Embed Size (px)
Citation preview

CONTINUING EDUCATION
Update on neuroimaging phenotypesof mid-hindbrain malformations
Patrice Jissendi-Tchofo & Mariasavina Severino &
Béatrice Nguema-Edzang & Cissé Toure &
Gustavo Soto Ares & Anthony James Barkovich
Received: 8 May 2014 /Accepted: 4 September 2014# Springer-Verlag Berlin Heidelberg 2014
AbstractPurpose Neuroimaging techniques including structural mag-netic resonance imaging (MRI) and functional positron emis-sion tomography (PET) are useful in categorizing variousmidbrain-hindbrain (MHB) malformations, both in allowingdiagnosis and in helping to understand the developmentalprocesses that were disturbed. Brain imaging phenotypes ofnumerous malformations are characteristic features that helpin guiding the genetic testing in case of direct neuroimaging-genotype correlation or, at least, to differentiate among MHBmalformations entities. The present review aims to provide thereader with an update of the use of neuroimaging applicationsin the fine analysis of MHB malformations, using a compre-hensive, recently proposed developmental and geneticclassification.Methods We have performed an extensive systematic reviewof the literature, from the embryology main steps of MHBdevelopment through the malformations entities, with regardto their molecular and genetic basis, conventional MRI fea-tures, and other neuroimaging characteristics.
Results We discuss disorders in which imaging features aredistinctive and how these features reflect the structural andfunctional impairment of the brain.Conclusion Recognition of specific MRI phenotypes, includ-ing advanced imaging features, is useful to recognize theMHBmalformation entities, to suggest genetic investigations,and, eventually, to monitor the disease outcome after support-ive therapies.
Keywords Cerebellum embryology .Mid-hindbrainmalformations . Posterior fossa malformations . Cerebellardysplasia . Pontine dysplasia
Introduction
Improvements in neuroimaging techniques over the last20 years, including structural magnetic resonance imaging(MRI) and functional positron emission tomography (PET),have helped to define the characteristic features of variousmidbrain-hindbrain (MHB) malformations (1–3). Correla-tions are beginning to be made between human genotypesand brain imaging phenotypes for numerous mutations asso-ciated with these developmental anomalies [1–6]. Therefore,the imaging features may help in guiding the genetic testing.Advanced MRI techniques are increasingly available in clin-ical settings and the proper use of these techniques in patientswith MHB malformations may provide additional insightconcerning such disorders and their various subtypes, as wellas alerting caretakers to potential motor and cognitive com-plications. The present review aims to provide the reader withan up-to-date inventory of the advanced neuroimaging tech-niques that can potentially aid in the diagnosis and assessmentof the MHB malformations addressed in a recently proposeddevelopmental and genetic classification of these disorders[6]. We focus on entities in which MR imaging features are
P. Jissendi-Tchofo (*) : B. Nguema-Edzang :C. Toure :G. Soto AresDepartment of Neuroradiology, MRI 3T Research, PlateformeImagerie du vivant, IMPRT-IFR 114, University Hospital of Lille(CHRU), 59037 Lille-Cedex, Francee-mail: [email protected]
P. Jissendi-TchofoRadiology Department, Pediatric Neuroradiology Section, CHUSaint-Pierre, Brussels, Belgium
M. SeverinoNeuroradiology Unit, Istituto Giannina Gaslini, Genoa, Italy
A. J. BarkovichNeuroradiology Section, Department of Radiology and BiomedicalImaging, University of California, San Francisco, CA, USA
NeuroradiologyDOI 10.1007/s00234-014-1431-2

described in the literature and discuss how more advancedtechniques may help in understanding the structural and func-tional impairment of the brain.
Midbrain-hindbrain development
Studies in mice have considerably helped to approach theMHB development in humans, even though several steps arestill not understood as well as the roles of many genes [7]. Thebrain is derived from the differentiation of the rostral neuraltube into the three primary vesicles: the prosencephalon, themesencephalon, and the rhombencephalon. The midbrain,which is the uppermost part of the brainstem, is derived fromthe mesencephalon. The primitive hindbrain or rhombenceph-alon is divided into segments known as rhombomeres (fromRh1 to Rh8); the more rostral is metencephalon and the caudalis myelencephalon. The metencephalon is the source of thecerebellum (from Rh1, the most rostral segment of the hind-brain) and the pons (rostral half of the hindbrain), while themyelencephalon gives the medulla (lower half of the hind-brain). The metencephalon-mesencephalon border is one ofthe fundamental divisions of the brain, which acts as an orga-nizing center (IsO) for patterning adjacent areas by secretingfibroblast growth factor 8 (Fgf8; Fig. 1). The main embryo-logical steps encompass (i) patterning of germinal zones andprogenitor domains during early neural tube development, (ii)cellular proliferation, and (iii) radial and tangential migrationsbetween the 9th and 13th gestational weeks. These steps arecontrolled by the expression of numerousmolecules that are, inturn, the transcription products of many genes.
Cerebellar patterning defines the zones and stripes thatdetermine the organization and function of the adult cerebel-lum as studied in mice [8]. Two germinal matrices are definedas follows: the dorsal rhombic lip (RL) and the ventricularzone (VZ) near the fourth ventricle. These zones are sites ofdifferent progenitors’ genesis, under the influence of varioustranscription factors. Pancreas transcription factor 1a (Ptf1a)acts in the VZ to induce formation of cerebellar GABAergicneurons, while Atoh1 expression in the upper RL stimulatesgenesis of glutamatergic excitatory neurons.
Among the cell lineages that will populate the adult cere-bellum, Purkinje cells (PKs) appear as the main determinant ofthe cerebellar stereotypical and topological organization. PKsare generated in the medial VZ from progenitors and arefurther differentiated in multiple subtypes [8]. Granule neuronprecursors are generated in the lateral VZ and migrate towardthe surface of the RL, where granule cells (GC) are producedand form the external granular layer (EGL) under the antago-nist expression of F3/contactin and TAG1 [9]. RLs also giverise to precerebellar (MHB junction, rostral hindbrain, andpons) and deep cerebellar nuclear neurons (glutamatergic)whose proliferation is stimulated by the expression of Pax6,
Tbr2, and Tbr1 factors [9]. Cerebellar interneurons (stellateand basket cells) production is induced by the expression ofAscl1 in the VZ [8].
PKs are still undergoing mitosis in the VZwhile the earliestpost-mitotic PKs migrate dorsally out of the VZ to form anirregular layer known as cerebellar plate, in which they arereorganized in a stereotyped array of PKs clusters under theinfluence of numerous molecular interactions, includingcalbindin, engrailed-2 (En2), cadherins, neurogenin (Nrgn),and Wnt7. PKs clusters are later dispersed over the cerebellarsurface by the action of reelin (secreted by the EGL) bindingtwo receptors on PKs: apolipoprotein E receptor 2 (Apoer2)and the very low-density lipoprotein receptor (Vldlr) [8]. TheEGL is a second zone of neurogenesis where GC neuronsprecursors proliferate and differentiate into GC neurons thatlater migrate radially inward along extensions of the Bergmanglial cells, deep to the PKs layer, to form the internal granularlayer (IGL), under the regulation of sonic hedgehog (SHH)and Notch signaling pathways [9]. Precerebellar neurons gen-erated in the RL reach their final destination via a tangentialmigration directed by complex molecular interactions of at-tractant (netrin-1 and Unc5 receptors) and repulsor (Slit andRobo receptors) systems [10]. Migration of both VZ and RLneurons to the deep cerebellar nuclei (DCN) is directed by thesequential expression of Pax6, Tbr2, and Tbr1 from the RL tothe nuclear transitory zone [11]. Neuronal and axonal path-finding errors lead to various MHB malformations.
Eventually, along with cellular proliferation and migrationstreams, a spatiotemporal movement changing the axes of thedeveloping cerebellum is required for its final shape. Therostral-caudal axis of Rh1 undergoes a 90° rotation that con-verts it into the medial-lateral axis of the cerebellum primor-dium, as shown in mice [12].
MHB malformations classification
Several imaging-based classification schemes have been ap-plied to MHB malformations [1–3]. The earlier ones werebased upon the characteristic MRI features analyzed fromgross images with relatively thick slices; these often poorlycorrelated with clinical presentations and neurological out-comes. Moreover, in numerous conditions, it was found thatdifferent imaging characteristics may result from mutations ofthe same gene, and patients with nearly identical MRI findingsoften had mutations of different genes [3–6]. The involvementof protein products of many genes in the same pathways, theinvolvement of some gene products in multiple pathways, thepotential for mosaicism, and the coexistence of multiplemalformations in the same patient also make classificationchallenging. Ultimately, classification will likely be based onthe specific molecular pathways affected but, currently, themost reasonable way to classify MHB malformations at this
Neuroradiology

time is to base it on the embryology. A recent classificationincludes molecular and genetic knowledge yielded by find-ings in humans (when available) and in animals [6], as fol-lows: (I) early anterior-posterior patterning defects, dorsoven-tral patterning defects, or misspecification of MHB germinalzones; (II) malformations associated with (a) later generalizeddevelopmental encephalopathies or abnormal neuronal migra-tion, (b) mesenchymal-neuroepithelial signaling defects in-volving or sparing the cerebellum, (c) abnormal cell (neuronaland glial) proliferation, and (d) defects in ciliary proteins; (III)localized brain malformations that predominantly affect thebrain stem and cerebellum; and (IV) combined hypoplasia andatrophy of putative prenatal onset. Chiari malformations areconsidered disorders of the craniocervical junction and willnot be discussed in this paper.
Neuroimaging techniques
The recent improvements of neuroimaging techniques in clin-ical settings have provided radiologists with advanced toolsfor fine structural analysis of the brain parenchyma, metabolicand functional brain assessment with precise anatomical cor-relations, and improved hardware and post-processing soft-ware for the analysis of these data. The benefit of brainimaging at higher magnetic fields has been extensively ad-dressed in the literature [13–15]. Unfortunately, 3 T imaging is
not commonly used for pediatric studies in many centers,possibly because of the increased expense of 3 T MRI scan-ners or because local customs or the absence of proper equip-ment do not allow anesthesia and monitoring of newborn andyoung children in the 3 T imaging [16, 17].
In addition to “conventional MRI (cMRI),” a term we willuse to refer to standard 2D T1- and T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences using slicethickness of 3–5 mm in at least two different planes, severalmore advanced MRI tools can be used for pediatric neuroim-aging [18]. (1) Structural analysis and anatomical correla-tions—three-dimensional (3D)-T1, T2, or FLAIR sequences(with water or fat suppression) with 1 mm or smaller isotropicvoxels allows assessment of the anatomical delineation, theshape, the size, and the 3D morphology of distinct brainstructures or parts of a structure. Additional software allowstissue segmentation, volume normalization, and coregistrationamong studies [19–23] [24]. (2) Microstructural analysis andconnectivity with fiber tracking using diffusion tensor imag-ing (DTI) or, better, diffusion spectral imaging [25] or q-ballimaging [26] assesses the local microscopic water motion,giving information about tissue microstructure, expressed asfractional anisotropy (FA), mean diffusivity (Dav), radial dif-fusivity (RD), and axial diffusivity (AD), with tractographythat can determine the predominant direction of axonal bun-dles in order to determine whether pathways are normal oraberrant [27–30]. (3) Vascular [31] morphology and
Fig. 1 a In the developing brain, signaling molecules are secreted atorganizing centers to pattern the anterior-posterior axis and to defineboundaries and polarity of segments. Fibroblast growth factors (FGFs),namely Fgf8 and Fgf17, are signaling molecules that act in the forebrainto guide formation of telencephalic structures (by regulating the expres-sion of the transcription factor Pax6) and at the mid-hindbrain (MHB)junction. TheMHB junction is determined by the isthmus organizer (Iso),which induces the secretion of Fgf8 and Fgf17 that regulate genes andtranscription factors expression to create differences among cells and toestablish the identity of each MHB segment. Therefore, the diencephalon
(Di)–mesencephalon (Mes) boundary is established by interaction ofPax6 (expressed in the diencephalon) and En1/Pax2 (expressed in therostral mesencephalon) while the Mes–rhombencephalon (Rhomb)boundary is established by formation of Gbx2 (in rostral Rhombomere1) due to repression of Otx2 expression (in caudal Mes) by Fgf8. (thefigure has been reprinted with the permission of the author, Barkovich AJ.Front Neuroanat. 2012 Jan 7; 67(67)). b The anatomical location of theIsO is shown. Gross appearance of the hindbrain, including the mesen-cephalon (A), the pons (B), the medulla (C), and the vermis (V), after thenormal MHB development
Neuroradiology

