert.
Abbildung 9 Beispiele Plazenten bei der Probeentnahme
Zur Vorbereitung der Genexpressionsanalyse wurde die RNA mittels RNAzol
isoliert. Das Plazentagewebe wurde homogenisiert: 50-70 mg des Gewebes,
jeweils von maternaler und fetaler Seite wird mit 1ml RNAzol/50 mg Gewebe in
2 ml Eppendorf Tubes mit einer Wolfram-Kugel auf Eis gelagert um mittels des
Tissue Lyser für 2x 30 Sekunden bei 30 Hz homogenisiert zu werden.

35
Die Separation erfolgte mit 0,1ml Chloroform pro 1ml RNAzol, die Proben
wurden 30 Sekunden gevortext, 5 min auf Eis gelagert und anschließend bei
4°C mit 12.000 g 15 min zentrifugiert.
Dadurch trennte sich das Homogenat in 3 Phasen auf: die obere, farblose
wässrige Phase, in der sich RNA befand; die weiße Interphase und die blaue
Phenol-Chloroform-Phase, in denen sich dann DNA und Proteine befanden.
Der nächste Schritt sah dann die RNA-Präziptiation vor: die obere farblose
Phase wurde in ein 1,5 ml Eppendorf Tube überführt und mit 0,5 ml
Isopropanol, das auf
-20°C heruntergekühlt ist, vermischt. Dieses Gemisch wurde bei
Raumtemperatur für 15 min inkubiert, anschließend für 5 min bei 12.000 g bei
4°C zentrifugiert. Im Anschluss lag ein weißlich-gelbes Pellet aus RNA vor.
Der Überstand wurde vorsichtig abpipettiert und verworfen. Das RNA-Pellet
wurde mit 1 ml/50 mg Gewebe 70%igen Ethanol - initial der RNAzol-Menge -,
welcher ebenfalls bei -20°C gelagert wurde, gewaschen. Das Eppendorf Tube
wurde gevortext um das Pellet zu lösen und erneut für 5 min bei 7500 g bei 4°C
zentrifugiert, der Ethanolüberstand wurde abgekippt.
Das restliche Ethanol wurde aus dem Tube herausgewischt. Nach einer 2
minütigen Trockenzeit wurde das Pellet in 15 µl DEPC-Wasser („Ultra Pure
Water“) resuspendiert, kurz gevortext und zentrifugiert.
Die RNA-Menge wurde mittels des Nanodrops photometrisch bestimmt und auf
ihre Reinheit überprüft, die OD-Ratio sollte bei 260/280 bei 1,6 - 2 liegen.
Im nächsten Schritt zur Expressionsanalyse wurde die RNA mittels dem
Fermentas Revert Aid first strand cDNA synthesis kit nach Protokoll in cDNA
umgeschrieben.
Der Master-Mix bestand aus 5 x Reaction Buffer (4 µl), RiboLock™Rnase
Inhibitor (20u/µl) (1 µl), 10 mM dNTP Mix (2µl) und RevertAid™ M-MuLV
Reverse Transcriptase (200u/µl) (1µl).
11 µl PCR-Wasser wurde in die Wells vorgelegt, plus das RNA Template in der
Konzentration von 1µg/µl. Außerdem wurde 1 µl Random hexamer primer, plus
8 µl des Master Mix hinzugefügt. Anschließend wurde es vermischt und
gevortext und 1 min zentrifugiert. Die Platte wurde in einem PCR -Cycler für 5

36
min bei 25°C, 60 min bei 42°C und 5 min bei 70°C erhitzt, danach wurde die
Platte auf 4°C abgekühlt und konnte bei -80°C gelagert werden.
Im Weiteren wurde dann mit vorgefertigten Taqman-Sonden ein quantitatives
Expressionsassay in Form einer Real-Time PCR durchgeführt. Zu Beginn der
Real-Time-PCR wurden die Proben erhitzt und so die Doppelstrang-cDNA
denaturiert. Zu diesem Zeitpunkt ist die Fluoreszenz des Farbstoffs am 5’-Ende
nicht detektierbar, da sie vom Quencher MBG am 3’-Ende verhindert wird. Im
nächsten Schritt wird die Reaktionstemperatur vermindert und die Probe mit
dazugehörigen Primer kann an die Zielsequenz binden. Anschließend bildet die
Taq-Polymerase einen komplementären DNA-Strang. Wenn die Polymerase die
Taqman Probe erreicht, spaltet die endogene 5’ Nukleaseaktivität den Quencher
von der Probe und das Fluoreszenzsignal kann detektiert werden. Mit jedem
weiteren Zyklus der PCR wird das Signal verstärkt und so die Intensität des
Signals proportional zu der Menge an amplifizierten DNA detektiert.
Abbildung 10 Taqman RT-PCR
Quelle: Applied Biosystems
Jede cDNA-Probe wurde mit Wasser 1:8 verdünnt: 2µl Probe wurden mit 14µl
Wasser gemischt, gevortext und kurz zentrifugiert.

37
Die Reaktionsansätze wurden in Reaktionsgefäßen gemischt, diese
beinhalteten 2x qPCR Master Mix, 20x TaqMan Probe und 8 µl Wasser. In der
Negativkontrolle wurde die cDNA durch Apothekenwasser ersetzt. Der
Reaktionsansatz wurde zunächst ohne Probe gevortext, kurz zentrifugiert und
dann wurden je 8µl auf Wells der PCR-Platte verteilt. Danach wurden 2 µl c-
DNA bzw. Standard zugefügt. Die PCR-Platte lagerte hierbei auf Eis. Für jede
Probe wurden das Zielgen und das Referenzgen, in diesem Fall GAPDH, immer
auf einer Platte gemessen. Die PCR-Platte wurde nun mit der Folie versiegelt
und für 1 Min bei 750 rpm zentrifugiert. Dann wurde die Realtime-PCR mittels
des ABI PRISM 7900 HT Thermocycler durchgeführt. Die Quantifizierung der
DNA wird mittels Fluoreszenzsignal in der exponentiellen Phase der PCR
vorgenommen. Im Anschluss führten wir eine relative Quantifizierung der
Expression der Zielgene im relativen Vergleich zu der Kontrollgruppe, nach
folgendem Rechenschema durch: ∆Ct = Ct Zielgen – Ct Referenzgen
∆∆Ct = ∆Ct Patienten – ∆ Ct Kontrollen
Ratio = 2 ^-∆∆Ct (Kenneth 2001)
3.6. Statistische Auswertung
Die statistische Auswertung wurde mit IBM SPSS Statistics 20® für Windows
(Superior Performing Software Systems Inc., Chicago, USA) und GraphPad Prism
5® (Graph Pad Software Inc., La Jolla, USA) durchgeführt. Das Signifikanzniveau
wurde auf p < 0,05 festgelegt. Der Chiquadrat-Test wurde zur Überprüfung von
Zusammenhängen zweier nominaler Variablen verwendet. Zum Vergleich von
Mittelwerten wurde der T-Test angewandt, im Fall von mehr als 2 Gruppen,
benutzten wir den ANOVA-Test. Um zwischen den Gruppen Unterschiede
darstellen zu können, wurde bei Signifikanz zusätzlich der Bonferroni-Posthoc-
Test durchgeführt. Die Odds Ratio wurde mittels einer logistischen Regression
berechnet. Die Korrelationen wurden bei parametrischen Daten mittels der
Pearson´s und bei nicht-parametrischen Daten mittels Spearman´s
Korrelationen berechnet.
Vor der Genotyp-Datenanalyse wurde getestet, ob Abweichungen von dem
Hardy-Weinberg-Gleichgewicht (Hardy-Weinberg-Equilibrium; HWE) vorlag.

38
4. Ergebnisse
4.1. Basischarakteristika
In der vorliegenden Studie wurden insgesamt 73 Patientinnen unter
Berücksichtigung der Ein- und Ausschlusskriterien nach Aufklärung und
Einwilligung aufgenommen. Es handelte sich um 37 Frauen, die bei der
Entbindung gesund waren und 36 Frauen, die während ihrer Schwangerschaft
definitionsgemäß an einer Präeklampsie oder einem HELLP-Syndrom
erkrankten.
Die Frauen der Kontrollgruppe waren im Mittel 32 Jahre alt, fünf entbunden
spontan, 20 wurden aufgrund von Beckenendlage des Feten, wegen Vasa
praevia oder aus psychologischen Gründen per Sectio entbunden.
Zwölf Patientinnen der Präeklampsiegruppe litten zum Studieneinschluss an
einer leichten, 16 an einer schweren Präeklampsie und acht Frauen
entwickelten im Laufe ihrer Schwangerschaft ein HELLP-Syndrom. Die Frauen
der Präeklampsiegruppe waren im Mittel 33 Jahre alt. Bei elf Patientinnen
wurde die Geburt mit Hilfe von Misoprostol (Cytotec) eingeleitet und sie konnten
spontan entbunden werden. Vierzehn Patientinnen wurden per Sectio aufgrund
der Fortschreitung der Erkrankung, bzw. wegen der Gefährdung des Fetus
entbunden. Von 23 der 73 Patientinnen standen nur Blutproben, jedoch keine
Probe der Plazenta zur Analyse zur Verfügung. Die Patientinnen unterschieden
sich aufgrund des Matchings weder in Alter, Gewicht vor der Schwangerschaft,
noch im BMI vor der Schwangerschaft signifikant von der Kontrollgruppe
(Tabelle 3).
Definitionsgemäß unterschieden sich die Frauen der Präeklampsiegruppe von
der Kontrollgruppe in Bezug auf die systolischen, bzw. diastolischen
Blutdruckwerte, die Proteinurie, sowie das Auftreten von Ödemen. Sowohl der
systolische, als auch der diastolische Blutdruck unterschieden sich ebenfalls
zwischen den Subgruppen leichte Präeklampsie, schwere Präeklampsie und
HELLP (p<0,01). Die systolischen und diastolischen Blutdruckwerte stiegen mit
Ausprägungsgrad der Erkrankung im Vergleich zu den gesunden Schwangeren
an: Frauen mit leichter Präeklampsie hatten im Mittel einen Blutdruck von

39
148/97 mmHg, Frauen mit einer schweren Präeklampsie Werte von 172/106
mmHg, die HELLP-Patientinnen wiesen im Mittel Werte von 167/110 mmHg auf
(p<0,01).
Etablierter Risikofaktoren für die Entwicklung einer Präeklampsie sind die
Gewichtszunahme während der Schwangerschaft und die Nulliparität dar.
Zwischen den Kontrollen (+15,3 ± 4,6 kg) und der Präeklampsiegruppe (+17,32
± 6,6 kg) fand sich kein Unterschied in der Gewichtszunahme während der
Schwangerschaft (p=0,13). Je schwerer die Ausprägung von leichter (+17,6 ±
6,7 kg) bis zu schwerer Präeklampsie (+19,0 ± 4,5 kg), je mehr stieg die
Gewichtszunahme an (p=0,06), dies traf nicht auf die HELLP-Gruppe zu (+13,6
± 9,14 kg).
75% der Präeklampsiepatientinnen hatten noch nie ein Kind geboren. Die
Frauen mit einer schweren Präeklampsie waren zu 81,2% nullipara, die Frauen
mit leichter Präeklampsie zu 75%, ebenso wie die HELLP-Patientinnen. Die
Patientinnen in der Kontrollgruppe waren zu 43,2% nullipara.
Die an Präeklampsie-erkrankten Frauen brachten im Mittel ihr Kind bereits in
der 36. Schwangerschaftswoche zur Welt, im Gegensatz zu den Kontrollen, die
aufgrund von geplanten Sectiones, später, d.h. erst in der 38.
Schwangerschaftswoche entbunden wurden. In Subgruppen aufgeteilt,
entbunden jene mit leichter Präeklampsie im Mittel in der 38. Schwanger-
schaftswoche, die Patientinnen mit schwereren Ausprägung ca. in der 37.
Schwangerschaftswoche, die Frauen mit einem HELLP-Syndrom bereits in der
34. Schwangerschaftswoche (p< 0,01). Entsprechend dem Entbindungs-
zeitpunkt verhielt sich auch das Geburtsgewicht der Kinder. Je schwerer die
Frauen erkrankt waren, umso früher wurden die Kinder geboren und desto
niedriger war in Folge das Geburtsgewicht, bzw. die altersadaptierten Gewichts-
perzentilen (p< 0,01).
Hinsichtlich der Vorerkrankungen der eingeschlossenen Patientinnen bestand
im Wesentlichen kein Unterschied zwischen den Patientinnengruppen: es
waren keine Herz- oder ZNS-Erkrankungen bekannt; eine Patientin aus der
Kontrollgruppe hatte eine Hepatits B, eine weitere aus dieser Gruppe hatte zum

40
Zeitpunkt der Schwangerschaft einen Harnstau. Alle anderen Patientinnen
hatten überdies keine weiteren Nieren- oder Lebererkrankungen.
Nach WHO-Definition waren zehn Patientinnen adipös, fünf jeweils aus beiden
Gruppen (p=0,96). Unter einer Hypertonie, die bereits vor der Schwangerschaft
vorlag, litten insgesamt drei Frauen, zwei in der Gruppe der Präeklampsie-
patientinnen und eine in der Kontrollgruppe (p=0,54). Eine diagnostizierte
Hypothyreose lag bei 14 Patientinnen vor, fünf in der Kontrollgruppe, neun unter
den Patientinnen der Präeklampsiegruppe (p=0,21), allerdings nahmen 17
Patientinnen während der Schwangerschaft L-Thyroxin ein (p=0,37).
Eine Patientin der Kontrollgruppe hatte einen vorbestehenden insulinpflichtigen
Diabetes. Vier Patientinnen haben während der Schwangerschaft geraucht, drei
in der Gruppe der Präeklampsie-Patientinnen, eine aus der Kontrollgruppe
(p=0,29).
In den dokumentierten Vorsorgeuntersuchungen zeigte sich bei neun
Patientinnen ein einseitig erhöhter Widerstand der Arteriae uterina, davon
sieben in der Präeklampsie-, zwei in der Kontrollgruppe. Bei sechs Patientinnen
wurde ein beidseitig erhöhter Widerstand gemessen, die alle zur Gruppe der
Präeklampsie-Patientinnen gehörten (p=0,04). Die beidseitig erhöhten uterinen
Wiederstände mit postsystolischer Inzisur (notch) in den Arteriae uterina, die als
Risikomarker für eine Minderversorgung der Plazenta gelten, traten nur in der
Gruppe der Frauen mit schwerer Präeklampsie (n=4) und HELLP-Syndrom
(n=2) auf (p=0,04).
Die Anamnese der Patientinnen bezüglich vorheriger Schwangerschaften
zeigte, dass 14 Patientinnen eine oder mehrere Fehlgeburten hatten: vier aus
der Präeklampsie- und neun aus der Kontrollgruppe (p=0,12). Vier Frauen
waren bereits in der Vorgeschichte an einer Präeklampsie erkrankt, jeweils zwei
aus den Gruppen (p=0,978). Fünf der Präeklampsie-Patientinnen hatten in der
vorherigen Schwangerschaft ein HELLP-Syndrom entwickelt (p=0,02). Elf
Patientinnen hatten einen Gestationsdiabetes in der vorherigen
Schwangerschaft, fünf aus der Kontrollgruppe, sechs aus der
Präeklampsiegruppe (p=0,71).

