Embed Size (px)
Citation preview

Untersuchung der Eigenschaften
reaktiver Gele im Kontext
der festphasengebundenen
DNA-Replikation
Dissertation
zur Erlangung des Doktorgrades
der Fakultät für Chemie
der Ruhr-Universität Bochum.
vorgelegt von
Malte Reimold aus Bochum
Bochum 2006

1. Referent: Prof. Dr. G. von Kiedrowski
2. Referent: Prof. Dr. M. Feigel
3. Referent: Prof. Dr. Ch. Herrmann
Tag der mündlichen Prüfung: 19.12.2006

Die vorliegende Arbeit wurde in der Zeit von Juni 2000 bis Oktober 2006 an der Fakultät für
Chemie der Ruhr-Universität Bochum unter der Leitung von Prof. Dr. Günter von Kiedrowski
angefertigt.
Ich danke Herrn Prof. Dr. von Kiedrowski für die interessante Themenstellung und die
Freiheit bei der Durchführung dieser Arbeit.

Publikationen
Towards replicatable, multifunctional, nano-scaffolded machines
- a chemical manifesto G. von Kiedrowski, L.-H. Eckardt, K. Naumann, W. M. Pankau, M. Reimold, M. Rein,
Pure Appl. Chem. 2003, 75(5), 609-619.
Systems chemistry: Kinetic and computational analysis of a nearly
exponential organic replicator M. Kindermann, I. Stahl, M. Reimold, W. M. Pankau, G. von Kiedrowski,
Angew. Chem. 2005, 117(41), 6908-6913.

V
Inhaltsverzeichnis
Allgemeiner Teil 1
1 Einleitung 2
1.1 Information in naturwissenschaftlichem Kontext 2
1.2 DNA-Computer 4
1.3 DNA als Informationsspeicher in biologischen Systemen 6
1.4 DNA-Chiptechnologie 7
1.4.1 Bedeutung in der Medizin 7
1.4.2 Herstellung von DNA-Chips 9
1.4.2 in situ Amplifikation 13
2 Aufgabenstellung 14
3 Vorüberlegungen 18
4 Eigenschaften unmodifizierter quervernetzter N,N-Dimethylacrylamid-Polymere 22
4.1 Polymerisationsgeschwindigkeit in Anwesenheit von DMF 22
4.2 Quellverhalten 25
4.2.1 Quellverhalten in reinen Lösungsmitteln 25
4.2.2 Theoretische Betrachtung des Quellverhaltens 28
4.2.2.1 Elastische Freie Enthalpie 29
4.2.2.2 Freie Mischungsenthalpie 30
4.2.2.3 Ionischer Anteil 34
4.2.2.4 Zusammenfassung der thermodynamischen Größen 34
4.2.3 Bestimmung des Quervernetzungsgrades von N,N-Dimethylacrylamid-Polymeren 35
4.2.4 Bestimmung der Flory-Huggins-Wechselwirkungsparameter 37
4.2.5 Quellverhalten in Lösungsmittelgemischen 39
4.2.5.1 Theoretische Betrachtung. 39
4.2.5.2 Quellverhalten in Alkohol/Wasser-Gemischen 42
4.2.5.3 Quellverhalten in Carbonsäure/Wasser-Gemischen 45
4.2.5.4 Quellverhalten in Formamid/Wasser-Gemischen 46
4.2.6. Quellverhalten in Salzlösungen 47

VI
4.2.6.1 Quellverhalten in Natriumchlorid-Lösungen 47
4.2.6.2 Quellverhalten in Natriumacetat-Lösungen 48
4.2.7 Quellverhalten in Harnstofflösungen 49
4.2.8 Vergleichende Zusammenfassung der Ergebnisse 51
5 Eigenschaften modifizierter quervernetzter N,N-Dimethylacrylamid-Polymere 52
5.1 Acrylamid -Copolymere 52
5.2. Beladungsbestimmungen 54
5.3 Copolymere mit Aldehyd-Modifikation 56
5.3.1 Synthesestrategie 56
5.3.2 Beladungsbestimmung 59
5.3.3 Immobilisierung von 5’-Hydrazid-Oligonukleotiden an Aldehyd-Gelen 60
5.3.3.1 5’-Hydrazid-Oligonukleotide 60
5.3.3.2 Immobilisierung von Hydrazid-Oligonukleotiden an Aldehyd-Gelen 61
5.3.3.3 Hybridisierungsexperimente 61
5.4 Copolymere mit Carboxylat-Modifikation 63
5.4.1 Synthesestrategie 63
5.4.2 Eigenschaften 63
5.5 Copolymere mit Amino-Modifikation 68
5.5.1 Synthesestrategie 68
5.6 Copolymere mit Hydrazid-Modifikation 69
5.6.1 Synthesestrategie 69
5.6.2 Beladungsbestimmung 69
5.6.3 Immobilisierung von 5’-Aldehyd-Oligonukleotiden an Hydrazid-Gelen 71
5.6.3.1 Darstellung von 5’-Aldehyd-Oligonukleotiden 71
5.6.3.2 Immobilisierung von 5’-Aldehyd-Oligonukleotiden an Hydrazid-Gelen 73
5.6.3.3 Hybridisierung und Denaturierung 73
5.7 Copolymere mit Phenylboronsäure-Modifikation 75
5.7.1 Synthesestrategie 75
5.7.2 Polymerisation 76
5.7.3 Anlagerung von Polyolen 76
5.7.3.1 Bistrispropan 80
5.7.3.2 Polyglycerin 86
5.8 Copolymere mit Thiol-Modifikation 89

VII
5.8.1 Synthesestrategie 89
6 Technische Aspekte 91
6.1 Möglicher Aufbau einer ElektroSpread-Apparatur 91
6.2 Sandwich-Gele 93
6.2.1 Hydrazid/Aldehyd-Hybrid-Gele 94
6.2.2 Capping von reaktiven Resten der N,N-Dimethylpolyacrylamid-Matrix 95
6.3 Kovalente Anbindung von N,N-Dimethylpolyacrylamiden an Glas-Oberflächen 98
7 Zusammenfassung und Ausblick 99
Experimenteller Teil 102
1 Material und Methoden 103
1.1 Spektroskopische Methoden 103
1.1.1 Magnetische Kernresonanzspektroskopie (NMR) 103
1.1.2 IR-Spektroskopie 103
1.1.3 UV/Vis – Spektroskopie 103
1.2 Massenspektrometrie 104
1.2.1 Fast-Atom-Bombardment (FAB) 104
1.2.2 Matrix-Assisted Laser Desorption Ionisation / Time Of Flight (MALDI-TOF) 104
1.3 Chromatographische Methoden 104
1.3.1 Dünnschichtchromatographie 104
1.3.2 Säulenchromatographie 104
1.3.3 High-Performance-Liquid-Chromatography (HPLC) 104
1.4 Automatisierte Oligonucleotidsynthesen 105
1.5 Imager 105
1.5 Chemikalien 105
1.5.1 Verwendete Lösungsmittel 105
1.5.2 Verwendete Feinchemikalien 105
1.5.3 Chemikalien für automatisierte Oligonucleotidsynthesen 105
1.6 Stammlösungen 106
1.6.1 Gelstammlösung A 106
1.6.1 Gelstammlösung B 106

VIII
2 Allgemeine Arbeitsvorschriften 107
AAV1.1 Lipophilisierung von Objektträgern aus Glas 107
AAV1.2 Acrylamid-Funktionalisierung von Objektträgern aus Glas 107
AAV2 Herstellung von Polymer-Gelen 107
AAV2.1 Herstellung von quervernetzten N,N-Dimethylacrylamid-Polymeren 108
AAV2.2 Herstellung von N,N-Dimethylacrylamid-Glycolacrylat-Copolymeren 108
AAV2.3 Herstellung von N,N-Dimethylacrylamid-Acrylsäure-Copolymeren 108
AAV2.4 Herstellung von Ethylendiaminmonoacrylamid-Copolymeren 108
AAV2.5 Herstellung von N,N-Dimethylacrylamid-Acrylsäurehydrazid-Copolymeren 109
AAV2.6 Herstellung von 3-Acrylamidophenylboronsäure-Copolymeren 109
AAV2.7 Herstellung von 5-Acrylamidovaleraldehyd-Copolymeren 109
AAV2.8 Herstellung von 2-Acrylamidoacetaldehyd-Copolymeren 109
AAV2.9 Herstellung von N-(2-Acrylamidoethyl)-4-formylbenzamid-Copolymeren 110
AAV2.10 Herstellung von N,N-Diacryloyl-L-cystindiethylester-Copolymeren 110
AAV3 Herstellung von Hybridpolymeren 110
AAV4 Anfärbung von Gelen 111
AAV4.1 Anfärbung von Aldehyd-Gelen mit 2,4-Dinitrophenylhydrazin 111
AAV4.2 Anfärbung von Hydrazid-Gelen mit 4-Nitrobenzaldehyd 112
AAV4.3 Anfärbung von Hydrazid-Gelen mit 2,4-Dinitrobenzaldehyd 112
AAV4.4 Kaiser-Test von Amino-Gelen 112
AAV4.5 Qualitative Anfärbung von Hybridgelen 112
AAV5 Capping von N,N-Dimethylacrylamid-Glycolacrylat-Copolymeren 113
AAV6 Durchführung des Stress-Strain-Experimentes 113
AAV7.1 Synthese von Oligonukleotiden 113
AAV7.2 Generierung von 5’-Hydrazid-Oligonukleotiden 113
AAV7.3 Festphasenextraktion 113
AAV7.4 Entschützung von Oligonukletiden 114
3 Synthesen 115
3.1 Synthese von 5,6-Isopropylidendioxyhexan-1-ol 115
3.2 Synthese von 1-Methansulfonyl-5,6-isopropylidendioxyhexan 116
3.3 Synthese von Lithiumazid 116
3.4 Synthese von 1-Azido-5,6-isopropylidendioxyhexan 117
3.5 Synthese von 5,6-Isopropylidendioxyhexylamin 118

IX
3.6 Synthese von N-(5,6-Isopropylidendioxyhexyl)acrylamid 119
3.7 Synthese von N-(2,2-Diethoxyethyl)acrylamid 120
3.8 Synthese von 4-Dimethoxymethylbenzoesäuremethylester 120
3.9 Synthese von N-(2-Aminoethyl)-4-dimethoxymethylbenzamid 121
3.10 Synthese von N-(2-Acrylamidoethyl)-4-dimethoxymethylbenzamid 122
3.12 Synthese von N-(2-Hydroxyethyl)-4-dimethoxymethylbenzamid 123
3.13 Synthese von 3-(Acrylamido)phenylboronsäure 124
3.14 Synthese von Cystaminbisacrylamid 125
3.15 Synthese von N,N-Diacryloyl-L-cystindiethylester 126
3.16 Synthese von 3-(Triethoxysilyl)propylacrylamid 127
Anhang 128
A: Daten 129
A.1 Quellverhalten und Dichten von unmodifizierten DMAA-Polymeren 129
A.1.1 Methanol-Wasser-Mischungen 130
A.1.2 Ethanol-Wasser-Mischungen 132
A.1.3 2-Propanol-Wasser-Mischungen 134
A.1.4 Butanol-Methanol-Wasser-Gemische 136
A.1.5 Essigsäure-Wasser-Mischungen 138
A.1.6 Propionsäure-Wasser-Mischungen 140
A.1.7 Formamid-Wasser-Mischungen 142
A.1.8 Natriumchloridlösungen 144
A.1.9 Natriumacetatlösungen 146
A.1.10 Harnstofflösungen 149
A.2 Eigenschaften von modifizierten N,N-Dimethylacrylamid-Polymeren 151
A.2.1 Acrylsäure-N,N-Dimethylacrylamid-Copolymere 151
A.2.2 N,N-Dimethylacrylamid-3-(Acrylamido)phenylboronsäure-Copolymere 151
A.2.2.1 Quellverhalten und Anreicherung von Bistrispropan 151
A.2.2.2 Quellverhalten und Anreicherung von PG02 153
A.3 Capping mit 2-Aminoethanol 154

X
B: Dreidimensionale Visualisierung chemischer Verbindungen 156
B.1 Motivation 156
B.2 Eigenschaften des pdb2blend-Skriptes 157
B.3 Quellcode 159
Abkürzungen und Akronyme 170
Verwendete Symbole 172
Literatur 174

Allgemeiner Teil

2
1 Einleitung
Eines der meistbeachteten wissenschaftlichen Projekte der letzten Jahre war die weitgehende
Entschlüsselung des menschlichen Genoms. Das breite öffentliche Interesse wurde sicherlich
durch die Erwartung hervorgerufen, dass durch die Kenntnis der gesamten Erbinformation des
Menschen auch der Mensch als solcher verstanden würde. Diese Erwartung ist insofern
begründet, als natürlich der Bauplan des Menschen im Genom kodiert ist. Die Schwierigkeit
liegt darin, dass nun die Strukturinformation bekannt ist, diese jedoch, um die Bedeutung
erfassen zu können, dekodiert werden muss. Die Sprache, in der das Genom verfasst ist, ist
jedoch nur teilweise bekannt.
1.1 Information in naturwissenschaftlichem Kontext
„Information ist ein potentiell oder tatsächlich nutzbares Muster von Materie und/oder
Energieformen, das für einen Betrachter in einem bestimmten Kontext relevant ist. (…) Das
verwendete Muster ändert den Zustand des Betrachters (…).“[1]
Genau genommen bedeutet diese Definition auch, dass alles auf der untersten Ebene die
Information über sich selbst trägt. Ein Atom hat Ortsinformationen, Zustandsinformationen,
die nur das Atom selbst betreffen. Diese Eigenschaften als Informationen zu begreifen, ist in
der Naturwissenschaft üblich.[2,3] Es ist evident, dass alles diese Informationen über sich
selbst trägt. Die Eigenschaften werden als Information begriffen. Auf dieser Ebene wird der
Zusammenhang zwischen Information und Unbestimmtheit von der Unschärferelation
bestimmt.
Für ein Ensemble entspricht diese unterste Ebene der Strukturinformation. Um eine
Bedeutungsinformation zu erhalten, muss die Strukturinformation dekodiert werden.
Dem kann beispielsweise ebenso das Lesen einer Zeitung sein, wie auch die Analyse
geologischer Zusammenhänge aus dem Aufbau von Sedimentgesteinen. Es gibt in diesem
Sinne eine nahezu unendliche Zahl von möglichen Codes, wobei angemerkt werden sollte,
dass die Informationsverarbeitung durch Menschen ohnehin eine lange Abfolge von
Dekodierungsprozessen darstellt, die z.B. auf neuronaler Ebene ablaufen. Die
Bedeutungsinformation stellt gegenüber der Strukturinformation eine höhere Information dar.
Nimmt man an, dass als Betrachter jedes informationsverarbeitende System in Frage kommt,
so wird klar, dass biologische Systeme eine Vielzahl höherer Informationssysteme besitzen.

3
Im Gegensatz dazu können zwar Lebewesen der unbelebten Welt Information entnehmen, die
unbelebte Welt selbst enthält jedoch kein in weiterem Sinne informationsverarbeitendes
System.
Vom strukturellen Gesichtspunkt aus gibt es im Menschen drei unterschiedliche Systeme des
Informationsaustausches, konzentrationsabhängige, topologische und sequenzabhängige,
wovon erstere am geringsten, letztere am stärksten konserviert sind, was den
Funktionsbereichen entspricht. Viele grundlegende Funktionen des Organismus ebenso wie
intrazelluläre Prozesse werden konzentrationsabhängig gesteuert, die Informationen sind
Zustandsinformationen. Kognitive Prozesse, die einen Datenabruf ebenso wie
Datenspeicherung erfordern, laufen im topologischen, neuronalen System ab. Diese Prozesse
benötigen eine hohe Flexibilität und Möglichkeit der Reorganisation. Grundlegende Baupläne
sind im Sequenzsystem des Genoms gespeichert. Hier führen Änderungen im schlimmsten
Fall dazu, dass der Organismus nicht mehr lebensfähig ist. Daher ist eine starke
Konservierung der Information hier lebenswichtig.
Wenn man die Art der Information betrachtet, dann bilden konzentrationsabhängige Systeme
analoge Signale, wobei die Amplitude der Konzentration entspricht. Neuronen zeigen sowohl
digitale als auch analoge Anteile, die Sequenzsysteme sind vollständig digital.
Die Informationsdichte pro Dateneinheit ist bei den konzentrationsabhängigen Systemen am
geringsten (Konzentrationsinformationen der Stoffe), bei den linearen Sequenzsystemen
höher, bei den neuronalen Systemen am höchsten. Es muss jedoch angemerkt werden, dass
die absolute Informationsdichte der Sequenzinformationen um etliche Größenordnungen
höher ist als die der neuronalen Netzwerke.
50 100 150 µm05 10 15 Å0 5 10 15 Å0
a) b) c)
Abb.1: Größen- und Strukturvergleich: a) Hormone (Insulin, Adrenalin & Estradiol), b)
DNA, c) Neuron des visuellen Cortex.[4]

4
1.2 DNA-Computer
Bei DNA handelt es sich um ein quarternäres (vier Basen) System mit linear angeordneten
Dateneinheiten (im Gegensatz dazu arbeiten Computer mit linear angeordneten binären
Dateneinheiten (Bits)).
Bei der Entwicklung von DNA-Computern wird DNA als Datenelement genutzt, wobei
jedoch nicht die einzelnen Basen Datenelemente darstellen sondern spezifische Sequenzen
(Wörter).
Im Hinblick auf die obigen Ausführungen sei angemerkt, dass die natürliche Funktion der
DNA der eines festen Speichers (ROM) entspricht. Als dynamisches Datenelement ist sie
weniger gut geeignet und wird auch in der Natur auf diese Weise nicht genutzt.
Die ersten erfolgreichen Berechnungen, die mit DNA durchgeführt wurden, basierten auf der
Selektion der richtigen Lösung aus einem Pool aller möglichen Lösungen. Auf diese Weise
konnte Leonard Adleman 1994 das Handelsreisenden-Problem lösen.[5] Auch wenn weitere,
komplexe NP-vollständige Probleme (Von einer nichtdeterministischen Turingmaschine in
polynomieller Zeit lösbare Probleme) gelöst werden konnten, erscheint eine Anwendung
jenseits eines proof of principle nur sinnvoll, wenn auch Probleme höherer Komplexität gelöst
werden können.[6,7]
Folgt man der geläufigen Definition von Computer, die voraussetzt, dass es sich um eine
Turing-vollständige Maschine handelt, so muss es neben der Möglichkeit, Eingabedaten von
definierten Positionen eines Bandes zu lesen, auch die Möglichkeit geben, Ausgabedaten an
definierte Positionen zu schreiben. Gerade letztere Funktion war bei frühen
Implementierungen nicht gegeben, ist aber für komplexe Prozessierungen unabdingbar.[6]
Neuere Veröffentlichungen zeigen, dass es in nicht autonomen Verfahren immerhin
Möglichkeiten gibt, Daten selektiv zu löschen
und zu ersetzen, wobei sich diese
Möglichkeiten jedoch immer auf das
Strangende beziehen.[8] Es konnte auch
gezeigt werden, dass z.B. unter Verwendung
von oberflächengebundener DNA die
Implementierung der logischen Funktion
NOR (NOT OR) gelingt (Abb.2).[6]
A B AND OR XOR NOR
0 0 0 0 0 1
0 1 0 1 1 0
1 0 0 1 1 0
1 1 1 1 0 0
Tab. 1: Wahrheitstabelle für logische
Operationen.

5
Aus dem NOR-Element lassen sich alle anderen logischen Funktionen ableiten:
NOT A = A NOR FALSE;
A OR B = NOT (A NOR B)
A AND B = (NOT A) NOR (NOT B);
A XOR B = (A AND (NOT B)) OR ((NOT A) AND B)
Dies entspricht demnach tatsächlich einer einfachen Implementierung der Turing-Maschine.
P
0
0
X
P
0
X
P
0
X
P
X
1
1
1
1
P
0
0
X
0
0
P
0
X
0
P
0
X
0
P
X
1
1
1
1
Ligation
P
P
P
P
0
0
X
0
0
P
0
X
0
P
0
X
0
P
X
1
1
1
1P
PP
P
0
0
X
0
0
P
0
X
P
0
X
P
X
1
1
1
1
P
1
2
3
4
CP
P
0
0
X
0
0
P
0
X
P
0
X
P
X
1
1
1
1
P
Verlängerung0
0
X
0
0
P
0
X
P
0
X
P
X
1
1
1
1
P
X
P
1
P
1
P
0
0
X
0
0
P
0
X
P
0
X
P
X
1
1
1
1
1
P
X
1
P
0
0
X
P
0
X
P
0
X
P
X
1
1
1
1
1
P
1
2
3
4
CP
0
0
X
P
0
X
P
0
X
P
X
1
1
1
1
1
P
1 1
11P
P P
X
0
X X
P
P
00
P
P
00
P
P
00
0
0
X
0
X
0
X X
1
1
1
1
1
P
1 1
11P
P P
X
0
X X
P
00
P
00
P
00
0
0
X
0
X
0
X X
1
1
1
1
1
P P
0
P
0
P
0
0
0
X
P
0
X
P
0
X
P
X
1
1
1
1
1
P
1 1
11P
P P
1
2
3
4
CP
(a) (b) (c)
(d) (h) (f)
(f) (e) (d)
(g)
Abb.2: Implementierung der logischen Funktion NOR durch eine oberflächengebundene
nicht autonome DNA-Recheneinheit.[6] Die Wörter in Position 1 und 2 werden miteinander
verglichen und das Ergebnis der logischen Operation in Position 4 geschrieben. Die
Sequenzen derselben Bedeutung in unterschiedlichen Positionen sind bei dieser
Implementierung nicht identisch und bestehen aus jeweils 16 Basen. Die Einzelschritte
bestehen aus sequenzieller Hybridisierung der Sequenzen (Position ist tiefgestellt) 01 und
02 (a) bzw. 11 und 12 (g), aus der enzymatischen Ligation (b) unter Zugabe der
Doppelstränge 0:04 (h) oder 1:14 (e), aus partieller (c) bzw. vollständiger Dehybridisierung
(f) sowie aus Verlängerung der hybridisierten Sequenzen durch Polymerase (d).

6
Interessant ist in diesem Zusammenhang auch die Fragestellung, ob es möglich ist, einen
universellen Computer im Sinne einer Von-Neumann-Architektur zu erhalten, der die
Fähigkeit besitzt, sein eigenes Programm zu verändern. Diesem Prinzip folgen heutige
Computer.
Die Entwicklung von DNA-Computern profitiert von der selektiven Paarung von
komplementären DNA-Sequenzen sowie der Möglichkeit einer großen Parallelisierung, da
jede Ausgangssequenz als eigener Nanoprozessor verstanden werden kann.[9] Jedoch führt die
Nutzung biochemischer Werkzeuge (Enzyme wie Ligasen, Exonucleasen, Polymerasen), die
für Schreibvorgänge im Sinne der Turing-Maschine benötigt werden, zu einer geringeren
Effizienz, die eine praktische Nutzung in vielen Fällen unmöglich macht.[6] Die Fehlerrate des
besten heute bekannten DNA-Computers liegt bei ca. 1:2500.[10]
Nimmt man von der Vorstellung eines DNA-Computers im Sinne eines klassischen, auf
logischen Funktionen basierenden Rechners Abstand, so können Mutationen auch als Chance
begriffen werden, Berechnungen auf der Basis von Mechanismen der Evolution
durchzuführen. Dies würde die Möglichkeiten der Silizium-basierten Rechner erweitern
anstatt sie zu ersetzen.[11]
1.3 DNA als Informationsspeicher in biologischen Systemen
Man unterscheidet üblicherweise zwischen kodierender und nicht kodierender DNA, was
jedoch vom informationstheoretischen Standpunkt aus problematisch ist.
Tatsächlich enthält das Genom nicht nur direkt Protein- oder ncRNA-kodierende
Informationen, sondern ebenso regulatorische Bereiche unterschiedlicher Art. Auch letztere
sind für die Funktion essentiell, ihr Code ist jedoch gegenüber der einfachen Syntax der
Protein-kodierenden Bereiche komplex, da er die Kenntnis der wechselwirkenden Faktoren
voraussetzt. Die kodierenden Bereiche können als unterste Ebene angesehen werden, sie
werden vom entsprechenden biochemischen Apparat sequentiell in RNA übertragen und in
den meisten Fällen in Proteine übersetzt. Die höheren Funktionen steuern im Prinzip, wann,
wie und in welchem Bereich diese Übersetzung stattfindet. Insofern stellt das Genom im
übertragenen Sinn nicht nur den Speicher sondern auch das Programm dar.
Auch wenn man zur absoluten Bestimmung der DNA-Basensequenz auf die auf Sanger
zurückgehende Sequenzierung der DNA angewiesen ist, ist die Kenntnis der vollständigen
Sequenz für viele Fragestellungen nicht notwendig. Da sich das Genom innerhalb einer Art

7
kaum variiert, liegt es nahe, lediglich die Bereiche, die für die jeweilige Fragestellung
relevant sind, mit einem Standard zu vergleichen. Dies erfolgt durch Hybridisierungsessays
unter Verwendung von DNA-Chips, wobei eine Vielzahl von Hybridisierungsexperimenten
simultan erfolgen kann. So lassen sich beispielsweise selektiv funktionelle Bereiche oder
Bereiche mit bekannten Variationen untersuchen.
1.4 DNA-Chiptechnologie
Als DNA-Chips bezeichnet man flache Träger, an denen in geordneter Form Oligonukleotide
gebunden sind. Je nach Feinheit der Strukturierung wird auch zwischen DNA-Microarrays
(spot-durchmesser < 200µm) und DNA-Macroarrays (spot-durchmesser > 300µm)
unterschieden.[12,13]
DNA Chips finden in Hybridisierungsassays Verwendung, wobei eine große Anzahl von
Hybridisierungsexperimenten simultan erfolgen kann. Ebenso ist die Verwendung von DNA-
Chips im Bereich der DNA-Computer beschrieben.[6] Beide Anwendungen machen sich die
Möglichkeit der Parallelisierung der Experimente zunutze. Im einen Fall erfolgt die
Hybridisierung parallel, im anderen Fall die Berechnung.
1.4.1 Bedeutung in der Medizin
Veränderungen des Erbguts können zu spezifischen Erkrankungen führen, z.B.
Mukoviszidose, Albinismus (autosomal-rezessiv), Neurofibromatose (autosomal-dominant
vererbt) oder Hämophilie (X-chromosomal rezessiv). Da Mutationen selten vorkommen, sind
die meisten Unterschiede zwischen dem Erbgut unterschiedlicher Menschen vererbt.
Ebenso sind auch Krankheitsdispositionen genetisch angelegt.
Es gibt darüber hinaus genetisch bedingte Erkrankungen, die nicht erblich sind, wie z.B. das
Down-Syndrom.
Eine Krankheitsdisposition führt nicht unbedingt zu einem Ausbruch der Krankheit, sondern
zu einem erhöhten Risiko, unter entsprechenden Einflüssen zu erkranken. Daher ist hier keine
strenge Vererbung sondern höchstens eine familiäre Häufung zu finden, wobei eine derartige
Häufung auch auf ähnlichen Lebensbedingungen beruhen kann.
Eine genetische Prädisposition für viele Krankheiten ist bekannt, z.B. für Adipositas,
Autoimmunerkrankungen, Krebserkrankungen, Hypertonie, Migräne oder Schizophrenie.
Darüber hinaus werden zahlreiche weitere genetisch festgelegte Eigenschaften, wie die
spezifische Reaktion auf Medikamente oder Umwelteinflüsse, angenommen.

8
Die Kenntnis dieser genetischen Parameter erlaubt es heute schon, bei Prädisposition für
bestimmte Krankheiten präventive Maßnahmen zu ergreifen bzw. eine entsprechend
angepasste Diagnostik durchzuführen.[14,15,16] Ebenso könnte man bei genauerer Kenntnis
dieser Parameter Therapien deutlich besser patientenspezifisch zuschneiden und damit
Unwirksamkeiten oder Nebenwirkungen von Medikamenten minimieren. Natürlich eröffnen
diese Informationen auch Möglichkeiten des Missbrauchs.[17]
Veränderungen des Genoms lassen sich strukturell in Veränderungen einzelner Basen
(Punktmutationen, Deletion oder Insertion einer Base) und in chromosomale Veränderungen
(Amplifikationen oder Deletionen chromosomaler Bereiche (z.B. Trisomie), Translokationen
etc.) unterteilen.
Während zweite als makroskopische Veränderungen gut nachweisbar sind, ist der Nachweis
von Änderungen einzelner Basen bei einer Gesamtgenomgröße von 3·109 Basenpaaren nicht
ohne weiteres möglich. Der Vergleich des genetischen Codes mehrerer Menschen fördert den
Befund zutage, dass gewisse Basen des Genoms eine erhöhte Variabilität (> 1% der
Population) aufweisen, die single nucleotide polymorphisms (SNP) genannt werden. Diese,
meist aus dem Austausch von Cytosin durch Thymin resultierenden Variationen, bedingen ca.
90% der genetischen Variabilität des Menschen.
Darüber hinaus eignen sich SNPs hervorragend zum Nachweis über Hybridisierungsassays
mit DNA-Chips, da eine einfache Fehlpaarung auf diese Art sehr gut nachzuweisen ist.
Die Häufigkeit und die gute Nachweisbarkeit bedingen den Fokus, der bzgl. genetisch
basierter Präventivmedizin auf die Untersuchung von SNPs gelegt wird.
Obwohl heute fast 1.8·106 SNPs bekannt sind,[18] sind nur wenige Krankheiten bekannt, die
auf dem Austausch einer einzigen Base beruhen. In den meisten Fällen muss davon
ausgegangen werden, dass Krankheiten oder Krankheitsprädispositionen durch eine Vielzahl
genetischer Faktoren beeinflusst werden.[19]
Durch die Zusammenführung aller vorhandenen SNPs entstehen die Haplotypen des
menschlichen Genoms, die die Muster der vorhandenen Variationen darstellen. Die
Korrelation der Haplotypen mit der Häufigkeit bestimmter Erkrankungen trägt dem
tatsächlichen Zusammenspiel der unterschiedlichen Variationen Rechnung.[20]
Es ist davon auszugehen, dass die Bedeutung der DNA-Chiptechnologie im diagnostischen
Bereich deutlich steigen wird, wenn entsprechende Daten vorhanden sind. Durch die
Möglichkeit, Tausende von SNPs gleichzeitig zu screenen, lassen sich durch ein
Hybridisierungsassay theoretisch Aussagen über eine Vielzahl von Prädispositionen
gleichzeitig gewinnen.

9
DNA-Chips spielen als Exon-Arrays auch eine Rolle bei der Genom-Analyse.[21]
1.4.2 Herstellung von DNA-Chips
Es ist eine Vielzahl unterschiedlicher Möglichkeiten, DNA-Chips herzustellen, bekannt, die
sich durch das Trägermaterial, die Immobilisierungschemie sowie die Fertigungstechnologie
unterscheiden. Trägermaterial und Immobilisierungschemie sind von den angestrebten
Anwendungen, wie auch von der Fertigungstechnologie abhängig. Einige gängige
Trägermaterialien sind in Tabelle 2 aufgelistet.[12]
synthetische Polymere anorganische Träger (modifizierte) Biopolymere
Polystyrol
Polyacrylamid
Polyethylenterephthalat
Polyurethan
Polyvinylalkohol
Nylon
Polypyrrol
Sephadex LH 20
Polypropylen
Gold
Glas
Titan
Aluminium
Metall-Chelate
Zellulose
Nitrozellulose
Latex
Carboxymethyldextran
Chitosan
Tab. 2: Gängige Trägermaterialien für DNA-Chips.[12]
Bezüglich der Fertigungstechnologie sind zwei grundsätzlich verschiedene Wege möglich, die
Immobilisierung präsynthetisierter Oligonukleotide sowie die Synthese auf dem Chip. Gerade
letztere Technologie eignet sich für die Herstellung großer kombinatorischer Bibliotheken auf
der Oberfläche mit mehr als 250 000 unterschiedlichen Sequenzen pro cm2. Dabei wird die
genaue Ortsadressierung bei hoher Auflösung durch die Verwendung photolabiler
Schutzgruppen und UV-Bestrahlung unter Verwendung photolithographischer Masken
erreicht.
Nur an den Positionen, die bestrahlt wurden, erfolgt die Abspaltung der photolabilen
5’-Schutzgruppe, im Kopplungsschritt kann daraufhin ein Nukleosid-3’-phosphoamidit mit
der deblockierten 5’-Hydroxyfunktion reagieren (Abb.3). So können durch die Verwendung
unterschiedlicher Masken sehr große Bibliotheken auf kleinem Raum hergestellt werden. Die
Länge der auf diese Art herstellbaren Oligonukleotide ist auf ca. 60 Basen begrenzt.[22]

10
Nachteile der Synthese auf dem Chip sind die Unmöglichkeit, Deletionsmutanten zu
entfernen, sowie die komplizierte Qualitätskontrolle.
Mit der Immobilisierung präsynthetisierter Oligonukleotide können nicht derart große
Diversitäten erzielt werden wie es mit der Synthese auf dem Chip möglich ist. Für die
Strukturierung kann ebenfalls auf photolitographische Techniken zurückgegriffen werden,[12]
wobei aber meist die Ortsadressierung mechanisch erfolgt, entweder durch contact printing
unter Verwendung einer Vielzahl von unterschiedlichen Nadeln,[23,24] oder durch non-contact
printing unter Verwendung von ink-jet Technologien (Abb.4).[25] Die Nachteile des contact
printing sind die physikalische Belastung der Oberfläche und die eingeschränkte Haltbarkeit
der Nadel. Der Nachteil des non contact printing die mögliche Bildung von Satellitenspots
durch Streuung.
APG
APG
APG
A
A APGPG
PGA
A APG
A
A
CPG
CPG
CPG
CPG
APG
CPG
APG
APG
A
A
CC C
CA
TPG
GPG
CPG
T
T
G
GGA
A A
A
C
C
PG PG PG
PGPGPG
Träger mit photolabilen Schutzgruppen; die Bestrahlung erfolgt unter Verwendung photolithographischer Masken.
UV-Strahlung
Es resultieren entschützte Bereiche. An die entschützten Bereiche werdenNukleosid-3'-phosphoamidite gekoppelt.
UV-Strahlung
Erneute Bestrahlung unter Verwendung einer anderen Maske
A T PGGCNukleosid-3'-phosphoamidite
photolabile 5'-Schutzgruppe,z.B. 4,5-Dimethoxy-2-nitro-benzyloxycarbonyl (NVOC)
multiple Belichtungs- und Kopplungsschritte Abb.3: Photolithographische Oberflächenstrukturierung und Oligonukleotidaufbau.[22]

11
Eine weitere Möglichkeit besteht in der Adressierung durch elektrische Felder, was durch die
Verwendung eines Elektroden-Arrays ermöglicht wird. Die Firma Nanogen verwendet diese
Technik, wobei biotinylierte Oligonucleotide an Spreptavidin-beschichtete Goldelektroden
gekoppelt werden.[25]
Eine weitere Möglichkeit der Fertigung von DNA tragenden Oberflächen bildet die
Übertragung der DNA von einer anderen Oberfläche im Sinne eines Drucks (nanoprinting).
Hier sind zwei grundsätzliche Verfahren gegeneinander abzugrenzen: Die eine Möglichkeit
besteht in der Übertragung ausgehend von einer hochbeladenen Oberfläche, die als Reservoir
dient. Dabei wird ein Teil der Oligonukleotide abgespalten und auf die Kopie übertragen. Die
andere Möglichkeit ist, ein komplementäres Oligonukleotid am Master-Chip zu
immobilisieren und nach Hybridisierung der Gegensequenzen diese auf eine andere
Oberfläche zu übertragen. Man kann diese beiden Verfahren als Postiv- und Negativ-Druck
verstehen.
Eine Möglichkeit des Positiv-Drucks besteht darin, im ersten Schritt das Oligonukleotid über
eine Disulfid-Bindung an einem Master-Chip zu immobilisieren. Die Übertragung erfolgt
dann bei erhöhter Temperatur auf ein Polyacrylamidpolymer, an dessen freie Acrylatreste die
Thiololigonukleotide binden können (Abb.5).[26]
ElektrodenPZT
VibrationsschichtProbenkammer Probenvorrat
TropfenDüse
micro spotting pin Piezo Ink-Jet
Abb.4: contact und non-contact printing. Als Beispiele schematisch ein micro spotting
pin[23,24] und eine Piezo Ink-Jet Düse. Bei letzterer wird durch Anlegen eines Strompulses
an den Piezokristall (PZT) eine Druckwelle erzeugt, die zur Freisetzung einer geringen
Probenmenge aus der Spitze führt.

12
2005 wurde im Sinne eines Negativ-Drucks von einer Methode berichtet, bei der unter
Verwendung eines Masters mit kovalent gebundenen Oligonukleotiden (5’-Amino-38mere an
NHS-funktionalisierten Polymer-beschichteten Glas-Slides) komplementäre Biotin-gelabelte
Sequenzen hybridisiert und durch Kontakt mit einer Streptavidin-funktionalisierten
Oberfläche bei einem Druck von 1.4 N cm-2 auf diese übertragen wurde.[28]
Aufgrund der Tatsache, dass die Übertragung nur möglich ist, wenn die Sequenzen direkt an
der Oberfläche lokalisiert sind, liegt die gefundene Übertragungsrate bei maximal 25%.
Außerdem zeigt sich, dass bei jeder weiteren Benutzung des Master-Chips die Beladung der
Kopie sinkt. Es kommt also durch die physikalische Belastung zu einer partiellen
Deaktivierung oder Zerstörung des Masters. Zudem ist das Verfahren nicht für einen Master-
Chip mit mehreren unterschiedlichen Sequenzen durchgeführt worden. Trotzdem zeigt dieses
Verfahren die grundsätzliche Möglichkeit, DNA-Chips im Sinne eines Negativ-Drucks zu
kopieren.
Leerer Glasträger mit Acrylamidsilan-Beschichtung
kopierter Chip
Master Chipa) Polymerisation
eines Acrylamid-Gels zwischen den Glasträgern; b) Erwärmen
führt zum Transfer der Oligonucleotide vom Master Chip in das Gel. Die Dauer wird mit der
Zeit gesteigert, wodurch trotz der abnehmenden Beladung des Master Chips eine gleichbleibende
Beladung der kopierten Chips erreicht wird.
Jeder Zyklus führt zu einer Abreicherungder DNA am Master Chip
Abb.5: Positiv-Printing Verfahren. Der Master-Chip dient als Reservoir und überträgt in
jedem Zyklus einen Teil seiner Beladung auf eine Kopie.[27]

13
1.4.2 in situ Amplifikation
Ein Verfahren, bei dem ebenfalls Oligonukleotide durch Kontakt übertragen werden, jedoch
durch in situ Amplifikation, wurde bereits 1999 beschrieben.[29]
Dabei wird die Polymerase-Kettenreaktion (PCR) innerhalb eines Polyacrylamid-Gels
durchgeführt, welches die entsprechenden PCR-Reagenzien enthält. Die PCR-Primer sind
dabei am 5’-Ende mit einer Methacrylamid-Modifikation (Acrydite) versehen und werden
somit kovalent mit dem Polymer verknüpft. Werden nun Spuren an DNA eingetragen, so
beginnt an dem Ort des Eintrags die Vervielfältigung. Da die Primer mit dem Polymer
verbunden sind, kommt es zu keiner ausgeprägten Diffusion, in erster Linie führt die lokale
Sättigung des Gels zu einer Verbreiterung der Spots. Es bilden sich ausgehend vom Initiator
Kolonien, die von den Autoren polonies genannt werden.
Wird auf ein so erhaltenes Gel eine weiteres gegossen, so erfolgt die Ausbreitung der
Kolonien auch in das neue Gel hinein und somit führt die Amplifikation zur Kopie von Orts-
und Sequenzinformation auf das neue Gel, welches vorsichtig vom Master getrennt werden
kann.

14
2 Aufgabenstellung
Mit dem 1998 veröffentlichten Spread-Verfahren wurde die nicht-enzymatische
oberflächengesteuerte Replikation von Oligonukleotiden (eigentlich von Oligonukleotid-
Analoga, da die Verknüpfung über eine Phosphoamidatbindung erfolgt) möglich.[30]
Bei dem Spread-Verfahren (Abb.6) geht man von Oligonukleotiden aus (14mere), die am
5’-Ende einen C6-Linker mit einem aktivierten Disulfid tragen. Die Immobilisierung erfolgt
durch Umsetzung mit Thiosepharose, wobei das in den folgenden Schritten als Templat
dienende Oligonukleotid A unter Freisetzung von Thiopyridon eine Disulfidbindung eingeht.
Durch Capping mit aktiviertem Mercaptoethanol werden die verbliebenen
Thiolfunktionalitäten der Festphase blockiert.
An das Templat A werden daraufhin zwei komplementäre Bausteine, X’ und Y’ hybridisiert,
wobei der Baustein X’ in 5’-Position ein aktiviertes Disulfid und in 3’-Position ein freies
Phosphat trägt. Der Baustein Y’ trägt in 5’-Position eine Aminofunktion. Die
templatgebundenen Bausteine werden unter Verknüpfung des 3’-Phosphats von X’ und der
Abb.6: Darstellung des Spread-Verfahrens.

15
5’-Aminogruppe von Y’ unter Ausbildung
einer Phosphoamidatbindung verknüpft,
wobei EDC als Aktivatorreagenz zum
Einsatz kommt. Das gebildete Molekül A’
wird dehybridisiert und kann an einer
weiteren Festphase immobilisiert werden,
an welcher die Verknüpfung von X und Y
zu A stattfinden kann. Die zyklische
Wiederholung des Vorgangs führt zu einer
exponentiellen Amplifikation der
Sequenzen A und A’.
Das Spread-Verfahren sollte grundsätzlich
auch auf weitere Oligonukleotidanaloga
übertragbar sein, ebenso wie auch auf
verzweigte Bausteine wie
Trisoligonukleotide unter Verwendung
eines CCC ähnlichen Verfahrens.[31,32]
Die Limitierungen des Spread-Verfahrens
ergeben sich aus der Verwendung einer
einzigen Immobilisierungsstrategie,
wodurch ein Capping der überschüssigen
freien Funktionalitäten an der Oberfläche
nötig wird. Daher wird für jeden
Immobilisierungsschritt eine neue
Festphase benötigt. Die Replikation ist
also mit einer entsprechenden Zunahme
der Festphasenmenge in jedem Schritt
gekoppelt (Abb.7). Dies erschwert ebenfalls eine mögliche Automatisierung des Verfahrens.
Um diese Beschränkungen zu überwinden, ist es nötig, zwei orthogonale
Immobilisierungsstrategien zu verwenden. Dadurch wird die grundsätzliche Möglichkeit
eröffnet, in einem zyklischen Verfahren eine Beladungserhöhung zu erzielen. Durch die
Kombination mit einem Transfer im elektrischen Feld zwischen zwei unterschiedlich
funktionalisierten Oberflächen ist so eine Amplifikation unter Erhalt der Sequenz- und
Ortsinformation denkbar. Dieses noch konzeptionelle Verfahren wird als ElektroSpread-
A
A'
A
A'
A
A'
A A
A
A'
A
A'
A'
A
A'
A
A'
A
A'
A'
A
A'
A
A'
A
A'
A
A'
A
A'
Abb.7: Schematische Darstellung des Spread-
Verfahrens. Das Verfahren führt zu einer
Zunahme der Festphasenmenge bei ähnlicher
Beladung.
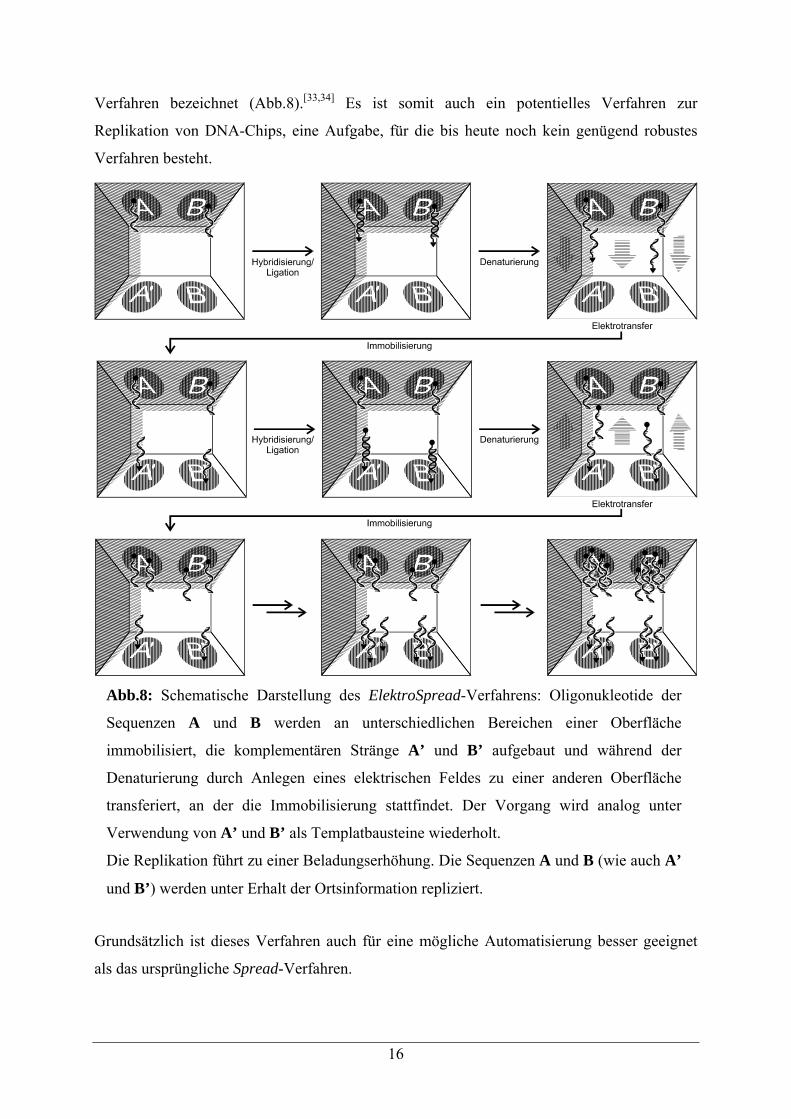
16
Verfahren bezeichnet (Abb.8).[33,34] Es ist somit auch ein potentielles Verfahren zur
Replikation von DNA-Chips, eine Aufgabe, für die bis heute noch kein genügend robustes
Verfahren besteht.
Grundsätzlich ist dieses Verfahren auch für eine mögliche Automatisierung besser geeignet
als das ursprüngliche Spread-Verfahren.
Abb.8: Schematische Darstellung des ElektroSpread-Verfahrens: Oligonukleotide der
Sequenzen A und B werden an unterschiedlichen Bereichen einer Oberfläche
immobilisiert, die komplementären Stränge A’ und B’ aufgebaut und während der
Denaturierung durch Anlegen eines elektrischen Feldes zu einer anderen Oberfläche
transferiert, an der die Immobilisierung stattfindet. Der Vorgang wird analog unter
Verwendung von A’ und B’ als Templatbausteine wiederholt.
Die Replikation führt zu einer Beladungserhöhung. Die Sequenzen A und B (wie auch A’
und B’) werden unter Erhalt der Ortsinformation repliziert.

17
Aufgrund der Tatsache, dass die für das Spread-Verfahren benutzte Thiosepharose in
käuflicher Form nicht für die Ausbildung zweidimensionaler Oberflächen geeignet ist sowie
ohnehin ein weiteres Immobilisierungsverfahren eingeführt werden muss, ist die Chemie des
Spread-Verfahrens nicht auf das ElektroSpread-Verfahren übertragbar, so dass hierfür andere
Trägermaterialien wie auch Immobilisierungsstrategien notwendig sind.
Ziel dieser Arbeit war die Untersuchung potentiell geeigneter Trägermaterialien und
Immobilisierungsverfahren im Hinblick auf die für das ElektroSpread-Verfahren gestellten
Anforderungen.

18
3 Vorüberlegungen
Aus dem Prinzip des ElektroSpread-Verfahrens ergeben sich die Anforderungen an
Oberflächen und Immobilisierungsstrategien:
Es ist die Kompatibilität mit Oligonukleotiden und den entsprechenden chemischen
Bedingungen zu gewährleisten. Ebenso muss der Ladungstransfer möglich sein.
Da der Transfer im elektrischen Feld zur Minimierung der diffusionsbedingten Verbreiterung
möglichst zügig erfolgen sollte, sollten auch potentielle Immobilisierungsverfahren schnell
verlaufen.
Die Immobilisierung direkt an der Elektrode ist für das Verfahren ungeeignet, weil es zum
einen zu Wechselwirkungen mit Oligonukleotiden kommen kann, zum anderen der Einfluss
durch Elektrodenprozesse entstandener Spezies schwer abzuschätzen ist. Besonders ist bei
den für den schnellen Transfer benötigten Spannungen eine elektrolytische Zersetzung von
Wasser und damit Gasbildung zu erwarten, die zu vermeiden ist. Die Zugabe von Redox-
Spezies verhindert wiederum die Nutzung entsprechender Systeme (z.B. Disulfidaustausch)
für die Immobilisierung.
Eine Trennung der immobilisierenden Schicht von der Elektrode erfordert die Wahl einer
Matrix, die die reaktiven Endgruppen trägt, wobei die Leitfähigkeit bzw. die
Ionendurchlässigkeit gewährleistet sein muss.
Abb.9: Schematische Darstellung der Anordnung funktioneller Gruppen an einer
Festphase (a), einem Copolymer-Gel (b) und (c) einer planen Oberfläche, wie z.B. einer
Elektrode.

19
Da mit der Gelelektrophorese und dem Blotting Verfahren bekannt sind, bei denen eine
Wanderung der Oligonukleotide im elektrischen Feld stattfindet, ist eine Betrachtung der
hierbei verwendeten Matrizes sinnvoll.
Als Gelbildner kommen Polyacrylamide sowie Polysaccharide (z.B. Agarose) zum Einsatz.
Während letztere bereits als Polymere vorliegen, die nach Auflösen bei höherer Temperatur
bei Abkühlung reversibel ein Gel bilden, werden Polyacrylamide irreversibel durch
radikalische Polymerisation hergestellt.
Zum Blotten kommen eine Vielzahl von speziellen Membranen zum Einsatz, z.B.
Nitrozellulose-, Nylon- oder PVDF (Polyvinyldifluorid)-Membranen, die häufig
Oligonukleotide unspezifisch binden und so ein Herauswandern aus der Membran verhindern.
Für die effektive Einführung funktioneller Gruppen ergibt sich somit für die Membranen wie
auch die Polysaccharide nur die Möglichkeit der Modifikation des bereits vorliegenden
Polymers. Dieser Weg beinhaltet jedoch gewisse Restriktionen. Die möglichen
Modifikationen sind beschränkt und eine genaue Beladungssteuerung ist in vielen Fällen nicht
möglich, ebensowenig wie die Analyse des erhaltenen Materials. Eventuell entstehende
Nebenprodukte oder nicht umgesetzte Vorstufen bei mehrstufigen Transformationen können
nicht aus dem Polymer entfernt werden.
Dagegen lassen sich funktionelle Gruppen bei Polyacrylamiden in Form von geeigneten
Monomeren einführen, was zur Bildung von entsprechend modifizierten Copolymeren führt.
Die durch chemische Synthese zugänglichen Monomere können in reiner Form hergestellt
werden, womit die Identität des Polymers gewährleistet ist. Zudem handelt es sich bei
Polyacrylamid um ein weitgehend inertes Trägermaterial, welches sowohl für native als auch
für denaturierende Gelelektrophorese genutzt werden kann, das demnach höchstens geringe
Wechselwirkungen mit Oligonukleotiden und der im Zuge des ElektroSpread-Verfahrens
zum Einsatz kommenden Chemie erwarten lässt.
Aufgrund dieser Vorüberlegungen konzentriert sich diese Arbeit auf die Darstellung und
Untersuchung modifizierter Polyacrylamide.
In der Literatur ist eine Vielzahl von N-substituierten Acrylamiden bekannt, die als potentielle
Monomere dienen können.[35] Die Geschwindigkeit der radikalischen Polymerisation hängt
jedoch vom Substitutionsgrad am Acrylamid-Stickstoff ab. Dabei ist die Geschwindigkeit
umso langsamer, je höher der Substitutionsgrad ist.[36] Daraus folgt, dass bei der
Copolymerisation von Acrylamid mit substituierten Acrylamiden in geringer Konzentration
die Gefahr besteht, dass diese Bausteine nicht in ausreichendem Maße oder nur zum Ende der
Polymerisation eingebaut werden, was zu einer heterogenen Verteilung führen würde.

20
Bei der Verwendung von N,N-Dimethylacrylamid als Trägermaterial wird ein eventuell
schnellerer Einbau der modifizierten Bausteine durch die geringere Konzentration teilweise
kompensiert.
Je nach beabsichtigter Modifikation ist es teilweise nicht möglich, diese in freier Form als
Monomer einzuführen. In diesen Fällen müssen Vorstufen bzw. geschützte Derivate
eingesetzt werden. Durch weitere chemische Transformationen erhält man das im Sinne der
angestrebten Immobilisierungsstrategie reaktive Gel (Abb.10).
Auch wenn chemische Transformationen nach der Polymerisation durchgeführt werden
müssen, ist diese Methode einer Modifizierung unmodifizierter Polymere überlegen, da hier
durch die Wahl der Monomere das Produkt bereits vorgebildet werden kann. Ebenso kann die
angestrebte Beladung durch die Konzentration der Monomere sehr gut kontrolliert werden.
Im Folgenden werden zunächst die Eigenschaften unmodifizierter N,N-Dimethylacryl-
amidpolymere bezüglich Polymerisation, Quellverhalten und Quervernetzungsgrad
beschrieben. Daraufhin werden die Betrachtungen auf modifizierte Systeme erweitert, wobei
Monomer-Lösung (Co)PolymerPolymerisation
chemischeTransformation
reaktives Gel Abb.10: Schema: Erstellung reaktiver Gele.
OONH
ONMe2
ONMe2
ONH
OO
ONMe2
ONH
ONMe2
ONMe2
ONH
O
OONMe2
NMe2
NMe2
NMe2
NMe2
OONH
ONMe2
ONMe2
ONMe2
ONH
ONMe2
OO
ONMe2
OONMe2
ONMe2
OO
OONH
ONMe2
NMe2
O
ONMe2
OONMe2
ONMe2
ONMe2
OONH
ONMe2
ONH
ONMe2
OONH
ONMe2
ONMe2
ONH
ONMe2
ONH
ONMe2
ONMe2
ONH
ONMe2
OO
ONMe2
OOONMe2
ONMe2
ONMe2
ONH
ONMe2
ONMe2
ONMe2
ONMe2
OONMe2
ONMe2
ONMe2
ONH
O
ONMe2
ONH
ONMe2
ONMe2
OONH
ONMe2
NMe2
NHO
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
NMe2
(P1)
Abb.11: Strukturausschnitt eines quervernetzten N,N-Dimethylacrylamid-Polymers (P1).

21
die Einführung der Funktionalitäten, Beladungen und Möglichkeiten der Immobilisierung
sowie einige funktionalitätsspezifische Eigenschaften betrachtet werden.
Zuletzt werden einige Fragen der technischen Implementierung untersucht.

22
4 Eigenschaften unmodifizierter quervernetzter N,N-
Dimethylacrylamid-Polymere
4.1 Polymerisationsgeschwindigkeit in Anwesenheit von DMF
Da die modifizierten Acrylamide teilweise eine stärkere Lipophilie als N,N-Dimethyl-
acrylamid und damit eine schlechtere Löslichkeit in Wasser aufwiesen, musste für die
Polymerisation teilweise auf die Verwendung alternativer Lösungsmittelgemische
ausgewichen werden.
Es ist aus der Literatur bekannt, dass die radikalische Polymerisation auch in organischen
Lösungsmitteln stattfinden kann.[37] Es zeigte sich jedoch, dass bei Verwendung von
Wasser/DMF-Gemischen unter Verwendung derselben Menge an Radikalstarter (APS) und
Überträger (TMEDA) die Polymerisation bei steigendem DMF-Gehalt deutlich langsamer
wurde und ab einem gewissen Mischungsverhältnis nicht mehr zur Ausbildung eines Geles
führte.
Um geeignete Bedingungen zur Copolymerisation hydrophober Bausteine zu ermitteln,
wurden diese Zusammenhänge systematisch untersucht.
Es zeigte sich, dass mechanische Bewegung den Polymersationsvorgang stören kann, was die
Vergleichbarkeit der Daten unmöglich machte. Daher wurde eine standardisierte
Vorgehensweise gewählt, bei der die Polymerisationslösungen bei konstanter Temperatur in
geschlossenen Eppendorf-Gefäßen gelagert wurden. Die Polymerisation wurde durch
Umdrehen in definierten und für alle Versuche identischen Zeitintervallen überprüft. Diese
Zeitintervalle betrugen zuerst eine Minute, wurden jedoch nach 15 bzw. 20 Minuten
verlängert. Der makroskopische Übergang von der flüssigen Phase zum Gel vollzog sich in
den meisten Fällen innerhalb eines kurzen Zeitintervalls (< 1Minute). Da dieser Übergang
einfach zu beobachten ist und die Beobachtung vieler kleiner Ansätze parallel ermöglicht,
wurde er als Messwert gewählt. Die Beobachtung des Brechungsindex, aus dem sich die
Kettenlänge ableiten lässt, würde zwar Rückschlüsse bezüglich des Beginns der
Polymerisation zulassen, jedoch den Übergang nicht erfassen können und wäre ohnehin nur
für den Beginn langsamer Polymerisationen sinnvoll durchführbar.[38]
Es kann angenommen werden, dass der Übergang von der Lösung zum Gel für die gleiche
Monomerzusammensetzung demselben Fortschritt der Polymerisation entspricht. Somit sollte

23
es sich bei der Zeit bis zu diesem Übergang um ein vergleichbares Maß für die
Polymerisationsgeschwindigkeit handeln.
Die Untersuchung der benötigten Menge an Radikalstarter und Überträger zeigt eindeutig den
erwarteten Zusammenhang, dass sowohl die Erhöhung der Konzentration des Radikalstarters
als auch des Überträgers zu einer
Beschleunigung führen (Abb.12). Für
APS-Konzentrationen von 2.2 mM
und TMEDA-Konzentrationen von
unterhalb 34 mM ist auch nach 24h
keine Gelbildung zu beobachten. Die
Konzentration an Hydrochinonmono-
methylether, welcher dem einge-
setzten Dimethylacrylamid als Radi-
kalfänger beigesetzt ist, beträgt 0.35
mM, die Konzentration an
Radikalstarter ist sonach selbst bei der
geringsten Konzentration um den
Faktor sechs größer. Aufgrund der
Tatsache, dass die Polymerisation hier
sehr langsam stattfindet, ist auch
denkbar, dass Radikale durch
Luftsauerstoff abgefangen werden
(4.25 µmol O2 in 0.5 ml Luftüber-
stand).
Eine zu schnelle Polymerisation ist aufgrund verschiedener Überlegungen zu vermeiden. Zum
einen führt die Reaktion zu einer stärkeren Wärmetönung, was zu einer mechanischen
Belastung des Gels führen kann,[39,40] zum anderen führt der Zusatz von zu viel Radikalstarter
zu einer niedrigeren Kettenlänge und der Vermehrung von Abbruchreaktionen, was zu einer
stärker heterogenen Struktur führen kann. Da bei den betrachteten Polymeren Quervernetzer
beigegeben sind, ist selbst bei einer geringeren Kettenlänge davon auszugehen, dass aufgrund
der Quervernetzung die Ketten miteinander verbunden sind, es sich also in der Näherung um
ein einziges Polymermolekül handelt. Üblicherweise angewendete Konzentrationen liegen im
Bereich von ca. 9 mM APS und 25 mM TMEDA, was in diesem Experiment zu einer Bildung
des Gels innerhalb von vier Minuten führt.[35]
Abb.12: Zeit bis zum Lösung/Gel-Phasenüber-
gang in Abhängigkeit von APS- und TMEDA-
Konzentration. Fehler sind in Form roter Balken
angegeben.
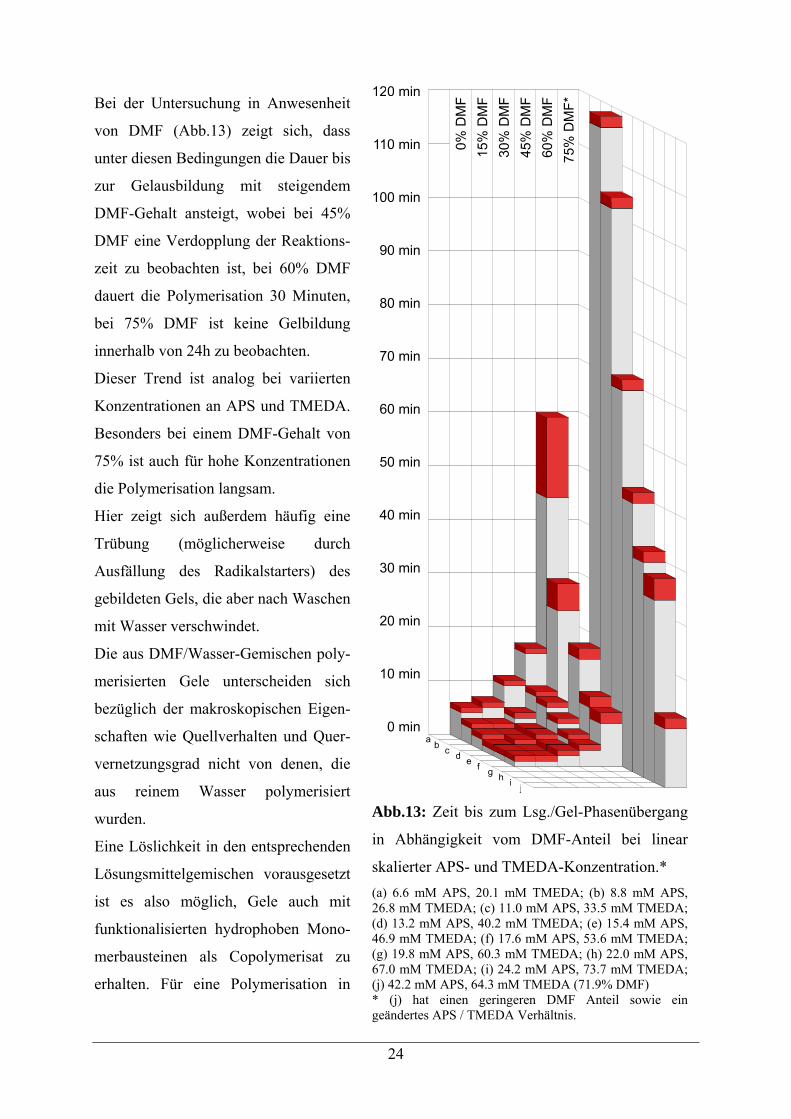
24
Bei der Untersuchung in Anwesenheit
von DMF (Abb.13) zeigt sich, dass
unter diesen Bedingungen die Dauer bis
zur Gelausbildung mit steigendem
DMF-Gehalt ansteigt, wobei bei 45%
DMF eine Verdopplung der Reaktions-
zeit zu beobachten ist, bei 60% DMF
dauert die Polymerisation 30 Minuten,
bei 75% DMF ist keine Gelbildung
innerhalb von 24h zu beobachten.
Dieser Trend ist analog bei variierten
Konzentrationen an APS und TMEDA.
Besonders bei einem DMF-Gehalt von
75% ist auch für hohe Konzentrationen
die Polymerisation langsam.
Hier zeigt sich außerdem häufig eine
Trübung (möglicherweise durch
Ausfällung des Radikalstarters) des
gebildeten Gels, die aber nach Waschen
mit Wasser verschwindet.
Die aus DMF/Wasser-Gemischen poly-
merisierten Gele unterscheiden sich
bezüglich der makroskopischen Eigen-
schaften wie Quellverhalten und Quer-
vernetzungsgrad nicht von denen, die
aus reinem Wasser polymerisiert
wurden.
Eine Löslichkeit in den entsprechenden
Lösungsmittelgemischen vorausgesetzt
ist es also möglich, Gele auch mit
funktionalisierten hydrophoben Mono-
merbausteinen als Copolymerisat zu
erhalten. Für eine Polymerisation in
Abb.13: Zeit bis zum Lsg./Gel-Phasenübergang
in Abhängigkeit vom DMF-Anteil bei linear
skalierter APS- und TMEDA-Konzentration.* (a) 6.6 mM APS, 20.1 mM TMEDA; (b) 8.8 mM APS, 26.8 mM TMEDA; (c) 11.0 mM APS, 33.5 mM TMEDA; (d) 13.2 mM APS, 40.2 mM TMEDA; (e) 15.4 mM APS, 46.9 mM TMEDA; (f) 17.6 mM APS, 53.6 mM TMEDA; (g) 19.8 mM APS, 60.3 mM TMEDA; (h) 22.0 mM APS, 67.0 mM TMEDA; (i) 24.2 mM APS, 73.7 mM TMEDA; (j) 42.2 mM APS, 64.3 mM TMEDA (71.9% DMF) * (j) hat einen geringeren DMF Anteil sowie ein geändertes APS / TMEDA Verhältnis.