hemodynamics of tissues including perfusion MRI, MR angi-ography (MRA, including MR arteriography and MR venog-raphy) techniques with or without contrast media administra-tion (such as 3D time of flight (TOF) and bolus track contrastenhancedMRA), and 2D or 3D phase-contrast arterial (PCA),or venous (PCV) encoding MRA allow assessment of theregional vessel development, whereas arterial spin labeling(ASL), dynamic susceptibility weighted sequences (SWI),and blood oxygen dependent level (BOLD) techniques allowthe assessment of relative or absolute local, regional, andoverall blood flow [32–35]; in addition, SWI can map thevenous drainage or depict hemosiderin punctate deposits [36,37]. (4) Metabolic and functional analyses can be performedusing PET to investigate the dynamic uptake of a radiophar-maceutical tracer [38], BOLD tissue response analysis(functional-fMRI) to assess resting state brain activity, local-ization of brain functions, and disturbances of localizationcaused by malformations [39], and MR spectroscopy (MRS)to assess neurochemical compounds of tissues. The latter isnot often useful in malformations but may provide additionalinformation in distinguishing signal abnormalities of develop-mental nature from those of evolving processes [40, 41].
MHB malformations—group I
This group includes malformations secondary to early pattern-ing defects with abnormal anteroposterior or dorsoventralsegmentation of the brainstem or cerebellum leading to gain,loss, or transformation/ectopia of segments at predefinedboundaries of the neural tube.
Anterior-posterior patterning defects
The early anteroposterior (AP) patterning defects or themisspecification of the MHB patterning determined by theso-called “isthmic” organizer (IsO, located at the MHB junc-tion as shown in Fig. 1) may result in such disorders. Thispatterning takes place through the activation of an extensivemolecular and genetic network involving the secretion ofWntand Fgf molecules and the expression of transcription factorsOtx2 (anteriorly) andGbx2 (posteriorly) [6]. An illustration ofAP patterning defect is given in Figs. 2 and 3.
Disconnection syndrome
Disconnection syndrome (DCS) refers to a malformation thatresults from segmental dysgenesis (segments of the midbrainand hindbrain do not develop), resulting in the absence ordiscontinuity of segments [42–44]. These are postulated toresult from a disruption or, perhaps, an impaired local geneticexpression as reported in about 13 cases. Mutation of the En2gene has been proposed [45, 46] as a cause, but no mutationshave been reported. The presence of associated intracranial
vascular abnormalities and extracranial anomalies in manycases suggest a more complex molecular basis, involving thevascular development or a disruption secondary to localizedhypoxic/ischemic or infectious damage, resulting in brainstemdisruption and cerebellar hypoplasia [46–48]. Affected neo-nates often present with symptoms ranging from severe cen-tral hypotonia and cranial neuropathies to spasticquadriparesis with hyperreflexia and respiratory drive failure.The condition is almost universally fatal [46–48].
The cMRI appearance of this malformation shows an over-all shortening of the brainstem as compared to normal; somesegments are lengthenedwhile others are shortened dependingon the location of the disconnection, which can be near themidbrain-pons junction or near the pontomedullary junction.The MHB junction is usually difficult to discern, the mesen-cephalon is enlarged with a midline cleft extending to the levelof the pons, and the medulla is thick (dysmorphic), giving insome cases the appearance of elongated pons due to theanterior flattening of the pontine-medulla junction [4, 6].There is either a profound narrowing, with just a few fiberspassing through, or a complete physical disconnection be-tween the upper and lower portions of the brain stem. Theclivus is small and dysmorphic. The cerebellum and thevermis show variable degrees of hypoplasia. Midlinesupratentorial malformations may include callosal anomaliesand periventricular nodular heterotopia [49]. Thin sectioncMRI and DTI can be useful in displaying a thin midline cordpassing from the upper segment to the lower segment of thebrainstem as found in neuropathological analyses [6, 45–48].Fractional anisotropy maps may show changes in axonalpathways in the MHB and cerebellar affected areas, similarto those reported in congenital central hypoventilation syn-drome (CCHS) [50]. MRA may demonstrate in the PF theabsence of vertebral arteries and the presence of bilateralproatlantal intersegmental arteries arising from the externalcarotid arteries [44, 49] and the prominence of persistentcervical occipital draining veins. The basilar artery may beabsent, dysmorphic, or even normal [42–45]. Ultra high-resolution 3D heavily T2-weighted (constructive interferenceof steady state, CISS; fast imaging with steady state acquisi-tion, FIESTA; or driven equilibrium, DRIVE) images mayshow cranial nerve (CN) hypoplasia or aplasia.
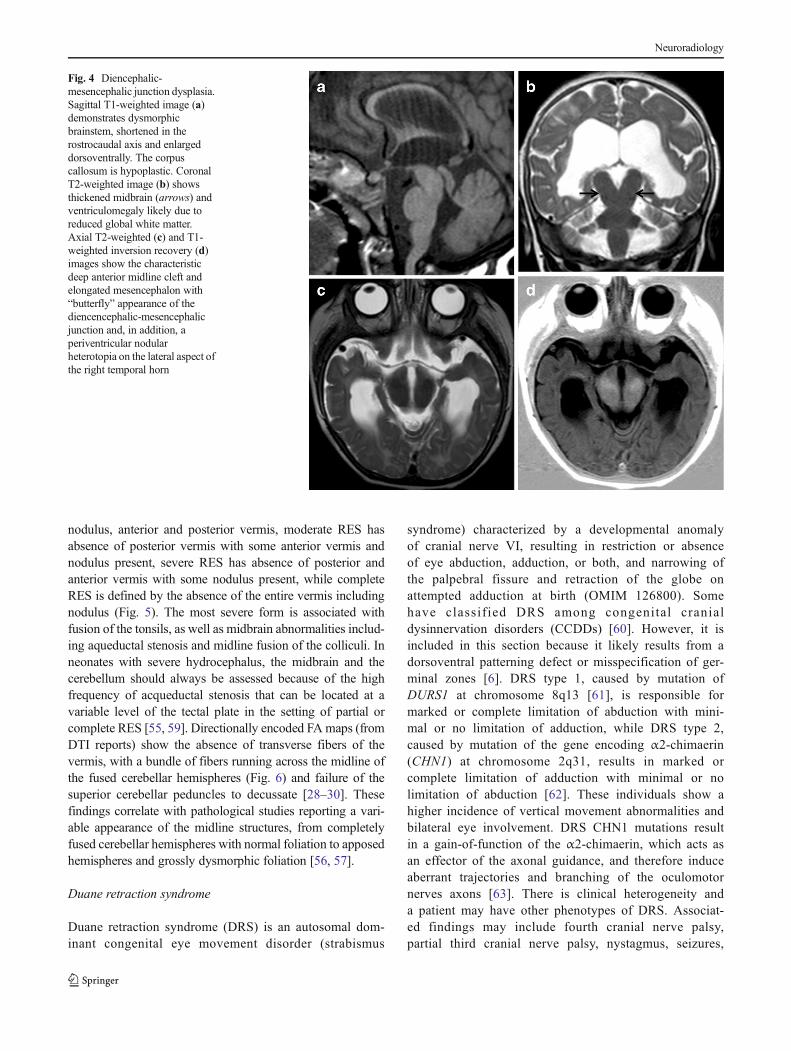
Diencephalic-mesencephalic junction dysplasia
Another malformation in this group is a newly describedentity involving the diencephalic-mesencephalic boundarycalled diencephalic-mesencephalic junction dysplasia(DMJD) [51], which is postulated to result from overexpres-sion of Pax6 in the diencephalon or underexpression of En1/Pax2 in the anterior mesencephalon. Clinical signs includepyramidal tract signs, speech and gait disturbances, autisticfeatures, and seizures.
Neuroradiology

On cMRI, the reported cases are heterogeneous, with thecommon feature being that the junction between the dienceph-alon and the midbrain is ill-defined. The midbrain appearsshortened in the A-P axis but shows an enlarged dorsoventralaxis and a ventral cleft is contiguous with the third ventricle,described by the authors as showing characteristic “butterflysign” on axial images [51] (Fig. 4). In some DMJD cases, DTIappears to show an abrupt arrest of the corticopontine andcorticospinal tracts at the level of the diencephalon as com-pared to normal controls, suggesting disturbed axonal path-finding, possibly due to misspecification or abnormal locationof guidance structures [51]. This disorder should not be mis-taken for the flattened midbrain seen in chronic intracranialhypotension [52].
Dorsoventral patterning defects
Rhombencephalosynapsis
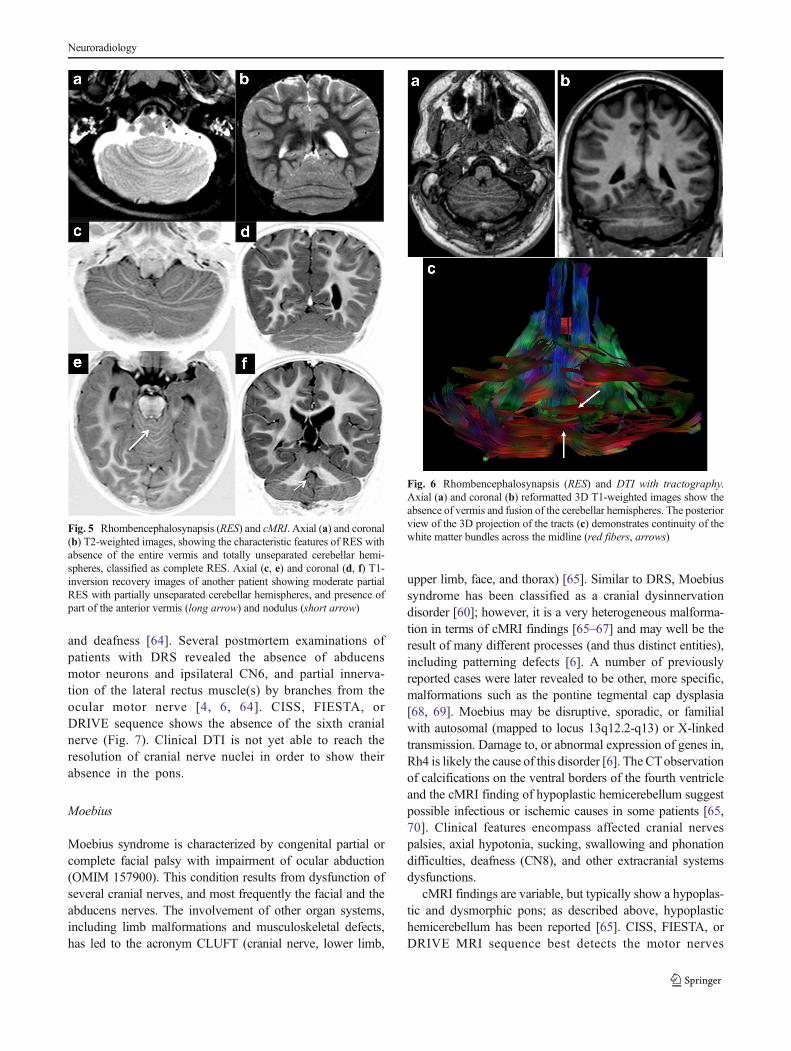
Rhombencephalosynapsis (RES) results from a presumed dor-soventral patterning defect in the rostral dorsal midline regions
of Rh1, just below the IsO and surrounding the upper fourthventricle. The molecular basis is still unknown but mutationsof Fgf8 and LMX1A have been suggested as possible causes[53]. Clinical findings are quite variable and depend upon theseverity of the cerebellar malformation and associatedsupratentorial anomalies, ranging from mild truncal ataxiaand normal cognitive abilities to severe cerebral palsy andintellectual disability. Severe hydrocephalus, acqueductal ste-nosis, and continuous colliculi across the midline were foundto be strongly correlated with poorer neurodevelopmental out-come [54–58]. Some patients have the Gomez-Lopez-Hernandez syndrome, defined as RES with parietal alopecia(consistently) and trigeminal anesthesia (variably), whileothers have features of VACTERL association [54–58].
Well-known typical cMRI features include unseparatedcerebellar hemispheres with white matter bands crossing themidline, rudimentary or absent vermis, and fused cerebellarnuclei arching in a horseshoe shape across the midline, with anarrow fourth ventricle [54] (Fig. 5). A spectrum of severityhas been recently proposed after the neuroimaging review of alarge population [55].Mild RES consists of partial absence of
Fig. 3 Anterior-posteriorpatterning defect and heavily T2-weighted DRIVE images. SagittalT2-weighted image (a) shows aslightly small pons (P) andelongated and thick medulla (m)with an abnormal pontomedullarytransition. Axial reformattedDRIVE images (b–e) revealmarked asymmetry of the ponswith abnormally fusedcochleovestibular nerves on theleft, forming a nodular structure atthe level of the left hypoplasticinternal auditory canal (b, arrow),the hypoplastic left facial nerve(arrow), and bilateral smalltrigeminal nerve (b, e)
Fig. 2 Anterior-posterior patterning defect and cMRI. Sagittal T1-weighted image (a) reveals elongation of the anterior aspect of themidbrain (arrowhead), long hypoplastic pons and thick medulla withmarkedly enlarged dorsal portion. Note the hypoplastic vermis and the
agenesis of the corpus callosum. Axial T2-weighted images (b, c) dem-onstrate the asymmetric enlarged medulla, the right cerebellar hypoplasiaand dysplasia, and the very small pons
Neuroradiology
nodulus, anterior and posterior vermis, moderate RES hasabsence of posterior vermis with some anterior vermis andnodulus present, severe RES has absence of posterior andanterior vermis with some nodulus present, while completeRES is defined by the absence of the entire vermis includingnodulus (Fig. 5). The most severe form is associated withfusion of the tonsils, as well as midbrain abnormalities includ-ing aqueductal stenosis and midline fusion of the colliculi. Inneonates with severe hydrocephalus, the midbrain and thecerebellum should always be assessed because of the highfrequency of acqueductal stenosis that can be located at avariable level of the tectal plate in the setting of partial orcomplete RES [55, 59]. Directionally encoded FAmaps (fromDTI reports) show the absence of transverse fibers of thevermis, with a bundle of fibers running across the midline ofthe fused cerebellar hemispheres (Fig. 6) and failure of thesuperior cerebellar peduncles to decussate [28–30]. Thesefindings correlate with pathological studies reporting a vari-able appearance of the midline structures, from completelyfused cerebellar hemispheres with normal foliation to apposedhemispheres and grossly dysmorphic foliation [56, 57].
Duane retraction syndrome
Duane retraction syndrome (DRS) is an autosomal dom-inant congenital eye movement disorder (strabismus
syndrome) characterized by a developmental anomalyof cranial nerve VI, resulting in restriction or absenceof eye abduction, adduction, or both, and narrowing ofthe palpebral fissure and retraction of the globe onattempted adduction at birth (OMIM 126800). Somehave classif ied DRS among congenital cranialdysinnervation disorders (CCDDs) [60]. However, it isincluded in this section because it likely results from adorsoventral patterning defect or misspecification of ger-minal zones [6]. DRS type 1, caused by mutation ofDURS1 at chromosome 8q13 [61], is responsible formarked or complete limitation of abduction with mini-mal or no limitation of adduction, while DRS type 2,caused by mutation of the gene encoding α2-chimaerin(CHN1) at chromosome 2q31, results in marked orcomplete limitation of adduction with minimal or nolimitation of abduction [62]. These individuals show ahigher incidence of vertical movement abnormalities andbilateral eye involvement. DRS CHN1 mutations resultin a gain-of-function of the α2-chimaerin, which acts asan effector of the axonal guidance, and therefore induceaberrant trajectories and branching of the oculomotornerves axons [63]. There is clinical heterogeneity anda patient may have other phenotypes of DRS. Associat-ed findings may include fourth cranial nerve palsy,partial third cranial nerve palsy, nystagmus, seizures,
Fig. 4 Diencephalic-mesencephalic junction dysplasia.Sagittal T1-weighted image (a)demonstrates dysmorphicbrainstem, shortened in therostrocaudal axis and enlargeddorsoventrally. The corpuscallosum is hypoplastic. CoronalT2-weighted image (b) showsthickened midbrain (arrows) andventriculomegaly likely due toreduced global white matter.Axial T2-weighted (c) and T1-weighted inversion recovery (d)images show the characteristicdeep anterior midline cleft andelongated mesencephalon with“butterfly” appearance of thediencencephalic-mesencephalicjunction and, in addition, aperiventricular nodularheterotopia on the lateral aspect ofthe right temporal horn
Neuroradiology
and deafness [64]. Several postmortem examinations ofpatients with DRS revealed the absence of abducensmotor neurons and ipsilateral CN6, and partial innerva-tion of the lateral rectus muscle(s) by branches from theocular motor nerve [4, 6, 64]. CISS, FIESTA, orDRIVE sequence shows the absence of the sixth cranialnerve (Fig. 7). Clinical DTI is not yet able to reach theresolution of cranial nerve nuclei in order to show theirabsence in the pons.
Moebius
Moebius syndrome is characterized by congenital partial orcomplete facial palsy with impairment of ocular abduction(OMIM 157900). This condition results from dysfunction ofseveral cranial nerves, and most frequently the facial and theabducens nerves. The involvement of other organ systems,including limb malformations and musculoskeletal defects,has led to the acronym CLUFT (cranial nerve, lower limb,
upper limb, face, and thorax) [65]. Similar to DRS, Moebiussyndrome has been classified as a cranial dysinnervationdisorder [60]; however, it is a very heterogeneous malforma-tion in terms of cMRI findings [65–67] and may well be theresult of many different processes (and thus distinct entities),including patterning defects [6]. A number of previouslyreported cases were later revealed to be other, more specific,malformations such as the pontine tegmental cap dysplasia[68, 69]. Moebius may be disruptive, sporadic, or familialwith autosomal (mapped to locus 13q12.2-q13) or X-linkedtransmission. Damage to, or abnormal expression of genes in,Rh4 is likely the cause of this disorder [6]. The CTobservationof calcifications on the ventral borders of the fourth ventricleand the cMRI finding of hypoplastic hemicerebellum suggestpossible infectious or ischemic causes in some patients [65,70]. Clinical features encompass affected cranial nervespalsies, axial hypotonia, sucking, swallowing and phonationdifficulties, deafness (CN8), and other extracranial systemsdysfunctions.
cMRI findings are variable, but typically show a hypoplas-tic and dysmorphic pons; as described above, hypoplastichemicerebellum has been reported [65]. CISS, FIESTA, orDRIVE MRI sequence best detects the motor nerves
Fig. 5 Rhombencephalosynapsis (RES) and cMRI. Axial (a) and coronal(b) T2-weighted images, showing the characteristic features of RES withabsence of the entire vermis and totally unseparated cerebellar hemi-spheres, classified as complete RES. Axial (c, e) and coronal (d, f) T1-inversion recovery images of another patient showing moderate partialRES with partially unseparated cerebellar hemispheres, and presence ofpart of the anterior vermis (long arrow) and nodulus (short arrow)
Fig. 6 Rhombencephalosynapsis (RES) and DTI with tractography.Axial (a) and coronal (b) reformatted 3D T1-weighted images show theabsence of vermis and fusion of the cerebellar hemispheres. The posteriorview of the 3D projection of the tracts (c) demonstrates continuity of thewhite matter bundles across the midline (red fibers, arrows)
Neuroradiology