41
Zehn Patientinnen entwickelten in der aktuellen Schwangerschaft einen
Gestationsdiabetes, vier aus der Kontrollgruppe und sechs aus der
Präeklampsiegruppe (drei mit leichter PE, zwei mit schwerer PE, eine der
HELLP-Patientinnen) (p= 0,35).
Elf Prozent (n=8) der eingeschlossenen Frauen, vier aus den jeweiligen
Gruppen, mussten in der aktuellen Schwangerschaft Insulin applizieren
(p=0,97).
19 der Präeklampsiepatientinnen mussten zum Zeitpunkt des Studien-
einschlusses oder im Verlauf wegen starken Bluthochdrucks mit α-Methyldopa
und zusätzlich bei Blutdruckspitzen mit Nifedipin behandelt werden.
Kontrollen
(n= 37)
Präeklampsie
(n=36)p-Wert
leichte
Präeklampsie
(n=12)
schwere
Präeklampsie
(n=16)
HELLP (n=8) p-Wert
Alter 32,49 ± 4,9 32,97 ± 4,1 0,65 33,3 ± 4,5 31,8 ± 3,4 35,0 ± 4,6 0,39
Gewicht vor der SS in kg 67,4 ± 14,8 68,8 ± 17,1 0,71 76,3 ± 22,0 63,9 ± 5,9 67,5 ± 21,7 0,23
BMI vor der SS in kg/m² 24,13 ± 5,2 24,64 ± 6,1 0,68 27,4 ± 7,9 22,9 ± 3,3 24,0 ± 6,9 0,20
Gewichtszunahme in der
SS in kg15,3 ± 4,6 17,32 ± 6,6 0,13 17,6 ± 6,7 19,0 ± 4,5 13,6 ± 9,14 0,06
Nulliparität n% 16 (43,2%) 28 (77,8%) - 9 (75,0%) 13 (81,2%) 6 (75,0%) -
RR sys mmHG bei
Aufnahme118,1 ± 12,7 163 ± 19,3 <0,01 147,8 ± 7,7 171,5 ± 16,7 167,9 ± 24,2 <0,01
RR dias mmHG bei
Aufnahme68 ± 11,5 103,7 ± 13,1 <0,01 97,0 ± 6,0 105,8 ± 10,8 109,5 ± 20,7 <0,01
Geburtsgewicht des
Kindes in g3297,8 ± 530,4 2510 ± 878,5 <0,01 2987,7 ± 678,3 2545,1 ± 686,1 1723,4 ± 1013,0 <0,01
Gewichtsperzentile des
Kindes bei Geburt52,49 ± 28,0 28,09 ± 26,25 <0,01 40,0 ± 31,9 25,3 ± 24,2 15,3 ± 11,7 <0,01
SSW bei Blutentnahme 37,41 ± 6,4 36,17 ± 4,1 0,33 37,9 ± 2,1 36,9 ± 2,5 32,0 ± 5,9 0,06
SSW bei Geburt 38 ± 1,2 36 ± 3,5 <0,01 38,1 ± 2,0 36,8 ± 2,5 33,0 ± 5,0 <0,01
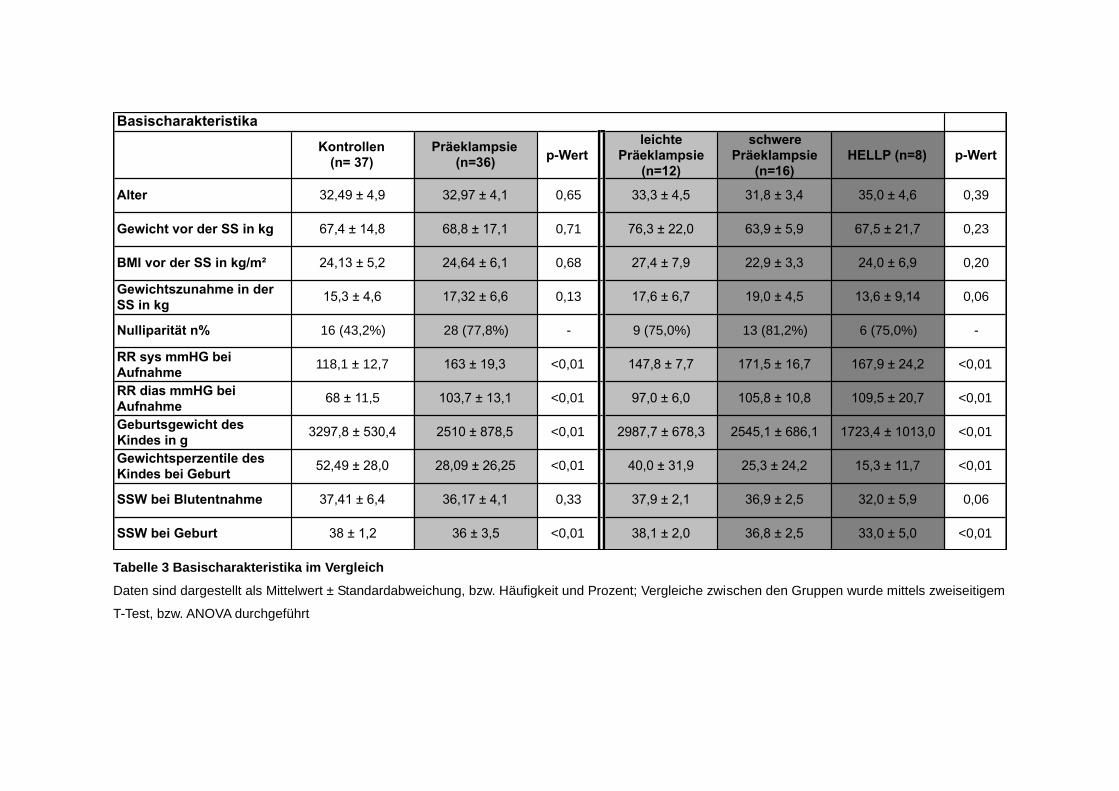
Basischarakteristika
Tabelle 3 Basischarakteristika im Vergleich
Daten sind dargestellt als Mittelwert ± Standardabweichung, bzw. Häufigkeit und Prozent; Vergleiche zwischen den Gruppen wurde mittels zweiseitigem
T-Test, bzw. ANOVA durchgeführt
43
Kontrollen Präeklampsie p
Adipostias 5 (13,5%) 5 (13,9%) 0,96
Blutungs-, bzw. Thromboseneigung 0 1 (2,8%) 0,31
Diabetes 1 (2,7%) 0 0,32
Einnahme L-Thyroxin 7 (18,9%) 10 (27,8%) 0,37
Einnahme von a-Methyldopa 0 19 (52,8%) <0,01
Eklampsie 0 1 (2,8%) 0,31
Fehlgeburten (eine oder mehrere) 9 (24,3%) 4 (13,9%) 0,12
Gestationsdiabetes 3 (8,3%) 6 (16,7%) 0,29
Gestationsdiabetes in vorherigen SS 5 (13,5%) 6 (16,7%) 0,71
HELLP in vorherigen SS 0 5 (13,9%) 0,02
Herzerkrankungen 0 0 -
Hypertonie 1 (2,7%) 2 (5,6%) 0,54
Hypothyreose 5 (13,5%) 9 (25%) 0,21
Insulinapplikation 4 (10,8%) 4 (11,1%) 0,97
Lebererkrankungen 1 (2,7%) 0 0,32
Nierenerkankungen 1 (2,7%) 0 0,32
Präeklampsie in vorherigen SS 2 (5,4%) 2 (5,6%) 0,98
Rauchen 1 (2,7%) 3 (8,3%) 0,29
ZNS-Erkankungen 0 0 -
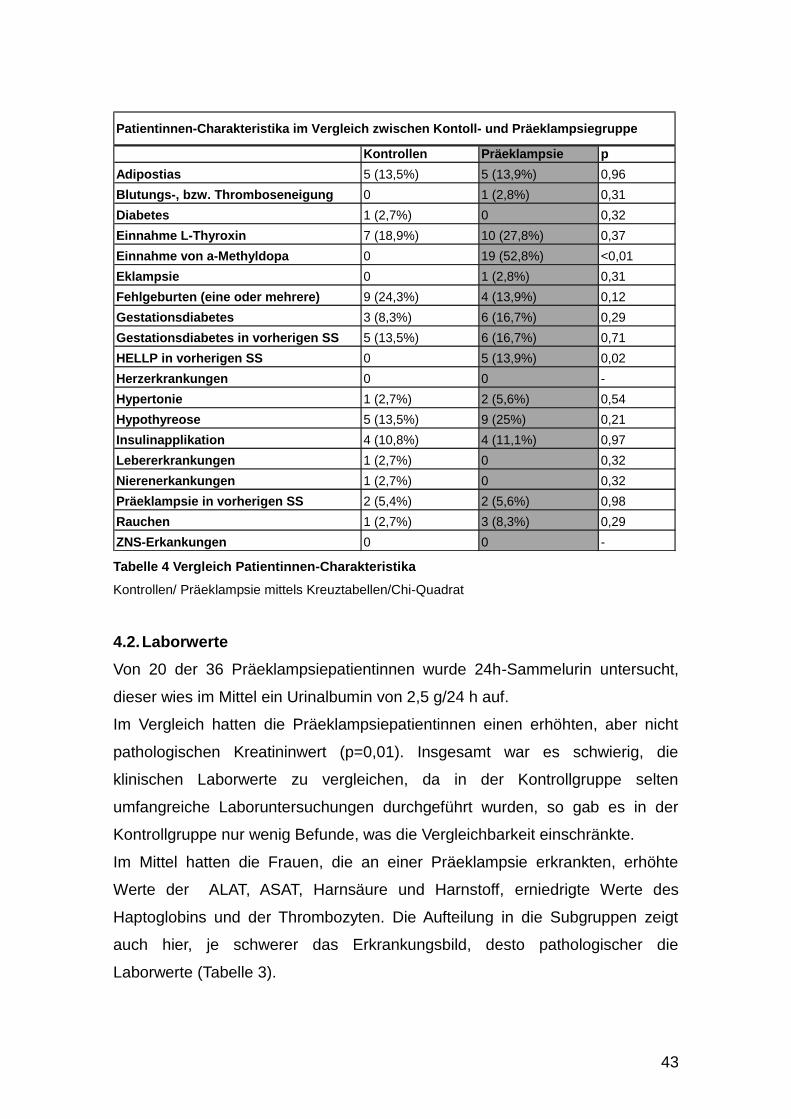
Patientinnen-Charakteristika im Vergleich zwischen Kontoll- und Präeklampsiegruppe
Tabelle 4 Vergleich Patientinnen-Charakteristika
Kontrollen/ Präeklampsie mittels Kreuztabellen/Chi-Quadrat
4.2. Laborwerte
Von 20 der 36 Präeklampsiepatientinnen wurde 24h-Sammelurin untersucht,
dieser wies im Mittel ein Urinalbumin von 2,5 g/24 h auf.
Im Vergleich hatten die Präeklampsiepatientinnen einen erhöhten, aber nicht
pathologischen Kreatininwert (p=0,01). Insgesamt war es schwierig, die
klinischen Laborwerte zu vergleichen, da in der Kontrollgruppe selten
umfangreiche Laboruntersuchungen durchgeführt wurden, so gab es in der
Kontrollgruppe nur wenig Befunde, was die Vergleichbarkeit einschränkte.
Im Mittel hatten die Frauen, die an einer Präeklampsie erkrankten, erhöhte
Werte der ALAT, ASAT, Harnsäure und Harnstoff, erniedrigte Werte des
Haptoglobins und der Thrombozyten. Die Aufteilung in die Subgruppen zeigt
auch hier, je schwerer das Erkrankungsbild, desto pathologischer die
Laborwerte (Tabelle 3).
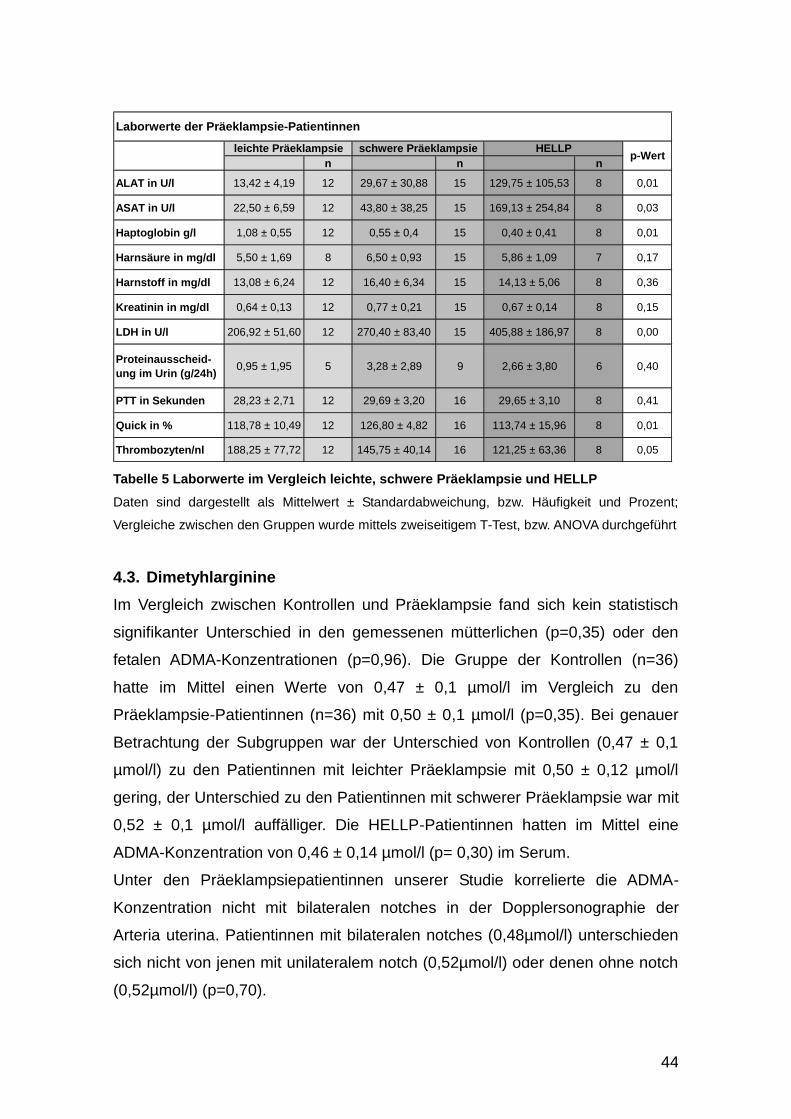
44
n n n
ALAT in U/l 13,42 ± 4,19 12 29,67 ± 30,88 15 129,75 ± 105,53 8 0,01
ASAT in U/l 22,50 ± 6,59 12 43,80 ± 38,25 15 169,13 ± 254,84 8 0,03
Haptoglobin g/l 1,08 ± 0,55 12 0,55 ± 0,4 15 0,40 ± 0,41 8 0,01
Harnsäure in mg/dl 5,50 ± 1,69 8 6,50 ± 0,93 15 5,86 ± 1,09 7 0,17
Harnstoff in mg/dl 13,08 ± 6,24 12 16,40 ± 6,34 15 14,13 ± 5,06 8 0,36
Kreatinin in mg/dl 0,64 ± 0,13 12 0,77 ± 0,21 15 0,67 ± 0,14 8 0,15
LDH in U/l 206,92 ± 51,60 12 270,40 ± 83,40 15 405,88 ± 186,97 8 0,00
Proteinausscheid-
ung im Urin (g/24h)0,95 ± 1,95 5 3,28 ± 2,89 9 2,66 ± 3,80 6 0,40
PTT in Sekunden 28,23 ± 2,71 12 29,69 ± 3,20 16 29,65 ± 3,10 8 0,41
Quick in % 118,78 ± 10,49 12 126,80 ± 4,82 16 113,74 ± 15,96 8 0,01
Thrombozyten/nl 188,25 ± 77,72 12 145,75 ± 40,14 16 121,25 ± 63,36 8 0,05
Laborwerte der Präeklampsie-Patientinnen
HELLPp-Wert
leichte Präeklampsie schwere Präeklampsie
Tabelle 5 Laborwerte im Vergleich leichte, schwere Präeklampsie und HELLP
Daten sind dargestellt als Mittelwert ± Standardabweichung, bzw. Häufigkeit und Prozent;
Vergleiche zwischen den Gruppen wurde mittels zweiseitigem T-Test, bzw. ANOVA durchgeführt
4.3. Dimetyhlarginine
Im Vergleich zwischen Kontrollen und Präeklampsie fand sich kein statistisch
signifikanter Unterschied in den gemessenen mütterlichen (p=0,35) oder den
fetalen ADMA-Konzentrationen (p=0,96). Die Gruppe der Kontrollen (n=36)
hatte im Mittel einen Werte von 0,47 ± 0,1 µmol/l im Vergleich zu den
Präeklampsie-Patientinnen (n=36) mit 0,50 ± 0,1 µmol/l (p=0,35). Bei genauer
Betrachtung der Subgruppen war der Unterschied von Kontrollen (0,47 ± 0,1
µmol/l) zu den Patientinnen mit leichter Präeklampsie mit 0,50 ± 0,12 µmol/l
gering, der Unterschied zu den Patientinnen mit schwerer Präeklampsie war mit
0,52 ± 0,1 µmol/l auffälliger. Die HELLP-Patientinnen hatten im Mittel eine
ADMA-Konzentration von 0,46 ± 0,14 µmol/l (p= 0,30) im Serum.
Unter den Präeklampsiepatientinnen unserer Studie korrelierte die ADMA-
Konzentration nicht mit bilateralen notches in der Dopplersonographie der
Arteria uterina. Patientinnen mit bilateralen notches (0,48µmol/l) unterschieden
sich nicht von jenen mit unilateralem notch (0,52µmol/l) oder denen ohne notch
(0,52µmol/l) (p=0,70).

45
12 der 36 der untersuchten Patientinnen der Präeklampsiegruppe brachten ein
Small-for-gestational-age-Baby zur Welt, d.h. mit einer Gewichtsperzentile <10.
Die ADMA-Konzentrationen unterschieden sich zwischen denen mit SGA (0,55
µmol/l) und ohne SGA (0,47 µmol/l) (p=0,04), allerdings nicht gleichsinnig der
Daten von Rizos et al. (Rizos et al. 2012).
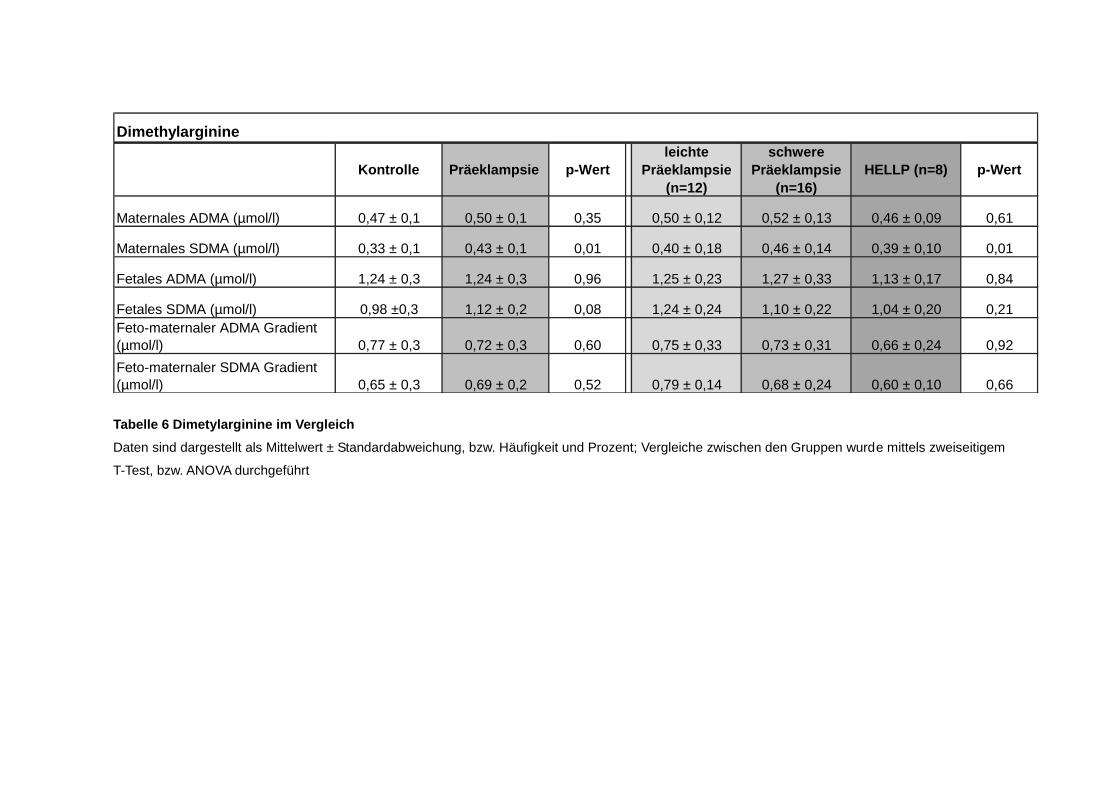
Kontrolle Präeklampsie p-Wert
leichte
Präeklampsie
(n=12)
schwere
Präeklampsie
(n=16)
HELLP (n=8) p-Wert
Maternales ADMA (µmol/l) 0,47 ± 0,1 0,50 ± 0,1 0,35 0,50 ± 0,12 0,52 ± 0,13 0,46 ± 0,09 0,61
Maternales SDMA (µmol/l) 0,33 ± 0,1 0,43 ± 0,1 0,01 0,40 ± 0,18 0,46 ± 0,14 0,39 ± 0,10 0,01
Fetales ADMA (µmol/l) 1,24 ± 0,3 1,24 ± 0,3 0,96 1,25 ± 0,23 1,27 ± 0,33 1,13 ± 0,17 0,84
Fetales SDMA (µmol/l) 0,98 ±0,3 1,12 ± 0,2 0,08 1,24 ± 0,24 1,10 ± 0,22 1,04 ± 0,20 0,21
Feto-maternaler ADMA Gradient
(µmol/l) 0,77 ± 0,3 0,72 ± 0,3 0,60 0,75 ± 0,33 0,73 ± 0,31 0,66 ± 0,24 0,92
Feto-maternaler SDMA Gradient
(µmol/l) 0,65 ± 0,3 0,69 ± 0,2 0,52 0,79 ± 0,14 0,68 ± 0,24 0,60 ± 0,10 0,66
Dimethylarginine
Tabelle 6 Dimetylarginine im Vergleich
Daten sind dargestellt als Mittelwert ± Standardabweichung, bzw. Häufigkeit und Prozent; Vergleiche zwischen den Gruppen wurde mittels zweiseitigem
T-Test, bzw. ANOVA durchgeführt
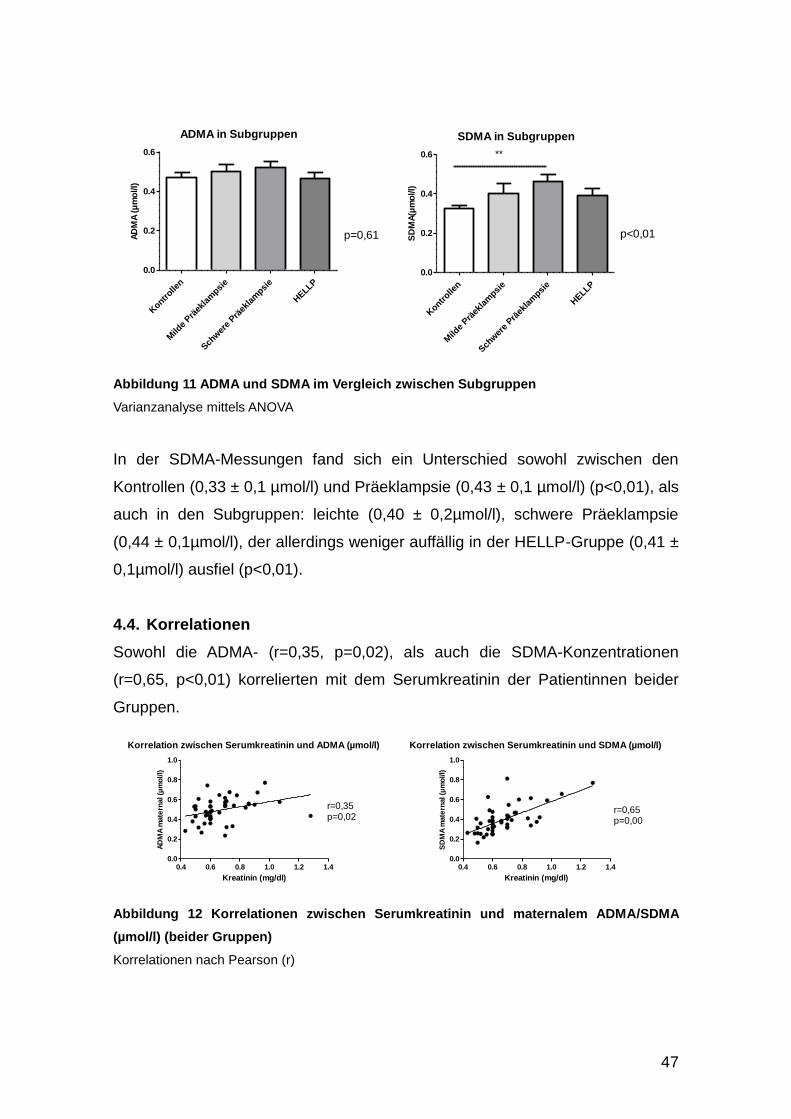
47
ADMA in Subgruppen
Kontr
ollen
Mild
e Prä
ekla
mpsi
e
Schwer
e Prä
ekla
mpsi
e
HELLP
0.0
0.2
0.4
0.6
p=0,61AD
MA
(µ
mo
l/l)
SDMA in Subgruppen
Kontr
ollen
Mild
e Prä
ekla
mpsi
e
Schwer
e Prä
ekla
mpsi
e
HELLP
0.0
0.2
0.4
0.6 **
p<0,01SD
MA
(µm
ol/l)
Abbildung 11 ADMA und SDMA im Vergleich zwischen Subgruppen
Varianzanalyse mittels ANOVA
In der SDMA-Messungen fand sich ein Unterschied sowohl zwischen den
Kontrollen (0,33 ± 0,1 µmol/l) und Präeklampsie (0,43 ± 0,1 µmol/l) (p<0,01), als
auch in den Subgruppen: leichte (0,40 ± 0,2µmol/l), schwere Präeklampsie
(0,44 ± 0,1µmol/l), der allerdings weniger auffällig in der HELLP-Gruppe (0,41 ±
0,1µmol/l) ausfiel (p<0,01).
4.4. Korrelationen
Sowohl die ADMA- (r=0,35, p=0,02), als auch die SDMA-Konzentrationen
(r=0,65, p<0,01) korrelierten mit dem Serumkreatinin der Patientinnen beider
Gruppen.
Korrelation zwischen Serumkreatinin und ADMA (µmol/l)
0.4 0.6 0.8 1.0 1.2 1.40.0
0.2
0.4
0.6
0.8
1.0
r=0,35p=0,02
Kreatinin (mg/dl)
AD
MA
mate
rnal (µ
mo
l/l)
Korrelation zwischen Serumkreatinin und SDMA (µmol/l)
0.4 0.6 0.8 1.0 1.2 1.40.0
0.2
0.4
0.6
0.8
1.0
r=0,65p=0,00
Kreatinin (mg/dl)
SD
MA
mate
rnal (µ
mo
l/l)
Abbildung 12 Korrelationen zwischen Serumkreatinin und maternalem ADMA/SDMA
(µmol/l) (beider Gruppen)
Korrelationen nach Pearson (r)