25
unpolaren Lösungsmitteln ist jedoch zusätzlich eine Modifikation der Radikalstarter nötig.
4.2 Quellverhalten
Da viele der hier vorgestellten Modifikationen weitere chemische Umsetzungen des Geles
voraussetzen oder nicht in rein wässrigen Lösungen eingeführt werden können, wurde
ebenfalls das Quellverhalten in unterschiedlichen Lösungsmitteln und Lösungs-
mittelgemischen untersucht sowie in Anwesenheit von Salzen und Harnstoff. Eine chemische
Umsetzung des Polymers setzt ein Quellen im gewählten Lösungsmittel voraus, da sonst die
Funktionalitäten im Inneren des Poly-
mers nicht zugänglich sind.
Die zusätzliche Bedeutung dieser
Untersuchung wird klar, wenn man
bedenkt, dass mit dem Quellen oder
Schrumpfen des Gels eine mecha-
nische Belastung verbunden ist. Ist das
Gel z.B. auf einer Oberfläche fixiert,
kann es sich nicht isotrop ausdehnen
oder schrumpfen, was zur Ablösung
oder zum Reißen des Polymers führt,
da die Ausdehnung parallel zur
Oberfläche gehindert ist (Abb.14).[35]
4.2.1 Quellverhalten in reinen Lösungsmitteln
Zur Ermittlung des Quellvolumens wurden Gelfragmente ähnlicher Form und Masse im
Vakuum über Phosphorpentoxid getrocknet. Nach Wägung ließ man die Fragmente 24h im
entsprechenden Lösungsmittel quellen. Daraufhin wurden die Fragmente abgetropft und
wiederum gewogen. Dieses Verfahren wurde möglichst einheitlich durchgeführt, was die
Reproduzierbarkeit der Ergebnisse ermöglicht.
Grundsätzlich ist anzumerken, dass quantitative Untersuchungen des Quellverhaltens recht
große statistische Fehler aufweisen, weshalb in manchen Fällen die Bestimmung indirekt über
durch Einlagerung von Kolloiden gebildete photonische Kolloidkristalle erfolgt, deren
Absorptionsmaxima sich abhängig vom Quellzustand verschieben.[41,42,43]
oberflächen-gebundenesGel
freies Gel
quellenschrumpfen
QV
quellenschrumpfen
Abb.14: Schematische Darstellung des
Verhaltens oberflächengebundener Gele bei
Änderung des Quellverhaltens.
26
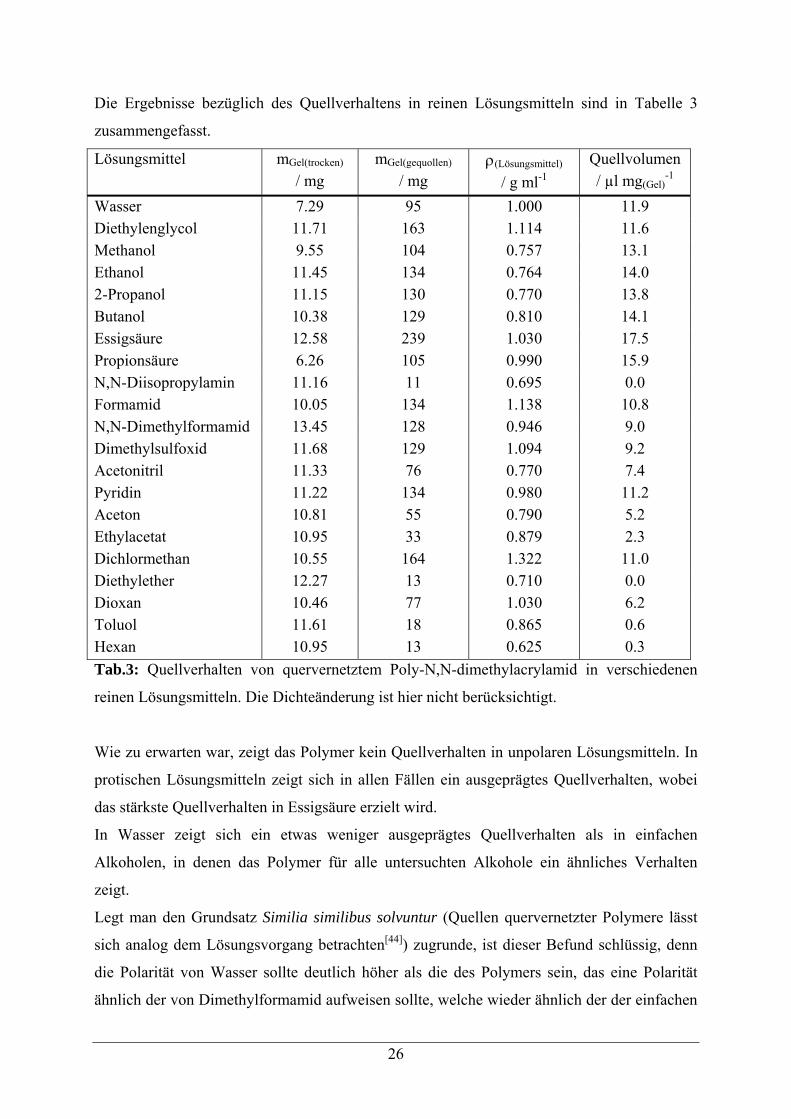
Die Ergebnisse bezüglich des Quellverhaltens in reinen Lösungsmitteln sind in Tabelle 3
zusammengefasst.
Lösungsmittel mGel(trocken) / mg
mGel(gequollen) / mg
ρ(Lösungsmittel) / g ml-1
Quellvolumen / µl mg(Gel)
-1 Wasser 7.29 95 1.000 11.9 Diethylenglycol 11.71 163 1.114 11.6 Methanol 9.55 104 0.757 13.1 Ethanol 11.45 134 0.764 14.0 2-Propanol 11.15 130 0.770 13.8 Butanol 10.38 129 0.810 14.1 Essigsäure 12.58 239 1.030 17.5 Propionsäure 6.26 105 0.990 15.9 N,N-Diisopropylamin 11.16 11 0.695 0.0 Formamid 10.05 134 1.138 10.8 N,N-Dimethylformamid 13.45 128 0.946 9.0 Dimethylsulfoxid 11.68 129 1.094 9.2 Acetonitril 11.33 76 0.770 7.4 Pyridin 11.22 134 0.980 11.2 Aceton 10.81 55 0.790 5.2 Ethylacetat 10.95 33 0.879 2.3 Dichlormethan 10.55 164 1.322 11.0 Diethylether 12.27 13 0.710 0.0 Dioxan 10.46 77 1.030 6.2 Toluol 11.61 18 0.865 0.6 Hexan 10.95 13 0.625 0.3 Tab.3: Quellverhalten von quervernetztem Poly-N,N-dimethylacrylamid in verschiedenen
reinen Lösungsmitteln. Die Dichteänderung ist hier nicht berücksichtigt.
Wie zu erwarten war, zeigt das Polymer kein Quellverhalten in unpolaren Lösungsmitteln. In
protischen Lösungsmitteln zeigt sich in allen Fällen ein ausgeprägtes Quellverhalten, wobei
das stärkste Quellverhalten in Essigsäure erzielt wird.
In Wasser zeigt sich ein etwas weniger ausgeprägtes Quellverhalten als in einfachen
Alkoholen, in denen das Polymer für alle untersuchten Alkohole ein ähnliches Verhalten
zeigt.
Legt man den Grundsatz Similia similibus solvuntur (Quellen quervernetzter Polymere lässt
sich analog dem Lösungsvorgang betrachten[44]) zugrunde, ist dieser Befund schlüssig, denn
die Polarität von Wasser sollte deutlich höher als die des Polymers sein, das eine Polarität
ähnlich der von Dimethylformamid aufweisen sollte, welche wieder ähnlich der der einfachen
27
Alkohole ist. Allerdings ist gerade das Quellverhalten in Dimethylformamid deutlich weniger
stark ausgeprägt als das in Wasser.
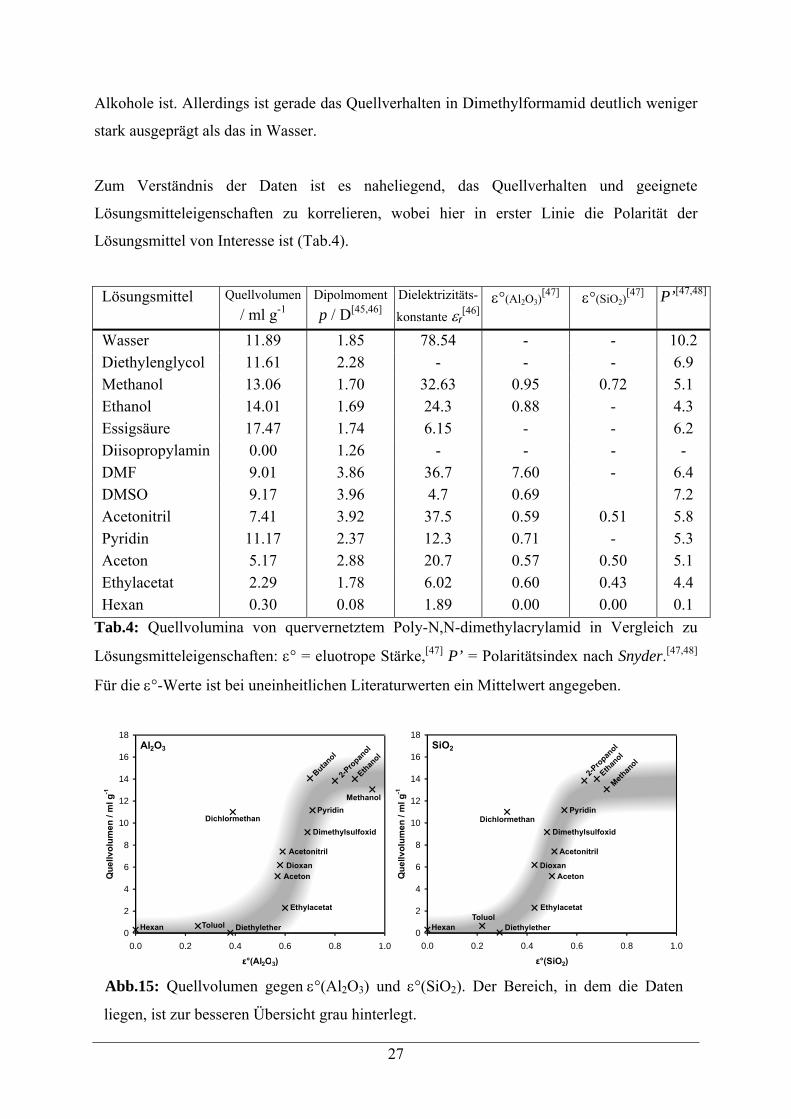
Zum Verständnis der Daten ist es naheliegend, das Quellverhalten und geeignete
Lösungsmitteleigenschaften zu korrelieren, wobei hier in erster Linie die Polarität der
Lösungsmittel von Interesse ist (Tab.4).
Lösungsmittel Quellvolumen / ml g-1
Dipolmoment p / D[45,46]
Dielektrizitäts-
konstante εr[46]
ε°(Al2O3)[47] ε°(SiO2)
[47] P’[47,48]
Wasser 11.89 1.85 78.54 - - 10.2 Diethylenglycol 11.61 2.28 - - - 6.9 Methanol 13.06 1.70 32.63 0.95 0.72 5.1 Ethanol 14.01 1.69 24.3 0.88 - 4.3 Essigsäure 17.47 1.74 6.15 - - 6.2 Diisopropylamin 0.00 1.26 - - - - DMF 9.01 3.86 36.7 7.60 - 6.4 DMSO 9.17 3.96 4.7 0.69 7.2 Acetonitril 7.41 3.92 37.5 0.59 0.51 5.8 Pyridin 11.17 2.37 12.3 0.71 - 5.3 Aceton 5.17 2.88 20.7 0.57 0.50 5.1 Ethylacetat 2.29 1.78 6.02 0.60 0.43 4.4 Hexan 0.30 0.08 1.89 0.00 0.00 0.1
Tab.4: Quellvolumina von quervernetztem Poly-N,N-dimethylacrylamid in Vergleich zu
Lösungsmitteleigenschaften: ε° = eluotrope Stärke,[47] P’ = Polaritätsindex nach Snyder.[47,48]
Für die ε°-Werte ist bei uneinheitlichen Literaturwerten ein Mittelwert angegeben.
0
2
4
6
8
10
12
14
16
18
0.0 0.2 0.4 0.6 0.8 1.0
ε°(Al2O3)
Que
llvol
umen
/mlg
-1
Al2O3
0
2
4
6
8
10
12
14
16
18
0.0 0.2 0.4 0.6 0.8 1.0
ε°(SiO2)
Que
llvol
umen
/mlg
-1
SiO2
Ethanol
Methanol
Hexan Diethylether
Ethylacetat
Toluol
AcetonDioxan
Acetonitril
Dichlormethan
Butanol
2-Pro
panol
Pyridin
Dimethylsulfoxid
Methan
ol
Hexan Diethylether
EthylacetatToluol
AcetonDioxan
Acetonitril
Dichlormethan
2-Pro
panol
Ethanol
Pyridin
Dimethylsulfoxid
Abb.15: Quellvolumen gegen ε°(Al2O3) und ε°(SiO2). Der Bereich, in dem die Daten
liegen, ist zur besseren Übersicht grau hinterlegt.

28
Eine Betrachtung der physikalischen Parameter Dipolmoment und Dieelektrizitätskonstante
führt zu keinem eindeutigen Trend. Im Gegensatz dazu scheint die eluotrope Reihe einen
besseren Anhaltspunkt zu liefern. Unterhalb von ε°(Al2O3) = 0.5 zeigt sich kein
Quellverhalten, im Bereich 0.5 < ε°(Al2O3) < 0.8 steigt das Quellvolumen an, um daraufhin
eine Sättigung zu erreichen. Trotz der großen Streuung scheint dieser Trend eindeutig zu sein,
er wird darüberhinaus auch von der Auftragung gegen ε°(SiO2) reproduziert (Abb.15).
Bei größeren Polaritäten (die von definitionsgemäß experimentellen ε° Werten aus
technischen Gründen nicht abgedeckt werden können) findet man wieder einen abfallenden
Trend.
Ein Vergleich der Quellvolumina mit dem Polaritätsindex P’ ergibt keine klare Korrelation.
4.2.2 Theoretische Betrachtung des Quellverhaltens
Das Quellverhalten quervernetzter Polymere lässt sich mit der Löslichkeit unvernetzter
Polymere vergleichen.[44] Im Falle der Quervernetzung ist eine Auflösung unmöglich, es
kommt zum Quellen der Polymere. Thermodynamisch entspricht dies einem
Mischungsvorgang, der durch die entsprechende Freie Mischungsenthalpie ∆GM
charakterisiert wird. Das Quellen des Polymers führt zu der Freien elastischen Enthalpie ∆GE,
die durch die Auslenkung des Polymers aus dem relaxierten Zustand resultiert. Für den Fall,
dass zusätzlich ionische Anteile zu berücksichtigen sind, muss zudem der ionische Anteil der
Freien Enthalpie ∆GIon berücksichtigt werden.
Man erhält für die Freie Gesamtenthalpie ∆GT also:[41]
∆GT = ∆GM + ∆GE + ∆GIon (1)
Ebenso lässt sich das Verhalten durch den osmotischen Druck Π beschreiben, was zu einem
analogen Ausdruck führt:
ΠT = ΠM + ΠE + ΠIon (2)
Der Zusammenhang zwischen osmotischem Druck und Freier Enthalpie ist durch Gleichung
(3) gegeben:
Π = - ∂∆G
∂V (3)
Im Gleichgewichtzustand, wie wir ihn hier betrachten, gilt sowohl ∆GT = 0 als auch ΠT = 0,
d.h. der osmotische Druck ist mit dem Druck, der durch die elastische Expansion des

29
Polymernetzwerkes erzeugt wird, im Gleichgewicht. Ohne Berücksichtigung ionischer
Anteile gilt dann:
ΠM = - ΠE (4)
4.2.2.1 Elastische Freie Enthalpie
Bei dem elastischen Anteil ist lediglich der Entropie-Term zu betrachten, da – abgesehen von
der Mischungsenthalpie, die gesondert behandelt wird – keine Änderung der inneren Energie
stattfindet. Nach der statistischen Theorie lassen sich die Entropieänderungen herleiten, wobei
unterschiedliche Modelle in der Literatur beschrieben werden.[44,49] Nach dem affine model
werden die Quervernetzungen als im Netzwerk eingebettet betrachtet. Damit erhält man als
Entropieänderung:
∆SE = -
kB νe
2αx
2 + αy2 + αz
2 - 3 - ln(αx αy αz)
(5)
Dabei beschreiben αx, αy, αz die Ausdehnungsfaktoren des Polymers in x, y und z – Richtung,
νe ist die effektive Gesamtzahl der Quervernetzungen. Geht man von einer einheitlichen
Expansion in alle Richtungen aus und drückt α durch die Volumina des gequollenen Polymers
V und das Volumen des Polymers im spannungsfreien Zustand Vm aus,
V
Vm
αs = αx = αy = αz =
1/3
(6)
so erhält man für die Entropieänderung:
V
Vm
V
Vm
∆SE = -3 kB νe
2- 1 - ln
1
3
2/3
(7)
Daraus folgt (mit ∆GE = -T∆SE):
V
Vm
V
Vm
∆GE =3 kB T νe
2- 1 - ln
1
3
2/3
(8)
Durch Ableitung nach V lässt sich gemäß Gleichung (3) der entsprechende Druck ΠE
berechnen:

30
kB T νe
Vm-
1
2
1/3Vm
V
Vm
VΠE = -
(9)
Werden die Quervernetzungen als frei fluktuierend betrachtet, so spricht man vom phantom
model[49] und erhält folgende Ausdrücke für ∆GE und ΠE:[42]
V
Vm
∆GE =3 kB T νe
4 - 1
2/3
(10)
kB T νe
2 Vm
1/3Vm
VΠE = -
(11)
4.2.2.2 Freie Mischungsenthalpie
Um die Freie Mischungsenthalpie ∆GM abschätzen zu können, müssen geeignete Ausdrücke
für den Enthalpie- sowie den Entropieterm gefunden werden.
∆GM = ∆HM - T∆SM (12)
Mischungsentropie
In der klassischen Thermodynamik ist die Mischungsentropie idealer Mischungen gegeben
als:
∆SM = - R(Σnixi)i (13)
(Ideale Mischung aus i Komponenten. ni = Stoffmenge, xi = Molenbruch der Komponente i)
Bei der Mischung von Polymeren mit niedermolekularen Lösungsmitteln kann die
Berechnung der Mischungsentropie jedoch nicht gemäß (13) erfolgen. Da hier die innere
Beweglichkeit des Polymers nicht berücksichtigt ist, liefern entsprechende Berechnungen
systematisch falsche Ergebnisse.[44]

31
Eine Möglichkeit der Abschätzung der
Mischungsentropie lässt sich aus der
statistischen Thermodynamik gewinnen,
nach der die Entropie eines Systems sich aus
der absoluten Anzahl möglicher
Anordnungen ergibt.
Betrachtet man die Mischung als ein Gitter
bestehend aus kleinen Volumeneinheiten
(Liquid-Lattice-Modell), die jeweils ein
Lösungsmittelmolekül oder ein Polymer-
segment enthalten können, und nimmt an,
dass jedes Polymermolekül eine zufällige
Anordnung annehmen kann, bei der die
Polymersegmente in benachbarten Volumen-
einheiten angeordnet sind, so erhält man als
Ausdruck für alle möglichen Anordnungen
Ω:[44]
Ω = n0!
(n0-xn2)!n2!
z-1
n0
n2(x-1)
(14)
Dabei sind n1 und n2 die Anzahl an Lösungsmittel- bzw. Polymermolekülen, wobei jedes
Polymer aus x Segmenten besteht. Die Gesamtanzahl n0 aller Volumenfragmente lässt sich
also auch durch n0 = n1 + x n2 angeben. Die Gitter-Koordinationsnummer z stellt die Anzahl
der einer Zelle benachbarten Zellen dar. (In einem kubischen Gitter bei Kontakten über die
Flächen wäre z.B. z = 6.)
In der statistischen Thermodynamik ist die Entropie Sc durch die Zahl der Mikrozustände
(bzw. das statistische Gewicht) Ω definiert:
Sc = kb lnΩ (15)
Durch Anwendung der Stirling-Formel zur Abschätzung von Fakultäten ( )
und Ersetzung von n0 erhält man aus (14) und (15):
Sc = -k n1 ln + n2 ln -n2(x - 1) ln[(z - 1) e-1] n2
n1 + xn2
n1
n1 + xn2
(16)
Abb.16: Liquid-Lattice-Modell.
(schematisch nach Flory[44])
Lösungsmittelmoleküle werden durch weiß
gefüllte Kreise, Polymersegmente durch
schwarz gefüllte Kreise symbolisiert.

32
Die Mischungsentropie kann durch Separation des Disorientierungsvorganges vom
Mischungsvorgang erhalten werden. Die Disorientierung entspricht dabei den
unterschiedlichen möglichen Anordnungen des Polymers.
Sc = SDis + SM (17)
Die reine Disorientierung des Polymers kann betrachtet werden, wenn das Lösungsmittel
eliminiert wird, d.h. n1 = 0. Damit erhält man (18):
SDis = kn2 [ln x + (x - 1) ln [(z - 1) e-1]] (18)
Anschaulich bedeutet das, dass im ersten Schritt das Polymer im leeren Gitter orientiert wird
(SDis), danach im zweiten Schritt die verbliebenen leeren Zellen mit Lösungsmittel gefüllt
werden (SM).
Aus (16) und (18) erhält man gemäß (17) die Mischungsentropie SM:
SM = - k(n1 ln υ1 + n2 ln υ2) (19)
In verallgemeinerter Form für mehrere Spezies (die sowohl unterschiedliche Lösungsmittel
als auch Polymere darstellen können) erhält man:
SM = - k ni ln υi Σi
(20)
Dieser Ausdruck ist dem klassischen Ausdruck (13) recht ähnlich, abgesehen davon, dass die
Molenbrüche (bzw. Aktivitäten) durch Volumenanteile ersetzt sind.
Mischungeenthalpie
Nachdem die Mischungsentropie abgeschätzt werden konnte, muss noch die
Mischungsenthalpie bestimmt werden. Der Enthalpieterm lässt sich ebenfalls aus dem Liquid-
Lattice-Modell abschätzen. Dabei werden die möglichen Kontakte einer Zelle zu der
Nachbarzelle betrachtet. Es besteht die Möglichkeit, dass es einen homogenen Kontakt
zwischen Lösungsmittel und Lösungsmittel [1,1] oder Polymersegment und Polymersegment
[2,2] gibt, wie auch die Möglichkeit des heterogenen Kontaktes zwischen Polymersegment
und Lösungsmittelmolekül [1,2].
Die stöchiometrische Gleichung für die Bildung eines heterogenen Kontaktes aus homogenen
Kontakten ist dann:
0.5 [1,1] + 0.5 [2,2] = [1,2] (21)
Werden die Energien der Kontakte als w11, w22 und w12 bezeichnet so erhält man als
Bildungsenergie eines heterogenen Kontaktes:

33
∆w12 = w12 - 0.5 (w11 + w22) (22)
Bei einer Anzahl von p12 heterogenen Kontakten ergibt sich für die Mischungsenthalpie:
∆HM = ∆w12 p12 (23)
Die Wahrscheinlichkeit, dass ein Polymersegment einen Kontakt zu einem
Lösungsmittelmolekül hat, entspricht (unter Vernachlässigung der Tatsache, dass man in der
Realität unterschiedliche Domänen berücksichtigen müsste) genau dem Volumenanteil υ1 des
Lösungsmittels. Die Gesamtzahl der Kontakte des Polymers ist (z - 2)x + 2. Bei großem x
lässt sich der Term durch zx ersetzen. Mit n1/n2 = xυ1/x1υ2 erhält man:
∆HM = ∆w12 z x1 n1 υ2 (24)
Dabei werden mit dem Faktor x1 mögliche Segmente des Lösungsmittels berücksichtigt.
Eine gebräuchliche Darstellung dieses Zusammenhangs ist:
∆HM = kT χ1 n1 υ2 (25)
Dabei ist χ1 der Flory-Huggins-Parameter, der die Wechselwirkung eines Lösungsmittels mit
einem Polymer beschreibt:
z ∆w12 x1
kTχ1 =
(26)
Freie Mischungenthalpie
Die Zusammenfassung von Entropieterm (19) und Enthalpieterm (25) führt für die binäre
Mischung zu (27):
∆GM = kBT (n1 ln(υ1) + n2 ln(υ2) + χ1 n1 υ2) (27)
Aufgrund der Tatsache, dass die Stoffmenge des Polymers verschwindend gering ist,
verschwinden die Terme, die n2 enthalten. So erhält man als Freie Mischungsenthalpie für den
binären Fall (Polymer und reines Lösungsmittel):
∆GM = kBT (n1 ln(υ1) + χ1 n1 υ2) (28)
Die verallgemeinerte Form für Lösungsmittelgemische (Komponenten i = 1,2,3...) ist dann:
∆GM = kBT ( ni ln υi + χi ni υPolymer )Σi
(29)
Dabei ist eine mögliche Wechselwirkung der Lösungsmittelmoleküle untereinander jedoch
vernachlässigt, d.h. es wird angenommen, dass diese Wechselwirkungen zwischen
Lösungsmittelmolekülen in Lösung und Gel gleich sind und dass das Partialvolumen des
Polymers gegenüber dem der Lösungsmittel klein ist.

34
Ersetzt man in (29) die Volumenanteile entsprechend υ1 = 1 - υ2 und υ2 = V0/V, wobei V0
dem Volumen des ungequollenen Polymers entspricht, so erhält man aus (29):
∆GM = kBT n1 ln 1 - + χ1 V0
V
V0
V
(30)
Wird die Teilchenzahl des Lösungsmittels n1 durch das Molvolumen v1 und die
Volumenfraktion des Lösungsmittels im Gel ersetzt ( n1 = (V - V0) / v1 ), so erhält man nach
Ableitung der freien Mischungsenthalpie nach dem Volumen den osmotischen Druck (31):
ΠM = - ln 1 - + + χ1 V0
V
V0
V
RT
v1
V0
V
2
(31)
4.2.2.3 Ionischer Anteil
Der osmotische Druck in Abhängigkeit von den Konzentrationen ci der Ionen mit einer
Ladung zi im Gel (gel), bzw. in Lösung (sol) ist bei genügender Verdünnung gegeben durch
(32): [41,44]
ΠIon = RT(Σci,gelzi,gel - Σci,solzi,sol)i i (32)
4.2.2.4 Zusammenfassung der thermodynamischen Größen
Im Quellgleichgewicht stehen die osmotischen Drücke im Gleichgewicht, es gilt ΠT = 0.
Damit lassen sich im Fall, dass keine ionischen Anteile zu betrachten sind (ΠIon = 0), gemäß
Gleichung (2) ΠM (31) und -ΠE (9) gleichsetzen. Auflösung nach χ1 führt zu:
V
V0
V0
V
V
V0
χ1 = - - - ln 1 - -1
2
v1νe V Vm
Vm V0 V
Vm
V
2 1/3 2
(33)
Um den Flory-Huggins-Parameter χ1 zu bestimmen, ist somit das Volumen des Polymers im
relaxierten gequollenen Zustand Vm, im ungequollenen (V0) wie auch im
Gleichgewichtszustand (V) zu bestimmen. Darüber hinaus muss der effektive
Quervernetzungsgrad bekannt sein.
Für Polymere mit einem bekannten Anteil an Quervernetzer liegt die Vermutung nahe, dass
dieser in erster Näherung dem Quervernetzungsgrad gleichzusetzen ist.
Tatsächlich zeigen Messungen, dass man teilweise tatsächlich große negative Abweichungen
findet.[42]

35
4.2.3 Bestimmung des Quervernetzungsgrades von N,N-Dimethylacrylamidpolymeren
Eine Möglichkeit der Bestimmung des effektiven Quervernetzungsgrades liegt in der
Untersuchung der Dehnung α bei Anwendung einer mechanischen Spannung τ.
Je nach Modell gilt näherungsweise für großes V/V0 Gleichung (34) bzw. (35)[42].
τ = RT(α - α-2)νe
Vm
affine model
(34)
τ = RT(α - α-2)νe
2Vm
phantom model
(35)
Für die Messung wurde in dieser Arbeit ein Aufbau gewählt, bei dem das frisch hergestellte
Polymergel definierter Dicke zwischen zwei Glasplatten mit unterschiedlichen Gewichten
belastet wurde. Die Dicke der Polymerschicht wurde durch Abfotografieren unter
Zuhilfenahme einer Referenzskala und nachträgliche digitale Auswertung bestimmt.
Die Spannung τ lässt sich aus der Masse des Gewichtes m und der Fläche der Polymerschicht
A bestimmen:
(36)
FG = g m ;
τ = FG A-1
mit g = 9.807 m s-2 (37)
y0 y
m F
Abb.17: Spannungs-Dehnungs-Experiment für ein unmodifiziertes N,N-
Dimethylacrylamidpolymer.
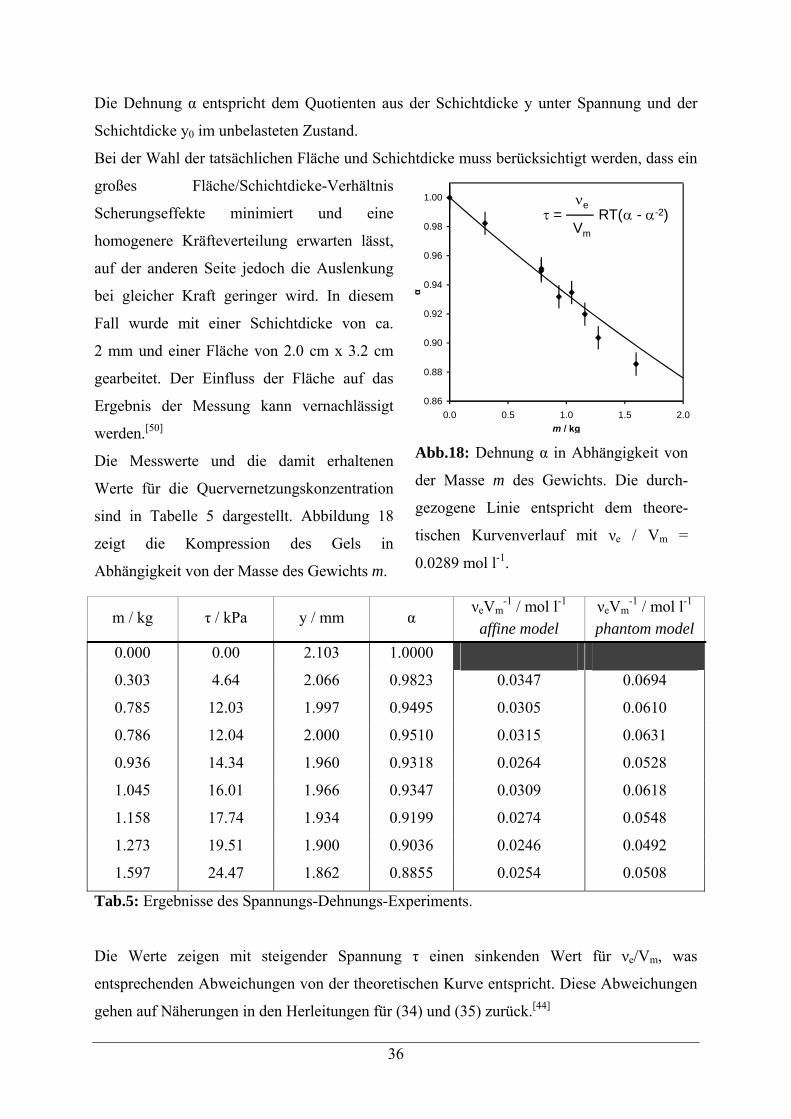
36
Die Dehnung α entspricht dem Quotienten aus der Schichtdicke y unter Spannung und der
Schichtdicke y0 im unbelasteten Zustand.
Bei der Wahl der tatsächlichen Fläche und Schichtdicke muss berücksichtigt werden, dass ein
großes Fläche/Schichtdicke-Verhältnis
Scherungseffekte minimiert und eine
homogenere Kräfteverteilung erwarten lässt,
auf der anderen Seite jedoch die Auslenkung
bei gleicher Kraft geringer wird. In diesem
Fall wurde mit einer Schichtdicke von ca.
2 mm und einer Fläche von 2.0 cm x 3.2 cm
gearbeitet. Der Einfluss der Fläche auf das
Ergebnis der Messung kann vernachlässigt
werden.[50]
Die Messwerte und die damit erhaltenen
Werte für die Quervernetzungskonzentration
sind in Tabelle 5 dargestellt. Abbildung 18
zeigt die Kompression des Gels in
Abhängigkeit von der Masse des Gewichts m.
m / kg τ / kPa y / mm α νeVm
-1 / mol l-1 affine model
νeVm-1 / mol l-1
phantom model 0.000 0.00 2.103 1.0000
0.303 4.64 2.066 0.9823 0.0347 0.0694
0.785 12.03 1.997 0.9495 0.0305 0.0610
0.786 12.04 2.000 0.9510 0.0315 0.0631
0.936 14.34 1.960 0.9318 0.0264 0.0528
1.045 16.01 1.966 0.9347 0.0309 0.0618
1.158 17.74 1.934 0.9199 0.0274 0.0548
1.273 19.51 1.900 0.9036 0.0246 0.0492
1.597 24.47 1.862 0.8855 0.0254 0.0508
Tab.5: Ergebnisse des Spannungs-Dehnungs-Experiments.
Die Werte zeigen mit steigender Spannung τ einen sinkenden Wert für νe/Vm, was
entsprechenden Abweichungen von der theoretischen Kurve entspricht. Diese Abweichungen
gehen auf Näherungen in den Herleitungen für (34) und (35) zurück.[44]
0.86
0.88
0.90
0.92
0.94
0.96
0.98
1.00
0.0 0.5 1.0 1.5 2.0m / kg
α
τ = RT(α - α-2)νe
Vm
Abb.18: Dehnung α in Abhängigkeit von
der Masse m des Gewichts. Die durch-
gezogene Linie entspricht dem theore-
tischen Kurvenverlauf mit νe / Vm =
0.0289 mol l-1.

37
Trotzdem bilden die erhaltenen Werte einen guten Anhaltspunkt für den tatsächlichen Grad
der Quervernetzung. Bei einer theoretischen Quervernetzungskonzentration von νe,theor / Vm =
0.02 mol l-1 entsprechend der Konzentration an Quervernetzer in der Gellösung fällt auf, dass
die Werte, die entsprechend des phantom-model erhalten werden, deutlich zu hoch sind,
während die Werte gemäß des affine-model im Bereich der theoretischen Konzentration
liegen.
Für die weiteren Berechnungen wird der Mittelwert des aus dem affine-model erhaltenen
Quervernetzungsgrades verwendet: νe / Vm = 0.0289 mol l-1.
4.2.4 Bestimmung der Flory-Huggins-Wechselwirkungsparameter
Nachdem der Quervernetzungsgrad bestimmt ist, lassen sich die Flory-Huggins-
Wechselwirkungsparameter für die hier untersuchten reinen Lösungsmittel mit N,N-Di-
methylacrylamid-Polymeren gemäß Gleichung (33) bestimmen.
V
V0
V0
V
V
V0
χ1 = - - - ln 1 - -1
2
v1νe V Vm
Vm V0 V
Vm
V
2 1/3 2
(33)
Dabei wird angenommen, dass sich das Polymer während der Polymerisation in relaxiertem
Zustand bildet. Dann entspricht Vm dem Volumen des Gels bei der Polymerisation. V ist das
tatsächliche Quellvolumen im jeweiligen Lösungsmittel, V0 das Volumen des trockenen
Polymers. Da als Messwerte für das trockene und gequollene Polymer die Massen ermittelt
wurden (vgl. Tab.3), muss die Umrechnung über die Dichten erfolgen.
Diese wurde in den meisten Fällen für die gequollenen Polymere bestimmt bzw. abgeschätzt
Lediglich das Volumen des ungequollenen Polymers ist schwierig zu bestimmen, weil die
getrockneten Gele eine sehr heterogene Struktur aufweisen. Es wurde eine Dichte von
ρ0 = 1 g cm-3 angenommen. (ρ(N,N-Dimethylacrylamid) = 0.962 g cm-3; bei der Polymerisation
erwartet man eine Volumenkontraktion und damit eine Zunahme der Dichte.)
Die Ergebnisse sind in Tabelle 6 angegeben.
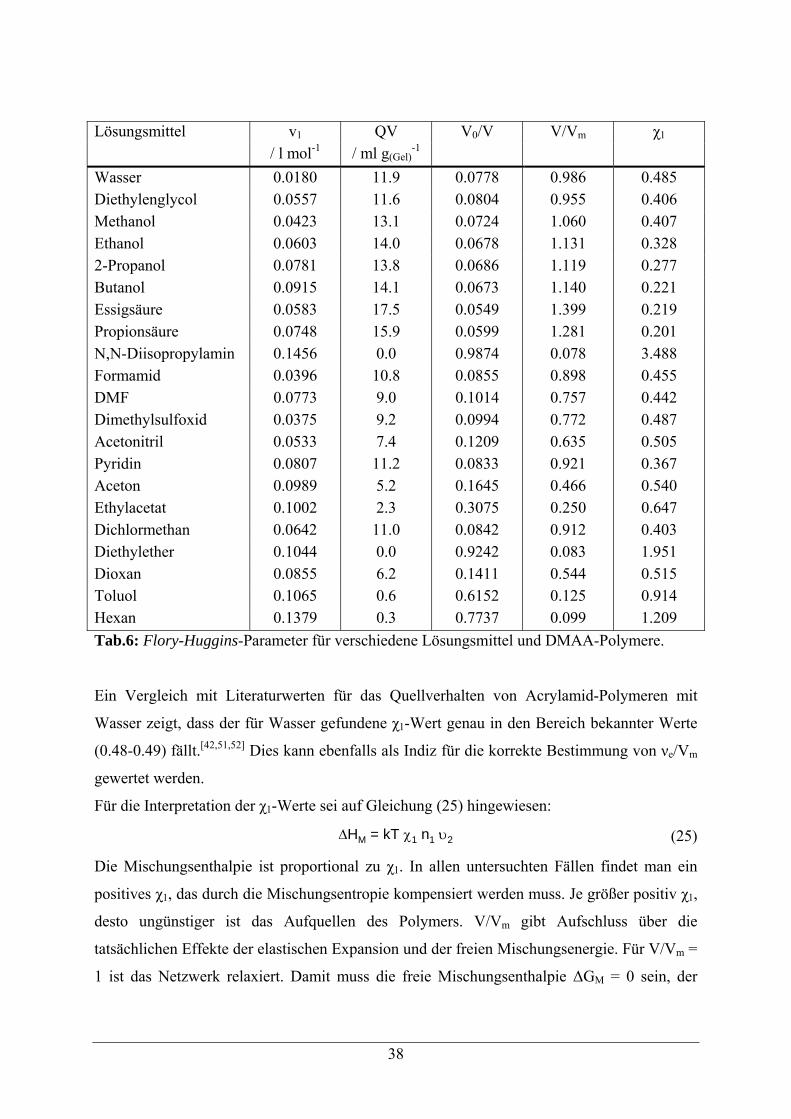
38
Lösungsmittel v1 QV V0/V V/Vm χ1 / l mol-1 / ml g(Gel)
-1 Wasser 0.0180 11.9 0.0778 0.986 0.485 Diethylenglycol 0.0557 11.6 0.0804 0.955 0.406 Methanol 0.0423 13.1 0.0724 1.060 0.407 Ethanol 0.0603 14.0 0.0678 1.131 0.328 2-Propanol 0.0781 13.8 0.0686 1.119 0.277 Butanol 0.0915 14.1 0.0673 1.140 0.221 Essigsäure 0.0583 17.5 0.0549 1.399 0.219 Propionsäure 0.0748 15.9 0.0599 1.281 0.201 N,N-Diisopropylamin 0.1456 0.0 0.9874 0.078 3.488 Formamid 0.0396 10.8 0.0855 0.898 0.455 DMF 0.0773 9.0 0.1014 0.757 0.442 Dimethylsulfoxid 0.0375 9.2 0.0994 0.772 0.487 Acetonitril 0.0533 7.4 0.1209 0.635 0.505 Pyridin 0.0807 11.2 0.0833 0.921 0.367 Aceton 0.0989 5.2 0.1645 0.466 0.540 Ethylacetat 0.1002 2.3 0.3075 0.250 0.647 Dichlormethan 0.0642 11.0 0.0842 0.912 0.403 Diethylether 0.1044 0.0 0.9242 0.083 1.951 Dioxan 0.0855 6.2 0.1411 0.544 0.515 Toluol 0.1065 0.6 0.6152 0.125 0.914 Hexan 0.1379 0.3 0.7737 0.099 1.209 Tab.6: Flory-Huggins-Parameter für verschiedene Lösungsmittel und DMAA-Polymere.
Ein Vergleich mit Literaturwerten für das Quellverhalten von Acrylamid-Polymeren mit
Wasser zeigt, dass der für Wasser gefundene χ1-Wert genau in den Bereich bekannter Werte
(0.48-0.49) fällt.[42,51,52] Dies kann ebenfalls als Indiz für die korrekte Bestimmung von νe/Vm
gewertet werden.
Für die Interpretation der χ1-Werte sei auf Gleichung (25) hingewiesen:
∆HM = kT χ1 n1 υ2 (25)
Die Mischungsenthalpie ist proportional zu χ1. In allen untersuchten Fällen findet man ein
positives χ1, das durch die Mischungsentropie kompensiert werden muss. Je größer positiv χ1,
desto ungünstiger ist das Aufquellen des Polymers. V/Vm gibt Aufschluss über die
tatsächlichen Effekte der elastischen Expansion und der freien Mischungsenergie. Für V/Vm =
1 ist das Netzwerk relaxiert. Damit muss die freie Mischungsenthalpie ∆GM = 0 sein, der

39
Enthalpieterm wird durch den Entropieterm kompensiert. Ist V/Vm > 1, so muss ∆GM < 0
sein, für V/Vm < 1 ist ∆GM > 0.
4.2.5 Quellverhalten in Lösungsmittelgemischen
Bei der Untersuchung des Quellverhaltens in reinen Lösungsmitteln waren auch 80%iges
Methanol und Ethanol untersucht worden, wobei sich zeigte, dass beide Mischungen ein
signifikant größeres Quellverhalten als die reinen Lösungsmittel zeigten.
Aufgrund der Tatsache, dass bei der chemischen Modifikation der Gele häufig mit wässrigen
Mischungen gearbeitet wird, bzw. diese beim Umspülen der Matrix auf ein anderes
Lösungsmittel automatisch entstehen, wurden auch diese Systeme untersucht.
Da die Unterschiede teilweise recht gering sind, wurden jeweils drei Messreihen zugrunde
gelegt, woraus sich die statistischen Fehler ergeben, die als Fehlerbalken angegeben sind,
wenn nicht anders erläutert.
Zusätzlich zum Quellverhalten wurden die Dichten der Gele bestimmt. Um eine möglichst
langsame Veränderung der inneren Zusammensetzung zu gewährleisten, wurden dazu, wenn
möglich, die Dichteunterschiede der Lösungsmittelgemische ausgenutzt. Die Dichte wurde
durch Schwebeexperimente abgeschätzt.
Die Dichten von Lösung und Gel sind, soweit sie bestimmt wurden, dargestellt. Bei einer
unabhängigen Bestimmung der Dichte des Gels wurde jene zur Berechnung des
Quellvolumens herangezogen. Daher unterscheiden sich die Werte teilweise von denen aus
Tabelle 3.
Es ist anzumerken, dass in jedem reinen Lösungsmittel, in dem das Polymer ein deutliches
Quellverhalten zeigt, die Dichte des Gels immer etwas größer als die der Lösung ist (∆ρ ≈ 10
- 20 mg cm-3). Zusätzlich zu dieser konstanten Abweichung findet sich teilweise ein
deutlicher Unterschied zwischen dem Dichteverlauf in Lösung und im Gel, was auf eine
unterschiedliche Zusammensetzung des Gemisches im Polymer hindeutet, da eine Zunahme
der Dichte des Gels auf eine Anreicherung der dichteren Lösungsmittelkomponente und
entsprechend eine Abnahme der Dichte auf eine Abreicherung der dichteren Komponente
hindeutet.
4.2.5.1 Theoretische Betrachtung.
Die theoretisch-thermodynamische Betrachtung der Quellvorgänge von Polymeren in
Lösungsmittelgemischen kann grundsätzlich durch Anwendung der aus der statistischen
Theorie erhaltenen Zusammenhänge erhalten werden.[44] Die elastischen Eigenschaften des
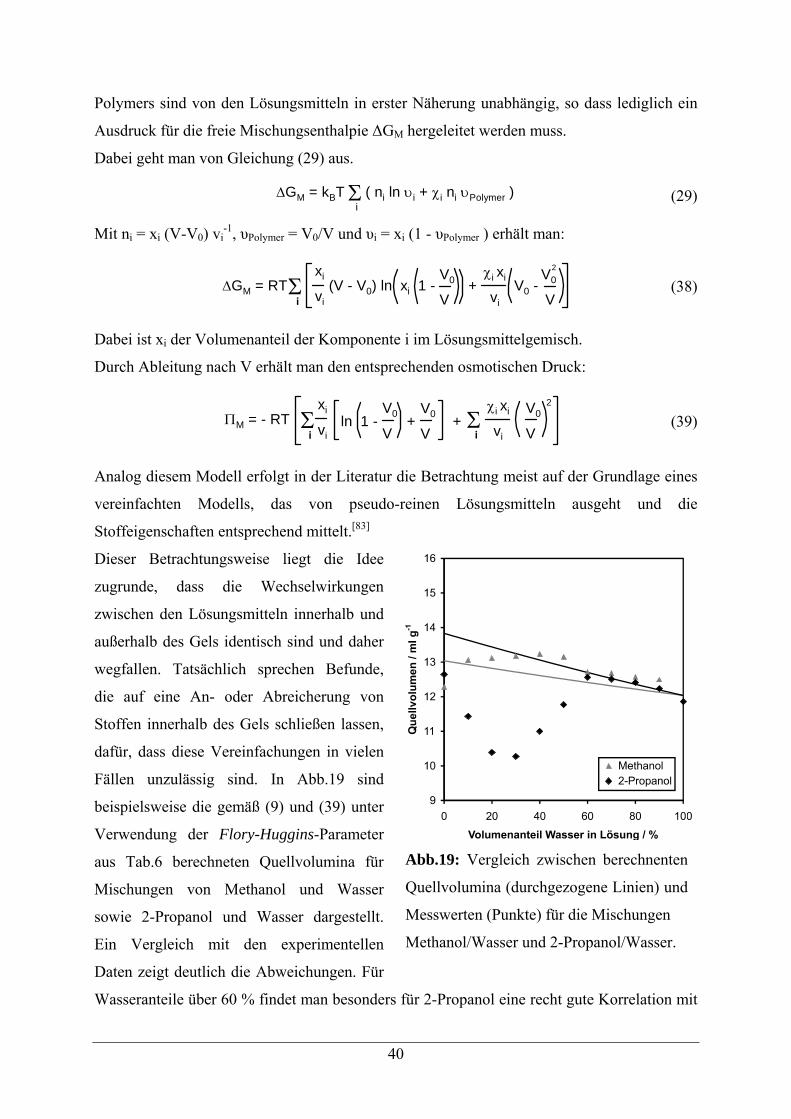
40
Polymers sind von den Lösungsmitteln in erster Näherung unabhängig, so dass lediglich ein
Ausdruck für die freie Mischungsenthalpie ∆GM hergeleitet werden muss.
Dabei geht man von Gleichung (29) aus.
∆GM = kBT ( ni ln υi + χi ni υPolymer )Σi
(29)
Mit ni = xi (V-V0) vi-1, υPolymer = V0/V und υi = xi (1 - υPolymer ) erhält man:
∆GM = RT (V - V0) ln xi 1 - + V0 -xi
vi
χi xi
vi
V0
VΣ
V0
Vi
2
(38)
Dabei ist xi der Volumenanteil der Komponente i im Lösungsmittelgemisch.
Durch Ableitung nach V erhält man den entsprechenden osmotischen Druck:
ΠM = - RTxi
vi
χi xi
vi
V0
VΣ
V0
Vi ln 1 - + +
V0
VΣ
i
2
(39)
Analog diesem Modell erfolgt in der Literatur die Betrachtung meist auf der Grundlage eines
vereinfachten Modells, das von pseudo-reinen Lösungsmitteln ausgeht und die
Stoffeigenschaften entsprechend mittelt.[83]
Dieser Betrachtungsweise liegt die Idee
zugrunde, dass die Wechselwirkungen
zwischen den Lösungsmitteln innerhalb und
außerhalb des Gels identisch sind und daher
wegfallen. Tatsächlich sprechen Befunde,
die auf eine An- oder Abreicherung von
Stoffen innerhalb des Gels schließen lassen,
dafür, dass diese Vereinfachungen in vielen
Fällen unzulässig sind. In Abb.19 sind
beispielsweise die gemäß (9) und (39) unter
Verwendung der Flory-Huggins-Parameter
aus Tab.6 berechneten Quellvolumina für
Mischungen von Methanol und Wasser
sowie 2-Propanol und Wasser dargestellt.
Ein Vergleich mit den experimentellen
Daten zeigt deutlich die Abweichungen. Für
Wasseranteile über 60 % findet man besonders für 2-Propanol eine recht gute Korrelation mit
Abb.19: Vergleich zwischen berechnenten
Quellvolumina (durchgezogene Linien) und
Messwerten (Punkte) für die Mischungen
Methanol/Wasser und 2-Propanol/Wasser.

41
der theoretischen Kurve, bei Wasseranteilen unter 60 % jedoch weichen die experimentellen
Werte deutlich von den theoretischen Kurven ab. Bei Methanol findet man eine leichte
positive Abweichung, bei 2-Propanol eine starke negative Abweichung.
Der einzige hier untersuchte Fall, bei dem das beobachtete Quellverhalten weitgehend dem
nach dem idealisierten Modell berechneten Quellverhalten entspricht, ist die Mischung aus
Wasser und Formamid (vgl. Kapitel 4.2.5.4).
Um nicht ideales Verhalten zu erklären, wurde in der Literatur die Möglichkeit von
unterschiedlichen Bereichen im somit heterogenen Gel diskutiert, die unterschiedliche
Eigenschaften aufweisen.[39,40,51] Allerdings konnte gezeigt werden, dass für Gele mit
geringem Quervernetzungsgrad keine derartige heterogene Struktur nachzuweisen ist.[53]
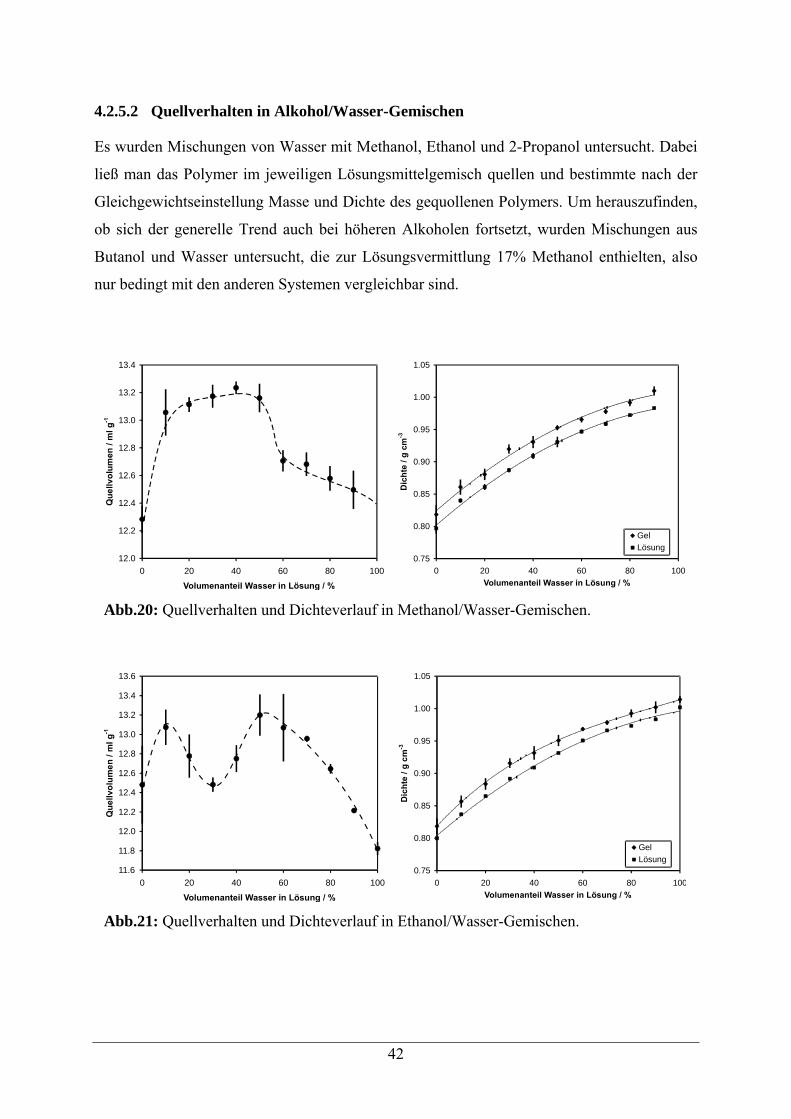
42
4.2.5.2 Quellverhalten in Alkohol/Wasser-Gemischen
Es wurden Mischungen von Wasser mit Methanol, Ethanol und 2-Propanol untersucht. Dabei
ließ man das Polymer im jeweiligen Lösungsmittelgemisch quellen und bestimmte nach der
Gleichgewichtseinstellung Masse und Dichte des gequollenen Polymers. Um herauszufinden,
ob sich der generelle Trend auch bei höheren Alkoholen fortsetzt, wurden Mischungen aus
Butanol und Wasser untersucht, die zur Lösungsvermittlung 17% Methanol enthielten, also
nur bedingt mit den anderen Systemen vergleichbar sind.
0.75
0.80
0.85
0.90
0.95
1.00
1.05
0 20 40 60 80 100Volumenanteil Wasser in Lösung / %
Dic
hte
/gcm
-3
GelLösung
12.0
12.2
12.4
12.6
12.8
13.0
13.2
13.4
0 20 40 60 80 100
Volumenanteil Wasser in Lösung / %
Que
llvol
umen
/mlg
-1
Abb.20: Quellverhalten und Dichteverlauf in Methanol/Wasser-Gemischen.
0.75
0.80
0.85
0.90
0.95
1.00
1.05
0 20 40 60 80 100Volumenanteil Wasser in Lösung / %
Dic
hte
/gcm
-3
GelLösung
11.6
11.8
12.0
12.2
12.4
12.6
12.8
13.0
13.2
13.4
13.6
0 20 40 60 80 100
Volumenanteil Wasser in Lösung / %
Que
llvol
umen
/mlg
-1
Abb.21: Quellverhalten und Dichteverlauf in Ethanol/Wasser-Gemischen.
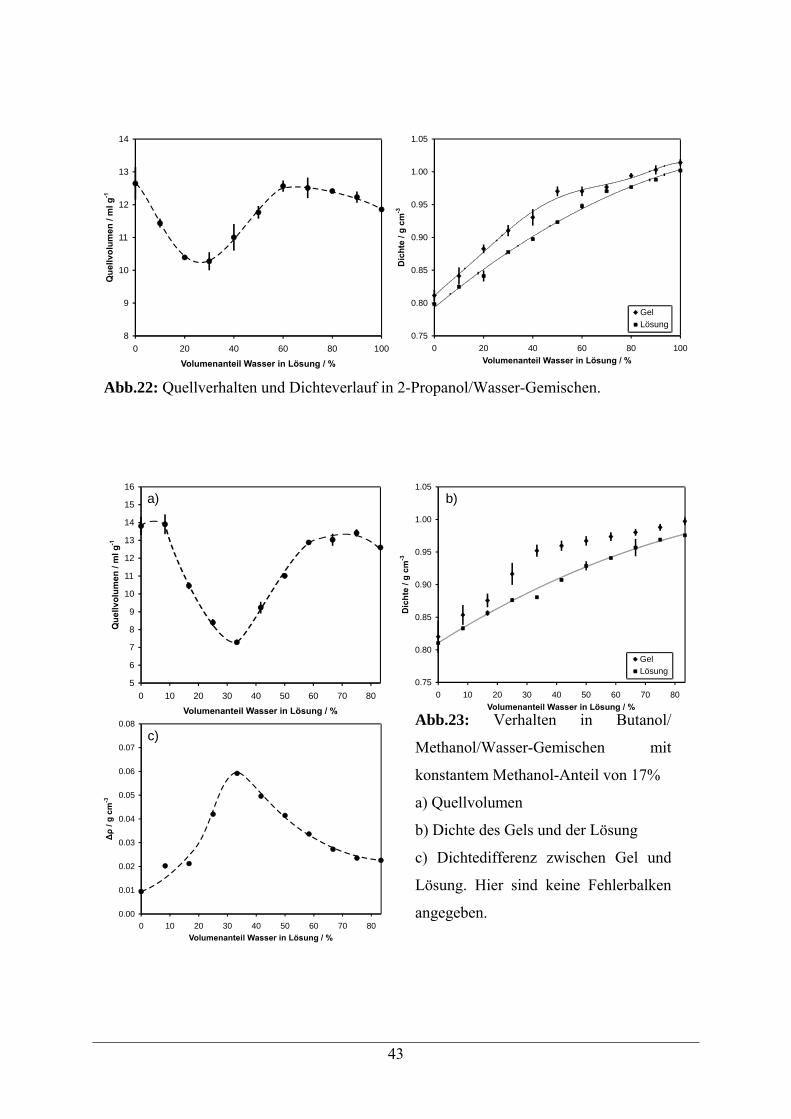
43
8
9
10
11
12
13
14
0 20 40 60 80 100
Volumenanteil Wasser in Lösung / %
Que
llvol
umen
/mlg
-1
0.75
0.80
0.85
0.90
0.95
1.00
1.05
0 20 40 60 80 100Volumenanteil Wasser in Lösung / %
Dic
hte
/gcm
-3
GelLösung
Abb.22: Quellverhalten und Dichteverlauf in 2-Propanol/Wasser-Gemischen.
5
6
7
8
9
10
11
12
13
14
15
16
0 10 20 30 40 50 60 70 80
Volumenanteil Wasser in Lösung / %
Que
llvol
umen
/mlg
-1
0.75
0.80
0.85
0.90
0.95
1.00
1.05
0 10 20 30 40 50 60 70 80Volumenanteil Wasser in Lösung / %
Dic
hte
/gcm
-3
GelLösung
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
0 10 20 30 40 50 60 70 80Volumenanteil Wasser in Lösung / %
∆ρ
/gcm
-3
a) b)
c)
Abb.23: Verhalten in Butanol/
Methanol/Wasser-Gemischen mit
konstantem Methanol-Anteil von 17%
a) Quellvolumen
b) Dichte des Gels und der Lösung
c) Dichtedifferenz zwischen Gel und
Lösung. Hier sind keine Fehlerbalken
angegeben.
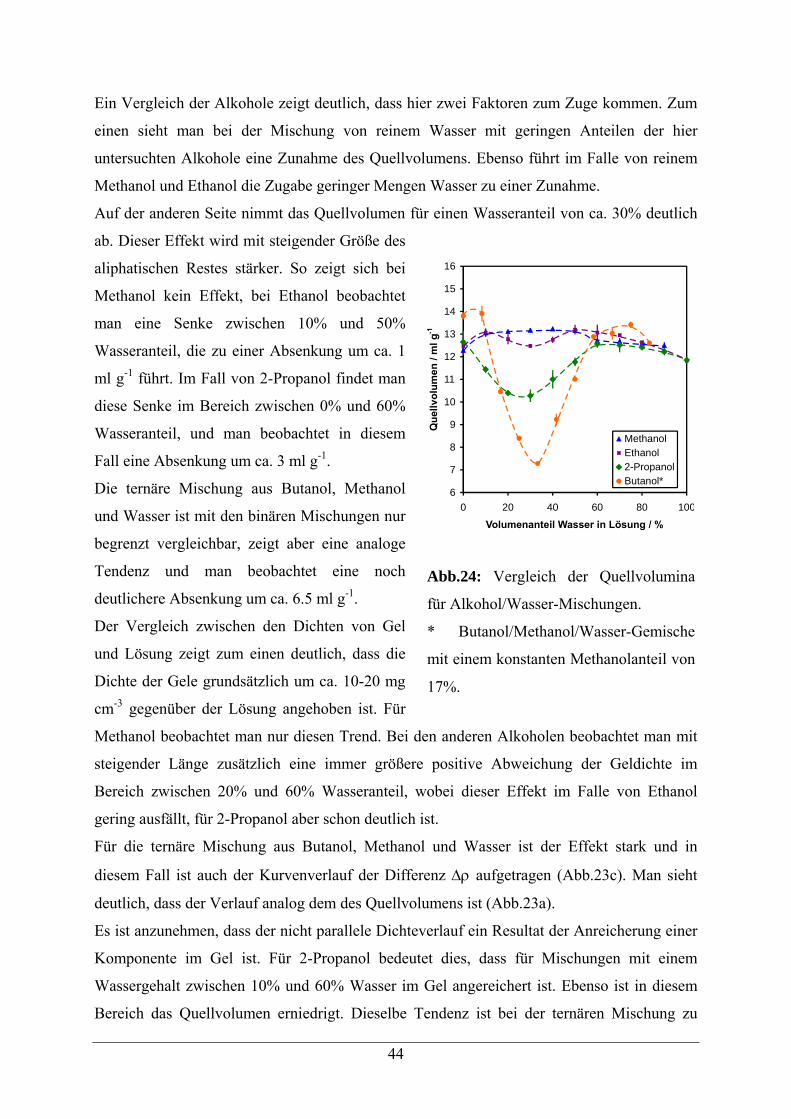
44
Ein Vergleich der Alkohole zeigt deutlich, dass hier zwei Faktoren zum Zuge kommen. Zum
einen sieht man bei der Mischung von reinem Wasser mit geringen Anteilen der hier
untersuchten Alkohole eine Zunahme des Quellvolumens. Ebenso führt im Falle von reinem
Methanol und Ethanol die Zugabe geringer Mengen Wasser zu einer Zunahme.
Auf der anderen Seite nimmt das Quellvolumen für einen Wasseranteil von ca. 30% deutlich
ab. Dieser Effekt wird mit steigender Größe des
aliphatischen Restes stärker. So zeigt sich bei
Methanol kein Effekt, bei Ethanol beobachtet
man eine Senke zwischen 10% und 50%
Wasseranteil, die zu einer Absenkung um ca. 1
ml g-1 führt. Im Fall von 2-Propanol findet man
diese Senke im Bereich zwischen 0% und 60%
Wasseranteil, und man beobachtet in diesem
Fall eine Absenkung um ca. 3 ml g-1.
Die ternäre Mischung aus Butanol, Methanol
und Wasser ist mit den binären Mischungen nur
begrenzt vergleichbar, zeigt aber eine analoge
Tendenz und man beobachtet eine noch
deutlichere Absenkung um ca. 6.5 ml g-1.
Der Vergleich zwischen den Dichten von Gel
und Lösung zeigt zum einen deutlich, dass die
Dichte der Gele grundsätzlich um ca. 10-20 mg
cm-3 gegenüber der Lösung angehoben ist. Für
Methanol beobachtet man nur diesen Trend. Bei den anderen Alkoholen beobachtet man mit
steigender Länge zusätzlich eine immer größere positive Abweichung der Geldichte im
Bereich zwischen 20% und 60% Wasseranteil, wobei dieser Effekt im Falle von Ethanol
gering ausfällt, für 2-Propanol aber schon deutlich ist.
Für die ternäre Mischung aus Butanol, Methanol und Wasser ist der Effekt stark und in
diesem Fall ist auch der Kurvenverlauf der Differenz ∆ρ aufgetragen (Abb.23c). Man sieht
deutlich, dass der Verlauf analog dem des Quellvolumens ist (Abb.23a).
Es ist anzunehmen, dass der nicht parallele Dichteverlauf ein Resultat der Anreicherung einer
Komponente im Gel ist. Für 2-Propanol bedeutet dies, dass für Mischungen mit einem
Wassergehalt zwischen 10% und 60% Wasser im Gel angereichert ist. Ebenso ist in diesem
Bereich das Quellvolumen erniedrigt. Dieselbe Tendenz ist bei der ternären Mischung zu
6
7
8
9
10
11
12
13
14
15
16
0 20 40 60 80 100
Volumenanteil Wasser in Lösung / %
Que
llvol
umen
/mlg
-1MethanolEthanol2-PropanolButanol*
Abb.24: Vergleich der Quellvolumina
für Alkohol/Wasser-Mischungen.
* Butanol/Methanol/Wasser-Gemische
mit einem konstanten Methanolanteil von
17%.
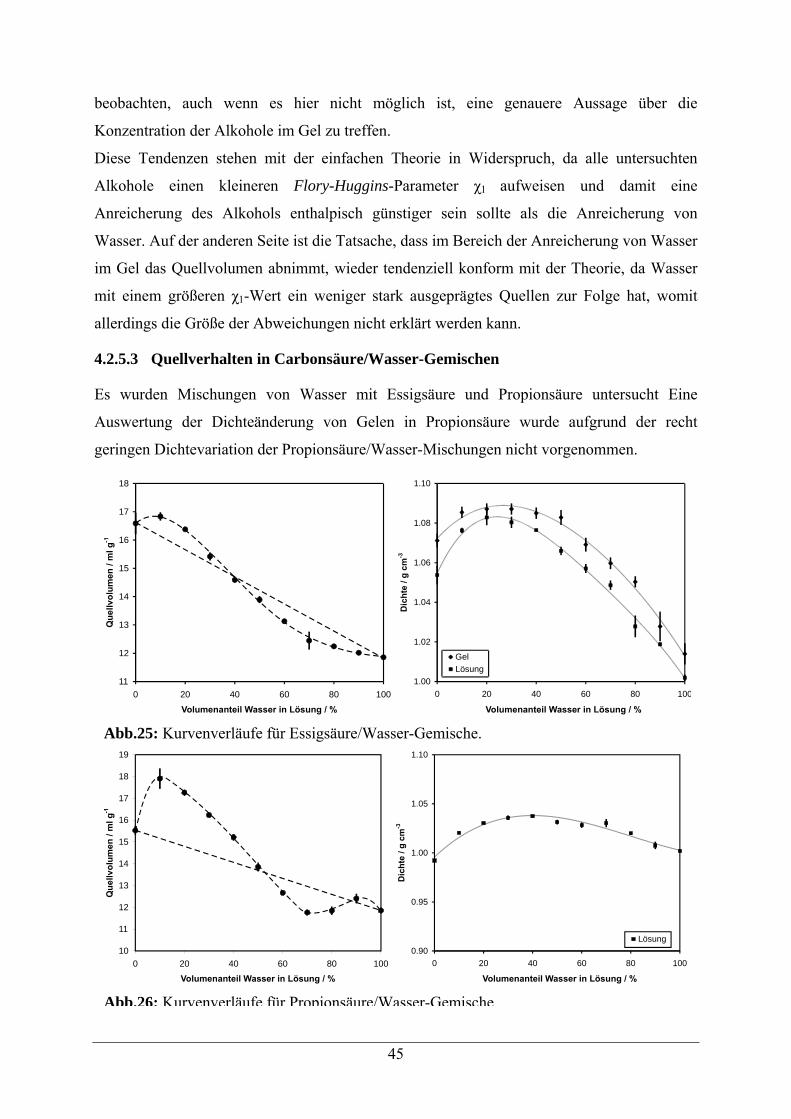
45
beobachten, auch wenn es hier nicht möglich ist, eine genauere Aussage über die
Konzentration der Alkohole im Gel zu treffen.
Diese Tendenzen stehen mit der einfachen Theorie in Widerspruch, da alle untersuchten
Alkohole einen kleineren Flory-Huggins-Parameter χ1 aufweisen und damit eine
Anreicherung des Alkohols enthalpisch günstiger sein sollte als die Anreicherung von
Wasser. Auf der anderen Seite ist die Tatsache, dass im Bereich der Anreicherung von Wasser
im Gel das Quellvolumen abnimmt, wieder tendenziell konform mit der Theorie, da Wasser
mit einem größeren χ1-Wert ein weniger stark ausgeprägtes Quellen zur Folge hat, womit
allerdings die Größe der Abweichungen nicht erklärt werden kann.
4.2.5.3 Quellverhalten in Carbonsäure/Wasser-Gemischen
Es wurden Mischungen von Wasser mit Essigsäure und Propionsäure untersucht Eine
Auswertung der Dichteänderung von Gelen in Propionsäure wurde aufgrund der recht
geringen Dichtevariation der Propionsäure/Wasser-Mischungen nicht vorgenommen.
1.00
1.02
1.04
1.06
1.08
1.10
0 20 40 60 80 100
Volumenanteil Wasser in Lösung / %
Dic
hte
/gcm
-3
GelLösung
11
12
13
14
15
16
17
18
0 20 40 60 80 100
Volumenanteil Wasser in Lösung / %
Que
llvol
umen
/mlg
-1
Abb.25: Kurvenverläufe für Essigsäure/Wasser-Gemische.
10
11
12
13
14
15
16
17
18
19
0 20 40 60 80 100
Volumenanteil Wasser in Lösung / %
Que
llvol
umen
/mlg
-1
0.90
0.95
1.00
1.05
1.10
0 20 40 60 80 100
Volumenanteil Wasser in Lösung / %
Dic
hte
/gcm
-3
Lösung
Abb.26: Kurvenverläufe für Propionsäure/Wasser-Gemische.
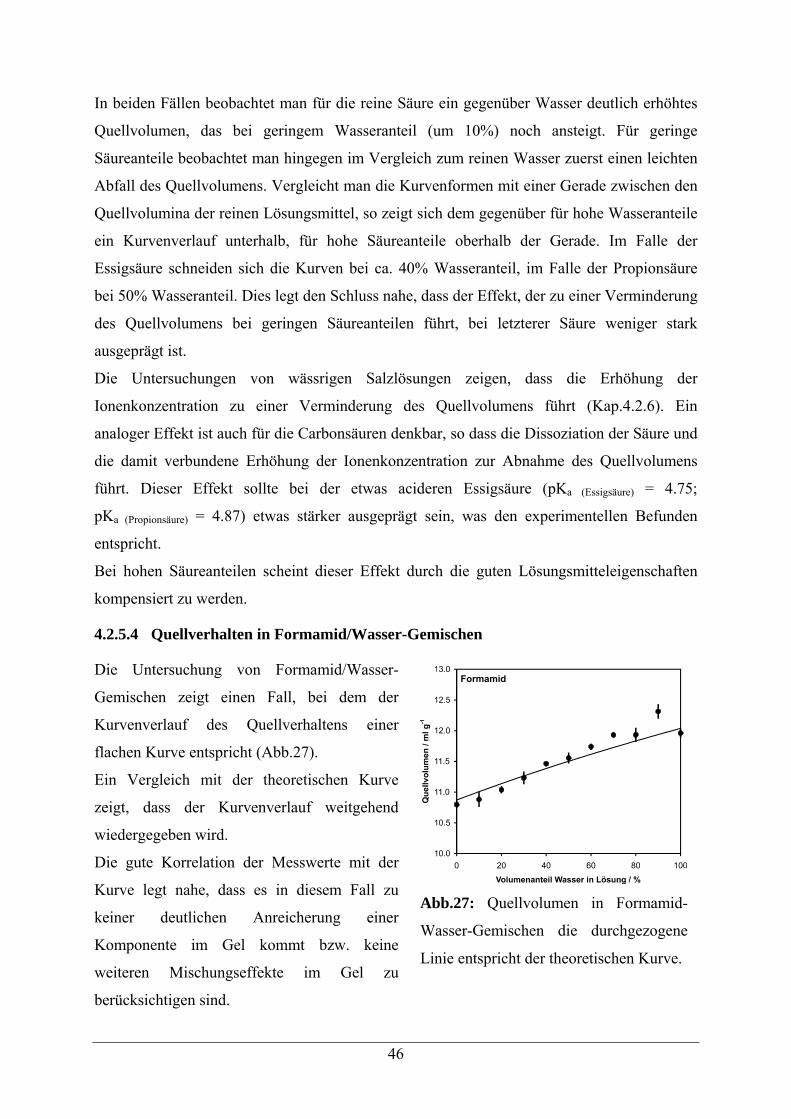
46
In beiden Fällen beobachtet man für die reine Säure ein gegenüber Wasser deutlich erhöhtes
Quellvolumen, das bei geringem Wasseranteil (um 10%) noch ansteigt. Für geringe
Säureanteile beobachtet man hingegen im Vergleich zum reinen Wasser zuerst einen leichten
Abfall des Quellvolumens. Vergleicht man die Kurvenformen mit einer Gerade zwischen den
Quellvolumina der reinen Lösungsmittel, so zeigt sich dem gegenüber für hohe Wasseranteile
ein Kurvenverlauf unterhalb, für hohe Säureanteile oberhalb der Gerade. Im Falle der
Essigsäure schneiden sich die Kurven bei ca. 40% Wasseranteil, im Falle der Propionsäure
bei 50% Wasseranteil. Dies legt den Schluss nahe, dass der Effekt, der zu einer Verminderung
des Quellvolumens bei geringen Säureanteilen führt, bei letzterer Säure weniger stark
ausgeprägt ist.
Die Untersuchungen von wässrigen Salzlösungen zeigen, dass die Erhöhung der
Ionenkonzentration zu einer Verminderung des Quellvolumens führt (Kap.4.2.6). Ein
analoger Effekt ist auch für die Carbonsäuren denkbar, so dass die Dissoziation der Säure und
die damit verbundene Erhöhung der Ionenkonzentration zur Abnahme des Quellvolumens
führt. Dieser Effekt sollte bei der etwas acideren Essigsäure (pKa (Essigsäure) = 4.75;
pKa (Propionsäure) = 4.87) etwas stärker ausgeprägt sein, was den experimentellen Befunden
entspricht.
Bei hohen Säureanteilen scheint dieser Effekt durch die guten Lösungsmitteleigenschaften
kompensiert zu werden.
4.2.5.4 Quellverhalten in Formamid/Wasser-Gemischen
Die Untersuchung von Formamid/Wasser-
Gemischen zeigt einen Fall, bei dem der
Kurvenverlauf des Quellverhaltens einer
flachen Kurve entspricht (Abb.27).
Ein Vergleich mit der theoretischen Kurve
zeigt, dass der Kurvenverlauf weitgehend
wiedergegeben wird.
Die gute Korrelation der Messwerte mit der
Kurve legt nahe, dass es in diesem Fall zu
keiner deutlichen Anreicherung einer
Komponente im Gel kommt bzw. keine
weiteren Mischungseffekte im Gel zu
berücksichtigen sind.
Abb.27: Quellvolumen in Formamid-
Wasser-Gemischen die durchgezogene
Linie entspricht der theoretischen Kurve.
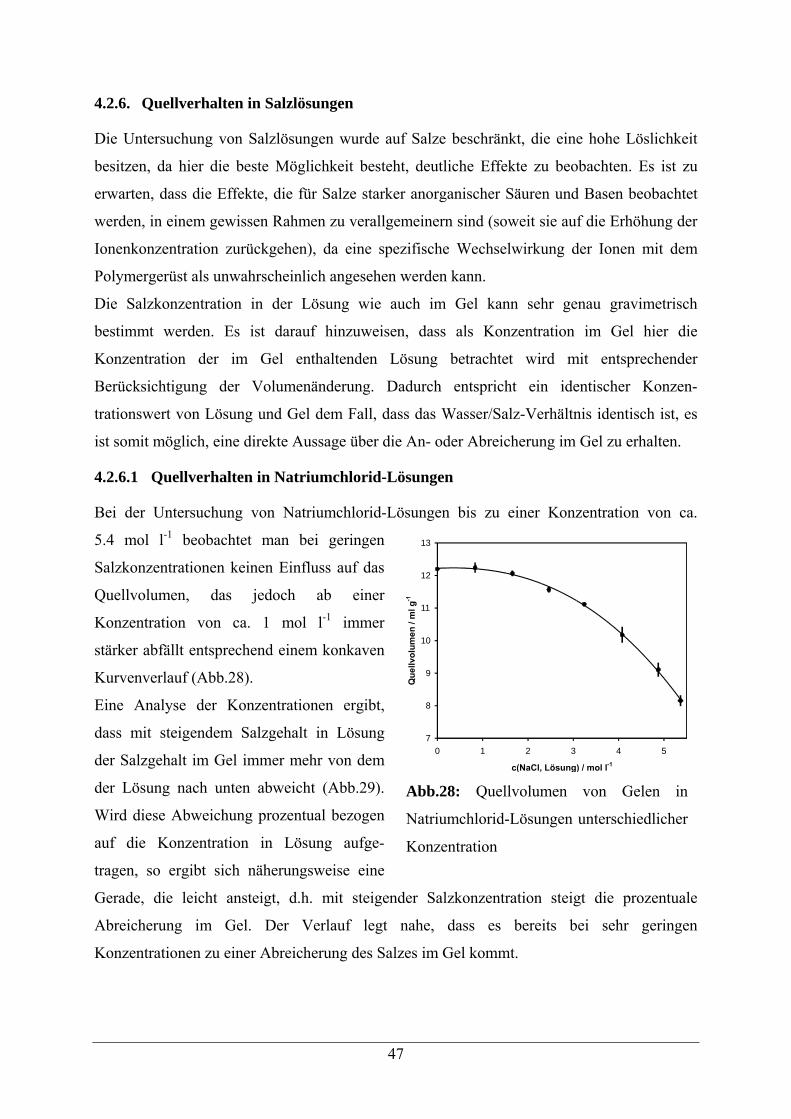
47
4.2.6. Quellverhalten in Salzlösungen
Die Untersuchung von Salzlösungen wurde auf Salze beschränkt, die eine hohe Löslichkeit
besitzen, da hier die beste Möglichkeit besteht, deutliche Effekte zu beobachten. Es ist zu
erwarten, dass die Effekte, die für Salze starker anorganischer Säuren und Basen beobachtet
werden, in einem gewissen Rahmen zu verallgemeinern sind (soweit sie auf die Erhöhung der
Ionenkonzentration zurückgehen), da eine spezifische Wechselwirkung der Ionen mit dem
Polymergerüst als unwahrscheinlich angesehen werden kann.
Die Salzkonzentration in der Lösung wie auch im Gel kann sehr genau gravimetrisch
bestimmt werden. Es ist darauf hinzuweisen, dass als Konzentration im Gel hier die
Konzentration der im Gel enthaltenden Lösung betrachtet wird mit entsprechender
Berücksichtigung der Volumenänderung. Dadurch entspricht ein identischer Konzen-
trationswert von Lösung und Gel dem Fall, dass das Wasser/Salz-Verhältnis identisch ist, es
ist somit möglich, eine direkte Aussage über die An- oder Abreicherung im Gel zu erhalten.
4.2.6.1 Quellverhalten in Natriumchlorid-Lösungen
Bei der Untersuchung von Natriumchlorid-Lösungen bis zu einer Konzentration von ca.
5.4 mol l-1 beobachtet man bei geringen
Salzkonzentrationen keinen Einfluss auf das
Quellvolumen, das jedoch ab einer
Konzentration von ca. 1 mol l-1 immer
stärker abfällt entsprechend einem konkaven
Kurvenverlauf (Abb.28).
Eine Analyse der Konzentrationen ergibt,
dass mit steigendem Salzgehalt in Lösung
der Salzgehalt im Gel immer mehr von dem
der Lösung nach unten abweicht (Abb.29).
Wird diese Abweichung prozentual bezogen
auf die Konzentration in Lösung aufge-
tragen, so ergibt sich näherungsweise eine
Gerade, die leicht ansteigt, d.h. mit steigender Salzkonzentration steigt die prozentuale
Abreicherung im Gel. Der Verlauf legt nahe, dass es bereits bei sehr geringen
Konzentrationen zu einer Abreicherung des Salzes im Gel kommt.
7
8
9
10
11
12
13
0 1 2 3 4 5
c(NaCl, Lösung) / mol l-1
Que
llvol
umen
/mlg
-1
Abb.28: Quellvolumen von Gelen in
Natriumchlorid-Lösungen unterschiedlicher
Konzentration
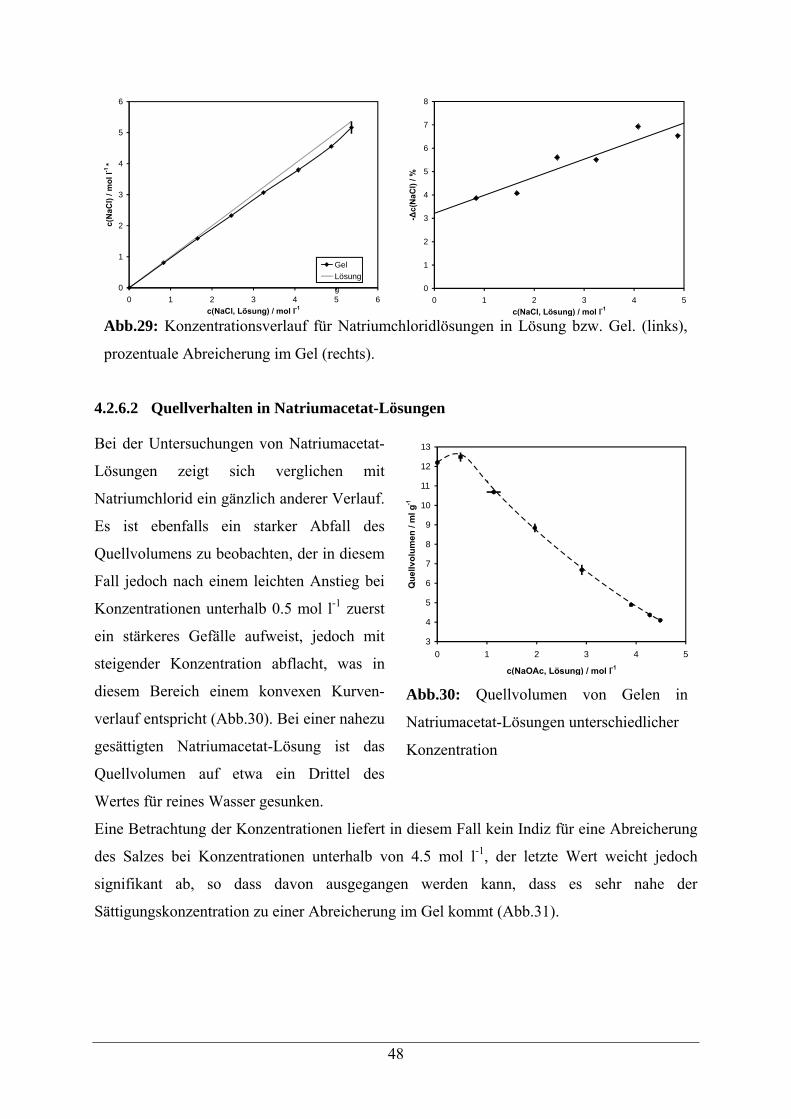
48
4.2.6.2 Quellverhalten in Natriumacetat-Lösungen
Bei der Untersuchungen von Natriumacetat-
Lösungen zeigt sich verglichen mit
Natriumchlorid ein gänzlich anderer Verlauf.
Es ist ebenfalls ein starker Abfall des
Quellvolumens zu beobachten, der in diesem
Fall jedoch nach einem leichten Anstieg bei
Konzentrationen unterhalb 0.5 mol l-1 zuerst
ein stärkeres Gefälle aufweist, jedoch mit
steigender Konzentration abflacht, was in
diesem Bereich einem konvexen Kurven-
verlauf entspricht (Abb.30). Bei einer nahezu
gesättigten Natriumacetat-Lösung ist das
Quellvolumen auf etwa ein Drittel des
Wertes für reines Wasser gesunken.
Eine Betrachtung der Konzentrationen liefert in diesem Fall kein Indiz für eine Abreicherung
des Salzes bei Konzentrationen unterhalb von 4.5 mol l-1, der letzte Wert weicht jedoch
signifikant ab, so dass davon ausgegangen werden kann, dass es sehr nahe der
Sättigungskonzentration zu einer Abreicherung im Gel kommt (Abb.31).
0
1
2
3
4
5
6
0 1 2 3 4 5 6c(NaCl, Lösung) / mol l-1
c(N
aCl)
/mol
l-1*
GelLösungg 0
1
2
3
4
5
6
7
8
0 1 2 3 4 5c(NaCl, Lösung) / mol l-1
-∆c(
NaC
l)/%
Abb.29: Konzentrationsverlauf für Natriumchloridlösungen in Lösung bzw. Gel. (links),
prozentuale Abreicherung im Gel (rechts).
3
4
5
6
7
8
9
10
11
12
13
0 1 2 3 4 5
c(NaOAc, Lösung) / mol l-1
Que
llvol
umen
/mlg
-1
Abb.30: Quellvolumen von Gelen in
Natriumacetat-Lösungen unterschiedlicher
Konzentration
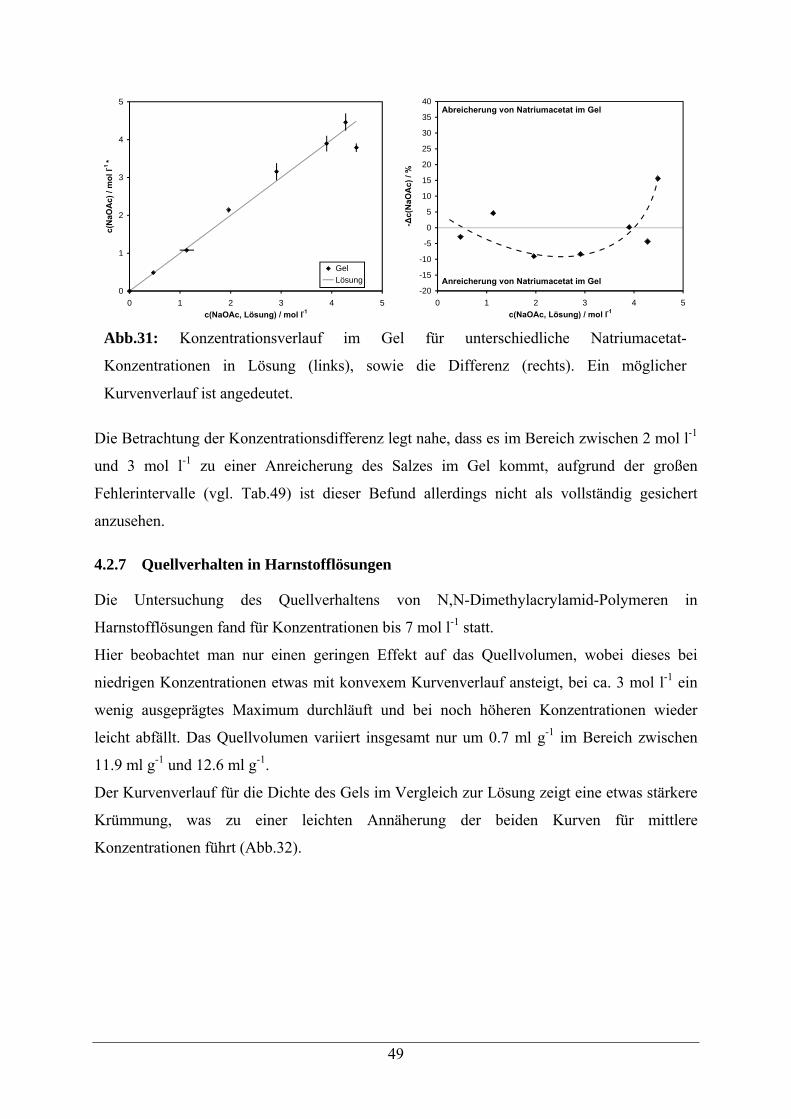
49
Die Betrachtung der Konzentrationsdifferenz legt nahe, dass es im Bereich zwischen 2 mol l-1
und 3 mol l-1 zu einer Anreicherung des Salzes im Gel kommt, aufgrund der großen
Fehlerintervalle (vgl. Tab.49) ist dieser Befund allerdings nicht als vollständig gesichert
anzusehen.
4.2.7 Quellverhalten in Harnstofflösungen
Die Untersuchung des Quellverhaltens von N,N-Dimethylacrylamid-Polymeren in
Harnstofflösungen fand für Konzentrationen bis 7 mol l-1 statt.
Hier beobachtet man nur einen geringen Effekt auf das Quellvolumen, wobei dieses bei
niedrigen Konzentrationen etwas mit konvexem Kurvenverlauf ansteigt, bei ca. 3 mol l-1 ein
wenig ausgeprägtes Maximum durchläuft und bei noch höheren Konzentrationen wieder
leicht abfällt. Das Quellvolumen variiert insgesamt nur um 0.7 ml g-1 im Bereich zwischen
11.9 ml g-1 und 12.6 ml g-1.
Der Kurvenverlauf für die Dichte des Gels im Vergleich zur Lösung zeigt eine etwas stärkere
Krümmung, was zu einer leichten Annäherung der beiden Kurven für mittlere
Konzentrationen führt (Abb.32).
0
1
2
3
4
5
0 1 2 3 4 5c(NaOAc, Lösung) / mol l-1
c(N
aOA
c)/m
oll-1
*
GelLösung
-20
-15
-10
-5
0
5
10
15
20
25
30
35
40
0 1 2 3 4 5c(NaOAc, Lösung) / mol l-1
-∆c(
NaO
Ac)
/%
Anreicherung von Natriumacetat im Gel
Abreicherung von Natriumacetat im Gel
Abb.31: Konzentrationsverlauf im Gel für unterschiedliche Natriumacetat-
Konzentrationen in Lösung (links), sowie die Differenz (rechts). Ein möglicher
Kurvenverlauf ist angedeutet.
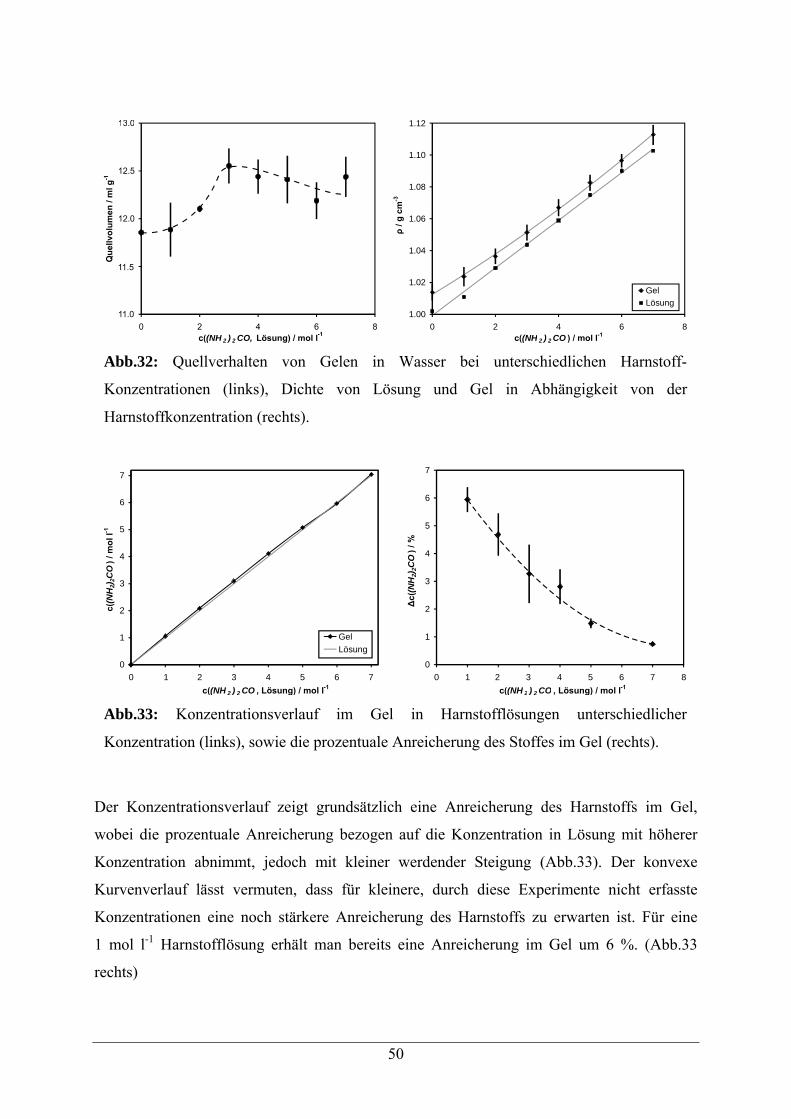
50
Der Konzentrationsverlauf zeigt grundsätzlich eine Anreicherung des Harnstoffs im Gel,
wobei die prozentuale Anreicherung bezogen auf die Konzentration in Lösung mit höherer
Konzentration abnimmt, jedoch mit kleiner werdender Steigung (Abb.33). Der konvexe
Kurvenverlauf lässt vermuten, dass für kleinere, durch diese Experimente nicht erfasste
Konzentrationen eine noch stärkere Anreicherung des Harnstoffs zu erwarten ist. Für eine
1 mol l-1 Harnstofflösung erhält man bereits eine Anreicherung im Gel um 6 %. (Abb.33
rechts)
1.00
1.02
1.04
1.06
1.08
1.10
1.12
0 2 4 6 8c((NH 2 ) 2 CO ) / mol l-1
ρ/g
cm-3
GelLösung
c((NH 2 ) 2 CO, Lösung) / mol l-1 Abb.32: Quellverhalten von Gelen in Wasser bei unterschiedlichen Harnstoff-
Konzentrationen (links), Dichte von Lösung und Gel in Abhängigkeit von der
Harnstoffkonzentration (rechts).
0
1
2
3
4
5
6
7
0 1 2 3 4 5 6 7c((NH 2 ) 2 CO , Lösung) / mol l-1
c((N
H2) 2
CO
)/m
oll-1
GelLösung
0
1
2
3
4
5
6
7
0 1 2 3 4 5 6 7 8
∆c(
(NH
2) 2C
O)/
%
c((NH 2 ) 2 CO , Lösung) / mol l-1 Abb.33: Konzentrationsverlauf im Gel in Harnstofflösungen unterschiedlicher
Konzentration (links), sowie die prozentuale Anreicherung des Stoffes im Gel (rechts).