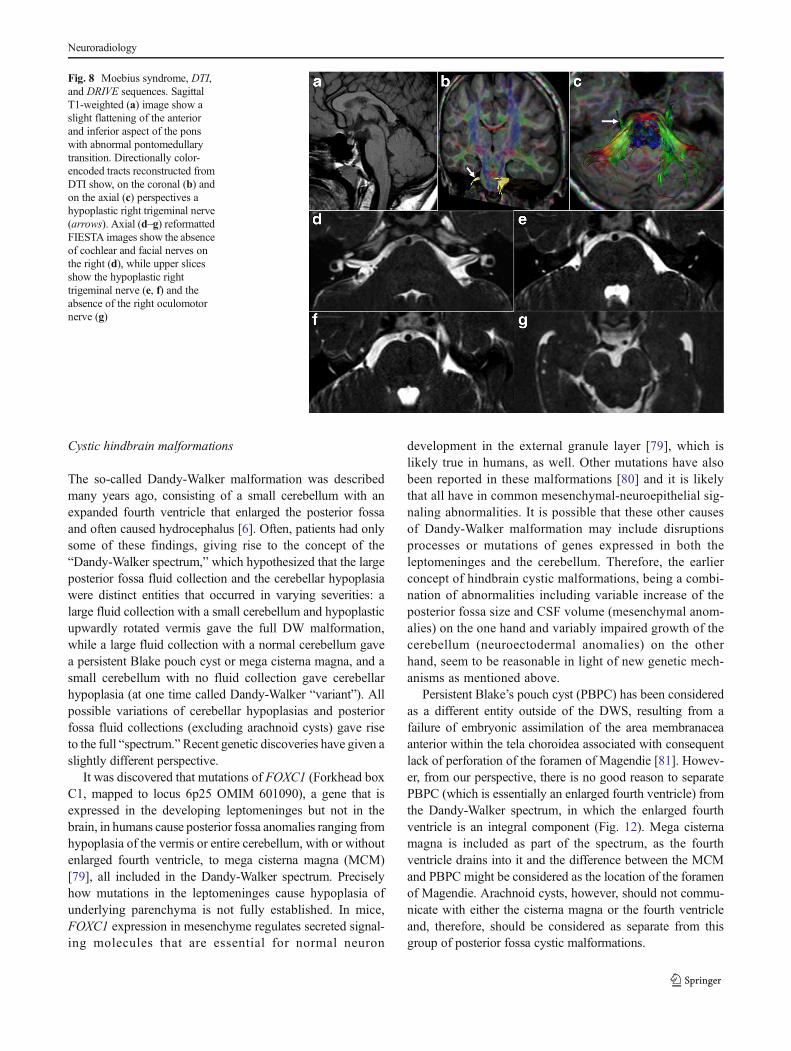
abnormalities (Fig. 8) and their consequences within the orbitsand the posterior fossa, demonstrating the absent or hypoplas-tic cisternal or intraorbital abducens nerves and aberrantintraorbital muscles innervation [67, 70]. DTI at high magnet-ic field allows tracking of some cranial nerve fibers through-out the midbrain and pons (Fig. 8); resolution is not yetsufficient to assess the thinnest nerves (CN6) or lower ones(CNs 9, 10, and 11) [71].
MHB malformations—group II
In this group are malformations that significantly affect thebrainstem and cerebellum, associated with (a) later generalizeddevelopmental encephalopathies or abnormal neuronal migra-tion that prominently affect the brainstem and the cerebellum(MHB hypoplasias), or resulting from (b) mesenchymal-neuroepithelial signaling defect involving or sparing the cere-bellum (cystic hindbrain malformations), (c) abnormal neuro-nal and glial proliferation that significantly affect the brainstemand cerebellum (Cowden syndrome and Lhermitte-Duclos,hemimegalencephaly with ipsilateral cerebellomegaly), and(d) defects in ciliary proteins (molar tooth malformations).
MHB hypoplasias
The term “hypoplasia” refers to the underdevelopment of astructure, or part of it, resulting in a size smaller than normal,regardless of whether its microarchitecture is disturbed or not.Cerebellar hypoplasia is a common finding in autopsy studiesand in clinical neuroimaging and has many causes [6]. MHBhypoplasia can be found in prenatal infections or exposure toteratogenic drugs, metabolic disorders, cystic and noncystichindbrain malformations, lissencephalies with cerebellar
hypoplasia, congenital muscular dystrophies, pontocerebellarhypoplasias, or as an isolated finding involving the vermisonly or variable amounts of the cerebellar hemispheres [2, 6,72]. A caudal displacement of the IsO likely due to overex-pression of Otx2 is expected to elongate the MB and shortenthe pons, potentially resulting in cerebellar hypoplasia thatwould particularly affect the vermis [7, 73]. PTF1A (pancreastranscription factor 1, mapped to locus 10p12.3) plays a majorrole in the patterning of the cerebellar ventricular zone wherePurkinje cells are generated as well as in specification andformation of the pancreas (OMIM 609069, [74, 75]). Inhumans, PTF1A deletions or missense mutations typicallyresult in profound cerebellar hypoplasia or near total absenceof the cerebellum with reduced pontine and PF size [6, 76, 77](Fig. 4a, b). Severe cerebellar hypoplasia might also be due toa prenatal disruption [78]. Incidental prenatal events, such asischemia, hemorrhage, or infection, may induce a disruptionof a developmental process. The spectrum of prenatal cere-bellar disruptions ranges from cerebellar agenesis or near totalabsence of the cerebellum, which is likely the most severeform, to bilateral hypoplasia or focal unilateral cerebellarhypoplasia. Therefore, unilateral cerebellar hypoplasias, iso-lated or associated with other brain or posterior fossamalformations, have to be considered first as resulting fromprenatal disruption of the cerebellar development [78]. In suchcases, along with cMRI features (Fig. 9), a 3D T1 acquisitionwith volume rendering shows a spatial topographic location ofundeveloped structures (Fig. 10). In addition, DTI enables thespecific tracking of descending white matter bundles andpontine fibers in order to better define those missing, thosemisdirected, and those preserved (Fig. 11); eventually, thistechnique may elucidate cerebrum-MHB connectivity in thiscondition.
Fig. 7 Duane retractionsyndrome (DRS), DTI, andDRIVE sequences. Sagittal T2-weighted (a) and colored FA mapfused with 3D T1-weighted (b)images show normal appearingbrainstem and tracts. Axialreformatted DRIVE sequence (c)reveals absence of the leftabducens nerve while thecontralateral is seen (arrow). Thecorpus callosum is very small
Neuroradiology
Cystic hindbrain malformations
The so-called Dandy-Walker malformation was describedmany years ago, consisting of a small cerebellum with anexpanded fourth ventricle that enlarged the posterior fossaand often caused hydrocephalus [6]. Often, patients had onlysome of these findings, giving rise to the concept of the“Dandy-Walker spectrum,” which hypothesized that the largeposterior fossa fluid collection and the cerebellar hypoplasiawere distinct entities that occurred in varying severities: alarge fluid collection with a small cerebellum and hypoplasticupwardly rotated vermis gave the full DW malformation,while a large fluid collection with a normal cerebellum gavea persistent Blake pouch cyst or mega cisterna magna, and asmall cerebellum with no fluid collection gave cerebellarhypoplasia (at one time called Dandy-Walker “variant”). Allpossible variations of cerebellar hypoplasias and posteriorfossa fluid collections (excluding arachnoid cysts) gave riseto the full “spectrum.”Recent genetic discoveries have given aslightly different perspective.
It was discovered that mutations of FOXC1 (Forkhead boxC1, mapped to locus 6p25 OMIM 601090), a gene that isexpressed in the developing leptomeninges but not in thebrain, in humans cause posterior fossa anomalies ranging fromhypoplasia of the vermis or entire cerebellum, with or withoutenlarged fourth ventricle, to mega cisterna magna (MCM)[79], all included in the Dandy-Walker spectrum. Preciselyhow mutations in the leptomeninges cause hypoplasia ofunderlying parenchyma is not fully established. In mice,FOXC1 expression in mesenchyme regulates secreted signal-ing molecules that are essential for normal neuron
development in the external granule layer [79], which islikely true in humans, as well. Other mutations have alsobeen reported in these malformations [80] and it is likelythat all have in common mesenchymal-neuroepithelial sig-naling abnormalities. It is possible that these other causesof Dandy-Walker malformation may include disruptionsprocesses or mutations of genes expressed in both theleptomeninges and the cerebellum. Therefore, the earlierconcept of hindbrain cystic malformations, being a combi-nation of abnormalities including variable increase of theposterior fossa size and CSF volume (mesenchymal anom-alies) on the one hand and variably impaired growth of thecerebellum (neuroectodermal anomalies) on the otherhand, seem to be reasonable in light of new genetic mech-anisms as mentioned above.
Persistent Blake’s pouch cyst (PBPC) has been consideredas a different entity outside of the DWS, resulting from afailure of embryonic assimilation of the area membranaceaanterior within the tela choroidea associated with consequentlack of perforation of the foramen of Magendie [81]. Howev-er, from our perspective, there is no good reason to separatePBPC (which is essentially an enlarged fourth ventricle) fromthe Dandy-Walker spectrum, in which the enlarged fourthventricle is an integral component (Fig. 12). Mega cisternamagna is included as part of the spectrum, as the fourthventricle drains into it and the difference between the MCMand PBPC might be considered as the location of the foramenof Magendie. Arachnoid cysts, however, should not commu-nicate with either the cisterna magna or the fourth ventricleand, therefore, should be considered as separate from thisgroup of posterior fossa cystic malformations.
Fig. 8 Moebius syndrome, DTI,and DRIVE sequences. SagittalT1-weighted (a) image show aslight flattening of the anteriorand inferior aspect of the ponswith abnormal pontomedullarytransition. Directionally color-encoded tracts reconstructed fromDTI show, on the coronal (b) andon the axial (c) perspectives ahypoplastic right trigeminal nerve(arrows). Axial (d–g) reformattedFIESTA images show the absenceof cochlear and facial nerves onthe right (d), while upper slicesshow the hypoplastic righttrigeminal nerve (e, f) and theabsence of the right oculomotornerve (g)
Neuroradiology

The main roles of imaging in the posterior fossa cysticmalformations are to detect hydrocephalus, if present, and todetermine the presence of brain anomalies, in particular cere-bellar dysgenesis and abnormal vermian lobulation, which isassociated with poorer neurodevelopmental outcome [82].MR midsagittal thin slices (2–3 mm) turbo spin echo T2-weighted or high-resolution steady-state sequences, possiblyassociated with MR cardiac-gated CSF flow sequences usingphase-contrast techniques [83], best demonstrate whether CSFflow is obstructed at the level of the aqueduct, at the fourthventricular outflow foramina, or elsewhere, which will deter-mine whether CSF will need to be diverted from the posteriorfossa collection as well as the lateral ventricles. These studiescan also be useful to determine the location of a posterior fossa
arachnoid cyst adjacent to the fourth ventricle, although thevast majority of posterior fossa arachnoid cysts cause noclinical signs or symptoms (Fig. 13). As mentioned above,in contrast to arachnoid cysts, the mega cisterna magna com-municates freely with the fourth ventricle through the foramenof Magendie and with the spinal subarachnoid spaces with nomembrane interposition.
Cowden syndrome and Lhermitte-Duclos disease
Lhermitte-Duclos disease (LDD) is caused by a very raremass-like cerebellar lesion alternatively named dysplastic cer-ebellar gangliocytoma. It may occur sporadically or, in nearly50 % of cases, in association with Cowden syndrome (CS),
Fig. 9 MHB hypoplasias. Thefirst patient (a, b) shows featuresof PTF1A mutation consisting innear total absence of thecerebellum and flattening of theanterior aspect of the pons withseverely hypoplastic vermis(arrow) in a small posterior fossa.The next patient (c, d) showsfeatures of prenatal cerebellardisruption, with unilateralcerebellar hypoplasia involvingthe right cerebellar hemisphere(CH) and vermis (long arrow), aswell as the ipsilateral middlecerebellar peduncle (short arrow).The third patient (e, f) shows asevere form of prenatal disruptionwith near total absence of the leftCH and severe hypoplasia of rightCH and vermis; the pons appearsthin and anteriorly flattened(arrow)
Neuroradiology

which is considered to be a phakomatosis responsible formultiple hamartomas throughout the body and caused bymutations of the PTEN gene at 10q23.31 (OMIM 158350)
[84]; it is sometimes called PTEN hamartomatous tumorsyndrome (PHTS). Affected patients have increased incidenceof thyroid (all patients), breast, and endometrial carcinoma
Fig. 10 Cerebellar hypoplasia,MRI, DTI, and 3D T1 volumerendering (VR). Coronalreformatted 3D T1 (a) andcoronal T1-inversion recovery (b)images showing left cerebellarhypoplasia; the left CH is reducedin size and its inferior aspect ismissing (long arrow), likely dueto prenatal disruption; note theobliquity of the vermis (fullyformed). This patient has inaddition a focal area of LDD(short arrow, detailed in Fig. 9).Anterior and posterior views (c,d) VR images show the brainstemright-left asymmetry due toreduced size of the left middlecerebellar peduncle. As comparedto the contralateral side, theinferior two thirds of the left CHincluding the tonsil did not form(arrow)
Fig. 11 Prenatal cerebellardisruption and DTI withtractography. Coronal (a) andsagittal (b) T2-weighted imagesshow bilateral cerebellarhypoplasia, more pronounced onthe left with marked flattening ofthe anterior aspect of the pons andsmall vermis. Note the corticalhemosiderin deposition on the leftinferior cerebellar hemisphere (a,arrows). RGB colored FA mapfused with 3D T1-weightedsagittal (c) and axial (d) images,and 3D tractography projections(e, f) reveal almost completeabsence of red transverse pontinefibers, hypoplasia of the rightmiddle cerebellar peduncle (MCP,arrows), and complete absence ofthe left MCP
Neuroradiology

(women). Dysplastic cells proliferate in the cerebellum,resulting in local areas of cellular overgrowth. A recent studyhas suggested that LDD results from activation of mammaliantarget of rapamycin (mTOR), a downstream effector in thePTEN/AKT pathway and a major regulator of cell prolifera-tion [85]. Clinically, there are no specific signs; most patientspresent with hydrocephalus or signs of local mass effect in theposterior fossa such as cranial nerve palsies, unsteadiness ofgait, ataxia, or headaches. The pathology of LD lesions re-veals enlarged, circumscribed cerebellar folia containing largeganglion cells in the granular cell layer and prominent mye-linated tracts in the outer molecular layer [6].
cMRI characteristic features include low T1- and mixedT2-signal intensities with linear striations parallel to the sur-face of the lesion (classic « tiger-striped appearance »), with orwithout local mass effect depending on the size of the areaaffected. Diminished volume of white matter, and widenedgranular cell and inner molecular layers contribute to the innerT2 hyperintensity, while an outer T2 iso/hypointense regioncomprises the outer dysplastic molecular layer and theleptomeninges; enhancement is uncommon [86]. Susceptibil-ity weighted imaging (SWI) shows abnormal venous-likestructures between the folia. MR perfusion discloses hetero-geneously increased relative cerebral blood volume (rCBV),while FDG PET studies show increased FDG uptake [87, 88].Di f fus ion weigh ted imag ing (DWI) may showhyperintensities within the lesion while ADC is decreased,likely due to cellular hyperdensity [89]. DTI demonstrates lossof anisotropy within the lesion and no connectivity withsurrounding structures (Fig. 14). Spectroscopy show de-creased choline/Cr (in contrast to astroglial tumors), dimin-ished NAA/Cr ratios, and may disclose lactate as reflecting theabnormal tissue glycosylation and neuronal dysfunction in thehamartomatous tissue [88] (Fig. 15). All these features accordwith the growing and dysplastic nature of LDD.
Hemimegalencephaly with ipsilateral cerebellomegaly
Hemimegalencephaly with ipsilateral cerebellomegaly(HME) is a rare brain malformation characterized by diffuseor focal hemispheric dysplastic enlargement often associatedwith neurocutaneous syndromes (epidermal nevus,hypomelanosis of Ito, and tuberous sclerosis). HME is thoughto result from abnormal neuronal and glial proliferation withfocal areas of overgrowth containing dysplastic cells [4,90–92]. Several very recent genetic studies have establishedthat HME can be caused by somatic mosaicism of activatingmutations of PI3K and AKT3 in the mTOR pathway [93–95].PI3K (phosphatidylinositol 3-kinase) is a signaling enzymeinvolved in a wide range of processes among which theregulation of cell growth and brain development associatedwith its predominant downstream effector, the AKT3 protein[93–95]. The reasons and clinical consequences of the
Fig. 12 Dandy-Walker spectrum. The typical appearance of DW mal-formation (DWM) includes increased posterior fossa size, dilatation of thefourth ventricle with hypoplastic vermis rotated upward, as seen in a fetus(a) (the vermian tail is always associated with the rotation, arrow) as wellas after birth (b). Other forms of DWM in the spectrum show enlargementof posterior fossa fluid spaces and widening of the fourth ventricle with asmall vermis, often called DW variant (c, in a fetus, arrow on the vermis)and after birth (d)
Fig. 13 Arachnoid cyst (AC) and 3D T2-weighted volume rendering(VR). The arachnoid cyst is a separate entity from the DWS since it doesnot communicate either with the surrounding free fluid spaces or with thefourth ventricle. A huge AC is shown on sagittal and coronal T2-wimages (a, b) with an important mass effect on posterior fossa structuresand hydrocephalus due to fourth ventricle and aqueduct compression. Onthe axial 3D CISS image (c), strips of the arachnoid membrane are visible(arrows). The VR (d) best shows the arachnoid membrane enclosing thefluid in a separate space. CSF derivation is therefore required
Neuroradiology