48
Bei der Korrelation von ADMA mit verschiedenen klinischen Parametern,
ergaben sich in der Kontrollgruppe signifikante Zusammenhänge zwischen
ADMA und BMI (r=0,35, p=0,04) und zwischen ADMA und dem Gewicht in kg
bei Studieneinschluss, bzw. bei Blutentnahme (r=0,39, p= 0,02). Außerdem
korrelierten in dieser Gruppe Geburtsgewicht und altersadaptierte
Gewichtsperzentile des Kindes positiv mit mütterlichem ADMA (r= 0,51, p<0,00
bzw. r=0,40, p<0,01).
In der Gruppe der Präeklampsiepatientinnen ergaben sich positive
Korrelationen von maternalem SDMA mit dem BMI zum Zeitpunkt der
Blutentnahme (r=0,41, p<0,01), bzw. Gewicht zum Zeitpunkt der Blutentnahme
(r=0,35, p<0,01). Bei der Korrelation von Kreatinin und maternalem ADMA
stellte sich in dieser Gruppe ein positiver Zusammenhang dar (r=0,35, p=0,02),
ebenso wie bei Kreatinin und SDMA (r=0,63, p<0,01), bei der Korrelation von
der GFR ergab sich zu SDMA ein negativer Zusammenhang (r=-0,56, p=0,04).
Die maternalen SDMA-Konzentrationen in der Präeklampsiegruppe korrelierten
mit negativ mit der Gewichtsperzentile der Kinder (r= -0,35, p=0,05).
Zwischen den fetalen ADMA-Konzentrationen und den fetalen
Gewichtsperzentilen konnten wir ebenfalls einen Zusammenhang feststellen
(r=0,56, p<0,01).
Die fetalen SDMA-Konzentrationen korrelierten zudem mit dem mütterlichen
BMI zum Zeitpunkt der Blutentnahme (r=0,52, p=0,02), bzw. Gewicht zum
Zeitpunkt der Blutentnahme (r=0,56, p<0,01).
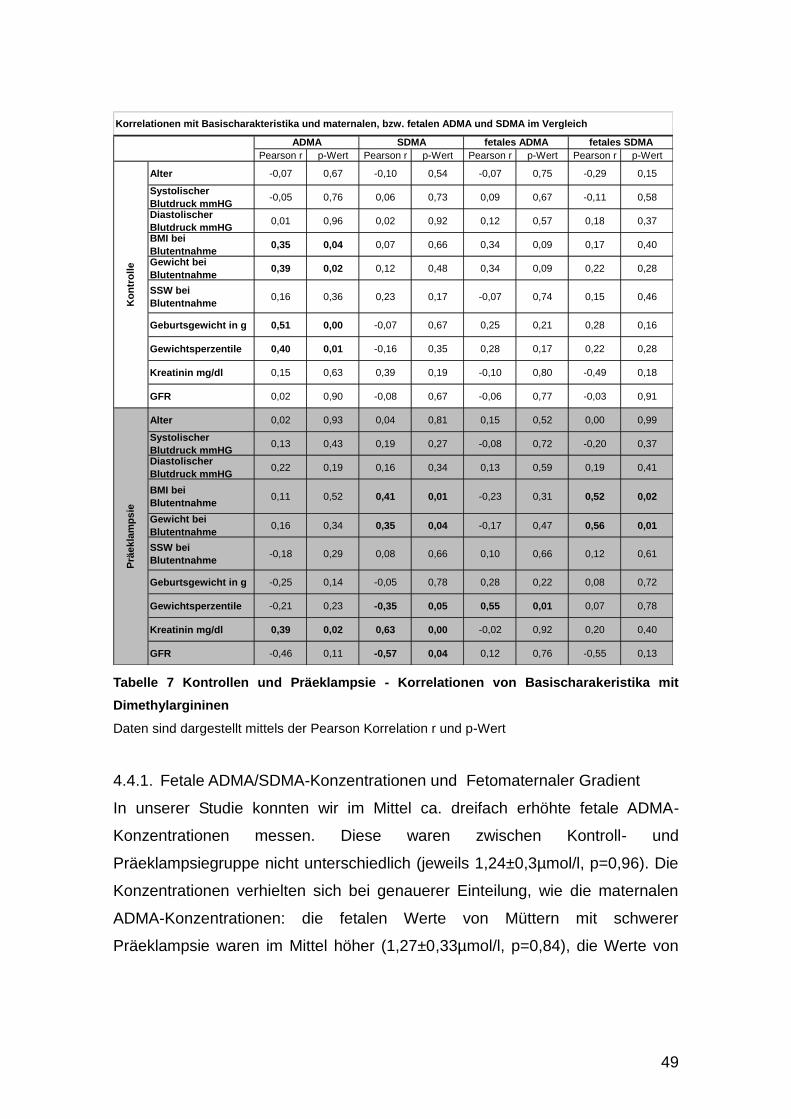
49
Pearson r p-Wert Pearson r p-Wert Pearson r p-Wert Pearson r p-Wert
Alter -0,07 0,67 -0,10 0,54 -0,07 0,75 -0,29 0,15
Systolischer
Blutdruck mmHG-0,05 0,76 0,06 0,73 0,09 0,67 -0,11 0,58
Diastolischer
Blutdruck mmHG0,01 0,96 0,02 0,92 0,12 0,57 0,18 0,37
BMI bei
Blutentnahme0,35 0,04 0,07 0,66 0,34 0,09 0,17 0,40
Gewicht bei
Blutentnahme0,39 0,02 0,12 0,48 0,34 0,09 0,22 0,28
SSW bei
Blutentnahme0,16 0,36 0,23 0,17 -0,07 0,74 0,15 0,46
Geburtsgewicht in g 0,51 0,00 -0,07 0,67 0,25 0,21 0,28 0,16
Gewichtsperzentile 0,40 0,01 -0,16 0,35 0,28 0,17 0,22 0,28
Kreatinin mg/dl 0,15 0,63 0,39 0,19 -0,10 0,80 -0,49 0,18
GFR 0,02 0,90 -0,08 0,67 -0,06 0,77 -0,03 0,91
Alter 0,02 0,93 0,04 0,81 0,15 0,52 0,00 0,99
Systolischer
Blutdruck mmHG0,13 0,43 0,19 0,27 -0,08 0,72 -0,20 0,37
Diastolischer
Blutdruck mmHG0,22 0,19 0,16 0,34 0,13 0,59 0,19 0,41
BMI bei
Blutentnahme0,11 0,52 0,41 0,01 -0,23 0,31 0,52 0,02
Gewicht bei
Blutentnahme0,16 0,34 0,35 0,04 -0,17 0,47 0,56 0,01
SSW bei
Blutentnahme-0,18 0,29 0,08 0,66 0,10 0,66 0,12 0,61
Geburtsgewicht in g -0,25 0,14 -0,05 0,78 0,28 0,22 0,08 0,72
Gewichtsperzentile -0,21 0,23 -0,35 0,05 0,55 0,01 0,07 0,78
Kreatinin mg/dl 0,39 0,02 0,63 0,00 -0,02 0,92 0,20 0,40
GFR -0,46 0,11 -0,57 0,04 0,12 0,76 -0,55 0,13
Prä
ekla
mp
sie
Ko
ntr
oll
e
ADMA
Korrelationen mit Basischarakteristika und maternalen, bzw. fetalen ADMA und SDMA im Vergleich
SDMA fetales ADMA fetales SDMA
Tabelle 7 Kontrollen und Präeklampsie - Korrelationen von Basischarakeristika mit
Dimethylargininen
Daten sind dargestellt mittels der Pearson Korrelation r und p-Wert
4.4.1. Fetale ADMA/SDMA-Konzentrationen und Fetomaternaler Gradient
In unserer Studie konnten wir im Mittel ca. dreifach erhöhte fetale ADMA-
Konzentrationen messen. Diese waren zwischen Kontroll- und
Präeklampsiegruppe nicht unterschiedlich (jeweils 1,24±0,3µmol/l, p=0,96). Die
Konzentrationen verhielten sich bei genauerer Einteilung, wie die maternalen
ADMA-Konzentrationen: die fetalen Werte von Müttern mit schwerer
Präeklampsie waren im Mittel höher (1,27±0,33µmol/l, p=0,84), die Werte von
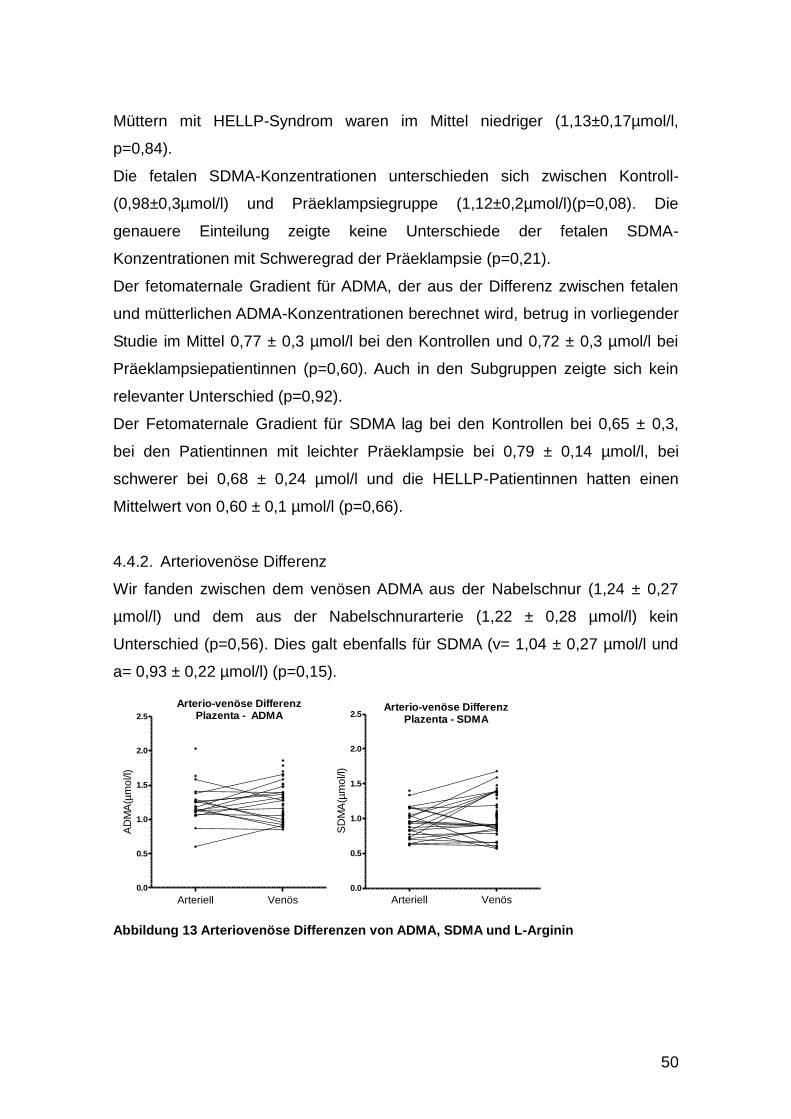
50
Müttern mit HELLP-Syndrom waren im Mittel niedriger (1,13±0,17µmol/l,
p=0,84).
Die fetalen SDMA-Konzentrationen unterschieden sich zwischen Kontroll-
(0,98±0,3µmol/l) und Präeklampsiegruppe (1,12±0,2µmol/l)(p=0,08). Die
genauere Einteilung zeigte keine Unterschiede der fetalen SDMA-
Konzentrationen mit Schweregrad der Präeklampsie (p=0,21).
Der fetomaternale Gradient für ADMA, der aus der Differenz zwischen fetalen
und mütterlichen ADMA-Konzentrationen berechnet wird, betrug in vorliegender
Studie im Mittel 0,77 ± 0,3 µmol/l bei den Kontrollen und 0,72 ± 0,3 µmol/l bei
Präeklampsiepatientinnen (p=0,60). Auch in den Subgruppen zeigte sich kein
relevanter Unterschied (p=0,92).
Der Fetomaternale Gradient für SDMA lag bei den Kontrollen bei 0,65 ± 0,3,
bei den Patientinnen mit leichter Präeklampsie bei 0,79 ± 0,14 µmol/l, bei
schwerer bei 0,68 ± 0,24 µmol/l und die HELLP-Patientinnen hatten einen
Mittelwert von 0,60 ± 0,1 µmol/l (p=0,66).
4.4.2. Arteriovenöse Differenz
Wir fanden zwischen dem venösen ADMA aus der Nabelschnur (1,24 ± 0,27
µmol/l) und dem aus der Nabelschnurarterie (1,22 ± 0,28 µmol/l) kein
Unterschied (p=0,56). Dies galt ebenfalls für SDMA (v= 1,04 ± 0,27 µmol/l und
a= 0,93 ± 0,22 µmol/l) (p=0,15).
Arterio-venöse DifferenzPlazenta - ADMA
Arteriell Venös
0.0
0.5
1.0
1.5
2.0
2.5
AD
MA
(µm
ol/l
)
Arteriell Venös0.0
0.5
1.0
1.5
2.0
2.5Arterio-venöse Differenz
Plazenta - SDMA
SD
MA
(µm
ol/l
)
Abbildung 13 Arteriovenöse Differenzen von ADMA, SDMA und L-Arginin