51
Diese Ergebnisse sind von Belang für die Frage nach einer geeigneten
Dehybridisierungsmethode für immobilisierte Oligonukleotide. Die Anreicherung von
Harnstoff im Gel hat zur Folge, dass Harnstoff schwierig durch Spülen wieder aus dem Gel
entfernt werden kann. Daher ist Harnstoff in diesem Kontext offenbar weniger gut geeignet.
4.2.8 Vergleichende Zusammenfassung der Ergebnisse
Das Quellverhalten quervernetzter N,N-Dimethylacrylamid-Polymere in unterschiedlichen
Lösungsmittelgemischen und Lösungen ergibt ein sehr vielfältiges und komplexes Bild,
wobei man sowohl positive als auch negative Abweichungen vom idealen Verhalten teilweise
recht großen Ausmaßes finden kann.
Für die untersuchten Lösungsmittelgemische wurde nur im Fall von Formamid/Wasser eine
gute Korrelation mit dem theoretisch vorhergesagten Kurvenverlauf gefunden. Von allen
untersuchten Lösungsmittelgemischen sind hier die χ1-Werte am ähnlichsten, was
möglicherweise als eine Erklärung für das Verhalten angesehen werden kann.
Gerade für die Carbonsäure-Wasser-Gemische kann das theoretische Modell ohnehin nur
äußerst eingeschränkt gelten, da die Deprotonierung nicht berücksichtigt wird.
Negative Abweichungen gehen in vielen aber nicht allen Fällen mit einer Anreicherung einer
Komponente im Gel gegenüber der Lösung einher, was sowohl für Salzlösungen als auch für
Lösungsmittelgemische beobachtet werden konnte.
Die Abweichungen bewegen sich in einem breiten Bereich. So konnte das Schrumpfen des
Gels auf die Hälfte des Volumens in relaxiertem Zustand Vm ebenso beobachtet werden wie
auch ein weiteres Quellen bis auf 180% von Vm. Eine weitere relevante Beobachtung ist die
An- bzw. Abreicherung bestimmter Komponenten im Gel. Gerade die unspezifische Bindung
kann zu signifikanten Konzentrationsänderungen im Gel und damit zu unerwünschten
Effekten führen. Eine Anreicherung von Harnstoff z.B. würde ebenso wie eine Abreicherung
von Natriumchlorid zu einer Herabsetzung der DNA-Schmelzpunkte führen.
Bezüglich der Frage nach geeigneten Danaturierungsreagenzien für Oligonukleotid-Dimere
erwies sich in dieser Untersuchung Formamid als am indifferentesten in Bezug auf die
Polymer-Matrix.

52
5 Eigenschaften modifizierter quervernetzter N,N-
Dimethylacrylamid-Polymere
5.1 Acrylamid -Copolymere
Die Einführung von Modifikationen am Gel erfolgt durch die Copolymerisation von N,N-
Dimethylacrylamid, einem Quervernetzer und geeigneten Acrylamiden oder Acrylsäureestern,
die die gewünschte Funktionalität tragen oder deren Funktionalitäten nach der Polymerisation
durch weitere chemische Transformationen in die gewünschten überführt werden können.[35]
Dabei kommen sowohl Schutzgruppen zum Einsatz, als auch Reste, die noch mehrstufige
Umsetzungen zum gewünschten End-
produkt erfordern.
Das allgemeine Schema ist in
Abbildung 34 aufgeführt.
Die Umsetzungen zum Monomer
erfolgen durch klassische Synthese,
während die Umsetzungen nach der
Polymerisation eher einer Festphasen-
synthese entsprechen - mit allen Vor-
und Nachteilen, die dies beinhaltet. Als
Vorteile sind in erster Linie die
einfache Entfernung der Reagenzien,
die auch in hohem Überschuss
eingesetzt werden können, zu nennen,
als auch die Tatsache, dass das Produkt
nicht isoliert werden muss. Die Nachteile sind die schlechte Überprüfbarkeit der Ausbeuten,
die Schwierigkeit, Nebenprodukte zu entfernen, als auch die Beschränkung der möglichen
Reaktionsführung: Es kommt nur ein kleiner Temperaturbereich in Frage, die Wahl des
Lösungsmittels ist auf jene beschränkt, in denen das Polymer quillt, ebenso sind nicht
homogene Systeme problematisch. Bezüglich der Quelleigenschaften des Polymers ist zu
berücksichtigen, dass gegenüber der klassischen Festphasensynthese die Funktionalitäten
nicht nur an der Oberfläche, sondern über das ganze Volumen des Polymers verteilt sind
O
R
O
N
O
NH
NH
O
R
R'
+ +
Polymer
Polymer
klassische Flüssigphasensynthese
Festphasensynthese
Copolymerisation
(1) (2) (3)
Abb.34: Allgemeines Schema zur Einführung
funktioneller Gruppen in N,N-Dimethylacryl-
amid-Copolymeren.

53
H
O
NH2
NH
R
O
R
O
NH2
SH
NH2R
H
N NH
RO
H
NH
R
NH
S
O
R
OH
O NH2R O
NH
R
NH2
NH
RO
RH
O
NH
R
NH
O
NH2
O
NH
NR
HR
H
O
R COOH
SH SS RR SH
NH
O O
O NH
O O
N RNH2R
Polymer
Aldehyd (aliphatisch, aromatisch)
a)
b) Reduktion
Polymer
Polymer
Polymer
Polymer
Carbonsäure
Kopplungs-reagenzien
Polymer
Polymer
primäres Amin
Kopplungs-reagenzien
Polymer
a)
b) ReduktionPolymer
Polymer
Hydrazid
Polymer
Polymer
Thiol
Polymera) PySSPy
b)
Polymer Polymer
Abb.35: Überblick über potentielle kovalente
Immobilisierungsstrategien.
(Abb.9). Dadurch gewinnt die Frage
nach den Quelleigenschaften eine
zusätzliche Relevanz, weil bei einem
geringen Quellvolumen nur geringe
Mengen eines Reaktanden in das
Innere des Polymers gelangen können.
Zusätzlich ist gegenüber den ober-
flächenfunktionalisierten Polymeren
der Transport der Reaktanden zu den
polymergebundenen reaktiven Zentren
deutlich verlangsamt, wodurch viele
typische Festphasenreaktionen mit
reaktiven Spezies und genau abge-
stimmten Reaktionszeiten wie z.B. das
Phosphoamiditverfahren hier nicht
ohne weiteres übertragbar sind. Durch
die Reaktion von Reagenzien mit
reaktiven Gruppen des Polymers
kommt es während des Transports in
das Innere des Polymers leicht zu einer
Abreicherung der Reagenzien, was zu
weiteren Problemen führen kann.
Es wurden in dieser Arbeit verschiedene Funktionalitäten untersucht, die in unterschiedlichen
Immobilisierungsverfahren eingesetzt werden können.
Zusätzlich zur chemisch kovalenten Immobilisierung (Abb.35) besteht auch die Möglichkeit,
Makromoleküle wie z.B. Avidin physikalisch im Polymernetzwerk einzubinden, indem die
Polymerisation in Anwesenheit dieser Makromoleküle stattfindet und das engmaschige
Polymernetzwerk eine Diffusion aus dem Polymer verhindert. Dies lässt die Immobilisierung
biotinylierter Moleküle zu.
Eine weitere Möglichkeit der nicht-kovalenten Immobilisierung besteht in der kooperativen
Nutzung mittelstarker Wechselwirkungen, wobei diese eine resultierende starke
Wechselwirkung ergeben, die eine Immobilisierung ermöglicht. Um dies zu erreichen,
werden mehrere Bindungspartner an dem zu immobilisierenden Molekül verankert, was z.B.
durch Zuhilfenahme einer dendritischen Struktur erreicht werden kann (Abb.36).

54
Die in dieser Arbeit untersuchten Polymer-Modifikationen und die möglichen darauf
basierenden Immobilisierungsstrategien sind im Weiteren dargelegt.
5.2. Beladungsbestimmungen
Bei der Beladungsbestimmung von
quervernetzten Copolymeren ist zu beachten,
dass diese nur in gequollenem Zustand
erfolgen kann, da die im Inneren liegenden
Funktionalitäten nur so ausreichend zugäng-
lich sind. Das gegenüber Festphasen viel
ausgeprägtere Quellverhalten beinhaltet
jedoch eine Anzahl von Fehlerquellen. Das
Prinzip der meisten Beladungsbestimmungen
ist, die Funktionalitäten A am Polymer mit
geeigneten Molekülen B quantitativ zur
Reaktion zu bringen. Im einfachsten Fall wird
die Abnahme der Konzentration an B in
Lösung gemessen. Entsprechend der Stöchio-
metrie der Reaktion kann die Beladung
bestimmt werden. Eine Bestimmung der
Beladung aus dieser Konzentrations-differenz
erwies sich im Fall der hier betrachteten
Polymer-Gele jedoch als sehr ungenau.
[B]Lsg.
[B]Lsg.
m(Gel)
m(Gel)
APolymer A BPolymerB+
APolymer A BPolymerB+
Abb.37: Schematische Kurvenverläufe
für die Beladungsbestimmung von Gelen.
(oben) irreversible Reaktion
(unten) Gleichgewichtsreaktion
c)a) b)
Abb.36: Möglichkeiten der Immobilisierung: a) klassische Immobilisierung, wie auch
Immobilisierung durch multiple Wechselwirkungen b) unter Verwendung einer
dendritischen Struktur, c) in linearer Anordnung.

55
Dies kann je nach betrachtetem System unterschiedliche Ursachen haben. Zum einen ist die
unspezifische An- oder Abreicherung des Moleküls B im Polymer-Gel möglich, dasselbe gilt
für die benutzten Lösungsmittel im Fall von Lösungsmittelgemischen, was Auswirkungen auf
die verwendeten Messsignale hat (z.B. sind Extinktionskoeffizienten lösungsmittelsensitiv).
Um trotz dieser teilweise nur schwer zu quantifizierenden Effekte eine genaue
Beladungsbestimmung zu erreichen, wurde ein Verfahren verwendet, bei dem Polymer-
Fragmente unterschiedlicher Masse mit Lösungen des Reagenzes B von identischem
Volumen und identischer Konzentration [B]° umgesetzt werden.
Im Idealfall bildet die Auftragung der Polymer-Masse gegen die Konzentration des
Reagenzes B eine Gerade. Die Beladung kann einfach aus dem Schnittpunkt mit der m(Gel)-
Achse ermittelt werden, da hier die absolute Beladung der eingesetzten Stoffmenge an B
entspricht (Abb.37).
Dieser Schnittpunkt kann auch dann recht genau bestimmt werden, wenn die Punkte stark
streuen oder aufgrund von unspezifischer Bindung systematisch von den theoretischen
abweichen.
Ein weiterer Vorteil dieser Methode ist darin zu sehen, dass sich aus dem Kurvenverlauf die
Eignung des verwendeten Systems ablesen lässt. Handelt es sich um eine Kurve, die für
höhere Polymer-Massen abflacht und sich asymptotisch der m(Gel)-Achse annähert, so kann
dies zwei Gründe haben. Zum einen ist es möglich, dass die Reaktionszeit zu kurz war um
eine vollständige Diffusion in das Gel zu ermöglichen. Eine andere Möglichkeit ist, dass es
sich um eine Gleichgewichtsreaktion handelt. Diese beiden Möglichkeiten sind durch
Variation der Reaktionszeit einfach zu unterscheiden, wodurch dieses Verfahren eine einfache
Methode zur ersten Validierung einer angestrebten Immobilisierungsstrategie darstellt.
Hierbei sei darauf hingewiesen, dass z.B. die Hydrazid-Aldehyd-Verknüpfung zwar eine
gängige und kommerziell erhältliche Methode zur Verknüpfung von Biomolekülen ist, jedoch
gezeigt werden konnte, dass ihre Anwendbarkeit durch Hydrolyse deutlich eingeschränkt ist,
wenn nicht ein aromatischer Aldehyd zum Einsatz kommt.[54]

56
5.3 Copolymere mit Aldehyd-Modifikation
5.3.1 Synthesestrategie
Für die Einführung einer Aldehyd-Modifikation wurden drei verschiedene geschützte
Acrylamide synthetisiert (Abb.38.).
Die Verwendung von (4) zu diesem Zweck ist in der Literatur beschrieben. Das Acrylamid
kann durch eine literaturbekannte Synthese ausgehend von 1,2,6-Hexantriol (7) dargestellt
werden (Abb.39).[35]
Dieses wird in einer ersten Stufe in das 1,3-Dioxolan (8) überführt, woraufhin die freie
Hydroxy-Funktion zum Mesylat (9) umgesetzt wird. Die nukleophile Substitution mit
Lithiumazid in DMF führt zum Azid (10), welches zum Amin (11) reduziert werden kann. Im
Gegensatz zu der in der Literatur beschriebenen Staudinger-Reduktion mit
Triphenylphosphan wurde hier eine katalytische Hydrierung angewendet, die eine deutlich
einfachere Aufarbeitung ermöglicht und nahezu quantitativ verläuft.
Das erhaltene Amin wird im letzten Schritt mit Acryloylchlorid zum Acrylamid (4)
umgesetzt, das in der Copolymerisation eingesetzt werden kann.
NH
OO
O
NH
NH
O
O
O
ONH
OO
O
(4) (5) (6) Abb.38: Acrylamide zur Einführung einer Aldehyfunktion in ein Acrylamid-Copolymer.
OHO
O
OHOH
OHO
OO
OSO
OMsCl
OO
N3
LiN3
OO
NH2O
O
NH
O Cl
O
H2, Pd/C 10%
(7) (8) (9)
(4) (11) (10)
(12)
pTsOH
Abb.39: Synthese von (4).

57
Zur Freisetzung der gewünschten Aldehydfunktion sind weitere Umsetzungen des Gels nötig
(Abb.40):
Die Umsetzung mit Säure führt zur Acetalspaltung und zur Freisetzung des vicinalen Diols
am Polymer sowie zur Freisetzung von Aceton. Das vicinale Diol wird in einem zweiten
Schritt durch Oxidation mit Periodat gespalten, wobei zum einen das freie Aldehyd-Gel (P4),
zum anderen Formaldehyd entsteht.
Sowohl Aceton als auch Formaldehyd müssen vor einer Verwendung vollständig aus dem
Polymer entfernt werden.
Im Experiment zeigte das mit (4) gebildete Copolymerisat nach Freisetzung der
Aldehydfunktion gute Immobilisierungseigenschaften bei der Anbindung von 5’-Hydrazid-
Oligonukleotiden (vgl. Kap.5.3.3), jedoch ist sowohl die Synthese des Monomers aufwendig
als auch die Umsetzung des Polymers umständlich, da mit Aceton und Formaldehyd zwei
potentiell bei der Immobilisierung störende Nebenprodukte entstehen.
Die einfachste Möglichkeit, ein geschütztes Aldehyd-Acrylamid darzustellen, besteht in einer
einstufigen Synthese ausgehend vom kommerziell erhältlichen Aminoacetaldehyd-
diethylacetal (13), welches durch Umsetzung mit Acryloylchlorid in 5%iger
Natriumhydrogencarbonatlösung in das gewünschte Acrylamid (5) überführt wird. Das
Produkt kann durch Extraktion mit Dichlormethan in reiner Form isoliert werden kann. Nach
HNH
O O
PolymerOH
OH
NH
O
PolymerO
O
NH
O
Polymer
O
H H
O
AcOH NaIO3
(P2) (P3) (P4)
Abb.40: Freisetzung der Aldehydfunktion aus dem Copolymer von (4) und N,N-
Dimethylacrylamid.
NH2
O
ONH
OO
O
Cl
O
ONH
O
O
ONH
O
H
5% NaHCO3 in H2O
Polymer Polymer
(13) (5)
(P5) (P6)
AcOH
(12)
Abb.41: Synthese von (5) und Freisetzung der Aldehydfunktion im Copolymer.

58
Copolymerisation erfolgt die Freisetzung der Aldehydfunktion durch Acetalspaltung im
Sauren (Abb.41).
Die Hydrazon-Bildung ist eine Gleichgewichtsreaktion. Auch wenn diese Chemie
kommerziell zur Immobilisierung oder Verknüpfung von Biomolekülen verwendet wird,
haben Untersuchungen gezeigt, dass die Hydrolyse in einem erheblichen Maße stattfindet.[54]
Da jedoch die Stabilität aromatischer Hydrazone deutlich größer ist als diejenige der
aliphatischen Vertreter, wurde neben den vorgestellten aliphatischen Aldehyden auch eine
aromatische Aldehyd-Gel-Modifikation entwickelt.
Dabei wird von (14) ausgegangen. Die Esterfunktion wird durch Aminolyse mit
Ethylendiamin (15) in ein Amid überführt. Die freie Aminofunktion in (16) kann in der
letzten Stufe mit Acryloylchlorid zum Acrylamid (6) umgesetzt werden, welches in Wasser
jedoch schlecht löslich ist.
Daher ist es notwendig, die Copolymerisation mit N,N-Dimethylacrylamid aus einem
DMF/Wasser-Gemisch (1:3 v/v) durchzuführen.
Nach Entfernung des organischen Lösungsmittels aus dem Polymer wird die Aldehydfunktion
durch Umsetzung mit Säure freigesetzt.
NH
NH
O
O
O
OO
O
O
ONH
NH2
O
O
O Cl
O
NH2
NH2
NH
O
O
H
NH
O
PolymerNH
O
O
O
NH
O
Polymer
O
O
O
H
(6)(17)(15)
(P7)
AcOH
(P8)
(16) (12)
(14)
HC(OMe)3p-TSOH
Abb.42: Synthese des aromatischen acetalgeschützten Aldehyd-Acrylamids und
Freisetzung der Funktionalität nach der Polymerisation.

59
5.3.2 Beladungsbestimmung
Die Beladung der Aldehyd-funktionalisierten Copolymere wurde durch die Reaktion mit
2,4-Dinitrophenylhydrazin (17) bestimmt. Da die Färbung des Polymer-Gels nicht ohne
weiteres ausgewertet werden kann, wurde für die Bestimmung die UV-Absorption der Lösung
herangezogen, entsprechend der oben beschriebenen Methode.
Da in diesen Experimenten - um eine eventuelle Veränderung des Polymers auszuschließen -
die Gelfragmente vor der Umsetzung nicht getrocknet wurden, ist trotz Korrektur mit
geringfügig unterschiedlichen Lösungsmittelzusammensetzungen und Konzentrationen zu
rechnen.
Zusätzlich ist bezüglich der Hydrazon-Bildung die Reversibilität zu beachten, wobei die
Hydrolyse besonders für aliphatische Aldehyde beobachtet wird. Ohne Zugabe von
Reduktionsmittel wurde in diesen Fällen keine Entfärbung der Lösung auch bei Überschuss
von Aldehydfunktionen gefunden. Durch Reduktion des Hydrazons mit
Natriumcyanoborhydrid kann das Gleichgewicht jedoch auf die Seite des Addukts verschoben
werden und man beobachtet bei Überschuss an Aldehydfunktionen eine vollständige
Entfärbung der Lösung.
N,N-Dimethylacrylamid-2,2-Diethoxyethylacrylamid-Copolymer (P6)
Die Analyse des Copolymers mit dem einfachsten untersuchten Aldehyd-Baustein (5) zeigte
den in Abb.44 abgebildeten Verlauf. Die theoretische Beladung des in Wasser gequollenen
Polymers beträgt 91 µmol g-1. Nach Zugabe von 100 µl ethanolischer 2,4-Dinitro-
phenylhydrazin-Lösung (c = 1.0045 g l-1) und 0.5 ml der Lösung von 75 mg Natrium-
cyanoborhydrid in 7.5 ml 3%iger Essigsäure (pH = 2.7)
NNH
NO2
NO2
PolymerO
H
Polymer NH
NH
NO2
NO2
Polymer
NH2
NH
NO2
NO22,4-DNPH
Na(CN)BH3
pH = 2,7(18)
Abb.43: Allgemeines Schema der Anfärbung von Aldehyd-funktionalisierten
Copolymeren mit 2,4-Dinitrophenylhydrazin.
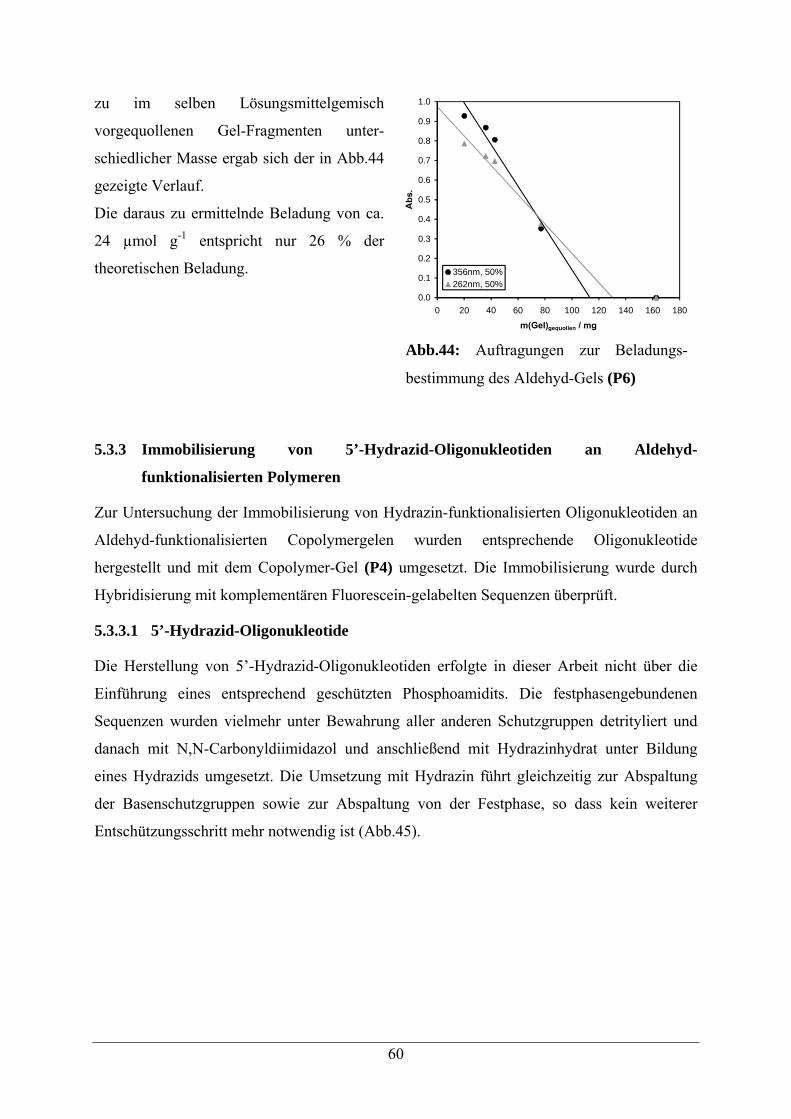
60
zu im selben Lösungsmittelgemisch
vorgequollenen Gel-Fragmenten unter-
schiedlicher Masse ergab sich der in Abb.44
gezeigte Verlauf.
Die daraus zu ermittelnde Beladung von ca.
24 µmol g-1 entspricht nur 26 % der
theoretischen Beladung.
5.3.3 Immobilisierung von 5’-Hydrazid-Oligonukleotiden an Aldehyd-
funktionalisierten Polymeren
Zur Untersuchung der Immobilisierung von Hydrazin-funktionalisierten Oligonukleotiden an
Aldehyd-funktionalisierten Copolymergelen wurden entsprechende Oligonukleotide
hergestellt und mit dem Copolymer-Gel (P4) umgesetzt. Die Immobilisierung wurde durch
Hybridisierung mit komplementären Fluorescein-gelabelten Sequenzen überprüft.
5.3.3.1 5’-Hydrazid-Oligonukleotide
Die Herstellung von 5’-Hydrazid-Oligonukleotiden erfolgte in dieser Arbeit nicht über die
Einführung eines entsprechend geschützten Phosphoamidits. Die festphasengebundenen
Sequenzen wurden vielmehr unter Bewahrung aller anderen Schutzgruppen detrityliert und
danach mit N,N-Carbonyldiimidazol und anschließend mit Hydrazinhydrat unter Bildung
eines Hydrazids umgesetzt. Die Umsetzung mit Hydrazin führt gleichzeitig zur Abspaltung
der Basenschutzgruppen sowie zur Abspaltung von der Festphase, so dass kein weiterer
Entschützungsschritt mehr notwendig ist (Abb.45).
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
0 20 40 60 80 100 120 140 160 180
m(Gel)gequollen / mg
Abs
.
356nm, 50%262nm, 50%
Abb.44: Auftragungen zur Beladungs-
bestimmung des Aldehyd-Gels (P6)

61
Es ist bekannt, dass die Umsetzung mit Hydrazin zur Abspaltung von Cytosin führen kann,
weshalb eine kurze und milde Reaktionsführung notwendig ist. Nach der Entfernung
niedermolekularer Bestandteile konnte für DNA1a kein Produkt mit abgespaltenem Cytosin
durch MALDI-TOF nachgewiesen werden.
5.3.3.2 Immobilisierung von Hydrazid-Oligonukleotiden an Aldehyd-Gelen
Die Immobilisierung zur Ermittlung der grundsätzlichen Eignung erfolgte in
Testexperimenten durch Pipettierung von wenigen µl der DNA-Lösungen in Aqua bidest. auf
das gequollene Polymer, sowohl unter Verwendung des Aldehyd-Gels als auch unter
Verwendung eines unmodifizierten Gels als Referenz. Nach der vollständigen Absorption
(30 min) des Flüssigkeitsüberstandes wurde das Polymer mit Phosphatpuffer (0.1 mol l-1,
pH 7, 1 mol l-1 NaCl) gespült.
5.3.3.3 Hybridisierungsexperimente
Die Hybridisierung erfolgte mit einem zu einem der beiden immobilisierten Oligonucleotiden
komplementären 5’-Fluorescein-funktionalisierten 14mer (3ml DNA2-Lösung (10 µmol l-1) in
0.1 mol l-1 Phosphatpuffer pH 7.0, 1 mol l-1 NaCl). Nach 2 h bei 8°C wurde die Fluoreszenz
der Gele überprüft (Abb.47a). Während man im Falle des unmodifizierten Gels nur eine
N N
O
N N
O
ODNA
3'5' OH
O
ODNA
3'5' O
NO N
OHDNA
3'5' O
NHO
NH2
DNA3'
5'
N2H4 * H2O
a b (DNA1a)(DNA1b)
Festphase Festphase
= 5'CTTACCA3' ; 5'TGGTAAG3'
(19)
Abb.45: Synthese von 5’-Hydrazid-modifizierten Oligonukleotiden.
DNA3'
5'
a b= 5'CTTACCA3' ; 5'TGGTAAG3'
OHDNAO
NH O
NH23'
5'
ONH
O H
NNH
NH
O HO
OOH
DNA
(DNA1a)(DNA1b)
(P4)
Polymer3'
5'
(P9a)(P9b)
Polymer
Abb.46: Immobilisierung von Hydrazid-Oligonukleotiden an Aldehyd-Gelen.

62
Hintergrundfluoreszenz fand, zeigte sich bei dem Aldehydgel erwartungsgemäß eine starke
Fluoreszenz an der Position von DNA1a und eine Abnahme der Hintergrundfluoreszenz an
der Position von DNA1b.
Nach der Denaturierung mit 6 mol l-1 Harnstoff findet man an der Position von DNA1a keine
erhöhte Fluoreszenz mehr, die Abnahme bei DNA1b ist jedoch immer noch zu beobachten
(Abb.47b).
DNA1aDNA1b
Aldehyd-Gel
Referenz-Gel
Aldehyd-Gel
b) Denaturierung
a) Hybridisierung
0 5 10 mm
DNA23'GAATGGTACCTAAG5' Fl
5'TGGTAAG3'DNA1b
DNA1a 5'CTTACCA3'
3'GAATGGTACCTAAG5' Fl
5'TGGTAAG3'DNA1b
DNA1a 5'CTTACCA3'
0 5 10 mm
3 ml -Lösung (10 µmol lDNA2 -1) in 0.1 mol l-1 Phosphatpuffer pH7.0, 1 mol l-1 NaCl, 2h bei 8°C
Das unmodifizierte Referenz-Gel wurde analog dem Aldehyd-Gel mit und behandelt.
Es ist keine unspezifische Anbindung an das Gel zu erkennen.DNA1a DNA1b
An den Positionen, an denen immobilisiert ist, erkennt man eineAbnahme der Grundfluoreszenz.
DNA1b
10 ml 6 mol l Harnstoff, 12h-1
Während keine erhöhte Fluoreszenz mehr an der Position von zu erkennen ist, beobachtet man immer noch die Abnahme der Grund-fluoreszenz an den Positionen von .
DNA1a
DNA1b
Abb.47: a) Hybridisierung des 5’-Fluorescein-markierten Oligonucleotids DNA2 mit
immobilisierten 5’Hydrazid-Oligonucleotiden. b) Denaturierung mit Harnstoff.
Fluoreszenzbilder wurden bei 490nm aufgenommen (Breitband-UV-Anregung).
Fluoreszenz ist dunkel dargestellt. Der Kontrast wurde zur besseren Erkennbarkeit deutlich
erhöht.

63
5.4 Copolymere mit Carboxylat-Modifikation
5.4.1 Synthesestrategie
Bei der Synthese von N,N-Dimethylacrylamid-Copolymeren mit Carboxylat-Modifikation ist
als einfachste Variante die Copolymerisation mit Acrylsäure denkbar. Um einen homogenen
Einbau zu gewährleisten, wurde hier jedoch auf die ebenfalls literaturbekannte
Copolymerisation von N,N-Dimethyl-
acrylamid (2) mit Glycolacrylat (20)
zurückgegriffen.[70] Die Esterfunktion
kann nach der Polymerisation durch eine
Reihe von Resten substituiert werden,
z.B. durch Aminolyse oder Hydra-
zinolyse (Abb.48).
Für die Überführung in die Carboxyl-
Funktion wird das Copolymer 16 h mit
10%iger Natronlauge behandelt, darauf-
hin mit Wasser neutral gewaschen,
weitere 16 h mit 1mol l-1 Salzsäure
behandelt und wieder neutral
gewaschen.
5.4.2 Eigenschaften
Das Acrylsäure-N,N-Dimthylacrylamid-Copolymer (P12) zeigt vor der Behandlung mit
Salzsäure in wässriger Lösung ein deutlich stärker ausgeprägtes Quellverhalten als in der
freien Säureform nach der Behandlung mit Salzsäure. Dies ist dadurch zu erklären, dass die
Kationen nicht das Gel verlassen können, da dies zu einer Ladungstrennung führen würde,
aber dennoch im Wasser gelöst sind, was zu einem Osmose-ähnlichen Prozess führt (ΠIon >>
0), der das starke Quellen des Gels bedingt.
Diese Eigenschaft von Gelen mit kovalent gebundenen Ladungsträgern wird durch den Term
ΠIon beschrieben:
ΠIon = RT(Σci,gelzi,gel - Σci,solzi,sol)i i (32)
An dieser Stelle sei angemerkt, dass es sich bei vielen technisch relevanten Super-Absorbern
um quervernetzte Polyacrylat-Polymere handelt.[55,56] Das Quellverhalten der Acrylsäure-
OHO
O
N
OO
OOH
NH
O
NH
O
OH
O
Polymer NH
ONH2Polymer
NH2NH
O
Polymer
NH2
NH2 N2H4 H2O.
Polymer
Copolymerisation
NaOH / H2O
(20)
(3)(2)
(P10)
(P11) (P12) (P13)
(16)
Abb.48: Einführung von Modifikationen
ausgehend vom Gylcolacrylatbaustein.

64
N,N-Dimethylacrylamid-Copolymere ist gegenüber dem der Acrylate deutlich weniger stark
ausgeprägt, was auf die unvollständige Dissoziation der Carboxylatfunktionalitäten
zurückgeführt werden kann.
So beträgt das Quellvolumen des Polymers 14.2 ml g-1 gegenüber 11.8 ml g-1 für das
unmodifizierte Polymer, ist also (verglichen mit den Quelleigenschaften der Acrylate) nicht
wesentlich größer. Wird angenommen, dass die Eigenschaften bezüglich der Wechselwirkung
des Polymers mit Wasser durch die N,N-Dimethylpolyacrylamidmatrix bestimmt werden und
dass die Carboxylatreste lediglich einen ionischen Beitrag entsprechend ihres
Deprotonierungsgrades beitragen, lässt sich der pKs-Wert der gelgebundenen
Säurefunktionen abschätzen. Da ein Gleichgewichtszustand vorliegt, gilt:
ΠIon = - (ΠM + ΠE) (40)
Mit den Werten für das unmodifizierte Polymer lässt sich ΠIon berechnen. Unter
Vernachlässigung der Autoprotolyse des Wassers gilt gemäß (32):
ΠIon = RT([H+]gel + [A-]gel) (41)
Der pKs-Wert ist definiert durch:
pKs = - log[H+][A-]
[HA]
(42)
Dabei entspricht A den Carboxylatresten im Gel.
Durch Kombination von (41) und (42) erhält man:
pKs = - log1
[HA]
ΠIon
2RT
2
(43)
Entnimmt man [HA] der Beladung der Gele umgerechnet auf den gequollenen Zustand und
berechnet ΠIon, so erhält man pKs = 4.16. Dieser Wert ist verhältnismäßig niedrig, da für
polymergebundene Carbonsäurefunktionen eine geringere Säurestärke verglichen mit
Carbonsäuren in Lösung erwartet wird.[42,57-60]
Die Tatsache, dass die Säurestärke nach dieser Berechnung verhältnismäßig hoch ist, geht
möglicherweise auf eine Verfälschung des Ergebnisses zurück, die auf der Vernachlässigung
der Wechselwirkungen der Säurefunktionen im protonierten Zustand mit Wasser beruhen.
Zur Untersuchung der Beladung wurde ein der Titration ähnliches Verfahren gewählt. Da eine
klassische Titration aufgrund der langsamen Gleichgewichtseinstellung nicht möglich ist,
wurden getrocknete Copolymere unterschiedlicher Masse mit Natronlauge identischer
Konzentration umgesetzt. Nach einer entsprechend langen Wartezeit wurde der pH-Wert der
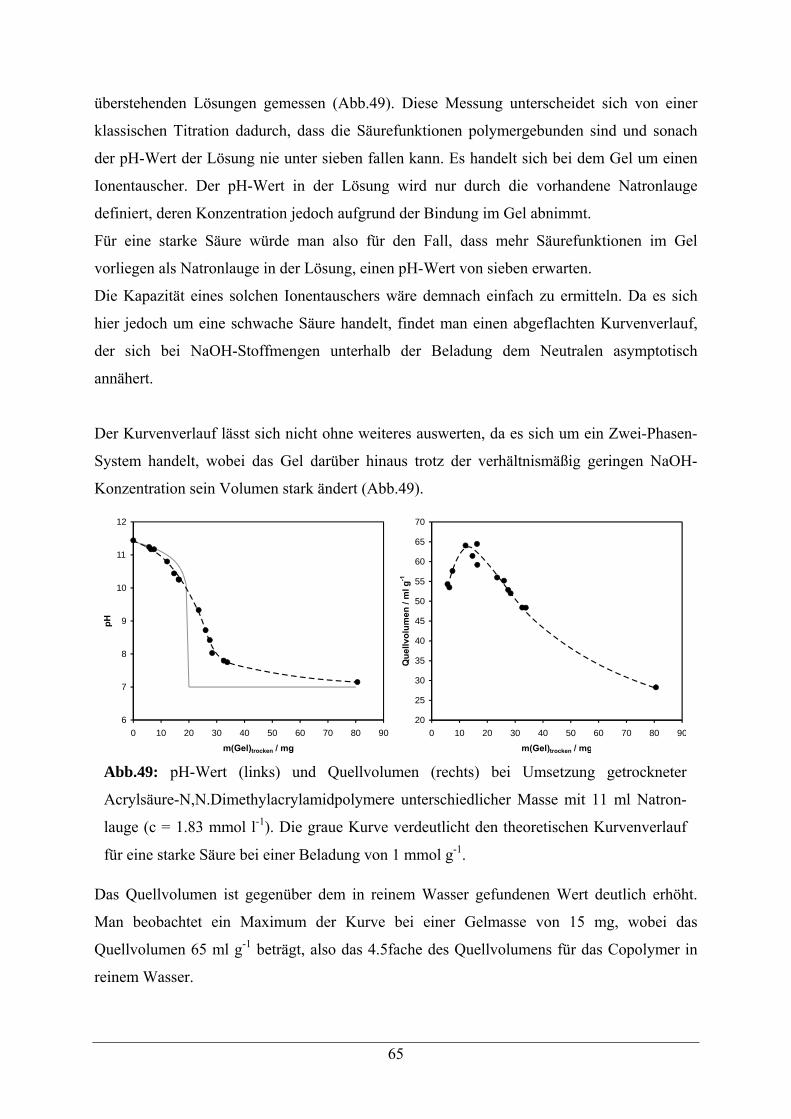
65
überstehenden Lösungen gemessen (Abb.49). Diese Messung unterscheidet sich von einer
klassischen Titration dadurch, dass die Säurefunktionen polymergebunden sind und sonach
der pH-Wert der Lösung nie unter sieben fallen kann. Es handelt sich bei dem Gel um einen
Ionentauscher. Der pH-Wert in der Lösung wird nur durch die vorhandene Natronlauge
definiert, deren Konzentration jedoch aufgrund der Bindung im Gel abnimmt.
Für eine starke Säure würde man also für den Fall, dass mehr Säurefunktionen im Gel
vorliegen als Natronlauge in der Lösung, einen pH-Wert von sieben erwarten.
Die Kapazität eines solchen Ionentauschers wäre demnach einfach zu ermitteln. Da es sich
hier jedoch um eine schwache Säure handelt, findet man einen abgeflachten Kurvenverlauf,
der sich bei NaOH-Stoffmengen unterhalb der Beladung dem Neutralen asymptotisch
annähert.
Der Kurvenverlauf lässt sich nicht ohne weiteres auswerten, da es sich um ein Zwei-Phasen-
System handelt, wobei das Gel darüber hinaus trotz der verhältnismäßig geringen NaOH-
Konzentration sein Volumen stark ändert (Abb.49).
Das Quellvolumen ist gegenüber dem in reinem Wasser gefundenen Wert deutlich erhöht.
Man beobachtet ein Maximum der Kurve bei einer Gelmasse von 15 mg, wobei das
Quellvolumen 65 ml g-1 beträgt, also das 4.5fache des Quellvolumens für das Copolymer in
reinem Wasser.
6
7
8
9
10
11
12
0 10 20 30 40 50 60 70 80 90
m(Gel)trocken / mg
pH
20
25
30
35
40
45
50
55
60
65
70
0 10 20 30 40 50 60 70 80 90
m(Gel)trocken / mg
Que
llvol
umen
/mlg
-1
Abb.49: pH-Wert (links) und Quellvolumen (rechts) bei Umsetzung getrockneter
Acrylsäure-N,N.Dimethylacrylamidpolymere unterschiedlicher Masse mit 11 ml Natron-
lauge (c = 1.83 mmol l-1). Die graue Kurve verdeutlicht den theoretischen Kurvenverlauf
für eine starke Säure bei einer Beladung von 1 mmol g-1.
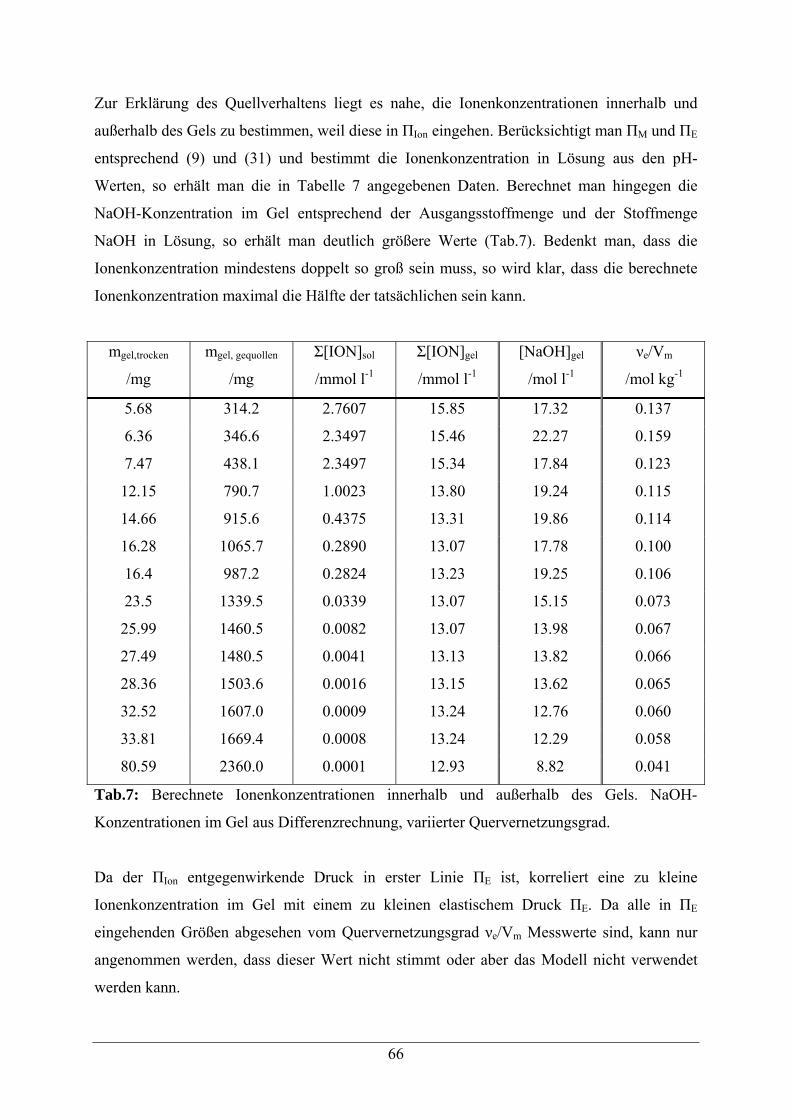
66
Zur Erklärung des Quellverhaltens liegt es nahe, die Ionenkonzentrationen innerhalb und
außerhalb des Gels zu bestimmen, weil diese in ΠIon eingehen. Berücksichtigt man ΠM und ΠE
entsprechend (9) und (31) und bestimmt die Ionenkonzentration in Lösung aus den pH-
Werten, so erhält man die in Tabelle 7 angegebenen Daten. Berechnet man hingegen die
NaOH-Konzentration im Gel entsprechend der Ausgangsstoffmenge und der Stoffmenge
NaOH in Lösung, so erhält man deutlich größere Werte (Tab.7). Bedenkt man, dass die
Ionenkonzentration mindestens doppelt so groß sein muss, so wird klar, dass die berechnete
Ionenkonzentration maximal die Hälfte der tatsächlichen sein kann.
mgel,trocken
/mg
mgel, gequollen
/mg
Σ[ION]sol
/mmol l-1
Σ[ION]gel
/mmol l-1
[NaOH]gel
/mol l-1
νe/Vm
/mol kg-1
5.68 314.2 2.7607 15.85 17.32 0.137
6.36 346.6 2.3497 15.46 22.27 0.159
7.47 438.1 2.3497 15.34 17.84 0.123
12.15 790.7 1.0023 13.80 19.24 0.115
14.66 915.6 0.4375 13.31 19.86 0.114
16.28 1065.7 0.2890 13.07 17.78 0.100
16.4 987.2 0.2824 13.23 19.25 0.106
23.5 1339.5 0.0339 13.07 15.15 0.073
25.99 1460.5 0.0082 13.07 13.98 0.067
27.49 1480.5 0.0041 13.13 13.82 0.066
28.36 1503.6 0.0016 13.15 13.62 0.065
32.52 1607.0 0.0009 13.24 12.76 0.060
33.81 1669.4 0.0008 13.24 12.29 0.058
80.59 2360.0 0.0001 12.93 8.82 0.041
Tab.7: Berechnete Ionenkonzentrationen innerhalb und außerhalb des Gels. NaOH-
Konzentrationen im Gel aus Differenzrechnung, variierter Quervernetzungsgrad.
Da der ΠIon entgegenwirkende Druck in erster Linie ΠE ist, korreliert eine zu kleine
Ionenkonzentration im Gel mit einem zu kleinen elastischem Druck ΠE. Da alle in ΠE
eingehenden Größen abgesehen vom Quervernetzungsgrad νe/Vm Messwerte sind, kann nur
angenommen werden, dass dieser Wert nicht stimmt oder aber das Modell nicht verwendet
werden kann.
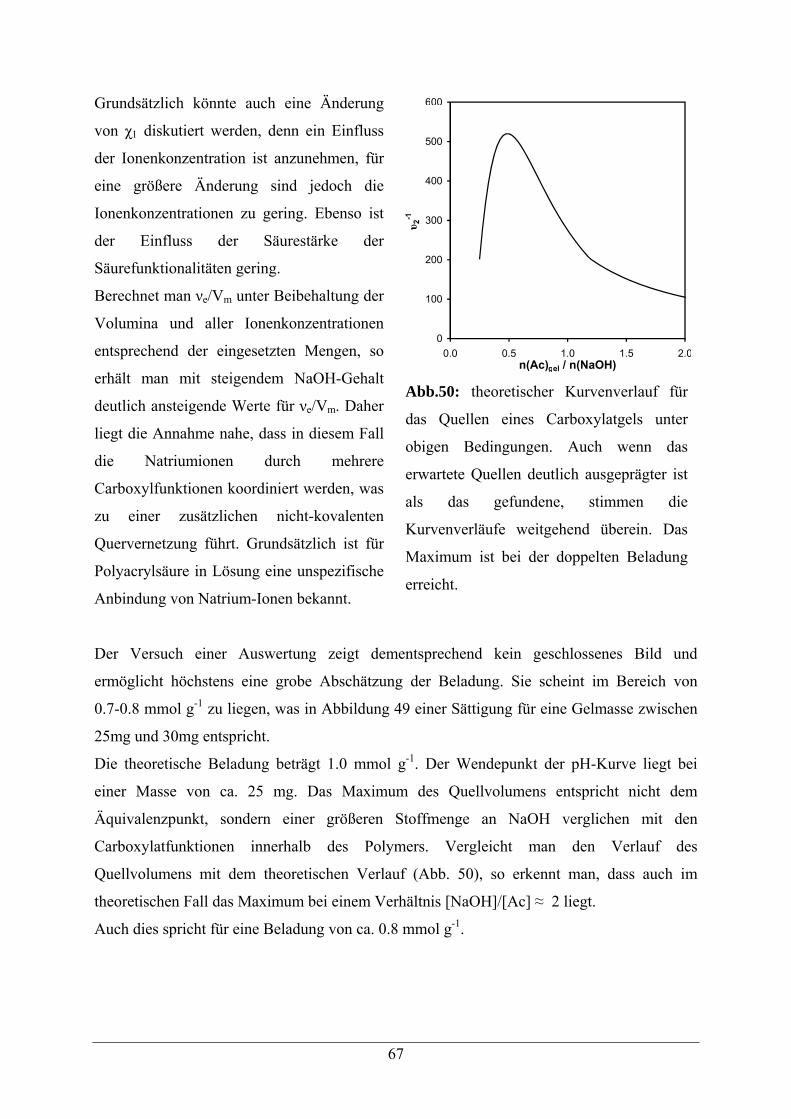
67
Grundsätzlich könnte auch eine Änderung
von χ1 diskutiert werden, denn ein Einfluss
der Ionenkonzentration ist anzunehmen, für
eine größere Änderung sind jedoch die
Ionenkonzentrationen zu gering. Ebenso ist
der Einfluss der Säurestärke der
Säurefunktionalitäten gering.
Berechnet man νe/Vm unter Beibehaltung der
Volumina und aller Ionenkonzentrationen
entsprechend der eingesetzten Mengen, so
erhält man mit steigendem NaOH-Gehalt
deutlich ansteigende Werte für νe/Vm. Daher
liegt die Annahme nahe, dass in diesem Fall
die Natriumionen durch mehrere
Carboxylfunktionen koordiniert werden, was
zu einer zusätzlichen nicht-kovalenten
Quervernetzung führt. Grundsätzlich ist für
Polyacrylsäure in Lösung eine unspezifische
Anbindung von Natrium-Ionen bekannt.
Der Versuch einer Auswertung zeigt dementsprechend kein geschlossenes Bild und
ermöglicht höchstens eine grobe Abschätzung der Beladung. Sie scheint im Bereich von
0.7-0.8 mmol g-1 zu liegen, was in Abbildung 49 einer Sättigung für eine Gelmasse zwischen
25mg und 30mg entspricht.
Die theoretische Beladung beträgt 1.0 mmol g-1. Der Wendepunkt der pH-Kurve liegt bei
einer Masse von ca. 25 mg. Das Maximum des Quellvolumens entspricht nicht dem
Äquivalenzpunkt, sondern einer größeren Stoffmenge an NaOH verglichen mit den
Carboxylatfunktionen innerhalb des Polymers. Vergleicht man den Verlauf des
Quellvolumens mit dem theoretischen Verlauf (Abb. 50), so erkennt man, dass auch im
theoretischen Fall das Maximum bei einem Verhältnis [NaOH]/[Ac] ≈ 2 liegt.
Auch dies spricht für eine Beladung von ca. 0.8 mmol g-1.
Abb.50: theoretischer Kurvenverlauf für
das Quellen eines Carboxylatgels unter
obigen Bedingungen. Auch wenn das
erwartete Quellen deutlich ausgeprägter ist
als das gefundene, stimmen die
Kurvenverläufe weitgehend überein. Das
Maximum ist bei der doppelten Beladung
erreicht.

68
5.5 Copolymere mit Amino-Modifikation
5.5.1 Synthesestrategie
Für die Einführung einer Amino-Modifikation wurden zwei unterschiedliche Wege
beschritten, die (bezüglich der Bausteine) zu einem analogen Polymer führen.
Der eine Weg basiert auf der Einführung des Acrylamids (21), das eine primäre
Aminofunktion als Hydrochlorid trägt. Der Baustein kann durch Reaktion von
Acryloylchlorid mit Ethylendiamin erhalten werden (Abb.51). Dieses Verfahren zur
Einführung einer Aminofunktion ist in der Literatur beschrieben.[70]
Allerdings lässt sich das einmal ausgefällte Salz schlecht wieder in wässrige Lösung bringen,
was den Einsatz als Monomer in einer Copolymerisation schwierig gestaltet.
Ein weiterer Weg, um zu einem analogen Polymer zu gelangen, besteht in der Aminolyse
eines N,N-Dimethylacrylamid-Glycolacrylat-Copolymers mit Ethylendiamin (Abb.52).
Diese Reaktion wird in Ethanol durchgeführt, um eine in Wasser mögliche basische
Verseifung des Esters als Nebenreaktion auszuschließen. Das erhaltene Polymer wird mit
Wasser bis zur neutralen Reaktion gespült.
Um einen quantitativen Kaisertest durchführen zu können, wurde das Gel getrocknet.[61] Die
Umsetzung mit den entsprechenden Reagenzien führt zu einer intensiv blauen Färbung, also
dem Nachweis der Aminfunktionalitäten. Jedoch verhindert die Tatsache, dass die Färbung zu
einem großen Teil im Gel lokalisiert ist, eine genaue quantitative Auswertung, weil die
relative Instabilität der Färbung eine langwierige Extraktion verhindert.[61]
Cl
ONH2
NH2 NH
NH3
OCl
+ -+
(12) (16) (21)
Abb.51: Synthese von N-Acryloyl-1,2-diaminoethan Hydrochlorid ()
NH2NH
O
Polymer
NH2
NH2
OHO
O
Polymer
(P10) (P11)
(16)
Abb.52: Generierung von Amino-funktionalisierten Gelen aus Glycolacrylat-
Copolymeren.