presence of ipsilateral brainstem and cerebellar hemisphericenlargement and dysplasia in some cases are much less un-derstood; presumably, activating mutations of different genesthat are equally active in forebrain and hindbrain are respon-sible. Whether the mid-hindbrain is involved or not, affectedpatients typically present with macrocephaly, developmentaldelay, and epilepsy [90, 91].
cMRI features encompass enlargement of all parts of onehemisphere with areas of heterogeneous high T2-signal
intensity; focal small or extensive calcification (better seenon CT); focal or diffuse cortical agyria, pachygyria, and/orpolymicrogyria for the cerebrum; and, in the posterior fossa,ipsilateral cerebellar and brainstem enlargement with abnor-mal folia pattern [90–96]. Applying DTI has permitted visu-alization of aberrant trajectories of midsagittal fibers, likelydue to the failure of callosal axons to cross the midline andmisspecification of cortical targets [97]. Further DTI studies inpatients’ harboring cerebellomegaly may help in investigatingthe corticopontine and corticocerebellar tracts as well as com-missural and pontine decussating fibers. The use of FDG-PETin patients with this condition displays the same results as inmost malformations of cortical development (MCD):hypometabolism in the areas that are abnormal on MRI [98].PET-MRI coregistration allows detection of functional meta-bolic disturbances in the cerebellum whether it is involved ornot in HME (Fig. 16).
Molar tooth malformation
The hallmark of molar tooth malformation (MTM) is a veryhypoplastic cerebellar vermis with thickened, elongated, andhorizontal superior cerebellar peduncles (SCP), resulting in a“molar tooth” appearance on axial MRI or CT images at thelower midbrain. There is a wide heterogeneity of presentationassociated, both clinically and on imaging, with this hindbrainmalformation [99, 100]. MTM is mandatory for the diagnosisof JS, which typically clinically presents with hypotonia orataxia, developmental delay, ocular motor apraxia, and breath-ing abnormalities, possibly associated with an involvement ofother organs such as the retina, kidneys, liver, and theorofacial sphere, the later relating to orofacial-digital syn-drome type VI (OFDVI). To best describe the heterogeneityof organs involvement, clinical presentations, and mutations,the term “Joubert syndrome and related disorders” (JSRD)was proposed, which include numerous syndromes, manywith anomalies of the eyes, kidneys, and distal limbs (mostcommonly polydactyl), which result from defects in ciliastructure or in cilia-localized proteins [101–103]. JSRD areassociated with mutations of 26 genes at the time of thiswriting, currently classified into several groups (OMIM213300). Other authors have suggested that MTM has nodifferential diagnoses but JS, to avoid the denomination ofJSRD and to group all the subgroups under the unique term“JS” [104]. However, we believe that this extreme simplifica-tion of the disorder does not reflect its real clinical diversity,with variable involvement of multiple organ systems resultingin very variable outcomes, as mentioned above. Clearly, theMRI phenotype ofMTM is mandatory for the diagnosis of JS,but evaluation of other organ systems and genotyping isessential for a definitive diagnosis.
The ciliary and centrosomal proteins that are affected bythe aforementioned mutations are very important in proper
Fig. 14 Lhermitte-Duclos-Cowden disease (LD-CD). Axial reformatted3D T1-w image showing a focal area of hypointense disorganized cere-bellar tissue, the classic “tiger-stripped appearance.” b Cross-section ofRGB colored FA map showing loss of anisotropy within the LDCanomaly, which seems to have no connection with the adjacent tissue,notably with the MCP fibers (arrows)
Neuroradiology
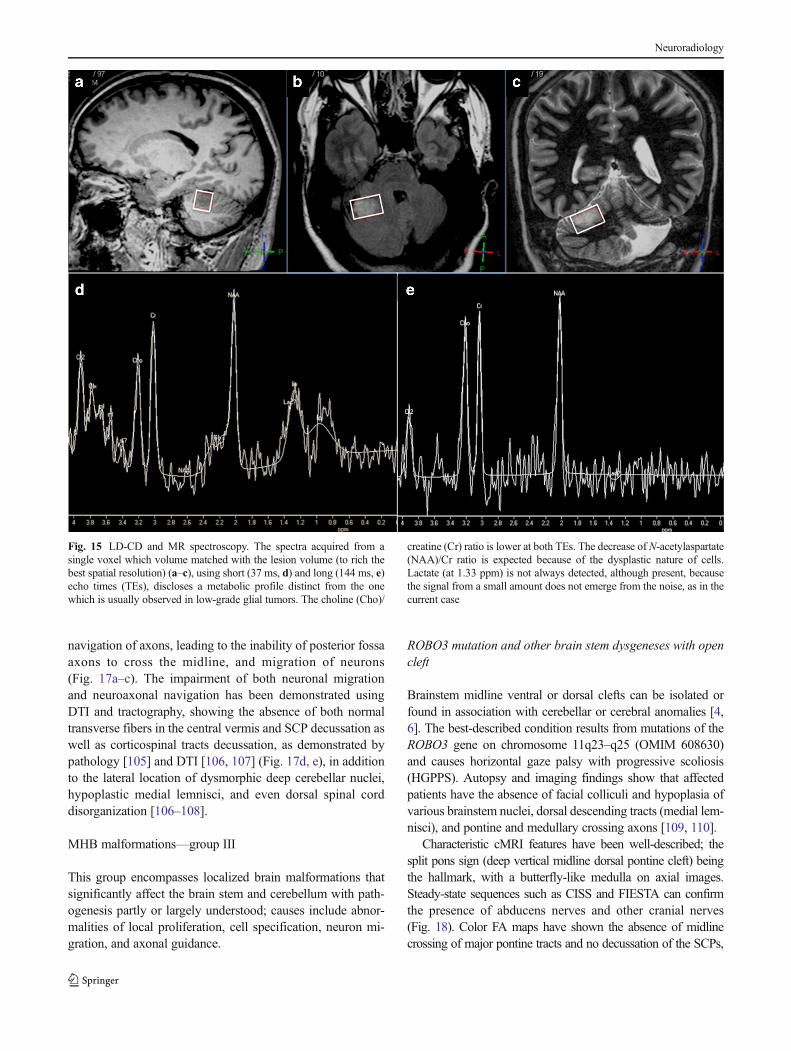
navigation of axons, leading to the inability of posterior fossaaxons to cross the midline, and migration of neurons(Fig. 17a–c). The impairment of both neuronal migrationand neuroaxonal navigation has been demonstrated usingDTI and tractography, showing the absence of both normaltransverse fibers in the central vermis and SCP decussation aswell as corticospinal tracts decussation, as demonstrated bypathology [105] and DTI [106, 107] (Fig. 17d, e), in additionto the lateral location of dysmorphic deep cerebellar nuclei,hypoplastic medial lemnisci, and even dorsal spinal corddisorganization [106–108].
MHB malformations—group III
This group encompasses localized brain malformations thatsignificantly affect the brain stem and cerebellum with path-ogenesis partly or largely understood; causes include abnor-malities of local proliferation, cell specification, neuron mi-gration, and axonal guidance.
ROBO3 mutation and other brain stem dysgeneses with opencleft
Brainstem midline ventral or dorsal clefts can be isolated orfound in association with cerebellar or cerebral anomalies [4,6]. The best-described condition results from mutations of theROBO3 gene on chromosome 11q23–q25 (OMIM 608630)and causes horizontal gaze palsy with progressive scoliosis(HGPPS). Autopsy and imaging findings show that affectedpatients have the absence of facial colliculi and hypoplasia ofvarious brainstem nuclei, dorsal descending tracts (medial lem-nisci), and pontine and medullary crossing axons [109, 110].
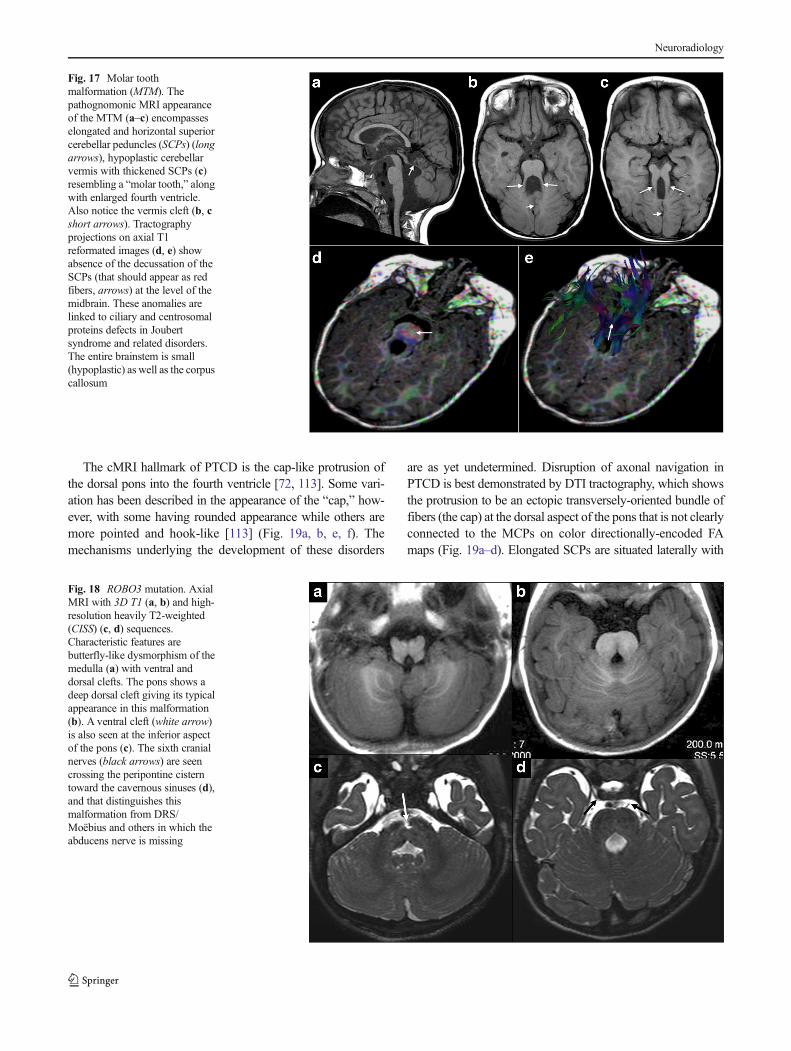
Characteristic cMRI features have been well-described; thesplit pons sign (deep vertical midline dorsal pontine cleft) beingthe hallmark, with a butterfly-like medulla on axial images.Steady-state sequences such as CISS and FIESTA can confirmthe presence of abducens nerves and other cranial nerves(Fig. 18). Color FA maps have shown the absence of midlinecrossing of major pontine tracts and no decussation of the SCPs,
Fig. 15 LD-CD and MR spectroscopy. The spectra acquired from asingle voxel which volume matched with the lesion volume (to rich thebest spatial resolution) (a–c), using short (37 ms, d) and long (144 ms, e)echo times (TEs), discloses a metabolic profile distinct from the onewhich is usually observed in low-grade glial tumors. The choline (Cho)/
creatine (Cr) ratio is lower at both TEs. The decrease of N-acetylaspartate(NAA)/Cr ratio is expected because of the dysplastic nature of cells.Lactate (at 1.33 ppm) is not always detected, although present, becausethe signal from a small amount does not emerge from the noise, as in thecurrent case
Neuroradiology

with normal-appearing ipsilateral ascending and descending con-nectivity in the brainstem. Supratentorially, normal interhemi-spheric connections via the corpus callosum are present [111,112]. Spine imaging shows progressive scoliosis but, using cur-rent clinical imaging techniques, a normal-appearing spinal cord.
Pontine tegmental cap dysplasia and other pontinedysgeneses
Pontine tegmental cap dysplasia (PTCD) has been iden-tified recently as a specific entity putatively secondary
to abnormal axonal pathfinding [69, 113]. At the timeof this writing, about 26 cases have been reported butno associated mutation has yet been disclosed. In addi-tion to facial dysmorphisms and extracranial abnormal-ities, typical clinical features include hypotonia, devel-opmental delay, ataxia, speech and hearing impairments,feeding difficulties, corneal anesthesia, and mirrormovements of the hands and feet [114]. These clinicalfeatures may cause death in the first year of life butfavorable developmental and cognitive outcomes have beenreported in three patients [115].
Fig. 16 Hemimegalencephaly with ipsilateral cerebellomegaly andcoregistrated FDG-PET-MRI 3D T1 images. From left to right (a) T1-inversion recovery (IR) and reformatted 3DT1 images show enlarged anddysplastic right cerebellar hemisphere (CH) with abnormal foliation,increased white matter volume, and dysplastic-appearing white matter.Some heterotopic gray matter is identified (arrow). The glucose uptakenative and coregistrated maps show diffuse hypometabolism of the rightCH indicating extensive cell dysfunction. b At the supratentorial level,the abnormality is limited to the right parietal lobe, which shows
thickening of the cortex with periventricular gray matter heterotopias(arrow), increased white matter, enlarged ventricle (distorted), and dimin-ished FDG uptake (arrows) correspond to parietal lobarhemimegalencephaly. c Coronal reformatted images help in viewingipsilateral parietal and cerebellar lower glucose uptake as compared tothe contralateral side (although the right cerebellar enlargement is not wellseen here because of slice orientation). d Surface mapping of thehypometabolic areas involving the right parietal lobe to thetemporoparietal junction and the right cerebellum
Neuroradiology
The cMRI hallmark of PTCD is the cap-like protrusion ofthe dorsal pons into the fourth ventricle [72, 113]. Some vari-ation has been described in the appearance of the “cap,” how-ever, with some having rounded appearance while others aremore pointed and hook-like [113] (Fig. 19a, b, e, f). Themechanisms underlying the development of these disorders
are as yet undetermined. Disruption of axonal navigation inPTCD is best demonstrated by DTI tractography, which showsthe protrusion to be an ectopic transversely-oriented bundle offibers (the cap) at the dorsal aspect of the pons that is not clearlyconnected to the MCPs on color directionally-encoded FAmaps (Fig. 19a–d). Elongated SCPs are situated laterally with
Fig. 17 Molar toothmalformation (MTM). Thepathognomonic MRI appearanceof the MTM (a–c) encompasseselongated and horizontal superiorcerebellar peduncles (SCPs) (longarrows), hypoplastic cerebellarvermis with thickened SCPs (c)resembling a “molar tooth,” alongwith enlarged fourth ventricle.Also notice the vermis cleft (b, cshort arrows). Tractographyprojections on axial T1reformated images (d, e) showabsence of the decussation of theSCPs (that should appear as redfibers, arrows) at the level of themidbrain. These anomalies arelinked to ciliary and centrosomalproteins defects in Joubertsyndrome and related disorders.The entire brainstem is small(hypoplastic) as well as the corpuscallosum
Fig. 18 ROBO3 mutation. AxialMRI with 3D T1 (a, b) and high-resolution heavily T2-weighted(CISS) (c, d) sequences.Characteristic features arebutterfly-like dysmorphism of themedulla (a) with ventral anddorsal clefts. The pons shows adeep dorsal cleft giving its typicalappearance in this malformation(b). A ventral cleft (white arrow)is also seen at the inferior aspectof the pons (c). The sixth cranialnerves (black arrows) are seencrossing the peripontine cisterntoward the cavernous sinuses (d),and that distinguishes thismalformation from DRS/Moëbius and others in which theabducens nerve is missing
Neuroradiology