51
4.5. SNPs
Homozygote Trägerinnen des minor allels des SNP rs2268667 der DDAH1
zeigten erhöhte ADMA-Konzentrationen (0,54µmol/l) im Vergleich zu den
homozygoten Trägerinnen des major Allels (0,44 µmol/l) und den heterozygote
Trägerinnen (0,51µmol/l) (p=0,03). Es zeigte sich jedoch keine Assoziation zur
Ausprägung der Präeklampsie. In unserer Studie gab es sieben homozygote
Trägerinnen des minor allels für diesen SNP (fünf der Kontrollgruppe und zwei
der Präeklampsiegruppe).
Die Trägerinnen einer Heterozygotie (CG) des SNP der DDAH2 rs805305
hatten ein erhöhtes Risiko der Erkrankung an einer Präeklampsie mit einer
Odds Ratio von 3,0 (CI 1,01-8,86) (p=0,05). 56,3% der an Präeklampsie-
erkrankten Patientinnen trugen diese Heterozygotie (n=18). Mit Trägerinnen des
major allels konnten wir keinen Zusammenhang darstellen, auch im Modell
unter der Annahme einer Dominanz des major allels nicht.
Der Einfluss von fetalen SNPs auf maternale ADMA-Konzentrationen ergab
einen Zusammenhang des SNPs rs11161606 der DDAH1. Feten, die
homozygot das minor allel (CC) trugen, waren mit erhöhten mütterlichen
ADMA-Konzentrationen assoziiert (0,61µmol/l, p= 0,08). Ähnliches war bei den
maternalen SNPs zu beobachten, Trägerinnen des minor allels CC hatten
erhöhte ADMA-Konzentrationen (0,51 µmol/l) im Unterschied zu homozygoten
Trägerinnen des major allels (0, 43 µmol/l) (p=0,08).
Ein signifikanter Zusammenhang ergab sich bei dem SNP rs506733 in
Zusammenhang mit dem Auftreten einer Präeklampsie, alle untersuchten
Feten, der Mütter mit Präeklampsie trugen eine Homozygotie für das major allel
TT)(Tabelle 8).
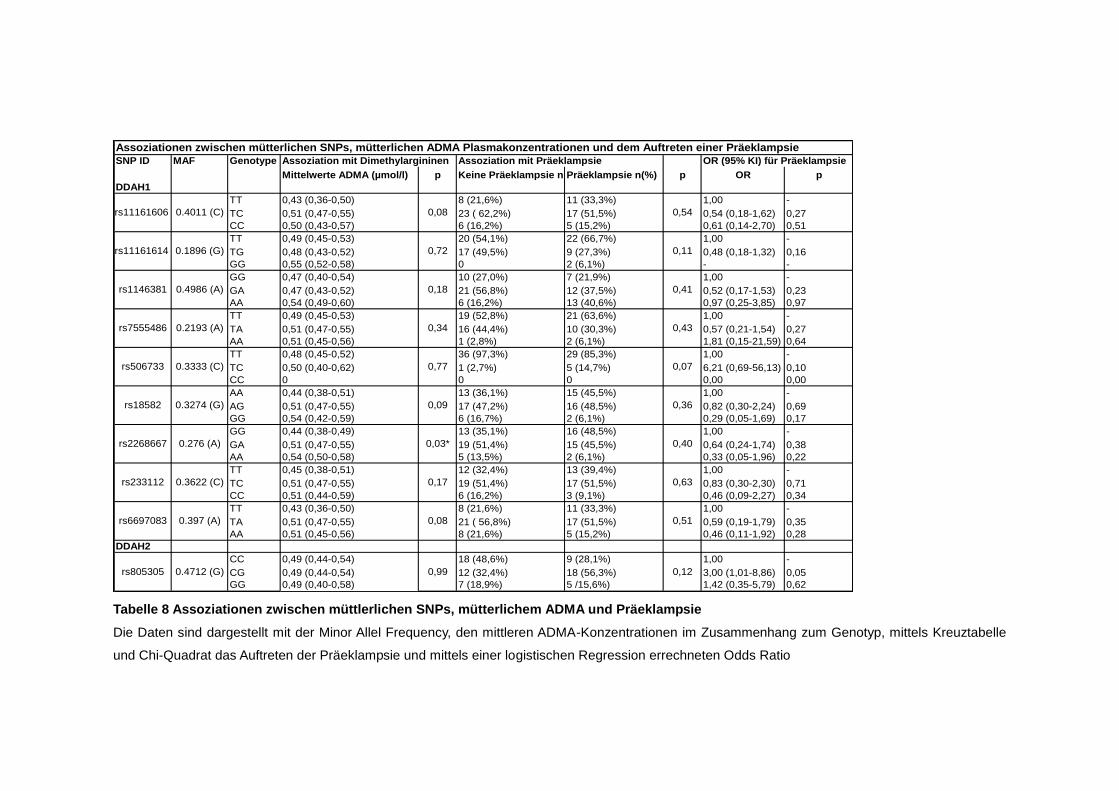
SNP ID MAF Genotype Assoziation mit Dimethylargininen Assoziation mit Präeklampsie OR (95% KI) für Präeklampsie
Mittelwerte ADMA (µmol/l) p Keine Präeklampsie n(%)Präeklampsie n(%) p OR p
DDAH1
TT 0,43 (0,36-0,50) 8 (21,6%) 11 (33,3%) 1,00 -
TC 0,51 (0,47-0,55) 23 ( 62,2%) 17 (51,5%) 0,54 (0,18-1,62) 0,27
CC 0,50 (0,43-0,57) 6 (16,2%) 5 (15,2%) 0,61 (0,14-2,70) 0,51
TT 0,49 (0,45-0,53) 20 (54,1%) 22 (66,7%) 1,00 -
TG 0,48 (0,43-0,52) 17 (49,5%) 9 (27,3%) 0,48 (0,18-1,32) 0,16
GG 0,55 (0,52-0,58) 0 2 (6,1%) - -
GG 0,47 (0,40-0,54) 10 (27,0%) 7 (21,9%) 1,00 -
GA 0,47 (0,43-0,52) 21 (56,8%) 12 (37,5%) 0,52 (0,17-1,53) 0,23
AA 0,54 (0,49-0,60) 6 (16,2%) 13 (40,6%) 0,97 (0,25-3,85) 0,97
TT 0,49 (0,45-0,53) 19 (52,8%) 21 (63,6%) 1,00 -
TA 0,51 (0,47-0,55) 16 (44,4%) 10 (30,3%) 0,57 (0,21-1,54) 0,27
AA 0,51 (0,45-0,56) 1 (2,8%) 2 (6,1%) 1,81 (0,15-21,59) 0,64
TT 0,48 (0,45-0,52) 36 (97,3%) 29 (85,3%) 1,00 -
TC 0,50 (0,40-0,62) 1 (2,7%) 5 (14,7%) 6,21 (0,69-56,13) 0,10
CC 0 0 0 0,00 0,00
AA 0,44 (0,38-0,51) 13 (36,1%) 15 (45,5%) 1,00 -
AG 0,51 (0,47-0,55) 17 (47,2%) 16 (48,5%) 0,82 (0,30-2,24) 0,69
GG 0,54 (0,42-0,59) 6 (16,7%) 2 (6,1%) 0,29 (0,05-1,69) 0,17
GG 0,44 (0,38-0,49) 13 (35,1%) 16 (48,5%) 1,00 -
GA 0,51 (0,47-0,55) 19 (51,4%) 15 (45,5%) 0,64 (0,24-1,74) 0,38
AA 0,54 (0,50-0,58) 5 (13,5%) 2 (6,1%) 0,33 (0,05-1,96) 0,22
TT 0,45 (0,38-0,51) 12 (32,4%) 13 (39,4%) 1,00 -
TC 0,51 (0,47-0,55) 19 (51,4%) 17 (51,5%) 0,83 (0,30-2,30) 0,71
CC 0,51 (0,44-0,59) 6 (16,2%) 3 (9,1%) 0,46 (0,09-2,27) 0,34
TT 0,43 (0,36-0,50) 8 (21,6%) 11 (33,3%) 1,00 -
TA 0,51 (0,47-0,55) 21 ( 56,8%) 17 (51,5%) 0,59 (0,19-1,79) 0,35
AA 0,51 (0,45-0,56) 8 (21,6%) 5 (15,2%) 0,46 (0,11-1,92) 0,28
DDAH2
CC 0,49 (0,44-0,54) 18 (48,6%) 9 (28,1%) 1,00 -
CG 0,49 (0,44-0,54) 12 (32,4%) 18 (56,3%) 3,00 (1,01-8,86) 0,05
GG 0,49 (0,40-0,58) 7 (18,9%) 5 /15,6%) 1,42 (0,35-5,79) 0,62
Assoziationen zwischen mütterlichen SNPs, mütterlichen ADMA Plasmakonzentrationen und dem Auftreten einer Präeklampsie
0,08 0,51
0,99 0,12
0,03* 0,40
0,17 0,63
0,77 0,07
0,09 0,36
0,18 0,41
0,34 0,43
rs18582 0.3274 (G)
0,08 0,54
0,72 0,11
rs11161606 0.4011 (C)
rs11161614 0.1896 (G)
rs1146381 0.4986 (A)
rs7555486 0.2193 (A)
rs506733 0.3333 (C)
rs805305 0.4712 (G)
rs2268667 0.276 (A)
rs233112 0.3622 (C)
rs6697083 0.397 (A)
Tabelle 8 Assoziationen zwischen müttlerlichen SNPs, mütterlichem ADMA und Präeklampsie
Die Daten sind dargestellt mit der Minor Allel Frequency, den mittleren ADMA-Konzentrationen im Zusammenhang zum Genotyp, mittels Kreuztabelle
und Chi-Quadrat das Auftreten der Präeklampsie und mittels einer logistischen Regression errechneten Odds Ratio
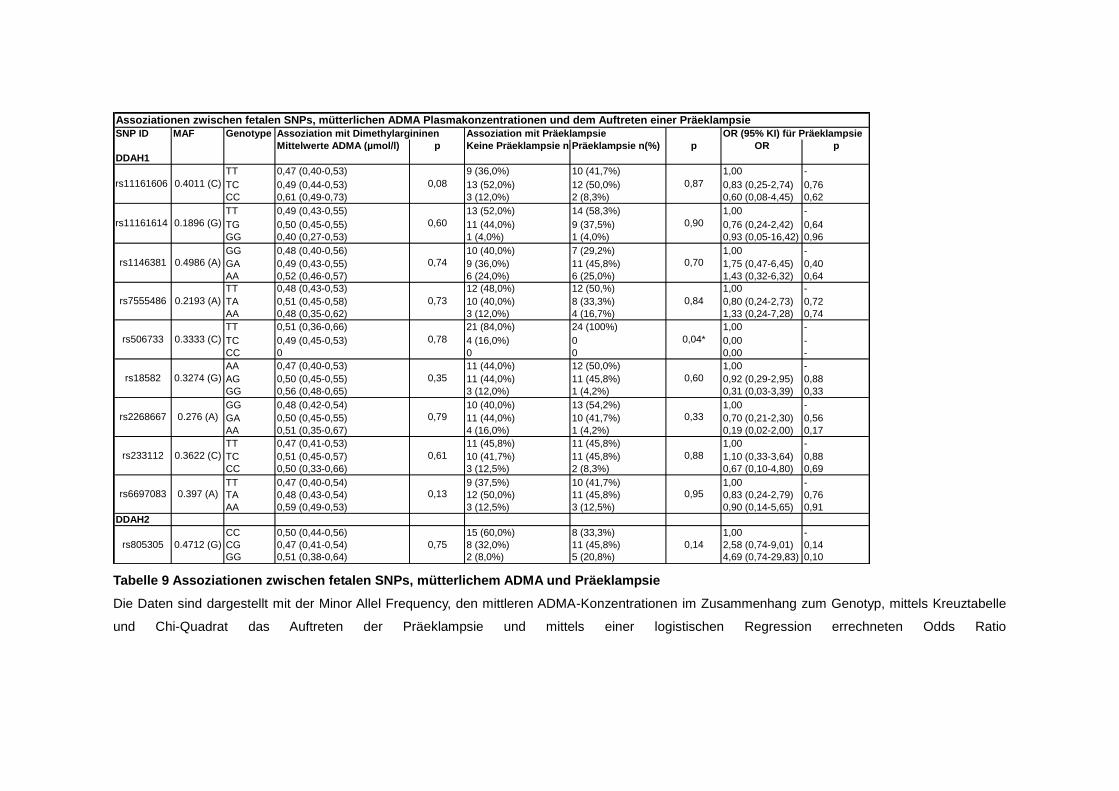
SNP ID MAF Genotype Assoziation mit Dimethylargininen Assoziation mit Präeklampsie OR (95% KI) für Präeklampsie
Mittelwerte ADMA (µmol/l) p Keine Präeklampsie n(%)Präeklampsie n(%) p OR p
DDAH1
TT 0,47 (0,40-0,53) 9 (36,0%) 10 (41,7%) 1,00 -
TC 0,49 (0,44-0,53) 13 (52,0%) 12 (50,0%) 0,83 (0,25-2,74) 0,76
CC 0,61 (0,49-0,73) 3 (12,0%) 2 (8,3%) 0,60 (0,08-4,45) 0,62
TT 0,49 (0,43-0,55) 13 (52,0%) 14 (58,3%) 1,00 -
TG 0,50 (0,45-0,55) 11 (44,0%) 9 (37,5%) 0,76 (0,24-2,42) 0,64
GG 0,40 (0,27-0,53) 1 (4,0%) 1 (4,0%) 0,93 (0,05-16,42) 0,96
GG 0,48 (0,40-0,56) 10 (40,0%) 7 (29,2%) 1,00 -
GA 0,49 (0,43-0,55) 9 (36,0%) 11 (45,8%) 1,75 (0,47-6,45) 0,40
AA 0,52 (0,46-0,57) 6 (24,0%) 6 (25,0%) 1,43 (0,32-6,32) 0,64
TT 0,48 (0,43-0,53) 12 (48,0%) 12 (50,%) 1,00 -
TA 0,51 (0,45-0,58) 10 (40,0%) 8 (33,3%) 0,80 (0,24-2,73) 0,72
AA 0,48 (0,35-0,62) 3 (12,0%) 4 (16,7%) 1,33 (0,24-7,28) 0,74
TT 0,51 (0,36-0,66) 21 (84,0%) 24 (100%) 1,00 -
TC 0,49 (0,45-0,53) 4 (16,0%) 0 0,00 -
CC 0 0 0 0,00 -
AA 0,47 (0,40-0,53) 11 (44,0%) 12 (50,0%) 1,00 -
AG 0,50 (0,45-0,55) 11 (44,0%) 11 (45,8%) 0,92 (0,29-2,95) 0,88
GG 0,56 (0,48-0,65) 3 (12,0%) 1 (4,2%) 0,31 (0,03-3,39) 0,33
GG 0,48 (0,42-0,54) 10 (40,0%) 13 (54,2%) 1,00 -
GA 0,50 (0,45-0,55) 11 (44,0%) 10 (41,7%) 0,70 (0,21-2,30) 0,56
AA 0,51 (0,35-0,67) 4 (16,0%) 1 (4,2%) 0,19 (0,02-2,00) 0,17
TT 0,47 (0,41-0,53) 11 (45,8%) 11 (45,8%) 1,00 -
TC 0,51 (0,45-0,57) 10 (41,7%) 11 (45,8%) 1,10 (0,33-3,64) 0,88
CC 0,50 (0,33-0,66) 3 (12,5%) 2 (8,3%) 0,67 (0,10-4,80) 0,69
TT 0,47 (0,40-0,54) 9 (37,5%) 10 (41,7%) 1,00 -
TA 0,48 (0,43-0,54) 12 (50,0%) 11 (45,8%) 0,83 (0,24-2,79) 0,76
AA 0,59 (0,49-0,53) 3 (12,5%) 3 (12,5%) 0,90 (0,14-5,65) 0,91
DDAH2
CC 0,50 (0,44-0,56) 15 (60,0%) 8 (33,3%) 1,00 -
CG 0,47 (0,41-0,54) 8 (32,0%) 11 (45,8%) 2,58 (0,74-9,01) 0,14
GG 0,51 (0,38-0,64) 2 (8,0%) 5 (20,8%) 4,69 (0,74-29,83) 0,10
rs233112 0.3622 (C)
rs6697083 0.397 (A)
rs805305 0.4712 (G)
0.2193 (A)
rs506733 0.3333 (C)
rs18582 0.3274 (G)
rs2268667 0.276 (A)
0,74
rs11161606 0.4011 (C) 0,08 0,87
rs11161614 0.1896 (G)
rs1146381
Assoziationen zwischen fetalen SNPs, mütterlichen ADMA Plasmakonzentrationen und dem Auftreten einer Präeklampsie
0,13 0,95
0,73
0,88
0,60 0,90
0,04*
0.4986 (A)
rs7555486
0,75 0,14
0,79 0,33
0,61
0,70
0,78
0,84
0,35 0,60
Tabelle 9 Assoziationen zwischen fetalen SNPs, mütterlichem ADMA und Präeklampsie
Die Daten sind dargestellt mit der Minor Allel Frequency, den mittleren ADMA-Konzentrationen im Zusammenhang zum Genotyp, mittels Kreuztabelle
und Chi-Quadrat das Auftreten der Präeklampsie und mittels einer logistischen Regression errechneten Odds Ratio

54
4.6. Genexpression
Die Daten der Genexpressionsanalyse der RT-PCR werteten wir im Vergleich
zwischen der Kontroll- und Präeklampsiegruppe, unterteilt in fetale und
maternale relative Expressionsdaten aus. Es fanden sich Unterschiede in der
relativen Genexpression der plazentaren eNOS-Expression im Vergleich der
fetalen und der maternalen Kontroll- und Präeklampsiegruppe (p<0,01). Im
Bonferroni-Test stellte sich nur ein Unterschied auf der fetalen Seite der
Plazenta zwischen Kontrollen und Präeklampsie dar. Ebenso ergab sich ein
Unterschied in der plazentaren DDHA2-Expression im Vergleich der fetalen und
maternalen Kontroll- und Präeklampsiegruppe, der ebenfalls nur auf der fetalen
Seite nachzuvollziehen war (p=0,05).
Die Expressionsanalyse von PRMT1 im Vergleich zwischen fetalen Kontroll-
und Präeklampsiegruppe war mittels ANOVA nicht unterschiedlich (p=0,15),
allerdings waren die Expressionsdaten der fetalen Seite mittels zweiseitigem T-
Test unterschiedlich zwischen den Gruppen (p<0,01). Dagegen war die von
DDAH1 nicht signifikant unterschiedlich zwischen den Gruppen
(p=0,52)(Abbildung 14).
Plazentare eNOS Expression
C fe
tal
PE fe
tal
C m
ater
nal
PE m
ater
nal
0
1
2
3
4
5 **
p<0,01
Rela
tive
Exp
ressio
n
Plazentare DDAH2 Expression
C fe
tal
PE fe
tal
C m
ater
nal
PE m
ater
nal
0
1
2
3
4
5
p=0,05
***
Rela
tive
Exp
ressio
n
Plazentare PRMT1 Expression
C fe
tal
PE fe
tal
C m
ater
nal
PE m
ater
nal
0
1
2
3
4
5
p=0,15
Rela
tive
Exp
ressio
n
Plazentare DDAH1 Expression
C fe
tal
PE fe
tal
C m
ater
nal
PE m
ater
nal
0
1
2
3
4
5
p=0,52
Rela
tive
Exp
ressio
n
Abbildung 14 Plazentare Expressionsanalysen von eNOS, PRMT1, DDAH 1 und 2
Vergleiche zwischen den Gruppen wurde mittels ANOVA durchgeführt
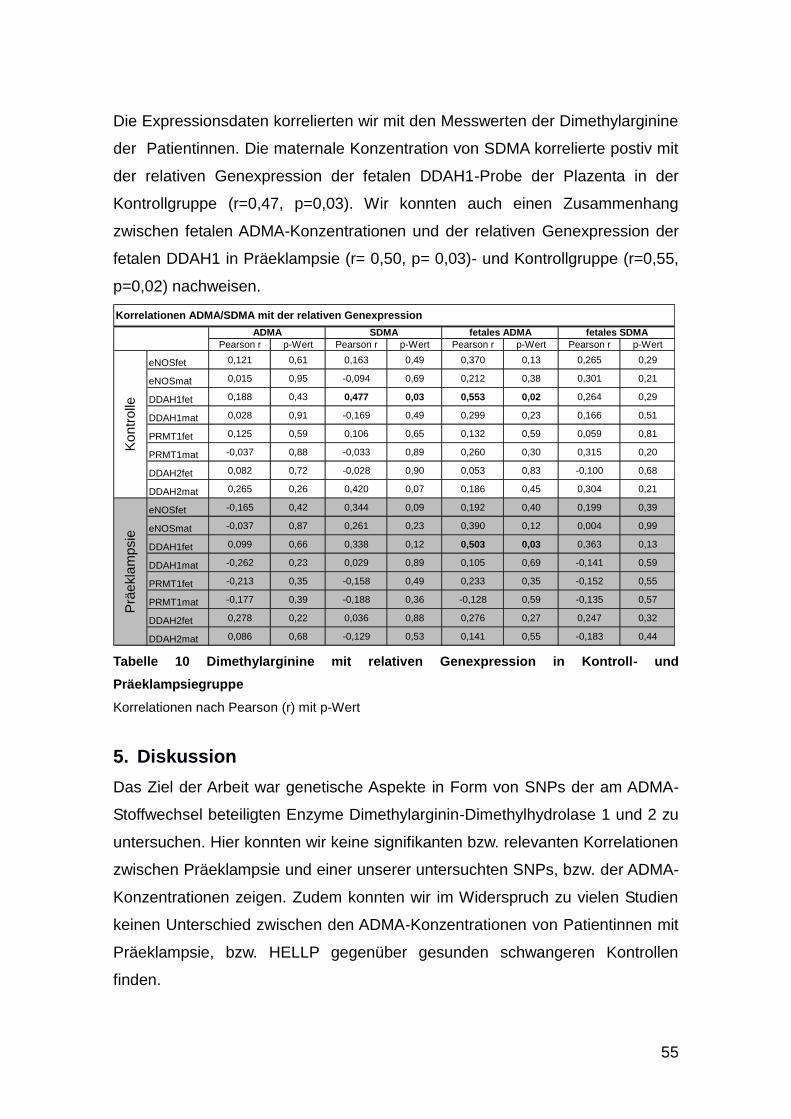
55
Die Expressionsdaten korrelierten wir mit den Messwerten der Dimethylarginine
der Patientinnen. Die maternale Konzentration von SDMA korrelierte postiv mit
der relativen Genexpression der fetalen DDAH1-Probe der Plazenta in der
Kontrollgruppe (r=0,47, p=0,03). Wir konnten auch einen Zusammenhang
zwischen fetalen ADMA-Konzentrationen und der relativen Genexpression der
fetalen DDAH1 in Präeklampsie (r= 0,50, p= 0,03)- und Kontrollgruppe (r=0,55,
p=0,02) nachweisen.
Pearson r p-Wert Pearson r p-Wert Pearson r p-Wert Pearson r p-Wert
eNOSfet 0,121 0,61 0,163 0,49 0,370 0,13 0,265 0,29
eNOSmat 0,015 0,95 -0,094 0,69 0,212 0,38 0,301 0,21
DDAH1fet 0,188 0,43 0,477 0,03 0,553 0,02 0,264 0,29
DDAH1mat 0,028 0,91 -0,169 0,49 0,299 0,23 0,166 0,51
PRMT1fet 0,125 0,59 0,106 0,65 0,132 0,59 0,059 0,81
PRMT1mat -0,037 0,88 -0,033 0,89 0,260 0,30 0,315 0,20
DDAH2fet 0,082 0,72 -0,028 0,90 0,053 0,83 -0,100 0,68
DDAH2mat 0,265 0,26 0,420 0,07 0,186 0,45 0,304 0,21
eNOSfet -0,165 0,42 0,344 0,09 0,192 0,40 0,199 0,39
eNOSmat -0,037 0,87 0,261 0,23 0,390 0,12 0,004 0,99
DDAH1fet 0,099 0,66 0,338 0,12 0,503 0,03 0,363 0,13
DDAH1mat -0,262 0,23 0,029 0,89 0,105 0,69 -0,141 0,59
PRMT1fet -0,213 0,35 -0,158 0,49 0,233 0,35 -0,152 0,55
PRMT1mat -0,177 0,39 -0,188 0,36 -0,128 0,59 -0,135 0,57
DDAH2fet 0,278 0,22 0,036 0,88 0,276 0,27 0,247 0,32
DDAH2mat 0,086 0,68 -0,129 0,53 0,141 0,55 -0,183 0,44
fetales SDMA
Korrelationen ADMA/SDMA mit der relativen Genexpression
SDMA
Ko
ntr
olle
Prä
ekla
mp
sie
fetales ADMAADMA
Tabelle 10 Dimethylarginine mit relativen Genexpression in Kontroll- und
Präeklampsiegruppe
Korrelationen nach Pearson (r) mit p-Wert
5. Diskussion
Das Ziel der Arbeit war genetische Aspekte in Form von SNPs der am ADMA-
Stoffwechsel beteiligten Enzyme Dimethylarginin-Dimethylhydrolase 1 und 2 zu
untersuchen. Hier konnten wir keine signifikanten bzw. relevanten Korrelationen
zwischen Präeklampsie und einer unserer untersuchten SNPs, bzw. der ADMA-
Konzentrationen zeigen. Zudem konnten wir im Widerspruch zu vielen Studien
keinen Unterschied zwischen den ADMA-Konzentrationen von Patientinnen mit
Präeklampsie, bzw. HELLP gegenüber gesunden schwangeren Kontrollen
finden.