69
5.6 Copolymere mit Hydrazid-Modifikation
5.6.1 Synthesestrategie
Die Herstellung eines Polymers mit Hydrazid-Endgruppen erfolgt analog der beschriebenen
aminolytischen Darstellung von Amino-funktionalisierten Polymeren.
Die Hydrazinolyse eines N,N-Dimethylacrylamid-Glycolacrylat-Copolymers mit Hydrazin-
hydrat führt nach Entfernung überschüssigen Hydrazins mit Essigsäure und Spülen mit
Wasser zu dem gewünschten Polymer.
Während der Hydrazinolyse ist eine deutliche Abnahme des Quellvolumens auf weniger als
die Hälfte zu beobachten, auch das gespülte Hydrazid-Copolymer hat ein gegenüber dem
Ausgangspolymer herabgesetztes Quellvolumen von ca. 8.8 ml g-1.
5.6.2 Beladungsbestimmung
Die Beladungsbestimmung des Hydrazid-funktionalisierten Copolymers erfolgt durch
Umsetzung mit 4-Nitrobenzaldehyd (22). Die ebenfalls untersuchte Umsetzung mit 2,4-
Dinitrobenzaldehyd (23), das einen etwas höheren Extinktionskoeffizienten aufweist, führt
unter gleichen Bedingungen nur zu einer unvollständigen Reaktion.
Da 4-Nitrobenzaldehyd in Lösung farblos ist, das Hydrazon indes gelb, lässt sich die Reaktion
mit den polymergebundenen Hydrazid-Funktionalitäten auch optisch gut verfolgen.
Die Messung wurde mit unterschiedlichen Stoffmengen an Aldehyd durchgeführt.
Als Kontrolle wurde auch das Polymer ohne Glycolacrylat der Umsetzung mit Hydrazin
unterzogen und seine Beladung wurde analog untersucht.
N2H4 H2O.OH
O
O
Polymer NH
ONH2Polymer
(P10) (P13) Abb.53: Generierung von Hydrazid-funktionalisierten Gelen durch Hydrazinolyse von
Glycolacrylat-Copolymeren.
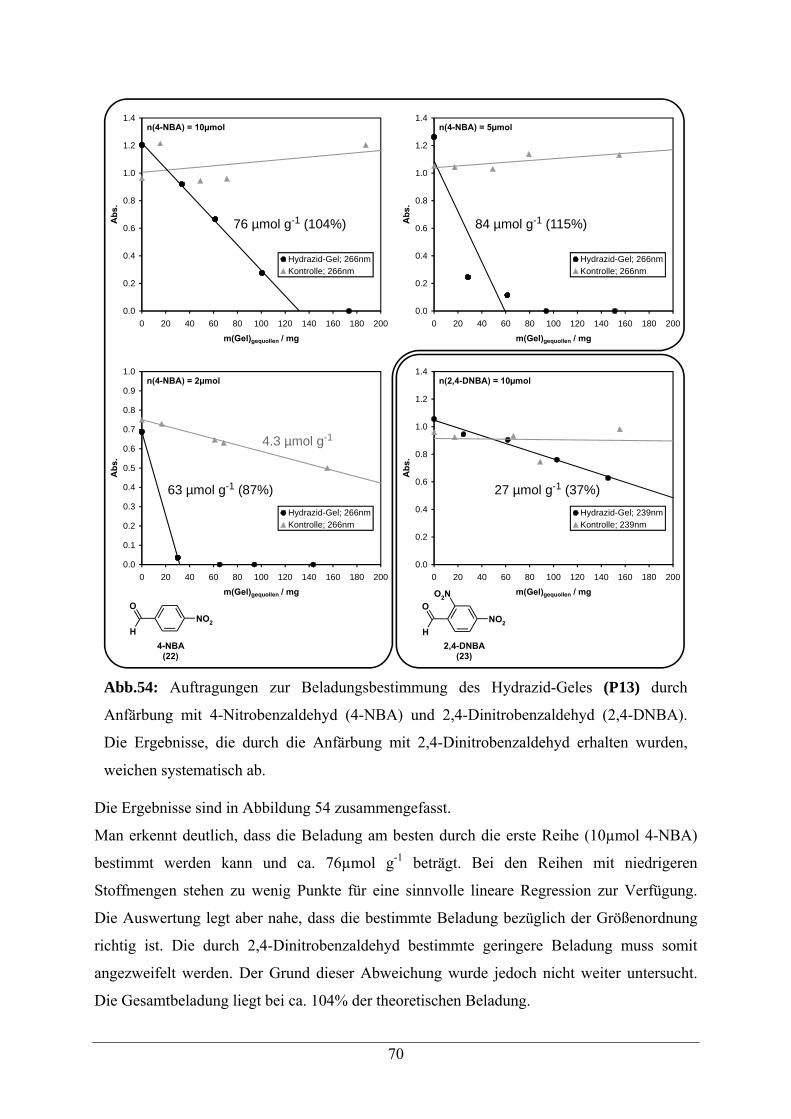
70
Die Ergebnisse sind in Abbildung 54 zusammengefasst.
Man erkennt deutlich, dass die Beladung am besten durch die erste Reihe (10µmol 4-NBA)
bestimmt werden kann und ca. 76µmol g-1 beträgt. Bei den Reihen mit niedrigeren
Stoffmengen stehen zu wenig Punkte für eine sinnvolle lineare Regression zur Verfügung.
Die Auswertung legt aber nahe, dass die bestimmte Beladung bezüglich der Größenordnung
richtig ist. Die durch 2,4-Dinitrobenzaldehyd bestimmte geringere Beladung muss somit
angezweifelt werden. Der Grund dieser Abweichung wurde jedoch nicht weiter untersucht.
Die Gesamtbeladung liegt bei ca. 104% der theoretischen Beladung.
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
0 20 40 60 80 100 120 140 160 180 200
m(Gel)gequollen / mg
Abs
.
Hydrazid-Gel; 239nmKontrolle; 239nm
n(2,4-DNBA) = 10µmol
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
0 20 40 60 80 100 120 140 160 180 200
m(Gel)gequollen / mg
Abs
.
Hydrazid-Gel; 266nmKontrolle; 266nm
n(4-NBA) = 2µmol
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
0 20 40 60 80 100 120 140 160 180 200
m(Gel)gequollen / mg
Abs
.
Hydrazid-Gel; 266nmKontrolle; 266nm
n(4-NBA) = 5µmol
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
0 20 40 60 80 100 120 140 160 180 200
m(Gel)gequollen / mg
Abs
.
Hydrazid-Gel; 266nmKontrolle; 266nm
n(4-NBA) = 10µmol
76 µmol g (104%)-1 84 µmol g (115%)-1
63 µmol g (87%)-1
4.3 µmol g-1
27 µmol g (37%)-1
O
HNO2
4-NBA(22)
O
H
O2N
NO2
2,4-DNBA(23)
Abb.54: Auftragungen zur Beladungsbestimmung des Hydrazid-Geles (P13) durch
Anfärbung mit 4-Nitrobenzaldehyd (4-NBA) und 2,4-Dinitrobenzaldehyd (2,4-DNBA).
Die Ergebnisse, die durch die Anfärbung mit 2,4-Dinitrobenzaldehyd erhalten wurden,
weichen systematisch ab.

71
Ein weiterer Befund ist, dass auch die als Kontrolle eingesetzten unmodifizierten Polymere
eine geringe Beladung aufweisen, die bei ca. 4.3µmol g-1 liegt. Diesem Befund entspricht
auch die Tatsache, dass auch diese Polymer-Gele sich gelb verfärbten, wobei diese Färbung
im Gegensatz zu den modifizierten Polymeren optisch nicht homogen erschien, sondern
vielmehr an den Ecken und Kanten intensiver war. Dies legt den Schluss nahe, dass die
Gruppen, die eine Modifikation des Polymers ermöglichen, in diesen Bereichen gehäuft
vorlagen. Diese makroskopische räumliche Anordnung kann aber nur ein Resultat der
Polymerisation sein, welche allen Beobachtungen nach im Inneren und nicht an den Kanten
der Kammer beginnt.
Von den Monomeren liegt N,N-Dimethylacrylamid in deutlichem Überschuss vor, so dass nur
eine An- oder Abreicherung des Quervernetzers an den Kanten denkbar wäre. Dies
wiederspricht jedoch der stärkeren Polymerisationsneigung niedriger N-substituierter
Acrylamide. Eine bessere Erklärung ist, dass im Laufe der Polymerisation alle Monomere mit
gleicher Wahrscheinlichkeit reagieren, mit dem Fortschreiten der Reaktion die
Monomerkonzentration aber immer mehr abnimmt. So ist es für einen zu Beginn
einpolymerisierten Quervernetzer sehr wahrscheinlich, dass die zweite Acrylamidfunktion
ebenfalls in eine Polymerkette eingebaut wird. Zum Ende der Reaktion stehen jedoch kaum
mehr freie Acrylamide zur Verfügung, so dass es wahrscheinlich ist, dass ein einmal
eingebauter Quervernetzer keinen weiteren Reaktionspartner findet. Dies widerspricht jedoch
wiederum dem experimentell bestimmten Quervernetzungsgrad.
Gegenüber Aminen weisen diese reaktiven Gruppen der Matrix eine höhere Reaktivität als die
Ester auf. Dies wäre nicht zu erwarten, wenn bei der Reaktion Dimethylamin verdrängt
würde, was teilweise in der Literatur als Begründung für die Reaktivität unmodifizierter
Polyacrylamide gegenüber Hydrazin angegeben wird.[35]
Eine Reduzierung der Hydrazinkonzentration bei der Reaktion (bis zu N2H2*H2O / Ethanol =
1:13 (v/v)) führt zu keiner Verhinderung der Matrixfunktionalisierung.
5.6.3 Immobilisierung von 5’-Aldehyd-Oligonukleotiden an Hydrazid-Gelen
5.6.3.1 Darstellung von 5’-Aldehyd-Oligonukleotiden
Die Darstellung von 5’-aldehydfunktionalisierten Oligonukleotiden erfolgt durch den Einbau
entsprechender Phosphoamidite beim Aufbau des Oligonukleotids im Rahmen der
automatisierten Festphasensynthese (Abb.55).[62-66]
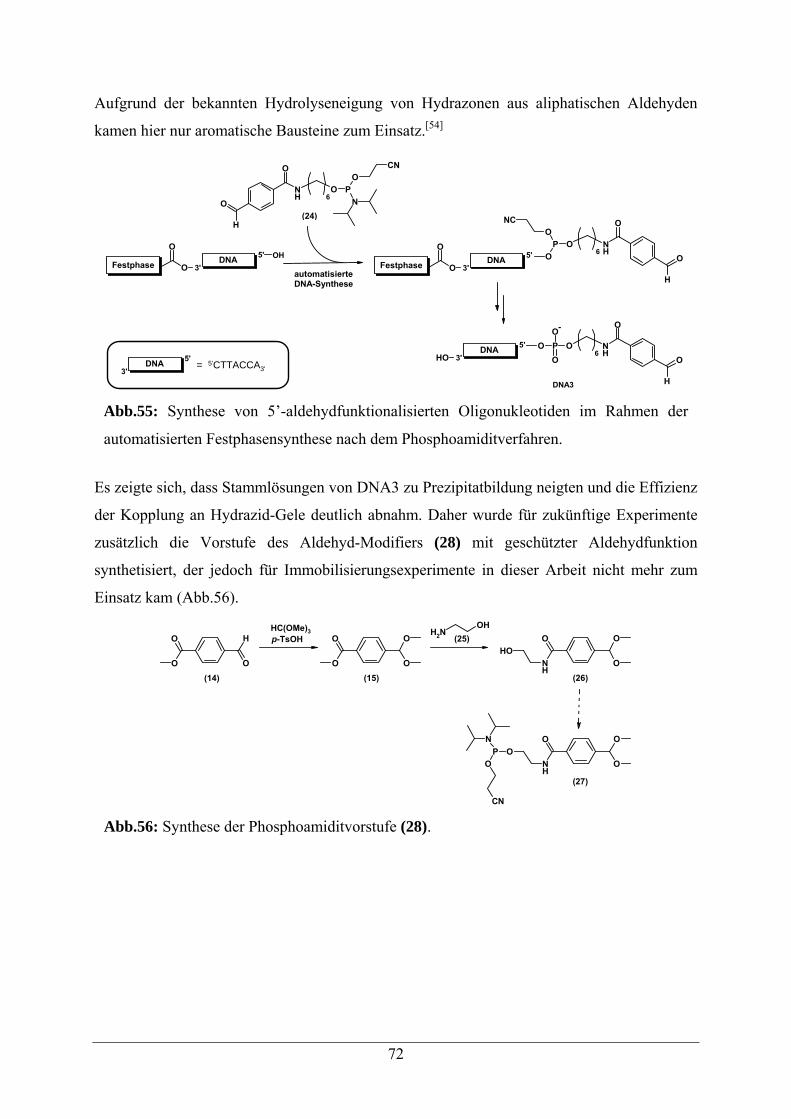
72
Aufgrund der bekannten Hydrolyseneigung von Hydrazonen aus aliphatischen Aldehyden
kamen hier nur aromatische Bausteine zum Einsatz.[54]
Es zeigte sich, dass Stammlösungen von DNA3 zu Prezipitatbildung neigten und die Effizienz
der Kopplung an Hydrazid-Gele deutlich abnahm. Daher wurde für zukünftige Experimente
zusätzlich die Vorstufe des Aldehyd-Modifiers (28) mit geschützter Aldehydfunktion
synthetisiert, der jedoch für Immobilisierungsexperimente in dieser Arbeit nicht mehr zum
Einsatz kam (Abb.56).
O
ODNA
3'5' OH
Festphase
PONH
O
O
H
N
OCN
6
O
ODNA
3'5'
P O NH
O
O
H
O
ONC
OHDNA
3'5' P O N
H
O
O
H
O
OO
6
-
DNA3'
5'
DNA3
Festphaseautomatisierte DNA-Synthese
6
(24)
= 5'CTTACCA3'
Abb.55: Synthese von 5’-aldehydfunktionalisierten Oligonukleotiden im Rahmen der
automatisierten Festphasensynthese nach dem Phosphoamiditverfahren.
O
HO
O O
OO
O O
OO
NH
OH
NH2OH
O
OO
NH
OPN
O
CN
HC(OMe)3p-TsOH
(14) (15) (26)
(27)
(25)
Abb.56: Synthese der Phosphoamiditvorstufe (28).

73
5.6.3.2 Immobilisierung von 5’-Aldehyd-Oligonukleotiden an Hydrazid-Gelen
Die Immobilisierung von 5’-Aldehyd-modifizierten Oligonukleotiden an Hydrazid-
funktionalisierte Gele erfolgte analog der Immobilisierung von 5’-Hydrazid-Oligonukleotiden
an Aldehyd-Gelen (Kap.5.3.3.2). Es wurden Volumina zwischen 0.5 und 2 µl von DNA3-
Lösungen mit Konzentrationen zwischen 0.06 und 1 mmol l-1 auf die Oberfläche des Gels
pipettiert. Nach der vollständigen Absorption (30 min) des Flüssigkeitsüberstandes wurde das
Polymer mit Phosphatpuffer (100 mM, pH 7, 1 M NaCl) gespült.
5.6.3.3 Hybridisierung und Denaturierung
Die Hybridisierung erfolgte mit einem 5’-Fluorescein-modifizierten 14mer (3ml DNA2-
Lösung (10 µmol l-1) in 0.1 mol l-1 Phosphatpuffer pH 7.0, 1 mol l-1 NaCl, 2h bei 20°C). Die
Bereiche, in denen DNA3 immobilisiert wurde, sind deutlich durch Fluoreszenz zu erkennen
(Abb. 58a).
Zur Untersuchung des zeitlichen Verlaufs der Denaturierung wurden hybridisierte
Gelfragmente mit Harnstofflösung (6 mol l-1) sowie mit Aqua bidest. versetzt und die
Veränderung verfolgt (Abb. 58b). Nach 1 h findet man im beiden Fällen verwaschene Ränder,
nach 2 h ist noch eine deutliche Fluoreszenz bei der Denaturierung mit Aqua bidest. zu
erkennen, im Fall von Harnstoff sind immer noch leichte Schatten erkennbar. Nach 16 h ist in
beiden Fällen keine lokalisierte Fluoreszenzerhöhung mehr zu erkennen.
Die Tatsache, dass die Dehybridisierung im Fall des reinen Wassers langsamer verläuft, ist
schon deshalb verständlich, weil hier erst die Verdünnung der im Gel vorhandenen Natrium-
Ionen soweit stattfinden muss, bis die verbliebene Salzkonzentration den DNA-Duplex nicht
mehr genügend stabilisiert. Demgegenüber fungiert Harnstoff als aktives
Denaturierungsreagenz, das auch bei höherer Salzkonzentration eine Denaturierung bewirkt.
OHDNAPON
H
O
O
H
O
OO
6
-
3'5'NH2N
H
O
OHDNAPON
H
O
N
H
O
OO
NH
O3'
5'6
-
Polymer DNA
DNA3
+Polymer
(P13)
(P14)
= 5'CTTACCA3'3'5'
Abb.57: Immobilisierung von 5’-Aldehyd-Oligonukleotiden an Hydrazid-Gelen.

74
Trotzdem ist auch bei der Dehybridisierung mit Harnstoff selbst nach 2 h noch eine lokale
Konzentrationserhöhung des Fluorescein-markierten Oligonukleotids nachweisbar.
Der Vorgang ist durch zwei Parameter bestimmt, zum einen die Diffusion des Salzes aus dem
Gel bzw. des Harnstoffs in das Gel, zum anderen durch die Diffusion des Fluorescein-
markierten Oligonukleotids aus dem Gel. Diese Diffusionsprozesse verlaufen offensichtlich
verhältnismäßig langsam, was eine der Voraussetzungen für die erfolgreiche Implementierung
b) Denaturierung
a) Hybridisierung
3 ml -Lösung (10 µmol lDNA2 -1) in 0.1 mol l-1Phosphatpuffer pH7.0, 1 mol l-1 NaCl, 2h bei 20°C
3 ml -Lösung (10 µmol lDNA2 -1) in 0.1 mol l-1 Phosphatpuffer pH7.0, 1 mol l-1 NaCl, 2h bei 20°C
0h
1h
2h
16h
Rehybridisierung:
Aqua bidest. Harnstoff(6 mol l )-1
t
0 5 10 mm
DNA23'GAATGGTACCTAAG5' Fl(P14)
DNA3 5'CTTACCA3'5'CTTACCA3'
3'GAATGGTACCTAAG5' FlDNA3
0.1 - 1 nmol DNA2 pro Spot
0h
1h
2h
16ht
Abb.58: a) Hybridisierung des 5’-Fluorescein-markierten Oligonucleotids DNA2 mit
immobilisierten 5’-Aldehyd-Oligonucleotiden. b) Zeitliche Verfolgung der Denaturierung.
Fluoreszenzbilder wurden bei 490 nm aufgenommen (Breitband-UV-Anregung).
Fluoreszenz ist dunkel dargestellt.

75
eines ElektroSpread-Verfahrens unter der Verwendung von modifizierten Polyacrylamidgelen
darstellt. Analog erschwert die langsame Diffusion die Nutzung von Hydrogelen als
Festphasen, wenn der Stofftransport geladener Teilchen nicht durch ein elektrisches Feld
unterstützt wird.
5.7 Copolymere mit Phenylboronsäure-Modifikation
Die Nutzung der spezifischen Bindung von Polyolen an Borsäure-Derivate für die Erkennung
und Trennung von Polyol-enthaltenden Molekülen ist seit langem bekannt. Polymere von
Phenylboronsäure-Derivaten werden in der Affinitätschromatographie verwendet, z.B. bei der
Trennung von DNA und RNA. Auf einem Boronsäure-modifizierten Trägermaterial wird
RNA aufgrund der enthaltenen Glycol-Einheiten stärker retardiert.[67,68]
Die Polymerisation in Gegenwart von Polyolen („Imprinting“) führt zur Ausbildung
spezifischer Bindungstaschen, die z.B. bei Verwendung chiraler Formen Enantiomere
unterschiedlich stark binden.[69,70,71]
Gegenstand aktueller Forschung ist die Nutzung von Phenylboronsäure-Acrylamid-
Copolymeren als Glucosesensoren.[41,43]
Die Tatsache, dass aliphatische Polyole eine gute Wasserlöslichkeit aufweisen und dabei
wenig Wechselwirkungen mit Oligonukleotiden erwarten lassen, prädestiniert das
Polyol/Phenylboronsäure-Hydrogel-System für die Anwendung im Rahmen einer
Immobilisierung durch kooperative schwache Wechselwirkungen.
5.7.1 Synthesestrategie
Für die Einführung von Phenylboronsäure-Modifikationen in N,N-Dimethylacrylamid-
Polymere wird ein entsprechend modifiziertes Boronsäure-Acrylamid durch die Umsetzung
von 3-Aminophenylboronsäure (28) mit Acryloylchlorid in wässriger Natriumh-
ydrogencarbonat-Lösung erhalten, wobei das Produkt nach einiger Zeit aus der Reaktions-
mischung ausfällt und nicht weiter gereinigt werden muss.
B NH2OH
OHB N
HOH
OH
OCl
O NH
BOH
OHO
Polymer+
(P15)(28) (12) (29) Abb.59: Synthese des Boronsäure-Acrylamids (29).

76
5.7.2 Polymerisation
Die Copolymerisation mit N,N-Dimethylacrylamid erfolgt in diesem Fall aufgrund der
schlechteren Löslichkeit der Boronsäureacrylamids aus einer Mischung von Wasser und DMF
(3:1 v/v), wobei zum Start der Polymerisation eine etwas höhere Konzentration von APS und
TMEDA eingesetzt wird (vgl. Kap.4.1). Das erhaltene Polymer zeigt in reinem Wasser ein
gegenüber dem nicht modifizierten Polymer schwächer ausgeprägtes Quellverhalten, was auf
den lipophileren Charakter des Boronsäure-Acrylamids zurückzuführen ist.
5.7.3Anlagerung von Polyolen
Grundsätzlich ist die Anlagerung von zwei Hydroxyfunktionen für die ungeladene
Boronsäure möglich, während die vollständige Anbindung eines Triols die Ausbildung eines
Borates erfordert. Es wird indes angenommen, dass auch die Ausbildung zweifach
koordinierender Polyole bevorzugt an den Borat-Ionen erfolgt.[41] Daher ist die
Komplexbildung bei höheren pH-Werten bevorzugt, allerdings sind basische Bedingungen für
die angestrebte Anwendung nur bedingt nutzbar, da höhere pH-Werte zu einer
Destabilisierung der DNA-Duplexe führen.
Da Boronsäuren schwache Säuren sind (pKs = 8.80 für Phenylboronsäure[72]), liegen diese im
Neutralen fast vollständig undissoziiert vor. Jedoch führt die Ausbildung der Komplexe zu
einer Erhöhung der Säurestärke.
OHOHOH
OHOOH
OH OHOHOH OHOH
OH
OH
OH
OH
CH2OH
H OHOH HH OHH OH
O H
CH2OH
OH HOH HH OHH OH
O H
CH2OH
H OHOH HOH H
O H
CH2OH
H OHOH HOH HH OH
O H CH2OH
CH2OH
OH HH OHH OH
OCH2OH
CH2OH
H OHOH HH OHH OH
CH2OH
CH2OH
H OHH OHOH HOH H
EthylenglycolK1 = 1.48 l mol-1K2 = 0.10 l2 mol-2
GylcerinKn = 67.6 ln mol-n(n = 1.25)
PropylenglycolK1 = 3.10 l mol-1K2 = 1.60 l2 mol-2
3-Methoxy-1,2-propandiolK1 = 18.8 l mol-1K2 = 13.4 l2 mol-2
PentaerythritolK1 = 240 l mol-1K2 = 1110 l2 mol-2
CatecholK1 = 7800 l mol-1K2 = 14200 l2 mol-2
D-GlucoseK1 = 80 l mol-1K2 = 770 l2 mol-2
D-MannoseK1 = 50 l mol-1K2 = 490 l2 mol-2
L-ArabinoseK1 = 130 l mol-1K2 = 675 l2 mol-2
D-GalactoseK1 = 127 l mol-1K2 = 298 l2 mol-2
SorbitolKn = 224000 ln mol-n(n = 1.95)
MannitolKn = 145000 ln mol-n(n = 2.27)
D-FructoseK1 K2 = 95000 l2 mol-2
Abb.60: Komplexbildungskonstanten entsprechend Gleichung (44) für die Anlagerung
von Polyolen an Borsäure.[73,74]

77
Dieser Zusammenhang kann genutzt werden, um aus Tritrationskurven der komplexierten
Boronsäure die Komplexbildungskonstanten zu ermitteln.[74]
Die Komplexbildungskonstanten von einigen Polyolen mit Borsäure sind in Abb. 60
zusammengefasst. Dabei sind in den meisten Fällen die Komplexbildungskonstanten für die
einfache wie auch die zweifache Komplexierung (n = 1, 2) entsprechend Gleichung (44)
angegeben.
[APn
(-)][A(-)][P]n
Kn = A = Borsäure / Borat-Ion; P = Polyol
(44)
K2 entspricht also nicht der Komplexbildungskonstante gemäß der Anlagerung eines zweiten
Polyols, sondern jener für die Anlagerung beider Polyole an das freie Borat.
Daraus ergibt sich in den meisten in Abbildung 60 gezeigten Fällen eine deutliche negative
Kooperativität für die Anbindung zweier Polyole.
Bei der Verwendung von Phenylboronsäuren kann keine doppelte Anlagerung von Polyolen
stattfinden, da nur drei Bindungen am Bor zur Verfügung stehen. Abgesehen davon sind die
a)
b)
BOH
OHOH
BOHOH
O
O
- BOO
O
O
-
OH OH
OH-
BOH
OHOHOH -
BO
OOH
OH-
OH OH OH OH
(-)A + P AP + P(-) AP2-AP(-)K1 K2 / K1
(-)A + 2P AP2-K2
Ks(AP)Ks(A)
A
P
AP
P P
-A AP- AP2-
Abb.61: Kompexbildung zwischen Borsäure und Polyolen, a) am Beispiel der
Komplexierung von Ethylenglycol, b) als allgemeines Schema. Bei der Angabe von
Komplexbildungskonstanten in der Literatur wird zwischen geladenen und ungeladenen
Spezies nicht unterscheiden. Die Säurestärke des AP-Komplexes Ks(AP) ist größer als die
der freien Borsäure Ks(A).

78
Abb.62: Berechnete Struktur (B3LYP, 6-
31G*) des Phenylboronsäure-Tri(hydroxy-
methyl)-methan-Komplexes[76] (bzgl. 3d-
Visualisierung siehe Anhang B).
Eigenschaften von Borsäure und Phenylboronsäuren bezüglich der Komplexbildung mit
Polyolen vergleichbar.[75] Die Acidität von Phenylboronsäure (pKs = 8.80) ist höher als die
der Borsäure (pKs = 9.14), was auf die Stabilisierung der Ladung durch den aromatischen
Ring zurückgeführt werden kann (eine entsprechende Erhöhung der Acidität wird bei
aliphatischen Boronsäuren nicht beobachtet).
Ein Vergleich der Komplexbildungskonstanten für einfache aliphatische Polyole zeigt
folgende Tendenzen: Die schwächsten Bindungen zeigen die aliphatischen Diole, wobei das
1,3-Diol Propylenglycol gegenüber dem 1,2-Diol Ethylenglycol eine doppelt so große
Komplexbildungskonstante aufweist. Bereits das Vorhandensein einer Methoxyfunktion, die
das Bor-Atom komplexieren kann, führt im Falle des 3-Methoxy-1,2-propandiols zu einer
enormen Stabilisierung um einen Faktor von 13, die Möglichkeit der Ausbildung einer dritten
Boronsäureester-Funktion führt im ansonsten strukturell gleichwertigen Glycerin zu einer
weiteren Stabilisierung um einen Faktor von 3.5. Analog zeigt Pentaerythritol, das ebenfalls
drei Bindungen zum Bor ausbilden kann, eine gegenüber Propylenglycol um den Faktor 77
erhöhte Komplexbildungskonstante.
Gegenüber Glycerin ist der Pentaerythritol-Borsäure-Komplex um einen Faktor von 3.5
stabilisiert.
Zusammenfassend ergibt sich somit das Bild, dass bei aliphatischen Polyolen die Ausbildung
dreifach koordinierender Komplexe zu recht stabilen Komplexen führt wohingegen Diole nur
wenig stabil sind.
Zum anderen ist eine generelle Bevorzugung von 1,3-Diolen gegenüber 1,2-Diolen zu
beobachten, was sich damit erklären lässt, dass in diesem Fall die Komplexierung zu der
Ausbildung von ungespannten Sechsringen führt. Dies ermöglicht eine tetraedrische
Koordinationsumgebung des Bors.
Im Falle des Tris(hydroxymethyl)methyl-
Fragmentes ist diese tetraedrische
Umgebung vorgebildet und es entsteht ein
stabiler und spannungsfreier Komplex
einer Bicyclo[2.2.2]oktan-Geometrie aus
drei Sechsringen (Abb.62).
Ein Molekül, das mehrere Polyol-
Funktionalitäten enthält, kann potentiell an
mehrere Boronsäurefunktionen gebunden
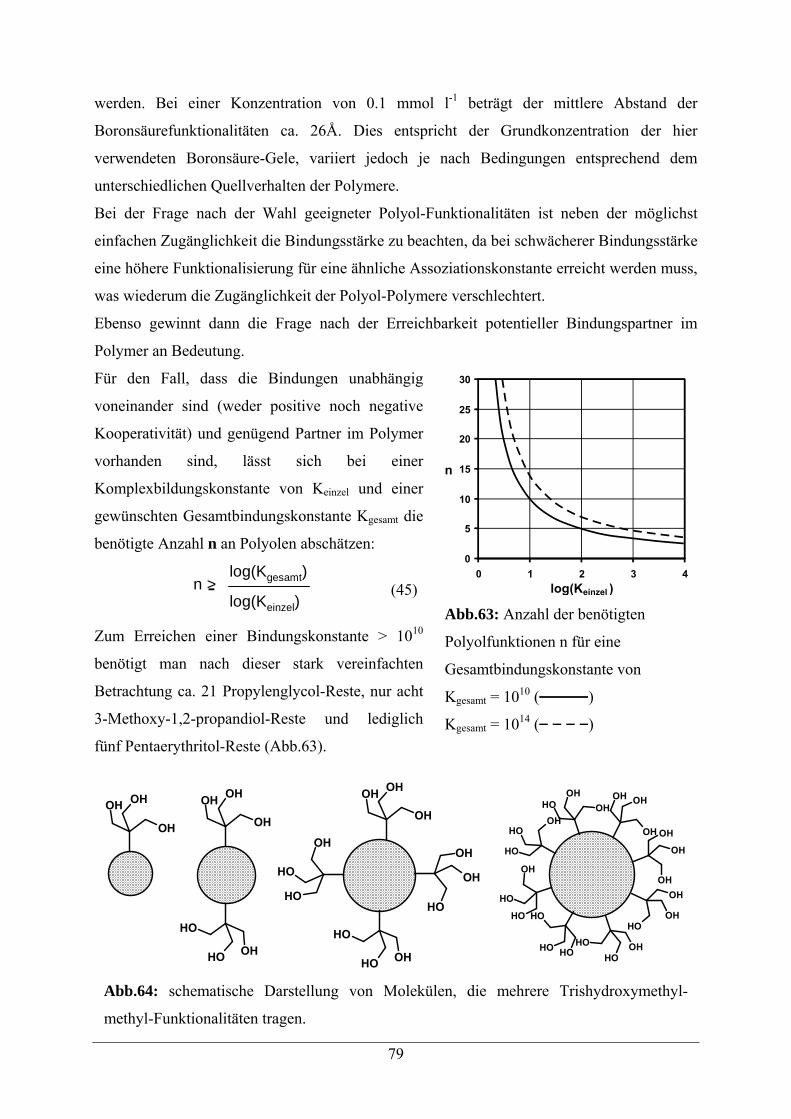
79
werden. Bei einer Konzentration von 0.1 mmol l-1 beträgt der mittlere Abstand der
Boronsäurefunktionalitäten ca. 26Å. Dies entspricht der Grundkonzentration der hier
verwendeten Boronsäure-Gele, variiert jedoch je nach Bedingungen entsprechend dem
unterschiedlichen Quellverhalten der Polymere.
Bei der Frage nach der Wahl geeigneter Polyol-Funktionalitäten ist neben der möglichst
einfachen Zugänglichkeit die Bindungsstärke zu beachten, da bei schwächerer Bindungsstärke
eine höhere Funktionalisierung für eine ähnliche Assoziationskonstante erreicht werden muss,
was wiederum die Zugänglichkeit der Polyol-Polymere verschlechtert.
Ebenso gewinnt dann die Frage nach der Erreichbarkeit potentieller Bindungspartner im
Polymer an Bedeutung.
Für den Fall, dass die Bindungen unabhängig
voneinander sind (weder positive noch negative
Kooperativität) und genügend Partner im Polymer
vorhanden sind, lässt sich bei einer
Komplexbildungskonstante von Keinzel und einer
gewünschten Gesamtbindungskonstante Kgesamt die
benötigte Anzahl n an Polyolen abschätzen:
n > log(Kgesamt)
log(Keinzel)-
(45)
Zum Erreichen einer Bindungskonstante > 1010
benötigt man nach dieser stark vereinfachten
Betrachtung ca. 21 Propylenglycol-Reste, nur acht
3-Methoxy-1,2-propandiol-Reste und lediglich
fünf Pentaerythritol-Reste (Abb.63).
OH
OH OH
OH
OHOH
OH
OH
OH
OH
OH
OH
OHOHOH
OH OHOH
OH
OH
OH
OH
OH
OH
OH
OH OH
OH
OHOH
OH
OH
OH
OH
OH
OHOH
OH OH
OH
OHOH
OH
OH OH
Abb.64: schematische Darstellung von Molekülen, die mehrere Trishydroxymethyl-
methyl-Funktionalitäten tragen.
0
5
10
15
20
25
30
0 1 2 3 4log(Keinzel )
n
Abb.63: Anzahl der benötigten
Polyolfunktionen n für eine
Gesamtbindungskonstante von
Kgesamt = 1010 ( )
Kgesamt = 1014 ( )

80
Für die angestrebte Anwendung im Rahmen orthogonaler Immobilisierungsstrategien ist
Brenzcatechin aufgrund der Wirkung als Reduktionsmittel nur bedingt geeignet, da es z.B.
nicht zusammen mit einer Disulfid-Chemie eingesetzt werden kann. Ebenso ist auch eine
Wechselwirkung mit den DNA-Basen nicht auszuschließen, wohingegen sich einfache
aliphatische Polyole inert verhalten.
Bei der Frage nach der Anordnung der Polyol-Funktionalitäten kommen dendritische genauso
wie kettenförmige Strukturen in Frage. Dabei muss bedacht werden, dass dendritische
Polymere mit wachsender Generation immer kompakter werden, was zu einer immer starreren
Struktur und immer kleineren Abständen der Endgruppen führt.
Gerade wenn möglichst viele dieser Reste mit polymergebundenen Funktionalitäten
wechselwirken sollen, erweist sich das als Problem, da keine ausreichende Menge dieser
Funktionalitäten in einem entsprechend kleinen Volumen zur Verfügung gestellt werden
können. Dieses Problem stellt sich bei der Verwendung kettenförmiger Polymere nicht in dem
Maße. Zudem ist hier bei größeren Polymeren eine deutlich höhere Flexibilität gegeben.
Darüber hinaus lassen sich Dendrimere an der Peripherie nur schlecht monofunktionalisieren,
eine Core-Funktionalisierung ist dagegen sterisch schlecht zugänglich.[77]
Aufgrund dieser Überlegungen erscheinen für diese Anwendung dendritische Polymere nicht
unbedingt von Vorteil zu sein.
Die Untersuchung der Wechselwirkungen von Polyolen mit Boronsäuregelen erfolgte ohne
Zusatz von Base bzw. Puffer, um eine gravimetrische Bestimmung der Polyole in Gel und
Lösung zu ermöglichen. Wie oben erläutert ist die Komplexbildung im Basischen begünstigt.
5.7.3.1 Bistrispropan
Bistrispropan (30) beinhaltet zwei Tris(hydroxymethyl)methyl-Fragmente, wodurch
grundsätzlich sowohl eine einfache als auch eine doppelte Anbindung an das Polymer
möglich ist. Eine Analyse der Struktur des Bistrispropans durch Geometrieoptimierung zeigt,
dass der maximale Abstand der Boratome ca. 13 Å beträgt. Wird angenommen, dass die
Phenylboronsäureamid-Reste frei beweglich sind, und lediglich das Polymergitter als starr
angesehen, so ergibt sich ein maximaler Abstand von 27 Å (Abb.65).
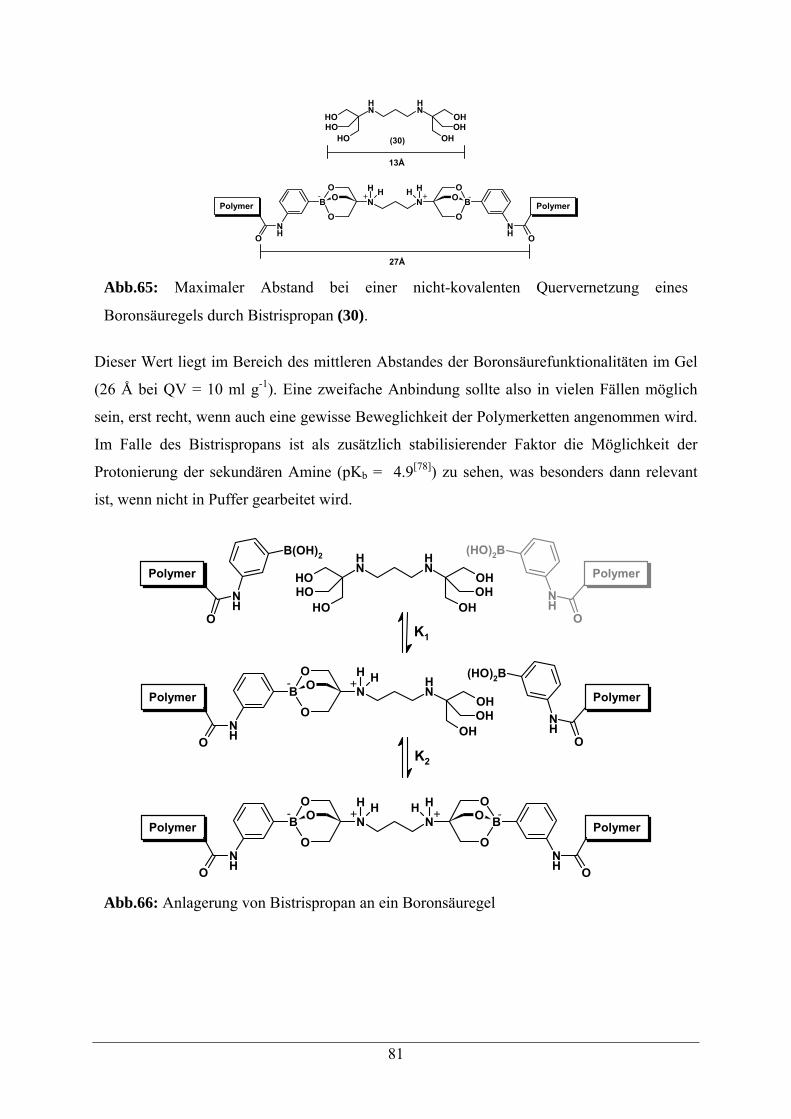
81
Dieser Wert liegt im Bereich des mittleren Abstandes der Boronsäurefunktionalitäten im Gel
(26 Å bei QV = 10 ml g-1). Eine zweifache Anbindung sollte also in vielen Fällen möglich
sein, erst recht, wenn auch eine gewisse Beweglichkeit der Polymerketten angenommen wird.
Im Falle des Bistrispropans ist als zusätzlich stabilisierender Faktor die Möglichkeit der
Protonierung der sekundären Amine (pKb = 4.9[78]) zu sehen, was besonders dann relevant
ist, wenn nicht in Puffer gearbeitet wird.
Polymer NH
OH
OHOH
NH
OH
OHOH
(HO)2B
NH
O
B(OH)2
NH
O
Polymer
Polymer PolymerN NH
O
OB O
NHO
H H
OH
OHOH
(HO)2B
NH
O
+-
Polymer PolymerO
OBON N
O
OB O
NH
NH OO
H HH H+- + -
K1
K2
Abb.66: Anlagerung von Bistrispropan an ein Boronsäuregel
NH
OH
OHOH
NH
OH
OHOH
O
OBON N
O
OB O
NH
NH OO
H HH H
13Å
27Å
+- + -Polymer Polymer
(30)
Abb.65: Maximaler Abstand bei einer nicht-kovalenten Quervernetzung eines
Boronsäuregels durch Bistrispropan (30).
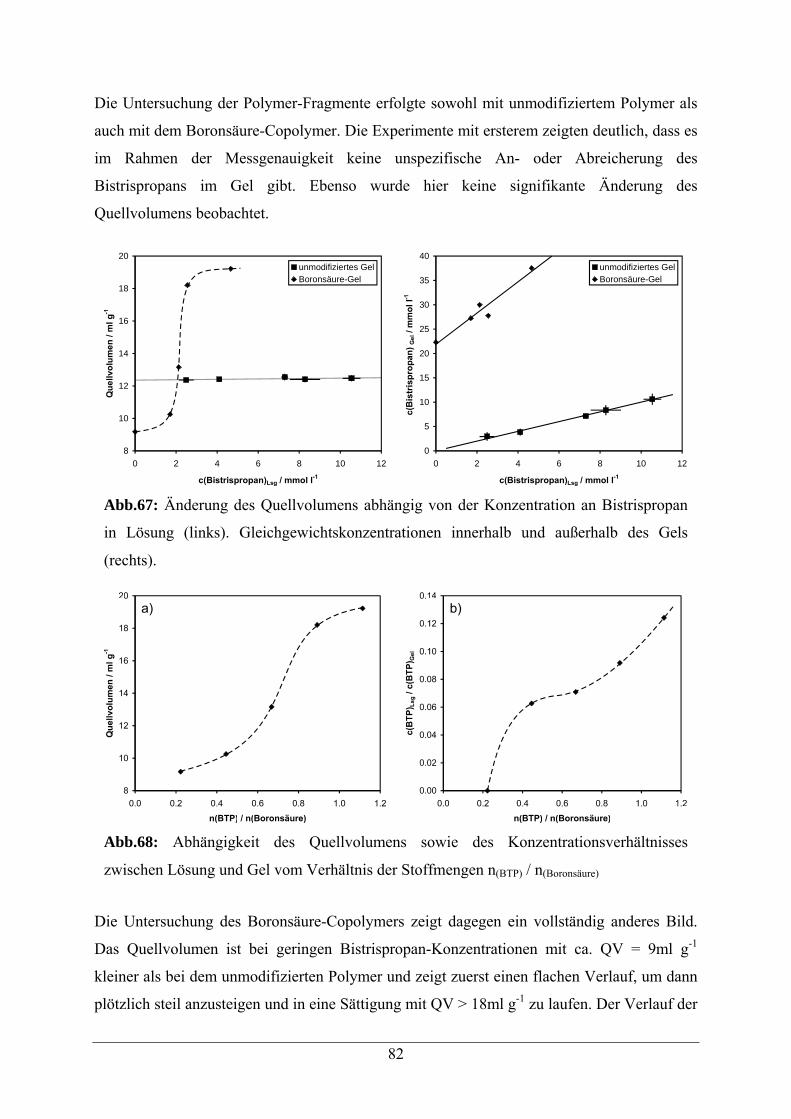
82
Die Untersuchung der Polymer-Fragmente erfolgte sowohl mit unmodifiziertem Polymer als
auch mit dem Boronsäure-Copolymer. Die Experimente mit ersterem zeigten deutlich, dass es
im Rahmen der Messgenauigkeit keine unspezifische An- oder Abreicherung des
Bistrispropans im Gel gibt. Ebenso wurde hier keine signifikante Änderung des
Quellvolumens beobachtet.
Die Untersuchung des Boronsäure-Copolymers zeigt dagegen ein vollständig anderes Bild.
Das Quellvolumen ist bei geringen Bistrispropan-Konzentrationen mit ca. QV = 9ml g-1
kleiner als bei dem unmodifizierten Polymer und zeigt zuerst einen flachen Verlauf, um dann
plötzlich steil anzusteigen und in eine Sättigung mit QV > 18ml g-1 zu laufen. Der Verlauf der
8
10
12
14
16
18
20
0 2 4 6 8 10 12
c(Bistrispropan)Lsg / mmol l-1
Que
llvol
umen
/mlg
-1
unmodifiziertes GelBoronsäure-Gel
0
5
10
15
20
25
30
35
40
0 2 4 6 8 10 12
c(Bistrispropan)Lsg / mmol l-1
c(B
istr
ispr
opan
) Gel
/mm
oll-1
unmodifiziertes GelBoronsäure-Gel
Abb.67: Änderung des Quellvolumens abhängig von der Konzentration an Bistrispropan
in Lösung (links). Gleichgewichtskonzentrationen innerhalb und außerhalb des Gels
(rechts).
Abb.68: Abhängigkeit des Quellvolumens sowie des Konzentrationsverhältnisses
zwischen Lösung und Gel vom Verhältnis der Stoffmengen n(BTP) / n(Boronsäure)
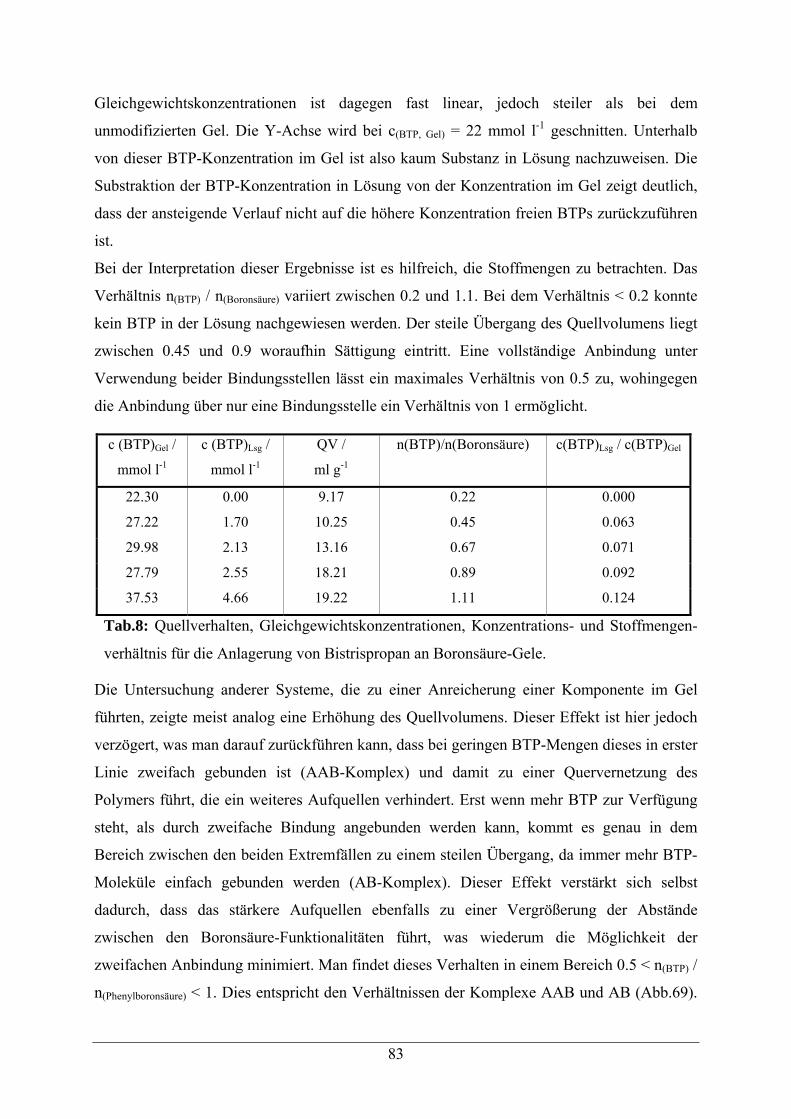
83
c (BTP)Gel /
mmol l-1
c (BTP)Lsg /
mmol l-1
QV /
ml g-1
n(BTP)/n(Boronsäure)
c(BTP)Lsg / c(BTP)Gel
22.30 0.00 9.17 0.22 0.000
27.22 1.70 10.25 0.45 0.063
29.98 2.13 13.16 0.67 0.071
27.79 2.55 18.21 0.89 0.092
37.53 4.66 19.22 1.11 0.124
Tab.8: Quellverhalten, Gleichgewichtskonzentrationen, Konzentrations- und Stoffmengen-
verhältnis für die Anlagerung von Bistrispropan an Boronsäure-Gele.
Gleichgewichtskonzentrationen ist dagegen fast linear, jedoch steiler als bei dem
unmodifizierten Gel. Die Y-Achse wird bei c(BTP, Gel) = 22 mmol l-1 geschnitten. Unterhalb
von dieser BTP-Konzentration im Gel ist also kaum Substanz in Lösung nachzuweisen. Die
Substraktion der BTP-Konzentration in Lösung von der Konzentration im Gel zeigt deutlich,
dass der ansteigende Verlauf nicht auf die höhere Konzentration freien BTPs zurückzuführen
ist.
Bei der Interpretation dieser Ergebnisse ist es hilfreich, die Stoffmengen zu betrachten. Das
Verhältnis n(BTP) / n(Boronsäure) variiert zwischen 0.2 und 1.1. Bei dem Verhältnis < 0.2 konnte
kein BTP in der Lösung nachgewiesen werden. Der steile Übergang des Quellvolumens liegt
zwischen 0.45 und 0.9 woraufhin Sättigung eintritt. Eine vollständige Anbindung unter
Verwendung beider Bindungsstellen lässt ein maximales Verhältnis von 0.5 zu, wohingegen
die Anbindung über nur eine Bindungsstelle ein Verhältnis von 1 ermöglicht.
Die Untersuchung anderer Systeme, die zu einer Anreicherung einer Komponente im Gel
führten, zeigte meist analog eine Erhöhung des Quellvolumens. Dieser Effekt ist hier jedoch
verzögert, was man darauf zurückführen kann, dass bei geringen BTP-Mengen dieses in erster
Linie zweifach gebunden ist (AAB-Komplex) und damit zu einer Quervernetzung des
Polymers führt, die ein weiteres Aufquellen verhindert. Erst wenn mehr BTP zur Verfügung
steht, als durch zweifache Bindung angebunden werden kann, kommt es genau in dem
Bereich zwischen den beiden Extremfällen zu einem steilen Übergang, da immer mehr BTP-
Moleküle einfach gebunden werden (AB-Komplex). Dieser Effekt verstärkt sich selbst
dadurch, dass das stärkere Aufquellen ebenfalls zu einer Vergrößerung der Abstände
zwischen den Boronsäure-Funktionalitäten führt, was wiederum die Möglichkeit der
zweifachen Anbindung minimiert. Man findet dieses Verhalten in einem Bereich 0.5 < n(BTP) /
n(Phenylboronsäure) < 1. Dies entspricht den Verhältnissen der Komplexe AAB und AB (Abb.69).

84
Damit lässt sich als Grenzfall für den
Übergang die Reaktion AAB + B → 2 AB
(Abb.69) formulieren.
Bei der Betrachtung der Gleichgewichts-
konzentrationen muss beachtet werden, dass
der Anstieg des Quellverhaltens zu einem
Anstieg des Volumens im Gel führt. Die
effektive Konzentration der Boronsäure-
funktionalitäten sinkt, während die
Stoffmengen an BTP im Gel stärker steigen
als die Konzentrationen. In einem weiten
Bereich, in dem sich die Stoffmenge des
zugegebenen Bistrispropans verdoppelt,
findet man sowohl innerhalb des Gels als
auch in der Lösung nur geringe
Konzentrationsvariationen (Abb.69). Die
Aufnahme des Bistrispropans wird fast
vollständig durch das Aufquellen des
Polymers erreicht. Dieses Verhalten
entspricht i.ü.S. dem eines Puffers.
Eine Beeinflussung der Quervernetzung
durch Zielmoleküle, die zweifach an das
Polymernetzwerk binden können, wird auch
für Zucker gefunden, allerdings sind dabei
die Effekte deutlich kleiner als die hier
gefundenen.[43]
Die Abhängigkeit der Quelleigenschaften
von der Anbindung von Zielmolekülen, wie
es bei Boronsäure-Hydrogelen beobachtet
werden kann, eröffnet die Möglichkeit, diese als Biosensoren zu benutzen, so wird z.B. die
Verwendung im Bereich minimal invasiver Glucosesensoren in der Therapie des Diabetes
mellitus (Typ 1) diskutiert. [41,43]
Das ausgeprägte Quellverhalten führt auch dazu, dass die Gleichgewichtskonstanten nicht
ohne weiteres bestimmt werden können. Für eine analytische Beschreibung durch Ausdrücke
0.4 0.6 0.8 1.0 1.2
n(BTP) / n(Boronsäure)
8
10
12
14
16
18
20
22
Que
llvol
umen
/mlg
-1
Polymer
Polymer
O OBO
N
N
O OBO
NH
NH
O
O
H
H
H
H
+
-
+
-
Polymer
N
NH
OOB
O
NH
O
HH
OHOH OH
+
-
BTP
Polymer
N
NH
OO
BO
NH
O
HH
OHOHOH
+
-
AAB
AB
AB
A + AAB AAB + AB AB + B
Abb.69: Quellverhalten von Boronsäure-
gelen bei unterschiedlichen Bistrispropan-
äquivalenten korreliert mit entsprechenden
Strukturen.

85
für ΠM und ΠE müssten statistische Ausdrücke für die nicht kovalente Quervernetzung
hergeleitet werden. Eine grobe Abschätzung unter Vernachlässigung von Wasser (was für
beide K-Werte einem Faktor von 3000 entsprechen würde) für geringe Konzentrationen lässt
auf Größenordnungen von K1 ≈ 400 M-1 und K2 > K1 schließen.

86
5.7.3.2 Polyglycerin
Bei der Untersuchung der Anbindung von
Polyolen an Boronsäuregele wurde auch das
Verhalten des dendritischen Polymers PG02
(31) untersucht. Dabei handelt es sich um ein
durch anionische Polymerisation von 2,3-
Epoxy-1-propanol gewonnenes dendritisches
Polymer[79] von enger Größenverteilung und
einer mittleren molaren Masse von
2000 g mol-1, was einer Anzahl von durch-
schnittlich 27 Glycerineinheiten entspricht,
die (wenn kein Zyklus ausgebildet wird) 29
Hydroxy-funktionalitäten zur Verfügung
stellen.
Diese können sowohl isoliert als auch in
Form von 1,2-Diolen oder 1,3-Diolen
vorkommen, wobei isolierte Hydroxy-
funktionen sich im Inneren des Polymers befinden, Diole an der Oberfläche. Die Art des
Aufbaus führt zu einer Überzahl an 1,2-Diolen, da die negative Ladung vom sekundären zum
primären Alkohol wandert. [79]
Eine Abschätzung der Wechselwirkung mit Boronsäuren kann auf der Grundlage der in Abb.
73 angegebenen Komplexbildungskonstanten erfolgen. Strukturell sind die Endgruppen dem
3-Methoxy-1,2-propandiol ähnlich, womit für jede Einheit ein Faktor von ca. Keinzel =
18 l mol-1 zur Gesamtbindungskonstante erwartet werden kann. Damit würde man für den Fall
der Ausbildung mehrerer Boronsäureaddukte eine deutliche Anreicherung wenn nicht
Immobilisierung erwarten. Eine maximale Anzahl von 14 3-Alkoxy-1,2-propandiolen ergäbe
bei Anbindung aller Funktionen an Boronsäurereste rechnerisch eine
Komplexbildungskonstante > 1017 l mol-1.
Die Untersuchung der Wechselwirkung von PG02 mit Polymer-Fragmenten erfolgte sowohl
mit unmodifiziertem Polymer als auch mit dem Boronsäure-Copolymer. Gegenüber
Bistrispropan sind die beobachteten Effekte deutlich schwächer ausgeprägt. Bezüglich des
unmodifizierten Polymers beobachtet man eine Verringerung des Quellvolumens bei
steigender PG02 Konzentration. Dem entspricht die deutliche Konzentrationsabnahme im
O
O
O
O
O
OH
OH
OO
OOH
OOO
OH
OOH
OH
OHO
OHOH
O
OH
O
OH
O
OH
O
OO
O
OO
O
O
OH
OH
OOH
OHOHOH
OH
OOH
OH OH
OH
OH
OH
OH
OH
OH
OH
(31)
Abb.70: Beispielstruktur eines dendri-
tischen Polyglycerin-Moleküls (PG02) mit
M = 2016 g mol-1
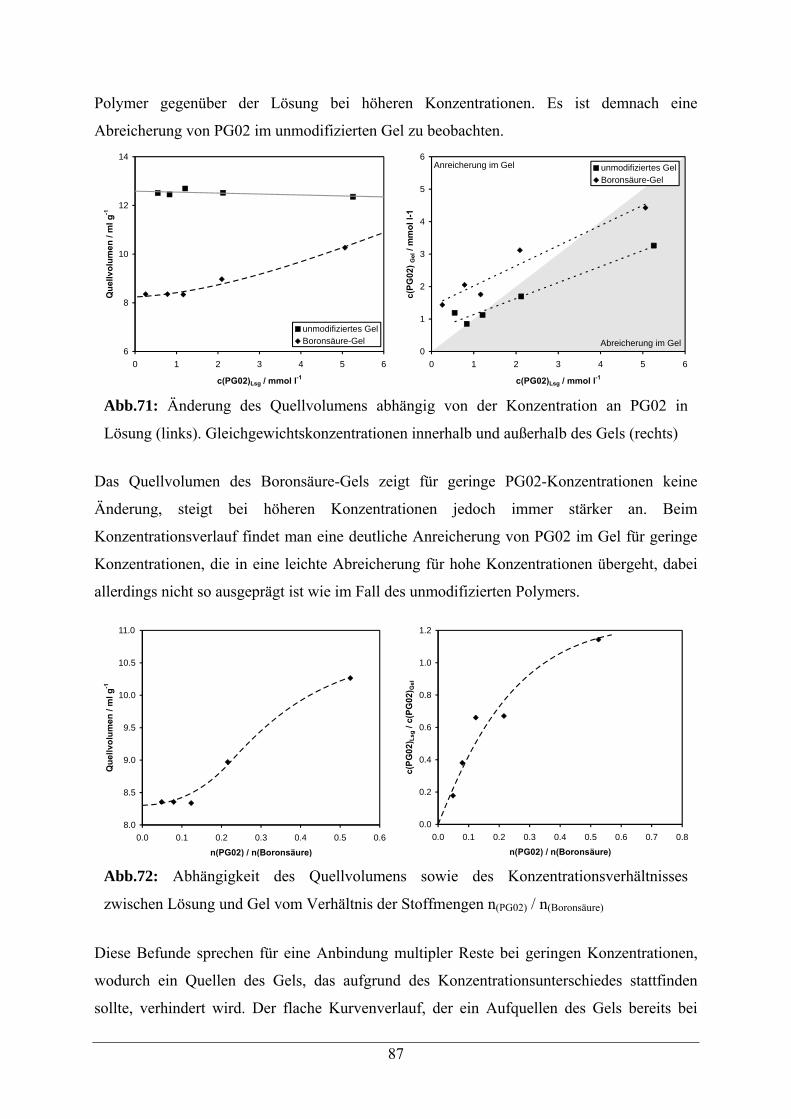
87
Polymer gegenüber der Lösung bei höheren Konzentrationen. Es ist demnach eine
Abreicherung von PG02 im unmodifizierten Gel zu beobachten.
Das Quellvolumen des Boronsäure-Gels zeigt für geringe PG02-Konzentrationen keine
Änderung, steigt bei höheren Konzentrationen jedoch immer stärker an. Beim
Konzentrationsverlauf findet man eine deutliche Anreicherung von PG02 im Gel für geringe
Konzentrationen, die in eine leichte Abreicherung für hohe Konzentrationen übergeht, dabei
allerdings nicht so ausgeprägt ist wie im Fall des unmodifizierten Polymers.
Diese Befunde sprechen für eine Anbindung multipler Reste bei geringen Konzentrationen,
wodurch ein Quellen des Gels, das aufgrund des Konzentrationsunterschiedes stattfinden
sollte, verhindert wird. Der flache Kurvenverlauf, der ein Aufquellen des Gels bereits bei
6
8
10
12
14
0 1 2 3 4 5 6
c(PG02)Lsg / mmol l-1
Que
llvol
umen
/mlg
-1
unmodifiziertes GelBoronsäure-Gel
0
1
2
3
4
5
6
0 1 2 3 4 5 6
c(PG02)Lsg / mmol l-1
c(PG
02) G
el/m
mol
l-1
unmodifiziertes GelBoronsäure-Gel
Anreicherung im Gel
Abreicherung im Gel
Abb.71: Änderung des Quellvolumens abhängig von der Konzentration an PG02 in
Lösung (links). Gleichgewichtskonzentrationen innerhalb und außerhalb des Gels (rechts)
8.0
8.5
9.0
9.5
10.0
10.5
11.0
0.0 0.1 0.2 0.3 0.4 0.5 0.6
n(PG02) / n(Boronsäure)
Que
llvol
umen
/mlg
-1
0.0
0.2
0.4
0.6
0.8
1.0
1.2
0.0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8
n(PG02) / n(Boronsäure)
c(PG
02) L
sg/c
(PG
02) G
el
Abb.72: Abhängigkeit des Quellvolumens sowie des Konzentrationsverhältnisses
zwischen Lösung und Gel vom Verhältnis der Stoffmengen n(PG02) / n(Boronsäure)

88
einem Verhältnis nPG02 : nBoronsäure = 1 : 8 erkennen lässt, weist auf eine eher schwache
Bindung hin. Die Tatsache, dass keine Anreicherung des Polyols im Gel ab nPG02 : nBoronsäure =
1 : 2 mehr zu beobachten ist, spricht dafür, dass hier die möglichen Bindungen an die
Boronsäurefunktionalitäten durch die im unmodifizierten Polymer beobachtete unspezifische
Abreicherung kompensiert wird.
Eine Abschätzung der Größe des dendritischen Polymers ergibt einen maximalen Radius von
ca. 15 Å, der experimentelle Trägheitsradius in D2O beträgt Rg = 13 Å. [80]
Bei einem maximalen Durchmesser von
30 Å und der Annahme der freien
Beweglichkeit der Boronsäurereste an einem
sonst starren Polymergerüst ergibt sich ein
Durchmesser von 44 Å. Die daraus
ermittelbare Anzahl von Boronsäure-
funktionalitäten in ausreichender Nähe zum
Polyol beträgt 3-4. Es ist somit nicht
anzunehmen, dass alle Polyolfunktionali-
täten des Polyglycerins mit Boronsäure-
funktionalitäten wechselwirken können und
die tatsächlich möglichen Wechsel-
wirkungen reichen offenbar für eine
effektive Immobilisierung nicht aus.
Ein Vergleich mit den Eigenschaften von
Bistrispropan kann jedoch nicht ohne
weiteres angestellt werden, da letzteres als mittelstarke Base die eigene Komplexierung
begünstigt. Um einen Vergleich anstellen zu können, müsste also der PG02-Lösung ein
entspechender Anteil an Base zugegeben werden.
In Hinblick auf eine mögliche Immobilisierung von DNA muss indes berücksichtigt werden,
dass ein Replikationsverfahren nicht unter stärker basischen Bedingungen durchgeführt
werden kann. Nach dieser Überlegung stellen TRIS-Derivate mit protonierbaren
Aminofunktionen eine Möglichkeit dar, den Boronsäurefunktionen lokal Protonenakzeptoren
zur Verfügung zu stellen. Ähnliche Konzepte unter Bereitstellung von Protonenakzeptoren,
benachbart den Boronsäurefunktionen im Polymer, sind für die effektive Anlagerung von
Zuckern aus der Literatur bekannt.
30 Å
44 Å
Boronsäure-Funktionalitäten
PG02
Abb.73: Radius des dendritischen Poly-
glycerins PG02.

89
5.8 Copolymere mit Thiol-Modifikation
5.8.1 Synthesestrategie
Zur Einführung einer Thiol-Funktion in das Polymer muss dieses in geeignet geschützter
Form als Acrylamid eingesetzt werden. Die einfachste Variante stellt das zweifache
Acrylamid des Cystamins dar, das durch Umsetzung des Cystaminhydrochlorids mit
Acryloylchlorid in wässriger Natriumhydrogencarbonatlösung dargestellt werden kann
(Abb.74a).
Die Löslichkeit der Verbindung in Wasser, DMF und DMSO ist jedoch zu schlecht, um sie
analog der hier vorgestellten Methodik einsetzen zu können.
Daher wurde auf Varianten zurückgegriffen, die eine etwas bessere Löslichkeit in Wasser
oder polaren Lösungsmitteln aufweisen.
Der Einsatz von (36) ermöglicht auf der einen
Seite aufgrund der etwas höheren Polarität die
Copolymerisation in DMF-Wasser-Gemischen
(5:3 v/v), auf der anderen Seite ist die
Lipophilie der Substanz so hoch, dass sie bei
der Synthese aus der wässrigen Reaktions-
mischung ausfällt.
Bei der Copolymerisation verhält sich diese
Verbindung wie ein Quervernetzer. Diese
Verknüpfungen können jedoch durch Reduktion
aufgebrochen werden, was zu zwei Thiol-
unktionalitäten pro eingesetztem Monomer
NH2
SS
NH2
OO
O O
NH
SS
NH
OO
O OO
O
NH2
SS
NH2
OOH
O OH
NH
SS
NH
O
OCl
O
Cl
O
NH3+ S
SNH3
+Cl
Cl
(34) (35) (36)
(33)
a)
b)
NaHCO3 / H2O(32)
Abb.74: Synthesen der Disulfid-Acrylamide (33) und (36).
Polymer PolymerNH
SS
NH
OO
O OO
O
Polymer PolymerNH
SH
OOO
SHNH
O OO
(P16)
(P17) (P17)
ReduktionOxidation
Abb.75: Redox-schaltbare Querver-
netzung von Disulfid/Thiol-Gelen.