no decussation, while the ventral and middle transverse pontinefibers are absent [113]. A recent report on high angular diffu-sion tensor imaging (HARDI) findings in PTCD demonstratethe presence of misoriented fibers connecting the basal pons tothe cerebellar hemispheres through the MCPs (peripontinearcuate fibers), some of which seem to join the dorsal ectopicband [116]. DTI is also useful to map the impaired anatomicalconnectivity in other pontine dysgeneses. In PTCD, CISS orFIESTA sequences may provide additional information such asthe absence or hypoplasia of the cochleovestibular and facialnerves, correlating with partial facial weakness and sensorineu-ral hearing loss (Fig. 19e–g). The presence of a normal-appearing acoustic nerve and positive auditory brainstem re-sponse testing are sometimes used to justify cochlear implan-tation, although this is controversial [117, 118].
Cerebellar dysplasia
The dysplasia may be focal or diffuse. Focal cerebellar corticaldysplasia (FCCD) is presumably due to a molecular defect
(integrin-linked kinase; ILK) or an injury (hemorrhage, virus-es, and toxins) during the proliferation or migration phases ofcerebellar development (starting at eighth week of gestationalage) with resulting disruption of cerebellar lobule morphogen-esis [119–123]. No specific causative mutation for FCCD hasbeen discovered in humans. In mice, mutant binding immu-noglobulin protein (BiP) alters the quality control in the en-doplasmic reticulum (ER), consequently affecting the expres-sion of several proteins in the Reelin pathway that are impor-tant in cerebral and cerebellar corticogenesis and, therefore,possibly resulting in FCCD [124, 125]. We distinguish thisentity from diffuse cerebellar dysplasias (DCD) associatedwith cobblestone complex lissencephalies and other musculardystrophy syndromes [6]. In this regard, GPR56-related cob-blestone phenotype with bilateral frontoparietalpolymicrogyria (BFPP) has been described as caused by mu-tations of GPR56, a protein involved in cell-cell and cell-extracellular matrix interactions [126]. In addition, diffusecerebellar dysplasia is also seen in patients with Chudley-McCollough syndrome (CMS) and GPSM2 mutations. These
Fig. 19 Pontine tegmental cap dysplasia (PTCD) and DTI tractography(a–d). a Sagittal T2-weighted image showing the dorsal cap (arrow)protruding into the fourth ventricle. b On the coronal reformatted 3DT1 image, the superior cerebellar peduncles (SCP) do not decussate,shifting from the midline (short arrows), while the dorsal band (DB)crosses the midline posteriorly (long arrow) to join the middle cerebellarpeduncles (MCP). Tractography (c, d reprinted with permission, posteriorviews) shows reduced corticospinal tracts as compared to normal withvery small MCPs, no crossing pontine fibers (which would appear red,
running from green SCP to green SCP; SCPs were not seeded in this view(d)) and the right-left DB. PTCD and axial FIESTA images (e–g). SomeMCP fibers that appear red distally on (d) might correspond to misori-entation of fibers connecting the basal pons to cerebellar hemispheres, asdemonstrated in a recent report [116]. Another case with similar typicalMRI features as the previous, on sagittal T1 (e, arrow) and coronal Flair(f, arrows). Axial and oblique reformatted FIESTA images (g) show theabsence of the acoustic and facial nerves on the right (R), hypoplastic onthe left (L). A anterior
Neuroradiology
patients have a generalized brain dysgenesis with dysmorphiccorpus callosum, dysmorphic cerebral hemispheres (sulcationabnormalities and heterotopia), as well as dysgenesis of de-veloping meninges (arachnoid cysts) and dysgenesis of thecerebellum. All of which could result from the abnormalleptomeningeal development.
FCCD typically shows normal cerebrum or very mildabnormalities such as white matter hypomyelination or dys-morphic corpus callosum [127] while DCD shows diffuseremodeling of the cerebellum including heterotopia, PMG,and cysts, as seen in cobblestone phenotypes [128].
FCCD is often disclosed when investigating children withlearning disabilities, sometimes associated with behavioraldisturbances [129]. The clinical spectrum of FCCD is veryheterogeneous and some children may have a normal neuro-psychological profile and education course, independent onthe location or the extent of the malformation [130, 131].
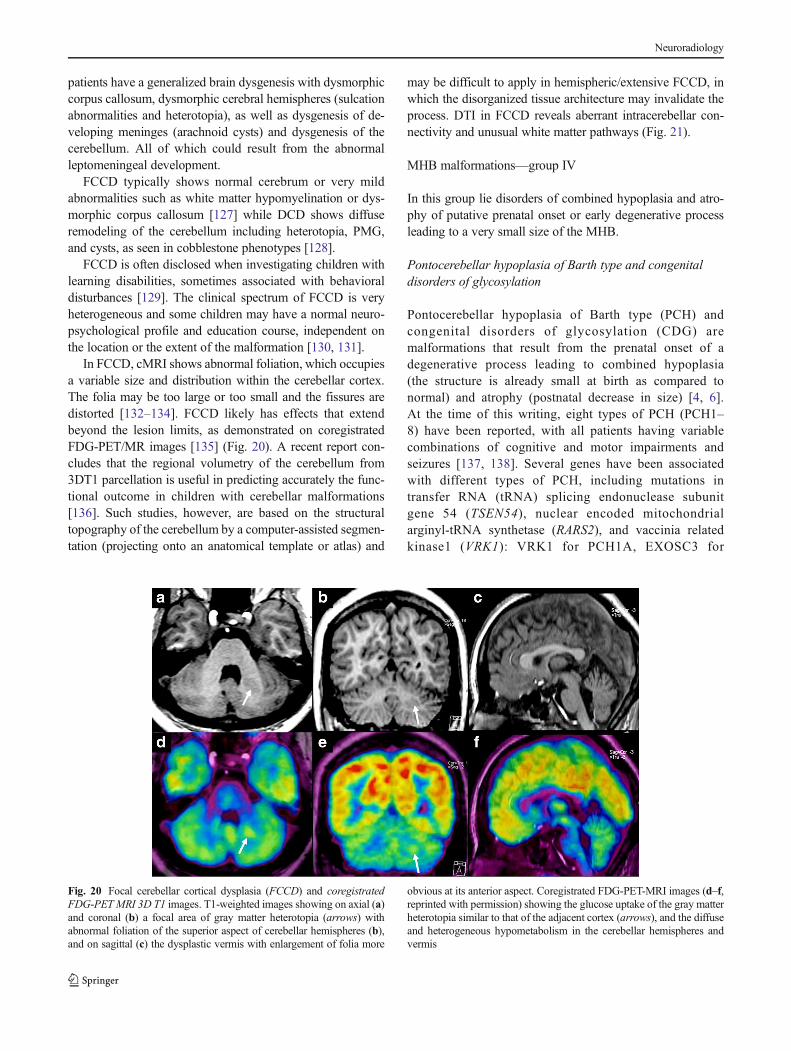
In FCCD, cMRI shows abnormal foliation, which occupiesa variable size and distribution within the cerebellar cortex.The folia may be too large or too small and the fissures aredistorted [132–134]. FCCD likely has effects that extendbeyond the lesion limits, as demonstrated on coregistratedFDG-PET/MR images [135] (Fig. 20). A recent report con-cludes that the regional volumetry of the cerebellum from3DT1 parcellation is useful in predicting accurately the func-tional outcome in children with cerebellar malformations[136]. Such studies, however, are based on the structuraltopography of the cerebellum by a computer-assisted segmen-tation (projecting onto an anatomical template or atlas) and
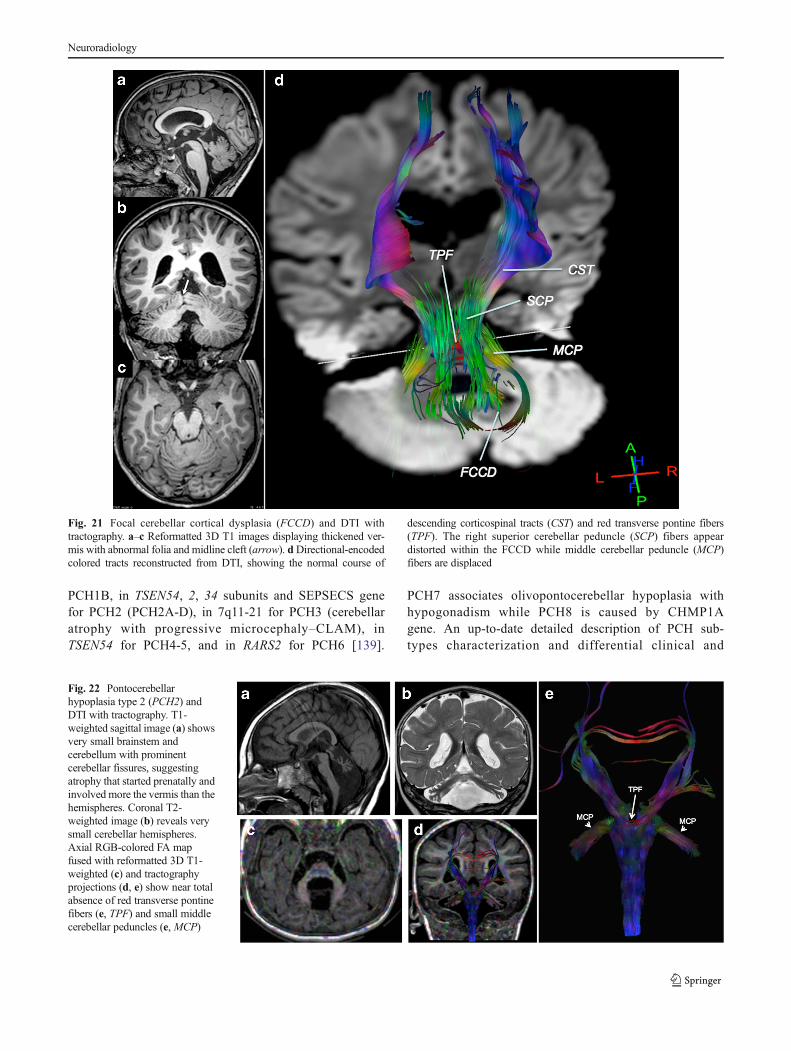
may be difficult to apply in hemispheric/extensive FCCD, inwhich the disorganized tissue architecture may invalidate theprocess. DTI in FCCD reveals aberrant intracerebellar con-nectivity and unusual white matter pathways (Fig. 21).
MHB malformations—group IV
In this group lie disorders of combined hypoplasia and atro-phy of putative prenatal onset or early degenerative processleading to a very small size of the MHB.
Pontocerebellar hypoplasia of Barth type and congenitaldisorders of glycosylation
Pontocerebellar hypoplasia of Barth type (PCH) andcongenital disorders of glycosylation (CDG) aremalformations that result from the prenatal onset of adegenerative process leading to combined hypoplasia(the structure is already small at birth as compared tonormal) and atrophy (postnatal decrease in size) [4, 6].At the time of this writing, eight types of PCH (PCH1–8) have been reported, with all patients having variablecombinations of cognitive and motor impairments andseizures [137, 138]. Several genes have been associatedwith different types of PCH, including mutations intransfer RNA (tRNA) splicing endonuclease subunitgene 54 (TSEN54), nuclear encoded mitochondrialarginyl-tRNA synthetase (RARS2), and vaccinia relatedkinase1 (VRK1): VRK1 for PCH1A, EXOSC3 for
Fig. 20 Focal cerebellar cortical dysplasia (FCCD) and coregistratedFDG-PET MRI 3D T1 images. T1-weighted images showing on axial (a)and coronal (b) a focal area of gray matter heterotopia (arrows) withabnormal foliation of the superior aspect of cerebellar hemispheres (b),and on sagittal (c) the dysplastic vermis with enlargement of folia more
obvious at its anterior aspect. Coregistrated FDG-PET-MRI images (d–f,reprinted with permission) showing the glucose uptake of the gray matterheterotopia similar to that of the adjacent cortex (arrows), and the diffuseand heterogeneous hypometabolism in the cerebellar hemispheres andvermis
Neuroradiology
PCH1B, in TSEN54, 2, 34 subunits and SEPSECS genefor PCH2 (PCH2A-D), in 7q11-21 for PCH3 (cerebellaratrophy with progressive microcephaly–CLAM), inTSEN54 for PCH4-5, and in RARS2 for PCH6 [139].
PCH7 associates olivopontocerebellar hypoplasia withhypogonadism while PCH8 is caused by CHMP1Agene. An up-to-date detailed description of PCH sub-types characterization and differential clinical and
Fig. 21 Focal cerebellar cortical dysplasia (FCCD) and DTI withtractography. a–c Reformatted 3D T1 images displaying thickened ver-mis with abnormal folia and midline cleft (arrow). dDirectional-encodedcolored tracts reconstructed from DTI, showing the normal course of
descending corticospinal tracts (CST) and red transverse pontine fibers(TPF). The right superior cerebellar peduncle (SCP) fibers appeardistorted within the FCCD while middle cerebellar peduncle (MCP)fibers are displaced
Fig. 22 Pontocerebellarhypoplasia type 2 (PCH2) andDTI with tractography. T1-weighted sagittal image (a) showsvery small brainstem andcerebellum with prominentcerebellar fissures, suggestingatrophy that started prenatally andinvolved more the vermis than thehemispheres. Coronal T2-weighted image (b) reveals verysmall cerebellar hemispheres.Axial RGB-colored FA mapfused with reformatted 3D T1-weighted (c) and tractographyprojections (d, e) show near totalabsence of red transverse pontinefibers (e, TPF) and small middlecerebellar peduncles (e, MCP)
Neuroradiology
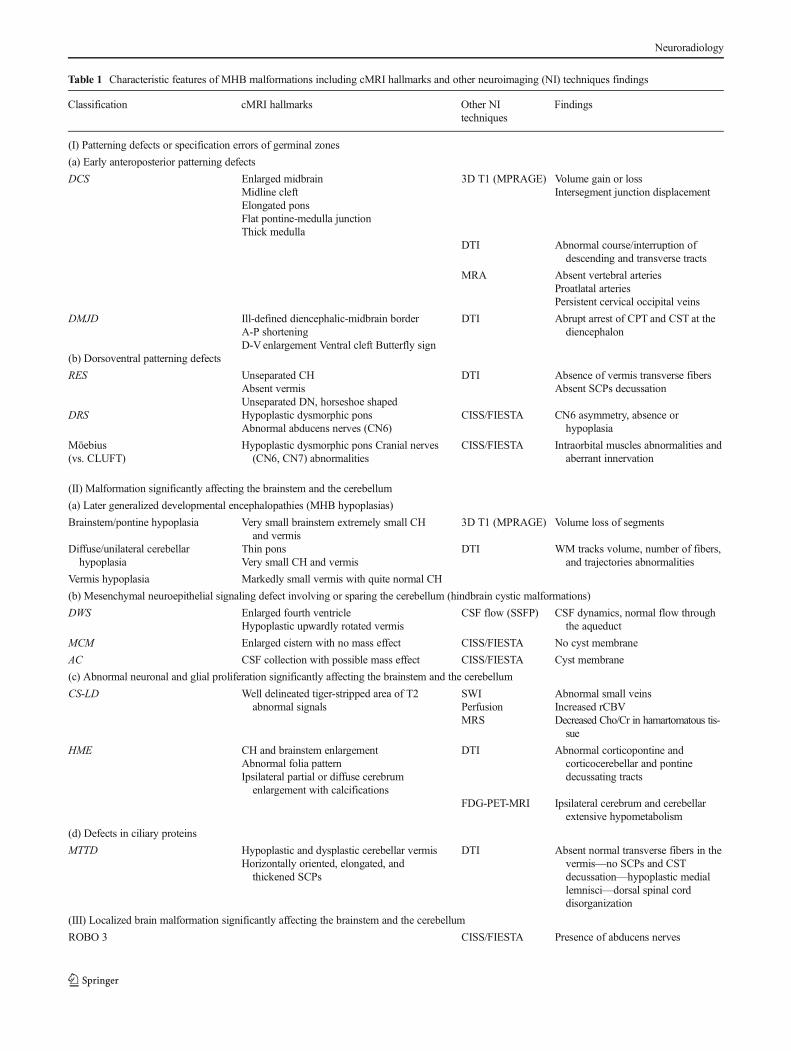
Table 1 Characteristic features of MHB malformations including cMRI hallmarks and other neuroimaging (NI) techniques findings
Classification cMRI hallmarks Other NItechniques
Findings
(I) Patterning defects or specification errors of germinal zones
(a) Early anteroposterior patterning defects
DCS Enlarged midbrainMidline cleftElongated ponsFlat pontine-medulla junctionThick medulla
3D T1 (MPRAGE) Volume gain or lossIntersegment junction displacement
DTI Abnormal course/interruption ofdescending and transverse tracts
MRA Absent vertebral arteriesProatlatal arteriesPersistent cervical occipital veins
DMJD Ill-defined diencephalic-midbrain borderA-P shorteningD-Venlargement Ventral cleft Butterfly sign
DTI Abrupt arrest of CPT and CST at thediencephalon
(b) Dorsoventral patterning defects
RES Unseparated CHAbsent vermisUnseparated DN, horseshoe shaped
DTI Absence of vermis transverse fibersAbsent SCPs decussation
DRS Hypoplastic dysmorphic ponsAbnormal abducens nerves (CN6)
CISS/FIESTA CN6 asymmetry, absence orhypoplasia
Möebius(vs. CLUFT)
Hypoplastic dysmorphic pons Cranial nerves(CN6, CN7) abnormalities
CISS/FIESTA Intraorbital muscles abnormalities andaberrant innervation
(II) Malformation significantly affecting the brainstem and the cerebellum
(a) Later generalized developmental encephalopathies (MHB hypoplasias)
Brainstem/pontine hypoplasia Very small brainstem extremely small CHand vermis
3D T1 (MPRAGE) Volume loss of segments
Diffuse/unilateral cerebellarhypoplasia
Thin ponsVery small CH and vermis
DTI WM tracks volume, number of fibers,and trajectories abnormalities
Vermis hypoplasia Markedly small vermis with quite normal CH
(b) Mesenchymal neuroepithelial signaling defect involving or sparing the cerebellum (hindbrain cystic malformations)
DWS Enlarged fourth ventricleHypoplastic upwardly rotated vermis
CSF flow (SSFP) CSF dynamics, normal flow throughthe aqueduct
MCM Enlarged cistern with no mass effect CISS/FIESTA No cyst membrane
AC CSF collection with possible mass effect CISS/FIESTA Cyst membrane
(c) Abnormal neuronal and glial proliferation significantly affecting the brainstem and the cerebellum
CS-LD Well delineated tiger-stripped area of T2abnormal signals
SWIPerfusionMRS
Abnormal small veinsIncreased rCBVDecreased Cho/Cr in hamartomatous tis-sue
HME CH and brainstem enlargementAbnormal folia patternIpsilateral partial or diffuse cerebrumenlargement with calcifications
DTI Abnormal corticopontine andcorticocerebellar and pontinedecussating tracts
FDG-PET-MRI Ipsilateral cerebrum and cerebellarextensive hypometabolism
(d) Defects in ciliary proteins
MTTD Hypoplastic and dysplastic cerebellar vermisHorizontally oriented, elongated, andthickened SCPs
DTI Absent normal transverse fibers in thevermis—no SCPs and CSTdecussation—hypoplastic mediallemnisci—dorsal spinal corddisorganization
(III) Localized brain malformation significantly affecting the brainstem and the cerebellum
ROBO 3 CISS/FIESTA Presence of abducens nerves
Neuroradiology