56
In unserer Studiengruppe wiesen die Präeklampsie-Patientinnen bestimmte
Risikofaktoren, bzw. Unterschiede zu der Kontrollgruppe auf, die mit bisherigen
Ergebnissen der Literatur übereinstimmten: Die Präeklampsie-Patientinnen
waren zu 75% im Unterschied zu den gesunden Kontrollen nullipara (43,2%
Nulliparität) (Misra 1997; Odegard 2000; Duckitt 2005). Im Mittel nahmen die
Frauen mit einer schweren Präeklampsie (19,0 kg) während der
Schwangerschaft mehr Gewicht im Verlauf zu, als die Kontrollen (15,3 kg)
(p=0,006) (Sibai 1997; Ornaghi 2013).
Hinsichtlich der bisher möglichen Risikoprofilerstellung durch die
Dopplersonographie der Arteriae uterina zeigte sich in dem eingeschlossenen
Patientinnenkollektiv ein beidseitig erhöhter Widerstand in Form eines
beidseitgen notches ausschließlich bei Patientinnen mit Präeklampsie (n=6),
und zwar jene mit schwerer Präeklampsie und HELLP-Syndrom (p=0,04). Unter
den Präeklampsie-Patientinnen unserer Studie konnten wir keinen
Zusammenhang zwischen der ADMA-Konzentration und dem Vorliegen von
bilateralen notches nachvollziehen (p=0,70). Prefumo et al. untersuchten 40
schwangere Frauen, 21 davon zeigten bilaterale notches der Aa. uterinae. Im
Verlauf entwickelten drei von ihnen eine Präeklampsie, er fand dabei ebenfalls
keine Korrelation mit der ADMA-Konzentration (Prefumo 2008). Savvidou et al.
fanden signifikant erhöhte ADMA-Konzentrationen bei Schwangeren mit
bilateralen notches. Diese standen aber nicht nur in Verbindung mit einer
Präeklampsie (Savvidou 2003). Insgesamt lässt sich so keine Verknüpfung
zwischen auffälligen Befunden in der Dopplersonographie und den ADMA-
Konzentrationen stellen.
5.1. Dimethylarginine
In unserer Studie konnte kein signifikanter Unterschied der ADMA-Plasma-
konzentrationen weder im Vergleich zwischen Kontroll- und
Präeklampsiegruppe (p=0,35), noch in den nach Schweregrad aufgegliederten
Subgruppen der Präeklampsie, bzw. HELLP-Syndrom (p=0,61) gemessen
werden. Auch nach Ausschluss der HELLP-Patientinnen ergibt sich kein

57
Unterschied (p=0,25). Auch das Ansteigen der ADMA-Konzentration mit der
Schwere der Präeklampsie konnten wir im Gegensatz zu Ellis et al. nicht
nachvollziehen (Ellis 2001).
Dies steht im Widerspruch zu einigen bisherigen Studien (Fickling 1993; Holden
1998; Speer 2008; Braekke 2009). Die derzeitige Studienlage zeigt allerdings
kein einheitliches Bild über die ADMA-Konzentrationen in Präeklampsie-
Patientinnen. Andere Studien zeigten, so wie wir, keine Unterschiede der
ADMA-Konzentrationen (López-Jaramillo 1996; Maas 2004; Kim 2006; Siroen
2006; Ehsanipoor 2013). Diese heterogenen Daten zu den Plasma-ADMA-
Konzentrationen bei Präeklampsie-Patientinnen der letzten Jahre könnten sich
unter anderem durch verschiedene Messtechniken erklären lassen (Tabelle 2).
Es gibt drei gängige Methoden sind die High-throughout Liqiud Chromatography
(HPLC) und die in unserem Institut verwendete Form der ADMA-
Konzentrationsmessung mittels Tandem Liquid Chromatograohy (LC-Ms/Ms)
als derzeitiger Goldstandard. Eine weitere Methode sind ELISA-Kits zur
Bestimmung der ADMA-Konzentration. Durch Vergleiche der einzelnen
Methoden konnte gezeigt werden, dass die HPLC-Methode innerhalb
unterschiedlicher Untersucher eine breite Streuung an ADMA-Konzentrationen
ergibt. Die neueren Kits für ELISA bemessen die ADMA-Konzentrationen
insgesamt zu hoch. Die LC-MS/MS weist dagegen eine hohe Spezifität auf
(Schwedhelm 2005; Horowitz 2007).
Tabelle 2 zeigt die Fallzahlen einzelner Studien mit den unterschiedlichen
Studien zu ADMA-Konzentrationen bei Präeklampsie. Vier der aufgeführten
Studien weisen ähnlich große Fallzahlen, bzw. größere als unsere (36
Präeklampsie-Patientinnen und 37 Kontrollen) auf. Drei von ihnen konnten
einen Unterschied der ADMA-Konzentrationen zwischen gesunden Kontrollen
und Präeklampsie-Patientinnen finden, davon wurden zwei mit HPLC
gemessen (Mao 2010; Sandrim 2010). Nur eine verwandte die LC-MS/MS
(Braekke 2009). Eine größere Studie mit gleicher Methode konnte ebenso wie
wir keinen Unterschied feststellen (Maas 2004).

58
Abgesehen von der Methode könnten zu kleine Fallzahlen, vor allem von
Patientinnen mit schwerer Präeklampsie, das Ergebnis unserer Studie
beeinflusst haben.
Ein weiterer Grund für die vorliegenden Ergebnisse könnte der Zeitpunkt der
Blutentnahme darstellen. Im Mittel wurde unseren Studienteilnehmerinnen in
der 36.-37. Schwangerschaftswoche, d.h. im 3. Trimenon, Blut abgenommen
um die ADMA-Konzentrationen zu bestimmen. Rizos et al. konnten kürzlich,
ebenso wie bereits Fickling et al. zeigen, dass die mittleren ADMA-
Konzentrationen in gesunden Schwangeren zum Ende der Schwangerschaft
ansteigen (Fickling 1993; Rizos 2012), so dass der Vergleich zu einem späten
Zeitpunkt der Schwangerschaft deutlich schwerer fällt. Allerdings fanden Rizos
et al. in allen 3 Tertialen erhöhte ADMA-Konzentrationen in zehn Präeklampsie-
Patientinnen, statistisch signifikant waren diese nur im 2. Trimenon gegenüber
den gesunden Schwangeren. Es handelt sich hierbei um eine retrospektive
Studie, in der eine bestimmte Anzahl an Schwangeren über die gesamte
Schwangerschaft beobachtet wurde. Die Blutentnahmen wurden im 1. Trimenon
(zwischen der 10.- 14. SSW), im 2. Trimenon (20.-24. SSW) und im 3. Trimenon
(28.-35. SSW) abgenommen.
Im Wesentlichen bestimmten die aufgeführten Studien die ADMA-Konzentration
im 3. Trimenon, was den Vergleich zwischen den Daten bei einem solchen
Zeitraum relativ ungenau macht, was die Betrachtung schwierig macht
Eine weitere Erklärung könnte darin begründet sein, dass nach Rizos et al.
Frauen mit Small-for-gestational-age (SGA-) Feten (d.h. mit einer
Gewichtsperzentile <10) signifikant geringere ADMA-Konzentrationen
aufwiesen. 12 der 36 der eingeschlossenen Präeklampsie-Patientinnen
brachten ein SGA-Neugeborenes zur Welt. Die ADMA-Konzentrationen
unterschieden sich zwischen denen mit SGA (0,55 µmol/l) und ohne SGA (0,47
µmol/l) (p<0,04), allerdings nicht gleichsinnig dieser Daten. Die Entstehung von
Wachstumsstillständen, bzw. Retardierungen ist eng verknüpft mit der
Ausprägung und dem Auftreten der Präeklampsie. Da die Frauen mit SGA-

59
Feten im Gegensatz eher von einer schweren Präeklampsie betroffen sind,
entspricht dies unserer Theorie nach erhöhten ADMA-Konzentrationen.
Eine weitere Einschränkung der Messwerte könnte durch die Medikation
bedingt sein. 19 der eingeschlossenen Präeklampsie-Patientinnen erhielten
bereits zum Zeitpunkt der Blutentnahme eine antihypertensive Therapie mit a-
Methyldopa und bei Blutdruckspitzen Nifedipin. Im Mittel waren die ADMA-
Konzentrationen unter der Einnahme von blutdrucksenkenden Mitteln deutlich
höher (0,55 µmol/l), als bei jenen, die keine Medikation erhielten (0,47 µmol/l)
(p=0,02). Dies könnte Zeichen des Schweregrades der Präeklampsie sein.
Effekte auf die ADMA-Plasmakonzentrationen antihypertensiver Medikamente
sind in Bezug auf die Behandlung bei Präeklampsie bisher nicht untersucht
worden. In vielen Untersuchungen in vitro konnte allerdings ein senkender
Effekt einer antihypertensiven Behandlung, beispielsweise durch Nebivolol und
Telmisartan, nachgewiesen werden (Tomiyama 2007; Wang 2011).
Die SDMA-Konzentrationen unterschieden sich signifikant zwischen Kontroll-
und Präeklampsiegruppe (p<0,01). Zudem korrelierten die SDMA-
Konzentrationen in den Präeklampsie-Patientinnen mit deren BMI, bzw.
Gewicht in kg zum Zeitpunkt der Blutentnahme.
Erhöhte SDMA-Konzentrationen in Präeklampsie-Patientinnen konnte auch in
anderen Studien nachvollzogen werden (Ellis 2001; Braekke 2009; Anderssohn
2012).
Kakimoto et al. vermutet eine rein renale Ausscheidung von SDMA. Es galt als
Nebenprodukt bei der Proteinmethylierung (Kakimoto 1970). Neuere Studien
zeigen eine mögliche Rolle als indirekter Inhibitor der NO-Synthase. SDMA
könnte indirekt durch eine verminderte Aufnahme von L-Arginin in die Zelle die
NO-Synthese verhindern (Schwedhelm; Böger 2011) und damit in Verbindung
mit der Pathogenese der Präeklampsie stehen. Da es ausschließlich renal
ausgeschieden wird, könnte jede Einschränkung der Nierenfunktion zu einer
erhöhten SDMA-Plasmakonzentration führen (Zoccali C. 2001). In unserem
Studienkollektiv konnten wir signifikant unterschiedliche Kreatinin-

60
Konzentrationen zwischen gesunden Kontrollen und Präeklampsie-Patientinnen
finden, auch wenn sich die Konzentration der Präeklampsie-Patientinnen in
einem höheren Bereich befanden, waren sie dennoch nicht pathologisch
(Kreatinin im Mittel 0,70 mg/dl).
Tanhäuserová et al. konnten in Patienten mit Diabetes mellitus eine positive
Korrelation zwischen SDMA und einer Mikroalbuminurie bzw. Proteinurie zeigen
(Tanhäuserová 2012). Da die Mehrzahl unserer eingeschlossenen
Präeklampsie-Patientinnen eine Proteinurie aufwies, könnte dies die Ursache
für signifikant erhöhte SDMA-Konzentrationen sein.
In unserer Studie korrelierten sowohl die ADMA-, als auch die SDMA-
Konzentrationen mit dem Serumkreatinin und der GFR.
SDMA wird zum Großteil, ADMA mit geringem Anteil renal ausgeschieden.
Obwohl ca. 90% der ADMA-Konzentrationen über die DDAH 1 metabolisiert
wird, scheint die Ausscheidung über die Niere Einfluss auf die
Serumkonzentration von ADMA zu haben (McDermott 1976, Ogawa et al.
1987). Zusätzlich wird die DDAH 1 vorrangig in der Niere und Leber exprimiert.
Unter dieser Annahme könnte eine Beschädigung der Nieren im Rahmen der
renaler Dysfunktion eine Einschränkung der DDAH 1-Aktivität zur Folge haben
(Schwedhelm; Xanthakis; Maas; Sullivan; Schulze; Riederer; Benndorf; Boger;
Vasan 2009). Im Rahmen der Präeklampsie konnte Stillmann et al. zeigen, dass
es histolgisch durch gezielte Nierenbiopsien zu einer endothelialen
Glomerulose, d.h. zu einer minimalen renalen Dysfunktion mit verminderter
GFR kommt (Stillman 2007).
In einer gesunden Schwangerschaft steigt die glomeruläre Filtrationsrate ab
dem 1. Trimenon um ca. 40-60% (Davison 1974). Bei Präeklampsie scheint es
durch eine verstärkte Vasokonstriktion zu einem verminderten renalen
Plasmafluss und somit zu einer Verringerung der GFR um ca. 30-40% zu
kommen, auch wenn die Retentionsparameter, wie das Kreatinin hoch normal
sind (Lafayette 1998; Moran 2003). Die Kreatinin-Konzentrationen in unserer
Studie korrelierten positiv sowohl mit den ADMA-, als auch den SDMA-
Konzentrationen in den Präeklampsie-Patientinnen.

61
Schon Vallance et al. konnten 1992 zeigen, dass sowohl ADMA- als auch
SDMA bei Patienten mit chronischer Niereninsuffizienz gleichsinnig mit dem
Plasma-Kreatinin steigt, während die Patienten ein ca. 9-fach erhöhten
Kreatininwert zeigten, waren die Konzentrationen von ADMA und SDMA um ein
8-faches erhöht (Vallance 1992).
Kielstein et al. zeigten in ihrem Review, dass SDMA eine Rolle als Marker für
die renale Funktion spielen könnte, da diese große Metaanalyse die Korrelation
von GFR und Serumkreatinin zu SDMA nachvollziehen konnte. (Kielstein 2011;
Schepers 2011). Kielstein konnte einen engen Zusammenhang zwischen der
GFR und SDMA herstellen. Indem er nach einseitiger Nephrektomie die GFR
und SDMA bestimmte und bezeichnete SDMA möglicherweise als frühen
Biomarker um Veränderungen der GFR anzuzeigen.
5.1.1. Fetale Dimethylarginine, fetomaternaler Gradient und arteriovenöse
Differenz
Erhöhte fetale ADMA-Konzentrationen könnten entweder durch ein hohes
Proteinturnover im Feten oder verminderter Elimination von ADMA entstehen.
Sie könnten auch als wichtige Mediatoren für den fetalen Blutkreislauf benötigt
werden. Tshukara et al. postulierten, dass ADMA eine physiologische Rolle zur
Aufrechterhaltung des Gefäßtonus und der Blutzufuhr in der Adaptationsphase
vom fetalen zum neonatalen Blutkreislauf einnehmen könne. Die fetalen ADMA-
Konzentrationen schienen weder vom Geburtsmodus, noch von einer
Präeklampsie beeinflusst zu sein (Tsukahara 2008). Vida et al. konnten das
Absinken der ADMA-Konzentration am zweiten post-partalen Tag nachweisen,
was auch der Hypothese entspräche, dass der fetale Kreislauf die Regulation
durch ADMA und NO benötigt, der neonatale jedoch nicht (Vida 2007 und
Braekke 2009).Wir fanden in unseren beiden Gruppen hohe fetale ADMA-
Konzentrationen. Sie waren zwar bis zu 4-fach gegenüber den maternalen
Konzentrationen erhöht, aber es stellten sich keine Unterschiede der fetalen
ADMA-Konzentrationen zwischen den Gruppen in den Plasmaproben der
Nabelschnur dar, gleich der Ergebnisse von Braekke et al.

62
Die Ursache erhöhter fetaler ADMA-Konzentrationen scheint also nicht an
ungenügender Metabolisierung von ADMA, bzw. einer Insuffizienz der Plazenta
bei Präeklampsie zu sein. Diese Hypothese unterstützend fanden wir keine
arteriovenöse Differenz der ADMA-Konzentrationen. Für diese Untersuchung
hatten wir nur eine geringe Anzahl an getrennten arteriellen und venösen
Proben. Eine erhöhte venöse umbilicale Konzentration, wie bei Braekke und
Vida et al., im Gegensatz zu arteriellen spräche wiederum für den größeren
Anteil von maternalem ADMA und gegebenenfalls doch für eine
Metabolisierungsstörung auf maternaler Seite (Braekke 2009; Vida 2007).
5.2. SNPs
Die Analyse der untersuchten SNPs in den Genen der DDAH1 und DDAH2,
sowohl in den maternalen, als auch fetalen genetischen Analyse, zeigte keinen
Zusammenhang mit Präeklampsie oder der ADMA-Konzentrationen.
Wir fanden in vorliegendem Studienkollektiv in Zusammenhang mit dem
DDAH2-Promoter-Polymorphismus 449 G/C (rs805305) einen signifikanten
Vorhersagewert, der zu einer Präeklampsie führen könnte. Das Risiko an einer
Präeklampsie zu erkranken war dreifach erhöht, wenn die Patientinnen einen
heterozygoten Genotypen (CG) trugen (p=0,05). Zwischen der ADMA-
Konzentration und den Genotypen des SNPs gab es allerdings keinen
Zusammenhang. Da es unwahrscheinlich ist, dass ein heterozygoter Genotyp
Einfluss auf das Auftreten einer Erkrankung zeigt, prüften wir dieses Modell
unter Annahme der Dominanz von Allel C ohne Ergebnis.
Ein weiterer der untersuchten SNPs war mit der ADMA-Konzentration
assoziiert.
Homozygote Trägerinnen des minor allels des SNP rs2268667 der DDAH1
zeigten erhöhte ADMA-Konzentrationen (0,54µmol/l) im Vergleich zu den
homozygoten Trägerinnen des major allels (0,44 µmol/l) und den heterozygote
Trägerinnen (0,51µmol/l) (p=0,03). Es zeigte sich jedoch keine Assoziation zur
Ausprägung der Präeklampsie. Homozygote Trägerinnen des minor allels für