90
führt. Das hat den Vorteil, dass keine weitere Schutzgruppe aus dem Polymer entfernt werden
muss. Grundsätzlich lassen sich die Quervernetzungen zu einem gewissen Masse durch
Oxidation wiederherstellen, wobei das Ausmaß davon abhängt, wie weit sich das Polymer
nach der Spaltung rekonfigurieren kann (Abb.75).
Tatsächlich findet man bei der Herstellung von Gelen, die außer (36) keinen Quervernetzer
enthalten, nach der Polymerisation keine deutlich anderes Quellverhalten, wird das Polymer
jedoch mit DTT reduziert, so quillt das Gel auf ein Vielfaches seines Ursprungsvolumens an.
Es ist auch denkbar, dass die potentielle Nachbarschaft der Thiolgruppen dazu führt, dass
durch Disulfidaustausch immobilisierte Substanzen abgespalten werden. Dies ist insbesondere
bei der Aktivierung der Thiole mit 2,2’-Dipyridinyldisulfid (PySSPy) zu erwarten, wenn die
diffusionsbedingte Konzentration im Inneren zu niedrig ist.
Die aktivierten Disulfide können mit Thiolen zu den modifizierten Gelen umgesetzt werden,
wobei sich die Reaktion aufgrund des freiwerdenden Thiopyridons gut über UV/Vis-
Spektroskopie verfolgen lässt.
Polymer NH
SH
OOO
Polymer NH
S
OOO
S N Polymer NH
S
OOO
SR
NH
S NH
S
(P17) (P18) (P19)PySSPy R-SH(37) (37)
(38) (38) Abb.76: Aktivierung und Funktionalisierung von Thiol-modifizierten N,N-
Dimethylacrylamidpolymeren.
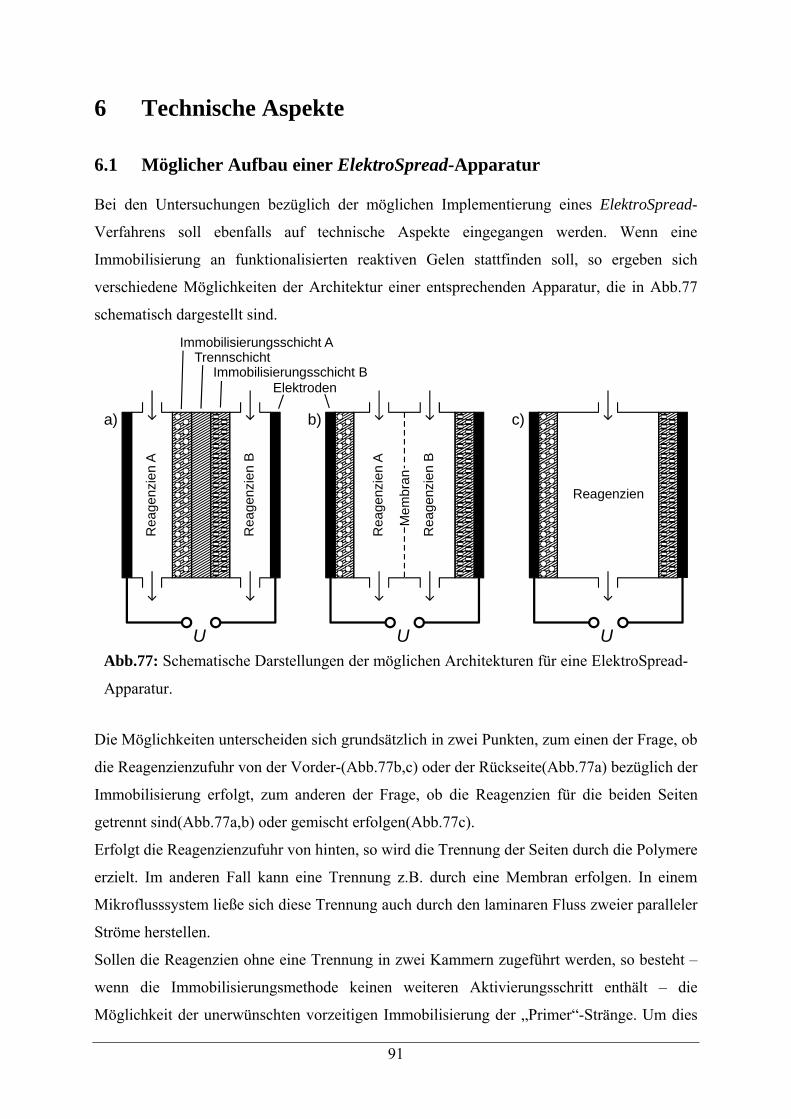
91
6 Technische Aspekte
6.1 Möglicher Aufbau einer ElektroSpread-Apparatur
Bei den Untersuchungen bezüglich der möglichen Implementierung eines ElektroSpread-
Verfahrens soll ebenfalls auf technische Aspekte eingegangen werden. Wenn eine
Immobilisierung an funktionalisierten reaktiven Gelen stattfinden soll, so ergeben sich
verschiedene Möglichkeiten der Architektur einer entsprechenden Apparatur, die in Abb.77
schematisch dargestellt sind.
Die Möglichkeiten unterscheiden sich grundsätzlich in zwei Punkten, zum einen der Frage, ob
die Reagenzienzufuhr von der Vorder-(Abb.77b,c) oder der Rückseite(Abb.77a) bezüglich der
Immobilisierung erfolgt, zum anderen der Frage, ob die Reagenzien für die beiden Seiten
getrennt sind(Abb.77a,b) oder gemischt erfolgen(Abb.77c).
Erfolgt die Reagenzienzufuhr von hinten, so wird die Trennung der Seiten durch die Polymere
erzielt. Im anderen Fall kann eine Trennung z.B. durch eine Membran erfolgen. In einem
Mikroflusssystem ließe sich diese Trennung auch durch den laminaren Fluss zweier paralleler
Ströme herstellen.
Sollen die Reagenzien ohne eine Trennung in zwei Kammern zugeführt werden, so besteht –
wenn die Immobilisierungsmethode keinen weiteren Aktivierungsschritt enthält – die
Möglichkeit der unerwünschten vorzeitigen Immobilisierung der „Primer“-Stränge. Um dies
U U U
Elektroden
Immobilisierungsschicht A Trennschicht Immobilisierungsschicht B
Mem
bran
Reagenzien
Rea
genz
ien
A
Rea
genz
ien
B
Rea
genz
ien
A
Rea
genz
ien
Ba) b) c)
Abb.77: Schematische Darstellungen der möglichen Architekturen für eine ElektroSpread-
Apparatur.

92
zu verhindern kann in diesem Fall z.B. eine Spannung angelegt werden, die einen Transport
der Oligonukleotide zu der gewünschten Seite bewirkt. Das Anlegen einer Spannung auch bei
der Reagenzienzufuhr ist ohnehin sinnvoll, um den vergleichsweise langsamen
diffusionsbedingten Transport in das Gel zu unterstützen.
Um mögliche Fehlerquellen auszuschließen, bietet sich indes eine Trennung der Kammern an.
Ein weiteres Problem kann die Querdiffusion während des Elektrotransfers darstellen. Daher
ist es sinnvoll, dass die beiden Immobilisierungsschichten möglichst nah angeordnet sind,
wobei ein direkter Kontakt wiederum verhindert werden muss.
Aufgrund der Tatsache, dass auch der direkte Kontakt der Gele mit den Elektroden aufgrund
der möglichen elektrolytischen Spaltung von Wasser problematisch ist, erscheint ein Aufbau
mit einer Sandwich-Struktur (Abb.77a), bei der die beiden Immobilisierungsschichten durch
eine inerte Trennschicht getrennt sind, am sinnvollsten. Darüber hinaus kann die Trennschicht
eine mechanisch stabilisierende Funktion übernehmen, wofür jedoch eine Fixierung der
Immobilisierungsschichten an der Trennschicht notwendig ist.
Zu diesem Zweck wurden Vorexperimente zur Einbettung fester Netzwerke in die Gelstruktur
während der Polymerisation unter Verwendung
von Nylon-Gewebe (Abb.78) als tragende
Struktur durchgeführt. Es zeigte sich jedoch,
dass bereits die mechanische Belastung durch
das Quellen beim Waschen der Gele zu einer
weitgehenden Zerstörung und damit zur
Loslösung des Gels vom Gewebe führt. Die
mechanische Einbettung ist offenbar nicht
ausreichend. Für zukünftige Experimente in
diesem Bereich sollten Gewebe mit der
Möglichkeit der kovalenten Anbindung an das
Gel verwendet werden.
0 5 10 mm
Abb.78: Vergrößerte Abbildung des
durch Behandlung einer Nytran-
Transfer-Membran[81] mit konzentrierter
Salzsäure erhaltenes Nylonnetzwerks.

93
6.2 Sandwich-Gele
Als Analogon zu Sandwich-Gelen wurden
zur Bewertung der Eigenschaften und
Möglichkeiten multifunktionale Gele
gefertigt, bei denen die unterschiedlich
funktionalisierten inselförmigen Bereiche
durch eine unmodifizierte Matrix getrennt
wurden. Der Aufbau erfolgte entsprechend
Abb.79. Zuerst wurden die modifizierten
Bereiche in die Taschen eines Teflonspacers
gegossen, nach der Polymerisation wurde
der Spacer entfernt, die polymerisierten
Bereiche wurden vorsichtig gewaschen und
getrocknet und danach wurde ein
unmodifiziertes Gel als umschließende
Matrix ergänzt.
Es zeigte sich, dass nur dann eine gute
Anbindung zwischen den Bereichen erfolgte,
wenn die Zugabe des unmodifizierten
Monomergemischs möglichst schnell nach
der Bildung der ersten Gele stattfand. Kam
es zu einer Alterung der modifizierten
Bereiche, fand keine ausreichende
Verknüpfung mehr zwischen den modi-
izierten und unmodifizierten Bereichen statt,
um den späteren Quellunterschieden der
Bereiche standzuhalten.
Dieser Befund lässt sich darauf zurück-
ühren, dass die Polymerisation mit dem
Phasenübergang noch nicht abgeschlossen ist und so direkt nach dem Phasenübergang noch
reaktionsfähige Reste existieren, die der kovalenten Verknüpfung zwischen den Bereichen
dienen können.
A B Immobilisierungsschicht
Teflonspacer
Monomer-Lösung
Nach Entfernung des Spacers verbleiben funktionalisierte Polymerinseln um die das unfunktionalisierte Trenngel gegossen wird.
Trennschicht
lipophilisierte Objektträger
Polymerisation
Abb.79: Aufbau von multifunktionalen
Hybridgelen.

94
6.2.1 Hydrazid/Aldehyd-Hybrid-Gele
Es wurden nach dem oben geschilderten Prinzip Hydrazid/Aldehyd-Hybrid-Gele angefertigt,
wobei als Monomere auf der einen Seite das Acetal-Acrylamid (5), auf der anderen Seite
Glycolacrylat zum Einsatz kamen. Nach der Polymerisation der Inseln wurde die
Dimethylacrylamid-Matrix gegossen. Beide eingesetzten Modifikationen mussten erst durch
weitere Umsetzungen zur gewünschten Funktionalität umgesetzt werden, d.h. das Acetal
musste sauer gespalten, der Ester hydrazinolysiert und sauer gewaschen werden. Hierbei muss
die Hydrazidfunktion zuerst eingeführt werden, um die Reaktion des Aldehyds mit freiem
Hydrazin zu verhindern. Es bot sich an, den ohnehin notwendigen Waschschritt unter
Bedingungen durchzuführen, unter denen auch das Acetal gespalten wird.
Zur Überprüfung der Umsetzung wurde nach dem Waschen das Hybridgel teilweise mit 2,4-
DNPH und teilweise mit 4-NBA angefärbt (Abb.80).
Dabei konnte festgestellt werden, dass mit 2,4-DNPH tatsächlich nur die Bereiche angefärbt
werden, die eine Aldehydfunktion tragen sollen. Mit 4-NBA findet man jedoch auch eine
schwächere Anfärbung der unmodifizierten Bereiche und lediglich die Aldehyd-tragenden
NH
NH2
O
NH
OO
H
NH
NH2
O
NH
OO
H
NH
NO
NO2
NH
OO
H
NH
NH2
O
NH
ON
HNH
NO2
NO22,4-DNPHNaCNBH3AcOH0.5 mol / lpH = 2.7
4-NBA
(P20)
A
B
A
B
(P21)
(P20)
A
B
A
B
(P22) 0 5 mm
Abb.80: Anfärbung des Hydrazid/Aldehyd-Hybridgels mit 2,4-DNPH und 4-NBA. Rechts
sind die Bilder der Gele vergrößert dargestellt, wobei die Grautönung der Intensität der
Gelbfärbung entspricht. Die einzelnen Bereiche sind zur Verdeutlichung nachträglich
schwarz umrandet worden. Man erkennt im oberen Fall deutlich die Anfärbung der Matrix
mit 4-NBA.

95
Inseln bleiben farblos. Diese Befunde entsprechen den bereits in Kap.5.6 besprochenen
Ergebnissen. Dass die Aldehydbereiche nicht davon betroffen sind, lässt sich daraus erklären,
dass hier zwar auch eine Funktionalisierung der Gelmatrix stattfindet, die Hydrazinreste
jedoch nach Freisetzung der Aldehydfunktionen mit letzteren unter Ausbildung weiterer
Quervernetzungen reagieren (Abb.81).
Während es für die alleinige Hydrazin-Funktionalisierung irrelevant ist, ob diese auch an der
Matrix stattfindet, ist dieser Befund für ein Sandwich-Gel sehr ungünstig. Daher wurde eine
Möglichkeit gesucht, die reaktiven Matrix-Reste in einem Capping-Schritt zu deaktivieren.
6.2.2 Capping von reaktiven Resten der N,N-Dimethylpolyacrylamid-Matrix
Als Capping-Reagenz wurde eine Verbindung gesucht, die eine Reaktivität gegenüber (wie
auch immer gearteten) reaktiven Resten aufweist und nach der Anbindung an das Polymer die
Eigenschaften des Gels nicht stark verändert. Grundsätzlich war in dieser Hinsicht die
Einführung eines kleinen, mäßig polaren Restes zweckmäßig. Zusätzlich durfte das
potentielle Reagenz die Esterfunktionalitäten nicht angreifen.
2-Aminoethanol wurde als ein potentielles derartiges Reagenz angesehen, wobei davon
ausgegangen wurde, dass ohne die Anwesenheit von Wasser keine Spaltung des Esters zu
erwarten ist.
NH
NH2
NH
NH2
O
NH
NH
ON H
NH
NH2
NH
NH2
O
NH
OO
H
NH
NH2
NH
N
H
NO2
NH
NO
H
NO2
NH
NH
ON H
4-NBA
A
B
A
B
A
B
(P20) (P20’) (P21) Abb.81: Chemische Erklärung für die unterschiedlich gefärbten Bereiche im
Hydrazid/Aldehyd-Hybridgel bei Anfärbung mit 4-NBA.
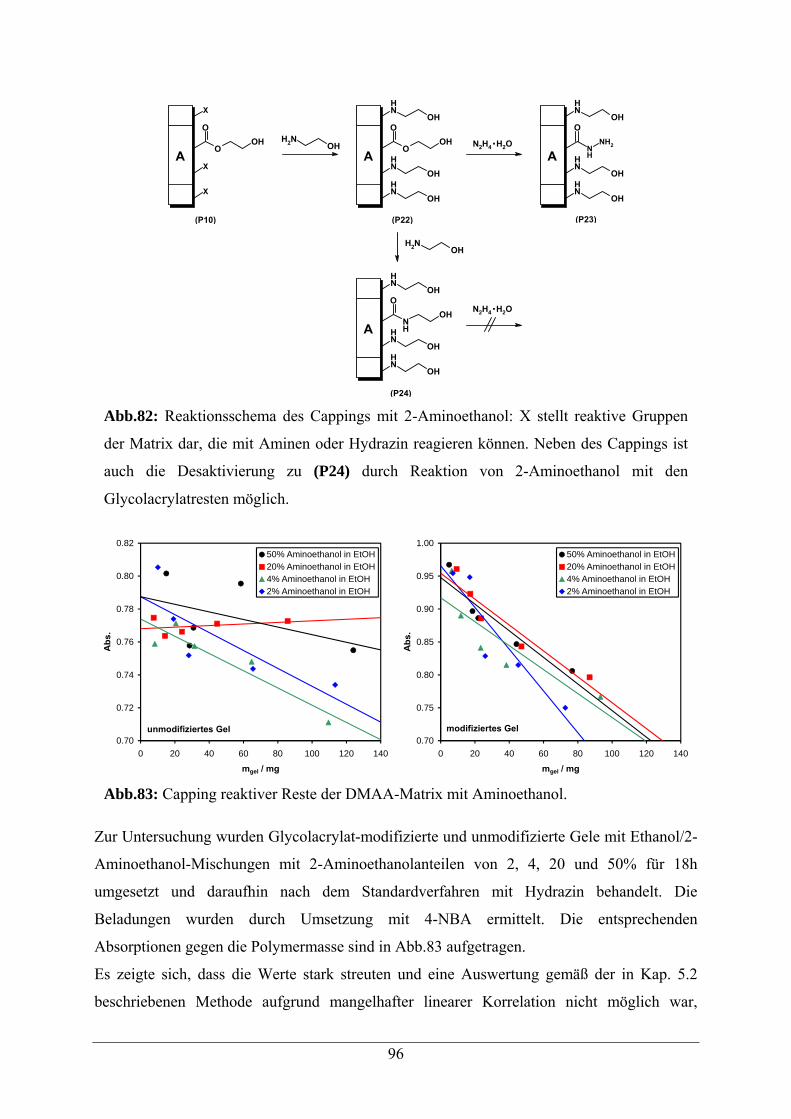
96
Zur Untersuchung wurden Glycolacrylat-modifizierte und unmodifizierte Gele mit Ethanol/2-
Aminoethanol-Mischungen mit 2-Aminoethanolanteilen von 2, 4, 20 und 50% für 18h
umgesetzt und daraufhin nach dem Standardverfahren mit Hydrazin behandelt. Die
Beladungen wurden durch Umsetzung mit 4-NBA ermittelt. Die entsprechenden
Absorptionen gegen die Polymermasse sind in Abb.83 aufgetragen.
Es zeigte sich, dass die Werte stark streuten und eine Auswertung gemäß der in Kap. 5.2
beschriebenen Methode aufgrund mangelhafter linearer Korrelation nicht möglich war,
0.70
0.75
0.80
0.85
0.90
0.95
1.00
0 20 40 60 80 100 120 140
mgel / mg
Abs
.
50% Aminoethanol in EtOH20% Aminoethanol in EtOH4% Aminoethanol in EtOH2% Aminoethanol in EtOH
modifiziertes Gel0.70
0.72
0.74
0.76
0.78
0.80
0.82
0 20 40 60 80 100 120 140
mgel / mg
Abs
.
50% Aminoethanol in EtOH20% Aminoethanol in EtOH4% Aminoethanol in EtOH2% Aminoethanol in EtOH
unmodifiziertes Gel
Abb.83: Capping reaktiver Reste der DMAA-Matrix mit Aminoethanol.
X
O
OOH
X
X
O
OOHOH
NH2
OHNH2
OHNH
OHNH
OHNH
O
NH
NH2
OHNH
OHNH
OHNH
N2H4 H2O.
NH
OOH
OHNH
OHNH
OHNH
N2H4 H2O.
A
(P10)
A A
A
(P22) (P23)
(P24) Abb.82: Reaktionsschema des Cappings mit 2-Aminoethanol: X stellt reaktive Gruppen
der Matrix dar, die mit Aminen oder Hydrazin reagieren können. Neben des Cappings ist
auch die Desaktivierung zu (P24) durch Reaktion von 2-Aminoethanol mit den
Glycolacrylatresten möglich.
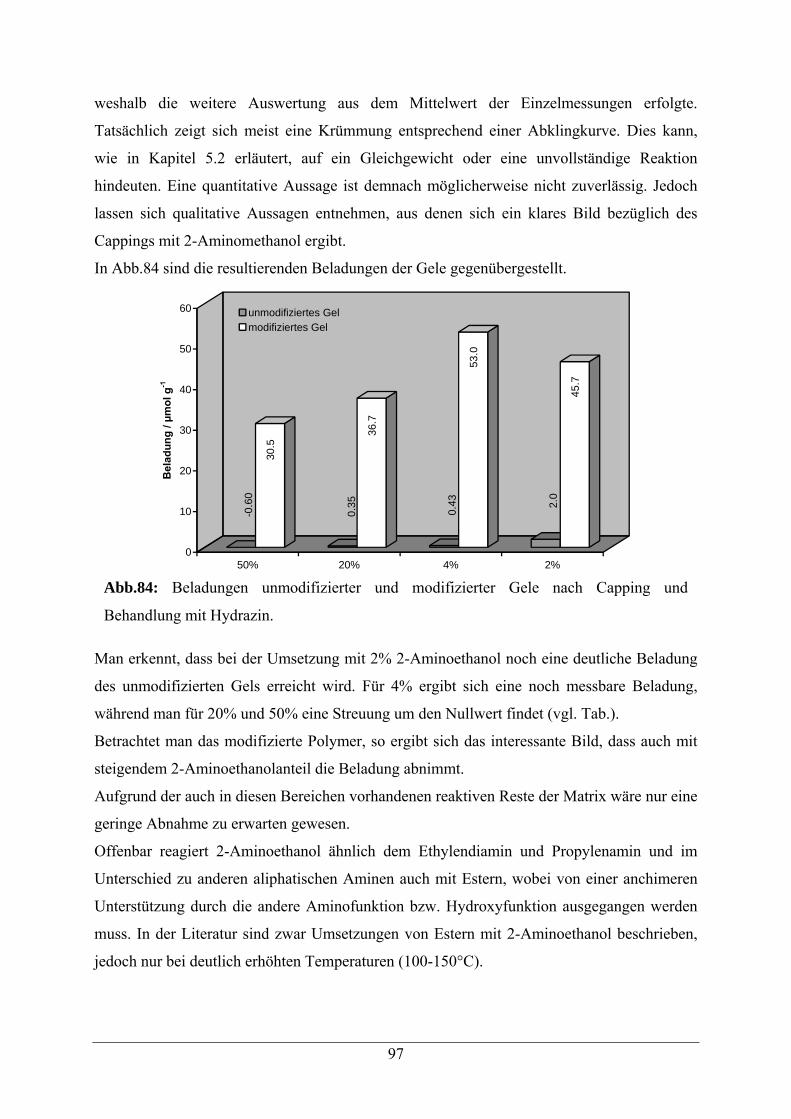
97
weshalb die weitere Auswertung aus dem Mittelwert der Einzelmessungen erfolgte.
Tatsächlich zeigt sich meist eine Krümmung entsprechend einer Abklingkurve. Dies kann,
wie in Kapitel 5.2 erläutert, auf ein Gleichgewicht oder eine unvollständige Reaktion
hindeuten. Eine quantitative Aussage ist demnach möglicherweise nicht zuverlässig. Jedoch
lassen sich qualitative Aussagen entnehmen, aus denen sich ein klares Bild bezüglich des
Cappings mit 2-Aminomethanol ergibt.
In Abb.84 sind die resultierenden Beladungen der Gele gegenübergestellt.
Man erkennt, dass bei der Umsetzung mit 2% 2-Aminoethanol noch eine deutliche Beladung
des unmodifizierten Gels erreicht wird. Für 4% ergibt sich eine noch messbare Beladung,
während man für 20% und 50% eine Streuung um den Nullwert findet (vgl. Tab.).
Betrachtet man das modifizierte Polymer, so ergibt sich das interessante Bild, dass auch mit
steigendem 2-Aminoethanolanteil die Beladung abnimmt.
Aufgrund der auch in diesen Bereichen vorhandenen reaktiven Reste der Matrix wäre nur eine
geringe Abnahme zu erwarten gewesen.
Offenbar reagiert 2-Aminoethanol ähnlich dem Ethylendiamin und Propylenamin und im
Unterschied zu anderen aliphatischen Aminen auch mit Estern, wobei von einer anchimeren
Unterstützung durch die andere Aminofunktion bzw. Hydroxyfunktion ausgegangen werden
muss. In der Literatur sind zwar Umsetzungen von Estern mit 2-Aminoethanol beschrieben,
jedoch nur bei deutlich erhöhten Temperaturen (100-150°C).
-0.6
0
30.5
0.35
36.7
0.43
53.0
2.0
45.7
0
10
20
30
40
50
60
Bel
adun
g/µ
mol
g-1
50% 20% 4% 2%
unmodifiziertes Gelmodifiziertes Gel
Abb.84: Beladungen unmodifizierter und modifizierter Gele nach Capping und
Behandlung mit Hydrazin.

98
Trotz dieser Abnahme der Beladung erscheint das Capping unter Verwendung von höheren
2-Aminoethanol-Anteilen als eine gute Möglichkeit zur Generierung von teil-Hydrazid-
funktionalisierten Hybrid-Gelen.
6.3 Kovalente Anbindung von N,N-Dimethylpolyacrylamiden an Glas-
Oberflächen
Für einige Anwendungen ist die kovalente Anbindung von N,N-Dimethylacrylamiden an
Glas-Oberflächen sinnvoll. Um diese Anbindung zu erreichen, muss die Glasoberfläche
entsprechend funktionalisiert werden. Ein Reagenz für diese Funktionalisierung stellt das
3-(Triethoxysilyl)propylacrylamid (41) dar, welches durch die Umsetzung von
3-(Triethoxysilyl)propylamin mit Acryloylchlorid erhalten wird (Abb.85).[35]
Die Funktionalisierung der Glas-Oberfläche erfolgt durch Behandlung mit einer 5%igen
Lösung von (41) in Ethanol. Nach Abtropfen der Flüssigkeit wird die Oberfläche kurz erhitzt
und gewaschen.
Die Polymerisation von N,N-Dimethylacrylamid auf der so behandelten Oberfläche führt zu
kovalent mit der Glasoberfläche verbundenen Polymeren (Abb.86).
Si NH
O
OO
O
Si NH2
O
OO
Cl
O+
(40) (12) (41) Abb.85: Synthese von 3-(Triethoxysilyl)propylacrylamid (41).
(EtO)3Si NH
O
OH
OH
OH
Glas
O
Si
Si NH
O
OO
O
NH
O
O
Si NH
O
O
O
O
NH
O
NH
O
Si
SiO
O
O
O
Si NH
O
O
O CONMe2
CONMe2
CONMe2
(41)
N,N-Dimethyl-acrylamid (2)Methylenbis-acrylamid (3)
TMEDA / APSGlas Glas
Abb.86: Kovalente Anbindung von N,N-Dimethylacrylamid-Polymeren an Glas-
Oberflächen.

99
7 Zusammenfassung und Ausblick
Während Methoden wie die Gelelektrophorese z.B. bei der Trennung von Oligonukleotiden
oder Peptiden eine breite Anwendung finden, wird das Polymergel selbst von vielen
Anwendern als inert angesehen. Auf der anderen Seite ist das Quellverhalten ein komplexes
Gebiet der Polymerforschung, wobei eine vollständige Beschreibung bis heute nicht erreicht
wurde.
In dieser Arbeit wurde versucht, dazu beizutragen, eine Verbindung zwischen diesen zwei
Bereichen zu schaffen. Es wurde gezeigt, dass sich N,N-Dimethylacrylamid-Copolymere, wie
sie z.B. bei der Herstellung von DNA-Chips Verwendung finden,[35] auch in Zusammenhang
mit in der Anwendung üblichen Reagenzien mitnichten inert verhalten.
Gerade bei kritischen Implementierungen, die schrittweise Prozeduren unter wechselnden
Bedingungen erfordern, wie z.B. DNA-Computer oder das ElektroSpread-Verfahren, können
diese Faktoren große Auswirkungen haben. Für die Planung derartiger Verfahren ist demnach
eine vorhergehende polymerchemische Untersuchung des Verhaltens der Matrizes
unumgänglich.
Es wurde das Quellverhalten von nicht funktionalisierten N,N-Dimethylacrylamid-Polymeren
in unterschiedlichen Lösungsmitteln und Lösungsmittelgemischen untersucht, wobei für reine
Lösungsmittel die Wechselwirkungen mit dem Polymer über die Flory-Huggins-
Wechselwirkungsparameter quantifiziert wurden. Bei der Untersuchung des Quellverhaltens
in Lösungsmittelgemischen wurden teilweise sehr starke Abweichungen vom idealen
Verhalten gefunden. Da in vielen Fällen starke Abweichungen auch bei Mischungen mit nur
geringem Anteil der einen Komponente gefunden wurden, sind diese Abweichungen auch für
Fälle relevant, in denen nur ein geringer Anteil eines Cosolvens beigemischt ist. Ebenso
wurden einige Fälle betrachtet, die den Bedingungen chemischer Umsetzungen modifizierter
Copolymere bzw. den Bedingungen beim Lösungsmittelwechsel entsprechen.
Die Abweichungen des Quellverhaltens in Lösungsmittelgemischen vom idealen Verlauf
zeigten ein sehr vielfältiges Bild. In vielen Fällen konnte eine An- oder Abreicherung von
Komponenten nachgewiesen werden, wobei dies nicht unbedingt mit den ermittelten Flory-
Huggins-Parametern korrelierte. Ein ebenso vielfältiges Bild ergab sich bei der Untersuchung
von wässrigen Lösungen. Während Natriumchlorid im Gel abgereichert ist, findet man im
Fall von Natriumacetat eine Anreicherung, in beiden Fällen jedoch eine Abnahme des

100
Quellvolumens. Eine Anreicherung im Gel wurde ebenfalls für Harnstofflösungen gefunden,
wobei das Quellvolumen weitgehend konstant blieb.
Ein Hauptaugenmerk dieser Arbeit lag auf der Untersuchung modifizierter Polymergele, die
zur Immobilisierung von Oligonukleotiden verwendet werden können. Dabei wurden
literaturbekannte ebenso wie neue Modifikationen untersucht. Für die Aldehyd- und
Hydrazid-Modifikation konnte die Immobilisierung von Oligonukleotiden und deren
Hybridisierung mit komplementären Sequenzen gezeigt werden. Denaturierungsexperimente
zeigten die langsame Diffusion im Polymer, was im Rahmen des angestrebten ElektroSpread-
Verfahrens günstig bezüglich des Erhalts der Ortsinformation, jedoch ungünstig für den
Transport ungeladener Bausteine und Reagenzien ist.
Die Eigenschaften von Polymergelen werden zu einem großen Teil von der Anzahl und Art
der Quervernetzungen bestimmt. Neben der Quervernetzung durch Copolymerisation wurden
in dieser Arbeit Möglichkeiten einer redox-schaltbaren Quervernetzung (Disulfid-verbrückte
N,N-Dimethylpolyacrylamide) sowie die effektive nicht-kovalente Quervernetzung bei
Boronsäuregelen untersucht. Derartige „intelligente“ Polymere mit schaltbaren Eigenschaften
eröffnen bei weiterer Entwicklung eine Vielzahl von interessanten Einsatzmöglichkeiten. Sind
die Kettenlängen kurz genug, so lässt sich der Sol/Gel-Übergang z.B. durch Redox-Prozesse
steuern, was beispielsweise die reversible Gelbildung in mikrofluidischen Strukturen
ermöglichen würde.
Einen weiteren Aspekt der Untersuchungen stellte die Herstellung von Hybridgelen dar, die
unterschiedlich funktionalisierte Bereiche aufweisen. Dabei wurde ein Aldehyd/Hydrazid-
Hybridgel hergestellt, wobei die funktionalisierten Bereiche durch nicht funktionalisierte
getrennt waren. Diese Anordnung stellt den Prototyp für eine potentielle Anwendung von
Sandwich-Strukturen im ElektroSpread-Verfahren dar. Das Problem der Matrix-
Funktionalisierung durch Hydrazin konnte durch ein Capping aktiver Funktionalitäten der
Gelmatrix gelöst werden.
Der Implementierung eines Spread-analogen Verfahrens auf der Basis von Hydrogelen ohne
Stofftransport im elektrischen Feld steht insbesondere entgegen, dass sich die Lösungen nicht
wie im Spread-Verfahren durch Zentrifugieren aus den Gelen entfernen lassen und somit
Edukte wie auch Produkte durch Diffusion in das Gel hinein und aus dem Gel heraus

101
transportiert werden müssen, was zur Folge hat, dass für die quantitative Entfernung aus dem
Gel sehr große Gegenvolumina und lange Zeiten zu erwarten sind.
Im Hinblick auf das ElektroSpread-Verfahren sind die chemischen Vorarbeiten somit
weitgehend abgeschlossen: Es wurden mit der Anbindung von Hydrazid-Oligonukleotiden an
Aldehyd-Gele und der inversen Anbindung von Aldehyd-Oligonukleotiden an Hydrazid-Gele
zwei orthogonale Immobilisierungsverfahren untersucht und ihre Anwendbarkeit für
Immobilisierungs- und Hybridisierungsexperimente getestet. Die Untersuchung der
Wechselwirkungen von N,N-Dimethylacrylamid-Copolymeren mit Lösungsmitteln und
Reagenzien zeigte, dass für die Denaturierung Formamid das am besten geeignete Reagenz
darstellt.
Die weitere Entwicklung beinhaltet hauptsächlich den Aufbau einer entsprechenden
Apparatur, in der das ElektroSpread-Verfahren durchgeführt werden kann.

Experimenteller Teil

103
1 Material und Methoden
1.1 Spektroskopische Methoden
1.1.1 Magnetische Kernresonanzspektroskopie (NMR)
Sämtliche NMR-Spektren wurden an den Geräten DPX 200 (200MHz) DRX 400 (400MHz)
sowie DRX 600 (600MHz) der Fa. Bruker aufgenommen.
Die Kalibrierung erfolgte auf die charakteristischen chemischen Verschiebungen der
Lösungsmittel.[82] Wurde das Lösungsmittelsignal überlagert, so wurde auf das TMS-Signal
kalibriert.
Die chemische Verschiebung δ wurde in ppm angegeben.
Kennzeichnung der Signalmultiplizitäten: s = Singulett, d = Dublett, t = Triplett, q = Quartett,
n = Quintett, x = Sextett, m = Multiplett. Zusammengesetzte Multiplizitäten werden durch
entsprechende Buchstabenreihen dargestellt, wobei diejenigen niederer Ordnung denen
höherer vorangestellt sind, z.B. dt = Dublett vom Triplett.
Die 13C- und 31P-Spektren wurden unter 1H-Breitbandentkopplung aufgenommen.
1.1.2 IR-Spektroskopie
Die IR-Spektren wurden mit dem Fourier-Transform-IR-Spektrometer Vector22 der Fa.
Bruker aufgenommen.
1.1.3 UV/Vis – Spektroskopie
Die Aufnahme der UV-Spektren erfolgte an dem Gerät Cary 13E der Fa. Varian.
Die Konzentrationsbestimmung der Oligonukleotide erfolgte durch Addition folgender
Extinktionskoeffizient-Inkremente ε254 bei λ = 254 nm und pH = 7.0 unter Anwendung des
Lambert Beer’schen Gesetzes:[83]
Chromophor ε254 / l mol-1 cm-1
Thymidinmonophosphat 7250
Cytidinmonophosphat 6541
Adenosinmonophosphat 13200
Guanosinmonophosphat 13679

104
1.2 Massenspektrometrie
1.2.1 Fast-Atom-Bombardment (FAB)
Die Messungen erfolgten an einem Massenspektrometer VG Autospec bei einer
Beschleunigungsspannung von 8 kV und Beschuss durch Cs+. Als Matrix wurden
3-Nitrobenzylalkohol und Glycerin sowie Milchsäure verwendet.
1.2.2 Matrix-Assisted Laser Desorption Ionisation / Time Of Flight (MALDI-TOF)
Die Spektren wurden an einem Voyager-DETM-Massenspektrometer der Fa. Perseptive
Biosystems aufgenommen. Die Messung erfolgte im Ultrahochvakuum bei ca. 10-7 Torr durch
Bestrahlung mit einem N2-Laser ( λ = 337 nm ), die Beschleunigungsspannung betrug 25 kV.
Als Matrix für Oligonukleotide diente 6-Aza-2-thiothymin + Glycerin (ATTG). Die Spektren
wurden im Linear- und Negative-Ion-Mode aufgenommen.
1.3 Chromatographische Methoden
1.3.1 Dünnschichtchromatographie
Es wurden Fertigfolien SIL G/UV254 der Fa. Macherey & Nagel ( 40 x 80 mm, Schichtdicke
0.25 mm Kieselgel, Fluoreszenzindikator) verwendet. Die Peaks wurden bei λ = 254 nm UV-
detektiert. Verbindungen mit primären Aminofunktionen wurden durch Tauchen in 10%ige
ethanolische Ninhydrin-Lsg. und anschließendes Erwärmen im Luftstrom detektiert.
Verwendete Laufmittelsysteme: (Angaben als v/v)
LM-A: CH2Cl2 / MeOH = 10:1
LM-B: CH2Cl2 / MeOH / NEt3 = 100:10:1
LM-C: CH2Cl2
1.3.2 Säulenchromatographie
Verwendetes Kieselgel: ICN Biomedicals, Silica 60 Å, Korngröße 0.032 – 0.063 mm, Lot
226.
1.3.3 High-Performance-Liquid-Chromatography (HPLC)
Es wurde ein gradientenfähiges Kontron-HPLC-System, bestehend aus zwei Pumpen 440 mit
Hinterkopfspülung (Fluss: 1 ml min-1), einem Autosampler 465 und einem Diode Array-
Detektor (190-798 nm) verwendet. Als funktionssteuernde Software stand das Kroma System

105
2000 zur Verfügung. Die verwendete HPLC-Säule war eine CC250/4-Nucleosil 120-5-C18-
Säule der Fa. Macherey&Nagel.
Die HPLC-Analysen wurden unter Verwendung eines 0.1 mol l-1 Triethylammonium-
acetatpuffers durchgeführt bei ansteigendem Gehalt an Acetonitril (auf 30% Acetonitril in
29min, auf 80% in 5min). Die Detektion erfolgte bei λ = 254 nm, λ = 273 nm (DNA) und
λ = 480 nm, λ = 490 nm (Fluorescein).
1.4 Automatisierte Oligonucleotidsynthesen
Automatisierte Oligonucleotidsynthesen wurden an einem Synthesizer Gene Assembler Plus
der Fa. Pharmacia Biosystems durchgeführt. Die Synthesen erfolgten nach dem
Phosphoamiditverfahren an CPG-Festphasen im 1.3 µmol - Maßstab.
Als Aktivierungsreagenzien wurden 1H-Tetrazol sowie 4,5-Dicyanoimidazol verwendet.
1.5 Imager
Die Geldokumentation erfolgte mittels Fluor-STM-Multimager der Fa. Biorad.
1.5 Chemikalien
1.5.1 Verwendete Lösungsmittel
Die Lösungsmittel wurden von Biosolve, J.T.Baker, Merck, Riedel-de Haën oder VWR
bezogen. Dabei kamen für Synthesen Lösungsmittel der Qualität p.a. zum Einsatz. DMF der
Fa. Biosolve kam in peptide synthesis grade zum Einsatz.
Die Trocknung der Lösungsmitteln erfolgte, soweit nötig, wie folgt:
Methylenchlorid zur Säulenchromatographie wurde ausgehend von technischem Methylen-
chlorid über Molsieb 4Å refluxiert und destilliert.
1.5.2 Verwendete Feinchemikalien
Die Chemikalien wurden von Acros, Aldrich, Fluka, Merck oder Riedel-de Haën bezogen.
Alle Chemikalien entsprachen dem Qualitätsstandard des Herstellers, zumindest jedoch „zur
Synthese“.
1.5.3 Chemikalien für automatisierte Oligonucleotidsynthesen
Phosphoamidite wurden von Glen Research bezogen. Tetrazol stammte von Amersham
Pharmacia Biotech, 4,5-Dicyanoimidazol von Proligo Biochemie GmbH.

106
Für das Fluoresceinlabelling diente das Fluorescein-Phosphoamidit FluoroPrimeTM der Fa.
Amersham Pharmacia Biotech.
Festphasen:
Primer-Support (1.3 µmol) CPG von Glen Research.
1.6 Stammlösungen
1.6.1 Gelstammlösung A
1.8 M N,N-Dimethylacrylamid
0.04 M N,N-Methylenbisacrylamid
in Aqua bidest.
1.6.1 Gelstammlösung B
3.6 M N,N-Dimethylacrylamid
0.08 M N,N-Methylenbisacrylamid
in Aqua bidest.

107
2 Allgemeine Arbeitsvorschriften
AAV1.1 Lipophilisierung von Objektträgern aus Glas
Objektträger aus Glas (76mm x 26mm) wurden zuerst mit Aceton und daraufhin mit Ethanol
jeweils mehrere Stunden gewaschen. Nach der Spülung mit Aqua bidest. wurden die
Objektträger zuerst 18h in Natronlauge (10%) gelagert, bis zum Neutralen mit Aqua bidest.
gewaschen und daraufhin 18h mit 1M Salzsäure behandelt. Nach dem Waschen mit Aqua
bidest bis zur neutralen Reaktion wurden die Objektträger im Trockenschrank getrocknet und
nach dem Abkühlen mehrere Stunden in eine 5%iger Lösung von Trimethoxyoctadecylsilan
in Methanol getaucht. Nach dem Tauchvorgang ließ man überschüssige Lösung abtropfen.
Man erhitzte kurzzeitig im Luftstrom bei 350°C, ließ abkühlen, spülte überschüssiges
Material mit Ethanol von der Glasoberfläche und trocknete.
Die Objektträger wurden für die Herstellung von Mikrogelen herangezogen, wenn sie optisch
keine matten Stellen aufwiesen und die Benetzung mit Wasser eine gleichmäßige Lipophilie
anzeigte.
AAV1.2 Acrylamid-Funktionalisierung von Objektträgern aus Glas
Objektträger aus Glas (76 mm x 26 mm) wurden zuerst mit Aceton und daraufhin mit Ethanol
jeweils mehrere Stunden gewaschen. Nach der Spülung mit Aqua bidest. wurden die
Objektträger zuerst 18h in Natronlauge (10%) gelagert, bis zum Neutralen mit Aqua bidest.
gewaschen und daraufhin 18h mit 1M Salzsäure behandelt. Nach dem Waschen mit Aqua
bidest bis zur neutralen Reaktion wurden die Objektträger im Trockenschrank getrocknet und
nach dem Abkühlen mehrere Stunden in eine 5%iger Lösung von 3-Triethoxy-
silylpropylacrylamid in Methanol getaucht. Nach dem Tauchvorgang ließ man überschüssige
Lösung abtropfen. Man erhitzte kurzzeitig im Luftstrom bei 150°C, ließ abkühlen, spülte
überschüssiges Material mit Ethanol von der Glasoberfläche und trocknete.
AAV2 Herstellung von Polymer-Gelen
Die Herstellung von den hier vorgestellten Polymergelen erfolgte in einer Gelkammer, deren
Ober- und Unterseite durch eine lipophilisierten Objektträger gebildet wurde. Die Seitenteile
bildeten jeweils rechteckige Teflonstücke eine Dicke von 1.5 mm. Die Kammer wurde jeweils
mit 1ml der Polymerisationslösung befüllt, was zu Abmessungen von ca. 1.5 mm x 15 mm x
45 mm bezüglich des Polymergels führte. Nach der Polymerisation wurde das Polymer

108
vorsichtig von den Glasplatten gelöst und in allen Fällen 16h mit einem großen Überschuss
Aqua bidest. gespült.
Die Trocknung von Polymergelen erfolgte im dynamischen Ölpumpenvakuum über
Phosphorpentoxid und dauerte je nach Größe der Polymerstücke einen oder auch mehrere
Tage. Die getrockneten Polymere zeigten an der Luft kein deutlich hygroskopisches
Verhalten.
AAV2.1 Herstellung von quervernetzten N,N-Dimethylacrylamid-Polymeren (P1)
Die Polymerisationsmischung bestand aus 0.5 ml Gelstammlösung, 0.5 ml Aqua bidest. und
20 µl einer 10%igen APS-Lösung. Die Polymerisation wurde durch Zugabe von 5 µl TMEDA
gestartet, woraufhin die Lösung nach gründlicher Durchmischung zügig in die Gelkammer
eingefüllt wurde.
Nach dem Phasenübergang wartete man mindestens 30min, bevor es der Kammer entnommen
wurde.
AAV2.2 Herstellung von N,N-Dimethylacrylamid-Glycolacrylat-Copolymeren (P10)
Die Polymerisationsmischung bestand aus 0.5 ml Gelstammlösung, 0.5 ml einer 0.2M Lösung
von Glycolacrylat in Aqua bidest. und 20 µl einer 10%igen APS-Lösung. Die Polymerisation
wurde durch Zugabe von 5 µl TMEDA gestartet, woraufhin die Lösung nach gründlicher
Durchmischung zügig in die Gelkammer eingefüllt wurde.
Nach dem Phasenübergang wartete man mindestens 30min, bevor es der Kammer entnommen
wurde.
AAV2.3 Herstellung von N,N-Dimethylacrylamid-Acrylsäure-Copolymeren (P12)
Das gemäß AAV2.2 hergestellte Copolymer (P10) wurde abgetropft und 24h mit 15 ml
Natronlauge (10%) ungesetzt. Daraufhin wurde mit Wasser neutral gewaschen, wobei das
Polymer-Gel deutlich aufquoll. Dies wurde im letzten Schritt mit HCl (1 mol l-1) 24 h
umgesetzt und wiederum mit Wasser neutral gewaschen.
AAV2.4 Herstellung von N,N-Dimethylacrylamid-Ethylendiaminmonoacrylamid-Co-
polymeren (P11)
Das gemäß AAV2.2 hergestellte Copolymer (P10) wurde abgetropft und zweimal 1 h mit je
18 ml Ethanol abs. zur vollständigen Entfernung des Wassers gespült. Daraufhin wurde 1g
Gel 24 h mit 18 ml der Mischung aus Ethylendiamin und Ethanol (1:1 v/v) unter

109
gelegentlichem Schwenken umgesetzt. Daraufhin wurde mehrmals 1 h mit je 18 ml Ethanol
abs. gespült. Das Polymer wurde in Ethanol gelagert.
AAV2.5 Herstellung von N,N-Dimethylacrylamid-Acrylsäurehydrazid-Copolymeren
(P13)
1 g des gemäß AAV2.2 hergestellten Copolymers (P10) wurde abgetropft und mit 10 ml
Hydrazinhydrat 18 h umgesetzt und18h mit 20 ml 5%iger Essigsäure gespült sowie mit Aqua
bidest. neutral gewaschen.
AAV2.6 Herstellung von N,N- Dimethylacrylamid-3-Acrylamidophenylboronsäure-
Copolymeren (P15)
Für die Herstellung von 1ml Gel wurden 19.2 mg 3-Acrylamidophenylboronsäure (29) in der
Mischung aus 0.25 ml DMF und 0.25 ml Aqua bidest. gelöst, mit 0.5 ml Gelstammlösung und
25 µl einer 10%igen APS-Lösung versetzt und durch Zugabe von 7.5 µl TMEDA zur
Polymerisation gebracht. Das Polymer wurde mit Aqua bidest. erschöpfend gespült.
AAV2.7 Herstellung von N,N-Dimethylacrylamid-5-Acrylamidovaleraldehyd-Copoly-
meren (P4)
Die Polymerisationsmischung bestand aus 0.5 ml Gelstammlösung, 0.5 ml einer 0.2 mol l-1
Lösung von N-(5,6-Isopropylidendioxyhexyl)acrylamid (4) in Aqua bidest. und 20 µl einer
10%igen APS-Lösung. Die Polymerisation wurde durch Zugabe von 5 µl TMEDA gestartet,
woraufhin die Lösung nach gründlicher Durchmischung zügig in die Gelkammer eingefüllt
wurde.
Nach dem Phasenübergang wartete man mindestens 30 min, bevor es der Kammer
entnommen wurde.
Das erhaltene Copolymer (P2) wurde mit 80% Essigsäure über Nacht bei RT behandelt,
daraufhin mit Aqua bidest. neutral gewaschen und 1 h mit 0.1 M NaIO4-Lösung oxidiert. Das
erhaltene Polymer wurde mit Aqua bidest. erschöpfend gespült.
AAV2.8 Herstellung von N,N-Dimethylacrylamid-2-Acrylamidoacetaldehyd-Copoly-
meren (P6)
Die Polymerisationsmischung bestand aus 0.5 ml Gelstammlösung, 0.5 ml einer 0.2 mol l-1
Lösung von N-(2,2-Diethoxyethyl)acrylamid (5) in Aqua bidest. und 20 µl einer 10%igen

110
APS-Lösung. Die Polymerisation wurde durch Zugabe von 5 µl TMEDA gestartet, woraufhin
die Lösung nach gründlicher Durchmischung zügig in die Gelkammer eingefüllt wurde.
Nach dem Phasenübergang wartete man mindestens 30 min, bevor es der Kammer
entnommen wurde.
Das erhaltene Copolymer (P5) wurde mit 80% Essigsäure über Nacht bei RT behandelt und
daraufhin mit Aqua bidest. neutral gewaschen.
AAV2.9 Herstellung von N,N-Dimethylacrylamid-N-(2-Acrylamidoethyl)-4-formylbenz-
amid-Copolymeren (P8)
Die Polymerisationsmischung bestand aus 0.5 ml Gelstammlösung, 0.5 ml einer 0.2 M
Lösung von N-(2,2-Diethoxyethyl)acrylamid (6) in Aqua bidest./DMF (1:1 v/v) und 25 µl
einer 10%igen APS-Lösung. Die Polymerisation wurde durch Zugabe von 7.5 µl TMEDA
gestartet, woraufhin die Lösung nach gründlicher Durchmischung zügig in die Gelkammer
eingefüllt wurde.
Nach dem Phasenübergang wartete man mindestens 30 min, bevor es der Kammer
entnommen wurde.
Das erhaltene Copolymer (P7) wurde mit 80% Essigsäure über Nacht bei RT behandelt und
daraufhin mit Aqua bidest. erschöpfend gewaschen.
AAV2.10 Herstellung von N,N-Dimethylacrylamid-N,N-Diacryloyl-L-cystindiethylester-
Copolymeren (P16)
Die Polymerisationsmischung bestand aus 0.25 ml Gelstammlösung B, 0.75 ml einer 0.075 M
Lösung von N,N-Diacryloyl-L-cystindiethylester (36) in DMF und 82 µl einer 10%igen APS-
Lösung. Die Polymerisation wurde durch Zugabe von 15 µl TMEDA gestartet, woraufhin die
Lösung nach gründlicher Durchmischung zügig in die Gelkammer eingefüllt wurde.
Nach dem Phasenübergang wartete man mindestens 30 min, bevor es der Kammer
entnommen wurde.
Das erhaltene Copolymer (P16) wurde mit Aqua bidest. erschöpfend gewaschen.
AAV3 Herstellung von Hybridpolymeren (P20)
Mithilfe eines 1.5 mm dicken Teflonkamms zwischen zwei lipophilisierten Glas-
Objektträgern wurden die Mischungen unterschiedlicher Monomerzusammensetzung
räumlich getrennt (A: 0.1 mol l-1 Glycolacrylat, 0.9 mol l-1 N,N-Dimethylacrylamid,
0.02 mol l-1 Methylenbisacrylamid; B: 0.1 mol l-1 N-(2,2-Diethoxyethyl)acrylamid (5), 0.9

111
mol l-1 N,N-Dimethylacrylamid, 0.02 mol l-1 Methylenbisacrylamid ) zur Polymerisation
gebracht. Direkt nach dem Phasenübergang zum Gel wurde der Kamm vorsichtig entfernt und
die Polymerinseln wurden zwischen den Glasscheiben mit Aqua bidest. gewaschen und im
Luftstrom oberflächlich getrocknet. Daraufhin erfolgte die Abdichtung an drei Seiten mit
Teflonstücken von 1.5 mm Dicke. Der Raum um die Inseln wurde zügig mit der
Polymerisationsmischung ohne Zugabe von funkionalisierten Monomeren (AAV2.1) unter
Vermeidung von Luftblasen befüllt.
Nach dem Phasenübergang wartete man mindestens 30 min, bevor das Hybridgel der
Kammer entnommen wurde.
Das Gel wurde 30 min mit 18 ml Ethanol gewaschen, daraufhin 1 h mit 4 ml Hydrazinhydrat
bzw. 4 ml der Mischung aus Hydrazinhydrat und Ethanol (1:1, 1:3, 1:7, 1:13 v/v) behandelt,
1 h mit 10 ml 5%iger Essigsäure und dreimal mit je 20 ml Aqua bidest. gewaschen. Daraufhin
erfolgte die Behandlung mit 10 ml Essigsäure für 18 h. Das Gel wurde viermal mit je 25 ml
Aqua bidest. gewaschen und daraufhin 30 min mit 18 ml Ethanol.
AAV4 Anfärbung von Gelen
Die Beladungsbestimmung erfolgte in den meisten Fällen durch das in Kap. erläuterte Prinzip.
Dabei wurde das Gel mit einem farbigen oder UV-aktiven Reagenz versetzt, der an die
funktionellen Gruppen des Gels bindet. Die verbliebene Extinktion in Lösung wurde
bestimmt. Durch Umsetzung von Gelfragmenten unterschiedlicher Masse mit denselben
Volumina gleich konzentrierter Reagenzlösungen konnte aus der Auftragung der Gelmasse
gegen die Extinktion auf die Beladung zurückgeschlossen werden.
AAV4.1 Anfärbung von Aldehyd-Gelen mit 2,4-Dinitrophenylhydrazin
Polymerfragmente der Massen zwischen 20 und 180 mg in gequollenem Zustand wurden mit
je 100 µl bzw. 500 µl der Lösung von 100.45 mg käuflichem 2,4-Dinitrophenylhydrazin
(enthält ca. 50% Wasser) in 100 ml Ethanol abs. und 500 µl der Lösung von 75 mg NaCNBH3
in 7.5 ml 3%iger Essigsäure (pH = 2.7) versetzt. Nach 18 h wurde die überstehende Lösung
verdünnt, so dass die Extinktion im linearen Bereich lag, und die Extinktion durch UV/Vis-
Spektroskopie (365 nm) ermittelt.
Die tatsächliche Konzentration der 2,4-Dinitrophenylhydrazinlösung wurde durch UV/Vis-
Spektroskopie unter Anwendung des Lambert-Beer-Gesetzes bestimmt (ε = 22000 l mol-1 cm-1
bei 365 nm) da der Wassergehalt käuflichen 2,4-Dinitrophenylhydrazins variiert.

112
AAV4.2 Anfärbung von Hydrazid-Gelen mit 4-Nitrobenzaldehyd
Gequollene Gelfragmente von Massen mGel zwischen 15 und 190 mg wurden zum Ausgleich
der Wasserkonzentration mit Wasser auf 190 mg aufgefüllt (mWasser = 190 mg - mGel) und mit
weiteren 300 µl H2O und 500 µl Ethanol abs. versetzt. Dazu wurden entweder 500 µl einer
20 mmol l-1 4-NBA-Lösung in Ethanol oder 250 µl einer 20 mmol l-1 4-NBA-Lösung in
Ethanol oder 500 µl einer 4 mmol l-1 4-NBA-Lösung in Ethanol gegeben.
Nach 18 h wurde die überstehende Lösung verdünnt, so dass die Extinktion im linearen
Bereich lag, und die Extinktion durch UV/Vis-Spektroskopie (266 nm) ermittelt.
(ε = 11002 l mol-1 cm-1 bei 266 nm)
AAV4.3 Anfärbung von Hydrazid-Gelen mit 2,4-Dinitrobenzaldehyd
Gequollene Gelfragmente von Massen mGel zwischen 15 und 190 mg wurden zum Ausgleich
der Wasserkonzentration mit Wasser auf 190 mg aufgefüllt (mWasser = 190 mg - mGel) und mit
500 µl einer 20 mmol l-1 2,4-DNBA-Lösung in Ethanol versetzt.
Nach 18 h wurde die überstehende Lösung verdünnt, so dass die Extinktion im linearen
Bereich lagt, und die Extinktion durch UV/Vis-Spektroskopie (239 nm) ermittelt.
(ε0 = 12550 l mol-1 cm-1 bei 239 nm)
AAV4.4 Kaiser-Test von Amino-Gelen
Ein getrocknetes Gelfragment der Masse von ca. 20 mg wurde mit 20µl der Lösungen A, B
und C versetzt und in einem verschraubbaren Eppendorfgefäß (1.5 ml) 10 min auf 70°C
erhitzt. Eine Blaufärbung des Polymers zeigte die Anwesenheit von Aminofunktionen an.
Lösung A: 0.5 mg Ninhydrin in 10 ml Ethanol
Lösung B: Mischung aus 8 g Phenol und 2 ml Ethanol
Lösung C: Mischung aus 1ml 1 mmol l-1 KCN in H2O und 49 ml Pyridin
AAV4.5 Qualitative Anfärbung von Hybridgelen
Zur Anfärbung der Hydrazidfunktionalitäten wurde für 18 h mit 18 ml gesättigter 4-NBA-
Lösung umgesetzt und mit Ethanol gespült bis keine weiter Entfärbung zu erkennen war.
Zur Anfärbung der Aldehydfunktionalitäten wurde das Gel 2 h mit 18 ml gesättigter 2,4-
DNPH-Lösung in Ethanol umgesetzt. Daraufhin wurde das Gel in die Lösung aus 27 mg
NaCNBH3 in 5 ml 0.5 mol l-1 Essigsäure überführt und nach 1 h erschöpfend mit Ethanol
gespült.

113
AAV5 Capping von N,N-Dimethylacrylamid-Glycolacrylat-Copolymeren ()
250 mg des gequollenen N,N-Dimethylacrylamid-Glycolacrylat-Copolymers wurden einmal
30min mit 4.5 ml Ethanol und einmal 2h mit 4.5 ml Ethanol gewaschen. Daraufhin wurde das
Polymer 18h mit 10 ml einer Ethanol/2-Aminoethanol-Mischung (1:1, 4:1, 24:1 oder 49:1
(v/v)) behandelt und zweimal je 1h mit 18 ml Ethanol gewaschen.
AAV6 Durchführung des Stress-Strain-Experimentes
Zur Durchführung des Stress-Strain-Experimentes wurde ein Gel gemäß AAV2 erstellt,
jedoch unter Verwendung von rechteckigen Teflon-Stücken von 2.0 mm Dicke. Nach der
Polymerisation wurden die Teflonstücke entfernt und das Gel ohne weiteren Waschschritt
verwendet. Das Gel wurde zwischen den Glasplatten waagerecht auf einer Waage positioniert
und senkrecht unter Zuhilfenahme von Metallringen als Führung mit an einer Metallstange
befestigten Gewichten belastet. Das Gesamtgewicht wurde mit Hilfe der Waage bestimmt.
Die Dicke des Gels wurde durch Fotographieren in der Waagerechten bestimmt.
AAV7.1 Synthese von Oligonukleotiden
Die Synthese der Oligonucleotide erfolgte am DNA-Synthesizer in 1.3 µmol-Ansätzen. Die
verwendeten Standard-Phosphoamidite wurden unmodifiziert als 0.1 mol l-1 Lösungen in
Acetonitril eingesetzt. Es wurde mit einer Kopplungszeit von 1 x 2min bei einem 6.25fachen
Überschuss an Phosphoamidit gearbeitet.
AAV7.2 Generierung von 5’-Hydrazid-Oligonukleotiden
Zur Entfernung des in der Festphase verbliebenen Lösungsmittels wurde einige Minuten bei
8000 Umdrehungen / min zentrifugiert. Die Festphase wurde entnommen und mit 1 ml der
1 mol l-1 N,N-Carbonyldiimidazol-Lösung in DMSO/Acetonitril 1:1(v/v) geschüttlet.
Daraufhin wurde die Festphase zweimal mit je 1 ml Acetonitril gewaschen und das in der
Festphase verbliebene Lösungsmittel jeweils durch Zentrifugieren entfernt.
Es wurden für 1-3h 500 µl der Mischung aus Hydrazinhydrat und Methanol 1:9 zugegeben.
Zur Entfernung des Hydrazins wurde die Lösung über eine Festphasen-Kartusche Oasis-HLB-
Carta (1 ml) gereinigt.
AAV7.3 Festphasenextraktion
Die Festphasen-Kartusche Oasis-HLB-Carta (1 ml) wurde zuerst mit 1 ml Methanol und
anschließend mit 1 ml Wasser gespült. Nach Einsinkenlassen der Probe wurde zweimal mit je

114
1 ml 5% igem Methanol gewaschen und danach mit 1 ml Methanol eluiert. Das Eluat wurde
in 1.5 ml Eppendorf-Gefäßen aufgefangen und am Vakuum-Konzentrator eingeengt.
AAV7.4 Entschützung von Oligonukletiden
Zur Entfernung des in der Festphase verbliebenen Lösungsmittels wurde einige Minuten bei
8000 Umdrehungen / min zentrifugiert. Die Festphase wurde entnommen und mit 1 ml
33%iger Ammoniaklösung 16h auf 55°C erhitzt.
Die Festphase wurde mehrmals mit je 200 µl Aqua bidest. gespült, bis die Spüllösung keine
gelbe Färbung mehr aufwies.
Die Lösungen wurden im Vakuum-Konzentrator bis zur Trockene eingeengt und je nach
Anwendung weiter aufgereinigt.

115
3 Synthesen
3.1 Synthese von 5,6-Isopropylidendioxyhexan-1-ol (8)
OHO
O
11.7 g Hexan-1,2,6-triol (87 mmol), 225 ml Chloroform, 75 ml Aceton (1.13 mol) sowie eine
katalytische Menge p-Toluolsulfonsäure wurden zusammengegeben und zum Sieden
gebracht. Durch abwechselndes Refluxieren und Destillieren des Azeotrops wurden innerhalb
von drei Stunden 100 ml Destillat entfernt bis keine Trübung des Destillats mehr festzustellen
war.
Die verbliebene Flüssigkeit wurde zuerst zur Entfernung der restlichen Lösungsmittel in
leichtem Vakuum destilliert, daraufhin im Ölpumpenvakuum fraktioniert.
Bei 74-84°C erhielt man 12.76 g (73 mmol) des Produktes als farblose Flüssigkeit.
Ausbeute: 84% der Theorie (Literatur[35]: 79%)
1H-NMR (400.13 MHz, DMSO-d6): δ = 1.25 (s, 3H; C(CH3)a(CH3)b), 1.25 – 1.55 (m, 6H;
HOCH2CH2CH2CH2-), 1.30 (s, 3H; C(CH3)a(CH3)b), 3.35 – 3.44 (m, 3H;
HOCH2CH2-, C(6)HaHb), 3.97 (t aa dd, 3J(C(6)HaHb, C(5)H) = 6.0 Hz, 2J(C(6)HaHb, C(6)HaHb) = 6.0 Hz, 1H; C(6)HaHb), 4.00 (n aa ddt, 3J(C(5)H,
C(6)HaHb) = 5.8 Hz, 3J(C(5)H, C(6)HaHb) = 5.8 Hz, 3J(C(5)H, C(4)H2) = 5.8 Hz,
1H; C(5)H), 4.32 ppm (t, 3J(-CH2OH, -CH2OH) = 5.3 Hz, 1H, -CH2OH). 13C-NMR (100.61 MHz, DMSO-d6): δ = 21.85 (C3), 25.63 (C(CH3)a(CH3)b), 26.84
(C(CH3)a(CH3)b), 32.43 (C4), 32.96 (C2), 60.53 (C1), 68.65 (C6), 75.41 (C5),
107.69 ppm (C(CH3)2).
MS (FAB) : m/z (%): 43.0 (90), 57.0 (45), 81.0 (57), 99.1 (100), 117.1 (25) [M+ -
(CH3)2CO], 157.1 (72) [M+ - OH], 157.1 (31) [M+ - CH3], 175.1 (36) [MH+].
C9H18O3
(174.24 g mol-1)

116
3.2 Synthese von 1-Methansulfonyl-5,6-isopropylidendioxyhexan (9)
OO
O
SO
O 10 g 5,6-Isopropylidendioxyhexan-1-ol (8) (57 mmol) wurden in 100 ml Pyridin gelöst,
woraufhin 4.5 ml Methansulfonsäurechlorid (58 mmol) zugegeben wurden.
Nach 18 h Rühren wurden 10 ml Wasser zugegeben, nach weiteren 10 min wurde das
Lösungsmittel evaporiert und der Rückstand in 100 ml Chloroform aufgenommen. Es wurde
dreimal mit jeweils 25 ml Wasser extrahiert. Die vereinigten wässrigen Phasen wurden mit
25 ml Chloroform extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat
getrocknet und evaporiert.
Man erhielt 7.81 g (31 mmol) eines gelblichen Öls als Produkt.
Ausbeute: 54% der Theorie (Literatur[35]: 83%)
1H-NMR (400.13 MHz, DMSO-d6): δ = 1.25 (s, 3H; C(CH3)a(CH3)b), 1.30 (s, 3H;
C(CH3)a(CH3)b), 1.32 – 1.78 (m, 6H; MeSO3-CH2CH2CH2CH2-), 3.14 (s,
2H(Austausch); CH3SO3-), 3.42 (t aa dd, 3J(C(6)HaHb, C(5)H) = 7.0 Hz, 2J(C(6)HaHb, C(6)HaHb) = 7.0 Hz, 1H; C(6)HaHb), 3.62 (t, 3J(MeSO3-CH2CH2-,
MeSO3-CH2CH2-) = 6.5 Hz, 2H; MeSO3-CH2CH2-), 3.95 – 4.05 ppm (m, 2H;
C(6)HaHb), C(5)H). 13C-NMR (100.61 MHz, DMSO-d6): δ = 22.69 (C3), 25.56 (C(CH3)a(CH3)b), 26.78
(C(CH3)a(CH3)b), 30.55 (C2), 32.27 (C4), 68.59 (C6), 70.24 (C1), 75.22 (C5),
107.80 ppm (C(CH3)2).
MS (FAB) : m/z (%): 43.0 (35), 81.1 (61), 99.1 (63), 145.0 (21), 163.0 (28), 236.2 (75) [M+ -
CH3], 253.1 (100) [MH+].
C10H20O5S
(252.33 g mol-1)
3.3 Synthese von Lithiumazid
26.00 g (0.40 mol) Natriumazid und 28.15 g (0.22 mol) Lithiumsulfatmonohydrat wurden
unter Rühren in 140 ml bidestilliertem Wasser gelöst. Zu der farblosen Lösung tropfte man

117
unter heftigem Rühren 700 ml Ethanol abs. langsam hinzu, wobei ein farbloser Niederschlag
ausfiel, der abgesaugt und mit wenig Ethanol abs. gewaschen wurde.
Das Lösungsmittel wurde evaporiert, der farblose Rückstand einmal mit Ethanol abs.
coevaporiert und erst über Phosphorpentoxid, später über Calciumhydrid im
Ölpumpenvakuum getrocknet.
Man erhielt 19.3 g (0.39 mol) eines farblosen Pulvers.
Ausbeute: 98.7% der Theorie
LiN3
(48.96 g mol-1)
3.4 Synthese von 1-Azido-5,6-isopropylidendioxyhexan (10)
OO
N3 Die Mischung aus 5.51 g Methansulfonyl-5,6-isopropylidendioxyhexan (9) (22 mmol) und
2.2 g Lithiumazid (44 mmol) in 5 ml DMF wurde 30 min auf 150°C unter Rückfluss erhitzt.
Nach Abkühlen der Mischung wurde diese in 50 ml Diethylether aufgenommen und mit
50 ml Wasser gewaschen. Die organsiche Phase wurde über Natriumsulfat getrocknet und
evaporiert.
Man erhielt 1.94 g (10 mmol) einer gelblichen Flüssigkeit als Produkt.
Ausbeute: 46% der Theorie (Literatur[35]: 88%)
Rf (LM-A): 0.72
1H-NMR (400.13 MHz, CDCl3): δ = 1.34 (s, 3H; C(CH3)a(CH3)b), 1.35 – 1.68 (m, 6H;
N3CH2CH2CH2CH2-), 1.40 (s, 3H; C(CH3)a(CH3)b), 3.28 (t, 3J(N3CH2CH2-,
N3CH2CH2-) = 7.0 Hz, 2H; N3CH2CH2-), 3.50 (t aa dd, 3J(C(6)HaHb, C(5)H) =
7.3 Hz, 2J(C(6)HaHb, C(6)HaHb) = 7.3 Hz, 1H; C(6)HaHb), 4.03 (t aa dd, 3J(C(6)HaHb, C(5)H) = 6.3 Hz, 2J(C(6)HaHb, C(6)HaHb) = 6.3 Hz, 1H;
C(6)HaHb), 4.07 ppm (dx aa dddt, 3J(C(5)H, C(6)HaHb) = 5.9 Hz, 3J(C(5)H,
C(6)HaHb) = 5.9 Hz, 3J(C(5)H, C(4)H2) = 5.9 Hz, 4J(C(5)H, C(3)H2) = 1.4 Hz,
1H; C(5)H). 13C-NMR (100.61 MHz, CDCl3): δ = 23.18 (C3), 25.83 (C(CH3)a(CH3)b), 27.08

118
(C(CH3)a(CH3)b), 29.00 (C4), 33.25 (C2), 51.43 (C1), 69.52 (C6), 75.94 (C5),
108.94 ppm (C(CH3)2).
MS (FAB) : m/z (%): 73.0 (100), 91.0 (23), 135.1 (17), 147.1 (51), 163.0 (35) [MNa+ -
(CH3)2CO], 207.0 (25), 221.1 (26) [MNa+], 235.1 (24), 281.1 (23).
C9H17N3O2
(199.25 g mol-1)
3.5 Synthese von 5,6-Isopropylidendioxyhexylamin (11)
OO
NH2 1 g 1-Azido-5,6-isopropylidendioxyhexan (10) (5.0 mmol) wurde in 100 ml Ethanol abs.
gelöst. Nach Zugabe einer Spatelspitze Pd/C-Katalysator (10%) wurde 17h bei Normaldruck
hydriert.
Der Katalysator wurde durch mehrfache Filtration entfernt. Nach Einengen des Filtrats erhielt
man 400 mg (2.3 mmol) eines leicht gelblichen Öls als Produkt.
Ausbeute: 46% der Theorie (Literatur[35]: 60%)
Rf (LM-A): 0.2 (breit)
1H-NMR (400.13 MHz, DMSO-d6): δ = 1.25 (s, 3H; C(CH3)a(CH3)b), 1.20 – 1.52 (m, 6H;
H2N-CH2CH2CH2CH2-), 1.30 (s, 3H; C(CH3)a(CH3)b), 2.52 (t, 3J(H2N-CH2CH2-,
H2N-CH2CH2-) = 6.8 Hz, 2H; H2N-CH2CH2-), 3.40 (t aa dd, 3J(C(6)HaHb,
C(5)H) = 5.8 Hz, 2J(C(6)HaHb, C(6)HaHb) = 5.8 Hz, 1H; C(6)HaHb), 3.97 (t aa
dd, 3J(C(6)HaHb, C(5)H) = 6.0 Hz, 2J(C(6)HaHb, C(6)HaHb) = 6.0 Hz, 1H;
C(6)HaHb), 4.00 ppm (n aa ddt, 3J(C(5)H, C(6)HaHb) = 6.4 Hz, 3J(C(5)H,
C(6)HaHb) = 6.4 Hz, 3J(C(5)H, C(4)H2) = 6.4 Hz, 1H; C(5)H). 13C-NMR (100.61 MHz, DMSO-d6): δ = 22.67 (C3), 25.61 (C(CH3)a(CH3)b), 26.82
(C(CH3)a(CH3)b), 33.01 (C2), 33.14 (C4), 41.44 (C1), 68.64 (C6), 75.37 (C5),
107.67 ppm (C(CH3)2).
MS (FAB) : m/z (%): 43.0 (28), 116.1 (33), 174.1 (92) [MH+], 216.1 (20) [MNa2+], 330.2
(100) [MCsNa+].
C9H19NO2
(173.26 g mol-1)