imaging phenotypes can be found in a recent paper byRudnik-Schöneborn et al [138].
CDG type 1a (CDG1A) is the best known carbohydrate-deficient glycoprotein (CDG) syndrome and is caused bymutations in the gene encoding phosphomannomutase-2(PMM2 mapped on locus 16p13.2–OMIM 212065), an en-zyme that acts in the synthesis of glycoproteins criticallyinvolved in multiple functions including metabolism, cellrecognition and adhesion, cell migration, protease resistance,host defense, and antigenicity [140].
PCH and CDG share characteristic cMRI features:hypoplasia/atrophy of MHB structures (Fig. 22), progressivemicrocephaly, and variable cerebral involvement, mainly char-acterized by progressive atrophy [141]. The most recognizableMRI phenotype of TSEN-related PCH is the so-called “drag-onfly” appearance (highly suggestive of TSEN54), resultingfrom the more severe involvement of the cerebellar hemi-spheres than the vermis, while PCH8 shows a severe atrophyof the vermis [138, 141]. Metabolic imaging using PET andspectroscopy may help in assessing the biochemical underly-ing disturbances. The 3D T1 acquisition can help in defining a3Dmorphological model of the differentMRI phenotypes, andmay permit the study of the relationship of alterations ofsupratentorial and subcortical structures by means of segmen-tation. As in other disorders mentioned above, DTI has poten-tial to study alterations of white matter pathways. In a recent
paper, Poretti et al. [72] reported on colored FA maps thepresence of an ectopic band of fibers at the dorsal aspect ofthe pons with reduced size of the middle cerebellar pedunclesin a POMGnT1 mutation muscle-eye-brain disease (Fig. 22).
Microcephalies with predominantly postnatal cerebellarhypoplasia
Two disorders have been described recently in whichpostmigrational microcephaly is accompanied by cerebellarand pontine hypoplasia that becomes more disproportionatelyhypoplastic over the first 6 months after birth. These disordersare caused by mutations of CASK at Xp11.4 [142] andCHMP1A at 16q24.3 [143]. CASK is involved in the regula-tion of gene expression, including Reelin, which is critical inbrain development [144] while CHMP1A a regulator of BMI1,which is a key regulator of stem cell renewal, and seems toparticularly affect cerebellar granule cell quantity [143].
Affected patients in both groups have low normal headcircumference at birth that progress significantly in earlyinfancy. Neurodevelopment, including motor and cognitivefunctions, is poor. Patients (mainly girls) with CASK muta-tions have frequent ophthalmologic anomalies including reti-nopathy, optic atrophy, and glaucoma [145]. In both groups,pontocerebellar hypoplasia is severe, with vermis and hemi-spheres fairly equally affected. The corpus callosum
Table 1 (continued)
Classification cMRI hallmarks Other NItechniques
Findings
(HGPPS and related syndromes) Split pons sign (deep midline dorsalpontine cleft)
Butterfly-like dysmorphism of the medullaDTI Absence of midline crossing of major
pontine tracts—no decussation ofthe SCPs
PTCD Cap sign (cap-like protrusion of the dorsalpons into the fourth ventricle)
Pontine WM bundle crossing the midlinedorsally
SCPs shift from the midline
DTI Dorsal transverse fibers within the capjoining the MCPs
No SCPs decussationAbsent transverse pontine fibers
CISS/FIESTA Absence or hypoplasia of thecochleovestibular and facial nerves
FCCD Localized or diffuse abnormal foliation/fissuration
DTI Aberrant intracerebellar connectivity—unusual white matter pathways
FDG-PET-MRI Glucose uptake changes that mayextend beyond the lesion limits
(IV) Combined hypoplasia and atrophy of putative prenatal onset (degenerative)
PCH–CDGand MPPCH–CASK
Hypoplasia/atrophy of MHB structuresProgressive atrophy*
3D T1 (MPRAGE) Segmental hypoplasia/atrophy
DTI Absent transverse pontine fibers andother white matter alterations
CH cerebellar hemispheres, DN dentate nuclei, CN cranial nerve, SCPs superior cerebellar peduncles, WM white matter. A-P anterior-posterior, D-Vdorso-ventral, CPT corticopontine tract, CST corticospinal tract. Asterisk please refer to the corresponding section in the text for more details
Neuroradiology

sometimes looks disproportionately large in girls with CASKmutations [146]. DTI may be useful in this condition todemonstrate abnormalities of pontine tracts as well ascerebellocortical connecting fibers.
Discussion and future directions
We have reviewed several of the more common MHBmalformations, according to a developmental and geneticclassification scheme recently proposed [6], with a particularattention to the contribution of advanced brain imaging tech-niques to better identify and understand the underlyingmalformative processes. We have not addressed the resultsof functional studies (fMRI) using BOLD techniques in anysection due to the fact that experiments reported in the currentliterature involved only very few of the entities discussedherein, namely cerebellar hypoplasia, often associated withautism spectrum [147, 148], attention deficits (ADHD) [149,150], and developmental dyslexia [151, 152]. The functionaltopography of the human cerebellum has been proposed [153,154]. However, the relationship between the anatomical loca-tion and type of MHB malformations and specific brain dys-functions is still unclear, and is therefore unpredictable. Theclinical impact of cerebellar malformations on brain functioninvolves motor skills (ranging from fine movements distur-bances to spastic quadriparesis) and cognition (from languageimpairment to autism spectrum), the latter known as“dysmetria of thought” or “cerebellar cognitive affective syn-drome” [155-157]. Thus, functional brain imaging in MHBmalformations is a largely unexplored field.
The continuing development of neuroimaging, the deliveryof new advanced techniques, and the early progress in highmagnetic field human imaging in research (4.7, 7, and 9 T)and clinical (3 T) settings have made possible reaching a levelof morphological and functional imaging close to the micro-scopic structure of the brain [156]. This development is verypromising for assessing further the disorders of brain forma-tion and, particularly, ofMHB genesis. Combining the knowl-edge of MHB embryology with advanced neuroimaging tech-niques may help in optimizing the protocol for any malforma-tion suspected on a clinical basis (Table 1). Recognition ofspecific MRI phenotypes, including advanced imaging fea-tures, is useful to recognize theMHBmalformation entities, tosuggest genetic investigations, and, eventually, to monitor thedisease outcome after supportive therapies.
Key points
& A classification ofMHBmalformations on the genetic andmolecular basis of embryology is helpful for the radiolo-gist to make the correct diagnosis on MRI.
& When genes mutations are known to cause specific MRIfeatures, the genetic analyses can be suggested from therecognition of these MRI phenotypes.
& Except in cases related to specific gene mutations, anyMHBmalformation can result either from a genemutationor a disruptive process such as infection, toxic or meta-bolic injury, trauma, and exposure to teratogenoussubstances.
& A multimodal MRI approach of MHB malformations,including notably the DTI technique, is the best way tounderstand which developmental phases failed.
Acknowledgments The authors are grateful to Mazen Ahmar (MD,Radiology, Alzahra Hospital, Sharjah, (UAE) for providing us withFig. 18.
Ethical standards and patient consent We declare that this manu-script does not contain clinical studies or patient data.
Conflict of interest We declare that we have no conflict of interest.
References
1. Niesen CE (2002) Malformations of the posterior fossa: currentperspectives. Semin Pediatr Neurol 9(4):320–34
2. Patel S, Barkovich AJ (2002) Analysis and classification of cere-bellar malformations. AJNR Am J Neuroradiol 23:1074–1087
3. Parisi MA, Dobyns WB (2003) Human malformations of the mid-brain and hindbrain: review and proposed classification scheme.Mol Genet Metab 80(1–2):36–53
4. Barkovich AJ, Millen KJ, Dobyns WB (2007) A developmentalclassification of malformations of the brainstem. Ann Neurol 62(6):625–39
5. Jissendi-Tchofo P, Kara S, Barkovich AJ (2009) Midbrain-hindbrain involvement in lissencephalies. Neurology 3;72(5):410–8
6. BarkovichAJ,Millen KJ, DobynsWB (2009) A developmental andgenetic classification for midbrain-hindbrain malformations. Brain132(Pt 12):3199–230
7. Chizhikov V, Millen KJ (2003) Development and malformations ofthe cerebellum in mice. Mol Genet Metab 80(1–2):54–65
8. Dastjerdi FV, Consalez GG, Hawkes R (2012) Pattern formationduring development of the embryonic cerebellum. Front Neuroanat6:10
9. Xenaki D, Martin IB, Yoshida L, Ohyama K, Gennarini G, GrumetM, Sakurai T, Furley AJ (2011) F3/contactin and TAG1 play antag-onistic roles in the regulation of sonic hedgehog-induced cerebellargranule neuron progenitor proliferation. Development 138(3):519–29
10. Fink AJ, Englund C, Daza RA, Pham D, Lau C, Nivison M,Kowalczyk T, Hevner RF (2006) Development of the deep cerebel-lar nuclei: transcription factors and cell migration from the rhombiclip. J Neurosci 15;26(11):3066–76
11. Bloch-Gallego E, Causeret F, Ezan F, Backer S, Hidalgo-SánchezM(2005) Development of precerebellar nuclei: instructive factors andintracellular mediators in neuronal migration, survival and axonpathfinding. Brain Res Brain Res Rev 49(2):253–66
12. Sgaier SK, Millet S, Villanueva MP, Berenshteyn F, Song C, JoynerAL (2005) Morphogenetic and cellular movements that shape themouse cerebellum; insights from genetic fate mapping. Neuron6;45(1):27–40
Neuroradiology