63
diesen SNP waren nur sieben Frauen insgesamt (fünf der Kontrollgruppe und
nur zwei der Präeklampsiegruppe).
Eine Einschränkung unserer Analysen besteht aufgrund der kleinen
Gruppengröße, so dass wir nur Tendenzen untersuchen konnten, zudem haben
wir aufgrund des explorativen Charakters unserer Untersuchung auf eine
Korrektur für multiples Testen verzichtet.
Die Betrachtung genetischer Analysen bezüglich der Präeklampsie macht
deutlich, dass die Präeklampsie eine noch weitestgehend ungeklärte
Pathogenese aufweist, die auch genetisch sehr vielschichtig zu sein scheint. Da
auch die fetalen Gene einen Einfluss auf maternale ADMA-Konzentrationen
haben könnten, testeten wir ebenfalls den Zusammenhang zwischen ADMA,
Präeklampsie und den fetalen Genotypen.
Es ergab sich dabei ein Trend für den Zusammenhang des SNPs rs11161606
der DDAH1. Feten, die homozygot das minor allel (CC) trugen, waren assoziiert
mit erhöhten maternalen ADMA-Konzentrationen (p=0,08). Ähnliches war bei
den maternalen SNPs zu beobachten, Trägerinnen des minor allels C hatten
einen Trend zu erhöhten ADMA-Konzentrationen im Vergleich mit homozygoten
Trägerinnen des major allels (p=0,08).
Ein signifikanter Zusammenhang ergab sich bei dem SNP rs506733 in
Zusammenhang mit dem Auftreten einer Präeklampsie, alle untersuchten
Feten, der Mütter mit Präeklampsie, trugen eine Homozygotie für das major
allel (TT). Allerdings konnte wir in der Genotypisierung nur homozygote
Trägerinnen für das major allel und heterozygote Trägerinnen (TC) und keine
der Studienteilnehmerinnen war homozygote Trägerin des minor allels, was mit
der recherchierten minor allel frequency (MAF) nicht übereinstimmt.
Eine mögliche Erklärung der erhöhten ADMA-Konzentrationen war die
Veränderung der DDAH-Expression durch bestimmte Polymorphismen, die
bereits in in vitro-Daten nachvollzogen werden konnten. Maas et al.
untersuchten im Rahmen der MONICA Studie in Augsburg zwei DDAH2-
Promoter-Polymorphismen (rs804304 und r805305) in einer Studie zum

64
Hypertonus. Sie fanden keinen Zusammenhang zu erhöhten ADMA-
Konzentrationen, allerdings zeigte sich eine erhöhte Prävalenz einer arteriellen
Hypertonie (Maas 2009).
Bisher konnte erst in einer Studie ein eventueller Zusammenhang zwischen
dem Auftreten von bestimmten DDAH1-Polymorphismen, bzw. daraus
resultierenden Haplotypen und der Erkrankung an einer Präeklampsie gestellt
werden. Eine finnische Arbeitsgruppe identifizierte in einer Studie mit 132
Präeklampsie-Patientinnnen und 112 gesunden Kontrollen acht DDAH1 SNPs,
aus denen vier gemeinsame DDAH1 Haplotypen resultierten. Zwei von den vier
Haplotypen korrelierten signifikant mit dem Auftreten einer Präeklampsie
während der Schwangerschaft. Patientinnen, die beide Risikoallele trugen,
hatten das höchste Risiko, während der Schwangerschaft an einer
Präeklampsie zu erkranken (Odds ratio: 3,93; 95% CI) (Akbar 2004).
In der gleichen Studie wurde ein DDAH1 SNP im kodierenden Bereich des
Exon 1identifiziert (Abbildung 3). Durch den Austausch der Base Cytosin an
Position 260gegen Thymin erfolgt ein Aminosäureaustausch von Threonin zu
Methionin. SNPs, die einen Aminosäureaustausch zur Folge haben führen zu
einer veränderten/fehlerhaften Proteinsequenz und können dadurch eine
starken Einfluss auf dieFunktion/ Aktivität des Proteins haben. Auf Grund dieser
möglichen Eigenschaft wurde dieser SNP zur Bestimmung in den klinischen
Studien, welche in dieser Arbeitbeschrieben sind, ausgewählt.
Es scheint schwierig zu sein, einzelne Polymorphismen für komplexe
Erkrankungen verantwortlich zu machen, vielmehr könnten SNPs Einfluss auf
die Anfälligkeit für Erkrankungen haben und vor allem Verknüpfungen mehrerer
Polymorphismen einen Einfluss ausüben.
Abschließend betrachtet, scheint nach Williams et al. die Großzahl an Studien
zur Charakterisierung von Genen in Zusammenhang mit der Präeklampsie zu
wenig Power zu haben, um genetische Varianten mit mildem Effekt zu ermitteln.
Zudem sind Studien dieser Art limitiert durch das unzureichende Wissen über
definitive pathogenetischen Hintergründe der Präeklampsie, die somit von
vornherein die Auswahl an Genen beschränkt (Williams 2012). Somit scheint es
schwierig bei einer Studiewie unserer mit explorativem Charakter naheliegende-

65
verantwortliche SNPs im Zusammenhang zur Präeklampsie bewerten zu
können.
5.3. Genexpressionsanalysen
Ursprung des vermehrten Anfalls von ADMA in Präeklampsie-Patientinnen
könnte die Plazenta sein. Veränderte Expressionsmustern der an Synthese,
Auf- und Abbau-beteiligten Enzyme von ADMA könnten einen Hinweis geben.
Wir gewannen aus der fetalen und maternalen Seite Proben der Plazenta,
isolierten RNA und untersuchten die relative Genexpression von eNOS, DDAH
1 und 2 und PRMT1.
In der Auswertung der eNOS-Expressionsdaten konnten wir Unterschiede,
sowohl zwischen maternalen Proben der Kontroll- und Präeklampsie-
Patientinnen, als auch zwischen den fetalen Proben finden. Das bedeutet, die
eNOS wurde in den Plazenten der an Präeklampsie-erkrankten Frauen deutlich
geringer exprimiert, als in den Kontrollen, was eine verminderte NO-
Konzentration in diesen Patientinnen und damit auf eine Funktionsstörung
hindeuten könnte. Kim et al. fand in der Untersuchung von Präeklampsie-
Patientinnen in Hinblick auf die eNOS-Expression ähnliche Ergebnisse: in
Präeklampsie-Patientinnen fand sich eine verminderte Expression von eNOS,
zudem vermindertes L-Arginin und ebenso kein Unterschied zwischen den
ADMA-Konzentrationen zwischen den Gruppen (Kim 2006). Myatt et. al fand
dagegen in Plazenten von Frauen mit Präeklampsie erhöhte eNOS-Expression,
er deutete es als Kompensationsmechanismus gegen den erhöhten Widerstand
in den Gefäßen. In dieser Studiengruppe wurden allerdings nur 9 betroffene
Patientinnen und 5 Kontrollen untersucht (Myatt; Eis; Brockman; Greer 1997).
Auch Schiessl et. al. untersuchte Patientinnen in Hinblick auf die eNOS
Expression, in Patientinnen mit IUGR- Feten maßen sie eine verminderte
Expression, aber in Präeklampsie-Patientinnen eine erhöhte Expression
(Schiessl B; Kuhn; Schulze; Kunze; Friese; Jeschke 2005).
Wir fanden keinen statistisch signifikanten Unterschied in der Expression von
DDAH 2 in den maternalen Proben der Präeklampsie-Patientinnen (p= 0,07).
Die fetalen Gruppen dagegen zeigten einen deutlicheren Unterschied in der

66
Expression der DDAH2 (p<0,01). Ebenso wie Sie konnten wir Unterschiede in
den PRMT 1- Expressionsdaten zwischen den fetalen Gruppen finden (p<0,01).
Die DDAH1-Expressionsraten unterschieden sich in unserem Studienkollektiv
nicht (p=0,9).
Anderssohn et al. fanden dagegen in ihrem Studienkollektiv 10-20-fache
Expressionsunterschiede der eNOS, DDAH1, DDAH2 und PRMT1 zwischen
Präeklampsie und Kontroll-Patientinnen (Anderssohn 2012).
Die Theorie veränderter Expressionswerte für DDAH1 und 2 konnten in einer
Studie von Ehsanipoor et. al nachvollzogen werden. Diese fanden ebenso eine
verminderte plazentare Expression von DDAH1- und 2 in Präeklampsie-
Patientinnen, die sie mit einem Western Blots gemessen hatten, ebenso wie
eine verminderte NO-Konzentration. Allerdings maßen Sie keine erhöhten,
sondern verminderte ADMA- Konzentrationen bei ihren eingeschlossenen
Patientinnen (Ehsanipoor 2013).
Zur Verbesserung der Analysen in unserer Studie hätten histologische
Nachweise, bzw. Western Blots verwendet werden sollen um eindeutiger
veränderte Expressionen der beteiligten Enzyme auch auf Proteinebene
nachweisen zu können.
Mögliche Ursachen für diese Ergebnisse könnten die unterschiedlichen
Geburtsmodi der eingeschlossenen Patientinnen ausgelöst haben, die damit
Einfluss auf das plazentare Gewebe gehabt haben könnten. Auch wenn
diesbezüglich gematcht wurde, könnte die Qualität der Plazenta erheblich vom
Geburtsmodus abhängen. Im Gegensatz zu einer spontanen Geburt ist die
Plazenta bei einer Sectio caesarea nicht mit einem vergleichbaren
Geburtsstress ausgesetzt und auch die manuelle Lösung der Plazenta während
der Operation könnte die Qualität der Plazenta beeinflussen.
Die Plazenta ist in unserem Studienkollektiv in Stickstoff gelagert worden, dies
unterlag geringen zeitlichen Schwankungen.

67
5.4. Klinische Relevanz der Ergebnisse und Pharmakologische Ziele
Ein genetischer Zusammenhang wurde bei der Präeklampsie anhand von
Metaanalysen schon früh erkannt (Lachmeijer 2002, Wilson 2003). Es scheint
jedoch nicht ein einzelner Genort für die Erkrankung verantwortlich zu sein. Viel
mehr scheint es die Suzeptibilität für die Erkrankung zu sein. Diese könnte sich
nicht nur durch die maternalen Gene, sondern durch die fetalen, bzw. einem
Zusammenspiel phänotypisch auswirken (Lachmeijer 2002, Williams 2012).
SNPs oder genetischen Analysen wie die Genonme wide assay study könnten
zur Prädikition und vor allem als pharmakologisches Ziel interessant sein. In
unserer Studie konnten wir keine Zusammenhänge zwischen den vermuteten
SNPs und Präeklampsie nachweisen. Trotz dieser fehlenden Zusammenhänge
bei unserer genetischen Analyse scheint die weitere Erforschung der Ursachen
der Präeklampsie, vor allem in Hinblick auf genetische Varianten relevant.
Dabei werden größere Studienpopulationen benötigt um signifikante
Zusammenhänge finden zu können. Denn insgesamt scheint diese Studien die
Power zu fehlen um genetische Varianten mit kleinem Effekt widerzuspiegeln.
Eine zusätzliche Limitation ist die unzureichend geklärte Pathogenese der
Präeklampsie (Williams 2012). Für eindeutige genetische
Vorhersagemöglichkeiten bedarf es der weiteren Klärung dieser, bzw. das
Finden von möglichen Prädiktoren. Nur so könnte man in Zukunft zur
Aufrechterhaltung der Schwangerschaft und Schutz von Mutter und Kind
beitragen. Zusätzlich würden erkannte genetische Varianten als Ziel von
pharmakologischer Prävention bzw. Behandlung dienen.

68
6. Zusammenfassung
Präeklampsie ist mit einer Prävalenz von ca. fünf bis acht 8 Prozent aller
Schwangerschaften eine der häufigsten Ursache maternaler und perinataler
Mortalität. Sie ist Ursache für 15 Prozent der Frühgeburten in den
industrialisierten Nationen und Nummer eins der ärztlichen Entscheidung zur
vorzeitigen Beendigung einer Schwangerschaft. Da die Präeklampsie eine
komplexe Pathogenese, auch in Hinblick auf genetische Varianten besitzt, ist es
wichtig, frühzeitige Screening-Methoden und pharmakologische
Ansatzmöglichkeiten zur Therapie zu implementieren.
Aktuell wird die Pathogenese der Präeklampsie als ein 2- Phasen-Modell
verstanden: eine inadäquaten Trophoblasteninvasion führt in der Plazenta zu
erhöhtem oxidativen Stress und endothelialer Dysfunktion. Dieser Zustand führt
zur Sekretion bisher nicht eindeutig identifizierter Stoffe, bzw. Hormone. Diese
werden für den Bluthochdruck, Proteinurie bis hin zu Leberfunktionsstörungen
und anderen Komplikationen verantwortlich gemacht.
Ausgehend von den Befunden vorausgegangener Studien, die erhöhte ADMA-
Konzentrationen als einer dieser Stoffe vermuten, wollen wir klären, in wieweit
genetischen Polymorphismen des abbauenden Enzym Dimethyarginin-
Dimethylaminohydrolase in der Plazenta hiermit in Verbindung stehen.
Die vorliegende Studie hatte zum Ziel, Polymorphismen und Expression der
abbauende Enzymen von ADMA, der Dimethylarginin-Dimethylaminohydrolase
1 und 2 zu analysieren, um einen Zusammenhang zwischen erhöhten ADMA-
Konzentrationen in Präeklampsie-Patientinnen und genetischer Disposition zu
untersuchen. In vorliegender Studie konnte kein Unterschied der ADMA-
Konzentrationen zwischen Präeklampsie-Patientinnen und Kontrollen, auch
nicht in der Aufteilung der Schweregrade nachgewiesen werden. Allerdings
konnte dies für die SDMA-Konzentration gezeigt werden, die eng verknüpft mit
der Nierenfunktion zu sein scheint, eine genauere Betrachtung könnte
Gegenstand neuer Forschungsvorhaben werden.
Es fanden sich zudem keine relevanten Zusammenhänge zu den untersuchten
maternalen und fetalen SNPs.

69
Es fanden sich auf RNA-Ebene Veränderungen der relativen Genexpression in
der plazentaren eNOS-, DDAH2- und PRMT1-Expression im Vergleich
zwischen der fetalen und maternalen Kontroll- und Präeklampsiegruppe.
Zusammenfassend sollte in Zukunft die genetische Analyse an Präeklampsie-
Patientinnen fortgeführt werden um bekannte genetische Varianten der
Dimethylarginin-Dimethylaminohydrolase als pharmakologisches Ziel und
Prädiktor nutzen zu können.

70
Anhang
7. Literaturverzeichnis
Abbasi F. Asagmi T, C. Plasma Concentrations of Asymmetric Dimethylarginine
Are Increased in Patients With Type 2 Diabetes Mellitus. Am J Cardiol. 2001(88
(10)):S. 1201–1203.
Achan, V. Broadhead, M.; Malaki, M.; Whitley, G.; Leiper, J.; MacAllister, R.;
Vallance, P. Asymmetric Dimethylarginine Causes Hypertension and Cardiac
Dysfunction in Humans and Is Actively Metabolized by Dimethylarginine
Dimethylaminohydrolase. Arteriosclerosis, Thrombosis, and Vascular Biology.
2003(23):S. 1455–1459.
Akbar, F. Heinonen, S.; Pirskanen, M.; Uimari, P.; Tuomainen, T.-P.; Salonen, J.
Haplotypic association of DDAH1 with susceptibility to pre-eclampsia. Molecular
Human Reproduction. 2004. 11(1):S. 73–77.
Aktories, K.; Förstermann, U.; Hofmann, F.B; Starke, K. (2009) Allgemeine und
spezielle Pharmakologie und Toxikologie, 10. Aufl., Urban & Fischer
Verlag/Elsevier GmbH [Online im Internet]München.
Altmann, K. Havemeyer, A.; Beitz, E.; Clement, B. Dimethylarginine-
Dimethylaminohydrolase-2 (DDAH-2) Does Not Metabolize Methylarginines.
ChemBioChem. 2012. 13(17):S. 2599–2604.
Altun, Z.; Uysal, S.; Guner, G.; Yilmaz, O.; Posaci, C. Effects of oral L‐arginine
supplementation on blood pressure and asymmetric dimethylarginine in
stress‐induced preeclamptic rats. Cell Biochem. Funct. 2008. 26(5):S. 648–653.
Anderssohn, M. Maaß, L.; Diemert, A.; Lüneburg, N.; Atzler, D.; Hecher, K.;
Böger, R. Severely decreased activity of placental dimethylarginine
dimethylaminohydrolase in pre-eclampsia. European Journal of Obstretrics ad
Gynecology and Reproductive Biology. 2012(161):S. 152–156.

71
AWMF-Leitline (2013) Diagnostik und Therapie hypertensiver
Schwangerschaftserkrankungen. URL:Online verfügbar unter
http://www.awmf.org/uploads/tx_szleitlinien/015-
018l_S1_Diagnostik_Therapie_hypertensiver_Schwangerschaftserkrankungen_
2014-01.pdf. [Stand: zuletzt aktualisiert am 01/2014].
AWMF-Leitline: Deutsche Gesellschaft für Gynäkologie und Geburtshilfe eV
(2012) Standards in der Perinatalmedizin - Dopplersonographie in der
Schwangerschaft. URL:Online verfügbar unter
www.awmf.org/uploads/tx_szleitlinien/015-
019l_S1_Doppplersonographie_in_der_Schwangerschaft_2013-03.pdf, [Stand:
zuletzt aktualisiert am 06/2012].
Ayling, L. Dimethylarginine dimethylaminohydrolase (DDAH) regulates
trophoblast invasion and motility through effects on nitric oxide. Human
Reproduction. 2006. 21(10):S. 2530–2537.
Becker, R.; Keller, T.; Kiesewetter, H.; Fangerau, H.; Bittner, U. Individual risk
assessment of adverse pregnancy outcome by multivariate regression analysis
may serve as basis for drug intervention studies: retrospective analysis of 426
high-risk patients including ethical aspects. Arch Gynecol Obstet. 2013.
288(1):S. 41–48.
Beinder, E. Mohaupt, M.; Schlembach, D.; Fischer, T.; Sterzel, R.; Lang, N.;
Baylis, C. Nitric Oxide Synthase Activity and Doppler Parameters in the
Fetoplacental and Uteroplacental Circulation in Preeclampsia. Hypertens
Pregnancy. 1999. 18(2):S. 115–127.
Bodnar, L. Ness, R.; Markovic, N.; Roberts, J. The Risk of Preeclampsia Rises
with Increasing Prepregnancy Body Mass Index. Annals of Epidemiology. 2005.
15(7):S. 475–482.

72
Böger, R. Bode-Boger, S.; Frölich, J. The I-arginine-nitric oxide pathway: role in
atherosclerosis and therapeutic implications. Atherosclerosis Supplements.
1996(127):S. 1–11.
Böger, R. Bode-Boger, S.; Szuba, A.; Tsao, P.; Chan, J.; Tangphao, O.;
Blaschke, T.; Cooke, J. Asymmetric Dimethylarginine (ADMA): A Novel Risk
Factor for Endothelial Dysfunction. Circulation. 1998. 98(18):S. 1842–1847.
Böger, R. Sydow, K.; Borlak, J.; Thum, T.; Lenzen, H.; Schubert, B. L
Cholesterol Upregulates Synthesis of Asymmetrical Dimethylarginine in Human
Endothelial Cells Involvement of S-Adenosylmethionine–Dependent
Methyltransferases. Circulation Research. 2000(87):S. 99–105.
Böger, R. Vallance, P.; Cooke, J. Asymmetric dimethylarginine (ADMA): a key
regulator of nitric oxide synthase. Atherosclerosis Supplements. 2003. 4(4):S.
1–3.
Böger, R. Asymmetric Dimethylarginine, an Endogenous Inhibitor of Nitric Oxide
Synthase, Explains the “L-Arginine Paradox” and Acts as a Novel
Cardiovascular Risk Factor. Journal of Nutrition. 2004aS. 2842–2847.
Böger, R. Asymmetrical methylarginine (ADMA) as a cardiovascular risk factor:
epidemiological and propective data. Dtsch med Wochenschr. 2004b.
129(15):S. 820–824.
Böger, R. Asymmetric dimethylarginine (ADMA): A novel risk marker in
cardiovascular medicine and beyond. Ann Med. 2006. 38(2):S. 126–136.
Böger, R. The Pharmacodynamics of L-Arginine. The Journal of Nutrition.
2007S. 1650S–1655S.