119
3.6 Synthese von N-(5,6-Isopropylidendioxyhexyl)acrylamid (4)
OO
NH
O
1.1 g 5,6-Isopropylidendioxyhexylamin (11) (5.9 mmol)) wurden in 5 ml Dichlormethan
gelöst und mit 0.8 ml Triethylamin (5.9 mmol) versetzt. Diese Lösung wurde langsam zu
einer stark gerührten, auf 0°C gekühlten Lösung von 0.53 ml Acryloylchlorid (6.5 mmol) in
5 ml Dichlormethan getropft. Nach 30min wurden dem Reaktionsgemisch 15 ml Hexan
zugegeben. Der ausgefallene Feststoff wurde abfiltriert und das Filtrat eingeengt.
Man erhielt eine viskose, farblose Flüssigkeit.
Die Produktausbeute betrug 1.44 g (6,3 mmol).
Ausbeute: 93% der Theorie (Literatur[35]: 97%)
1H-NMR (200.13 MHz, CDCl3): δ = 1.34 (s, 3H; C(CH3)a(CH3)b), 1.39 – 1.68 (m, 6H;
CONHCH2CH2CH2CH2-), 1.39 (s, 3H; C(CH3)a(CH3)b), 3.34 (q aa dt, 3J(CONHCH2CH2-, CONHCH2CH2-) = 6.3 Hz, 3J(CONHCH2-, CONHCH2-) =
6.3 Hz, 2H; CONHCH2CH2-), 3.51 (t aa dd, 3J(C(6)HaHb, C(5)H) = 6.3 Hz, 3J(C(6)HaHb, C(6)HaHb) = 6.3 Hz, 1H; C(6)HaHb), 4.02 – 4.07 (m, 2H; C(5)H,
C(6)HaHb), 5.62 (dd, 2J(CH2=CHCO, CH2=CHCO) = 1.8 Hz, 3J(CH2=CHCO,
CH2=CHCO)cis = 10.0 Hz, 1H; CH2=CHCO), 6.06 (dd, 3J(CH2=CHCO,
CH2=CHCO)cis = 9.9 Hz, 3J (CH2=CHCO, CH2=CHCO)trans = 17.0 Hz, 1H;
CH2=CHCO), 6.27 ppm (dd, 2J(CH2=CHCO, CH2=CHCO) = 1.9 Hz, 3J
(CH2=CHCO, CH2=CHCO)trans = 16.9 Hz, 1H; CH2=CHCO).
MS (FAB) : m/z (%): 152.1 (58), 170 (100) [M+ - (CH3)2CO], 212.1 (23) [M+ - CH3], 228.2
(64) [MH+], 250.1 (65) [MNa+].
C12H21NO3
(227.31 g mol-1)

120
3.7 Synthese von N-(2,2-Diethoxyethyl)acrylamid (5)
NH
OO
O
0.5 ml 2,2-Diethoxyethylamin (3.4 mmol) wurden in 12 ml 5%iger NaHCO3-Lösung
aufgelöst und im Eisbad abgekühlt, woraufhin 0.28 ml Acryloylchlorid (3.4 mmol) zugegeben
wurden.
Nach 2h wurde die Mischung mit 1 g NaCl versetzt und dreimal mit je 50 ml Dichlormethan
extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet und im
Vakuum eingeengt.
Man erhielt 0.626 g (3.35 mmol) eines gelblichen Öls als Produkt.
Ausbeute: 98% der Theorie
1H-NMR (400.13 MHz, CDCl3): δ = 1.20 (t, 3J(CH3-CH2-O-, CH3-CH2-O-) = 7.1 Hz, 6H;
CH3-CH2-O-), 3.35 - 3.80 (m, 6H; CH3-CH2-O-, -CH2-CH(OEt)2), 4.52 (t, 3J(-
CH2-CH(OEt)2, -CH2-CH(OEt)2) = 5.2 Hz, 1H; -CH2-CH(OEt)2), 5.63 (dd, 3J(CH2=CH-R, CH2=CH-R)cis = 9.9Hz, 2J(CH2=CH-R, CH2=CH-R) = 1.8Hz,
1H; CH2=CH-R), 6.09 (dd, 3J(CH2=CH-R, CH2=CH-R)trans = 16.9Hz, 3J(CH2=CH-R, CH2=CH-R)cis = 9.9Hz, 1H; CH2=CH-R), 6.28 ppm (dd, 3J(CH2=CH-R, CH2=CH-R)trans = 17.3Hz, 2J(CH2=CH-R, CH2=CH-R) = 1.8Hz,
1H; CH2=CH-R).
MS (FAB) : m/z (%): 100.1 (44), 130.1 (51), 229.1 (27) [M(H2O)Na+], 321.2 (46) [MCs+],
343.2 (100) [MCsNa+], 530.4 (30) [M2CsNa+].
C9H17NO3
(187.24 g mol-1)
3.8 Synthese von 4-Dimethoxymethylbenzoesäuremethylester (15)
O
O O
O Zu der Lösung von 13.12 g p-Formylbenzoesäuremethylester (80 mmol) in 250 ml Methanol
wurden 9.6 ml Trimethylorthoformiat (88 mmol) und 0.72 g p-Toluolsulfonsäure gegeben.
Die Mischung wurde über Nacht zum Sieden erhitzt. Die Lösungsmittel sowie vebliebenes

121
Trimethylorthoformiat wurden am Rotationsverdampfer entfernt. Das verbliebene gelbe Öl
wurde im Ölpumpenvakuum fraktioniert destilliert.
15.20 g (72.3 mmol) des Produktes wurde bei einer Kopftemperatur von 109-116°C als
Destillat erhalten.
Die farblose Flüssigkeit erstarrte nach einigen Tagen unter Ausbildung grober Kristalle.
Ausbeute: 90% der Theorie
Rf (LM-C): 0.61
1H-NMR (400.13 MHz, CDCl3): δ = 3.32 (s, 6H; -CH(OCH3)2), 3.90 (s, 3H; -COOCH3),
5.41 (s, 1H; -CH(OCH3)2), 7.51 (d, 3J(H3/H5, H2/H6) = 8.0 Hz, 2H; H3/H5),
8.03 ppm (d, 3J(H2/H6, H3/H5) = 8.5 Hz, 2H; H2/H6). 13C-NMR (100.61 MHz, CDCl3): δ = 52.2 (-COOCH3), 52.77 (-CH(OCH3)2), 102.50 (-
CH(OCH3)2), 126.92 (C3/C5), 129.62 (C2/C6), 130.35 (C1), 143.09 (C4), 166.95
ppm (-COOMe).
MS (EI) : m/z (%): 59 (32), 75 (25), 179 (100) [M – (OCH3)], 210 (2) [M+].
C11H14O4
(210.23 g mol-1)
3.9 Synthese von N-(2-Aminoethyl)-4-dimethoxymethylbenzamid (17)
O
O O
NH
NH2
3.04 g 4-Dimethoxymethylbenzoesäuremethylester (15) (14.5 mmol) wurden mit 12 ml
Ethylendiamin (180 mmol) 5h auf 120°C erhitzt. Das überschüssige Ethylendiamin wurde
daraufhin im Vakuum destilliert und die verbleibende viskose Masse im Vakuum getrocknet.
Die Reinigung erfolgte durch Säulenchromatographie (Kieselgel, Eluent: CH2Cl2 / MeOH /
NEt3 100:10:1 (v/v)).
Man erhielt 600 mg (2.6 mmol) einer viskosen Masse als Produkt.
Ausbeute: 19% der Theorie
Rf (LM-B): 0.15
1H-NMR (400.13 MHz, DMSO-d6): δ = 2.82 (t, 3J(-CH2-CH2-NH2, -CH2-CH2-NH2) = 6.3
Hz, 2H; -CH2-CH2-NH2), 3.25 (s, 6H; -CH(OCH3)2), 3.37 (dt, 3J(CONH-CH2-

122
CH2-NH2, CONH-CH2-CH2-NH2) = 6.0 Hz, 3J(CONH-CH2-CH2-NH2, CONH-
CH2-CH2-NH2) = 6.0 Hz, 2H; CONH-CH2-CH2-NH2), 5.43 (s, 1H; -
CH(OCH3)2), 7.45 (d, 3J(H3/H5, H2/H6) = 8.0 Hz, 2H; H3/H5), 7.89 (d, 3J(H2/H6, H3/H5) = 8.5 Hz, 2H; H2/H6), 8.63 ppm (t, 3J(CONH-CH2-CH2-NH2,
CONH-CH2-CH2-NH2) = 5.5 Hz, 1H; CONH-CH2-CH2-NH2). 13C-NMR (100.61 MHz, DMSO-d6): δ = 40.11 (-CH2-NH2), 40.58 (-CONH-CH2-), 52.50 (-
CH(OCH3)2), 102.14 (-CH(OCH3)2), 126.31 (C3/C5), 127.15 (C2/C6), 134.41
(C1), 141.00 (C4), 166.18 ppm (-CONH-).
MS (FAB) : m/z (%): 45.0 (16), 91.0 (15), 163.1 (18), 207.1 (15) [M – OCH3], 222.1 (20)
[M – CH3], 239.2 (100) [MH+].
C12H18N2O3
(238.29 g mol-1)
3.10 Synthese von N-(2-Acrylamidoethyl)-4-dimethoxymethylbenzamid (6)
O
O O
NH
NH
O 262 mg N-(2-Aminoethyl)-4-dimethoxymethylbenzamid (17) (1.1 mmol) wurden in 4 ml
5%iger NaHCO3-Lösung aufgelöst und im Eisbad abgekühlt, woraufhin 90 µl
Acryloylchlorid (1.1 mmol) zugegeben wurden.
Nach 40min wurde die Mischung zweimal mit je 25 ml Diethylether extrahiert. Die
Etherphasen wurden einmal mit 2 ml 10%iger NaOH-Lösung extrahiert, über Natriumsulfat
getrocknet und im Vakuum eingeengt.
Man erhielt 90 mg (0.37 mmol) eines farblosen Feststoffes als Produkt.
Ausbeute: 34% der Theorie
Rf (LM-A): 0.52
1H-NMR (400.13 MHz, CDCl3): δ = 3.25 (s, 6H; -CH(OCH3)2), 3.53-3.65 (m, 4H; CONH-
CH2-CH2-NHCO), 5.40 (s, 1H; -CH(OCH3)2), 5.63 (d, 3J(CH2=CH-R, CH2=CH-
R)cis = 10.1Hz, 1H; CH2=CH-R), 6.13 (dd, 3J(CH2=CH-R, CH2=CH-R)cis =
9.9Hz, 3J(CH2=CH-R, CH2=CH-R)trans = 17.4Hz, 1H; CH2=CH-R), 6.13 (d, 3J(CH2=CH-R, CH2=CH-R)trans = 16.3Hz, 1H; CH2=CH-R), 7.49 (d, 3J(H3/H5,
H2/H6) = 8.1 Hz, 2H; H3/H5), 7.80ppm (d, 3J(H2/H6, H3/H5) = 8.1 Hz, 2H;

123
H2/H6). 13C-NMR (100.61 MHz, CDCl3): δ = 39.82 (Ar-CONH-CH2-), 40.88 (-CH2-NH-Acr),
52.55 (-CH(OCH3)2), 102.31 (-CH(OCH3)2), 126.63 (CH2=CHCONH), 126.84
(C3/C5), 126.87 (C2/C6), 130.46 (CH2=CHCONH), 133.83 (C1), 141.45 (C4),
166.18 (Ar-CONH-), 40.88ppm (CH2=CHCONH).
MS (FAB) : m/z (%): 45.0 (16), 91.0 (15), 163.1 (18), 207.1 (15) [M – OCH3], 222.1 (20)
[M – CH3], 239.2 (100) [MH+].
C12H18N2O3
(238.29 g mol-1)
3.12 Synthese von N-(2-Hydroxyethyl)-4-dimethoxymethylbenzamid (26)
O
O O
NH
OH
1.377 g 4-Dimethoxymethylbenzoesäuremethylester (15) (6.56 mmol) wurden mit 10 ml
Ethanolamin (158 mmol) und 10 ml Ethanol über Nacht unter Rückfluss zum Sieden erhitzt.
Das Lösungsmittel wurde daraufhin am Rotationsverdampfer entfernt, überschüssiges
Ethanolamin im Ölpumpenvakuum destilliert. Man erhielt ein orange-gelbes viskoses Öl, das
in 40 ml Dichlormethan aufgenommen und durch mehrfache Extraktion mit jeweils 10 ml
Wasser neutral gewaschen wurde. Die organische Phase wurde mit 10 ml gesättigter NaCl-
Lösung extrahiert und evaporiert. Weiteres Produkt erhielt man, indem man die vereinigten
wässrigen Phasen mit NaCl sättigte und zweimal mit je 40 ml Dichlormethan extrahierte. Die
vereinigten organischen Phasen wurden über Natriumsulfat getrocknet und evaporiert.
Man erhielt insgesamt 1.28 g (5.36 mmol) eines leicht gelblichen kristallinen Feststoffes als
Produkt.
Ausbeute: 82% der Theorie
Rf (LM-A): 0.48
1H-NMR (400.13 MHz, DMSO-d6): δ = 3.25 (s, 6H; -CH(OCH3)2), 3.34 (dt aa q,
3J(CONH-CH2-CH2-OH, CONH-CH2-CH2-OH) = 6.0 Hz, 3J(CONH-CH2-CH2-
OH, CONH-CH2-CH2-OH) = 6.0 Hz, 2H; CONH-CH2-CH2-OH), 3.52 (dt aa q, 3J(-CH2-CH2-OH, -CH2-CH2-OH) = 6.0 Hz, 3J(-CH2-CH2-OH, -CH2-CH2-OH) =
6.0 Hz, 2H; -CH2-CH2-OH), 4.96 (t, 3J(-CH2-OH, -CH2-OH) = 5.8 Hz, 1H; -CH2-

124
OH), 5.42 (s, 1H; -CH(OCH3)2), 7.46 (d, 3J(H3/H5, H2/H6) = 8.3 Hz, 2H;
H3/H5), 7.87 (d, 3J(H2/H6, H3/H5) = 8.3 Hz, 2H; H2/H6), 8.41 ppm (t, 3J(CONH-CH2-CH2-OH, CONH-CH2-CH2-OH) = 5.4 Hz, 1H; CONH-CH2-CH2-
OH). 13C-NMR (100.61 MHz, DMSO-d6): δ = 42.17 (-CONH-CH2-), 52.56 (-CH(OCH3)2),
59.75 (-CH2-OH), 102.17 (-CH(OCH3)2), 126.35 (C3/C5), 127.06 (C2/C6),
134.58 (C1), 140.93 (C4), 166.02 ppm (-CONH-).
MS (FAB) : m/z (%): 105.0 (51), 136.0 (42), 179.1 (92), 208.1 (75) [M – OCH3], 240.1 (80)
[MH+], 262.1 (100) [MNa+].
C12H17NO4
(239.27 g mol-1)
3.13 Synthese von 3-(Acrylamido)phenylboronsäure (29)
BOH
OHNH
O
100 mg 3-Aminophenylboronsäure (0.73 mmol) werden unter Erwärmen in 5 ml 5%iger
NaHCO3-Lösung gelöst und im Eisbad abgekühlt, woraufhin 100 µl Acryloylchlorid
(1.23 mmol) zugetropft wurden.
Nach kurzer Zeit wurde die Lösung trüb, wobei eine Gasentwicklung zu beobachten war.
Nach 10min wurde das Eisbad entfernt, nach 2.5 h das ausgefallene Produkt abgesaugt, mit
wenig Wasser gewaschen und im Vakuum getrocknet.
Man erhielt 50 mg (0.26 mmol) eines farblosen Feststoffes als Produkt.
Ausbeute: 36% der Theorie
Rf (LM-A): 0.50
1H-NMR (400.13 MHz, DMSO-d6): δ = 3.30 (s, 2H; H2O), 5.73 (dd, 3J(CH2=CH-R,
CH2=CH-R)cis = 10.0Hz, 2J(CH2=CH-R, CH2=CH-R) = 2.0Hz, 1H; CH2=CH-R),
6.25 (dd, 3J(CH2=CH-R, CH2=CH-R)trans = 17.1Hz, 2J(CH2=CH-R, CH2=CH-R)
= 2.0Hz, 1H; CH2=CH-R), 6.45 (dd, 3J(CH2=CH-R, CH2=CH-R)cis = 10.0Hz, 3J(CH2=CH-R, CH2=CH-R)trans = 17.1Hz, 1H; CH2=CH-R), 7.28 (dd, 3J(arom-
C5-H, arom-C4-H) = 7.8Hz, 3J(arom-C5-H, arom-C6-H) = 7.8Hz, 1H; arom-C5-
H), 7.49 (d, 3J(arom-C6-H, arom-C5-H) = 7.5Hz, 1H; arom-C6-H), 7.82 (d,

125
3J(arom-C4-H, arom-C5-H) = 8.0Hz, 1H; arom-C4-H), 7.88 (s, 1H; arom-C2-H),
7.98 (s, 2H; B(OH)2), 10.04 ppm (s, 1H; R-CO-NH-R).
C9H10BNO3
(191.00 gmol-1)
3.14 Synthese von Cystaminbisacrylamid (33)
NH
SS
ONH
O
Zu der im Eisbad gekühlten Lösung von 2 g Cystamindihydrochlorid (8.9 mmol) in 200 ml
5%iger NaHCO3-Lösung wurden 3 ml Acryloylchlorid (37 mmol) langsam zugetropft.
Nach zweistündigem Rühren wurde die Mischung fünfmal mit je 200 ml Dichlormethan
extrahiert. Die vereinigten organischen Phasen wurden über Natriumsulfat getrocknet und
evaporiert.
Man erhielt 0.5 g (1.9 mmol) eines farblosen Feststoffes als Produkt.
Ausbeute: 21% der Theorie
1H-NMR (400.13 MHz, CDCl3): δ = 2.89 (t, 3J(NHCH2CH2S, NHCH2CH2S) = 6.5 Hz, 4H;
NHCH2CH2S), 3.67 (dt, 3J(NHCH2CH2S, NHCH2CH2S) = 6.4 Hz, 3J(NHCH2CH2S, NHCH2CH2S) = 6.3 Hz, 4H; NHCH2CH2S), 5.67 (dd, 3J(CH2=CH-R, CH2=CH-R)cis = 10.0 Hz, 2J(CH2=CH-R, CH2=CH-R) = 2.0 Hz,
2H; CH2=CH-R), 6.20 (dd, 3J(CH2=CH-R, CH2=CH-R)trans = 16.6 Hz, 3J(CH2=CH-R, CH2=CH-R)cis = 10.0 Hz, 2H; CH2=CH-R), 6.32 (dd, 3J(CH2=CH-R, CH2=CH-R)trans = 16.8 Hz, 2J(CH2=CH-R, CH2=CH-R) = 1.8
Hz, 2H; CH2=CH-R), 6.51 ppm (bs, 2H; CH2NHCO). 13C-NMR (100.61 MHz, CDCl3): δ = 38.62 (NHCH2CH2S), 126.84 (CH2=CHCONH),
130.66 (CH2=CHCONH), 171.91 ppm (CONH).
MS (FAB) : m/z (%): 73.0 (60), 136.0 (75), 154.0 (49), 176.0 (72), 207.0 (35), 261.1 (67)
[MH+], 283.1 (100) [MNa+], 329 (15).
C10H16N2O2S2
(260.38 g mol-1)

126
3.15 Synthese von N,N-Diacryloyl-L-cystindiethylester (36)
NH
SS
OO O
NH
OOO
1 g L-Cystindiethylester (3.4 mmol) wurden in 10 ml Wasser gelöst und mit 1 ml
Triethylamin versetzt. Bei langsamem Zutropfen von 0.55 ml Acryloylchlorid (6.8 mmol)
bildete sich sofort ein farbloser Niederschlag. Es wurde über Nacht gerührt.
Der Niederschlag wurde abgesaugt, mit wenig kaltem Wasser gewaschen und im Vakuum
getrocknet.
Man erhielt 240 mg (0.59 mmol) eines farblosen Feststoffes als Produkt.
Ausbeute: 18% der Theorie
1H-NMR (400.13 MHz, DMSO-d6): δ = 1.20 (t, 3J(CH3-CH2-O-, CH3-CH2-O-) = 7.0 Hz,
6H; CH3-CH2-O-), 2.99 (dd, 3J(SCHaHbCH, SCHaHbCH) = 8.8 Hz, 2J(SCHaHbCH, SCHaHbCH) = 13.8 Hz, 2H; SCHaHbCH), 3.17 ((dd, 3J(SCHaHbCH, SCHaHbCH) = 5.0 Hz, 2J(SCHaHbCH, SCHaHbCH) = 13.6 Hz,
2H; SCHaHbCH)), 4.12 (q, 3J(CH3-CH2O-, CH3-CH2-O-) = 7.0 Hz, 4H; CH3-
CH2-O-), 4.63 (ddd, 3J(SCHaHbCHNH, SCHaHbCHNH) = 8.3 Hz, 3J(SCHaHbCHNH, SCHaHbCHNH) = 8.3 Hz, 3J(SCHaHbCHNH, SCHaHbCHNH)
= 5.4 Hz, 2H; SCHaHbCHNH), 5.65 (dd, 3J(CH2=CH-R, CH2=CH-R)cis = 10.3
Hz, 2J(CH2=CH-R, CH2=CH-R) = 2.3 Hz, 2H; CH2=CH-R), 6.12 (dd, 3J(CH2=CH-R, CH2=CH-R)trans = 17.1 Hz, 2J(CH2=CH-R, CH2=CH-R) = 2.0
Hz, 2H; CH2=CH-R), 6.29 (dd, 3J(CH2=CH-R, CH2=CH-R)trans = 17.1 Hz, 3J(CH2=CH-R, CH2=CH-R)cis = 10.0 Hz, 2H; CH2=CH-R), 8.62 ppm (t, 3J(CHNHCO, CHNHCO) = 8.3 Hz, 2H; CHNHCO).
13C-NMR (100.61 MHz, DMSO-d6): δ = 13.94 (OCH2CH3), 39.15 (α-C), 51.34 (β-C),
60.95 (OCH2CH3), 126.24 (CH2=CHCONH), 130.84 (CH2=CHCONH), 164.61
(COOEt), 170.26 ppm (CONH).
MS (FAB) : m/z (%): 55.0 (42), 102.1 (43), 170.0 (40), 202.0 (85) [1/2 M], 405.0 (100)
[MH+], 427 (55) [MNa+].
C16H24N2O6S2
(404.51 g mol-1)

127
3.16 Synthese von 3-(Triethoxysilyl)propylacrylamid (41)
Si NH
O
OO
O
Zu der im Eisbad abgekühlten Lösung von 4.25 ml Acryloylchlorid (52 mmol) in 125 ml
Dichlormethan wurde die Lösung von 11.75 ml 3-Aminopropyltriethoxysilan (50 mmol) und
7.5 ml Triethylamin in 25ml Dichlormethan getropft.
Nach 2h wurde bei Normaldruck der Großteil des Lösungsmittels destilliert, woraufhin ein
farbloser Feststoff ausfiel. Die Mischung wurde filtriert und das Filtrat im Ölpumpenvakuum
bei 165°C destilliert. Da sich im Destillat als Verunreinigung ein feiner Niederschlag von
mitgeschlepptem Triethylammoniumchlorid bildete, wurde es filtriert.
Man erhielt 2.87 g (10.4 mmol) einer viskosen, leicht gelblichen Flüssigkeit.
Ausbeute: 21% der Theorie (Literatur[35]: 77%)
1H-NMR (400.13 MHz, DMSO-d6): δ = 0.62 (t, 3J((Et3O)3SiCH2-CH2-R, (Et3O)3SiCH2-
CH2-R) = 8.0 Hz, 2H; (Et3O)3SiCH2-CH2-R), 1.19 (t, 3J(CH3-CH2-O-, CH3-CH2-
O-) = 7.0 Hz, 9H; CH3-CH2-O-), 1.65 (tt, 3J((Et3O)3SiCH2-CH2-CH2-NH-R,
(Et3O)3SiCH2-CH2- CH2-NH-R) = 7.8 Hz, 3J((Et3O)3SiCH2-CH2-CH2-NH-R,
(Et3O)3SiCH2-CH2- CH2-NH-R) = 7.8 Hz, 2H; (Et3O)3SiCH2-CH2- CH2-NH-R),
3.31 (t, 3J((Et3O)3SiCH2-CH2-CH2-NH-R, (Et3O)3SiCH2-CH2- CH2-NH-R) = 6.7
Hz, 2H; (Et3O)3SiCH2-CH2- CH2-NH-R), 3.78 (q, 3J(CH3-CH2-O-, CH3-CH2-O-)
= 7.0 Hz, 6H; CH3-CH2-O-), 5.57 (d, 3J(CH2=CH-R, CH2=CH-R)cis = 10.0Hz,
1H; CH2=CH-R), 6.11 (dd, 3J(CH2=CH-R, CH2=CH-R)trans = 17.1Hz, 3J(CH2=CH-R, CH2=CH-R)cis = 10.0Hz, 1H; CH2=CH-R), 6.24 (d, 3J(CH2=CH-
R, CH2=CH-R)trans = 16.1Hz, 1H; CH2=CH-R), 6.51 ppm (s, 1H; R-NH-CO-R’). 13C-NMR (100.61 MHz, DMSO-d6): δ = 7.91 ((Et3O)3Si-CH2-CH2-CH2-NH-R), 18.47 (R-
O-CH2-CH3), 22.82 ((Et3O)3Si-CH2-CH2-CH2-NH-R), 42.11 ((Et3O)3Si-CH2-
CH2-CH2-NH-R), 58.50 (R-O-CH2-CH3), 126.46 (CH2=CH-CO-R), 130.86
(CH2=CH-CO-R), 165.98 ppm (CO).
MS (FAB) : m/z (%): 55.0 (100), 136.0 (95), 154.0 (70), 176.0 (15), 309.1 (20).
C12H25NO4Si
(275.42 g mol-1)