13. Frayne R, Goodyear BG, Dickhoff P, Lauzon ML, Sevick RJ(2003) Magnetic resonance imaging at 30 Tesla: challenges andadvantages in clinical neurological imaging. Invest Radiol38(7):385–402
14. Runge VM, Case RS, Sonnier HL (2006) Advances in clinical 3-tesla neuroimaging. Invest Radiol 41(2):63–7
15. Moseley ME, Liu C, Rodriguez S, Brosnan T (2009) Advances inmagnetic resonance neuroimaging. Neurol Clin 27(1):1–19
16. Ment LR, Bada HS, Barnes P, Grant PE, Hirtz D, Papile LA, Pinto-Martin J, Rivkin M, Slovis TL (2002) Practice parameter: neuroim-aging of the neonate: report of the Quality Standards Subcommitteeof the American Academy of Neurology and the PracticeCommittee of the Child Neurology Society. Neurology 58(12):1726–38
17. Zimmerman RA, Bilaniuk LT, Pollock AN, Feygin T, Zarnow D,Schwartz ES, Harris C (2006) 3.0 T versus 1.5 T pediatric brainimaging. Neuroimaging Clin N Am 16(2):229–39
18. Hoon AH Jr, Melhem ER (2000) Neuroimaging: applications indisorders of early brain development. J Dev Behav Pediatr 21(4):291–302
19. Mugler JP III, Brookeman JR (1991) Rapid three-dimensional T1-weighted MR imaging with the MP-RAGE sequence. J MagnReson Imaging 1:561–567
20. Mugler JP III, Spraggins TA, Brookeman JR (1991) T2-weightedthree-dimensional MP-RAGEMR imaging. J Magn Reson Imaging1:731–737
21. Conklin J, Winter JD, Thompson RT, Gelman N (2008) High-contrast 3D neonatal brain imaging with combined T1- and T2-weighted MP-RAGE. Magn Reson Med 59(5):1190–6
22. Srinivasan L, Dutta R, Counsell SJ, Allsop JM, Boardman JP,Rutherford MA, Edwards AD (2007) Quantification of deep graymatter in preterm infants at term-equivalent age using manualvolumetry of 3-tesla magnetic resonance images. Pediatrics119(4):759–65
23. Prastawa M, Gilmore JH, Lin W, Gerig G (2005) Automatic seg-mentation of MR images of the developing newborn brain. MedImage Anal 9:457–466
24. Barkovich AJ (2005) Pediatric Neuroimaging, 4th edn. LippincottWilliams & Wilkins, Philadelphia, pp 294–339
25. Wedeen VJ, Hagmann P, Tseng WY, Reese TG, Weisskoff RM(2005) Mapping complex tissue architecture with diffusion spec-trum magnetic resonance imaging. Magn Reson Med 54(6):1377–86
26. Tuch DS (2004) Q-ball imaging. Magn Reson Med 52(6):1358–7227. Mukherjee P, McKinstry RC (2006) Diffusion tensor imaging and
tractography of human brain development. Neuroimaging Clin NAm 16(1):19–43
28. Widjaja E, Blaser S, Raybaud C (2006) Diffusion tensor imaging ofmidline posterior fossa malformations. Pediatr Radiol 36(6):510–7
29. Wahl M, Barkovich AJ, Mukherjee P (2010) Diffusion imaging andtractography of congenital brain malformations. Pediatr Radiol40(1):59–67
30. Poretti A, Meoded A, Rossi A, Raybaud C, Huisman TA (2013)Diffusion tensor imaging and fiber tractography in brainmalformations. Pediatr Radiol 43(1):28–54
31. Patil S, Biassoni L, Borgwardt L (2007) Nuclear medicine inpediatric neurology and neurosurgery: epilepsy and brain tumors.Semin Nucl Med 37(5):357–81
32. Cha S (2003) Perfusion MR imaging: basic principles and clinicalapplications. Magn Reson Imaging Clin N Am 11(3):403–13
33. Cha S (2006) Dynamic susceptibility-weighted contrast-enhancedperfusion MR imaging in pediatric patients. Neuroimaging Clin NAm 16(1):137–47
34. Ivancevic MK, Geerts L, Weadock WJ, Chenevert TL (2009)Technical principles of MR angiography methods. Magn ResonImaging Clin N Am 17(1):1–11
35. Wang J, Licht DJ (2006) Pediatric perfusion MR imagingusing arterial spin labeling. Neuroimaging Clin N Am 16(1):149–67
36. Sehgal V, Delproposto Z, Haacke EM, Tong KA, Wycliffe N, KidoDK, Xu Y, Neelavalli J, Haddar D, Reichenbach JR (2005) Clinicalapplications of neuroimaging with susceptibility-weighted imaging.J Magn Reson Imaging 22(4):439–50
37. Niwa T, de Vries LS, Benders MJ, Takahara T, Nikkels PG,Groenendaal F (2011) Punctate white matter lesions in infants:new insights using susceptibi l i ty-weighted imaging.Neuroradiology 53(9):669–79
38. Shi Y, Jin RB, Zhao JN, Tang SF, Li HQ, Li TY (2009) Brainpositron emission tomography in preterm and term newborn infants.Early Hum Dev 85(7):429–32
39. Leach JL, Holland SK (2010) Functional MRI in children: clinicaland research applications. Pediatr Radiol 40(1):31–49
40. Vigneron DB (2006) Magnetic resonance spectroscopic imaging ofhuman brain development. Neuroimaging Clin N Am 16(1):75–85
41. Xu D, Vigneron D (2010) Magnetic resonance spectroscopy imag-ing of the newborn brain: a technical review. Semin Perinatol 34(1):20–7
42. Mamourian AC, Miller G (1994) Neonatal pontomedullary discon-nection with aplasia or destruction of the lower brain stem: a case ofpontoneocerebellar hypoplasia. AJNR Am J Neuroradiol 15(8):1483–5, 49-53
43. Barth PG, de Vries LS, Nikkels PG, Troost D (2008) Congenitalbrainstem disconnection associated with a syrinx of the brainstem.Neuropediatrics 39(1):1–7
44. Poretti A, Boltshauser E, Plecko B (2007) Brainstem disconnection:case report and review of the literature. Neuropediatrics 38(4):210–2
45. Sarnat HB, Benjamin DR, Siebert JR, Kletter GB, CheyetteSR (2002) Agenesis of the mesencephalon and metencepha-lon with cerebellar hypoplasia: putative mutation in the EN2gene: report of 2 cases in early infancy. Pediatr Dev Pathol5(1):54–68
46. Scholpp S, Lohs C, Brand M (2003) Engrailed and Fgf8 act syner-gistically to maintain the boundary between diencephalon and mes-encephalon. Development 130(20):4881–93
47. Duffield C, Jocson J, Wootton-Gorges SL (2009) Brainstem dis-connection. Pediatr Radiol 39(12):1357–60
48. Jurkiewicz E, Dobrzańska A, Nowak K, Pleskaczyńska (2010) AMRI findings in the young infant with brainstem disconnection andextracerebral features Report of one case and review of the litera-ture. Brain Dev 32(6):495–8
49. Okumura A, Lee T, Shimojima K, Hisata K, Shoji H, Takanashi J,Yamamoto T, Shimizu T, Barkovich AJ (2009) Brainstem discon-nection associated with nodular heterotopia and proatlantal arteries.Am J Med Genet A 149A(11):2479–83
50. Kumar R, Macey PM, Woo MA, Alger JR, Harper RM (2008)Diffusion tensor imaging demonstrates brainstem and cerebellarabnormalities in congenital central hypoventilation syndrome.Pediatr Res 64(3):275–80
51. Zaki MS, Saleem SN, Dobyns WB, Barkovich AJ, Bartsch H, DaleAM, Ashtari M, Akizu N, Gleeson JG, Grijalvo-Perez AM (2012)Diencephalic-mesencephalic junction dysplasia: a novel recessivebrain malformation. Brain 135(Pt 8):2416–27
52. Savoiardo M, Minati L, Farina L, De Simone T, Aquino D, Mea E,Filippini G, Bussone G, Chiapparini L (2007) Spontaneous intra-cranial hypotension with deep brain swelling. Brain 130(Pt 7):1884–93
53. Chizhikov VV, Millen KJ (2005) Roof plate-dependent patterningof the vertebrate dorsal central nervous system. Dev Biol 277(2):287–95
54. Toelle SP, Yalcinkaya C, Kocer N, Deonna T, Overweg-PlandsoenWC, Bast T, Kalmanchey R, Barsi P, Schneider JF, CaponeMori A,
Neuroradiology

Boltshauser E (2002) Rhombencephalosynapsis: clinical findingsand neuroimaging in 9 children. Neuropediatrics 33:209–214
55. Ishak GE, Dempsey JC, Shaw DW, Tully H, Adam MP, Sanchez-Lara PA, Glass I, Rue TC, Millen KJ, Dobyns WB, Doherty D(2012) Rhombencephalosynapsis: a hindbrain malformation asso-ciated with incomplete separation of midbrain and forebrain, hydro-cephalus and a broad spectrum of severity. Brain 135(Pt 5):1370–86
56. McAuliffe F, Chitayat D, Halliday W, Keating S, Shah V, Fink M,N e v o O , Ry a n G , S h a n n o n P, B l a s e r S ( 2 0 0 8 )Rhombencephalosynapsis: prenatal imaging and autopsy findings.Ultrasound Obstet Gynecol 31(5):542–8
57. Pasquier L, Marcorelles P, Loget P, Pelluard F, Carles D, Perez MJ,Bendavid C, de La Rochebrochard C, Ferry M, David V, Odent S,Laquerrière A (2009) Rhombencephalosynapsis and related anom-alies: a neuropathological study of 40 fetal cases. Acta Neuropathol117(2):185–200
58. Sukhudyan B, Jaladyan V,MelikyanG, Schlump JU, Boltshauser E,Poretti A (2010) Gómez-López-Hernández syndrome: reappraisalof the diagnostic criteria. Eur J Pediatr 169(12):1523–8
59. Whitehead MT, Choudhri AF, Grimm J, Nelson MD (2014)Rhombencephalosynapsis as a cause of aqueductal stenosis: anunder-recognized association in hydrocephalic children. PediatrRadiol 44(7):849–56
60. Engle EC (2007) Oculomotility disorders arising from disruptionsin brainstem motor neuron development. Arch Neurol 64(5):633–7
61. Calabrese G, Telvi L, Capodiferro F, Morizio E, Pizzuti A, StuppiaL, Bordoni R, Ion A, Fantasia D, Mingarelli R, Palka G (2000)Narrowing the Duane syndrome critical region at chromosome 8q13down to 40 kb. Eur J Hum Genet 8:319–324
62. Appukuttan B, Gillanders E, Juo SH, Freas-Lutz D, Ott S, Sood R,Van Auken A, Bailey-Wilson J, Wang X, Patel RJ, Robbins CM,Chung M, Annett G, Weinberg K, Borchert MS, Trent JM,Brownstein MJ, Stout JT (1999) Localization of a gene for Duaneretraction syndrome to chromosome 2q31. Am J Hum Genet 65(6):1639–46
63. Engle EC (2010) Human genetic disorders of axon guidance. ColdSpring Harb Perspect Biol 2(3):a001784
64. Khan AO, Oystreck D (2006) Clinical characteristics of bilateralDuane syndrome. J AAPOS 10(3):198–201
65. Abramson DL, Cohen M, Mulliken JB (1998) Moëbius syndrome:classification and grading system. Plast Reconstr Surg 102:961–967
66. Verzijl HT, Valk J, de Vries R, Padberg GW (2005) Radiologicevidence for absence of the facial nerve in Möbius syndrome.Neurology 64(5):849–55
67. Pedraza S, Gámez J, Rovira A, Zamora A, Grive E, Raguer N,Ruscalleda J (2000) MRI findings in Möbius syndrome: correlationwith clinical features. Neurology 55(7):1058–60
68. Ouanounou S, Saigal G, Birchansky S (2005) Möbius syndrome.AJNR Am J Neuroradiol 26(2):430–2
69. Barth PG, Majoie CB, Caan MW, Weterman MA, Kyllerman M,Smit LM, Kaplan RA, Haas RH, Baas F, Cobben JM, Poll-The BT(2007) Pontine tegmental cap dysplasia: a novel brain malformationwith a defect in axonal guidance. Brain 130(Pt 9):2258–66
70. Denis D, Dauletbekov D, Girard N (2008) Duane retraction syn-drome: type II with severe abducens nerve hypoplasia on magneticresonance imaging. J AAPOS 12(1):91–3
71. Hodaie M, Quan J, Chen DQ (2010) In vivo visualization of cranialnerve pathways in humans using diffusion-based tractography.Neurosurgery 66(4):788–95
72. Poretti A, Boltshauser E, Doherty D (2014) Cerebellar hypoplasia:differential diagnosis and diagnostic approach. Am J Med Genet C:Semin Med Genet 166C(2):211–26
73. Barkovich AJ (2012) Developmental disorders of the midbrain andhindbrain. Front Neuroanat 6:7
74. Broccoli V, Boncinelli E, Wurst W (1999) The caudal limit of Otx2expression positions the isthmic organizer. Nature 401(6749):164–8
75. Sellick GS, Barker KT, Stolte-Dijkstra I, Fleischmann C, ColemanRJ, Garrett C, Gloyn AL, Edghill EL, Hattersley AT, Wellauer PK,Goodwin G, Houlston RS (2004) Mutations in PTF1A cause pan-creatic and cerebellar agenesis. Nat Genet 36(12):1301–5
76. Hoshino M, Nakamura S, Mori K, Kawauchi T, Terao M,Nishimura YV, Fukuda A, Fuse T, Matsuo N, Sone M, WatanabeM, Bito H, Terashima T, Wright CV, Kawaguchi Y, Nakao K,Nabeshima Y (2005) Ptf1a, a bHLH transcriptional gene, definesGABAergic neuronal fates in cerebellum. Neuron 47(2):201–13
77. Millen KJ, Steshina EY, Iskusnykh IY, Chizhikov VV (2014)Transformation of the cerebellum into more ventral brainstem fatescauses cerebellar agenesis in the absence of Ptf1a function. ProcNatl Acad Sci USA 111(17):E1777–86
78. Poretti A, Prayer D, Boltshauser E (2009) Morphological spectrumof prenatal cerebellar disruptions. Eur J Paediatr Neurol 13(5):397–407
79. Aldinger KA, LehmannOJ, Hudgins L, Chizhikov VV, BassukAG,Ades LC, Krantz ID, Dobyns WB, Millen KJ (2009) FOXC1 isrequired for normal cerebellar development and is a major contrib-utor to chromosome 6p253Dandy-Walker malformation. Nat Genet41(9):1037–42
80. Calabrò F, Arcuri T, Jinkins JR (2000) Blake's pouch cyst: an entitywithin the Dandy-Walker continuum. Neuroradiology 42(4):290–5
81. Yildiz H, Yazici Z, Hakyemez B, Erdogan C, Parlak M (2006)Evaluation of CSF flow patterns of posterior fossa cysticmalformations using CSF flow MR imaging. Neuroradiology48(9):595–605
82. Boddaert N, Klein O, Ferguson N, Sonigo P, Parisot D, Hertz-Pannier L, Baraton J, Emond S, Simon I, Chigot V, Schmit P,Pierre-Kahn A, Brunelle F (2003) Intellectual prognosis of theDandy-Walker malformation in children: the importance of vermianlobulation. Neuroradiology 45(5):320–4
83. Robinson S, Cohen AR (2000) Cowden disease and Lhermitte-Duclos disease: characterization of a new phakomatosis.Neurosurgery 46(2):371–83
84. Abel TW, Baker SJ, Fraser MM, Tihan T, Nelson JS, Yachnis AT,Bouffard JP, Mena H, Burger PC, Eberhart CG (2005) Lhermitte-Duclos disease: a report of 31 cases with immunohistochemicalanalysis of the PTEN/AKT/mTOR pathway. J Neuropathol ExpNeurol 64(4):341–9
85. Kulkantrakorn K, Awwad EE, Levy B, Selhorst JB, Cole HO,Leake D, Gussler JR, Epstein AD, Malik MM (1997) MRI inLhermitte-Duclos disease. Neurology 48(3):725–31
86. Thomas B, Krishnamoorthy T, Radhakrishnan VV, Kesavadas C(2007) Advanced MR imaging in Lhermitte-Duclos disease: mov-ing closer to pathology and pathophysiology. Neuroradiology 49(9):733–8
87. Padma MV, Jacobs M, Sequeira P, Adineh M, Satter M, Kraus G,Mantil JC (2004) Functional imaging in Lhermitte-Duclos disease.Mol Imaging Biol 6(5):319–23
88. Klisch J, Juengling F, Spreer J, Koch D, Thiel T, Büchert M, ArnoldS, Feuerhake F, Schumacher M (2001) Lhermitte-Duclos disease:assessment with MR imaging, positron emission tomography,single-photon emission CT, and MR spectroscopy. AJNR Am JNeuroradiol 22(5):824–30
89. Moonis G, Ibrahim M, Melhem ER (2004) Diffusion-weightedMRI in Lhermitte-Duclos disease: report of two cases.Neuroradiology 46(5):351–4
90 . Sene r RN (1997 ) MR demons t r a t i on o f ce r eb r a lhemimegalencephaly associated with cerebellar involvement (totalhemimegalencephaly). Comput Med Imaging Graph 21(3):201–4
91. Flores-Sarnat L (2002) Hemimegalencephaly: part 1 genetic, clini-cal, and imaging aspects. J Child Neurol 17(5):373–84
92. Flores-Sarnat L, Sarnat HB, Dávila-Gutiérrez G, Alvarez A (2003)Hemimegalencephaly: part 2 Neuropathology suggests a disorder ofcellular lineage. J Child Neurol 18(11):776–85
Neuroradiology