73
Böger, R. Diemert, A.; Schwedhelm, E.; Luneburg, N.; Maas, R.; Hecher, K. The
Role of Nitric Oxide Synthase Inhibition by Asymmetric Dimethylarginine in the
Pathophysiology of Preeclampsia. Gynecol Obstet Invest. 2010. 69(1):S. 1–13.
Braekke, K. Ueland, P.; Harsem, N.; Staff, A. Asymmetric Dimethylarginine in
the Maternal and Fetal Circulation in Preeclampsia. Pediatric Research. 2009.
66(4):S. 411–415.
Brahmarshi, D. Santa, S.-R.; Anindya, D.; Kumar, L.; Nath, D. Assessment of
Placental Oxidative Stress in Pre-Eclampsia. J Obstet Gynecol India. 2012.
62(1):S. 39–42.
Brosens I.A. Robertson W.B.; Dixon H.G. The role of the spiral arteries in the
pathogenesis of preeclampsia. Obstet Gynecol Annu. 1972(1):S. 177–191.
Calver, A. Collier, J.; Leone, A.; Moncada, S.; Vallance, P. Effect of local intra-
arterial asymmetric dimethylarginine (ADMA) on the forearm arteriolar bed of
healthy volunteers. J Hum Hypertens. 1993(7):S. 193–194.
Cartwright, J. Tse W. K., W. Hepatocyte Growth Factor Induced Human
Trophoblast Motility Involves Phosphatidylinositol-3-Kinase, Mitogen-Activated
Protein Kinase, and Inducible Nitric Oxide Synthase. Experimental Cell
Research. 2002. 279(2):S. 219–226.
CLASP Study CLASP: a randomised trial of low-dose aspirin for the prevention
and treatment of pre-eclampsia among 9364 pregnant women. CLASP
(Collaborative Low-dose Aspirin Study in Pregnancy) Collaborative Group. The
Lancet. 1994(343):S. 619–629.
Davison, J. Hytten, F. Glomerular filtration during and after pregnancy. J Obstet
Gynaecol Br Commonw. 1974. 81(8):S. 588–595.

74
Di Sessa T.G. Moretti ML, K. Cardiac function in fetuses and newborns exposed
to low-dose aspirin during pregnancy. Am J Obstet Gynecol. 1994(171 (4)):S.
892–900.
Duckitt, K. Risk factors for pre-eclampsia at antenatal booking: systematic
review of controlled studies. BMJ. 2005. 330(7491):S. 565–0.
Duley L. Henderson-Smart D.J.; Meher S.; King J.F. Antiplatelet agents for
preventing pre-eclampsia and its complications. The Cochrane Library.
2007(2):S. 1–150.
Duley, L. Henderson-Smart D.; Knight M.; King J. Antiplatelet drugs for
prevention of preeclampsia and its consequences: systematic review. British
Medical Journal. 2001(322):S. 329–333.
Duley, L. The Global Impact of Pre-eclampsia and Eclampsia. Seminars in
Perinatology. 2009. 33(3):S. 130–137.
Ehsanipoor, R. Fortson, W.; Fitzmaurice, L.; Liao, W.-X.; Wing, D.; Chen, D.-b.;
Chan, K. Nitric Oxide and Carbon Monoxide Production and Metabolism in
Preeclampsia. Reproductive Sciences. 2013. 20(5):S. 542–548.
Ellis, J. Wennerholm, U.-B.; Lilja, H.; Pettersson, A.; Sultan, B.; Wennergren,
M.; Hagberg, H. Levels of dimethylarginines and cytokines in mild and severe
preeclampsia. Acta Clinica Belgica. 2001(80):S. 602–608.
Fickling, S. Williams, D.; Vallance, P.; Nussey, S.; Whitley, G. Plasma
concentrations of endogenous inhibitor of nitric oxide synthesis in normal
pregnancy and pre-eclampsia. The Lancet. 1993. 342S. 242–243.

75
Fischer, T. Krause M.; Beinder, E.; Schlembach, D.; Rabenbauer B.; Wildt L.;
Lang, N. Schwangerschaftsverlängerung bei Patientinnen mit HELLP-Syndrom.
Geburtsh Frauenheilk. 1999(59):S. 345–355.
Gaugler-Senden, I.; Huijssoon, A.; Visser, W.; Steegers, E.; Groot, C. de
Maternal and perinatal outcome of preeclampsia with an onset before 24 weeks’
gestation. European Journal of Obstetrics & Gynecology and Reproductive
Biology. 2006. 128(1-2):S. 216–221.
Haig D Genetic conflicts in human pregnancy. Q Rev Biol. 1993. 68(4):S. 495–
532.
Holden, D. Fickling, S.; Whitley, G. Plasma concentrations of asymmetric
dimethylarginine, a natural inhibitor of nitric oxide synthase, in normal
pregnancy and preeclampsia. American Journal of Obstetrics and Gynecology.
1998(178):S. 551–556.
Holland, P. Abramson, R.; Watson, R.; Gefland, D. Detection of specific
polymerase chain reaction product by utilizing the 5' -3' exonuclease activity of
Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA. 1991(88):S.
7276–7280.
Horowitz, J. Heresztyn, T. An overview of plasma concentrations of asymmetric
dimethylarginine (ADMA) in health and disease and in clinical studies:
Methodological considerations. Journal of Chromatography B. 2007. 851(1-2):S.
42–50.
Kakimoto, Y. Akazawa, S. Isolation and identification of NG,NG- and NG,NG-
dimethyl-arginine, N-epsilon-mono-, di-, and trimethyllysine, and
glucosylgalactosyl- and galactosyl-delta-hydroxylysine from human urine. J Biol
Chem. 1970(245):S. 5751–5758.

76
Kenneth, J. Livak and Thomas D. Schmittgen Analysis of Relative Gene
Expression Data Using Real-Time Quantitative PCR and the 2 ^-∆∆Ct Method.
METHODS 25 Elsevier Science. 2001S. 402–408.
Khan, F. Changes in Endothelial Function Precede the Clinical Disease in
Women in Whom Preeclampsia Develops. Hypertension. 2005. 46(5):S. 1123–
1128.
Kiechle, M. (2007) Gynäkologie und Geburtshilfe, 1; S. 225, Elesevier GmbH
München.
Kielstein, J. Veldink, H.; Martens-Lobenhoffer, J.; Haller, H.; Burg, M.; Lorenzen,
J.; Lichtinghagen, R.; Bode-Böger, S.; Kliem, V. SDMA is an early marker of
change in GFR after living-related kidney donation. Nephrol Dial Transplant.
2011(26):S. 324–328.
Kim, Y. Park, H.; Lee, H.; Ha, E.; Suh, S.; Oh, S.; Yoo, H.-S. Reduced l-arginine
Level and Decreased Placental eNOS Activity in Preeclampsia. Placenta. 2006.
27(4-5):S. 438–444.
Kim, Y. Pathogenesis and Promising non-invasive markers for Preeclampsia.
Obstet Gynecol. Sci. 2013(56):S. 2–7.
Kirkpatrick, C. The HELLP Syndrome. Acta Clinica Belgica. 2010S. 65–72.
Kul, M. Demirkaya, E.; İpçioğlu, O.; Karadeniz, R.; Tunc, T.; Vurucu, S.; Yesil, F.;
Öztin, H.; Cakir, E. Perinatal risk factors affecting the maternal and fetal
asymmetric dimethylarginine levels. The Turkish Journal of Pediatrics. 2009S.
141–145.

77
Lachmeijer, A. M. A., Dekker, G. A., Pals, G., Aarnoudse,J. G., ten Kate, L. P.
and Arngrimsson, R. (2002), Searching for preeclampsia genes: the current
position. Eur. J. Obstet. Gyn. 105, 94–113
Lafayette, R. Druzin M, S. Nature of glomerular dysfunction in pre-eclampsia.
Kidney International. 1998(54):S. 1240–1249.
Lees MM, Scott DB, Kerr MG, Taylor SH.The circulatory effects of recumbent
postural change in late pregnancy. Clin Sci 1967;32:453-65.
Lees, C. Valensise, H.; Black, R.; Harrington, K.; Byiers, S.; Romanini, C.;
Campbell, S. The efficacy and fetal-maternal cardiovascular effects of
transdermal glyceryl trinitrate in the prophylaxis of pre-eclampsia and its
complications: a randomized double-blind placebo-controlled trial. Ultrasound
Obstet Gynecol. 1998S. 334–338.
Leiper, J. Santa Maria J, C. Identification of two human dimethylarginine
dimethylaminohydrolases Identification of two human dimethylarginine
dimethylaminohydrolases with distinct tissue distributions and homology with
microbial arginine deiminases. Biochem J. 1999(343):S. 209–214.
Leitich H. Egarter C, H. A meta-analysis of low dose aspirin for the prevention of
intrauterine growth retardation. Br J Obstet Gynaecol. 1997(104):S. 450–459.
Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF,
Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA,
Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004
Feb 12;350(7):672-83.
Lin S. Shimizu I, S. Uterine artery Doppler velocimetry in relation to trophoblast
migration into the myometrium of the placental bed. Obstet Gynecol.
1995(86):S. 760–765.

78
López-Jaramillo, P. Narváez, M.; Calle, A.; Rivera, J.; Jácome, P.; Ruano, C.;
Nava, E. Cyclic guanosine 3’3’ monophosphate concentrations in pre-eclampsia
: effects of hydralazine. BJOG 1996. 103(1):S. 33–38.
Lüneburg, N. Lieb, W.; Schwedhelm, E.; Zeller, T.; Carter, A.; Sullivan, L.; Chen,
M.; Maas, R.; Xanthakis, V.; Schillert, A.; Ziegler, A.; Wild, P.; Blankenberg, S.;
Erdmann, J.; Grant, P.; Muenzel, T.; Vasan, R.; Böger, R. Genome-Wide
Association Study of Dimethylarginines Reveals Novel Metabolic Pathway for
SDMA. Circulation. 2011(124):
Lyall F. Jablonka-Shariff A, J. Gene expression of nitric oxide synthase in
cultured human term placental trophoblast during in vitro differentiation.
Placenta. 1998(19):S. 253–260.
Maas, R. Böger, R.; Schwedhelm, E. Maas_ADMA-Columbian-(ADMA) in
Colombian Women With Pre-eclampsia Plasma Concentrations of Asymmetric
Dimethylarginine (ADMA) in Colombian Women with Preeclampsia. Journal of
American Medical Association. 2004. 291(7):S. 823–824.
Maas, R. Baschat A., H. Asymmetrisches Dimethylarginin (ADMA): Ein
endogener Hemmstoff der NO-Synthase – und auch ein Risikomarker der
Präeklampsie? Geburtsh Frauenheilk. 2007(67):S. 611–619.
Maas, R. Erdmann J; Lüneburg N.; Stritzke J.; Schwedhelm E.; Meisinger C.;
Peters A.; Weil J.; Schunkert H. Polymorphisms in the promoter region of the
dimethylarginine dimethylaminohydrolase 2 gene are associated with
prevalence of hypertension. Pharmacological Research. 2009(60):S. 488–493.
Maeda T. Yoshimura T.; Okamura H. Asymmetric Dimethylarginine, an
Endogenous Inhibitor of Nitric Oxide Synthase, in Maternal and Fetal

79
Circulation. Journal of the Society for Gynecologic Investigation. 2003(10):S. 2–
4.
Magann, E. Bass D, C., JR. Antepartum corticosteroids: disease stabilization in
patients with the syndrome of hemolysis, elevated liver enzymes, and low
platelets (HELLP). Am J Obstet Gynecol. 1994(171):S. 1148–1153.
Many, A. Carl A. Hubel Susan J. Fisher James M. Roberts and Yan Zhou
Invasive Cytotrophoblasts Manifest Evidence of Oxidative Stress in
Preeclampsia. American Journal of Pathology. 2000. 156S. 321–331.
Mao, D. Che, J.; Li, K.; Han, S.; Yue, Q.; Zhu, L.; Zhang, W.; Li, L. Association
of homocysteine, asymmetric dimethylarginine, and nitric oxide with
preeclampsia. Arch Gynecol Obstet. 2010. 282(4):S. 371–375.
Meekins J.W. van Pijnenborg R, H. A study of placental bed spiral arteries and
trophoblast invasion in normal and severe pre-eclamptic pregnancies. Br J
Obstet Gynaecol. 1994(101):S. 669–674.
Misra, D. Kiely, J. The Association Between Nulliparity and Gestational
Hypertension. J Clin Epidemiol. 1997. 50(7):S. 851–855.
Mistry, H. Kurlak L.O.; Pipkin F.B. The placental renin-angiotensin system and
oxidative stress in pre-eclampsia. Placenta. 2013(34):S. 182–186.
Mittermayer, F. Prusa, A.-R.; Pollak, A.; Wolzt, M. Umbilical vein plasma
concentrations of asymmetrical dimethylarginine are increased in male but not
female neonates delivered preterm: A pilot study. Early Human Development.
2006. 82(7):S. 421–424.

80
Moran, P. Baylis PH, L. Glomerular Ultrafiltration in Normal and Preeclamptic
Pregnancy. Journal of the American Society of Nephrology. 2003. 14(3):S. 648–
652.
Myatt, L.; Eis, A.; Brockman, D.; Greer, I. Endothelial nitric oxide synthase in
placental villous tissue from normal, pre-eclamptic and intrauterine growth
restricted pregnancies*. Human Reproduction. 1997. 12(1):S. 167–172.
Nakajima, T. Matsuoka, Y.; Kakimoto, Y. Isolation and Identification of NG-
Monomethyl, NG, NG Dimethyl- and NG, NG, Dimethylarginine from the
hydrolysate of proteins of bovine brain. Biochim Biophys Acta. 1971(23):S. 212–
222.
O´Dwyer M.J. Dempsey F, C. Septic shock is correlated with asymmetrical
dimethyl arginine levels, which may be influenced by a polymorphism in the
dimethylarginine dimethylaminohydrolase II gene: a prospective observational
study. Crit Care. 2006. 10(5):S. R139.
Odegard, R. Vatten, L.; Nilsen, S.; Salvesen, K.; Austgulen, R. Risk factors and
clinical manifestations of pre-eclampsia. British Journal of Obstetrics and
Gynaecology. 2000. 197S. 1410–1416.
Ogawa T, K. Metabolism of NG,NG-and NG,N'G-dimethylarginine in rats. Arch
Biochem Biophys. 1987. 252(2):S. 526–537.
Ogawa T, K. Kimoto, M.; Sasaoka, K. Dimethylarginine:pyruvate
aminotransferase in rats. Purification, properties, and identity with
alanine:glyoxylate aminotransferase 2. The Journal of Biological Chemistry.
1990(34):S. 20938–20945.

81
Ornaghi, S. Tyurmorezova, A.; Algeri, P.; Giardini, V.; Ceruti, P.; Vertemati, E.;
Vergani, P. Influencing factors for late-onset preeclampsia. J Matern Fetal
Neonatal Med. 2013. 26(13):S. 1299–1302.
Papageorghiou, A. Yu, C.; Bindra, R.; Pandis, G.; Nicolaides, K. Multicenter
screening for pre-eclampsia and fetal growth restriction by transvaginal uterine
artery Doppler at 23 weeks of gestation. Ultrasound Obstet Gynecol. 2001. 18S.
441–449.
Phyllis A Clinical features, d. (2010) Clinical features, diagnosis, and long-term
prognosis of preeclampsia. URL online verfügbar unter
http://www.uptodate.com/contents/preeclampsia-clinical-features-and-diagnosis.
Pijnenborg, R. Dixon, G.; Robertson, W.; Brosens, I. Trophoblastic Invasion of
Human Decidua From 8 to 18 Weeks of Pregnancy. Placenta. 1980. 1(3-19):
Prefumo, F. Thilaganathan B.; Whitley G. S. First-trimester uterine artery
resistance and maternal serum concentration of asymmetric dimethylarginine.
Ultrasound Obstet Gynecol. 2008. 31(2):S. 153–157.
Redman, C. Latest Advances in Understanding Preeclampsia. Science. 2005.
308(5728):S. 1592–1594.
Rey, E. Garneau, P.; David, M.; Gauthier, R.; Leduc, L.; Michon, N.; Morin, F.;
Demers, C.; Kahn, S.; Magee, L.; Rodger, M. Dalteparin for the prevention of
recurrence of placental-mediated complications of pregnancy in women without
thrombophilia: a pilot randomized controlled trial. Journal of Thrombosis and
Haemostasis. 2009. 7(1):S. 58–64.
Rizos, D. Eleftheriades, M.; Batakis, E.; Rizou, M.; Haliassos, A.; Hassiakos, D.;
Botsis, D. Levels of asymmetric dimethylarginine throughout normal pregnancy

82
and in pregnancies complicated with preeclampsia or had a small for
gestational age baby. J Matern Fetal Neonatal Med. 2012. 25(8):S. 1311–1315.
Roberts, J. Hubel C.A. The Two Stage Model of Preeclampsia: Variations on the
Theme. Placenta. 2009(30):S. 32–37.
Rodionov, R. Murry, D.; Vaulman, S.; Stevens, J.; Steven R. Human Alanine-
Glyoxylate Aminotransferase 2 Lowers Asymmetric Dimethylarginine and
Protects from Inhibition of Nitric Oxide Production. The Journal of Biological
Chemistry 2010(285):S. 5385–5391.
Sandrim, V. Palei, A.; Metzger, I.; Cavalli, R.; Duarte, G.; Tanus-Santos, J.
Interethnic differences in ADMA concentrations and negative association with
nitric oxide formation in preeclampsia. Clinica Chimica Acta. 2010. 411(19-
20):S. 1457–1460.
Savvidou, M. Hingorani, A.; Tsikas, D.; Frölich, J.; Vallance, P.; Nicolaides, K.
Endothelial dysfunction and raised plasma concentrations of asymmetric
dimethylarginine in pregnant women who subsequently develop pre-eclampsia.
The Lancet. 2003. 361(9368):S. 1511–1517.
Schepers, E. Barreto DV, L. Symmetric Dimethylarginine as a Proinflammatory
Agent in Chronic Kidney Disease. Clin J Am Soc Nephrol. 2011. 6(10):S. 2374–
2383.
Schiessl B, M.; Kuhn, C.; Schulze, S.; Kunze, S.; Friese, K.; Jeschke, U.
Expression of Endothelial NO Synthase, Inducible NO Synthase, and Estrogen
Receptors Alpha and Beta in Placental Tissue of Normal, Preeclamptic, and
Intrauterine Growth-restricted Pregnancies. Journal of Histochemistry and
Cytochemistry. 2005. 53(12):S. 1441–1449.