Anhang

129
A: Daten
A.1 Quellverhalten und Dichten von unmodifizierten N,N-
Dimethylacrylamid-Polymeren
Die Messwerte und abgeleiteten Größen, die den in Kap. angegebenen Kurven zugrunde
liegen, sind hier tabellarisch zusammengefasst.
Bei durch Differenzwägung ermittelten Größen sind nicht die zugrundeliegenden Messwerte
angegeben.
Das Quellvolumen (QV) wurde folgendermaßen bestimmt:
m(Gel, gequollen) - m(Gel, trocken)
m(Gel, trocken) ρGelQV = .
Diese Art der Definition des Quellvolumens bietet den Vorteil, dass man ein tatsächliches
Maß für das Volumen der gebundenen Flüssigkeit erhält. Auf eine prozentuale Angabe der
Volumenänderung bezogen auf das trockene Polymer wurde verzichtet, da die Bestimmung
des Volumens des trockenen Ausgangspolymers aufgrund der unregelmäßigen
dreidimensionalen Struktur stark fehlerbehaftet ist.
Die Dichte des Gels wurde in vielen Fällen experimentell ermittelt, wenn sie jedoch nicht
vorlag, wurde angenommen, dass ρGel ≈ ρLösung. In diesen Fällen wurden die entsprechenden
Werte für ρLösung den Berechnungen zugrunde gelegt.
Um die Streuung der abgeleiteten Größen zu verringern, wurde in vielen Fällen eine
Ausgleichskurve (Polynom zweiter oder dritter Ordnung bzw. eine Gerade im Fall der
Salzlösungen) durch die experimentellen Dichtewerte abhängig von Konzentration bzw.
Volumenanteil gelegt und die entsprechend berechneten Dichten in die weitere Auswertung
übernommen. Der Verlauf der Ausgleichskurven lässt sich den Abbildungen entnehmen. Die
erhaltenen Polynome sind bei den Auswertungen angegeben.
Experimentelle Dichten wurden - soweit möglich - durch Schwebeexperimente in ähnlichen
Lösungsmittelgemischen ermittelt, wobei auch die Steig- und Sinkgeschwindigkeit durch
Wichtungsfaktoren mit in die Abschätzung einbezogen wurde.
In allen Fällen wurden die Experimente in mehreren Datenreihen durchgeführt. In die
statistische Auswertung wurden stark abweichende Werte nicht aufgenommen. Diese sind in
den jeweiligen Tabellen dunkel hinterlegt.
130
Bei der Angabe der Konzentrationen von Salzen bzw. Harnstoff im Gel wurde die
Konzentration der gebundenen Lösung unter Berücksichtigung der Dichte der ungebundenen
Lösung berechnet. Es handelt sich also nicht um die tatsächliche Konzentration im Gel,
sondern um die der gebundenen Lösung in freier Form. Dies ermöglicht die direkte
Vergleichbarkeit der Werte, denn eine identische Konzentration in Gel und Lösung bedeutet
in diesem Fall, dass das Verhältnis zwischen Lösungsmittel und gelöstem Stoff in Lösung und
Gel gleich ist also ein Konzentrationsgleichgewicht vorliegt.
A.1.1 Methanol-Wasser-Mischungen
Exp. Vol-Anteil H2O
/ % ρLösung / g ml-1
ρGel, calc. / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen/ ml g-1
1 0 0.8059 0.8242 9.57 105.8 12.20 2 10 0.8444 0.8554 9.50 113.5 12.80 3 20 0.8541 0.8838 10.10 127.8 13.19 4 30 0.8902 0.9093 10.32 134.5 13.23 5 40 0.9049 0.9321 9.19 122.7 13.25 6 50 0.9439 0.9520 7.72 105.2 13.26 7 60 0.9514 0.9691 8.92 119.0 12.73 8 70 0.9576 0.9834 8.48 114.5 12.71 9 80 0.9736 0.9948 10.40 141.7 12.69
10 90 0.9842 1.0035 9.90 132.5 12.34 11 0 0.7837 0.8242 8.49 93.9 12.21 12 10 0.8395 0.8554 8.53 105.3 13.26 13 20 0.8670 0.8838 9.86 124.2 13.12 14 30 0.8815 0.9093 9.49 126.9 13.61 15 40 0.9055 0.9321 7.31 97.0 13.16 16 50 0.9256 0.9520 9.05 121.0 12.99 17 60 0.9426 0.9691 9.41 126.2 12.80 18 70 0.9636 0.9834 10.87 145.0 12.55 19 80 0.9700 0.9948 10.01 133.9 12.44 20 90 0.9829 1.0035 8.95 120.6 12.43 21 0 0.8006 0.8242 7.87 88.6 12.45 22 10 0.8362 0.8554 7.68 93.8 13.11 23 20 0.8615 0.8838 10.56 132.2 13.03 24 30 0.8892 0.9093 8.92 115.3 13.12 25 40 0.9165 0.9321 7.38 98.8 13.29 26 50 0.9223 0.9520 9.89 134.4 13.22 27 60 0.9466 0.9691 9.82 129.6 12.59 28 70 0.9549 0.9834 8.96 121.6 12.78 29 80 0.9732 0.9948 9.63 130.4 12.61 30 90 0.9829 1.0035 8.72 120.0 12.72
Tab.9: Messwerte (grau hinterlegt) und Quellvolumina für N,N-Dimethylacrylamidpolymere
in Methanol-Wasser-Mischungen. ρGel, calc. wurde nach Gleichung (46) berechnet, welche die
Ausgleichskurve durch die experimentell ermittelten Dichten (Tab.11) darstellt:
ρGel = ( -0.1410 x2 + 0.3261 x + 0.8242 ) g ml-1 (46)
x bezeichnet den Volumenanteil Wasser in Lösung.
131
Vol.-Anteil H2O / % 0 10 20 30 40 50 60 70 80 90 100
ρLösung / g cm-3 0.797 0.840 0.861 0.887 0.909 0.931 0.947 0.959 0.972 0.983 1.000 Exp
1 ↓ ↑ 2 → 3 ↓ 4 ↓ ↑ 5 ↔ 6 ↓ ↑ 7 ↓ ↑ 8 ↓ ↑ 9 ↓ ↑
10 ↓ ↑ : steigt; : steigt langsam; ↔ : schwebt; : sinkt langsam; ↓ : sinkt
Tab.10: Abschätzung von ρGel für Methanol-Wasser-Mischungen.
Vol.-Anteil H2O / % ρLösung / g ml-1 ρGel / g cm-3 0 0.7967 ± 0.0082 0.8184 ± 0.0144 10 0.8400 ± 0.0029 0.8609 ± 0.0117 20 0.8609 ± 0.0046 0.8804 ± 0.0087 30 0.8869 ± 0.0034 0.9198 ± 0.0072 40 0.9089 ± 0.0047 0.9306 ± 0.0095 50 0.9306 ± 0.0082 0.9528 ± 0.0039 60 0.9469 ± 0.0031 0.9655 ± 0.0045 70 0.9587 ± 0.0032 0.9778 ± 0.0037 80 0.9723 ± 0.0014 0.9917 ± 0.0055 90 0.9834 ± 0.0005 1.0100 ± 0.0070
Tab.11: Statistische Auswertung von ρGel (aus Tab.10) und ρLösung (aus Tab.9) für Methanol-
Wasser-Mischungen.
Vol.-Anteil Quellvolumen / ml g-1 H2O / % Reihe1 Reihe2 Reihe3 Mittelwert Standardabweichung
0 12.20 12.21 12.45 12.28 0.10 10 12.80 13.26 13.11 13.06 0.17 20 13.19 13.12 13.03 13.11 0.05 30 13.23 13.61 13.12 13.17 0.08 40 13.25 13.16 13.29 13.24 0.05 50 13.26 12.99 13.22 13.16 0.10 60 12.73 12.80 12.59 12.71 0.08 70 12.71 12.55 12.78 12.68 0.09 80 12.69 12.44 12.61 12.58 0.09 90 12.34 12.43 12.72 12.50 0.14
Tab.12: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in Methanol-Wasser-Mischungen.
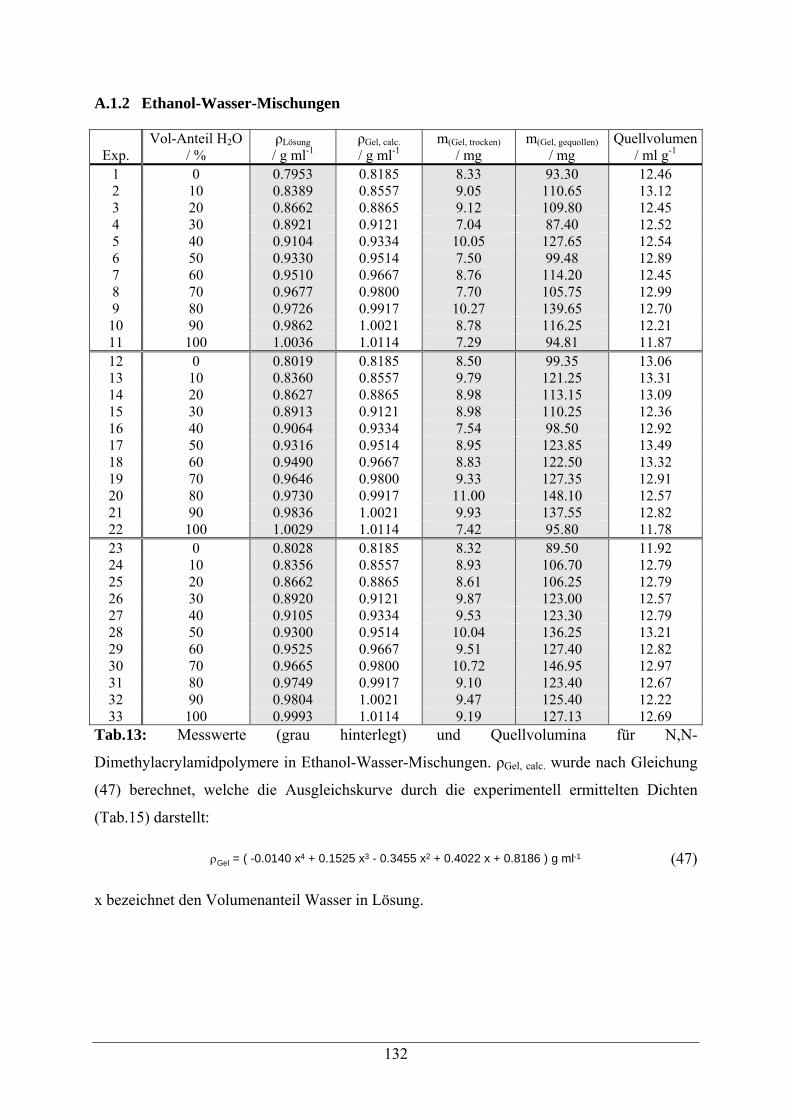
132
A.1.2 Ethanol-Wasser-Mischungen
Exp. Vol-Anteil H2O
/ % ρLösung / g ml-1
ρGel, calc. / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen/ ml g-1
1 0 0.7953 0.8185 8.33 93.30 12.46 2 10 0.8389 0.8557 9.05 110.65 13.12 3 20 0.8662 0.8865 9.12 109.80 12.45 4 30 0.8921 0.9121 7.04 87.40 12.52 5 40 0.9104 0.9334 10.05 127.65 12.54 6 50 0.9330 0.9514 7.50 99.48 12.89 7 60 0.9510 0.9667 8.76 114.20 12.45 8 70 0.9677 0.9800 7.70 105.75 12.99 9 80 0.9726 0.9917 10.27 139.65 12.70
10 90 0.9862 1.0021 8.78 116.25 12.21 11 100 1.0036 1.0114 7.29 94.81 11.87 12 0 0.8019 0.8185 8.50 99.35 13.06 13 10 0.8360 0.8557 9.79 121.25 13.31 14 20 0.8627 0.8865 8.98 113.15 13.09 15 30 0.8913 0.9121 8.98 110.25 12.36 16 40 0.9064 0.9334 7.54 98.50 12.92 17 50 0.9316 0.9514 8.95 123.85 13.49 18 60 0.9490 0.9667 8.83 122.50 13.32 19 70 0.9646 0.9800 9.33 127.35 12.91 20 80 0.9730 0.9917 11.00 148.10 12.57 21 90 0.9836 1.0021 9.93 137.55 12.82 22 100 1.0029 1.0114 7.42 95.80 11.78 23 0 0.8028 0.8185 8.32 89.50 11.92 24 10 0.8356 0.8557 8.93 106.70 12.79 25 20 0.8662 0.8865 8.61 106.25 12.79 26 30 0.8920 0.9121 9.87 123.00 12.57 27 40 0.9105 0.9334 9.53 123.30 12.79 28 50 0.9300 0.9514 10.04 136.25 13.21 29 60 0.9525 0.9667 9.51 127.40 12.82 30 70 0.9665 0.9800 10.72 146.95 12.97 31 80 0.9749 0.9917 9.10 123.40 12.67 32 90 0.9804 1.0021 9.47 125.40 12.22 33 100 0.9993 1.0114 9.19 127.13 12.69
Tab.13: Messwerte (grau hinterlegt) und Quellvolumina für N,N-
Dimethylacrylamidpolymere in Ethanol-Wasser-Mischungen. ρGel, calc. wurde nach Gleichung
(47) berechnet, welche die Ausgleichskurve durch die experimentell ermittelten Dichten
(Tab.15) darstellt:
ρGel = ( -0.0140 x4 + 0.1525 x3 - 0.3455 x2 + 0.4022 x + 0.8186 ) g ml-1
(47)
x bezeichnet den Volumenanteil Wasser in Lösung.
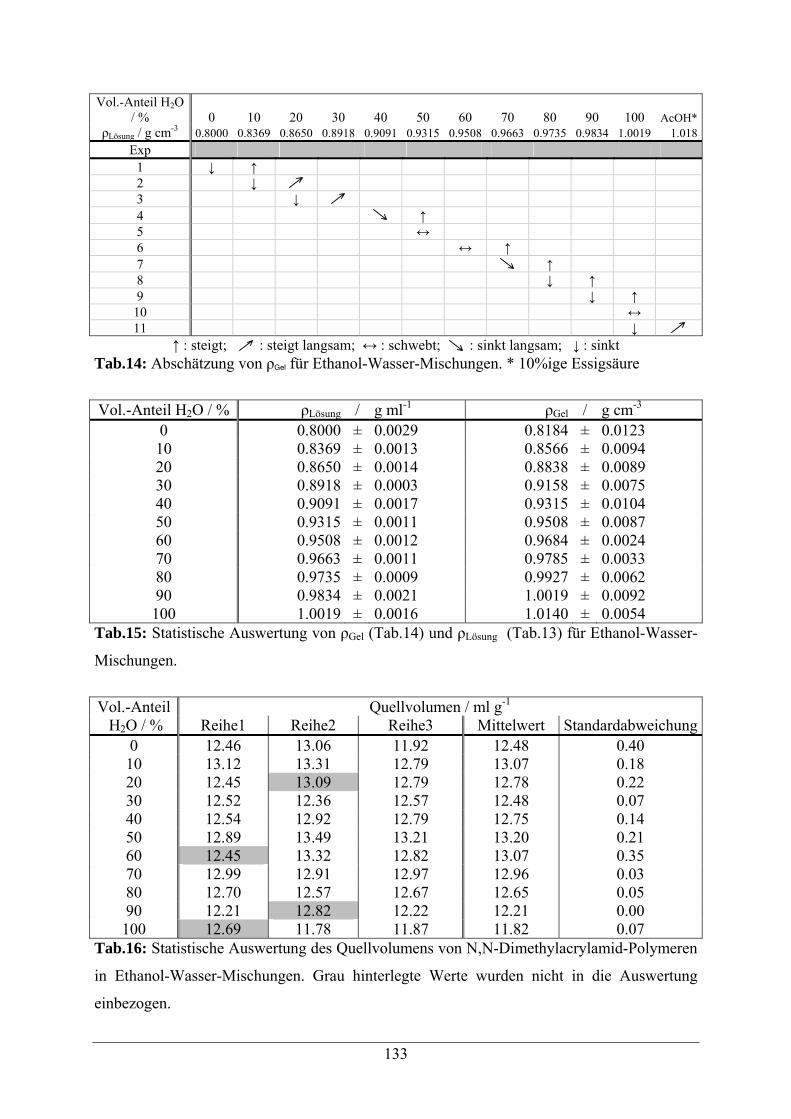
133
Vol.-Anteil H2O / % 0 10 20 30 40 50 60 70 80 90 100 AcOH*
ρLösung / g cm-3 0.8000 0.8369 0.8650 0.8918 0.9091 0.9315 0.9508 0.9663 0.9735 0.9834 1.0019 1.018Exp
1 ↓ ↑ 2 ↓ 3 ↓ 4 ↑ 5 ↔ 6 ↔ ↑ 7 ↑ 8 ↓ ↑ 9 ↓ ↑
10 ↔ 11 ↓
↑ : steigt; : steigt langsam; ↔ : schwebt; : sinkt langsam; ↓ : sinkt Tab.14: Abschätzung von ρGel für Ethanol-Wasser-Mischungen. * 10%ige Essigsäure
Vol.-Anteil H2O / % ρLösung / g ml-1 ρGel / g cm-3 0 0.8000 ± 0.0029 0.8184 ± 0.0123 10 0.8369 ± 0.0013 0.8566 ± 0.0094 20 0.8650 ± 0.0014 0.8838 ± 0.0089 30 0.8918 ± 0.0003 0.9158 ± 0.0075 40 0.9091 ± 0.0017 0.9315 ± 0.0104 50 0.9315 ± 0.0011 0.9508 ± 0.0087 60 0.9508 ± 0.0012 0.9684 ± 0.0024 70 0.9663 ± 0.0011 0.9785 ± 0.0033 80 0.9735 ± 0.0009 0.9927 ± 0.0062 90 0.9834 ± 0.0021 1.0019 ± 0.0092 100 1.0019 ± 0.0016 1.0140 ± 0.0054
Tab.15: Statistische Auswertung von ρGel (Tab.14) und ρLösung (Tab.13) für Ethanol-Wasser-
Mischungen.
Vol.-Anteil Quellvolumen / ml g-1 H2O / % Reihe1 Reihe2 Reihe3 Mittelwert Standardabweichung
0 12.46 13.06 11.92 12.48 0.40 10 13.12 13.31 12.79 13.07 0.18 20 12.45 13.09 12.79 12.78 0.22 30 12.52 12.36 12.57 12.48 0.07 40 12.54 12.92 12.79 12.75 0.14 50 12.89 13.49 13.21 13.20 0.21 60 12.45 13.32 12.82 13.07 0.35 70 12.99 12.91 12.97 12.96 0.03 80 12.70 12.57 12.67 12.65 0.05 90 12.21 12.82 12.22 12.21 0.00 100 12.69 11.78 11.87 11.82 0.07
Tab.16: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in Ethanol-Wasser-Mischungen. Grau hinterlegte Werte wurden nicht in die Auswertung
einbezogen.
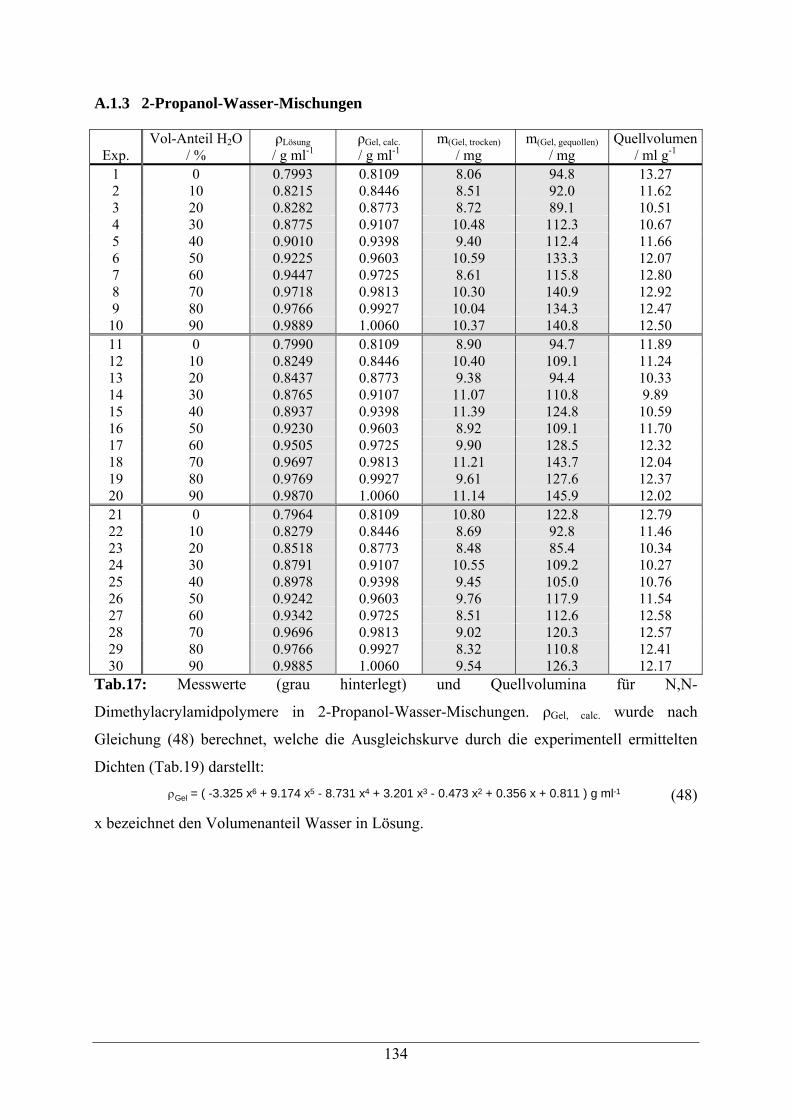
134
A.1.3 2-Propanol-Wasser-Mischungen
Exp. Vol-Anteil H2O
/ % ρLösung / g ml-1
ρGel, calc. / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen/ ml g-1
1 0 0.7993 0.8109 8.06 94.8 13.27 2 10 0.8215 0.8446 8.51 92.0 11.62 3 20 0.8282 0.8773 8.72 89.1 10.51 4 30 0.8775 0.9107 10.48 112.3 10.67 5 40 0.9010 0.9398 9.40 112.4 11.66 6 50 0.9225 0.9603 10.59 133.3 12.07 7 60 0.9447 0.9725 8.61 115.8 12.80 8 70 0.9718 0.9813 10.30 140.9 12.92 9 80 0.9766 0.9927 10.04 134.3 12.47
10 90 0.9889 1.0060 10.37 140.8 12.50 11 0 0.7990 0.8109 8.90 94.7 11.89 12 10 0.8249 0.8446 10.40 109.1 11.24 13 20 0.8437 0.8773 9.38 94.4 10.33 14 30 0.8765 0.9107 11.07 110.8 9.89 15 40 0.8937 0.9398 11.39 124.8 10.59 16 50 0.9230 0.9603 8.92 109.1 11.70 17 60 0.9505 0.9725 9.90 128.5 12.32 18 70 0.9697 0.9813 11.21 143.7 12.04 19 80 0.9769 0.9927 9.61 127.6 12.37 20 90 0.9870 1.0060 11.14 145.9 12.02 21 0 0.7964 0.8109 10.80 122.8 12.79 22 10 0.8279 0.8446 8.69 92.8 11.46 23 20 0.8518 0.8773 8.48 85.4 10.34 24 30 0.8791 0.9107 10.55 109.2 10.27 25 40 0.8978 0.9398 9.45 105.0 10.76 26 50 0.9242 0.9603 9.76 117.9 11.54 27 60 0.9342 0.9725 8.51 112.6 12.58 28 70 0.9696 0.9813 9.02 120.3 12.57 29 80 0.9766 0.9927 8.32 110.8 12.41 30 90 0.9885 1.0060 9.54 126.3 12.17
Tab.17: Messwerte (grau hinterlegt) und Quellvolumina für N,N-
Dimethylacrylamidpolymere in 2-Propanol-Wasser-Mischungen. ρGel, calc. wurde nach
Gleichung (48) berechnet, welche die Ausgleichskurve durch die experimentell ermittelten
Dichten (Tab.19) darstellt:
ρGel = ( -3.325 x6 + 9.174 x5 - 8.731 x4 + 3.201 x3 - 0.473 x2 + 0.356 x + 0.811 ) g ml-1 (48)
x bezeichnet den Volumenanteil Wasser in Lösung.
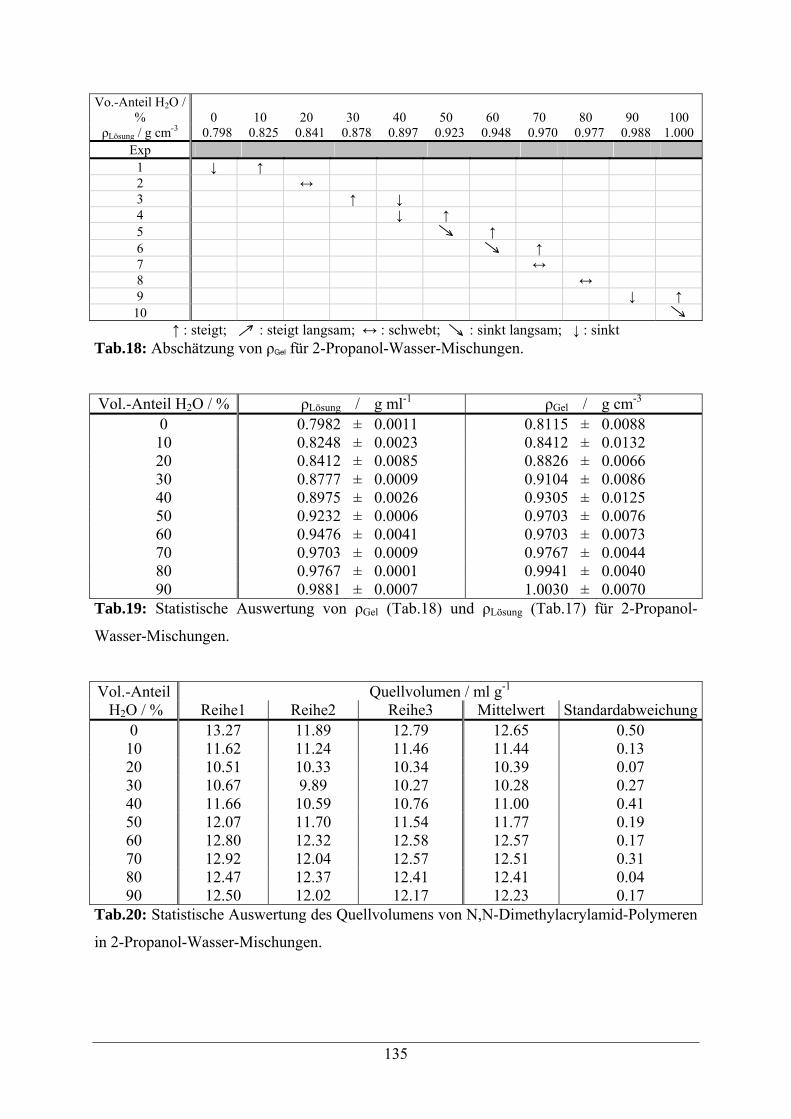
135
Vo.-Anteil H2O / % 0 10 20 30 40 50 60 70 80 90 100
ρLösung / g cm-3 0.798 0.825 0.841 0.878 0.897 0.923 0.948 0.970 0.977 0.988 1.000 Exp
1 ↓ ↑ 2 ↔ 3 ↑ ↓ 4 ↓ ↑ 5 ↑ 6 ↑ 7 ↔ 8 ↔ 9 ↓ ↑
10 ↑ : steigt; : steigt langsam; ↔ : schwebt; : sinkt langsam; ↓ : sinkt
Tab.18: Abschätzung von ρGel für 2-Propanol-Wasser-Mischungen.
Vol.-Anteil H2O / % ρLösung / g ml-1 ρGel / g cm-3 0 0.7982 ± 0.0011 0.8115 ± 0.0088 10 0.8248 ± 0.0023 0.8412 ± 0.0132 20 0.8412 ± 0.0085 0.8826 ± 0.0066 30 0.8777 ± 0.0009 0.9104 ± 0.0086 40 0.8975 ± 0.0026 0.9305 ± 0.0125 50 0.9232 ± 0.0006 0.9703 ± 0.0076 60 0.9476 ± 0.0041 0.9703 ± 0.0073 70 0.9703 ± 0.0009 0.9767 ± 0.0044 80 0.9767 ± 0.0001 0.9941 ± 0.0040 90 0.9881 ± 0.0007 1.0030 ± 0.0070
Tab.19: Statistische Auswertung von ρGel (Tab.18) und ρLösung (Tab.17) für 2-Propanol-
Wasser-Mischungen.
Vol.-Anteil Quellvolumen / ml g-1 H2O / % Reihe1 Reihe2 Reihe3 Mittelwert Standardabweichung
0 13.27 11.89 12.79 12.65 0.50 10 11.62 11.24 11.46 11.44 0.13 20 10.51 10.33 10.34 10.39 0.07 30 10.67 9.89 10.27 10.28 0.27 40 11.66 10.59 10.76 11.00 0.41 50 12.07 11.70 11.54 11.77 0.19 60 12.80 12.32 12.58 12.57 0.17 70 12.92 12.04 12.57 12.51 0.31 80 12.47 12.37 12.41 12.41 0.04 90 12.50 12.02 12.17 12.23 0.17
Tab.20: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in 2-Propanol-Wasser-Mischungen.
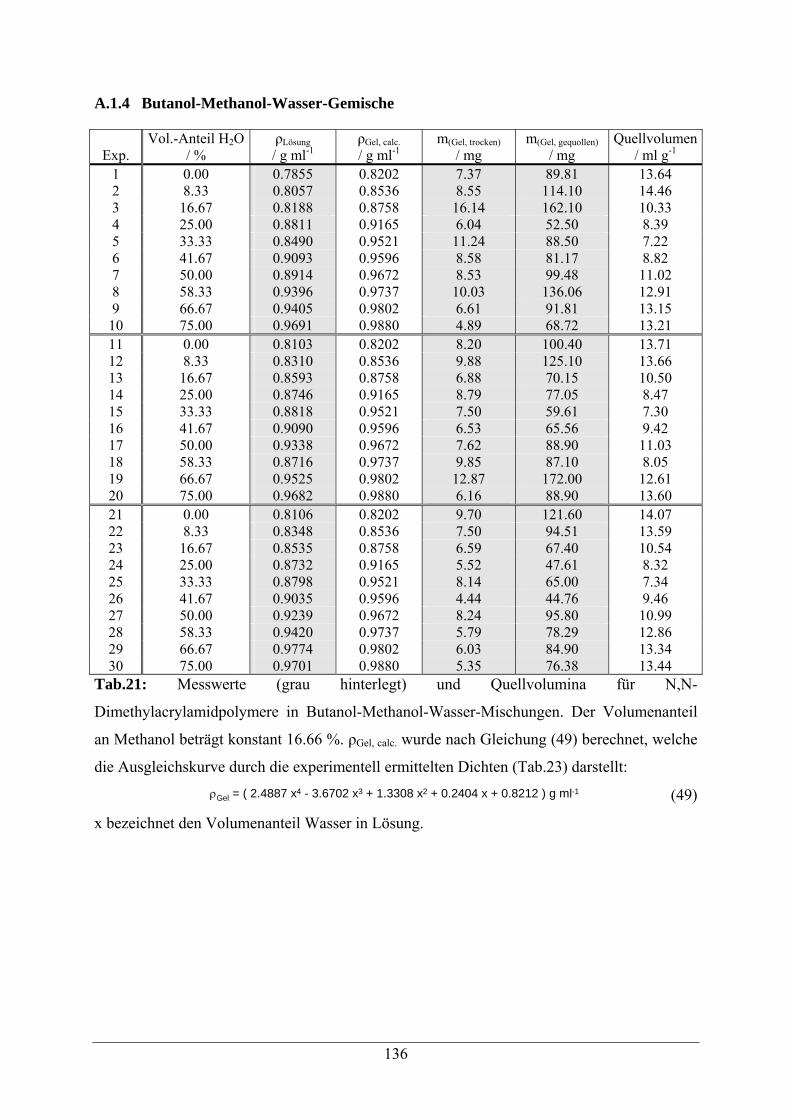
136
A.1.4 Butanol-Methanol-Wasser-Gemische
Exp. Vol.-Anteil H2O
/ % ρLösung / g ml-1
ρGel, calc. / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen/ ml g-1
1 0.00 0.7855 0.8202 7.37 89.81 13.64 2 8.33 0.8057 0.8536 8.55 114.10 14.46 3 16.67 0.8188 0.8758 16.14 162.10 10.33 4 25.00 0.8811 0.9165 6.04 52.50 8.39 5 33.33 0.8490 0.9521 11.24 88.50 7.22 6 41.67 0.9093 0.9596 8.58 81.17 8.82 7 50.00 0.8914 0.9672 8.53 99.48 11.02 8 58.33 0.9396 0.9737 10.03 136.06 12.91 9 66.67 0.9405 0.9802 6.61 91.81 13.15
10 75.00 0.9691 0.9880 4.89 68.72 13.21 11 0.00 0.8103 0.8202 8.20 100.40 13.71 12 8.33 0.8310 0.8536 9.88 125.10 13.66 13 16.67 0.8593 0.8758 6.88 70.15 10.50 14 25.00 0.8746 0.9165 8.79 77.05 8.47 15 33.33 0.8818 0.9521 7.50 59.61 7.30 16 41.67 0.9090 0.9596 6.53 65.56 9.42 17 50.00 0.9338 0.9672 7.62 88.90 11.03 18 58.33 0.8716 0.9737 9.85 87.10 8.05 19 66.67 0.9525 0.9802 12.87 172.00 12.61 20 75.00 0.9682 0.9880 6.16 88.90 13.60 21 0.00 0.8106 0.8202 9.70 121.60 14.07 22 8.33 0.8348 0.8536 7.50 94.51 13.59 23 16.67 0.8535 0.8758 6.59 67.40 10.54 24 25.00 0.8732 0.9165 5.52 47.61 8.32 25 33.33 0.8798 0.9521 8.14 65.00 7.34 26 41.67 0.9035 0.9596 4.44 44.76 9.46 27 50.00 0.9239 0.9672 8.24 95.80 10.99 28 58.33 0.9420 0.9737 5.79 78.29 12.86 29 66.67 0.9774 0.9802 6.03 84.90 13.34 30 75.00 0.9701 0.9880 5.35 76.38 13.44
Tab.21: Messwerte (grau hinterlegt) und Quellvolumina für N,N-
Dimethylacrylamidpolymere in Butanol-Methanol-Wasser-Mischungen. Der Volumenanteil
an Methanol beträgt konstant 16.66 %. ρGel, calc. wurde nach Gleichung (49) berechnet, welche
die Ausgleichskurve durch die experimentell ermittelten Dichten (Tab.23) darstellt:
ρGel = ( 2.4887 x4 - 3.6702 x3 + 1.3308 x2 + 0.2404 x + 0.8212 ) g ml-1 (49)
x bezeichnet den Volumenanteil Wasser in Lösung.
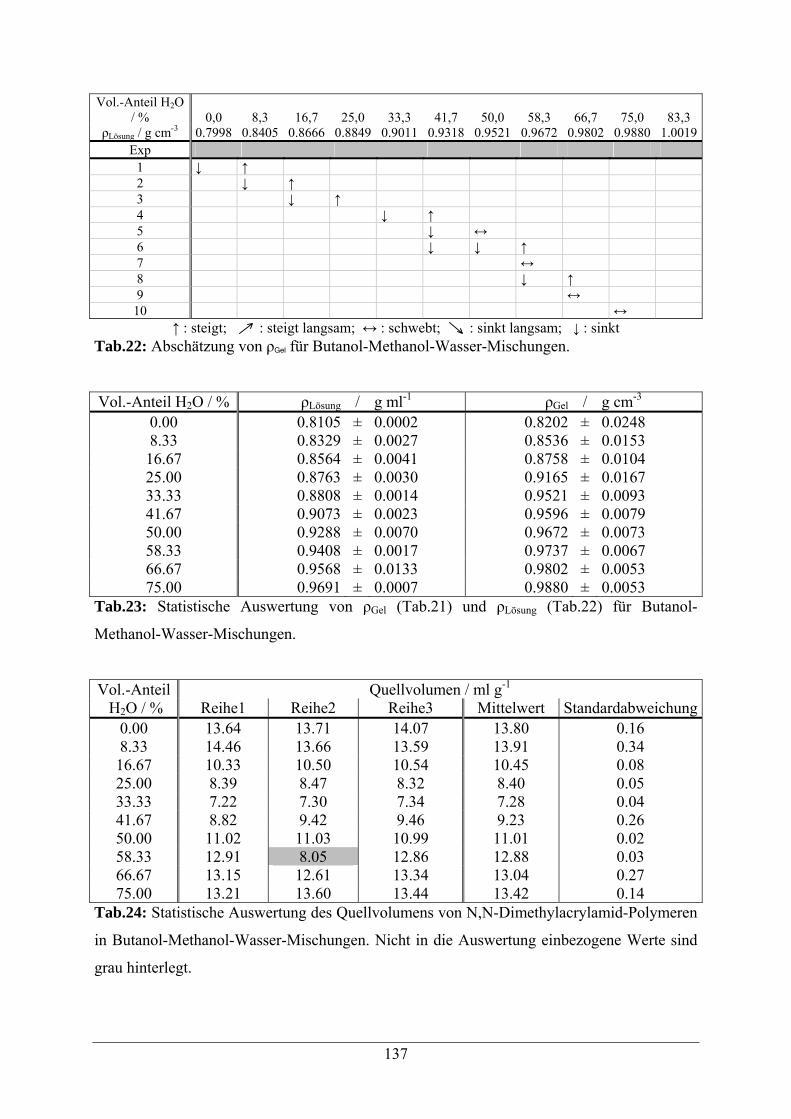
137
Vol.-Anteil H2O / % 0,0 8,3 16,7 25,0 33,3 41,7 50,0 58,3 66,7 75,0 83,3
ρLösung / g cm-3 0.7998 0.8405 0.8666 0.8849 0.9011 0.9318 0.9521 0.9672 0.9802 0.9880 1.0019Exp
1 ↓ ↑ 2 ↓ ↑ 3 ↓ ↑ 4 ↓ ↑ 5 ↓ ↔ 6 ↓ ↓ ↑ 7 ↔ 8 ↓ ↑ 9 ↔
10 ↔ ↑ : steigt; : steigt langsam; ↔ : schwebt; : sinkt langsam; ↓ : sinkt
Tab.22: Abschätzung von ρGel für Butanol-Methanol-Wasser-Mischungen.
Vol.-Anteil H2O / % ρLösung / g ml-1 ρGel / g cm-3 0.00 0.8105 ± 0.0002 0.8202 ± 0.0248 8.33 0.8329 ± 0.0027 0.8536 ± 0.0153 16.67 0.8564 ± 0.0041 0.8758 ± 0.0104 25.00 0.8763 ± 0.0030 0.9165 ± 0.0167 33.33 0.8808 ± 0.0014 0.9521 ± 0.0093 41.67 0.9073 ± 0.0023 0.9596 ± 0.0079 50.00 0.9288 ± 0.0070 0.9672 ± 0.0073 58.33 0.9408 ± 0.0017 0.9737 ± 0.0067 66.67 0.9568 ± 0.0133 0.9802 ± 0.0053 75.00 0.9691 ± 0.0007 0.9880 ± 0.0053
Tab.23: Statistische Auswertung von ρGel (Tab.21) und ρLösung (Tab.22) für Butanol-
Methanol-Wasser-Mischungen.
Vol.-Anteil Quellvolumen / ml g-1 H2O / % Reihe1 Reihe2 Reihe3 Mittelwert Standardabweichung
0.00 13.64 13.71 14.07 13.80 0.16 8.33 14.46 13.66 13.59 13.91 0.34 16.67 10.33 10.50 10.54 10.45 0.08 25.00 8.39 8.47 8.32 8.40 0.05 33.33 7.22 7.30 7.34 7.28 0.04 41.67 8.82 9.42 9.46 9.23 0.26 50.00 11.02 11.03 10.99 11.01 0.02 58.33 12.91 8.05 12.86 12.88 0.03 66.67 13.15 12.61 13.34 13.04 0.27 75.00 13.21 13.60 13.44 13.42 0.14
Tab.24: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in Butanol-Methanol-Wasser-Mischungen. Nicht in die Auswertung einbezogene Werte sind
grau hinterlegt.
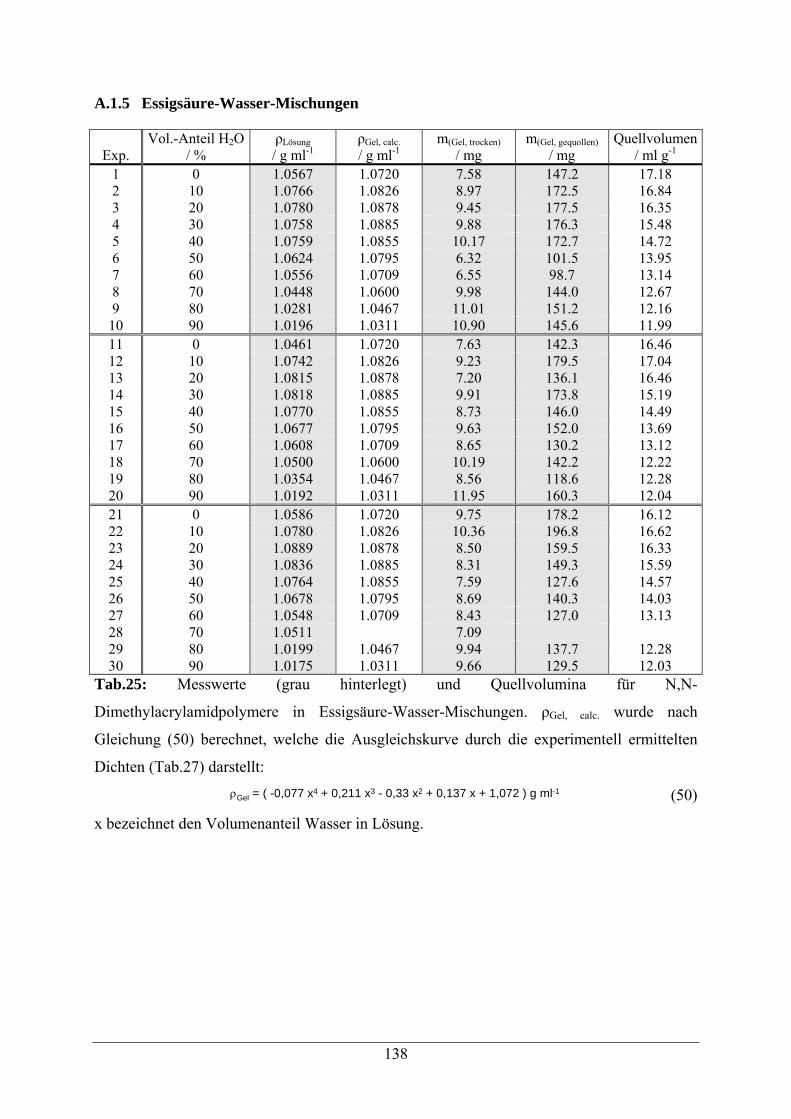
138
A.1.5 Essigsäure-Wasser-Mischungen
Exp. Vol.-Anteil H2O
/ % ρLösung / g ml-1
ρGel, calc. / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen/ ml g-1
1 0 1.0567 1.0720 7.58 147.2 17.18 2 10 1.0766 1.0826 8.97 172.5 16.84 3 20 1.0780 1.0878 9.45 177.5 16.35 4 30 1.0758 1.0885 9.88 176.3 15.48 5 40 1.0759 1.0855 10.17 172.7 14.72 6 50 1.0624 1.0795 6.32 101.5 13.95 7 60 1.0556 1.0709 6.55 98.7 13.14 8 70 1.0448 1.0600 9.98 144.0 12.67 9 80 1.0281 1.0467 11.01 151.2 12.16
10 90 1.0196 1.0311 10.90 145.6 11.99 11 0 1.0461 1.0720 7.63 142.3 16.46 12 10 1.0742 1.0826 9.23 179.5 17.04 13 20 1.0815 1.0878 7.20 136.1 16.46 14 30 1.0818 1.0885 9.91 173.8 15.19 15 40 1.0770 1.0855 8.73 146.0 14.49 16 50 1.0677 1.0795 9.63 152.0 13.69 17 60 1.0608 1.0709 8.65 130.2 13.12 18 70 1.0500 1.0600 10.19 142.2 12.22 19 80 1.0354 1.0467 8.56 118.6 12.28 20 90 1.0192 1.0311 11.95 160.3 12.04 21 0 1.0586 1.0720 9.75 178.2 16.12 22 10 1.0780 1.0826 10.36 196.8 16.62 23 20 1.0889 1.0878 8.50 159.5 16.33 24 30 1.0836 1.0885 8.31 149.3 15.59 25 40 1.0764 1.0855 7.59 127.6 14.57 26 50 1.0678 1.0795 8.69 140.3 14.03 27 60 1.0548 1.0709 8.43 127.0 13.13 28 70 1.0511 7.09 29 80 1.0199 1.0467 9.94 137.7 12.28 30 90 1.0175 1.0311 9.66 129.5 12.03
Tab.25: Messwerte (grau hinterlegt) und Quellvolumina für N,N-
Dimethylacrylamidpolymere in Essigsäure-Wasser-Mischungen. ρGel, calc. wurde nach
Gleichung (50) berechnet, welche die Ausgleichskurve durch die experimentell ermittelten
Dichten (Tab.27) darstellt:
ρGel = ( -0,077 x4 + 0,211 x3 - 0,33 x2 + 0,137 x + 1,072 ) g ml-1 (50)
x bezeichnet den Volumenanteil Wasser in Lösung.
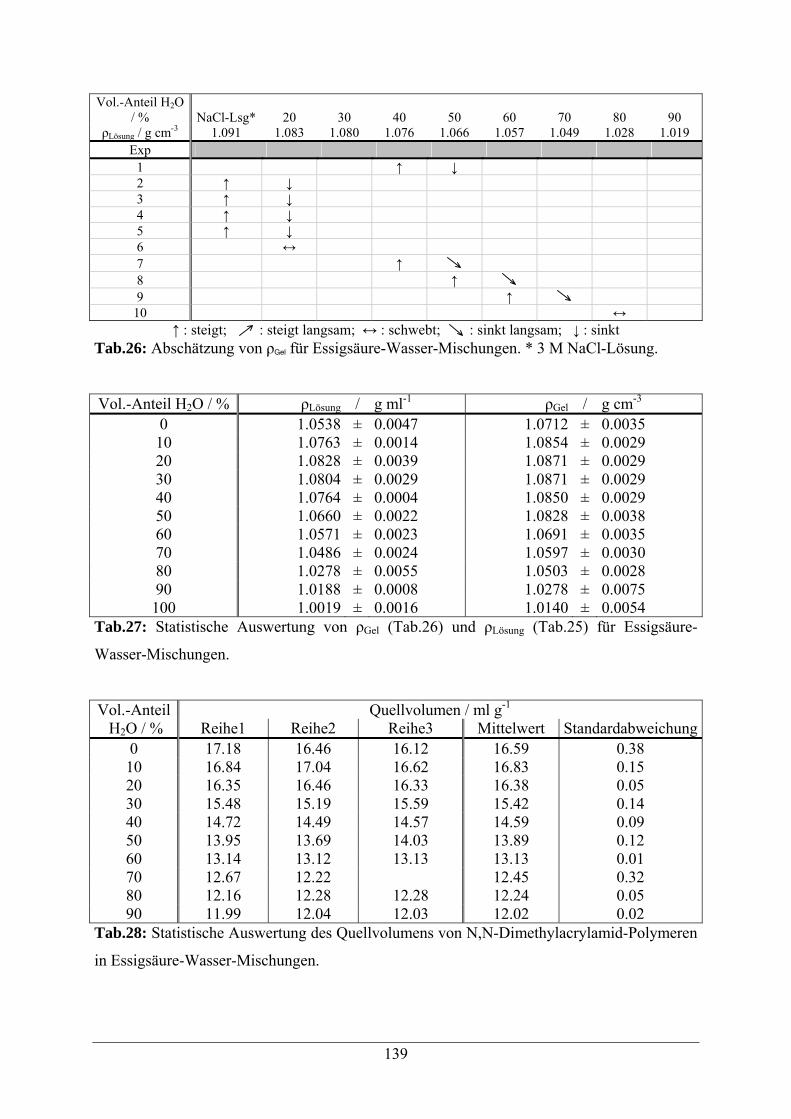
139
Vol.-Anteil H2O / % NaCl-Lsg* 20 30 40 50 60 70 80 90
ρLösung / g cm-3 1.091 1.083 1.080 1.076 1.066 1.057 1.049 1.028 1.019 Exp
1 ↑ ↓ 2 ↑ ↓ 3 ↑ ↓ 4 ↑ ↓ 5 ↑ ↓ 6 ↔ 7 ↑ 8 ↑ 9 ↑
10 ↔ ↑ : steigt; : steigt langsam; ↔ : schwebt; : sinkt langsam; ↓ : sinkt
Tab.26: Abschätzung von ρGel für Essigsäure-Wasser-Mischungen. * 3 M NaCl-Lösung.
Vol.-Anteil H2O / % ρLösung / g ml-1 ρGel / g cm-3 0 1.0538 ± 0.0047 1.0712 ± 0.0035 10 1.0763 ± 0.0014 1.0854 ± 0.0029 20 1.0828 ± 0.0039 1.0871 ± 0.0029 30 1.0804 ± 0.0029 1.0871 ± 0.0029 40 1.0764 ± 0.0004 1.0850 ± 0.0029 50 1.0660 ± 0.0022 1.0828 ± 0.0038 60 1.0571 ± 0.0023 1.0691 ± 0.0035 70 1.0486 ± 0.0024 1.0597 ± 0.0030 80 1.0278 ± 0.0055 1.0503 ± 0.0028 90 1.0188 ± 0.0008 1.0278 ± 0.0075 100 1.0019 ± 0.0016 1.0140 ± 0.0054
Tab.27: Statistische Auswertung von ρGel (Tab.26) und ρLösung (Tab.25) für Essigsäure-
Wasser-Mischungen.
Vol.-Anteil Quellvolumen / ml g-1 H2O / % Reihe1 Reihe2 Reihe3 Mittelwert Standardabweichung
0 17.18 16.46 16.12 16.59 0.38 10 16.84 17.04 16.62 16.83 0.15 20 16.35 16.46 16.33 16.38 0.05 30 15.48 15.19 15.59 15.42 0.14 40 14.72 14.49 14.57 14.59 0.09 50 13.95 13.69 14.03 13.89 0.12 60 13.14 13.12 13.13 13.13 0.01 70 12.67 12.22 12.45 0.32 80 12.16 12.28 12.28 12.24 0.05 90 11.99 12.04 12.03 12.02 0.02
Tab.28: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in Essigsäure-Wasser-Mischungen.
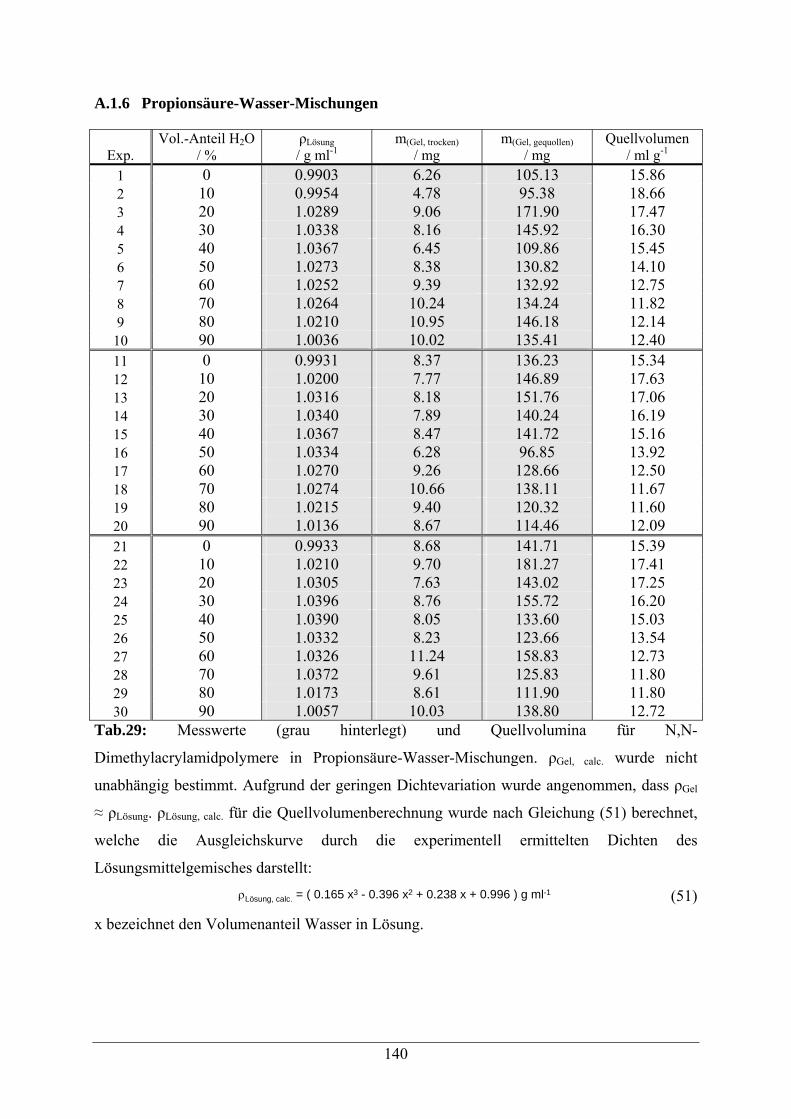
140
A.1.6 Propionsäure-Wasser-Mischungen
Exp. Vol.-Anteil H2O
/ % ρLösung / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen / ml g-1
1 0 0.9903 6.26 105.13 15.86 2 10 0.9954 4.78 95.38 18.66 3 20 1.0289 9.06 171.90 17.47 4 30 1.0338 8.16 145.92 16.30 5 40 1.0367 6.45 109.86 15.45 6 50 1.0273 8.38 130.82 14.10 7 60 1.0252 9.39 132.92 12.75 8 70 1.0264 10.24 134.24 11.82 9 80 1.0210 10.95 146.18 12.14
10 90 1.0036 10.02 135.41 12.40 11 0 0.9931 8.37 136.23 15.34 12 10 1.0200 7.77 146.89 17.63 13 20 1.0316 8.18 151.76 17.06 14 30 1.0340 7.89 140.24 16.19 15 40 1.0367 8.47 141.72 15.16 16 50 1.0334 6.28 96.85 13.92 17 60 1.0270 9.26 128.66 12.50 18 70 1.0274 10.66 138.11 11.67 19 80 1.0215 9.40 120.32 11.60 20 90 1.0136 8.67 114.46 12.09 21 0 0.9933 8.68 141.71 15.39 22 10 1.0210 9.70 181.27 17.41 23 20 1.0305 7.63 143.02 17.25 24 30 1.0396 8.76 155.72 16.20 25 40 1.0390 8.05 133.60 15.03 26 50 1.0332 8.23 123.66 13.54 27 60 1.0326 11.24 158.83 12.73 28 70 1.0372 9.61 125.83 11.80 29 80 1.0173 8.61 111.90 11.80 30 90 1.0057 10.03 138.80 12.72
Tab.29: Messwerte (grau hinterlegt) und Quellvolumina für N,N-
Dimethylacrylamidpolymere in Propionsäure-Wasser-Mischungen. ρGel, calc. wurde nicht
unabhängig bestimmt. Aufgrund der geringen Dichtevariation wurde angenommen, dass ρGel
≈ ρLösung. ρLösung, calc. für die Quellvolumenberechnung wurde nach Gleichung (51) berechnet,
welche die Ausgleichskurve durch die experimentell ermittelten Dichten des
Lösungsmittelgemisches darstellt:
ρLösung, calc. = ( 0.165 x3 - 0.396 x2 + 0.238 x + 0.996 ) g ml-1 (51)
x bezeichnet den Volumenanteil Wasser in Lösung.
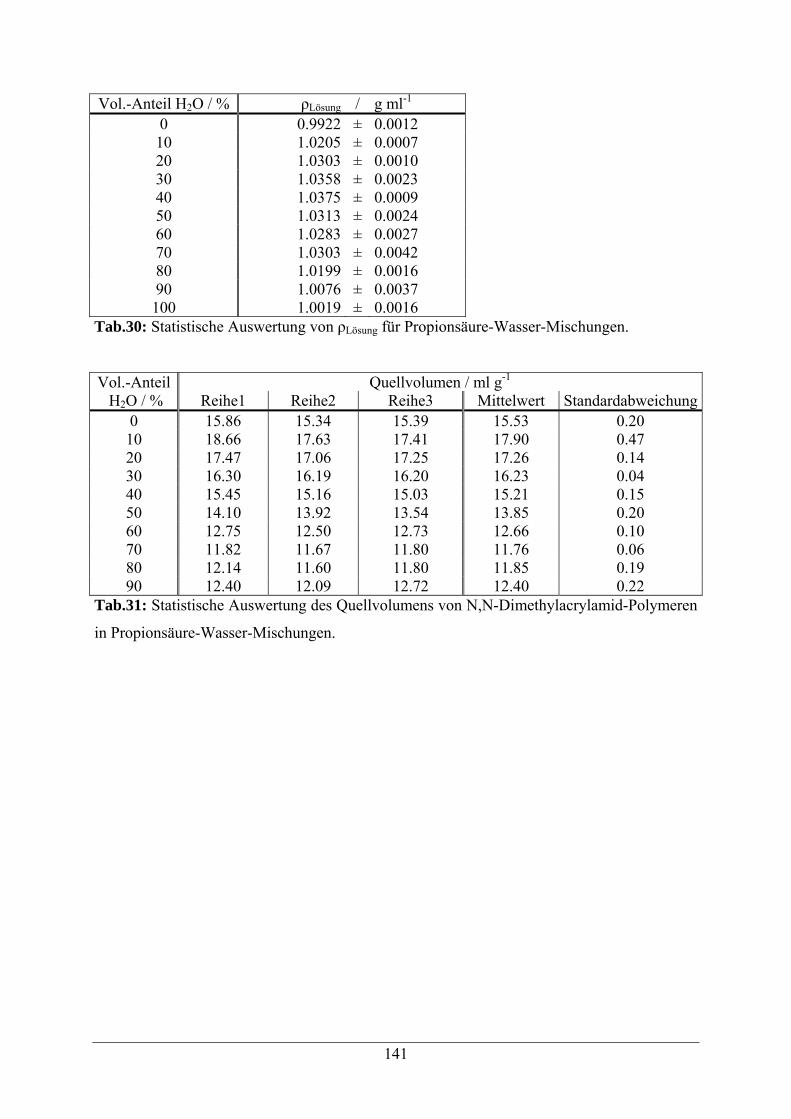
141
Vol.-Anteil H2O / % ρLösung / g ml-1 0 0.9922 ± 0.0012 10 1.0205 ± 0.0007 20 1.0303 ± 0.0010 30 1.0358 ± 0.0023 40 1.0375 ± 0.0009 50 1.0313 ± 0.0024 60 1.0283 ± 0.0027 70 1.0303 ± 0.0042 80 1.0199 ± 0.0016 90 1.0076 ± 0.0037 100 1.0019 ± 0.0016
Tab.30: Statistische Auswertung von ρLösung für Propionsäure-Wasser-Mischungen.
Vol.-Anteil Quellvolumen / ml g-1 H2O / % Reihe1 Reihe2 Reihe3 Mittelwert Standardabweichung
0 15.86 15.34 15.39 15.53 0.20 10 18.66 17.63 17.41 17.90 0.47 20 17.47 17.06 17.25 17.26 0.14 30 16.30 16.19 16.20 16.23 0.04 40 15.45 15.16 15.03 15.21 0.15 50 14.10 13.92 13.54 13.85 0.20 60 12.75 12.50 12.73 12.66 0.10 70 11.82 11.67 11.80 11.76 0.06 80 12.14 11.60 11.80 11.85 0.19 90 12.40 12.09 12.72 12.40 0.22
Tab.31: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in Propionsäure-Wasser-Mischungen.
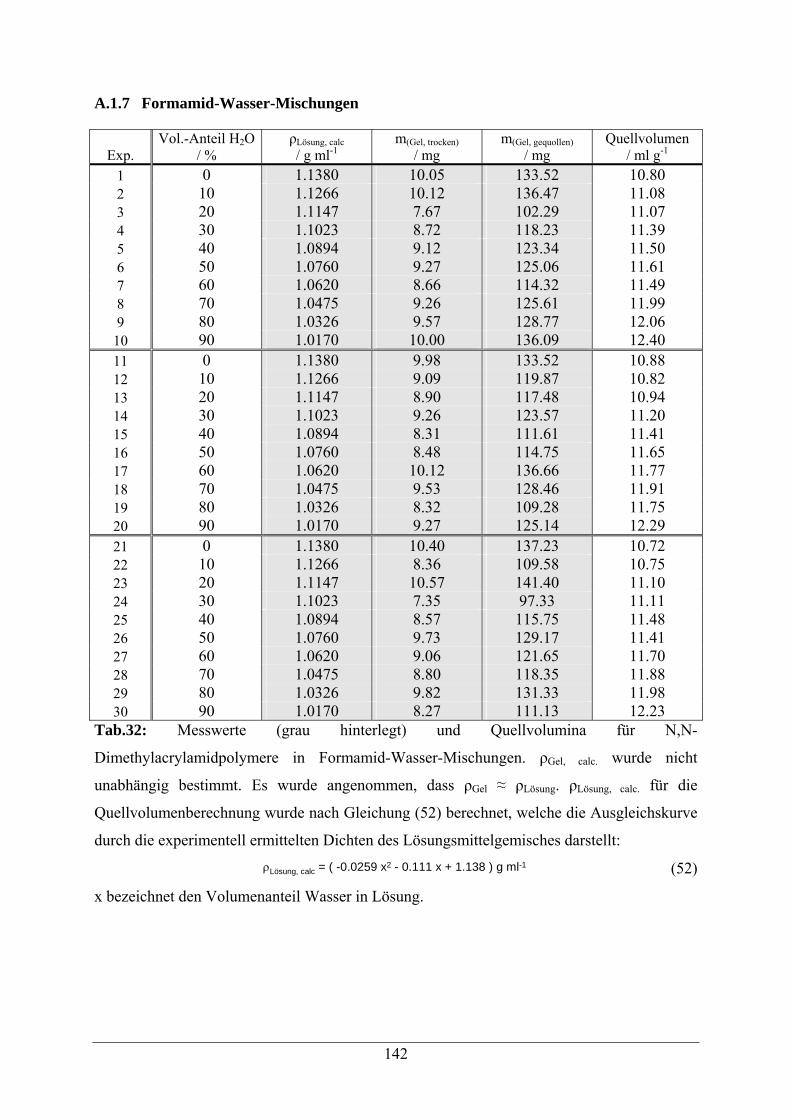
142
A.1.7 Formamid-Wasser-Mischungen
Exp. Vol.-Anteil H2O
/ % ρLösung, calc / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen / ml g-1
1 0 1.1380 10.05 133.52 10.80 2 10 1.1266 10.12 136.47 11.08 3 20 1.1147 7.67 102.29 11.07 4 30 1.1023 8.72 118.23 11.39 5 40 1.0894 9.12 123.34 11.50 6 50 1.0760 9.27 125.06 11.61 7 60 1.0620 8.66 114.32 11.49 8 70 1.0475 9.26 125.61 11.99 9 80 1.0326 9.57 128.77 12.06
10 90 1.0170 10.00 136.09 12.40 11 0 1.1380 9.98 133.52 10.88 12 10 1.1266 9.09 119.87 10.82 13 20 1.1147 8.90 117.48 10.94 14 30 1.1023 9.26 123.57 11.20 15 40 1.0894 8.31 111.61 11.41 16 50 1.0760 8.48 114.75 11.65 17 60 1.0620 10.12 136.66 11.77 18 70 1.0475 9.53 128.46 11.91 19 80 1.0326 8.32 109.28 11.75 20 90 1.0170 9.27 125.14 12.29 21 0 1.1380 10.40 137.23 10.72 22 10 1.1266 8.36 109.58 10.75 23 20 1.1147 10.57 141.40 11.10 24 30 1.1023 7.35 97.33 11.11 25 40 1.0894 8.57 115.75 11.48 26 50 1.0760 9.73 129.17 11.41 27 60 1.0620 9.06 121.65 11.70 28 70 1.0475 8.80 118.35 11.88 29 80 1.0326 9.82 131.33 11.98 30 90 1.0170 8.27 111.13 12.23
Tab.32: Messwerte (grau hinterlegt) und Quellvolumina für N,N-
Dimethylacrylamidpolymere in Formamid-Wasser-Mischungen. ρGel, calc. wurde nicht
unabhängig bestimmt. Es wurde angenommen, dass ρGel ≈ ρLösung. ρLösung, calc. für die
Quellvolumenberechnung wurde nach Gleichung (52) berechnet, welche die Ausgleichskurve
durch die experimentell ermittelten Dichten des Lösungsmittelgemisches darstellt:
ρLösung, calc = ( -0.0259 x2 - 0.111 x + 1.138 ) g ml-1 (52)
x bezeichnet den Volumenanteil Wasser in Lösung.
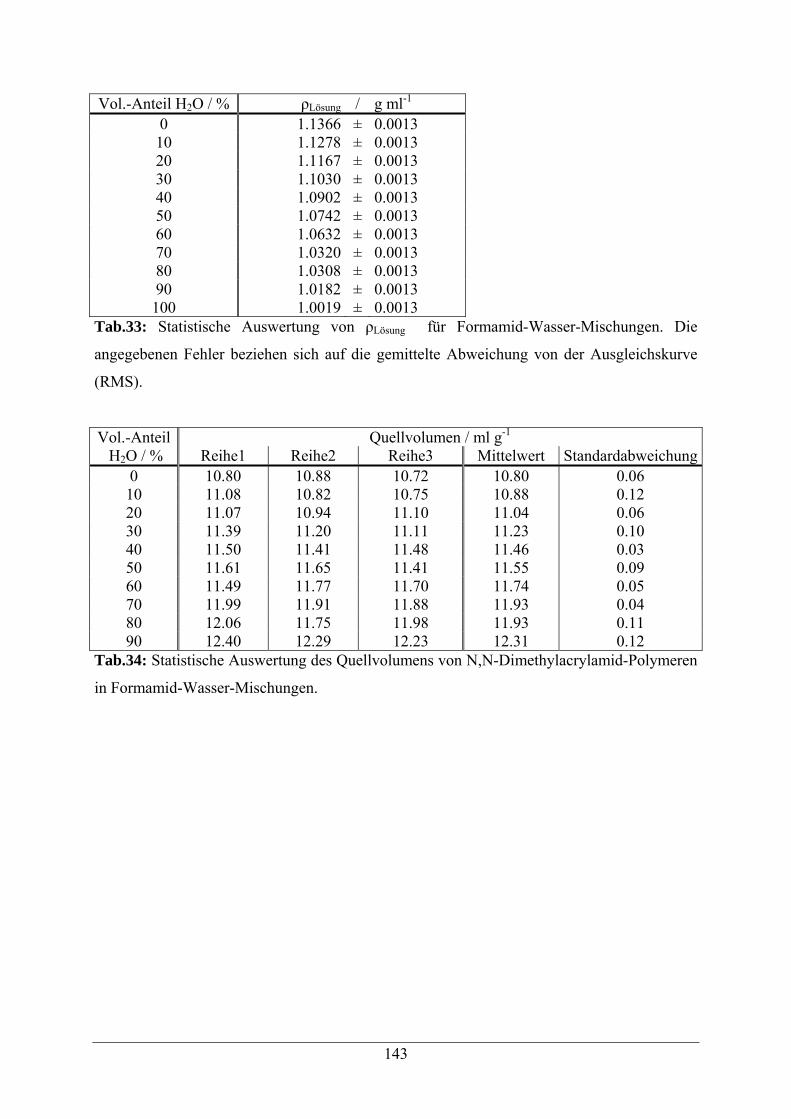
143
Vol.-Anteil H2O / % ρLösung / g ml-1 0 1.1366 ± 0.0013 10 1.1278 ± 0.0013 20 1.1167 ± 0.0013 30 1.1030 ± 0.0013 40 1.0902 ± 0.0013 50 1.0742 ± 0.0013 60 1.0632 ± 0.0013 70 1.0320 ± 0.0013 80 1.0308 ± 0.0013 90 1.0182 ± 0.0013 100 1.0019 ± 0.0013
Tab.33: Statistische Auswertung von ρLösung für Formamid-Wasser-Mischungen. Die
angegebenen Fehler beziehen sich auf die gemittelte Abweichung von der Ausgleichskurve
(RMS).
Vol.-Anteil Quellvolumen / ml g-1 H2O / % Reihe1 Reihe2 Reihe3 Mittelwert Standardabweichung
0 10.80 10.88 10.72 10.80 0.06 10 11.08 10.82 10.75 10.88 0.12 20 11.07 10.94 11.10 11.04 0.06 30 11.39 11.20 11.11 11.23 0.10 40 11.50 11.41 11.48 11.46 0.03 50 11.61 11.65 11.41 11.55 0.09 60 11.49 11.77 11.70 11.74 0.05 70 11.99 11.91 11.88 11.93 0.04 80 12.06 11.75 11.98 11.93 0.11 90 12.40 12.29 12.23 12.31 0.12
Tab.34: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in Formamid-Wasser-Mischungen.
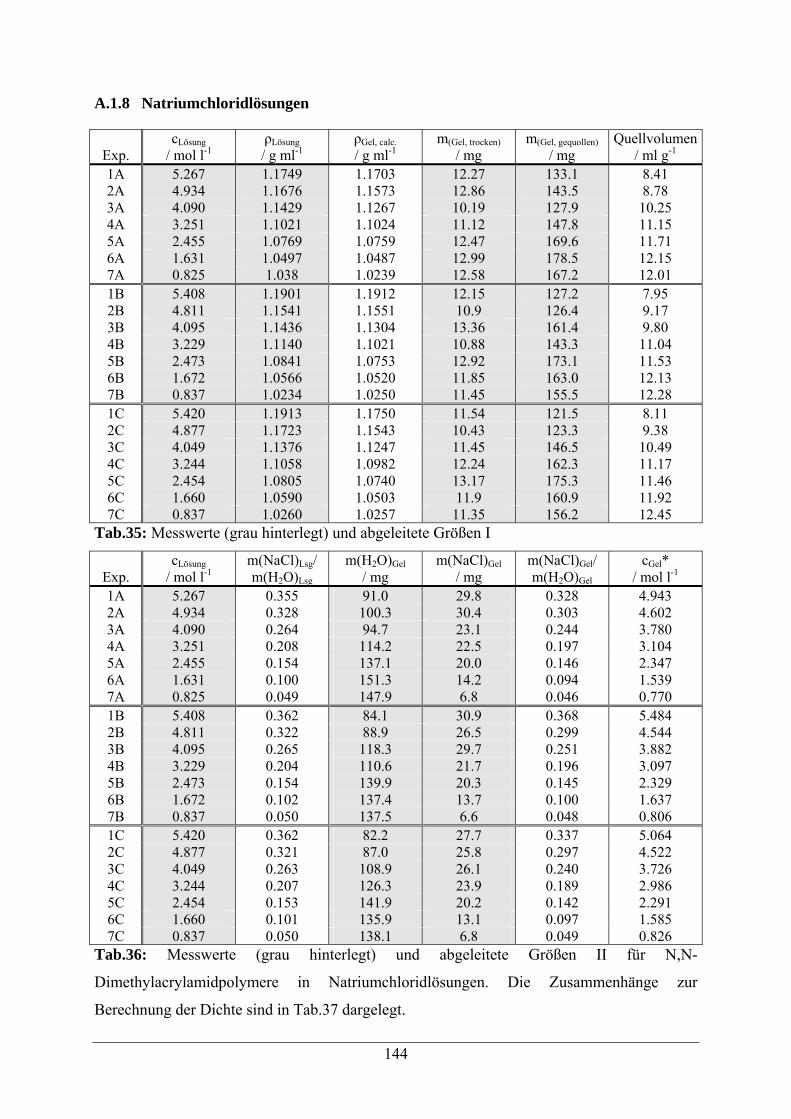
144
A.1.8 Natriumchloridlösungen
Exp. cLösung
/ mol l-1 ρLösung / g ml-1
ρGel, calc. / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen/ ml g-1
1A 5.267 1.1749 1.1703 12.27 133.1 8.41 2A 4.934 1.1676 1.1573 12.86 143.5 8.78 3A 4.090 1.1429 1.1267 10.19 127.9 10.25 4A 3.251 1.1021 1.1024 11.12 147.8 11.15 5A 2.455 1.0769 1.0759 12.47 169.6 11.71 6A 1.631 1.0497 1.0487 12.99 178.5 12.15 7A 0.825 1.038 1.0239 12.58 167.2 12.01 1B 5.408 1.1901 1.1912 12.15 127.2 7.95 2B 4.811 1.1541 1.1551 10.9 126.4 9.17 3B 4.095 1.1436 1.1304 13.36 161.4 9.80 4B 3.229 1.1140 1.1021 10.88 143.3 11.04 5B 2.473 1.0841 1.0753 12.92 173.1 11.53 6B 1.672 1.0566 1.0520 11.85 163.0 12.13 7B 0.837 1.0234 1.0250 11.45 155.5 12.28 1C 5.420 1.1913 1.1750 11.54 121.5 8.11 2C 4.877 1.1723 1.1543 10.43 123.3 9.38 3C 4.049 1.1376 1.1247 11.45 146.5 10.49 4C 3.244 1.1058 1.0982 12.24 162.3 11.17 5C 2.454 1.0805 1.0740 13.17 175.3 11.46 6C 1.660 1.0590 1.0503 11.9 160.9 11.92 7C 0.837 1.0260 1.0257 11.35 156.2 12.45
Tab.35: Messwerte (grau hinterlegt) und abgeleitete Größen I
Exp. cLösung
/ mol l-1 m(NaCl)Lsg/ m(H2O)Lsg
m(H2O)Gel / mg
m(NaCl)Gel / mg
m(NaCl)Gel/ m(H2O)Gel
cGel* / mol l-1
1A 5.267 0.355 91.0 29.8 0.328 4.943 2A 4.934 0.328 100.3 30.4 0.303 4.602 3A 4.090 0.264 94.7 23.1 0.244 3.780 4A 3.251 0.208 114.2 22.5 0.197 3.104 5A 2.455 0.154 137.1 20.0 0.146 2.347 6A 1.631 0.100 151.3 14.2 0.094 1.539 7A 0.825 0.049 147.9 6.8 0.046 0.770 1B 5.408 0.362 84.1 30.9 0.368 5.484 2B 4.811 0.322 88.9 26.5 0.299 4.544 3B 4.095 0.265 118.3 29.7 0.251 3.882 4B 3.229 0.204 110.6 21.7 0.196 3.097 5B 2.473 0.154 139.9 20.3 0.145 2.329 6B 1.672 0.102 137.4 13.7 0.100 1.637 7B 0.837 0.050 137.5 6.6 0.048 0.806 1C 5.420 0.362 82.2 27.7 0.337 5.064 2C 4.877 0.321 87.0 25.8 0.297 4.522 3C 4.049 0.263 108.9 26.1 0.240 3.726 4C 3.244 0.207 126.3 23.9 0.189 2.986 5C 2.454 0.153 141.9 20.2 0.142 2.291 6C 1.660 0.101 135.9 13.1 0.097 1.585 7C 0.837 0.050 138.1 6.8 0.049 0.826
Tab.36: Messwerte (grau hinterlegt) und abgeleitete Größen II für N,N-
Dimethylacrylamidpolymere in Natriumchloridlösungen. Die Zusammenhänge zur
Berechnung der Dichte sind in Tab.37 dargelegt.
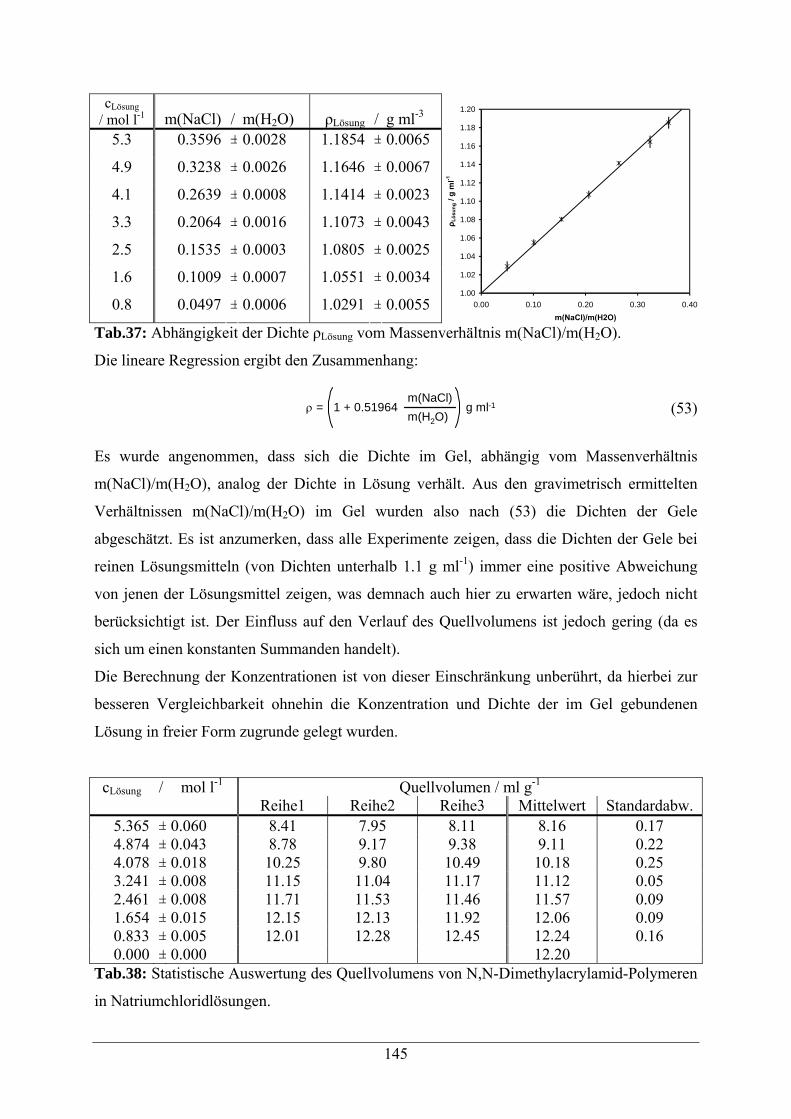
145
cLösung / mol l-1 m(NaCl) / m(H2O) ρLösung / g ml-3
5.3 0.3596 ± 0.0028 1.1854 ± 0.0065
4.9 0.3238 ± 0.0026 1.1646 ± 0.0067
4.1 0.2639 ± 0.0008 1.1414 ± 0.0023
3.3 0.2064 ± 0.0016 1.1073 ± 0.0043
2.5 0.1535 ± 0.0003 1.0805 ± 0.0025
1.6 0.1009 ± 0.0007 1.0551 ± 0.0034
0.8 0.0497 ± 0.0006 1.0291 ± 0.0055 1.00
1.02
1.04
1.06
1.08
1.10
1.12
1.14
1.16
1.18
1.20
0.00 0.10 0.20 0.30 0.40m(NaCl)/m(H2O)
ρ Lös
ung
/gm
l-1
Tab.37: Abhängigkeit der Dichte ρLösung vom Massenverhältnis m(NaCl)/m(H2O).
Die lineare Regression ergibt den Zusammenhang:
ρ = 1 + 0.51964 g ml-1m(NaCl)
m(H2O)
(53)
Es wurde angenommen, dass sich die Dichte im Gel, abhängig vom Massenverhältnis
m(NaCl)/m(H2O), analog der Dichte in Lösung verhält. Aus den gravimetrisch ermittelten
Verhältnissen m(NaCl)/m(H2O) im Gel wurden also nach (53) die Dichten der Gele
abgeschätzt. Es ist anzumerken, dass alle Experimente zeigen, dass die Dichten der Gele bei
reinen Lösungsmitteln (von Dichten unterhalb 1.1 g ml-1) immer eine positive Abweichung
von jenen der Lösungsmittel zeigen, was demnach auch hier zu erwarten wäre, jedoch nicht
berücksichtigt ist. Der Einfluss auf den Verlauf des Quellvolumens ist jedoch gering (da es
sich um einen konstanten Summanden handelt).
Die Berechnung der Konzentrationen ist von dieser Einschränkung unberührt, da hierbei zur
besseren Vergleichbarkeit ohnehin die Konzentration und Dichte der im Gel gebundenen
Lösung in freier Form zugrunde gelegt wurden.
cLösung / mol l-1 Quellvolumen / ml g-1 Reihe1 Reihe2 Reihe3 Mittelwert Standardabw.
5.365 ± 0.060 8.41 7.95 8.11 8.16 0.17 4.874 ± 0.043 8.78 9.17 9.38 9.11 0.22 4.078 ± 0.018 10.25 9.80 10.49 10.18 0.25 3.241 ± 0.008 11.15 11.04 11.17 11.12 0.05 2.461 ± 0.008 11.71 11.53 11.46 11.57 0.09 1.654 ± 0.015 12.15 12.13 11.92 12.06 0.09 0.833 ± 0.005 12.01 12.28 12.45 12.24 0.16 0.000 ± 0.000 12.20
Tab.38: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in Natriumchloridlösungen.
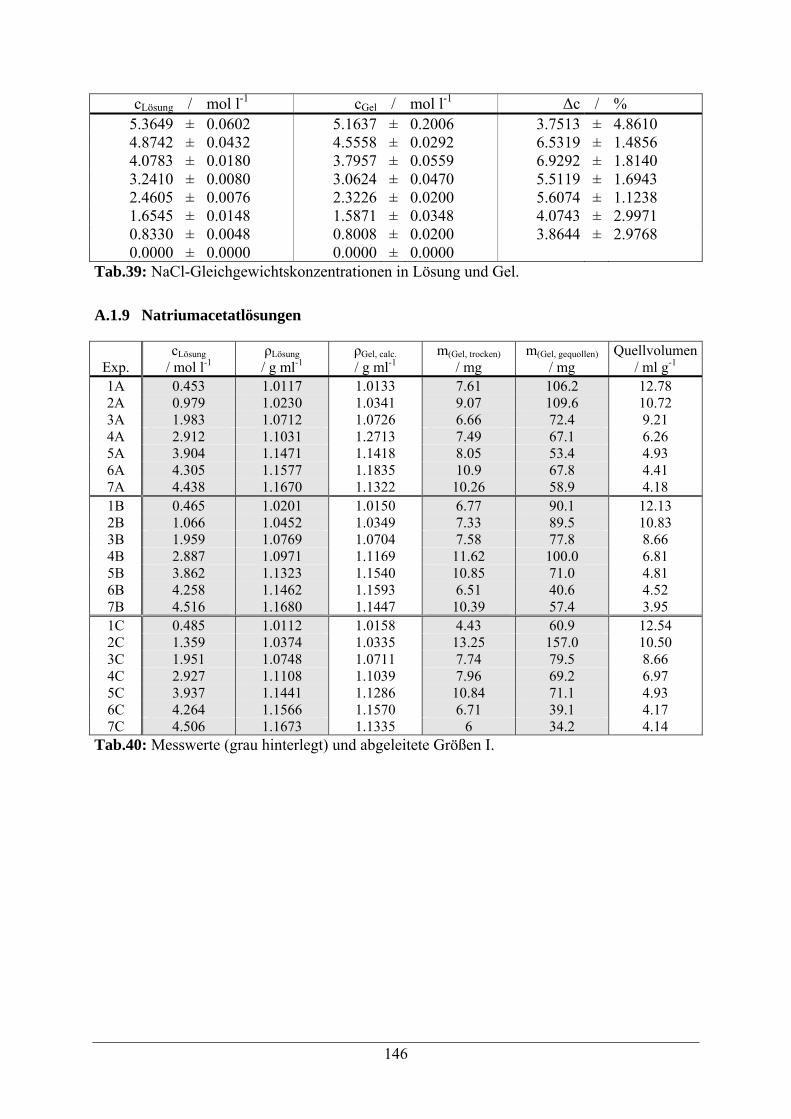
146
cLösung / mol l-1 cGel / mol l-1 ∆c / % 5.3649 ± 0.0602 5.1637 ± 0.2006 3.7513 ± 4.8610 4.8742 ± 0.0432 4.5558 ± 0.0292 6.5319 ± 1.4856 4.0783 ± 0.0180 3.7957 ± 0.0559 6.9292 ± 1.8140 3.2410 ± 0.0080 3.0624 ± 0.0470 5.5119 ± 1.6943 2.4605 ± 0.0076 2.3226 ± 0.0200 5.6074 ± 1.1238 1.6545 ± 0.0148 1.5871 ± 0.0348 4.0743 ± 2.9971 0.8330 ± 0.0048 0.8008 ± 0.0200 3.8644 ± 2.9768 0.0000 ± 0.0000 0.0000 ± 0.0000
Tab.39: NaCl-Gleichgewichtskonzentrationen in Lösung und Gel.
A.1.9 Natriumacetatlösungen
Exp. cLösung
/ mol l-1 ρLösung / g ml-1
ρGel, calc. / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen/ ml g-1
1A 0.453 1.0117 1.0133 7.61 106.2 12.78 2A 0.979 1.0230 1.0341 9.07 109.6 10.72 3A 1.983 1.0712 1.0726 6.66 72.4 9.21 4A 2.912 1.1031 1.2713 7.49 67.1 6.26 5A 3.904 1.1471 1.1418 8.05 53.4 4.93 6A 4.305 1.1577 1.1835 10.9 67.8 4.41 7A 4.438 1.1670 1.1322 10.26 58.9 4.18 1B 0.465 1.0201 1.0150 6.77 90.1 12.13 2B 1.066 1.0452 1.0349 7.33 89.5 10.83 3B 1.959 1.0769 1.0704 7.58 77.8 8.66 4B 2.887 1.0971 1.1169 11.62 100.0 6.81 5B 3.862 1.1323 1.1540 10.85 71.0 4.81 6B 4.258 1.1462 1.1593 6.51 40.6 4.52 7B 4.516 1.1680 1.1447 10.39 57.4 3.95 1C 0.485 1.0112 1.0158 4.43 60.9 12.54 2C 1.359 1.0374 1.0335 13.25 157.0 10.50 3C 1.951 1.0748 1.0711 7.74 79.5 8.66 4C 2.927 1.1108 1.1039 7.96 69.2 6.97 5C 3.937 1.1441 1.1286 10.84 71.1 4.93 6C 4.264 1.1566 1.1570 6.71 39.1 4.17 7C 4.506 1.1673 1.1335 6 34.2 4.14
Tab.40: Messwerte (grau hinterlegt) und abgeleitete Größen I.
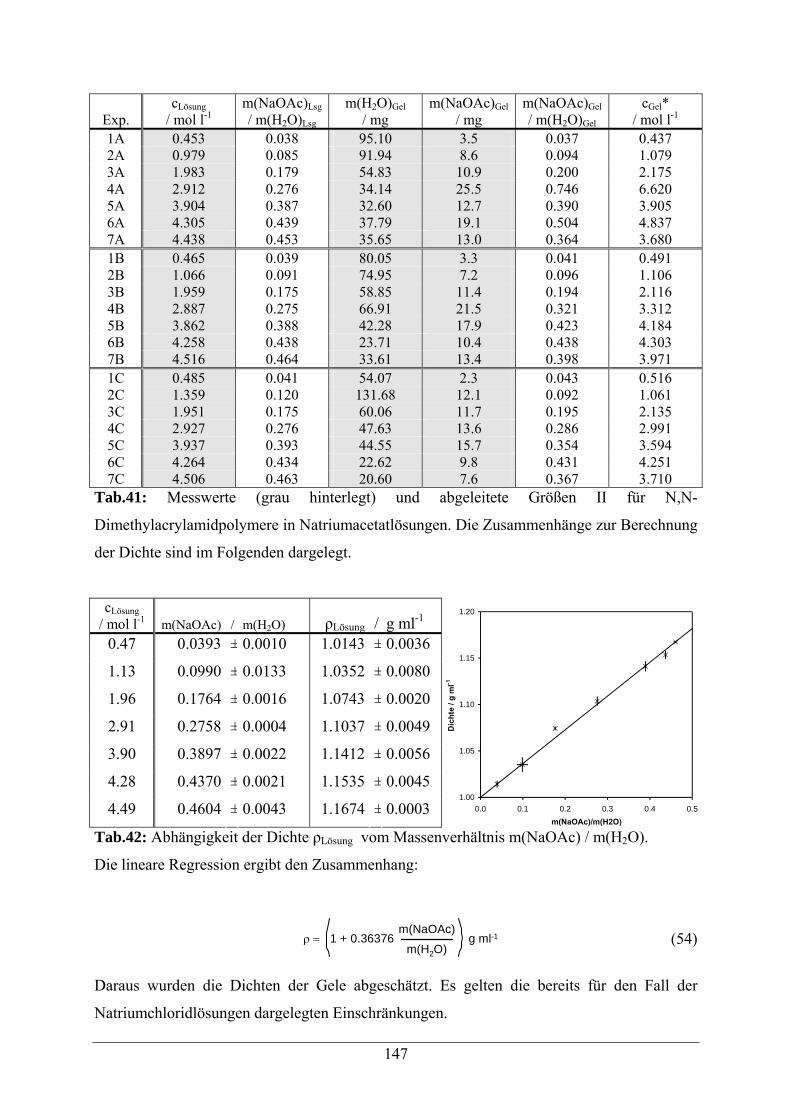
147
Exp. cLösung
/ mol l-1 m(NaOAc)Lsg / m(H2O)Lsg
m(H2O)Gel / mg
m(NaOAc)Gel / mg
m(NaOAc)Gel / m(H2O)Gel
cGel* / mol l-1
1A 0.453 0.038 95.10 3.5 0.037 0.437 2A 0.979 0.085 91.94 8.6 0.094 1.079 3A 1.983 0.179 54.83 10.9 0.200 2.175 4A 2.912 0.276 34.14 25.5 0.746 6.620 5A 3.904 0.387 32.60 12.7 0.390 3.905 6A 4.305 0.439 37.79 19.1 0.504 4.837 7A 4.438 0.453 35.65 13.0 0.364 3.680 1B 0.465 0.039 80.05 3.3 0.041 0.491 2B 1.066 0.091 74.95 7.2 0.096 1.106 3B 1.959 0.175 58.85 11.4 0.194 2.116 4B 2.887 0.275 66.91 21.5 0.321 3.312 5B 3.862 0.388 42.28 17.9 0.423 4.184 6B 4.258 0.438 23.71 10.4 0.438 4.303 7B 4.516 0.464 33.61 13.4 0.398 3.971 1C 0.485 0.041 54.07 2.3 0.043 0.516 2C 1.359 0.120 131.68 12.1 0.092 1.061 3C 1.951 0.175 60.06 11.7 0.195 2.135 4C 2.927 0.276 47.63 13.6 0.286 2.991 5C 3.937 0.393 44.55 15.7 0.354 3.594 6C 4.264 0.434 22.62 9.8 0.431 4.251 7C 4.506 0.463 20.60 7.6 0.367 3.710
Tab.41: Messwerte (grau hinterlegt) und abgeleitete Größen II für N,N-
Dimethylacrylamidpolymere in Natriumacetatlösungen. Die Zusammenhänge zur Berechnung
der Dichte sind im Folgenden dargelegt.
cLösung / mol l-1 m(NaOAc) / m(H2O) ρLösung / g ml-1
0.47 0.0393 ± 0.0010 1.0143 ± 0.0036
1.13 0.0990 ± 0.0133 1.0352 ± 0.0080
1.96 0.1764 ± 0.0016 1.0743 ± 0.0020
2.91 0.2758 ± 0.0004 1.1037 ± 0.0049
3.90 0.3897 ± 0.0022 1.1412 ± 0.0056
4.28 0.4370 ± 0.0021 1.1535 ± 0.0045
4.49 0.4604 ± 0.0043 1.1674 ± 0.0003 1.00
1.05
1.10
1.15
1.20
0.0 0.1 0.2 0.3 0.4 0.5m(NaOAc)/m(H2O)
Dic
hte
/gm
l-1
Tab.42: Abhängigkeit der Dichte ρLösung vom Massenverhältnis m(NaOAc) / m(H2O).
Die lineare Regression ergibt den Zusammenhang:
ρ = 1 + 0.36376 g ml-1m(NaOAc)
m(H2O)
(54)
Daraus wurden die Dichten der Gele abgeschätzt. Es gelten die bereits für den Fall der
Natriumchloridlösungen dargelegten Einschränkungen.
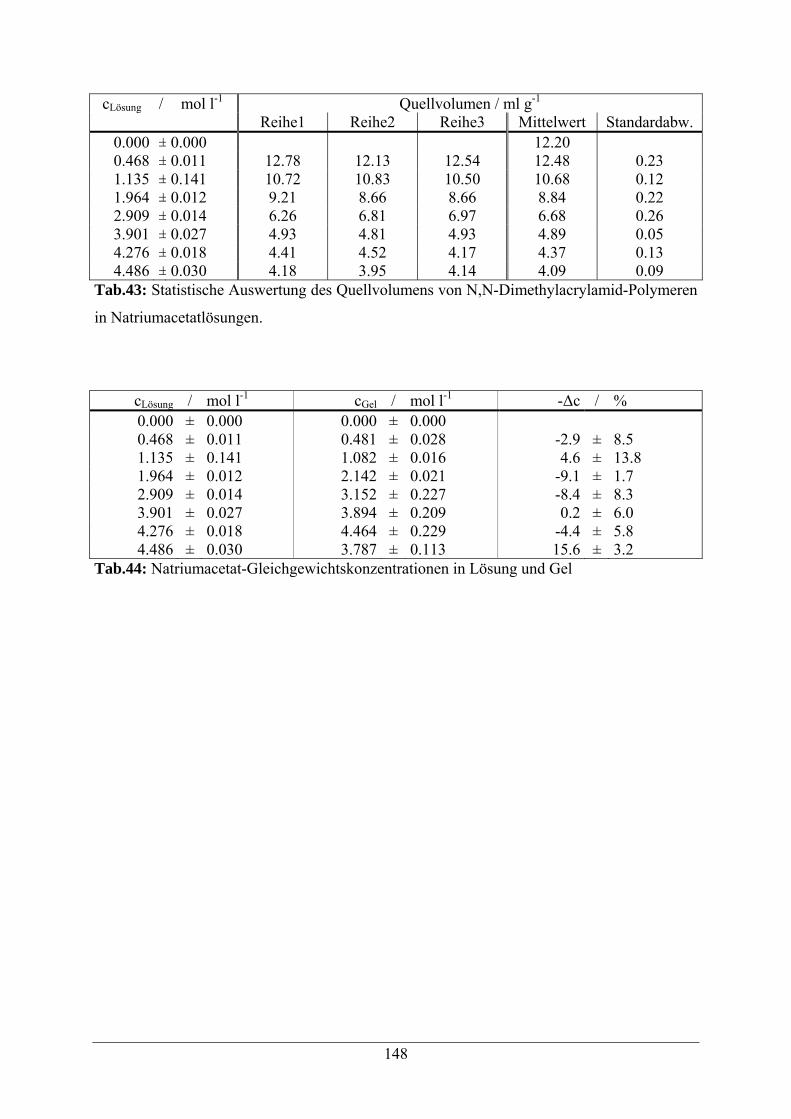
148
cLösung / mol l-1 Quellvolumen / ml g-1 Reihe1 Reihe2 Reihe3 Mittelwert Standardabw.
0.000 ± 0.000 12.20 0.468 ± 0.011 12.78 12.13 12.54 12.48 0.23 1.135 ± 0.141 10.72 10.83 10.50 10.68 0.12 1.964 ± 0.012 9.21 8.66 8.66 8.84 0.22 2.909 ± 0.014 6.26 6.81 6.97 6.68 0.26 3.901 ± 0.027 4.93 4.81 4.93 4.89 0.05 4.276 ± 0.018 4.41 4.52 4.17 4.37 0.13 4.486 ± 0.030 4.18 3.95 4.14 4.09 0.09
Tab.43: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in Natriumacetatlösungen.
cLösung / mol l-1 cGel / mol l-1 -∆c / % 0.000 ± 0.000 0.000 ± 0.000 0.468 ± 0.011 0.481 ± 0.028 -2.9 ± 8.5 1.135 ± 0.141 1.082 ± 0.016 4.6 ± 13.8 1.964 ± 0.012 2.142 ± 0.021 -9.1 ± 1.7 2.909 ± 0.014 3.152 ± 0.227 -8.4 ± 8.3 3.901 ± 0.027 3.894 ± 0.209 0.2 ± 6.0 4.276 ± 0.018 4.464 ± 0.229 -4.4 ± 5.8 4.486 ± 0.030 3.787 ± 0.113 15.6 ± 3.2
Tab.44: Natriumacetat-Gleichgewichtskonzentrationen in Lösung und Gel
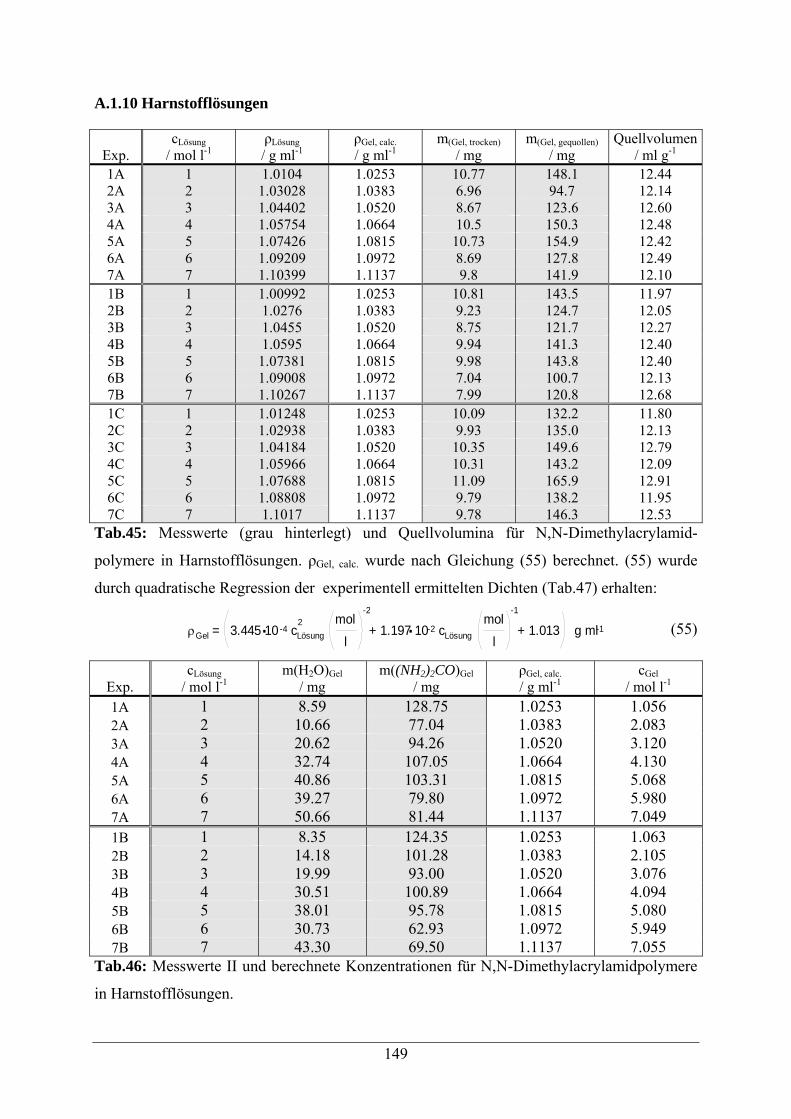
149
A.1.10 Harnstofflösungen
Exp. cLösung
/ mol l-1 ρLösung / g ml-1
ρGel, calc. / g ml-1
m(Gel, trocken) / mg
m(Gel, gequollen) / mg
Quellvolumen/ ml g-1
1A 1 1.0104 1.0253 10.77 148.1 12.44 2A 2 1.03028 1.0383 6.96 94.7 12.14 3A 3 1.04402 1.0520 8.67 123.6 12.60 4A 4 1.05754 1.0664 10.5 150.3 12.48 5A 5 1.07426 1.0815 10.73 154.9 12.42 6A 6 1.09209 1.0972 8.69 127.8 12.49 7A 7 1.10399 1.1137 9.8 141.9 12.10 1B 1 1.00992 1.0253 10.81 143.5 11.97 2B 2 1.0276 1.0383 9.23 124.7 12.05 3B 3 1.0455 1.0520 8.75 121.7 12.27 4B 4 1.0595 1.0664 9.94 141.3 12.40 5B 5 1.07381 1.0815 9.98 143.8 12.40 6B 6 1.09008 1.0972 7.04 100.7 12.13 7B 7 1.10267 1.1137 7.99 120.8 12.68 1C 1 1.01248 1.0253 10.09 132.2 11.80 2C 2 1.02938 1.0383 9.93 135.0 12.13 3C 3 1.04184 1.0520 10.35 149.6 12.79 4C 4 1.05966 1.0664 10.31 143.2 12.09 5C 5 1.07688 1.0815 11.09 165.9 12.91 6C 6 1.08808 1.0972 9.79 138.2 11.95 7C 7 1.1017 1.1137 9.78 146.3 12.53
Tab.45: Messwerte (grau hinterlegt) und Quellvolumina für N,N-Dimethylacrylamid-
polymere in Harnstofflösungen. ρGel, calc. wurde nach Gleichung (55) berechnet. (55) wurde
durch quadratische Regression der experimentell ermittelten Dichten (Tab.47) erhalten:
mol
l
-2mol
l
-1
ρGel = 3.445 10-4 cLösung + 1.197 10-2 cLösung + 1.013 g ml-12. .
(55)
Exp. cLösung
/ mol l-1 m(H2O)Gel
/ mg m((NH2)2CO)Gel
/ mg ρGel, calc. / g ml-1
cGel / mol l-1
1A 1 8.59 128.75 1.0253 1.056 2A 2 10.66 77.04 1.0383 2.083 3A 3 20.62 94.26 1.0520 3.120 4A 4 32.74 107.05 1.0664 4.130 5A 5 40.86 103.31 1.0815 5.068 6A 6 39.27 79.80 1.0972 5.980 7A 7 50.66 81.44 1.1137 7.049 1B 1 8.35 124.35 1.0253 1.063 2B 2 14.18 101.28 1.0383 2.105 3B 3 19.99 93.00 1.0520 3.076 4B 4 30.51 100.89 1.0664 4.094 5B 5 38.01 95.78 1.0815 5.080 6B 6 30.73 62.93 1.0972 5.949 7B 7 43.30 69.50 1.1137 7.055
Tab.46: Messwerte II und berechnete Konzentrationen für N,N-Dimethylacrylamidpolymere
in Harnstofflösungen.
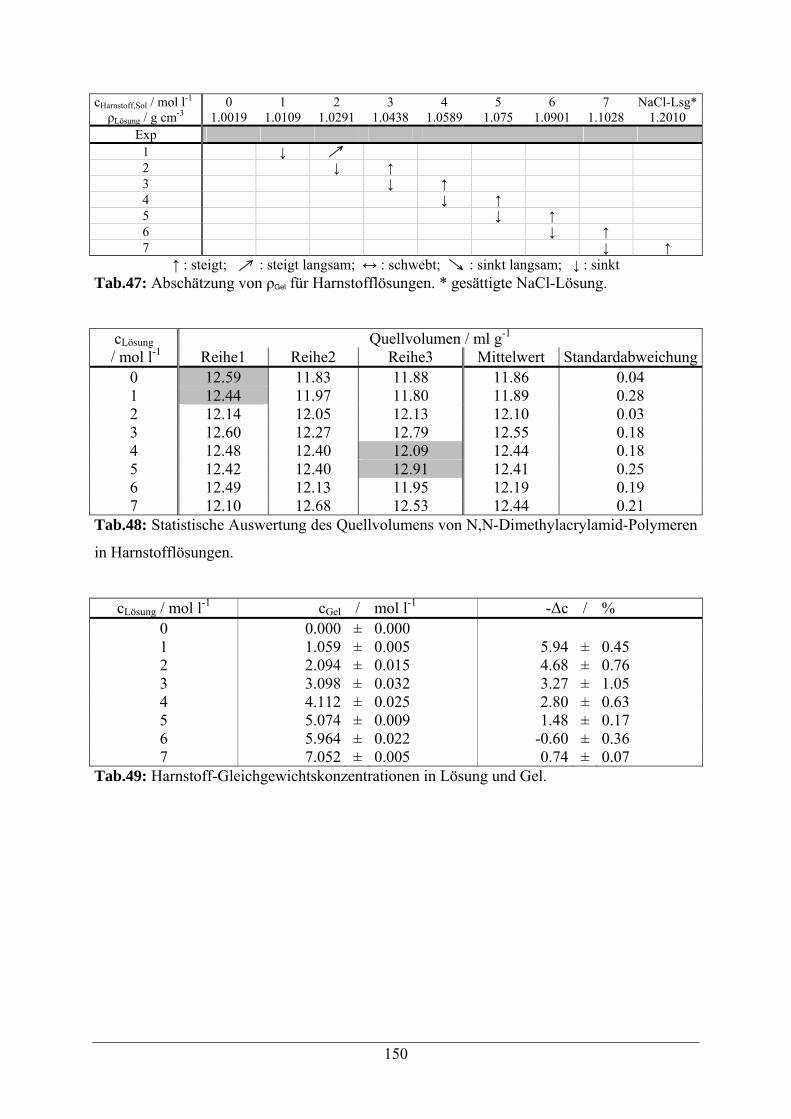
150
cHarnstoff,Sol / mol l-1 0 1 2 3 4 5 6 7 NaCl-Lsg*ρLösung / g cm-3 1.0019 1.0109 1.0291 1.0438 1.0589 1.075 1.0901 1.1028 1.2010
Exp 1 ↓ 2 ↓ ↑ 3 ↓ ↑ 4 ↓ ↑ 5 ↓ ↑ 6 ↓ ↑ 7 ↓ ↑
↑ : steigt; : steigt langsam; ↔ : schwebt; : sinkt langsam; ↓ : sinkt Tab.47: Abschätzung von ρGel für Harnstofflösungen. * gesättigte NaCl-Lösung.
cLösung Quellvolumen / ml g-1 / mol l-1 Reihe1 Reihe2 Reihe3 Mittelwert Standardabweichung
0 12.59 11.83 11.88 11.86 0.04 1 12.44 11.97 11.80 11.89 0.28 2 12.14 12.05 12.13 12.10 0.03 3 12.60 12.27 12.79 12.55 0.18 4 12.48 12.40 12.09 12.44 0.18 5 12.42 12.40 12.91 12.41 0.25 6 12.49 12.13 11.95 12.19 0.19 7 12.10 12.68 12.53 12.44 0.21
Tab.48: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-Polymeren
in Harnstofflösungen.
cLösung / mol l-1 cGel / mol l-1 -∆c / % 0 0.000 ± 0.000 1 1.059 ± 0.005 5.94 ± 0.45 2 2.094 ± 0.015 4.68 ± 0.76 3 3.098 ± 0.032 3.27 ± 1.05 4 4.112 ± 0.025 2.80 ± 0.63 5 5.074 ± 0.009 1.48 ± 0.17 6 5.964 ± 0.022 -0.60 ± 0.36 7 7.052 ± 0.005 0.74 ± 0.07
Tab.49: Harnstoff-Gleichgewichtskonzentrationen in Lösung und Gel.
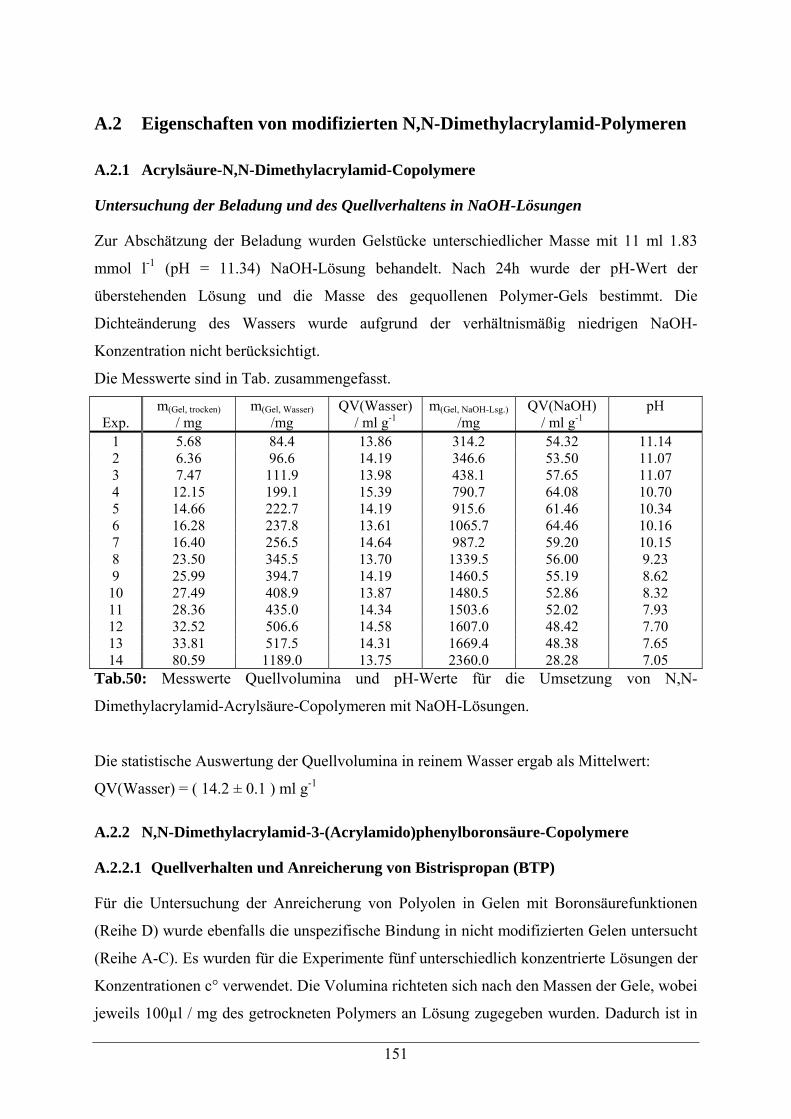
151
A.2 Eigenschaften von modifizierten N,N-Dimethylacrylamid-Polymeren
A.2.1 Acrylsäure-N,N-Dimethylacrylamid-Copolymere
Untersuchung der Beladung und des Quellverhaltens in NaOH-Lösungen
Zur Abschätzung der Beladung wurden Gelstücke unterschiedlicher Masse mit 11 ml 1.83
mmol l-1 (pH = 11.34) NaOH-Lösung behandelt. Nach 24h wurde der pH-Wert der
überstehenden Lösung und die Masse des gequollenen Polymer-Gels bestimmt. Die
Dichteänderung des Wassers wurde aufgrund der verhältnismäßig niedrigen NaOH-
Konzentration nicht berücksichtigt.
Die Messwerte sind in Tab. zusammengefasst.
Exp. m(Gel, trocken)
/ mg m(Gel, Wasser)
/mg QV(Wasser)
/ ml g-1 m(Gel, NaOH-Lsg.)
/mg QV(NaOH)
/ ml g-1 pH
1 5.68 84.4 13.86 314.2 54.32 11.14 2 6.36 96.6 14.19 346.6 53.50 11.07 3 7.47 111.9 13.98 438.1 57.65 11.07 4 12.15 199.1 15.39 790.7 64.08 10.70 5 14.66 222.7 14.19 915.6 61.46 10.34 6 16.28 237.8 13.61 1065.7 64.46 10.16 7 16.40 256.5 14.64 987.2 59.20 10.15 8 23.50 345.5 13.70 1339.5 56.00 9.23 9 25.99 394.7 14.19 1460.5 55.19 8.62
10 27.49 408.9 13.87 1480.5 52.86 8.32 11 28.36 435.0 14.34 1503.6 52.02 7.93 12 32.52 506.6 14.58 1607.0 48.42 7.70 13 33.81 517.5 14.31 1669.4 48.38 7.65 14 80.59 1189.0 13.75 2360.0 28.28 7.05
Tab.50: Messwerte Quellvolumina und pH-Werte für die Umsetzung von N,N-
Dimethylacrylamid-Acrylsäure-Copolymeren mit NaOH-Lösungen.
Die statistische Auswertung der Quellvolumina in reinem Wasser ergab als Mittelwert:
QV(Wasser) = ( 14.2 ± 0.1 ) ml g-1
A.2.2 N,N-Dimethylacrylamid-3-(Acrylamido)phenylboronsäure-Copolymere
A.2.2.1 Quellverhalten und Anreicherung von Bistrispropan (BTP)
Für die Untersuchung der Anreicherung von Polyolen in Gelen mit Boronsäurefunktionen
(Reihe D) wurde ebenfalls die unspezifische Bindung in nicht modifizierten Gelen untersucht
(Reihe A-C). Es wurden für die Experimente fünf unterschiedlich konzentrierte Lösungen der
Konzentrationen c° verwendet. Die Volumina richteten sich nach den Massen der Gele, wobei
jeweils 100µl / mg des getrockneten Polymers an Lösung zugegeben wurden. Dadurch ist in
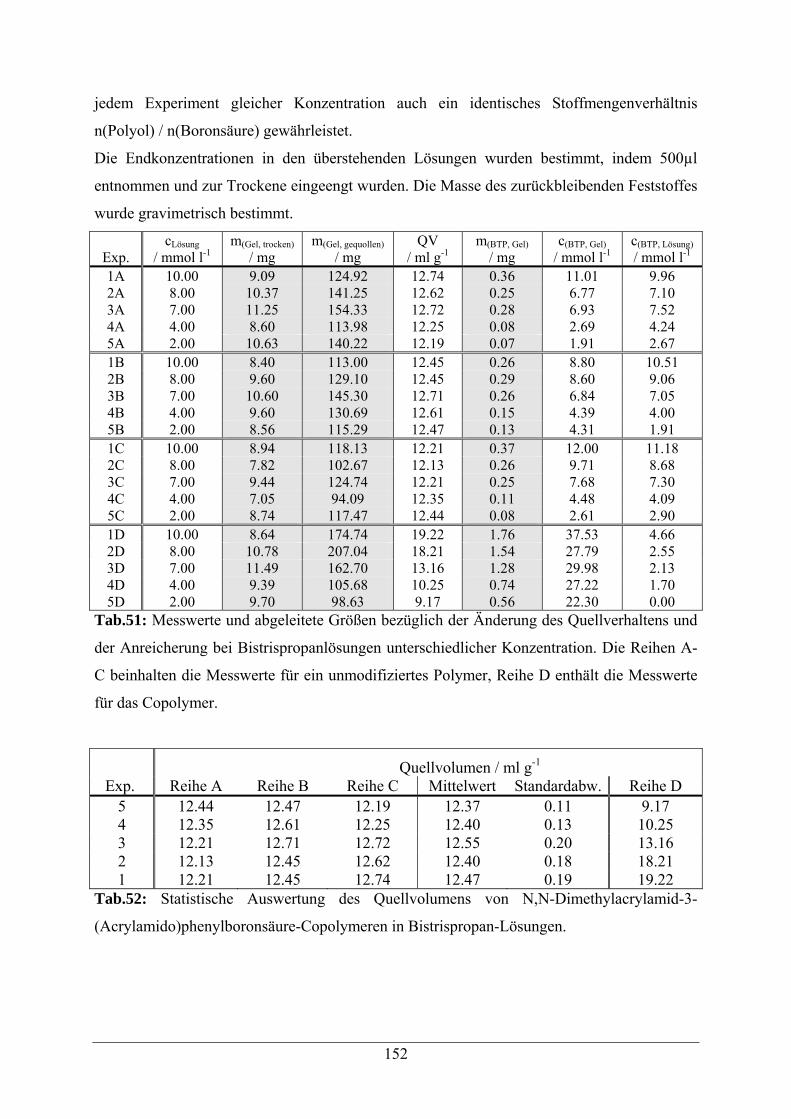
152
jedem Experiment gleicher Konzentration auch ein identisches Stoffmengenverhältnis
n(Polyol) / n(Boronsäure) gewährleistet.
Die Endkonzentrationen in den überstehenden Lösungen wurden bestimmt, indem 500µl
entnommen und zur Trockene eingeengt wurden. Die Masse des zurückbleibenden Feststoffes
wurde gravimetrisch bestimmt.
Exp. cLösung
/ mmol l-1 m(Gel, trocken)
/ mg m(Gel, gequollen)
/ mg QV
/ ml g-1 m(BTP, Gel)
/ mg c(BTP, Gel)
/ mmol l-1 c(BTP, Lösung) / mmol l-1
1A 10.00 9.09 124.92 12.74 0.36 11.01 9.96 2A 8.00 10.37 141.25 12.62 0.25 6.77 7.10 3A 7.00 11.25 154.33 12.72 0.28 6.93 7.52 4A 4.00 8.60 113.98 12.25 0.08 2.69 4.24 5A 2.00 10.63 140.22 12.19 0.07 1.91 2.67 1B 10.00 8.40 113.00 12.45 0.26 8.80 10.51 2B 8.00 9.60 129.10 12.45 0.29 8.60 9.06 3B 7.00 10.60 145.30 12.71 0.26 6.84 7.05 4B 4.00 9.60 130.69 12.61 0.15 4.39 4.00 5B 2.00 8.56 115.29 12.47 0.13 4.31 1.91 1C 10.00 8.94 118.13 12.21 0.37 12.00 11.18 2C 8.00 7.82 102.67 12.13 0.26 9.71 8.68 3C 7.00 9.44 124.74 12.21 0.25 7.68 7.30 4C 4.00 7.05 94.09 12.35 0.11 4.48 4.09 5C 2.00 8.74 117.47 12.44 0.08 2.61 2.90 1D 10.00 8.64 174.74 19.22 1.76 37.53 4.66 2D 8.00 10.78 207.04 18.21 1.54 27.79 2.55 3D 7.00 11.49 162.70 13.16 1.28 29.98 2.13 4D 4.00 9.39 105.68 10.25 0.74 27.22 1.70 5D 2.00 9.70 98.63 9.17 0.56 22.30 0.00
Tab.51: Messwerte und abgeleitete Größen bezüglich der Änderung des Quellverhaltens und
der Anreicherung bei Bistrispropanlösungen unterschiedlicher Konzentration. Die Reihen A-
C beinhalten die Messwerte für ein unmodifiziertes Polymer, Reihe D enthält die Messwerte
für das Copolymer.
Quellvolumen / ml g-1 Exp. Reihe A Reihe B Reihe C Mittelwert Standardabw. Reihe D
5 12.44 12.47 12.19 12.37 0.11 9.17 4 12.35 12.61 12.25 12.40 0.13 10.25 3 12.21 12.71 12.72 12.55 0.20 13.16 2 12.13 12.45 12.62 12.40 0.18 18.21 1 12.21 12.45 12.74 12.47 0.19 19.22
Tab.52: Statistische Auswertung des Quellvolumens von N,N-Dimethylacrylamid-3-
(Acrylamido)phenylboronsäure-Copolymeren in Bistrispropan-Lösungen.
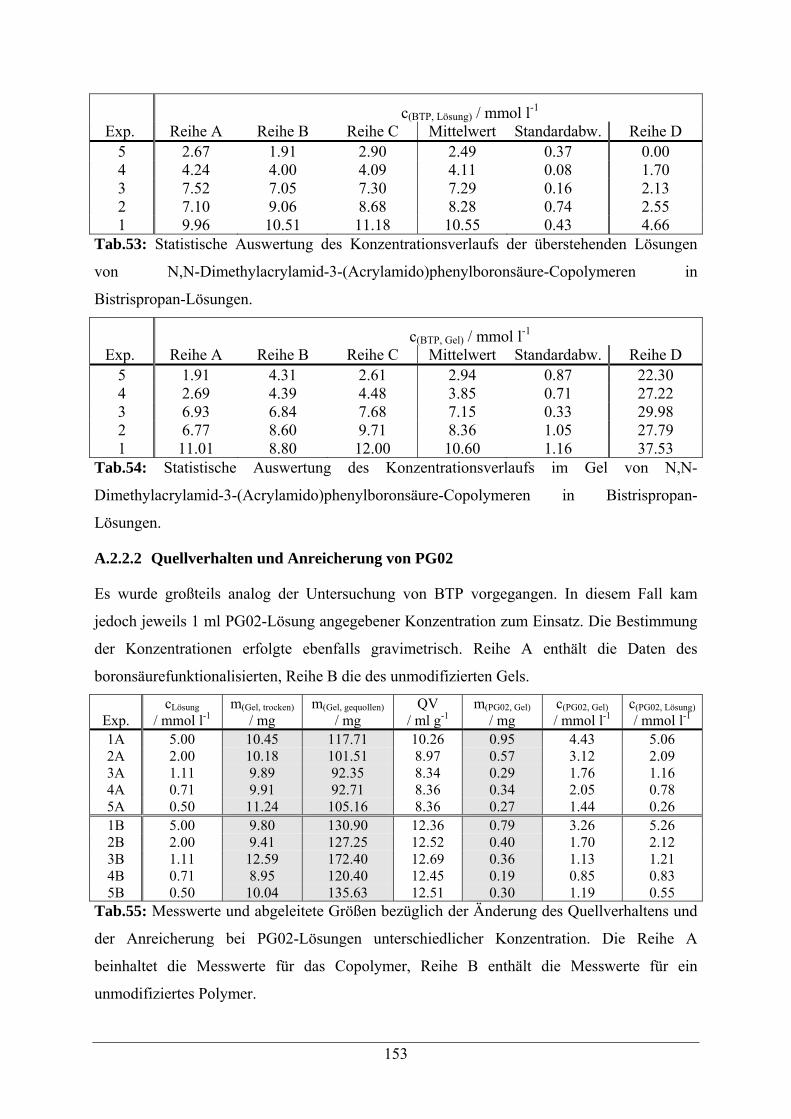
153
c(BTP, Lösung) / mmol l-1 Exp. Reihe A Reihe B Reihe C Mittelwert Standardabw. Reihe D
5 2.67 1.91 2.90 2.49 0.37 0.00 4 4.24 4.00 4.09 4.11 0.08 1.70 3 7.52 7.05 7.30 7.29 0.16 2.13 2 7.10 9.06 8.68 8.28 0.74 2.55 1 9.96 10.51 11.18 10.55 0.43 4.66
Tab.53: Statistische Auswertung des Konzentrationsverlaufs der überstehenden Lösungen
von N,N-Dimethylacrylamid-3-(Acrylamido)phenylboronsäure-Copolymeren in
Bistrispropan-Lösungen.
c(BTP, Gel) / mmol l-1 Exp. Reihe A Reihe B Reihe C Mittelwert Standardabw. Reihe D
5 1.91 4.31 2.61 2.94 0.87 22.30 4 2.69 4.39 4.48 3.85 0.71 27.22 3 6.93 6.84 7.68 7.15 0.33 29.98 2 6.77 8.60 9.71 8.36 1.05 27.79 1 11.01 8.80 12.00 10.60 1.16 37.53
Tab.54: Statistische Auswertung des Konzentrationsverlaufs im Gel von N,N-
Dimethylacrylamid-3-(Acrylamido)phenylboronsäure-Copolymeren in Bistrispropan-
Lösungen.
A.2.2.2 Quellverhalten und Anreicherung von PG02
Es wurde großteils analog der Untersuchung von BTP vorgegangen. In diesem Fall kam
jedoch jeweils 1 ml PG02-Lösung angegebener Konzentration zum Einsatz. Die Bestimmung
der Konzentrationen erfolgte ebenfalls gravimetrisch. Reihe A enthält die Daten des
boronsäurefunktionalisierten, Reihe B die des unmodifizierten Gels.
Exp. cLösung
/ mmol l-1 m(Gel, trocken)
/ mg m(Gel, gequollen)
/ mg QV
/ ml g-1 m(PG02, Gel)
/ mg c(PG02, Gel) / mmol l-1
c(PG02, Lösung)/ mmol l-1
1A 5.00 10.45 117.71 10.26 0.95 4.43 5.06 2A 2.00 10.18 101.51 8.97 0.57 3.12 2.09 3A 1.11 9.89 92.35 8.34 0.29 1.76 1.16 4A 0.71 9.91 92.71 8.36 0.34 2.05 0.78 5A 0.50 11.24 105.16 8.36 0.27 1.44 0.26 1B 5.00 9.80 130.90 12.36 0.79 3.26 5.26 2B 2.00 9.41 127.25 12.52 0.40 1.70 2.12 3B 1.11 12.59 172.40 12.69 0.36 1.13 1.21 4B 0.71 8.95 120.40 12.45 0.19 0.85 0.83 5B 0.50 10.04 135.63 12.51 0.30 1.19 0.55
Tab.55: Messwerte und abgeleitete Größen bezüglich der Änderung des Quellverhaltens und
der Anreicherung bei PG02-Lösungen unterschiedlicher Konzentration. Die Reihe A
beinhaltet die Messwerte für das Copolymer, Reihe B enthält die Messwerte für ein
unmodifiziertes Polymer.
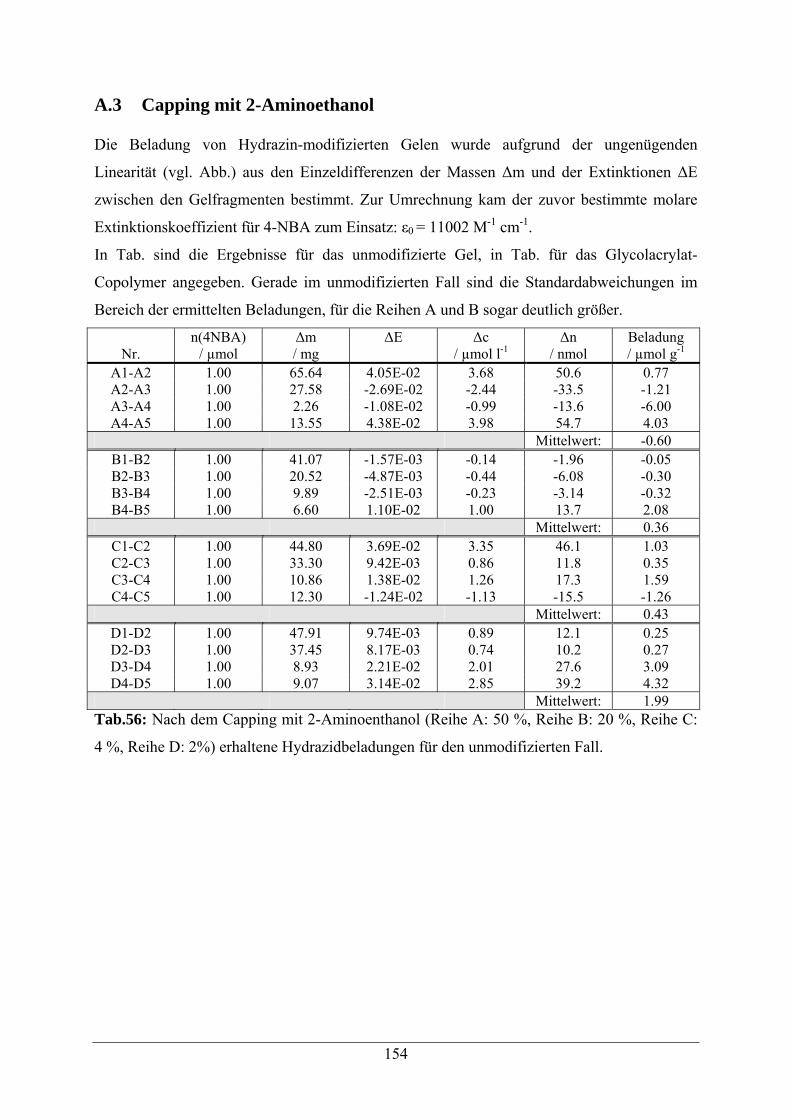
154
A.3 Capping mit 2-Aminoethanol
Die Beladung von Hydrazin-modifizierten Gelen wurde aufgrund der ungenügenden
Linearität (vgl. Abb.) aus den Einzeldifferenzen der Massen ∆m und der Extinktionen ∆E
zwischen den Gelfragmenten bestimmt. Zur Umrechnung kam der zuvor bestimmte molare
Extinktionskoeffizient für 4-NBA zum Einsatz: ε0 = 11002 M-1 cm-1.
In Tab. sind die Ergebnisse für das unmodifizierte Gel, in Tab. für das Glycolacrylat-
Copolymer angegeben. Gerade im unmodifizierten Fall sind die Standardabweichungen im
Bereich der ermittelten Beladungen, für die Reihen A und B sogar deutlich größer.
Nr. n(4NBA)
/ µmol ∆m / mg
∆E
∆c / µmol l-1
∆n / nmol
Beladung / µmol g-1
A1-A2 1.00 65.64 4.05E-02 3.68 50.6 0.77 A2-A3 1.00 27.58 -2.69E-02 -2.44 -33.5 -1.21 A3-A4 1.00 2.26 -1.08E-02 -0.99 -13.6 -6.00 A4-A5 1.00 13.55 4.38E-02 3.98 54.7 4.03
Mittelwert: -0.60 B1-B2 1.00 41.07 -1.57E-03 -0.14 -1.96 -0.05 B2-B3 1.00 20.52 -4.87E-03 -0.44 -6.08 -0.30 B3-B4 1.00 9.89 -2.51E-03 -0.23 -3.14 -0.32 B4-B5 1.00 6.60 1.10E-02 1.00 13.7 2.08
Mittelwert: 0.36 C1-C2 1.00 44.80 3.69E-02 3.35 46.1 1.03 C2-C3 1.00 33.30 9.42E-03 0.86 11.8 0.35 C3-C4 1.00 10.86 1.38E-02 1.26 17.3 1.59 C4-C5 1.00 12.30 -1.24E-02 -1.13 -15.5 -1.26
Mittelwert: 0.43 D1-D2 1.00 47.91 9.74E-03 0.89 12.1 0.25 D2-D3 1.00 37.45 8.17E-03 0.74 10.2 0.27 D3-D4 1.00 8.93 2.21E-02 2.01 27.6 3.09 D4-D5 1.00 9.07 3.14E-02 2.85 39.2 4.32
Mittelwert: 1.99 Tab.56: Nach dem Capping mit 2-Aminoenthanol (Reihe A: 50 %, Reihe B: 20 %, Reihe C:
4 %, Reihe D: 2%) erhaltene Hydrazidbeladungen für den unmodifizierten Fall.
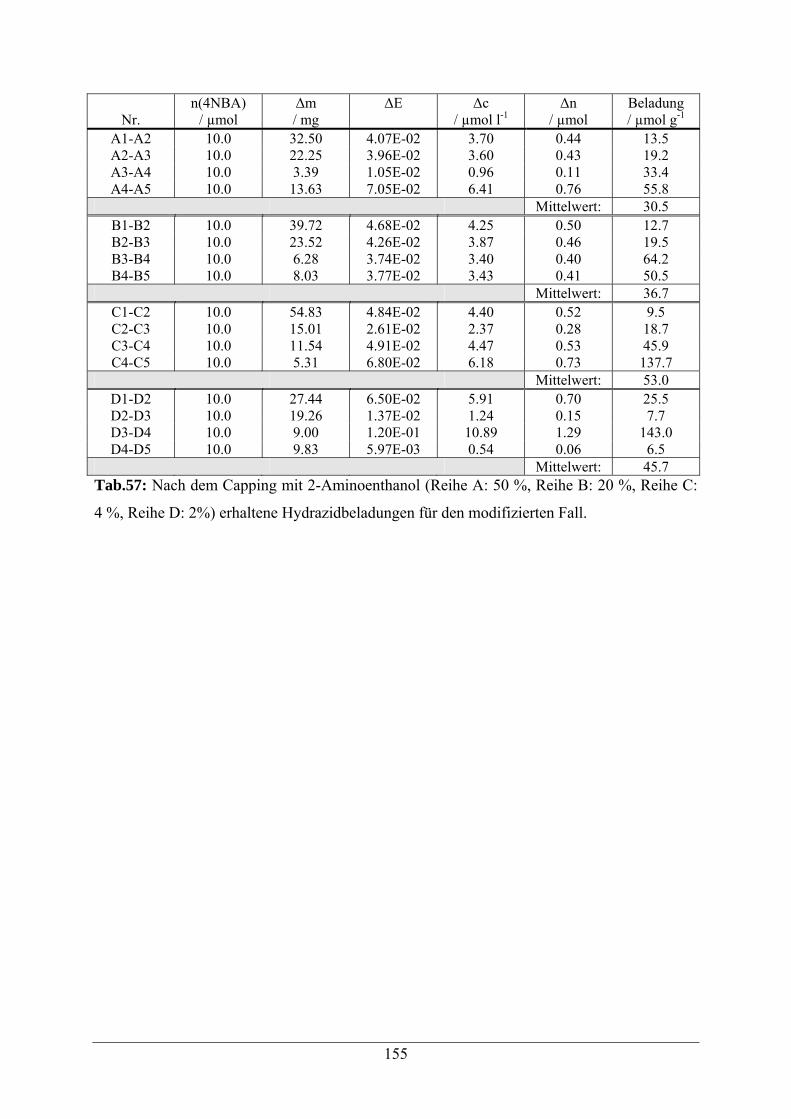
155
Nr. n(4NBA)
/ µmol ∆m / mg
∆E
∆c / µmol l-1
∆n / µmol
Beladung / µmol g-1
A1-A2 10.0 32.50 4.07E-02 3.70 0.44 13.5 A2-A3 10.0 22.25 3.96E-02 3.60 0.43 19.2 A3-A4 10.0 3.39 1.05E-02 0.96 0.11 33.4 A4-A5 10.0 13.63 7.05E-02 6.41 0.76 55.8
Mittelwert: 30.5 B1-B2 10.0 39.72 4.68E-02 4.25 0.50 12.7 B2-B3 10.0 23.52 4.26E-02 3.87 0.46 19.5 B3-B4 10.0 6.28 3.74E-02 3.40 0.40 64.2 B4-B5 10.0 8.03 3.77E-02 3.43 0.41 50.5
Mittelwert: 36.7 C1-C2 10.0 54.83 4.84E-02 4.40 0.52 9.5 C2-C3 10.0 15.01 2.61E-02 2.37 0.28 18.7 C3-C4 10.0 11.54 4.91E-02 4.47 0.53 45.9 C4-C5 10.0 5.31 6.80E-02 6.18 0.73 137.7
Mittelwert: 53.0 D1-D2 10.0 27.44 6.50E-02 5.91 0.70 25.5 D2-D3 10.0 19.26 1.37E-02 1.24 0.15 7.7 D3-D4 10.0 9.00 1.20E-01 10.89 1.29 143.0 D4-D5 10.0 9.83 5.97E-03 0.54 0.06 6.5
Mittelwert: 45.7 Tab.57: Nach dem Capping mit 2-Aminoenthanol (Reihe A: 50 %, Reihe B: 20 %, Reihe C:
4 %, Reihe D: 2%) erhaltene Hydrazidbeladungen für den modifizierten Fall.