93. Finding of Rare Disease Genes (FORGE) Canada Consortium,Rivière JB, Mirzaa GM, O'Roak BJ, Beddaoui M, Alcantara D,Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, NikkelSM, Worthylake T, Sullivan CT, Ward TR, Butler HE, Kramer NA,Albrecht B, Armour CM, Armstrong L, Caluseriu O, CytrynbaumC, Drolet BA, Innes AM, Lauzon JL, Lin AE, Mancini GM,Meschino WS, Reggin JD, Saggar AK, Lerman-Sagie T, UyanikG, Weksberg R, Zirn B, Beaulieu CL, Majewski J, Bulman DE,O'Driscoll M, Shendure J, Graham JM Jr, Boycott KM, DobynsWB (2012) De novo germline and postzygotic mutations in AKT3,PIK3R2 and PIK3CA cause a spectrum of related megalencephalysyndromes. Nat Genet 44(8):934–40
94. Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T,Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, FunariV, Russ C, Gabriel SB, Mathern GW, Gleeson JG (2012) Denovo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet 44(8):941–5
95. Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, LehtinenMK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, BourgeoisBF, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, Walsh CA(2012) Somatic activation of AKT3 causes hemispheric develop-mental brain malformations. Neuron 74(1):41–8
96. Sato N, Yagishita A, Oba H, Miki Y, Nakata Y, Yamashita F,Nemoto K, Sugai K, Sasaki M (2007) Hemimegalencephaly: astudy of abnormalities occurring outside the involved hemisphere.AJNR Am J Neuroradiol 28(4):678–82
97. Sato N, Ota M, Yagishita A, Miki Y, Takahashi T, Adachi Y, NakataY, Sugai K, Sasaki M (2008) Aberrant midsagittal fiber tracts inpatients with hemimegalencephaly. AJNR Am J Neuroradiol 29(4):823–7
98. Colombo N, Salamon N, Raybaud C, Ozkara C, Barkovich AJ(2009) Imaging of malformations of cortical development.Epileptic Disord 11(3):194–205
99. Parisi MA (2009) Clinical and molecular features of Joubert syn-drome and related disorders. Am JMed Genet C: SeminMed Genet151C(4):326–40
100. Bolduc ME, Limperopoulos C (2009) Neurodevelopmental out-comes in children with cerebellar malformations: a systematic re-view. Dev Med Child Neurol 51(4):256–67
101. Goetz SC, Anderson KV (2010) The primary cilium: a signal-ling centre during vertebrate development. Nat Rev Genet11(5):331–44
102. Sattar S, Gleeson JG (2011) The ciliopathies in neuronal develop-ment: a clinical approach to investigation of Joubert syndrome andJoubert syndrome-related disorders. Dev Med Child Neurol 53(9):793–8
103. Valente EM, Dallapiccola B, Bertini E (2013) Joubert syndrome andrelated disorders. Handb Clin Neurol 113:1879–88
104. Romani M, Micalizzi A, Valente EM (2013) Joubert syndrome:congenital cerebellar ataxia with the molar tooth. Lancet Neurol12(9):894–905
105. Friede RL, Boltshauser E (1978) Uncommon syndromes of cere-bellar vermis aplasia. I: Joubert syndrome. Dev Med Child Neurol20(6):758–63
106. Lee SK, Kim DI, Kim J, Kim DJ, Kim HD, Kim DS,Mori S (2005)Diffusion-tensorMR imaging and fiber tractography: a new methodof describing aberrant fiber connections in developmental CNSanomalies. Radiographics 25(1):53–65
107. Poretti A, Boltshauser E, Loenneker T, Valente EM, Brancati F,Il'Yasov K, Huisman TAGM (2007) Diffusion tensor imaging inJoubert Syndrome. AJNR 28:1929–1933
108. Juric-Sekhar G, Adkins J, Doherty D, Hevner RF (2012) Joubertsyndrome: brain and spinal cord malformations in genotyped casesand implications for neurodevelopmental functions of primary cilia.Acta Neuropathol 123(5):695–709
109. Rossi A, Catala M, Biancheri R, Di Comite R, Tortori-Donati P (2004)MR imaging of brain-stem hypoplasia in horizontal gaze palsy withprogressive scoliosis. AJNR Am J Neuroradiol 25(6):1046–8
110. Bomfim RC, Távora DG, Nakayama M, Gama RL (2009)Horizontal gaze palsy with progressive scoliosis: CT and MRfindings. Pediatr Radiol 39(2):184–7
111. Sicotte NL, Salamon G, Shattuck DW, Hageman N, Rüb U,Salamon N, Drain AE, Demer JL, Engle EC, Alger JR, BalohRW, Deller T, Jen JC (2006) Diffusion tensor MRI shows abnormalbrainstem crossing fibers associated with ROBO3 mutations.Neurology 67(3):519–21
112. Avadhani A, Ilayaraja V, Shetty AP, Rajasekaran S (2010) Diffusiontensor imaging in horizontal gaze palsy with progressive scoliosis.Magn Reson Imaging 28(2):212–6
113. Jissendi-Tchofo P, Doherty D, McGillivray G, Hevner R, Shaw D,Ishak G, Leventer R, Barkovich AJ (2009) Pontine tegmental capdysplasia: MR imaging and diffusion tensor imaging features ofimpaired axonal navigation. AJNR Am J Neuroradiol 30(1):113–9
114. Rudaks LI, Patel S, Barnett CP (2012) Novel clinical features inpontine tegmental cap dysplasia. Pediatr Neurol 46(6):393–6
115. CBCD Study Group, Briguglio M, Pinelli L, Giordano L, FerrarisA, Germanò E, Micheletti S, Severino M, Bernardini L, Loddo S,Tortorella G, Ormitti F, Gasparotti R, Rossi A, Valente EM (2011)Pontine tegmental cap dysplasia: developmental and cognitive out-come in three adolescent patients. Orphanet J Rare Dis 6:36
116. Caan MW, Barth PG, Niermeijer JM, Majoie CB, Poll-The BT(2014) Ectopic peripontine arcuate fibres, a novel finding in pontinetegmental cap dysplasia. Eur J Paediatr Neurol 18(3):434–8
117. Bacciu A, Ormitti F, Pasanisi E, Vincenti V, Zanetti D, Bacciu S(2010) Cochlear implantation in pontine tegmental cap dysplasia.Int J Pediatr Otorhinolaryngol 74(8):962–6
118. Desai NK, Young L, Miranda MA, Kutz JW Jr, Roland PS, BoothTN (2011) Pontine tegmental cap dysplasia: the neurotologic per-spective. Otolaryngol Head Neck Surg 145(6):992–8
119. ten Donkelaar HJ, Lammens M, Wesseling P, Thijssen HO, RenierWO (2003) Development and developmental disorders of the hu-man cerebellum. J Neurol 250:1025–1036
120. Ten Donkelaar HJ, Lammens M (2009) Development of the humancerebellum and its disorders. Clin Perinatol 36(3):513–30
121. Carletti B, Rossi F (2008) Neurogenesis in the cerebellum.Neuroscientist 14(1):91–100
122. Manto MU, Jissendi P (2012) Cerebellum: links between develop-ment, developmental disorders and motor learning. Front Neuroanat6:1
123. Demaerel P (2002) Abnormalities of cerebellar foliation andfissuration: classification, neurogenetics and clinicoradiologicalcorrelations. Neuroradiology 44(8):639–46
124. Mimura N, Yuasa S, Soma M, Jin H, Kimura K, Goto S, Koseki H,Aoe T (2008) Altered quality control in the endoplasmic reticulumcauses cortical dysplasia in knock-in mice expressing a mutant BiP.Mol Cell Biol 28(1):293–301
125. Miyata T, Ono Y, Okamoto M, Masaoka M, Sakakibara A,Kawaguchi A, Hashimoto M, Ogawa M (2010) Migration, earlyaxonogenesis, and reelin-dependent layer-forming behavior ofearly/posterior-born Purkinje cells in the developing mouse lateralcerebellum. Neural Dev 5:23
126. Quattrocchi CC, Zanni G, Napolitano A, Longo D, Cordelli DM,Barresi S, Randisi F, Valente EM, Verdolotti T, Genovese E,Specchio N, Vitiello G, Spiegel R, Bertini E, Bernardi B (2013)Conventional magnetic resonance imaging and diffusion tensorimaging studies in children with novel GPR56 mutations: furtherdelineation of a cobblestone-like phenotype. Neurogenetics 14(1):77–83
127. Demaerel P (2002) Abnormalities of cerebellar foliation andfissuration: classification, neurogenetics and clinicoradiologicalcorrelations. Neuroradiology 44(8):639–46,15
Neuroradiology

128. Devisme L, Bouchet C, Gonzalès M, Alanio E, Bazin A, BessièresB, Bigi N, Blanchet P, Bonneau D, BonnièresM, Bucourt M, CarlesD, Clarisse B, Delahaye S, Fallet-Bianco C, Figarella-Branger D,Gaillard D, Gasser B, Delezoide AL, Guimiot F, Joubert M, LaurentN, Laquerrière A, Liprandi A, Loget P, Marcorelles P, Martinovic J,Menez F, Patrier S, Pelluard F, Perez MJ, Rouleau C, Triau S, Attié-Bitach T, Vuillaumier-Barrot S, Seta N, Encha-Razavi F (2012)Cobblestone lissencephaly: neuropathological subtypes and corre-lations with genes of dystroglycanopathies. Brain 135(Pt 2):469–82
129. Misciagna S (2011) Cerebellar contribution to cognitive, emotional,and behavioural functions in children with cerebellar abnormalities.Dev Med Child Neurol 53(12):1075–6
130. Jissendi-Tchofo P, Pandit F, Soto-Ares G, Vallee L (2011)Neuropsychological evaluation and follow-up of children with cere-bellar cortical dysplasia. Dev Med Child Neurol 53(12):1119–27
131. Bolduc ME, Du Plessis AJ, Sullivan N, Khwaja OS, Zhang X,Barnes K, Robertson RL, Limperopoulos C (2011) Spectrum ofneurodevelopmental disabilities in children with cerebellarmalformations. Dev Med Child Neurol 53(5):409–16
132. Soto-Ares G, Delmaire C, Deries B, Vallee L, Pruvo JP (2000)Cerebellar cortical dysplasia: MR findings in a complex entity.AJNR Am J Neuroradiol 21(8):1511–9
133. Demaerel P, Lagae L, Casaer P, Baert AL (1998) MR of cerebellarcortical dysplasia. AJNR Am J Neuroradiol 19(5):984–6
134. Soto-Ares G, Devisme L, Jorriot S, Deries B, Pruvo JP, RuchouxMM (2002) Neuropathologic and MR imaging correlation in aneonatal case of cerebellar cortical dysplasia. AJNR Am JNeuroradiol 23(7):1101–4
135. Jissendi-Tchofo P, Pandit F, Vallée L, Vinchon M, Pruvo JP,Baleriaux D, Soto Ares G (2012) Brain regional glucose uptakechanges in isolated cerebellar cortical dysplasia: qualitative assess-ment using coregistrated FDG-PET/MRI. Cerebellum 11(1):280–8
136. Bolduc ME, du Plessis AJ, Sullivan N, Guizard N, Zhang X,Robertson RL, Limperopoulos C (2012) Regional cerebellar vol-umes predict functional outcome in children with cerebellarmalformations. Cerebellum 11(2):531–42
137. PCHConsortium, Namavar Y, Barth PG, Kasher PR, van Ruissen F,Brockmann K, Bernert G, Writzl K, Ventura K, Cheng EY, FerrieroDM, Basel-Vanagaite L, Eggens VR, Krägeloh-Mann I, DeMeirleirL, King M, Graham JM Jr, von Moers A, Knoers N, Sztriha L,Korinthenberg R, Dobyns WB, Baas F, Poll-The BT (2011)Clinical, neuroradiological and genetic findings in pontocerebellarhypoplasia. Brain 134(Pt 1):143–56
138. Rudnik-Schöneborn S, Barth PG, Zerres K (2014) Pontocerebellarhypoplasia. Am J Med Genet C: Semin Med Genet 166(2):173–83
139. Maricich SM, Aqeeb KA, Moayedi Y, Mathes EL, Patel MS,Chi tayat D, Lyon G, Leroy JG, Zoghbi HY (2011)Pontocerebellar hypoplasia: review of classification and genetics,and exclusion of several genes known to be important for cerebellardevelopment. J Child Neurol 26(3):288–94
140. Di Costanzo S, Balasubramanian A, Pond HL, Rozkalne A,Pantaleoni C, Saredi S, Gupta VA, Sunu CM, Yu TW, Kang PB,Salih MA, Mora M, Gussoni E, Walsh CA, Manzini MC (2014)POMK mutations disrupt muscle development leading to a spec-trum of neuromuscular presentations. Hum Mol Genet. Jun 11
141. Namavar Y, Chitayat D, Barth PG, van Ruissen F, de Wissel MB,Poll-The BT, Silver R, Baas F (2011) TSEN54 mutations causepontocerebellar hypoplasia type 5. Eur J Hum Genet 19(6):724–6
142. Najm J, Horn D, Wimplinger I, Golden JA, Chizhikov VV, Sudi J,Christian SL, Ullmann R, Kuechler A, Haas CA, Flubacher A,Charnas LR, Uyanik G, Frank U, Klopocki E, Dobyns WB,Kutsche K (2008) Mutations of CASK cause an X-linked brainmalformation phenotype with microcephaly and hypoplasia of thebrainstem and cerebellum. Nat Genet 40(9):1065–7
143. Mochida GH, Ganesh VS, de Michelena MI, Dias H, Atabay KD,Kathrein KL, Huang HT, Hill RS, Felie JM, Rakiec D, Gleason D,Hill AD, Malik AN, Barry BJ, Partlow JN, Tan WH, Glader LJ,Barkovich AJ, Dobyns WB, Zon LI, Walsh CA (2012) CHMP1Aencodes an essential regulator of BMI1-INK4A in cerebellar devel-opment. Nat Genet 44(11):1260–4
144. Förster E, Bock HH, Herz J, Chai X, Frotscher M, Zhao S(2010) Emerging topics in Reelin function. Eur J Neurosci31(9):1511–8
145. Burglen L, Chantot-Bastaraud S, Garel C, Milh M, Touraine R,Zanni G, Petit F, Afenjar A, Goizet C, Barresi S, Coussement A,Ioos C, Lazaro L, Joriot S, Desguerre I, Lacombe D, des Portes V,Bertini E, Siffroi JP, de Villemeur TB, Rodriguez D (2012)Spectrum of pontocerebellar hypoplasia in 13 girls and boys withCASK mutations: confirmation of a recognizable phenotype andfirst description of a male mosaic patient. Orphanet J Rare Dis 7:18
146. Takanashi J, Arai H, Nabatame S, Hirai S, Hayashi S, Inazawa J,Okamoto N, Barkovich AJ (2010) Neuroradiologic features ofCASK mutations. AJNR Am J Neuroradiol 31(9):1619–22
147. Courchesne E (1997) Brainstem, cerebellar and limbic neuroana-tomical abnormalities in autism. Curr Opin Neurobiol 7(2):269–78
148. Philip RC, Dauvermann MR, Whalley HC, Baynham K, LawrieSM, Stanfield AC (2012) A systematic review and meta-analysis ofthe fMRI investigation of autism spectrum disorders. NeurosciBiobehav Rev 36(2):901–42
149. Frodl T, Skokauskas N (2012) Meta-analysis of structural MRIstudies in children and adults with attention deficit hyperactivitydisorder indicates treatment effects. Acta Psychiatr Scand 125(2):114–26
150. Paloyelis Y, Mehta MA, Kuntsi J, Asherson P (2007) FunctionalMRI in ADHD: a systematic literature review. Expert RevNeurother 7(10):1337–56
151. Stoodley CJ, Stein JF (2011) The cerebellum and dyslexia. Cortex47(1):101–16
152. Shaywitz BA, Lyon GR, Shaywitz SE (2006) The role of functionalmagnetic resonance imaging in understanding reading and dyslexia.Dev Neuropsychol 30(1):613–32
153. Stoodley CJ, Schmahmann JD (2010) Evidence for topographicorganization in the cerebellum of motor control versus cognitiveand affective processing. Cortex 46(7):831–44
154. Buckner RL, Krienen FM, Castellanos A, Diaz JC, Yeo BT (2011)The organization of the human cerebellum estimated by intrinsicfunctional connectivity. J Neurophysiol 106(5):2322–45
155. Tavano A, Borgatti R (2010) Evidence for a link among cognition,language and emotion in cerebellar malformations. Cortex 46:907–18
156. Prats-Galino A, Soria G, de Notaris M, Puig J, Pedraza S (2012)Functional anatomy of subcortical circuits issuing from or integrat-ing at the human brainstem. Clin Neurophysiol 123(1):4–12
157. Schmahmann JD (2004) Disorders of the cerebellum: ataxia,dysmetria of thought, and the cerebellar cognitive affective syn-drome. J Neuropsychiatry Clin Neurosci 16:367–78
Neuroradiology