83
Schneider, H. (2006) Präimplantation, Implantation und Plazentation:
Bedeutung für den Schwangerschaftsverlauf, Die Geburtshilfe, 2011, pp 3-17
Springer.
Schulze, F.; Lenzen, H.; Hanefeld, C.; Bartling, A.; Osterziel, K.; Goudeva, L. et
al. Asymmetric dimethylarginine is an independent risk factor for coronary heart
disease: Results from the multicenter Coronary Artery Risk Determination
investigating the Influence of ADMA Concentration (CARDIAC) study. American
Heart Journal. 2006. 152(3):S. 493.e1–493.e8.
Schwedhelm, E. Quantification of ADMA: analytical approaches. Vasc med.
2005. 10(2):S. 89–95.
Schwedhelm, E. Maas, R.; Tan-Andresen, J.; Schulze, F.; Riederer, U.; Böger,
R. High-throughput liquid chromatographic-tandem mass spectrometric
determination of arginine and dimethylated arginine derivatives in human and
mouse plasma. Journal of Chromatography B. 2007. 851(1-2):S. 211–219.
Schwedhelm, E.; Böger, R. The role of asymmetric and symmetric
dimethylarginines in renal disease. Nat. Rev. Nephrology. 2011(7):S. 275–285.
Schwedhelm, E.; Xanthakis, V.; Maas, R.; Sullivan, L.; Schulze, F.; Riederer, U.
et al. Asymmetric Dimethylarginine Reference Intervals Determined with Liquid
Chromatography-Tandem Mass Spectrometry: Results from the Framingham
Offspring Cohort. Clinical Chemistry. 2009. 55(8):S. 1539–1545.
Seck, K. Fischer, T. Empfehlungen zur Diagnostik und Behandlung der
Präeklampsie im internationalen Vergleich: Was ist neu? Z Geburtshilfe
Neonatol. 2009. 213(03):S. 106–112.
Sibai, B. Ewell, M.; Levine, R.; Klebanoff, M.; Esterlitz, J.; Catalano, P.;
Goldenberg, R.; Joffe, G. Risk factors associated with preeclampsia in healthy

84
nulliparous women. American Journal of Obstetrics and Gynecology. 1997.
177(5):S. 1003–1010.
Sibai, B. Dekker, G.; Kupferminc, M. Pre-eclampsia. The Lancet. 2005.
365(9461):S. 785–799.
Siroen, M. Teerlink, T.; Bolte, A.; van Elburg, R.; Richir, M.; Nijveldt, R.; van der
Hoven, B.; van Leeuwen, P. No Compensatory Upregulation of Placental
Dimethylarginine Dimethylaminohydrolase Activity in Preeclampsia. Gynecol
Obstet Invest. 2006. 62(1):S. 7–13.
Slaghekke, F. Dekker, G.; Jeffries, B. Endogenous inhibitors of nitric oxide and
preeclampsia: A review. J Matern Fetal Neonatal Med. 2006. 19(8):S. 447–452.
Speer, P. Powers, R.; Frank, M.; Harger, G.; Markovic, N.; Roberts, J. Elevated
asymmetric dimethylarginine concentrations precede clinical preeclampsia, but
not pregnancies with small-for-gestational-age infants. American Journal of
Obstetrics and Gynecology. 2008. 198(1):S. 112.e1.
Steegers, E. Peter von Dadelszen; Johannes J Duvekot; Robert Pijnenborg Pre-
eclampsia. The Lancet. 2010(376):S. 631–644.
Steinhard, J. Klockenbusch, W. Schwangerschaftsinduzierte Hypertonie und
Präeklampsie: Risikofaktoren und Vorhersagemöglichkeiten. Gynäkologe. 1999.
32S. 753-760.
Stephan, H. Faber, R. Die uterine Perfusionsstörung im zweiten Trimenon -
aktuelle pathophysioogische, diagnostische und therapeutische Aspekte. Z
Geburtshilfe Neonatol. 2004S. 205–209.
Stillman, I. Karumanchi, S. The Glomerular Injury of Preeclampsia. Journal of
the American Society of Nephrology. 2007. 18(8):S. 2281–2284.

85
Surdacki A. Nowicki M, S. Reduced urinary excretion of nitric oxide metabolites
and increased plasma levels of asymmetric dimethylarginine in men with
essential hypertension. J Cardiovasc Pharmacol. 1999(33 (4)):S. 652–658.
Tanhäuserová, V. Tomandl, J.; Pácal, L.; Klepárník, M.; Malúšková, D.;
Bartáková, V.; Kuricová, K.; Rehořová, J.; Stěpánková, S.; Svojanovský, J.;
Olšovský, J.; Bělobrádková, J.; Krusová D; Jurajda, M.; Mužík, J.; Pavlík, T.;
Kaňková, K. ADMA, SDMA and L-arginine/ADMA Ratio but not DDAH Genetic
Polymorphisms are Reliable Predictors of Diabetic Nephropathy Progression as
Identified by Competing Risk Analysis. Kidney Blood Press Res. 2012. 36(1):S.
200–208.
Thaler I. Manor D, R. Hemodynamic evaluation of the female pelvic vessels
using a high-frequency transvaginal image-directed Doppler system. J Clin
Ultrasound. 1990(18):S. 364–369.
The GOPEC Consortium Disentangling Fetal and Maternal Susceptibility for
Pre-Eclampsia: A British Multicenter Candidate-Gene Study. Am. J. Hum.
Genet. 2005. 77S. 127–131.
Tomiyana, H. Yamada, J.; Koji, Y.; Shiina, K.; Yoshida, M.; Yamashina, A. Effect
of Telmisartan on Forearm Postischemic Hyperemia and Serum Asymmetric
Dimethylarginine Levels. American Journal of Hypertension. 2007. 20(12):S.
1305–1311.
Tsukahara, H. Ohta, N.; Tokuriki, S.; Nishijima, K.; Kotsuji, F.; Kawakami, H.;
Ohta, N.; Sekine, K.; Nagasaka, H.; Mayumi, M. Determination of asymmetric
dimethylarginine, an endogenous nitric oxide synthase inhibitor, in umbilical
blood. Metabolism. 2008. 57(2):S. 215–220.

86
Valkonen, V.-P. Tuomainen, T.-P.; Laaksonen, R. DDAH gene and
cardiovascular risk. Vasc med. 2005. 10(2):S. 45–48.
Vallance, P. Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an
endogenous inhibitor of nitric oxide synthesis in chronic renal failure. The
Lancet. 1992. 339S. 572–575.
Vallance, P. Leiper, J. Cardiovascular Biology of the Asymmetric
Dimethylarginine:Dimethylarginine Dimethylaminohydrolase Pathway.
Arteriosclerosis, Thrombosis, and Vascular Biology. 2004. 24(6):S. 1023–1030.
Vida, G. Sulyok, E.; Ertl, T.; Martens-Lobenhoffer, J.; Bode-Böger, S. Plasma
Asymmetric Dimethylarginine Concentration during the Perinatal Period.
Neonatology. 2007. 92(1):S. 8–13.
Wang Y. Walsh SW. Placental mitochondria as a source of oxidative stress in
pre-eclampsia. Placenta. 1998(19 (8)):S. 581–586.
Wang, Y.; Zhang, M.; Liu, Y.; Liu, Y.; Chen, M. The effect of nebivolol on
asymmetric dimethylarginine system in spontaneously hypertension rats.
Vascular Pharmacology. 2011. 54(1-2):S. 36–43.
Williams, P.; Morgan, L. The role of genetics in pre-eclampsia and potential
pharmacogenomic interventions. PGPM. 2012, S. 37.
Wilson, M. L., Goodwin, T. M., Pan, V. L. and Ingles, S. A. (2003) Molecular
epidemiology of preeclampsia. Obstet. Gynecol. Surv. 58, 39–66
Xu, A. Xu RM; Lu CQ; Li DD; Xu QF; Guo J.; Fu X.; Zhao W.; Yao MY
Association study of dimethylarginine dimethylaminohydrolase 2 gene
polymorphisms and coronary heart disease. Mol Med Rep. 2012. 6(5):S. 1103–
1105.

87
Yu, C. Smith Gordon C.S; Papageorghiou Aris T.; Cacho Ana Maria; Nicolaides
Kypros H. An integrated model for the prediction of preeclampsia using
maternal factors and uterine artery Doppler velocimetry in unselected low-risk
women. American Journal of Obstetrics and Gynecology. 2005. 193(2):S. 429–
436.
Yu, C.; Papageorghiou, A.; Parra, M.; Palma Dias, R.; Nicolaides, K.
Randomized controlled trial using low-dose aspirin in the prevention of pre-
eclampsia in women with abnormal uterine artery Doppler at 23 weeks'
gestation. Ultrasound Obstet Gynecol. 2003. 22(3):S. 233–239.
Zoccali C. Bode-Böger S, M. Plasma concentration of asymmetrical
dimethylarginine and mortality in patients with end-stage renal disease: a
prospective study. The Lancet. 2001. 358(9299):S. 2113–2117.

88
Anhang
8. Abkürzungsverzeichnis
A
ADMA: Asymmetrisches Dimethylarginin
ALAT Alanin-Aminotransferase
ANOVA analysis of variance Varianzanalyse
ASS Acetylsalicylsäure
ASAT Aspartat-Aminotransferase
AWMF Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fach
gesellschaften e.V.
B
BMI Body Mass Index in kg/m2
C
cDNA Complementary DNA = komplementäre Desoxyribonukleinsäure
cGMP Zyklisches Guanosinmonophosphat
D
DDAH 1 und 2 Dimethylarginin-Dimethylaminohydrolase 1 und 2
DGGG Deutsche Gesellschaft für Gynäkologie und Geburtshilfe e.V.
DIC Disseminated Intravasal Coagulation = Disseminierte Intravasale
Gerinnung
DNA Desoxyribonukleinsäure
E
EDRF Endothelium Derived Relaxing Factor (= NO)
EDTA Ethylendiamintetraacetat
ELISA Enzyme Linked Immunosorbent Assay
eNOS Endotheliale Sickstoffmonoxid-Synthase
F
FAM 6-Carboxy-Fluorescein
G
GTN Glyceroltrinitrat
GFR Glomeruläre Filtrationsrate
GMP Guanosinmonophosphat

89
GSNO S-Nitrosoglutathion
GTP Guanosintriphosphat
H
HELLP Haemolysis, elevated liver enzymes, low platelet count/ HELLP-
Syndrom
HGF Hepatocyte Growth Factor
HPLC High-performance liquid chromatography
I
iNOS Induzierbare Sickstoffmonoxid-Synthase
ISDN Iso Isosorbidnitrat
Isosorbidmononitrat
IUGR Intrauterine Growth Restriction
K
K Kontrollen
L
LC-MS/MS Liquid chromatography mass spectrometry
LDH Lactatdehydrogenase
L-NMMA N-Monomethyl-L-Arginin
M
MGB minor groove binder
MRM Multiple Reaction Monitoring
mRNA Messenger Ribonukleinsäure
N
NO Stickstoffmonoxid
NOS NO-Synthase = Stickstoffmonoxid-Synthase
P
PBS Phosphate buffered saline = Phosphatgepufferte Salzlösung
PCR Polymerase chain reaction = Polymerase-Kettenreaktion
PE Präeklampsie
PI Pulsatilitäts-Index (in der Dopplersonografie)
PRMT Protein-N-Arginin-Methyltransferase
PTT Partial Thromboplastin Time (partielle Thromboplastinzeit)

90
R
RNA Ribonukleinsäure
Rpm Rounds per minute
RR Blutdruck (nach Riva Rocci) sowie Relative Risikoreduktion
RT-PCR RealTime PCR
S
SAM S-Adenosylmethinonin
SDMA Symmetrisches Dimethylarginin
SGA Small for gestational age
SNPs Single nucleotide polymorphisms
SS Schwangerschaft
SSW Schwangerschaftswoche
T
TXA Thromboxan
U
UKE Universitätsklinikum Hamburg-Eppendorf
V
VEGF Vascular endothelial growth factor (Wachstumsfaktor)
W
WHO World Health Organisation
Z
ZNS Zentrales Nervensystem

91
9. Abbildungsverzeichnis
Abbildung 1 Dopplerhüllkurven der Aa. ueterinae ..................................................... 11
Abbildung 2 Trophoblasteninvasion im Vergleich bei Nichtschwangeren,
Präeklampsie und einer gesunden Schwangerschaft ................................................ 16
Abbildung 3 NO-Synthese im Gefäßendothel............................................................ 16
Abbildung 4 Methylariginine ...................................................................................... 18
Abbildung 5 Plasmakonzentrationen von ADMA in Nicht-Schwangeren,
gesunden Schwangeren und Präeklampsie-Patientinnen ......................................... 20
Abbildung 6 NucleoSpin Blood Quick Pure ............................................................... 31
Abbildung 7 Beispiel eines Allelic Discrimnator Plots beispielhaft für den SNP
DDAH1 rs1146381 .................................................................................................... 33
Abbildung 8 Untersuchte SNPs ................................................................................. 33
Abbildung 9 Beispiele Plazenten bei der Probeentnahme ......................................... 34
Abbildung 10 Taqman RT-PCR.................................................................................. 36
Abbildung 11 ADMA und SDMA im Vergleich zwischen Subgruppen ........................ 47
Abbildung 12 Korrelationen zwischen Serumkreatinin und maternalem
ADMA/SDMA (µmol/l) (beider Gruppen) .................................................................... 47
Abbildung 13 Arteriovenöse Differenzen von ADMA, SDMA und L-Arginin ............... 50
Abbildung 14 Plazentare Expressionsanalysen von eNOS, PRMT1, DDAH 1
und 2 ......................................................................................................................... 54

92
10. Tabellenverzeichnis
Tabelle 1 Formen der Gestationshypertonie ....................................................... 8
Tabelle 2 Messmethoden von Asymmetrischem Dimethylarginin in
Präeklampsie-Patientinnen ............................................................................... 23
Tabelle 3 Basischarakteristika im Vergleich ...................................................... 42
Tabelle 4 Vergleich Patientinnen-Charakteristika ............................................. 43
Tabelle 5 Laborwerte im Vergleich leichte, schwere Präeklampsie und HELLP 44
Tabelle 6 Dimetylarginine im Vergleich ............................................................. 46
Tabelle 8 Kontrollen und Präeklampsie - Korrelationen von Basischarakeristika
mit Dimethylargininen ....................................................................................... 49
Tabelle 9 Assoziationen zwischen müttlerlichen SNPs, mütterlichem ADMA und
Präeklampsie .................................................................................................... 52
Tabelle 10 Assoziationen zwischen fetalen SNPs, mütterlichem ADMA und
Präeklampsie .................................................................................................... 53
Tabelle 11 Dimethylarginine mit relativen Genexpression in Kontroll- und
Präeklampsiegruppe ........................................................................................ 55

93
11. Materialien
11.1. Chemikalien und Reagenzien
Acetat-Klebefolie
Astec Chirobiotic T Vorsäule 20x1.0 mm, Alltech (spec Astec) 12101
10 nM dNTP Mix
384-well PCR-Platten von Saarstedt
Abdeckfolie für die Platten
Abdeckmatte für MegaBlock 1.2 mL (rund), Sarstedt 95.1990.002
Acetonitril, gradient grade
ADMA
Apothekenwasser
autoklavierte Pipettenspitzen
autoklavierte Reaktionsgefäße 1,5 ml
Brand Multipette
Butanolische Salzsäure
Chloroform
Chloroform 99+%, Sigma, C2432-500ML
DEPC-Wasser
Deuteriertes [2H7]-Arginin, Cambridge Isotope Labs, EURISO-Top, DLM-541
Eis mit Metallblock
Eppendorf 8-Kanal Transferpipette 20 bis 200 uL
Eppendorf epTIPS 100 uL
Ethanol 70 %ig (Ethanol abs, verdünnt), Merck, 1009832500
Fermentas RevertAid first strand cDNA synthesis kit
flüssigen Stickstoff
Greiner PP Mikrotiterplatten 650201
Hydroxy-nor-L-arginin (nor-NOHA) Calbiochem #399275wN-
Isopropanol (2-Propanol), Merck, 1096342500
L-Arginin
Master Mix Taqman
Maxima Probe/ROX qPCR Master Mix(2x)(Fermentas K0232)
Methanol, gradient grade

94
Millipore 96well-Filterplatten 0.22 µm MultiScreen MSGVN 2210
Nucleo Spin® Blood Quick Pure Kit (Macherey-Nage)
PBS
Probenpuffer
Reaction Buffer
RevertAid™M-MuLV Reverse -Transcriptase (200u/µl)
RiboLock™Rnase Inhibitor (20u/µl)
RNAzol® B - RNA Isolation Reagent, WAK-Chemie, WAK-CS105
Sonden:
C_31397494_10, DDAH1, rs 11161606, 07/2016
C_1406511_10, DDAH1, rs11161614, 07/2016
C_1406524_10, DDAH1, rs1146381, 07/2016
C_2246286_10, DDAH1, rs18582, 08/2015
C_2518313_1, DDAH1, rs2268667, 08/2015
C_2246275_10, DDAH1, rs233112, 08/2015
C_1406535_10, DDAH1, rs506733, 07/2016
C_11907905_10, DDAH1, rs6697083, 12/2014
C_29938550_10, DDAH1, rs7555486, 07/2016
C_3233673_10, DDAH2, rs805305, 02/2012
SDMA
Skalpell (für Gewebe)
sterile 2 mL Eppendorfgefäße
sterile Spitzen für Eppendorf Pipetten
TaqMan Probes (Assay on Demand, Applied Biosystems)
Tissue Lyser Kugeln
Wasser, gradient grade (Baker)
Wattestäbchen
Geräte
Eppendorf Zentrifuge 5810R
Autoklav Wesarg, Medizintechnik, Deutschland
Bio-Rad SmartSpec 3000 Bio-Rad-Laboratories GmbH, München
Elektroporationsküvette BioRad Laboratories, USA

95
Eppendorf Mastercycler gradient Eppendorf AG, Hamburg
Eppendorf Safe Lock Reaktionsgefäße Eppendorf AG, Hamburg
Eppendorf Thermomixer 5436 Eppendorf AG, Hamburg
Eppendorf Thermomixer compact Eppendorf AG, Hamburg
Feinwaage, Sartorius CP225D
Heizblock
HPLC Varian ProStar
Kühlzentrifuge, Eppendorf 5415 R
NanoDrop 3300, Thermo Scientific
Reagenzglasschüttler (Vortexer)
Sartorius Laborwaage CP 225D Sartorius AG, Göttingen
Taqman
Tissue Lyser, QIAGEN
Varian L1200 MS/MS inkl. Computer/Drucker

96
12. Studienorganisation
12.1. Einverständnis Studienteilnahme

97
12.2. Einverständnis Gewebe- und Nabelschnurblutentnahme

98
12.3. Einverständniserklärung Genetische Untersuchung

99
12.4. Patientinnen-Information Seite 1

100
12.5. Patientinnen-InformationSeite 2-

101
12.6. Patientinnenerfassungsbogen

102

103
13. Danksagung
Mein großer Dank geht an Herrn Prof. Dr. Böger und Dr. Maike Anderssohn für
die Bereitstellung des Themas meiner Doktorarbeit und die große
Unterstützung, die ich von Ihnen erhalten habe!
Herr Prof. Böger sagte zu Beginn, dass die Zeit der Doktorarbeit mit vielen
Höhen und Tiefen einhergehen wird und dass ich erst danach wissen werde,
was Frustrationstoleranz bedeutet! Wie wahr!
Bedanken möchte ich mich zudem bei Herrn Prof. Dr. Hecher und Herrn Prof.
Dr. Hackeloer für die Ermöglichung der klinischen Studie in ihren Abteilungen
und für die Mitarbeit aller Ärztinnen und Ärzten, besonders bei Frau Dr. Anke
Diemert und Herrn PD Dr. Lars Hellmeyer. Ohne die Hilfe der Hebammen und
Krankenschwestern der Kreißsäle und der Pränatalstationen am Uniklinikum
Hamburg-Eppendorf und in der Asklepiosklinik Barmbek wäre der klinische
Anteil meiner Doktorarbeit nie gelungen wäre- vielen Dank!
Für den experimentellen Teil meiner Doktorarbeit hatte ich große Unterstützung
durch das Labor der Klinischen Pharmakologie, vielen herzlichen Dank, liebe
Anna, liebe Mariola, liebe Sandra und liebe Waltraud.
Nicht zuletzt bekam ich viele Anregungen von all den Kolleginnen und Kollegen
der Klinischen Pharmakologie, besonders von Nicole Lüneburg, Susann
Groschke und Isabell Bernges!
Durch unseren gemeinsamen Schwerpunkt habe ich diese Zeit besonders
intensiv mit Ninja Hoffmann geteilt, vielen lieben Dank Ninja, das ich immer auf
Dich zählen konnte, geteiltes Leid ist halbes Leid!!
Großer Dank geht an meine Familie und meine Freunden, auf deren
Unterstützung ich mich immer verlassen kann.

104
14. Lebenslauf
Entfällt aus datenschutzrechtlichen Gründen

105
15. Eidesstattliche Versicherung
Ich versichere ausdrücklich, dass ich die Arbeit selbständig und ohne fremde
Hilfe verfasst, andere als die von mir angegebenen Quellen und Hilfsmittel nicht
benutzt und die aus den benutzten Werken wörtlich oder inhaltlich
entnommenen Stellen einzeln nach Ausgabe (Auflage und Jahr des
Erscheinens), Band und Seite des benutzten Werkes kenntlich gemacht habe.
Ferner versichere ich, dass ich die Dissertation bisher nicht einem Fachvertreter
an einer anderen Hochschule zur Überprüfung vorgelegt oder mich anderweitig
um Zulassung zur Promotion beworben habe.
Ich erkläre mich einverstanden, dass meine Dissertation vom Dekanat der
Medizinischen Fakultät mit einer gängigen Software zur Erkennung von
Plagiaten überprüft werden kann.
Unterschrift Pia Schmidt-Ropertz





