156
B: Dreidimensionale Visualisierung chemischer Verbin-
dungen
B.1 Motivation
Einen entscheidenden Beitrag zum Verständnis molekularer und supramolekularer
Zusammenhänge liefert die dreidimensionale Darstellung. Dabei haben sich bestimmte
Darstellungsarten für Moleküle, besonders im biochemischen Bereich auch abstrahierende
Darstellungen für Peptide und Nukleotide, etabliert. Während diese Darstellungen im Rahmen
von molecular modelling- und
Visualisierungsprogrammen implementiert
sind, bestehen kaum Möglichkeiten zur
Kombination dieser dreidimensionalen Dar-
stellungen mit dreidimensionalen Objekten,
die nicht aus molekularem Kontext
stammen.
Die abstrahierende Visualisierung von DNA-
Nanostrukturen auf der Basis von
Trisoligonucleotiden erfolgte daher bisher in
erster Linie durch den Aufbau
dreidimensionaler DNA-Modelle in 3d-
Modelling Programmen (Abb.87), da
Molekül-visualisierungsprogramme zwar die
abstra-hierende Darstellung von DNA, nicht
jedoch z.B. eine schematische Darstellung der Trislinker ermöglichen.[84-87]
Es ist also wünschenswert, eine Möglichkeit zu erhalten, dreidimensionale Modelle von
Molekülen in hoher Qualität in eine 3d-Modelling-Umgebung zu importieren und in dieser
Umgebung bearbeiten zu können.
Abb.87: Schematische 3d-Abbildung eines
DNA-Tetraders.[84] Die Trislinker sind
durch Kegel symbolisiert.

157
B.2 Eigenschaften des pdb2blend-Skriptes
Zur Visualisierung dreidimensionaler Strukturen ausgehend von Moleküldaten wurde im
Zuge dieser Arbeit ein Skript zum Importieren von pdb-Dateien, die die dreidimensionale
Strukturinformation von Molekülen beinhalten, in das 3d-Modelling Programm Blender
erstellt.[88,89] Das Skript wurde insbesondere für die vereinfachte Darstellung von
dreidimensionalen Strukturen kombiniert
mit geometrischen Modellen für zukünftige
Veröffentlichungen im Bereich der DNA-
Nanostruktur-Forschung aufgebaut,
weshalb Funktionalitäten zur abstrahierten
Darstellung von DNA-Phosphatgerüsten
implementiert sind.
Die mit dem Skript möglichen
Darstellungsvarianten sind Balls, Sticks
and Balls und Sticks, womit die üblichen
3d-Moleküldarstellungen abgedeckt sind.
Die Einstellungen werden können über eine graphische Benutzeroberfläche vorgenommen
werden (Abb.88). Die Atomtypen, Koordinaten und Konnektivitäten werden aus den pdb-
Dateien extrahiert (Abb.89).
Für die Darstellung der Bälle werden die literaturbekannten Atomradien zugrunde gelegt und
mit Modifikationsparametern skaliert. Für die Darstellung der Sticks werden die
HETATM 1 O 1 -0.391 0.314 -0.088HETATM 2 H 2 -0.136 -0.506 0.319HETATM 3 H 3 -1.083 0.697 0.440CONECT 1 2 3CONECT 2 1CONECT 3 1END
ball Größe, Material
ball Mittelpunktkoordinaten
stick Länge, Winkel
InformationstypAtomnummer
Atomart
Konnektivitätstabelle
x,y,z-Koordinaten
Abb.89: Datenextraktion aus pdb-Dateien.[89]
Abb.88: graphische Benutzeroberfläche des
pdb2blend-Skriptes.

158
Atomabstände und Winkel berechnet und daraufhin durch abgerundete Stäbe verbunden. Die
Dicken der Stäbe können frei bestimmt werden.
Die Berechnung der Bälle und Stäbe erfolgt nach bekannten trigonometrischen Funktionen.
Die absoluten Abhängigkeiten sind in Abb.90 dargestellt.
Die Materialien, die den farblichen Darstellungen der Atome entsprechen, sind vordefiniert,
können aber beliebig geändert werden.
Beispiele der Abbildung von Molekülen durch die Darstellungsmodi Sticks, Sticks and Balls
und Balls sind in Abb.91 gegeben.
Als Beispiel für die Möglichkeiten der Kombination von Moleküldaten und 3d-Modellen ist
in Abb.92 die Kombination der Struktur eines DNA-Dodekaeders zusammen mit dem
einfachen 3d-Modell eines Dodekaeders dargestellt. Zur besseren Erkennbarkeit der
Doppelhelix-Struktur wurden lediglich die Phosphoratome in den DNA-Strängen miteinander
verbunden. Diese Darstellung ermöglicht es, das zugrunde liegende Prinzip in dem relativ
unübersichtlichen Molekül sichtbar zu machen.
Kugeln:
Stäbe:
Atomkoordinaten + Radius (Atomtypen + Einstellungen) + Kugelqualität Punkte Flächen
Konnektivitäten + Atomkoordinaten + Radius (Einstellungen) + Stabqualität Punkte Flächen
Abb.90 Abhängigkeiten der Punkte und Flächen von Atom- und Einstellungsparametern.
Abb.91: Durch pdb2blend in Blender erstellte dreidimensionale Abbildungen von
Molekülen am Beispiel von Thymidin in den Darstellungsarten a) Sticks, b) Sticks and
Balls und c) Balls.

159
B.3 Quellcode
Der Quellcode ist in der Programmiersprache PYTHON verfasst, die von Blender interpretiert
werden kann.[90] #################################################################################################### # # # ####### ####### ####### ##### ####### #### ####### ### ### ####### # # ## ## ## ## ## ## ## ## ## ## ## ## ## ### ## ## ## # # ## ## ## ## ## ## ## ## ## ## ## #### ## ## ## # # ###### ## ## ###### ### ###### ## ##### ## ## ## ## ## # # ## ## ## ## ## ## ## ## ## ## ## #### ## ## # # ## ## ## ## ## ## ## ## ## ## ## ## ## ## ### ## ## # # #### ####### ####### ####### ####### ####### ####### ### ### ####### V1.2 # # # #################################################################################################### # # pdb Molecule 2 Blender 3d Model Converter by Malte Reimold 2006 # # Modes: # 1. Ball # creates a ball model with overlapping spheres when using the standard parameters # the ball sizes can be adjusted by choosing a constant scaling factor (atom scale) # as well as a constant summand (atom sum) that is added to the ball size. # With atom scale = 1 and atom sum = 0 one will get the covalent radius for any atom defined. # With atom scale = 0 and atom sum > 0 the model will have uniform ball sizes. # 2. Sticks # creates a stick model. # The stick diameter can be chosen here (Stick Thickness). # Until now the regular stick mode (as well as stick and ball) does only work for less than # 10000 atoms. # 3. Sticks and Balls # A combination of ball and stick mode. When using standard parameters the atoms are scaled # down a little bit. # 4. DNA Backbone Follow # When turning on this option a stick model will be formed connecting the phosphorous atoms # of oligonucleotide backbones. This works even for systems with more than 10000 atoms. # # Model refinement: # The refinement of spheres and sticks can be chosen. With higher values the model becomes more # detailed.
Abb.92 Mit pdb2blend erstelltes Modell eines DNA-Dodekaeders zusammen mit einem
Dodekaeder-Modell.

160
# # The pdb 2 blender converter is still experimental. Please report problems and errors to me. # # Version History # v1.2 Fixed some problems concerning pdb-file conventions. # v1.1 Fixed some minor bugs. # ##############################################################################(c)2006MalteReimold import Blender from Blender import * from Blender.Draw import * from math import * editmode = Window.EditMode() if editmode: Window.EditMode(0) scene = Blender.Scene.getCurrent () matlist = str(Material.Get ()) if not ('[Material "C"]' in matlist): mat = Material.New('C') mat.R = 0.8 mat.G = 0.8 mat.B = 0.8 if not ('[Material "H"]' in matlist): mat = Material.New('H') mat.R = 0.6 mat.G = 0.6 mat.B = 0.6 if not ('[Material "B"]' in matlist): mat = Material.New('B') mat.R = 0.8 mat.G = 0.6 mat.B = 0.1 if not ('[Material "P"]' in matlist): mat = Material.New('P') mat.R = 0.9 mat.G = 0.95 mat.B = 0.1 if not ('[Material "N"]' in matlist): mat = Material.New('N') mat.R = 0.2 mat.G = 0.1 mat.B = 0.9 if not ('[Material "O"]' in matlist): mat = Material.New('O') mat.R = 1.0 mat.G = 0.2 mat.B = 0.1 if not ('[Material "anyatom"]' in matlist): mat = Material.New('anyatom') mat.R = 0.8 mat.G = 1.0 mat.B = 1.0 if not ('[Material "sticks"]' in matlist): mat = Material.New('sticks') mat.R = 0.8 mat.G = 0.8 mat.B = 0.8 def ball(name, type, x, y, z): global structmode, scatom, sumatom, refineballs, refinesticks, balls, sticks, scsticks # Objekt erstellen ob = Object.New('Mesh', name) # Mesh erstellen me = Mesh.New(name) # Ball erstellen a = 0.0000 b = 0.0000 vcount = 0 da = 360.00 / refineballs.val db = 360.00 / refineballs.val #da = 30.0 #db = 30.0 dx = 0.0 dy = 0.0

161
dz = 0.0 pi = asin(1) if type == "C": radius = 0.772 mat = Material.Get('C') elif type == "H": radius = 0.373 mat = Material.Get('H') elif type == "B": radius = 0.83 mat = Material.Get('B') elif type == "N": radius = 0.71 mat = Material.Get('N') elif type == "O": radius = 0.604 mat = Material.Get('O') elif type == "P": radius = 0.93 mat = Material.Get('P') else: radius = 0.8 mat = Material.Get('anyatom') me.materials = [mat] radius = radius * scatom.val + sumatom.val # erster Punkt me.verts.extend(0, 0, radius) a = a + da # Kappe while b < 360: dz = radius*cos(a/90*pi) dx = radius*sin(a/90*pi)*sin(b/90*pi) dy = radius*sin(a/90*pi)*cos(b/90*pi) b = b + db me.verts.extend(dx, dy, dz) vcount = vcount + 1 if vcount > 1: me.faces.extend([me.verts[0],me.verts[vcount],me.verts[vcount-1]]) me.faces.extend([me.verts[0],me.verts[1],me.verts[vcount]]) b = 0.00 kcount = 0 vvcount = 0 a = a + da while a < 180: while b < 360: dz = radius*cos(a/90*pi) dx = radius*sin(a/90*pi)*sin(b/90*pi) dy = radius*sin(a/90*pi)*cos(b/90*pi) me.verts.extend(dx, dy, dz) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]]) b = b + db kcount = kcount + 1 b = 0 vvcount = 0 a = a + da # letzter Punkt me.verts.extend(0, 0, -radius) vcount = vcount + 1 b = 0 while b < 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(b/db)-2],me.verts[vcount-int(b/db)-1]]) b = b + db me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int((360-db)/db)-1]]) eins = 1 for face in me.faces: face.smooth=1

162
ob.link (me) ob.loc = (x,y,z) scene.link (ob) def stick1(x1, y1, z1, x2, y2, z2, radius): global structmode, scatom, sumatom, refineballs, refinesticks, balls, sticks, scsticks # Objekt erstellen ob = Object.New('Mesh') # Mesh erstellen me = Mesh.New() mat = Material.Get('sticks') me.materials = [mat] # Stick erstellen a = 0.0000 b = 0.0000 vcount = 0 da = 360.00 / refinesticks.val db = 360.00 / refinesticks.val dx = 0.0 dy = 0.0 dz = 0.0 pi = asin(1) laenge = sqrt((x1-x2)*(x1-x2)+(y1-y2)*(y1-y2)+(z1-z2)*(z1-z2)) wsz = atan2((y1-y2), (x1-x2)) wz = acos((z1-z2)/laenge) # erster Punkt me.verts.extend(0, 0, laenge/2+radius) a = a + da # Kappe while b < 360: dz = radius*cos(a/90*pi) dx = radius*sin(a/90*pi)*sin(b/90*pi) dy = radius*sin(a/90*pi)*cos(b/90*pi) b = b + db me.verts.extend(dx, dy, dz+laenge/2) vcount = vcount + 1 if vcount > 1: me.faces.extend([me.verts[0],me.verts[vcount],me.verts[vcount-1]]) me.faces.extend([me.verts[0],me.verts[1],me.verts[vcount]]) b = 0.00 kcount = 0 vvcount = 0 a = a + da while a < 90: while b < 360: dz = radius*cos(a/90*pi) dx = radius*sin(a/90*pi)*sin(b/90*pi) dy = radius*sin(a/90*pi)*cos(b/90*pi) me.verts.extend(dx, dy, dz+laenge/2) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]]) b = b + db kcount = kcount + 1 b = 0 vvcount = 0 a = a + da a = 90 b = 0.00 vvcount = 0 while b < 360: dx = radius*sin(b/90*pi) dy = radius*cos(b/90*pi) me.verts.extend(dx, dy, laenge/2) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]])

163
b = b + db kcount = kcount + 1 b = 0.00 vvcount = 0 while b < 360: dx = radius*sin(b/90*pi) dy = radius*cos(b/90*pi) me.verts.extend(dx, dy, -laenge/2) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]]) b = b + db kcount = kcount + 1 b = 0.00 vvcount = 0 while a < 180: while b < 360: dz = radius*cos(a/90*pi) dx = radius*sin(a/90*pi)*sin(b/90*pi) dy = radius*sin(a/90*pi)*cos(b/90*pi) me.verts.extend(dx, dy, dz-laenge/2) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]]) b = b + db kcount = kcount + 1 b = 0.00 vvcount = 0 a = a + da # letzter Punkt me.verts.extend(0, 0, -laenge/2-radius) vcount = vcount + 1 b = 0 while b < 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(b/db)-2],me.verts[vcount-int(b/db)-1]]) b = b + db me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int((360-db)/db)-1]]) eins = 1 for face in me.faces: face.smooth=1 ob.link (me) x1 = float(x1) y1 = float(y1) z1 = float(z1) x2 = float(x2) y2 = float(y2) z2 = float(z2) ob.loc = (float(x1+x2)/2,(y1+y2)/2,(z1+z2)/2) ob.RotY = wz ob.RotZ = wsz scene.link (ob) def stick2(x1, y1, z1, x2, y2, z2, radius): global structmode, scatom, sumatom, refineballs, refinesticks, balls, sticks, scsticks # Objekt erstellen ob = Object.New('Mesh') # Mesh erstellen me = Mesh.New() mat = Material.Get('sticks') me.materials = [mat] # Stick erstellen b = 0.0000 vcount = -1 db = 360.00 / refinesticks.val dx = 0.0 dy = 0.0 dz = 0.0 pi = asin(1) laenge = sqrt((x1-x2)*(x1-x2)+(y1-y2)*(y1-y2)+(z1-z2)*(z1-z2)) wsz = atan2((y1-y2), (x1-x2)) wz = acos((z1-z2)/laenge)

164
# erster Punkt # Kappe while b < 360: dx = radius*sin(b/90*pi) dy = radius*cos(b/90*pi) me.verts.extend(dx, dy, laenge/2) me.verts.extend(dx, dy, -laenge/2) vcount = vcount + 2 if vcount > 2: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-3],me.verts[vcount-2]]) if b == 360-db: me.faces.extend([me.verts[0],me.verts[vcount-1],me.verts[vcount],me.verts[1]]) b = b + db for face in me.faces: face.smooth=1 ob.link (me) x1 = float(x1) y1 = float(y1) z1 = float(z1) x2 = float(x2) y2 = float(y2) z2 = float(z2) ob.loc = (float(x1+x2)/2,(y1+y2)/2,(z1+z2)/2) ob.RotY = wz ob.RotZ = wsz scene.link (ob) def stick3(x1, y1, z1, type1, x2, y2, z2, type2, radius): global structmode, scatom, sumatom, refineballs, refinesticks, balls, sticks, scsticks # Objekt erstellen ob = Object.New('Mesh') # Mesh erstellen me = Mesh.New() if type1 in ['C', 'H', 'B', 'N', 'O', 'P']: mat = Material.Get(type1) else: mat = Material.Get('anyatom') me.materials = [mat] # Stick erste Haelfte erstellen a = 0.0000 b = 0.0000 vcount = 0 da = 360.00 / refinesticks.val db = 360.00 / refinesticks.val dx = 0.0 dy = 0.0 dz = 0.0 pi = asin(1) laenge = sqrt((x1-x2)*(x1-x2)+(y1-y2)*(y1-y2)+(z1-z2)*(z1-z2)) wsz = atan2((y1-y2), (x1-x2)) wz = acos((z1-z2)/laenge) # erster Punkt me.verts.extend(0, 0, laenge/2+radius) a = a + da # Kappe while b < 360: dz = radius*cos(a/90*pi) dx = radius*sin(a/90*pi)*sin(b/90*pi) dy = radius*sin(a/90*pi)*cos(b/90*pi) b = b + db me.verts.extend(dx, dy, dz+laenge/2) vcount = vcount + 1 if vcount > 1: me.faces.extend([me.verts[0],me.verts[vcount],me.verts[vcount-1]]) me.faces.extend([me.verts[0],me.verts[1],me.verts[vcount]]) b = 0 kcount = 0 vvcount = 0 a = a + da while a < 90: while b < 360: dz = radius*cos(a/90*pi) dx = radius*sin(a/90*pi)*sin(b/90*pi)

165
dy = radius*sin(a/90*pi)*cos(b/90*pi) me.verts.extend(dx, dy, dz+laenge/2) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]]) b = b + db kcount = kcount + 1 b = 0 vvcount = 0 a = a + da a = 90 b = 0 vvcount = 0 while b < 360: dx = radius*sin(b/90*pi) dy = radius*cos(b/90*pi) me.verts.extend(dx, dy, laenge/2) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]]) b = b + db kcount = kcount + 1 b = 0 vvcount = 0 while b < 360: dx = radius*sin(b/90*pi) dy = radius*cos(b/90*pi) me.verts.extend(dx, dy, 0) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]]) b = b + db kcount = kcount + 1 b = 0 vvcount = 0 for face in me.faces: face.smooth=1 ob.link (me) x1 = float(x1) y1 = float(y1) z1 = float(z1) x2 = float(x2) y2 = float(y2) z2 = float(z2) ob.loc = (float(x1+x2)/2,(y1+y2)/2,(z1+z2)/2) ob.RotY = wz ob.RotZ = wsz scene.link (ob) #zweite Haelfte vom Stick ob = Object.New('Mesh') # Mesh erstellen me = Mesh.New() if type2 in ['C', 'H', 'B', 'N', 'O', 'P']: mat = Material.Get(type2) else: mat = Material.Get('anyatom') me.materials = [mat] vcount = -1 while b < 360: dx = radius*sin(b/90*pi) dy = radius*cos(b/90*pi) me.verts.extend(dx, dy, 0) vcount = vcount + 1 b = b + db

166
kcount = kcount + 1 b = 0 vvcount = 0 while b < 360: dx = radius*sin(b/90*pi) dy = radius*cos(b/90*pi) me.verts.extend(dx, dy, -laenge/2) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]]) b = b + db kcount = kcount + 1 b = 0 vvcount = 0 while a < 180: while b < 360: dz = radius*cos(a/90*pi) dx = radius*sin(a/90*pi)*sin(b/90*pi) dy = radius*sin(a/90*pi)*cos(b/90*pi) me.verts.extend(dx, dy, dz-laenge/2) vcount = vcount + 1 vvcount = vvcount + 1 if vvcount > 1: me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int(360/db)-1],me.verts[vcount-int(360/db)]]) if b == 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(360/db)],me.verts[vcount-2*int(360/db)+1],me.verts[vcount-int(360/db)+1]]) b = b + db kcount = kcount + 1 b = 0 vvcount = 0 a = a + da # letzter Punkt me.verts.extend(0, 0, -laenge/2-radius) vcount = vcount + 1 b = 0 while b < 360-db: me.faces.extend([me.verts[vcount],me.verts[vcount-int(b/db)-2],me.verts[vcount-int(b/db)-1]]) b = b + db me.faces.extend([me.verts[vcount],me.verts[vcount-1],me.verts[vcount-int((360-db)/db)-1]]) eins = 1 for face in me.faces: face.smooth=1 ob.link (me) x1 = float(x1) y1 = float(y1) z1 = float(z1) x2 = float(x2) y2 = float(y2) z2 = float(z2) ob.loc = (float(x1+x2)/2,(y1+y2)/2,(z1+z2)/2) ob.RotY = wz ob.RotZ = wsz scene.link (ob) def import_pdb(path): global structmode, scatom, sumatom, refineballs, refinesticks, scsticks, balls, sticks, hydros Blender.Window.WaitCursor(1) x, y, z, atom = [0], [0], [0], [0] file = open(path, 'r') for line in file.readlines(): if len(line) == 0 or line == ('END'): pass elif line[:6] == 'HETATM' or line[:4] == 'ATOM': name = line[7:11] while name[:1]==' ': name = name[1:] if len(line) > 76: atom0 = line[77:(len(line)-1)] while atom0[-1] == ' ': atom0 = atom0[:-1] else: atom0 = line[13:16] while atom0[-1] == ' ': atom0 = atom0[:-1] esym = [' ','1','2','3','4','5','6','7','8','9']

167
for a in esym: if atom0[-1] == a: atom0 = atom0[:-1] if atom0[0] == a: atom0 = atom0[1:] atom.append (atom0) x0 = line[31:38] while x0[:1]==' ': x0 = x0[1:] y0 = line[39:46] while y0[:1]==' ': y0 = y0[1:] z0 = line[47:54] while z0[:1]==' ': z0 = z0[1:] x.append (float(x0)) y.append (float(y0)) z.append (float(z0)) if balls: if atom[int(name)] == 'H': if hydros: ball(name, atom[int(name)], x[int(name)],y[int(name)],z[int(name)]) else: ball(name, atom[int(name)], x[int(name)],y[int(name)],z[int(name)]) elif line[:3] == 'TER': name = 'platzhalter' atom.append ('platzhalter') x.append (0) y.append (0) z.append (0) elif line[:6] == 'CONECT': if sticks: con0 = line[6:11] while con0[:1]==' ': con0 = con0[1:] for i in range(0,4): if len(line) > (15 + 5 * i): if not line[(11+5*i):(16+5*i)] == ' ': con1 = line[(11+5*i):(16+5*i)] while con1[:1]==' ': con1 = con1[1:] if int(con1)> int(con0): if hydros: stick3(x[int(con0)], y[int(con0)], z[int(con0)], atom[int(con0)], x[int(con1)], y[int(con1)], z[int(con1)], atom[int(con1)], scsticks.val) else: if not (atom[int(con0)]=='H' or atom[int(con1)]=='H'): stick3(x[int(con0)], y[int(con0)], z[int(con0)], atom[int(con0)], x[int(con1)], y[int(con1)], z[int(con1)], atom[int(con1)], scsticks.val) scene.update() Blender.Redraw() Blender.Window.WaitCursor(0) def import_p_follow(path): global structmode, scatom, sumatom, refineballs, refinesticks, balls, sticks, scsticks Blender.Window.WaitCursor(1) x, y, z = 0, 0, 0 pcount = 0 file = open(path, 'r') for line in file.readlines(): if len(line) == 0 or line[:3] == ('END') or line[:6] == ('CONECT'): pass elif line[:6] == 'HETATM' or line[:4] == 'ATOM': if len(line) > 76: atom0 = line[77:(len(line)-1)] while atom0[-1] == ' ': atom0 = atom0[:-1] else: atom0 = line[13:16] while atom0[-1] == ' ': atom0 = atom0[:-1] esym = [' ','1','2','3','4','5','6','7','8','9'] for a in esym: if atom0[-1] == a: atom0 = atom0[:-1] if atom0[0] == a: atom0 = atom0[1:] if atom0 == 'P': xold = x yold = y zold = z x0 = line[31:38] while x0[:1]==' ': x0 = x0[1:] y0 = line[39:46] while y0[:1]==' ': y0 = y0[1:] z0 = line[47:54] while z0[:1]==' ': z0 = z0[1:] x = float(x0) y = float(y0) z = float(z0) if line[30] == '-': x = 0 - x if line[38] == '-': y = 0 - y if line[46] == '-': z = 0 - z abstandq = (x - xold) * (x - xold) + (y - yold) * (y - yold) + (z - zold)*(z - zold)

168
if pcount > 0 and abstandq < 100.00: stick1(xold, yold, zold, x, y, z, scsticks.val) pcount = pcount + 1 elif line[:3] == 'TER': pcount = 0 scene.update() Blender.Redraw() Blender.Window.WaitCursor(0) def gui(): global structmode, scatom, sumatom, refineballs, refinesticks, balls, sticks, scsticks, hydros, bbut, sbbut, sbut, pbut bbut = Toggle('Balls',3, 40, 240, 80, 19, bbut.val) sbbut = Toggle('Sticks and Balls',4, 120, 240, 150, 19, sbbut.val) sbut = Toggle('Sticks',5, 270, 240, 80, 19, sbut.val) pbut = Toggle('DNA Backbone Follow',12, 40, 220, 310, 19, pbut.val) if hydros: Button('Hydrogens on',11, 40, 190, 310, 19) else: Button('Hydrogens off',11, 40, 190, 310, 19) if balls: scatom = Slider('Atom Scale :', 6, 40, 160, 310,19, scatom.val, 0.00, 4.00) if balls: sumatom = Slider('Atom Sum :', 7, 40, 140, 310,19, sumatom.val, 0.00, 2.00) if balls: refineballs = Slider('Atom Refinement :', 8, 40, 120, 310,19, refineballs.val, 4, 36) if sticks or pbut.val: scsticks = Slider('Stick Thickness :', 9, 40, 90, 310,19, scsticks.val, 0.00, 4.00) if sticks or pbut.val: refinesticks = Slider('Stick Refinement :', 10, 40, 70, 310,19, refinesticks.val, 4, 36) Button('Import',1, 40, 40, 155, 19) Button('Cancel',2, 195, 40, 155, 19) def event(evt, val): if (evt == ESCKEY and not val): Exit() def bevent(evt): global structmode, scatom, sumatom, refineballs, refinesticks, balls, sticks, scsticks, hydros, bbut, sbbut, sbut, pbut if evt == 2: Exit() elif evt == 1: if structmode == 'Backbone': Blender.Window.FileSelector(import_p_follow, 'Import') else: Blender.Window.FileSelector(import_pdb, 'Import') elif evt == 3: structmode = "balls" scatom.val = 1.0 sumatom.val = 0.4 refineballs.val = 24 balls = 1 sticks = 0 bbut.val = 1 sbbut.val = 0 sbut.val = 0 pbut.val = 0 Redraw() elif evt == 4: structmode = "sticks and balls" scatom.val = 0.75 sumatom.val = 0.0 refineballs.val = 20 refinesticks.val = 12 balls = 1 scsticks.val = 0.16 sticks = 1 bbut.val = 0 sbbut.val = 1 sbut.val = 0 pbut.val = 0 Redraw() elif evt == 5: structmode = "sticks" scsticks.val = 0.24 refinesticks.val = 12 balls = 0 sticks = 1 bbut.val = 0 sbbut.val = 0 sbut.val = 1 pbut.val = 0 Redraw() elif evt == 12: structmode = "Backbone" scsticks.val = 1.00 refinesticks.val = 8 balls = 0 sticks = 1 bbut.val = 0 sbbut.val = 0 sbut.val = 0 pbut.val = 1

169
Redraw() elif evt == 11: if hydros: hydros = 0 else: hydros = 1 Redraw() elif evt == 8: while not ((int(360/refineballs.val) == float(360.00/refineballs.val)) and (int(refineballs.val)/2) == (float(refineballs.val)/2)): refineballs.val = refineballs.val + 1 Redraw() elif evt == 10: while not ((int(360/refinesticks.val) == float(360.00/refinesticks.val)) and (int(refinesticks.val)/2) == (float(refinesticks.val)/2)): refinesticks.val = refinesticks.val + 1 Redraw() def initialize(): global structmode, scatom, sumatom, refineballs, refinesticks, balls, sticks, scsticks, hydros, bbut, sbbut, sbut, pbut structmode = Create("balls") scatom = Create(1.0) sumatom = Create(0.4) scsticks = Create(0.24) refineballs = Create(24) refinesticks = Create(12) balls = Create(1) sticks = Create(0) hydros = Create(1) bbut = Create(1) sbbut = Create(0) sbut = Create(0) pbut = Create(0) Register(gui, event, bevent) initialize() Blender.Redraw()

170
Abkürzungen und Akronyme
A Adenosin
abs. absolut
αCN α-Cyanozimtsäure
APS Ammoniumperoxodisulfat
ATTG 6-Aza-2-thiothymin + Glycerin
bidest. bidestilliert
BTP Bistrispropan
C Cytosin
CDI N,N-Carbonyldiimidazol
CPG controlled pore glass
DC Dünnschichtchromatographie
DMAA N,N-Dimethylacrylamid
DMF Dimethylformamid
DMSO Dimethylsulfoxid
DNA Desoxyribonucleinsäure
2,4-DNBA 2,4-Dinitrobenzaldehyd
2,4-DNPH 2,4-Dinitrophenylhydrazin
DTT Dithiothreitol
EDC 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid
EDU 1-Ethyl-3-(3-dimethylaminopropyl)harnstoff
eq Äquivalent(e)
FAB Fast Atom Bombardement
Fl. Fluorescein
G Guanosin
HPLC High-Performance-Liquid-Chromatography
IR Infrarot
Lit. Literatur
MALDI-TOF Matrix-Assisted Laser Desorption Ionisation / Time-Of-Flight
MS Massenspektrometrie
4-NBA 4-Nitrobenzaldehyd
NHS N-Hydroxysuccinimid

171
NMR Nuclear Magnetic Resonance
NVOC 4,5-Dimethoxy-2-nitrobenzyloxycarbonyl
PAGE Polyacrylamidgelelektrophorese
PCR Polymerase Kettenreaktion
QV Quellvolumen
RNA Ribonukleinsäure
ROM read only memory
RT Raumtemperatur
SNP single nucleotide polymorphism
T Thymidin
TMEDA N,N,N’,N’-Tetramethylethylendiamin
TMS Tetramethylsilan
TRIS Tris(hydroxymethyl)methylamin
UV Ultraviolett
Vis Visual

172
Verwendete Symbole
Einige der verwendeten Größen werden sowohl in molarer Form, als auch in nicht molarer
Form verwendet, z.B. ni und νe. Die Verwendung geht jeweils aus dem Kontext hervor.
α(i) Linearer Ausdehnungskoeffizient (in Richtung i)
A belastete Fläche im stress-strain-Experiment
χi Flory-Huggins-Wechselwirkungsparameter eines Polymers mit der Komponente i
ε°(i) Eluotrope Stärke (i = stationäre Phase)
εr Dielektrizitätskonstante
FG Gewichtskraft
g Erdbeschleunigung
∆GE Elastische Freie Enthalpie
∆GIon Ionische Freie Enthalpie
∆GM Freie Mischungsenthalpie
∆GT Gesamtänderung der Freien Enthalpie
∆HM Mischungsenthalpie
k(B) Boltzmann-Konstante
m Masse
ni Stoffmenge der Komponente i
νe effektive (molare) Gesamtzahl der Quervernetzungen im Polymer
Ω statistisches Gewicht
p Dipolmoment
P’ Polaritätsindex nach Snyder[48]
ΠE Druck durch elastische Expansion/Kompression des Netzwerks
ΠIon Osmotischer Druck (ionisch)
ΠM Osmotischer Druck (Mischung)
ΠT Gesamtdruck
QV Quellvolumen
R allgemeine Gaskonstante
Sc statistische Gesamtentropie
SDis Disorientierungsentropie
∆SE Entropieänderung bei elastischer Expansion/Kompression des Netzwerks

173
∆SM Mischungsentropie
τ mechanische Spannung
V Volumen des gequollenen Polymers
V0 Volumen des Polymers in ungequollenem Zustand
vi Molvolumen der Komponente i
Vm Volumen des Polymers in spannungsfreiem Zustand (ΠE = 0)
υi Volumenanteil der Komponente i
wij Energie eines i-j-Kontaktes nach dem liquid-lattice Modell
x Zahl der Segmente im liquid-lattice Modell
xi Molenbruch der Komponente i
y Schichtdicke des Polymers
y0 Schichtdicke des Polymers im relaxierten Zustand
z Gitterkoordinationsnummer (Zahl der benachbarten Zellen) im liquid-lattice
Modell

174
Literatur
[1] http://de.wikipedia.org/wiki/Information Stand 18.09.2006.
[2] C. Floyd, C. Fuchs, W. Hofkirchner (Hrsg.), Stufen zur Informationsgesellschaft, Peter
Lang Verlag Frankfurt 2002, 241-281.
[3] H. Krapp, T. Wägenbaur (Hrsg.), Komplexität und Selbstorganisation – „Chaos“ in
Natur- und Kulturwissenschaften, Wilhelm Fink Verlag München 1997, 99-129.
[4] W.-C.A. Lee, H. Huang, G. Feng, J.R. Sanes, E.N. Brown, P.T. So, E. Nedivi, PLoS
Biol. 2006, 4(2), e29.
[5] L.M. Adleman, Science 1994, 266, 1021-1024.
[6] X. Su, L.M. Smith, Nucleic Acid Res. 2004, 32(10), 3115-3123.
[7] R.S. Braich, N. Chelyapov, C. Johnson, P.K.W. Rothemund, L.M. Adelman, Science
2002, 296, 499-502.
[8] Q. Liu, L. Wang, A. Frutos, A.E. Condon, L.M. Smith, Nature 2000, 413, 175-179.
[9] K.A. Schmidt, C.V. Henkel. G. Rozenberg, H.P. Spaink, Nucleic Acid Res. 2004,
32(17), 4962-4968.
[10] J.J. Tabor, M. Levy, A.D. Ellington, Nucleic Acid Res. 2006, 34(8), 2166-2172.
[11] J. Chem, D.H. Wood, PNAS 2000, 97, 1328-1330.
[12] C. Heise, F.F. Bier, Top. Curr. Chem. 2006, 261, 1-25.
[13] C.M. Niemeyer, D. Blohm, Angew. Chem. 1999, 111(19), 3039-3043.
[14] K.-D. Bachmann, C.R. Bartram, J. Chang-Claude, C. Fonatsch, P. Propping,
Deutsches Ärzteblatt 1998, 75(22), A1396-A1403.
[15] O. Vierimaa, M. Georgitsi, R. Lehtonen, P. Vahteristo, A. Kokko, A. Raitila, K.
Tuppurainen, T.M.L. Ebeling, P.I. Salmela, R. Paschke, S. Gündogdu, E. De Menis,
M.J. Mäkinen, V. Launonen, A. Karhu, L.A. Aaltonen, Science 2006, 312, 1228-1230.
[16] S. Marsh, H.L. McLeod, Human Molecular Genetics 2006, 15, R89-R93.
[17] F. S. Collins, V. A. McKusick, JAMA 2001, 285, 540-544.
[18] TSC: The SNP Consortium Website: http://snp.cshl.org/ Stand 10.8.2006
[19] R. Sachidanandam, D. Weissman, S.C. Schmidt, J.M. Kakoi, L.D. Stein, G. Marth, S.
Sherry, J.C. Mullikin, B.J. Mortimore, D.L. Willey, S.E. Hunt, C.G. Cole, P.C.
Coggill, C.M. Rice, Z. Ning, J. Rogers, D.R. Bentley, P. Kwok, E.R. Mardis, R.T.
Yeh, B. Schutlz, L. Cook, R. Davenport, M. Dante, L. Fulton, L. Hillier, R.H.
Waterston, J. McPherson, B. Gilman, S. Schaffner, W.J. Van Etten, D. Reich, J.

175
Higgins, M.J. Daly, B. Blumenstiel, J. Baldwin, N. Stange-Thomann, M.C. Zody, L.
Linton, E.S. Lander, D. Altshuler, Nature 2001, 409 (6822), 928-933.
[20] R.A. Gibbs, H. Yang, S.B. Gabriel, D. Atlshuler, Y. Shen, W. Huang, M.M.Y. Waye,
H. Xue, L.M. Galver, M.S. Chee, A. Montpetit, T.J. Hudson, K.A. Frazer, P. Kwok, L.
Tsui, Y. Nakamura, P. Deloukas, D.R. Bentley, M.J. Daly, D. Altshuler, David; L.D.
Stein, A. Chakravarti, G.R. Abecasis, G. McVean, P. Donnelly, L.R. Cardon, T.
Tsunoda, J.C. Mullikin, S.T. Sherry, M. Feolo, H. Zahng, C. Zeng, H. Zhao, I.
Matsuda, C.N. Rotimi, M.F. Leppert, R. Qui, A. Kent, K. Kato, N. Niikawa, I.F.
Adewole, B.M. Knoppers, M.W. Foster, E.W. Clayton, J. Watkin, S.B. Gabriel, R.K.
Wilson, J. Rogers, Z. Chen, H. Han, L. Kang, M. Godbout, J.C. Wallenburg, P.L.
Archeveque, G. Bellemare, K. Saeki, H. Wang, D. An, H. Fu, Q. Li, Z. Wang, R.
Wang, A.L. Holden, L.D. Brooks, J.E. McEwen, C.R. Bird, M.S. Guyer, P.J. Nailer,
V.O. Wang, J.L. Peterson, M. Shi, J. Spiegel, L.M. Sung, J. Witonsky, L.F. Zacharia,
F.S. Collins, K. Kennedy, R. Jamieson, J. Stewart, Nature 2005, 437(7063), 1299-
1320.
[21] D.D. Shoemaker, E.E. Schadt, C.D. Armour, Y.D. He, P. Garrett-Engele, P.D.
McDonagh, P.M. Loerch, A. Leonardson, P.Y. Lum, G. Cavet, L.F. Wu, S.J.
Altschuler, S. Edwards, J. King, J.S. Tsang, G. Schimmack, J.M. Schelter, J. Koch, M.
Ziman, M.J. Marton, B. Li, P. Cundiff, T. Ward, J. Castle, M. Krolewski, M.R. Meyer,
M. Mao, J. Burchard, M.J. Kidd, H. Dai, J.W. Phillips, P.S. Linsley, R. Stoughton, S.
Scherer, M.S. Boguski, Nature 2001, 409, 922-927.
[22] S.P. Fodor, J.L. Read, M.C. Pirrung, Science 1991, 767-773.
[23] R.S.Martinsky, United States Patent 6,101,946; 2000.
[24] http://www.arrayit.com/Products/Printing/Stealth/stealth.html Stand 10.8.2006.
[25] L.R. Allain, D.N. Stratis-Cullum, T. Vo-Dinh, Analytica Chimica Acta 2004, 518, 77-
78.
[26] M.J. Heller, E. Tu,WO9512808, US5605662, EP0727045.
[27] Q. Du, O. Larsson, H. Swerdlow, Z. Liang, Top. Curr. Chem. 2006, 261, 45-61.
[28] H. Lin, L. Sun, R.M. Crooks, JACS 2005, 127(32), 11210-11211.
[29] R.D. Mitra, G.M. Church, Nucleic Acid Res. 1999, 27(24), e34.
[30] A. Luther, R. Brandsch, G. von Kiedrowski, Nature 1998, 396, 245-248.
[31] L.H. Eckardt, K. Naumann, M.W. Pankau, M. Rein, M. Schweitzer, N. Windhab, G.
von Kiedrowski, Nature 2002, 420, 286.

176
[32] G. von Kiedrowski, L.-H. Eckardt, K. Naumann, W. M. Pankau, M. Reimold, M.
Rein, Pure Appl. Chem. 2003 75(5), 609-619.
[33] J.P. Fürste, S. Klussman, T. Klein, G. von Kiedrowski, Cloning and copying on
surfaces, EP1135527, WO0032809, US2002022276.
[34] M. Reimold, Diplomarbeit, Bochum 2000.
[35] E. Timofeev, S.V. Kochetkova, A.D. Mirzabekov, V.L. Florentiev, Nucleic Acids Res.
1996, 24(16), 3142–3148.
[36] A.M. North, A.M. Scallan, Polymer 1964, 5(9), 447-455.
[37] D.B. Thomas, A.J. Convertine, L.J. Myrick, C.W. Scales, A.E. Smith, A.B. Lowe,
Y.A. Vasilieva, N. Ayres, C.L. McCormick, Macromolecules 2004, 27, 8941-8950.
[39] M. Marchetti, S. Prager, E.L. Cussler, Macromolecules 1990, 23, 1760-1765.
[40] M. Marchetti, S. Prager, E.L. Cussler, Macromolecules 1990, 23, 3445-3450.
[41] S.A. Asher, V.L. Alexeev, A.V. Goponenko, A.C. Sharma, I.K. Lednev, C.S. Wilcox,
D.N. Finegold, J. Am. Chem. Soc. 2003, 125(11), 3322-3329.
[42] K. Lee, S.A Asher, J. Am. Chem. Soc. 2000, 122(39), 9534-9537.
[43] V.L. Alexeev, A.C. Sharma. A.V. Goponenko, S. Das, I.K. Lednev, C.S. Wilcox, D.N.
Finegold, S.A. Asher, Anal. Chem. 2003, 75, 2316-2323.
[44] P.J. Flory, Principles of polymer chemistry, Cornell University Press Ithaca 1953.
[45] B. Krishna, B. Brakash, Australian Journal of Chemistry 1969, 22(12), 2679-2680.
[46] D.R. Lide (ed.), CRC Handbook of Chemistry and Physics, 74th Edition, CRC Press
Boca Raton, 1993.
[47] P.C. Sadek, The HPCL Solvent Guide, John Wiley & Sons, Inc. 1996.
[48] L.R. Snyder, J. Chromatogr. Sci. 1978, 16, 223-234.
[49] P.J. Flory, The Journal of Chemistry and Physics 1977, 66 (12), 5720-5729.
[50] R.E. Melley, J.E. Stuckey, J. Appl. Polym. Sc. 1970, 14, 2327-2331.
[51] M. Ilavsky, Macromolecules 1982, 15, 782-788.
[52] V.F. Janas, F. Rodriguez, C. Cohen, Macromolecules 1980, 13, 977-983.
[53] L. Benguigui, F. Boué, Eur. Phys. J. B 1999, 11, 439-444.
[54] K. Achilles, G. v. Kiedrowski, Bioorganic and Medicinal Chemistry Letters 2005, 15,
1229-1233.
[55] F.L. Buchholz, Superabsorbent Science and Technology, ACS symposium Series 573,
American Chemical Society Washington, D.C. 1998.
[56] Y.M. Mohan, P.S.K. Maurthy, K.M. Raju, Journal of Applied Polymer Science 2006,
101, 3202-3214.

177
[57] G.S.Hammond, D.H. Hogle, JACS 1954, 77, 338-340.
[58] S. Fisher, R. Kunin, Journal of Physical Chemistry 1956, 60, 1030-1032.
[59] H.P. Gregor, M. Frederick, Journal of Polymer Science 1957, 23, 451-465.
[60] H.G. Spencer, Journal of Polymer Science 1962, 56(163), S25-S28.
[61] V.K. Sarin, S.B.H. Kent, J.P. Tam, R.B. Merrifield, Analytical Biochemistry 1981,
117, 147-157.
[62] S. L. Beaucage, R. P. Iyer, Tetrahedron 1992, 48, 2223-2311.
[63] L. J. McBride, M. H. Caruthers, Tetrahedron Lett. 1983, 24, 245-248.
[64] M. D. Matteucci, M. H. Caruthers, Tetrahedron Lett. 1980, 21, 3243-3246.
[65] M. D. Matteucci, M. H. Caruthers, J. Am. Chem. Soc. 1981, 103, 3185-3191.
[66] M. J. Gait, Oligonucleotide Chemistry – a practical approach, IRL Press Oxford
1984.
[67] J.X. Khym, Methods Enzymol. Part 1 1967, 12, 93-101.
[68] H. Schott, Angew. Chem. 1972, 84(17), 819-820.
[69] G. Wulff, A. Sarhan, Angew. Chem. 1972, 84(8), 364.
[70] G. Wulff, G. Kirstein, Angew. Chem. 1990, 102(6), 706-708.
[71] R.A. Potyrailo, Angew. Chem. 2006, 118(5), 718-738.
[72] L.I. Bosch, T.M. Fyles, T.D. James, Tetrahedron 2004, 60(49), 11175-11190.
[73] G.L. Roy, A.L. Laferriere, J.O. Edwards, J. Inorg. Nucl. Chem. 1957, 4, 106-114.
[74] G. Dawber, D.H. Matusin, J. Chem. Soc., Faraday Trans I 1982, 78, 2521-2528.
[75] C. D’Silva, D. Green J. Chem. Soc., Chem. Commun. 1991, (4), 227-228.
[76] Windows Titan 1.0.5, Wavefunction, Inc., Schrödinger, Inc. 2000.
[77] G.R. Newkome, C.N. Moorefield, F. Vögtle, Dendritic Molecules, VCH Weinheim
1996.
[78] Y. Kitamura, T. Itoh, J. Solution Chem. 1987, 16(9), 715-726.
[79] A. Sunder, R. Hanselmann, H. Frey, R. Mülhaupt, Macromolecules 1999, 32, 4240-
4246.
[80] V.M. Garamus, T.V. Maksimova, H. Kautz, E. Barriau, H. Frey, U. Schlotterbeck, S.
Mecking, W. Richtering, Macromolecules 2004, 37, 8394-8399.
[81] Schleicher & Schuell Nytran ® SPC Nylon Transfer Membran.
[82] H. E. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem. 1997, 62, 7512 – 7515.
[83] P.L. Biochemicals, ICN, Firmenschrift, Circular No. OR-10.
[84] DFG-Kalender 2003, Deutsche Forschungsgemeinschaft 2003, 63.
[85] M. Reimold, Nachrichten aus der Chemie 2003, 51(6), Titelbild.

178
[86] COST D27 Chembiogenesis 2005, Konferenzposter.
[87] I. Reimold-Stahl, G. von Kiedrowski, Rubin 2006, 16(1), 57-64.
[88] Blender 2.42a, Blender Foundation 2006.
[89] Protein Data Bank Contents Guide: Atomic Coordinate Entry Format Description
Version 2.2, 1996.
[90] Python 2.4.3, Python Software Foundation 2006.

Danksagung
Ich danke Prof. Dr. Rainer Haag für eine Probe des dendritischen Polyglycerins PG02.
Ich danke allen, die mich während der Zeit meiner Promotion unterstützt und begleitet haben.
Mein Dank gilt den aktuellen und ehemaligen Mitarbeitern des Lehrstuhls für Organische
Chemie I, Karin Achilles, Sergey Antsypovitsch, Alexander Azzawi, Rolf Breuckmann, Jan
Bülle, Frank Buch, Martin Cebulla, Arne Dieckmann, Axel Dorenbeck, Rainer Döring, Lars
Eckardt, Nils Hellwig, Frank Hühnerschulte, Aloys Hüttermann, Johannes Jäger, Florian
Kaschuba, Maik Kindermann, Klaus Körner, Hans-Werner Lennartz, Karl Lenßen, Carsten
Lodwig, Beate Materne, Sven Mönninghoff, Kai Naumann, Matthias Pankau, Volker Patzke,
Tobias Plöger, Maya Radeva, Insa Reimold-Stahl, Michael Rein, Holger Schöneborn,
Johanna Stankiewicz, Olga Taran, Oliver Thoennessen, Dagmar Vössing, Tim Wilmsen,
Marcel Wollenweber, Michael Wüstefeld und Jan Zimmermann, für das gute Arbeitsklima
und die nette gemeinsame Zeit.
Besonders danke ich Insa Reimold-Stahl für die bedingungslose Unterstützung und den
Glauben an diese Arbeit. Ich danke ihr für das Korrekturlesen und die aufwändige
Überprüfung der Formeln.
Ebenso danke ich Matthias Pankau für das Korrekturlesen.
Ich danke Michael Wüstefeld für die Versorgung mit DNA und Kaffee.
Ich danke Rolf Breuckmann für die Unterstützung bei MALDI- und ESI-Messungen, ich
danke ihm und ebenso Margot Bock und Stefanie Wittmann für die Unterstützung in vielen
verwaltungstechnischen Angelegenheiten.
Ich danke Kai Naumann für manche interessante Anregung aus der Ferne.
Ich danke Maik Kindermann für viele synthetische Ratschläge, ich danke Jan Zimmermann
für die Hilfe bei der postsynthetischen 5’-Hydrazidmodifizierung von Oligonukleotiden.

Ich danke Oliver Thoennessen und Florian Kaschuba für die Hilfe bei computertechnischen
und Netzwerk-Problemen.
Ich danke Martin Cebulla, der in seinem Vertiefungspraktikum an der Immobilisierung von
Hydrazid-Oligonukleotiden an Aldehyd-Gelen mitgearbeitet hat.
Ich danke meiner Familie für die Unterstützung während all der Jahre meines Studiums.

Lebenslauf
Persönliche Angaben
Name: Malte Reimold
Geburtstag: 25. März 1975
Geburtsort: Bochum
Staatsangehörigkeit: deutsch
Familienstand: verheiratet
Schulbildung
1981 - 1985 Grundschule Salzbergen
1985 - 1994 Gymnasium Dionysianum in Rheine
7. Juni 1994 Allgemeine Hochschulreife am Gymnasium Dionysia-
num in Rheine
Ersatzdienst
Oktober 1995 - November 1996 Caritas-Sozialstation Emsbüren-Salzbergen
Studium
September 1994 - Mai 2000 Studium der Chemie an der Ruhr-Universität Bochum
Wahlpflichtfach: Analytische Chemie
06. Oktober 1997 Vordiplom an der Ruhr-Universität Bochum
Note: gut
November 1999 - Mai 2000 Diplomarbeit bei Prof. Dr. Günter von Kiedrowski am
Lehrstuhl für Organische Chemie I der Ruhr-Universi-
tät Bochum mit dem Thema "Synthese von fluoreszenz-
markierten Dendrimeren für die 5’-Modifikation von
Oligonucleotiden"
26. Mai 2000 Diplom an der Ruhr-Universität Bochum
Note: sehr gut
Promotion
Juni 2000 - Dezember 2006 Promotion bei Prof. Dr. Günter von Kiedrowski am
Lehrstuhl für Organische Chemie I der Ruhr-Univer-
sität Bochum





























